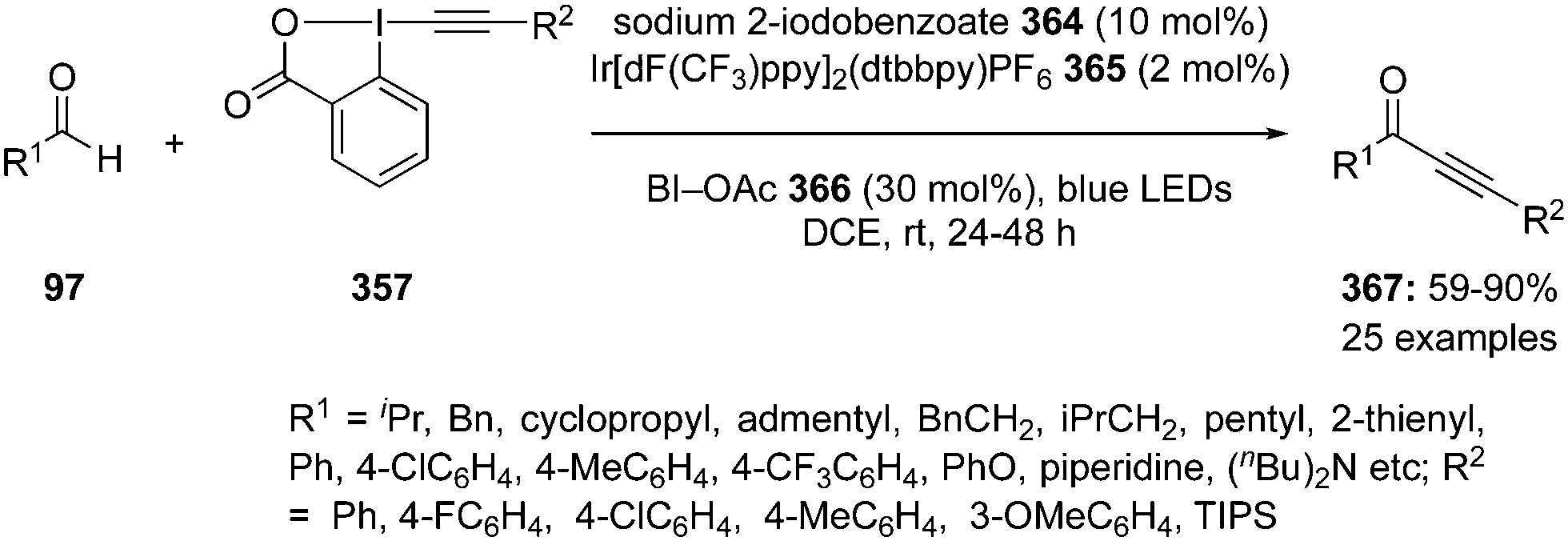DOI:
10.1039/D2CS00206J
(Review Article)
Chem. Soc. Rev., 2022,
51, 8102-8139
Progress in organocatalysis with hypervalent iodine catalysts
Received
26th June 2022
First published on 5th September 2022
Abstract
Hypervalent iodine compounds as environmentally friendly and relatively inexpensive reagents have properties similar to transition metals. They are employed as alternatives to transition metal catalysts in organic synthesis as mild, nontoxic, selective and recyclable catalytic reagents. Formation of C–N, C–O, C–S, C–F and C–C bonds can be seamlessly accomplished by hypervalent iodine catalysed oxidative functionalisations. The aim of this review is to highlight recent developments in the utilisation of iodine(III) and iodine(V) catalysts in the synthesis of a wide range of organic compounds including chiral catalysts for stereoselective synthesis. Polymer-, magnetic nanoparticle- and metal organic framework-supported hypervalent iodine catalysts are also described.

Fateh V. Singh
| Fateh V. Singh was born in Ravani Katiry, Bulandshahr, UP, India in 1976. He completed his Masters in Chemistry at SSV College, Hapur, UP, India in 1998. He received his PhD in 2007 under the supervision of Dr Atul Goel, CSIR-CDRI, Lucknow, India. After the completion of his doctoral studies, he was in Prof. H. A. Stefani's research group at USP, São Paulo, Brazil for more than two years. In 2010, he joined as a Marie Curie postdoctoral fellow with Prof. Thomas Wirth at Cardiff University, UK and learned various new reactions regarding organoselenium and hypervalent iodine chemistry. He subsequently stayed with Prof. G. Mugesh at IISc Bangalore, India for more than one year. In 2014, he started his independent career and joined VIT Chennai as an Assistant Professor. His research group is interested in new organoselenium and hypervalent iodine catalysts for organic synthesis. |

Samata E. Shetgaonkar
| Samata E. Shetgaonkar was born in Morjim, Pernem, Goa, India in 1992. After completing her MSc in Chemistry at Goa University, Goa, India, in 2015, she completed her PhD at VIT Chennai under the supervision of Dr Fateh V. Singh. She has published more than 15 research papers during her doctoral research studies. Her research interest mainly involves the synthesis of novel hypervalent reagents and their application in organic synthesis. |

Manjula Krishnan
| Manjula Krishnan obtained her MSc degree in Chemistry (2007) from Bangalore University and MPhil in Chemistry (2009) from Cochin University of Science and Technology. In 2011, she joined Sree Sankara College, Kalady affiliated to Mahatma Gandhi University (Kottayam, India) as an Assistant Professor. She is currently working as an Internal Full Time (IFT) research scholar to pursue her PhD at VIT Chennai under the supervision of Dr Fateh V. Singh. Her research focus is mainly associated with application of hypervalent iodine reagents in organic synthesis. |

Thomas Wirth
| Thomas Wirth is a professor of organic chemistry at Cardiff University. He received his PhD after studying chemistry at Bonn and the Technical University of Berlin. After a postdoctoral stay at Kyoto University as a JSPS fellow, he worked independently at the University of Basel before taking up his current position at Cardiff University in 2000. He has been invited as a visiting professor to a number of places such as Toronto, Tokyo, Osaka and Kyoto. He was awarded the Werner Prize from the New Swiss Chemical Society, the Furusato award from JSPS London, the Wolfson Research Merit Award from the Royal Society and the Bader Award from the Royal Society of Chemistry and was elected as a Fellow of The Learned Society of Wales. His main interests of research concern stereoselective electrophilic reactions, oxidative transformations with hypervalent iodine reagents and flow chemistry performed in microreactors. |
1. Introduction
The major challenge of synthetic organic chemistry in the 21st century is the selective synthesis of target compounds in an efficient and economical way using mild reaction conditions. The most striking approach in environmentally benign reactions is the development of catalytic strategies in the synthesis of organic molecules. Over the past few decades, hypervalent iodine reagents have emerged as efficient organocatalysts for the oxidative transformations of a wide range of organic substrates.1–5 These reagents are mild, non-toxic, moisture resistant, inexpensive and often recyclable. These properties make them ideal eco-friendly reagents to be employed for various organic transformations.6,7 Several reviews,6,8–22 book chapters23–27 and books28–31 have been published in the past years emphasising the progress and development of hypervalent iodine chemistry.
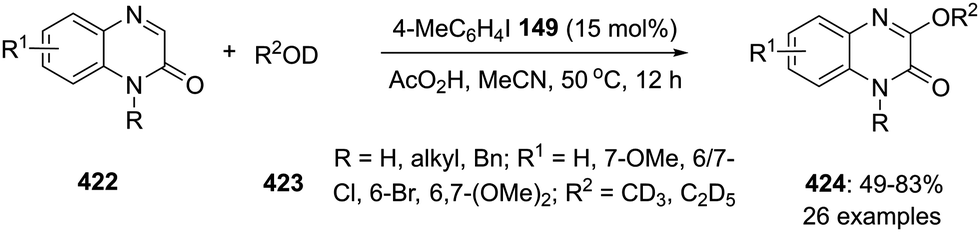
Prominent features of hypervalent iodine compounds are their oxidising properties and their electrophilic nature. They are commonly used as stoichiometric oxidants which makes them attractive candidates for the replacement of toxic heavy-metal oxidants.6 Moreover, hypervalent iodine reagents have been extensively used in the total synthesis of natural products and their intermediates.32 Representative examples of various hypervalent iodine reagents are shown in Fig. 1. For example, (diacetoxyiodo)benzene 1 and [bis(trifluoroacetoxy)iodo]benzene 2 are used as efficient oxidants in many organic transformations such as oxidation of alcohols, alkenes or organosulfides,33,34 rearrangements,35 cyclisations36,37 and transition metal-catalysed C–H bond functionalisations38,39 and alkene difunctionalisations.40,41
 |
| | Fig. 1 Examples of hypervalent iodine(III)/(V) reagents 1–14. | |
(Dichloroiodo)benzene 3 is a chlorinating reagent7 while [hydroxy(tosyloxy)iodo]benzene 4 (Koser's reagent) can be used for α-oxytosylations of ketones.42–44 Owing to the electrophilic and excellent leaving nature of diaryliodonium salts 5, they are employed as versatile arylating agents in coupling reactions by reacting with suitable nucleophile.45 Cyclic hypervalent iodine(III) reagents such as 2-iodosobenzoic acid 6 (IBA) is synthesised by the oxidation of 2-iodobenzoic acid and less explored due to its poor reactivity.46 Trifluoromethyl benziodoxolone 7 was developed by Togni's research group as trifluoromethylation reagent for the transfer of CF3 moiety to the organic molecules.47,48 Zhdankin and coworkers reported the synthesis of 1-[(triisopropylsilyl)ethynyl]-1λ3,2-benziodoxol-3(1H) TIPS-EBX 8 as an efficient alkyne transfer reagent to various substrates.49 Later on Waser and coworkers published a review article in which they have compiled the application of other benziodoxole-based reagents.50
Chiral hypervalent iodine reagents in stereoselective synthesis have made an impressive developments in recent times.12 The first chiral reagent was prepared by Pribram51 in 1907 followed by many more optically active iodine(III)/(V) compounds which have been employed in asymmetric transformations.12 For example, Ishihara and his team reported the preparation of lactate-based C2-symmetric chiral iodine(III) reagents 9a–b and their use in enantioselective spirolactonisations of 1-naphthol derivatives with high selectivities.52 Furthermore, pseudocyclic hypervalent iodine compounds is another interesting class containing additional non-covalent coordination at the iodine center.53
Iodine(V) reagents such as 2-iodoxybenzoic acid (IBX) 10a and FIBX 10b, 2-iodoxybenzenesulfonic acid (IBS) 11 and Dess–Martin periodinane (DMP) 12 are routinely used as oxidising agents in a variety of oxidative transformations including oxidation of alcohol moieties and other functional groups.54,55 Recently, Wirth and his team synthesised pseudocyclic iodine(III) reagents containing furan and thiophene units 13a,b and proved their oxidising nature in various oxidative transformations.56 Very recently, Zhdankin and co-workers reported the synthesis of a powerful iodine(V) oxidant, 2-iodoxybenzoic acid bistriflate 14, by reacting IBX with trifluoromethanesulfonic acid and illustrated its potential application in the direct oxidation of hydrocarbons.57
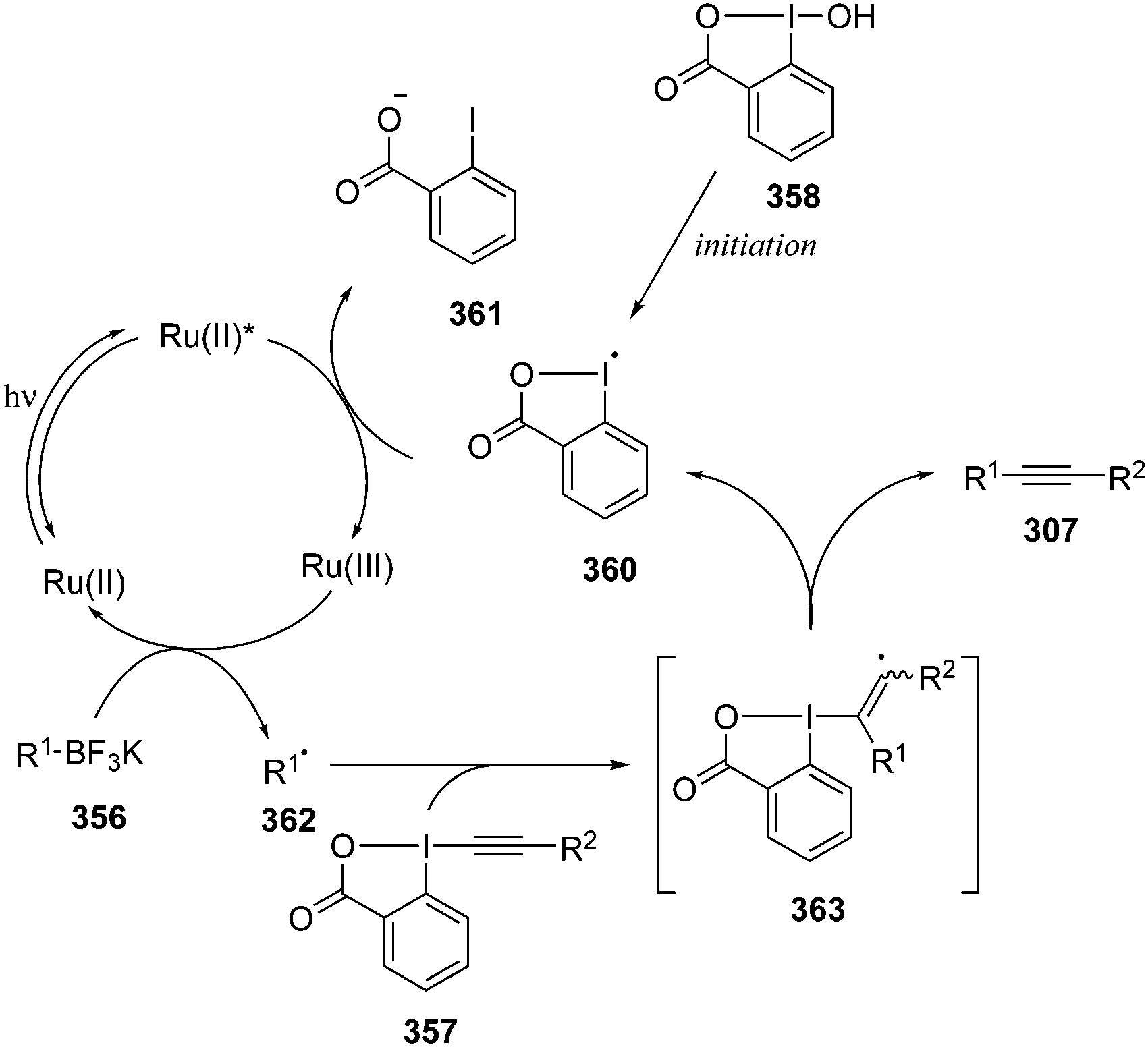
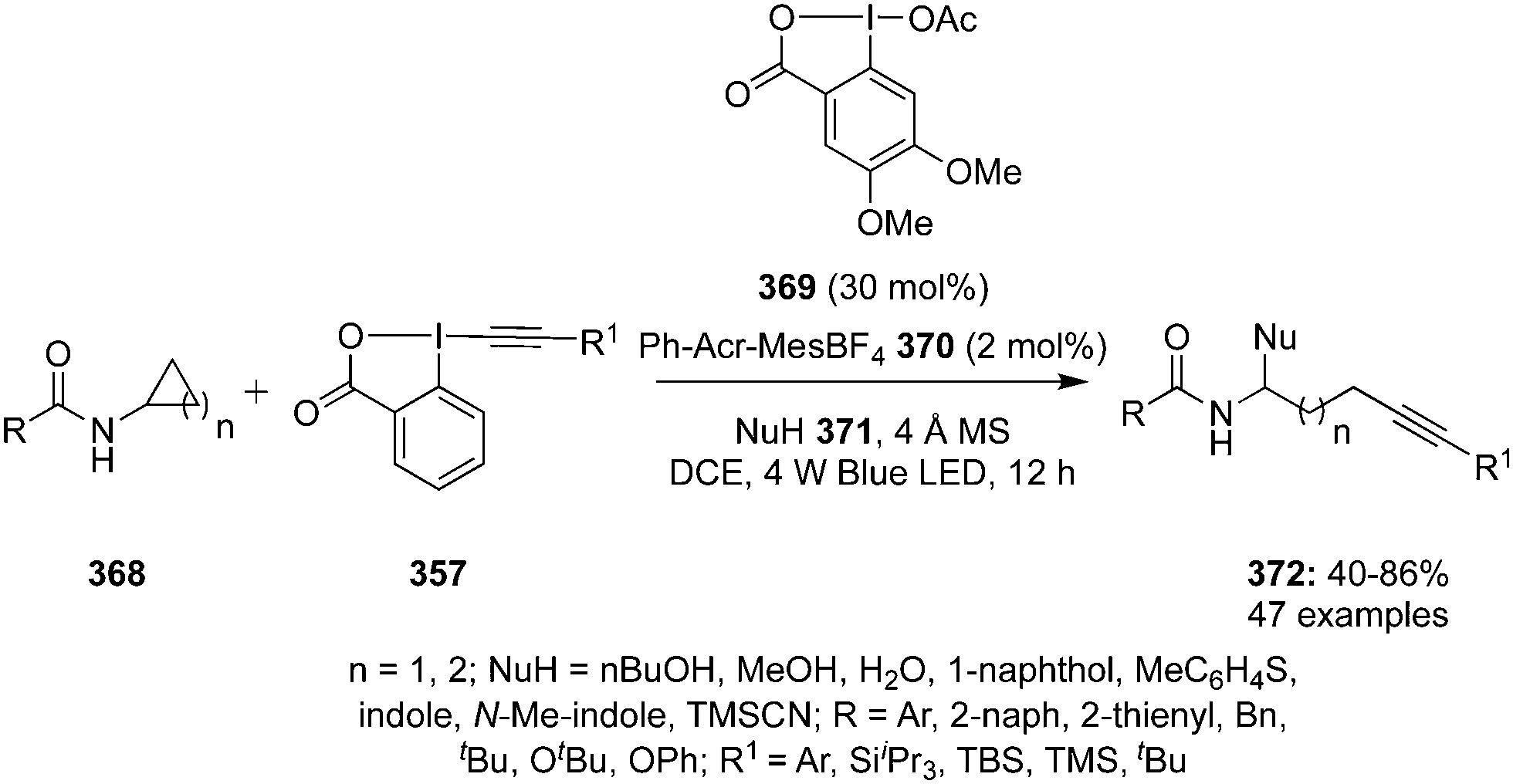
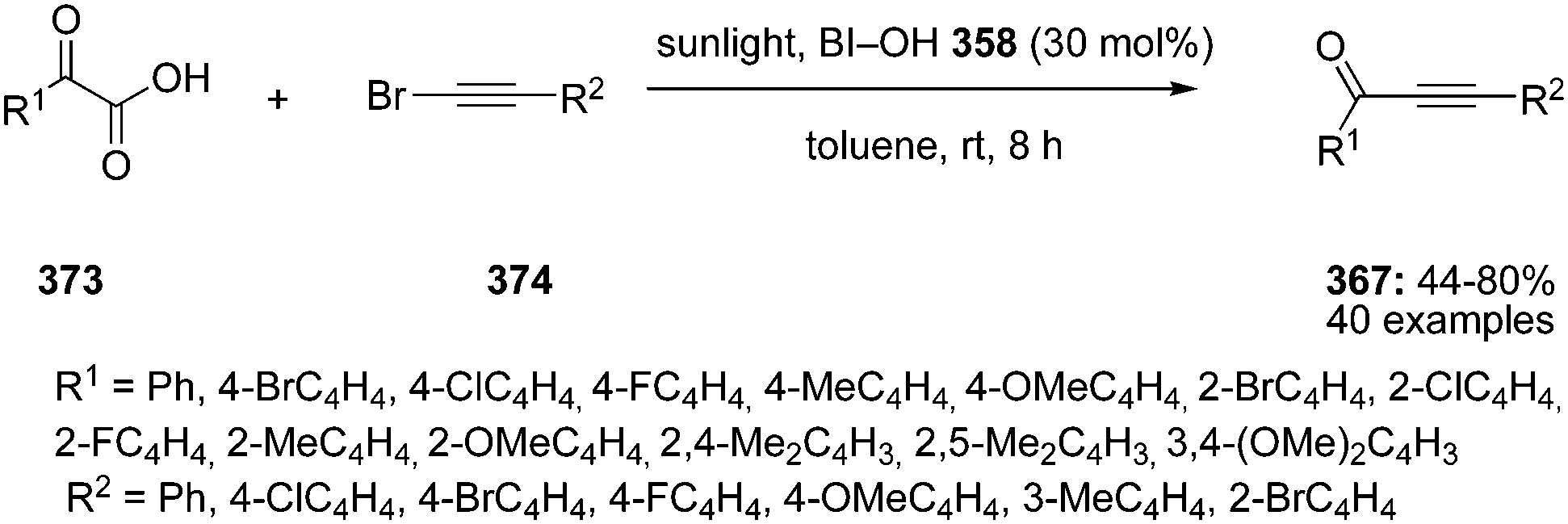
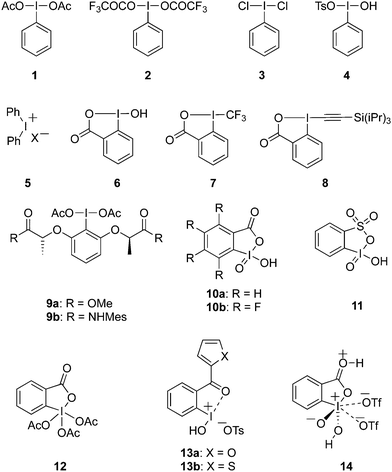
Hypervalent iodine reagents have properties resembling those of transition metals and can be employed as environmentally sustainable alternatives to transition metal catalysts such as mercury, lead and thallium reagents.58,59 Within this context, copious synthetic procedures have been developed using achiral or chiral iodine(III)/(V) pre-catalysts in the presence of stoichiometric oxidants such as mCPBA, oxone, peracetic acid and molecular O2, etc. which play a significant role in the in situ generation of active catalytic species such as hypoiodite, trivalent, or pentavalent hypervalent iodine species.3 The utility of hypervalent iodine reagents as catalyst was introduced by Fuchigami and Fujita in 1994 during the electrocatalytic fluorination of dithioacetals (Fig. 2).60 Initially, the progress of hypervalent iodine catalysis was not quite encouraging as it relied mainly on electrocatalysis that required a particular relation in the oxidation potential of catalyst and substrate to generate the active hypervalent iodine catalyst.61 To overcome the limitation, some inorganic oxidants were tested to generate the catalytic species but success was quite limited.62–64 Later on a remarkable success of hypervalent iodine catalysis was achieved with the use of organic oxidants to generate the catalytic species in situ.3 As a sequel to the review published by our group in 2014,3 we highlight herein the use of hypervalent iodine catalysts in synthetic transformations since 2014.
 |
| | Fig. 2 Iodine(III)-catalysed gem-difluorination of dithioketals III using in situ generated iodine(III) catalytic species II by anodic oxidation of 4-methoxyiodobenzene I. | |
2. Aminations
Amines have widespread uses in many facets of our lives and are present not only in natural products, but also play a vital role in medicinal chemistry. Metal-free approaches for the synthesis of amines through C–H aminations using hypervalent iodine reagents as catalysts were initially developed by the research groups of Chang,65 DeBoef66 and Antonchick.67
2.1. C–H amination of arenes
2.1.1. Intermolecular C–H amination of arenes.
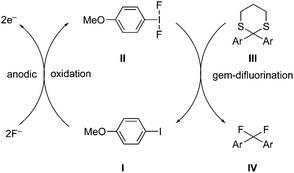
C–H Aminations of arenes is a common reaction catalysed by an active hypervalent iodine catalytic species under mild reaction conditions. A novel metal-free route for the amination of simple electron-rich arenes 16 with 3-aminopyridine derivatives 15 using iodobenzene 17 as catalyst in the presence of peracetic acid as oxidant was developed by Antonchick and co-workers (Scheme 1).68 The desired arylated 3-aminopyridines 18 were formed in good yields. Notably, the amination of electron-deficient arenes was not observed.
 |
| | Scheme 1 Iodine(III)-catalysed amination of arenes 16 with 3-aminopyridine derivatives 15 using peracetic acid as an oxidant. | |
Later, Muñiz and co-workers employed 1,2-diiodobenzene 20 as the pre-catalyst for an intermolecular C–H amination of substituted arenes 16 using N-disubstituted amines 19 as nitrogen sources and peracetic acid as oxidant (Scheme 2).69 The reaction performed remarkably well even at reduced catalyst loadings of 3–4 mol%. The interesting feature of this catalytic approach is the successful amination of electron-deficient arenes. However, the amination products of electron-deficient arenes 16 were obtained in poor yields compared to other arenes. Mechanistic studies revealed the formation of μ-oxo-bridged bisiodine(III) derivative 22 upon oxidation of 1,2-diiodobenzene with peracetic acid.
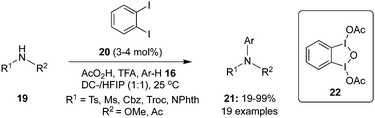 |
| | Scheme 2 Iodine(III)-catalysed C–H amination of arenes 16 with 19 using 20 as precatalyst. | |
The concept of soft–hard acid–base (SHAB) theory was introduced in the N–H arylation of sulfanilides 23 by Mal and Maiti. The catalytic system comprises of iodobenzene 17 as the pre-catalyst and mCPBA as oxidant (Scheme 3).70 Sulfanilides with electron donating and sulfonyl groups like –SO2Ph, –Ts and –Ns were tolerated and amination products 24 were isolated in moderate to excellent yields.
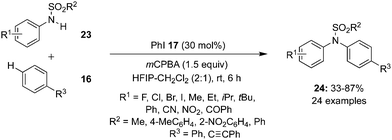 |
| | Scheme 3 Iodine(III)-catalysed N-arylation of 23 with 16 using iodobenzene 17 as pre-catalyst. | |
The proposed catalytic cycle for the N-arylation of 23 with 16 is shown in Scheme 4. Nitrenium ion 25 is a soft electrophile and generated from the interaction between sulfanilide 23 and iodine(III) species [A].71 The newly generated nitrenium ion 25 interacts with electron rich arenes and undergoes an electrophilic aromatic substitution to furnish arylated amines 24via generating carbocation intermediate 26. The regenerated iodobenzene 17 is further oxidised to the iodine(III) compound [A] re-entering the catalytic cycle.
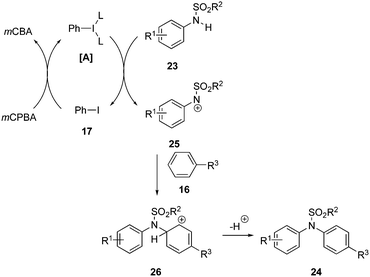 |
| | Scheme 4 Catalytic cycle for the iodine(III)-catalysed N-arylation of 23 with 16 to 24 using iodobenzene 17 as pre-catalyst. | |
2.1.2. Intramolecular C–H amination of arenes.
Tanimori's research group reported an iodine(III)-catalysed intramolecular oxidative C–H amination of aryl hydrazones 27 to provide N-arylsubstituted 1H-indazoles 28 with iodobenzene 17 as pre-catalyst in the presence of Oxone® and TFA (Scheme 5).72 Aryl hydrazones 27 with electron-withdrawing as well electron-donating groups afforded the desired products 28 in moderate to good yields. However, hydrazones with terminal nitrogen atom without substituents or with substituents that destabilize the nitrenium ion intermediate were not tolerated. They also successfully demonstrated a one pot process for the synthesis of indazoles without isolating the intermediate hydrazone 27 using benzophenone and phenyl hydrazine as precursors in the presence of acid catalyst under the optimal conditions.
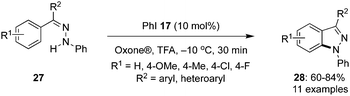 |
| | Scheme 5 Iodine(III)-catalysed intramolecular oxidative C–H amination of aryl hydrazones 27 to 28. | |
In 2022, Kita and co-workers designed an efficient method to synthesize benzolactams 32via an intramolecular C–H amination of aryl amides 29 using biaryl-based iodoarene precatalyst 30.73 The reaction involves the in situ generation of μ-oxo hypervalent iodine compound 31 through the oxidation of iodoarene 30 using mCPBA as oxidant (Scheme 6). When 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was used as solvent, the desired benzolactams 32 were obtained in higher yields. Addition of CF3COOH (2.0 eq.) significantly boosted the production of 32. Notably, other pre-catalysts such as iodobenzene, 4-iodotoluene and 4-iodoanisole were found unsuitable to catalyse this oxidative C–H amination reaction. Aryl amides 29 substituted with nitro, trifluoromethyl or ester functionalities were tolerant to the catalytic conditions. Moreover, the synthesis of five-, seven-, and eight-membered benzolactams was achieved successfully in excellent to good yields using this protocol.
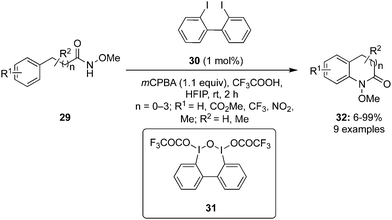 |
| | Scheme 6 Hypervalent iodine-catalysed C–H amination of aryl amides 29 for the synthesis of benzolactams 32. | |
An interesting intramolecular C–H amination of aryl substituted amides 33 to functionalized lactams 35 was developed by involving chiral diiodospirobiindane precatalyst 34 in the presence of mCPBA by Cai and co-workers (Scheme 7).74 The key feature of the reaction was the amination of amides along with desymmetrisation. Amides with cyclopentoxy substituents on the nitrogen gave the desired lactams 35 with better enantioselectivities than with other alkoxy substituents. The substrate scope was further corroborated with other functional groups R2 on the aryl moiety.
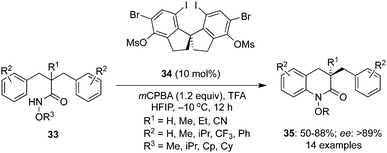 |
| | Scheme 7 Intramolecular C–H amination of aryl substituted amides 33 followed by desymmetrisation using 34 as chiral pre-catalyst. | |
2.2. Amination of alkenes
2.2.1. Intermolecular amination of alkenes.
The amination of alkenes is a useful reaction providing easy access to different aliphatic amines. In 2022, a methodology of group-assisted purification (GAP) was adopted by Ali and co-workers for the regioselective and stereoselective synthesis of vicinal chloroamines 39 from electron-deficient cinnamates and cinnamamides 36 tethered with benzyldiphenylphosphine oxide (Bndpp) group as the GAP candidate (Scheme 8).75 The reaction was carried out by refluxing GAP anchored substrates 36 in the presence of 4 Å molecular sieves, PhI(OAc)21 as catalyst, 4-TsNH237 and 4-TsNCl238 as the nitrogen and chlorine source, respectively, in dichloromethane under argon atmosphere. This protocol tolerated an array of functional groups providing products 39 in good yields. The benefits of this method are the simple and cost-effective purification technique which requires only a wash of the crude mixture with inexpensive solvents such as petroleum ether, as well as the recyclability and reusability of GAP auxiliary.
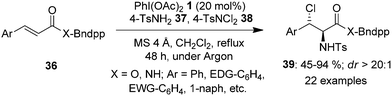 |
| | Scheme 8 Iodine(III)-catalysed stereoselective amino chlorination of GAP anchored cinnamates and cinnamamides 36. | |
Vicinal diamines are a significant class of compounds in the biopharmaceutical field. Enantioselective diaminations of alkenes is typically performed with palladium, copper and titanium catalysts,76–78 and lately Muñiz and colleagues have established an inexpensive route for the intermolecular diamination of styrenes 40 with bissulfonimides 41 as nitrogen source utilizing achiral as well as chiral aryl iodides as catalysts (Scheme 9).79,80 They described the first iodine(III)-catalysed enantioselective intermolecular diamination of styrenes 40 using chiral aryl iodide 42 as catalyst.79anti-Diamines 44 were obtained in moderate to good yields with high enantiomeric excess from both terminal as well as substituted styrenes. Using achiral aryliodines 43a or 43b, irrespective of the position of substituents, styrenes 40 with various electron donating and electron withdrawing groups afforded diamine products 45 in good yields.80 In addition to styrenes, diamination of (E)-stilbene proceeded to afford diamines in moderate yield while allylbenzene produced the corresponding diamine in excellent yield.
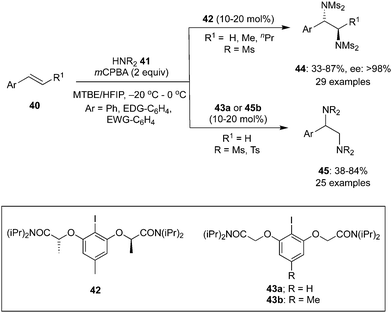 |
| | Scheme 9 Iodine(III)-catalysed vicinal diamination of styrenes 40 to 44 and 45 using pre-catalysts 42 and 43, respectively. | |
Later, the same group developed a scale-up protocol for the synthesis of aryliodine precatalysts 47 and successfully applied it to the diamination of functionalised terminal styrenes 46 using HNMs248 as nucleophile and mCPBA as oxidant (Scheme 10).81 Amination products 49 were obtained in moderate to good yields with high enantiomeric excess (up to 99% ee).
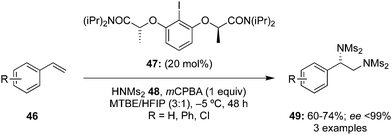 |
| | Scheme 10 Iodine(III)-catalysed vicinal diamination of terminal styrenes 46 using C2-symmetric chiral iodoarene 47 as pre-catalyst and HNMs248 as nitrogen source. | |
2.2.2. Intramolecular amination of alkenes.
Wirth and co-workers employed a novel pyridine-based chiral iodine(I) catalyst 51 in the enantioselective intramolecular diamination of homoallylic guanidine and diaminosulfone derivatives 50 to bicyclic products 52 in the presence of sodium perborate and acetic acid in acetonitrile (Scheme 11).82 Lactate-based catalysts of type 9 were found to be inefficient in this reactions. The protecting group in 52 could be removed to provide free diamines through reduction using lithium aluminium hydride.
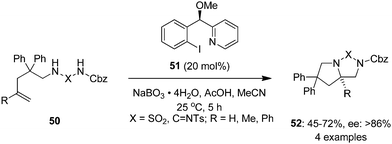 |
| | Scheme 11 Enantioselective intramolecular diamination reactions of 50 to yield 52 using chiral precatalyst 51. | |
2.3. Oxyamination of alkenes
2.3.1. Intermolecular oxyamination of alkenes.
During the last years, attention has been focused on the development of oxyamination reactions using hypervalent iodine catalysis. Wata and Hashimoto developed a protocol for an enantioselective oxyamination of aryl- or alkyl-substituted alkenes 40 using organoiodine(I/III) catalysis.83 The use of N-(fluorosulfonyl)carbamate 53 as bifunctional N,O-nucleophile was considered as a critical element in this reaction. Chiral organoiodine catalyst 54 was found indispensable to achieve good turnover and high enantioselectivity. Notably, the use of magnesium monoperoxyphthalate hexahydrate (MMPP) as oxidant gave high product yields for electronically neutral or slightly electron-poor vinylarenes whereas Selectfluor was found optimal for electron-deficient or ortho-halogenated vinylarenes. The reaction proceeds via formation of intermediates 56–58 (Scheme 12). Carbamate 53 reacts with in situ generated hypervalent iodine to form intermediate 56, which converts into intermediate 58via formation of an alkene-coordinated iodonium intermediate 57. Finally, the intermediate 58 cyclises intramolecularly to yield product 55 by the nucleophilic attack of oxygen and regenerates the chiral iodoarene 54.
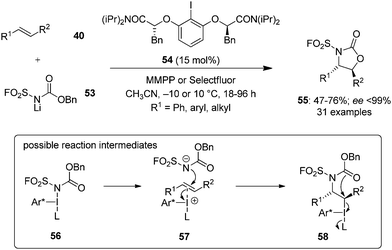 |
| | Scheme 12 Hypervalent iodine-catalysed enantioselective oxyamination of aryl- or alkyl-substituted alkenes 40. | |
The oxyaminated products 55 can be easily deprotected to yield free β-amino alcohols 59 in good yields without loss of enantioselectivity (Scheme 13).83 Chiral β-amino alcohols are interesting reaction intermediates in the field of organic synthesis.
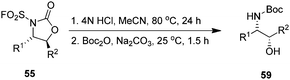 |
| | Scheme 13 Acid-mediated deprotection and ring opening of cyclic product 55 to amino alcohol 59. | |
Lei and others designed a regiodivergent and regioselective intermolecular oxyamination of styrene 46 using the combination of chiral C2-symmetric iodoarene 47 and Selectfluor as oxidant (Scheme 14).84 Oxidation of chiral iodoarene 47 by Selectfluor and subsequent salt metathesis with LiBF4 leads to the in situ generation of the active hypervalent iodine(III) reagent. Using amide 60 as O- and N-source and nitromethane as solvent, the desired regioisomeric oxazoline product 61 was obtained in 60% yield with 86% ee. On the other hand, the regioisomeric addition product 62 was obtained in 23% yield (74% ee) by employing acetonitrile and water as the nucleophiles.
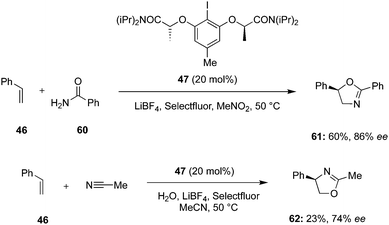 |
| | Scheme 14 Iodine(III)-catalysed intermolecular amination of styrene 46. | |
2.3.2. Intramolecular oxyamination of alkenes.
In 2021, Deng et al. reported an iodine(III)-catalysed intramolecular oxyamination of alkenes 63 containing an amide functionality using Ts2NH 41 as an external nitrogen source. Optimisation results showed that 2,6-dimethoxy iodobenzene 64 provided the best catalytic activity in the presence of mCPBA as an oxidant (Scheme 15).85 A variety of N-aryl, N-benzyl and N-methoxyl substituted pentenamides smoothly underwent this transformation, affording desired oxyamination products 65 in good yields and with high regioselectivity. Additionally, substrates with cycloalkyl rings provided spiro-tetrahydrofuranyl methanamine products in high yields.
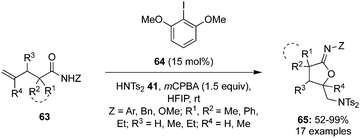 |
| | Scheme 15 Iodine(III)-catalysed intramolecular oxyamination of terminal alkenes 63 to functionalized furans 65 using HNTs241 as a nitrogen source in the presence of precatalyst 64. | |
An intramolecular oxyamination of γ,δ- and δ,ε-unsaturated esters 66 and N-allyl amides 70 was developed with benzyl N-(fluorosulfonyl)carbamate 67 as an exogenous nitrogen source using hypervalent iodine catalysis (Scheme 16).86 Selectfluor was found as the best oxidant for these aminations. Various functional groups were tolerated under the given reaction conditions and the corresponding lactones 69 and oxazolines 71 in good yields with up to 77% enantiomeric excess. Moreover, the protecting group of the aminated products was removed to make free amino compounds under acidic conditions without losing any selectivity.
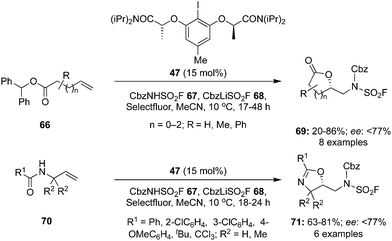 |
| | Scheme 16 Iodine(III)-catalysed intramolecular oxyamination of unsaturated esters 66 and N-allyl amides 70 using benzyl N-(fluorosulfonyl)carbamate 67 as an exogenous nitrogen source. | |
2.4. C–H amination at sp3 carbon
Shi and co-workers developed an intramolecular sp3 C–H amination of ortho-substituted N-methoxy benzamides 72 for the synthesis of γ-lactams 74 catalysed by an iodine(III) species generated in situ by using catalytic amounts of iodoarene 73 in the presence of mCPBA (Scheme 17).87 Among the various iodoarenes investigated, 2-iodobiphenyl 73 emerged as a good pre-catalyst. The reaction proceeded smoothly with electron-neutral and electron-deficient substrates, while electron-rich substrates gave poor yield. The amination reaction worked well for cyclic as well as acyclic tertiary C–H bonds and due to the high energy barrier, a direct amination of secondary C–H bonds was not observed. Notably, the amination at chiral centres worked smoothly and it was found to be stereospecific.
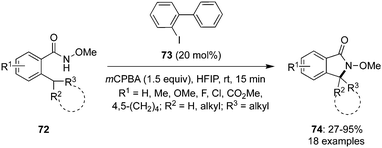 |
| | Scheme 17 Iodine(III)-catalysed intramolecular sp3 C–H amination of N-methoxy benzamides 72 to compounds 74. | |
The mechanism for the amination of ortho-substituted N-methoxy benzamides 72 to γ-lactams 74 is given in Scheme 18. The mechanism was proposed based on the DFT calculations for the reaction of benzamides 72a with PIDA 1.87 Reaction is initiated with the formation of an iodonium intermediate 75 which converts into the protonated lactam 76via the transition state TS 76 involving a hydride shift, followed by C–N bond formation. Finally, the protonated lactam 76 undergoes deprotonation to give amination product 74a.
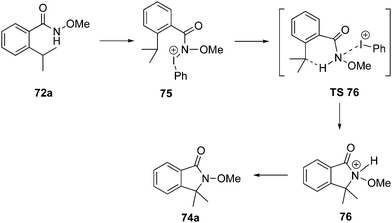 |
| | Scheme 18 Proposed mechanism for iodine(III)-catalysed intramolecular sp3 C–H amination of N-methoxy benzamides 72 to compounds 74. | |
2.5. Imination of benzylic C–H
In 2019, Mal and co-workers reported an intramolecular oxidative C–N bond formation via C–H imination reaction at sp3 carbon centre. During these imination reactions, the synthesis of 1,2-disubstituted benzimidazoles 78 was achieved from dibenzyl amines 77 using two different catalytic systems, one with the conventional iodobenzene 17 as precatalyst88 (Scheme 19, Method A) and the second with tetrabutylammonium iodide as precatalyst (Scheme 19, Method B).89 The amination reaction proceeded through hydrogen elimination, two hydrogens from the highly acidic benzylic C(sp3) and the remaining two from the aryl-N(sp3). Symmetrical dibenzyl amines afforded a single isomer of benzimidazoles but unsymmetrical dibenzyl amines produced a mixture of isomers as major product being the imination at benzylic centre substituted with electron rich arenes.
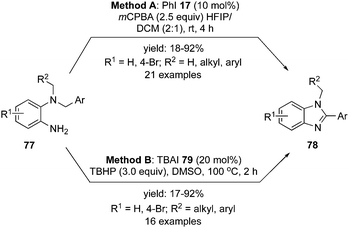 |
| | Scheme 19 Iodine(III)-catalysed C–H imination reaction of amines 77 to benzimidazoles 78. | |
3. Oxidation reactions
3.1. Oxidation of alcohols
3.1.1. Oxidation of primary and secondary alcohols.
Oxidation of alcohols is traditionally an indispensable reaction of organic synthesis as it provides synthetically valuable carbonyl compounds. Hypervalent iodine catalysis was employed for the oxidation of alcohols for the first time in 2005.90 Since that first report, various iodine(III) and iodine(V) catalysts have been developed for the oxidation of alcohols.3 An eco-friendly protocol for the oxidation of primary and secondary alcohols was developed at room temperature by using 2-iodo-N-isopropyl-5-methoxybenzamide 80 as a catalyst with Oxone® and Bu4NHSO4.91 Secondary benzylic and aliphatic alcohols 81 afforded the corresponding ketones 82 in good to excellent yields and primary alcohols 83 were converted to the corresponding carboxylic acids 84 in moderate to excellent yields (Scheme 20). During the oxidation of primary alcohols, the corresponding aldehydes were not observed probably due to the presence of water in the reaction.
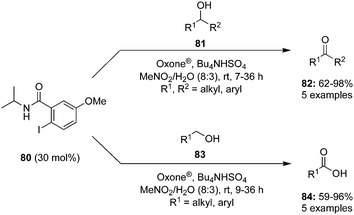 |
| | Scheme 20 Oxidation of secondary 81 and primary 83 alcohols using 80 as precatalyst. | |
In another report, an iodine(V)-catalysed aerobic oxidation of secondary alcohols 81 to the corresponding ketones 82 was achieved in good to excellent yields catalysed by 2-tert-butylsulfonyl-iodobenzene 85 in the presence of n-butyraldehyde 86 and CoCl2·6H2O (Scheme 21).92 The role of CoCl2·6H2O was to initiate the aldehyde-promoted aerobic oxidation of precatalyst 85 to generate the iodine(V) species in situ. In case of aromatic secondary alcohols, both electron rich and electron deficient derivatives were tolerated. The oxidation of primary alcohols 83 gave the carboxylic acids 84.
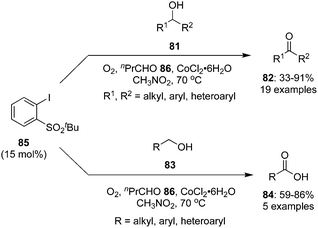 |
| | Scheme 21 Iodine(V)-catalysed oxidation of alcohols 81 and 83 to corresponding carbonyl compounds 82 and 84, respectively using 85 as precatalyst. | |
The proposed catalytic cycle for the above oxidation process is shown in Scheme 22. Initially, the aldehyde-promoted aerobic oxidation of precatalyst 85 occurred to form iodosylbenzene 87 followed by disproportionation to generate iodylbenzene 88. Iodine(V) intermediate 88 then oxidises the alcohol 81 to ketone 82 and regenerates 87, which on disproportionation forms the active catalytic iodine(V) species 88.91
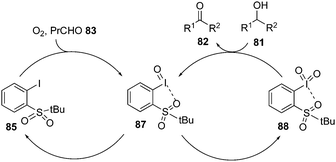 |
| | Scheme 22 Catalytic cycle for the iodine(V)-catalysed aerobic oxidation of alcohols 81 to ketones 82 using 85 as precatalyst. | |
Polymer-supported hypervalent iodine pre-catalysts 89 and 90 were synthesised by Kirsch and Ballaschk and revealed their potential application in the oxidation of secondary alcohols 81 to corresponding ketones 82 (Scheme 23).93 The primacy of this green synthesis is the multiple reusability of the catalyst without losing much catalytic activity and easy work-up. A wide range of secondary alcohols including cyclic, bicyclic and benzylic alcohols were tolerated. Phenols and amines are vulnerable to these catalytic conditions. Furthermore, the catalytic oxidation with IBS-derived catalyst 90 proceeds faster and cleaner compared to IBX-derived catalyst 89.
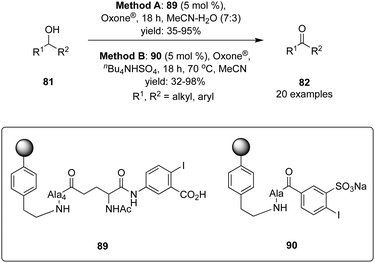 |
| | Scheme 23 Iodine(V)-catalysed oxidation of alcohols 81 to corresponding ketones 82 using pre-catalyst 89 or 90. | |
In 2020, Enderlin and co-workers demonstrated a simple and extremely efficient iodine(V)-catalysed gram scale synthesis of 2,5-diformylfuran 93 (87% yield) by the partial oxidation of 5-hydroxymethylfurfural (HMF) 91 using sodium 2-iodobenzenesulfonate 92 as precatalyst, Oxone® as oxidant and nitromethane as solvent. A notable feature of this process is its simple work-up procedure involving only filtrations and extractions to obtain 93 in high purity (Scheme 24).94
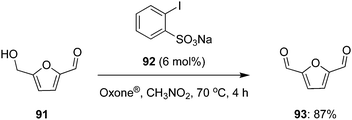 |
| | Scheme 24 Iodine(V)-catalysed selective oxidation of HMF 91 to 93 using 92 as precatalyst. | |
Nemati and co-workers designed and developed a hypervalent iodine(III) based heterogeneous nano-catalyst using magnetic polyiodoaniline nano-composite, Fe3O4-PANI-I(OAc)295 for the selective oxidation of functionalised benzyl alcohols 94 to corresponding aldehydes 97 in the presence of TEMPO 96 as oxidant and acetonitrile as solvent (Scheme 25).95 A wide range of electron-withdrawing and donating groups were tolerated to give the corresponding benzaldehydes in desirable yields without the formation of any by-product. The key feature of this nano-composite precatalyst is its stability and reusability for five consecutive cycles.
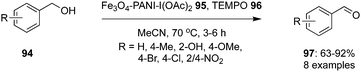 |
| | Scheme 25 Iodine(III)-catalysed selective oxidation of benzylalcohols 94 to aldehydes 97 in the presence of Fe3O4-PANI-I(OAc)2 nano-composite 95. | |
3.1.2. Oxidation of allylic alcohols.
Rao and co-workers described the catalytic use of 2-iodoxybenzoic acid (IBX) 11 generated in situ by the oxidation of 2-iodosobenzoic acid (IBA) 6 using Oxone® as an oxidant. During these oxidations, allylic alcohols 98 are oxidised to the corresponding ketones 99.96 Various electron-withdrawing and donating substituents on the aromatic moiety of the secondary alcohol were tolerated. Even the internal allylic alcohols 100 afforded the cinnamyl aldehydes 101 in excellent yields under the same reaction conditions (Scheme 26). The benchmark of this green reaction is that the precatalyst can be recovered by simple filtration and there was no side product observed during these oxidations.
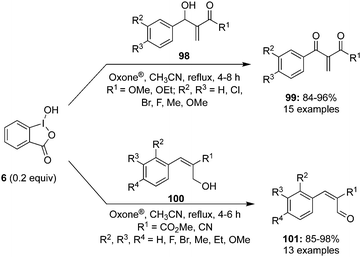 |
| | Scheme 26 Iodine(V)-catalysed oxidation of terminal allylic alcohols 98 and internal allylic alcohols 100 to corresponding carbonyl compounds 99 and 101, respectively. | |
3.1.3. Oxidation of 1,2-diols.
Hypervalent iodine catalysis in the presence of molecular oxygen was used for glycol scission of 1,2 diols 102 by Uchiyama and coworkers.97 By optimizing the reaction conditions, isobutyraldehyde 104, pentamethyliodobenzene 103 and acetonitrile emerged as the best O2 mediator, catalyst and solvent, for the cleavage of diols. Mono- and di-substituted diols 102 (R1 = H; R2 = aryl, alkyl, H) and various dihydrobenzoins were smoothly cleaved to give the corresponding carboxylic acids 84. Notably, tri- and tetra-substituted diols 102 (R1, R2 = aryl, alkyl) afforded desired ketones 82 even in air or in the dark (Scheme 27). The efficiency of the reaction can be enhanced by premixing the aldehyde and O2 before the addition of the substrate.
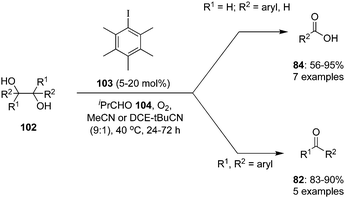 |
| | Scheme 27 Iodine(III)-catalysed oxidation of 1,2-diols 102 using pentamethyl iodobenzene 103 as precatalyst in the presence of molecular O2 as oxidant. | |
3.2. Oxidation of phenols
The oxidation of phenolic compounds is usually known as the dearomatisation of phenols. The oxidative dearomatisation of phenols is one of the common reaction of hypervalent iodine(III) reagents. Both internal and external nucleophiles have been employed during these oxidation reactions which lead to dearomatised products such as highly functionalised quinones, quinols and spirolactones.21,38,98
A general reaction pathway for the hypervalent iodine mediated oxidation of phenols is shown in Scheme 28. The phenolic compound 105 reacts with the hypervalent iodine(III) compound 1 or 2 through a ligand exchange and form intermediate 106, which then undergoes a nucleophilic attack by an external nucleophile and forms either ortho-cyclohexadienone 107 or para-cyclohexadienone 108via a dearomatisation process. In case of phenols 105 having an internal nucleophile, dearomatisation to ortho-spirocycles 109 and para-spirocycles 110 are taking place.
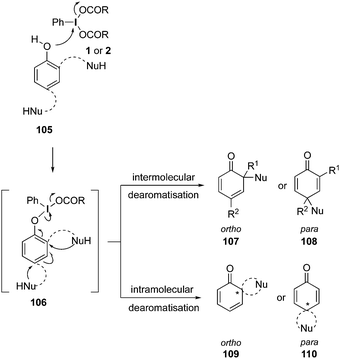 |
| | Scheme 28 Hypervalent iodine-mediated dearomatisation of phenols 105via formation of intermediate 106. | |
3.2.1. Intermolecular dearomatisation of phenols.
In recent years, the iodine(III)-catalysed dearomatisation of phenols has received a particular attention by various hypervalent iodine chemists around the world.3,21 Dearomatisations of phenols have been developed using hypervalent iodine catalysis in past two decades.3 An eco-friendly protocol was reported by Yakura and co-workers for the oxidation of 4-alkoxyphenols 111 to p-benzoquinones 113 in good yields using magnetic nanoparticle-supported iodoarene catalyst 112 in the presence of Oxone® (Scheme 29).99,100 The catalyst consists of phosphonate groups connecting magnetite (Fe3O4) nanoparticles to the iodoarene catalyst. After the completion of reaction, the catalyst can be easily separated by applying an external magnetic field and reused several times.
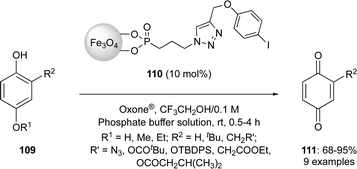 |
| | Scheme 29 Oxidation of 4-alkoxyphenols 111 to p-benzoquinones 113 using magnetic nanoparticle-supported iodoarene precatalyst 112. | |
Muñiz and Fra described an enantioselective hydroxylative dearomatisation of 4-substituted phenols 105 to afford the corresponding p-quinols 115 using the lactic amide motif-based chiral aryliodide catalyst 114 in the presence of mCPBA as an oxidant (Scheme 30).101 Two different solvent mixtures (A and B) were used during these dearomatisation reactions. The dearomatised products 115 were obtained in almost similar yields in both reaction conditions. However, the chiral catalyst could not transfer the chirality successfully and products were obtained in only up to 5% enantiomeric excess.
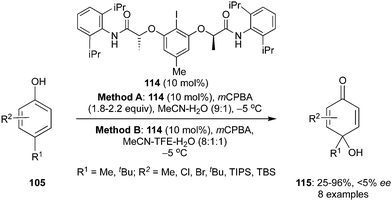 |
| | Scheme 30 Hydroxylative dearomatisation of phenols 105 to p-quinols 115 using chiral aryliodide catalyst 114. | |
Hypervalent iodine(V)-mediated hydroxylative dearomatisation of 2-substituted phenols 105 to their cyclodimers 117via [4+2] cycloaddition was developed by Ishihara and co-workers.102 The catalytic system comprises of precatalyst 116a or 116b which generates the catalytic species 2-iodoxybenzenesulfonic acid in situ in the presence of Oxone® as oxidant (Scheme 31). Inclusion of a trialkylsilylmethyl substituent at the ortho-position of phenols facilitates the reaction and use of buffered Oxone® suppresses silanol elimination. The reaction was performed with various (2-(silylmethyl)phenols) 105 (R2 = CH2SiMe3) requiring the addition of K2CO3 (0.375 eq.). Under similar catalytic conditions, oxidation of o-substituted 1- or 2-naphthols 118 provided ortho naphthoquinols 119 in excellent yields (Scheme 31). The same catalytic approach was employed for the synthesis of the natural products biscarvacrol and lacinilene C methyl ether in high yields.
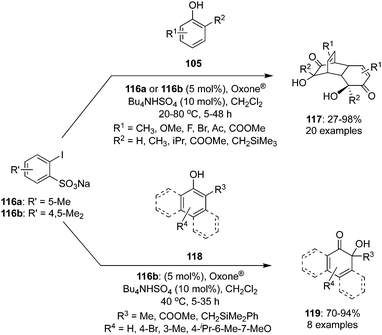 |
| | Scheme 31 Iodine(V)-catalysed dearomatisation of functionalised phenols 105 and naphthols 118. | |
Over the past few years, metal–organic frameworks (MOFs) have emerged as a support to catalyse organic reactions by offering high reactant selectivity and reusability. Various multivariate Al and Zr-MOF supported iodine catalysts 120 and 121, that can be recovered and recycled several times, were developed by Cozzolino and co-workers for the oxidation of hydroquinones 111 to p-quinones 113 in the presence of mCPBA and MeNO2 (Scheme 32).103 These catalysts were prepared by treating the appropriate amount of linkers with zirconium(IV)chloride or aluminiumchloride and catalysts with 25% linkers were found to be ideally suited to achieve the optimal balance between catalyst loading and catalyst accessibility. 2-Iodoterephthalic acid was used as iodine linker in UiO-66 25%-I 120 and MIL-53 25%-I 121. The same group prepared two novel expanded-pore iodine-functionalized UiO-67 (Zr) 122 and DUT-5 (Al) 123 catalysts and employed them in the oxidation of hydroquinones 111 to p-quinones 113 and catechol derivatives 124 to o-quinones 125, respectively (Scheme 32).104 2-Iodo-[1,1′-biphenyl]-4,4′-dicarboxylic acid was used as linker in the UiO-67-25%-I 122 and DUT-5 25%-I 123 catalysts. Like other oxidations, mCPBA was used to regenerate the active iodine(V) catalytic species.
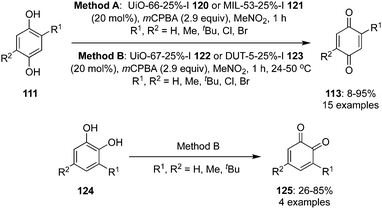 |
| | Scheme 32 Iodine(V)-catalysed oxidation of hydroquinones 111 and catechols 124 to p-quinones 113 and o-quinones 125, respectively using Al and Zr-MOF supported iodine catalysts 120–123. | |
Hashimoto and co-workers introduced a coherent procedure for the asymmetric catalysis of para-hydrative intermolecular dearomatisation of functionalised phenols 105 to 115 through in situ generation of a chiral iodine(III) catalyst by oxidation of indanol-based precatalyst 126 in the presence of mCPBA and butanol-H2O as solvent mixture (Scheme 33).105 A variety of functional groups are tolerated and p-quinols 115 were obtained in good yields with up to 84% ee.
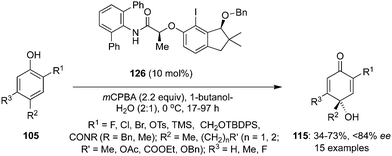 |
| | Scheme 33 Iodine(III)-catalysed enantioselective dearomatisation of functionalized phenols 105 to p-quinols 115 using 126 as precatalyst. | |
An environmentally friendly hypervalent iodine-catalysed oxidation of alkoxyarenes 127a,b to p-quinones 129 was developed using 2-iodobenzoic acid 128 as precatalyst and Oxone® as oxidant in acetonitrile–water (Scheme 34).106 This approach provides p-quinones 129 in excellent yields in a short reaction time at room temperature. Usually, these oxidation reactions suffer from the formation of o-quinones as side products, but this reaction provides p-quinones exclusively. Earlier studies107 using Oxone®-generated hypervalent iodine oxidants for the dearomatisation of phenols have indicated a preference for p-quinones 129 formation over o-quinones 130, which made Oxone® as the oxidant of choice in this procedure (Scheme 34). Notably, the in situ generated cyclic iodine(III) compound IBA 6 was acting as catalytic species.
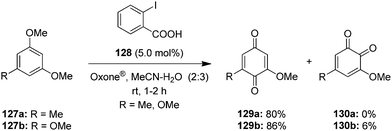 |
| | Scheme 34 Iodine(III)-catalysed oxidation of alkoxyarenes 127 to p-quinones 129 using 128 as precatalyst. | |
3.2.2. Intramolecular dearomatisation of phenols.
Intramolecular dearomatisation of phenols using hypervalent iodine reagents provides various biologically active cyclic and spirocyclic scaffolds.38 Several hypervalent iodine-catalysed approaches are now available for the intramolecular dearomatisation of phenols.3,21
The first hypervalent iodine-catalysed intramolecular dearomatisation of phenols was investigated in 2005 by Kita and co-workers.108 Kita and few other research groups employed hypervalent iodine catalysis to construct different spirocyclic scaffolds via intramolecular dearomatisation of phenols and naphthols.109–116 All these reports are covered in our previous review on hypervalent iodine catalysis published in 2014.3
Ishihara and co-workers developed the spirolactonisation of phenols 131 with a propionic acid functionality in the ortho-position to enantiomerically rich ortho-dioxolanones 133 using a conformationally flexible chiral iodoarene 132 as precatalyst in the presence of mCPBA (Scheme 35).117 Oxidation products 133 were obtained comparatively with low ee when 10% of methanol was used as an additive (Method A). Interestingly, the ee was improved up to 96% in case of 60 equivalents of methanol (Method B). Probably, methanol suppresses the dissociative pathway of ligand(III) and might improve the selectivity.118,119 The reaction required longer reaction time as methanol deactivates the regeneration of iodine(I) to iodine(III). The absolute configuration of products (S-isomers) were assigned based on single crystal X-ray analysis.
