
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The integration of bio-catalysis and electrocatalysis to produce fuels and chemicals from carbon dioxide
Xinyi
Tan
a and
Jens
Nielsen
 *bc
*bc
aDepartment of Materials Science and Engineering, Beijing Institute of Technology, Beijing, 100081, China. E-mail: monica950521@126.com
bDepartment of Biology and Biological Engineering, Chalmers University of Technology, SE41296 Gothenburg, Sweden. E-mail: nielsenj@chalmers.se
cBioInnovation Institute, Ole Maaløes Vej 3, DK2200 Copenhagen N, Denmark
First published on 18th May 2022
Abstract
The dependence on fossil fuels has caused excessive emissions of greenhouse gases (GHGs), leading to climate changes and global warming. Even though the expansion of electricity generation will enable a wider use of electric vehicles, biotechnology represents an attractive route for producing high-density liquid transportation fuels that can reduce GHG emissions from jets, long-haul trucks and ships. Furthermore, to achieve immediate alleviation of the current environmental situation, besides reducing carbon footprint it is urgent to develop technologies that transform atmospheric CO2 into fossil fuel replacements. The integration of bio-catalysis and electrocatalysis (bio-electrocatalysis) provides such a promising avenue to convert CO2 into fuels and chemicals with high-chain lengths. Following an overview of different mechanisms that can be used for CO2 fixation, we will discuss crucial factors for electrocatalysis with a special highlight on the improvement of electron-transfer kinetics, multi-dimensional electrocatalysts and their hybrids, electrolyser configurations, and the integration of electrocatalysis and bio-catalysis. Finally, we prospect key advantages and challenges of bio-electrocatalysis, and end with a discussion of future research directions.
 Xinyi Tan | Xinyi Tan received her PhD degree in Chemical and Biomolecular Engineering of University of California Los Angeles (UCLA), where she had an in-depth study of energy storage materials for lithium-ion and sodium-ion batteries. Tan received her bachelor degree in polymer materials and engineering in Jilin University. Now she is a post doc in Materials Science and Engineering of Beijing Institute of Technology, and focused on the research of energy storage and CO2 reduction that based on the integration of electrocatalysis and bio-catalysis. |
 Jens Nielsen | Jens Nielsen is a professor at Chalmers University of Technology, Sweden, and CEO of the BioInnovation Institute in Denmark. Prof. Nielsen is member of several academies, including the National Academy of Engineering and National Academy of Science in USA, Chinese Academy of Engineering, the Royal Swedish Academy of Science, the Royal Danish Academy of Science and Letters, and the Royal Swedish Academy of Engineering Sciences. Prof. Nielsen has published so far more than 850 papers that have been cited more than 90 |
1. Introduction
Fossil fuels assume a key position in human society. However, the total amount of fossil fuels on earth is limited, and it is crucial to find substitutes and sustainable energy sources to replace them in the future. Furthermore, the dependence on fossil fuels has caused excessive emissions of greenhouse gases, intensifying the risk of climate change and global warming. To circumvent these severe consequences and avoid the point of no return, “2-degree scenario” should be met. It requires that increases in global temperature are less than 2 °C above ‘pre-industrial’ levels. Major efforts have been directed towards the capture and utilization of CO2. However, capture and storage of CO2 (mostly underground) is energy intensive, cost prohibitive, and incapable of producing value-added compounds. Transformation of CO2 as cheap, nontoxic, and naturally abundant carbon-based resource into fuels and chemicals is desirable from both ecological and economic viewpoints, and is therefore attractive.Current CO2 utilization approaches include photocatalysis, chemical catalysis, bio-catalysis and electrocatalysis.1 Among these strategies, electrocatalysis is particularly appealing because it simply makes use of clean and renewable electricity (with lower costs deriving from growing abundant sources) generated at (or near) room temperature and ambient pressure, enabling a sustainable future with a low-carbon footprint. During this process, the required hydrogen is generated in situ by water electrolysis, thus eliminating the use of blue hydrogen from fossil fuels. The application of electrocatalysis is suitable to decentralization and holds great economic promise with a foreseen surplus of cheap electrical energy from a well-distributed and intermittent renewable source that would otherwise be difficult to store. Nevertheless, it remains a challenge to obtain long-chain carbon products with high selectivity via direct CO2 electrocatalysis, due to the high C–C coupling energy barrier, the linear scaling relations, as well as the concurrent occurrence of the competing hydrogen evolution reaction (HER).
Considering that bio-catalysis can produce a wide range of C2+ products (containing two or more carbon atoms) with high efficiency and specificity, the integration of bio-catalysis with electrocatalysis through rational design of electrode and catalyst materials using CO2 and renewably generated electricity has spurred increased research interest. Classic electrocatalysis for CO2 fixation is efficient in many ways, however, the most feasible products remain CO, methane, methanol and formic acid, while ethanol and other C2+ chemicals are difficult to obtain.2 In addition, there are different catalytic pathways for CO2 reduction leading to multiple compounds at similar overpotentials, which affects the selectivity of the reaction.3 By combining advantages of both bio-catalysis and electrocatalysis, bio-electrocatalysis aims to produce C2+ products from CO2 with high conversion efficiency and selectivity.4,5
Great achievements have been made in bio-electrocatalysis. For example, the cost of electricity from renewable sources has reduced substantially.6 Advanced devices, complex three-dimensional electrode materials and their hybrids have been developed to enhance the electron transfer efficiency and the CO2 utilization rate.7,8 Furthermore, the suitable candidates for electrocatalysis have been identified, involving efficient C1 utilizing enzymes, pathways and microbial cell factories,9,10 as demonstrated by the production and commercialization of several C2+ products.11–13 In this review, we first provide a fundamental explanation of CO2 utilizing classic electrocatalysis and bio-electrocatalysis. Next, key aspects for bio-electrocatalysis are discussed. A summary of the most important advances in the design of electron transfer materials, catalysts, electrolytes, electrolysers, promising CO2 fixation and pathways, as well as compelling microbial hosts is presented. Strategies to further boost the integration of bio-catalysis and CO2 electrocatalysis are highlighted. Finally, future challenges and prospects of CO2 conversion via coupling of bio-catalysis and electrocatalysis are outlined.
2. Electrocatalysis-based CO2 reduction
CO2 transformation cannot be carried out spontaneously due to thermodynamic constraints, and external energy is required to drive the reduction. Many efforts have been attempted to elucidate the mechanisms of CO2 reduction in both theoretical and experimental research. However, due to the complexity of these pathways and difficulties in capturing intermediates of the reactions, many of the molecular mechanisms have only been confirmed partially for a few steps. The transfer of electrons and protons provides the driving force for CO2 reductions. Various products such as CO, formic acid, methane, methanol, ethylene, ethanol, and other hydrocarbons can be acquired, and different numbers of electrons and protons are transferred to carbon dioxide. Equations 1–10 display the Gibbs free energy (at 298 K, 1 atm) and the reduction potential versus standard hydrogen electrode (SHE) at pH = 7 of various CO2 reduction reactions with different products.14 Gibbs free energies for all these reactions are higher than zero, suggesting that they cannot take place without external energy input. It is obvious that long-chain carbon products require more energy input to proceed the reactions and therefore are harder to produce than C1 chemicals. In electrocatalysis-based CO2 reduction, electricity that is generated by renewable energy can be introduced as the driving force, and catalysts that can reduce energy barriers during the reactions can achieve high conversion efficiency for the products. Due to the clean and sustainable energy source of CO2 electrocatalysis and the high selectivity for long-chain carbon products of bio-catalysis, the integration of electrocatalysis and bio-catalysis can combine the merits of both transformations.2.1 Classic electrocatalysis for CO2 reduction
During classic electrocatalysis for CO2 reduction, electrons and protons that participate in reactions usually derive from the electrolytes (Fig. 1a).15 Dispersed CO2 gas will first dissolve in the electrolyte solution and then adsorbed by binding sites or defects on the surface of catalysts to form a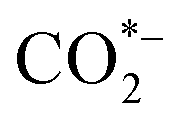 radical anion. The
radical anion. The 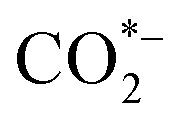 has short lifetime and is rapidly protonated to either form *COOH or *OCHO. After the second electron transfer and protonation, *COOH is reduced to CO*, which are key intermediates in forming hydrocarbons or oxygenates in CO2 electroreduction. For example, the CO* intermediate can form COH* or CHO* through electron-proton pair transfer, and these two intermediates can be reduced to methanol in subsequent steps. During the formation of C2 compounds, the C–C bond is coupled between surface-bound C1 oxygenates, through enol-like surface species to other C2 compounds with further oxidation-reduction reaction. Classic electrocatalysis of CO2 can occur through these reactions and produce various products, including formic acid, CO, methanol, methane, ethanol, and ethylene.16,17 However, their most feasible products remain C1 products, with C2+ chemicals being difficult to produce.
has short lifetime and is rapidly protonated to either form *COOH or *OCHO. After the second electron transfer and protonation, *COOH is reduced to CO*, which are key intermediates in forming hydrocarbons or oxygenates in CO2 electroreduction. For example, the CO* intermediate can form COH* or CHO* through electron-proton pair transfer, and these two intermediates can be reduced to methanol in subsequent steps. During the formation of C2 compounds, the C–C bond is coupled between surface-bound C1 oxygenates, through enol-like surface species to other C2 compounds with further oxidation-reduction reaction. Classic electrocatalysis of CO2 can occur through these reactions and produce various products, including formic acid, CO, methanol, methane, ethanol, and ethylene.16,17 However, their most feasible products remain C1 products, with C2+ chemicals being difficult to produce.| CO2 + 2H+ + 2e− → CO + H2O, ΔG° = 102.274 kJ mol−1, E°′ redox = −0.53 V | (1) |
| CO2 + 2H+ + 2e− → HCOOH, ΔG° = 117.712 kJ mol−1, E°′ redox = −0.61 V | (2) |
| 2CO2 + 2H+ + 2e− → H2C2O4, ΔG° = 176.182 kJ mol−1, E°′ redox = −0.91 V | (3) |
| CO2 + 4H+ + 4e− → HCHO + H2O, ΔG° = 185.251 kJ mol−1, E°′ redox = −0.48 V | (4) |
| CO2 + 6H+ + 6e− → CH3OH + H2O, ΔG° = 219.986 kJ mol−1, E°′ redox = −0.38 V | (5) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O, ΔG° = 185.251 kJ mol−1, E°′ redox = −0.24 V | (6) |
| 2CO2 + 12H+ + 12e− → CH2H4 + 4H2O, ΔG° = 404.079 kJ mol−1, E°′ redox = −0.35 V | (7) |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O, ΔG° = 380.923 kJ mol−1, E°′ redox = −0.33 V | (8) |
| 2CO2 + 14H+ + 14e− → C2H6 + 4H2O, ΔG° = 364.713 kJ mol−1, E°′ redox = −0.27 V | (9) |
| 3CO2 + 18H+ + 18e− → C3H7OH + H2O, ΔG° = 538.386 kJ mol−1, E°′ redox = −0.31 V | (10) |
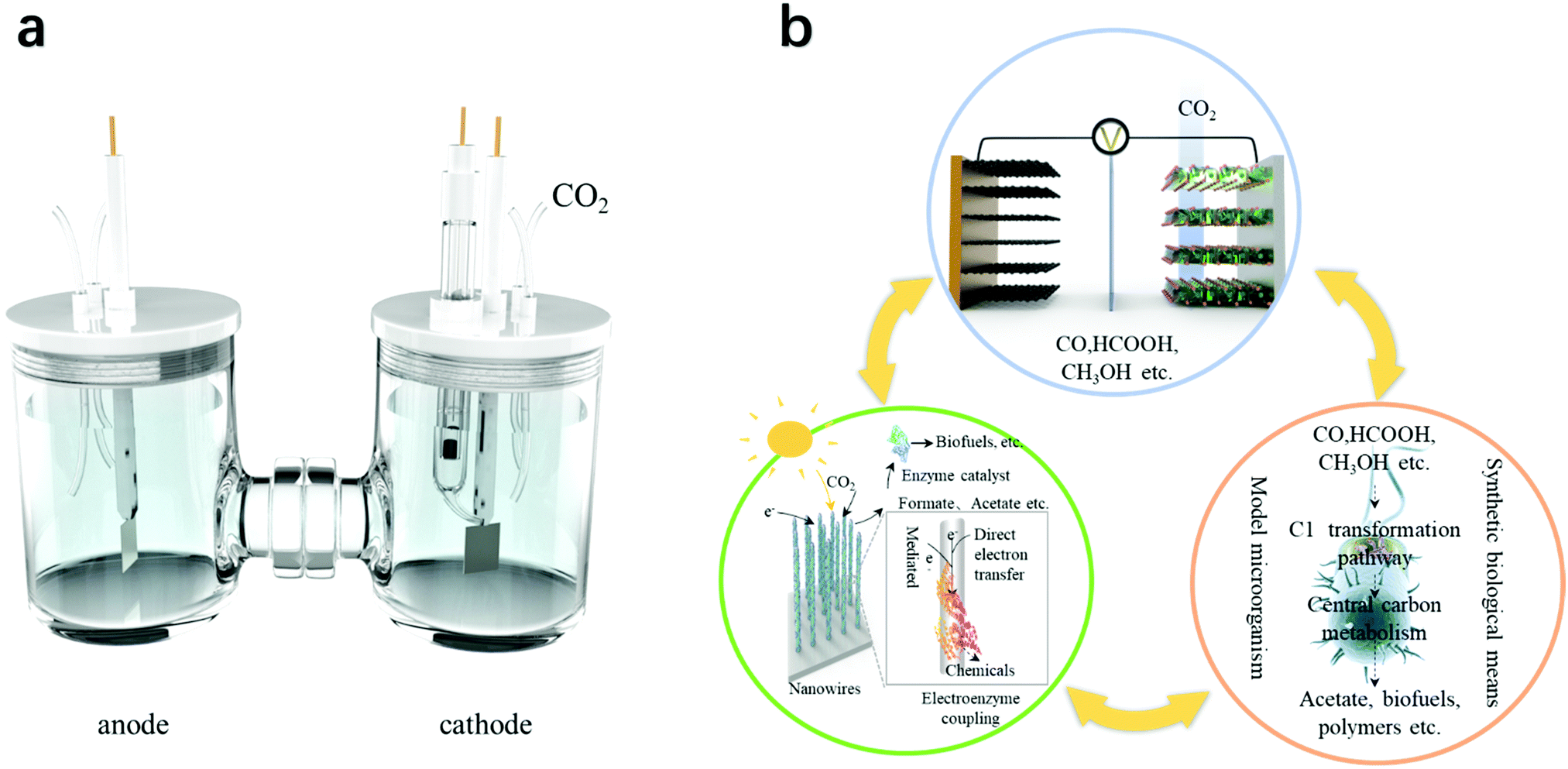 | ||
| Fig. 1 The two generations of electrocatalysis used for CO2 conversion. (a) Classic electrocatalysis that utilizes electricity to produce mainly C1 and C2 compounds. (b) Bio-electrocatalysis that could efficiently utilize electricity from renewable sources together with bio-catalysis to produce C2+ compounds. | ||
2.2 Volcano plot and scaling relationship
Nobel laureate Paul Sabatier stated that an “ideal” catalyst should interact with a substrate neither too weak (i.e., strong binding of reactants) nor too strong (i.e., weak binding of products ensuring their desorption at ease), which is known as the Sabatier's principle. A too strong binding strength leads to surface poisoning, while a too weak adsorption entails insufficient coverage and thus poor reaction rate. Plotting the catalytic performance against the binding energy of reactants yields a “volcano” plot.18 This can be used as an intuitive tool to compare the thermodynamics among catalysts, assisting understanding and predicting active candidates that may approach the peak or plateau of the volcano. The volcano slopes indicate regions where the catalyst/intermediates binding is either too strong (left slope) or too weak (right slope).18 For CO2 reduction reactions, the crucial intermediate for the volcano plot varies according to the type of product.Given that analogous surface–C bonds are formed regardless of different CO2 reduction intermediates (e.g., *CO, *COOH, and *COH, * denotes the adsorbed site), it remains a formidable challenge to optimize the binding strength of the key intermediate without impacting the other products. This phenomenon is referred to as scaling relationship. As a proof-of-principle example, the hydrogenation of *CO to *CHO is inferred to be the potential-determining step (PDS) of CO2 reduction to CH4.19 The high free energy change of this step (0.74 eV) is plausibly attributed to the weak adsorption of *CHO as opposed to *CO. However, achieving stronger binding of *CHO (i.e., more negative) than *CO for selective CH4 formation is limited by the scaling relationship.20 By analogy, the weak binding of *COOH caused by the linear scaling between *COOH and *CO results in large overpotentials and low turnover frequencies of Ag and Au electrodes for CO2 conversion to CO.21 Conceptually resembling the general scaling relationships delineated in heterogeneous catalysts, a decrease in thermodynamic effective overpotential ηeff (the difference between redox potential of a molecular catalyst and the thermodynamic CO2 reduction potential) is usually correlated with drop in the kinetic maximum catalytic turnover frequency TOFmax (as reflected by the plateaued catalytic current) for molecular catalysts. This general trend is often described as a “molecular scaling relationship” (i.e., the trade-off between ηeff and TOFmax). From these scenarios, optimization of the binding energy of a specific intermediate by overcoming the energetic linearity constrains enables one to lower the overpotential and improve selectivity. To this end, several useful protocols based on alloying, tethering active ligands, introducing promoters, and creating multiple functional sites may be attempted.22 Exploration of mixed solvents may be another avenue.