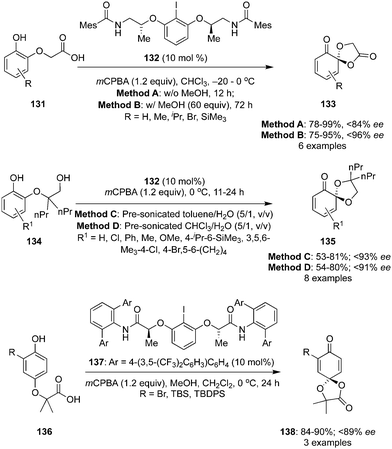 |
| | Scheme 35 Hypervalent iodine(III)-catalysed enantioselective dearomatisation of phenolic compounds 131, 134 and 136. | |
It was observed that few of the dioxolanones were not stable at room temperature while the same catalytic system was employed for the oxidation of phenols 134 to spiroketals 135 with excellent selectivity (up to 93% ee) (Scheme 35).117 Interestingly, same precatalyst 132 was not found suitable for the lactonisation of phenols 136 substituted with acetic acid in the para-position to para-dioxolanones 138. Another conformationally flexible chiral iodoarene based precatalyst 137 was used for the lactonisation of phenols 136 and para-dioxolanones 138 were obtained with up to 89% ee (Scheme 35).117
The same group accomplished an enantioselective hypervalent iodine(III)-catalysed intramolecular oxidative dearomatisation of naphthols 139 by generating conformationally flexible λ3 iodine catalysts in situ from 2-aminoalcohol based aryl iodide 132 in the presence of mCPBA as oxidant (Scheme 36).120 Highly functionalized spirolactones 140 were obtained in moderate to high yields with up to 98% ee. Use of HFIP as an additive along with the solvent DCE facilitated the oxidation of less reactive 2-naphthols whereas ethanol was used as additive in the case of 1-naphthols. The current protocol tolerates electron-donating as well as electron-withdrawing substituents in 139.
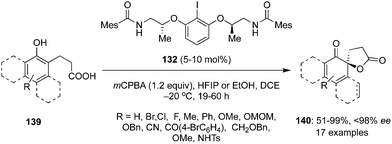 |
| | Scheme 36 Iodine(III)-catalysed enantioselective spirolactonisation of naphthols 139 to 140 using conformationally flexible chiral iodoarene 132 as precatalyst. | |
Similarly, an iodine(III)-catalysed enantioselective spirolactonisation of 4-substituted 1-naphthols 139a was developed by Nachtsheim et al. using a novel triazole-based chiral iodoarene precatalyst 141 in the presence of mCPBA (Scheme 37).121 During these oxidations, spirolactones 140a with electron withdrawing and donating groups were prepared in moderate to good yields. Notably, the precatalyst 141 was not found equally effective compare to the C2-symmetric chiral precatalyst 132 for the same reaction and enantiomeric excess was reduced to <72% (Scheme 37).121
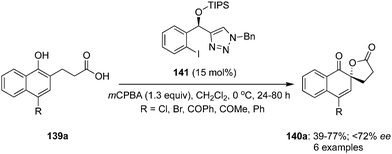 |
| | Scheme 37 Iodine(III)-catalysed enantioselective spirolactonisation of 4-substituted 1-naphthols 139a to spirolactones 140a. | |
Atropisomers play a crucial role as catalyst in asymmetric catalysis. In 2017, Ogasawara and co-workers developed a novel C2-symmetric conformationally rigid atropisomeric chiral diiododiene 142 from 1,2-bis(4,4-dimethyl-2-pentynyl)benzene and Cp2ZrCl2/Mg.122 Design and synthesis of low-cost and reliable chiral iodoarene reagents for asymmetric catalysis is of tremendous interest nowadays, two research groups recently succeeded in constructing novel chiral organoiodanes based on carbohydrates and helicenes. Ziegler and Imrich reported D-glucose-based chiral iodoarene 143.123 Helicine-based chiral iodoarene catalyst 144 was designed and synthesised by Quideau and co-workers through a double Wittig olefination followed by the double photo-cyclisation from inexpensive starting materials.124 These novel chiral aryl iodide reagents served as interesting catalysts for the spirolactonisation of naphthols 139b to afford chiral spirolactone 140b employing mCPBA as oxidant (Scheme 38). The reaction catalysed by 142 yielded 140b as (S)-isomer in 73% ee whereas 143 or 144 provided 140b as (R)-isomer with up to 60% ee. Notably, the lower reaction temperature resulted in longer reaction times with improved yield and enantioselectivity.
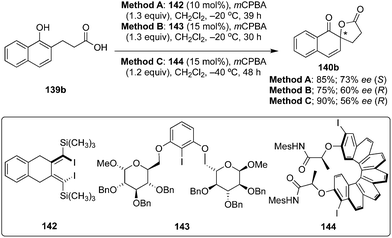 |
| | Scheme 38 Iodine(III)-catalysed enantioselective spirolactonisation of naphthols 139 to spirolactones 140 using precatalysts 142–144 in the presence of mCPBA. | |
In 2017, Ciufolini and co-workers described an enantioselective intramolecular oxidative spiroetherification of naphthoic alcohols 145 employing chiral aryl iodide 146 as precatalyst in the presence of mCPBA (Scheme 39).125 A wide range of spirocyclic ethers 147 bearing electron donating and withdrawing substituents were synthesized in high yields with up to 93% enantiomeric excess. Like in other spirocyclisations, the active iodine(III) catalytic species was generated in situ by oxidation of the chiral precatalyst 146 with 3-chloroperbenzoic acid. The absolute configuration of product 147 was assigned as (R)-isomer by its single crystal X-ray analysis.
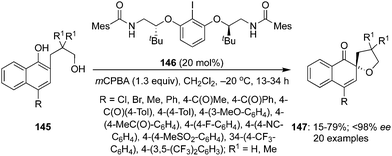 |
| | Scheme 39 Iodine(III)-catalysed enantioselective oxidative spirocycloetherification of naphthoic alcohols 145 to 147 using 146 as precatalyst. | |
In 2020, Tariq and Moran performed an oxidative dearomatisation of amide-tethered phenols 148 mediated by λ3-iodanes generated in situ from the 4-iodotoluene 149/m-CPBA catalytic system (Scheme 40).126 The intramolecular dearomatisation protocol furnished spirooxazolines 151 in 30–94% yields with excellent functional group compatibility. The reaction scope was investigated with a range of aryl, alkyl and heteroaryl amide-based phenols under optimised conditions. Notably, methoxy and alkyl substituted phenyl amides 148 yielded spirocycles 151 in moderate yields whereas the fluoro-substituted substrates led to the higher yields of the products. It was suggested that the activation of phenolic oxygen by λ3-iodane and subsequent cyclisation of pendent amide on to the aromatic ring results in the formation of dearomatised product 151. Further oxidative dearomatisation of naphthols 152 was performed with 40 mol% of 4-iodotoluene 149 to produce spirocycles 153 in moderate yields. Moreover, triptycene based pre-catalysts 154–156 were also employed in the same reaction but could achieved only very limited success.127
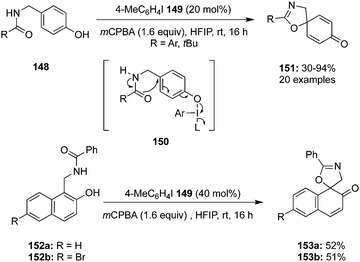 |
| | Scheme 40 Iodine(III)-catalysed synthesis of spirooxazolines 151 and 153 by dearomatisation of phenolic compounds 148 and 149. | |
Wirth and co-workers developed the synthesis of novel iodotriptycenes 154–156 and employed them as precatalyst for the intramolecular dearomatisation of naphthols 152. The spirocyclic product 153 was obtained in moderate yields with only up to 6% ee (Scheme 41).127
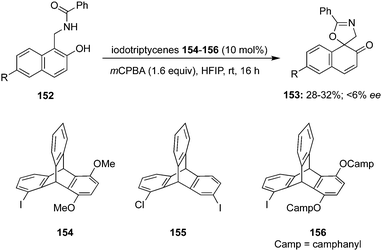 |
| | Scheme 41 Iodine(III)-catalysed dearomatisation of phenolic compound 152 using triptycene based pre-catalysts 154–156. | |
Gong and co-workers described an elegant method for the construction of spirooxindoles 159 by an intramolecular dearomatisation of 1-hydroxy-N-aryl-2-naphthamides 158 using chiral iodoarene 157 as precatalyst (Scheme 42).128 This is the first example of an enantioselective dearomatisation of 1-hydroxy-N-aryl-2-naphthamides 158 providing a facile access to a library of spirooxindoles 159 in good yields with up to 92% ee. The dearomatisation involves the oxidation of chiral iodoarene 157 to generate the active chiral hypervalent λ3-iodane in situ, which catalyses the oxidative of spirocyclisation. Notably, an all-carbon stereogenic centre in the products is generated during these oxidation reactions.
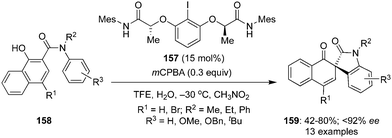 |
| | Scheme 42 Iodine(III)-catalysed intramolecular dearomatisation of 1-hydroxy-N-aryl-2-naphthamides 158 to spirooxindoles 159. | |
In another report, the same research group developed a highly enantioselective approach for the spirocarbocyclisation of N1,N3-diphenylmalonamides 160 using hypervalent iodine catalysis.128 Oxidation reactions were performed by using (S)-proline-derived chiral iodoarene 161 as precatalyst in the presence of peracetic acid leading to the synthesis of spirooxindoles 162 in variable yields with up to 90% ee (Scheme 43).129 Once again a quaternary carbon stereogenic centre is generated.
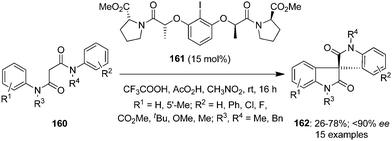 |
| | Scheme 43 Iodine(III)-catalysed enantioselective synthesis of spirooxindoles 162 from N1,N3-diphenylmalonamides 160 using (S)-proline-derived chiral iodoarene 161 as precatalyst. | |
In 2020, Xiong and co-workers developed an enantioselective intramolecular alkoxy-oxylactonisation followed by dearomatisation of 3′-hydroxy-[1,1′-biphenyl]-2-carboxylic acids 163 employing chiral C2-symmetric iodoarene 164 as precatalyst using mCPBA and MeOH (Scheme 44).130 Functionalised cyclohexadienones 165 were prepared in moderate yields in up to 52% ee. The size of the alkyl group in the alcohols played a significant role as the yields of the products were decreased with an increased size while the ee was improved significantly. Moreover, the selectivity was influenced by the position of substituents. Selectivity was quite similar in case of o- and m-substituted phenols while ee was improved significantly with p-fluoro substituted phenols due to an increased nucleophilic character of the carboxylate. This is one of the rare reports where oxylactonisation is achieved alongside the dearomatisation.
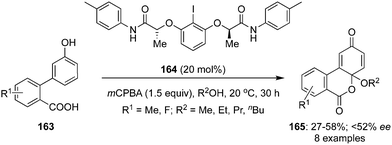 |
| | Scheme 44 Iodine(III)-catalysed intramolecular dearomatising alkoxy-oxylactonisation of 3′-hydroxy-[1,1′-biphenyl]-2-carboxylic acids 163 to cyclohexadienones 165. | |
3.3. Oxidation of aromatic amines
Hypervalent iodine reagents have been employed successfully for the dearomatisation of aromatic amines but there is paucity of the literature to achieve similar oxidations using hypervalent iodine catalysis.23 In 2021, Shimazaki et al. developed a highly enantioselective hydrative para-dearomatisation of anilides 166 with water as nucleophile using indanol-based chiral organoiodine precatalyst 167. This oxidation approach offers functionalised p-quinol imines 168 in poor to excellent yields with up to 95% enantiomeric excess (Scheme 45).131 In general, 4-methyl sulfonanilides with different 2-substituents such as chloro, bromo, methyl, phenyl, silyl and amide groups were tolerated. Moreover, the dearomatisation of 2-bromoanilides 166 substituted with ethyl, methyl, acetoxy and methoxycarbonyl functional groups at para position of the phenyl ring were also accomplished successfully under these conditions.
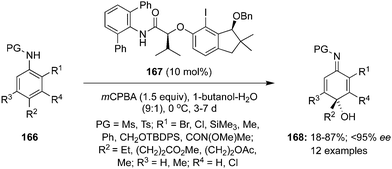 |
| | Scheme 45 Iodine(III)-catalysed highly enantioselective hydrative para-dearomatisation of anilides 166 using indanol-based chiral organoiodine precatalyst 167. | |
3.4. Oxidation of sulfides and sulfenamides
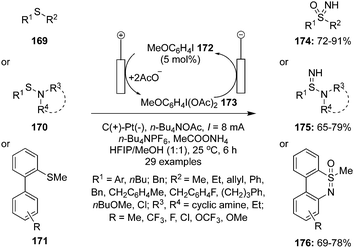
Various hypervalent iodine reagents have been used as oxidants for the oxidation of organosulfur compounds under mild reaction conditions.21 The role of hypervalent iodine catalysis in the oxidation of organosulfur compounds is very limited.3 In 2021, Kong and co-workers reported an environmental friendly approach for the oxidation of sulfides 169 to NH-sulfoximines 174 by the use of hypervalent iodine(III) catalyst 173 generated via anodic oxidation of 4-iodoanisole 172 (Scheme 46).132 During these oxidations, n-Bu4NPF6 was employed as a supporting electrolyte and AcONH4 as an ammonium source. The combination of graphite (C) anode and Pt cathode led to the highest product yields. Methyl phenyl sulfides 169 substituted with electron-withdrawing and -donating groups gave the desired products 174 in good to excellent yields. Additionally, the synthesis of NH-sulfonimidamides 175 from variety of sulfenamides 170 was achieved under standard conditions (Scheme 46).130 Furthermore, an electrochemical oxidation of [1,1′-biaryl]-2-sulfonamides 171 provided the corresponding dibenzothiazines 176 in good yields (Scheme 46).132
 |
| | Scheme 46 Iodine(III)-catalysed electrochemical oxidation of sulfides 169, sulfenamides 170 and [1,1′-biaryl]-2-sulfonamides 171. | |
3.5. Oxidation of alkenes
Oxidation of alkenes with hypervalent iodine reagents is one of the key reaction of hypervalent iodine reagents.21 Usually, hypervalent iodine reagents activates the olefinic double bond and lead to different oxidations such as epoxidations, hydroxylations, acetoxylations or oxidative cleavages.23 Oxidation of alkenes achieved by involving hypervalent iodine catalysis until 2014 are compiled in our previous review article.3
3.5.1. Acetoxylation of alkenes.
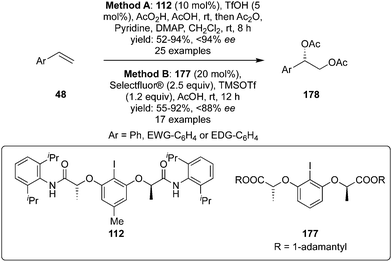
Muñiz and co-workers developed iodine(III)-catalysed enantioselective diacetoxylation of styrenes 48 to 178 using C2-symmetric chiral iodoarene 112 as precatalyst, peracetic acid as an oxidant and acetic anhydride as the acetylating agent (Method A, Scheme 47).133 Various substituted styrenes gave the desired diacetoxylation products 178 in good yields with high enantioselectivities (up to 94% ee). Another iodine(III)-catalysed approach was developed by using chiral precatalyst 177 in the presence of Selectfluor as a terminal oxidant and diacetoxylation of styrenes 48 was achieved in high yields with up to 88% ee (Method B, Scheme 47).134
 |
| | Scheme 47 Iodine(III)-catalysed enantioselective diacetoxylation of styrenes 48 using chiral precatalyst 112 and 177. | |
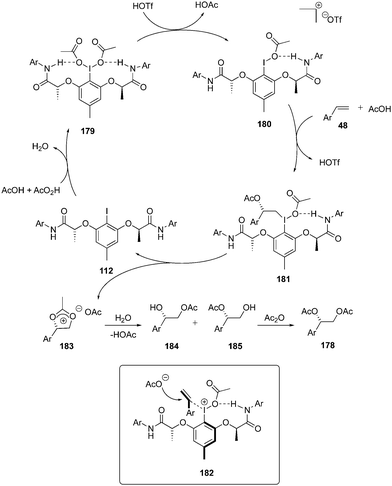
A proposed mechanism for the diacetoxylation of styrenes 48 using chiral iodoarene 112 is shown in Scheme 48. Peracetic acid oxidises the iodoarene 112 to the iodine(III) species 179. One of the acetate group in 179 dissociates to create a free coordination site at iodine(III) in the presence of triflic acid while the other acetate group participates in hydrogen bonding to generate intermediate 180. Subsequently, styrene 48 coordinates to intermediate 180 followed by nucleophilic attack of acetate to the exposed re-face of 180 to form intermediate 181. Intramolecular nucleophilic addition of the acetyl group provides Woodward dioxolonium intermediate 183 and regenerates the iodine(I) catalyst 112. Dioxolonium intermediate 183 gives two regioisomeric alcohols 184 and 185 on hydrolysis, which on further treatment with acetic anhydride generates the desired product 178.133
 |
| | Scheme 48 Catalytic cycle for iodine(III)-catalysed enantioselective diacetoxylation of styrenes 48. | |
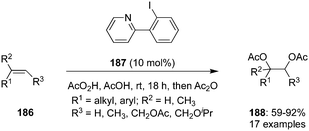
Pyridine-based iodoarene 187 was developed and employed as precatalyst for the iodine(III)-catalysed vicinal diacetoxylation of trisubstituted alkenes 186 in the presence of peracetic acid (Scheme 49).135 The acetoxylation exhibited good functional group tolerance and afforded vicinal diacetoxylation products 188 in good yields. This catalyst acts as a kinetically excellent catalyst due to the Lewis base adduct formation between the pyridine nitrogen and electrophilic iodine(III) centre.
 |
| | Scheme 49 Iodine(III)-catalysed vicinal diacetoxylation of trisubstituted alkenes 186 to 188 using 187 as precatalyst. | |
3.5.2. Fluorination of alkenes.
In recent years, several research groups have established different methodologies for vicinal and geminal difluorinations of alkenes by involving hypervalent iodine catalysis.64 Kitamura and co-workers developed the geminal difluorination of functionalised styrenes 48 with 4-iodotoluene 149 in the presence of mCPBA as oxidant and pyridine·HF as fluorine source to afford the 1,1-difluorinated compounds 189 in 31–66% yield (Scheme 50).136 An increase in reaction time or temperature did not influence the yield of the products. The reaction was applicable for styrenes substituted with alkyl groups and halides.
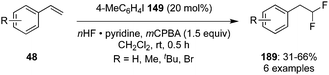 |
| | Scheme 50 Iodine(III)-catalysed difluorination of styrenes 48 to 189 using 4-iodotoluene 149 as precatalyst. | |
4-Iodotoluene 149 was also employed for the vicinal difluorination of terminal olefins 190 using Selectfluor as an oxidant. Vicinal fluorinated products 191 can be obtained in good yields and different functional groups were successfully tolerated under the given reaction conditions (Scheme 51).137 Notably, alcohol containing alkenes were fluorinated with the protection of alcohol functionality. The combination of amine and HF was used as source of fluorine and their ratio is playing a vital role during the progress of this reaction. Fluorination reactions were influenced significantly by the ratio of amine and HF and the best results were obtained with using amine–HF in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.5. Attempts were also made to develop the stereoselective version of the reaction using C2-symmetric chiral iodoarene 136 but only low selectivities were observed. Different reaction conditions and reagents can lead to either a vicinal or geminal difluorination, details are shown below (Scheme 58).
:4.5. Attempts were also made to develop the stereoselective version of the reaction using C2-symmetric chiral iodoarene 136 but only low selectivities were observed. Different reaction conditions and reagents can lead to either a vicinal or geminal difluorination, details are shown below (Scheme 58).
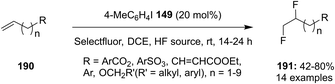 |
| | Scheme 51 Iodine(III)-catalysed 1,2-difluorination of terminal olefins 190 to 191. | |
Later on, the same research group reported vicinal difluorinations of styrenes 48 using the precatalyst 149 in the presence of Selectfluor as the oxidant and in combination of an amine and HF as fluorine source (Scheme 52).138 Fluorinations proceeded smoothly and vicinal difluorinated products 192 were obtained in good yields. The percentage of geminal fluorination was enhanced on increasing the ratio of HF/amine and the geminal fluorination was observed when amine–HF was used in a 1:9.2 ratio. Additionally, an enantioselective catalytic fluorination of styrenes 48 to 192 was also developed by employing the chiral iodoarene 193 (Scheme 52).138 Similar to racemic fluorinations, enantioselective fluorinations proceeded smoothly and vicinal difluorinated products are obtained in good yields in up to 88% enantiomeric excess. The major enantiomer was assigned to have syn configuration based on X-ray analysis. Brønsted acidity of the HF-amine source and deactivating groups in the aromatic ring are significant factors which favours the formation of vicinal difluorination over the geminal by subduing the 1,2-aryl shift. The effect of electronic factors were further validated by computing correlations of the enantioselectivity versus the 13C NMR shift of ipso carbon of the aryl ring and log(ee) versus the Hammett value σ.
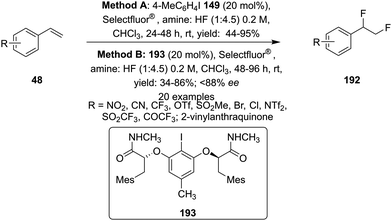 |
| | Scheme 52 Iodine(III)-catalysed vicinal difluorination of alkenes 48 using achiral precatalyst 149 (Method A) and chiral precatalyst 193 (Method B). | |
Jacobsen's research group reported a novel method for the catalytic 1,2-difluorination of trisubstituted olefins 186 using aryl iodide catalyst 194 in the presence of HF–pyridine as the nucleophilic fluoride source and mCPBA as the stoichiometric oxidant (Scheme 53).139 Terminal and internal alkenes, especially with substituents such as amino and nitrogen containing heterocycles, were tolerated under the reaction conditions. anti-Difluorination products 197 and 199 were observed with o-nitro styrenes 196 and acrylamides 198 due to the anchimeric assistance of Lewis basic groups adjacent to the reaction site (Scheme 53).139
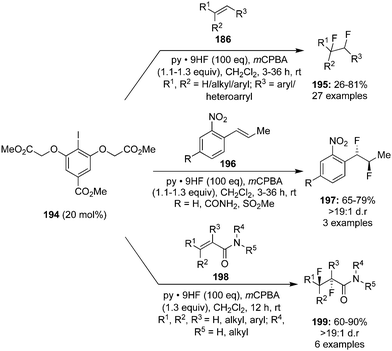 |
| | Scheme 53 Iodine(III)-catalysed 1,2-difluorination of alkenes 186, 196 and 198 using 194 as precatalyst. | |
A stereoselective version of this reaction was endeavoured by using the chiral precatalyst 200 with the same oxidant and fluorine source to afford vicinal anti-difluorination product 199 in 51% yield with 93% ee (Scheme 54).139 The reaction using the chiral precatalyst was much slower compared to the achiral precatalyst.
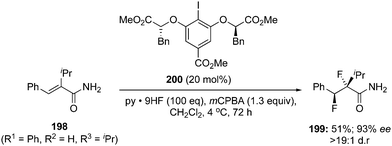 |
| | Scheme 54 Iodine(III)-catalysed enantioselective 1,2-difluorination of alkenes 198 using chiral precatalyst 200 in the presence of mCPBA. | |
The ester moiety of 200 was modified to form another precatalyst 202 for an enantioselective geminal difluorination of tetra-substituted olefins 201 to 203.140 Reactions were performed at relatively at low temperate (−50 °C to −20 °C) and fluorinated products 203 were obtained in good yields with up to 97% ee (Scheme 55).140 Introducing a substituent at benzylic position of the styrenes provided high enantioselectivity. Notably, the cinnamamides and cinnamate esters afforded the desired products 203 in excellent yields (Scheme 55). Tertiary and quaternary stereocenters were generated during these geminal difluorination and cation⋯π interactions played a vital role in achieving the high selectivity. Absolute configuration of the product was assigned based on the single crystal X-ray analysis.
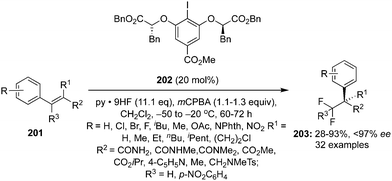 |
| | Scheme 55 Geminal difluorination of olefins 201 to 203 using chiral iodoarene 202 as precatalyst. | |
The same research group reported an enantiocontrolled synthesis of vicinal 1,2-difluorinated products 205 from secondary cinnamamides 204 using hypervalent iodine catalysis.141 Vicinal difluorinated products were obtained in moderate to high yields with up to 98% ee (Scheme 56). Interestingly, anchimeric assistance by the neighbouring tert-butyl amide group suppresses the competing 1,1-difluorination reaction via a rearrangement pathway thereby increasing chemoselectivity towards the formation of vicinal 1,2-difluorinated products 206. Notably, the ratio of geminal difluorination was increased in case of neighbouring Me or Et instead of tBu in the substrates.
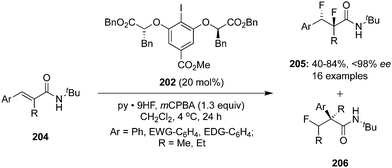 |
| | Scheme 56 Iodine(III)-catalysed enantioselective difluorination of 204 to 205 using C2-symmetric chiral precatalyst 202. | |
Another novel chiral precatalyst 208 was synthesized by Jacobsen and co-workers and employed in the iodine(III)-catalysed enantioselective geminal difluorination of α-bromostyrenes 207 to afford β,β-difluoroalkyl bromides 209 in moderate to excellent yields with up to 93% enantiomeric excess (Scheme 57).142 Electron-deficient bromo styrenes with meta- and para-substituents were tolerated whereas ortho-substituted as well as electron-rich styrenes were not tolerated due to the proclivity of the substrates to engage in selective π interactions with the catalyst in the enantio-determining transition state as revealed by SAPT studies.
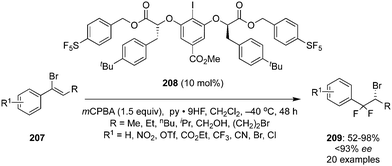 |
| | Scheme 57 Iodine(III)-catalysed enantioselective geminal difluorination of 207 to β,β-difluoroalkyl bromides 209 using chiral iodoarene 208 as precatalyst. | |
A general mechanism for iodine(III)-catalysed vicinal and geminal difluorination of alkenes is shown in Scheme 58.138,143 The catalytic cycle is initiated by the iodoarene oxidation to the active catalytic species 210. This activates the olefinic substrate 48 to afford an iodonium intermediate 211. Iodonium intermediate 211 undergoes ring opening by the nucleophilic attack of fluoride ion to form a common intermediate 212. Intermediate 212 leads the formation of two different reaction products through path ‘a’ and ‘b’. In path ‘a’, intermediate 212 undergoes a nucleophilic substitution reaction with fluoride ion to yield vicinal difluorination product 192. In path ‘b’, intermediate 212 undergoes an aryl migration via formation of phenonium intermediate 213 to provide the geminal fluorination product 189. The mechanism is well supported by theoretical studies.143
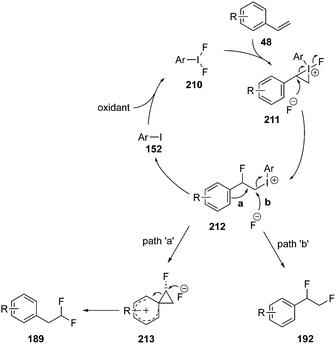 |
| | Scheme 58 General mechanism for vicinal and geminal difluorination of olefins 48 using iodine(III) catalysis. | |
3.5.3. Fluoroaziridination of alkenes.
Hypervalent iodine catalysis can also be efficiently used for the development of aziridinations of electron deficient alkenes.144 Previously, chiral aryl iodide 202 was employed for the iodine(III)-catalysed fluoroaziridination of cinnamyl amines 214 to enantiomerically enriched fluoroaziridines 215 in moderate to good yields (Scheme 59).143
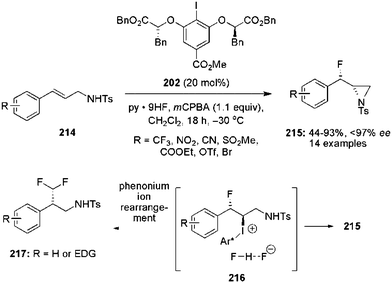 |
| | Scheme 59 Iodine(III)-catalysed anti-β-fluoroaziridination of cinnamyl amines 214 to fluoroaziridines 215 using precatalyst 202. | |
The absolute configuration of the fluoroaziridines 215 was assigned as anti-isomer based on single crystal X-ray analysis. Cinnamyl amines with electron-withdrawing substituents were tolerated well, but electron-rich substrates afforded 1,1-difluoromethylated products 217 instead of the desired fluoroaziridines 215via a phenonium ion rearrangement pathway.138 Moreover, the aziridine ring of anti-fluoroaziridines 215 was opened by using different nucleophiles to obtain enantiomerically enriched anti-fluoroamines.
3.5.4. Epoxidation of alkenes.
Hypervalent iodine catalysis was employed for the epoxidation of electron deficient alkenes 218 using iodobenzene 17 in the presence of terminal oxidant Oxone® using TFA as an additive. Reactions were completed in a short reaction time by using ultrasound as energy source and afforded the epoxides 219 in good yields (Scheme 60).145 Oxone® was particularly selected as oxidant to generate the hypervalent iodine catalytic species in this reaction because of its inertness towards the alkene epoxidation. Substrates with electron-donating substituents gave excellent yields compared to hindered substrates and arenes with electron-withdrawing groups. The scope of the reaction was also extended to styrenes with ester functionality.
 |
| | Scheme 60 Iodine(III)-catalysed epoxidation of β-cyanostyrenes 218 to 219. | |
A possible catalytic cycle for the epoxidation of alkenes 218 to epoxides 219 is given in Scheme 61.145 The catalytic cycle is initiated with the formation of an iodine(III) species 2 by in situ oxidation of iodobenzene 17. The iodine(III) species 2 activates the olefinic double bond and forms an iodonium intermediate 220 which converts to intermediate 221 on the ring opening by the nucleophilic attack by trifluoroacetoxy anion. Finally, intermediate 221 cyclizes intramolecularly to epoxides 219 along with the formation of precatalyst 17. The active catalytic hypervalent iodine species 2 is regenerated by the oxidation of precatalyst 17 to continue the catalytic cycle.
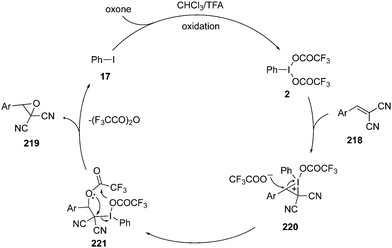 |
| | Scheme 61 Possible catalytic cycle for iodine(III)-catalysed epoxidation of electron deficient alkenes 218 to epoxides 219. | |
3.5.5. Epoxidation of hydroperoxides.
Hypervalent iodine catalysis was used for the dehydration of hydroperoxides.146 Hydroperoxides are useful substrates and provide the corresponding carbonyl compounds through oxidation. Hydroperoxides 222 were oxidized to α,β-unsaturated carbonyl compounds 223 when treated with 10 mol% of IBX 11 as dehydration catalyst in the presence of p-TsOH hydrate in DMSO at room temperature (Scheme 62).147 The iodine(V)-catalysed dehydration of hydroperoxides worked well and both acyclic and cyclic allylic hydroperoxides are successfully converted during these catalytic reactions. In most of the oxidation reactions, enones were obtained in good yields except with acetal based hydroperoxides, which decomposed and formed the products in low yields. It is quite challenging to use allylic hydroperoxides due to their explosive nature.147 To overcome this issue, a promising one pot methodology was developed for the direct conversion of alkenes 224 into enones 225 using C60 as a photosensitizer in an O2 atmosphere using a fluorescent lamp in the presence of IBX 11 (Scheme 62).147 Enones were obtained in good yields except the amino based cyclic olefins. Acyclic enones were obtained in only moderate yields probably due to the low selectivity of singlet oxygen in ene reactions of acyclic alkenes. Low concentration of hydroperoxides in 1H NMR studies clearly supports the better safety profile of this one pot oxidation compared to the direct oxidation of hydroperoxides generated by singlet oxygen.147
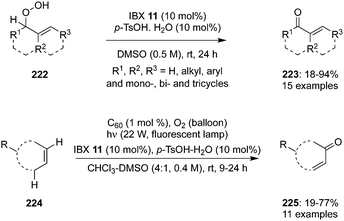 |
| | Scheme 62 Iodine(V)-catalysed oxidation of allylic hydroperoxides 222 and alkenes 224 to enones 223 and 225. | |
3.6. Oxidation of benzylic C–H bonds
Oxidation of benzylic C–H bonds is an important reaction in organic synthesis and a number of hypervalent iodine mediated approaches have been used to achieve these oxidation reactions.148,149 In 2020, Maruoka and co-workers reported the application of a hypervalent iodine(III) reagent as redox-neutral catalyst for the selective benzylic C–H oxidation of various arenes 226 to the corresponding carbonyl derivatives 228 at room temperature by employing polymeric iodosyl-benzene (PhIO)n86 as catalyst and AlNO3 as oxidant.150 Monomeric PhIO, the active iodine(III) species, is generated in situ by the depolymerisation of (PhIO)n228 using aluminium nitrate as reagent (Scheme 63). Interestingly, only arenes that are moderately activated by electron-donating groups were reactive in these oxidations. The current protocol is inefficient for arenes which are strongly activated with electron-rich groups and N-heterocycles, the former leads the over-oxidation products (benzoic acids) and the latter was unreactive due to the Lewis basicity of nitrogen.
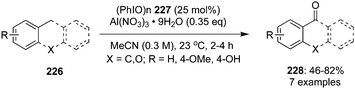 |
| | Scheme 63 Benzylic oxidation of arenes 226 using hypervalent iodine catalysis. | |
Another catalytic approach for benzylic C–H oxidation was developed by Nachtsheim and co-workers.151 An enantioselective hydroxylation of alkyl arenes 229 to 331 with a newly designed triazole substituted chiral precatalyst 230 was communicated, which acts not only as a halogen donor for the non-stereoselective radical halogenation but also as a chiral ligand during these enantioselective oxidations. This methodology involved irradiation of alkyl arenes 229 with blue LED's in the presence of chiral aryl iodide 230 (15 mol%), mCPBA (2.5 eq.), CuBr (20 mol%) in combination with NaBr (1.5 eq.) in acetonitrile at room temperature (Scheme 64).151 Various substrates were found to be compatible with this current protocol to afford the corresponding benzylic alcohols in moderate to good yields with excellent enantioselectivities.
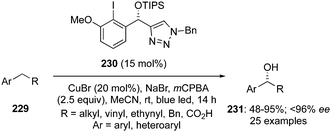 |
| | Scheme 64 Iodine(III)-catalysed enantioselective benzylic hydroxylation of 229 to 231 using chiral precatalyst 230. | |
4. α-Functionalisations of ketones
Functionalisation of carbonyl compounds in the α-position employing hypervalent iodine reagents is another key reactions of organic synthesis.21 Additionally, hypervalent iodine catalysis has been proved a quite useful approach for the developing these reactions.3 In this section, various α-functionalisations of carbonyl compounds will be discussed.
4.1. α-Oxytosylation of ketones
Basdevant and Legault achieved the α-oxytosylation of acetyl enol ether 232 using hypervalent iodine catalysis in the presence of PhI 17, mCPBA and p-TsOH·H2O. It was required to add the precursor 232 in portions and only then α-tosyloxy ketone 233 was obtained in 74% yield (Scheme 65).152 It was observed that the catalytic reaction was quite slow compared to the stoichiometric reaction. Under catalytic reaction conditions, acetyl enol ethers 232 served as suitable substrates due to their low nucleophilicity and easy availability.151
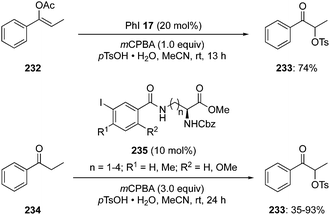 |
| | Scheme 65 Iodine(III)-catalysed α-oxytosylation of acetyl enol ether 232 and propiophenone 234 to α-tosyloxy ketone 233. | |
Furthermore, Whitehead and coworkers designed and synthesised a series of iodoarenes 235 coupled with diamino acids and the reactivity of these catalysts was assessed for α-oxytosylation of propiophenone 234 (Scheme 65). These pre-catalysts 235 allow the α-oxytosylation of propiophenone 234 in 35–93% yield. Notably, there was no asymmetric induction observed during these reactions.153
In the continuation of searching for high selectivities during these reactions, a novel C2-symmetric chiral iodoarene 236 was synthesised and used as precatalyst to transfer the chirality in the presence of terminal oxidant during the α-oxytosylation of acetyl enol ether 232.154 Once again, the catalytic reaction was found slower than the stoichiometric reaction, but the α-oxytosylated product 233 was obtained as (S)-isomer in 90% ee (Scheme 66). Notably, the same precatalyst 236 showed moderate selectivity in the direct oxytosylation of propiophenone 234.154
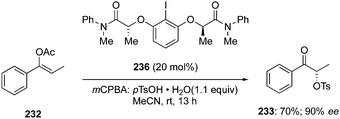 |
| | Scheme 66 Iodine(III)-catalysed α-oxytosylation of acetyl enol ether 232 to enantiomerically enriched α-tosyloxy ketone 233. | |
Recently, the concept of “hypervalent twist” was effectively used to develop more reactive hypervalent iodine reagents.155–159 In case of twisted hypervalent iodine reagents, the presence of ortho-substituents leads to an out-of-plane distortion that destabilises the hypervalent iodine reagents.160
The same concept was used to design and synthesis of N-heterocyclic substituted iodoarene precatalyst (NHIA) 238 which was used for α-oxytosylation of ketones 237 in the presence of mCPBA (Scheme 67).161 Notably, very low catalytic loadings (1 mol%) were efficient the catalyse these reactions successfully and in good yields. The reaction showed tolerance for both aromatic and aliphatic ketones but α-oxytosylation of aromatic ketones provided better yields. Aromatic ketones bearing electron donating functionalities exhibited lower yields during these transformations. Moreover, the same reaction condition was applied for the α-oxytosylation of cyclic ketones.
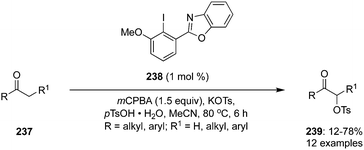 |
| | Scheme 67 Iodine(III)-catalysed α-oxytosylation of ketones 237 by using N-heterocyclic substituted iodoarene precatalyst (NHIA) 238. | |
4.2. α-Acetoxylation of ketones
In 2020, Wirth and Hokamp reported an iodine(III)-catalysed enantioselective α-acetoxylation of acetyl enol ethers 232 with high enantioselectivities (up to 88% ee) using hypervalent iodine catalysis (Scheme 68).162 This methodology required resorcinol/lactamide-based chiral aryl iodide 112 as precatalyst in the presence of mCPBA and the additive BF3·OEt2. Aromatic moieties with substituents such as halogens, nitro, alkyl and methoxy groups were well tolerated but substrates having electron withdrawing groups at the aromatic ring gave higher yields. The enantioselectivity was influenced by the nature and position of the functional group present in the aromatic ring. High selectivities were observed in case of substrates with electron withdrawing groups while enantiomeric excess was reduced drastically when the substitution was present at the sterically more demanding ortho-position.
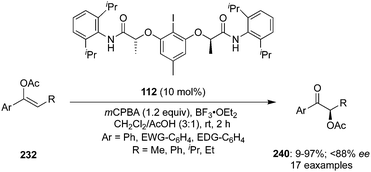 |
| | Scheme 68 Enantioselective α-acetoxylation of acetyl enol ethers 232 to α-acetoxylated ketones 240 using iodine(III) catalysis. | |
A plausible mechanism for the iodine(III)-catalysed α-acetoxylation of acetyl enol ethers 232 is shown in Scheme 69. The catalytic cycle is initiated by the formation of an active iodine(III) catalytic species 241 through oxidation of the chiral aryl iodide catalyst 112 with the terminal oxidant. The catalytic species 241 is further activated by boron trifluoride etherate and undergoes a ligand exchange with the substrate 232 to form another intermediate 242. The subsequent SN2 displacement provides α-acetoxylated ketones 240 with regeneration of precatalyst 112.162
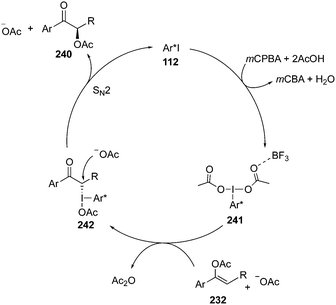 |
| | Scheme 69 Catalytic cycle for an iodine(III)-catalysed α-acetoxylation of acetyl enol ethers 232 using precatalyst 112. | |
4.3. α-Fluorination of ketones
Fluorination of carbonyl compounds is an interesting reaction for the formation of C–F bonds.43 Shibata and co-workers reported the α-fluorination of 1,3-dicarbonyl compounds 243 catalysed by 4-iodotoluene 149 using mCPBA as oxidant and HF/pyridine as fluorine source (Scheme 70).163 The targeted tertiary α-fluorinated compounds 244 were obtained in moderate to excellent yields. In the case of α-fluorination, aromatic compounds with electron-withdrawing as well as electron-donating substituents, aliphatic and heteroaromatics substrates were tolerated. In addition, the reaction of cyclic/acyclic tertiary β-ketoesters provided α-fluorinated-β-ketoesters with a quaternary stereogenic centre in good yields.
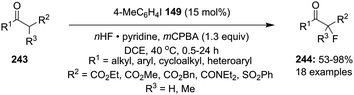 |
| | Scheme 70 Iodine(III)-catalysed fluorination of β-ketocarbonyl compounds 243 using 149 as precatalyst. | |
A versatile asymmetric α-fluorination of cyclic β-keto esters 245 using the C2-symmetric aryl iodide catalyst 246a or 246b in the presence of mCPBA and triethylamine pentafluoride as fluoride source was developed by Rueping and co-workers.164 Enantiomerically enriched α-fluorinated carbonyl compounds 247 bearing quaternary stereocenter were obtained in good yields with up to 83% ee (Scheme 71). It was observed that precatalyst 246b exhibited better selectivities compare to 246a, probably due to the presence of the electron-withdrawing functionality at C3 position. Interestingly, the precatalysts can be recovered and reused without loss of selectivity and catalytic potential. The reaction mechanism includes in situ formation of chiral hypervalent iodine difluoride source which is supported by the computational studies. Different cyclic esters with electron-withdrawing and electron-donating substituents, irrespective of their position in the aromatic ring, were tolerated. Furthermore, a similar approach was developed by Zheng and co-workers using a novel planar chiral iodoarene 248 with a [2.2]paracyclophane motif for the enantioselective α-fluorination of β-ketoesters and β-ketoamides 245, affording the enantiomerically enriched α-fluorinated carbonyl compounds 247 (Scheme 71).165 It was observed that the enantioselectivity increases with the size of the ester group of the β-ketoesters.
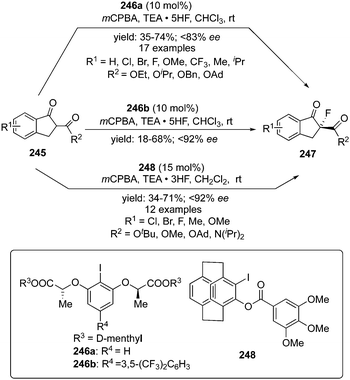 |
| | Scheme 71 Iodine(III)-catalysed enantioselective fluorination of β-keto esters 245 to the corresponding α-fluorinated chiral compounds 247 using chiral precatalysts 246 and 248. | |
5. Cyclisation reactions
In past decades, a number of cyclisation reactions have been developed using hypervalent iodine reagents.23 These reactions constitute an integral part of organic synthesis as they lead to the formation of several biologically important heterocycles.15 More importantly, hypervalent iodine catalysis has played a significant role in the progress of these reactions.3 In this section, both intramolecular cyclisations and intermolecular annulations with hypervalent iodine catalysis are highlighted.
5.1. Intramolecular cyclisations
Intramolecular cyclisation reactions have been extensively used to achieve different oxygen- and nitrogen-containing heterocyclic scaffolds. Synthesis of three-membered N- and O-heterocycles144,145 was already discussed in the oxidation of alkenes (Section 3.5). Herein, the application of hypervalent iodine catalysis for the construction of five- and six-membered heterocycles will be discussed.
5.1.1. Synthesis of O-heterocycles.
5.1.1.1. Synthesis of lactones.
The synthesis of chiral 4-fluoroisochromanones 251 was achieved by Jacobsen and co-workers with excellent enantio- and diastereoselectivities.166 The reaction involves catalytic fluorolactonisation of vinyl benzoates 249 with the aid of chiral aryl iodide 250 in the presence of mCPBA by taking HF·py in the ratio 1:9 as the fluoride source (Scheme 72). A syn diastereoisomer with 35–86% yield and up to 96% ee is formed in this reaction by the nucleophilic displacement of the aryliodonium group in the intermediate 252 with the aid of anchimeric assistance of the carboxylate functionality. Various electron-withdrawing and electron-donating groups at the aryl moiety are tolerated.
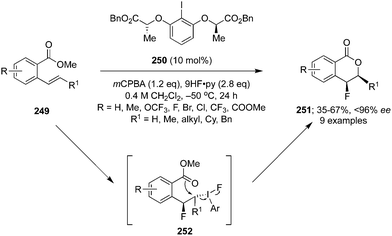 |
| | Scheme 72 Iodine(III)-catalysed fluorolactonisation of vinyl benzoates 249 to 251. | |
Furthermore, an enantioselective sulfonyloxylactonisation and phosphoryloxylactonatisation of 4-pentenoic acid derivatives 253 was reported by Masson and co-workers using C2-symmetric chiral iodoarene 254 as precatalyst in the presence of mCPBA as oxidant (Scheme 73).167 This method enabled a straightforward synthesis of sulfonyloxy- and phosphoryloxy-γ-butyrolactones 256 and 258 in good yields with moderate to high enantioselectivities. Both, 4-pentenoic acids and 1-allylcycloalkane carboxylic acids afforded γ-lactones and spirolactones, respectively, in good yields.
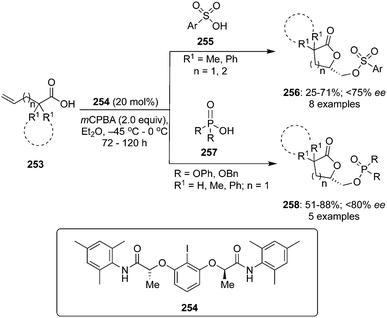 |
| | Scheme 73 Iodine(III)-catalysed sulfonyloxylactonisation and phosphoryloxylactonisation of 253 using 254 as precatalyst in the presence of mCPBA. | |
A possible catalytic cycle for the sulfonyloxylactonisation of iodine(III) catalysis is shown in Scheme 74. The catalytic cycle is initiated with the oxidation of chiral aryl iodide 254 to iodine(III) species 259 by mCPBA in the presence of sulfonic acid 255. The iodine(III) intermediate 259 activates the double bond of olefinic acid 253 to form the chiral iodonium intermediate 260. Iodonium intermediate 260 undergoes an intramolecular cyclisation to form the lactone intermediate 261. Finally, lactone intermediate 261 provides sulfonylated lactones 256 and regenerates the catalyst to continue the catalytic cycle.167
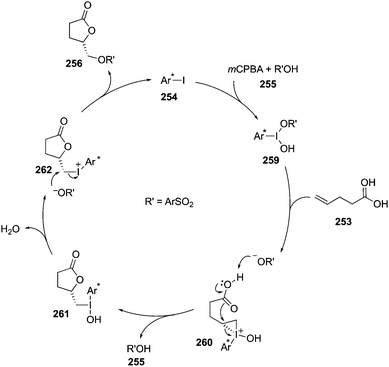 |
| | Scheme 74 Catalytic cycle for the hypervalent iodine(III)-catalysed sulfonyloxylactonisation of 4-pentenoic acid derivatives 253 using C2-symmetric chiral iodoarene 254 as precatalyst. | |
Later, Hilt and co-workers developed an electrochemical approach for the lactonisation of vinyl benzoates 249 mediated by hypervalent iodine(III) catalysis using PhI 17 as precatalyst in the presence of lithium perchlorate as electrolyte and trifluoroacetic acid to form trifluoroethoxy-substituted isochromanones 263 in moderate to good yields (Scheme 75).168 The scope of the reaction was expanded by changing the steric and electronic components of the substrates; only functional groups labile to oxidative conditions show low yields. Moreover, N-heterocyclic substituted iodoarene precatalyst (NHIA) 238 was also employed to achieve similar lactonisations.161
 |
| | Scheme 75 Iodine(III)-catalysed lactonisation of vinyl benzoates 249 using iodobenzene 17 as precatalyst. | |
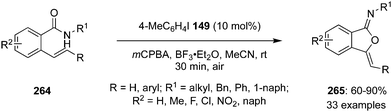 |
| | Scheme 76 Iodine(III)-catalysed intramolecular oxy-cyclisation of 2-vinylbenzamides 264 to benzoiminolactones 265 using 149 as precatalyst. | |
An environmental friendly synthesis of 2-arylbenzofurans 267 was developed by indine(III)-catalysed intramolecular cyclisation of o-hydroxystilbenes 266 with the help of PIDA 1 as catalyst (Scheme 77).171 The reaction is performed at room temperature and required a longer time to complete as unwanted side products were formed at higher temperatures. These challenges were overcome by performing the reaction using ultrasound and within a short reaction time, the desired products were obtained in good to excellent yields. The same cyclisations were developed by in situ generated active catalysts.172
 |
| | Scheme 77 Iodine(III)-catalysed intramolecular cyclisation of o-hydroxystilbenes 266 to 2-arylbenzofurans 267 using PhI(OAc)21 as catalyst in the presence of mCPBA. | |
In 2020, Wang et al. developed an oxidative fluorocyclisation of 1,1-disubstituted styrenes 268 with an internal oxygen nucleophile using in situ generated chiral iodine(III)-catalysts. The reaction employed C2-symmetric aryl iodides 269 as catalyst with HF–pyridine as fluorine source and mCPBA as oxidant. The fluorinated cyclised products 270 were obtained in good yields with up to 96% ee (Scheme 78).173 The fluorocyclisation of para-substituted styrenes 269 proceeded with high selectivity compared to meta-substituted derivatives, whereas low selectivities and a decreased reactivity was observed in ortho substituted styrenes. Optimisation of various chiral catalysts revealed that increasing the steric demand of the α-substituent R1 in 269 improved the stereoselectivity of the fluorocyclisation. Both catalysts 269 were able to induce high enantioselectivities, particularly the newly developed 1-naphthyllactate catalyst (R,R)-269b yielded higher ee values than the mesityl analogue (R,R)-269a. Additionally, the synthesis of fluorinated pyrrolidines via an aminofluorination of styrenes was achieved under similar conditions.
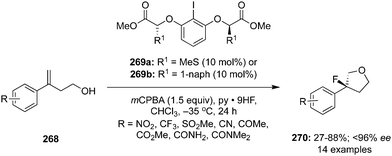 |
| | Scheme 78 Iodine(III)-catalysed enantioselective fluorocyclisation 1,1-disubstituted styrenes 268 to fluorinated cyclised products 270 using precatalysts 269. | |
5.1.2. Synthesis of N-heterocycles.
5.1.2.1. Synthesis of piperidines.
Hypervalent iodine catalysis has been successfully used to synthesise fluorinated piperidines 272 by involving an intramolecular aminofluorination of ω-aminoalkenes 271 using 4-MeC6H4I 149/nHF·pyridine/mCPBA catalytic system (Scheme 79).163 Cyclisations proceeded smoothly and various fluorinated piperidines 272 with alkyl, aryl and cyclic substituents were obtained in 31–77% yield. Notably, efforts were made to develop an asymmetric variant of this reaction, but only moderate enantiomeric excesses were obtained (not shown).
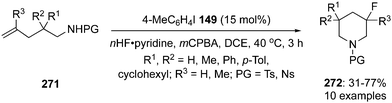 |
| | Scheme 79 Iodine(III)-catalysed synthesis of fluorinated piperidines 272 by involving an intramolecular aminofluorination of alkenes 271. | |
The proposed catalytic cycle for the hypervalent iodine-catalysed cyclisation of alkenes 271 to fluorinated piperidines 272 is shown in Scheme 80. The oxidant oxidises the precatalyst 149 to ArIO 273 which, in turn, reacts with HF to form the electrophilic hypervalent iodine species ArIF2274. ArIF2274 reacts with alkenes 271 to form intermediate 275 with the liberation of HF. Furthermore, the intermediate 275 converts into aziridinium intermediate 277via formation of intermediate 276. Final nucleophilic attack of fluoride ion to aziridinium intermediate 277 affords the fluoropiperidine 272 along with the regeneration of precatalyst 149 to continue the catalytic cycle. To improve the yield, HF has to be used in excess because of the reversible nature of the reaction from ArIF2274 to ArIO 273 and then due to a competitive hydroxylation reaction.163
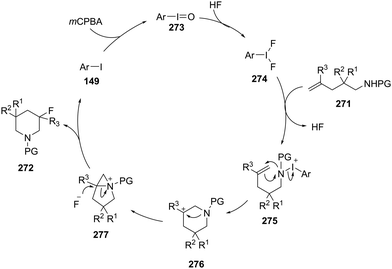 |
| | Scheme 80 Catalytic cycle for the hypervalent iodine-catalysed cyclisation of alkenes 271 to piperidines 272 using 149 as precatalyst. | |
Furthermore, N-heterocyclic substituted iodoarene precatalyst (NHIA) 238 was employed for the hypervalent iodine-catalysed cyclisation of ortho-phenyl acetanilide 278 to N-acyl carbazole 280 (Scheme 81).161 Notably, the ‘twisted’ hypervalent iodine species was generated during the progress of this reaction and N-acyl carbazole 280 was obtained in 77% yield. Moreover, another precatalyst 279 of the same series was also used and gave carbazole 280 in 49% yield (Scheme 81).161 Peracetic was used as terminal oxidant to generate active catalytic species.
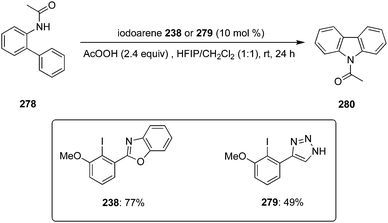 |
| | Scheme 81 Iodine(III)-catalyzed cyclisation of ortho-phenyl acetanilide 278 to N-acyl carbazole 280 using N-heterocyclic substituted iodoarene precatalyst (NHIA) 238 and 279. | |
5.1.3. Synthesis of O,N-heterocycles.
5.1.3.1. Synthesis of oxazoles.
Punniyamurthy and co-workers developed a simple and efficient catalytic procedure for the synthesis of benzoxazoles 283 and benzothiazoles 285 by intramolecular cyclisation of arylanilides 281 and arylthioanilides 284, respectively (Scheme 82).174 This transformation employed 1-iodo-4-nitrobenzene 282 as precatalyst, Oxone® as oxidant in the presence of HFIP at room temperature. Anilides with halogen substituents afforded the desired products in good to excellent yields whereas nitro substituents were not compatible with the reaction. Amide groups with aryl, heteroaryl and alkyl substituents were tolerated. This protocol was successfully extended to gram scale.
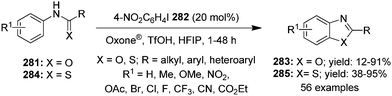 |
| | Scheme 82 Iodine(III)-catalysed synthesis of benzoxazoles 283 and benzothiazoles 285. | |
The proposed catalytic cycle for the iodine(III)-catalysed cyclisation of arylanilides 281 to benzoxazoles 284 is shown in Scheme 83. As usual, the active hypervalent iodine species 286 is generated by the oxidation of aryl iodide 282 which reacts with substrate 281 to form new hypervalent iodine species 287. Furthermore, species 287 undergo an intramolecular cyclisation to form a cationic intermediate 288. Finally, the cationic intermediate 288 gave benzoxazoles 283 through deprotonation and releases aryl iodide 282 to re-enter into the catalytic cycle.174
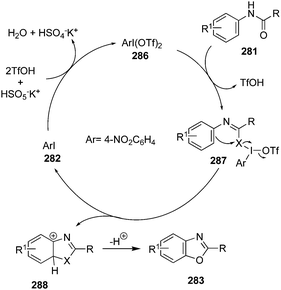 |
| | Scheme 83 The proposed catalytic cycle for iodine(III)-catalysed intramolecular cyclisation of arylanilides 281 to benzoxazoles 283. | |
In 2015, Moran and co-workers developed an intramolecular cyclisation of N-alkenylamides 289 for the synthesis of five to seven membered ring systems 291 containing both nitrogen and oxygen atoms (Scheme 84).175 The catalytic system employed 2-iodoanisole 290 as precatalyst, Selectfluor as oxidant and TFA as additive. The cyclisation was not effective when mCPBA or Oxone® were used as oxidants. An array of electron rich and electron poor aryl amides were cyclised to obtain the corresponding products in good yields. Moreover, an enantioselective synthesis of isoxazoline 291a from 289a (R1 = Ph; R2 = H and n = 1) was accomplished by the use of 47 as chiral iodine precatalyst, but the product was obtained in low yield with 69% ee (Scheme 84).175
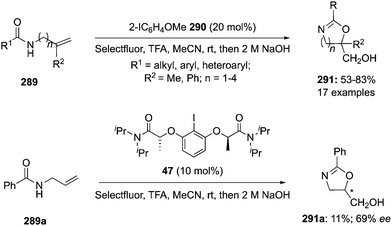 |
| | Scheme 84 Iodine(III)-catalysed intramolecular cyclisation of N-alkenylamides 289 to heterocycles 291 using achiral 290 and chiral 47 precatalyst. | |
Similar substrates 289 were cyclised to 2-oxazolines 292 with an exocyclic fluoromethyl group in the presence of p-methyliodobenzene 149 as precatalyst, Selectfluor and a mixture of triethylamine tris(hydrogenfluoride) (Et3N·3HF) and Olah's reagent (Py·HF) as fluoride source (Scheme 85).176 This cyclisation reaction was compatible with several functional groups and extended to prepare six-membered rings.
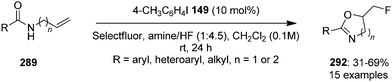 |
| | Scheme 85 Iodine(I)/iodine(III) catalysis for the cyclisation of 289 to 2-oxazolines 292. | |
N-Propargyl amides 293 were cyclised to oxazoles 295 in good yields by iodine(III) species ArIF2, generated in situ from 4-iodoanisole 294 in the presence of Selectfluor and HF–pyridine as the fluoride source (Scheme 86).177 Aromatic as well as aliphatic amides were tolerated. Internal alkynes and amides containing haloarenes were found futile as substrates. Later, similar cyclisations were achieved in moderate yields by treating N-propargyl amides 293 with bisulfonyl(imides) 41 using PhI 17 as the precatalyst, Oxone® as oxidant and TBAHSO4 (TBA: tetra-n-butylammonium) as a phase transfer reagent178 or by using precatalyst (2-IC6H4OMe) 290 in combination with mCPBA.179
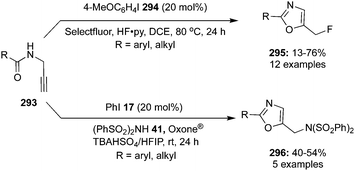 |
| | Scheme 86 Iodine(III)-catalysed cyclisation of N-propargyl amides 293 to oxazoles 295 and 296 using 294 and 17 as precatalyst. | |
An one-pot protocol for the synthesis of oxazole-5-carbaldehydes 297 was developed by the cyclisation of N-propargylamides 293 using the PIDA/LiI catalytic system in the presence of oxygen under irradiation with visible light. This process involves an iodocyclisation followed by oxidative deiodination and cyclised products 297 were isolated in good to excellent yields (Scheme 87).180
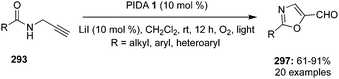 |
| | Scheme 87 PIDA-catalysed cyclisation of N-propargylamides 290 to oxazole-5-carbaldehydes 294. | |
The mechanism for PIDA-catalysed cyclisation of N-propargylamides 293 to oxazole-5-carbaldehydes 297 is given in Scheme 88. The reaction is initiated by the PIDA mediated oxidation of iodide to iodine monoacetate which induced the cyclisation of substrate 293 to cyclic intermediate 299. Under the visible light, C–I cleaves homolytically and forms radical 300 along with an iodine radical. The radical 300 reacts with oxygen to form peroxy radical intermediate 301 which is subsequently converted to another radical intermediate 302. Intermediate 302 rearranges to radical species 303 that gave the final aldehyde product 297 along with a hydroxyl radical. The hydroxyl radical reacts with iodine radical to form HIO that produced iodine on decomposition to continue the catalytic cycle.180 Moreover, the tetrabutylammonium iodide (TBAI) has been also employed as precatalyst in the presence of terminal oxidant to develop the synthesis of similar oxazole scaffolds.181
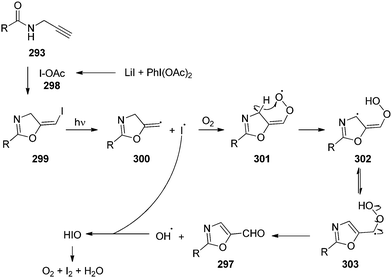 |
| | Scheme 88 Mechanism for PIDA-catalysed cyclisation of N-propargylamides 293 to oxazole-5-carbaldehydes 297. | |
5.1.4. Synthesis of carbocycles.
Polycyclic aromatic hydrocarbons 305 were easily prepared by Murphy and others via oxidative intramolecular C–H coupling of styrenes 304 containing arene and alkene functionalities. These cyclisations were achieved using 4-iodotoluene 149 as precatalyst, mCPBA as oxidant, BF3·OEt2 as additive and chlorobenzene as solvent (Scheme 89).182 Polysubstituted phenanthrene derivatives were successfully prepared in moderate to high yields. Among the various functional groups, only very strong electron-withdrawing substituents such as NO2, Ac, COOMe and CF3 on the vinyl as well as arene moiety were not found suitable during these cyclisations. The scope of the reaction was also expanded for the formation of tetra and pentacyclic aromatics.
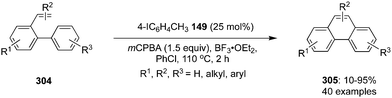 |
| | Scheme 89 Iodine(III)-catalysed oxidative intramolecular C–H coupling of styrenes 304 to polycyclic aromatic hydrocarbons 305. | |
5.2. Intermolecular annulations
Like intramolecular cyclisations, intermolecular reactions have been extensively used for developing the synthesis of various heterocycles under metal-free reaction conditions. Applications of hypervalent iodine catalysis to achieve intermolecular annulations until 2013 are reviewed in our previous article.3
5.2.1. Synthesis of N-heterocycles.
Various hypervalent iodine-catalysed intermolecular annulations have been used to construct nitrogen-containing heterocycles. Isoquinolones 308 were synthesised by the cycloaddition of alkynes 307 and benzamides 306 using catalytic amounts of iodobenzene 17 in HFIP in the presence of peracetic acid (Scheme 90).183 Notably, a significant increase in the yield was observed by the portionwise addition of the oxidant. Electron-withdrawing and electron-donating groups in alkynes were tolerated and some regioselectivity was witnessed in the case of unsymmetrically substituted diarylacetylenes. Irrespective of the position of electron-donating and electron neutral substituents on the aryl ring, various N-alkoxy benzamides gave the isoquinolones 308 in 46–85% yield.183
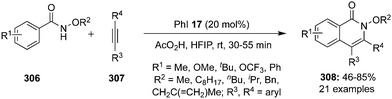 |
| | Scheme 90 Iodine(III)-catalysed cycloaddition of benzamides 306 with alkynes 307 to isoquinolones 308 using PhI 17 as precatalyst. | |
In 2017, Mal and co-workers reported the synthesis of multi-substituted carbazoles 310 in moderate to good yields via an intermolecular dehydrogenative annulation reaction of anilides 309 and arenes 16 (Scheme 91).184 A trace amount of minor isomer 311 was also observed. Both, a stoichiometric and an organocatalytic version of this reaction were developed. The catalytic condition involved the use of iodobenzene 17 as the precatalyst, mCPBA as the oxidant and HFIP/dichloromethane as solvent at room temperature. Various anilides with electron-withdrawing and electron-donating substituents at para-position and arenes with alkyl/aryl/alkoxy groups provided the desired products and sulfonyl or carbonyl groups on nitrogen atoms of the anilides were also tolerated. The catalytic pathway was found to be less effective compared to the stoichiometric one.
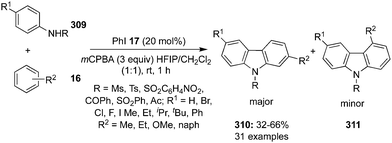 |
| | Scheme 91 Iodine(III)-catalysed intermolecular dehydrogenative annulation reaction of anilides 309 and arenes 16. | |
The synthesis of benzimidazoles 313 and quinoxalines 315 was developed by Kamal and co-workers using hypervalent iodine catalysis.185 The condensation of o-phenyldiamines 312 with various aryl or heteroaryl aldehydes 97 afforded the corresponding benzimidazoles 313 in good to excellent yields (Scheme 92). Moreover, the condensation of o-phenyldiamines 312 with benzil 314 provides quinoxalines 315 in excellent yields (Scheme 92). During these annulations, very low catalytic loading of Koser's reagent 4 (1 mol%) was sufficient to achieve the products in high yields.
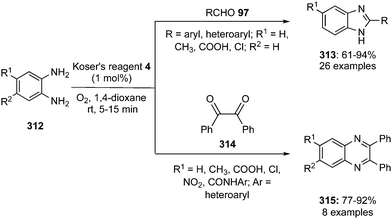 |
| | Scheme 92 Iodine(III)-catalysed synthesis of benzimidazoles 313 and quinoxalines 315 using Koser's reagent 4 as catalyst. | |
5.2.2. Synthesis of N,O-heterocycles.
In the past years, organocatalysis involving hypervalent iodine catalysts has been used to construct various N,O-heterocycles.3 Zhdankin and co-workers developed a catalytic system using hypervalent iodine(III) reagents for the synthesis of pyrrolo isoxazolines 320via the oxidative cycloaddition of aldoximes 316 with maleimide 319 (Scheme 93).186 This catalytic protocol involves an in situ generation of the cyclic iodine(III) species 318 (IBA-OTf) by oxidation of 2-iodobenzoic acid 317 with mCPBA in the presence of trifluoromethanesulfonic acid. Various substituted aromatic aldoximes 316 with electron-rich and electron-poor aryl rings were tolerated. The same research group reported also a similar catalytic protocol for the oxidative cycloaddition of aldoximes 316 with organonitriles 321 to prepare 1,2,4-oxadiazoles 322 (Scheme 93).187 Moreover, similar substrates were employed in the annulations using modified reaction conditions.188
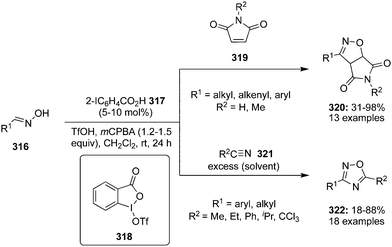 |
| | Scheme 93 Iodine(III)-catalysed oxidative cyclisation of aldoximes 316 with maleimide 319 and organonitriles 321 to N,O-heterocycles 320 and 322. | |
2,4-Disubstituted and 2,4,5-trisubstituted oxazoles 325 were synthesised regioselectively through a [2+2+1] addition of internal as well as terminal alkynes 307, nitriles 321 and oxygen atoms employing iodine(III) catalysis. The scope of the reaction was examined by using two different precatalyst PhI 17 and 4-ClC6H4I 323 in the presence of mCPBA and Tf2NH. All the cyclisation reactions were performed at room temperature and cyclised products were obtained in moderate to good yields (Scheme 94).189 Mechanistic studies showed the involvement of iodine(III) species in the catalytic cycle and the active catalytic iodine(III) intermediate PhI(OH)NTf2324 was isolated. The hypervalent iodine catalysis was also employed for the construction of isoxazole systems.190 Moreover, a few other catalytic systems have been used to build similar scaffolds.191
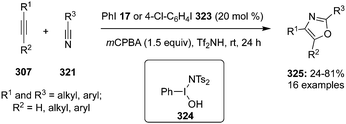 |
| | Scheme 94 Iodine(III)-catalysed cycloaddition of alkynes 307 with nitriles 321 to trisubstituted oxazoles 325. | |
5.2.3. Synthesis of N,S-heterocycles.
Wang and co-workers established a catalytic protocol for the preparation of various thiadiazole scaffolds 328 through an intermolecular oxidative annulation via the formation of an intermediate 327 formed from aldehydes 97 and thiosemicarbazide 326 employing a catalytic method consisting of iodobenzene 17, H2O2 and AcOH (Scheme 95).192 Mono- and di-substituted aryl aldehydes irrespective of the position of functional groups gave moderate to excellent yields. Naphthyl, heteroaryl and alkyl aldehydes were also tolerated.
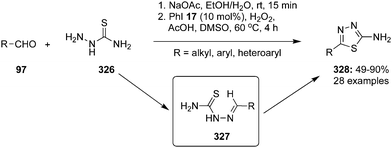 |
| | Scheme 95 Iodine(III)-catalysed oxidative cyclisation of thiosemicarbazides 326 with aldehydes 97 to thiadiazoles 328 using iodobenzene 17 as precatalyst. | |
Kamal and co-workers described the synthesis of benzothiazoles 330 by the condensation of 2-aminothiophenol 329 with aromatic aldehydes 97 using Koser's reagents as catalyst in the presence of molecular oxygen. Reactions were completed in short reaction time and afforded the benzothiazoles 330 in excellent yields (Scheme 96).185 During these cyclisations, a very low catalytic loading (1 mol%) was sufficient for catalysis to the cyclised products in high yields. Notably, aromatic aldehydes bearing electron donating groups showed slightly better yields compare to substrates with electron withdrawing groups.
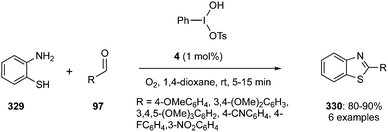 |
| | Scheme 96 Iodine(III)-catalysed synthesis of benzothiazoles 330 using Koser's reagent 4 as catalyst in the presence of molecular oxygen. | |
6. Oxidative rearrangements
Hypervalent iodine reagents are known for activating the olefinic double bonds and later they behave as good leaving groups.23 Additionally, they can participate in the formation of cationic intermediates that lead to variety of rearrangement reactions.35 Initially, the focus of hypervalent iodine chemists was on the developments of oxidative rearrangements using hypervalent iodine reagents in stoichiometric amounts but later rearrangements have been developed using hypervalent iodine catalysis.3 Some rearrangements have already been described in Section 3.5.2 discussing the geminal difluorination of alkenes.136,142
6.1. 1,2-Aryl/alkyl migration reactions
In 2018, Mal and co-workers developed an iodine(III)-catalysed C–H functionalisation of N-(5-bromo-2′,4′,6′-triethyl-[1,1′-biphenyl]-2-yl)methanesulfonamide 331 to carbazole 332 along with an 1,2-migration of an ethyl group.193 The reaction was carried out by using 20 mol% of iodobenzene 17 in the presence of mCPBA in trifluoroethanol and the rearranged product 332 was obtained in 48% yield (Scheme 97). Notably, the stoichiometric version of the same reaction was also developed and rearranged products were obtained in better yields compare to the catalytic reaction.
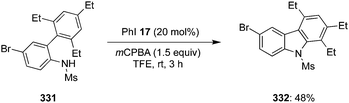 |
| | Scheme 97 Iodine(III)-catalysed C–H functionalisation of biphenylsulfonamide 331 to carbazole 332 along with 1,2-ethyl group migration. | |
An enantioselective rearrangement of allylic alcohols 333 using (S)-proline-derived chiral iodoarene 334 as precatalyst assisted by the Brønsted acid p-TsOH was described by Gong and co-workers.194 Brønsted acids promote the formation of ethers from allylic alcohols whereas the chiral aryl iodide catalyses the 1,2-aryl migration to afford chiral α-arylated-β-alkoxylated ketones 335 in good yields and with excellent enantiomeric excess (Scheme 98).194 The presence of electron withdrawing and electron donating groups at para- or meta-position of the phenyl rings of allylic alcohols are well tolerated. Moreover, N-heterocyclic substituted chiral iodoarene precatalyst (NHIA) 230 was also employed to perform these rearrangements under almost similar catalytic reaction conditions and the selectivity was increased to up to 98% ee.195 The same rearrangement was also achieved by using another N-heterocyclic substituted achiral precatalyst 238 in moderate yields.160
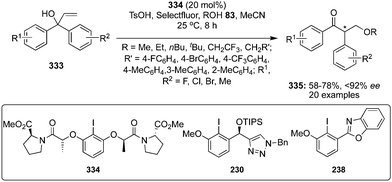 |
| | Scheme 98 Iodine(III)-catalysed enantioselective 1,2-aryl migration of allylic alcohols 333 to ketones 335 using C2-symmetric iodoarene 334 and N-heterocyclic substituted chiral iodoarene 230 and achiral iodoarene 238 as precatalysts. | |
A plausible mechanistic pathway for the enantioselective 1,2-aryl migration in allylic alcohols 333 catalysed by in situ generated chiral iodine(III) reagent is shown in Scheme 99.193 Initially, the allylic alcohol 333 reacts with ROH 83 to form an alkoxylated product 337. Simultaneously, aryl iodide 334 is oxidized to iodine(III) 336 which activates the double bond of diarylalkene 337 in presence of p-TsOH to give complex 338. Nucleophilic attack on 338 by H2O generates the intermediate 339 which undergoes a semipinacol-type rearrangement to furnish intermediate 340 along with regeneration of the precatalyst 334. Finally, deprotonation of intermediate 340 gives the product 335.
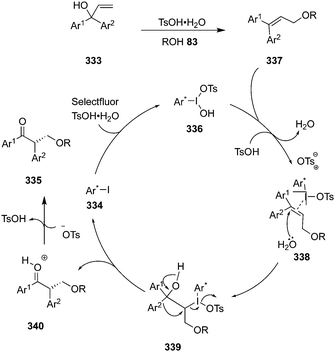 |
| | Scheme 99 Plausible catalytic cycle for the (S)-proline-derived chiral iodoarene-catalysed 1,2-aryl migration in allylic alcohols 333. | |
In 2020, Tiwari and co-workers reported an iodine(III)-catalysed enantioselective 1,2-tolyl group migration with geminal diacetoxylation of aromatic alkene 342.196 The reaction was performed at low temperature with the rearranged product 344 obtained in 27% yield with up to 92% ee (Scheme 100). The catalytic system involved 20 mol% chiral iodine(III) catalyst 343 and mCPBA in CHCl3:AcOH (1:1). Some research groups have achieved a similar 1,2-aryl migration reactions by using ammonium iodide197 and molecular iodine198 as catalysts. Hypervalent iodine catalytic species are not generated during these two rearrangements but these transformations may be quite useful for the readers who are working in hypervalent iodine catalysis.
 |
| | Scheme 100 Iodine(III)-catalysed 1,2-tolyl group migration with geminal diacetoxylation of alkene 342 using chiral iodine(III) catalyst 343. | |
In 2015, Gulder and co-workers reported a novel rearrangement of imides 345 using catalytic amounts of iodobenzamide 346 in the presence of N-bromosuccinimide (NBS) 347 as oxidant in hexafluoro-2-propanol (HFIP) at room temperature.199 This metal-free route lead to the facile preparation of valuable α,α-disubstituted-α-hydroxycarboxylamides 349 in good to excellent yields (Scheme 101).199 The reaction involved the formation of cyclic hypervalent iodine(III) species (bromo benziodoxole) 348 by oxidation of iodobenzamide 346 with NBS 347. Notably, none of these reactions exhibited aryl bromination and longer reaction times were observed with reduced catalyst loading and when bromo benziodoxole 348 was used instead of NBS 347.
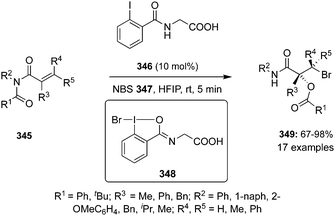 |
| | Scheme 101 Hypervalent iodine-catalysed rearrangement of imides 345 to 349 by involving cyclic iodine(III) reagent 348 as catalyst. | |
In 2020, an iodine(III)-catalysed enantioselective Wagner–Meerwein rearrangement of β-substituted styrenes 350 involving aryl, alkyl and hydride migrations was published affording 1,3-difluorinated products 351 and 352 in good to excellent yields with an excellent enantiomeric excess (Scheme 102).200 The catalytic system comprises of chiral aryl iodide 200 as precatalyst, mCPBA as the oxidant and py·9HF as the fluoride source. Notably, the 1,2-anti-diastereomers 351 were obtained when aryl is the migrating group and 1,2-syn diastereomers 352 were obtained when methyl is the migrating group (Scheme 102).
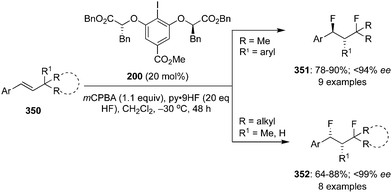 |
| | Scheme 102 Iodine(III)-catalysed Wagner–Meerwein rearrangements of β-substituted styrenes 350 to 1,3-difluorinated products 351 and 352 using C2-symmetric chiral iodoarene 200 as precatalyst. | |
6.2. Hofmann rearrangements
Also Hofmann rearrangements have been developed by using hypervalent iodine reagents in stoichiometric amounts.36,37 The first report on the Hofmann rearrangement appeared in 2012 by Ochiai and his research group.201 Later in the same year, the application of hypervalent iodine catalysis in Hofmann rearrangements was extended by Zhdankin and co-workers.202 In 2017, Hofmann rearrangements of primary amides 353 to carbamates 355 was successfully achieved by Miyamoto and co-workers using precatalyst 103, molecular oxygen as an oxidant and isobutyraldehyde as the O2 mediator.97
The rearranged products were obtained in moderate to excellent yields (Scheme 103). A variety of aliphatic as well as aryl amides were tolerated.
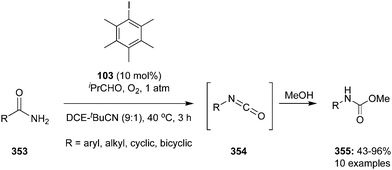 |
| | Scheme 103 Iodine(III)-catalysed Hofmann rearrangement of primary amides 353 to carbamates 355via the formation of isocyanate intermediate 354. | |
7. Photoredox catalysis
Hypervalent iodine reagents have a unique property of producing free radicals that makes these reagents suitable for photochemical reactions.203 In past decade, a number of hypervalent iodine reagents have been successfully employed in photoredox catalysis.19 In the majority of these hypervalent iodine induced photoredox reactions, the iodine reagents have been used in stoichiometric amounts, but there are few photoredox reactions where these reagents play a role of co-catalysts in combination with photoredox catalysts. In this section, the photoredox reactions catalysed by both hypervalent iodine and photoredox catalysts are highlighted.
7.1. Alkynylations
In 2014, Chen and others reported the deboronative alkynylation of potassium alkyl trifluoroborates 356 with EBX (ethynylbenziodoxole) 357 as alkynyl source under visible-light catalysis conditions (Scheme 104).204 The ruthenium complex [Ru-(bpy)3](PF6)2359 (2 mol%) was employed as photoredox catalyst in the presence of a catalytic amount of hydroxybenziodoxole (BI–OH) 358 as radical initiator. Blue light irradiation was critical to drive the photoredox reaction and the anticipated 1,3-disubstituted alkynes 307 were obtained in good yields. This reaction was highly chemoselective and tolerated a wide range of functional groups.
 |
| | Scheme 104 Iodine(III)-catalysed deboronative alkynylation of potassium alkyl trifluoroborates 356 with alkynyl benziodoxole 357 to alkynes 307 using photoredox catalysis in the presence of catalyst BI–OH 358. | |
Scheme 105 depicts the proposed catalytic cycle for the photoredox-catalysed deboronative alkynylation of potassium alkyl trifluoroborates 356 with EBX 357. Initially, photo-excitation of Ru(bpy)32+ takes place to give  which is further oxidized to Ru(bpy)33+ either by the benziodoxole radical 360 or its precursor BI–OH 358. Eventually, Ru(bpy)33+ oxidises alkyl trifluoroborate 356 to an alkyl R1 radical 362 and regenerates Ru(bpy)32+. Finally, α-addition of alkyl R1 radical 362 to EBX 357 provides the desired alkynes 307via formation of intermediate 363 and eliminates benziodoxole radical 360 which later oxidizes
which is further oxidized to Ru(bpy)33+ either by the benziodoxole radical 360 or its precursor BI–OH 358. Eventually, Ru(bpy)33+ oxidises alkyl trifluoroborate 356 to an alkyl R1 radical 362 and regenerates Ru(bpy)32+. Finally, α-addition of alkyl R1 radical 362 to EBX 357 provides the desired alkynes 307via formation of intermediate 363 and eliminates benziodoxole radical 360 which later oxidizes  to Ru(bpy)33+ and forms ortho-iodobenzoic acid.
to Ru(bpy)33+ and forms ortho-iodobenzoic acid.
 |
| | Scheme 105 Proposed catalytic cycle for iodine(III)-catalysed alkynylation of potassium alkyl trifluoroborates 356 to alkynes 307 using photoredox catalysis. | |
Glorius and co-workers developed a hydrogen atom transfer (HAT) method for the selective alkynylation of sp2 C(O)–H bond of aldehydes 97via photoredox catalysis (Scheme 106).205 This process delineates effective synthesis of ynones 367 by treating aldehydes 97 with ethynylbenziodoxole (EBX) 357 using catalytic amounts of sodium 2-iodobenzoate 364 and BI–OAc 366 in the presence of photocatalyst 365. Notably, sodium benzoate as HAT catalyst reductively quench the excited Ir(III)* to Ir(II) and generates a 2-iodobenzoyloxyl radical, which selectively abstracts a hydrogen and forms the carbonyl radical of the substrates. The scope of the reaction was widely explored with different aliphatic and aromatic aldehydes and the corresponding alkynylation products were obtained in decent yields.
 |
| | Scheme 106 Photoredox-catalysed alkynylation of aldehydes 97 with alkynyl benziodoxole 357 to ynones 367 using a catalytic amount of BI–OAc 366. | |
In 2021, Chen and others reported the synthesis of aminoalkynes 372 from cycloalkylamides 368via a selective C(sp3)–C(sp3) cleavage/alkynylation strategy using photoredox catalysis.206 The reaction employed Ph-Acr-MesBF4370 as photocatalyst and catalytic amounts of cyclic iodine(III) reagent 3,4-OMe-BI-OAc 369 (Scheme 107). The photoredox catalyst 370 was quite effective at low catalyst loading. Notably, 369 non-covalently activates cycloalkylamide 368 thereby facilitating the single-electron oxidation and ring-opening alkynylation as governed by various mechanistic probing experiments. A variety of nucleophiles 371 such as n-butanol, methanol, water, p-toluenethiol, 1-naphthol, TMSCN, indole and N-Me-indole were used to trap the iminium intermediate to give the desired aminoalkyne products in decent yields. Additionally, the bifunctional aminoalkynes were used to prepare indolizidine-fused azacycles via metal-catalysed cyclisations.
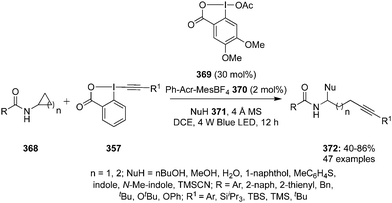 |
| | Scheme 107 Photoredox-catalysed synthesis of aminoalkynes 372 from cycloalkylamides 368via a selective C(sp3)–C(sp3) cleavage/alkynylation strategy. | |
7.2. Decarboxylative coupling of α-ketoacids
Wang and co-workers described the decarboxylative alkynylation of α-ketoacids 373 with functionalised bromoacetylenes 374 using BI–OH 358 as catalyst under sunlight irradiation. This method tolerated a wide range of functional groups and led to the energy-efficient synthesis of ynones 367 in good yields (Scheme 108).207 The substrate scope showed that bromoacetylenes functionalised with electron-withdrawing groups provided higher product yields while those with electron-donating groups showed inferior yields. Notably, the results of sunlight-driven reaction were comparable to those obtained by using blue light (λ = 450–455 nm).
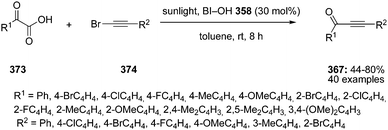 |
| | Scheme 108 Photoredox catalysed decarboxylative coupling of α-ketoacids 373 with bromoacetylenes 374 to ynones 367 using BI–OH 358 as catalyst in the presence of sunlight. | |
The possible catalytic cycle for the decarboxylative coupling reaction is summarized in Scheme 109. Initially, BI–OH 358 reacts with α-ketoacid 373 to form the intermediate 375, which upon sunlight irradiation generates iodanyl radical 359 and acyl radical 376. The iodanyl radical 359 reacts with bromoacetylene 374 to give BI–alkyne intermediate 379 along with the formation of a Br radical. Subsequently, the addition of acyl radical 377 to the intermediate 379 forms intermediate 380, which eventually releases the coupling product 367 and regenerates intermediate 359. Finally, coupling of iodanyl radical 359 with bromine radical produces bromobenziodoxole 378, which undergoes hydrolysis to regenerate BI–OH 358.
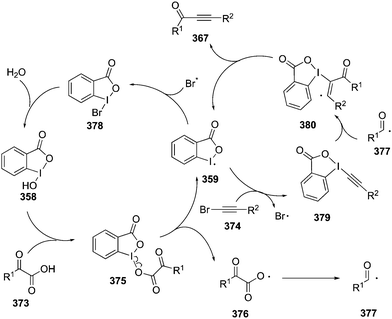 |
| | Scheme 109 Proposed catalytic cycle for the sunlight-driven decarboxylative coupling of α-ketoacids 373 with bromoacetylenes 374 to ynones 367. | |
7.3. Synthesis of phenols
In 2015, an organo-photoredox catalysed activation of PhI(OAc)21 was reported by Yadav and co-workers for the conversion of arylboronic acids 381 to phenols 383.208 This transformation was performed with 1.0 mol% of Eosin Y 382 as photoredox catalyst and K2CO3 as base in acetonitrile under visible light irradiation (Scheme 110). The reaction proceeded smoothly with substrates bearing electron-donating or electron-withdrawing substituents and the corresponding phenols 383 were isolated in excellent yields. Notably, the photo-chemically excited Eosin Y activates PhI(OAc)21 to form a methyl radical, which plays a key role for conversion of arylboronic acids to phenols.
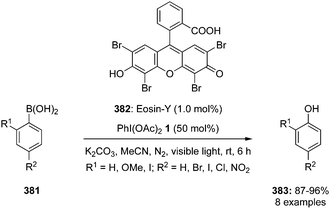 |
| | Scheme 110 Photoredox-catalysed synthesis of phenols 383 from arylboronic acids 381. | |
7.4. Dearomatisation
Furthermore, photoredox catalysis was employed for the dearomatisation of p-substituted anisole derivatives 384 to spirolactams 387 under blue light irradiation.209 Photoredox catalyst 386 and iodoarene 385 used were mesityl-2,6-diphenylpyrylium tetrafluoroborate (MDPT) and Kita's catalyst, respectively. The substrates with electron-withdrawing groups and those groups capable of stabilising a putative radical intermediate on nitrogen were found ineffective while electron-rich groups were tolerated successfully. All dearomatisation reactions required longer reaction time and the products were isolated in reasonable yields (Scheme 111).
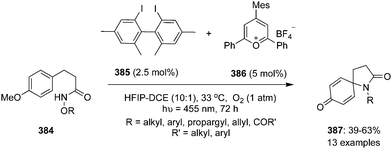 |
| | Scheme 111 Photoredox catalysed dearomatisation of p-substituted anisole derivatives 384 to spirolactams 387 using Kita's catalyst 385 under blue light irradiation. | |
8. Photochemical reactions
There are several hypervalent iodine mediated reactions driven by light.19 Moreover, there are hypervalent iodine catalysed organic reactions that require light to proceed. The visible-light driven decarboxylative acylarylation of acrylamides 388 with α-ketoacids 373 was developed using hypervalent iodine(III)-catalysis. This method led to the energy-efficient synthesis of 3,3-disubstituted 2-oxindoles 389 in good yields without using any photoredox catalyst (Scheme 112).210 Hypervalent iodine reagent BI–OAc 366 was employed as catalyst, which generates the radical species by cleavage of oxygen–iodine bond in the presence of blue LED (450–455 nm). The course of the reaction was examined with a diverse array of N-methyl-N-arylmethacrylamides 388, functionalised with electron-donating or withdrawing groups at the benzene ring and produced 2-oxindoles 389 in good yields. Notably, ketoacids 373 with electron-donating substituents in the aryl ring provided slightly higher yields.
 |
| | Scheme 112 Visible-light driven decarboxylative acylarylation of acrylamides 388 with α-ketoacids 373 using hypervalent iodine(III)-catalysis. | |
A photocatalytic approach towards the synthesis of chiral ketones 392 was introduced by Maruoka and co-workers using hypervalent iodine catalysis.211,212 In this study, the diastereoselective radical hydroacylation of alkylidenemalonates 390 was developed with various linear and branched chain aldehydes 97 under UV light irradiation using hypervalent iodine 391 as catalyst. Chiral ketones 392 were obtained in good yields with high diastereoselectivity accomplished by employing (−)-8-phenylmenthol as chiral auxiliary (Scheme 113). Acyl radical addition preferably takes place at the less sterically hindered face of the alkenes and thereby (S)-isomers forms predominantly because of the effective shielding of Re face by the phenyl group of the chiral auxiliary.211 Moreover, the same approach was successfully applied for the synthesis of (−)-methyleneolactocin.212
 |
| | Scheme 113 Iodine(III)-catalysed diastereoselective hydroacylation of alkylidenemalonates 390 with aldehydes 97 under UV irradiation using iodine(III) catalyst 391. | |
9. Miscellaneous reactions
Hypervalent iodine catalysis is used to achieve many different organic transformation and it is not possible to categorise all of the published reactions. Yakura et al. developed a mild, efficient and eco-friendly iodine(V)-catalysed oxidative cleavage of tetrahydrofuran-2-methanols 393 to form γ-lactones 395 using 2-iodobenzamide 394 as precatalyst and Oxone® as co-oxidant (Scheme 114).213 The corresponding γ-lactones 395 were obtained in moderate to good yields. This protocol provides an alternate route to access functionalized γ-lactones 395 at room temperature under metal-free conditions.
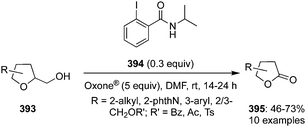 |
| | Scheme 114 Iodine(V)-catalysed oxidative cleavage of tetrahydrofuran-2-methanols 393 to γ-lactones 395. | |
The catalytic cycle for an iodine(V)-catalysed oxidative cleavage of tetrahydrofuran-2-methanols 393 to γ-lactones 395 is explained in Scheme 115. The reaction begins with the oxidation of iodobenzamide 394 by Oxone® to give iodine(V) species 397via formation of iodine(III) intermediate 396. Iodine(V) species 397 oxidises the alcohol 393 to generate key aldehyde intermediate 398 along with the regeneration of iodine(III) species for the next cycle. Eventually, the aldehyde 398 reacts with Oxone® to form formate 399via a Baeyer–Villiger type rearrangement which is oxidised to the desired lactone 395. Notably, the aldehyde 398 did not oxidise to the corresponding carboxylic acid 400.
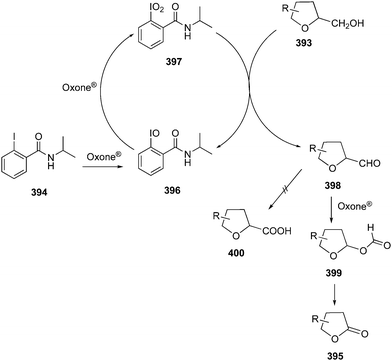 |
| | Scheme 115 Catalytic cycle for an iodine(V)-catalysed oxidative cleavage of tetrahydrofuran-2-methanols 393 to γ-lactones 395. | |
Hu and colleagues developed an iodine(III)-catalysed Balz–Schiemann fluorination of aromatic diazonium salts 401 under mild reaction conditions where the iodine(III) species lowered the energy activation barrier of the reaction.214 A wide range of aromatic fluorides 405 were prepared in good yields in the presence of 402–404 as iodine(III) catalysts and BF3·Et2O as additive (Scheme 116).
 |
| | Scheme 116 Iodine(III)-catalysed Balz–Schiemann fluorination of aromatic diazonium salts 401 to aryl fluorides 405 using iodine(III) catalysts 402–404. | |
Cyclopropanes behave often similar to olefins and can be activated by hypervalent iodine compounds.21 In 2017, hypervalent iodine catalysis has been used for the oxidative ring opening of substituted cyclopropanes 407 to obtain 1,3-difluorinated compounds 408 using PhI 17 as precatalyst and Py·9HF as source of fluoride in the presence of mCPBA (Scheme 117).215 Arylcyclopropanes 407 with electron-withdrawing substituents were tolerated while those with electron-donating substituents were not. With increased catalytic loadings (20 mol%), non-conjugated mono-substituted cyclopropanes 407 bearing ether and amine functionalities afforded difluorinated products 408 in good to excellent yields. The reaction involves the formation of key intermediate 409 that converts into a fluoroiodine(III) species 410. Eventually, iodine(III) intermediate 410 gave the final product 408 either through a SN1 route involving the formation of a tertiary carbocation or through a concerted backside fluoride substitution, the latter affords diastereomerically enriched products.
 |
| | Scheme 117 Iodine(III)-catalysed ring opening of cyclopropanes 407 to 1,3-difluorinated compounds 408. | |
The acidic PIFA 2 was explored as catalyst for nucleophilic substitutions of internal and terminal propargylic alcohols 411.216 Aromatic and heteroaromatic propargyl alcohols 411 with electron-donating substituents reacted faster with allyl silyl ethers 412 to afford 1,5-enynes 413 in good yields using PIFA 2 as catalyst in the absence of any oxidant (Scheme 118). Various acid and transition metal sensitive groups such as halogens, methoxy, silyl and heterocyclic rings, and cyano groups were tolerated. Additionally, Friedel–Crafts type propargylation went smoothly with aromatic compounds, O, S and N nucleophiles 371 to afford propargylic arenes/ethers/sulfides/amides 414 in moderate to excellent yields (Scheme 118).
 |
| | Scheme 118 Iodine(III)-catalysed nucleophilic substitution of propargyl alcohols 411 using PIFA 2 as catalyst without using any terminal oxidant. | |
The intermediate propargylic cation was generated by the reversible equilibrium between propargyl alcohol 417 and the PIFA 2 catalyst. Scheme 119 shows the proposed mechanism for the formation of an adduct 415 from the ligand exchange of PIFA 2 with propargyl alcohol 411, which decomposes into iodosobenzene 416 and progarylic carbocation 417. Propargylic carbocation 417 undergoes a nucleophilic substitution with allyl silyl ethers 412 to form substituted product 413. Iodosobenzene 416 binds with the eliminated TFA to regenerate the catalyst 2. The use of a stronger nucleophile would decrease the product yield due to the competition at the active site of PIFA 2 catalyst by the nucleophile and propargyl alcohol and anilines cannot be used as nucleophiles due to their Lewis basicity.215
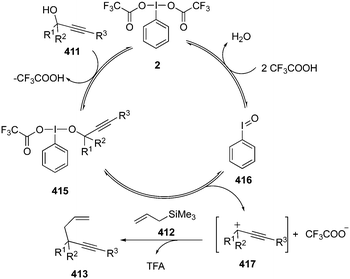 |
| | Scheme 119 Mechanism for the PIFA-mediated nucleophilic substitution of propargyl alcohols 411 using PIFA 2 as catalyst. | |
Synthetically important E-vinyl boronates 420 were prepared by Wei and co-workers by the hydroboration of terminal alkynes 418 with bis(pinacolato)diboron (B2pin2) 419 using catalytic amounts of PhI(OAc)21 in the presence of tBuONa and EtOH as the hydrogen donor in air (Scheme 120).217 Aromatic as well as aliphatic terminal alkynes 418 gave moderate to good yields with good regio- and stereoselectivity.
 |
| | Scheme 120 Iodine(III)-catalysed hydroboration of alkynes 418 with 419 to 420 using PhI(OAc)21 as catalyst. | |
A facile and effective iodine(III)-catalysed synthesis of pharmacologically important trideuteroalkoxylated quinoxalinones 424 was established by Shen and co-workers using a cross-dehydrogenative coupling of quinoxalinones 422 with deuterated alcohols 423 and 4-iodotoluene 149 as precatalyst in the presence of peracetic acid (Scheme 121).218 Irrespective of the nature and position of different substituents on the quinoxaline rings, several N-substituted quinoxalin-2(1H)-ones gave the corresponding products in good yields.
 |
| | Scheme 121 Iodine(III)-catalysed synthesis of trideutero alkoxylated quinoxalinones 424 using 149 as catalyst. | |
10. Conclusions
In this review article, we highlight recent applications of organocatalysis involving hypervalent iodine catalysts in organic synthesis. Hypervalent iodine catalysis has been used for the preparation of various synthetic intermediates such as amines, carbonyl compounds, alcohols, acetals and organofluorine derivatives. Some of these catalytic approaches have been used to construct biologically active heterocyclic and spirocyclic scaffolds. Additionally, hypervalent iodine catalysis has been used significantly in asymmetric synthesis with high stereoselectivity. Mainly, hypervalent iodine catalytic species were generated in situ by the oxidation of iodoarene precatalysts using stoichiometric oxidants such as Oxone® and peroxyacids like mCPBA. In a few catalytic reactions, the catalysts were reused several times without losing their catalytic potential. In this era of ever growing interest in green chemistry, the readily available environmentally benign hypervalent iodine compounds with their ease of handling are in increasing demand as green and environmentally sustainable alternatives to heavy metals in synthetic organic chemistry. Improving the catalytic attributes of hypervalent iodine is continuing to be a challenging goal. In addition, the improvements on the stoichiometric oxidant needed for all the transformations has to be considered in the future. Recent developments such as the use of oxygen (air) or electricity seem to be highly promising in advancing this area. The often delicate balance between substrate and iodine reactivity has to be further understood and investigated as there is the need of more general applicable catalysts, especially when considering stereoselective reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
F. V. S., S. E. S. and M. K. acknowledge the support of VIT Chennai, Chennai. F. V. S. is thankful to CSIR New Delhi [Grant No. 02/(0330)/17-EMR-II] for the financial support. S. E. S. is thankful to CSIR New Delhi for providing her fellowship. M. K. acknowledges Sree Sankara College, Kalady, Kerala for providing an opportunity to carry out her research studies at VIT, Chennai. TW acknowledges the support of Cardiff University.
References
-
V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds, Wiley, Chichester, I, 2013 Search PubMed.
- T. Dohia and Y. Kita, Chem. Commun., 2009, 2073 RSC.
- F. V. Singh and T. Wirth, Chem. – Asian J., 2014, 9, 950 CrossRef CAS PubMed and references are cited therein.
- R. Narayan, S. Manna and A. P. Antonchick, Synlett, 2015, 1785 CAS.
- A. Maity and D. C. Powers, Synlett, 2019, 257 CAS.
- M. S. Yusubov and V. V. Zhdankin, Curr. Org. Synth., 2012, 9, 247 CrossRef CAS.
- F. V. Singh, J. Rehbein and T. Wirth, ChemistryOpen, 2012, 1, 242 CrossRef PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed.
- S. E. Shetgaonkar and F. V. Singh, Front. Chem., 2020, 8, 1 CrossRef PubMed.
- M. Elsherbini and T. Wirth, Chem. – Eur. J., 2018, 24, 13399 CrossRef CAS PubMed.
- I. F. D. Hyatt, L. Dave, N. David, K. Kaur, M. Medard and C. Mowdawalla, Org. Biomol. Chem., 2019, 17, 7822 RSC.
- A. Parra, Chem. Rev., 2019, 119, 12033 CrossRef CAS PubMed.
- S. Maiti, M. T. Alam, A. Bal and P. Mal, Adv. Synth. Catal., 2019, 361, 4401 CrossRef CAS.
- A. Flores, E. Cots, J. Berges and K. Muñiz, Adv. Synth. Catal., 2019, 361, 2 CrossRef CAS.
- S. E. Shetgaonkar and F. V. Singh, ARKIVOC, 2020, 86 Search PubMed.
- X.-G. Yang, Z.-N. Hu, M.-C. Jia, F.-H. Du and C. Zhang, Synlett, 2021, 1289 CAS.
- Z. Wang, New J. Chem., 2021, 45, 509 RSC.
- C. Mairhofer, L. Stockhammer and M. Waser, ARKIVOC, 2021, 112 Search PubMed.
- F. V. Singh and T. Wirth, ARKIVOC, 2021, 12 Search PubMed.
- S. Rimi, S. Soni, B. Uttam, H. China, T. Dohi, V. V. Zhdankin and R. Kumar, Synthesis, 2022, 2731 Search PubMed.
- R. Kumar, F. V. Singh, N. Takenaga and T. Dohi, Chem. – Asian J., 2022, 17, e202101115 CAS.
- K. N. Parida and J. N. Moorthy, Synlett, 2022 DOI:10.1055/a-1813-7319.
-
F. V. Singh and T. Wirth, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. Molander, Elsevier, 2014, p. 880 Search PubMed.
-
F. V. Singh and T. Wirth, in Science of Synthesis: Catalytic Oxidation in Organic Synthesis, ed. K. Muñiz, Thieme, Stuttgart, 2017, p. 29 Search PubMed.
-
F. V. Singh and T. Wirth, in The Chemistry of Hypervalent Halogen Compounds, ed. B. Olofsson, M. Ilan and R. Zvi, John Wiley & Sons, Ltd, Chichester, 2018, p. 809 Search PubMed.
-
T. Dohi and Y. Kita, in Iodine Catalysis in Organic Synthesis, ed. K. Ishihara and K. Muñiz, John Wiley & Sons, Ltd, Chichester, Ltd, Chichester, 2022, p. 211 Search PubMed.
-
F. Ballaschk and S. F. Kirsch, in Iodine Catalysis in Organic Synthesis, ed. K. Ishihara and K. Muñiz, John Wiley & Sons, Ltd, Chichester, Ltd, Chichester, 2022, p. 299 Search PubMed.
-
Hypervalent Iodine Chemistry Modern Developments in Organic Synthesis, ed. T. Wirth, Springer, Verlag, 2003 Search PubMed.
-
Hypervalent Iodine Chemistry. Topics in Current Chemistry, ed. T. Wirth, Springer, Cham, 2016, vol. 373 Search PubMed.
-
B. Olofsson, I. Marek and Z. Rappoport, The Chemistry of Hypervalent Halogen Compounds, John Wiley & Sons, Ltd, Chichester, 2019 Search PubMed.
-
K. Ishihara and K. Muñiz, in Iodine Catalysis in Organic Synthesis, ed. K. Ishihara and K. Muñiz, John Wiley & Sons, Ltd, Chichester, Ltd, Chichester, 2022 Search PubMed.
- L. F. Silva Jr. and B. Olofsson, Nat. Prod. Rep., 2011, 28, 1722 RSC.
- F. V. Singh, S. Mangaonkar and P. B. Kole, Synth. Commun., 2018, 48, 2169 CrossRef CAS.
- S. R. Mangaonkar, P. B. Kole and F. V. Singh, Synlett, 2018, 199 CAS.
- S. E. Shetgaonkar, M. Krishnan and F. V. Singh, Mini. Rev. Org. Chem., 2021, 18, 138 CrossRef CAS.
- F. V. Singh, P. B. Kole, S. R. Mangaonkar and S. E. Shetgaonkar, Beilstein J. Org. Chem., 2018, 14, 1778 CrossRef CAS PubMed.
- F. V. Singh and T. Wirth, Synthesis, 2012, 1171 CAS.
- N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924 CrossRef CAS PubMed.
- F. C. Sousa e Silva, A. F. Tierno and S. E. Wengryniuk, Molecules, 2017, 22, 780 CrossRef PubMed.
- S. V. Kohlhepp and T. Gulder, Chem. Soc. Rev., 2016, 45, 6270 RSC.
- J. H. Lee, S. Choi and K. B. Hong, Molecules, 2019, 24, 2634 CrossRef.
- G. F. Koser, R. H. Wettach, J. M. Troup and B. A. Frenz, J. Org. Chem., 1976, 41, 3609 CrossRef CAS.
- E. Merritt and B. Olofsson, Synthesis, 2011, 517 CAS.
- D.-Q. Dong, S.-H. Hao, Z.-L. Wang and C. Chen, Org. Biomol. Chem., 2014, 12, 4278 RSC.
- E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed.
- M. Ochiai, A. Nakanishi and T. Suefuji, Org. Lett., 2000, 2, 2923 CrossRef CAS PubMed.
- P. Eisenberger, S. Gischig and A. Togni, Chem. – Eur. J., 2006, 12, 2579 CrossRef CAS PubMed.
- I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., 2007, 119, 768 CrossRef.
- V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz and A. J. Simonsen, J. Org. Chem., 1996, 61, 6547 CrossRef CAS PubMed.
- J. P. Brand, D. F. González, S. Nicolai and J. Waser, Chem. Commun., 2011, 47, 102 RSC.
- R. Pribram, Justus Liebigs Ann. Chem., 1907, 351, 481 CrossRef CAS.
- M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2010, 49, 2175 CrossRef CAS PubMed.
- A. Yoshimura, M. S. Yusubovb and V. V. Zhdankin, Org. Biomol. Chem., 2016, 14, 4771 RSC.
- H. Tohma and Y. Kita, Adv. Synth. Catal., 2004, 346, 111 CrossRef CAS.
- R. D. Richardson, J. M. Zayed, S. Altermann, D. Smith and T. Wirth, Angew. Chem., Int. Ed., 2007, 46, 6529 CrossRef CAS PubMed.
- J. Qurban, M. Elsherbini, H. Alharbia and T. Wirth, Chem. Commun., 2019, 55, 7998 RSC.
- M. S. Yusubov, N. S. Soldatova, P. S. Postnikov, R. R. Valiev, A. Yoshimura, T. Wirth, V. N. Nemykine and V. V. Zhdankin, Chem. Commun., 2019, 55, 7760 RSC.
- A. Sreenithya, K. Surya and R. B. Sunoj, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1299 Search PubMed.
- M. S. Yusubov and V. V. Zhdankin, Resour.-Effic. Technol., 2015, 1, 49 Search PubMed.
- T. Fuchigami and T. Fujita, J. Org. Chem., 1994, 59, 7190 CrossRef CAS.
- T. Fujita and T. Fuchigami, Tetrahedron Lett., 1996, 37, 4725 CrossRef CAS.
- P. Kazmierczak, L. Skulski and L. Kraszkiewicz, Molecules, 2001, 6, 881 CrossRef CAS.
- A. Zielinska and L. Skulski, Molecules, 2002, 7, 806 CrossRef CAS.
- A. Zielinska and L. Skulski, Molecules, 2005, 10, 190 CrossRef CAS PubMed.
- H. J. Kim, J. Kim, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2011, 133, 16382 CrossRef CAS PubMed and references cited therein.
- A. A. Kantak, S. Potavathri, R. A. Barham, K. M. Romano and B. DeBoef, J. Am. Chem. Soc., 2011, 133, 19960 CrossRef CAS PubMed.
- R. Samanta, J. Lategahn and A. P. Antonchick, Chem. Commun., 2012, 48, 3194 RSC.
- S. Manna, P. O. Serebrennikova, I. A. Utepova, A. P. Antonchick and O. N. Chupakhin, Org. Lett., 2015, 17, 4588 CrossRef CAS PubMed.
- N. Lucchetti, M. Scalone, S. Fantasia and K. Muñiz, Adv. Synth. Catal., 2016, 358, 2093 CrossRef CAS.
- S. Maiti and P. Mal, J. Org. Chem., 2018, 83, 1340 CrossRef CAS PubMed.
- E. Miyazawa, T. Sakamoto and Y. Kikugawa, J. Org. Chem., 2003, 68, 5429 CrossRef CAS PubMed.
- M. Kashiwa, M. Sonoda and S. Tanimori, Eur. J. Org. Chem., 2014, 4720 CrossRef CAS.
- H. Sasa, K. Mori, K. Kikushima, Y. Kita and T. Dohi, Chem. Pharm. Bull., 2022, 70, 106 CrossRef CAS PubMed.
- Q. Ding, H. He and Q. Cai, Org. Lett., 2018, 20, 4554 CrossRef CAS.
- A. U. Rahman, N. Zarshad, P. Zhou, W. Yang, G. Li and A. Ali, Front. Chem., 2020, 8, 523 CrossRef CAS PubMed.
- F. Cardona and A. Goti, Nat. Chem., 2009, 1, 269 CrossRef CAS PubMed.
- Y. Zhu, R. G. Cornwall, H. Du, B. Zhao and Y. Shi, Acc. Chem. Res., 2014, 47, 3665 CrossRef CAS.
- K. Muñiz and C. Martínez, J. Org. Chem., 2013, 78, 2168 CrossRef PubMed.
- K. Muñiz, L. Barreiro, R. M. Romero and C. Martínez, J. Am. Chem. Soc., 2017, 139, 4354 CrossRef.
- E. Cots, A. Flores, R. M. Romero and K. Muñiz, ChemSusChem, 2019, 12, 3028 CrossRef CAS PubMed.
- E. Cots, J. Rintjema, F. Bravo and K. Muñiz, Org. Lett., 2021, 23, 6429 CrossRef CAS PubMed.
- P. Mizar, A. Laverny, M. El-Sherbini, U. Farid, M. Brown, F. Malmedy and T. Wirth, Chem. – Eur. J., 2014, 20, 9910 CrossRef PubMed.
- C. Wata and T. Hashimoto, J. Am. Chem. Soc., 2021, 143, 1745 CrossRef CAS PubMed.
- F. Wu, N. Kaur, N.-E. Alom and W. Li, JACS Au, 2021, 1, 734 CrossRef CAS.
- X.-J. Deng, H.-X. Liu, L.-W. Zhang, G.-Y. Zhang and Z. Xiang, J. Org. Chem., 2021, 86, 235 CrossRef CAS PubMed.
- C. Wata and T. Hashimoto, Synthesis, 2021, 2594 CAS.
- C. Zhu, Y. Liang, X. Hong, H. Sun, W.-Y. Sun, K. N. Houk and Z. Shi, J. Am. Chem. Soc., 2015, 137, 7564 CrossRef CAS.
- A. Bose, S. Maiti, S. Sau and P. Mal, Chem. Commun., 2019, 55, 2066 RSC.
- A. Bose, S. Sau and P. Mal, Eur. J. Org. Chem., 2019, 4105 CrossRef CAS.
- A. P. Thottumkara, M. S. Bowsher and T. K. Vinod, Org. Lett., 2005, 7, 2933 CrossRef CAS PubMed.
- T. Yakura, T. Fujiwara, A. Yamada and H. Nambu, Beilstein J. Org. Chem., 2018, 14, 971 CrossRef CAS PubMed.
- A. Maity, S. Hyun, A. K. Wortman and D. C. Powers, Angew. Chem., 2018, 130, 7323 CrossRef.
- F. Ballaschk and S. F. Kirsch, Green Chem., 2019, 21, 5896 RSC.
- N. Ayoub, C. Bergère, J. Toufaily, E. Guénin and G. Enderlin, New J. Chem., 2020, 44, 11577 RSC.
- Z. Shahamat, F. Nemati and A. Elhampour, React. Funct. Polym., 2020, 146, 104415 CrossRef CAS.
- R. Bikshapathi, P. S. Prathima and V. J. Rao, New J. Chem., 2016, 40, 10300 RSC.
- K. Miyamoto, J. Yamashita, S. Narita, Y. Sakai, K. Hirano, T. Saito, C. Wang, M. Ochiai and M. Uchiyama, Chem. Commun., 2017, 53, 9781 RSC.
-
S. Quideau, L. Pouységu, P. A. Peixoto and D. Deffieux, in Hypervalent Iodine Chemistry. Topics in Current Chemistry, ed. T. Wirth, Springer, Cham., 2016, vol. 373, p. 25 Search PubMed.
- H. Nambu, I. Shimokawa, T. Fujiwara and T. Yakura, Asian J. Org. Chem., 2016, 5, 486 CrossRef CAS.
-
H. Nambu, I. Shimokawa, T. Fujiwara and T. Yakura, in New Horizons of Process Chemistry: Scalable Reactions and Technologies, ed. K. Tomioka, T. Shioiri and H. Sajiki, Springer, Singapore, 2017, p. 121 Search PubMed.
- K. Muñiz and L. Fra, Synthesis, 2017, 2901 CrossRef.
- M. Uyanik, T. Mutsuga and K. Ishihara, Angew. Chem., Int. Ed., 2017, 56, 3956 CrossRef CAS PubMed.
- B. Tahmouresilerd, P. J. Larson, D. K. Unruh and A. F. Cozzolino, Catal. Sci. Technol., 2018, 8, 4349 RSC.
- B. Tahmouresilerd, M. Moody, L. Agogo and A. F. Cozzolino, Dalton Trans., 2019, 48, 6445 RSC.
- T. Hashimoto, Y. Shimazaki, Y. Omatsu and K. Maruoka, Angew. Chem., 2018, 130, 7318 CrossRef.
- H. China, K. Tanihara, H. Sasa, K. Kikushima and T. Dohi, Catal. Today, 2020, 348, 2 CrossRef CAS.
- T. Yakura, M. Omoto, Y. Yamauchi, Y. Tian and A. Ozono, Tetrahedron, 2010, 66, 5833 CrossRef CAS.
- T. Dohi, A. Maruyama, M. Yoshimura, K. Morimoto, H. Tohma and Y. Kita, Angew. Chem., Int. Ed., 2005, 44, 6193 CrossRef CAS PubMed.
- T. Dohi, A. Maruyama, Y. Minamitsuji, N. Takenaga and Y. Kita, Chem. Commun., 2007, 1224 RSC.
- T. Dohi, Y. Minamitsuji, A. Maruyama, S. Hirose and Y. Kita, Org. Lett., 2008, 10, 3559 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, Bioorg. Med. Chem. Lett., 2009, 19, 3848 CrossRef CAS PubMed.
- T. Dohi, N. Takenaga, K. Fukushima, T. Uchiyama, D. Kato, M. Shiro, H. Fujioka and Y. Kita, Chem. Commun., 2010, 46, 7697 RSC.
- Y. Minamitsuji, D. Kato, H. Fujioka, T. Dohi and Y. Kita, Aust. J. Chem., 2009, 62, 648 CrossRef CAS.
- Z. Yu, X. Ju, J. Wang and W. Yu, Synthesis, 2011, 860 CrossRef CAS.
- T. Dohi, T. Nakae, Y. Ishikado, D. Kato and Y. Kita, Org. Biomol. Chem., 2011, 9, 6899 RSC.
- M. Ngatimin, R. Frey, C. Andrews, D. W. Lupton and O. E. Hutt, Chem. Commun., 2011, 47, 11778 RSC.
- M. Uyanik, N. Sasakura, M. Mizuno and K. Ishihara, ACS Catal., 2017, 7, 872 CrossRef CAS.
- M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2013, 52, 9215 CrossRef CAS PubMed.
- C. Bosset, R. Coffinier, P. A. Peixoto, M. El Assal, K. Miqueu, J.-M. Sotiropoulos, L. Pouységu and S. Quideau, Angew. Chem., Int. Ed., 2014, 53, 9860 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, J. Org. Chem., 2017, 82, 11946 CrossRef CAS.
- C. Hempel, C. Maichle-Mössmer, M. A. Pericàs and B. J. Nachtsheim, Adv. Synth. Catal., 2017, 359, 2931 CrossRef CAS.
- M. Ogasawara, H. Sasa, H. Hu, Y. Amano, H. Nakajima, N. Takenaga, K. Nakajima, Y. Kita, T. Takahashi and T. Dohi, Org. Lett., 2017, 19, 4102 CrossRef CAS.
- M. R. Imrich and T. Ziegler, Tetrahedron Lett., 2019, 60, 150954 CrossRef CAS.
- K. Antien, L. Pouységu, D. Deffieux, S. Massip, P. A. Peixoto and S. Quideau, Chem. – Eur. J., 2019, 25, 2852 CrossRef CAS.
- N. Jain, S. Xu and M. A. Ciufolini, Chem. – Eur. J., 2017, 23, 4542 CrossRef CAS PubMed.
- M. U. Tariq and W. J. Moran, Eur. J. Org. Chem., 2020, 153 Search PubMed.
- N. Khan, K. Itaya and T. Wirth, ChemistryOpen, 2022, 11, e202200145 CAS.
- D.-Y. Zhang, L. Xu, H. Wu and L.-Z. Gong, Chem. – Eur. J., 2015, 21, 10314 CrossRef CAS PubMed.
- H. Wu, Y.-P. He, L. Xu, D.-Y. Zhang and L.-Z. Gong, Angew. Chem., Int. Ed., 2014, 53, 3466 CrossRef CAS.
- Q. Deng, W. Xia, M. I. Hussain, X. Zhang, W. Hu and Y. Xiong, J. Org. Chem., 2020, 85, 3125 CrossRef CAS PubMed.
- Y. Shimazaki, C. Wata, T. Hashimoto and K. Maruoka, Asian J. Org. Chem., 2021, 10, 1638 CrossRef CAS.
- X. Kong, L. Lin, X. Chen, Y. Chen, W. Wang and B. Xu, ChemSusChem, 2021, 14, 3277 CrossRef CAS.
- S. Haubenreisser, T. H. Wöste, C. Martínez, K. Ishihara and K. Muñiz, Angew. Chem., Int. Ed., 2016, 55, 413 CrossRef CAS.
- T. H. Wöste and K. Muñiz, Synthesis, 2016, 816 Search PubMed.
- K. Aertker, R. J. Rama, J. Opalach and K. Muñiz, Adv. Synth. Catal., 2017, 359, 1290 CrossRef CAS.
- T. Kitamura, K. Muta and J. Oyamada, J. Org. Chem., 2015, 80, 10431 CrossRef CAS PubMed.
- I. G. Molnár and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 5004 CrossRef.
- F. Scheidt, M. Schäfer, J. C. Sarie, C. G. Daniliuc, J. J. Molloy and R. Gilmour, Angew. Chem., 2018, 130, 16669 CrossRef.
- S. M. Banik, J. W. Medley and E. N. Jacobsen, J. Am. Chem. Soc., 2016, 138, 5000 CrossRef CAS PubMed.
- S. M. Banik, J. W. Medley and E. N. Jacobsen, Science, 2016, 353, 51 CrossRef CAS PubMed.
- M. K. Haj, S. M. Banik and E. N. Jacobsen, Org. Lett., 2019, 21, 4919 CrossRef CAS PubMed.
- M. D. Levin, J. M. Ovian, J. A. Read, M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 14831 CrossRef CAS PubMed.
- B. Zhou, M. K. Haj, E. N. Jacobsen, K. N. Houk and X.-S. Xue, J. Am. Chem. Soc., 2018, 140, 15206 CrossRef CAS PubMed.
- K. M. Mennie, S. M. Banik, E. C. Reichert and E. N. Jacobsen, J. Am. Chem. Soc., 2018, 140, 4797 CrossRef CAS.
- S. R. Mangaonkar and F. V. Singh, Synthesis, 2019, 4473 CAS.
- T. Kuga, Y. Sasano and Y. Iwabuchi, Chem. Commun., 2018, 54, 798 RSC.
- T. Dohi, N. Takenaga, A. Goto, H. Fujioka and Y. Kita, J. Org. Chem., 2008, 73, 7365 CrossRef CAS PubMed.
- V. A. Telvekar and K. A. Sasane, Synth. Commun., 2012, 42, 1325 CrossRef CAS.
- K. C. Nicolaou, P. S. Baran and Y. Zhong, J. Am. Chem. Soc., 2001, 123, 3183 CrossRef CAS PubMed.
- B. Yahuaca-Juárez, G. González, M. A. Ramírez-Morales, C. Alba-Betancourt, M. A. Deveze-Álvarez, C. L. Mendoza-Macías, R. Ortiz-Alvarado, K. A. Juárez-Ornelas, C. R. Solorio-Alvarado and K. Maruoka, Synth. Commun., 2020, 50, 539 CrossRef.
- A. H. Abazid, N. Clamor and B. J. Nachtsheim, ACS Catal., 2020, 10, 8042 CrossRef CAS.
- B. Basdevant and C. Y. Legault, J. Org. Chem., 2015, 80, 6897 CrossRef CAS PubMed.
- T. R. Lex, M. I. Swasy, S. Panda, B. R. Brummel, L. N. Giambalvo, K. G. Gross, C. D. McMillen, K. Kobra, W. T. Pennington and D. C. Whitehead, Tetrahedron Lett., 2020, 61, 151723 CrossRef CAS.
- B. Basdevant and C. Y. Legault, Org. Lett., 2015, 17, 4918 CrossRef CAS PubMed.
- J. T. Su and W. A. Goddard, J. Am. Chem. Soc., 2005, 127, 14146 CrossRef CAS PubMed.
- J. N. Moorthy, N. Singhal and K. Senapati, Tetrahedron Lett., 2008, 49, 80 CrossRef CAS.