2.3 Competition between CO2 reduction and hydrogen evolution
CO2 activation and reduction process is always plagued by the concomitant HER (from proton and/or water reduction) in protic solvents. HER has a much faster kinetics and proceeds at a similar or even more positive potential compared with the CO2 reductions. Meanwhile, the simultaneous binding of *H with CO2 reduction intermediates influences their adsorption energies, thus profoundly affecting reaction kinetics. Therefore, to boost the CO2 reductions, parameters such as the properties of electrode, local availability of protons/water, operating conditions, and composition of electrolyte can be tailored to combat the undesired HER. This can be done by (1) using an electrolyte with diminished proton donor activity (e.g. organic solvents with a low dissociation constant, ionic liquids, and mixed solvents);23 (2) optimizing reaction conditions (with comparatively lower local alkalinity near the electrode surface and enhanced mass transport);24 (3) engineering electrode-electrolyte interface to construct aprotic and hydrophobic protection layers;25 or by (4) modifying electrocatalysts to favour adsorption and binding of CO2 rather than hydrogen.262.4 Bio-electrocatalysis for CO2 reduction
Bio-electrocatalysis employs biocatalysts to enhance the productivity and selectivity in production of C2+ products, and can be divided into enzymatic electrocatalysis and microbial electrocatalysis (Fig. 1b). Classic electrocatalysis is characterized by high electron transfer efficiency, while bio-catalysis has the ability to produce more valuable and complex products with high specificity, stereoselectivity and regioselectivity.27 Moreover, biocatalysts including oxidoreductases and microbes that can utilize C1 and C2 substrates are diverse and abundant. By using substrates produced through classic electrocatalysis, bio-electrocatalysis has the potential to produce a wide range of C2+ fuels and chemicals.28 Microbes used in bio-catalysis could also be considered as self-regenerating catalysts with long-term stability, and sustainability.29 Bio-electrocatalysis can be divided into direct-electron-transfer strategies and indirect-electron-transfer strategies. During direct-electron-transfer bio-electrocatalysis, electrocatalysis and bio-catalysis could be carried out in one pot, and electrons generated from renewable sources can be consumed by biocatalysts directly or indirectly through electron carriers, such as hydrogen, ammonia, sulphur species and ferrous salts, to convert CO2 into desired products.30,31 During indirect-electron-transfer bio-electrocatalysis, CO2 is first converted into C1 and C2 compounds through classic electrocatalysis. These C1 and C2 compounds can then be used as substrates, mostly in separate devices, for biocatalysts to produce desired products with higher chain lengths. Formate dehydrogenase (FDH),29 formaldehyde dehydrogenase (FaldDH),32 acetogens,33 methanogens,34,35Clostridia,36 and other biocatalysts capable of efficiently utilizing C1 and C2 compounds can be used in indirect-electron-transfer strategies, and a wide range of chemicals including secondary metabolites can hereby be produced.3. Crucial factors in bio-electrocatalysis
Bio-electrocatalysis aims to improve product range and efficiency through interlinking classic electrocatalysis with bio-catalysis. Crucial factors in bio-electrocatalysis include electron transfer materials, catalysts, electron transfer and catalytical materials complexes with one-dimensional, two-dimensional, or three-dimensional composites, electrolyser configurations, as well as promising CO2 fixation pathways and attractive microbial hosts.3.1 Electron transfer materials
Electron transfer materials are used to transport and conduct electrical currents. Electron transfer materials include metals, semi-conductors, carbon-based materials, metal-organic frameworks (MOFs) and other composites. Key factors that govern the performance of electron transfer materials are Ohmic resistance, concentration polarization potential and the driving force required by the reaction itself. A good electron transfer material should have small Ohmic resistance and low concentration polarization potential. Silver, copper, gold, and platinum are typical metal materials with good electrical conductivity, while silicon and germanium are representative materials in the family of semi-conductors. Detailed information of metal and semi-conductor materials can be found in recent reviews.37,38Carbon-based materials are divided into several categories, such as carbon nanotubes, graphene, and reduced graphene oxide. Carbon nanotubes (CNTs) are composed by carbon hexagons that are arranged in a concentric manner.39 CNTs are widely used by catalysis due to their high specific area (1315 m2 g−1 for single-walled CNTs),40 extraordinary electrical conductivity (5000 S cm−1) and high chemical stability.41 As the most common one-dimensional material with outstanding electrical conductivity, CNTs can be utilized as the electron transfer to mediate and support catalytically active sites during the CO2 transformation process.42,43 For example, Wu et al. reported the utilization of N-doped CNTs in CO2 electrocatalysis and obtained CO as the major product with faradaic efficiency of 80% at −0.78 V vs. RHE (reversible hydrogen electrode).44 The defects in N-doped CNTs can provide more catalytically active sites for CO2 reduction, and lead to a lower free energy barrier for the adsorption of COOH which is a potential rate-limiting step in CO formation. In addition, the pyrrolic-N and pyridinic-N in N-doped CNTs can also enable favourable CO desorption with suitable binding energy that can be beneficial to CO production.
Another typical carbon-based material is graphene, with large specific surface area (2630 m2 g−1), good thermal conductivity (5000 W m−1 K−1), high electrical conductivity (106 S cm−1) and high mechanical strength.45,46 Han et al. described a defective graphene that can be used in CO2 electrocatalysis to acquire CO as the major products with a faradaic efficiency of 84% at −0.6 V vs. RHE, the topological defects in graphene can expose abundant catalytically active sites for CO2 reduction and provide strong adsorption capacity for CO2 which can improve the conversion efficiency for reduction products.47 Chae et al. compared different types of composite materials, and managed to produce 40.6% methane using the Ni nanoparticles doped granular activated carbon composite.48 The granular activated carbon with large surface area can function as a conductive bridge to transfer electrons and protons between different microbes and provide multiple catalytically active sites, and Ni catalysts in the composites exhibit high catalytic activity and low toxicity to microbes that can be favourable in methanogenesis.
MOFs are also attractive materials with diverse structures and stable porosity.49 The specific framework formed by metal and organic coordination not only has metal activity, but also obtain the flexibility of organic ligands, the selectivity of functional groups, the special spatial structure, and other physical and chemical properties from organic coordination at the same time.50 The porous structure of MOFs can provide a large amount of catalytically active sites and immense pore volume to accommodate CO2 molecules which has a significant influence on the improvement of conversion efficiency of CO2 reduction.51,52 Deng et al. employed bismuth-based MOFs to fabricate Bi2O3@C-800 composites to achieve formate production with a 92% faradaic efficiency and a partial current density of 7.5 mA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−2 at −0.9 V vs. RHE in CO2 electroreduction, the MOFs in the composites act as a carbon matrix with high electrical conductivity and abundant adsorption sites for CO2, while Bi2O3 contributes to the high selectivity and conversion efficiency of formate during the electroreduction.53 Qiu et al. reported MOFs composites PcCu–Cu–O which can transform CO2 into C2H4 with a faradaic efficiency of 50%, and it has been shown that the abundant C2H4-producing and CO-producing sites in this MOFs composites can be favourable in C2H4 formation, and the high crystallinity and strong π–π interaction between MOFs layers can be helpful in limiting the distortion of Cu ions from tetragonal to tetrahedral which guarantee the electrode durability for electro-conversion of CO2 to C2H4.54
cm−2 at −0.9 V vs. RHE in CO2 electroreduction, the MOFs in the composites act as a carbon matrix with high electrical conductivity and abundant adsorption sites for CO2, while Bi2O3 contributes to the high selectivity and conversion efficiency of formate during the electroreduction.53 Qiu et al. reported MOFs composites PcCu–Cu–O which can transform CO2 into C2H4 with a faradaic efficiency of 50%, and it has been shown that the abundant C2H4-producing and CO-producing sites in this MOFs composites can be favourable in C2H4 formation, and the high crystallinity and strong π–π interaction between MOFs layers can be helpful in limiting the distortion of Cu ions from tetragonal to tetrahedral which guarantee the electrode durability for electro-conversion of CO2 to C2H4.54
Different from the traditional electron transfer materials in CO2 electroreduction, cofactors such as NAD/NADH+, ATP/ADP, FMNH2/FADH2 serve as typical electron transfer media in biocatalytic reduction of CO2.55,56 Cofactors can provide protons and electrons during the oxidation–reduction process in microbial systems, and enzymes function as catalysts in the bio-catalysis process. Eelectron transfer in enzymatic and microbial electrocatalysis can be divided into a direct electron transfer (DET) scheme and a mediated electron transfer (MET) scheme (Fig. 2).57–59 During DET schemes, electrons could be utilized directly by oxidoreductase containing active centres and microbes through membrane-bound redox proteins.60 During MET schemes, electrons are transferred by external soluble mediators between electrodes and biocatalysts. Potential mediators for bio-electrocatalysis include NAD(P)H, flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), pyrroloquinoline, methyl viologen, anthraquinone-2,6-disulfonate, neutral red (NR), haem and other transition metals.58,61 MET may result in the principal thermodynamic losses, but will enable large communication distance and cascade reactions.62
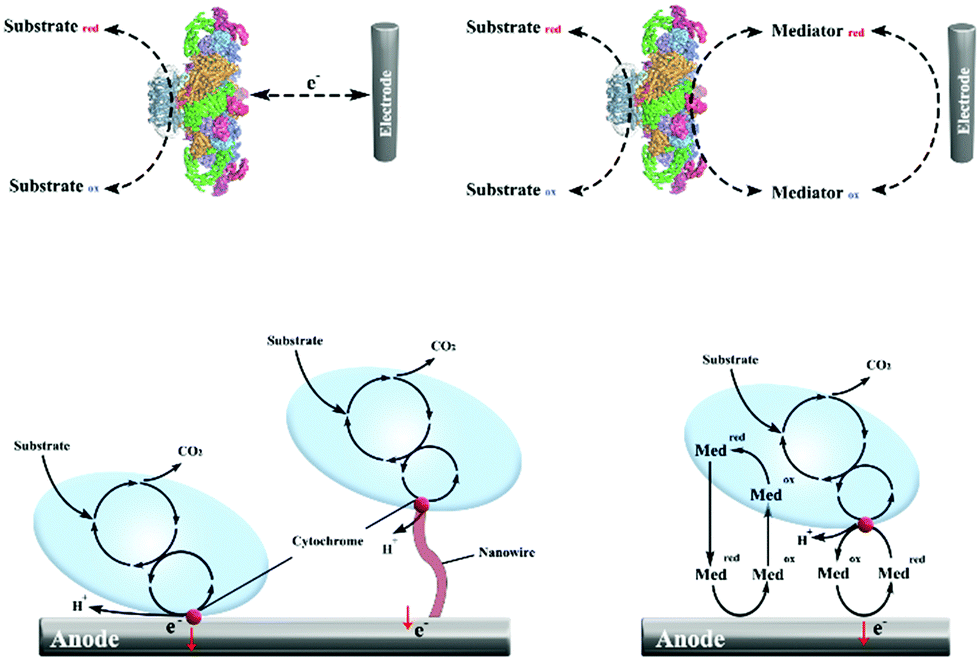 | ||
| Fig. 2 Schemes of DET and MET in enzymatic electrocatalysis and microbial electrocatalysis. (a) DET in enzymatic electrocatalysis. (b) MET in enzymatic electrocatalysis. (c) DET in microbial electrocatalysis. (d) MET in microbial electrocatalysis. | ||
3.2 Catalysts
In electrocatalysis, catalysts are used to accelerate CO2 reductions, and increase the specificity towards CO2 over H+. Two major types of catalysts used in electrocatalysis are metals and enzymes. Even further, metals can be classified as metals, metal oxides and alloys. Table 1 provides an overview of widely used catalysts.| Type | Materials | Main product | Potential | Faradaic efficiency | Ref. |
|---|---|---|---|---|---|
| NPs, nanoparticles. | |||||
| Metal | Au | CO | −0.6 V vs. RHE | 83% | 186 |
| Au25 nanosphere | CO | −0.57 V vs. RHE | 73.7% | 187 | |
| h-Zn | CO | −0.95 V vs. RHE | 85.4% | 188 | |
| Pd NPs | CO | −0.89 V vs. RHE | 91.2% | 109 | |
| Ag nano-corals | CO | −0.6 V vs. RHE | 95% | 189 | |
| β PdHx@Pd | CO | −0.7 V vs. RHE | 93.4% | 190 | |
| β PdHx@Pd | Formic acid | −0.1 V vs. RHE | 98.9% | 190 | |
| Pd | Formic acid | −0.25 V vs. RHE | 94% | 191 | |
| 3D tin | Formic acid | −1.2 V vs. RHE | 90% | 192 | |
| Cu foam | Ethylene | −0.8 V vs. RHE | 55% | 70 | |
| Cu | Ethylene | −0.54 V vs. RHE | 70% | 68 | |
| Cu hollow fibers | CO | −0.4 V vs. RHE | 75% | 193 | |
| Ag 3D foam | CO | −0.99 V vs. RHE | 94.7% | 194 | |
| Cu-HNs foam | CO | −0.5 V vs. RHE | 60% | 195 | |
| Zn dendrite | CO | −1.1 V vs. RHE | 70% | 196 | |
| Bi nanotubes | Formic acid | −1.0 V vs. RHE | 97% | 197 | |
| Bi | Formic acid | −0.9 V vs. RHE | 92% | 198 | |
| Polycrystalline Cu | Ethylene | −0.9 V vs. RHE | 15% | 69 | |
| Polycrystalline | Ethanol | −0.9 V vs. RHE | 7.6% | 69 | |
| Metal-oxide catalysts | RuO2/TiO2 NTs | Methanol | −0.8 V vs. SCE | 60.5% | 199 |
| RuO2/TiO2 NPs | Methanol | −0.8 V vs. SCE | 40.2% | 199 | |
| Au–CeOx/C | CO | −0.89 V vs. RHE | 89.1% | 200 | |
| Oxide-derived Au | CO | −0.35 V vs. RHE | 96% | 201 | |
| SnO2/NSC | Formic acid | −1.15 V vs. RHE | 94.4% | 202 | |
| Mo–ZnO | CO | −1.0 V vs. RHE | 39.8% | 203 | |
| Bi/Bi2O3 | Formic acid | −0.87 V vs. RHE | 90.4% | 204 | |
| Alloy catalysts | Pd–Pt NPs | Formic acid | −0.4 V vs. RHE | 88% | 205 |
| Au–Cu NPs | CO | −0.73 V vs. RHE | 65% | 206 | |
| Pd@Au | CO | −0.5 V vs. RHE | 80% | 207 | |
| PdAg | CO | −0.75 V vs. RHE | 87.5% | 208 | |
| Au-bipy-Cu | Acetaldehyde | −0.9 V vs. RHE | 25% | 209 | |
| Cu–Pt NCs | Methane | −1.6 V vs. SCE | 21% | 210 | |
| Cu–Sn | CO | −0.6 V vs. RHE | 90% | 211 | |
| Tungsten diselenide nanoflakes | CO | −0.164 V vs. RHE | 24% | 212 | |
| Au NPs-coupled ZnTe/ZnO array | CO | −0.7 V vs. RHE | 97% | 213 | |
| CuAg wire | Ethylene | −0.7 V vs. RHE | 60% | 214 | |
| CuAg wire | Ethanol | −0.7 V vs. RHE | 25% | 214 | |
| Cu4Zn | Ethanol | −1.05 V vs. RHE | 29.1% | 215 | |
As the most distinctive metal material having the potential to convert CO2 into C2 and C3 products, Cu has attracted intense interests. The major C2 products of the Cu-catalysed CO2 reductions are ethylene and ethanol.68,69 Modification of the surface and design of the nanostructure of metal electrodes is significant. Flat surfaces of the bulk metal electrode show limited activity in CO2 reduction, while electrodes consisting of porosity nanoparticles on the surface showed increased activity. For instance, Cu foam can be used to convert CO2 into ethylene and ethane with a 55% faradaic efficiency due to the porous surface that helps to expose more active sites during catalysis, while Cu nanoparticles without any modification can only lead to the formation of methane with 57% selectivity.70 In addition, the crystal facet of a metal electrode can also influence the catalytic performances.71 Heijne et al. tested biocompatible and costless cathode materials including heat-treated stainless steel felt, untreated stainless steel felt and graphite felt, and reported a 60.8% current efficiency and 21.9% energy efficiency using the heat-treated stainless steel felt biocathode.67
The morphology of electrodes can influence the electrocatalytic performances for the CO2 reduction. Compared with electrodes with a smooth surface, electrodes with a rougher surface exhibit high density of under-coordinated sites and display enhanced hydrocarbon selectivity.77 Sargent et al. reported a catalytic trend in which sharper structures such as nanoneedles, nanowhiskers and nanodendrites favour C2+ production.78 Sharp tips have been reported to improve bubble nucleation, stabilize cations, and increase local pH to limit the protonation of bound CO, and improve production specificity of multi-carbon products.79–82 Kim and Palmore designed Cu(I)–halide-derived catalysts that promote adsorption of carbon intermediates and C–C coupling reactions, improve the faradaic efficiency of C2+ products to 72.6%.83 Anwer et al. reported the development of a reduced graphene oxide/tungsten oxide modified cathode and the production of acetate with the 86% coulombic efficiency.84
3.3 Integration of electron transfer and catalytical materials
Carbon dioxide electroreduction requires electron transfer as the driving force and catalytical sites to bind with the CO2 molecules. Hence, good electrocatalysts should integrate electron transfer materials and materials with catalytic activity. Depending on the structures, electron transfer and catalytical materials complexes can be divided into one-dimensional, two-dimensional, or three-dimensional composites (Fig. 3).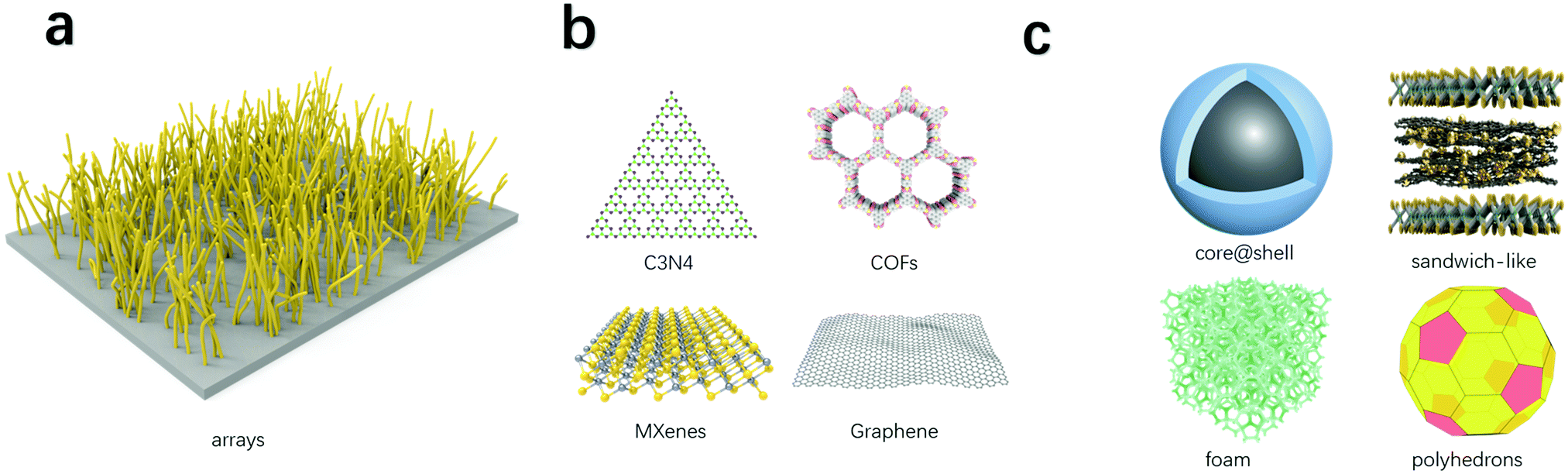 | ||
| Fig. 3 Electron transfer and catalytical materials complexes. (a) One-dimensional composites. (b) Two-dimensional composites. (c) Three-dimensional composites. | ||
Major products of one-dimensional composites electrode materials for CO2 electrocatalysis are usually methane, CO, and formic acid. Several improvements have been attributed to one-dimensional electrode materials for CO2 reduction, such as heteroatom doping, composite with other high active catalysts, and catalysis on crystal facets. For instance, nitrogen-doped CNTs have better catalytic activity due to the introduction of nitrogen defects that can mediate the electronic structure and regulate the atomic arrangement on the complexes surface. Sun et al. reported In-doped SnO2 hollow nanofibers which can improve the electrocatalytic performance by limiting the grain size of SnO2 and increasing the electron density of Sn to produce formic acid with a faradaic efficiency of 86.2%.91 Liang et al. fabricated Ag nanowires which can expose a large amount of Ag(200) facets that can provide more electroactive sites on the surface of Ag nanowires to facilitate the conversion from CO2 to CO with a faradaic efficiency of 81%.92 Wang et al. constructed a N/Ni nanoparticles (NPs) @CNT/Graphene hybrid electrode which encapsulated Ni nanoparticles and N dopants in CNTs, which could achieve a high faradaic efficiency of 97.7% to convert CO2 into CO.93 Zhu et al. developed protoporphyrin IX cobalt (CoPP) @CNT composites that achieve a 98.3% faradaic efficiency for CO products in CO2 electroreduction.94 Xu et al. reported the production of 80.9 mL L−1 methane using the hybrid graphite felt biocathode at −1.4 V vs. Ag/AgCl cathode potential.95 Details of more widely used one-dimensional composites are summarized in Table 2.