- J. N. Moorthy, K. Senapati, K. N. Parida, S. Jhulki, K. Sooraj and N. N. Nair, J. Org. Chem., 2011, 76, 9593 CrossRef CAS PubMed.
- Y. A. Vlasenko, P. S. Postnikov, M. E. Trusova, A. Shafir, V. V. Zhdankin, A. Yoshimura and M. S. Yusubov, J. Org. Chem., 2018, 83, 12056 CrossRef CAS PubMed.
- A. Boelke, E. Lork and B. J. Nachtsheim, Chem. – Eur. J., 2018, 24, 18653 CrossRef CAS PubMed.
- A.-A. Guilbault and C. Y. Legault, ACS Catal., 2012, 2, 219 CrossRef CAS.
- A. Boelke and B. J. Nachtsheim, Adv. Synth. Catal., 2020, 362, 184 CrossRef CAS.
- T. Hokamp and T. Wirth, Chem. – Eur. J., 2020, 26, 10417 CrossRef CAS PubMed.
- S. Suzuki, T. Kamo, K. Fukushi, T. Hiramatsu, E. Tokunaga, T. Dohi, Y. Kita and N. Shibata, Chem. Sci., 2014, 5, 2754 RSC.
- R. Pluta, P. E. Krach, L. Cavallo, L. Falivene and M. Rueping, ACS Catal., 2018, 8, 2582 CrossRef CAS.
- Y. Wang, H. Yuan, H. Lu and W.-H. Zheng, Org. Lett., 2018, 20, 2555 CrossRef CAS PubMed.
- E. M. Woerly, S. M. Banik and E. N. Jacobsen, J. Am. Chem. Soc., 2016, 138, 13858 CrossRef CAS PubMed.
- C. Gelis, A. Dumoulin, M. Bekkaye, L. Neuville and G. Masson, Org. Lett., 2017, 19, 278 CrossRef CAS PubMed.
- R. Möckel, E. Babaoglu and G. Hilt, Chem. – Eur. J., 2018, 24, 15781 CrossRef PubMed.
- H. Liu, X. Deng, X. Huang, N. Ji and W. He, Org. Biomol. Chem., 2020, 18, 3654 RSC.
- J. Häfliger, O. O. Sokolova, M. Lenz, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2022, 61, e202205277 CrossRef PubMed.
- F. V. Singh and S. R. Mangaonkar, Synthesis, 2018, 4940 CrossRef CAS.
- S. R. Mangaonkar, S. E. Shetgaonkar, A. A. Vernekar and F. V. Singh, ChemistrySelect, 2020, 5, 10754 CrossRef CAS.
- Q. Wang, M. Lübcke, M. Biosca, M. Hedberg, L. Eriksson, F. Himo and K. J. Szabo, J. Am. Chem. Soc., 2020, 142(47), 20048 CrossRef CAS PubMed.
- S. K. Alla, P. Sadhu and T. Punniyamurthy, J. Org. Chem., 2014, 79, 7502 CrossRef CAS PubMed.
- A. Alhalib, S. Kamouka and W. J. Moran, Org. Lett., 2015, 17, 1453 CrossRef CAS PubMed.
- F. Scheidt, C. Thiehoff, G. Yilmaz, S. Meyer, C. G. Daniliuc, G. Kehr and R. Gilmour, Beilstein J. Org. Chem., 2018, 14, 1021 CrossRef CAS PubMed.
- N. Asari, Y. Takemoto, Y. Shinomoto, T. Yagyu, A. Yoshimura, V. V. Zhdankin and A. Saito, Asian J. Org. Chem., 2016, 5, 1314 CrossRef CAS.
- Y. Okamura, D. Sato, A. Yoshimura, V. V. Zhdankin and A. Saito, Adv. Synth. Catal., 2017, 359, 3243 CrossRef CAS.
- S. Kamouka and W. J. Moran, Beilstein J. Org. Chem., 2017, 13, 1823 CrossRef CAS PubMed.
- W. Yi, Q.-Y. Liu, X.-X. Fang, S.-C. Lou and G.-Q. Liu, Org. Biomol. Chem., 2018, 16, 7012 RSC.
- S. S. K. Boominathan, W.-P. Hu, G. C. Senadi, J. K. Vandavasia and J.-J. Wang, Chem. Commun., 2014, 50, 6726 RSC.
- Z. Zhao, L. H. Britt and G. K. Murphy, Chem. – Eur. J., 2018, 24, 17002 CrossRef CAS.
- S. Manna and A. P. Antonchick, Angew. Chem., Int. Ed., 2014, 53, 7324 CrossRef CAS.
- S. Maiti, T. K. Achar and P. Mal, Org. Lett., 2017, 19, 2006 CrossRef CAS.
- P. V. Sri Ramya, S. Angapelly, R. S. Rani, C. S. Digwal, C. Ganesh Kumar, B. Nagendra Babu, L. Guntuku and A. Kamal, Arab. J. Chem., 2020, 13, 120 CrossRef CAS.
- A. Yoshimura, K. C. Nguyen, G. T. Rohde, A. Saito, M. S. Yusubov and V. V. Zhdankin, Adv. Synth. Catal., 2016, 358, 2340 CrossRef CAS.
- A. Yoshimura, K. C. Nguyen, S. C. Klasen, P. S. Postnikov, M. S. Yusubov, A. Saito, V. N. Nemykin and V. V. Zhdankin, Asian J. Org. Chem., 2016, 5, 1128 CrossRef CAS.
- I. A. Mironova, V. G. Nenajdenko, P. S. Postnikov, A. Saito, M. S. Yusubov and A. Yoshimura, Molecules, 2022, 27, 3860 CrossRef CAS PubMed.
- T. Yagyu, Y. Takemoto, A. Yoshimura, V. V. Zhdankin and A. Saito, Org. Lett., 2017, 19, 2506 CrossRef CAS PubMed.
- M. Waheed, N. Ahmed, M. A. Alsharif, M. I. Alahmdi and S. Mukhtar, ChemistrySelect, 2019, 4, 1872 CrossRef CAS.
- A. Matsuzawa, S. Takeuchi and K. Sugita, Chem. – Asian J., 2016, 11, 2863 CrossRef CAS PubMed.
- Y. Han, Y. Sun, A. Abdukader, B. Liu and D. Wang, Catal. Lett., 2018, 148, 3486 CrossRef CAS.
- A. Bal, S. Maiti and P. Mal, J. Org. Chem., 2018, 83, 11278 CrossRef CAS PubMed.
- D.-Y. Zhang, Y. Zhang, H. Wu and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 7450 CrossRef CAS PubMed.
- A. H. Abazid and B. J. Nachtsheim, Angew. Chem., Int. Ed., 2020, 59, 1479 CrossRef CAS PubMed.
- C. B. Pandey, T. Azaz, R. S. Verma, M. Mishra, J. L. Jat and B. Tiwari, J. Org. Chem., 2020, 85, 10175 CrossRef CAS.
- P. Swamy, M. Naresh, M. M. Reddy, K. Srujana, C. Durgaiah, S. Prabhakar and N. Narender, RSC Adv., 2015, 5, 73732 RSC.
- S. Kodumuri, S. Peraka, N. Mameda, D. Chevella, R. Banothu and N. Nama, RSC Adv., 2016, 6, 6719 RSC.
- A. Ulmer, M. Stodulski, S. V. Kohlhepp, C. Patzelt, A. Pöthig, W. Bettray and T. Gulder, Chem. – Eur. J., 2015, 21, 1444 CrossRef CAS.