| Materials | Main product | Potential | Faradaic efficiency | Ref. |
|---|---|---|---|---|
| PEI/NGCNT | Formic acid | −1.8 V vs. SCE | 87% | 216 |
| Pd/PANI-CNT | Formic acid | −0.8 V vs. SCE | 83% | 217 |
| NiSA-N-CNT | CO | −0.7 V vs. RHE | 91.3% | 218 |
| Cu/SnOx-CNT-#7 | CO | −0.99 V vs. RHE | 89% | 219 |
| Cu/SnOx-CNT-#12 | Formic acid | −1.09 V vs. RHE | 79% | 219 |
| AuCNT | CO | −0.55 V vs. RHE | 70% | 220 |
| g-C3N4/MWCNT | CO | −0.75 V vs. RHE | 60% | 221 |
| NCNTs-ACN-850 | CO | −1.05 V vs. RHE | 80% | 222 |
| CN-H-CNTs | CO | −0.5 V vs. RHE | 88% | 223 |
| CoPP@CNT | CO | −0.6 V vs. RHE | 98.3% | 94 |
| NFe-CNT/CNS | CO | −0.59 V vs. RHE | 69% | 224 |
| Ni@NCNTs | CO | −0.8 V vs. RHE | 99.1% | 225 |
| CdS-CNT | CO | −1.2 V vs. RHE | 92% | 226 |
| Ni-CNT-CC | CO | −0.53 V vs. RHE | 90% | 227 |
| MWNT/PyPBI/Au | CO | −1.4 V vs. Ag/AgCl | 90% | 228 |
| CoPc-py-CNT | CO | −0.63 V vs. RHE | 98% | 229 |
Different from one-dimensional composites that can only lead to the formation of C1 products, two-dimensional composites have the possibility in forming C2+ products such as ethanol and propanol. For example, Rinaldi et al. reported a flower-like composite Graphene/ZnO/Cu2O with the combination of graphene, ZnO nanoparticles and Cu2O nanoparticles, and produced propanol with a faradaic efficiency of 30%.97 Cu2O has the ability to stabilize the adsorption of CO which is a key step for the further formation of C2 or C2+ species, while ZnO is able to stabilize Cu atoms to strengthen Cu–CO− links, graphene in this composite serves as a highly electronically conductive matrix to support the catalysts, so Graphene/ZnO/Cu2O took advantage of the synergic effect of these three materials to achieve chain-growth and acquire propanol as the final product. Rezazi et al. designed Pt@ reduced graphene oxide and achieved the formation of methanol at −0.3 V vs. Ag/AgCl with a faradaic efficiency of 41%.98 The pyridine structure in rNGO can accept a proton to form pyridinium ion which has a synergic effect with Pt to stabilize the adsorption of PyH+–COOH that can lead to the formation of methanol. Yuan reported Graphene oxide-VB6-Cu-2 composites that can convert CO2 into ethanol with a faradaic efficiency of 56.3% over 24 hours.99 Pyridoxine (VB6) is an N-containing material that has high catalytical activity to ethanol,100 while graphene oxide nanosheets can provide a large amount of edge sites such as carboxyl groups to facilitate the CO2 electroreduction, and Cu nanoparticles in this composite can serve as high performance catalysts to improve the conversion efficiency of ethanol from CO2. Sekar developed N-doped graphene-Co3O4-30 nanohybrid catalyst that can transform CO2 into formic acid with a faradaic efficiency of 83% at an onset potential of −0.82 V vs. SCE.101 The pyridinic defects in NG-Co3O4-30 hybrids are able to convert CO2 into CO at a low overpotential, and Co3O4 has the selectivity to catalyse CO2 into formic acid. Details of widely used two-dimensional composites are summarized in Table 3.
| Materials | Structure | Main product | Potential | Faradaic efficiency | Ref. |
|---|---|---|---|---|---|
| HNCM/CNT | Membrane | Formic acid | −0.9 V vs. RHE | 81% | 230 |
| Ni–N-Gr | Nanosheet | CO | −0.7 to −0.9V vs. RHE | 90% | 231 |
| Fe–N-C | Nanosheet | CO | −0.9 V vs. NHE | 80% | 232 |
| p-NG-Cu-7 | Nanosheet | Ethylene | −0.9 V vs. RHE | 19% | 233 |
| NG-Co3O4-30 | Nanosheet | Formic acid | −0.95 V vs. SCE | 83% | 101 |
| Ag2S/N-S-doped rGO | Nanosheet | CO | −0.76 V vs. RHE | 87.4% | 234 |
| In2O3-rGO | Nanobelt | Formic acid | −1.2 V vs. RHE | 84.6% | 235 |
| GO-VB6-4 | Nanosheet | Ethanol | −0.4 V vs. RHE | 36.4% | 100 |
| GO-VB6-4 | Nanosheet | Acetone | −0.4 V vs. RHE | 8.9% | 100 |
| 30.87 wt% PO-5 nm Co/SL-NG | Nanosheet | Methanol | −0.9 V vs. SCE | 71.4% | 236 |
| SnO2/0.14@N-rGO | Nanosheet | Formic acid | −0.8 V vs. RHE | 78% | 237 |
| NapCo@SNG | Nanosheet | CO | −0.8 V vs. RHE | 97% | 238 |
| w-CCG/CoPc-A Hybrid | Nanosheet | CO | −0.79 V vs. RHE | 91.5% | 239 |
| FeN5 | Nanosheet | CO | −0.46 V vs. RHE | 97% | 240 |
| FePGF | Nanosheet | CO | −0.54 V vs. RHE | 98.7% | 241 |
| Ni2+@NG | Nanosheet | CO | −0.68 V vs. RHE | 92% | 242 |
| Pt@rNGO/GCE | Nanosheet | Methanol | −0.3 V, 2.0 V vs. Ag/AgCl | 41% | 98 |
| Phen-Cu/G | Nanosheet | CO | −0.6 V vs. RHE | 90% | 243 |
| Fe/NG-750 | Flake-like sheet | CO | −0.57 V vs. RHE | 80% | 244 |
| Ni-N4 | Nanosheet | CO | −0.81 V vs. RHE | 99% | 245 |
| AuNP-GNR | Nanoribbon | CO | −0.66 V vs. RHE | 92% | 246 |
| Materials | Structure | Main product | Potential | Faradaic efficiency | Ref. |
|---|---|---|---|---|---|
| SnO2/CC | Hierarchical with SnO2 nanosheets | Formic acid | −1.6 V vs. Ag/AgCl | 87% | 7 |
| NG-800 | Foam | CO | −0.58 V vs. RHE | 85% | 8 |
| FePGH-H | Porous hydrogels | CO | −0.49 V vs. RHE | 92.1% | 247 |
| Cu/C | Octahedron | Ethanol | −0.5 V vs. RHE | 34.8% | 248 |
| Cu/C | Octahedron | Methanol | −0.3 V vs. RHE | 43.2% | 248 |
| NGQDs | Quantum dots | Ethanol | −0.78 V vs. RHE | 16% | 249 |
| NGQDs | Quantum dots | Ethylene | −0.75 V vs. RHE | 31% | 249 |
| GN/Sn | Sandwich-like | Formic acid | −1.8 V vs. RHE | 89% | 103 |
| GN/ZnO/Cu2O | Flower-like | Propanol | −0.9 V vs. Ag/AgCl | 30% | 97 |
| Co–N5/HNPCSs | Core@shell | CO | −0.73 V vs. RHE | 99.2% | 250 |
| Fe–N–C | Rhombic dodecahedron | CO | −0.47 V vs. RHE | 93% | 251 |
| NPC-Pt | Copper skeletons and platinum shells | Dimethyl carbonate | −2.24 V vs. Ag/AgCl | 81% | 252 |
Array nanostructures composed of a series of nanowires that are vertically grown on a substrate. Vertical channels in array nanostructures can shorten the diffusion path, accelerate the charge transfer efficiency, and composite with catalyst nanoparticles inside or outside the channels to help improve the overall performances of the electrodes. Cai et al. fabricated Cu nanoarrays as the electrode for CO2 reduction, and the Cu arrays function as rapid pathways for the transport of charge and CO2 molecules, the high electrocatalytic activity of Cu also provide the selectivity for the formation of ethanol and propanol, so this array-structure electrode can finally achieve a 67% faradaic efficiency of the CO2 electroreduction.102
For core–shell nanostructures, the shell protects the extremely active nanoparticles inside to avoid deactivation during the reduction process, as well as hybridize electronic structure to improve the catalytic performance of the catalyst. In addition, the shell is relatively more active than the core, and the surface modification of the shell is easier to operate than other nanostructures. Polyhedron nanostructures are widely used in metal electrodes because of their specific crystal facets. Sun designed a unique sandwich-like electrocatalyst for CO2 reduction, the ultrathin tin metal layers that confined in graphene can provide enough space for the diffusion of electrolyte and expose abundant surface atoms as catalytically active sites for CO2 adsorption.103 Wang developed Sn–Cu/SnOx with a core–shell nanostructure for CO2 reduction to achieve a high faradaic efficiency above 98% with formic acid as the main product, the in situ reconstructed Sn/SnOx interface can facilitate the binding of the HCOO* intermediate during the reduction process and thus result in an extremely high faradaic efficiency.104 Yakobson et al. developed a three-dimensional N-doped graphene foam for CO2 reduction, this hierarchical foam provides a large interfacial area for the penetration of electrolyte and offers copious catalytically active sites to adsorb CO2, the maximum faradaic efficiency for CO production of this foam-like catalyst can reach 85% at a lower overpotential (−0.47 V) for at least 5 h.8
Three-dimensional composites with arrays, sandwich-like, core–shell and foam nanostructures can improve the catalytic performance with its huge surface area, and thus expose more catalytically active sites during the reactions. In the design of three-dimensional catalysts for CO2 electrochemical reduction, there is a trend to construct hierarchical structures, including foam, flower-like, sandwich-like, and other three-dimensional structures, to improve surface areas and pore volumes. Large surface areas and pore volumes can provide increased catalytically active sites for CO2 reduction, decrease contact resistance, facilitate electron transfer and mass transfer, as well as suppress the aggregation of active sites. Ma and Han synthesized a three-dimensional dendritic Cu–Cu2O composite with minimal contacting resistance between electrocatalysts and substrates, increased exposed active sites and suitable CuI/Cu0 ratios, and managed to convert CO2 into acetic acid and ethanol at a low potential of −0.4 V vs. RHE with 80% faradaic efficiency.105 Nwanebu et al. developed a three-dimensional polyactic acid lattice cathode with Ni/Fe/Mn-based coatings, and exhibited stable production of 50 ml d−1 methane and 185 mg d−1 acetate at ∼100% Coulombic efficiency.106
To further improve the catalytic efficiency in electrocatalysis, it is crucial to design and construct new electrode materials. Based on the current state of the art, we propose that the next generation of electrode materials should consist of the composite of catalysts and electron transfer materials with spatial structures. To optimize the catalytic activity and product selectivity, the catalyst of the composite should be filtered from nano sizes and even crystal planes. Different crystal planes of the same nanoparticle may have very different catalytic properties. In-depth studies evaluating the impact from various crystal planes in catalysis should be performed. As for electron transfer materials, the most significant point is to improve the electron transfer efficiency. The improvement of electron transfer efficiency, design and modification of catalyst materials will become the direction for future development of CO2 electroreductions.
Size effect has a significant impact on the catalytical activity and products selectivity of electrode materials. For instance, Au nanoparticles with size <5 nm tend to produce H2, while Au nanoparticles with sizes ∼8 nm have better selectivity in the formation of CO, these results can be explained as the number of low-coordinated sites increasing on small size nanoparticles and these low-coordinated sites suppress the formation of CO.63 Nanosized particles such as Bi, Sn, Pb can be well-dispersed in the electrodes, and nano structure can provide larger surface area since the particles have closer contact than catalysts with macro size. Higher surface area usually means the catalysts can expose more active sites for CO2 to adsorb and then reduce to other chemical products, and hence improve the overall catalytical activity of the electrode materials. Liu et al. developed an N-doped PC61BM ([6,6]-phenyl-C61-butyric acid methyl ester) (N-C61) nanostructure electrocatalyst, and reported stable production of formate with 91.2% faradaic efficiency at a moderate overpotential of 700 mV (Fig. 4).107,108 Their result showed better performance for CO2 reduction by the nano structure. Sn particles can have closer contact with electrolyte when their size in the nano scale, and this property will be beneficial in improving the efficiency of electron transfer. Gao et al. also described the size-dependent catalytic activity of Pd nanoparticles in CO2 electroreduction, the faradaic efficiency for CO production can be increased from 5.8% to 91.2% with the size of Pd nanoparticles decreasing from 10.3 nm to 3.7 nm.109 Small size nanoparticles can expose more edge catalytic sites to adsorb key intermediates in CO2 electroreduction, and size control can change the ratio of edge and terrace sites as well as the electronic structure of nanoparticles to improve the electrocatalytic performances needed for CO2 reduction.
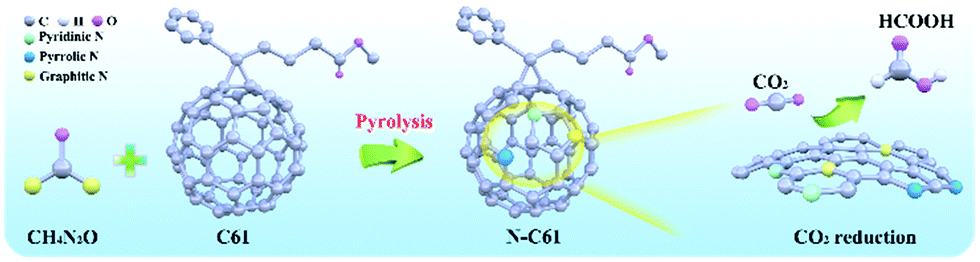 | ||
| Fig. 4 N-C61 electrocatalysts exhibit efficient formate production. (a) The synthesis procedure for N-C61 nanostructure electrocatalyst. (b) Comparison of maximum faradaic efficiency of N-C61 for formate production with other metal electrocatalysts. | ||
Tuning the crystal facets can be regarded as another feasible approach to achieve high electrocatalytic activity and products selectivity for CO2 reduction. Different crystal facets of the same catalysts may lead to the formation of different products. Rosen et al. found that Ag electrodes with Ag(211) facets have stronger adsorption of COOH intermediates which can improve CO production rates and selectivity in CO2 electroreduction.64 Zhang et al. adjusted the Cu-based catalysts with abundant Cu(100) facets which can lead to the formation of C2+ products with 40.5% conversion efficiency in CO2 electroreduction.110 It has been proven that Cu(100) facets can lower the dimerization energy barrier,111 provide strong stabilization for CO intermediates on the surface of catalysts, and thus lead to the growth of carbon chain. Wang et al. found that Cu(110) facets in Cu nanocubes can have better selectivity for C2 and C2+ products in CO2 electroreduction, while Cu(111) facets only have little electrocatalytic activity.112 Kim et al. reported that the Zn(101) facet is more favourable in producing CO in CO2 electroreduction, while the Zn(002) facet tends to produce H2.113 According to DFT calculated ions, Zn(101) facet has better ability to stabilize COOH intermediates due to its lower reduction potential from CO2 to CO, and this feature can improve the electrocatalytic performance of the electrode material.
Interfaces modification has gained increasing attention since most of the CO2 electrocatalysis have involved reactions at the liquid–solid–gas three phases. As a result, the modification of interfaces can optimize the electrocatalytic performance of the electrode materials. CO2 electrocatalysis usually occurs in the presence of aqueous electrolytes, and hydrogen evolution reaction will compete with the formation of products due to their low overpotential. To achieve better interface control in electrocatalysis, surface modification has been applied in some of the electrode materials. For instance, Li et al. covered a thin layer of nitrogen-doped carbon on the Cu surface, this thin carbon layer can provide abundant active sites due to the nitrogen defects, and function as a protective shell to resist the morphological change of the catalytical active Cu to achieve a high faradaic efficiency of 90% from CO2 to C2 products.114 Cai et al. modified the Cu nanoarray electrode with polytetrafluoroethylene (PTFE) on the surface to achieve a 67% faradaic efficiency for ethanol and propanol products in CO2 electroreduction, the PTFE here can change the electrode surface from aerophobic to aerophilic state which improves the electrocatalytic performance for the electrode.102Fig. 5 shows that carbon nanotube (CNT) was modified with epichlorohydrin dimethylamine copolymer (EDC) and the performance of electrocatalysis was improved.108
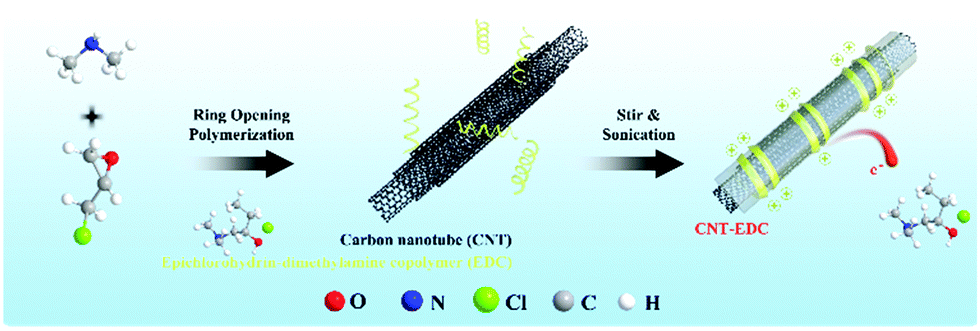 | ||
| Fig. 5 EDC-modified CNTs as metal-free carbonaceous electrocatalysts. | ||
We propose that next-generation electrode materials for bio-electrocatalysis should combine the nanostructure design, control of size effect, selectivity in crystal facets and modification of interfaces to improve the electrocatalytic performance. To optimize the catalytic activity and product selectivity, the electrode materials should be filtered from nano sizes and crystal facets to achieve high conversion efficiency for value-added products. In-depth studies of impacts from various crystal planes in catalysis should be performed. Modification of interfaces should focus on the adjustment of hydrophobic/hydrophilic or aerophobic/aerophilic properties of the electrode surface to optimize the performance.
3.4 Electrolysers
In bio-electrocatalysis, the electrolyser is a system using electricity for reduction-oxidation reactions, typically including CO2 reduction reactions and oxygen evolution reactions, and determines the local environment around the electrode. Common configurations of electrolysers include H-cell, flow-cell and membrane electrode assembly cell (MEA-cell) electrolysers.115 The H-cell electrolyser has been widely applied because of its robust and simple operation over a wide range of catalysts and electrodes. However, carbon dioxide first has to be dissolved into the electrolyte and then to the cathode to be reduced. Further, the poor CO2 mass transport rate limits current density, and restricts its commercial applications.116 The flow-cell electrolyser and MEA-cell electrolyser both utilize gas diffusion electrodes and a continuous flow of electrolyte through the electrolyser, with improved mass transport and current densities, as well as the opportunity of online adjustment of operating procedures.117 However, flow-cell electrolysers may exhibit potential risk of crossover of cathodic and anodic products, and poor stability in alkaline electrolytes, which reduces the overall productivity and energy efficiency.118 The MEA-cell electrolyser, on the other hand, assembles the catalytic layer of cathodic gas diffusion electrode closely with the bipolar membrane without catholyte, and reduces ohmic losses and catalyst contamination, while increasing overall energy efficiency and stability.119 Thus, MEA-cell electrolysers have attracted increased attentions, especially for C2+ products and commercialization purposes. Li et al. coated the Cu catalyst with N-aryl-substituted tertrahydro-4,4′-bipyridine organic films, and managed to produce ethylene with a 64% faradaic efficiency and a 20% energy efficiency at the 80 mA cm−2 current density for 190 h.120 Endorödi et al. developed a multilayer electrolyser for CO production, with serial connections of MEA units to increase the electrochemically active surface area, and achieved a 40% CO2 conversion efficiency, a 95% faradaic efficiency and >250 mA cm−2 current densities.121 Details of MEA-cell representatives are provided in Table 5.| Catalyst | Anolyte | Main product | Cell voltage | Faradaic efficiency | Current density | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| GDE-PTFE|GDE-PTFE | DI water | CO | −3.6 V | 4.3% | 755 mA cm−2 | — | 253 |
| GDE-Ag/Ir nanoparticles|Ti | — | CO | −3 V | 95% | >250 mA cm−2 | — | 254 |
| Cobalt phthalocyanine|Ni | 1 M KOH | CO | −2.2 V | >90% | 50 mA cm−2 | 8 h | 255 |
| Ni-NCB|IrO2 | 0.1 M KHCO3 | CO | −2.8 V | >90% | ∼8 A | 6 h | 256 |
| Ni–N/C|IrO2 | 0.1 M KHCO3 | CO | −3 V | >93% | 113.6 mA cm−2 | — | 257 |
| GDE-Ag|IrO2/C | 0.1 M KHCO3 | CO | −3.3 V | >90% | 200 mA cm−2 | — | 258 |
| Bi0.1Sn/PTFE|IrOx/Ti | 0.1 M KHCO3 | Formic acid | −4.4 V | 90% | 60 mA cm−2 | 100 h | 259 |
| 2D-Bi|IrO2–C | DI water | Formic acid | −3 V | 80% | 30 mA cm−2 | 100 h | 260 |
| GDE-CuPc | 0.05 M KHCO3 | Methane | −4.4 V | 62% | 190 mA cm−2 | 110 h | 261 |
| FeTPP [Cl]/Cu|IrOx/Ti | 0.1 M KHCO3 | Ethanol | −3.7 V | 41% | 124 mA cm−2 | 12 h | 262 |
| N–C/Cu|IrOx/Ti | 0.2 M KHCO3 | −3.67 V | 52% | 156 mA cm−2 | 15 h | 263 | |
| Molecule/Cu/PTFE|IrOx/Ti | 0.1 M KHCO3 | Ethylene | −3.65 V | 64% | 600 mA | 190 h | 264 |
| Cu|IrO2 on Ti mesh | 0.1 M KHCO3 | Ethylene | −3.75 V | 40% | 120 mA cm−2 | 100 h | 119 |
| CTPI|Ti | 0.1 M KHCO3 | Ethylene | −3.9 V | 66% | 208 mA cm−2 | 100 h | 265 |
| Cu-PTFE|IrO2 | 0.1 M KHCO3 | Ethylene | −4.1 V | 65% | 215 mA cm−2 | 50 h | 266 |
| Ti-Based bipolar plate|IrOx/Ti mesh | 0.1 M KHCO3 | Ethanol/ethylene | −2.41 V | 85.6% | 12 A | 4.5 h | 110 |
Further improvement of MEA-cell electrolysers includes continuous upgrading of gas diffusion electrodes to sustain high ion mobility while buffering the local environment, improving membrane ion mobilities and robustness to increase process stability and avoid product crossover, increases interface densities of carbon dioxide/catalyst/electrolyte to improve mass transfer efficiency and production rate.118,122
3.5 Enzymatic electrocatalysis
In classic electrocatalysis, metal catalysts may exhibit low availability, low selectivity, and high overpotentials. Enzymatic electrocatalysis integrates electrodes for electrons and fast cofactor regeneration with oxidoreductase enzymes or isolated organelles for high selectivity and self-regeneration, and is a valuable addition and even promising alternatives for noble metal catalysts.123 Both metalloenzymes and non-metalloenzymes can be utilized in enzymatic electrocatalysis and produce a range of products including CO by CO dehydrogenases, formate by formate dehydrogenases, methane and methanol by formate dehydrogenase and formaldehyde dehydrogenase (Fig. 6).29,32 For example, Reisner et al. utilized formate dehydrogenase and metal oxidase TiO2, and established selective and reversible conversion of CO2 to formate with a turnover frequency of 11 s−1.124 Recently, nitrogenases have drawn increasing interest as it has been shown to also reduce CO2 to produce CO, formate, ethylene, and propene.32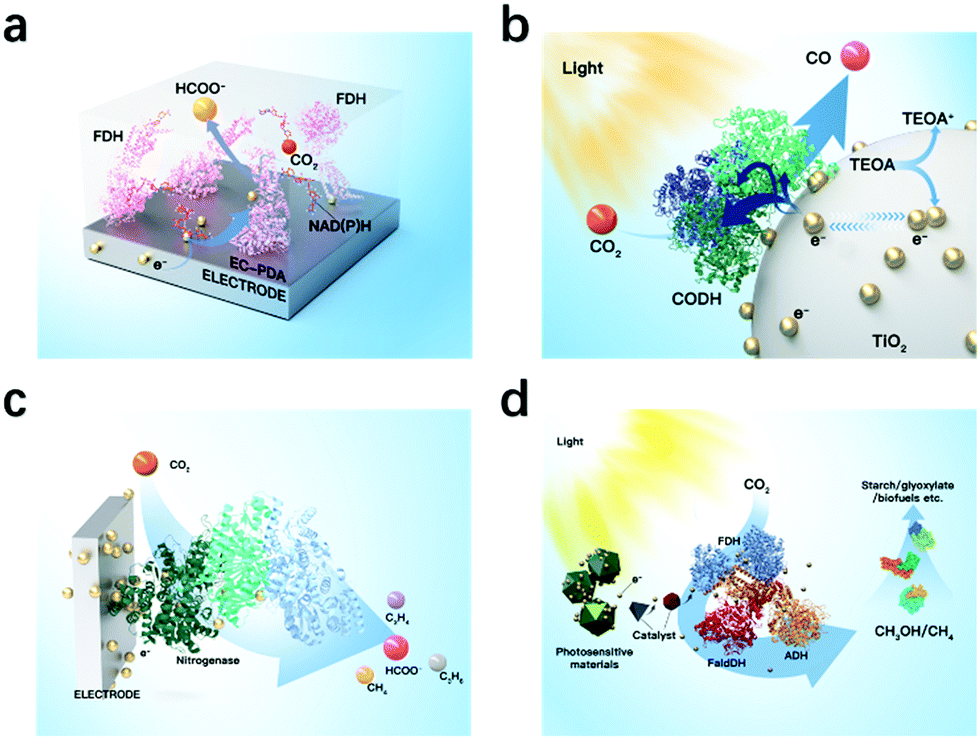 | ||
| Fig. 6 Representative enzymes used in enzymatic electrocatalysis for fixation. (a) Formate dehydrogenase that converts CO2 to formate. (b) CO dehydrogenase that converts CO2 to CO. (c) Nitrogenases. (d) Enzyme cascade of formate dehydrogenase and formaldehyde dehydrogenase that converts CO2 to methane and methanol. | ||
Key factors regarding enzymatic electrocatalysis include the activity and stability of enzymes through electrosynthesis environments, the electron transfer efficiency between electrodes and enzymes, as well as the distance between electron donors and enzyme's redox cofactors.125 Electron mediators and heterogeneous materials can be applied to ensure long distance electron transfers or to achieve cascade reactions. Mediators must be carefully selected and sometimes co-immobilized based on the reduction potentials required for the target reaction. Ideally the potential difference between the electron mediator and the enzymatic cofactor should be 50–170 mV.126 For cascade reactions, uncontrolled intermediates concentrations and distributions may cause mismatched enzymatic kinetics and product inhibition.127 A co-immobilization of cascade enzymes and their mediators sequentially with precise control of the enzyme ratio and inter-enzyme distance, also called electrostatic substrate channelling, have been employed. Zhang et al. encapsulated formate dehydrogenase, formaldehyde dehydrogenase, the organometallic mediator [Cp*Rh(bpy)Cl]Cl and photosensitizer using poly(allylamine hydrochloride)-doped nanofibers, and produced methanol with 90% yield from CO2.128 Lv utilized Amine-functionalized MIL-101(Cr) and two-HKUST-1-layer MOFs for CO2 storage and immobilization of carbonic anhydrase (CA), formate dehydrogenase and glutamate dehydrogenase, and exhibited high catalytic efficiency with the formate yield of 179% (Fig. 7).129
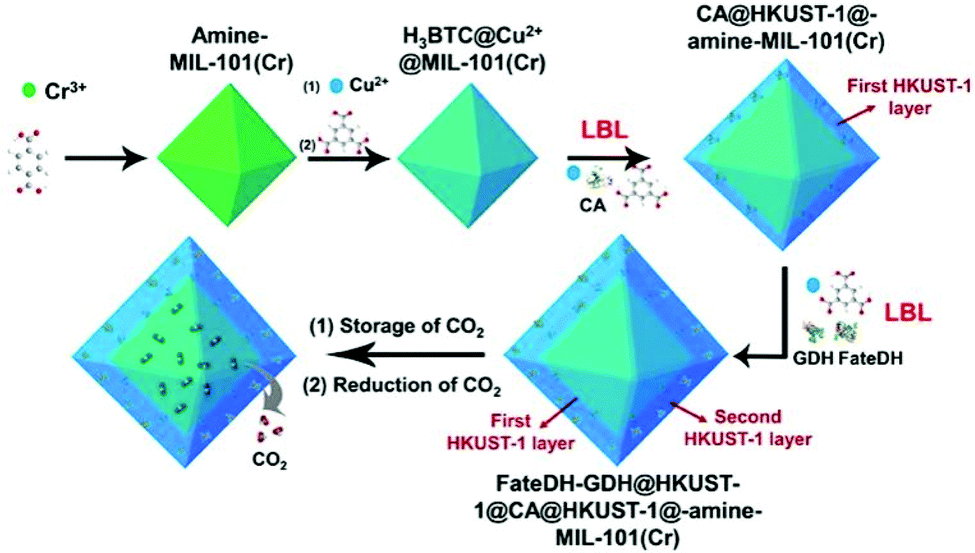 | ||
| Fig. 7 Schematic illustration of the preparation of HKUST-1@amine-MIL-101(Cr)-based multienzymes for the reduction of adsorbed CO2. Reproduced with permission from ref. 129, copyright 2019 to Frontiers Media S.A. | ||
While enzymatic electrocatalysis advances with high reaction specificity, robust to co-solvents, and simple workflows, it is limited by the need for enzyme purification, degradation, and regeneration.32 In addition, many efficient CO2 reducing enzymes are oxygen sensitive, e.g. CO dehydrogenases from Methanosarcina bakerii, Moorella thermoacetica, and Carboxythermus hydrogenoformas, as well as Mo and W-containing formate dehydrogenases, and may require substantial enzyme and process engineering for application.127,130
3.6 Bio-catalysis strains and pathways
Several microbes have been described that are able to tolerate low voltage of electricity, and may also conduct electricity and utilize the electrons as the energy source. Thus, microbial electrocatalysis has been developed for CO2 utilization. Microbial electrocatalysis has many advantages, including self-regeneration, good stability, and the ability to produce a wide range of products. However, compared with enzymatic electrocatalysis it requires a continuous supply of nutrients to keep the microbes functional, and because of its complex metabolic networks, microbial electrocatalysis may have less specificity and efficiency.57 In microbial electrocatalysis, the identification of suitable host strains and carbon utilization pathways are important. Suitable host strains for bio-electrocatalysis should have robustness and the ability to perform efficient electron transfer, an appropriate metabolic environment for productions of desired products, and more importantly, the compatibility with current reported C1 utilization pathways. Holtmann et al. compared different methanogens, including Methanococcus vannielii, Methanococcus maripaludis, Methanolacinia petrolearia, Methanobacterium congolense, and Methanoculleus submarinus, and demonstrated that M. maripaludis represents a promising methane electrocatalysis producer capable of producing methane with an 8.81 mmol m−2 d−1 productivity and a 58.9% coulombic efficiency.131 Host strains that are widely used in CO2-based bio-electrocatalysis include methanogens, acetogens, Synechocystis species, Clostridia species, Microcystis aeruginosa, and Acidithiobacillus ferrooxidans.57,123Current C1 utilization pathways include the reductive glycine pathway,9 the Wood-Ljungdahl pathway,132 the Calvin-Benson-Bassham cycle (CBB cycle),133 the dicarboxylate/4-hydroxybutyrate (DC/HB) cycle,134 the reductive TCA cycle,135 the 3-hydroxypropionate/4-hydroxybutyrate (HP/HB) cycle,136 the 3-hydroxypropionate (3-HP) bicycle,137 the crotonyl–coenzyme A (CoA)/ethylmalonyl-CoA/hydroxybutyryl-CoA (CETCH) cycle,138 the reductive glyoxylate and pyruvate synthesis-malyl-CoA-glycerate (rGPS-MCG) cycle,139 the PYC-OAH-ACS-PFOR (POAP) cycle,140 the half-Wood-Ljungdahl-formolase (HWLS) pathway,141 the ribulose monophosphate (RuMP) Cycle,142 the xylulose monophosphate (XuMP) cycle,143 and the serine cycle (Fig. 8).144 Among these pathways, the CBB cycle, the Wood-Ljungdahl pathway and the reductive glycine pathway attracted increased attentions for CO2 bio-electrocatalysis. The CBB cycle is widely used by plants, algae, cyanobacteria and proteobacteria,145 and converts CO2 into glyceraldehyde 3-phosphate using NADPH as the electron donor.146 Milo et al. introduced the complete CBB cycle in Escherichia coli and enabled cell growth solely from CO2 and formate.147 The Wood-Ljungdahl pathway can be found in euryarchaeota, proteobacteria, planctomycetes and spirochaetes,145 and can convert CO2 and a variety of C1 and C2 compounds into acetyl-CoA. Within microorganisms utilizing the Wood-Ljungdahl pathway, Methylorubrum extorquens, Cupriavidus necator and Clostridium species have attracted increased attention and are able to produce a wide range of products, including butanol, 2-oxobutyrate and 3-butanediol.30,36 The reductive glycine pathway is found in Candidatus Phosphitivorax anaerolimi, and can convert CO2 and formic acid into pyruvate.9 Lee et al. reported the introduction of the reductive glycine pathway in E. coli and recovered cell growth solely from CO2 and formate.148