- H. A. Sharma, K. M. Mennie, E. E. Kwan and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 16090 CrossRef CAS PubMed.
- K. Miyamoto, Y. Sakai, S. Goda and M. Ochiai, Chem. Commun., 2012, 48, 982 RSC.
- A. Yoshimura, K. R. Middleton, M. W. Luedtke, C. Zhu and V. V. Zhdankin, J. Org. Chem., 2012, 77, 11399 CrossRef CAS PubMed.
- L. Wang and J. Liu, Eur. J. Org. Chem., 2016, 1813 CrossRef CAS.
- H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280 CrossRef CAS PubMed.
- S. Mukherjee, R. A. Garza-Sanchez, A. Tlahuext-Aca and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 14723 CrossRef CAS PubMed.
- Z. Liu, S. Wu and Y. Chen, ACS Catal., 2021, 11, 10565 CrossRef CAS.
- H. Tan, H. Li, W. Ji and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 8374 CrossRef CAS PubMed.
- A. Paul, D. Chatterjee, Rajkamal, T. Halder, S. Banerjee and S. Yadav, Tetrahedron Lett., 2015, 56, 2496 CrossRef CAS.
- L. Habert and K. Cariou, Angew. Chem., Int. Ed., 2020, 60, 171 CrossRef PubMed.
- W. Ji, H. Tan, M. Wang, P. Li and L. Wang, Chem. Commun., 2016, 52, 1462 RSC.
- S. Selvakumar, R. Sakamoto and K. Maruoka, Chem. – Eur. J., 2016, 22, 6552 CrossRef CAS PubMed.
- S. Selvakumar, Q. K. Kang, N. Arumugam, A. I. Almansour, R. S. Kumar and K. Maruoka, Tetrahedron, 2017, 73, 5841 CrossRef CAS.
- T. Yakura, Y. Horiuchi, Y. Nishimura, A. Yamada, H. Nambu and T. Fujiwara, Adv. Synth. Catal., 2016, 358, 869 CrossRef CAS.
- B. Xing, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2018, 57, 9896 CrossRef CAS PubMed.
- S. M. Banik, K. M. Mennie and E. N. Jacobsen, J. Am. Chem. Soc., 2017, 139, 9152 CrossRef CAS PubMed.
- S.-S. Weng, K.-Y. Hsieh and Z.-J. Zeng, Tetrahedron, 2015, 71, 2549 CrossRef CAS.
- S. Chen, L. Yang, D. Yi, Q. Fu, Z. Zhang, W. Liang, Q. Zhang, J. Ji and W. Wei, RSC Adv., 2017, 7, 26070 RSC.
- J. Jin, J. Tong, W. Yu, J. Qiao and C. Shen, Catal. Commun., 2020, 141, 106008 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Samata E.
Shetgaonkar
a,
Manjula
Krishnan
*a,
Samata E.
Shetgaonkar
a,
Manjula
Krishnan




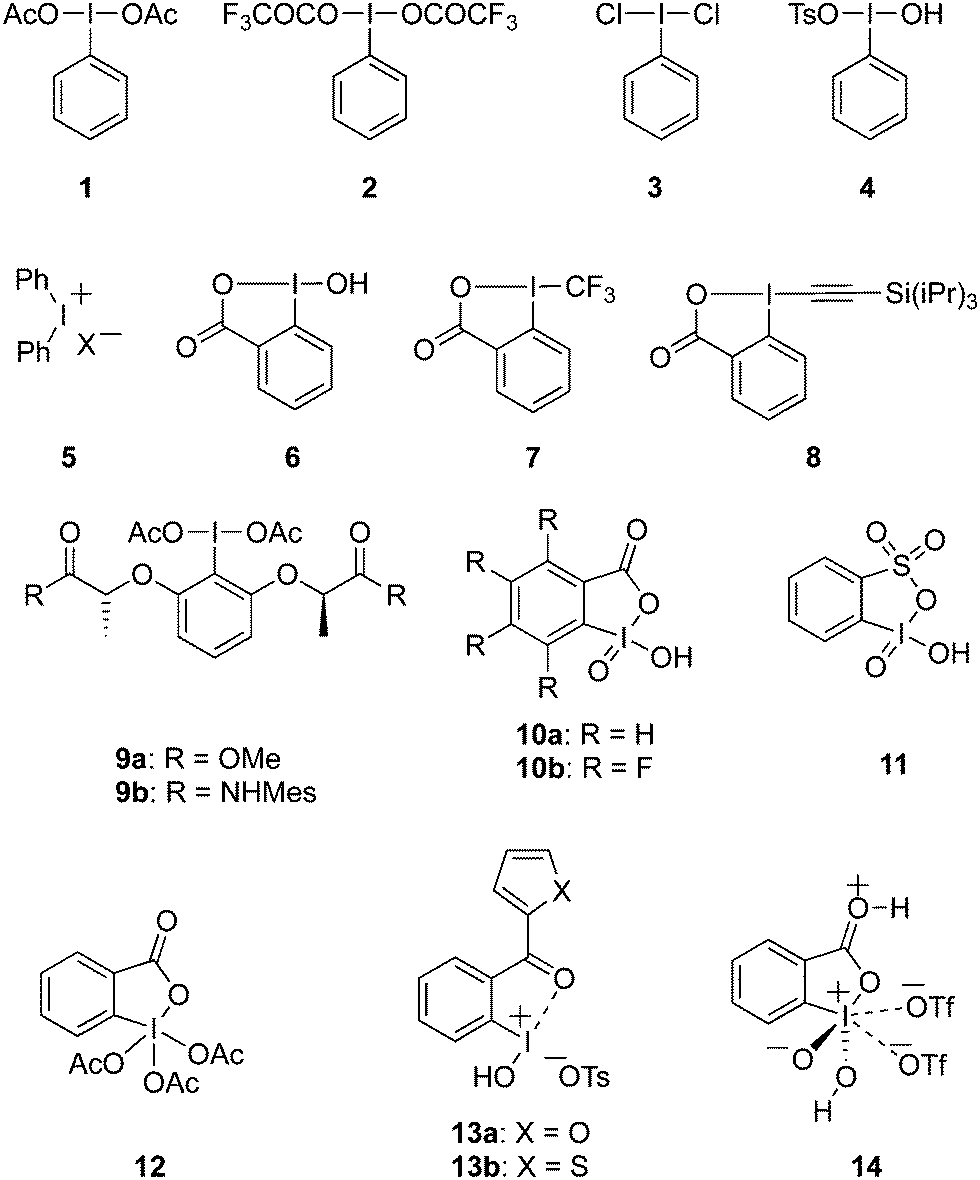
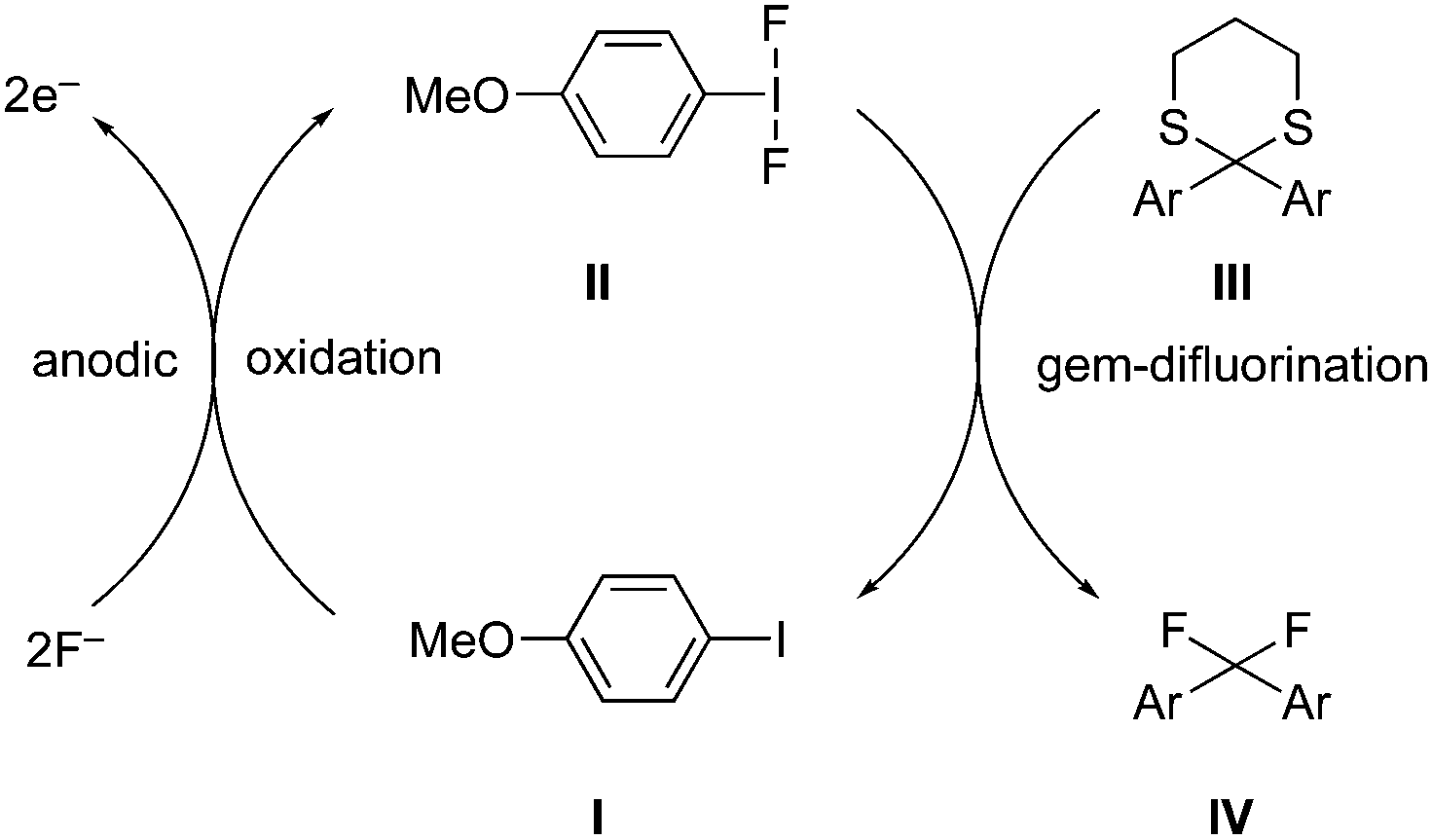
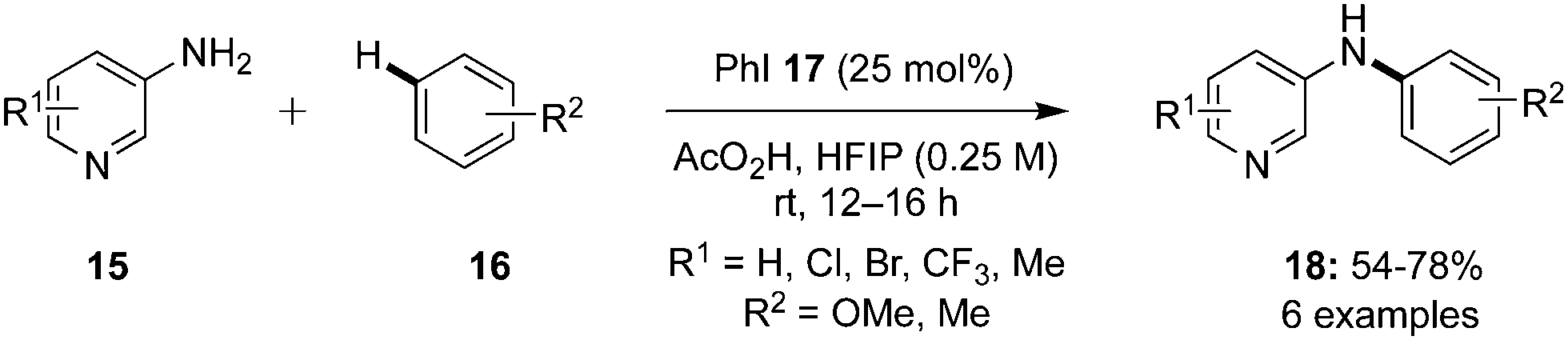
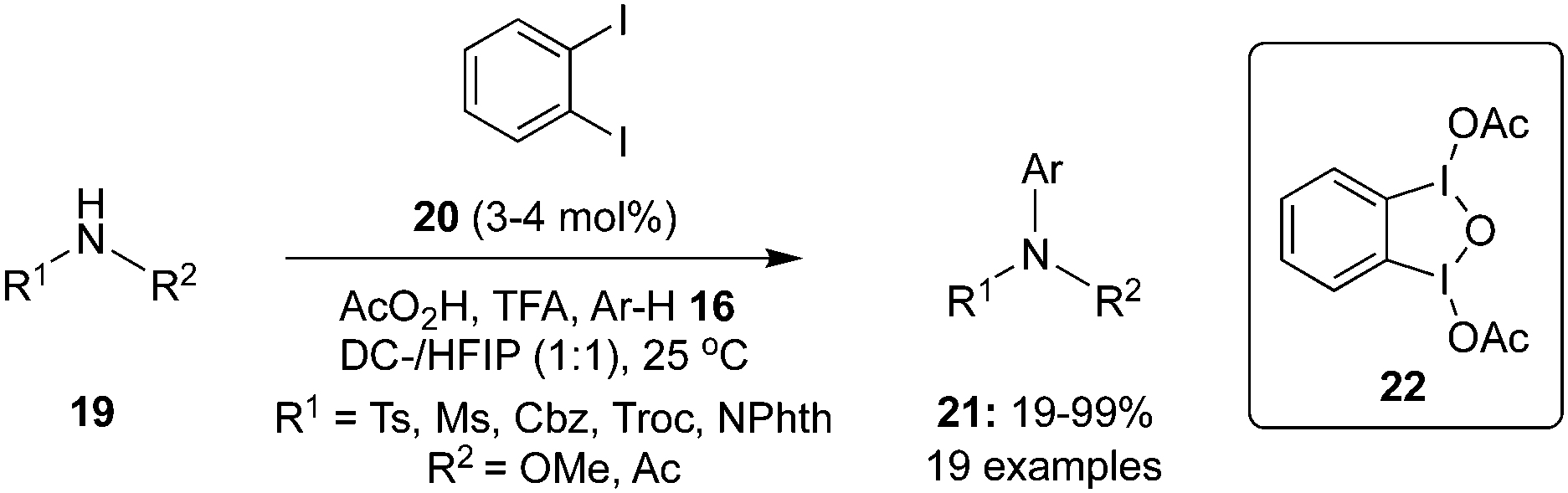
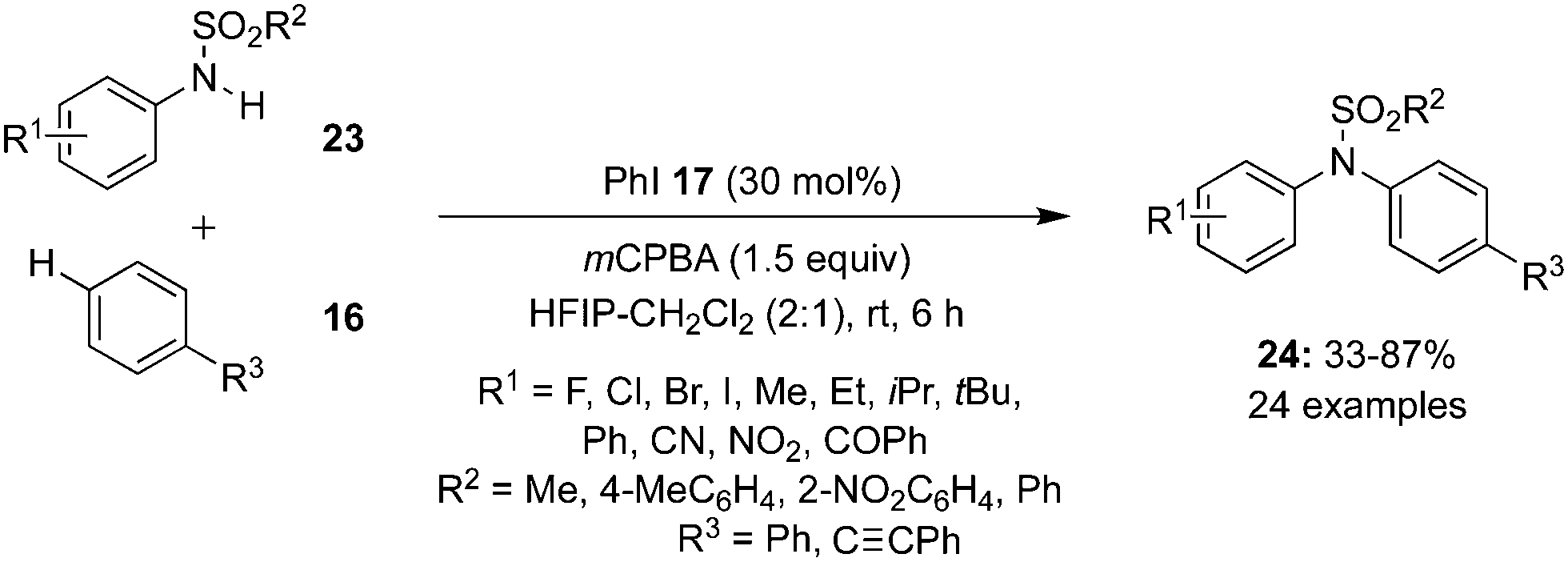
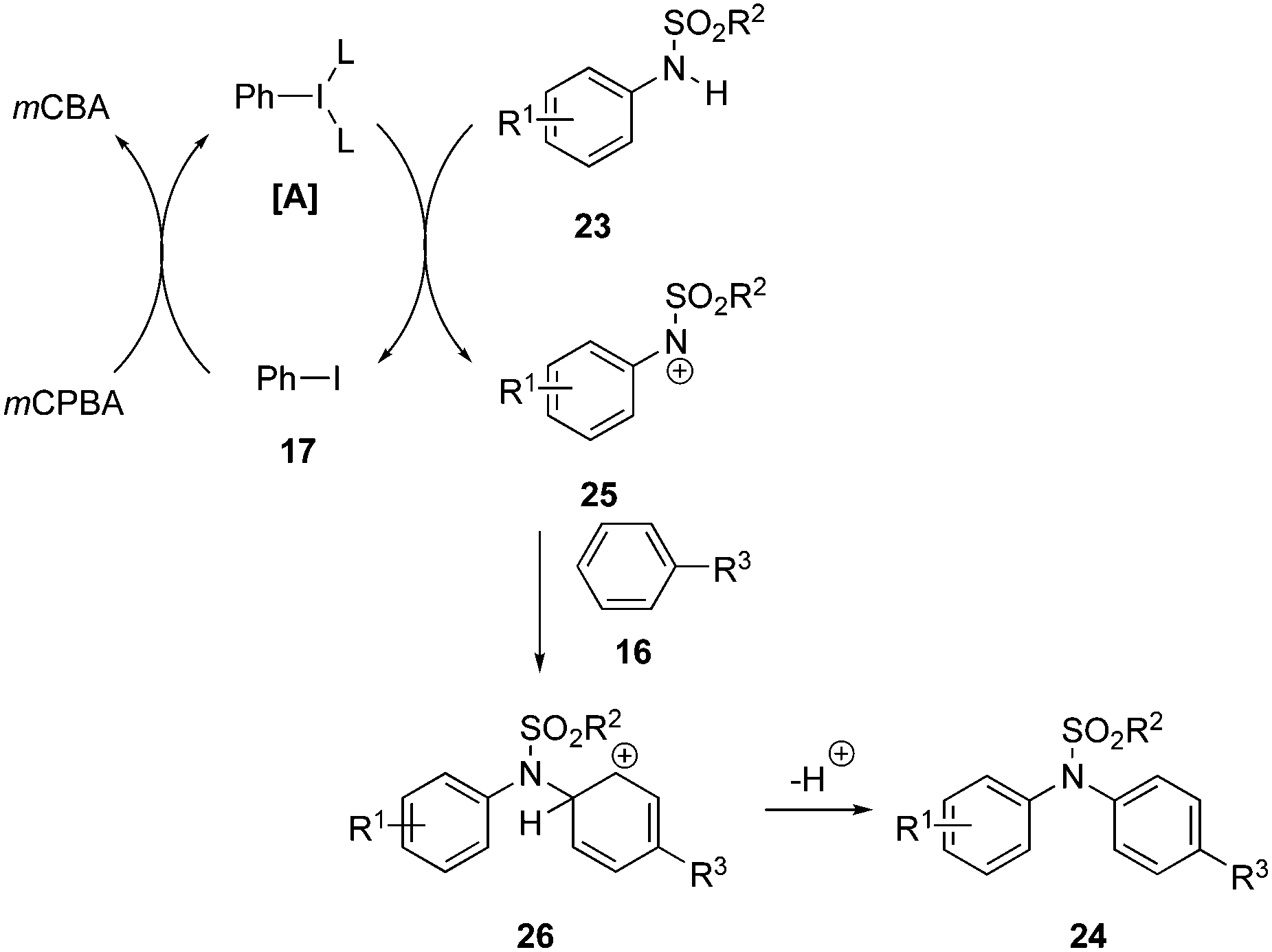
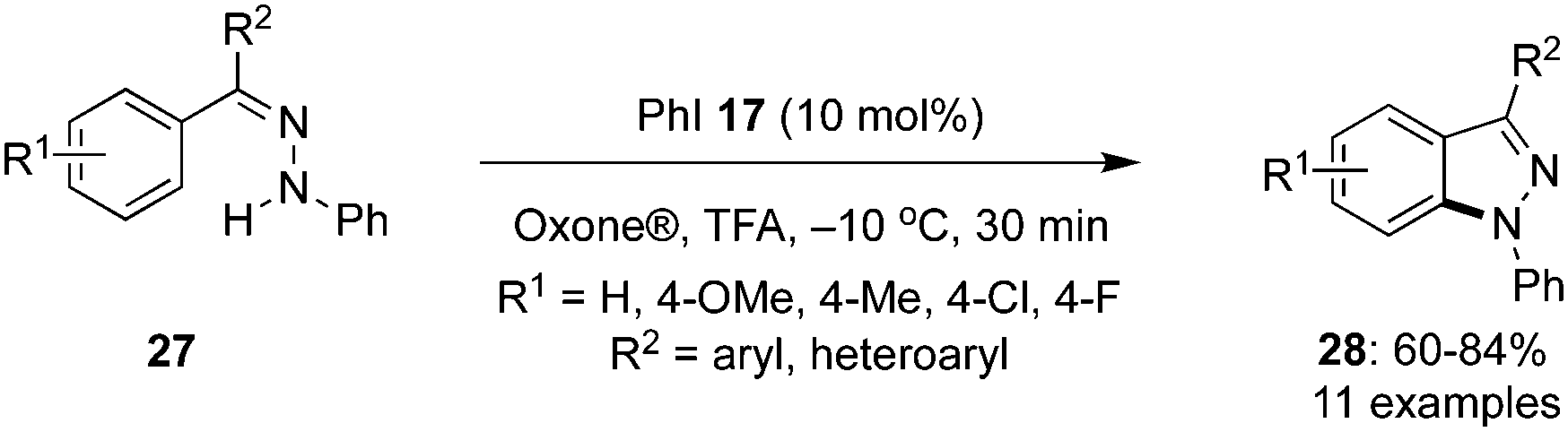