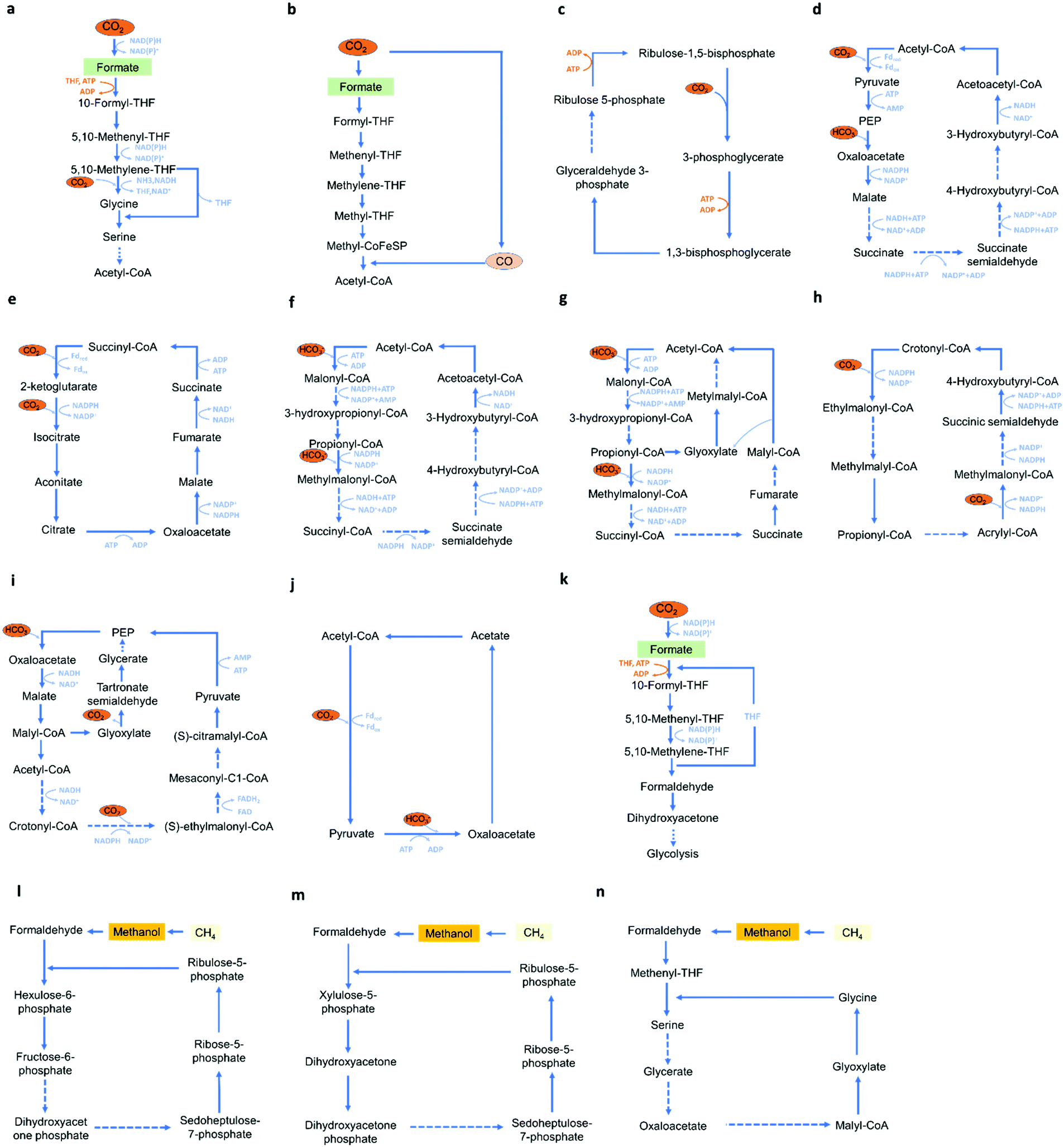 | ||
| Fig. 8 Representations of current C1 utilization pathways. (a) The reductive glycine pathway. (b) The Wood-Ljungdahl pathway. (c) The CBB cycle. (d) The DC/HB cycle. (e) The reductive TCA cycle. (f) The HP/HB cycle. (g) 3-HP bicycle. (h) The CETCH cycle. (i) The rGPS–MCG cycle. (j) The POAP cycle. (k) The HWLS pathway. (l) The RuMP cycle. (m) The XuMP cycle. (n) The serine cycle. | ||
4. Integration of electrocatalysis and bio-catalysis
The integration of classic electrocatalysis and bio-catalysis will enable production of a wide range of chemicals from CO2. The chemically inert nature of CO2 severely limited its reduction to C1 compounds, especially through bio-catalysis that requires mild operational conditions. Electrocatalysis has the advantages of high electron transfer efficiency that can accelerate bio-catalysis processes with sufficient and rapid electron supply. Meanwhile, bio-catalysis can solve the side-product problem of electrocatalysis with higher specificity, longer stability, and longer chain length.A number of C2+ products, such as isopropanol,11 butyric acid,149 and even C6+ products, such as α-humulene,150L-erythrulose151 and terpenes,152 have been produced (Fig. 9). For example, Chen et al. reported the development of the Cu–Ag tandem catalyst, and improved the production of ethylene, ethanol and acetate by 3–6 fold compared with the Cu catalyst.153 Song et al. employed formate dehydrogenase, NADH and NR, to produce formate, and together with Ralstonia eutropha was able to produce 485 mg L−1 poly(3-hydroxybutyrate) from CO2.10 Wu et al. reported the production of 1.73 mg L−1 lycopene from the flue gas of coal-fired power plant through the direct-electron-transfer strategies, using H2 as the electron carrier.154 Bolognesi et al. integrated a bio-electrochemical system converting CO2 to acetate and Auxenochlor CO2ella protothecoides converting acetate to the diesel compatible algae oil.155 More applications of bio-electrocatalysis in production of fuels and chemicals are shown in Table 6. With the rapid development of electrocatalysis, material science and synthetic biology, we foresee in the near future, that bio-electrocatalysis will manage to produce custom-designed, energy-rich products with even longer chain length, such as polymers, and fatty acid derived products.
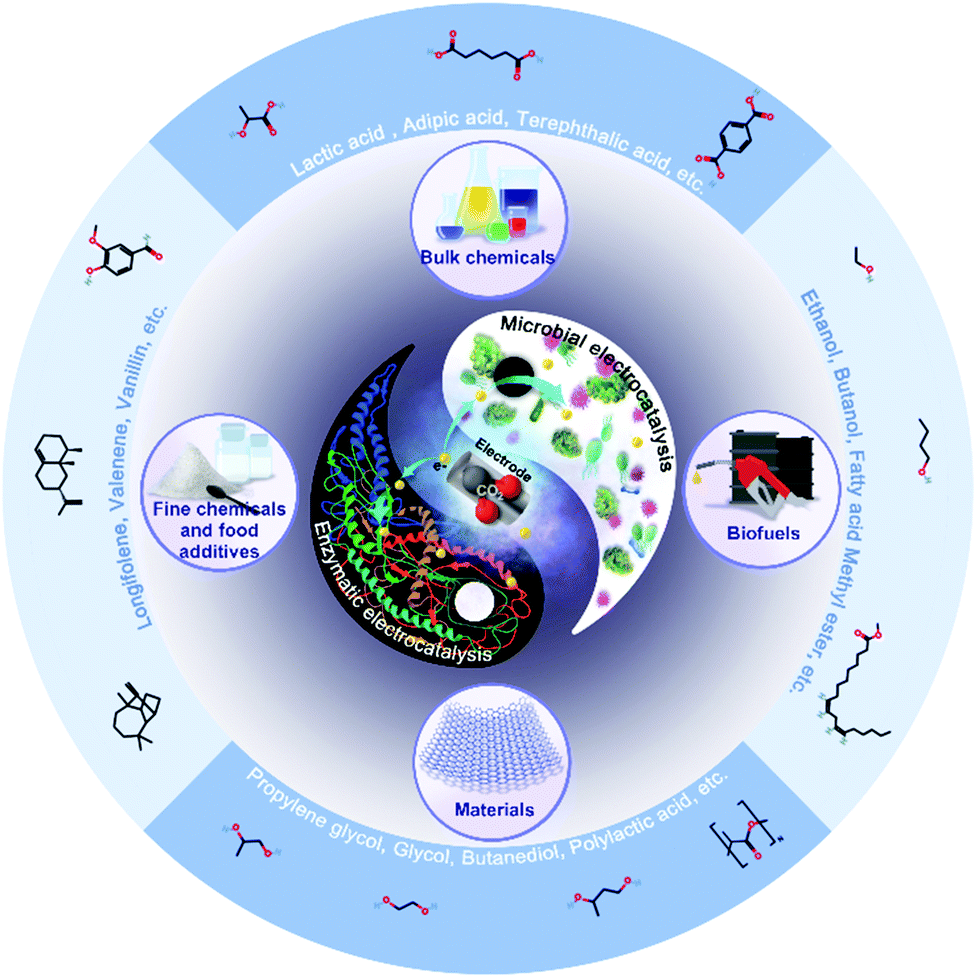 | ||
| Fig. 9 Applications of bio-electrocatalysis for CO2 utilizations. | ||
| Carbon source | Production method | Chain length of products | Production host | Product | Production levela | Catalyst | Ref. |
|---|---|---|---|---|---|---|---|
| Note.a Here, we calculate the average value according to the final output in the respective literature. If there is no specific value, it cannot be calculated because it is not given in the article. | |||||||
| CO2 | Microbial transformation | C1–C6 | Methanosarcina barkeri | Methane | 15.71 mM d−1 | — | 183 |
| C. autoethanogenum | Acetate | 2.42 mM L−1 d−1 | Cds | 267 | |||
| Acetogen co-culture | Acetate | 23.50 mg d−1 | — | 268 | |||
| Sporomusa ovata | Acetate | 6.66 mM d−1 | — | 269 | |||
| M. thermoacetica | Acetate | Milligram level per h | Cds | 270 | |||
| M. thermoacetica | Acetate | Milligram level per h | AuNCs | 5 | |||
| C. scatologenes | Ethanol | 2.14 mg L−1 d−1 | rGO/biofilm | 271 | |||
| Synechococcus elongatus | 1,3-Propanediol | 21 mg L−1 d−1 | — | 272 | |||
| Synechocystis sp. | 3-Hydroxybutyrate | 25.4 mg L−1 d−1 | — | 273 | |||
| C. eticum | 2-Oxobutyrate | 2 mg L−1 d−1 | — | 274 | |||
| Clostridium co-culture | Butyrate | 1.82 mMC d−1 | Carbon cloth | 149 | |||
| C. necator | Isobutanol | ∼8.5 mg L−1 h−1 | Indium foil | 275 | |||
| 3-Methyl-1-butanol | ∼5.7 mg L−1 h−1 | ||||||
| Microbiome | Acid alcohol mixture | Milligram level per d | Graphite granules | 276 | |||
| Microbiome | Alcohols | 0.34 kg m−3 day−1 | VITO-Core® GDE | 277 | |||
| C6+ | C. necator | α-Humulene | 36 mg L−1 d−1 | Patinum late | 150 | ||
| Synechocystis sp. | Hydrocarbon fuel | 11.12 mg L−1 d−1 | — | 278 | |||
| Enzymatic synthesis | C1–C6 | FDH | Formic acid | 0.92 μM h−1 | Graphene-based photocatalyst | 279 | |
| CA, FDH | Formic acid | 48.6 μM h−1 | g-C3N4/ZIF-8 | 280 | |||
| FDH | Formic acid | 225.81 mg L−1 h−1 | Plain graphite rod | 281 | |||
| FDH, FaldDH and alcohol dehydrogenase (ADH) | Methanol | 0.4 mM h−1 | UF membranes | 282 | |||
| Glyoxylate carboligase | Ethylene glycol | 3.5 mM h−1 | — | 283 | |||
| C6+ | Multienzyme cascade | Terpenes | 0.2 mg L−1 h−1 | — | 152 | ||
| C1 utilization | Microbial transformation | C1–C6 | Methylomonas sp | Succinate | 1.54 mg L−1 h−1 | — | 34 |
| C. carboxidivorans | Alcohols | Milligram level per d | — | 284 | |||
| Clostridia | 2,3-Butnanediol | 0.25 mM d−1 | — | 285 | |||
| M. extorquens | Itaconic acid | 2.63 mg L−1 d−1 | — | 286 | |||
| C6+ | M. extorquens | α-Humulene | 15.87 mg L−1 h−1 | — | 35 | ||
| M. extorquens | Mevalonate | 6.6 mg L−1 h−1 | — | 287 | |||
| Enzymatic synthesis | C1–C6 | Multienzyme cascade | Glyoxylate | 26.7 mg L−1 h−1 | — | 288 | |
| Multienzyme cascade | L-Erythrulose | 126 g L−1 h−1 | — | 151 | |||
| C6+ | Multienzyme cascade | Starch | 17.2 nmol C min−1 mg−1 | ZnO-ZrO2 | 289 | ||
5. Future research directions
5.1 Improving the compatibility between electrodes and biocatalysts
Bio-catalysis can be highly active under mild conditions. Although this requirement can be sustainable and cost-effective, it also restricts the operational conditions of bio-electrocatalysis. Enzymes with their active sites exposed, such as ferredoxin, peroxidase, and cytochrome C, are suitable for direct electron transfer.156 However, it is relatively difficult to keep enzymes close to the electrode while maintaining their stability and activity. Mediators can be used to improve electron transfer efficiencies; however, they also have limitations such as their potential toxicity to biocatalysts, especially enzymes. The utilization of redox polymers and biofilms to co-assemble biocatalysts and electron mediators can enhance the communication between biocatalysts and electrodes on the nanometre and micrometre scale, while improving the biocompatibility, conductivity, and mass transfer rate. For example, Milton et al. reported the synthesis of the cobaltocene-functionalized poly(allylamine) to immobilize formate dehydrogenase while mediating electron transfers from the electrode, and produce formate with a 99% faradaic efficiency.157Furthermore, microbial communities have also been described that can improve the electric tolerance and bio-electrocatalysis production. Hogan reported the methanogenesis rate at 0.72 mg methane L−1 day−1 using the microbial community, and demonstrated that the abundance of Euyarcheota and the Desulfovibrionaceae phylotype positively corelated with methane production.158 They also demonstrated that for some species in the community that do not produce methane themselves they may help improve the electron transfer and the overall synthesis.158
Optimization of the electrode composition and the electrolyte can also increase the compatibility of electrocatalysis and bio-catalysis.57,123 For example, Nocera et al. developed a Reactive oxygen species-resistant Co–P cathode and substantially reduced the release of toxic metals during aerobic electrocatalysis.159,160 Liao et al. applied a porous ceramic cup to shield the anode to allow efficient diffusion of chemicals while reducing exposure of the cell to toxic reactive oxygen and nitrogen compounds, and produced 140 mg L−1 biofuels.161 Cornejo et al. utilized an ultrathin silica membrane to separate abiotic and biotic components while maintaining their electrochemical intereactions.162 Hass et al. developed a gas diffusion cathode allowing a direct interaction with gaseous CO2, and substantially increased the mass transferring rate of CO2 in salt-based electrolytes.163 Rodrigues et al. utilized a biocompatible perfluorocarbon nano-emulsion as the H2 carrier and increased acetate production by 190% to the highest reported productivity (1.1 mM h−1).164 Photoelectrodes, which directly harvest solar energy to provide electrons, have also been developed for CO2 fixation and conversion. Current reports integrated photoelectrocatalysis with bio-catalysis mostly used NAD(P)H as the electron mediator and carrier.165 For example, Guo et al. reported the integration of yeast cell factories with indium phosphide based light-harvesting nanoparticles using a polyphenol-based assembly method, and enabled carbon-and energy-efficient production of shikimic acid.166 Similarly, Hu et al. incorporated CdS nanoparticles and a synthetic half-Wood–Ljungdahl-formolase CO2 fixation pathway in E. coli, and improved the anaerobic production of L-malate and butyrate approaching to the theoretical yields.141
Future research directions include improvement on the electrochemical surface area, absorption of bio-electrocatalysts, intrinsic conductivity, electron transfer rates, the operational performance and stability, as well as metabolic engineering of microbes to better tolerate electrocatalysis conditions, such as pH, temperature, electrolyte composition and toxic byproducts.166,167
5.2 Optimizing the electron transfer efficiency
The electron transfer rate and multi-electron transfer efficiency in bio-electrocatalysis need to be optimized.168 For enzymatic electrocatalysis, oxidoreductases generally hide active sites in the centre and strictly regulate intra-enzyme electron transfers, that reduces electron transfer efficiency between electrodes and enzymes.169 By surveying existing electron transfer mechanisms, Dutton et al. reported that electrons can travel up to 14 Å within the protein medium without significantly reducing the transfer rate and efficiency.170 Enzyme engineering has been utilized to improve the communications between electrode and oxidoreductases, including protein truncation to delete unessential peptides and expose active sites, surface modifications (e.g. de-glycosylation) to downsize the dimensions of the protein and facilitate electron transfer, and modification of active sites to improve electron channeling.171 The addition of the redox polymer and the docking motif to immobilize oxidoreductases to the electrode surface and even in the optimal orientation may reduce the distance while improving localized enzyme concentrations, however, the requirement of an monolayer of enzymes may restrict the overall catalyst loading.167In microbial electrocatalysis, most cell membranes are non-conductive.172 Moreover, microbial pathways can be complicated in a microbial system since it requires several cofactors to transfer electrons as the driving force. Strategies to efficiently bridge electronic devices and microbial cell factories include direct electron transfer though cytochromes and conductive nanowires, and indirect electron transfer through redox mediators or energy carriers. Currently used protein nanowires include curli fibers,173,174 pilin-based nanowires,175 cytochrome wires,176,177 and conductive fibres of cable bacteria.178,179 Clarke et al. reported the atomic structure of the electron conducting MtrAB protein complex spanning outer membranes of Shewanella species, that could connect cells with extracellular electrodes and conduct electrons.180 Malvankar et al. also described the production of cytochrome OmcZ nanowires with high conductivity and stiffness from electric stimulated Geobacter sulfurreducens biofilms.177 Similarly, when analysing the structure of widely spread coronavirus, we identified a number of protein receptors similar to antenna on its surface that accelerate the electron transfer and information exchange of the virus. Future research direction may indicate whether the incorporation of these protein nanowires on the cell surface of microbial cell factories may act as quick and efficient electron transfer channels. One could also build artificial channels that possess effective metabolic pathways to produce high value-added products that enable direct electron harvest for biosynthesis (Fig. 10). Hereby the dependence on cofactors and overall energy consumption could be greatly reduced.
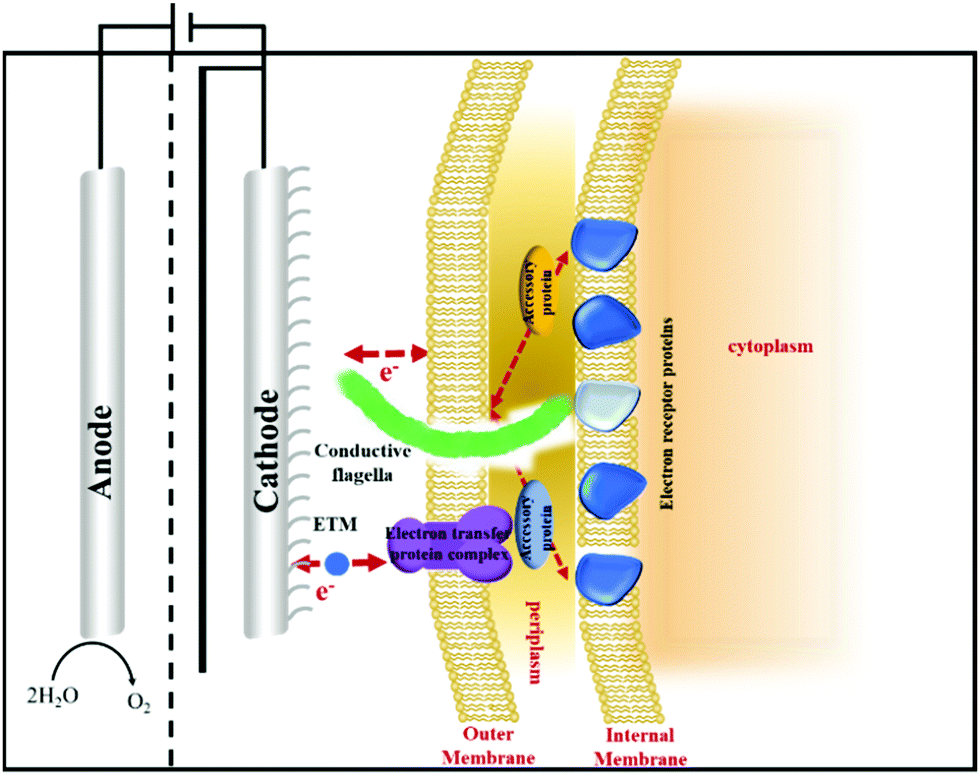 | ||
| Fig. 10 Illustration of the artificial cell with antenna electron transfer channels on its surface. | ||
Common mediators and electron carriers of in-direct electron transfer processes include native mediators such as flavins and quinine, electron carriers such as cofactors, H2 and formate, natural and artificial substances such as humic acid and neutral red, as well as enzyme like hydrogenases and formate hydrogenases.181 It has been suggested that optimizing the bound flavin cofactor mechanisms could substantially increase the cross-membrane electron transfer.182 Moreover, Nichols introduced the Pt and Ni based Hydrogen evolution reaction electrocatalysts into Methanosarcina barkeri, and achieved conversion of CO2 to methane with over 81% faradaic efficiency.183 Liu integrated Mn based H2 catalysis into C. necator, and achieved efficient production of cell mass, fusel alcohols and PHB.159 Li et al. discovered three gene modules and five key genes regarding the NAD+ generation in Shewanella oneidensis, and increased the intracellular NAD(H) concentration by 2.1-fold.184 Similarly, Yang et al. integrated the flavin-generation pathway from Bacillus subtilis to S. oneidensis, and increased the inward power density by 15.5 fold.185
Future research directions include the optimization of the uptake rates of electron donors and electron carriers, as well as the intracellular transportation of electrons, the oxidation of electron donors to release electrons, the reduction of the use of costly catalysts, the development of synthetic biology and metabolic engineering tools of less-characterized electroactive enzymes and microbes, deepening the understanding of cross-membrane and intracellular electron transport mechanisms, and harmonizing heterologous pathways and endogenous metabolisms.123,181
6. Outlook
The growing concerns about elevated GHGs emissions calls for immediate shift of fossil fuel dependent industries. Simple capture of CO2 is no longer enough to hold back the negative effect of greenhouse gases. As a result, transformation of CO2 into fuels and chemicals has attracted increased attention. Bio-electrocatalysis for CO2 utilization offers solutions to this challenge through the following aspects. (i) Use of electricity from renewable sources, including solar, wind, tidal, thermal, and hydro. In combination with bio-electrocatalysis, carbon neutral (or in some cases even carbon negative) production of valuable products can be acquired. (ii) Compared with classic electrocatalysis, bio-electrocatalysis has a lower environmental impact, higher selectivity, longer stability, more flexibility for custom-designed products, and a wider product range, especially for products with higher chain length, such as biofuels and biopolymers. (iii) Bio-electrocatalysis showed the superiorities than the bio-catalysis in the two critical steps, including the CO2 fixation efficiency and the energy utilization efficiency, and bio-electrocatalysis could exhibit greater potentials for producing energy-dense compounds, such as jet fuels.Future research directions include the characterisation and re-construction of biologically produced electron transferring nanowires and extracellular respiratory modules to improve the energy conversion efficiency and specific productivity. In addition, the identification of more efficient fixation pathways, in terms of conversion rates for the identification of appropriate microbial cell factories having efficient carbon fixation rates, high growth rates, and electron tolerance and utilization efficiency. Other advances could involve the use of complex materials and advanced devices. Two limiting factors of electrocatalysis include the electrons transfer rate and the CO2 mass transport rate. Bio-electrocatalysis aims to enhance the electron transfer efficiency and the CO2 utilization rates through the optimisation of three-dimensional electrode materials and their hybrids, e.g., arrays, core@shell, sandwich-like, foam and polyhedrons. Meanwhile, the continuous development of MEA-cell electrolysers will further enhance bio-electrocatalysis towards commercialisation.
In conclusion, with the advancement of the technologies presented in this review, we expect that bio-electrocatalysis will be able to play a significant role in the future energy and chemical sector (Fig. 11).
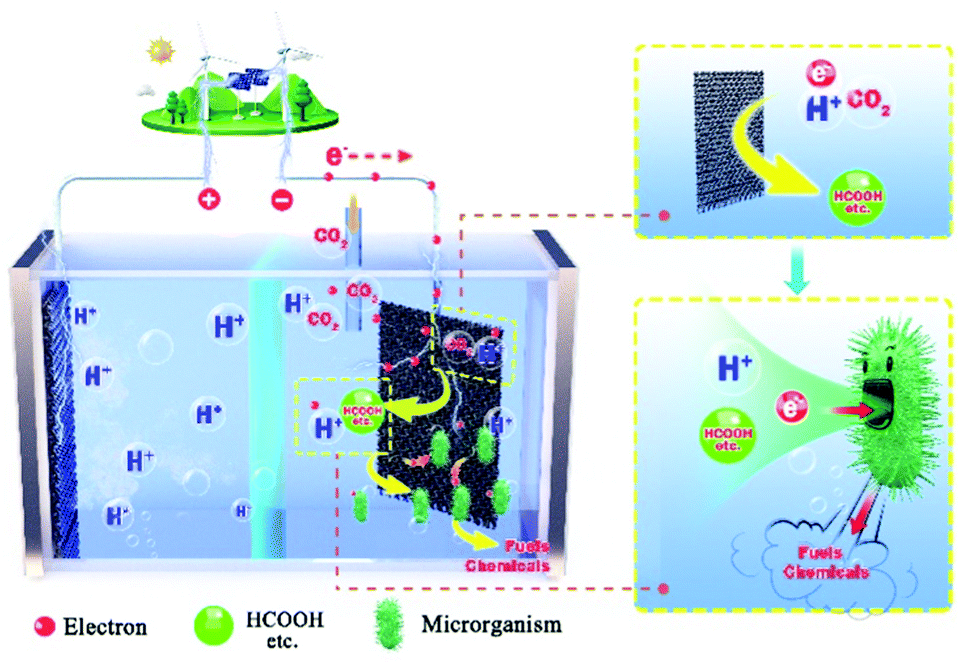 | ||
| Fig. 11 Illustration of bio-electrocatalysis enabled bio-economy. | ||
Author contributions
X. T. drafted the outline. X. T. and J. N. wrote the manuscript. J. N. supervised the research. All the authors have read and approved the final manuscript.Conflicts of interest
The authors declare no competing interests.Notes and references
- Z. Liu, K. Wang, Y. Chen, T. Tan and J. Nielsen, Nat. Catal., 2020, 3, 274–288 CrossRef CAS.
- N. J. Claassens, C. A. Cotton, D. Kopljar and A. Bar-Even, Nat. Catal., 2019, 2, 437 CrossRef CAS.
- L. V. Teixeira, L. F. Moutinho and A. S. Romão-Dumaresq, Biofuels Bioprod. Biorefining., 2018, 12(6), 1103–1117 CrossRef CAS.
- S. Cestellos-Blanco, H. Zhang, J. M. Kim, Y.-X. Shen and P. Yang, Nat. Catal., 2020, 3, 245–255 CrossRef CAS.
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS PubMed.
- D. Karamanev, V. Pupkevich, K. Penev, V. Glibin, J. Gohil and V. Vajihinejad, Energy, 2017, 129, 237–245 CrossRef CAS.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X.-D. Zhou, R. Vajtai and B. I. Yakobson, Nano Lett., 2016, 16, 466–470 CrossRef CAS PubMed.
- I. A. Figueroa, T. P. Barnum, P. Y. Somasekhar, C. I. Carlström, A. L. Engelbrektson and J. D. Coates, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 92–101 CrossRef PubMed.
- X. Chen, Y. Cao, F. Li, Y. Tian and H. Song, ACS Catal., 2018, 8, 4429–4437 CrossRef CAS.
- S. Bajracharya, K. Vanbroekhoven, C. J.-N. Buisman, D. Strik and D. Pant, Faraday Discuss., 2017, 202, 433–449 RSC.
- L. Jourdin, S. M. Raes, C. J. Buisman and D. P. Strik, Front. Energy Res., 2018, 6, 7 CrossRef.
- T. Krieg, A. Sydow, S. Faust, I. Huth and D. Holtmann, Angew. Chem., Int. Ed., 2018, 57, 1879–1882 CrossRef CAS PubMed.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- P.-L. Tremblay, L. T. Angenent and T. Zhang, Trends Biotechnol., 2017, 35, 360–371 CrossRef CAS PubMed.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- M. D. Wodrich, B. Sawatlon, M. Busch and C. Corminboeuf, Acc. Chem. Res., 2021, 54, 1107–1117 CrossRef CAS PubMed.
- Y. Li and Q. Sun, Adv. Energy Mater., 2016, 6, 1600463 CrossRef.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- W. Zhu, R. Michalsky, O. N. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- Y. Lai, N. B. Watkins, A. Rosas-Hernández, A. Thevenon, G. P. Heim, L. Zhou, Y. Wu, J. C. Peters, J. M. Gregoire and T. Agapie, ACS Cent. Sci., 2021, 7, 1756–1762 CrossRef CAS PubMed.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS.
- C. J. Bondue, M. Graf, A. Goyal and M. T. Koper, J. Am. Chem. Soc., 2020, 143, 279–285 CrossRef PubMed.
- H.-Q. Liang, S. Zhao, X.-M. Hu, M. Ceccato, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2021, 11, 958–966 CrossRef CAS.
- H. Tao, X. Sun, S. Back, Z. Han, Q. Zhu, A. W. Robertson, T. Ma, Q. Fan, B. Han and Y. Jung, Chem. Sci., 2018, 9, 483–487 RSC.
- A. Schmid, J. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364(6438), eaav3506 CrossRef CAS PubMed.
- S. Schlager, A. Fuchsbauer, M. Haberbauer, H. Neugebauer and N. S. Sariciftci, J. Mater. Chem. A, 2017, 5, 2429–2443 RSC.
- N. J. Claassens, I. Sánchez-Andrea, D. Z. Sousa and A. Bar-Even, Curr. Opin. Biotechnol, 2018, 50, 195–205 CrossRef CAS PubMed.
- D. E. Holmes, D. R. Bond and D. R. Lovley, Appl. Environ. Microbiol., 2004, 70, 1234–1237 CrossRef CAS PubMed.
- M. Yuan, M. J. Kummer and S. D. Minteer, Chem. – Eur. J., 2019, 25, 14258–14266 CrossRef CAS PubMed.
- E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt and F. Thompson, The Prokaryotes, Springer, New York, 2006 Search PubMed.
- D. T.-N. Nguyen, O. K. Lee, S. Hadiyati, A. N. Affifah, M. S. Kim and E. Y. Lee, Metab. Eng., 2019, 54, 170–179 CrossRef CAS PubMed.
- F. Sonntag, C. Kroner, P. Lubuta, R. Peyraud, A. Horst, M. Buchhaupt and J. Schrader, Metab. Eng., 2015, 32, 82–94 CrossRef CAS PubMed.
- C. M. Humphreys and N. P. Minton, Curr. Opin. Biotechnol, 2018, 50, 174–181 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E.-L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- A. Peigney, C. Laurent, E. Flahaut, R. Bacsa and A. Rousset, Carbon, 2001, 39, 507–514 CrossRef CAS.
- M. Dresselhaus, Phys. World, 1996, 9, 18 CrossRef.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 53, 8709–8713 CrossRef CAS PubMed.
- H. Zhao, Y. Zhang, B. Zhao, Y. Chang and Z. Li, Environ. Sci. Technol., 2012, 46, 5198–5204 CrossRef CAS PubMed.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson and J. Lou, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nanosci. Technol., World Scientific, 2010, pp. 11–19 Search PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- P. Han, X. Yu, D. Yuan, M. Kuang, Y. Wang, A. M. Al-Enizi and G. Zheng, J. Colloid Interface Sci., 2019, 534, 332–337 CrossRef CAS PubMed.
- K.-R. Kim, J. Kang and K.-J. Chae, Int. J. Hydrogen Energ., 2017, 42, 27623–27629 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341(6169), 1230444 CrossRef PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- D.-H. Nam, O. S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C.-T. Dinh, F. P. García de Arquer, Y. Wang, Z. Liang and A. H. Proppe, J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef CAS PubMed.
- P. Deng, F. Yang, Z. Wang, S. Chen, Y. Zhou, S. Zaman and B. Y. Xia, Angew. Chem., 2020, 132, 10899–10905 CrossRef.
- X.-F. Qiu, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed.
- V. Uppada, S. Bhaduri and S. B. Noronha, Curr. Sci., 2014, 946–957 CAS.
- H. Zhao and W. A. Van Der Donk, Curr. Opin. Biotechnol, 2003, 14, 583–589 CrossRef CAS PubMed.
- H. Chen, F. Dong and S. D. Minteer, Nat. Catal., 2020, 3, 225–244 CrossRef.
- R. D. Milton and S. D. Minteer, J. R. Soc., Interface, 2017, 14, 20170253 CrossRef PubMed.
- U. Schröder, Phys. Chem. Chem. Phys., 2007, 9, 2619–2629 RSC.
- M. T. Matsena and E. M.-N. Chirwa, Biofuels and Bioenergy, Elsevier, 2022, pp. 321–358 Search PubMed.
- X. Liu and X. Yu, ACS Energy Lett., 2020, 5, 867–878 CrossRef CAS.
- M. Muthuvel, X. Jin and G. Botte, Encyclopedia of electrochemical power sources, Elsevier, 2009, pp. 158–171 Search PubMed.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS.
- F. Yang, A. O. Elnabawy, R. Schimmenti, P. Song, J. Wang, Z. Peng, S. Yao, R. Deng, S. Song and Y. Lin, Nat. Commun., 2020, 11, 1–8 CrossRef CAS PubMed.
- R. Zhang, W. Lv and L. Lei, Appl. Surf. Sci., 2015, 356, 24–29 CrossRef CAS.