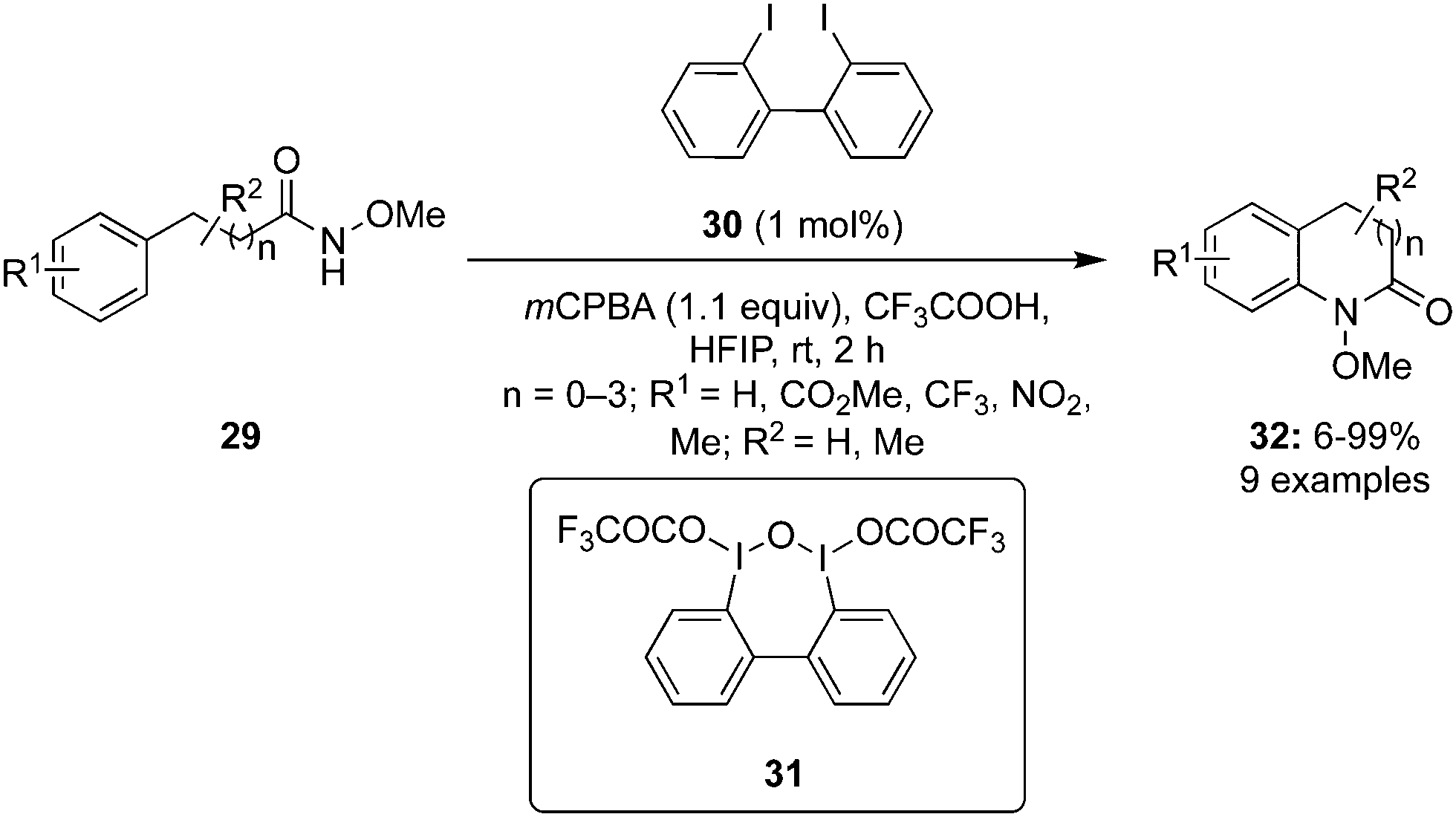
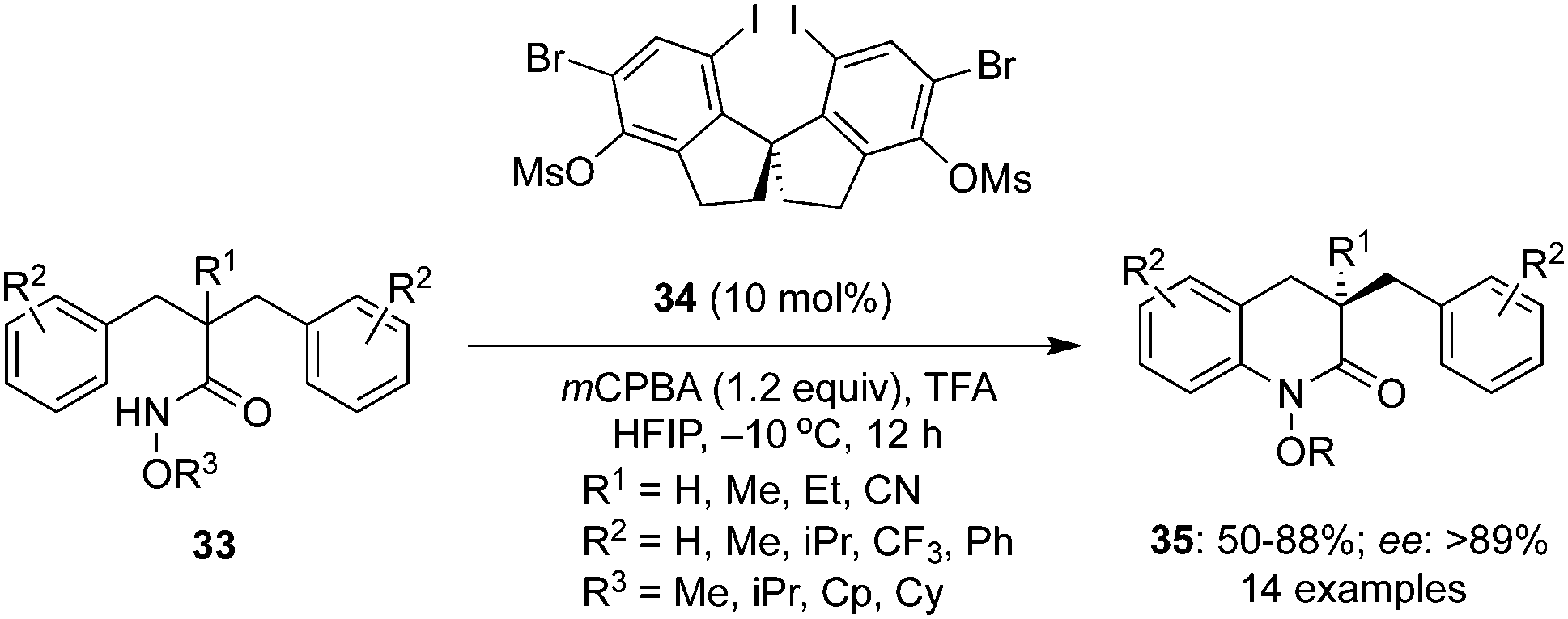
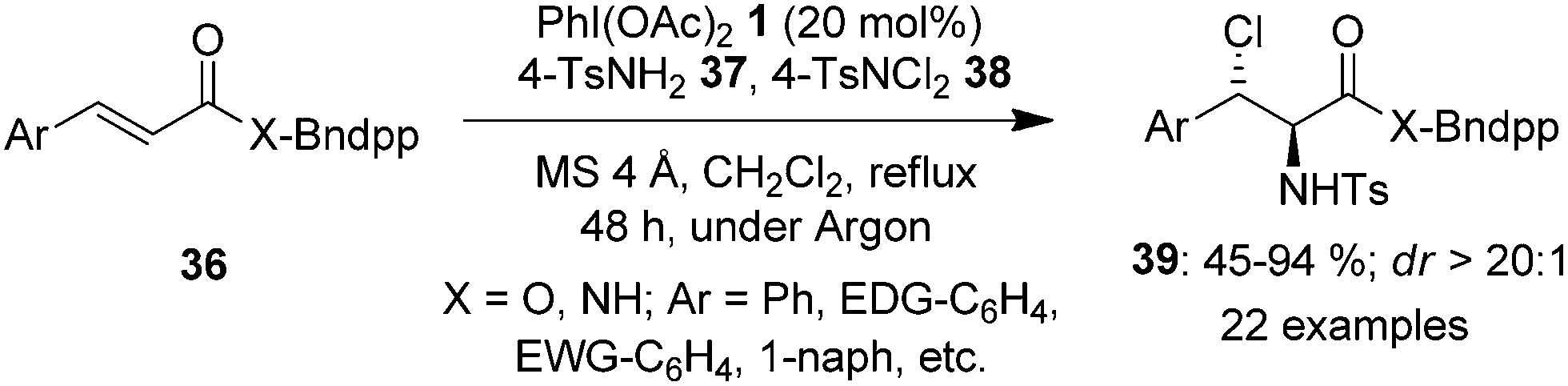

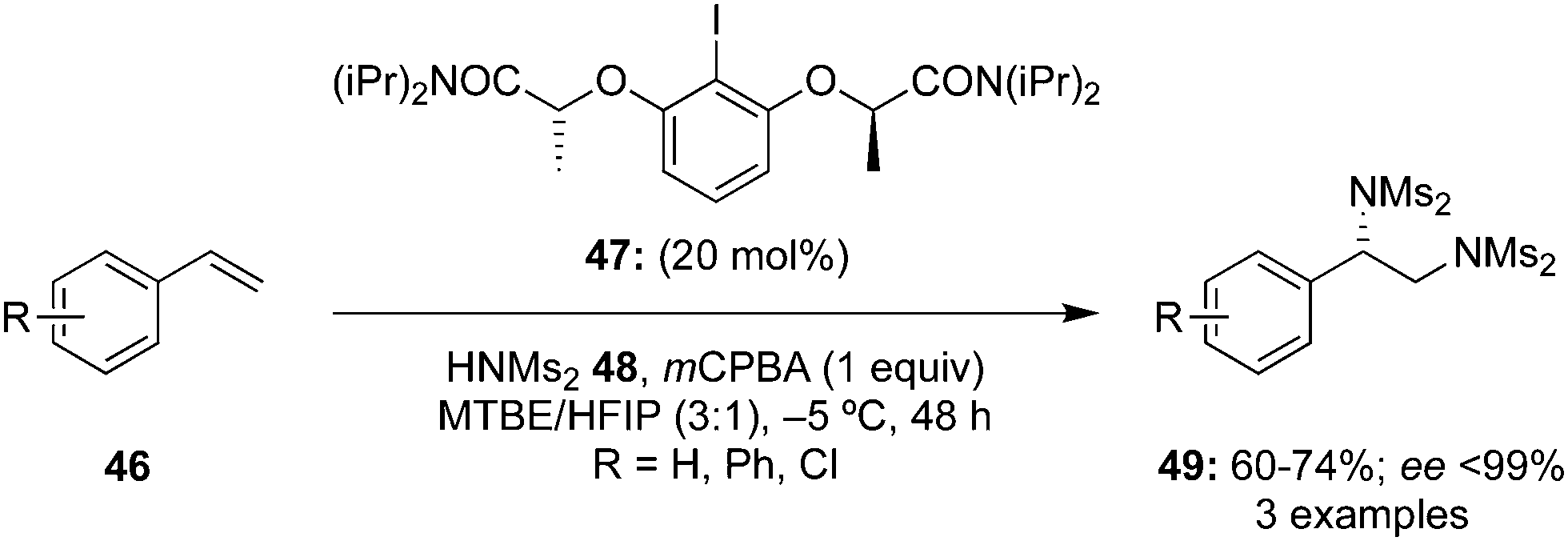
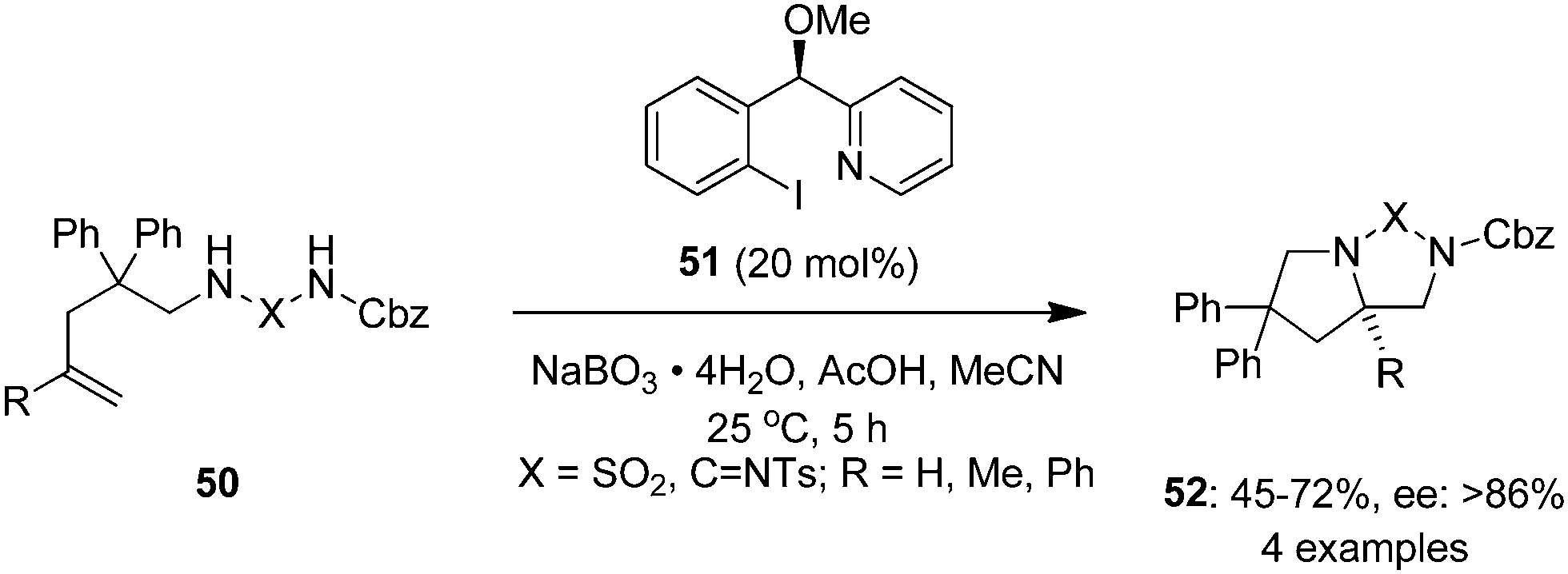
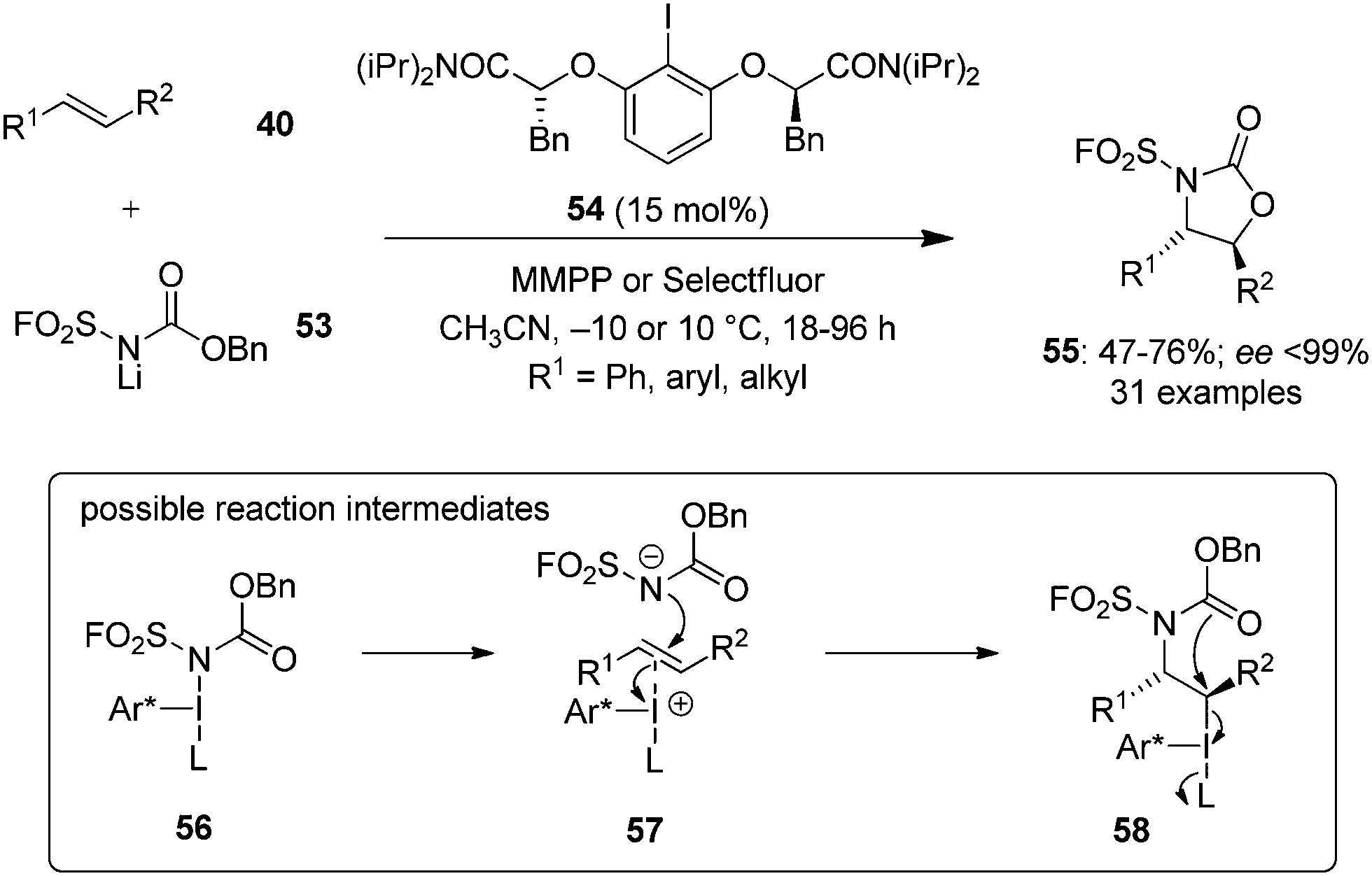
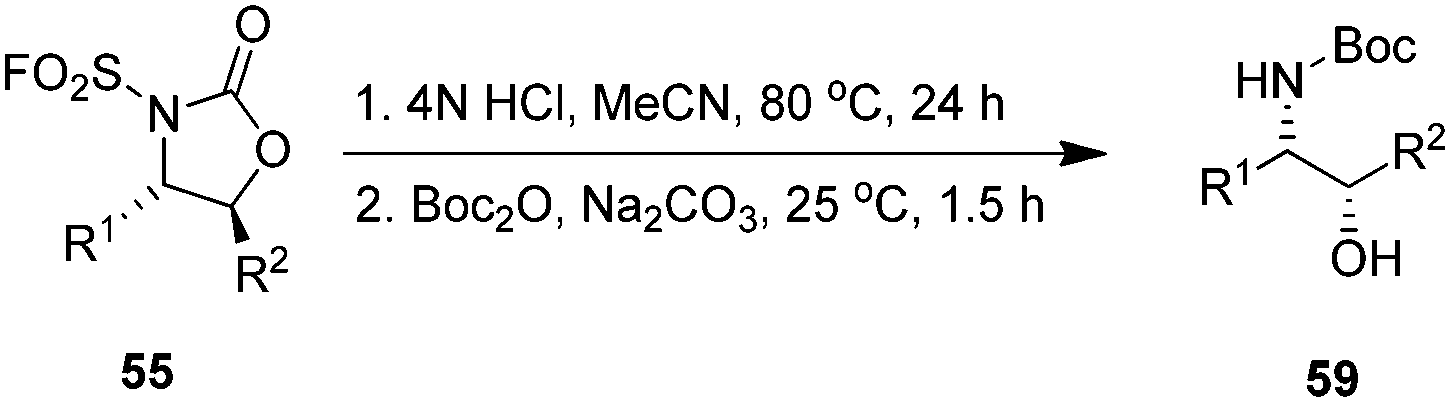
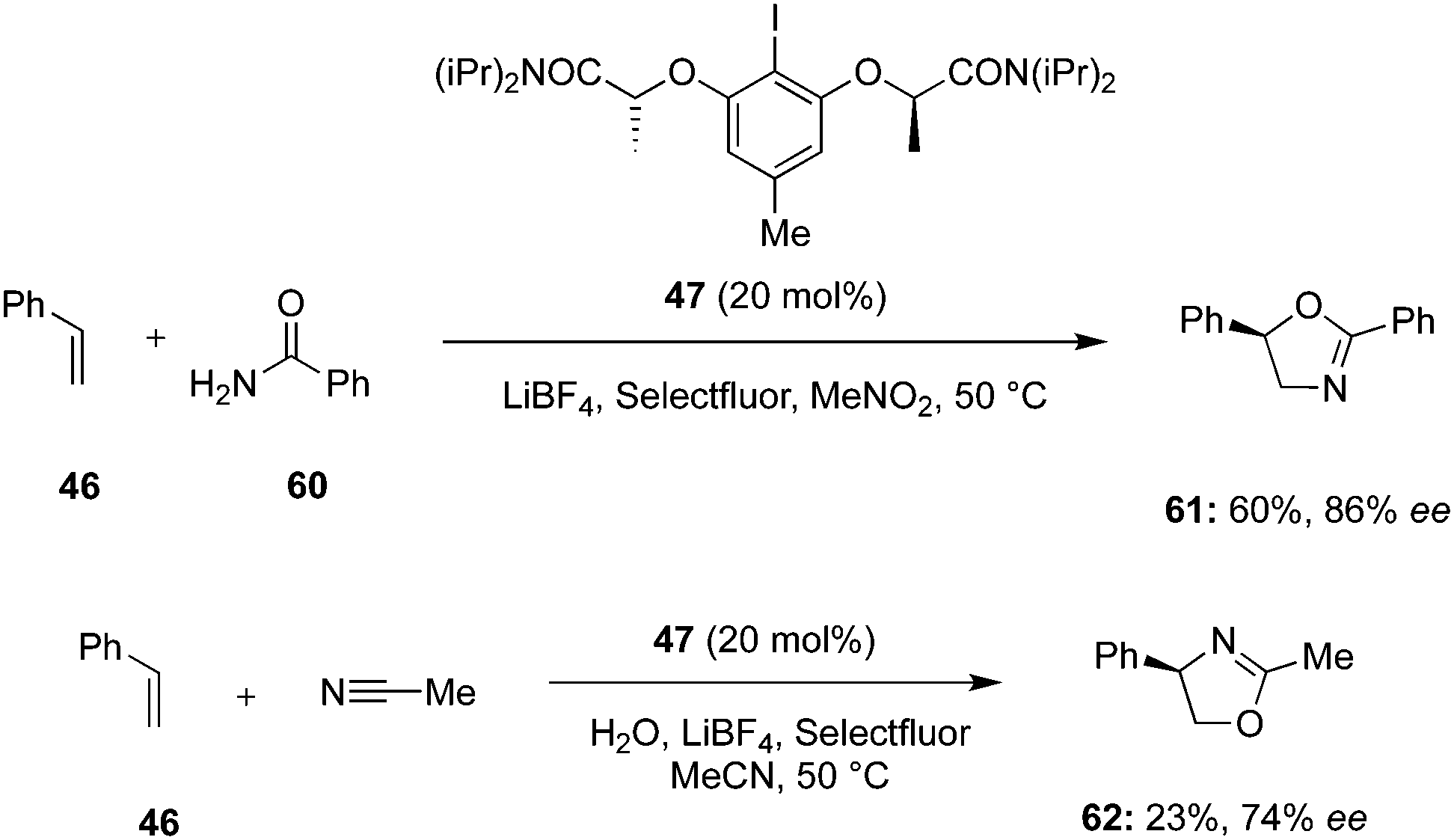
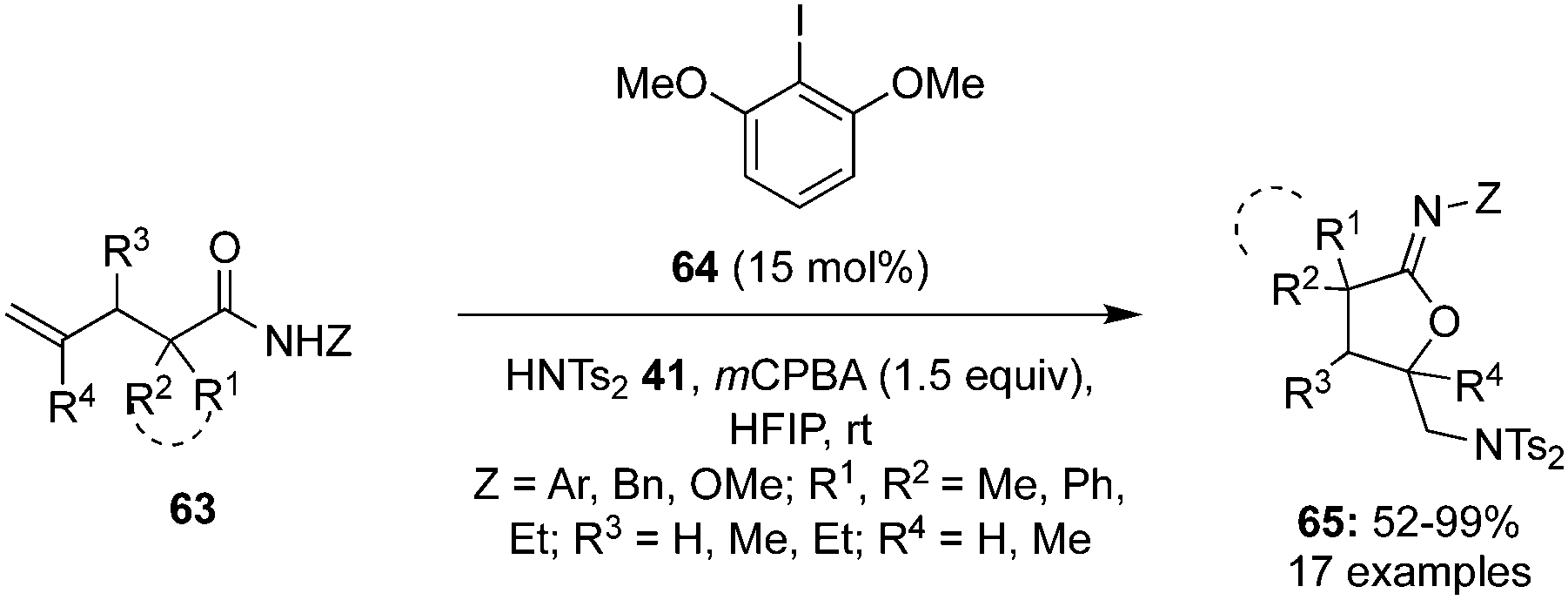
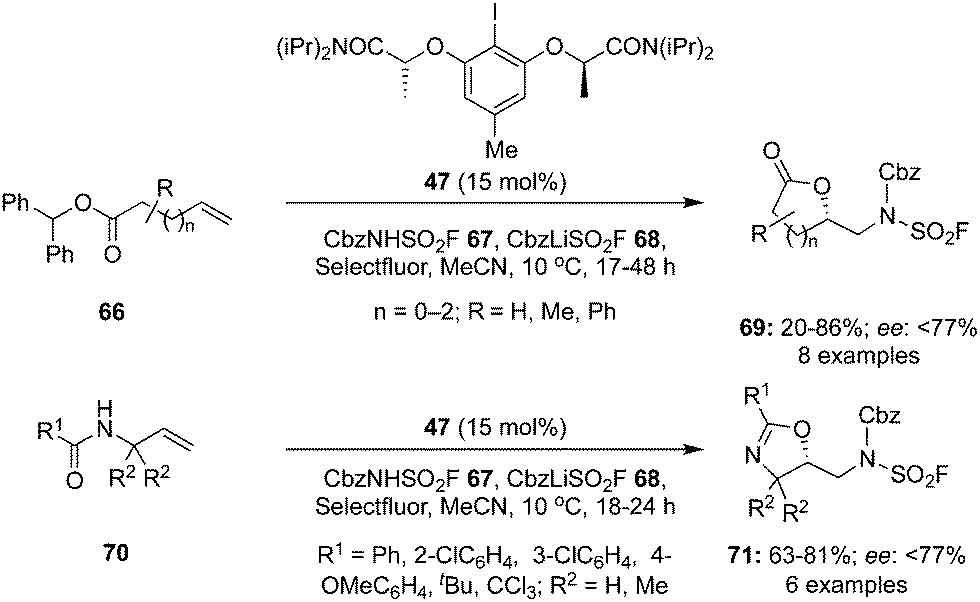
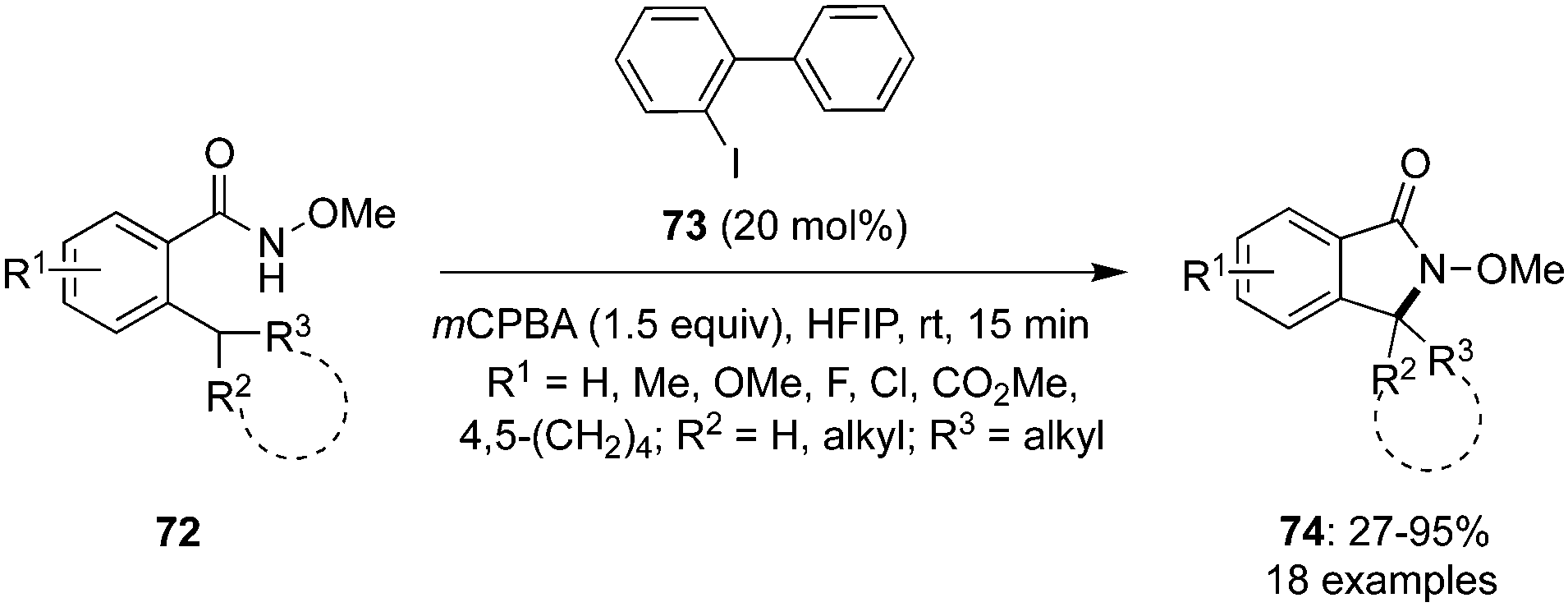
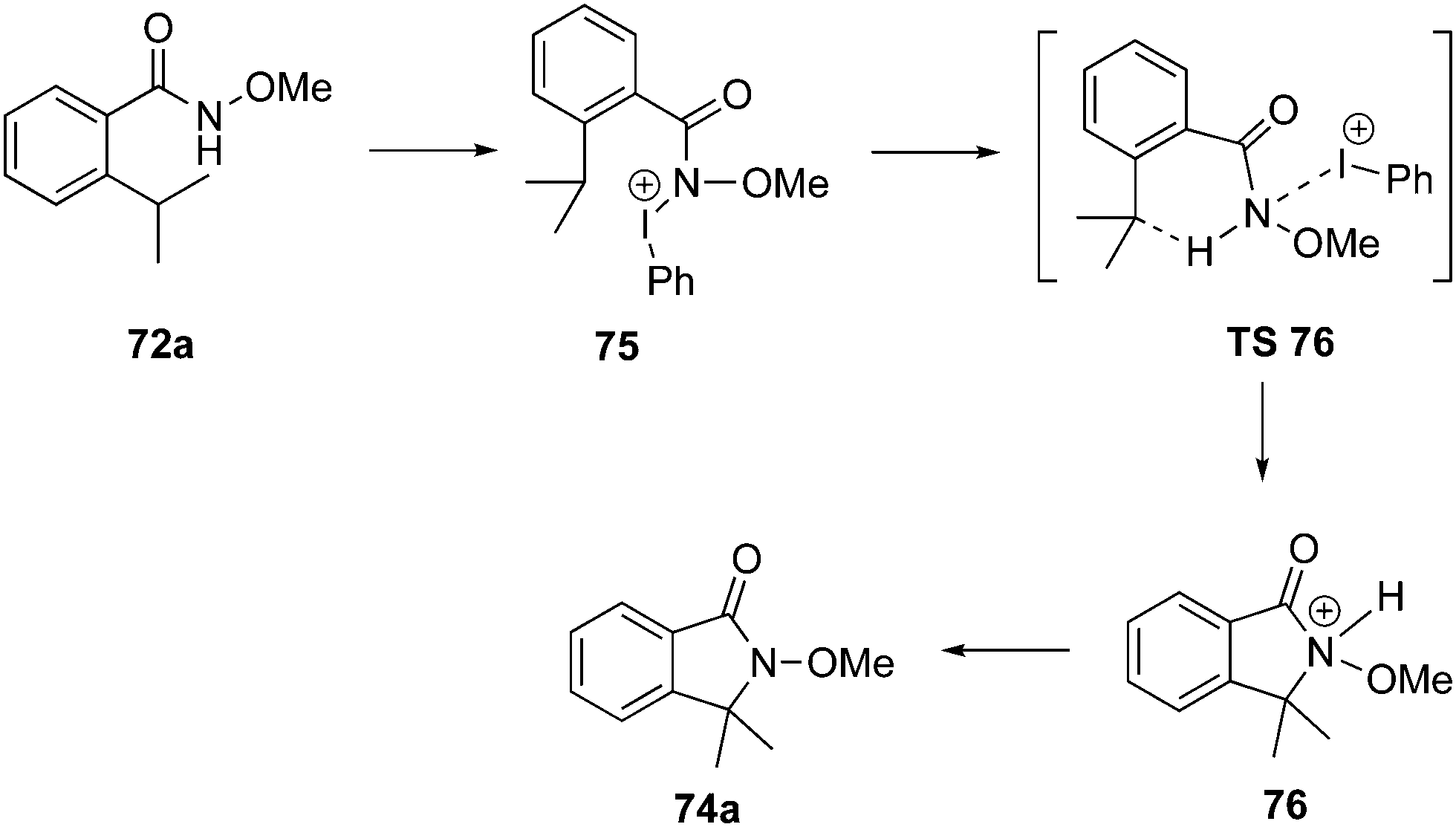
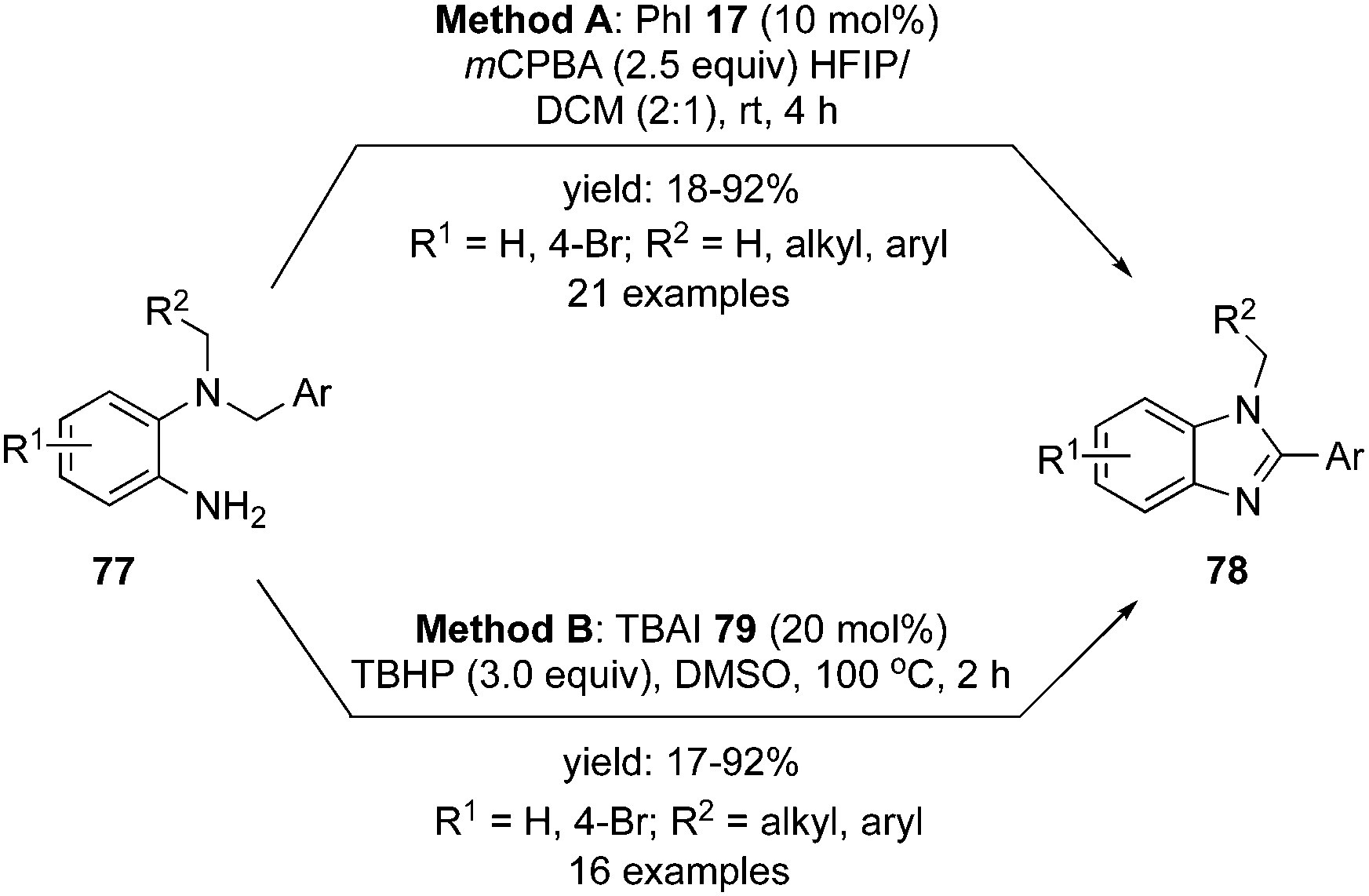
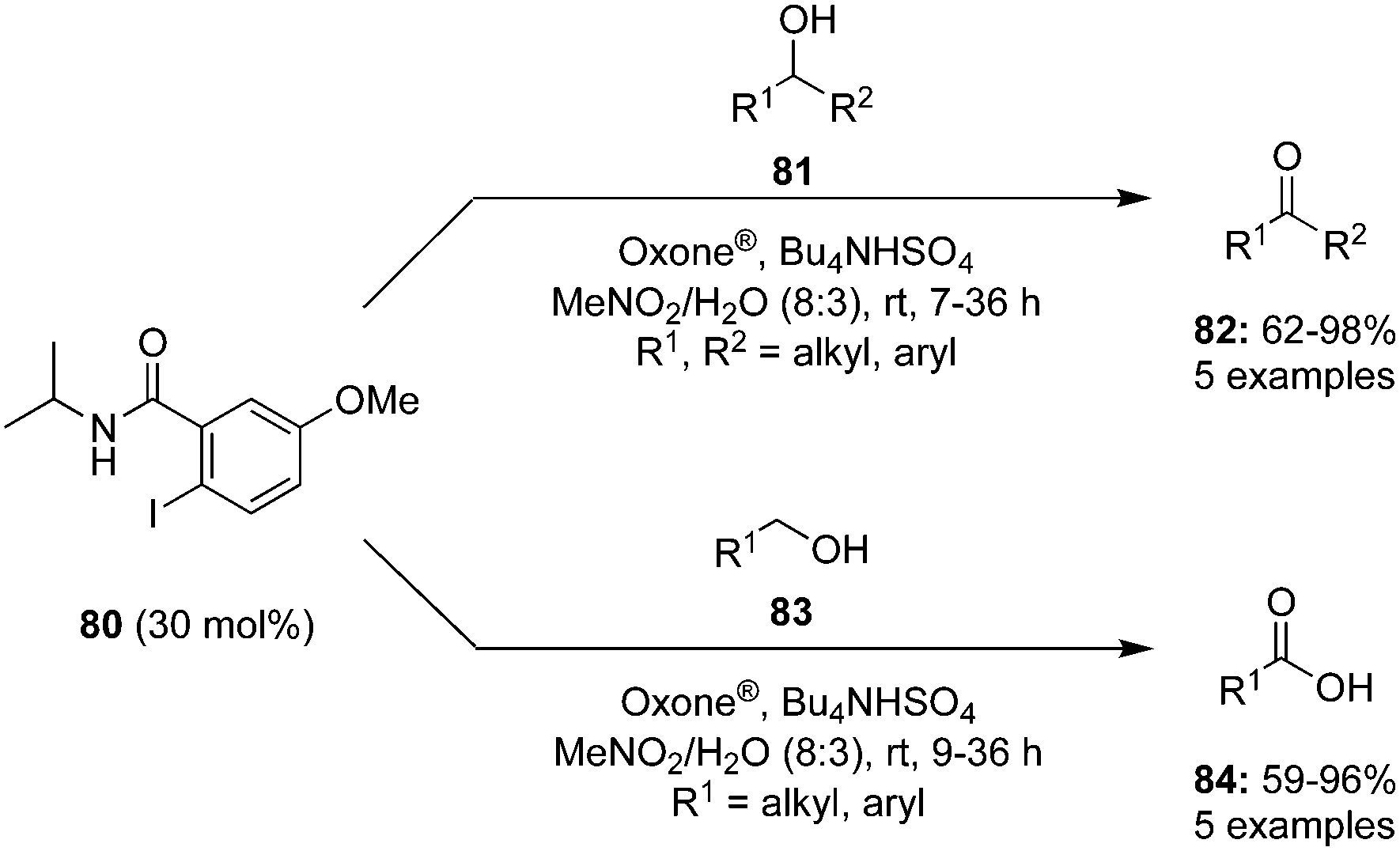
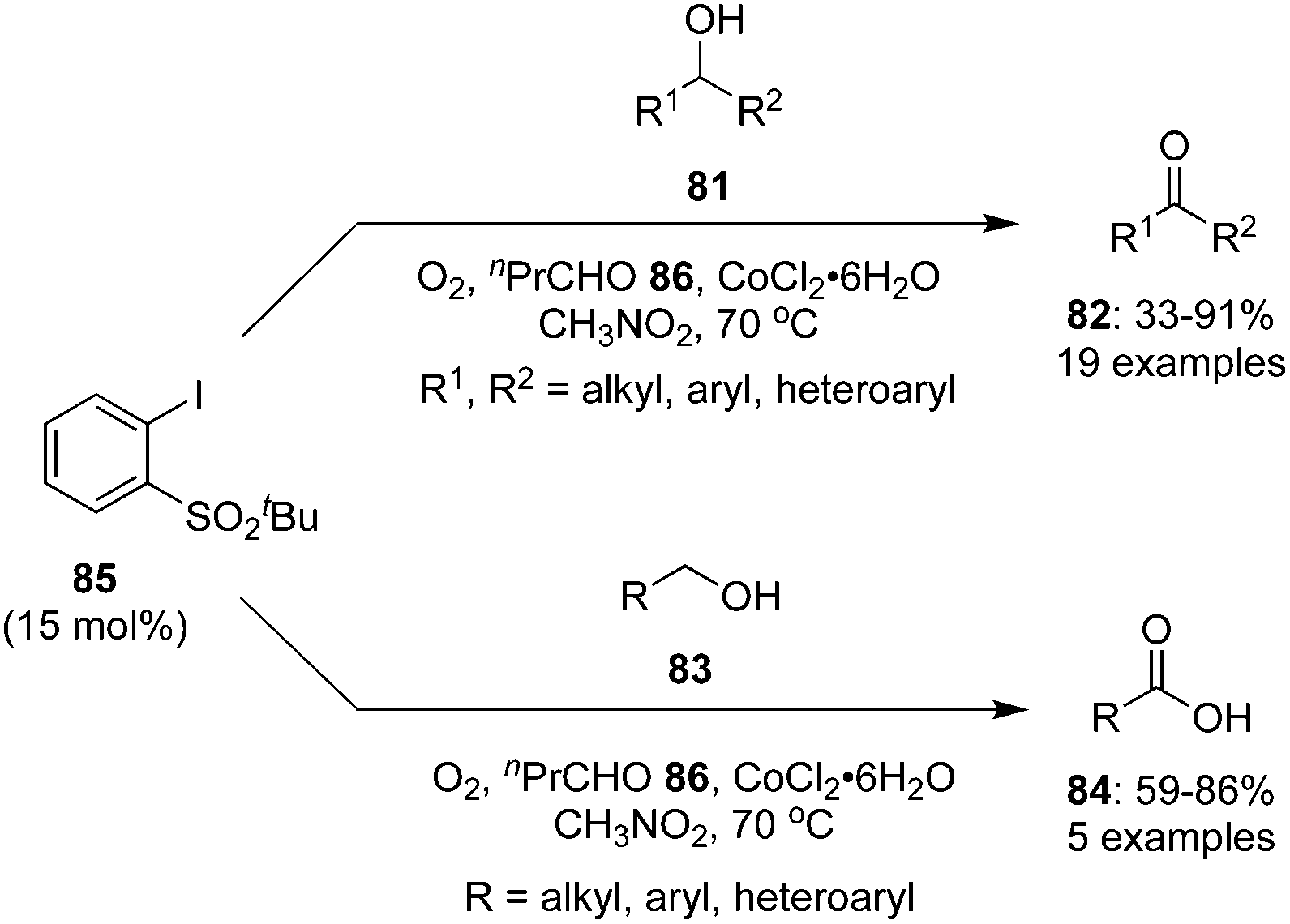
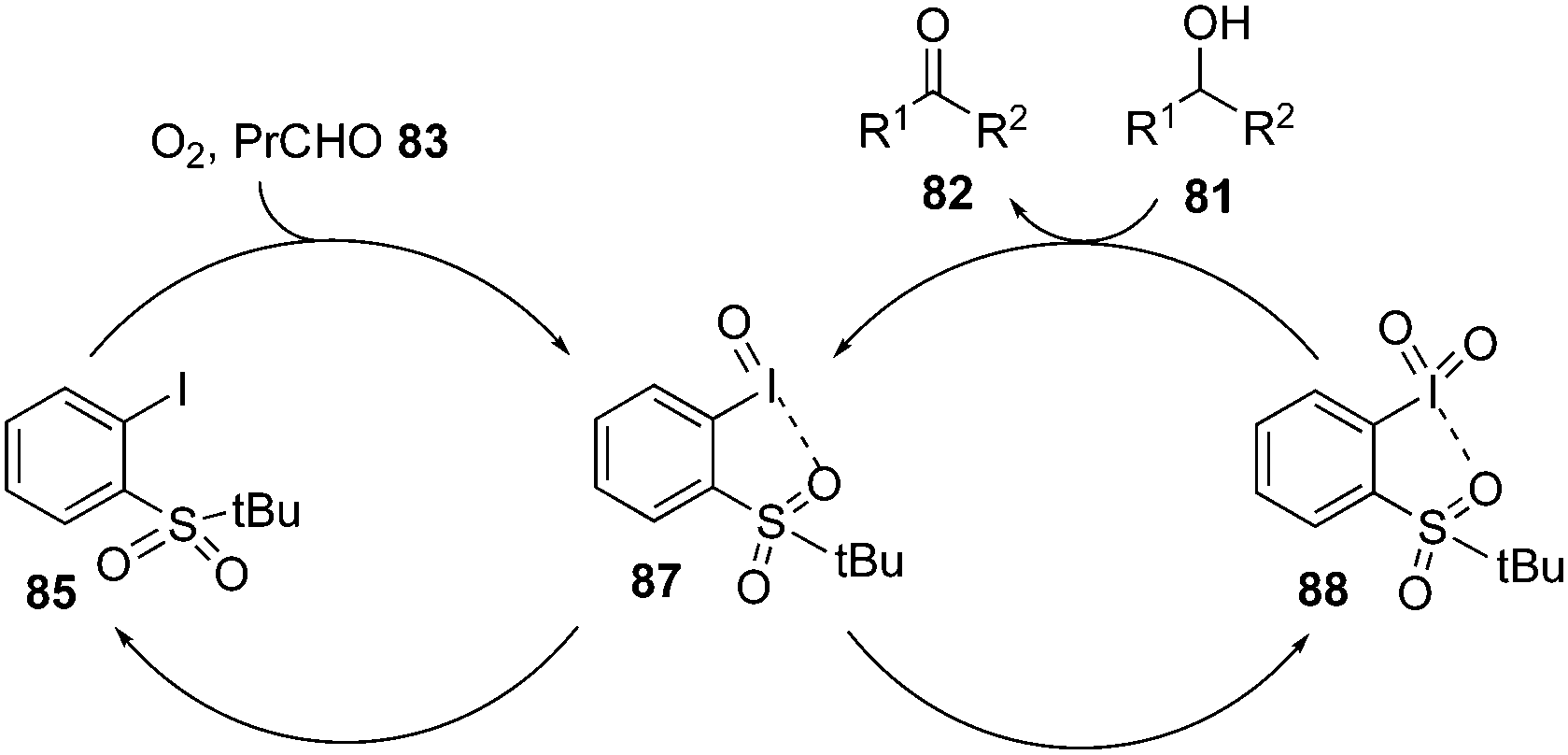
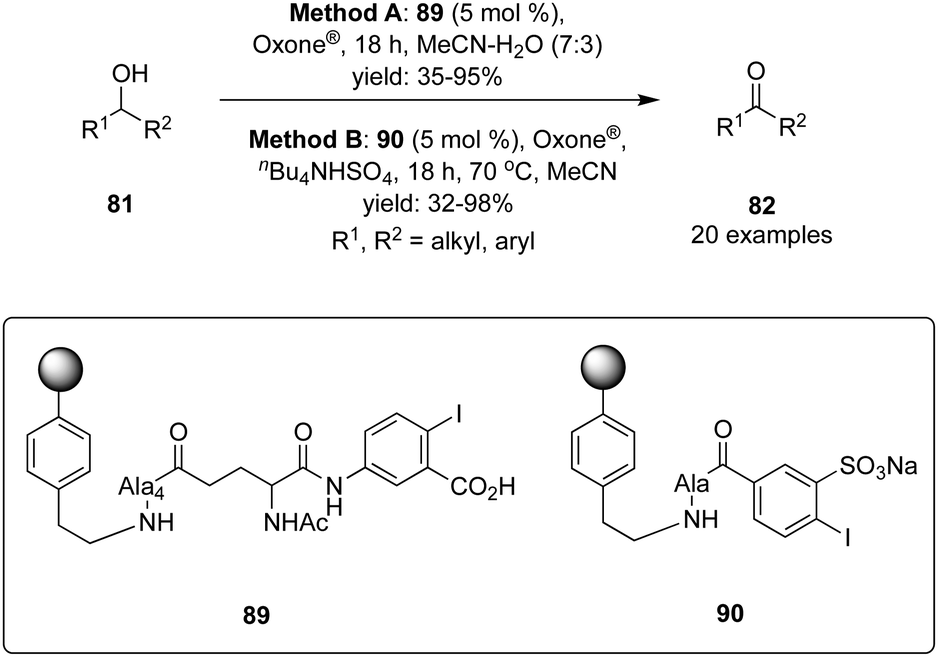
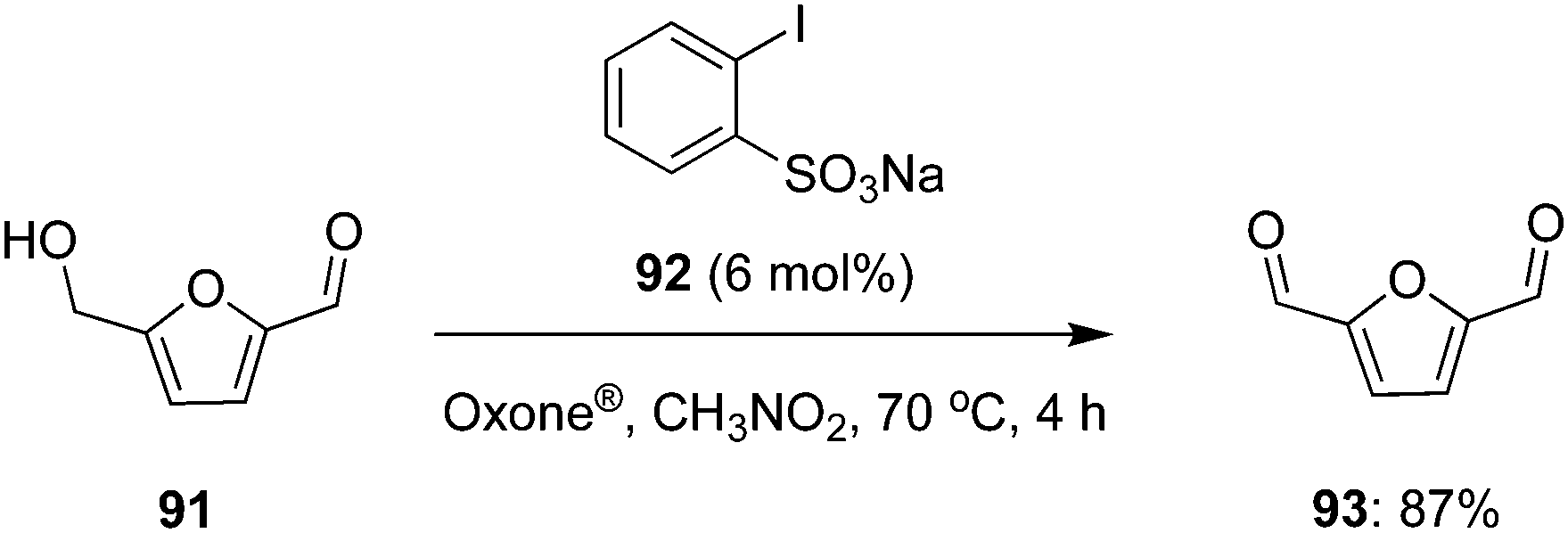
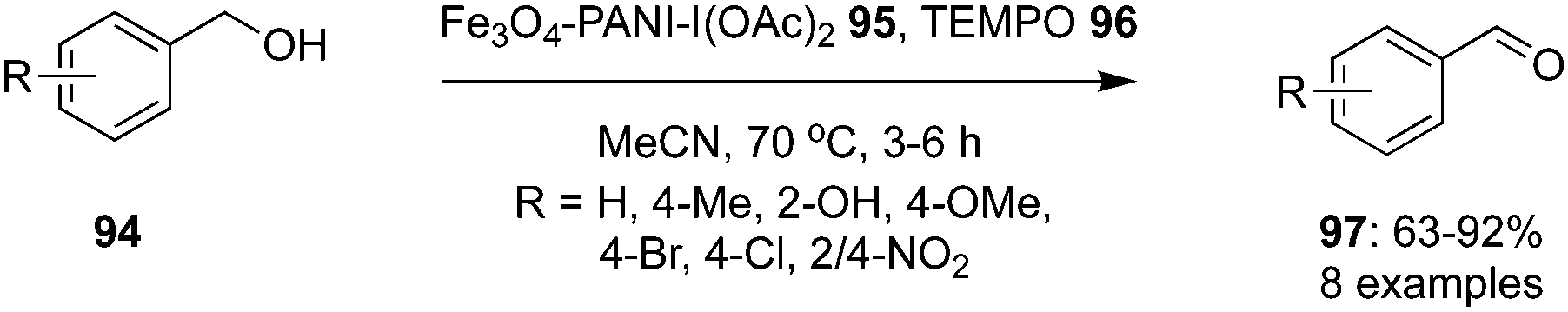
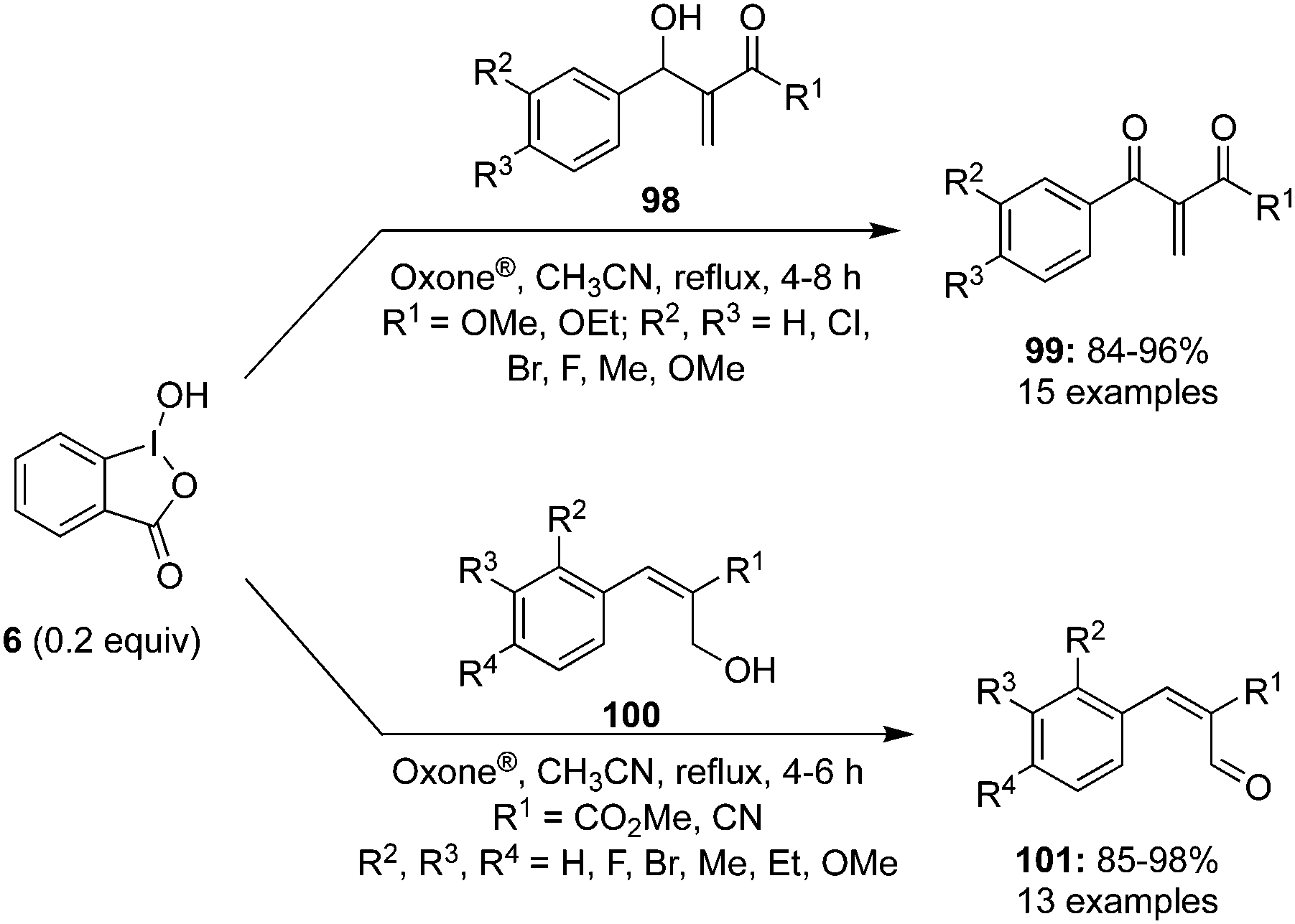
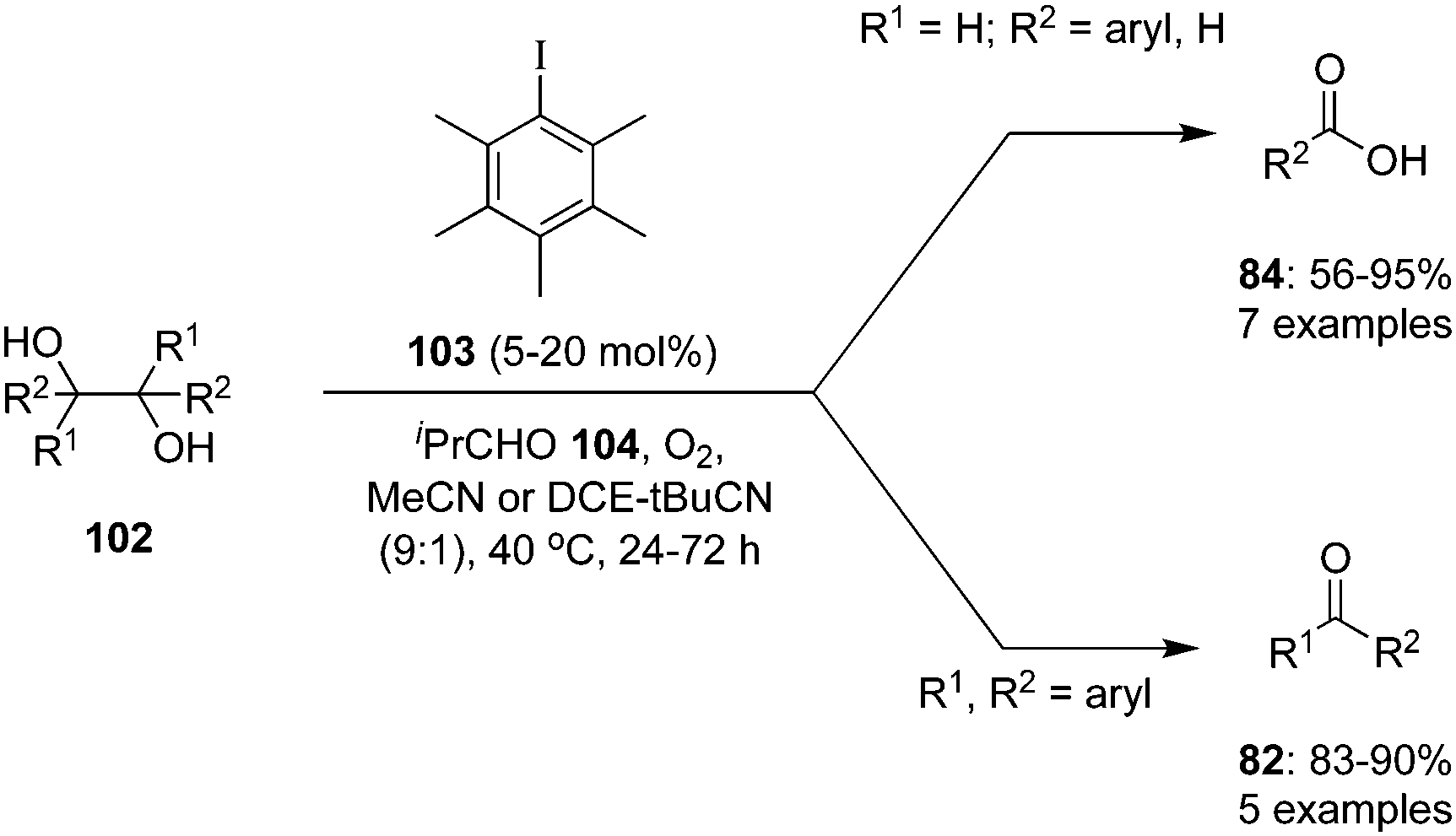
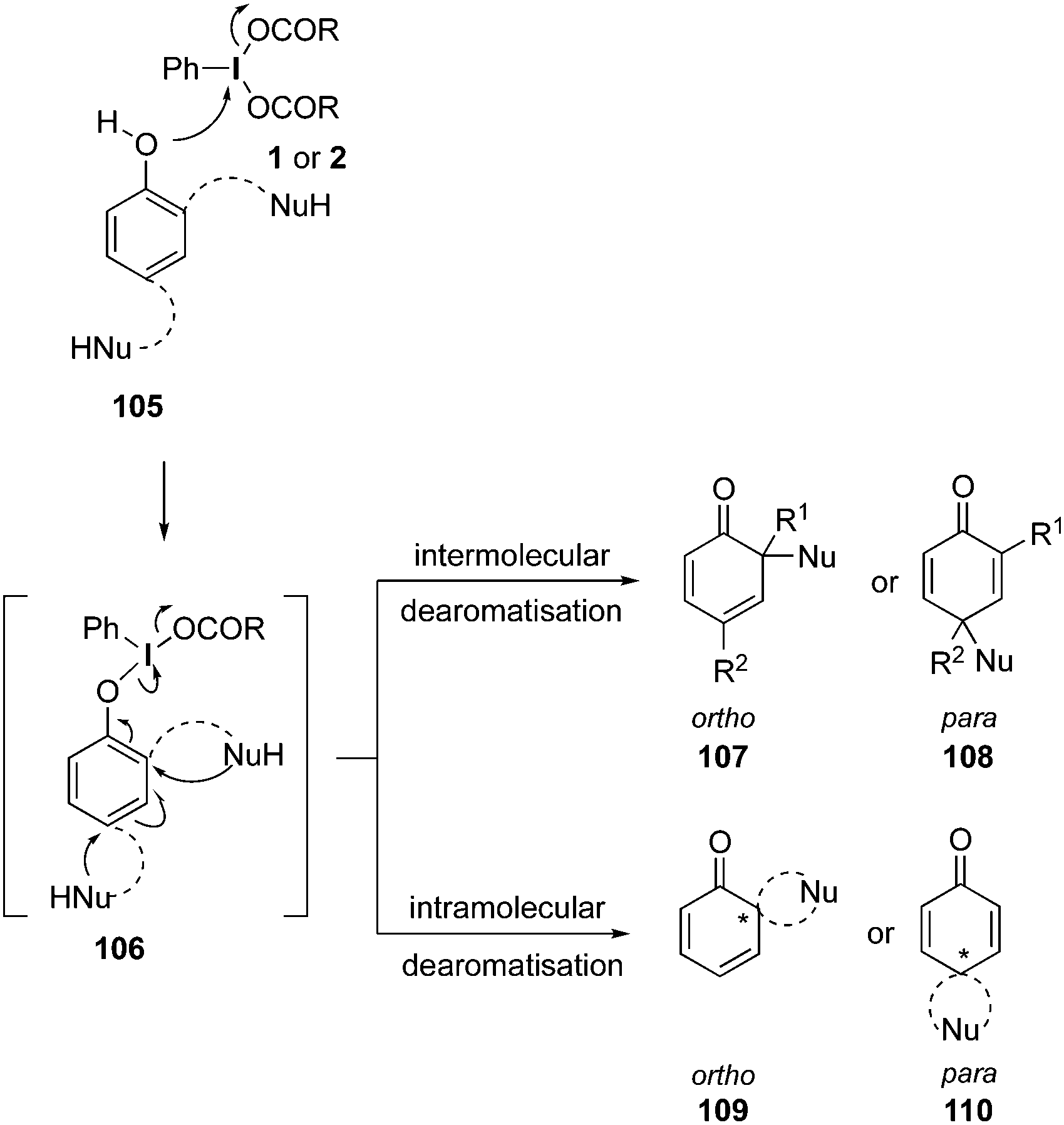
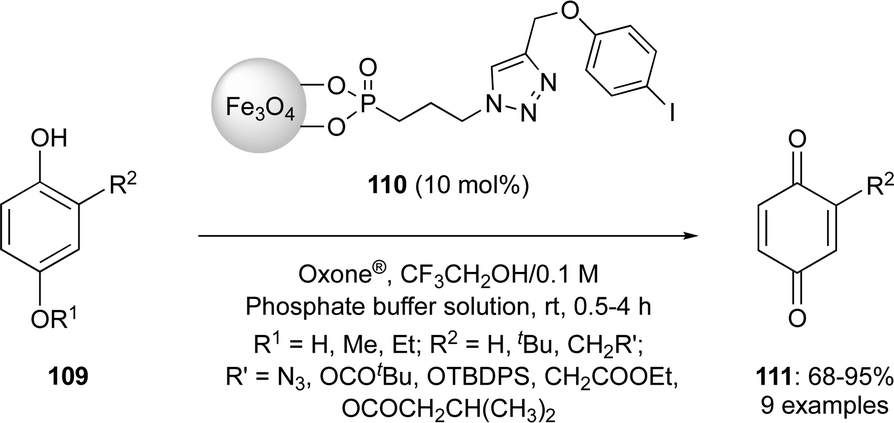
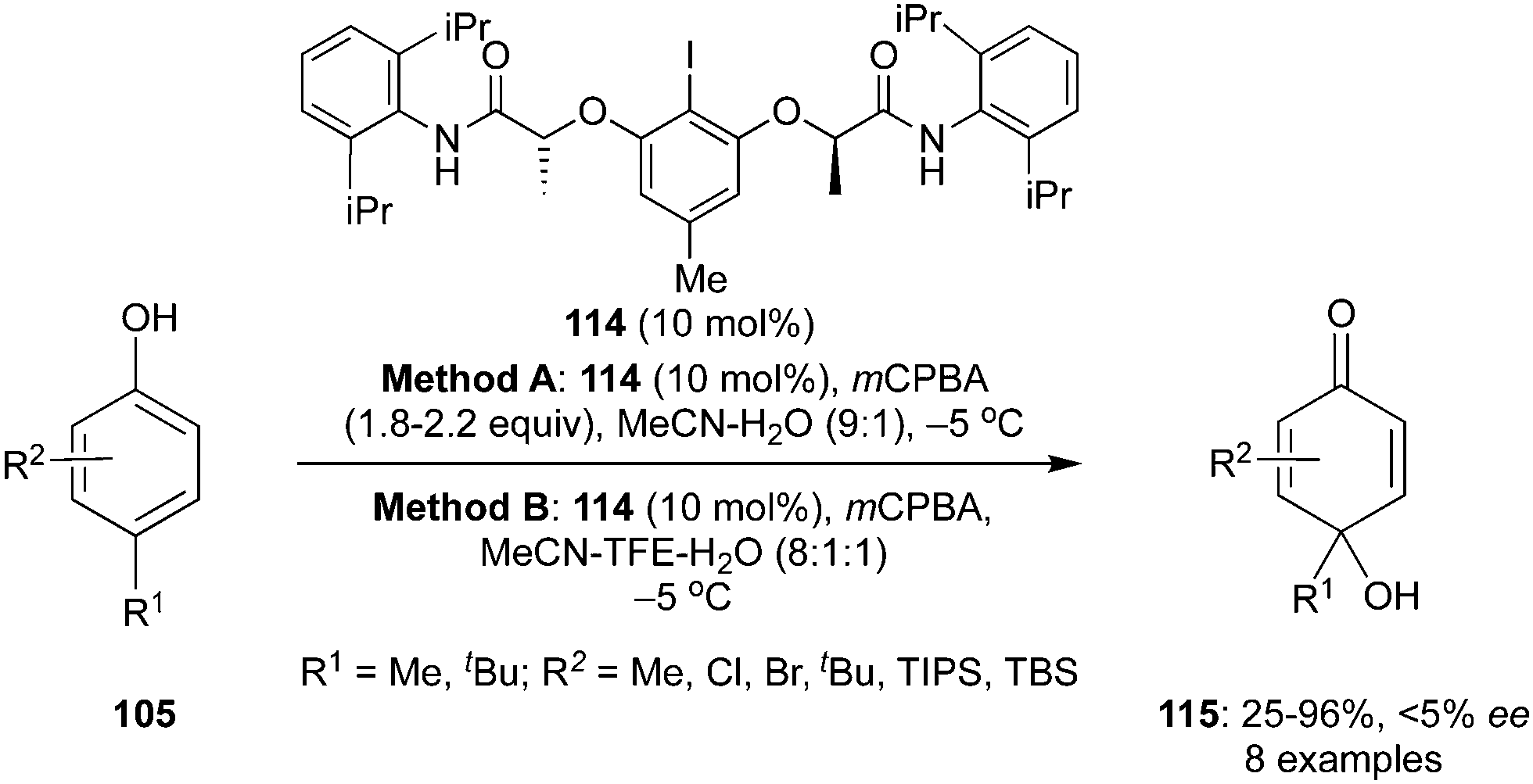
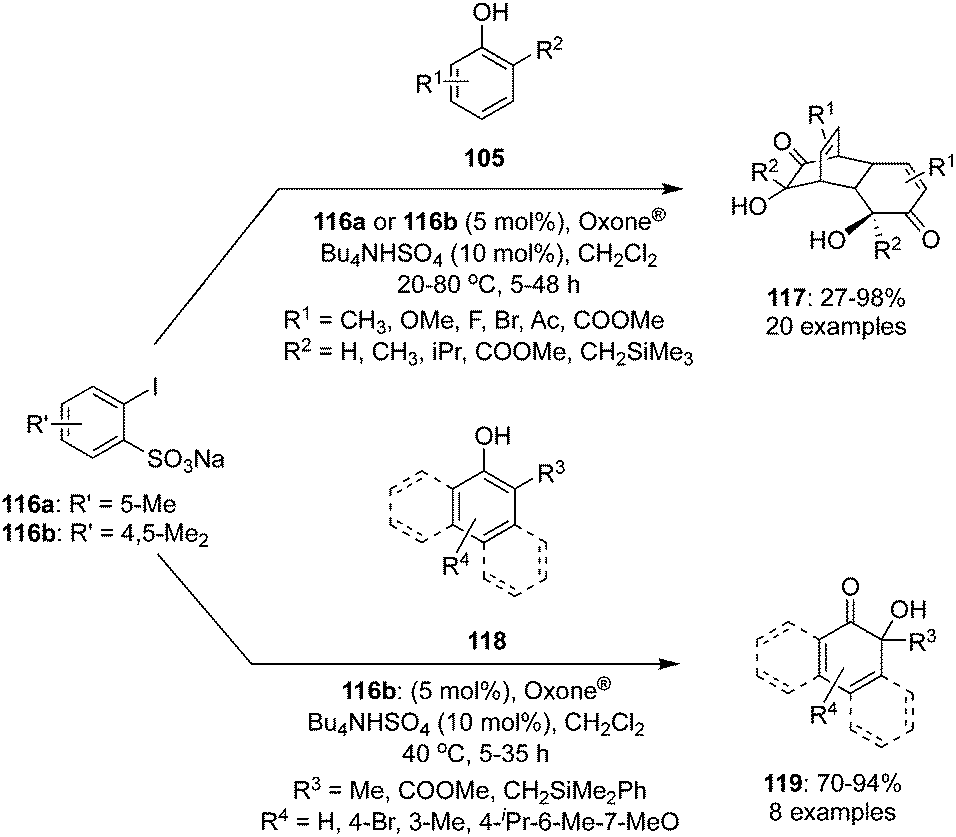
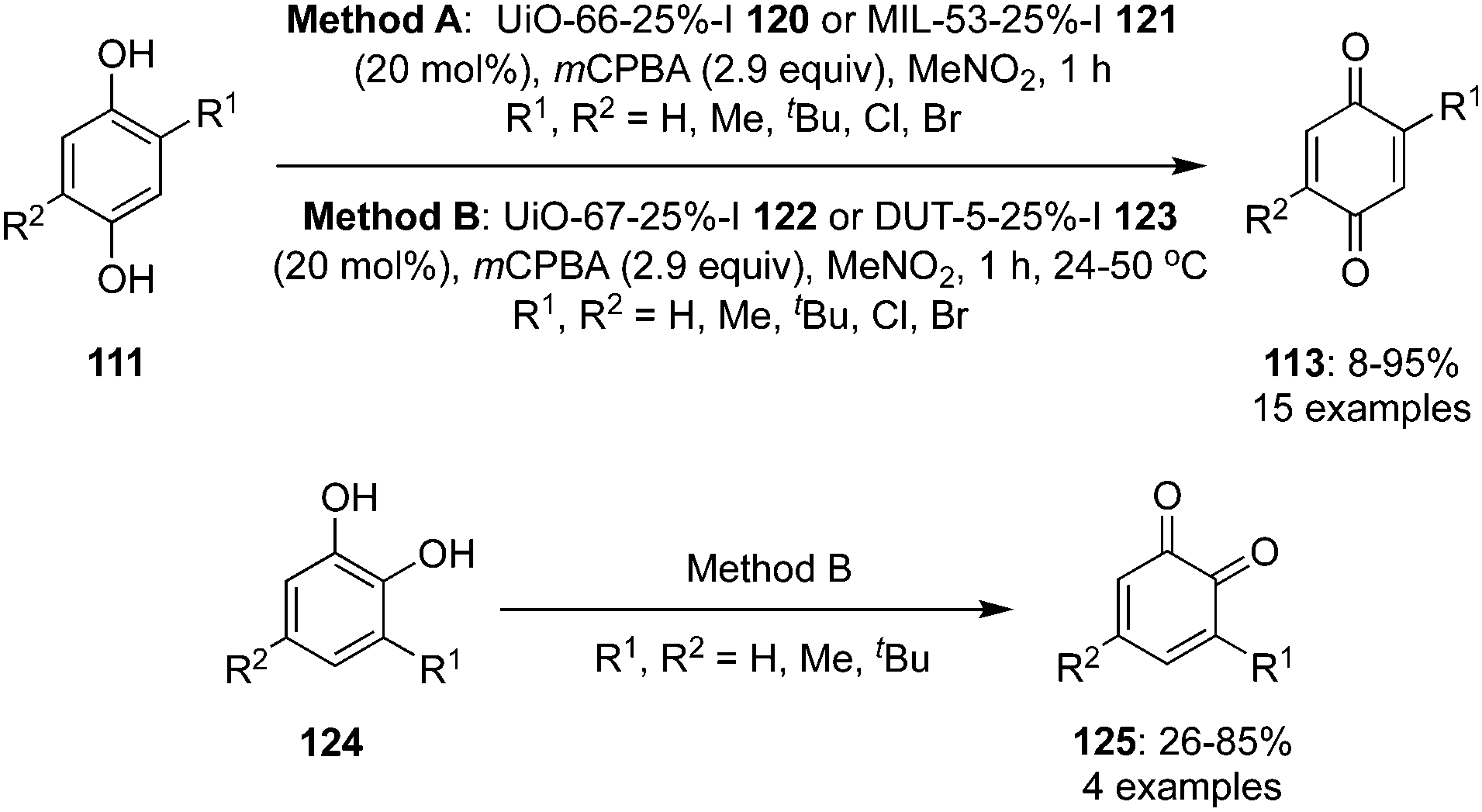
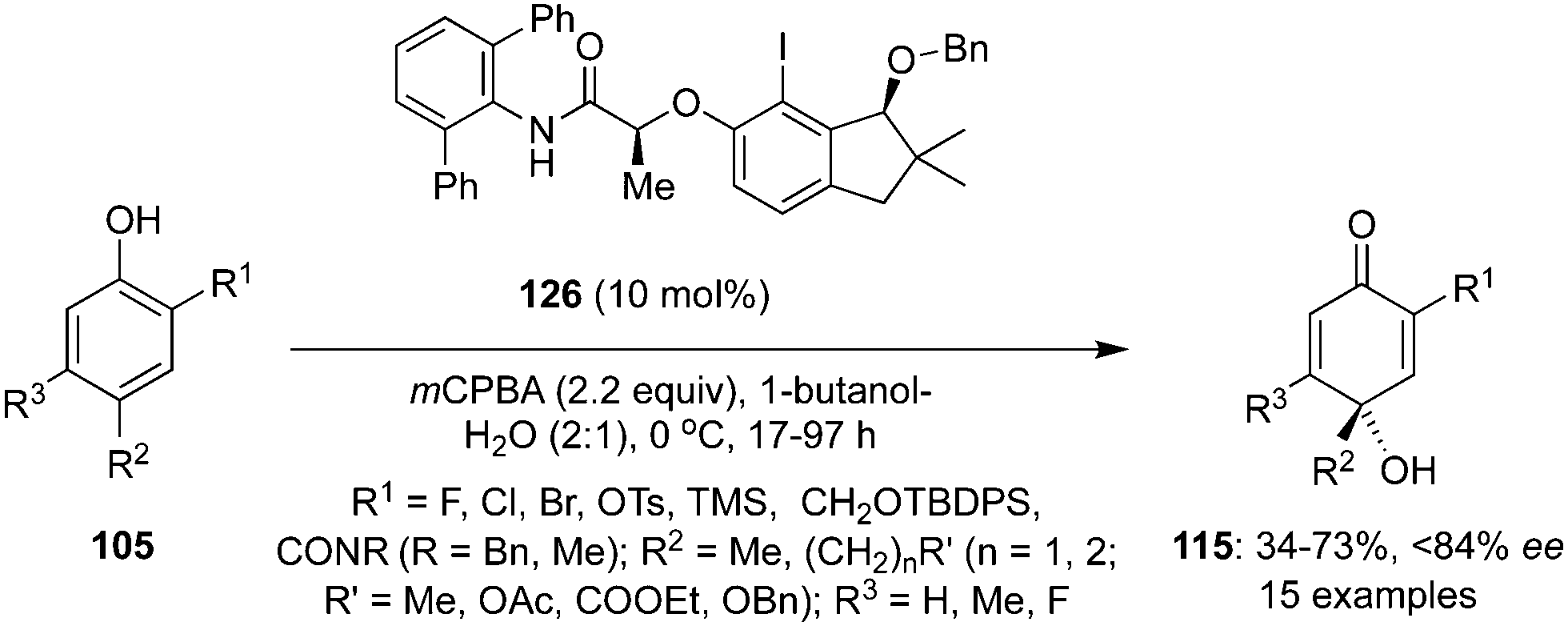
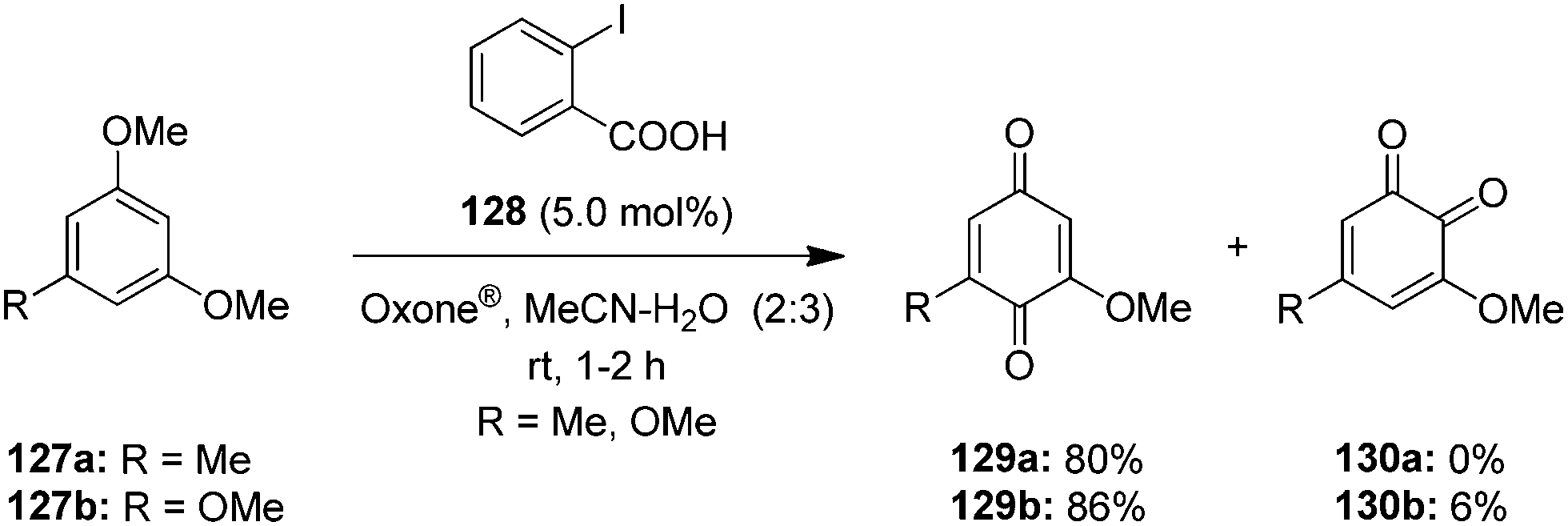
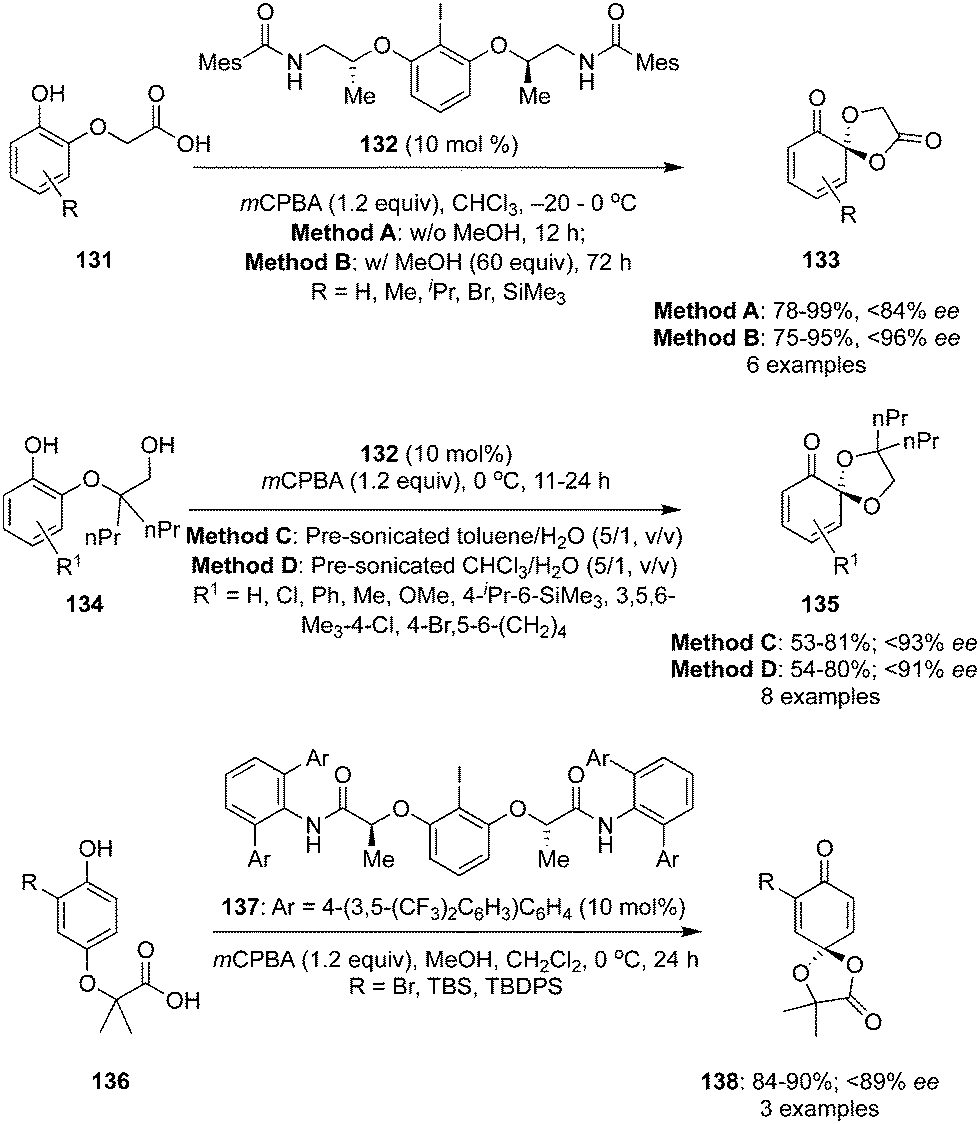
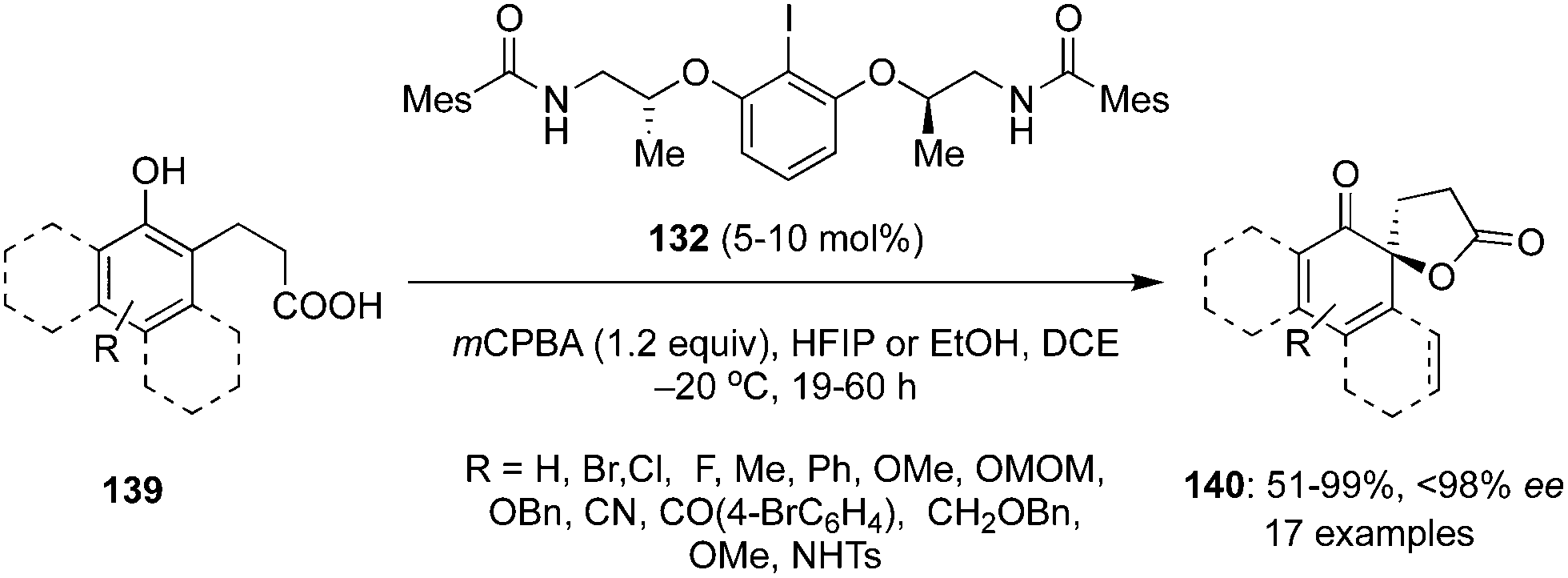
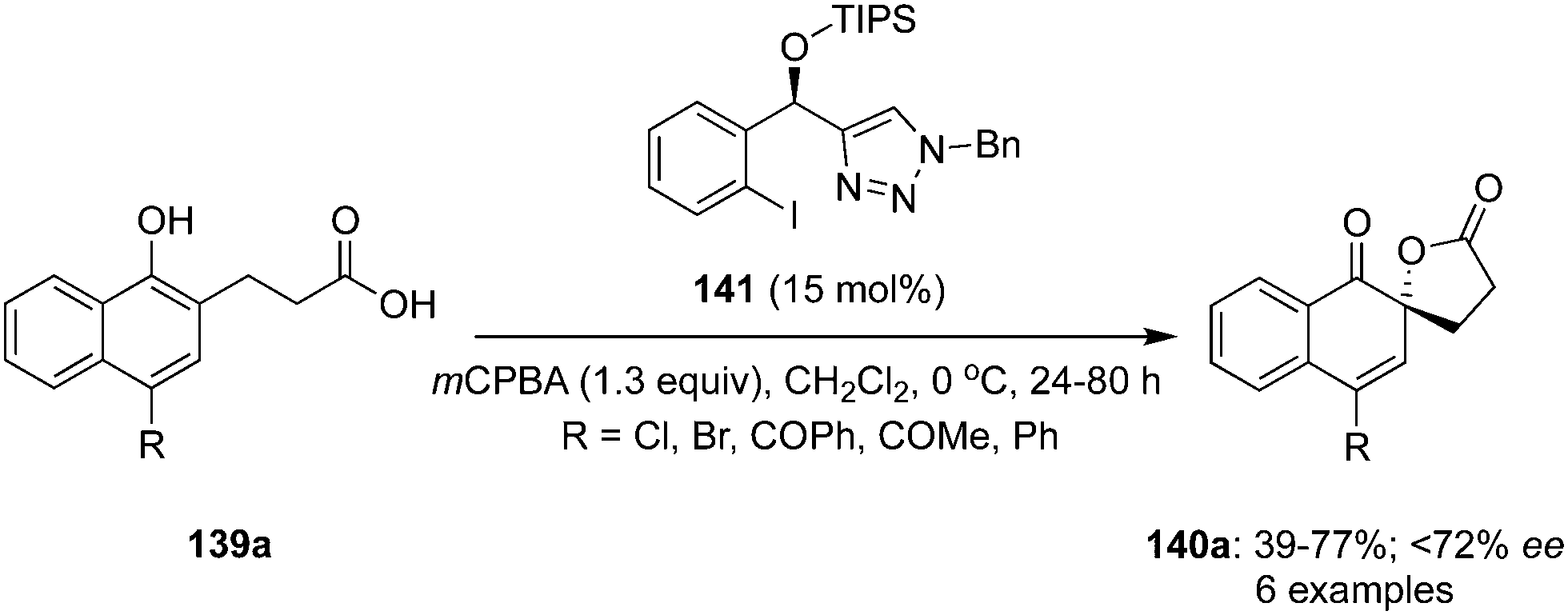
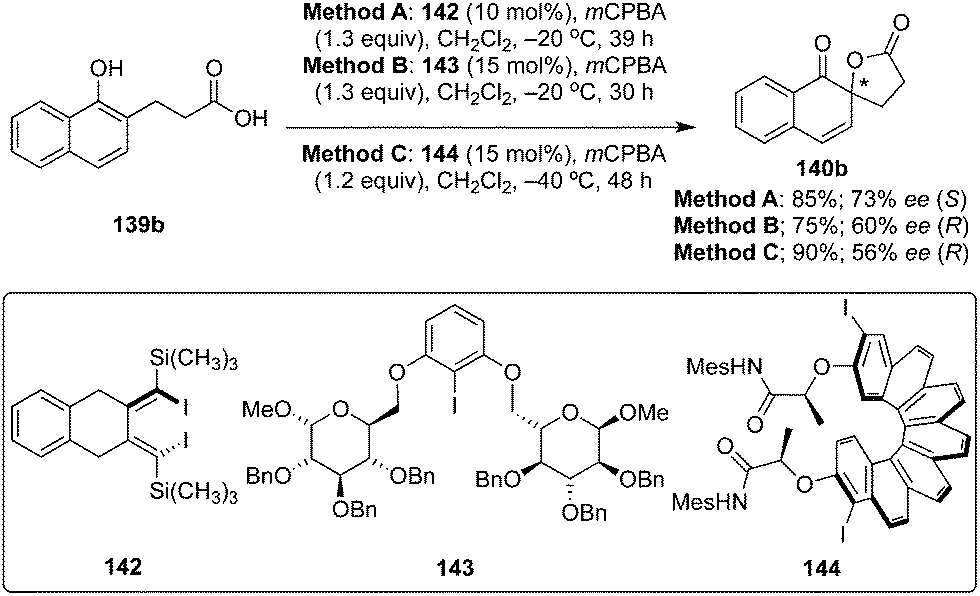
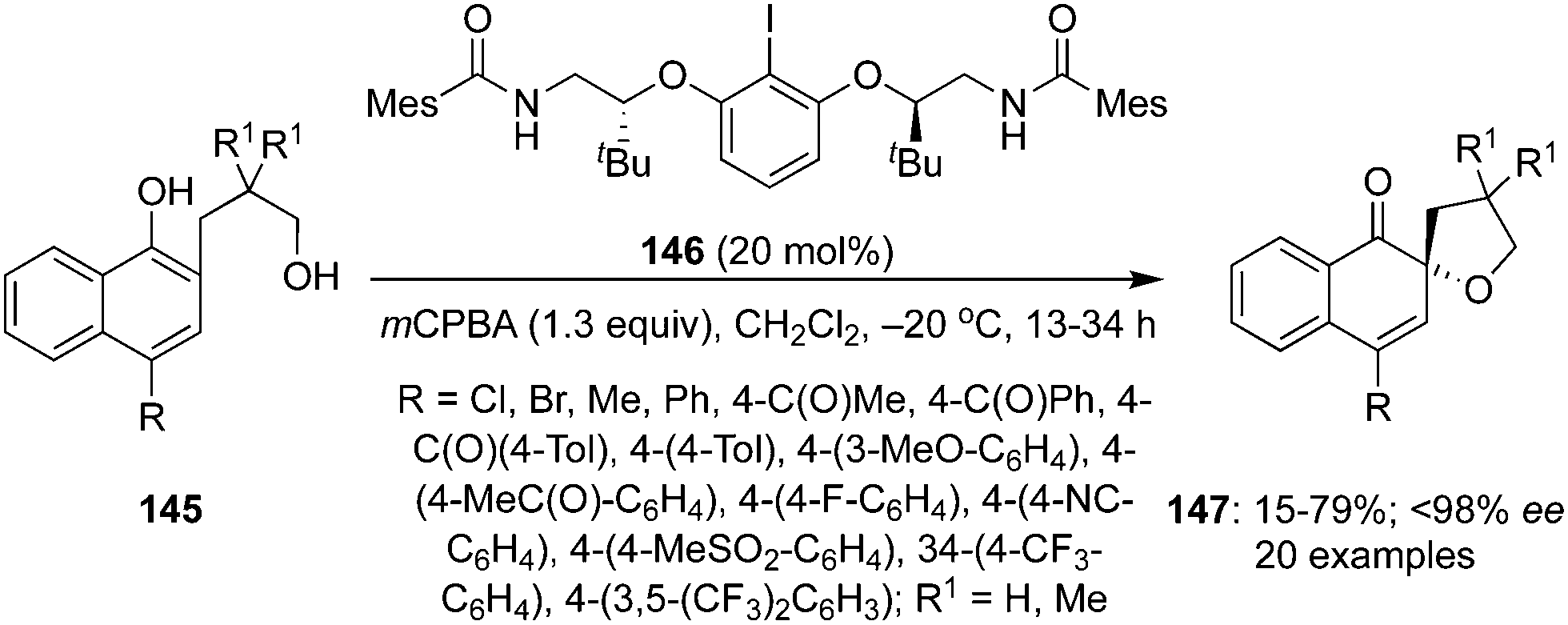
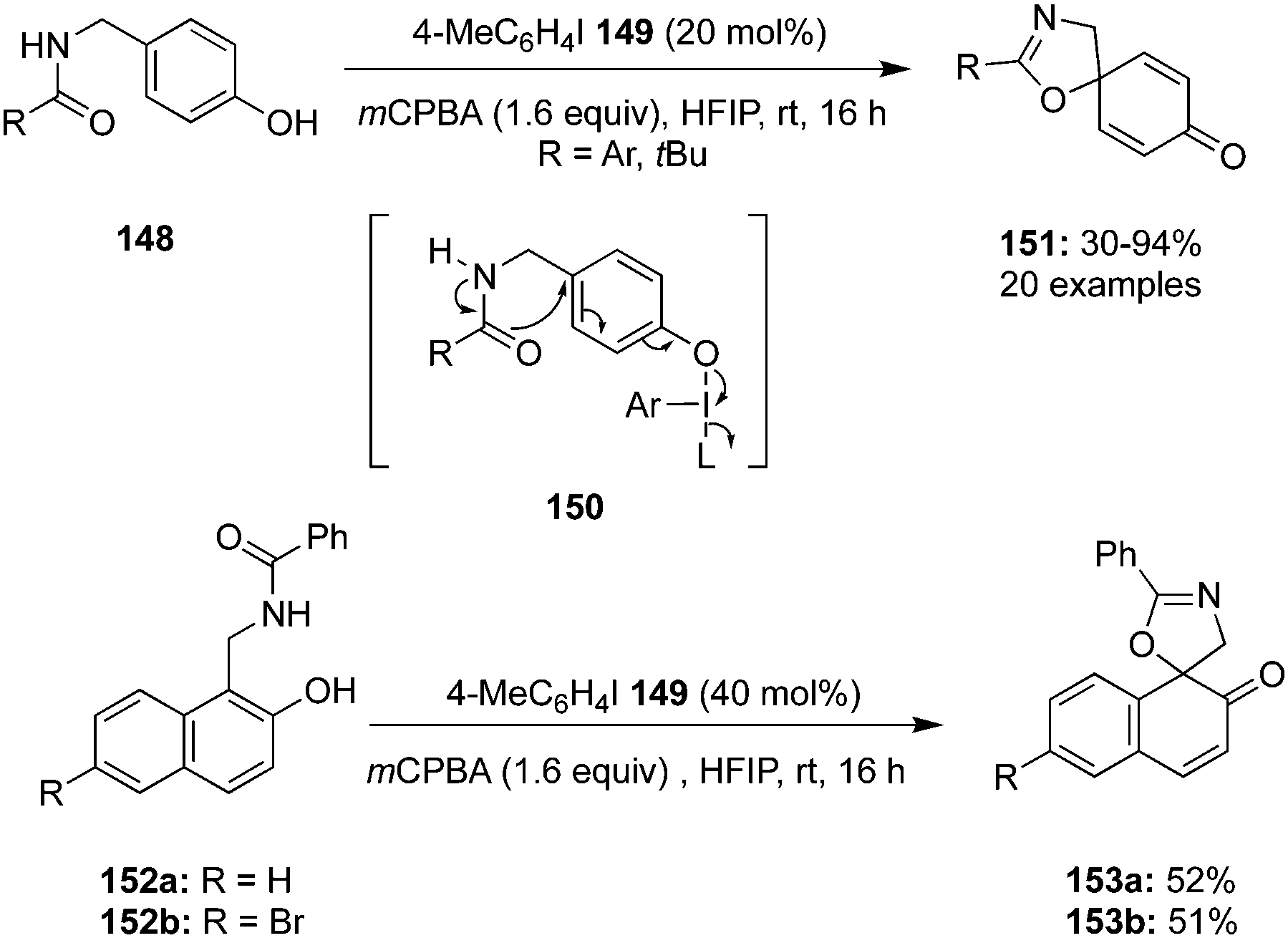
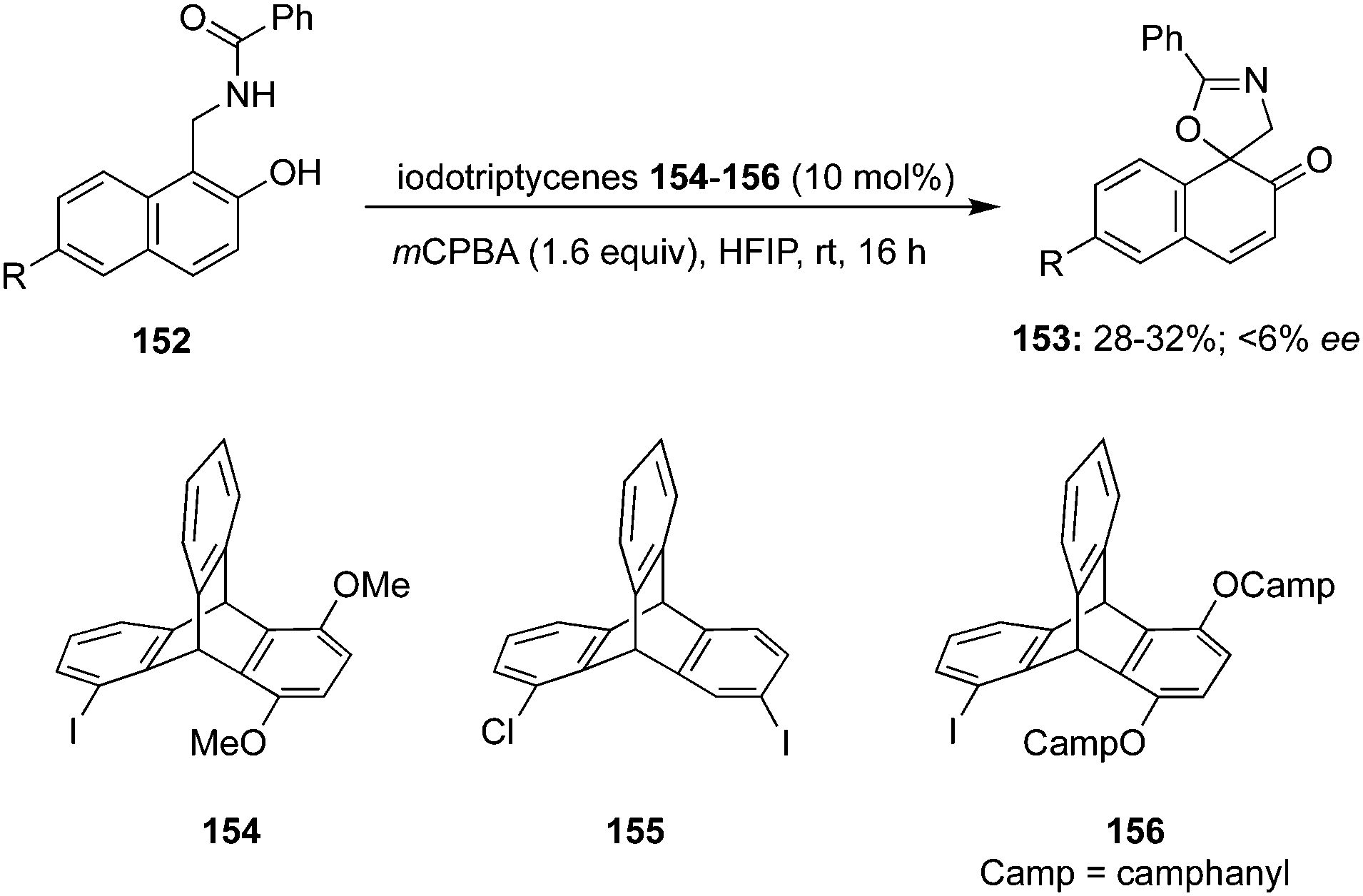
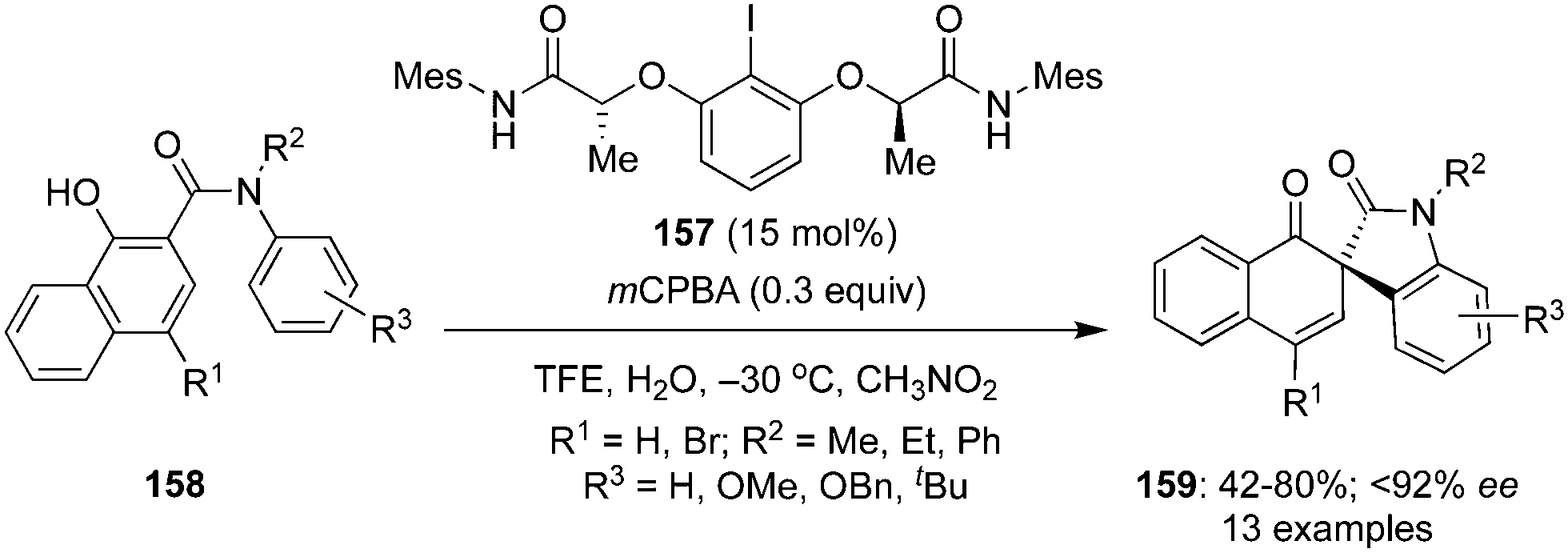
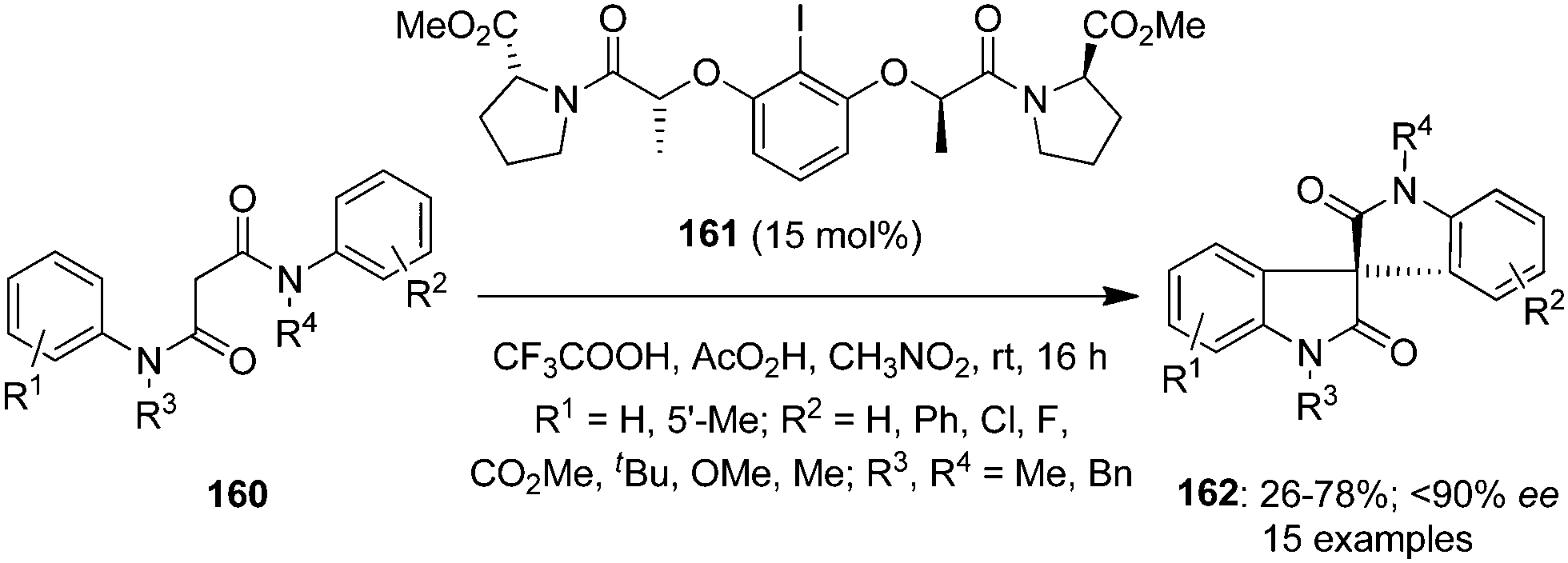
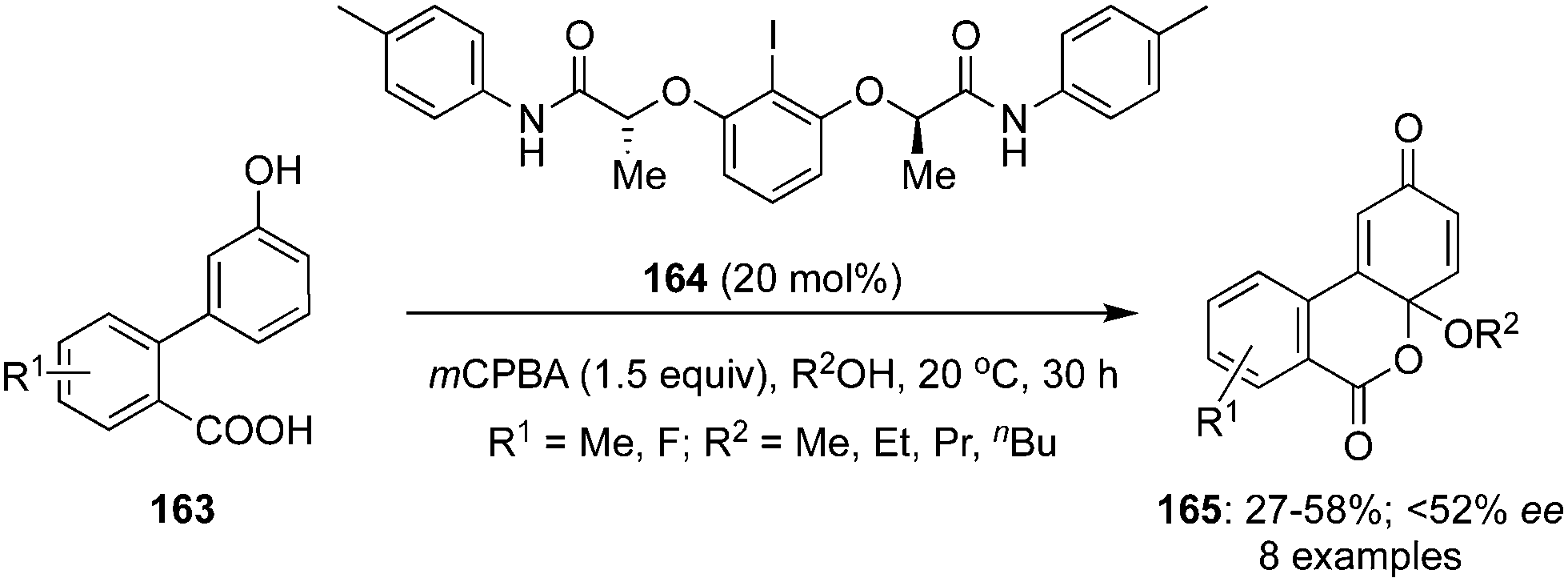
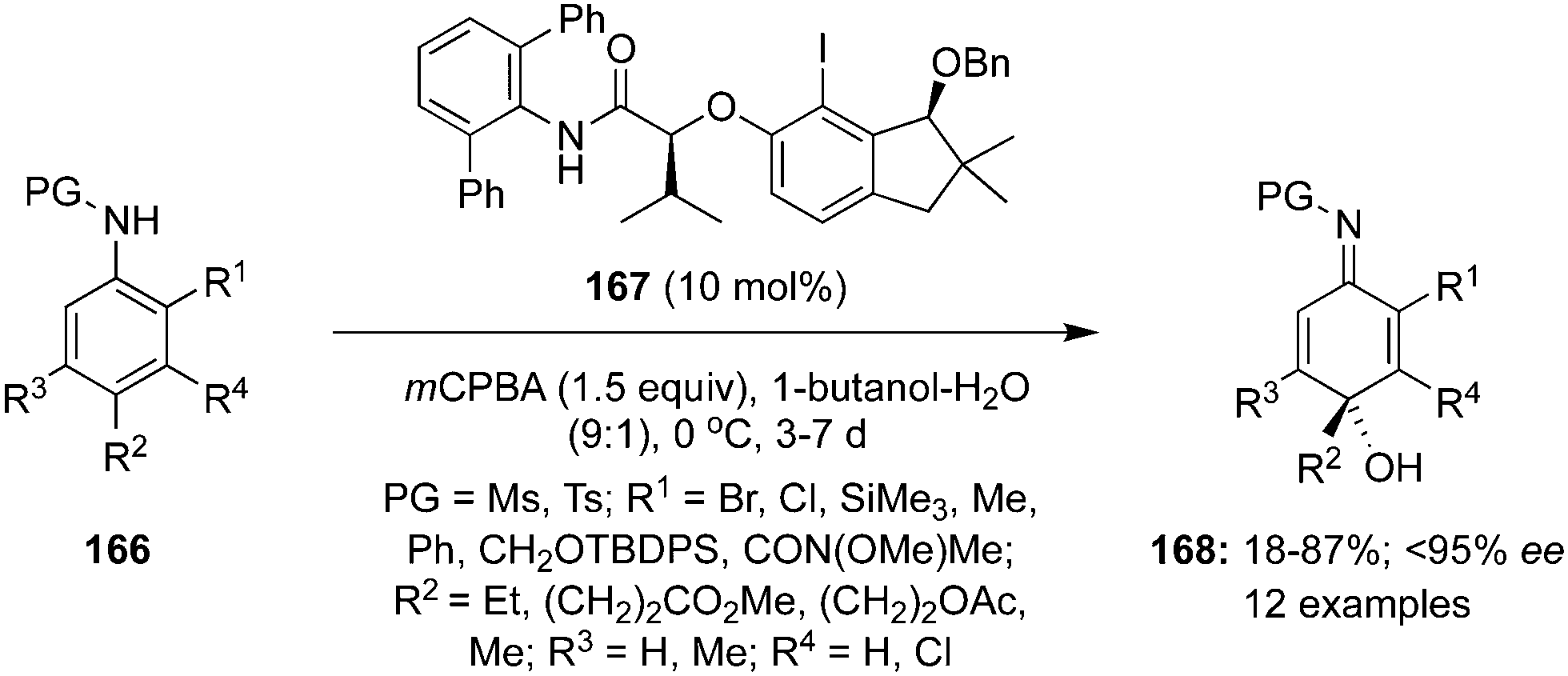
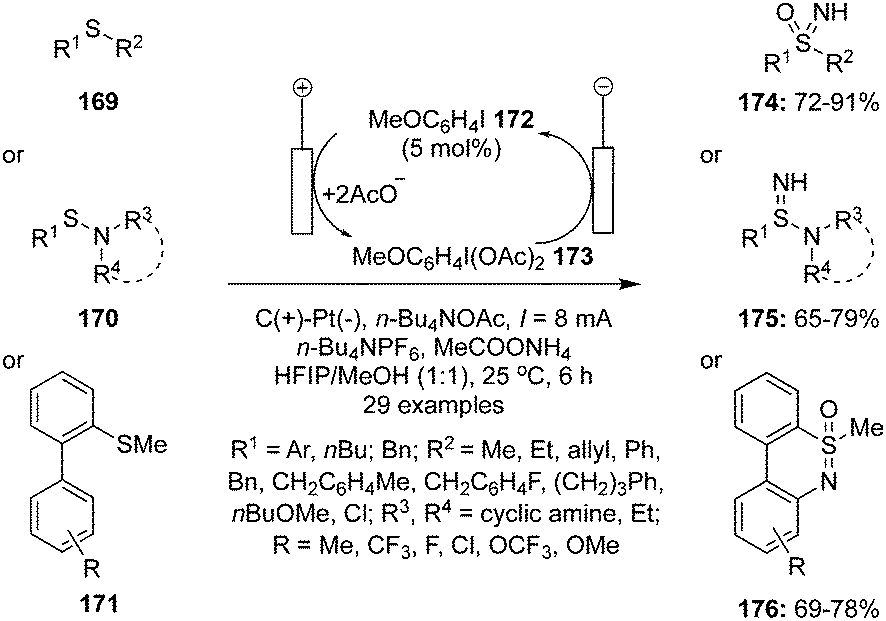
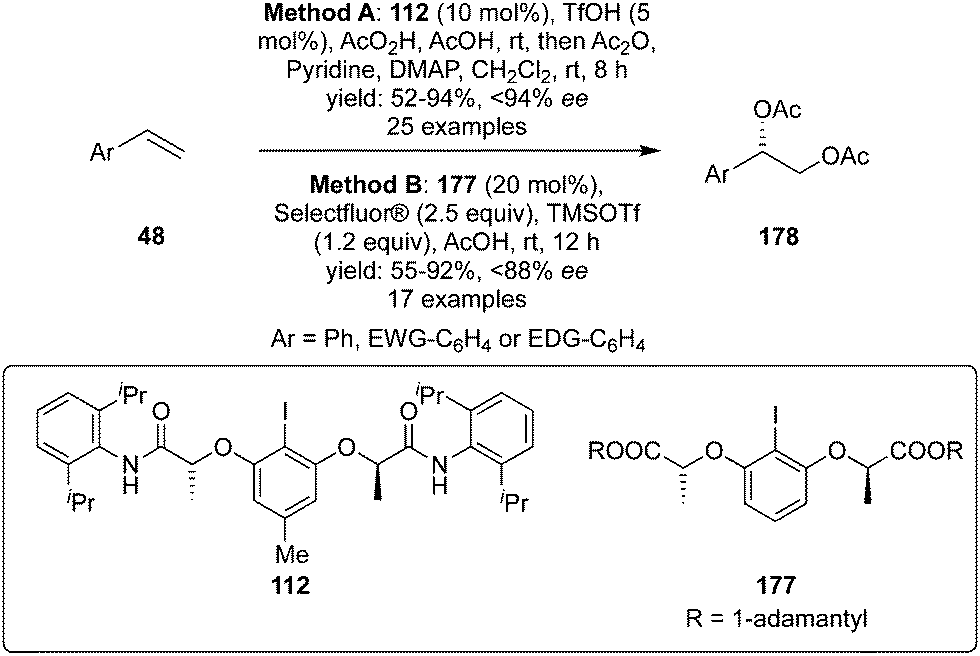
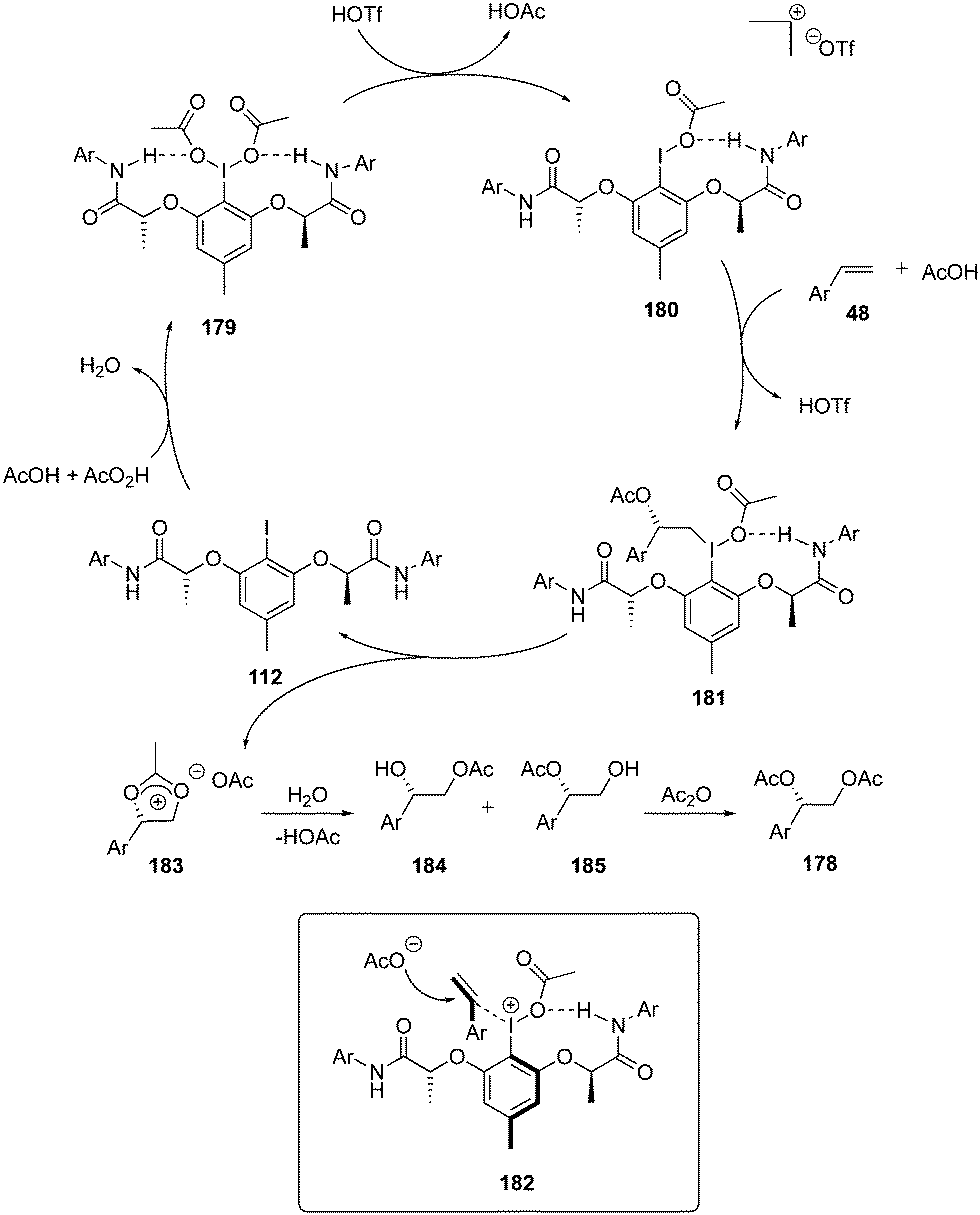
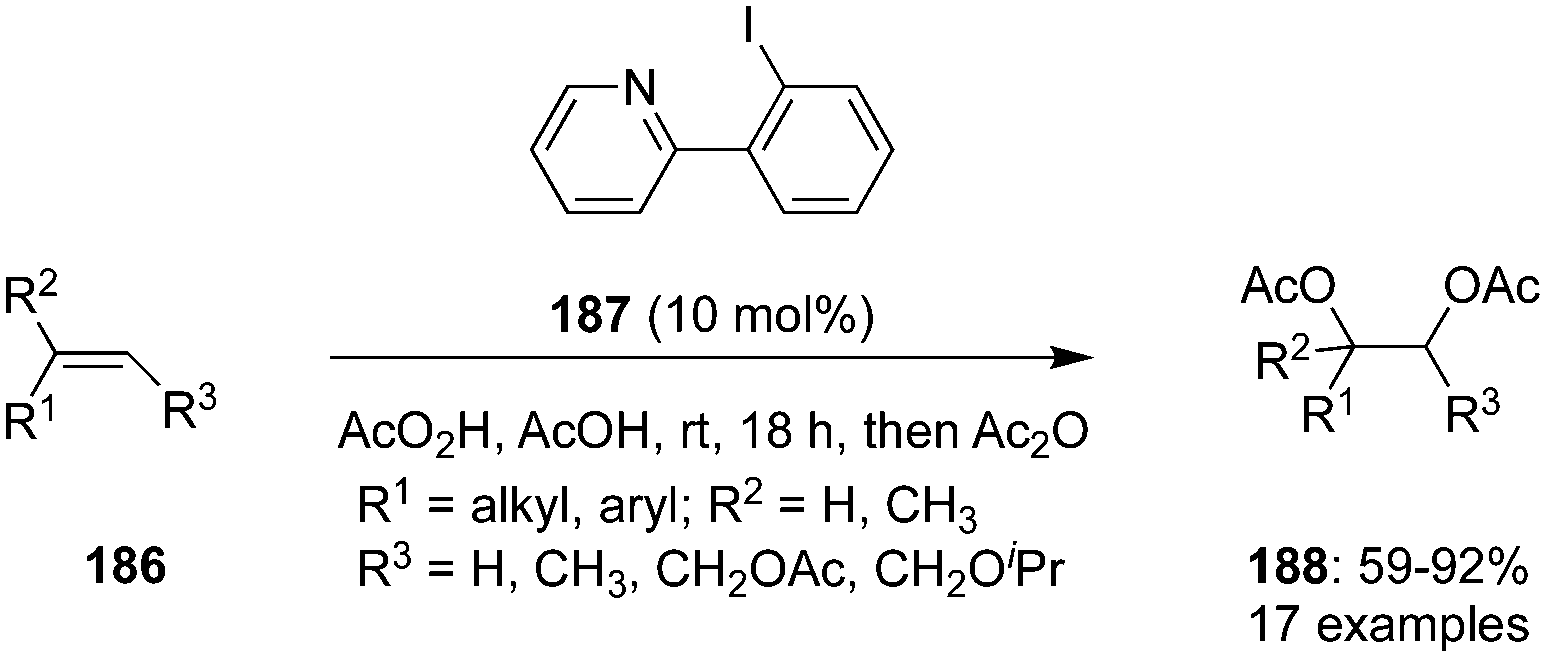
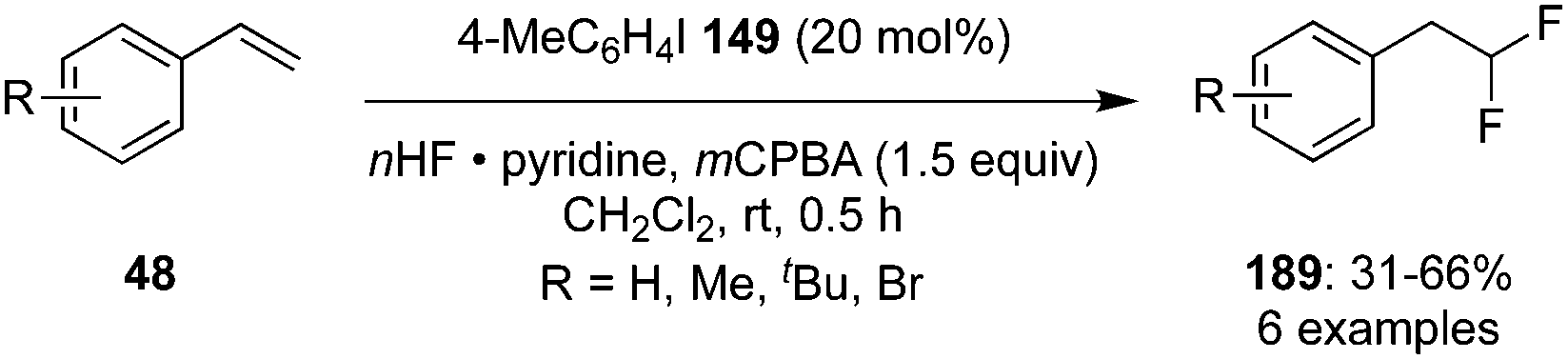
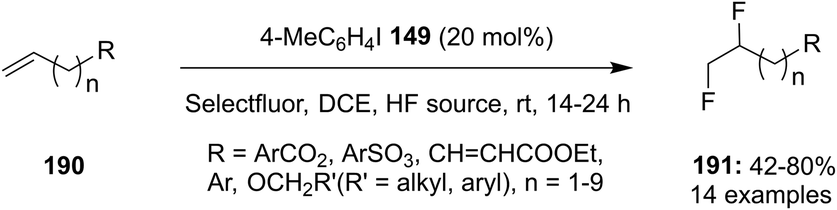
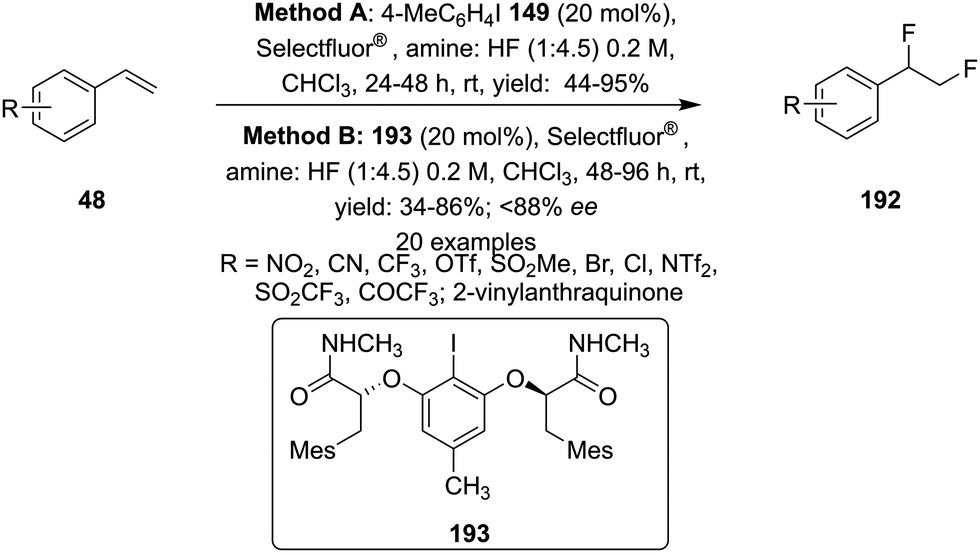
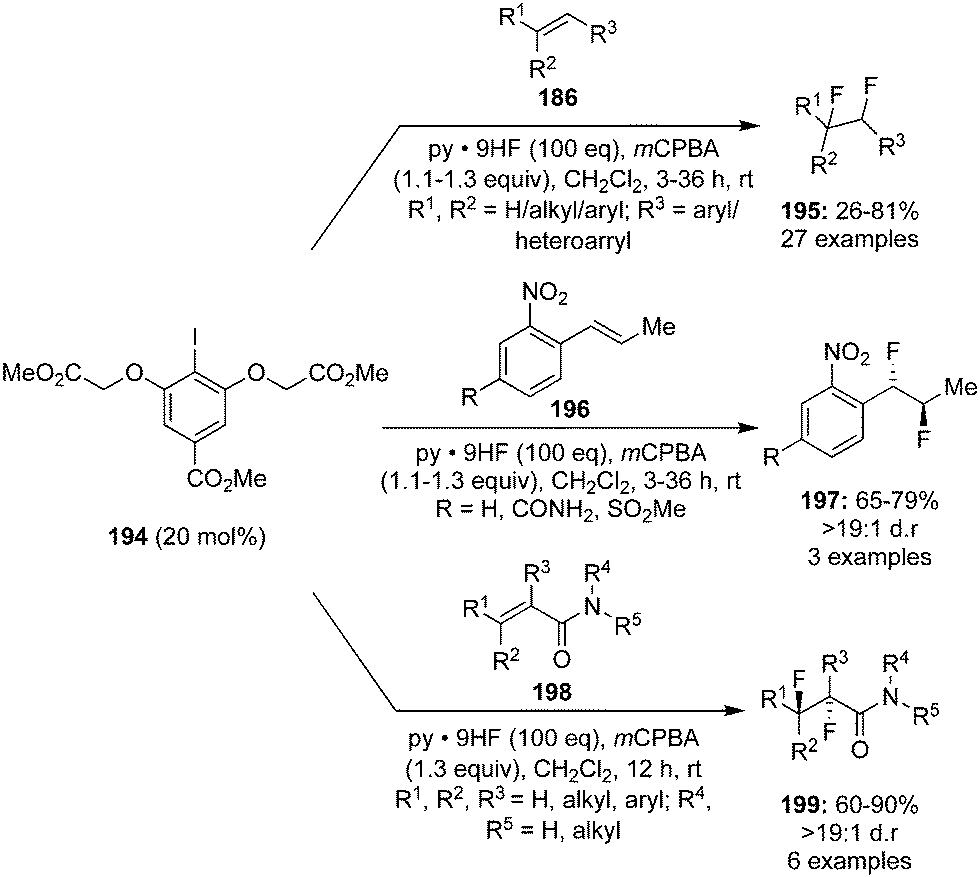
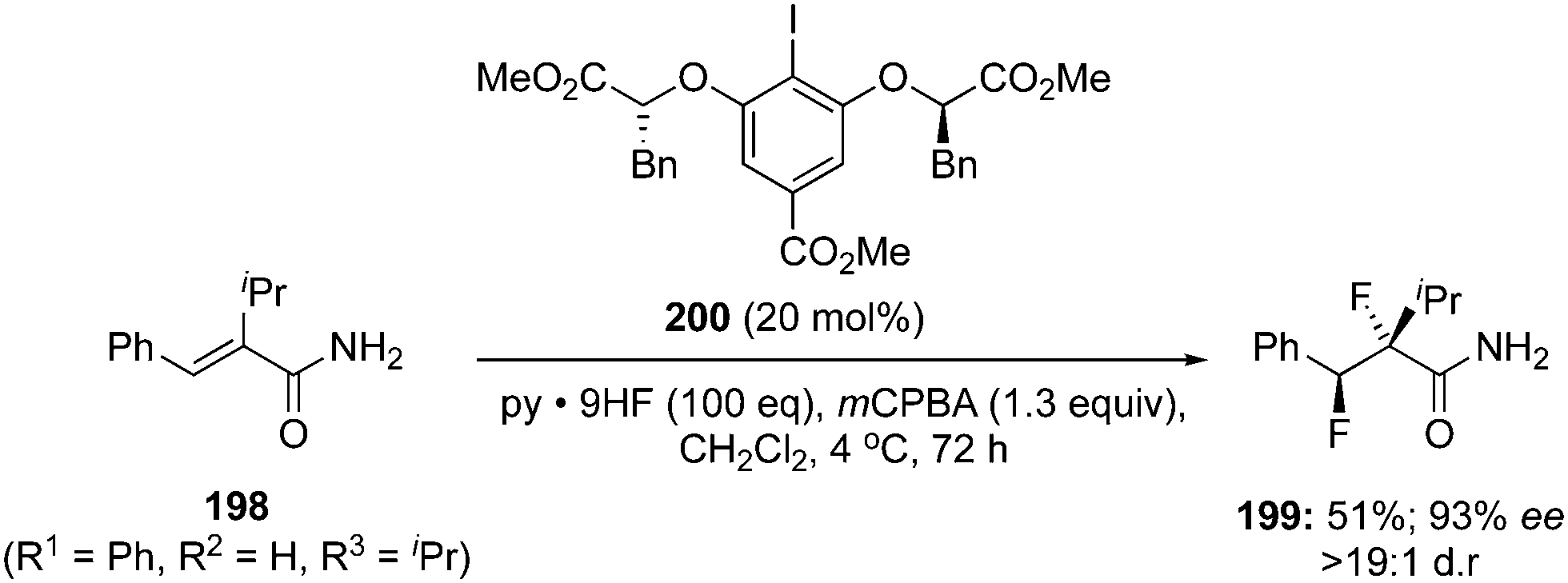
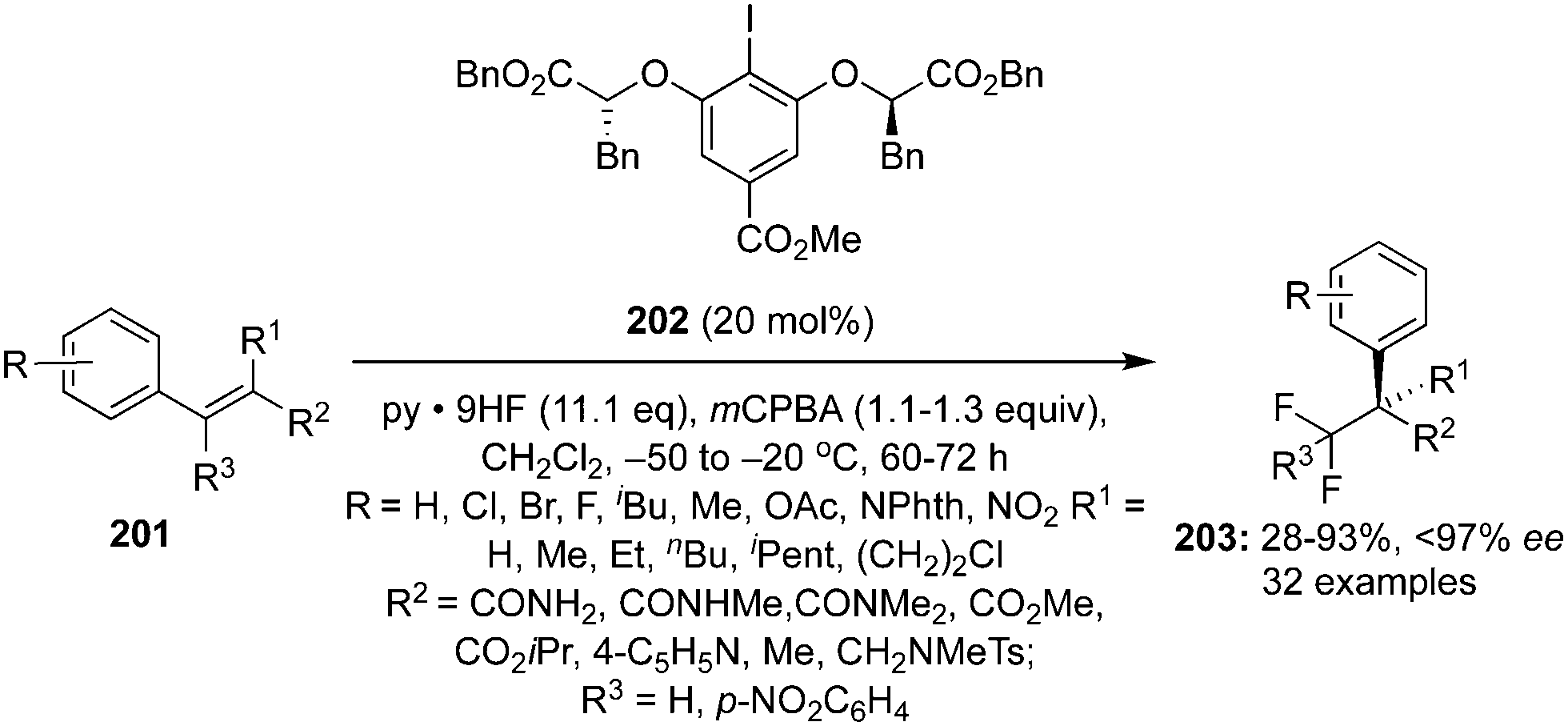
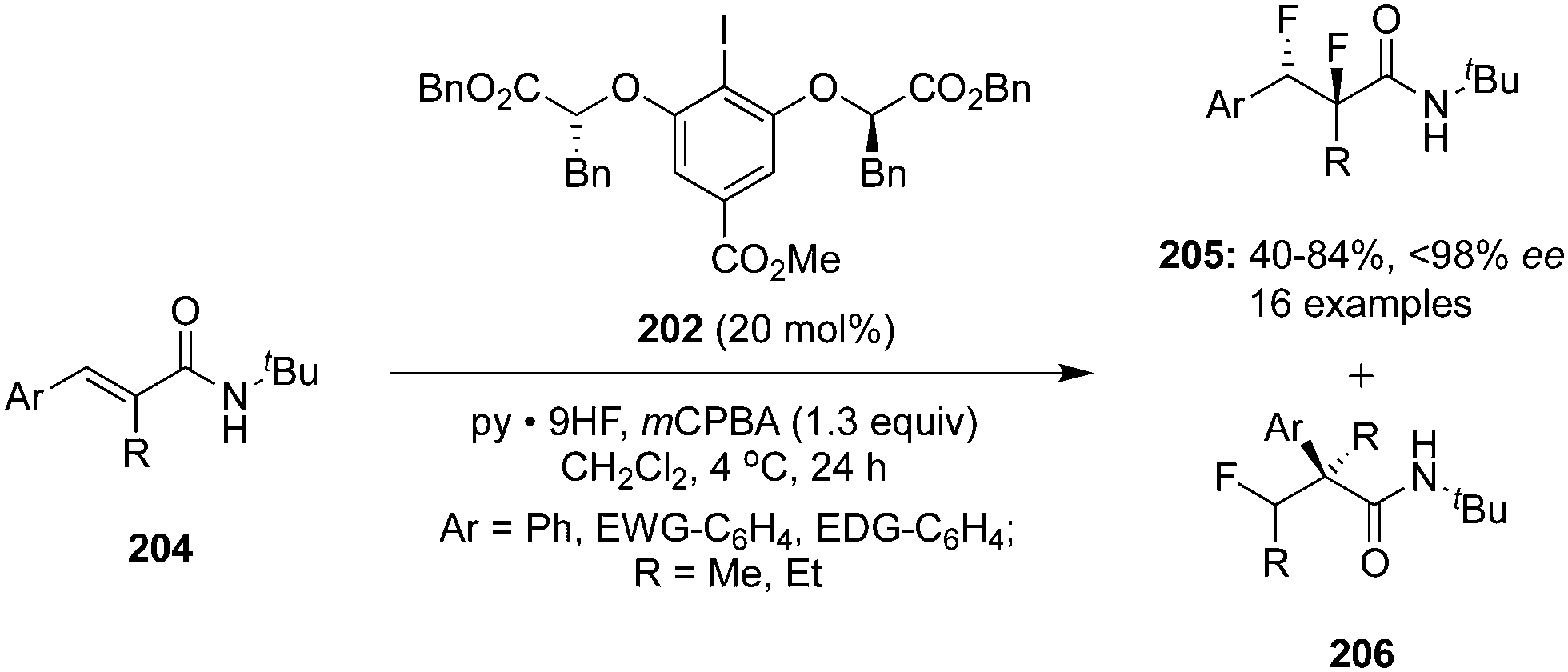
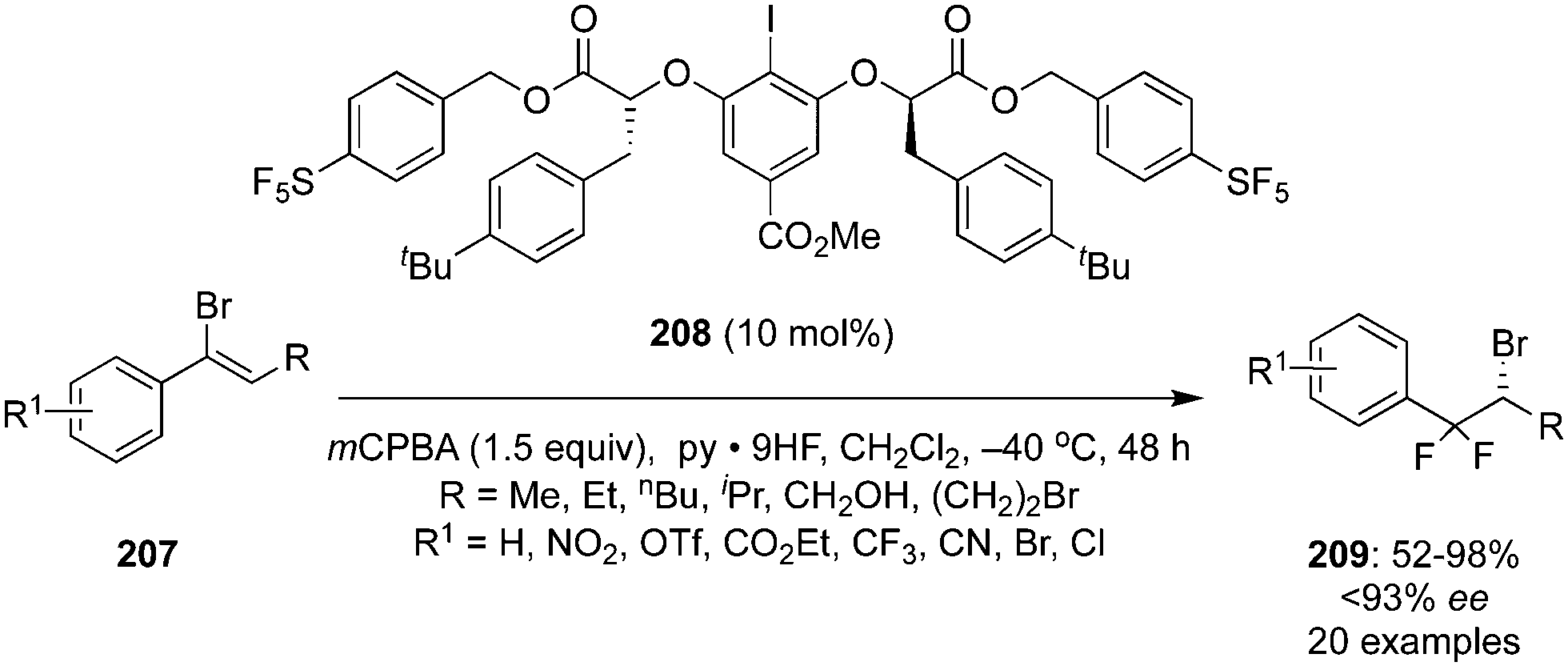
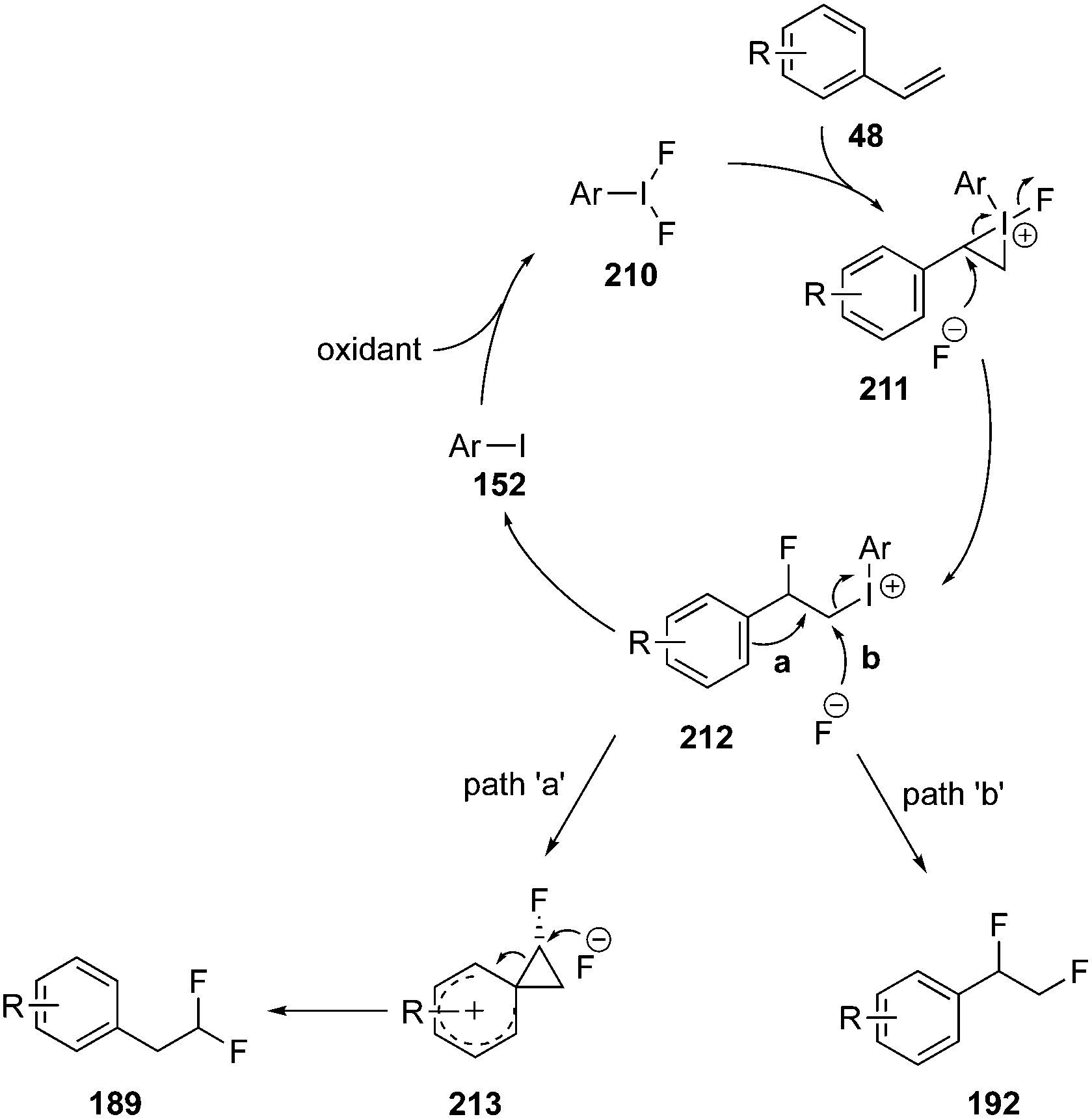
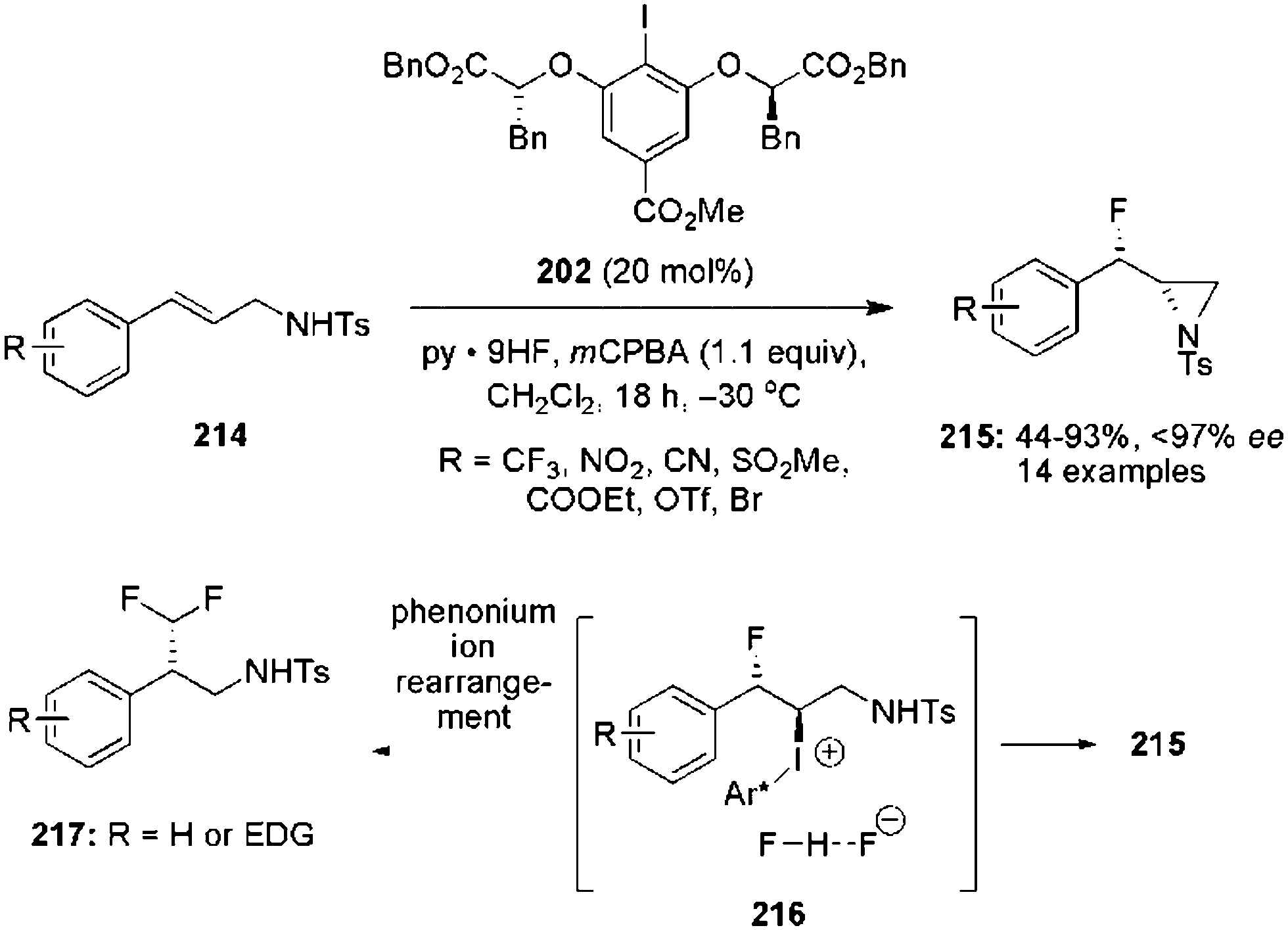
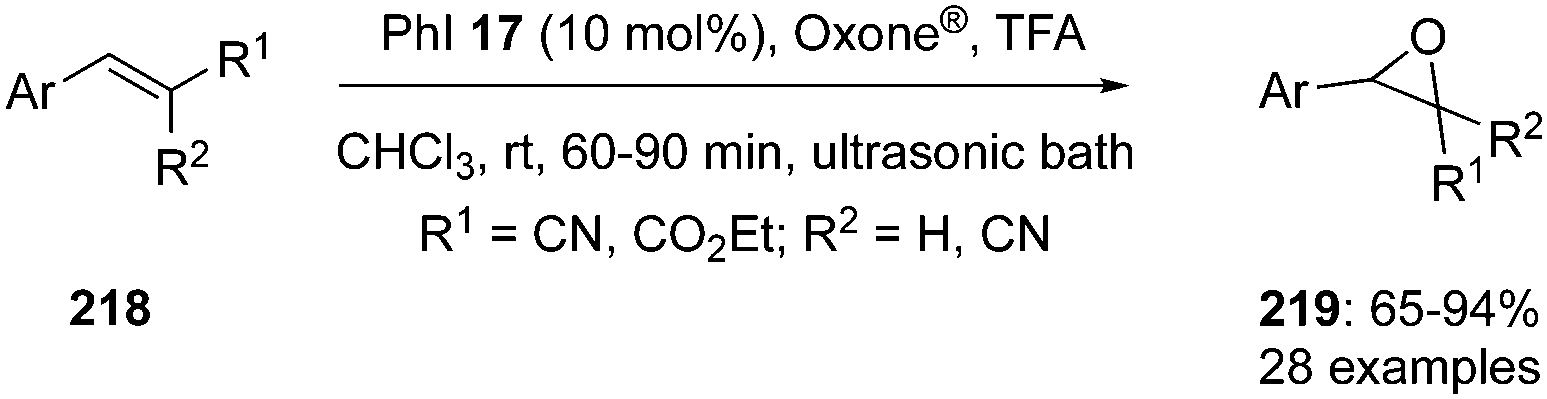
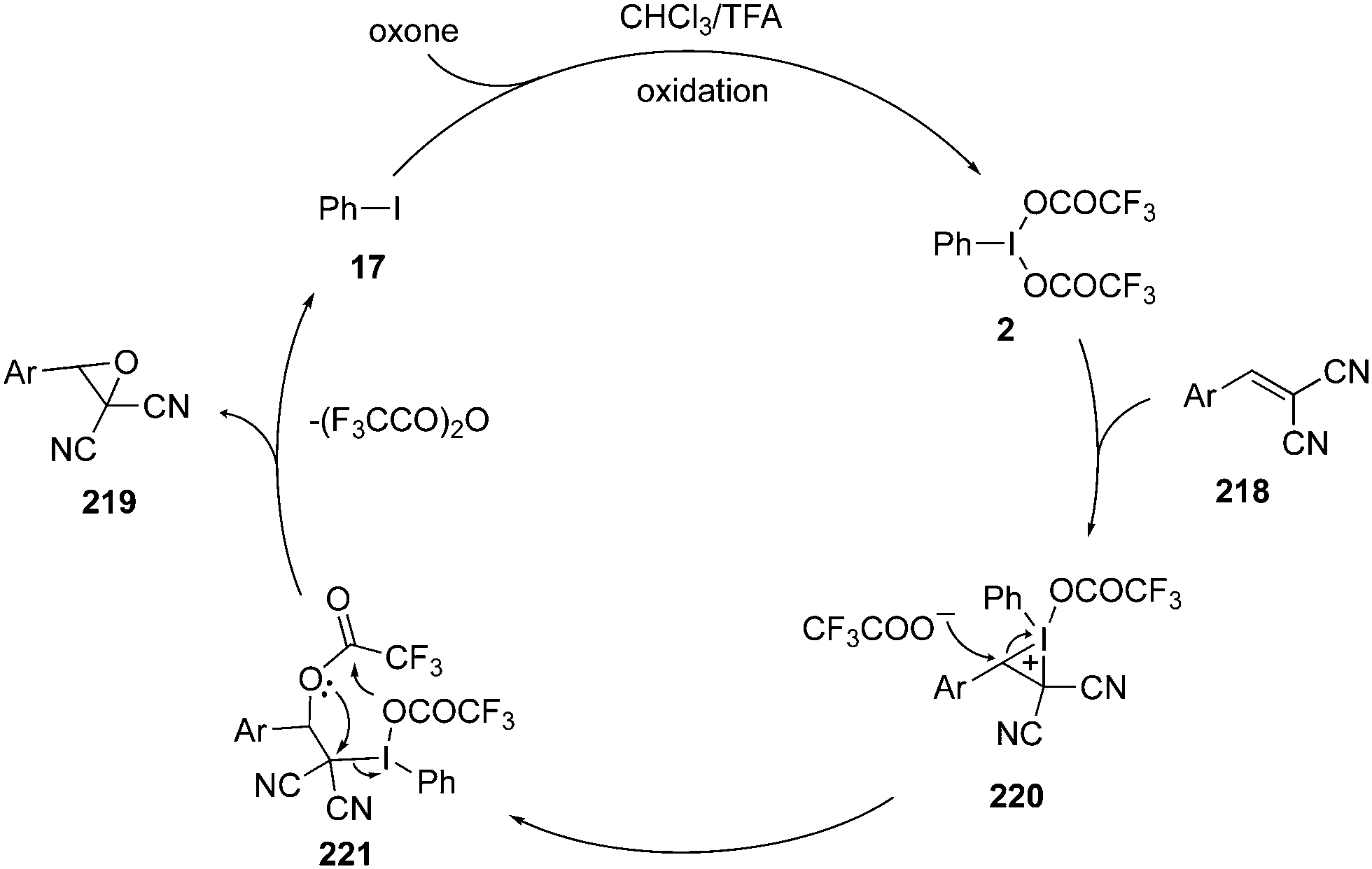
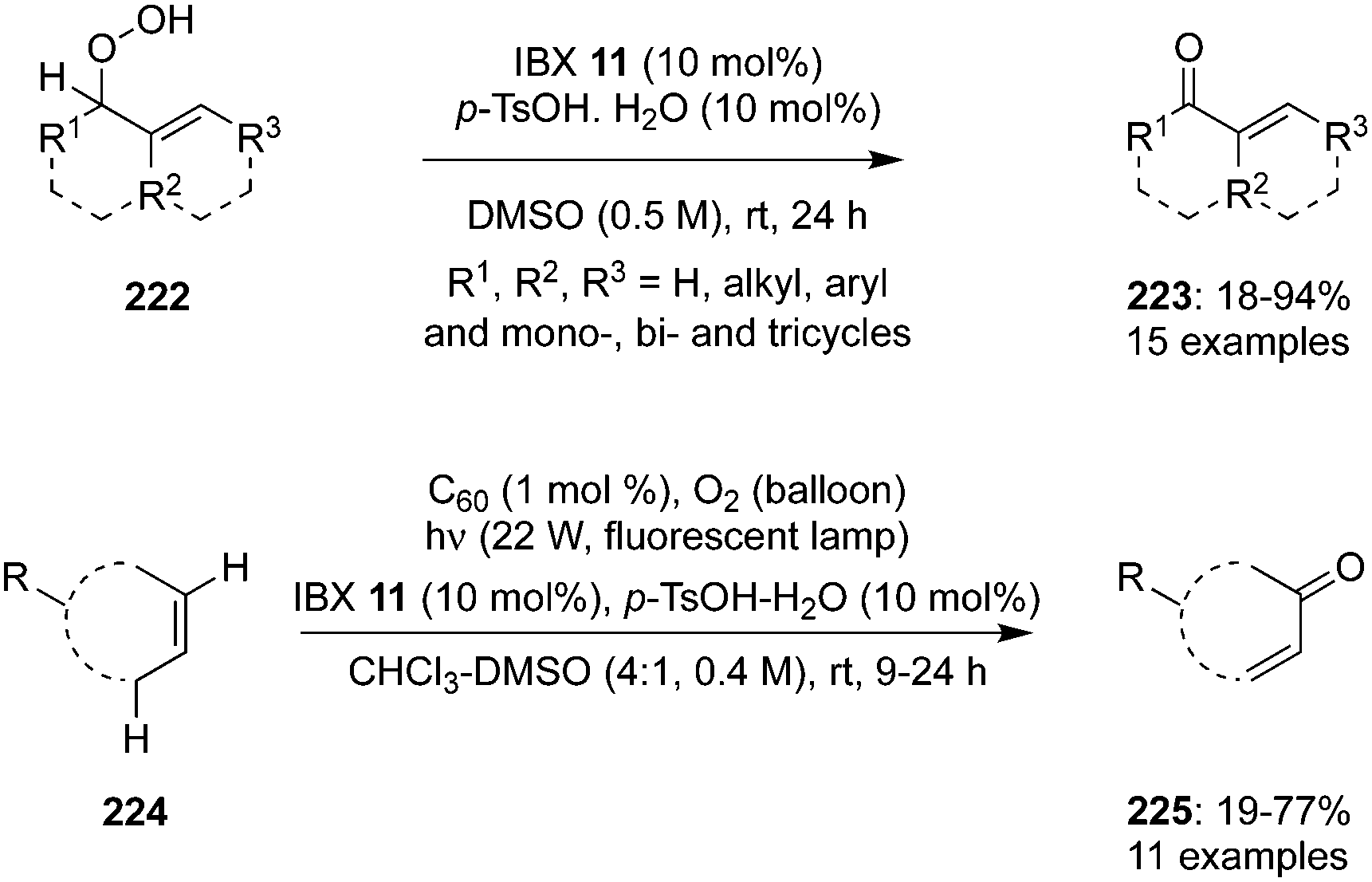
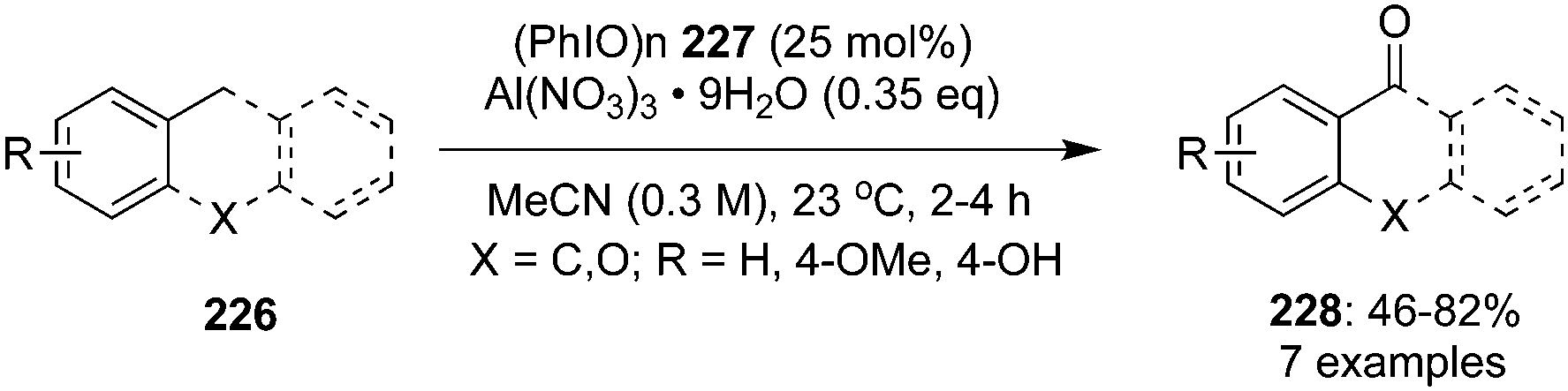
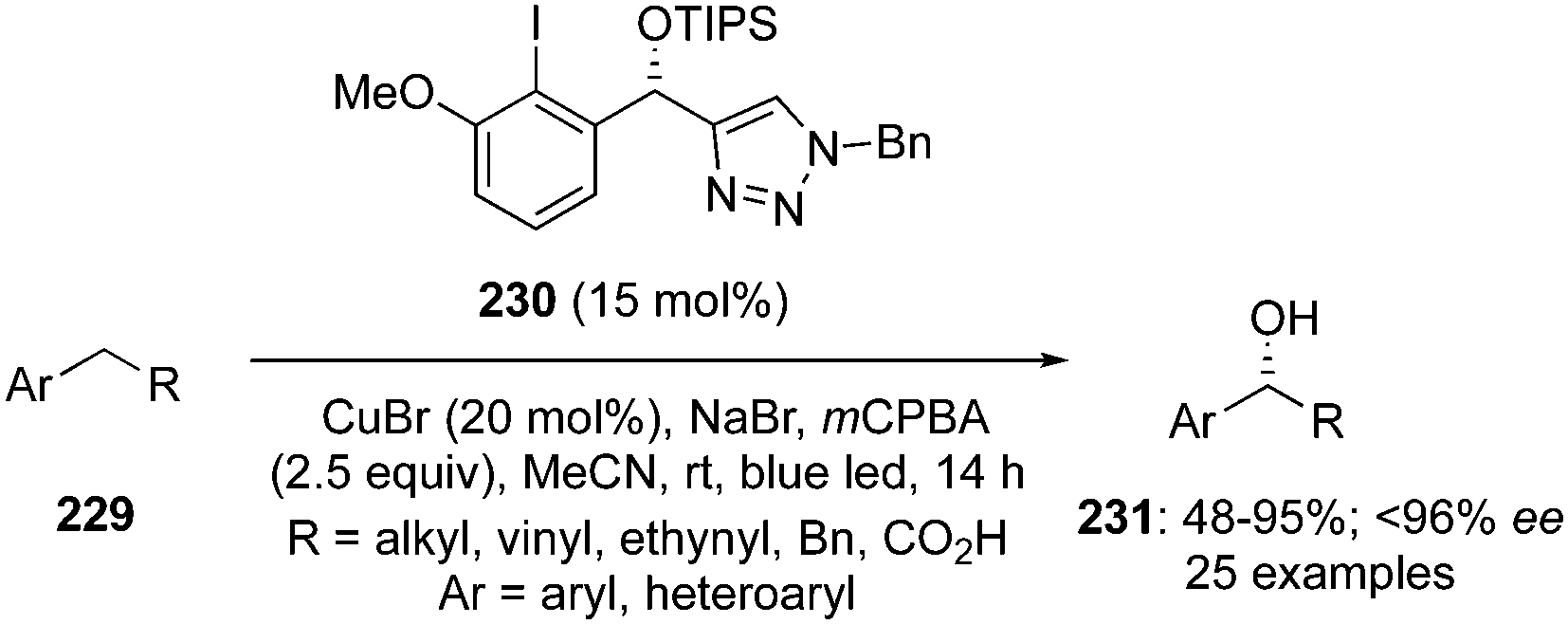
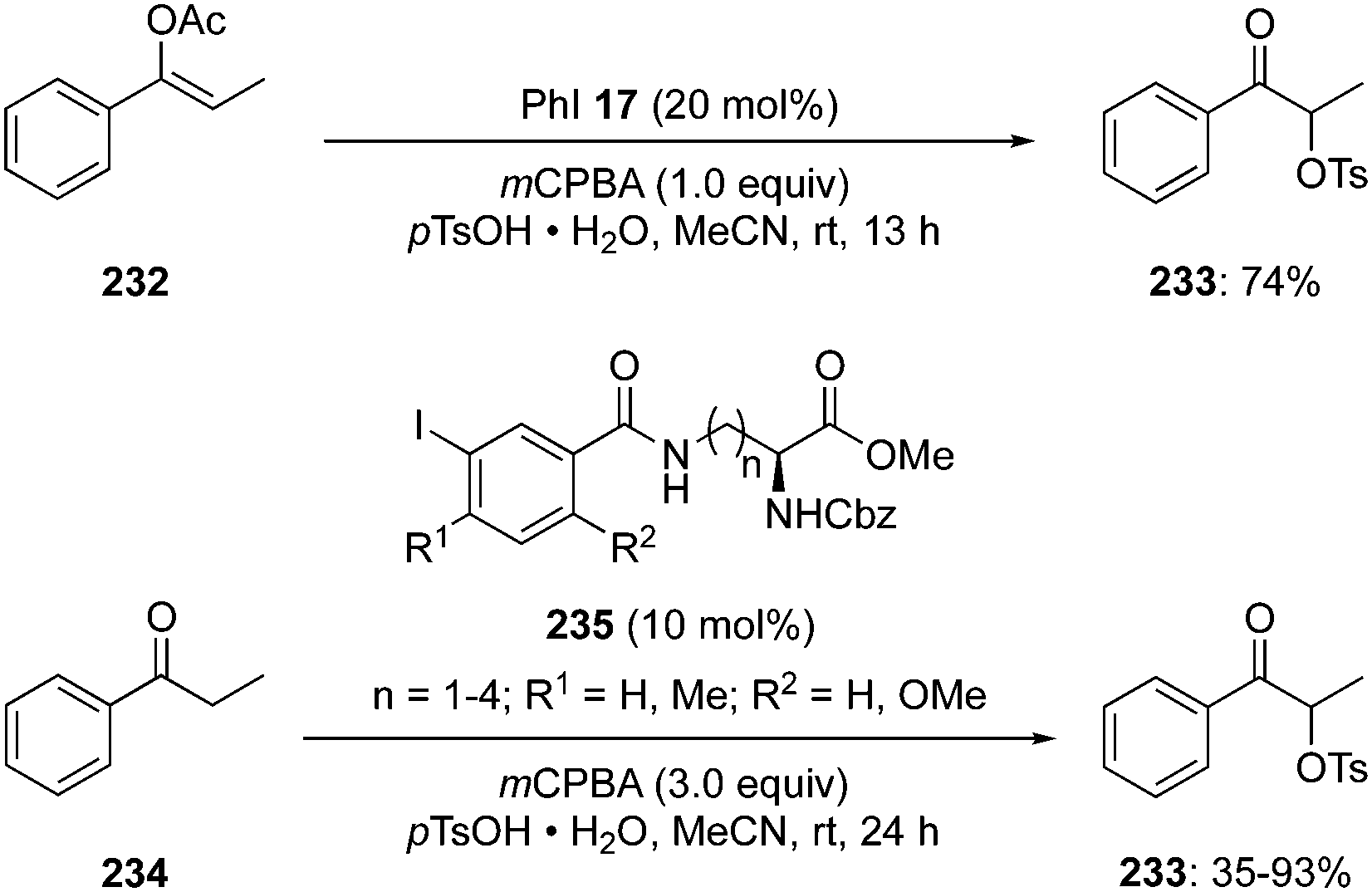
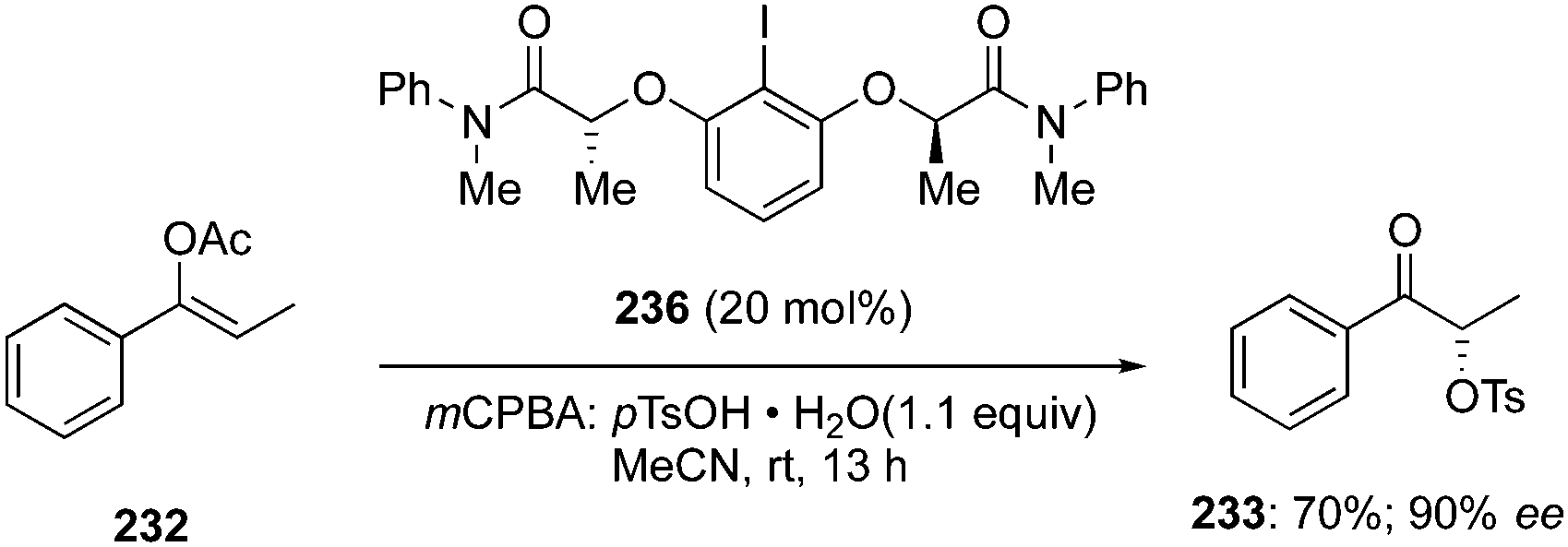
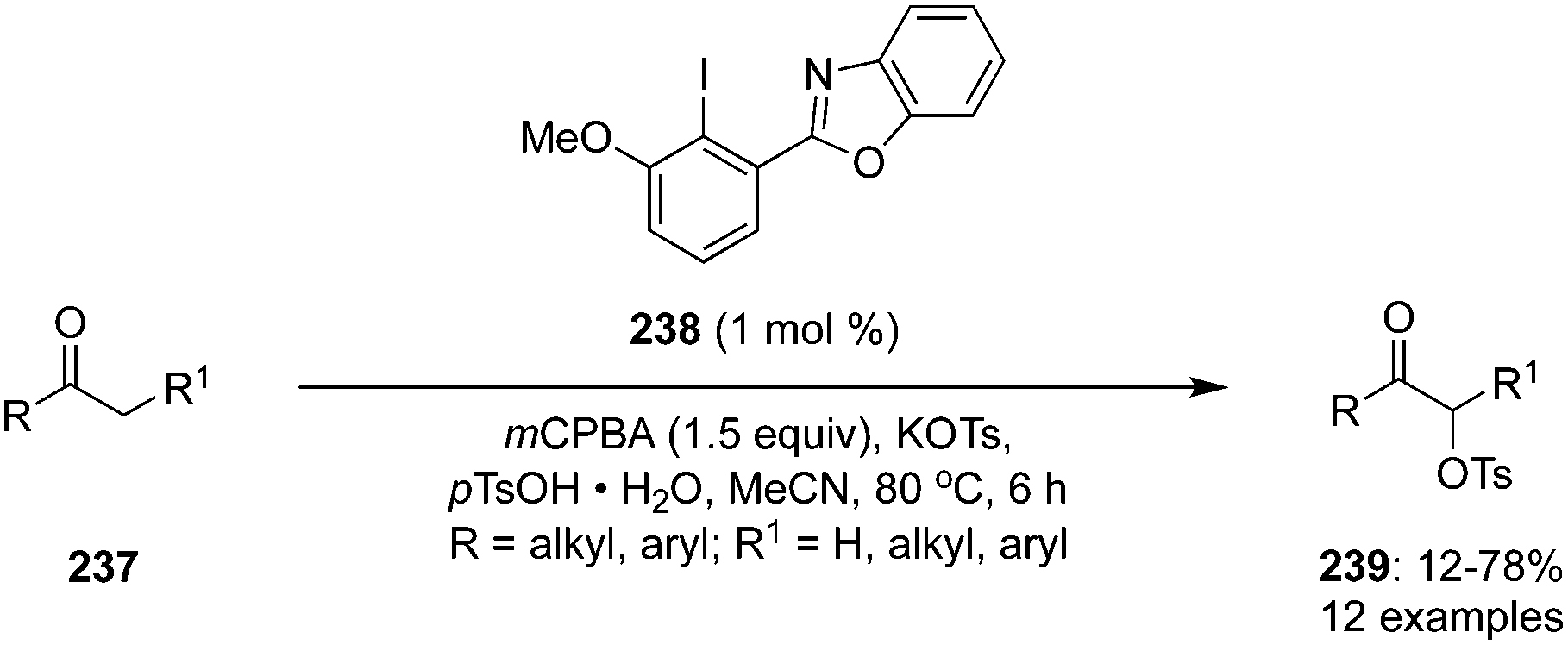
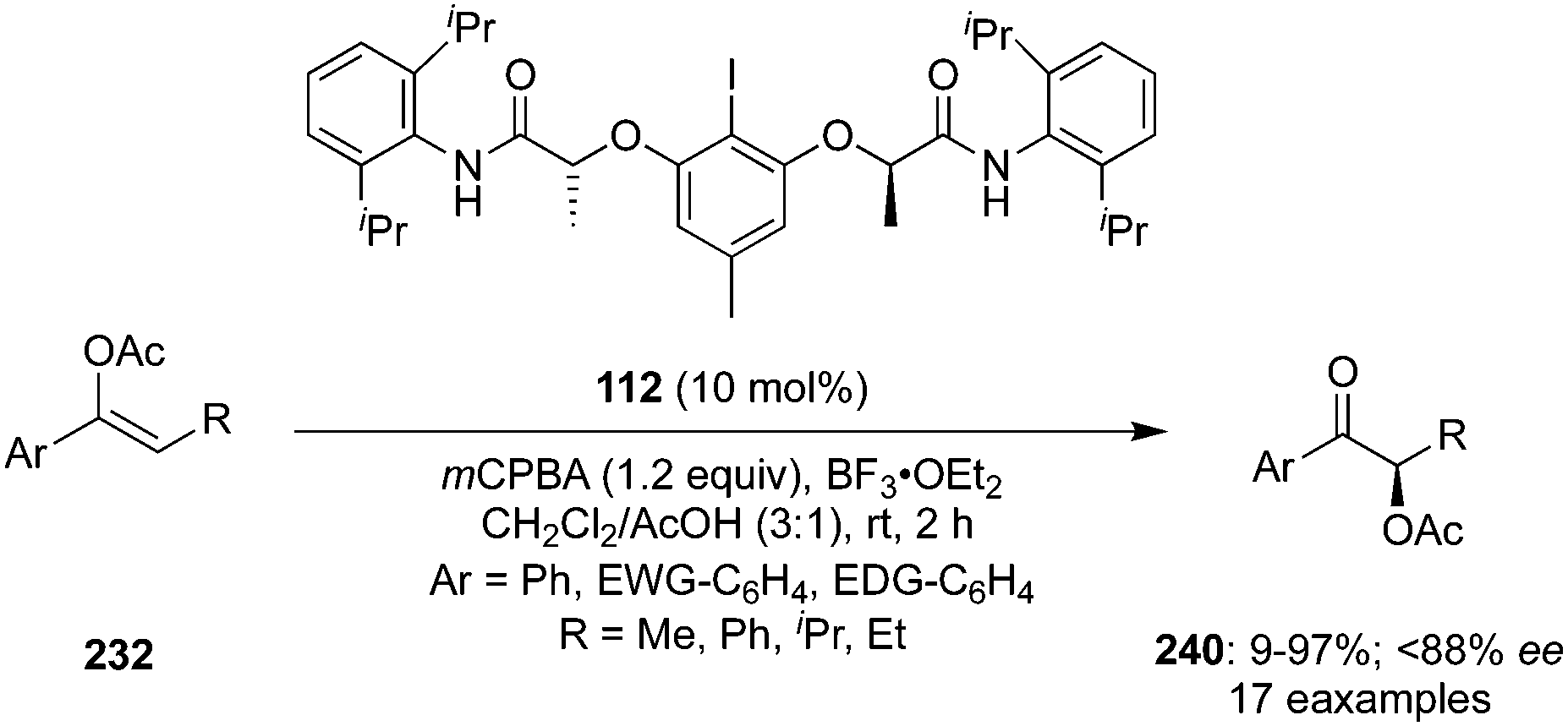
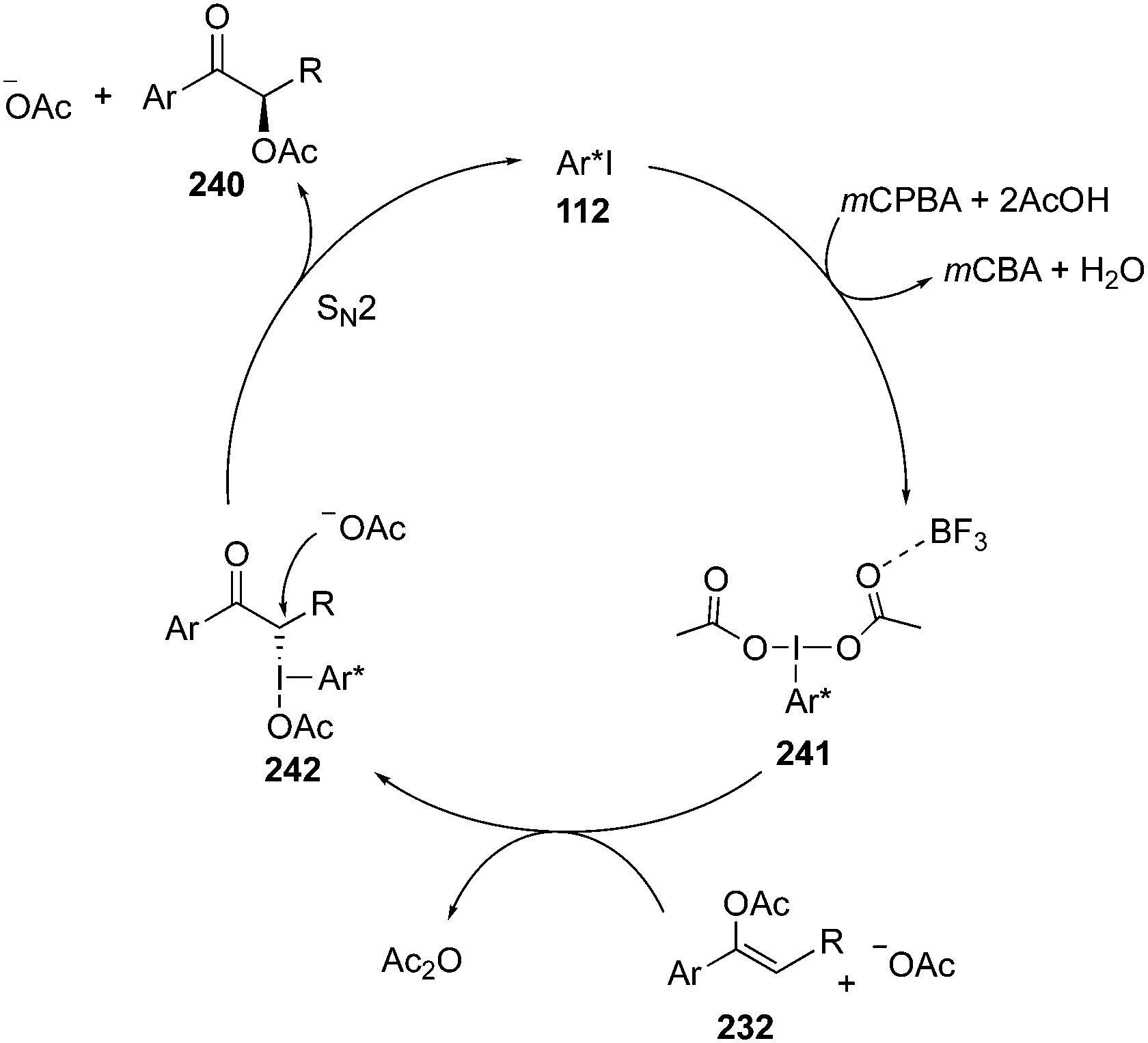
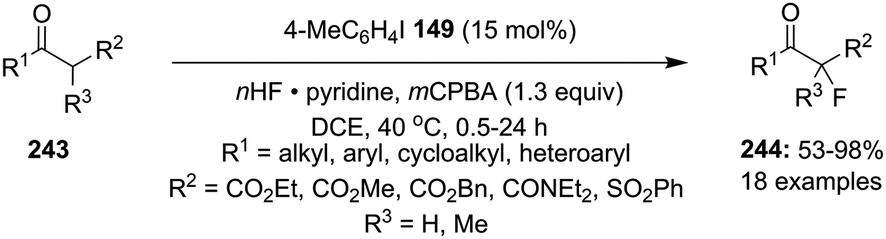
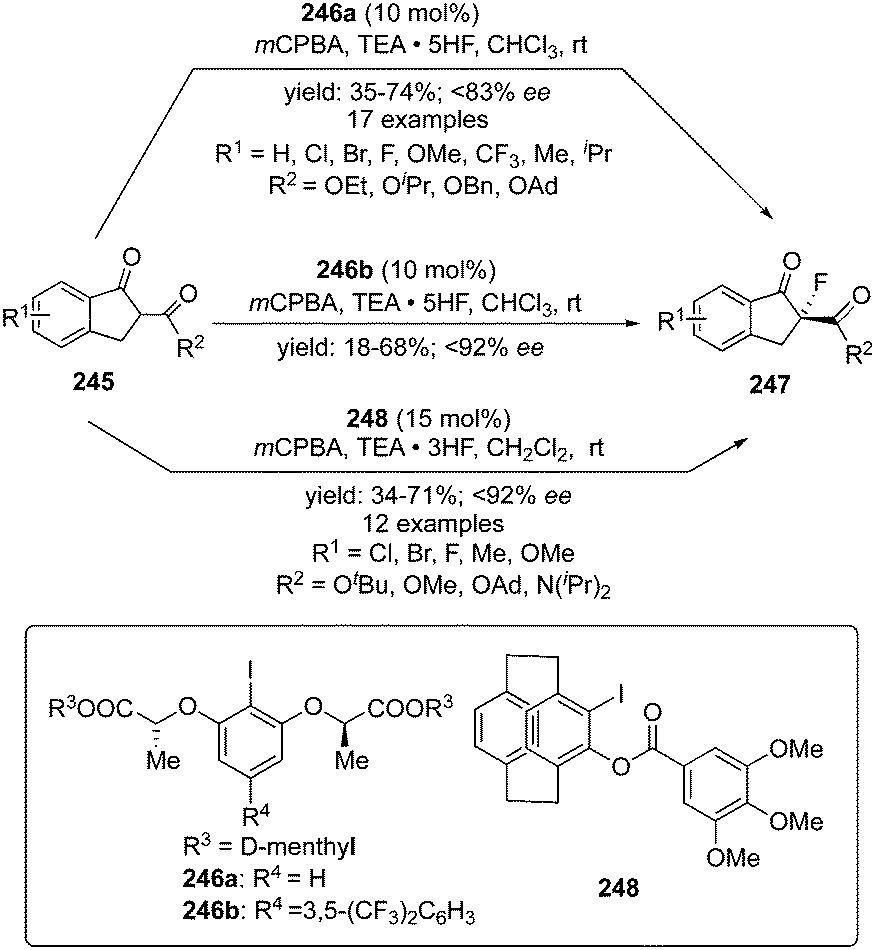
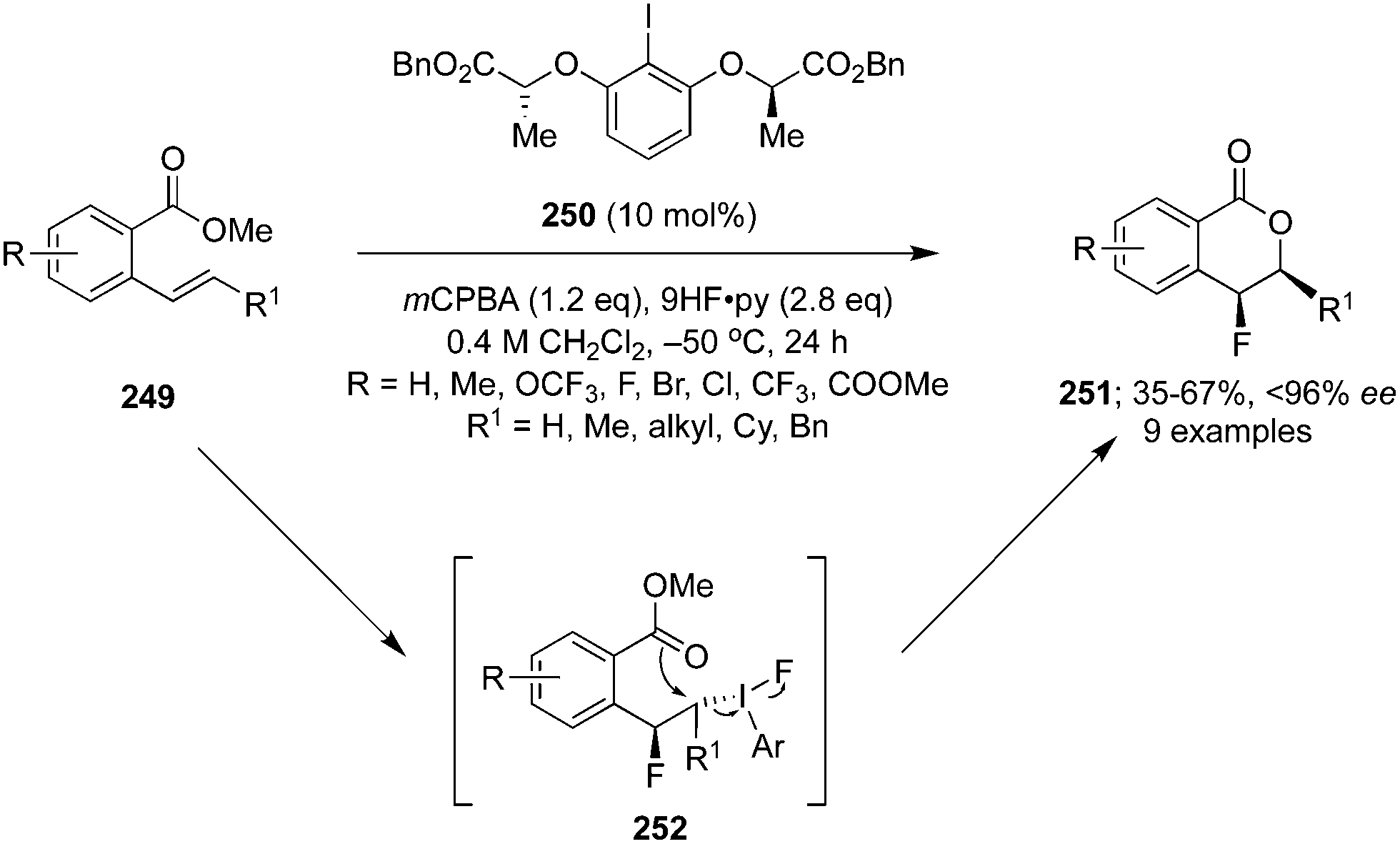
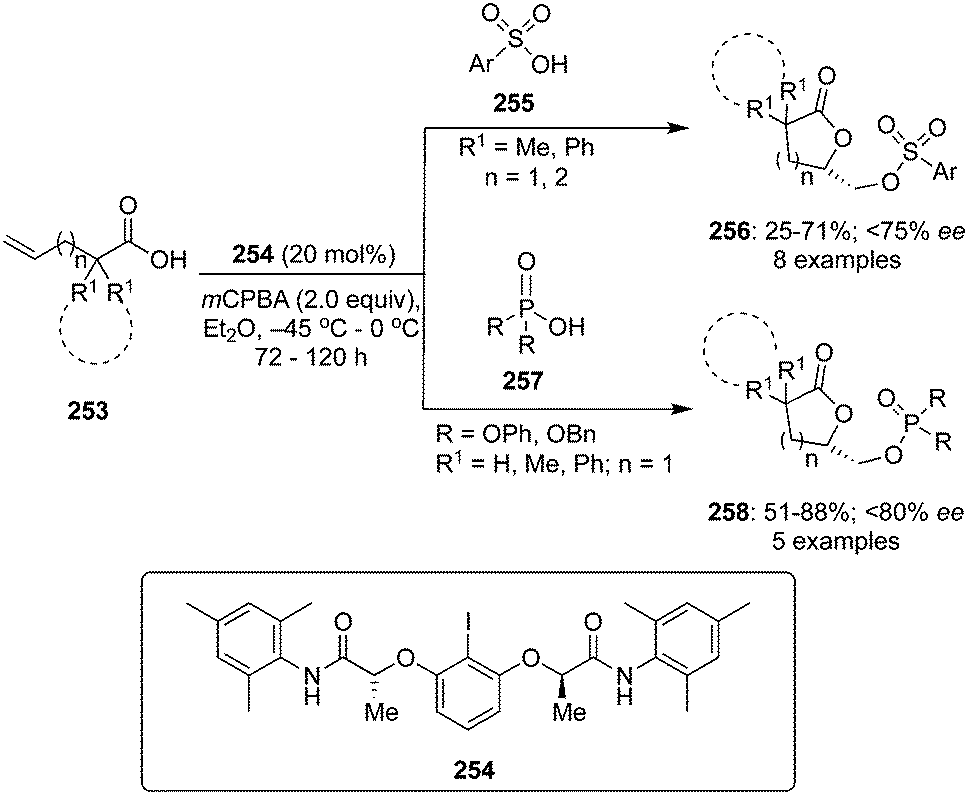
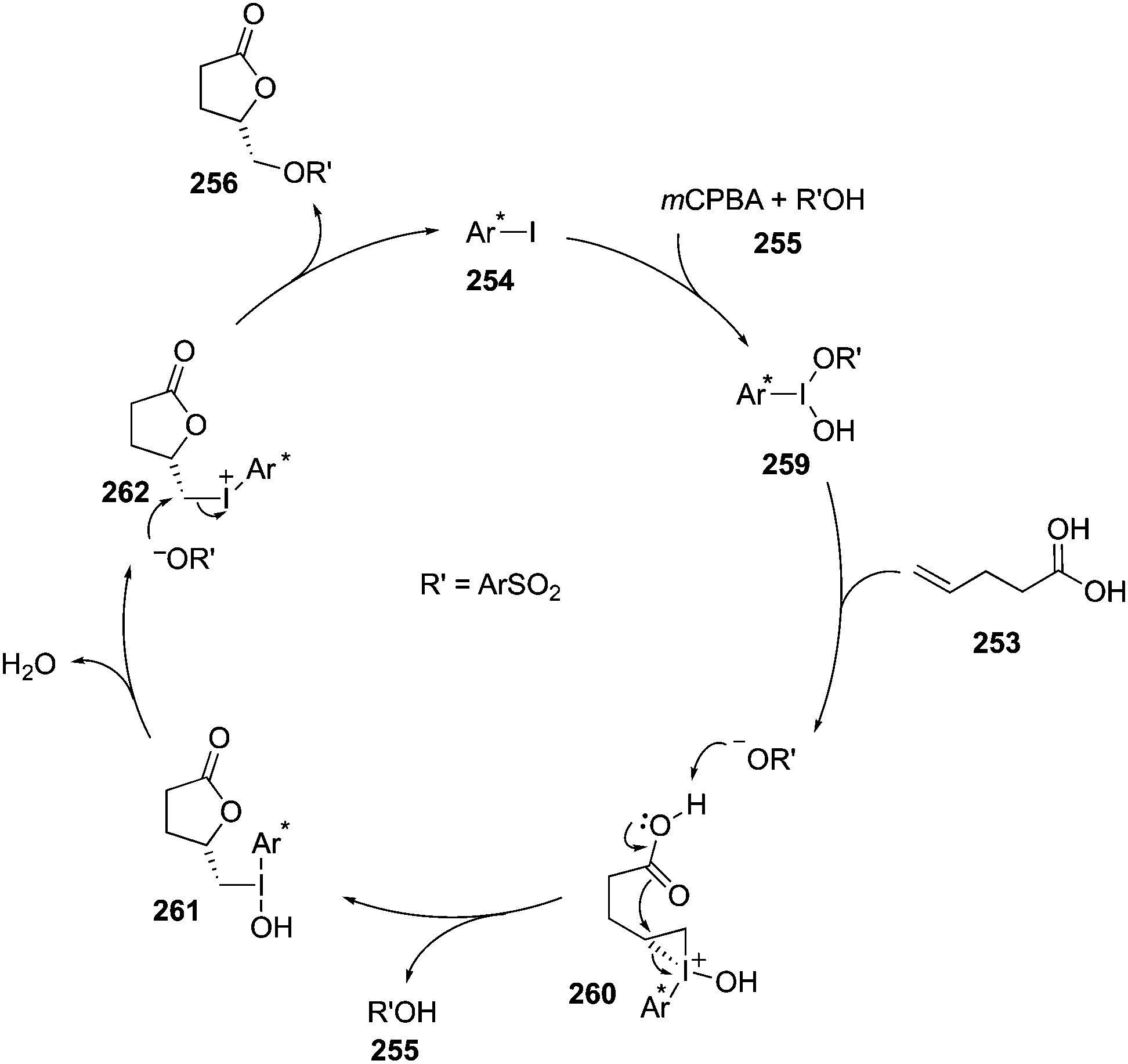
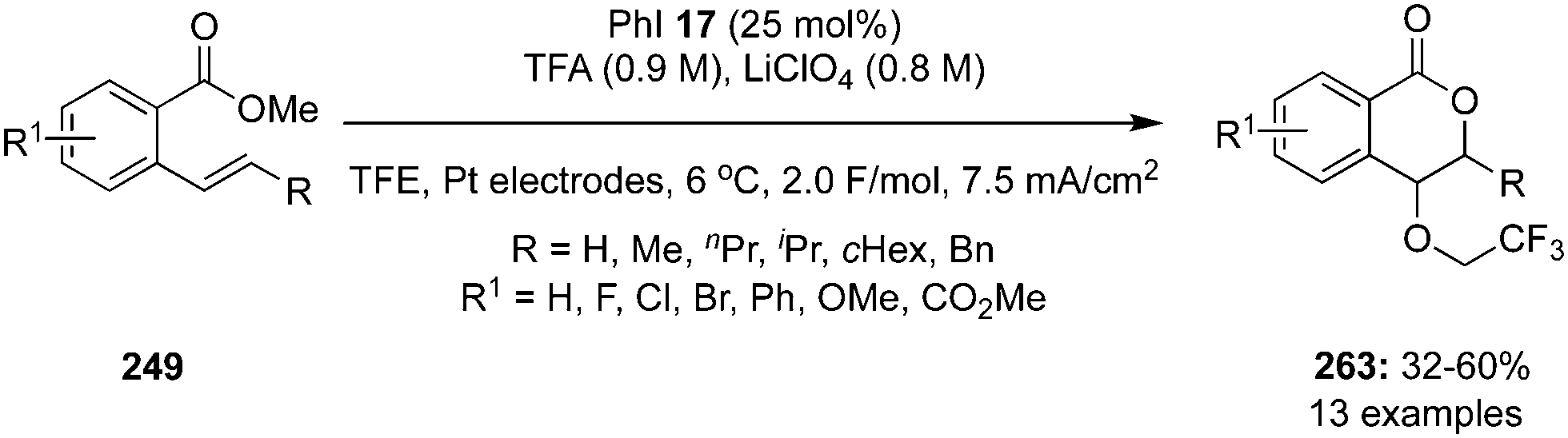
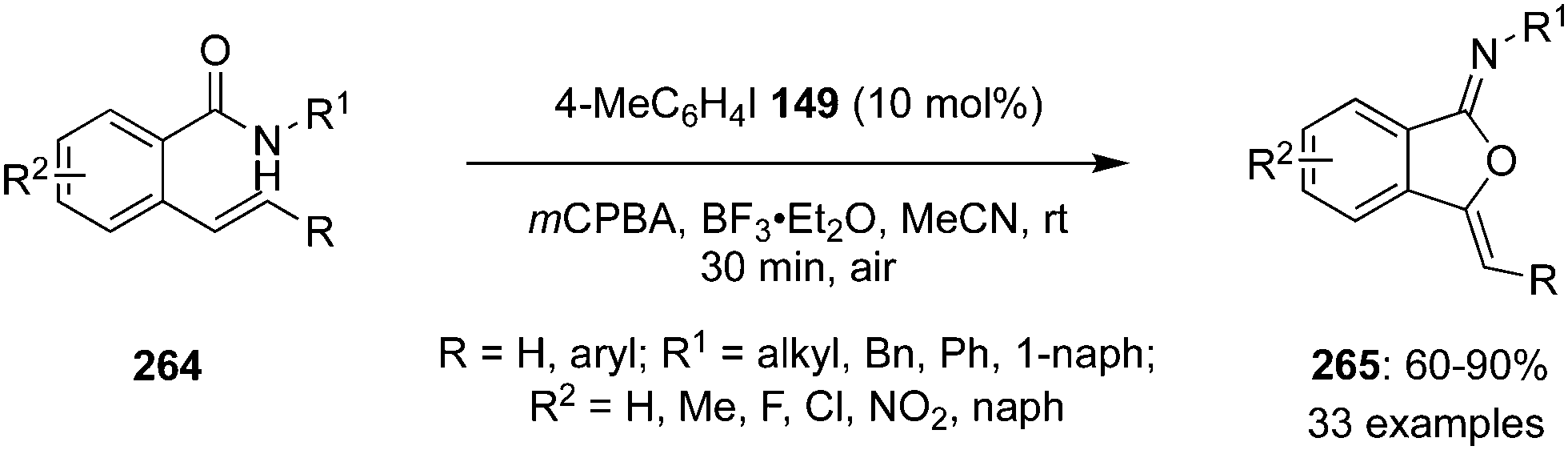
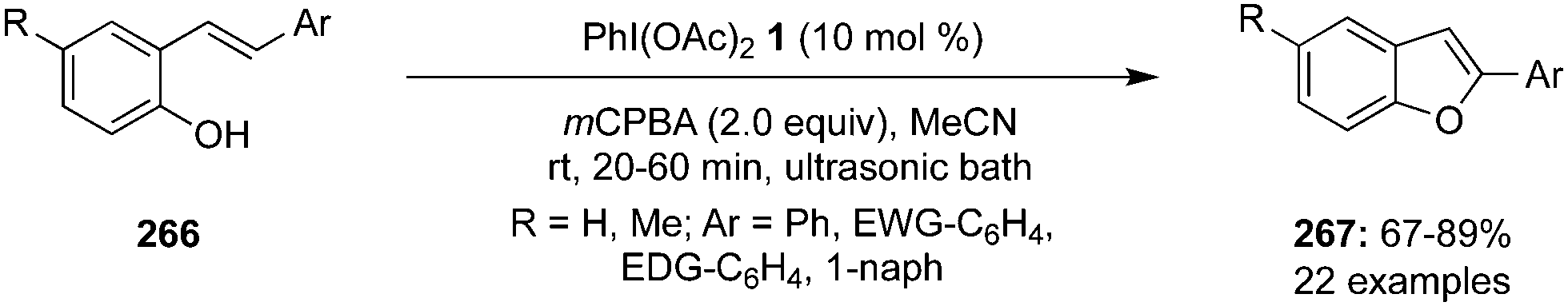
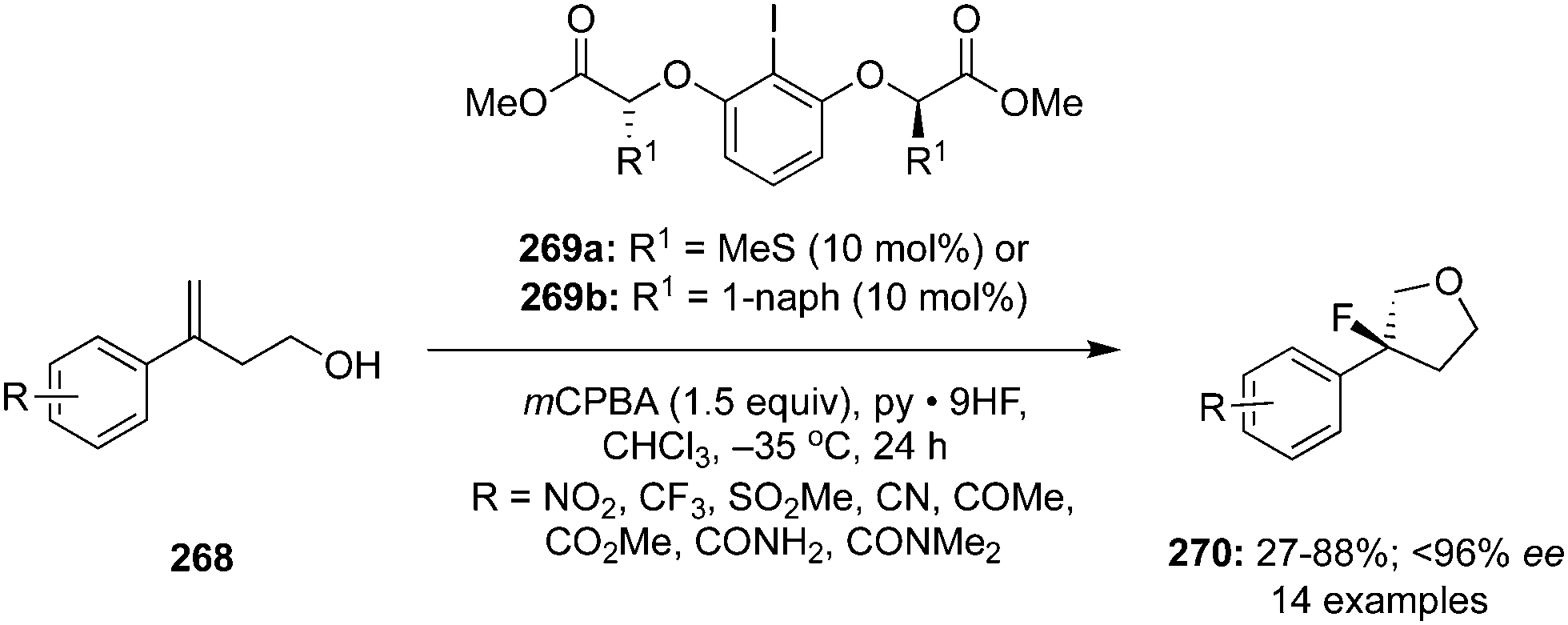
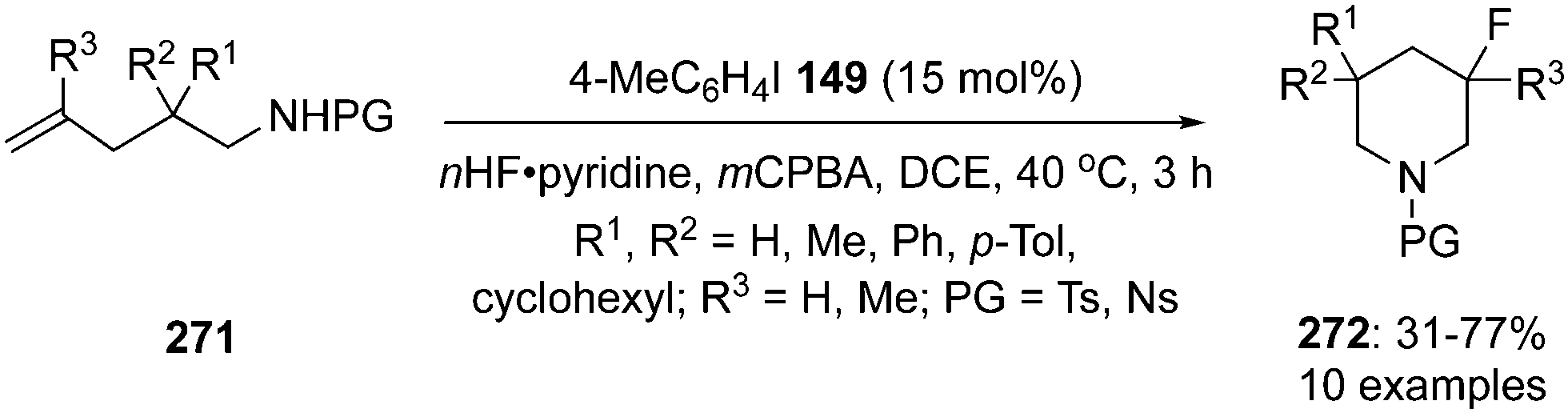
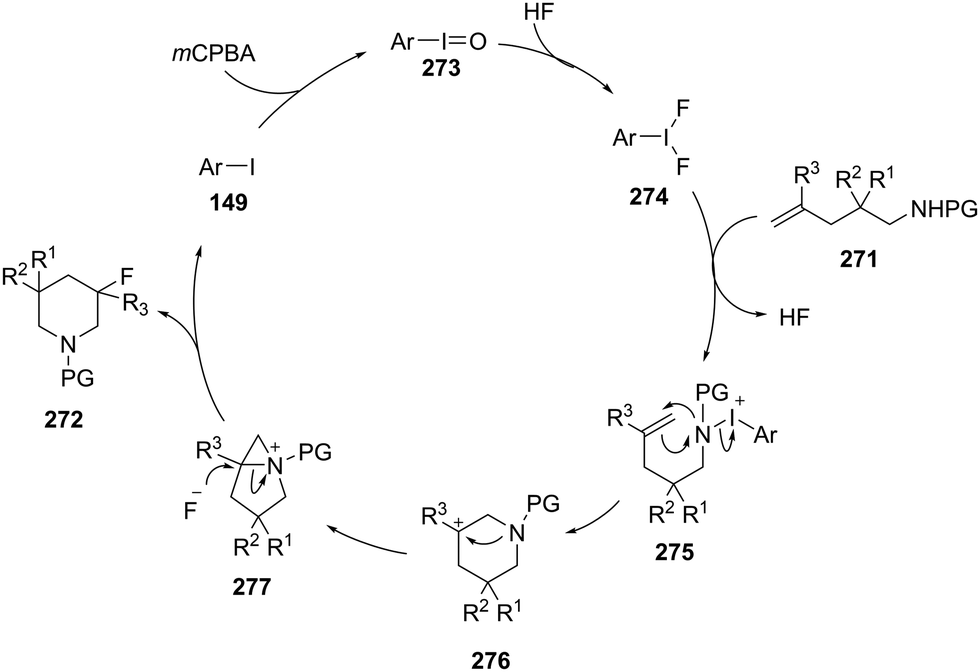
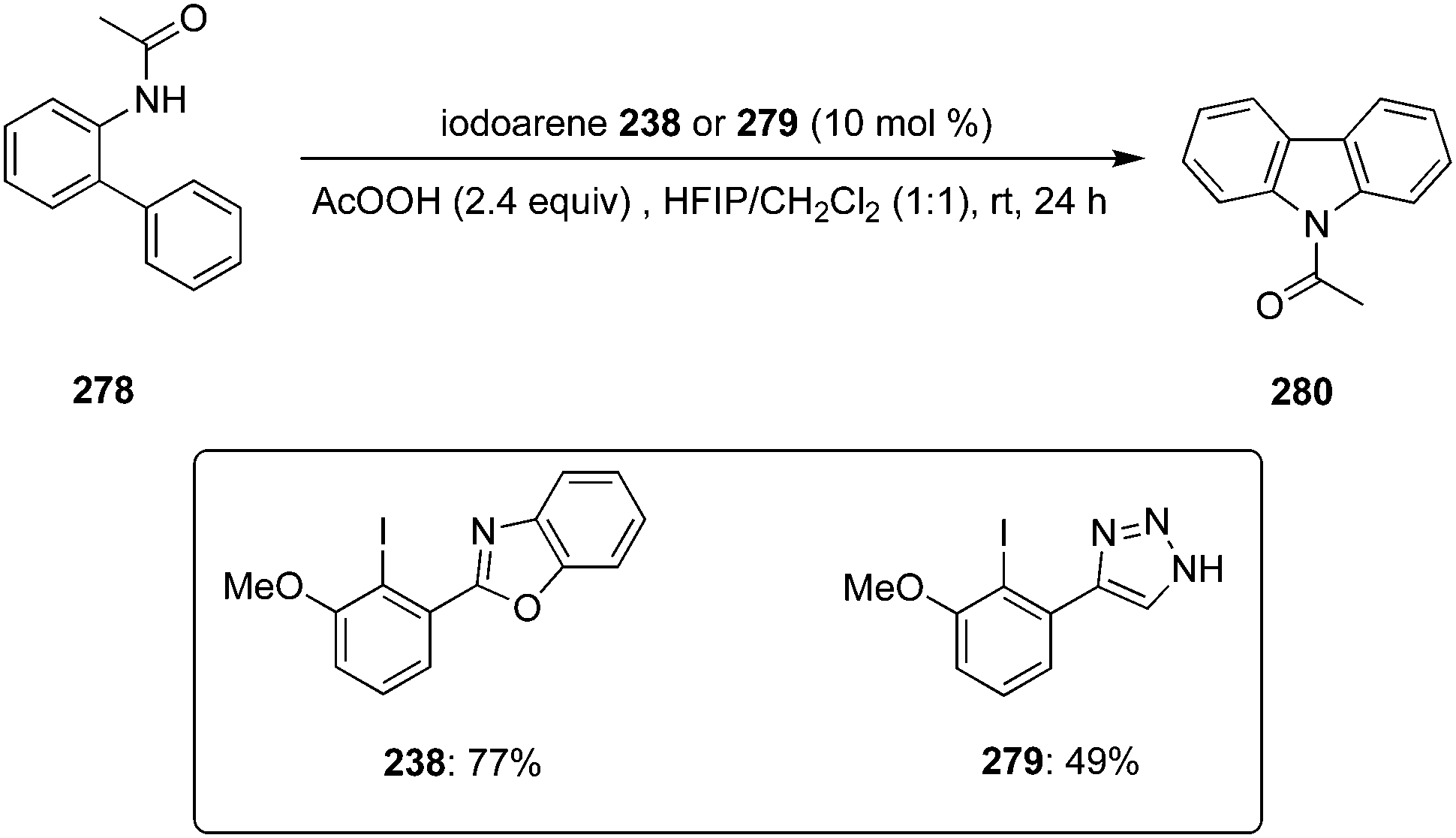
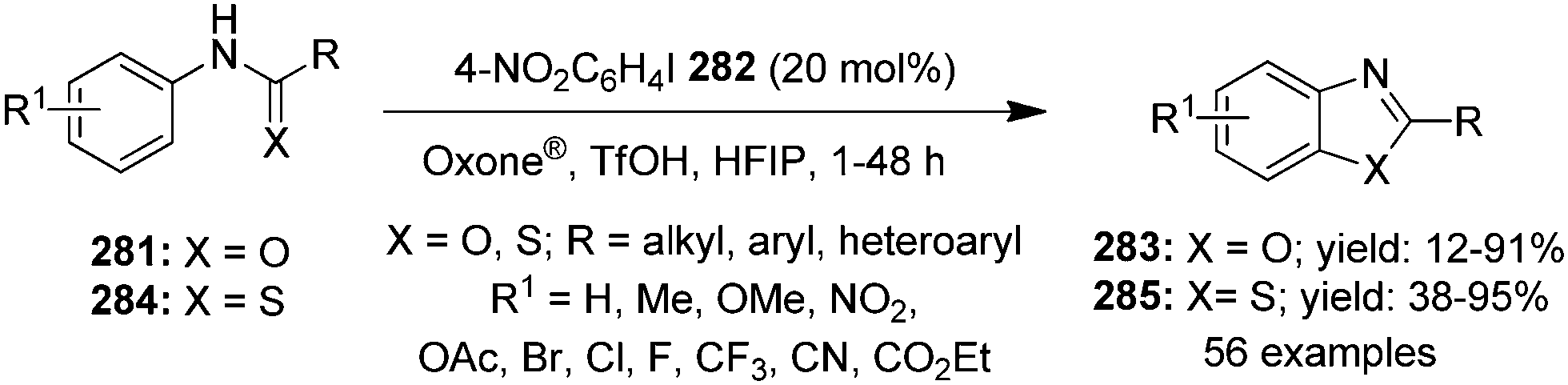
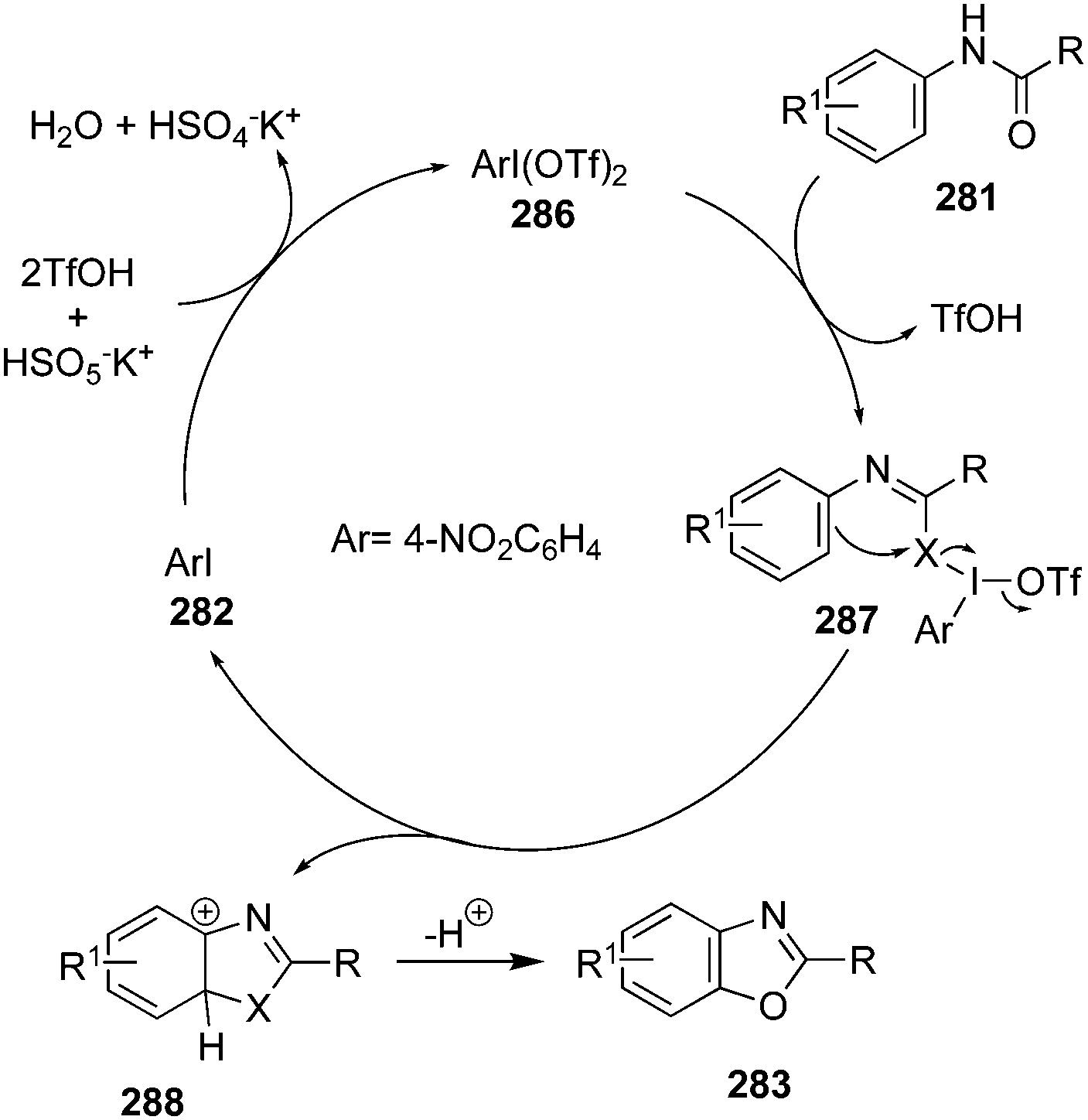
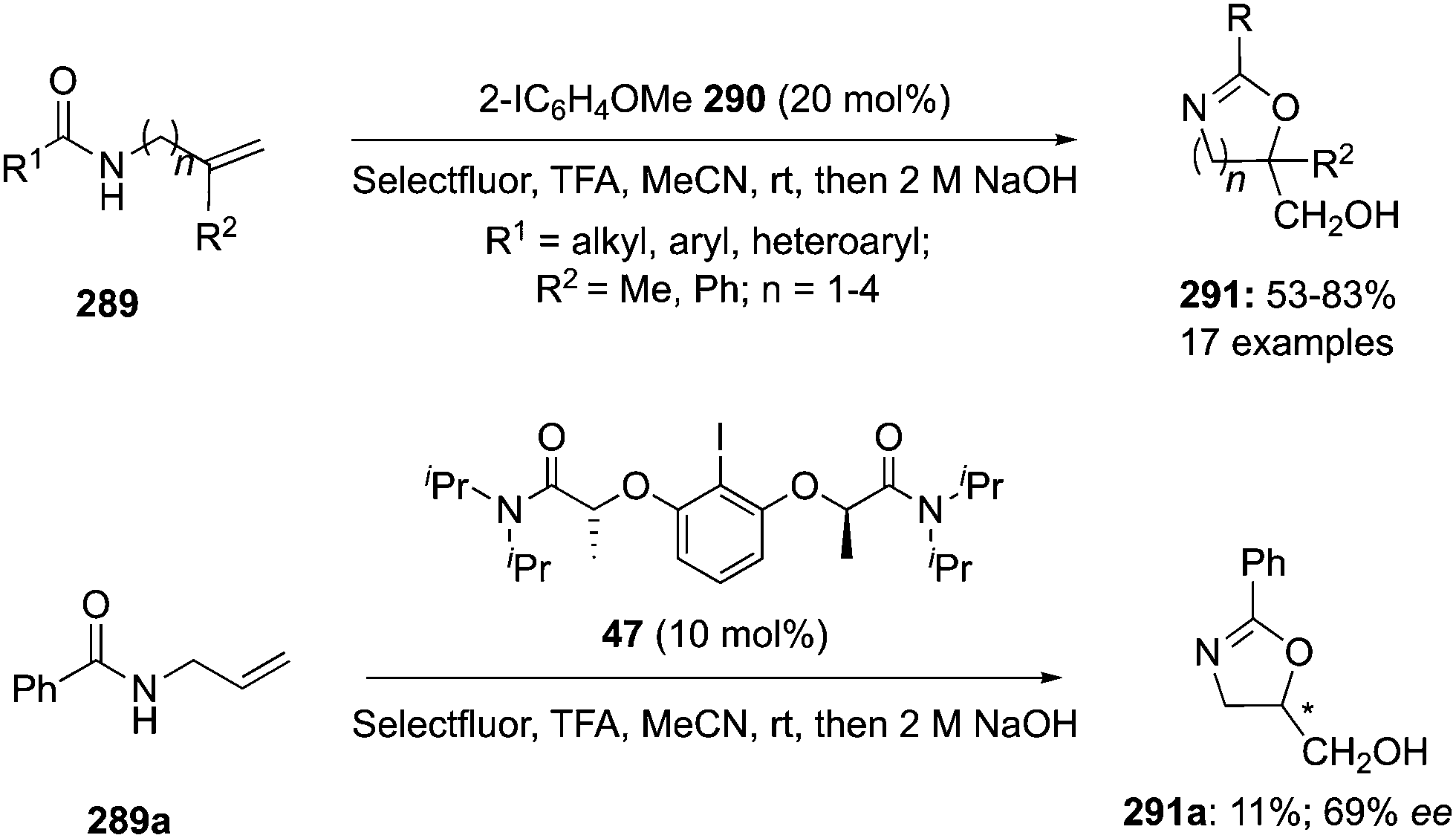
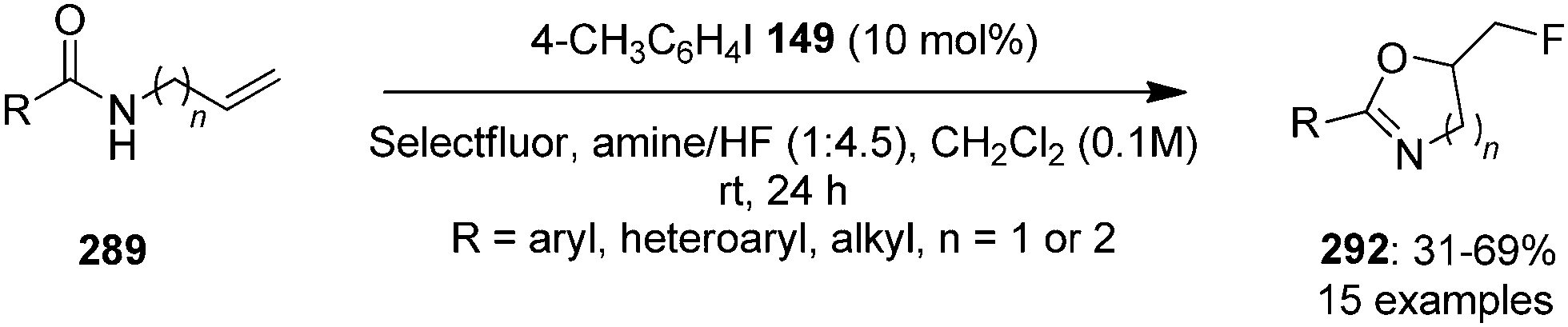
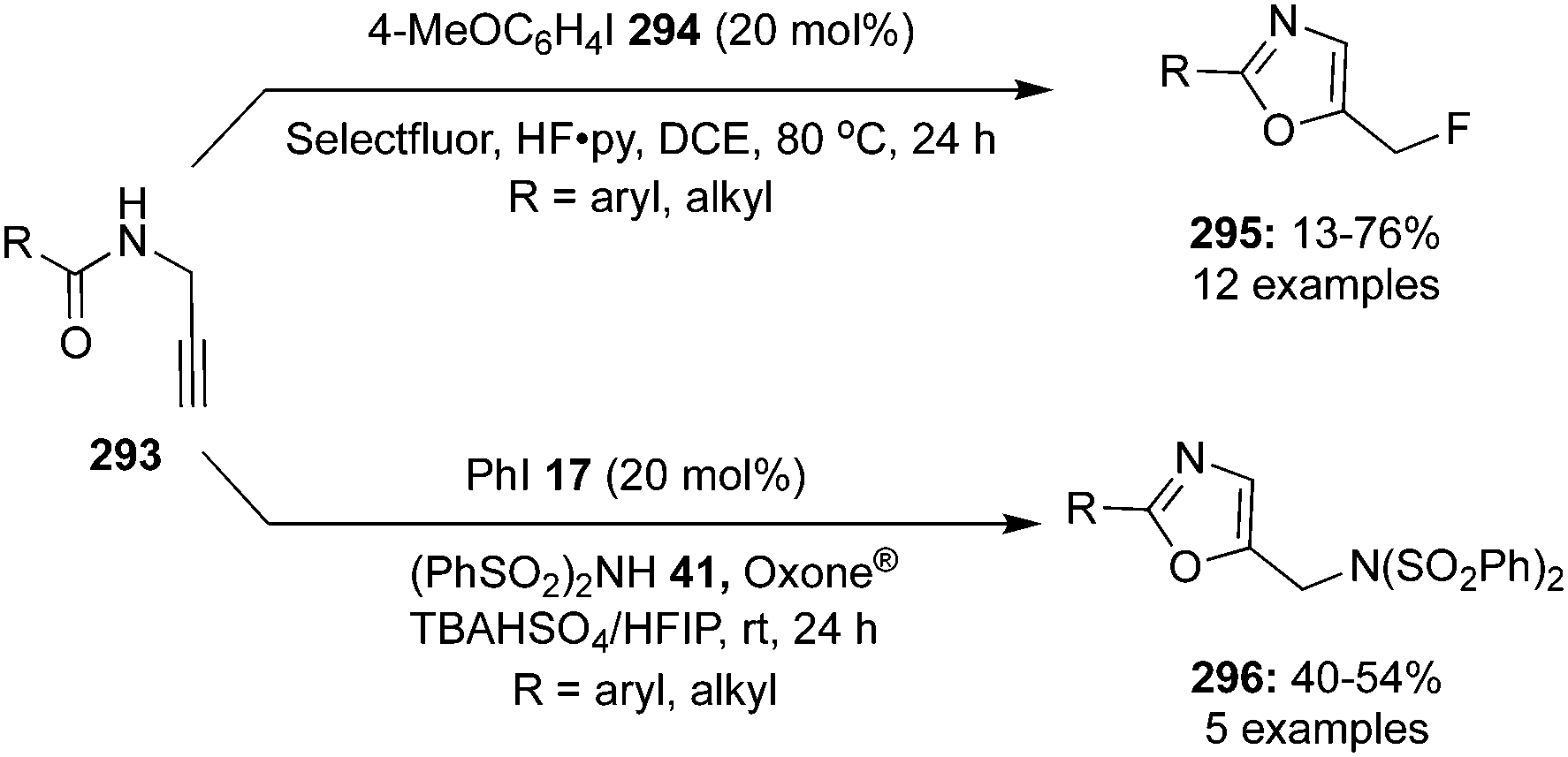
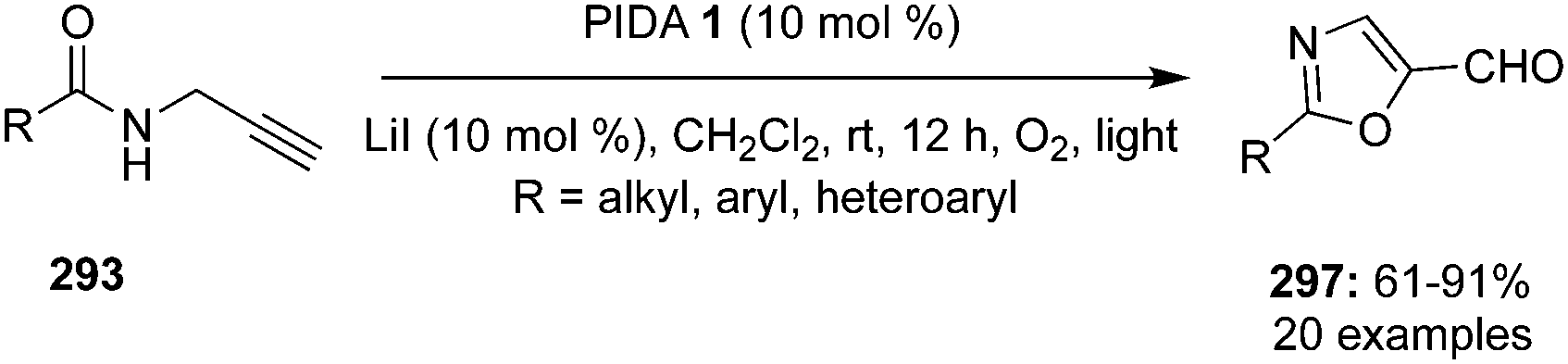
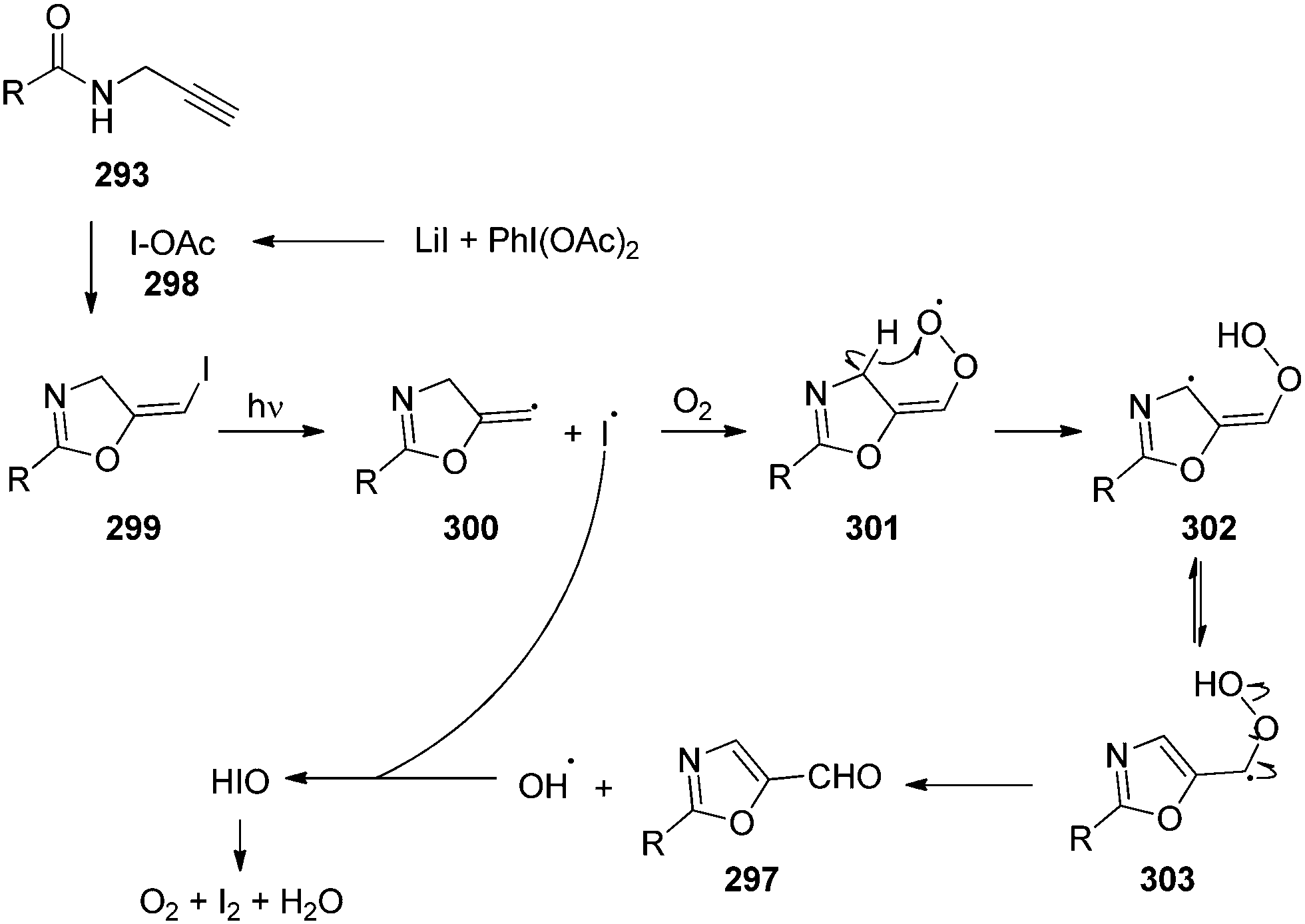
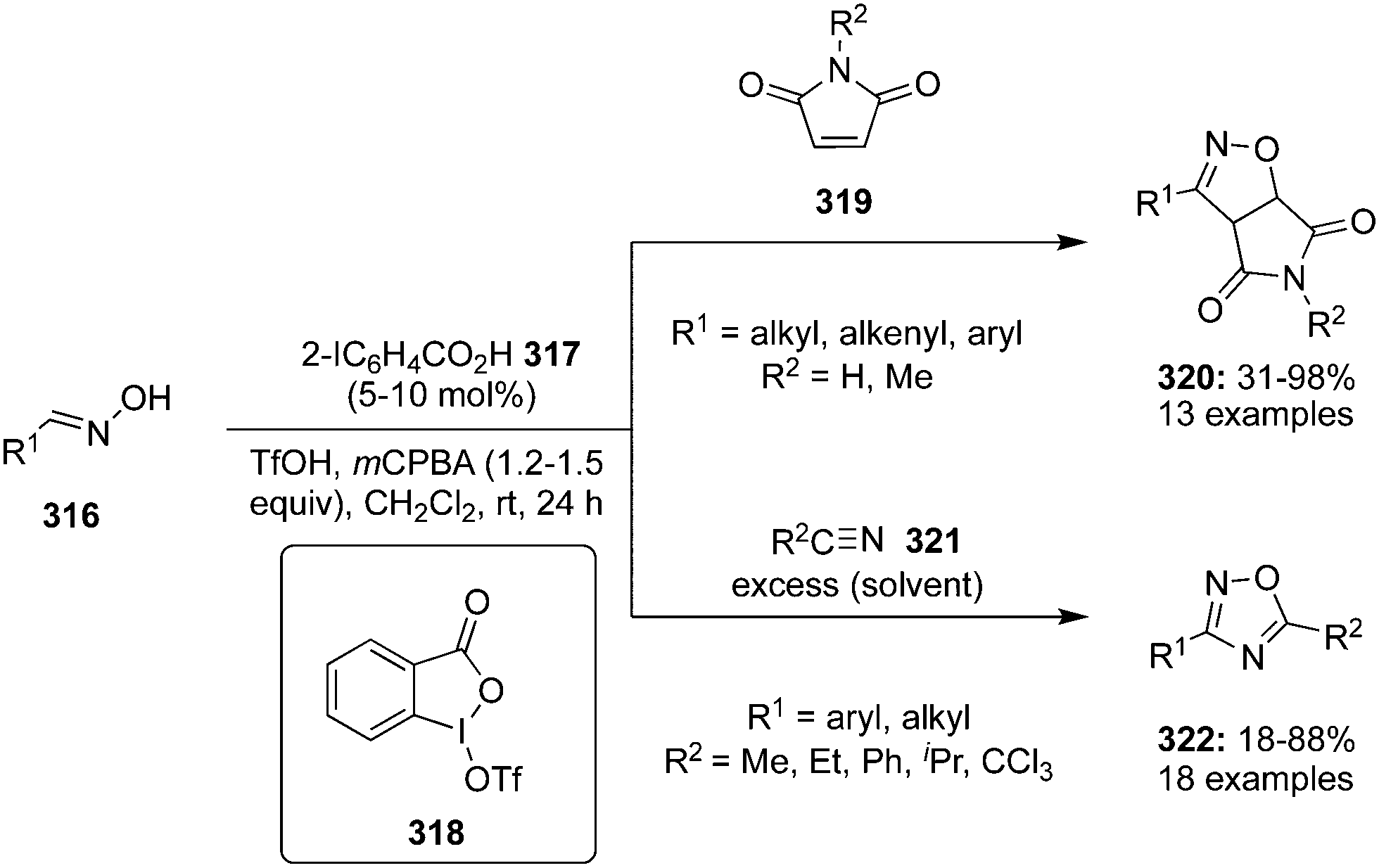
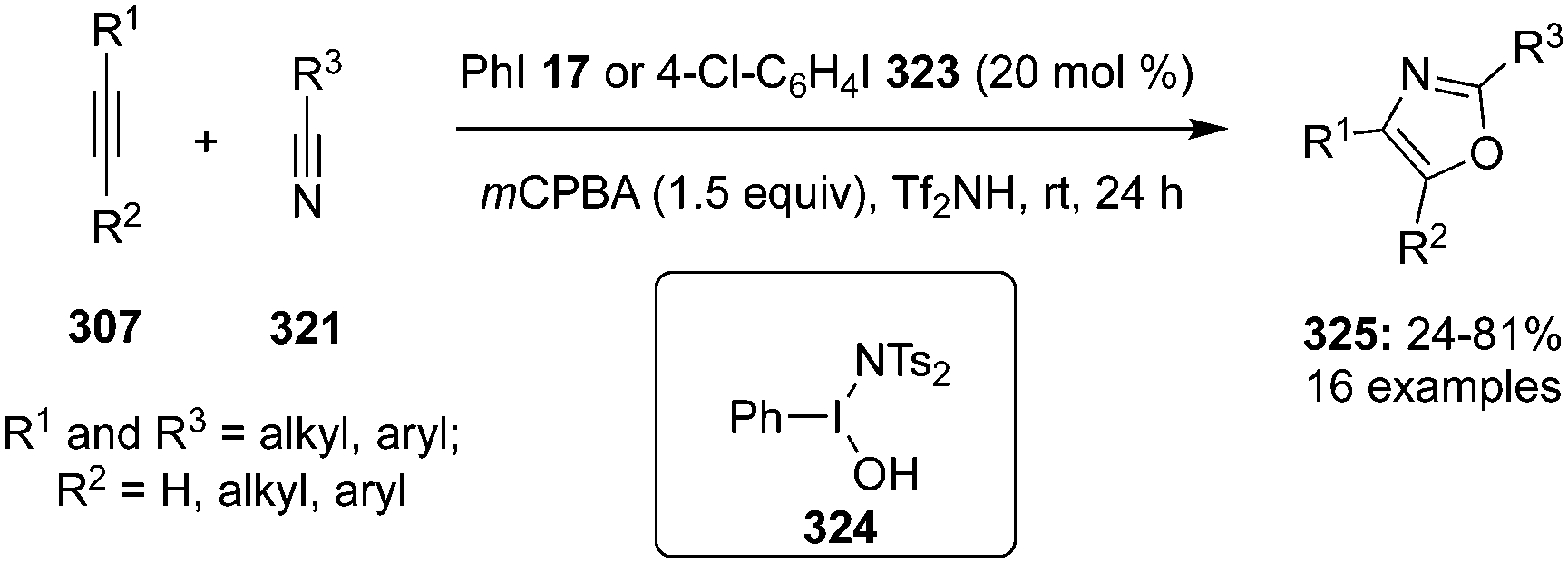
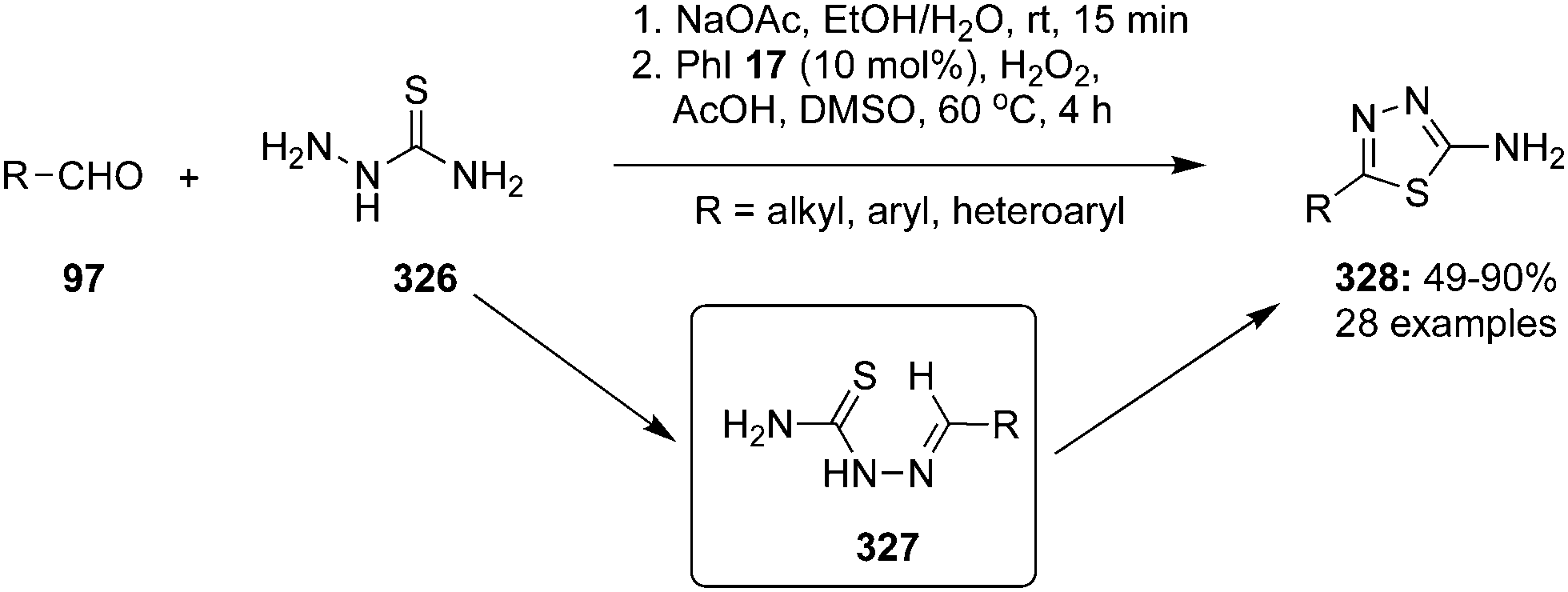
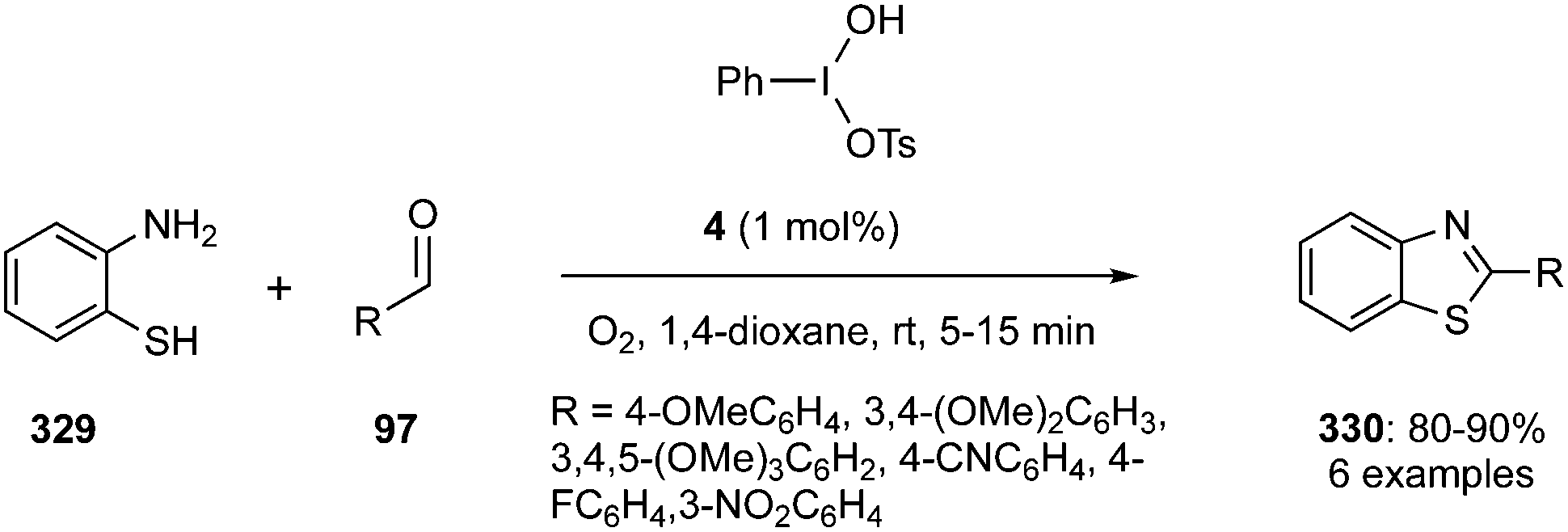
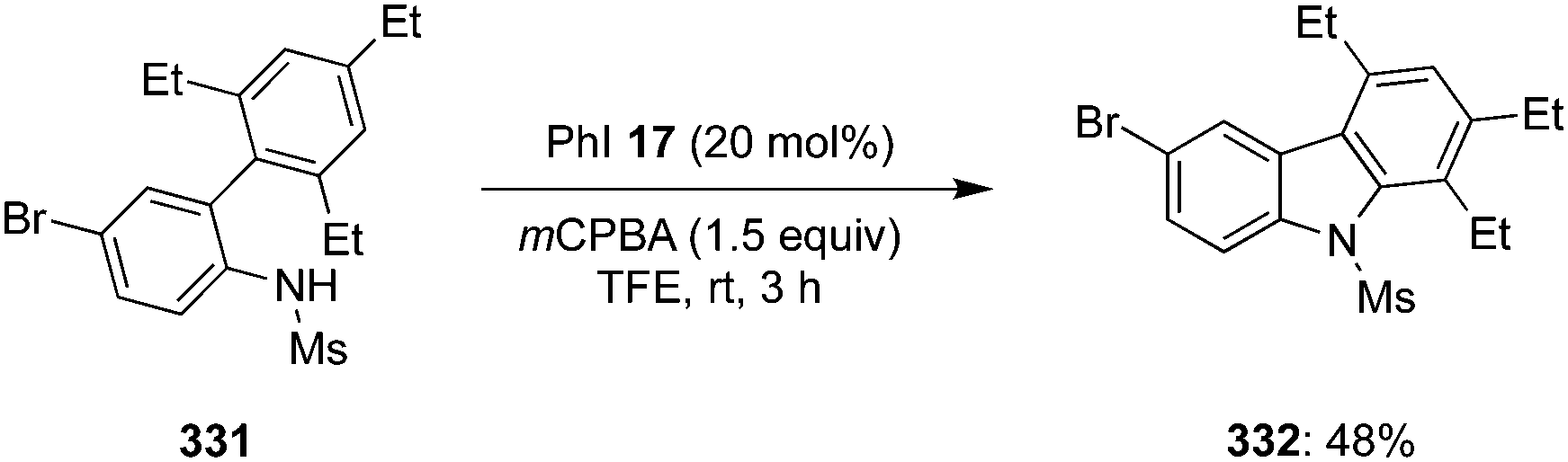
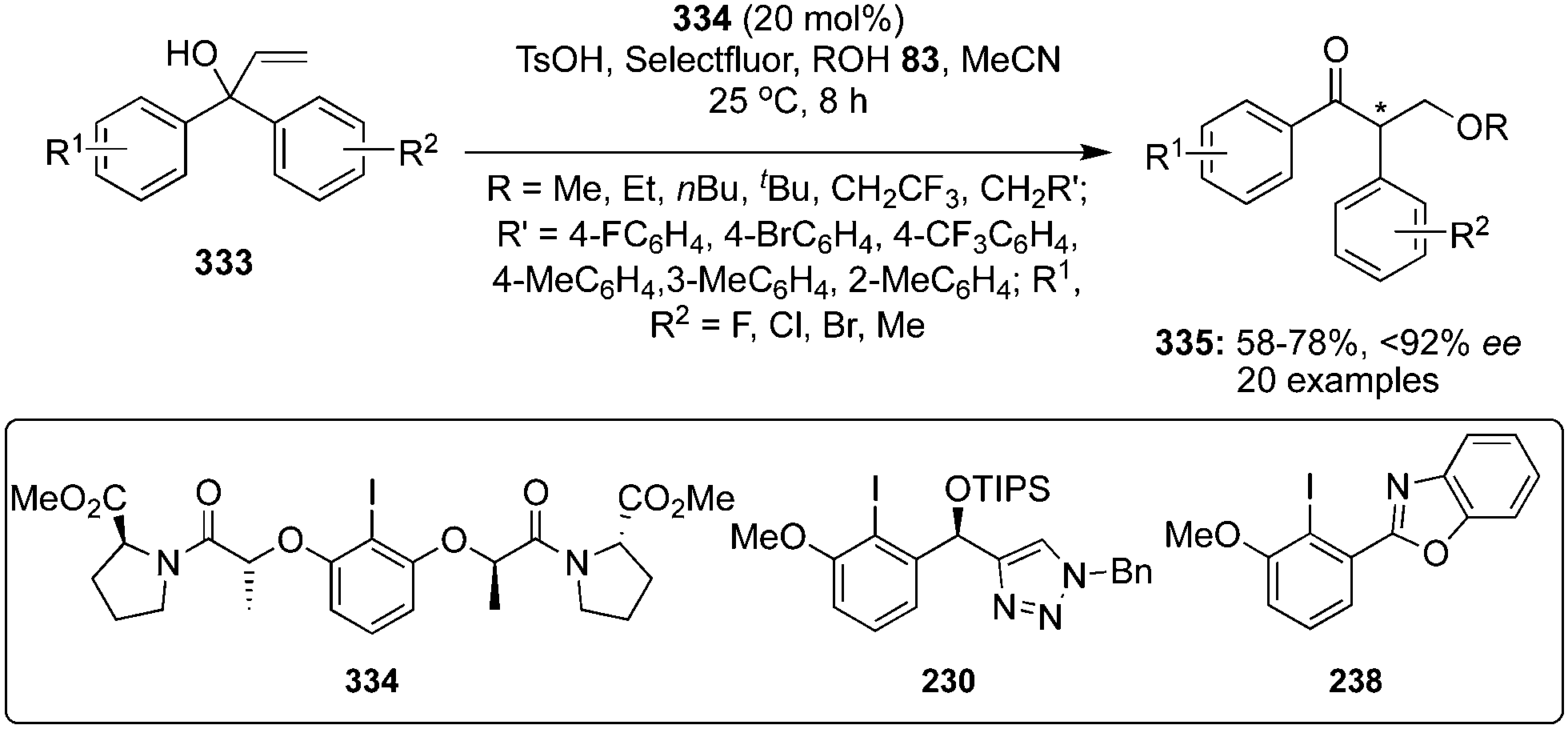
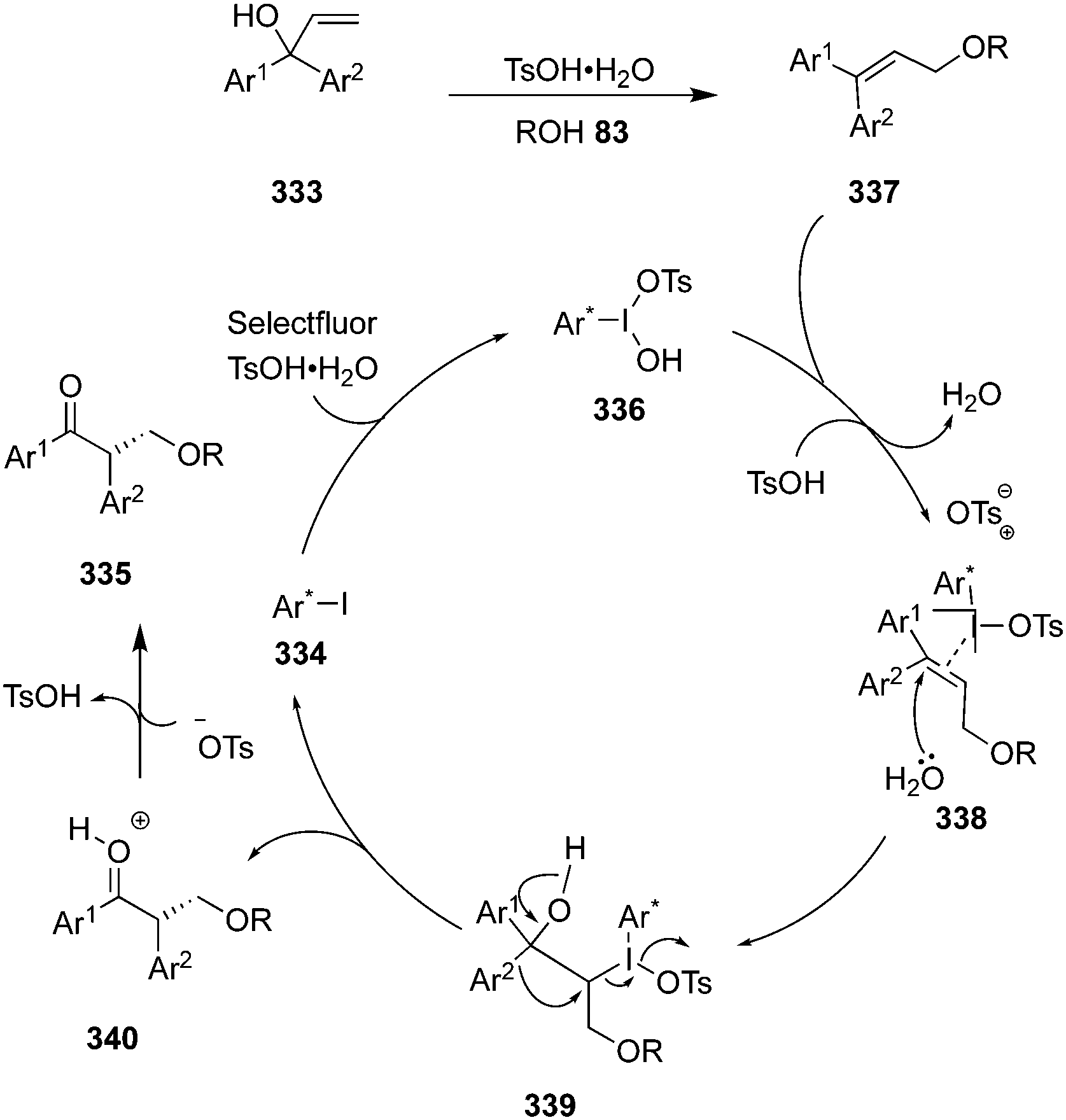
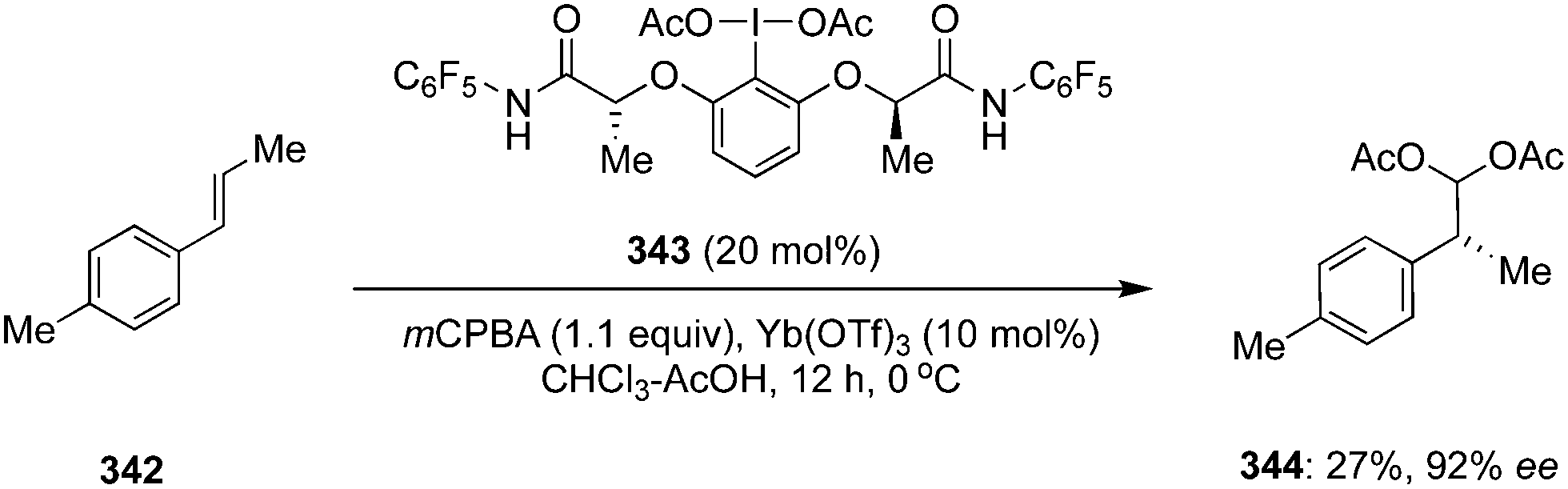
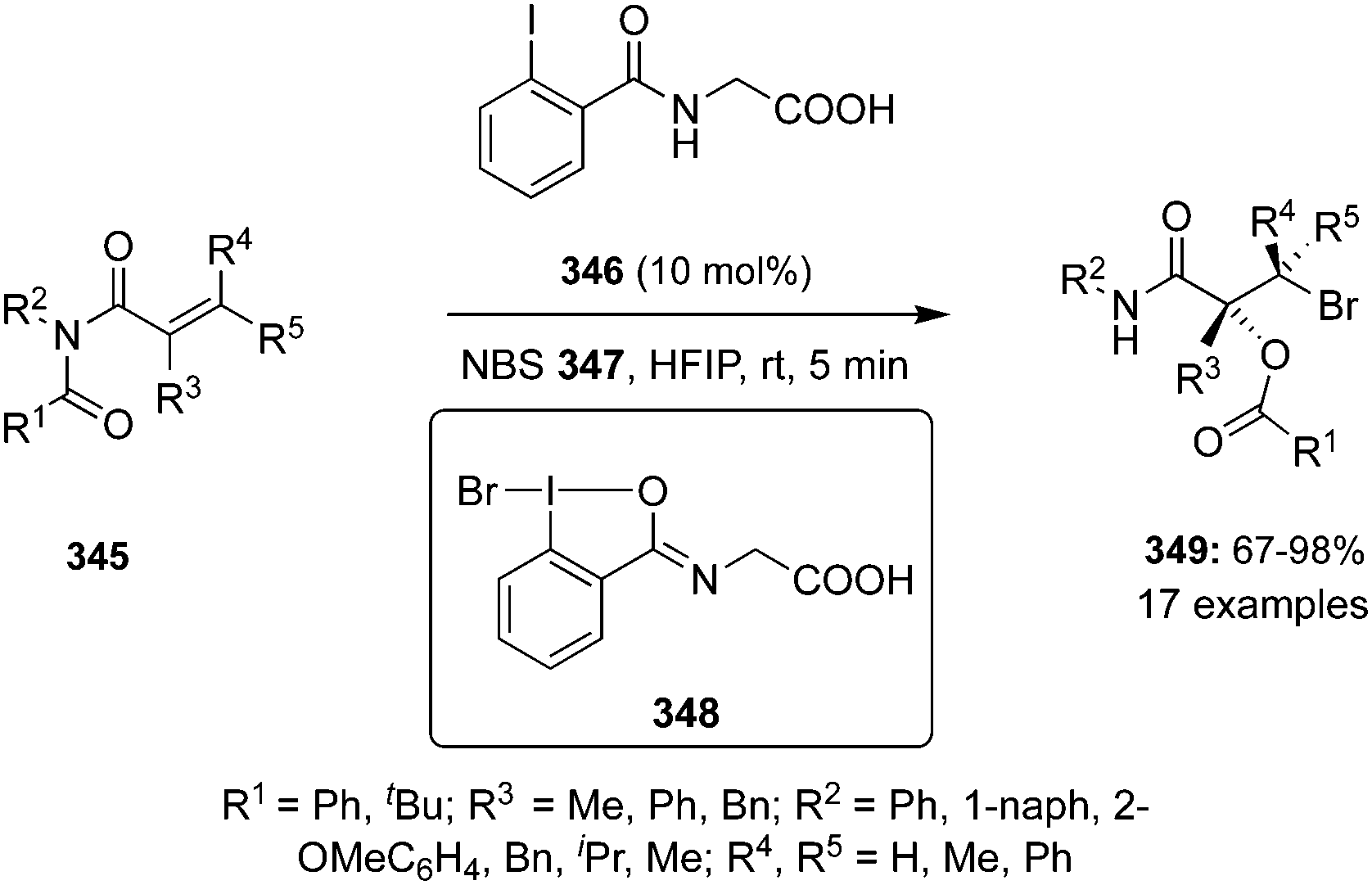
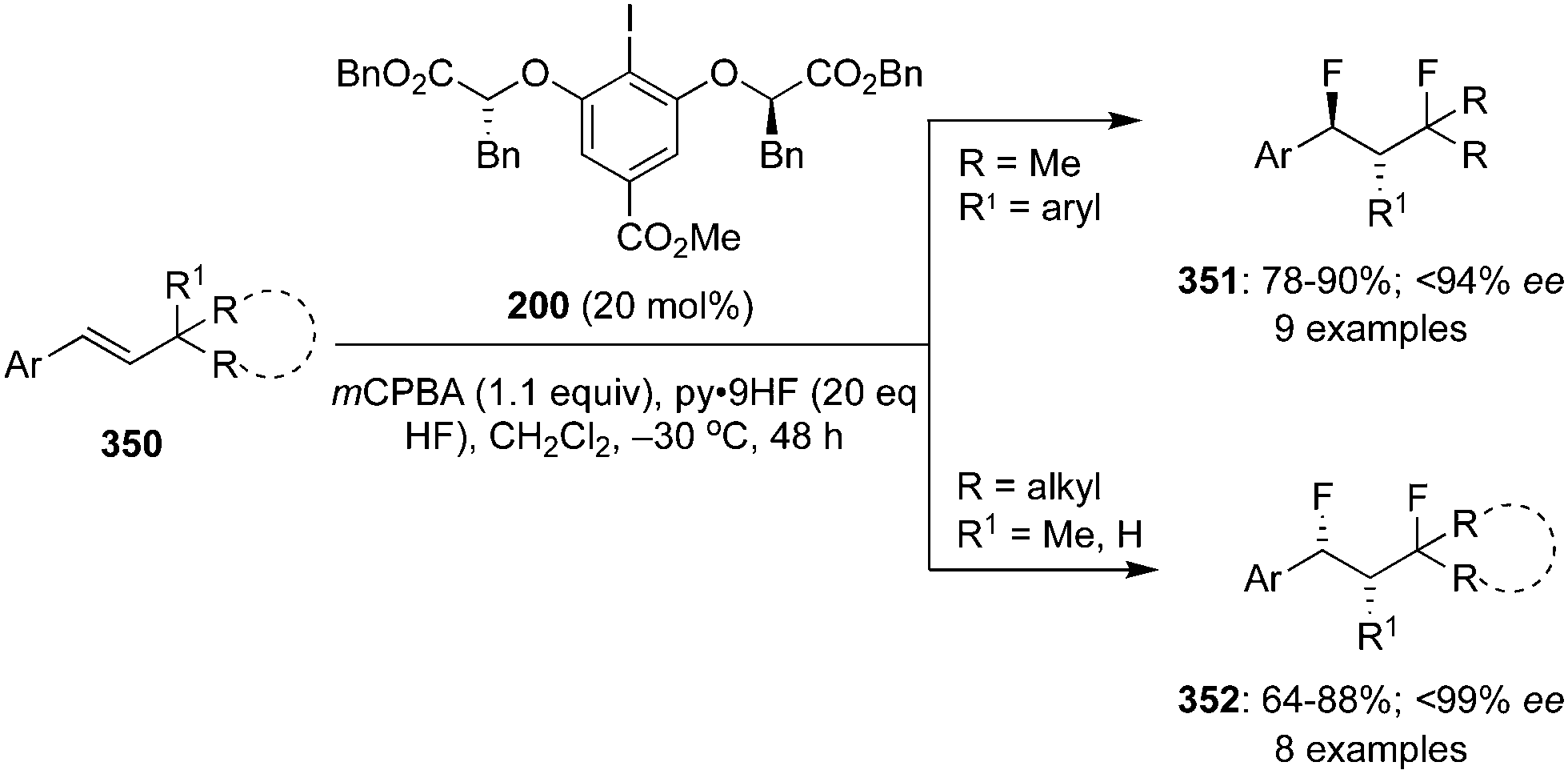
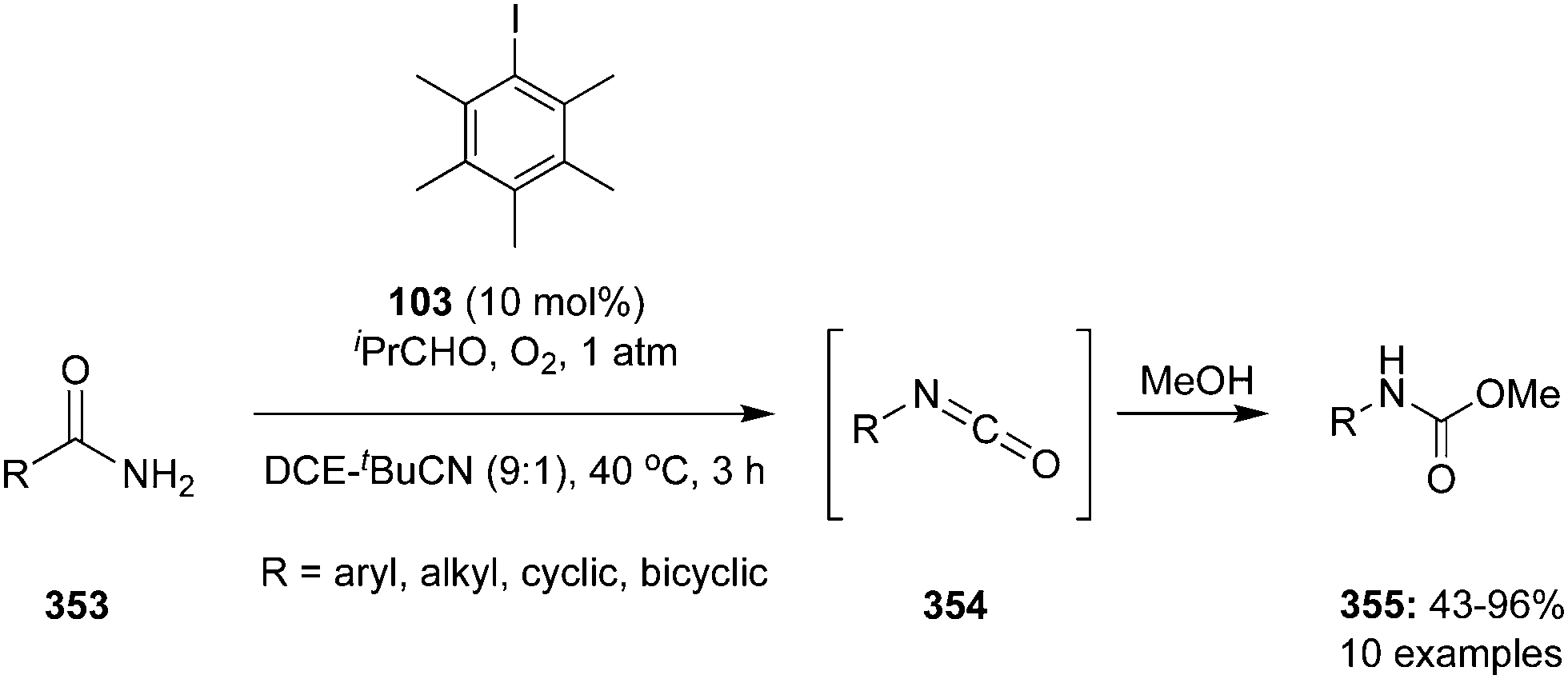
 which is further oxidized to Ru(bpy)33+ either by the benziodoxole radical 360 or its precursor BI–OH 358. Eventually, Ru(bpy)33+ oxidises alkyl trifluoroborate 356 to an alkyl R1 radical 362 and regenerates Ru(bpy)32+. Finally, α-addition of alkyl R1 radical 362 to EBX 357 provides the desired alkynes 307via formation of intermediate 363 and eliminates benziodoxole radical 360 which later oxidizes
which is further oxidized to Ru(bpy)33+ either by the benziodoxole radical 360 or its precursor BI–OH 358. Eventually, Ru(bpy)33+ oxidises alkyl trifluoroborate 356 to an alkyl R1 radical 362 and regenerates Ru(bpy)32+. Finally, α-addition of alkyl R1 radical 362 to EBX 357 provides the desired alkynes 307via formation of intermediate 363 and eliminates benziodoxole radical 360 which later oxidizes  to Ru(bpy)33+ and forms ortho-iodobenzoic acid.
to Ru(bpy)33+ and forms ortho-iodobenzoic acid.