- D. Liu, T. Zheng, C. Buisman and A. Ter Heijne, ACS Sustainable Tain. Chen. Eng., 2017, 5, 11346–11353 CrossRef CAS PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P.-G. de Arquer, A. Kiani, J. P. Edwards, P. De Luna and O. S. Bushuyev, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- Y. Kwon, Y. Lum, E. L. Clark, J. W. Ager and A. T. Bell, ChemElectroChem, 2016, 3, 1012–1019 CrossRef CAS.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- N. Hoshi, M. Kato and Y. Hori, J. Electroanal. Chem., 1997, 440, 283–286 CrossRef CAS.
- Q. Zhang, W. Xu, J. Xu, Y. Liu and J. Zhang, Catal. Today, 2018, 318, 15–22 CrossRef CAS.
- M. Li, S. Garg, X. Chang, L. Ge, L. Li, M. Konarova, T. E. Rufford, V. Rudolph and G. Wang, Small Methods, 2020, 4, 2000033 CrossRef CAS.
- W. Ye, X. Guo and T. Ma, Chem. Eng. J., 2021, 128825 CrossRef CAS.
- S. Lee, D. Kim and J. Lee, Angew. Chem., 2015, 127, 14914–14918 CrossRef.
- D. Cheng, Z.-J. Zhao, G. Zhang, P. Yang, L. Li, H. Gao, S. Liu, X. Chang, S. Chen, T. Wang, G. A. Ozin, Z. Liu and J. Gong, Nat. Commun., 2021, 12, 395 CrossRef CAS PubMed.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Nørskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- T. Burdyny, P. J. Graham, Y. Pang, C.-T. Dinh, M. Liu, E. H. Sargent and D. Sinton, ACS Sustainable Tain. Chen. Eng., 2017, 5, 4031–4040 CrossRef CAS.
- A. Klinkova, P. De Luna, C.-T. Dinh, O. Voznyy, E. M. Larin, E. Kumacheva and E. H. Sargent, ACS Catal., 2016, 6, 8115–8120 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan and C. Cao, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- T. Saberi Safaei, A. Mepham, X. Zheng, Y. Pang, C.-T. Dinh, M. Liu, D. Sinton, S. O. Kelley and E. H. Sargent, Nano Lett., 2016, 16, 7224–7228 CrossRef CAS PubMed.
- T. Kim and G. T.-R. Palmore, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- A. H. Anwer, N. Khan, M. D. Khan, M. Shahadat and M. Z. Khan, J. Environ. Chem. Eng., 2021, 106650 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang and H. Xie, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- J. Christophe, T. Doneux and C. Buess-Herman, Electrocatalysis, 2012, 3, 139–146 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J.-A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- Z. Huang, J. Gong and Z. Nie, Acc. Chem. Res., 2019, 52, 1125–1133 CrossRef CAS PubMed.
- H. Li, S. Zha, Z.-J. Zhao, H. Tian, S. Chen, Z. Gong, W. Cai, Y. Wang, Y. Cui, L. Zeng, R. Mu and J. Gong, ACS Catal., 2018, 8, 5526–5532 CrossRef CAS.
- A. Guan, C. Yang, Y. Quan, H. Shen, N. Cao, T. Li, Y. Ji and G. Zheng, Chem. – Asian J., 2019, 14, 3969–3980 CrossRef CAS PubMed.
- S. Sun, H. Cheng, X. Li, X. Wu, D. Zhen, Y. Wang, R. Jin and G. He, Ind. Eng. Chem. Res., 2021, 60, 1164–1174 CrossRef CAS.
- L. Hu, Y. Zhang and W. Han, New J. Chem., 2019, 43, 3269–3272 RSC.
- T. Wang, J. Yang, J. Chen, Q. He, Z. Li, L. Lei, J. Lu, M. K. Leung, B. Yang and Y. Hou, Chin. Chem. Lett., 2020, 31, 1438–1442 CrossRef CAS.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y. F. Han, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS PubMed.
- G. Zhen, X. Lu, T. Kobayashi, G. Kumar and K. Xu, Chem. Eng. J., 2016, 284, 1146–1155 CrossRef CAS.
- X. Li, S. Wang, L. Li, X. Zu, Y. Sun and Y. Xie, Acc. Chem. Res., 2020, 53, 2964–2974 CrossRef CAS PubMed.
- R. Geioushy, M. M. Khaled, K. Alhooshani, A. S. Hakeem and A. Rinaldi, Electrochim. Acta, 2017, 245, 456–462 CrossRef CAS.
- A. A. Ensafi, H. A. Alinajafi and B. Rezaei, J. Electroanal. Chem., 2016, 783, 82–89 CrossRef CAS.
- J. Yuan, M.-P. Yang, W.-Y. Zhi, H. Wang, H. Wang and J.-X. Lu, J. CO2 Util., 2019, 33, 452–460 CrossRef CAS.
- J. Yuan, W.-Y. Zhi, L. Liu, M.-P. Yang, H. Wang and J.-X. Lu, Electrochim. Acta, 2018, 282, 694–701 CrossRef CAS.
- P. Sekar, L. Calvillo, C. Tubaro, M. Baron, A. Pokle, F. Carraro, A. Martucci and S. Agnoli, ACS Catal., 2017, 7, 7695–7703 CrossRef CAS.
- Z. Cai, Y. Zhang, Y. Zhao, Y. Wu, W. Xu, X. Wen, Y. Zhong, Y. Zhang, W. Liu and H. Wang, Nano Res., 2019, 12, 345–349 CrossRef CAS.
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- K. Ye, Z. Zhou, J. Shao, L. Lin, D. Gao, N. Ta, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 4814–4821 CrossRef CAS PubMed.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- E. Nwanebu, S. Omanovic, S. Hrapovic, A. G. Vidales and B. Tartakovsky, Int. J. Hydrogen Energy, 2021, 47(1), 203–215 CrossRef.
- Z. Chen, K. Mou, S. Yao and L. Liu, J. Mater. Chem. A, 2018, 6, 11236–11243 RSC.
- C. Hu, R. Paul, Q. Dai and L. Dai, Chem. Soc. Rev., 2021, 50, 11785–11843 RSC.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- G. Zhang, Z. J. Zhao, D. Cheng, H. Li, J. Yu, Q. Wang, H. Gao, J. Guo, H. Wang, G. A. Ozin, T. Wang and J. Gong, Nat. Commun., 2021, 12, 5745 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- Z. Wang, G. Yang, Z. Zhang, M. Jin and Y. Yin, ACS Nano, 2016, 10, 4559–4564 CrossRef CAS PubMed.
- D. H. Won, H. Shin, J. Koh, J. Chung, H. S. Lee, H. Kim and S. I. Woo, Angew. Chem., 2016, 128, 9443–9446 CrossRef.
- Z. Li, Y. Yang, Z. Yin, X. Wei, H. Peng, K. Lyu, F. Wei, L. Xiao, G. Wang and H. D. Abruna, ACS Catal., 2021, 11, 2473–2482 CrossRef CAS.
- G. Dunfeng, W. Pengfei, L. Hefei, L. Long, W. Guoxiong and B. Xinhe, Acta Phys. Sin., 2021, 37(5), 2009021 Search PubMed.
- S. Verma, B. Kim, H. R.-M. Jhong, S. Ma and P. J. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- D. T. Whipple, E. C. Finke and P. J. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109 CrossRef CAS.
- Y. Yang and F. Li, Curr. Opin. Green Sustain., 2021, 27, 100419 CrossRef.
- C. M. Gabardo, C. P. O’Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li and Y. Wang, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- B. Endrodi, E. Kecsenovity, A. Samu, F. Darvas, R. Jones, V. Török, A. Danyi and C. Janáky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef CAS PubMed.
- X. Tan, C. Yu, Y. Ren, S. Cui, W. Li and J. Qiu, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- H. Chen, O. Simoska, K. Lim, M. Grattieri, M. Yuan, F. Dong, Y. S. Lee, K. Beaver, S. Weliwatte and E. M. Gaffney, Chem. Rev., 2020, 120, 12903–12993 CrossRef CAS PubMed.
- M. Miller, W. E. Robinson, A. R. Oliveira, N. Heidary, N. Kornienko, J. Warnan, I. A. Pereira and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 4601–4605 CrossRef CAS PubMed.
- C. Cadoux and R. D. Milton, ChemElectroChem, 2020, 7, 1974–1986 CrossRef CAS.
- D. Leech, P. Kavanagh and W. Schuhmann, Electrochim. Acta, 2012, 84, 223–234 CrossRef CAS.
- D. P. Hickey, E. M. Gaffney and S. D. Minteer, Top Curr. Chem. (Cham), 2018, 376, 43 CrossRef PubMed.
- X. Ji, Z. Su, P. Wang, G. Ma and S. Zhang, Small, 2016, 12, 4753–4762 CrossRef CAS PubMed.
- Y. Li, L. Wen, T. Tan and Y. Lv, Front. Bioeng. Biotechnol., 2019, 7, 394 CrossRef PubMed.
- S. Amanullah, P. Saha, A. Nayek, M. Ahmed and A. Dey, Chem. Soc. Rev., 2021, 50, 3755–3823 RSC.
- F. Mayer, F. Enzmann, A. M. Lopez and D. Holtmann, Bioresour. Technol., 2019, 289, 121706 CrossRef CAS PubMed.
- M. Schulman, D. Parker, L. G. Ljungdahl and H. G. Wood, J. Bacteriol. Parasitol, 1972, 109, 633–644 CrossRef CAS PubMed.
- M. Calvin and A. A. Benson, Science, 1948, 107, 476 CrossRef CAS PubMed.
- H. Huber, M. Gallenberger, U. Jahn, E. Eylert, I. A. Berg, D. Kockelkorn, W. Eisenreich and G. Fuchs, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7851–7856 CrossRef CAS PubMed.
- M. Evans, B. B. Buchanan and D. I. Arnon, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 928–934 CrossRef CAS PubMed.
- M. Hügler, H. Huber, K. O. Stetter and G. Fuchs, Arch. Microbiol., 2003, 179, 160–173 CrossRef PubMed.
- H. Holo, Arch. Microbiol., 1989, 151, 252–256 CrossRef CAS.
- T. Schwander, L. Schada von Borzyskowski, S. Burgener, N. S. Cortina and T. J. Erb, Science, 2016, 354, 900 CrossRef CAS PubMed.
- S. Luo, P. P. Lin, L.-Y. Nieh, G.-B. Liao, P.-W. Tang, C. Chen and J. C. Liao, Nat. Catal., 2022, 5, 154–162 CrossRef CAS.
- L. Xiao, G. Liu, F. Gong, H. Zhu, Y. Zhang, Z. Cai and Y. Li, ACS Catal., 2021, 12, 799–808 CrossRef.
- G. Hu, Z. Li, D. Ma, C. Ye, L. Zhang, C. Gao, L. Liu and X. Chen, Nat. Catal., 2021, 4, 395–406 CrossRef CAS.
- W. B. Whitaker, J. A. Jones, R. K. Bennett, J. E. Gonzalez, V. R. Vernacchio, S. M. Collins, M. A. Palmer, S. Schmidt, M. R. Antoniewicz and M. A. Koffas, Metab. Eng., 2017, 39, 49–59 CrossRef CAS PubMed.
- F. Guo, Z. Dai, W. Peng, S. Zhang, J. Zhou, J. Ma, W. Dong, F. Xin, W. Zhang and M. Jiang, Biotechnol. Bioeng., 2021, 118, 357–371 CrossRef CAS PubMed.
- N. J. Claassens, H. He and A. Bar-Even, Curr. Issues Mol. Biol., 2019, 33, 237–248 CrossRef PubMed.
- M. Kumar, S. Sundaram, E. Gnansounou, C. Larroche and I. S. Thakur, Bioresour. Technol., 2018, 247, 1059–1068 CrossRef CAS PubMed.
- M. Calvin and A. A. Benson, Science, 1949, 109, 140–142 CrossRef CAS PubMed.
- S. Gleizer, R. Ben-Nissan, Y. M. Bar-On, N. Antonovsky, E. Noor, Y. Zohar, G. Jona, E. Krieger, M. Shamshoum, A. Bar-Even and R. Milo, Cell, 2019, 179, 1255–1263 CrossRef CAS PubMed.
- J. Bang, C. H. Hwang, J. H. Ahn, J. A. Lee and S. Y. Lee, Nat. Microbiol., 2020, 5, 1459–1463 CrossRef CAS PubMed.
- R. Ganigué, S. Puig, P. Batlle-Vilanova, M. D. Balaguer and J. Colprim, Chem. Commun., 2015, 51, 3235–3238 RSC.
- T. Krieg, A. Sydow, S. Faust, I. Huth and D. Holtmann, Angew. Chem., Int. Ed. Engl., 2018, 57, 1879–1882 CrossRef CAS PubMed.
- J. Yang, S. Sun, Y. Men, Y. Zeng, Y. Zhu, Y. Sun and Y. Ma, Catal. Sci. Technol., 2017, 7, 3459–3463 RSC.
- S. Sundaram, C. Diehl, N. S. Cortina, J. Bamberger, N. Paczia and T. J. Erb, Angew. Chem., Int. Ed. Engl., 2021, 60, 16420–16425 CrossRef CAS PubMed.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- H. Wu, H. Pan, Z. Li, T. Liu, F. Liu, S. Xiu, J. Wang, H. Wang, Y. Hou and B. Yang, Chem. Eng. J., 2022, 430, 132943 CrossRef CAS.
- S. Bolognesi, L. Bañeras, E. Perona-Vico, A. G. Capodaglio, M. D. Balaguer and S. Puig, Sustain. Energy Fuels, 2022, 6, 150–161 RSC.
- A. L. Ghindilis, P. Atanasov and E. Wilkins, Electroanalysis, 1997, 9, 661–674 CrossRef CAS.
- M. Yuan, S. Sahin, R. Cai, S. Abdellaoui, D. P. Hickey, S. D. Minteer and R. D. Milton, Angew. Chem., 2018, 130, 6692–6696 CrossRef.
- O. Bretschger, K. Carpenter, T. Phan, S. Suzuki, S. I. Ishii, E. Grossi-Soyster, M. Flynn and J. Hogan, Bioresour. Technol, 2015, 195, 254–264 CrossRef CAS PubMed.
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef CAS PubMed.
- H. M. Woo, Curr. Opin. Biotechnol, 2017, 45, 1–7 CrossRef CAS PubMed.
- H. Li, P. H. Opgenorth, D. G. Wernick, S. Rogers, T.-Y. Wu, W. Higashide, P. Malati, Y.-X. Huo, K. M. Cho and J. C. Liao, Science, 2012, 335, 1596 CrossRef CAS PubMed.
- J. A. Cornejo, H. Sheng, E. Edri, C. Ajo-Franklin and H. Frei, Nat. Commun., 2018, 9, 2263 CrossRef PubMed.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32 CrossRef CAS.
- R. M. Rodrigues, X. Guan, J. A. Iñiguez, D. A. Estabrook, J. O. Chapman, S. Huang, E. M. Sletten and C. Liu, Nat. Catal., 2019, 2, 407–414 CrossRef CAS.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
- J. Guo, M. Suástegui, K. K. Sakimoto, V. M. Moody, G. Xiao, D. G. Nocera and N. S. Joshi, Science, 2018, 362, 813–816 CrossRef CAS PubMed.
- A. Ruff, F. Conzuelo and W. Schuhmann, Nat. Catal., 2020, 3, 214–224 CrossRef CAS.
- L. Xu, Y. Xiu, F. Liu, Y. Liang and S. Wang, Molecules, 2020, 25, 3653 CrossRef CAS PubMed.
- R. E. Sharp and S. K. Chapman, Biochim. Biophys. Acta, 1999, 1432, 143–158 CrossRef CAS.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
- J. T. Holland, C. Lau, S. Brozik, P. Atanassov and S. Banta, J. Am. Chem. Soc., 2011, 133, 19262–19265 CrossRef CAS PubMed.
- K. Xiang, Y. Qiao, C. B. Ching and C. M. Li, Electrochem. Commun., 2009, 11, 1593–1595 CrossRef CAS.
- N.-M. D. Courchesne, E. P. DeBenedictis, J. Tresback, J. J. Kim, A. Duraj-Thatte, D. Zanuy, S. Keten and N. S. Joshi, Nanotechnology, 2018, 29, 454002 CrossRef PubMed.
- P. Q. Nguyen, Z. Botyanszki, P. K.-R. Tay and N. S. Joshi, Nat. Commun., 2014, 5, 1–10 Search PubMed.
- D. R. Lovley and J. Yao, Trends Biotechnol., 2021, 39, 940–952 CrossRef CAS PubMed.
- F. Wang, Y. Gu, J. P. O’Brien, M. Y. Sophia, S. E. Yalcin, V. Srikanth, C. Shen, D. Vu, N. L. Ing and A. I. Hochbaum, Cell, 2019, 177, 361–369.e310 CrossRef CAS PubMed.
- S. E. Yalcin, J. P. O’Brien, Y. Gu, K. Reiss, S. M. Yi, R. Jain, V. Srikanth, P. J. Dahl, W. Huynh, D. Vu, A. Acharya, S. Chaudhuri, T. Varga, V. S. Batista and N. S. Malvankar, Nat. Chem. Biol., 2020, 1136–1142 CrossRef CAS PubMed Medium: ED; Size.
- F. J. Meysman, R. Cornelissen, S. Trashin, R. Bonné, S. H. Martinez, J. van der Veen, C. J. Blom, C. Karman, J.-L. Hou and R. T. Eachambadi, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- K. U. Kjeldsen, L. Schreiber, C. A. Thorup, T. Boesen, J. T. Bjerg, T. Yang, M. S. Dueholm, S. Larsen, N. Risgaard-Petersen and M. Nierychlo, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19116–19125 CrossRef CAS PubMed.
- M. J. Edwards, G. F. White, J. N. Butt, D. J. Richardson and T. A. Clarke, Cell, 2020, 181, 665–673.e610 CrossRef CAS PubMed.
- J. Zhao, F. Li, Y. Cao, X. Zhang, T. Chen, H. Song and Z. Wang, Biotechnol. Adv., 2020, 107682 Search PubMed.
- M. A. Thirumurthy and A. K. Jones, Nanotechnology, 2020, 31, 124001 CrossRef CAS PubMed.
- E. M. Nichols, J. J. Gallagher, C. Liu, Y. Su, J. Resasco, Y. Yu, Y. Sun, P. Yang, M. C.-Y. Chang and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11461–11466 CrossRef CAS PubMed.
- F. Li, Y.-X. Li, Y.-X. Cao, L. Wang, C.-G. Liu, L. Shi and H. Song, Nat. Commun., 2018, 9, 1–13 CrossRef PubMed.
- Y. Yang, Y. Ding, Y. Hu, B. Cao, S. A. Rice, S. Kjelleberg and H. Song, ACS Synth. Biol., 2015, 4, 815–823 CrossRef CAS PubMed.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- S. Zhao, N. Austin, M. Li, Y. Song, S. D. House, S. Bernhard, J. C. Yang, G. Mpourmpakis and R. Jin, ACS Catal., 2018, 8, 4996–5001 CrossRef CAS.
- S. Chu, S. Fan, Y. Wang, D. Rossouw, Y. Wang, G. A. Botton and Z. Mi, Angew. Chem., Int. Ed., 2016, 55, 14262–14266 CrossRef CAS PubMed.
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- D. Gao, H. Zhou, F. Cai, D. Wang, Y. Hu, B. Jiang, W.-B. Cai, X. Chen, R. Si and F. Yang, Nano Res., 2017, 10, 2181–2191 CrossRef CAS.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- B. Qin, H. Wang, F. Peng, H. Yu and Y. Cao, J. CO2 Util., 2017, 21, 219–223 CrossRef CAS.
- R. Kas, K. K. Hummadi, R. Kortlever, P. De Wit, A. Milbrat, M. W. Luiten-Olieman, N. E. Benes, M. T. Koper and G. Mul, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- R. Daiyan, X. Lu, Y. H. Ng and R. Amal, ChemistrySelect, 2017, 2, 879–884 CrossRef CAS.
- D. Raciti, Y. Wang, J. H. Park and C. Wang, ACS Appl. Energy Mater., 2018, 1, 2392–2398 CrossRef CAS.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS.
- K. Fan, Y. Jia, Y. Ji, P. Kuang, B. Zhu, X. Liu and J. Yu, ACS Catal., 2019, 10, 358–364 CrossRef.
- P. Lu, D. Gao, H. He, Q. Wang, Z. Liu, S. Dipazir, M. Yuan, W. Zu and G. Zhang, Nanoscale, 2019, 11, 7805–7812 RSC.
- J. Qu, X. Zhang, Y. Wang and C. Xie, Electrochim. Acta, 2005, 50, 3576–3580 CrossRef CAS.
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu and F. Yang, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- L.-P. Yuan, W.-J. Jiang, X.-L. Liu, Y.-H. He, C. He, T. Tang, J. Zhang and J.-S. Hu, ACS Catal., 2020, 10, 13227–13235 CrossRef CAS.
- J. Wang, G. Wang, J. Zhang, Y. Wang, H. Wu, X. Zheng, J. Ding, X. Han, Y. Deng and W. Hu, Angew. Chem., Int. Ed., 2021, 60, 7602–7606 CrossRef CAS PubMed.
- D. Wu, G. Huo, W. Chen, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2020, 271, 118957 CrossRef CAS.
- R. Kortlever, I. Peters, S. Koper and M. T. Koper, ACS Catal., 2015, 5, 3916–3923 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- Y. Wang, L. Cao, N. J. Libretto, X. Li, C. Li, Y. Wan, C. He, J. Lee, J. Gregg and H. Zong, J. Am. Chem. Soc., 2019, 141, 16635–16642 CrossRef CAS PubMed.
- R. Lin, X. Ma, W.-C. Cheong, C. Zhang, W. Zhu, J. Pei, K. Zhang, B. Wang, S. Liang and Y. Liu, Nano Res., 2019, 12, 2866–2871 CrossRef CAS.
- J. Fu, W. Zhu, Y. Chen, Z. Yin, Y. Li, J. Liu, H. Zhang, J. J. Zhu and S. Sun, Angew. Chem., 2019, 131, 14238–14241 CrossRef.
- X. Guo, Y. Zhang, C. Deng, X. Li, Y. Xue, Y.-M. Yan and K. Sun, Chem. Commun., 2015, 51, 1345–1348 RSC.
- S. Sarfraz, A. T. Garcia-Esparza, A. Jedidi, L. Cavallo and K. Takanabe, ACS Catal., 2016, 6, 2842–2851 CrossRef CAS.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato and R. Haasch, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- Y. J. Jang, J.-W. Jang, J. Lee, J. H. Kim, H. Kumagai, J. Lee, T. Minegishi, J. Kubota, K. Domen and J. S. Lee, Energy Environ. Sci., 2015, 8, 3597–3604 RSC.
- T. T. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
- C. Zhao, Z. Yin and J. Wang, ChemElectroChem, 2015, 2, 1974–1982 CrossRef CAS.
- Y. Cheng, S. Zhao, B. Johannessen, J. P. Veder, M. Saunders, M. R. Rowles, M. Cheng, C. Liu, M. F. Chisholm and R. De Marco, Adv. Mater., 2018, 30, 1706287 CrossRef PubMed.
- S. Huo, Z. Weng, Z. Wu, Y. Zhong, Y. Wu, J. Fang and H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 28519–28526 CrossRef CAS PubMed.
- T. N. Huan, P. Prakash, P. Simon, G. Rousse, X. Xu, V. Artero, E. Gravel, E. Doris and M. Fontecave, ChemSusChem, 2016, 9, 2317–2320 CrossRef CAS PubMed.
- X. Lu, T. H. Tan, Y. H. Ng and R. Amal, Chem. – Eur. J., 2016, 22, 11991–11996 CrossRef CAS PubMed.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X. D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 CrossRef CAS PubMed.
- X. Cui, Z. Pan, L. Zhang, H. Peng and G. Zheng, Adv. Energy Mater., 2017, 7, 1701456 CrossRef.
- F. Pan, H. Zhao, W. Deng, X. Feng and Y. Li, Electrochim. Acta, 2018, 273, 154–161 CrossRef CAS.
- W. Zheng, C. Guo, J. Yang, F. He, B. Yang, Z. Li, L. Lei, J. Xiao, G. Wu and Y. Hou, Carbon, 2019, 150, 52–59 CrossRef CAS.
- B. Qin, Y. Li, H. Wang, G. Yang, Y. Cao, H. Yu, Q. Zhang, H. Liang and F. Peng, Nano Energy, 2019, 60, 43–51 CrossRef CAS.
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren and Z. Kuang, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- H. R.-M. Jhong, C. E. Tornow, C. Kim, S. Verma, J. L. Oberst, P. S. Anderson, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. Kenis, ChemPhysChem, 2017, 18, 3274–3279 CrossRef CAS PubMed.
- M. Zhu, J. Chen, R. Guo, J. Xu, X. Fang and Y.-F. Han, Appl. Catal., B, 2019, 251, 112–118 CrossRef CAS.
- H. Wang, J. Jia, P. Song, Q. Wang, D. Li, S. Min, C. Qian, L. Wang, Y. F. Li and C. Ma, Angew. Chem., Int. Ed., 2017, 56, 7847–7852 CrossRef CAS PubMed.
- P. Su, K. Iwase, S. Nakanishi, K. Hashimoto and K. Kamiya, Small, 2016, 12, 6083–6089 CrossRef CAS PubMed.
- A. S. Varela, M. Kroschel, N. D. Leonard, W. Ju, J. Steinberg, A. Bagger, J. Rossmeisl and P. Strasser, ACS Energy Lett., 2018, 3, 812–817 CrossRef CAS.
- Q. Li, W. Zhu, J. Fu, H. Zhang, G. Wu and S. Sun, Nano Energy, 2016, 24, 1–9 CrossRef CAS.
- L. Zeng, J. Shi, J. Luo and H. Chen, J. Power Sources, 2018, 398, 83–90 CrossRef CAS.
- Z. Zhang, F. Ahmad, W. Zhao, W. Yan, W. Zhang, H. Huang, C. Ma and J. Zeng, Nano Lett., 2019, 19, 4029–4034 CrossRef CAS PubMed.
- J. Huang, X. Guo, G. Yue, Q. Hu and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 44403–44414 CrossRef CAS PubMed.
- B. Zhang, Z. Guo, Z. Zuo, W. Pan and J. Zhang, Appl. Catal., B, 2018, 239, 441–449 CrossRef CAS.
- J. Wang, X. Huang, S. Xi, J. M. Lee, C. Wang, Y. Du and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 13532–13539 CrossRef CAS PubMed.
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. MacFarlane, G. G. Wallace and D. L. Officer, ACS Energy Lett., 2019, 4, 666–672 CrossRef CAS.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang and J. Lu, Angew. Chem., 2019, 131, 15013–15018 CrossRef.
- J. Choi, P. Wagner, R. Jalili, J. Kim, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Adv. Energy Mater., 2018, 8, 1801280 CrossRef.
- W. Bi, X. Li, R. You, M. Chen, R. Yuan, W. Huang, X. Wu, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2018, 30, 1706617 CrossRef PubMed.
- J. Wang, L. Gan, Q. Zhang, V. Reddu, Y. Peng, Z. Liu, X. Xia, C. Wang and X. Wang, Adv. Energy Mater., 2019, 9, 1803151 CrossRef.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie and A. S. Jalilov, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu and C. Wu, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- C. Rogers, W. S. Perkins, G. Veber, T. E. Williams, R. R. Cloke and F. R. Fischer, J. Am. Chem. Soc., 2017, 139, 4052–4061 CrossRef CAS PubMed.
- J. Choi, J. Kim, P. Wagner, S. Gambhir, R. Jalili, S. Byun, S. Sayyar, Y. M. Lee, D. R. MacFarlane and G. G. Wallace, Energy Environ. Sci., 2019, 12, 747–755 RSC.
- K. Zhao, Y. Liu, X. Quan, S. Chen and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 5302–5311 CrossRef CAS PubMed.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh and R. Vajtai, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong and Y. Wang, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- Q. Feng, S. Liu, X. Wang and G. Jin, Appl. Surf. Sci., 2012, 258, 5005–5009 CrossRef CAS.
- H. W. Shafaque, C. Lee, K. F. Fahy, J. K. Lee, J. M. LaManna, E. Baltic, D. S. Hussey, D. L. Jacobson and A. Bazylak, ACS Appl. Mater. Interfaces, 2020, 12, 54585–54595 CrossRef CAS PubMed.
- B. Endrodi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Torok, A. Danyi and C. Janaky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef CAS PubMed.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and P. Berlinguette Curtis, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- D. Kim, W. Choi, H. W. Lee, S. Y. Lee, Y. Choi, D. K. Lee, W. Kim, J. Na, U. Lee, Y. J. Hwang and D. H. Won, ACS Energy Lett., 2021, 6, 3488–3495 CrossRef CAS.
- G. O. Larrazabal, P. Strom-Hansen, J. P. Heli, K. Zeiter, K. T. Therkildsen, I. Chorkendorff and B. Seger, ACS Appl. Mater. Interfaces, 2019, 11, 41281–41288 CrossRef CAS PubMed.
- L. Li, A. Ozden, S. Guo, A. D.-A. F.-P. Garci, C. Wang, M. Zhang, J. Zhang, H. Jiang, W. Wang, H. Dong, D. Sinton, E. H. Sargent and M. Zhong, Nat. Commun., 2021, 12, 5223 CrossRef CAS PubMed.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- Y. Xu, F. Li, A. Xu, J. P. Edwards, S. F. Hung, C. M. Gabardo, C. P. O'Brien, S. Liu, X. Wang, Y. Li, J. Wicks, R. K. Miao, Y. Liu, J. Li, J. E. Huang, J. Abed, Y. Wang, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2932 CrossRef CAS PubMed.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2019, 3, 75–82 CrossRef.
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O’Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- A. Ozden, F. Li, F. P. García de Arquer, A. Rosas-Hernández, A. Thevenon, Y. Wang, S.-F. Hung, X. Wang, B. Chen, J. Li, J. Wicks, M. Luo, Z. Wang, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, ACS Energy Lett., 2020, 5, 2811–2818 CrossRef CAS.
- J. Li, A. Ozden, M. Wan, Y. Hu, F. Li, Y. Wang, R. R. Zamani, D. Ren, Z. Wang, Y. Xu, D. H. Nam, J. Wicks, B. Chen, X. Wang, M. Luo, M. Graetzel, F. Che, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2808 CrossRef CAS PubMed.
- S. Jin, Y. Jeon, M. S. Jeon, J. Shin, Y. Song, S. Kang, J. Bae, S. Cho, J. K. Lee, D. R. Kim and B. K. Cho, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020552118 CrossRef CAS PubMed.
- H. Luo, J. Qi, M. Zhou, G. Liu, Y. Lu, R. Zhang and C. Zeng, Bioresour. Technol., 2020, 309, 123322 CrossRef CAS PubMed.
- F. Ammam, P.-L. Tremblay, D. M. Lizak and T. Zhang, Biotechnol. Biofuels, 2016, 9, 163 CrossRef PubMed.
- K. K. Sakimoto, A. B. Wong and P. Yang, Science, 2016, 351, 74–77 CrossRef CAS PubMed.
- H. Liu, T. Song, K. Fei, H. Wang and J. Xie, Bioresour. Bioprocess., 2018, 5, 7 CrossRef.
- Y. Hirokawa, Y. Maki, T. Tatsuke and T. Hanai, Metab. Eng., 2016, 34, 97–103 CrossRef CAS PubMed.
- B. Wang, S. Pugh, D. R. Nielsen, W. Zhang and D. R. Meldrum, Metab. Eng., 2013, 16, 68–77 CrossRef CAS PubMed.
- K. P. Nevin, S. A. Hensley, A. E. Franks, Z. M. Summers, J. Ou, T. L. Woodard, O. L. Snoeyenbos-West and D. R. Lovley, Appl. Environ. Microbiol., 2011, 77, 2882–2886 CrossRef CAS PubMed.
- H. Li, P. H. Opgenorth, D. G. Wernick, S. Rogers, T. Y. Wu, W. Higashide, P. Malati, Y. X. Huo, K. M. Cho and J. C. Liao, Science, 2012, 335, 1596 CrossRef CAS PubMed.
- I. Vassilev, P. A. Hernandez, P. Batlle-Vilanova, S. Freguia, J. O. Krömer, J. R. Keller, P. Ledezma and B. Virdis, ACS Sustainable Tain. Chen. Eng., 2018, 6, 8485–8493 CrossRef CAS.
- S. Srikanth, D. Singh, K. Vanbroekhoven, D. Pant, M. Kumar, S. K. Puri and S. S.-V. Ramakumar, Bioresour. Technol., 2018, 265, 45–51 CrossRef CAS PubMed.
- I. S. Yunus, J. Wichmann, R. Wordenweber, K. J. Lauersen, O. Kruse and P. R. Jones, Metab. Eng., 2018, 49, 201–211 CrossRef CAS PubMed.
- R. K. Yadav, J.-O. Baeg, G. H. Oh, N.-J. Park, K.-J. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455–11461 CrossRef CAS PubMed.
- S. Yu, P. Lv, P. Xue, K. Wang, Q. Yang, J. Zhou, M. Wang, L. Wang, B. Chen and T. Tan, Chem. Eng. J., 2021, 420, 127649 CrossRef CAS.
- S. Srikanth, M. Maesen, X. Dominguez-Benetton, K. Vanbroekhoven and D. Pant, Bioresour. Technol., 2014, 165, 350–354 CrossRef CAS PubMed.
- J. Luo, A. S. Meyer, R. V. Mateiu and M. Pinelo, N. Biotechnol., 2015, 32, 319–327 CrossRef CAS PubMed.
- H.-J. Jo, J.-H. Kim, Y.-N. Kim, P.-W. Seo, C.-Y. Kim, J.-W. Kim, H.-N. Yu, H. Cheon, E. Y. Lee, J.-S. Kim and J.-B. Park, Green Chem., 2022, 24, 218–226 RSC.
- J. R. Phillips, H. K. Atiyeh, R. S. Tanner, J. R. Torres, J. Saxena, M. R. Wilkins and R. L. Huhnke, Bioresour. Technol., 2015, 190, 114–121 CrossRef CAS PubMed.
- M. Kopke, C. Mihalcea, F. Liew, J. H. Tizard, M. S. Ali, J. J. Conolly, B. Al-Sinawi and S. D. Simpson, Appl. Environ. Microbiol., 2011, 77, 5467–5475 CrossRef CAS PubMed.
- C. K. Lim, J. C. Villada, A. Chalifour, M. F. Duran, H. Lu and P. K.-H. Lee, Front. Microbiol., 2019, 10, 1027 CrossRef PubMed.
- W. F. Liang, L. Y. Cui, J. Y. Cui, K. W. Yu, S. Yang, T. M. Wang, C. G. Guan, C. Zhang and X. H. Xing, Metab. Eng., 2017, 39, 159–168 CrossRef CAS PubMed.
- T. Schwander, L. Schada von Borzyskowski, S. Burgener, N. S. Cortina and T. J. Erb, Science, 2016, 354, 900–904 CrossRef CAS PubMed.
- T. Cai, H. Sun, J. Qiao, L. Zhu, F. Zhang, J. Zhang, Z. Tang, X. Wei, J. Yang, Q. Yuan, W. Wang, X. Yang, H. Chu, Q. Wang, C. You, H. Ma, Y. Sun, Y. Li, C. Li, H. Jiang, Q. Wang and Y. Ma, Science, 2021, 373, 1523–1527 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |