
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porphyrins and phthalocyanines as biomimetic tools for photocatalytic H2 production and CO2 reduction
Emmanouil
Nikoloudakis
 a,
Ismael
López-Duarte
b,
Georgios
Charalambidis
*a,
Kalliopi
Ladomenou
*c,
Mine
Ince
*d and
Athanassios G.
Coutsolelos
*ae
a,
Ismael
López-Duarte
b,
Georgios
Charalambidis
*a,
Kalliopi
Ladomenou
*c,
Mine
Ince
*d and
Athanassios G.
Coutsolelos
*ae
aUniversity of Crete, Department of Chemistry, Laboratory of Bioinorganic Chemistry, Voutes Campus, Heraklion, Crete, Greece. E-mail: acoutsol@uoc.gr; gcharal@uoc.gr
bDepartamento de Química en Ciencias Farmacéuticas, Universidad Complutense de Madrid, 28040 Madrid, Spain
cInternational Hellenic University, Department of Chemistry, Laboratory of Inorganic Chemistry, Agios Loucas, 65404, Kavala Campus, Greece. E-mail: kladomenou@chem.ihu.gr
dDepartment of Natural and Mathematical Sciences, Faculty of Engineering, Tarsus University, Mersin, Turkey. E-mail: mine.ince@tarsus.edu.tr
eInstitute of Electronic Structure and Laser (IESL) Foundation for Research and Technology - Hellas (FORTH), Vassilika Vouton, Heraklion, Crete, Greece
First published on 10th June 2022
Abstract
The increasing energy demand and environmental issues caused by the over-exploitation of fossil fuels render the need for renewable, clean, and environmentally benign energy sources unquestionably urgent. The zero-emission energy carrier, H2 is an ideal alternative to carbon-based fuels especially when it is generated photocatalytically from water. Additionally, the photocatalytic conversion of CO2 into chemical fuels can reduce the CO2 emissions and have a positive environmental and economic impact. Inspired by natural photosynthesis, plenty of artificial photocatalytic schemes based on porphyrinoids have been investigated. This review covers the recent advances in photocatalytic H2 production and CO2 reduction systems containing porphyrin or phthalocyanine derivatives. The unique properties of porphyrinoids enable their utilization both as chromophores and as catalysts. The homogeneous photocatalytic systems are initially described, presenting the various approaches for the improvement of photosensitizing activity and the enhancement of catalytic performance at the molecular level. On the other hand, for the development of the heterogeneous systems, numerous methods were employed such as self-assembled supramolecular porphyrinoid nanostructures, construction of organic frameworks, combination with 2D materials and adsorption onto semiconductors. The dye sensitization on semiconductors opened the way for molecular-based dye-sensitized photoelectrochemical cells (DSPECs) devices based on porphyrins and phthalocyanines. The research in photocatalytic systems as discussed herein remains challenging since there are still many limitations making them unfeasible to be used at a large scale application before finding a large-scale application.
 Emmanouil Nikoloudakis | Emmanouil Nikoloudakis has recently finished his PhD degree in the Laboratory of Bio-Inorganic Chemistry, in the Department of Chemistry at the University of Crete. He got his Bachelor and his Master's degrees from the Department of Chemistry at the University of Crete in 2017 and 2019, respectively. In 2017 and 2020 he visited collaborating laboratories in France, in Grenoble and Nantes respectively, as a post-graduate student. In 2021, he visited the University of North Texas, in Denton in USA, under the Fulbright program as a visiting research student where he worked for 4 months. |
 Ismael López-Duarte | Ismael López-Duarte received his PhD (2010) from the Universidad Autónoma de Madrid under the supervision of Profs. T. Torres and M. V. Martínez-Díaz. He carried out postdoctoral work at the University of Oxford as Marie Curie Fellow (Prof. H. L. Anderson FRS), the Imperial College London (Prof. M. K. Kuimova) and the Queen Mary University of London (Prof. L. Milanesi). Currently, he is Assistant Professor in the Department of Chemistry in Pharmaceutical Sciences at Universidad Complutense de Madrid. His research interests include the design and synthesis of porphyrinoid chromophores for energy and biomedical applications. |
 Georgios Charalambidis | Georgios Charalambidis received his Bachelor's (2002), Master's (2005), and PhD (2007) degrees from the University of Crete. Currently his is the Principal Investigator in the project entitled “Development of Bio-Inspired Photocatalytic Systems for H2 Production and CO2 Reduction” funded from the Hellenic Foundation for Research (HFRI), at the Chemistry Department of the University of Crete. His current research interests include the synthesis of porphyrin-related compounds for their application in solar cells, photocatalytic hydrogen production and CO2 reduction, artificial photosynthesis molecular materials and self-assembling nanostructures. |
 Kalliopi Ladomenou | Assistant Professor Kalliopi Ladomenou received her BSc (1998) and MSc from the University of Crete. She obtained her PhD (2002) from the University of Liverpool and worked as a Post-Doctoral Researcher at the University of Edinburgh (2002–2004). Then she joined the University of Crete as a Researcher. In 2022 she was elected as an Assistant Professor at the Chemistry Department of the International Hellenic University. During her research activity she has been involved in several national and European projects. Her research interests include the synthesis of chromophores and catalysts for applications in solar cells, H2 production, CO2 reduction, artificial photosynthesis and biomimetic catalysis. |
 Mine Ince | Prof. Mine Ince received her PhD (2012) from the Universidad Autónoma de Madrid (UAM) under the supervision of Profs. T. Torres and M.V. Martínez-Díaz. She, then, joined the group of Prof. Michael Grätzel at EPFL (Switzerland) as a postdoctoral researcher (2013). She is currently a full professor in the Department of Natural and Mathematical Sciences at Tarsus University (Turkey). Her current research interests include the preparation of novel phthalocyanine-based materials for their application in organic photovoltaic devices and in photocatalysis (CO2 reduction and water splitting). |
 Athanassios G. Coutsolelos | Professor Athanassios G. Coutsolelos received his PhD from the University of Dijon in France (1985) with Prof. R. Guilard. After postdoctoral research (1985–1986), at the University of Texas in Houston with Prof. K. M. Kadish, he joined the University of Crete where he obtained a full Professor position in 1999. He was invited as a Visiting Professor at the University of Notre Dame (USA) in 1996, as well as in Orsay (1994, 2000 and 2016) and in Strasbourg (2010), France. He is the director of the Laboratory of Bioinorganic Chemistry and his primary research interests focuses on the development of new materials based on porphyrin derivatives for artificial photosynthesis. |
1. Introduction
The use of fossil fuels for the increasing energy demand indicates that the global sources will be depleted in the near future. The relentless use of fossil fuels as the primary energy source at this pace will force us to face two major global challenges: increasing energy costs due to limited fossil fuel resources and climate change caused by greenhouse gas emissions. Solar energy is an inexhaustible source of energy and the sunlight reaching the earth daily is enough to meet all the energy needs of humanity. Of all the renewable energy sources, solar power has the highest potential to become the energy source of the future.1 However, the major problem is related to the storage of sunlight which remains an important scientific and technological challenge. In fact, photovoltaic technologies that produce electrical energy from solar light have shown great development in the last decades compared to other renewable energy sources and generate electricity at a considerable scale.2 Though the widespread use of photovoltaic technology is a promising approach to curb the demand for fossil fuels. Today's photovoltaic technologies, especially for the transport sector, are not able to fulfil societal needs. Considering that currently, transportation is one of the sectors with the highest energy supply, the conversion of solar energy into storable energy carriers and solar fuels appears as a promising solution to produce cost-effective fuels and ensure the sustainability of the energy supply. As previously mentioned, one additional problem arising from the use of fossil fuels is global warming which causes many uncontrollable problems such as climate change, depletion of biodiversity, and change of the water cycle. CO2 is the main greenhouse gas causing global warming.3 In this sense, technologies for converting CO2 and water into sustainable fuels and/or high value-added chemicals are of particular interest as they have the potential to address challenges of the dependency on fossil fuels and greenhouse emissions. Undoubtedly, the conversion of CO2 into chemical fuel and raw materials will not only contribute to the CO2 emission balance but will also have a significant environmental and economic impact.4On the other hand, as a zero-emission energy carrier, H2 is an ideal alternative to carbon-based fossil fuels that can be used to produce both fuel and electricity as well as value-added feedstock such as methanol, methane, etc.5 However, most of the H2 production is currently based on the conventional reform of fossil fuel that has been causing greenhouse gas emissions. The contradiction of using fossil fuels to produce hydrogen has been addressed over the last decades by the development of low-cost and environmentally friendly alternative methods such as photocatalytic H2 production.6 As a consequence, from an environmental, economic, and social point of view, the generation of H2 from water and reduction of CO2 to solar fuels by using sunlight could be an optimal solution for the sustainability of energy supply and fuel sources.
Sunlight can be converted into storable chemicals and fuels with three main processes: natural photosynthesis, artificial photosynthesis and thermochemical method.7,8 Photosynthesis is the process in which plants and living organisms convert energy from sunlight into chemical energy in the form of sugar, which acts as an energy source. In this process, the sunlight absorbed by the light-harvesting pigments, i.e., antenna molecules, is transformed into chemical energy in the reaction center by cascades of energy and electron transfer between organic pigments. Artificial photosynthesis aims to mimic the photochemical process of natural photosynthesis and consists of light absorption and charge-transfer processes together with catalytic oxidation and reduction of H2O and catalytic reduction of CO2.9 Artificial photosynthesis can play a crucial role, without a doubt, in tackling global energy and environmental problems, not only in producing renewable fuels such as H2 but also in reducing atmospheric CO2 emissions by converting CO2 into valuable chemicals. In the thermochemical method, water and CO2 can be converted into solar fuels including H2 by using sunlight as the heat source to drive endothermic chemical reactions.
CO2 is a thermodynamically extremely stable molecule and breaking the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (750 kJ mol−1) requires high energy. Consequently, CO2 reduction is a notably challenging process due to the large conversion energy barrier, since any reduction process demands high energy input.10 An encouraging approach to overcome the energy sufferings of CO2 conversion has been the direct use of solar energy by mimicking natural photosynthetic systems. Indeed, compared to the CO2 reduction methods such as thermochemical conversion,11 electrochemical reduction12–15 or biochemical technology,16 the photocatalytic conversion of CO2 based on artificial photosynthesis is the most attractive route in terms of using direct solar energy and water at room temperature and atmospheric pressure.17
O bonds (750 kJ mol−1) requires high energy. Consequently, CO2 reduction is a notably challenging process due to the large conversion energy barrier, since any reduction process demands high energy input.10 An encouraging approach to overcome the energy sufferings of CO2 conversion has been the direct use of solar energy by mimicking natural photosynthetic systems. Indeed, compared to the CO2 reduction methods such as thermochemical conversion,11 electrochemical reduction12–15 or biochemical technology,16 the photocatalytic conversion of CO2 based on artificial photosynthesis is the most attractive route in terms of using direct solar energy and water at room temperature and atmospheric pressure.17
Photocatalytic conversion is the process in which a certain photocatalyst, i.e., semiconductors or molecular chromophores, uses the energy from sunlight to drive the reduction reactions of CO2 into carbon-based products and the splitting of water into H2 and O2. The photocatalytic process can be divided into homogenous and heterogeneous systems depending on the reaction phase. In a photochemical system three components are essential (i) a photosensitizer (PS) able to absorb visible light generating excited species PS˙ with useful redox properties (ii) a sacrificial electron donor (SED) that can be reduced or oxidized by quenching of the excited species PS˙ in electron transfer reactions and (iii) a catalyst, able to collect several electrons facilitating the exchange of two or four electrons with water. All the above components can be in the same (homogenous) or in different phases (heterogeneous).
Each system has its own set of benefits and drawbacks. In homogeneous photocatalysis, the molecular structure of the components is usually well-defined, and this enables the determination of the corresponding catalytic mechanism. The chemical versatility of the molecular catalysts provides control of the physical, optical, and electronic properties by chemical modifications. Furthermore, these systems present higher selectivity and the identification of the structural characteristics that impact catalytic activity is easier. However, homogeneous catalysts suffer from high costs, difficult recovery/separation process from the reaction, and lack of long-term use. Although heterogeneous systems have the advantage of efficient recovery and recyclability of photocatalysts, other factors such as low light-harvesting capacity and low reactivity limit the overall efficiency. A hybrid catalyst, on the other hand, is a potential way to combine the advantages of homogeneous and heterogeneous catalysts by immobilizing a homogeneous molecular catalyst on solids.
The operating principle of the photocatalytic systems based on a single semiconductor or a molecular PS for water splitting and CO2 reduction are very similar as shown in Fig. 1.
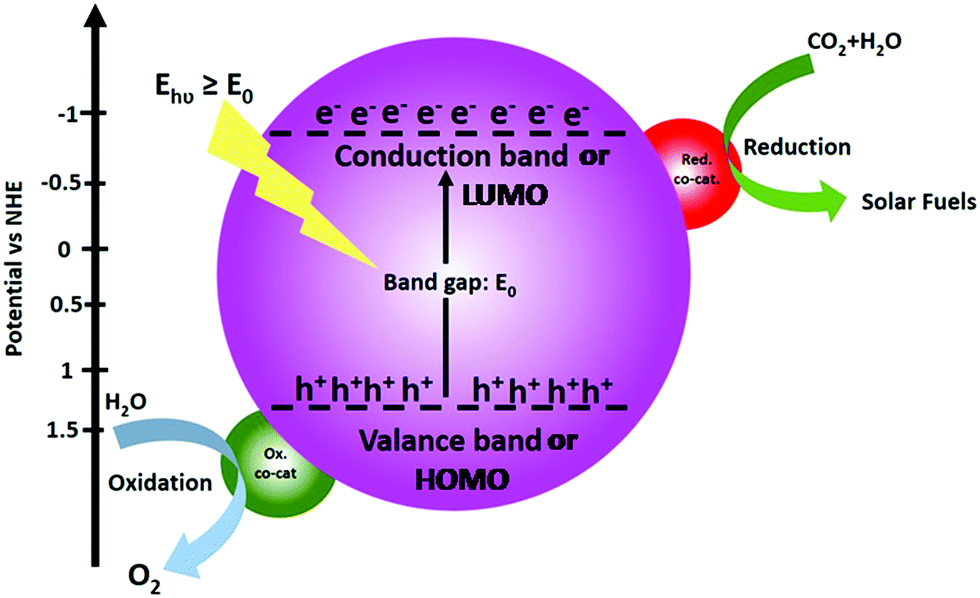 | ||
| Fig. 1 Schematic energy diagram of photocatalytic water splitting and CO2 reduction reactions. | ||
There are four critical processes in the operation of photocatalytic reduction including, light-harvesting, charge separation, redox reactions, and product desorption. In this process, the absorption of light by the photosensitizer generates electron–hole pairs which can either undergo the desired charge or turn back to the ground state by the radiative or non-radiative process (recombination). The photo-excited electron is used in H2O and CO2 reduction reactions, meanwhile, the holes in the valence band are consumed by oxidation of H2O to produce O2.18 For both photocatalytic H2 production from H2O and CO2 reduction, the CB level/LUMO orbital must be more negative than the H2 evolution potential and the reduction potential of the desired CO2 conversion products (Fig. 1). In addition, the VB level/HOMO orbital in both systems should be more positive than the oxygen generation potential.
As far as the reduction products are concerned, CO2 can be converted to a variety of chemicals such as formic acid, carbon monoxide, methanol, C2+ alcohols, methane etc. (Table 1). The product diversity of CO2 reductions largely depends on the reaction conditions, types of catalyst and the number of electron and proton transfers. Since the selectivity problem of the CO2 reduction product is much more pronounced due to the diversity of CO2 reduction products compared to H2O reduction, photocatalysts with high selectivity are required for photocatalytic CO2 reduction. One of the most important aspects governing catalytic efficiency and product selectivity is the rational design of catalysts with a proper structure since the optimum CB band edge position/HOMO–LUMO gap provide sufficient driving force for photo-driven redox processes. However, the product selectivity and inhibition of H2 formation are governed by several factors such as active sites of catalysts, adsorption of the reactants, desorption/dissociation of the products etc. Meanwhile, the splitting of water consists of two steps of reactions namely oxidation and reduction of water to produce O2 and H2, respectively. Hydrogen and oxygen production from water by visible light requires one or several intermediates able to absorb visible light, to convert the excitation energy to redox energy (charges) and to transfer several electrons to water leading to the formation of H2 and O2. The reaction is a multi-electron transfer process, where the hydrogen requires two electrons reaction (g), Table 1) while oxygen provides four electrons (reaction (f), Table 1).
| CO2 + 2e− + 2H+ → CO + H2O E0 = −0.53 V | (a) |
| CO2 + 2e− + 2H+ → HCOOH E0 = −0.61 V | (b) |
| CO2 + 4e− + 4H+ → CH2O + H2O E0 = −0.48 V | (c) |
| CO2 + 6e− + 6H+ → CH3OH + H2O E0 = −0.38 V | (d) |
| CO2 + 8e− + 8H+ → CH4 + 2H2O E0 = −0.24 V | (e) |
| 2H2O + 4h+ → O2 + 4H+ E0 = 0.81 V | (f) |
| 2H+ + 2e− → H2 E0 = −0.42 V | (g) |
Homogeneous photocatalysis is used to convert solar energy into accessible and clean fuel in a molecular accessible form. The difficulty in this type of catalysis is the separation of the catalyst after the completion of the reaction and the low stability of molecular catalysts. The first homogeneous photocatalytic H2 production was reported by Lehn et al.19 In this review, we will only present the most recent reports of this category. In typical examples of photocatalytic H2 production, an organometallic complex is a catalyst and an organic or organometallic compound is the PS. In order to achieve effective H2 production, the PS must be able to absorb photons from a light source to reach an excited state. Then the excited state is quenched by electrons from an electron donor. This procedure can be achieved in two different ways, a reductive or an oxidative path. During the reductive quenching, an electron is transferred from the SED to the PS. The photosensitizer is reduced and an electron is transferred to the catalyst. In the case of oxidative quenching, an electron from the excited PS is transferred to the catalyst and the oxidized form of the PS oxidizes the SED. Then the catalyst reduces the protons, resulting to H2 production.
Heterogeneous photocatalysis usually use photocatalytic system with fulfilling fundamental requirements: (1) high light-harvesting efficiency; (2) fast charge separation; (3) slow charge recombination rate, could have the potential to address these challenges. Since the first breakthroughs came with a photoreduction of CO2 in an aqueous suspension catalyzed by diverse semiconductors20 and an evolution of H2via photoelectrochemical water splitting on TiO2,21 researchers have concentrated their efforts on developing effective photocatalysts. Most common photocatalysts are based on transition metals or semiconductors such as TiO2, ZnO, BiVO4, ZrO2, WO3, ZnS, CdS, etc.22,23 However, the photocatalytic performance of these semiconductors in their pristine form is substantially limited by insufficient absorption of visible light with wide bandgap, poor charge separation and concomitant low conversion efficiency. An ideal photocatalyst should match key requirements such as a suitable energy band structure that efficiently absorbs light throughout a large portion of the solar emission spectrum and efficient charge transfer. Reports have indicated that structural and chemical modifications of photocatalyst including metal/ion doping, semiconductor/co-catalyst coupling, dye-sensitization, creating surface disordering, and defects, controlled morphology, etc., are effective ways to increase the photocatalytic performance of photocatalysts and to tune the photocatalytic selectivity.24,25
Among them, heterojunction-based system (p–n junction), Z scheme configuration and dye sensitization of semiconductors are of particular interest in the point of having the potential to address the challenge of the charge recombination rate, solar light harvesting, and selectivity and stability towards the desired products.18 The additional benefit of these structures is to provide an extra photoexcited electron which is advantageous in terms of yield of reduction product. Similar to those proposed these structure modifications, the incorporation of co-catalyst (generally noble metal nanoparticles) which serve as active catalytic centers play an important role in reducing recombination by extracting excited electrons.23
Artificial antenna systems based on organic sensitizers have attracted much attention as light-harvesting photosensitizers (PSs) in artificial energy conversion systems, such as dye-sensitized solar cells (DSSCs).26 Attachment of the photosensitizers on the semiconductor surface increases the light-harvesting ability and facilitates the charge transport properties without altering the catalytic activity of the semiconductor. Since the key process of this system is photo-induced charge separation and transfer, the dye is the pivotal component in artificial photosynthetic devices. The role of dye is multiple as it absorbs light and then directs the charge transfer kinetics and the dye regeneration. The dyes must, therefore, fulfil some essential requirements such as high LUMO energy level than CB of semiconductor, more positive oxidation potential than SED, broad absorption spectrum cover the whole visible region and even the part of near-infrared (NIR) and an anchoring group to bind strongly onto a semiconductor.27
In order to maximize the productivity of the photocatalyst, particularly in the function of solar irradiation, the utilization of photosensitizer to form dye-sensitized photocatalyst (DSP) is an alternative strategy to increase the yield of solar to fuel conversion.27 Considering that nearly 50% of the solar energy lies in the visible-NIR region, a light-harvesting system capable of absorbing in this region is required not only to avoid loss of photons but also to increase charge transfer rates without altering the catalytic activity of the semiconductor. Using a dye sensitizer to enhance the functionality of a photocatalyst has proven to be a highly successful approach to panchromatic sensitization of semiconductors, and it is widely utilized in artificial energy conversion systems such as DSSCs.26 The operating mechanism of DSP is very similar to DSSCs, that is, the excitation of the dye upon visible-light irradiation is followed by charge separation by electron injection from excited dye to the CB of semiconductor and leaving the photogenerated holes in dye sensitizer (Fig. 2). In contrast to DSSCs, the excited electron is used to reduce CO2 or H2O on the semiconductor surface while the oxidized dye sensitizer is regenerated by electron transfer from the SED component.28
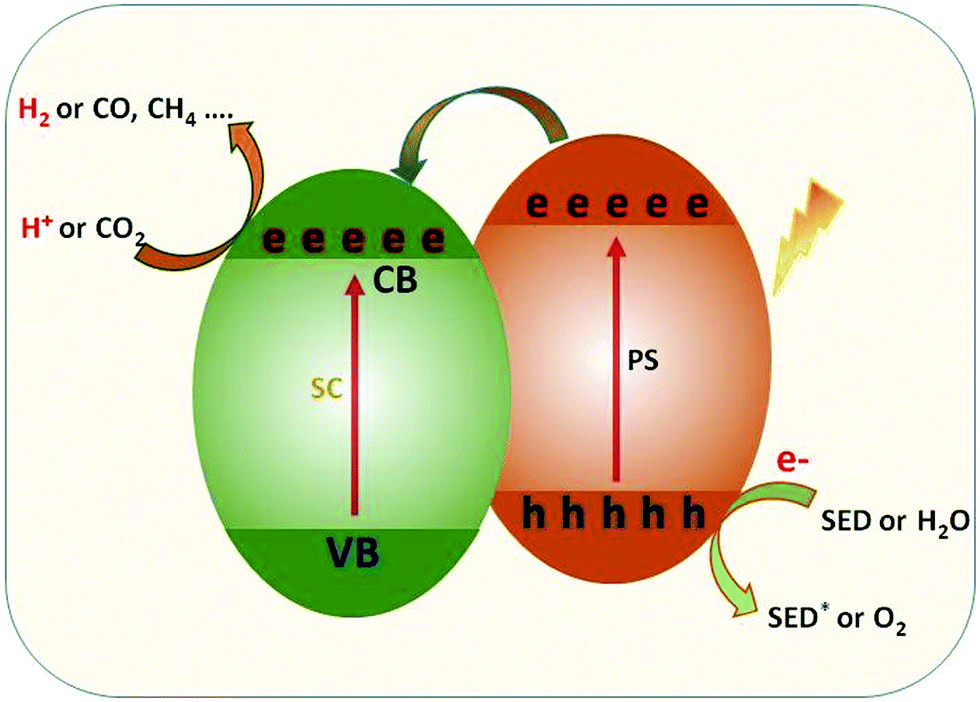 | ||
| Fig. 2 Dye-sensitized photocatalyst, SC: semiconductor, PS: photosensitizer. | ||
Apart from the desired pathway of electron transfer (injection and dye regeneration) processes, there are also undesirable competing processes, namely the recombination of injected electrons with oxidized dye and/or the relaxation of excited electrons in the ground state which are critical for the function of the DSP.25 In this electron-transfer process, SED plays an important role, not only restricting the recombination between the injected electrons and dye cation but also suppressing the photocatalytic O2 production half-reaction. Similarly, the incorporation of co-catalyst on the semiconductor appears as a possible way to improve the photocatalytic activity by increasing charge separation and suppressing the charge recombination.27 From the H2 production performance point of view, the loading of semiconductors with a noble metal-based co-catalyst, which serves as an active centre, such as Pt, Au, Pd, Rh etc. is required to increase the photocatalytic H2 production efficiencies. The large work functions and better conductivity of noble metals accelerate photocatalytic reduction reactions by extracting photoinduced electrons from the semiconductor surface.29
Dye-sensitized photoelectrochemical cells (DSPECs) towards proton as well as CO2 reduction have attracted extensive scientific attention.30–33 The development of molecular-based DSPECs opened the way to the utilization of porphyrins and phthalocyanines in this field. These systems are designed to perform a reduction catalytic reaction at the cathode like H2 production or CO2 conversion while an oxidation process like O2 production is performed at the anode. One target of such systems is to create water-splitting devices with the benefit of separately H2 and O2 production.34,35 Moreover, the big advantage of DSPEC schemes is that no sacrificial agent is needed since the necessary electrons for the reduction are provided from the oxidation process through the external circuit. On the other hand, a common drawback of these systems is the need to apply external electrical bias to accomplish the desired conversions, however, there are examples where this issue was overwhelmed and only the energy input from sunlight was necessary. Noteworthy, the nature of the dye sensitizer in all the aforementioned systems strongly affects the activity of the catalysts.
Porphyrins and related compounds occur in nature and are essential for life on Earth as they play a crucial role in relevant biological processes. In natural photosynthesis, chlorophylls are primary components of the light-harvesting antenna systems which mission is to collect most of the sunlight and then efficiently transfer the resulting singlet excitation energy to the reaction centers. Porphyrins (Pors) are macrocycles composed of four pyrrole units interconnected by methine bridges, presenting a highly conjugated 18-π electron aromatic system that accounts for their unique optical and physicochemical properties. Phthalocyanines (Pcs) are synthetic analogous of porphyrins and are both a family of pigments that present similarities between their structures. Pcs are composed of four isoindole units linked by four aza-nitrogen bridges. The Pc macrocycle is also a fully aromatic 18-π electron system. Because of their extraordinary properties, it is not surprising that porphyrins, phthalocyanines and other porphyrinoid derivatives have been the candidate of choice for constructing artificial molecule-based reaction centers that mimic the light-harvesting and charge-separation functions in the photosynthetic reaction center, with the aim to shed light on the basic principles that govern photoinduced charge separation and electron transfer processes.36 Pors and Pcs are thermally and photochemically stable chromophores whose optical and electronic properties can be easily tailored by systematic modification of the peripheral and non-peripheral substituents and the metal centre. Pors and Pcs possess efficient light absorption features in the visible and NIR regions of the solar spectrum, which makes them ideal components of light-harvesting systems in artificial photosynthesis and photovoltaics. In particular, Pors have a very intense absorption in the visible region centered around 400–450 nm (Soret band) and moderate absorption around 500–650 nm (Q band). While Pcs possess a very strong absorption at 300–400 nm (Soret band) and 700 nm (Q band). The possibilities of engineering novel functional materials based on Pcs and Pors macrocycles for applications in artificial photosynthesis are almost endless because of the extraordinary synthetic versatility and robustness of these macrocycles. Over the years, a myriad of porphyrins and analogues have been extensively studied as model compounds that mimic the light-harvesting and charge-separation functions in the photosynthetic reaction center and relevant results have been summarised in excellent reviews.37–40
This review highlights recent advances in the development of porphyrinoid-based photocatalytic H2 production and CO2 reduction schemes. In particular, the roles of porphyrin and phthalocyanine derivatives as dye sensitizers, catalysts and co-catalysts and the influence of their structural aspects on the photocatalytic H2 production and CO2 reduction activity are systematically discussed. A section of this review will also pay particular attention to the use of supramolecular assemblies based on phthalocyanines or porphyrins to promote the visible-light-driven photocatalytic CO2/H2O conversion. The recent advances in the effects of the integration of Pc and Pors as catalytically active building blocks and/or photosensitizers in both homogeneous and heterogeneous photocatalytic systems on CO2 conversion and H2 production efficiency will be highlighted.
2. Porphyrins for photocatalytic H2 production
2.1 Homogeneous systems
In these homogeneous systems all components, the reactants and the catalyst are in the same phase (solution) and are uniformly distributed. In this section, recent examples of homogeneous photocatalytic H2 production will be discussed, based on porphyrin molecules that have been used either as PSs or as catalysts. In the first photocatalytic systems, different porphyrins are utilized as PS and cobalt-based complexes are mainly employed as catalysts. There are also a few examples where the PS and the catalyst are covalently linked. The porphyrins that are used as catalysts are water-soluble cobalt-based molecules.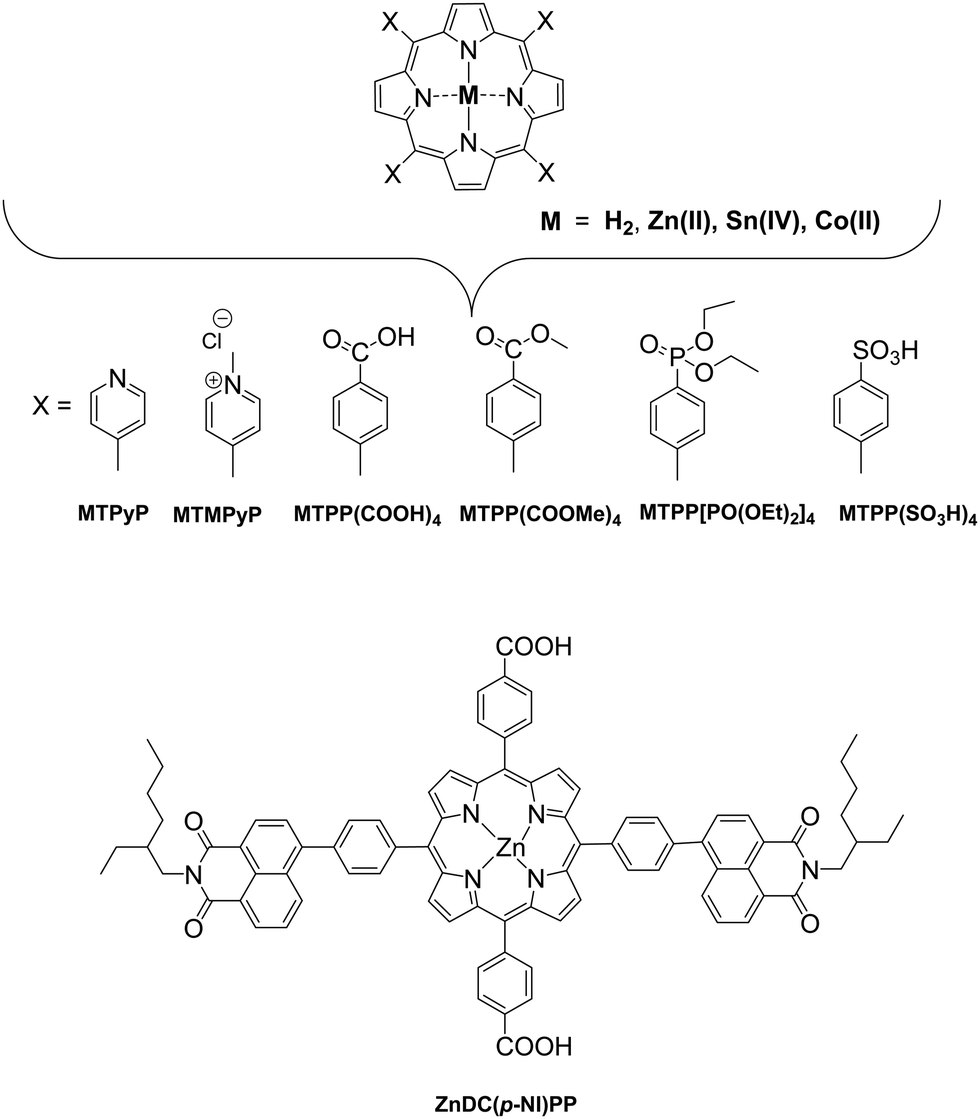 | ||
| Fig. 3 Chemical structures of porphyrins used as photosensitizers. | ||
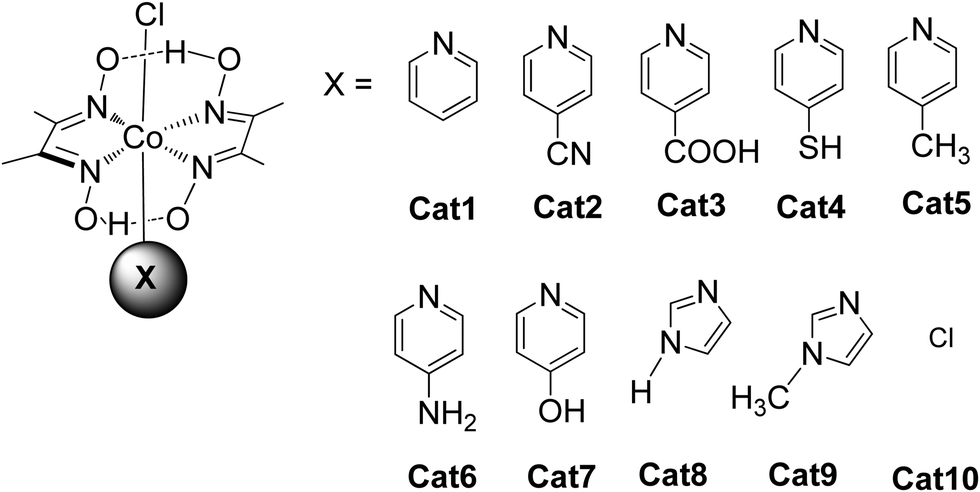
Coutsolelos and co-workers used the Zn porphyrin, ZnTMPyP as photosensitizer and a cobaloxime complex [CoIII(dmgH)2(py)Cl] Cat1 as a catalyst (Fig. 4).41 This system upon visible photo-irradiation (λ > 400 nm) of light produced H2 in MeCN–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with 10% triethanolamine (TEOA) as SED. A maximum of 280 turn over numbers (TONs) was obtained after 25 h of irradiation. Next, the same research group used the same water-soluble porphyrin as PS, ZnTMPyP and a series of cobaloxime catalysts bearing different axial groups Cat1-10 (Fig. 4).42
:1) with 10% triethanolamine (TEOA) as SED. A maximum of 280 turn over numbers (TONs) was obtained after 25 h of irradiation. Next, the same research group used the same water-soluble porphyrin as PS, ZnTMPyP and a series of cobaloxime catalysts bearing different axial groups Cat1-10 (Fig. 4).42
 | ||
| Fig. 4 Chemical structures of various cobaloxime-based catalysts. | ||
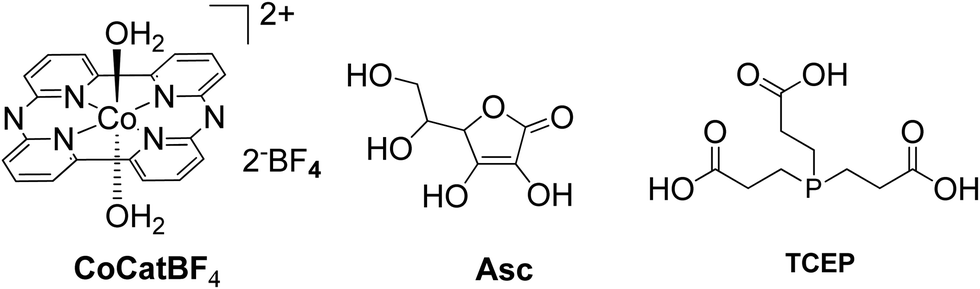
The catalysts were synthesized altering the pyridine and imidazole axial ligands. All photocatalytic experiments were done under similar experimental conditions with their previous work as shown in (Table 2, entries 1a, b–11). More specifically, N-methyl imidazole Cat9 achieved 1135 TONs for H2 production upon 50 h of irradiation, while Cat8 with an imidazole as axial ligand reached 565 TONs. In the case of the catalysts that have pyridine as an axial ligand, the ones with an electron-donating group were proved to be more effective towards H2 production (Table 2, entries 9 and 10). Moreover, upon addition of TiO2 nanoparticles to the less active catalyst Cat3 (40 TON) the activity of the system increased to 223 TONs (Table 2, entry 4). This enhanced activity in the presence of TiO2 may be attributed to the lower electron-withdrawing properties of the new complex or due to the role of TiO2 as an electron reservoir. In a similar work, Bodedla et al. synthesized a meso-substituted porphyrin conjugated with two naphthalimide (NI) moieties (ZnDC(p-NI)PP, Fig. 3) and utilized as photosensitizer in combination with cobaloxime catalyst Cat1.43 ZnDC(p-NI)PP exhibited a H2 evolution rate of 35.70 mmol g−1 h−1 and reached a TON of 5958 (Table 2, entry 11), outperforming the reference systems lacking the NI moieties. The enhanced activity of this system was attributed to the intramolecular energy transfer from the NI donor to the porphyrin acceptor that would promote long-lived photoexcitation states. In addition, Coutsolelos and coworkers extended their research and used Sn metallated water-soluble porphyrin, SnTMPyP as photosensitizer with different cobaloximes as catalysts Cat1, Cat3, Cat7 in a mixture of 1:1 (v/v) MeCN and aqueous TEOA (10%) solution (Fig. 3 and 4).44 The samples were irradiated with a 500 W Xenon lamp at λ > 440 nm and H2 production occurred. The best result obtained with Cat 1 that reached a TON of 150 at pH = 7 after 100 h of irradiation, while when Cat3 was used 53 TON was obtained. Also, when TiO2 was added at the catalyst Cat3 the hydrogen production was increased to 131 TON. In the case of Cat7, no H2 was produced under the same experimental conditions (Table 2, entries 12–15). Exploring more cobalt-based water soluble catalysts Chavarot-Kerlidou and Coutsolelos studied the H2 production of a novel polypyridyl cobalt catalyst CoCatBF4, with the water-soluble photosensitizer ZnTMPyP (Fig. 5).45 The system was able to evolve H2 (443 TONs) under visible light irradiation at pH 4.5 and in the presence of ascorbic acid (AA)/tris-(2-carboxyethyl)phosphine (TCEP) 0.1 M each, as sacrificial electron donor system (Table 2, entry 16).
| Entry | PS (C) | Catalyst (C) | SED (C) | Solvent | Light source | Irr. time (h) | TON | TOF (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1a | ZnTMPyP (4.0 × 10−5 M) | Cat1 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 400 nm | 25 | 280 | 41 | |
| 1b | ZnTMPyP (4.0 × 10−5 M) | Cat1 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 63 | 320 | 8 | 42 |
| 2 | ZnTMPyP (4.0 × 10−5 M) | Cat2 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 11 | 77 | 10 | 42 |
| 3 | ZnTMPyP (4.0 × 10−5 M) | Cat3 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 26 | 40 | 2 | 42 |
| 4 | ZnTMPyP (4.0 × 10−5 M) | Cat3-TiO2 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 29 | 223 | 9 | 42 |
| 5 | ZnTMPyP (4.0 × 10−5 M) | Cat4 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 43 | 425 | 12 | 42 |
| 6 | ZnTMPyP (4.0 × 10−5 M) | Cat5 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 40 | 443 | 9 | 42 |
| 7 | ZnTMPyP (4.0 × 10−5 M) | Cat6 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | — | — | — | 42 |
| 8 | ZnTMPyP (4.0 × 10−5 M) | Cat7 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | — | — | — | 42 |
| 9 | ZnTMPyP (4.0 × 10−5 M) | Cat8 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 50 | 565 | 11 | 42 |
| 10a | ZnTMPyP (4.0 × 10−5 M) | Cat9 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 50 | 1135 | 23 | 42 |
| 10b | ZnTMPyP (4.0 × 10−5 M) | Cat9 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 23 | 290 | 10 | 42 |
| 11 | ZnDC(p-NI)PP (1.0 × 10−5 M) | Cat1 (2.0 × 10−3 M) | AA 0.4 M | THF/H2O (1:9 v/v), pH = 7.4 |
148.5 W LED light | 5 | 5958 | — | 43 |
| 12 | SnTMPyP (8.0 × 10−5 M) | Cat1 (9.8 × 10−4 M) | TEOA 10% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 100 | 150 | 44 | |
| 13 | SnTMPyP (8.0 × 10−5 M) | Cat3 (9.8 × 10−4 M) | TEOA 10% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 100 | 53 | 44 | |
| 14 | SnTMPyP (8.0 × 10−5 M) | Cat3-TiO2 (9.8 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7 |
500 W Xenon lamp λ > 440 nm | 100 | 131 | 44 | |
| 15 | SnTMPyP (8.0 × 10−5 M) | Cat7 (9.8 × 10−4 M) | TEOA 10% (v/v) | MeCN/H2O (1:1 v/v), pH = 77 |
500 W Xenon lamp λ > 440 nm | — | — | 44 | |
| 16 | ZnTMPyP (4.0 × 10−5 M) | CoCatBF4 (4.9 × 10−4 M) | AA/TCEP (100 mM each) | H2O, pH = 4.5 | White LED | 25 | 443 | 45 | |
| 17 | SnTMPyP (4.9 × 10−4 M) | Cat9 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7.0 |
White LED | 96 | 303 | 46 | |
| 18 | SnTPP(COOMe)4 (4.0 × 10−5 M) | Cat9 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7.0 |
White LED | 48 | 128 | 46 | |
| 19 | SnTPP[PO(OEt)2]4 (4.0 × 10−5 M) | Cat9 (4.9 × 10−4 M) | TEOA 5% (v/v) | MeCN/H2O (1:1 v/v), pH = 7.0 |
White LED | 48 | 48 | 46 | |
| 20 | SnTPP(COOH)4 (1.0 × 10−3 M) | Cat1 (1.0 × 10−4 M) | TEOA 60 mM | H2O, pH = 8.5 | White LED | 20 | 1.3 | 47 | |
| 21 | ZnTPP(SO3)4− (8.3 × 10−4 M) | — | TEOA 10% (v/v) | H2O, pH = 7.0 | Mercury lamp, 100 W, λ > 400 nm filter | 70 | 0.03 | 48 | |
| 22 | ZnTPP(SO3)4− (6.7 × 10−5 M) | — | Na2SO3 800 mM | H2O, pH = 8.0 | Mercury lamp, 100 W, λ > 400 nm | 29 | 0.11 | 48 | |
| 23 | ZnTPP(SO3)4− (2.3 × 10−4 M) | — | TEA 2% (v/v) | H2O, pH = 10.0 | Mercury lamp, 100 W, λ > 400 nm | 21 | 1.15 | 48 | |
| 24 | Fe4(ZnL)6 (8.0 × 10−5 M) | — | 4-Mercapto benzoic acid 200 mM | MeCN, TFA 0.03 mM | LED, 40 mW, λ = 470 nm | 2 | 0.4 | 49 | |
| 25 | MPtX2 | — | TEA 30% | MeCN/TEA/H2O (6:3:1 v/v) |
LED, 40 mW, λ = 470 nm | 24 | 15.6 | 0.66 | 50 |
| 26 | Mo2–N(CH3)3 (5.0 × 10−5 M) | — | TEOA 188 mM | THF/H2O (1:1 v/v), pH = 9.0 |
Xenon arc lamp (AM 1.5) | 4 | 640 | 160 | 52 |
| 27 | Mo2–OCH3 (5.0 × 10−5 M) | — | TEOA 188 mM | THF/H2O (1:1 v/v), pH = 9.0 |
Xenon arc lamp (AM 1.5) | 4 | 586 | 146 | 52 |
| 28 | Mo2–OCF3 (5.0 × 10−5 M) | — | TEOA 188 mM | THF/H2O (1:1 v/v), pH = 9.0 |
Xenon arc lamp (AM 1.5) | 4 | 459 | 115 | 52 |
| 29 | MZnP–Ir (1.0 × 10−5 M) | Cat1 (4.0 × 10−5 M) | TEA 800 mM | MeCN/H2O (2:1 v/v) |
White OLED, 148.5 mW cm−2 | 40 | 82 | 53 | |
| 30 | TZnP–Ir (1.0 × 10−5 M) | Cat1 (4.0 × 10−5 M) | TEA 800 mM | MeCN/H2O (2:1 v/v) |
White OLED, 148.5 mW cm−2 | 40 | 246 | 53 | |
| 31 | ZnP–T–Ir (1.0 × 10−5 M) | — | TEOA 800 mM | MeCN/H2O (1:9 v/v) |
White OLED, 148.5 mW cm−2 | 5 | 1.42 mmol g−1 h−1 | 54 | |
| 32 | Ru(bpy)32+ (1.0 × 10−3 M) | CoTMPyP (2.5 × 10−6 M) | AA 100 mM | phosphate buffer 1 M, pH = 7.0 | Xenon arc lamp, 175 W, λ > 400 nm | 4 | 725 | 8.8 | 55 |
| 33 | [Ru(bpy)3]2+ (1.2 × 10−3 M) | CoTPP(SO3H4 (1.5 × 10−6 M) | AA 300 mM | phosphate buffer 1 M, pH = 6.8 | LED, λ = 420 nm | 1.5 | 6410 | 120.8 min−1 | 56 |
 | ||
| Fig. 5 Chemical structure of the cobalt-based catalyst, and the employed SEDs. | ||
All the previous work that was conducted in our group, inspired us to explore further positively, negatively charged and neutral zinc and tin porphyrins as photosensitizers with cobaloximes as catalysts Cat1 and Cat9 for H2 evolution (Fig. 3 and 4).46 We wanted to investigate if different charges on the photosensitizer can influence its ability to produce H2. The optimum conditions of this type of system were found and the positively charged ZnTMPyP reached 1135 TON, where the neutral tin porphyrins SnTMPyP, SnTPP(COOMe)4 and SnTPP[PO(OEt)2]4 reached maximum TONs of 303, 128 and 48, respectively. In all cases, the catalyst that gave the best results was Cat9 at pH 7 in 1:1 MeCN/H2O and TEOA [5% (v/v)] (Table 2, entries 17–19). Moreover, after photophysical and electrochemical studies it was found that the photocatalysis was proceeding mostly via oxidative quenching of the PS. In another report, a water soluble Sn(IV) porphyrin SnTCPP was used as photosensitizer and as catalyst a cobalt-based molecule Cat1.47 The system was able to produce H2 with 1.3 TON under visible light irradiation at pH 8.5 and with TEOA as SED (Table 2, entry 20). Additionally, a single water-soluble porphyrin ZnTPP(SO3)4− was reported to produce H2 in the presence of different sacrificial electron donors and in the absence of a catalyst.48 The photocatalytic system produced H2 with 0.03 TON in TEOA at pH 7, in the presence of an inorganic salt Na2SO3 with 0.11 TON. The best result was obtained upon visible light irradiation in the presence of triethylamine (TEA) with 1.15 TON (Table 2, entries 21–23). In this case, the water-soluble porphyrin had a dual role; it could act as a light-harvesting chromophore and as a hydride supplying co-catalyst, with the formation of a chlorin–phlorin anion intermediate.
In an attempt to prepare more effective systems for H2 evolution, researchers prepared a photocatalytic system that was able to encapsulate the catalyst in a cage-like structure. More specific, a pyridyl phosphole FeFe hydrogenase mimic was encapsulated in a metal–organic cage structure based on zinc porphyrin.49 More specifically, the trihedral cage Fe4(ZnL)6 was composed of six metallated porphyrins that were connected with four Fe(II) corners (Fig. 6). The complex was soluble in organic solvents, therefore upon irradiation in MeCN, using trifluoroacetic acid (TFA) as a proton source in and 4-mercaptobenzoic acid as SED, the system produced low amount of H2 with no decomposition (Table 2, entry 24). When TEA was used as SED H2 was produced, but decomposition of the catalyst was observed. It was found that fast photoinduced electron transfer (0.5 ps) occurred from the cage to the catalyst that was present inside the cage, but the charge recombination of this system was also fast (37 ps).
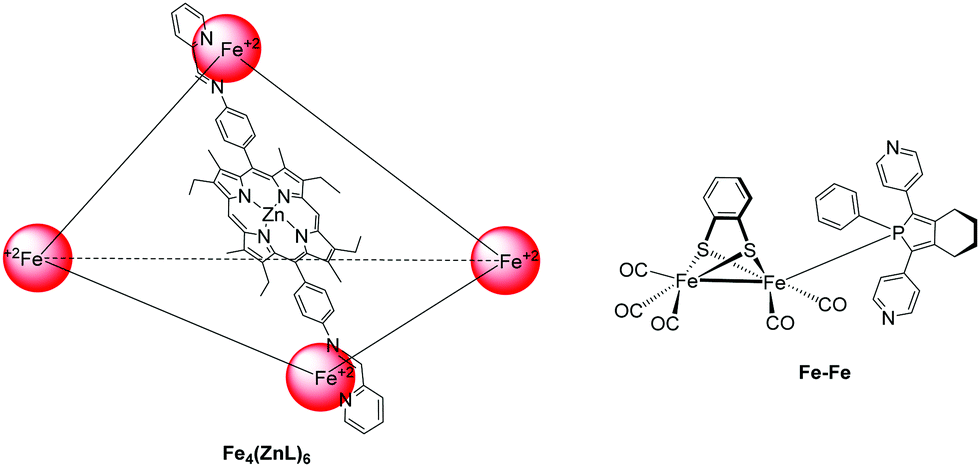 | ||
| Fig. 6 Chemical structure of the cage Fe4(ZnL)6 and hydrogenase mimic FeFe. | ||
A different approach in homogenous photocatalytic H2 production was the preparation of complexes where the photosensitizer and the catalyst are covalently linked. According to this approach, a bimetallic porphyrin MPtX2 was synthesized with Zn, Co or Cu and with platinum at the phenanthroline side with either chloride or iodide ligands (Fig. 7).50 The photoreduction of MPtX2 was performed with a monochromatic LED irradiation at 470 nm and with the use of trimethylamine as SED in the presence of 10% H2O as a proton source. The zinc complex ZnPtX2 was able to produce H2, while Cu and Co derivatives showed no activity. The ZnPtCl2 photocatalyst with the chloride ligands proved to be seven times more effective towards H2 evolution compared to its iodine analogue ZnPtI2, 15.6 versus 2.1 TON, respectively, after 24 h of light irradiation (Table 2, entry 25).
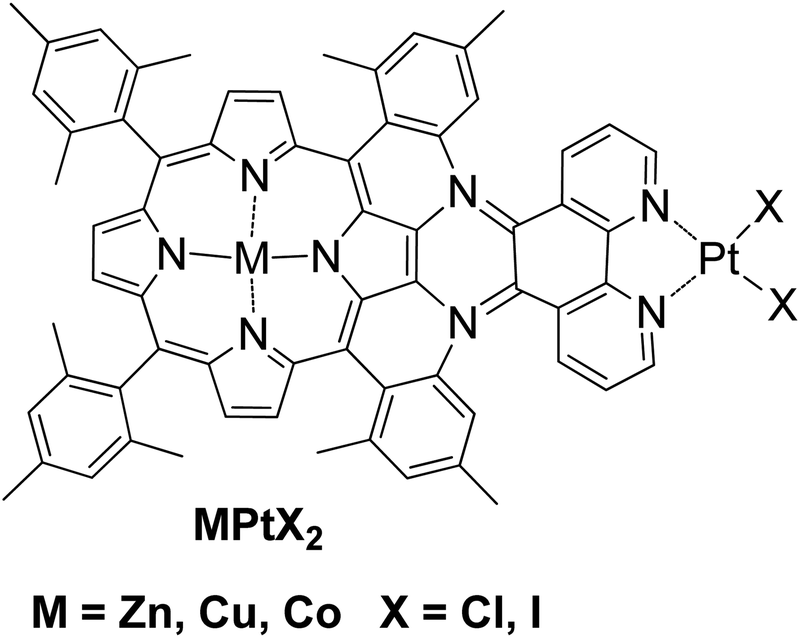 | ||
| Fig. 7 Chemical structure of dinuclear π-extended porphyrin MPtX2. | ||
Manton et al. synthesized a free base and Zn porphyrin–cobaloxime complex MPyCat where the porphyrin was linked to a cobaloxime via a pyridyl cobalt bond (Fig. 8).51 After 20 h of irradiation with different LED light sources in THF/H2O with 33% v/v TEA as SED, no H2 production was detected. In contrary, both complexes MPyCat were able to produce H2 upon electrochemical conditions.
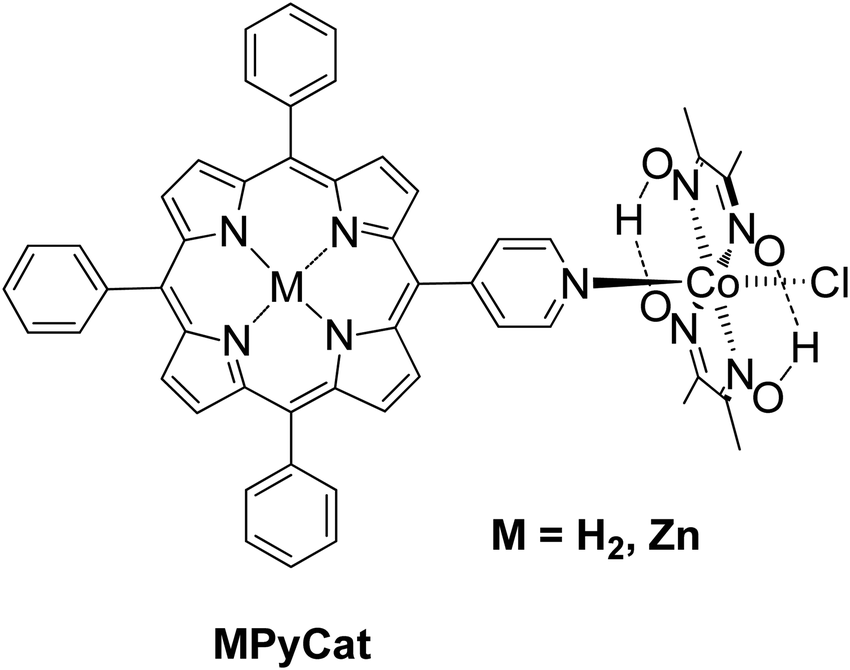 | ||
| Fig. 8 Chemical structures of porphyrin–cobaloxime complex. | ||
Moreover, a series of three photocatalysts were able to reduce protons to H2 and synthesized by combining a zinc porphyrin with a Mo2 quadruply-bonded unit.52 The three complexes differ in the X unit of the aryl group in the para position of the ligand (Fig. 9). These X units were N(CH3)3 as electron-donating groups, OCH3 as intermediate groups or OCF3 as electron-withdrawing groups. Photocatalytic experiments were done with TEOA as SED donor in THF/H2O (1:1 v/v). After irradiation with a Xenon lamp, H2 was produced and its production ability was increased with the electron-donating ability of the X groups, 640 TON for Mo2–N(CH3)3, 586 TON for Mo2–OCH3, 459 TON for Mo2–OCF3 (Table 2, entries 26–28).
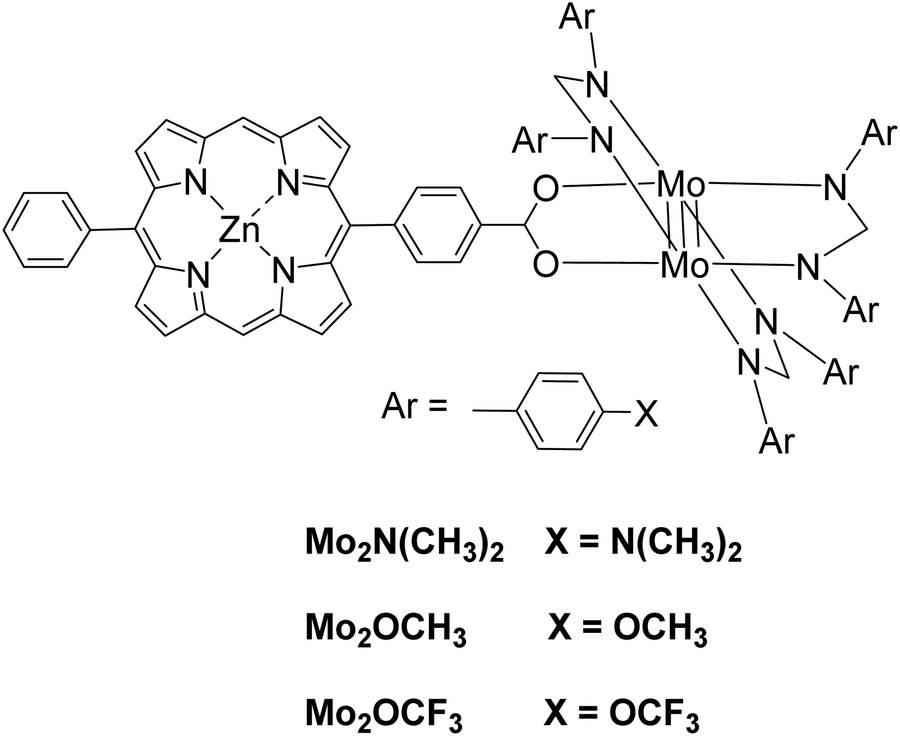 | ||
| Fig. 9 Chemical structure of photocatalytic complexes Mo2X. | ||
In a different approach, a bimetallic Zn–porphyrin covalently linked to Ir(III) was used as PS and a chloro(pyridine)cobaloxime molecule Cat1 as a catalyst in homogenous H2 evolution.53 In this report, two different porphyrins were synthesized, a mono iridium linked MZnP–Ir and a tetra iridium linked TZnP–Ir, where the tetra iridium displayed the highest H2 production 246 TON, upon 40 h of irradiation (Fig. 10). This could be attributed to the stabilization of the triplet state of the porphyrin by efficient intramolecular energy transfer from the excited state of the porphyrin to the cobaloxime catalyst (Table 2, entries 29 and 30). Moreover, the complexes with the iridium were photostable. In a similar work, Zheng et al. synthesized a porphyrin iridium dyad (ZnP–T–Ir, Fig. 10) and applied it in photocatalytic H2 evolution without any co-catalyst.54 The researchers observed a H2 production rate of 1.42 mmol g−1 h−1, which is much higher than the control porphyrin lacking the Ir moiety (Table 2, entry 31).
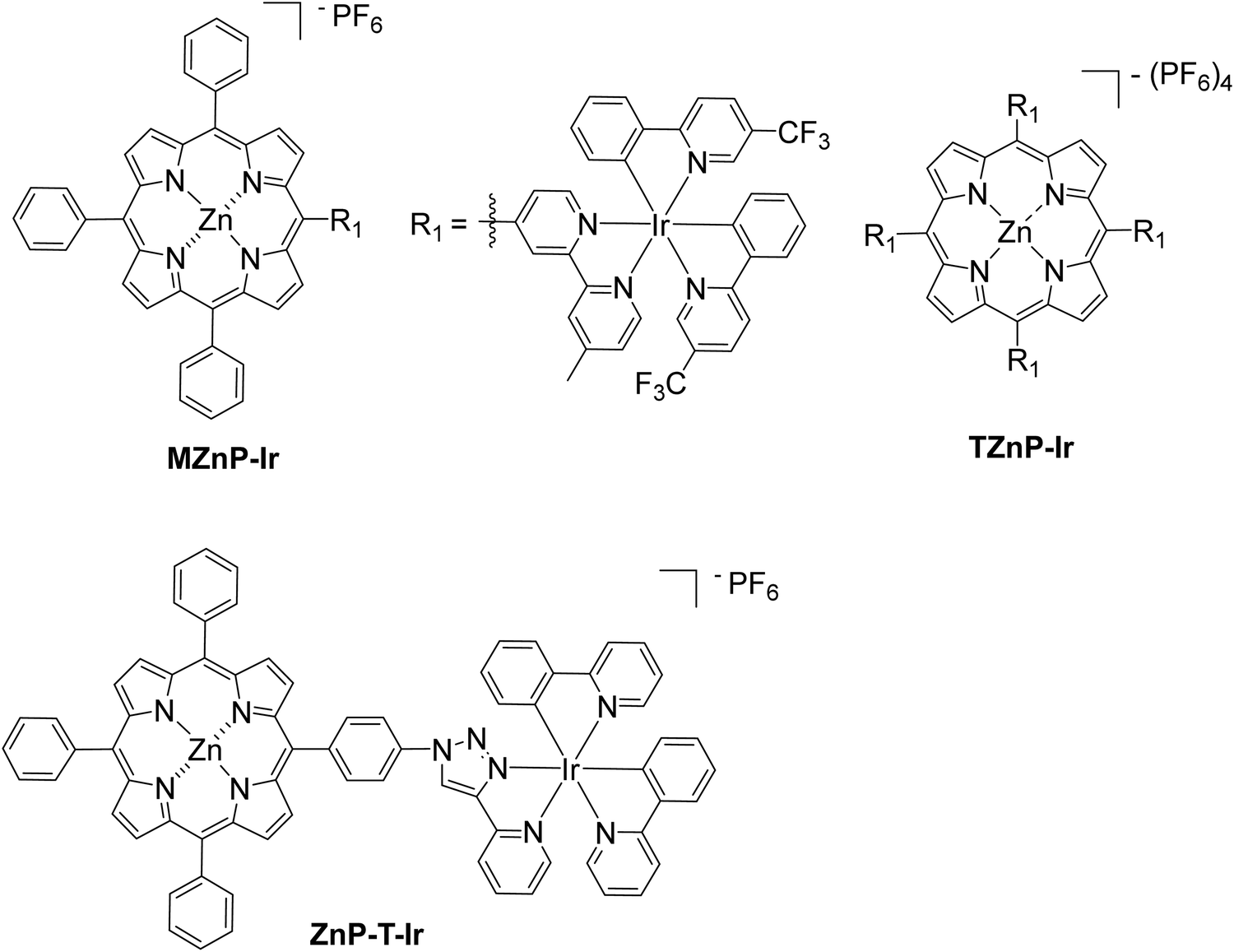 | ||
| Fig. 10 Chemical structure of bimetallic Ir complexes. | ||
 | ||
| Fig. 11 Chemical structure of a ruthenium based photosensitizer. | ||
2.2 Heterogeneous systems
The following section covers all the recent reports of light-driven H2 production in heterogeneous systems where one or more of the fundamental components of the photocatalytic system (SED, catalyst, electron donor, electron acceptor) are not in the same phase as the others. Initially, we present the utilization of self-assembled porphyrin nanostructures in photocatalytic H2 evolution, which was shown as a promising strategy to improve the overall performance of the system. Another form of self-assembly can be considered the metal–organic frameworks (MOFs)57 since they are formed via the self-assembling of organic ligands and metal cations. Porphyrin MOFs will be discussed next followed by the covalent organic frameworks (COFs) towards light-driven H2 generation. Subsequently, we describe reports which combine porphyrins with 2D materials or metal oxides. All the above systems use, as an unavoidable necessity, a sacrificial electron donor. This issue can be overcome by the construction of a photoelectrochemical device where the necessary electrons are provided by an external circuit and H2 is generated at the photocathode.Porphyrin and phthalocyanine molecules are ideal self-assembly building blocks since they possess symmetric molecular structures, planar geometry, chemical and thermal stability, are easily modified and can utilize plenty of supramolecular interactions such as hydrogen bonding, π–π stacking, axial coordination and noncovalent interactions.61 There are various protocols to induce self-assembly to chromophores, including reprecipitation or “good” and “bad” solvent method, ionic self-assembly, peptide-assisted self-assembly, acid-base neutralization assisted surfactant self-assembly, microemulsion-assisted self-assembly and other methods.62 Although the self-assembly of porphyrins is known from the 80's, self-assembled nanostructures of porphyrins had not been employed in photocatalytic H2 evolution for sever years.63 In the following reports, several symmetrical and unsymmetrical porphyrins formed distinctive architectures via their self-assembly and subsequently applied in light-driven H2 production.
Bai and Fan and co-workers64 developed an acid-base surfactant-assisted self-assembly process to fabricate porphyrin nanoparticles of the zinc-tetra(4-pyridyl) porphyrin (ZnTPyP, Fig. 3). For this purpose, they used the cetyltrimethyl ammonium bromide (CTAB) surfactant dissolved in a basic aqueous solution. An acidic solution of the free-base porphyrin derivative (H2TPyP) was injected into the solution of the surfactant and led to the formation of 3D octahedral architectures. By the addition of Zn2+ cations in the self-assembly solution, Zn was inserted into the porphyrin core. Further self-assembly of ZnTPyP was achieved due to the axial interaction between the Zn center and the pyridyl groups, resulting into nanowires (Fig. 12a). These well-defined nanostructures with different morphologies were investigated for their photocatalytic activity using K2PtCl4 as co-catalyst. ZnTPyP nanowires exhibited the highest H2 production activity (47.1 mmol g−1 h−1, Table 3, entry 1), while the H2TPyP nanooctahedra exhibited negligible performance (0.8 mmol g−1 h−1). Noteworthy the nanowires showed stable H2 evolution rate for 75 hours of irradiation and the morphology of the nanostructures was maintained after the photocatalytic reaction.
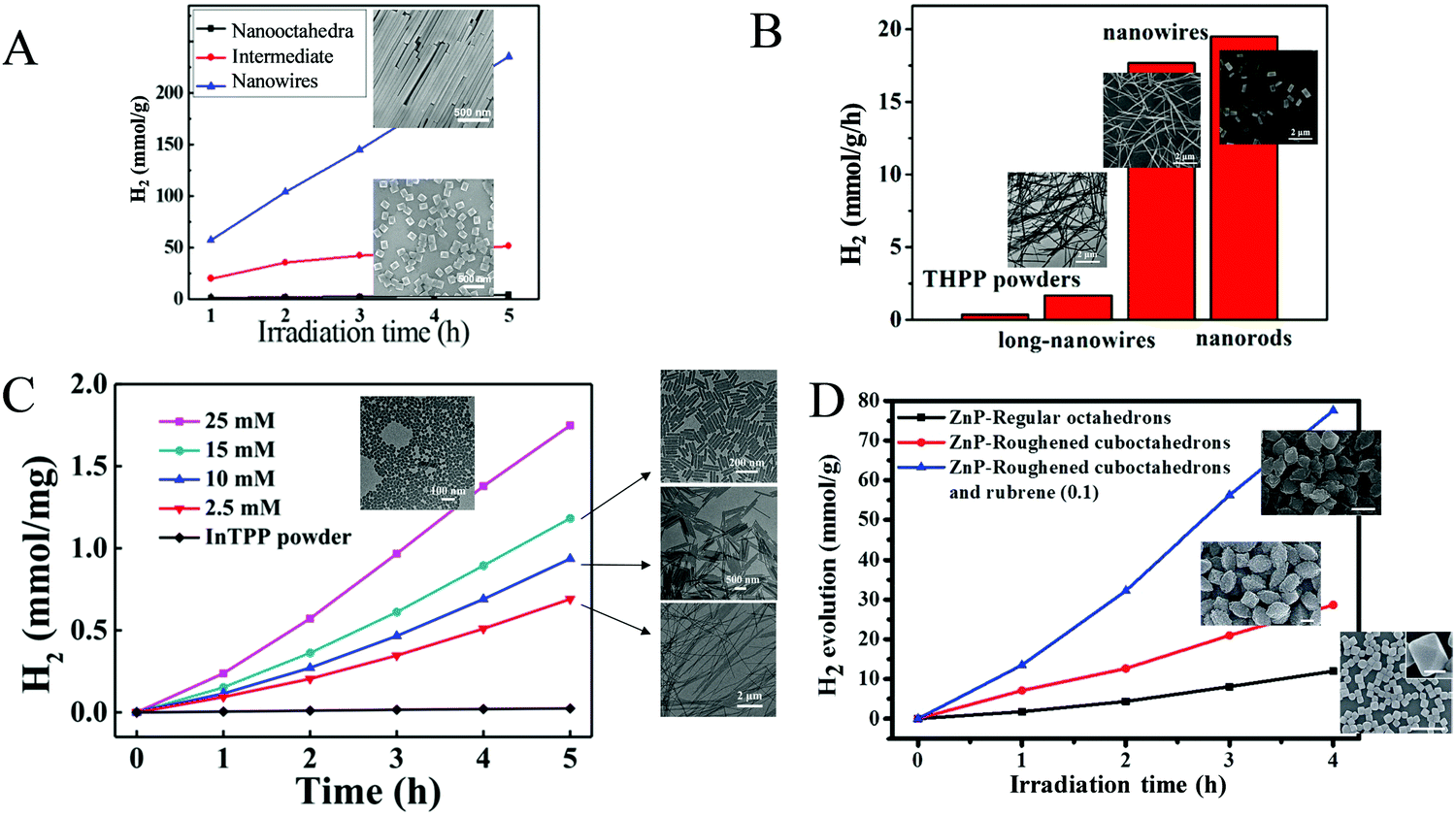 | ||
| Fig. 12 (A) H2 evolution plots of ZnTPyP self-assemblies. Reproduced from ref. 64, with permission from American Chemical Society, Copyright 2016. (B) Amount of H2 evolution of THPP nanowires. Reproduced from ref. 66, with permission from American Chemical Society, Copyright 2018. (C) H2 production experiments of different InTPP nanocrystals. Reproduced from ref. 67, with permission from American Chemical Society, Copyright 2019. (D) H2 evolution plots of ZnTPP self-assemblies. Reproduced from ref. 68, with permission from Royal Society of Chemistry, Copyright 2017. | ||
| Entry | Photocatalysts | Cocatalyst | SED | Solvent and ingredients | Light source | H2 production rate | Ref. |
|---|---|---|---|---|---|---|---|
| a Estimated from the graph the authors presented. | |||||||
| 1 | ZnTPyP nanowires | 1 wt% Pt | AA (0.2 M) | H2O, pH = 3.5 | 300 W Xe lamp, λ > 420 nm | 47.1 mmol g−1 h−1 | 64 and 65 |
| 2 | H2THPP nanorods | 5 wt% Pt | AA (0.2 M) | H2O, pH = 4 | 300 W Xe lamp, λ > 420 nm | 19.5 mmol g−1 h−1 | 66 |
| 3 | InTPP nanorods | 1 wt% Pt | AA (0.2 M) | H2O, pH = 4 | 300 W Xe lamp, λ > 400 nm | 845.4 mmol g−1 h−1 | 67 |
| 4 | ZnTPP cuboctahedrons | — | TEOA 10% | H2O, pH = 8 | 300 W Xe lamp, λ > 365 nm | 0.28 mmol g−1 h−1 | 68 |
| 5 | ZnTPP cuboctahedrons | PVP–Pt (2 × 10−6 M) | TEOA 10% | H2O, pH = 8 MV (3 × 10−4 M) | 300 W Xe lamp, λ > 365 nm | 7.16 mmol g−1 h−1 | 68 |
| 6 | ZnTPP cuboctahedrons/rubrene | PVP–Pt (2 × 10−6 M) | TEOA 10% | H2O, pH = 8 MV (3 × 10−4 M) | 300 W Xe lamp, λ > 365 nm | 19.39 mmol g−1 h−1 | 68 |
| 7 | FeTPP–OH H2O nanoflakes | — | TEOA 10% | H2O | 300 W Xe lamp | 0.75 mmol g−1 h−1 | 69 |
| 8 | TCPP lamellas | — | TEOA 10% | H2O | 300 W Xe lamp | 0.0408 mmol g−1 h−1 | 70 |
| 9 | TCyPP | — | TEOA 10% | H2O | 300 W Xe lamp | 0 mmol g−1 h−1 | 70 |
| 10 | TPyP | — | TEOA 10% | H2O | 300 W Xe lamp | 0.0008 mmol g−1 h−1 | 70 |
| 11 | ZnTCPP nanosheets | 3 wt% Pt | AA (0.2 M) | H2O, pH = 4 | 300 W Xe lamp | 3.4873 mmol g−1 h−1 | 71 |
| 12 | ZnTCPP powder | 3 wt% Pt | AA (0.2 M) | H2O, pH = 4 | 300 W Xe lamp | 0.6543 mmol g−1 h−1 | 71 |
| 13 | TCPP powder | 3 wt% Pt | AA (0.2 M) | H2O, pH = 4 | 300 W Xe lamp | 0.0147 mmol g−1 h−1 | 71 |
| 14 | MnTCPP assemblies | 3 wt% Pt | AA (0.2 M) | H2O, pH = 4 | 300 W Xe lamp | 0.0231 mmol g−1 h−1 | 71 |
| 15 | FeTCPP assemblies | 3 wt% Pt | AA (0.2 M) | H2O, pH = 4 | 300 W Xe lamp | 0.2033 mmol g−1 h−1 | 71 |
| 16 | CoTCPP assemblies | 3 wt% Pt | AA (0.2 M) | H2O, pH = 4 | 300 W Xe lamp | 0.0358 mmol g−1 h−1 | 71 |
| 17 | NiTCPP assemblies | 3 wt% Pt | AA (0.2 M) | H2O, pH = 4 | 300 W Xe lamp | 0.0589 mmol g−1 h−1 | 71 |
| 18 | TCPP/THPP gels | Pt (0.0386 M) | AA (0.15 M) | H2O | 300 W Xe lamp, λ = [440–780 nm] | 1.3 mmol g−1 h−1 | 72 |
| 19 | TCPP gels | Pt (0.0386 M) | AA (0.15 M) | H2O | 300 W Xe lamp, λ = [440–780 nm] | 0.087 mmol g−1 h−1 | 72 |
| 20 | TSPP fibers | Pt (5 × 10−6 M) | AA (0.1 M) | H2O | 350 W Xe lamp | 80 nmol h−1 | 76 |
| 21 | TSPP microspheres | Pt (20 × 10−6 M) | AA (0.1 M) | H2O | 450 W Xe lamp, λ > 400 nm | 80 nmol h−1 | 77 |
| 22 | PNA–TPP nanospheres | Pt (5 × 10−6 M) | AA (0.1 M) | H2O | 300 W Xe lamp, λ > 400 nm | 16.5 nmol h−1 | 78 |
| 23 | FmocFF-ZnTPP fibers | 5 wt% Pt | AA (1 M) | H2O, pH = 4 | LED lamp ring of 40 W | 1.96 mmol g−1 h−1 | 87 |
| 24 | SnPy3P-FF | [Co(dmgH)2(Cl)(Py)] | TEOA 5% | H2O, pH = 7 | LED lamp ring of 40 W | 7 mmol g−1 h−1 | 88 |
| 25 | GdH(TPyP)2 | 5 wt% Pt | AA (1 M) | H2O, pH = 4 | LED lamp ring of 40 W | 13.8 mmol g−1 h−1 | 90 |
| 26 | ZnT(p-NI)PP | 3 wt% Pt | TEOA 10% | H2O | 500 W Xe lamp | 0.973 mmol g−1 h−1 | 91 |
| 27 | ZnT(p-NI)PP nanospheres | 3 wt% Pt | TEOA 10% | H2O | OLED white light | 1.50 mmol g−1 h−1 | 92 and 93 |
| 28 | ZnT(p-NI)TP | 3 wt% Pt | TEOA 10% | H2O | OLED white light | 4.28 mmol g−1 h−1 | 92 |
| 29 | ZnD(p-NI)PP nanowires | 3 wt% Pt | TEOA 10% | H2O | OLED white light | 5.40 mmol g−1 h−1 | 93 |
| 30 | Zn–Mesoporphyrin IX | Pt (20 × 10−6 M) | TEOA (0.1 M) | H2O, MV (0.002 M), NaCl (0.5 M), pH = 6 | 300 W halogen lamp | 222.5 nmol h−1 | 94 |
| 31 | Zn-Protoporphyrin IX | Pt (30 × 10−6 M) | AA (0.002 M) | H2O, MOPS (0.050 M), pH = 7.4 | 300 W halogen lamp | 950 nmol h−1 | 95 |
| 32 | H4DPP2+(Cl−)2 | PVP–Pt (0.15 mg mL−1) | AcrH2 (0.020 M) | MeOH/MeCN 1:1 v/v, TsOH (0.030 M) |
300 W Xe lamp, λ > 710 nm | 53a mmol g−1 h−1 | 96 |
Further investigation of the same porphyrin was conducted using the same self-assembling protocol towards different architectures.65 The researchers employed different surfactants (CTAB, MTAB, SDS) and accomplished to obtained various shapes of self-assembled nanostructures namely hexagonal, nanodisc, nanorod and tetragonal assemblies. By altering the pH of the self-assembling solution, they were able to obtain nanorods of different sizes. These nanostructures were utilized for photocatalytic hydrogen evolution and displayed morphology-dependent (Khexagonal > Knanodisc > Knanorod > Ktetragonal) and size-dependent activity (higher activity for higher aspect ratio length/diameter). An interesting correlation was that the trend of the fluorescence decay time of the different morphologies is consistent with the trend of light driven H2 production performance, indicating that the electron transfer between the zinc porphyrin molecules in the self-assemblies is more efficient.
The same research group utilized the CTAB surfactant with a different free-base porphyrin, namely 5,10,15,20-tetrakis(4-(hydroxyl)phenyl)porphyrin (THPP).66 They achieved the self-assembly of the porphyrin into nanorods and nanowires through nucleation and growth process inside the surfactant micelles. When they applied these nanostructures for photocatalytic H2 evolution using Pt as catalyst, they observed significantly boosted photocatalytic H2 production performance compared to the initial porphyrin powders under visible light irradiation. Notably, the photocatalytic H2 evolution rate of the self-assembled nanostructures was nearly 20 times improved than that of the THPP powder (Fig. 12b). More specifically the nanorods and the nanowires reached 19.5 and 17.6 mmol g−1 h−1 H2 evolution rate respectively (Table 3, entry 2), while the powder exhibited only 0.7 mmol g−1 h−1.
Bai and Fan and co-workers67 modified the self-assembling protocol by introducing an organic solvent to dissolve the porphyrin chromophore while keeping the use of CTAB as surfactant. In detail, they dissolved In(III) meso-tetraphenylporphine chloride (InTPP) in chloroform and then mixed it with an aqueous solution of CTAB to form microemulsion. After the evaporation of the oil phase, InTPP was self-assembled into uniform nanocrystals through noncovalent π–π stacking and hydrophobic interactions. The morphology of the assemblies depended on the concentration of CTAB and they demonstrated that the self-assembled nanocrystals presented enriched photocatalytic H2 evolution within 5 h of irradiation (Fig. 12c), compared to the commercial porphyrin powder which showed negligible activity. The photocatalytic performance is influenced by the aspect ratio (length/diameter), with the highest H2 production rate of 845.4 mmol g−1 h−1 obtained from the nanorods with the smallest aspect ratio of 1.6 (Table 3, entry 3). The higher H2 evolution rate was attributed to the larger active surface areas of these nanorods.
In another report, Liu et al. reported the self-assembly of ZnTPP into quasi-octahedral nanoparticles starting from zinc porphyrin perchlorate (ZnTPP·ClO4).68 After injecting a MeCN solution of ZnTPP·ClO4 in water and let the reaction proceed for different experimental time (from 30 minutes till 20 days) they observed different morphologies regarding the surface of the nanostructures (from smooth octahedrons to rough cuboctahedrons). The researchers showed that the rough ZnTPP cuboctahedrons exhibited higher photocatalytic activity than the regular octahedrons, with the first ones reaching 0.28 mmol g−1 h−1 in the presence of TEOA as sacrificial electron donor and in the absence of other cocatalysts (Fig. 12d). After these ZnTPP nanoparticles were utilized in a ternary system containing TEOA, methyl viologen (MV) as the electron mediator and colloidal platinum (PVP–Pt) as catalyst the photocatalytic performance was significantly enhanced since it reached 7.16 mmol g−1 h−1. The presence of another chromophore, namely rubrene, further improved the activity to 19.39 mmol g−1 h−1 (Table 3, entries 4–6).
Another approach from the same research team utilized again a metallated tetra-phenyl porphyrin and the CTAB surfactant.69 In detail they used iron porphyrin perchlorate (FeTPP·ClO4) to prepare organic nanocrystals (ONCs) via the hydrolysis of FeTPP·ClO4 in the presence of CTAB. They achieved to prepare uniform and regular 0-dimensional FeTPP–Cl, ultrafine 1 dimensional [FeTPP]2O and ultrathin 2D FeTPP–OH H2O ONCs by tuning the pH, and the concentrations of the monomer and of CTAB. After applying these nanostructures in a visible light-driven catalytic system containing TEOA and lacking any other cocatalyst, the scientists revealed the dependence of the photocatalytic performance on the size, the shape and axial ligand of the porphyrin nanocrystals. As it is depicted on Fig. 13a, the FeTPP–OH H2O nanoflakes displayed the highest production frequency (0.75 mmol g−1 h−1, Table 3, entry 7), followed by the FeTPPCl octahedrons (0.48 mmol g−1 h−1) and the [FeTPP]2O nanorods (0.20 mmol g−1 h−1).
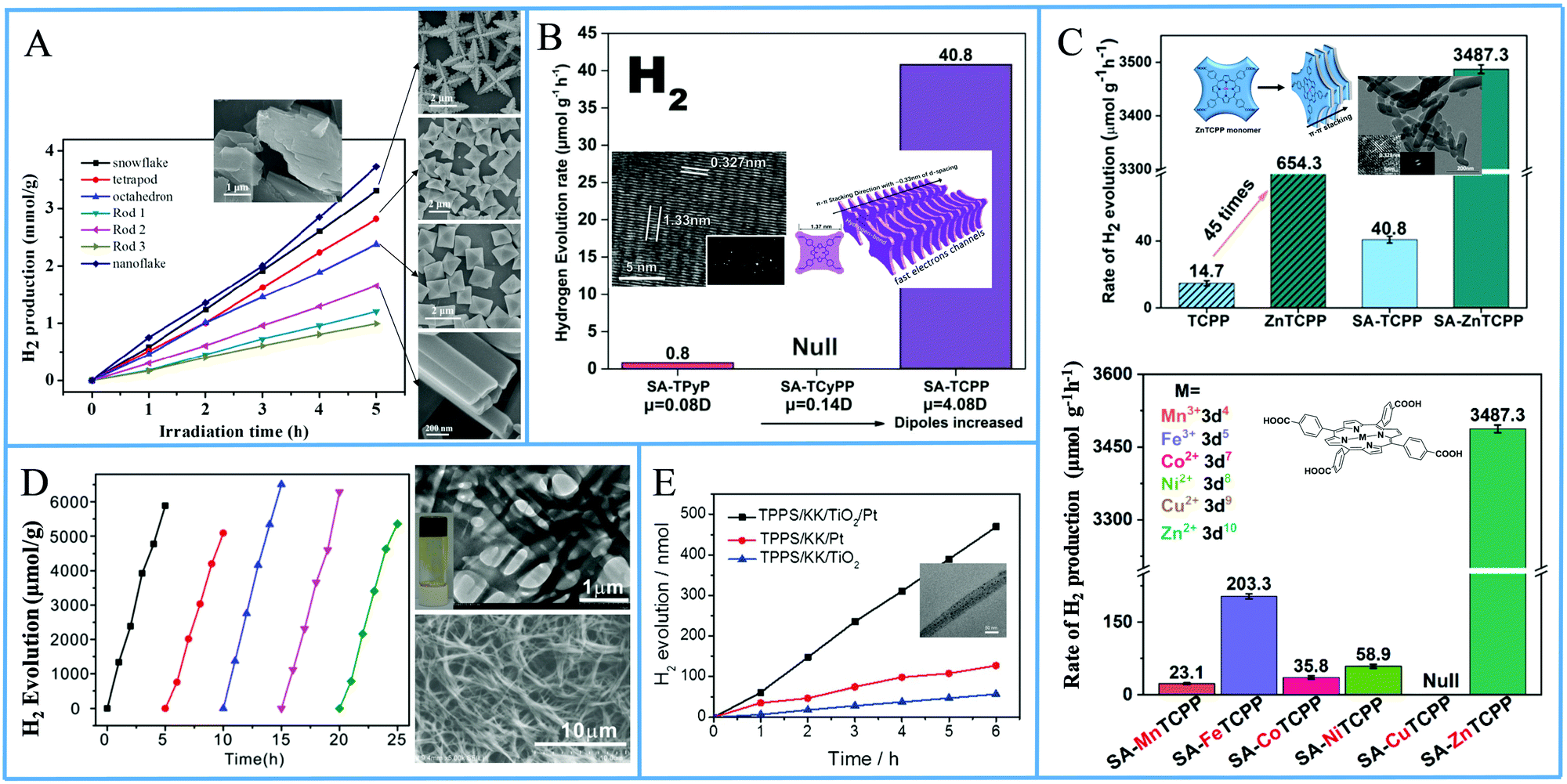 | ||
| Fig. 13 (A) H2 production of different FeTPP nanostructures. Reproduced from ref. 69, with permission from American Chemical Society, Copyright 2019. (B) Amount of H2 evolution of TCPP self-assemblies compared with TPyP and TCyP. Reproduced from ref. 70, with permission from John Wiley and Sons, Copyright 2019. (C) H2 production using self-assembled nanostructures metallated MTCPP porphyrins. Reproduced from ref. 71, with permission from John Wiley and Sons, Copyright 2019. (D) Recycling H2 production experiments of LBG/TCPP/THPP/Pt DMF gels, Reproduced from ref. 72, with permission from Royal Society of Chemistry, Copyright 2020. (E) H2 evolution of TSPP/KK/TiO2/Pt fibers. Reproduced from ref. 76, with permission from John Wiley and Sons, Copyright 2016. | ||
From the above reports, it is clear that symmetric meso-substituted porphyrins with the ability to form self-assembled nanostructures have attracted a lot of scientific interest towards H2 evolution. In this content, Zhang et al. described the ability of self-assembled free-base porphyrins to produce H2 photocatalytically without the addition of any cocatalysts.70 In detail, they prepared self-assembled nanostructures of 5,10,15,20-tetrakis(4-carboxylphenyl)porphyrin (TCPP) and 5,10,15,20-tetrakis(4-pyridyl)porphyrin (TPyP) using a simple base-acid solution method, while the self-assembled 5,10,15,20-tetrakis(4-cyanophenyl)porphyrin (TCyPP) was prepared by the “good–bad” solvent method. TCPP exhibited thin lamellar nanostructures which were able to reduce the aqueous protons to H2 in the presence of TEOA with a hydrogen production rate of 0.0408 mmol g−1 h−1, whereas the TPyP and TCyPP derivatives displayed negligible and null activity respectively (Fig. 13b and Table 3, entries 8–10). Noteworthy the self-assemblies of TCPP were also capable of water oxidation to O2 in the presence of AgNO3, so they achieved to perform both water splitting half reactions.
In a recent publication by the same research group, the free-base porphyrin TCPP was metallated with various metals (Mn, Fe, Co, Ni, Cu and Zn) and self-assembled nanostructures of these porphyrins were prepared afterwards using CTAB (SA-MTCPP).71 The authors applied both the self-assembled derivatives as well as the commercial porphyrin powders in photocatalytic H2 evolution with in situ photo-deposited Pt as cocatalyst and AA as SED at pH = 4. ZnTCPP nanosheets presented efficient H2 evolution reaching 3.4873 mmol g−1 h−1, which was 5 times higher than the performance of the unassembled ZnTCPP powder (0.6543 mmol g−1 h−1) under full-spectrum irradiation. The other metallated porphyrin assemblies, bearing different 3d orbital electron configuration, exhibited significantly lower activity and this was attributed to the differences in the reduction potential. In detail, the best performing ZnTCPP assemblies had the strongest reduction potential while the self-assembled CuTCPP (SA-CuTCPP), which had the lowest reduction potential, presented no H2 evolution (Fig. 13c). The photocatalytic activity of the remaining SA-MTCPP followed the reduction potential trend, showing that the position of the CB of porphyrin self-assemblies determines the H2 production performance (Table 3, entries 11–17).
In a recent publication, Yang et al. employed two different free-base porphyrins and an amphiphilic surfactant to create supramolecular gels which were capable of photocatalytic H2 generation in the presence of Pt co-catalyst.72 More specifically, 5,10,15,20-tetrakis(4-hydroxyphenyl)porphyrin (THPP) and tetra(4-carboxylphenyl)porphyrin (TCPP) were dissolved in DMF along with the known gelator N,N′-bis(octadecyl)-L-Boc-glutamic diamide (LBG) via heating the solution. After cooling to room temperature, gels consisting of fibrous nanostructures were obtained, which were subsequently dried for the photocatalytic experiments. The LBG/TCPP/THPP/Pt system obtained from DMF gels was able to produce H2 with AA as SED, and the total photocatalytic H2 generation after 5 h of visible light irradiation was 6.5 mmol g−1 (Fig. 13d). This performance is translated to 1.3 mmol g−1 h−1 H2 evolution rate and is 15 times higher than the LBG/TCPP/Pt system with only one porphyrin chromophore (Table 3, entries 18 and 19). Emission studies in solution revealed that the TCPP/THPP mixture in DMF solution shows a broadened and red-shifted peak in comparison with that of TCPP or THPP alone, indicating the formation of the TCPP/THPP exciplex in DMF solution. This exciplex was also observed in the gels obtained from DMF and it provides enhanced optical properties for efficient photocatalytic H2 evolution.
In continuation of the presentation of self-assembled porphyrins towards visible light-driven H2 production, another approach is the co-assembly with peptides. Yan's group, which has extensively investigated porphyrin–peptide co-assembly,73–75 moved one step further and applied a co-assembling fiber system composed of a dipeptide and a porphyrin into photocatalytic H2 production.76 Aiming to simulate natural hydrogen-producing photobacteria, they designed an acidic, hot, and mineral-containing (Na+, Ti4+, Pt2+, and so forth) prebiotic soup including L-Lys–L-Lys (KK) and 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin, (TSPP). This bioinspired system begins with the spontaneous self-assembly of the porphyrin and the dipeptide into ordered hybrid hierarchical fibers, which are subsequently self-mineralized with TiO2 and Pt. The authors explained how the positively charged peptide facilitates the TiO2 and Pt NPs self-mineralization on the surface of the fibers and that the mineralized TiO2/Pt nanoparticles constitute the reaction center for H2 production. Under visible-light irradiation, the TSPP/KK/TiO2/Pt fibers exhibited the highest H2 production rate of approximately 80 nmol h−1, while noticeable H2 bubbles appeared in the test tube (Fig. 13e and Table 3, entry 20).61,76
One year later the same group employed the same porphyrin (TSPP) with the Fmoc-protected lysine (Fmoc-L-Lys) instead of the unprotected dipeptide, in order to fabricate self-assembled architectures through the one-step co-assembly of the amphiphilic amino acid and the porphyrin chromophore.77 Fmoc-L-Lys was able to self-assemble into nanofibers via π–π stacking and hydrogen bonding. The negatively charged TSPP co-assembled on the surface of the nanofibers through electrostatic interactions, and these nanofibers were further self-assembled into microspheres. The co-assembled microspheres demonstrated enriched light-harvesting ability and were able to produce H2 photocatalytically using Pt as co-catalyst under visible light illumination (Fig. 14a and Table 3, entry 21). The reaction center was located on the platinum nanoparticles which funneled the energy from the excited porphyrin chromophores and performed the light-induced reduction of protons to molecular hydrogen.
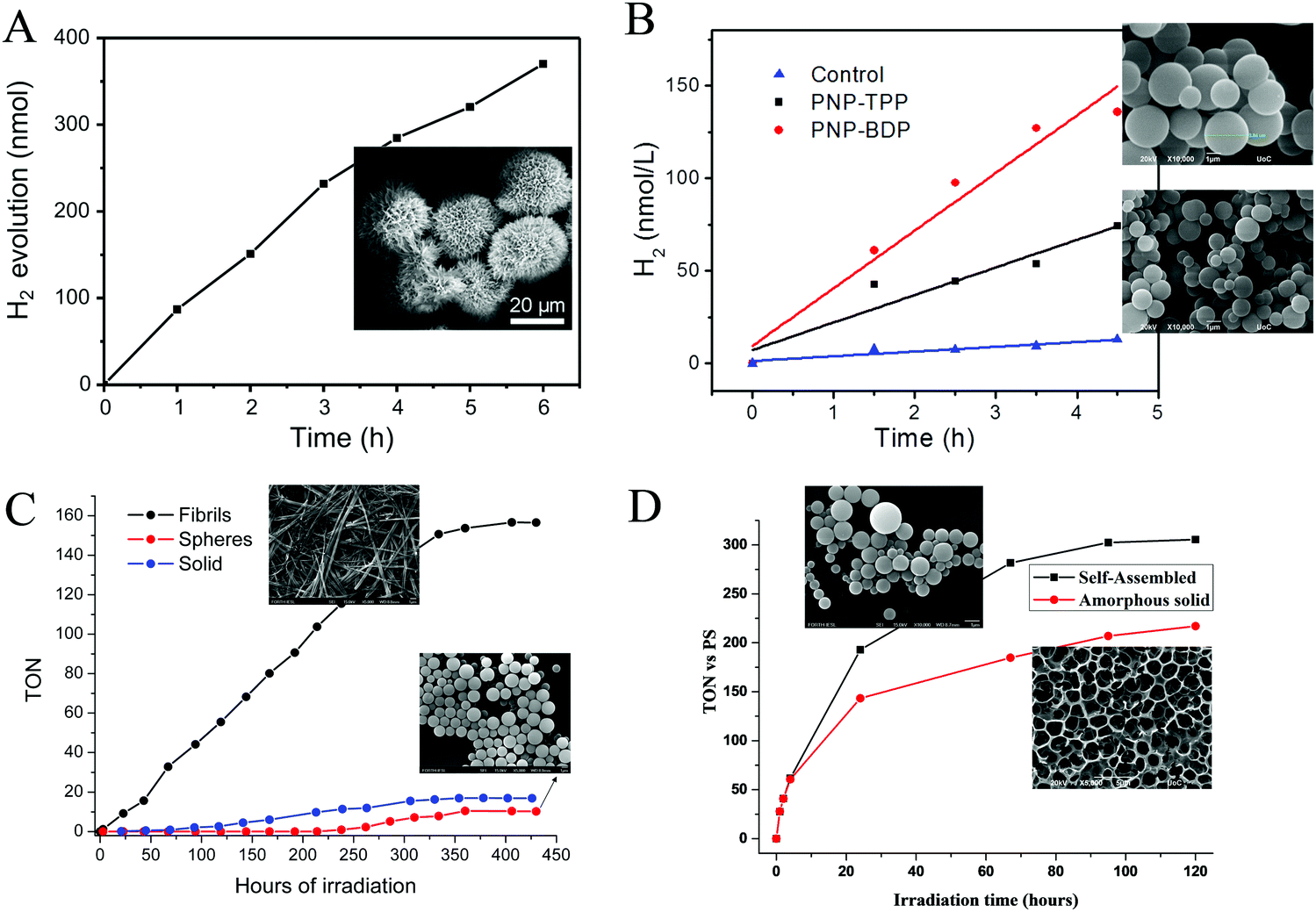 | ||
| Fig. 14 H2 production plots of (A) TSPP microspheres, Reproduced from ref. 77, with permission from Elsevier, Copyright 2017. (B) PNA–TPP and PNA–BDP microspheres, Reproduced from ref. 78, with permission from Royal Society of Chemistry, Copyright 2019. (C) FmocFF-ZnTPP fibers and nanospheres Reproduced from ref. 87, with permission from Royal Society of Chemistry, Copyright 2021 and (D) self-assembled SnPy3P-FF. Reproduced from ref. 88, with permission from American Chemical Society, Copyright 2021. | ||
Following the methodology of peptide co-assembly with porphyrin chromophores, Nikoloudakis et al. utilized a peptide nucleic acid (PNA) covalently linked to meso-tetraphenylporphyrin (PNA–TPP) and a boron-dipyrromethene (BODIPY) molecule (PNA–BDP) with the purpose of conveying self-assembling properties to the chromophores.78 Indeed, the hybrid molecules were able to self-assemble into spherical nanostructures with enhanced light-harvesting properties, using the “good–bad” solvent self-assembling protocol.78,79 Subsequently these nanospheres were applied into a photocatalytic system with in situ mineralized Pt nanoparticles as photocatalysts and AA as electron donor. Noteworthy the PNA–BDP assemblies exhibited higher activity reaching 30.1 nmol h−1 compared to the PNA–TPP nanostructures (16.5 nmol h−1, Table 3, entry 22) as it is presented in Fig. 14b.
The most extensively studied small peptide, famous for its self-assembling properties, is diphenylalanine (Phe–Phe, FF).80,81 Coutsolelos and co-workers have extensively investigated chromophores such as BODIPYs,82 porphyrins,83,84 corroles,85 and polyoxometalates (POMs)86 covalently linked with Phe–Phe and the resulting conjugates retained the characteristic of self-assembly. In this concept, we synthesized FmocFF-ZnTPP and examined its well-defined nanostructures as photocatalysts in H2 production.87 In detail, following the “good” and “poor” solvent method we obtained spherical and fibrillar self-assembling architectures from the same porphyrin molecule, which presented much different catalytic activity towards H2 evolution. After photo-deposition of Pt NPs on the fibrillar nanostructures in the presence of AA as SED, H2 generation was achieved with a rate of 1.96 mmol g−1 h−1 (Table 3, entry 23). The spherical and the amorphous solid displayed negligible photocatalytic activity and verified that the self-assembly mode controls H2 production in this system since fibrils were more efficient than spheres or amorphous solid (Fig. 14c).
In another work of Nikoloudakis et al.88 we covalently connected the diphenylalanine dipeptide with tris(pyridyl)porphyrin in order to transfer self-assembling properties to the resulting hybrid. The pyridyl substitution was chosen since it led to superior photocatalytic activity in previous reports from the same group.44,89 So we prepared the SnPy3P-FF, ZnPy3P-FF and the free-base hybrid and we verified that the metal inside the porphyrin influences the self-assembly mode of the porphyrin–peptide hybrids, since it induces different nanostructures: nanospheres, irregular aggregation and fibrils, respectively. The self-assembled nanostructures and the amorphous solids were employed in photocatalytic hydrogen production using a known cobaloxime complex as molecular catalyst and TEOA as SED. The zinc and free base hybrids did not display any photocatalytic activity, whereas the SnPy3P-FF was able to act as a photosensitizer and produced H2. Notably, the performance of the self-assembled SnPy3P-FF nanospheres was greater compared to the amorphous chromophore, demonstrating the significance of self-assembly towards improving the light-harvesting ability and the photocatalytic properties of the porphyrin. The best performing system exhibited 305 TON after 120 hours of visible light irradiation, while the H2 evolution rate at 24 hours was 7 mmol g−1 h−1 (Fig. 14d and Table 3, entry 24).
Another report from the same team concerned the photocatalytic H2 production of a series of modified Gd-porphyrins in the presence of Pt NPs.90 The Gd bisporphyrinate double decker with pyridyl peripheral groups (GdH(TPyP)2) exhibited the highest activity reaching 13.8 mmol g−1 h−1 (Table 3, entry 25), while the corresponding Gd monoporphyrinate with the same peripheral substitution (Gd(TPyP)acac) was totally inactive. In this study, the authors were able to obtain different supramolecular architectures by altering the solvent mixture and the peripheral substitution, however, the different nanostructures had no effect on the photocatalytic performance of the complexes.
Porphyrin photosensitizers in combination with Pt nanoparticles have been also reported without the formation of self-assemblies or aggregates. Bodedla et al. synthesized meso-substituted porphyrins conjugated with four naphthalimide (NI) moieties.91 The light harvesting properties of the isomeric porphyrins varied with the position of NI onto the phenyl attached to the meso-position of porphyrin ring, which affected the H2 production activity in the presence of Pt co-catalyst. In the photocatalytic system, the porphyrin was suspended in an aqueous TEOA solution and Pt was loaded by the in situ photoreduction deposition method. It was found that the para-substituted porphyrin isomer ZnT(p-NI)PP exhibited the higher H2 production rate of 0.973 mmol g−1 h−1 (Fig. 15a and Table 3, entry 26), which was 200 times enhanced compared to ZnTPP, showing the importance of intramolecular energy transfer from the naphthalimide moiety. Moreover the corresponding free base derivative T(p-NI)PPH2 displayed only 0.013 mmol g−1 h−1 rate, verifying that the zinc metalation was also essential.
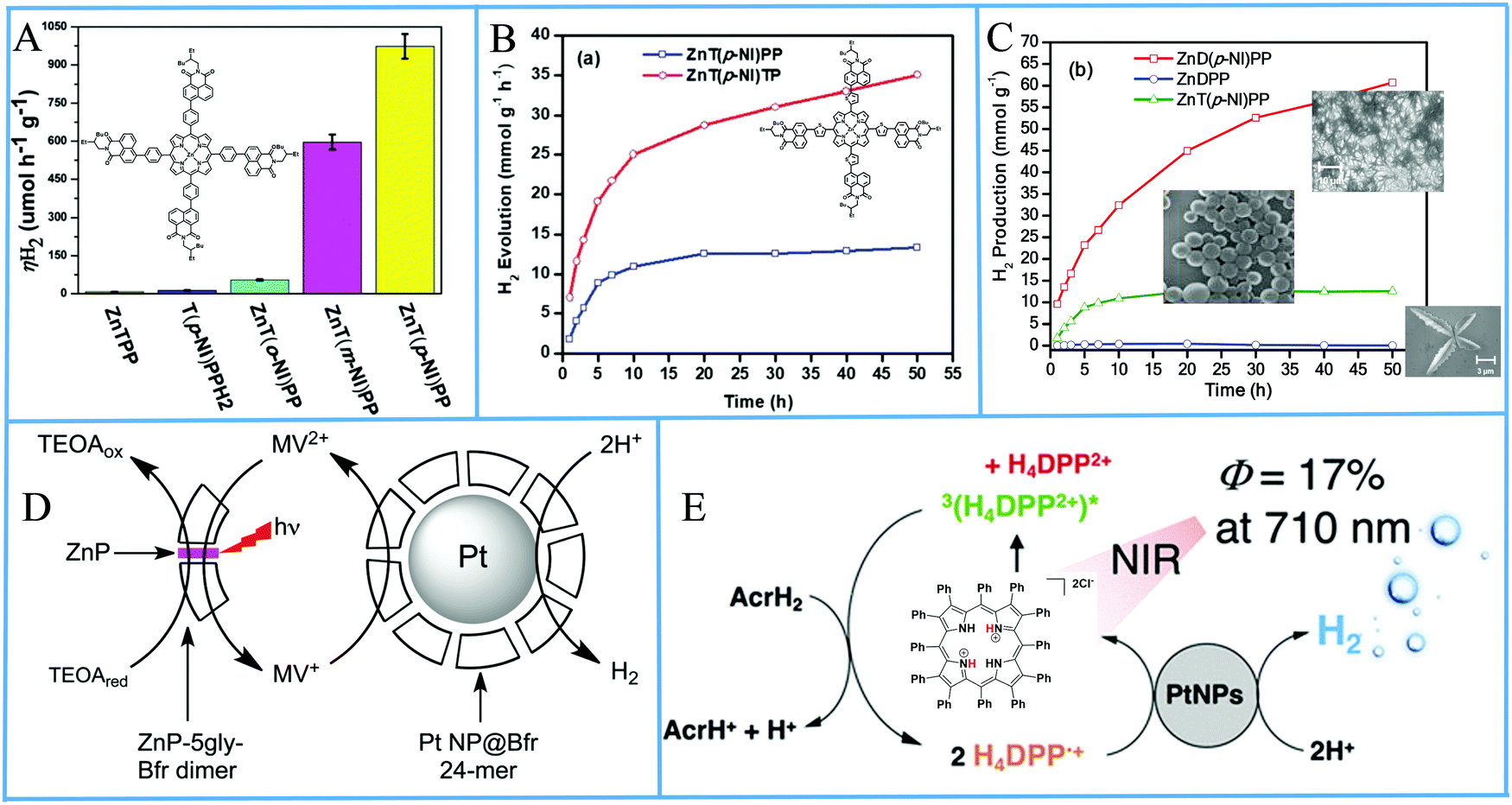 | ||
| Fig. 15 (A) amount of H2 evolution of tetra-naphthalimide substituted porphyrins, Reproduced from ref. 91, with permission from Royal Society of Chemistry, Copyright 2018. (B) H2 evolution plots of tetra-naphthalimide porphyrin with thiophene π-linker, Reproduced from ref. 92, with permission from Royal Society of Chemistry, Copyright 2020. (C) H2 evolution plots of NI-porphyrin self-assemblies, Reproduced from ref. 93, with permission from American Chemical Society, Copyright 2020. (D) Proposed H2 production diagram of the Bfr–porphyrin dimer and Pt NPs-Bfr system, Reproduced from ref. 94, with permission from Royal Society of Chemistry, Copyright 2016. (E) Schematic representation of photocatalytic H2 production by H4DPP2+(Cl−)2. Reproduced from ref. 96, with permission from American Chemical Society, Copyright 2020. | ||
The same research group modified the π-linker between the porphyrin and the naphthalimide moieties by inserting an electron-rich thiophene group instead of a phenyl linker.92 They synthesized the ZnT(p-NI)TP which showed superior light-harvesting ability in comparison with ZnT(p-NI)PP. After applying these porphyrins in the photocatalytic system following a similar Pt co-catalyst deposition as above, the scientists observed increased photocatalytic activity with a ∼2.9 times higher H2 evolution rate (4.28 mmol g−1 h−1) for the ZnT(p-NI)TP (Fig. 15b and Table 3, entries 27 and 28). Noteworthy this derivative exhibited also improved stability since it continued to be active for more than 50 hours, while ZnT(p-NI)PP reached a plateau after 20 h.
Bodedla et al. continued modifying their porphyrin-naphthalimide systems and recently utilized the self-assembly of their complexes in order to further improve the overall photocatalytic performance.93 More specifically they synthesized the linear shaped di-substituted zinc porphyrin bearing two naphthalimide moieties (ZnD(p-NI)PP) which was able to self-assemble into nanowires. The scientists mention that in their previous reports they did not study the solid phase morphology of the porphyrins and that ZnT(p-NI)PP self-assembled into nanospheres in the photocatalytic system. The ZnD(p-NI)PP nanowires clearly outperformed the tetra- naphthalimide substituted porphyrins because it exhibited a hydrogen evolution rate of 5.4 mmol g−1 h−1 (Table 3, entry 29) in the presence of Pt co-catalyst, which was 3.6 times higher than the ZnT(p-NI)PP nanospheres (Fig. 15c).
Kurtz's group utilized a protein scaffold that incorporates bioinspired porphyrins in order to promote the nucleation of Pt NPs.94 In detail, they incorporated zinc–protoporphyrin IX and zinc–mesoporphyrin IX in the place of heme in the iron storage protein bacterioferritin (Bfr) by adding the porphyrins to the E. coli expression medium, resulting into Bfr–porphyrin dimers with two protein moieties sandwiching a zinc porphyrin. Subsequently, Pt nanoparticles were merged into the Bfrs by reduction using K2PtCl4 and NaBH4, with half of the dimers surrounding platinum nanoparticles (Fig. 15d). The best performing system, containing both Pt-free Bfr–porphyrin dimer and Pt NPs-Bfr along with TEOA as electron donor and MV as electron relay, reached 0.89 μmol H2 after 4 hours of white light irradiation which is translated into a TON of 60 (Table 3, entry 30). The researchers showed that the activity of the Zn–mesoporphyrin IX was superior to that of zinc–protoporphyrin IX.
The same group improved their photocatalytic system one year later, by using citrate coated Pt nanoparticles which afforded H2 production without the need of an electron relay.95 Indeed their modified approach reached 5.7 μmol H2 after 6 hours of white light irradiation with the Bfr– Zn–protoporphyrin IX dimer, which is 3 fold higher than the Pt NPs-Bfr–porphyrin system (Table 3, entry 31). The scientists also reported that ZnTMPyP was inactive as photosensitizer following their new protocol with citrate coated Pt NPs and ascorbate as SED, while the use of MV in combination with ethylenediaminetetraacetic acid (EDTA) showed noticeable H2 generation.
From all above examples, one understands that various protocols have been used in order to combine porphyrin PSs along with Pt nanoparticles co-catalysts. Although self-assembly of porphyrins has been extensively studied, there are other ways to construct photocatalytic systems that combine these two constituents. Kotani et al. synthesized a diprotonated saddle-distorted dodecaphenylporphyrin [H4DPP2+(Cl−)2], which absorbs in NIR, and poly(vinylpyrrolidone)-protected Pt NPs (PVP–Pt) via a known procedure.96 Afterwards, the chromophore, the Pt NPs co-catalyst, 10-methyl-9,10-dihydroacridine (AcrH2) as SED and p-toluenesulfonic acid (TsOH) as proton source were applied towards photocatalytic H2 production in 1:1 MeOH/MeCN using λ > 710 nm light irradiation (Fig. 15e). Indeed, the researchers observed NIR light driven H2 evolution and verified the necessity of each component. The photocatalytic performance was proved to be dependent on the concentration of the SED and the amount of Pt NPs while the concentration of the proton source didn’t vary the overall activity. The best performing system reached a TON of 1500 after 3 hours of irradiation and showed an initial H2 production rate of 53 mmol g−1 h−1 (Table 3, entry 32) (estimated from the graph the authors presented).
The first example of porphyrin MOFs (PMOFs) applied in photocatalytic H2 production was reported in 2012 by Rosseinsky and coworkers.98 The researchers prepared a water-stable, microcrystalline porous MOF by reacting AlCl3·6H2O with the free-base TCPP and also synthesized the zinc metallated porphyrin MOF with the same procedure. Colloidal platinum nanoparticles were utilized as catalyst for the photocatalytic experiments in a system that contained the porphyrin MOF, EDTA as SED and MV as electron acceptor which mediates electrons to Pt (Table 4, entries 1 and 2). Two years after this report, Sasan et al. utilised again the same chromophore, ZnTCPP to create a robust zirconium-porphyrin metal–organic framework (ZrPF).99 Noteworthy, this is one of the rare examples where no platinum co-catalyst was used. Instead, the scientists incorporated an organometallic [Fe2S2] complex inside the MOF, with the zinc center of the porphyrin providing binding sites for this complex and stabilizing the resulting material. Photocatalytic H2 evolution experiments were conducted in the presence of AA as SED at pH = 5 and reached 3.5 μmol of H2 production after 120 minutes of visible light irradiation (Fig. 16a and Table 4, entry 3). The di-iron complex acted as a catalyst while the coordination to the porphyrin inside the stable PMOF afforded also high stability to the catalyst.
| Entry | MOF name | Metal node | Linker | Cocatalyst | SED | Solvent and ingredients | Light source | Hydrogen production rate | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Al-PMOF | Al | TCPP | Pt NPs (1 × 10−5 M) | EDTA (0.015 M) | H2O, MV (8 × 10−3 M), pH = 5.5 | 300 W Xe lamp λ > 420 nm | 0.200 mmol g−1 h−1 | 98 |
| 2 | Al-PMOF | Al | ZnTCPP | Pt NPs (1 × 10−5 M) | EDTA (0.015 M) | H2O, MV, pH = 5.5 | 300 W Xe lamp, λ > 420 nm | 0.100 mmol g−1 h−1 | 98 |
| 3 | ZrPF | Zr | ZnTCPP | [Fe2S2] (2 × 10−6 M) | AA (0.02 M) | H2O, Acetate (1.0 M) pH = 5 | 300 W Xe lamp, λ > 420 nm | 1750 nmol h−1 | 99 |
| 4 | Al–TCPP–Pt | Al | PtTCPP | — | TEOA 5% | MeCN, H2O 5% | 300 W Xe lamp, λ > 380 nm | 0.129 mmol g−1 h−1 | 100 |
| 5 | USTC-8(In) | In | InTCPP | 1.5 wt% Pt NPs | TEA 8% | MeCN, H2O 2% | 300 W Xe lamp, λ > 380 nm | 0.341 mmol g−1 h−1 | 101 |
| 6 | HNTM-Ir | Zr | IrTCPP | — | TEOA 8% | MeCN, H2O 2% | 300 W Xe lamp, λ > 400 nm | 0.0076 mmol g−1 h−1 | 102 |
| 7 | HNTM-Pt | Zr | PtTCPP | — | TEOA 8% | MeCN, H2O 2% | 300 W Xe lamp, λ > 400 nm | 0.0567 mmol g−1 h−1 | 102 |
| 8 | HNTM-Ir/Pt | Zr | IrTCPP–PtTCPP | — | TEOA 8% | MeCN, H2O 2% | 300 W Xe lamp, λ > 400 nm | 0.2019 mmol g−1 h−1 | 102 |
| 9 | Ru-TBP | Ru | TCPP/RuTCPP | — | TEOA 19% | MeCN, H2O 4% | 230 W lamp, λ > 400 nm | 0.13 mmol g−1 h−1 | 103 |
| 10 | Ru-TBP-Zn | Ru | ZnTCPP | — | TEOA 19% | MeCN, H2O 4% | 230 W lamp, λ > 400 nm | 0.24 mmol g−1 h−1 | 103 |
| 11 | Pd-PCN-222(Hf) | Hf | PdTCPP | 0.92 wt% Pt NPs | TEOA 20% | MeCN, H2O 2% | 300 W Xe lamp λ > 420 nm | 22.674 mmol g−1 h−1 | 104 |
| 12 | Pd-PCN-222(Hf) | Hf | PdTCPP | — | TEOA 20% | MeCN, H2O 2% | 300 W Xe lamp λ > 420 nm | 2.476 mmol g−1 h−1 | 104 |
| 13 | PCN-222(Hf) | Hf | TCPP | 0.92 wt% Pt NPs | TEOA 20% | MeCN, H2O 2% | 300 W Xe lamp λ > 420 nm | 0.441 mmol g−1 h−1 | 104 |
| 14 | PCN-22 | Ti | TCPP | 3 wt% Pt NPs | AA (0.037 M) | H2O | 300 W Xe lamp λ > 420 nm | 8.52 mmol g−1 h−1 | 106 |
| 15 | PtSA-MNS | Cu | PtTCPP (12 wt% Pt) | — | AA (0.1 M) | H2O, pH = 4 | 300 W Xe lamp λ > 420 nm | 11.320 mmol g−1 h−1 | 107 |
| 16 | PtSA-MNS | Cu | TCPP | — | AA (0.1 M) | H2O, pH = 4 | 300 W Xe lamp λ > 420 nm | 0.002 mmol g−1 h−1 | 107 |
| 17 | ZrTTA-6SH | Zr | H3TTA-6SH | ZnTFPP with 3 wt% Pt NPs | TEOA 10% | H2O | 300 W Xe lamp | 0.110 mmol g−1 h−1 | 108 |
| 18 | ZrTTA-6SH | Zr | H3TTA-6SH | NiTFPP with 3 wt% Pt NPs | TEOA 10% | H2O | 300 W Xe lamp | 0.071 mmol g−1 h−1 | 108 |
| 19 | ZrTTA-6SH | Zr | H3TTA-6SH | FeTFPP with 3 wt% Pt NPs | TEOA 10% | H2O | 300 W Xe lamp | 0.017 mmol g−1 h−1 | 108 |
| 20 | HER-MOF | Hf | ZnTCPP–PtTCPP | — | Phenol (∼0.1 M) | H2O | LED lamp λ = 400 nm | 12000 h−1 |
109 |
| 21 | LP–HER-WOR-MOF | Hf and Zr | ZnTCPP–PtTCPP | — | — | H2O | LED lamp λ = 400 nm | 0.0116 mmol g−1 h−1 | 109 |
| 22 | PCN-223 | Hf | ZnTCPP–PtTCPP | — | CA (0.005 M) | H2O | LED lamp λ = 420 nm | 521 h−1 | 110 |
| 23 | ZnIn2S4@PCN-224 | Zr | TCPP | — | Na2S (0.35 M), Na2SO3 (0.25 M) | H2O | 300 W Xe lamp λ > 420 nm | 0.284 mmol h−1 | 111 |
| 5.675 mmol g−1 h−1 | |||||||||
| 24 | ZnIn2S4@PCN-224 | Zr | TCPP | 8 wt% Pt | Na2S (0.35 M), Na2SO3 (0.25 M) | H2O | 300 W Xe lamp λ > 420 nm | 3.41 mmol h−1 | 111 |
| 25 | PCN-222 | Zr | PtTCPP | — | TEOA 10% | MeCN, H2O 2% | 300 W Xe lamp λ > 400 nm | 0.35108 mmol g−1 h−1 | 112 |
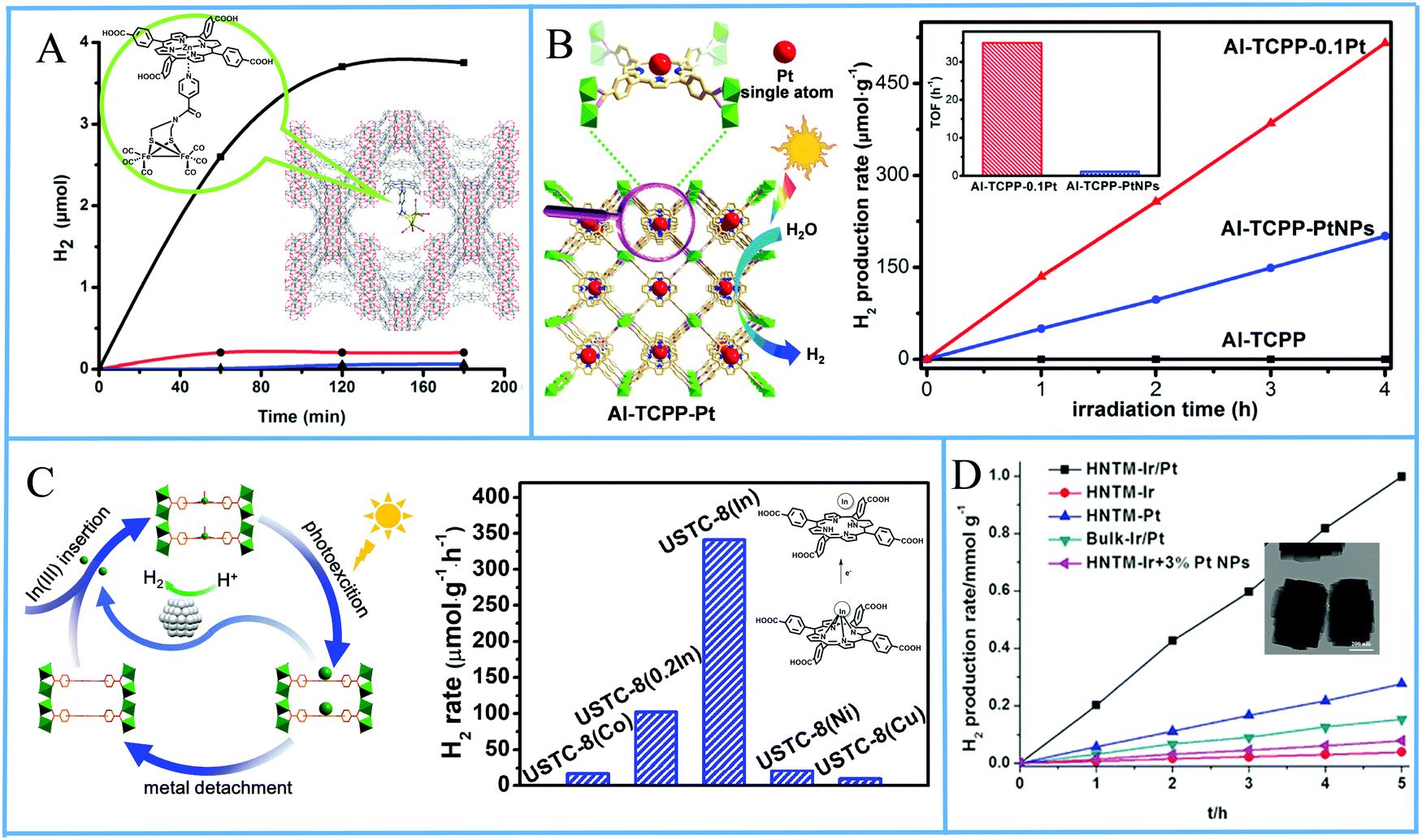 | ||
| Fig. 16 (A) H2 evolution plots of ZrPF with [Fe2S2] complex, Reproduced from ref. 99, with permission from Royal Society of Chemistry, Copyright 2014. (B) Proposed H2 evolution diagram of Al–TCPP–Pt along with the production rate, Reproduced from ref. 100, with permission from John Wiley and Sons, Copyright 2018. (C) Amount of H2 evolution of USTC-8(In) and other USTC-8(M), Reproduced from ref. 101, with permission from American Chemical Society, Copyright 2018. (D) H2 evolution plots of HNTM-M. Reproduced from ref. 102, with permission from John Wiley and Sons, Copyright 2018. | ||
The following years there was no report on light-driven H2 production based on porphyrin MOFs until 2018 when Jiang's groups synthesized an aluminum-based porphyrin MOF which included single platinum atoms and applied it in photocatalytic hydrogen evolution. For this purpose, the scientists utilized the TCPP and AlCl3·6H2O and prepared the Al–TCPP PMOF.100 Subsequently, the PMOF was heated in water along with K2PtCl4 in order to incorporate Pt ions in the porphyrin core to afford Al–TCPP–Pt(II) and then it was reduced at 180 °C in H2 atmosphere to obtain single Pt atoms inside Al–TCPP. For comparison reasons, PVP-protected Pt nanoparticles were also synthesized and applied in the photocatalytic system. The single Pt atoms confined in the porphyrin MOF were able to produce hydrogen under visible light irradiation in the presence of TEOA as SED with a TOF of 35 h−1 which was ∼30 times superior to the performance of Pt NPs stabilized by the same PMOF (Fig. 16b). As far as the H2 production rate is concerned the rate of Al–TCPP–PtNPs was 0.05 mmol g−1 h−1 while the system with single Pt atoms (Al–TCPP–Pt) exhibited 0.129 mmol g−1 h−1, highlighting the potential of such single-atom catalysts in MOFs (Table 4, entry 4).
The same research group created another PMOF based on TCPP and In(III) metal nodes, which was able to act as photosensitizer in the presence of platinum nanoparticles as co-catalysts and produce hydrogen photocatalytically.101 The authors managed to obtain single crystal of the metal organic framework where In(III) had also completely occupied the porphyrin centers and proved that the metal is out-of-plane (∼0.4 Å above the porphyrin plane). Moreover, they synthesized the corresponding In-PMOFs using the metallated CoTCPP, NiTCPP and CuTCPP and they verified from the achieved single crystals that in these MOFs the metal ions stay in-plane of the porphyrin. Interestingly, the InTCPP porphyrin metal organic framework displayed the highest photocatalytic performance (0.341 mmol g−1 h−1, Table 4, entry 5) under visible-light irradiation which is much greater than the activities of the other MTCPP (M = Co, Cu, Ni) PMOFs (Fig. 16c). The In-PMOF that had low occupation of the porphyrin centers with In(III) presented ∼3.4 times decreased photocatalytic activity. The researchers explained that the out-of-plane In(III) ions are eagerly detached from the porphyrin after light excitation and therefore avoid the fast back electron transfer. This characteristic leads to greatly enhanced photocatalytic performance in comparison with the equivalent in-plane metallated PMOFs.
He et al. further explored the immobilization of single metal atoms on PMOFs towards photocatalytic H2 evolution.102 For this purpose, they synthesized the known Zr-porphyrinic MOF hollow nanotubes (HNTM) using TCPP and subsequently incorporated various single noble metal atoms (Ru, Au, Pd, Ir, Pt) in the porphyrin cores. The HNTM-M PMOFs were tested for photocatalytic H2 production from water splitting under visible light irradiation using TEOA as SED and the reaction solvent was MeCN. The non-metallated HNTM exhibited negligible activity and the HNTM-Ir PMOF showed poor photocatalytic performance (0.0076 mmol g−1 h−1). The platinum metallated HNTM presented moderate activity (0.0567 mmol g−1 h−1), while the most efficient system was HNTM-Ir/Pt where both noble metals were utilized and reached 0.2019 mmol g−1 h−1 (Fig. 16d and Table 4, entries 6–8). The last system was stable for three catalytic runs and the morphology of the hollow nanotubes was retained, while no metal NPs were formed.
Lin's group prepared two new PMOFs (Ru-TBP and Ru-TBP-Zn) built from Ru2 paddlewheel building units and TCPP porphyrin.103 The Ru2 units were utilized as catalysts for visible-light-driven H2 production while the porphyrin moieties played the role of the photosensitizer. The photocatalytic activities of Ru-TBP and Ru-TBP-Zn were studied in MeCN with H2O as proton source and TEOA as SED and showed linear H2 productivity with a rate of 0.13 mmol g−1 h−1 and 0.24 mmol g−1 h−1 respectively (Fig. 17a and Table 4, entries 9 and 10). The authors also measured the performance of a homogeneous control system with Ru2 paddlewheels (Ru2-PD) and ZnTCPP which exhibited negligible activity, highlighting the significance the hierarchical organization of photosensitizing ligands and the catalytic units towards photocatalytic H2 production.
 | ||
| Fig. 17 (A) H2 evolution plots and proposed H2 evolution diagram of Ru-TBP PMOFs, Reproduced from ref. 103, with permission from American Chemical Society, Copyright 2018. (B) H2 evolution plots and schematic illustration of Pd-PCN-222(Hf), Reproduced from ref. 104, with permission from Royal Society of Chemistry, Copyright 2019. (C) Amount of H2 evolution of PCN-22 PMOFs, Reproduced from ref. 106, with permission from Elsevier, Copyright 2019. (D) H2 evolution plots of and schematic illustration of PtSA-MNS, Reproduced from ref. 107, with permission from John Wiley and Sons, Copyright 2019. (E) Cross-linking between ZrTTA-6SH MOF and MTFPP porphyrins. Reproduced from ref. 108, with permission from American Chemical Society, Copyright 2020. | ||
Li et al. utilized a known PMOF, the PCN-222(Hf), based on TCPP porphyrin linker metallated with Pd resulting in Pd-PCN-222 through the self-assembly of HfCl4 and PdTCPP. Pt nanorods with a width of ∼3 nm were successfully immobilized into the hexagonal channels (with diameters of 3.7 nm) of Pd-PCN-222(Hf) and the resulting material was employed in photocatalytic hydrogen production.104 The authors selected PdTCPP porphyrin as photosensitizer due to its long lived phosphorescence and visible-light absorption, and encapsulated the Pt NP co-catalyst in the PMOF for efficient electron–hole separation. Indeed, they achieved a synergetic effect between these components and accomplished a H2 evolution rate of 22.674 mmol g−1 h−1 under visible light irradiation (Fig. 17b). The PMOF without the Pt NPs was also examined and exhibited good photocatalytic activity 2.476 mmol g−1 h−1 indicating that the Pd-porphyrin itself has a critical role in the photocatalytic reaction. Noteworthy the isostructural with PCN-222(Hf) was inactive without Pt NPs and exhibited 0.441 mmol g−1 h−1 in their presence (Table 4, entry 11–13).
Wang et al. utilized analogous PMOF nanosheets, which instead of Hf contained Ti metal nodes,105 in photocatalysis with Pt NPs as co-catalyst and water as reaction medium.106 In detail, they synthesized PCN-22 with Ti–oxo clusters and subsequently photodeposited Pt NPs under 300 W Xe lamp irradiation in the presence of AA. The resulting material was able to act as photocatalyst for visible light driven H2 evolution and exhibited a rate of 8.52 mmol g−1 h−1 (Table 4, entry 14). This performance was significantly higher than the system of Pt@TCPP which contained platinum NPs photodeposited on TCPP porphyrin as shown in Fig. 17c. The authors examined the catalytic mechanism and explained that after visible light excitation, the photo-induced electrons are transferred via ligand to metal charge transfer from the LUMO orbital of porphyrin linkers to the conduction band of the Ti–oxo clusters and then to Pt NPs, where photocatalytic hydrogen evolution takes place.
Zhou and Mai and co-workers synthesized ultrathin PMOF nanosheets using PtTCPP porphyrin linker and Cu2–(COO)4 paddle-wheel clusters as metal node in order to prepare a 2-dimensional single-atom-catalyst (SAC) system for photocatalytic H2 production.107 Following this approach, which varies from previous post-modification strategies, the authors created the first example of 2D MOF-based SACs and achieved an ultrahigh Pt loading of 12.0 wt%. The resulting nanosheets exhibited great photocatalytic activity for H2 production (11.320 mmol g−1 h−1) in water under visible-light irradiation and AA as SED (Table 4, entries 15 and 16). Furthermore, the 2D PMOF was drop-casted onto a solid surface, forming thin films which kept their photocatalytic activity and exhibited H2 evolution visible by naked (Fig. 17d).
Diao et al. reported one year later the utilization of a 2D, lamellar Zr-MOF (ZrTTA-6SH) as a host for porphyrin guests towards photocatalytic H2 evolution.108 Notably in this example the porphyrin is not the linker between the metal nodes. The H3TTA-6SH linker bear nucleophilic thiol groups and the porphyrin guests (ZnTFPP, NiTFPP, and FeTFPP), on the other hand, contain fluoro groups to enable the sulfide crosslinking as shown in Fig. 17e, resulting in the encapsulation of the porphyrins inside the MOF. Photocatalytic H2 generation experiments under visible light irradiation in the presence of TEOA as SED and 3 wt% Pt NPs as co-catalyst showed that the material with the ZnTFPP anchored guests exhibit the highest activity (0.110 mmol g−1 h−1) followed by the NiTFPP and then the FeTFPP derivatives (Table 4, entries 17–19).
Wang and co-workers109 constructed an overall photocatalytic water splitting system using MOFs. To achieve this goal, the authors prepared HER-PMOF nanosheets based on ZnTCPP and PtTCPP linkers and WOR-MOF flakes and subsequently integrated these MOFs into liposome (LP) vesicles. The lipid bilayer was selected to separate the oxidative and reductive parts and avoid charge recombination. The HER-MOF consisted of light-harvesting ZnTCPP and catalytic PtTCPP porphyrin linkers and Hf metal nodes, which were initially tested for photocatalytic H2 evolution in the presence of phenol as SED and exhibited a really high TOF (1.2 × 104 h−1, Table 4, entries 20 and 21). The WOR-MOF comprised of appropriately modified [Ru(bpy)3]2+ photosensitizers and Ir-bpy catalytic centres and was incorporated in the hydrophilic part of the LP, while the HER-PMOF was localized in the hydrophobic lipid bilayer of the LP. Noteworthy, the final liposome–MOF assembly (LP-HER-WOR-MOF) accomplished overall photocatalytic water splitting under visible light irradiation without any sacrificial reagents, reaching catalytic activity for H2 836 μmol g−1 over 72 h (0.0116 mmol g−1 h−1).
The same group presented another work,110 where they utilized the same PMOF towards visible light-driven H2 evolution. In their study, the authors compared the molecular catalyst PtTCPP with the corresponding PCN-223 PMOF containing PtTCPP linkers, as well as with the PMOF including both PtTCPP catalytic and ZnTCPP photosensitizing sites (Fig. 18a). Under optimal reaction conditions, the molecular system (in DMF–H2O 8:2) exhibited a TON of 60 after 96 h of visible irradiation using chloranilic acid (CA) as SED, while the PtTCPP PCN-223 showed a TON = 78 in water. Interestingly the PMOF comprising of both PtTCPP and ZnTCPP units displayed much higher photocatalytic activity (TON = 5 × 104), performance which was attributed to the antenna effect between the photosensitizer and the catalyst (Table 4, entry 22).
 | ||
| Fig. 18 (A) schematic illustration of PCN-223 PMOFs containing only Pt or both Pt and Zn sites, along with their photocatalytic performance diagrams, Reproduced from ref. 110, with permission from Elsevier, Copyright 2021. (B) H2 evolution plots of ZnIn2S4@PCN-224, Reproduced from ref. 111, with permission from Elsevier, Copyright 2021. (C) Amount of H2 production of PMOF PCN-222 containing different ratios of PtTCPP and free-base TCPP. Reproduced from ref. 112, with permission from American Chemical Society, Copyright 2021. | ||
Jin et al.111 presented another PMOF example with photocatalytic properties, where they achieved both H2 production as well as degradation of tetracycline hydrochloride pollutant. More specifically the authors prepared ZnIn2S4@PCN-224 comprising of TCPP porphyrin linkers, Zr metal nodes and ZnIn2S4 nanocomposites, which exhibited a hydrogen evolution rate of 0.284 mmol h−1 in absence of Pt, whereas the presence of platinum increased the performance approximately 12 times (3.41 mmol h−1) (Fig. 18b and Table 4, entries 23 and 24). The researchers advertise that their Pt free system possesses a much higher photocatalytic activity than many ZnIn2S4− based photocatalysts even in presence of Pt cocatalyst, result that could be attributed to the hierarchical heterostructure of ZnIn2S4@PCN-224, in which the photoexcited carriers are effectively separated and migrated.
The commonly used PtTCPP porphyrin linker has been investigated also in a work by Lin et al., where the authors prepared the zirconium-based PMOF PCN-222 containing different ratios of PtTCPP and free-base TCPP.112 In detail four different ratios of TCPP/PtTCPP were studied (4:1, 3:2, 2:3, and 0:1) with the best performing system being the one with the fully Pt metallated porphyrin linkers (Fig. 18c). Under visible light irradiation, this system reached 0.35108 mmol g−1 h−1 in the presence of TEOA as SED (Table 4, entry 25). Lowering the ratio of Pt caused a decrease in the overall activity, result that was attributed to the uniformly dispersed catalytic Pt(II) ions.
In a recent report, Chen et al.113 designed a series of porphyrin-based conjugated polymers as depicted in Fig. 19a, where metallated porphyrin monomers were connected through a triple bond via Sonogashira coupling and contained two metal ions, namely Zn(II) and Co(II). The synthesized materials were utilized in photocatalytic H2 production with Pt as co-catalyst and TEOA as the best performing SED. The photocatalytic activity was increased by replacing Zn with Co, while the best performing system was the one with both metal centers (ZnZnP < CoCoP < ZnCoP) (Table 5, entries 1–3). The authors attributed the observed differences in the overall performance to the Co–porphyrin units in the ZnCoP system, which acted as an electron acceptor, promoting the charge transfer from the Zn–porphyrin to the Co–porphyrin moieties and resulting in a more effective charge separation.
 | ||
| Fig. 19 (A) Molecular structures of ZnZnP, CoCoP, ZnCoP and ZnCoP–F COFs, (B) molecular structure of the DhaTph COF, (C) molecular structures of MPor-DETH-COF where M: H2, Co, Ni, Zn, (D) molecular structures of M-2DP where M: = Co, Ni, Zn, Co:Ni 3:1, (E) molecular structures of fused Cu(II) porphyrins, (F) molecular structures and schematic illustration of the Co–porphyrin polymers with different side-chain groups, Reproduced from ref. 120, with permission from John Wiley and Sons, Copyright 2020. (G) Molecular structures and SEM image of the imine gels based on porphyrin molecular precursors, Reproduced from ref. 121, with permission from Royal Society of Chemistry, Copyright 2018. (H) Schematic illustration of porphyrin conjugated polymers with quaternary ammonium side chains towards H2 production. Reproduced from ref. 122, with permission from American Chemical Society, Copyright 2021. | ||
| Entry | COF/polymer name | Cocatalyst | SED | Solvent and ingredients | Light source | Hydrogen production rate | Ref. |
|---|---|---|---|---|---|---|---|
| a Estimated from the graph the authors presented. | |||||||
| 1 | ZnCoP | 1.0 wt% Pt | TEOA 15% | H2O | 300 W Xe lamp λ > 400 nm | 43 μmol h−1 | 113 |
| 2 | CoCoP | 1.0 wt% Pt | TEOA 15% | H2O | 300 W Xe lamp λ > 400 nm | 33 μmol h−1 | 113 |
| 3 | ZnZnP | 1.0 wt% Pt | TEOA 15% | H2O | 300 W Xe lamp λ > 400 nm | 8 μmol h−1 | 113 |
| 4 | ZnCoP–F | — | TEOA 15% | H2O | 300 W Xe lamp λ > 400 nm | 58 μmol h−1 | 114 |
| 5 | ZnCoP | — | TEOA 15% | H2O | 300 W Xe lamp λ > 400 nm | 27 μmol h−1 | 114 |
| 6 | DhaTph | Pt/RGO | EDTA (0.5 M) | H2O | HAL-320W solar | 0.75a μmol h−1 | 115 |
| Simulator λ > 420 nm | |||||||
| 7 | DhaTph e-CONs | Pt/RGO | EDTA (0.5 M) | H2O | HAL-320W solar | 1.25a μmol h−1 | 115 |
| Simulator λ > 420 nm | |||||||
| 8 | CoPor-DETH-COF | 8 wt% H2PtCl6 | TEOA 1% | H2O | 300 W Xe lamp λ > 400 nm | 25 μmol g−1 h−1 | 116 |
| 9 | H2Por-DETH-COF | 8 wt% H2PtCl6 | TEOA 1% | H2O | 300 W Xe lamp λ > 400 nm | 80 μmol g−1 h−1 | 116 |
| 10 | NiPor-DETH-COF | 8 wt% H2PtCl6 | TEOA 1% | H2O | 300 W Xe lamp λ > 400 nm | 211 μmol g−1 h−1 | 116 |
| 11 | ZnPor-DETH-COF | 8 wt% H2PtCl6 | TEOA 1% | H2O, phosphate buffer 0.1 M, pH = 7.0 | 300 W Xe lamp λ > 400 nm | 413 μmol g−1 h−1 | 116 |
| 12 | Co-2DP | 1 wt% H2PtCl6 | TEOA 1% | H2O | Direct sunlight | 1.25a μmol h−1 | 117 |
| 13 | CoNi-2DP | — | TEOA 30% | H2O | Direct sunlight | 75 μmol g−1 h−1 | 118 |
The same research group improved the aforementioned system in a recent work, by introducing perfluorophenyls groups to the porphyrin moieties metallated with Co (19a).114 The resulting ZnCoP–F conjugated polymer displayed greater photocatalytic performance than ZnCoP, which contained only phenyl groups. The authors attributed this improvement to the higher electron-withdrawing capacity of perfluorophenyls in the ZnCoP–F system (Table 5, entries 4 and 5). Moreover, the new system was sufficiently active in the absence of Pt cocatalyst, with the Co ions in the Co–porphyrin moieties serving as single atom catalytic sites. To prove this point the authors synthesized a Co-free analogue (ZnH2P–F conjugated polymer) and found no photocatalytic activity, while both ZnH2P–F and ZnCoP–F conjugated polymers exhibit much better H2 production rate after loading with 1.0 wt% Pt.
In another report, Osakada and coworkers synthesized a covalent organic framework (Fig. 19b) and subsequently prepared 2D-porphyrin polymer nanodisks (of 1 nm thickness) via an exfoliation method.115 In order to achieve this exfoliation the authors utilized the incorporation of metal ions (Mg2+ and Cu2+) in combination with pyridine axial ligands to disrupt the π–π stacking of the porphyrin covalent framework through steric interactions. Both materials, the initial COF (DhaTph) and the exfoliated covalent organic nanodisks (e-CONs), were applied in photocatalytic H2 generation under visible and near-infrared light irradiation using Pt nanoparticles/reduced-graphene oxide (Pt/RGO) as cocatalysts. The later exhibited enhanced photocatalytic activity compared to the initial material, result that might be attributed to the higher surface area between e-CON chromophore and the Pt/RGO cocatalyst (Table 5, entries 6 and 7).
Wang, Wu and co-workers116 synthesized another covalent organic framework (Fig. 19c), having structural similarities with the COF in the previous report. Metalation with Co, Ni and Zn followed and all four 2D PCOFs (MPor-DETH-COF) were evaluated for their activity in photocatalytic hydrogen production. In detail, the authors employed H2PtCl6 as a co-catalyst, TEOA as SED and under visible light irradiation they observed H2 evolution rates of corresponding MPor-DETH-COF, with the following order of M: Co < H2 < Ni < Zn (Table 5, entries 8–11). An effort to explain this trend was provided via DFT calculations based on the tailored charge-carrier dynamics of the synthesized materials.
Babu and coworkers117 designed and synthesized a different 2D polymer (Co-2DP) based on cobalt porphyrin monomers, as depicted in Fig. 19d. This material was utilized as water splitting photocatalyst, since it was able to perform both H2 and O2 evolution reactions under visible light irradiation. Regarding the hydrogen production, the authors used the known Pt co-catalyst and TEOA as SED. Photocatalytic evolution of H2 and O2 was observed for 20 catalytic cycles with insignificant decrease in the efficiency, hence proving the stability of this new material. Control experiments with porphyrin monomers demonstrated the importance of the hydroxyl groups for the bifunctional water splitting ability (Table 5, entry 12).
This material was further developed by the same research team in the following years.118 The authors used different metal ions to metallate the porphyrin 2D-polymer, namely Co(II), Ni(II) and Zn(II), and synthesized the four derivatives described in Fig. 19d. These porphyrin polymers exhibited bifunctional photocatalytic water splitting ability towards both H2 and O2 evolution. Like their previous studies, the best light source was proven the direct sunlight and the best performing material was the one, which contained both Co and Ni in 3:1 ratio inside the porphyrin cores. This bimetallic (CoNi-2DP) porphyrin polymer displayed long-term stability for 15 cycles of H2 and O2 production, it was active for sea and river waters besides pure water and presented significant photocatalytic activity even in the absence of Pt (Table 5, entry 13).
Another type of porphyrin conjugated polymers that were employed in photocatalytic HER were the fused Cu(II) porphyrins reported by Boscher and coworkers.119 The authors synthesized thin films of fused Cu(II) porphyrin which were proved to be active both in electrocatalytic and photocatalytic H2 evolution (Fig. 19e). In detail, the synthesized materials showed superior hydrogen production under visible light irradiation in the presence of EDTA as SED, in comparison to the reference non-fused porphyrin thin films. Noteworthy for the photocatalytic experiments the porphyrin-based thin films were deposited on FTO substrates before irradiation by a solar simulator. The most efficient system was the one which contained fused phenyl substituents reaching 9.2 μmol cm−2 h−1.
In another report that focused both on electro- and photo-catalysis, Xie et al. prepared three water-soluble porphyrin polymers with Co metal cores and different side-chain groups (Fig. 19f).120 The varied side chains were mimicking the enzyme residue groups, which are located close to reaction centers and play significant role in the overall activity. In their photocatalytic studies, the authors used CdSe as photosensitizer, the Co–porphyrin polymers were the catalytic part while ascorbic acid was the SED. Co-2 (Fig. 19f) displayed the highest efficiency reaching a TON = 2.7 × 104 at neutral pH and this result was attributed to the strong interactions of its protonated amino side-chain groups with the negatively charged CdSe photosensitizer.
In another work, Liao et al. prepared a series of imine gels based on porphyrin molecular precursors and investigated their photocatalytic activity towards hydrogen production.121 In detail the imine gels were synthesized by reacting the free-base porphyrin 5,10,15,20-tetrakis(p-aminophenyl)porphyrin (H2TAPP) and its Pd- and Sn- metallated derivatives with the 1,3,5-tris-(4-formyl-phenyl)triazine (TFPT) linker (Fig. 19g). The best performing system was the Pt-doped PdTAPP–TFPT gel, which achieved 1.0744 × 105 μmol g−1 of hydrogen productivity for 120 h. In this work, ascorbate was utilized as SED while water was the proton source. Interestingly, the Pd based wet gel exhibited considerably higher H2 evolution efficiency and stability than the dried aerogel, the free base and the Sn-metallated photocatalytic systems. The authors showed that the PdTAPP–TFPT gel was also electrocatalytically activity towards H2 production after coated on FTO electrodes.
Zhao et al. designed porphyrin conjugated polymers with different coordinated metals and applied them in photocatalytic hydrogen evolution (Fig. 19h).122 The side chains contained quaternary ammonium salts in order to provide good dispersibility in water and allow the formation of nanoscale aggregates, while the porphyrin core was metallated with Ni, Cu, and Pt. The Pt metallated polymers enabled efficient photocatalytic hydrogen evolution of up to 5.39 mmol g−1 h−1 without the assistance of co-catalysts and reached 37.9 mmol g−1 h−1 in the presence of Pt co-catalyst.
 | ||
| Fig. 20 Photocatalytic hydrogen production of a biomimetic system of a [Fe–Fe]-hydrogenases model as a catalyst and a covalently linked graphene oxide with a porphyrin TPP–GO and cystine. | ||
 | ||
| Fig. 21 Chemical structures of [Fe–Fe]-hydrogenase model catalysts. | ||
| Entry | PS (C) | Catalyst (C) | SED (C) | Solvent | Light source | Irr. time (h) | H2 | TOF (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | TPP–GO (4.4 × 10−5 M based on TPP) | [Fe–Fe]1 (4.8 × 10−6 M) | Cystine (1.8 × 10−3 M) | EtOH–water (1:24), pH = 1.5 |
Hg lamp (450 W) (λ > 380 nm) | 5 | 1.61TON | 0.32 | 123 |
| 2 | TPP–GO (4.4 × 10−5 M based on TPP) | [Fe–Fe]2 (4.8 × 10−6 M) | Cystine (1.8 × 10−3 M) | EtOH–water (1:24), pH = 1.5 |
Hg lamp (450 W) (λ > 380 nm) | 5 | 1.80 TON | 0.36 | 123 |
| 3 | TPP–GO (4.4 × 10−5 M based on TPP) | [Fe–Fe]3 (4.8 × 10−6 M) | Cystine (1.8 × 10−3 M) | EtOH–water (1:24), pH = 1.5 |
Hg lamp (450 W) (λ > 380 nm) | 5 | 2.82 TON | 0.56 | 123 |
| 4 | TPP–GO (4.4 × 10−5 M based on TPP) | NiPPh2 (3.0 × 10−5 M) | Ascorbic acid (0.1 M) | EtOH–water (1:24), pH = 4.0 |
Hg lamp (450 W) (λ > 380 nm) | 5 | 1.2 μl H2 | 124 | |
| 5 | TPP–GO (4.4 × 10−5 M based on TPP) | — | TEA (10% vol) | Water | UV-Vis | 6 | 3.8 μmol mg−1 | 125 | |
| 6 | TPP–GO (4.4 × 10−5 M based on TPP) | Pt | TEA (10% vol) | Water | UV-Vis | 6 | 4.6 μmol mg−1 | 125 | |
| 7 | TPP–GO/PVP (4.4 × 10−5 M based on TPP) | Pt | TEA (10% vol) | Water | UV-Vis | 6 | 5.2 μmol mg−1 | 125 | |
| 8 | GO (10 mg in 60 ml) | — | TEOA (10% vol) | Water | 300 W Xe lamp | 8 | 137 μmol g−1 | 126 | |
| 9 | TPPy–GO (10 mg in 60 ml) | — | TEOA (10% vol) | Water | 300 W Xe lamp | 8 | 686 μmol g−1 | 126 | |
| 10 | DPyP–Cr3+–GO (10 mg in 60 ml) | — | TEOA (10% vol) | Water | 300 W Xe lamp | 8 | 928 μmol g−1 | 126 | |
| 11 | DPyP–Co2+–GO (10 mg in 60 ml) | — | TEOA (10% vol) | Water | 300 W Xe lamp | 6 | 1677 μmol g−1 | 127 | |
| 12 | DPyP–Co–GO (10 mg in 60 ml) | — | TEOA (10% vol) | Water | 300 W Xe lamp | 2 | 1093 μmol g−1 | 128 | |
| 13 | GO–Sm–DPyP | — | TEOA (16.7% vol) | Water | 300 W Xe lamp | 8 | 4.39 mmol g−1 | 129 | |
| 14 | [ZnTMPyP]4+/MoS2/RGO | — | TEOA | Water, pH = 7.0 | 300 W Xe lamp | 2560 μmol g−1 h−1 | 130 | ||
| 15 | GO/THPP/PSA | — | TEA | Water | light at λ = 450 nm | 8 | 44.3 μmol g−1 h−1 | 131 | |
| 16 | pTCPP/Pt/g-C3N4 | Pt | TEOA (10% vol) | Water | Hg lamp (450 W) with a cut off filter (λ > 380 nm) | 1208 μmol g−1 h−1 | 132 | ||
| 17 | mTCPP/Pt/g-C3N4 | Pt | EDTA | Water | Light irradiation (λ > 400 nm) | 6 | 48.4 μmol | 133 | |
| 18 | ZnMT3PyP/Pt/g-C3N4 | Pt | AA (50 mM) | Water | Light irradiation (λ > 420 nm) | 8 | 437 μmol g−1 | 134 | |
| 19 | ZnMT4PyP/Pt/g-C3N4 | Pt | AA (50 mM) | Water | Light irradiation (λ > 420 nm) | 8 | 524 μmol h−1 | 135 | |
| 20 | 2%NP/g-C3N4 | Pt | H2O:TEOA (9:1 vol%) |
Water | LED monochromatic lamp (3 W, 420 nm) | 3 | 2297 μmol g−1 h−1 | 137 | |
| 21 | g-C3N4–Cu-TCPP | — | TEOA (10 vol%) | Water | 300 W Xe lamp | 2 | 10 mmol g−1 | 138 | |
| 22 | g-C3N4–Cu-THPP | — | TEOA (16.7 vol%) | Water | 300 W Xe lamp | 6 | 7.50 μmol h−1 | 139 | |
| 23 | g-C3N4–ZnPy-5 | Pt | AA (50 mM) | Water | 300 W Xe lamp (λ >420 nm) | 418 mmol h−1 | 140 | ||
| 24 | g-C3N4–ZnPy-6 | Pt | AA (50 mM) | Water | 300 W Xe lamp (λ >420 nm) | 585 mmol h−1 | 140 | ||
| 25 | g-CNU-TDPP | Pt | TEOA (10%) | Water | 300 W Xe lamp | 7.6 mmol g−1 h−1 | 141 |
The same group performed photocatalytic studies using the same TPP–GO as photosensitizer and a Ni-based catalyst, NiPPh2 in an ethanol aqueous solution (Fig. 22).124 Upon light irradiation with an Hg lamp and a cutoff filter (λ > 380 nm) in the presence of ascorbic acid as a sacrificial electron donor almost 1.2 μl H2 were obtained upon 5 h of irradiation (Table 6, entry 4). The presence of GO and in this case enhances the hydrogen production and the present of all components are necessary to be present for effective hydrogen evolution.
 | ||
| Fig. 22 Chemical structure of a mononuclear nickel catalyst (NiPPh2). | ||
In another report, a MnTPP porphyrin was covalently linked to graphene oxide (MnTPP–GO) and the material was used as photosensitizer in hydrogen evolution experiments (Fig. 23).125 Upon UV-Vis irradiation in the presence of TEA, for 6 h, 3.8 μmol mg−1 of H2 was produced and this amount increased to 4.6 μmol mg−1 in the presence of Pt nanoparticles, under the same conditions (Table 6, entries 5–7). Moreover, the performance of the system was improved further 5.2 μmol mg−1 when a surfactant PVP was added, possibly due to prevention of the aggregation of the photocatalyst. Recycling experiments proved the stability of this type of system.
 | ||
| Fig. 23 Chemical structure of MnTPP–GO photocatalyst. | ||
Instead of altering the surface of GO through covalent bonding, another way is to graft GO with metal ions that can act as interfacial linkers. This type of material can then interact with chromophores such as porphyrins with electrostatic interactions or with coordination bonds. Therefore, different metals (M = K+, Ca2+, Zn2+, Cu2+, Co2+, Cr3+) were added onto GO in order to form the M–GO material where the content of ions was about 10%.126 Then, by adding DPyP porphyrin the DPyP–M–GO material was prepared and by mixing just GO with DPyP the DPyP–GO nanocomposite was synthesized (Fig. 24 and 25). The photocatalytic studies were performed upon irradiation with 300 W Xe lamp as the light source, in the presence of TEA as a sacrificial electron donor. GO, DPyP–GO and DPyP–Cr3+–GO after 8 h of visible irradiation produced 137, 686 and 928 μmol g−1, respectively (Table 6, entries 8–10). The highest photocatalytic activity was performed from DPyP–Cr3+–GO photocatalyst possibly due to the stronger coordination interaction between metal ions and the porphyrin. The porphyrin on the material was used as an antenna system, able to absorb light quite efficiently, while the GO acted as the electron transfer support. The metal ions promote the transfer of the photogenerated electrons and finally improve the photocatalytic performance of the system. Moreover, the same group prepared another metal implanted bispyridyl porphyrin with graphene oxide DPyP–Co2+–GO photocatalyst.127 This material with the absence of a cocatalyst under visible irradiation produced 1677 μmol g−1, compared to DPyP–GO that evolved 1131 μmol g−1 (Table 6, entry 11). The presence of Co2+ ions between the porphyrin and the graphene oxide promotes efficient electron transfer and therefore improves the photocatalytic activity of the material. Next, since metal nanoparticles have a better activity as co-catalysts compared to the metal ions, a photocatalyst DPyP–Co–GO was prepared.128 The photocatalytic activity towards hydrogen production of DPyP–Co–GO was 2 times higher 1093 μmol g−1 compared to DPyP–Co2+–GO after 2 h of irradiation (Table 6, entry 12). This improved activity is attributed to the higher light absorption and the strong interaction between the components. The same group in a later report combined the photoactive porphyrin DPyP with a rare earth ion (Ln) and graphene oxide in order to improve the performance of the photocatalyst (Fig. 26).129 Therefore, GO–Sm–DPyP nanohybrid was prepared and its hydrogen evolution performance was studied. In this structure, Sm3+ is combined with the oxygen groups of GO, when porphyrin was introduced Sm3+ was coordinated with the N atoms of the pyridyl group. Upon, photocatalytic studies the hybrid material GO–Sm–DPyP performed the maximum hydrogen production 4.39 mmol g−1 compared to GO-DPyP 3.09 mmol g−1 and GO 2.58 mmol g−1 after 8 h of irradiation under a Xenon lamp (Table 6, entry 13). This enhanced evolution and stability of the system is attributed to the improved communication and efficient electron transfer between the GO and the porphyrin due to the presence of Sm3+. In another report, a water-soluble porphyrin [ZnTMPyP]4+ was used as a photosensitizer in combination with a MoS2-reduced graphene oxide (MoS2/RGO) as catalyst in order to form a hybrid system for effective hydrogen evolution (Fig. 24).130 The system was irradiated with a Xe lamp in the presence of triethanolamine as a sacrificial electron donor and showed a high H2 evolution rate of 2560 μmol g−1 h−1 at pH 7 in the case of 5:1 ratio of MoS2 to RGO (Table 6, entry 14). According to the mechanistic studies the exited sate of the porphyrin is quenched by the catalyst (MoS2/RGO) through an oxidative quenching.
 | ||
| Fig. 24 Chemical structures of DPyP and [ZnTMPyP]4+ porphyrins. | ||
 | ||
| Fig. 25 Chemical structure of DPyP and DPyP–GO. | ||
 | ||
| Fig. 26 Chemical structure of GO–Sm–DPyP. | ||
Another research group in order to enhance the light absorption combined two different dyes onto GO, a porphyrin molecule (THPP) and pyrene sulfonic acid (PSA) (Fig. 27).131 Therefore, the dyes were non covalently attached onto GO and a nanohybrid material (GO/THPP/PSA) was constructed. This nanohybrid material showed better photocatalytic activity for H2 evolution compared to all other combinations. More specifically, H2 production was 7 times, 2.8 and 1.4 times greater than GO, GO/THPP and GO/PSA, respectively. After monochromatic irradiation at λ = 450 nm for 8 h, GO/THPP/PSA evolved hydrogen with a rate of 44.3 μmol g−1 h−1, in the presence of TEA, as sacrificial electron donor (Table 6, entry 15). This improved hydrogen production of the hybrid material is attributed mainly to the complementary absorption efficiency of THPP and PSA and to the strong interaction of GO with the two dyes.
 | ||
| Fig. 27 Chemical structure of THP and PSA. | ||
As discussed in the introduction part, another efficient photocatalyst for hydrogen evolution is graphitic carbon nitride (g-C3N4). Sensitization of this catalyst with porphyrin dyes can enhance the hydrogen production and improve the stability of the system. In this report, 4-tetracarboxyphenyl porphyrin (pTCPP) was combined with g-C3N4via π–π stacking interactions and studied towards hydrogen evolution in 10% vol TEOA aqueous solution under visible light irradiation (Fig. 28).132 The hydrogen evolution rate of pTCPP/Pt/g-C3N4 was 1208 μmol g−1 h−1 that is higher compared to pTCPP/g-C3N4 and Pt/g-C3N4 that reached 30 and 564 μmol g−1 h−1 respectively (Table 6, entry 16). Hence, the combination of Pt nanoparticles as co-catalyst and pTCPP as a chromophore are important factors for improving the photocatalytic activity of g-C3N4, due to the electron transfer acceleration and the prevention of the recombination of electrons and holes in g-C3N4. Another research group investigated the influence of the position of the carboxylic acid on the porphyrin phenyl ring (pTCPP and mTCPP, Fig. 28).133 The hybrid material mTCPP/Pt/g-C3N4 showed the highest H2 evolution of 48.4 μmol under 6 h of UV-vis and visible light (λ > 400 nm) irradiation in the presence of EDTA (Table 6, entry 17). The system was quite stable, since after four consecutive recycles, the amount of hydrogen evolution hardly changed. In another example, an asymmetric zinc porphyrin ZnMT3PyP, bearing three pyridines at the phenyl rings and a benzoic acid was sensitized onto Pt/g-C3N4 in order to produce hydrogen upon visible light irradiation.134 For comparison another asymmetric porphyrin was used with three phenyl rings. The dye bearing the three pyridine groups showed greater photocatalytic hydrogen production with a rate of 437 μmol g−1 compared to ZnMTPP/Pt/g-C3N4 (293 μmol g−1) under visible light irradiation in the presence of ascorbic acid (Table 6, entry 18). This difference is due to efficient electron transfer from ZnMT3PyP to Pt/g-C3N4. Moreover, ZnMT3PyP/Pt/g-C3N4 had a greater stability during the photoreaction compared to ZnMTPP/Pt/g-C3N4 due to the presence of N atoms, that are combined with ascorbic acid and it is easier to regenerate the electrons. The same research group prepared a series of asymmetric zinc porphyrins and studied the influence of the N atom position on the photoactivity.135 Under the same condition, in the presence of ascorbic acid as SED and upon visible light irradiation they concluded that the N atom position affects the photocatalytic hydrogen production. The best hydrogen production activity was obtained of 524 μmol h−1 was exhibited with ZnPy-4-Pt/g-C3N4, where the N atom is at the para position of the phenyl ring (Table 6, entry 19). This nanohybrid material has the lowest steric hindrance therefore, it is easier to combine with g-C3N4 and AA.
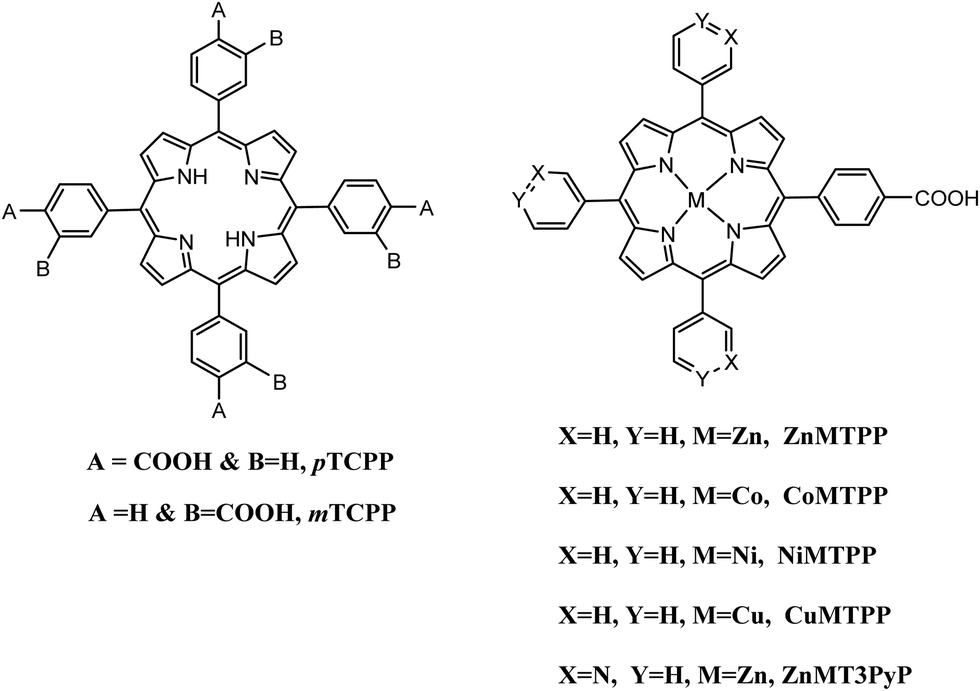 | ||
| Fig. 28 Chemical structure of TCPP, ZnMTPP, CoMTPP, NiMTPP, CuMTPP and ZnMT3PyP. | ||
Subsequently, the effect on hydrogen evolution of different metals on the photosensitizer that is loaded onto carbon nitride (C3N4) was examined.136 The sensitizers were a Co, Ni, or a Cu metallated porphyrin with three 4-pyridines and a benzoic acid at the meso position of the phenyl ring (CoMTPP, NiMTPP, CuMTPP, Fig. 28). The best hydrogen evolution remained the ZnMTPP/C3N4 material under λ > 420 nm light irradiation. The other metallated dyes produced hydrogen following the sequence of CoMTPP < NiMTPP< CuMTPP. The different hydrogen production ability of the porphyrins may be attributed to the different light absorption of the dyes and the diverse electron transfer between the porphyrin and C3N4. A novel hybrid material comprised of a naphthalimide porphyrin (NP) absorbed through π–π stacking onto a two-dimensional graphitic carbon nitride (g-C3N4) (Fig. 29).137 The hybrid material with 2% weight ratio of NP (2%NP/g-C3N4), exhibited a photocatalytic hydrogen production of 2297 μmol g−1 h−1, with a good stability and reproducibility (Table 6, entry 20). In comparison with pristine g-C3N4 and 2%ZnTCPP/g-C3N4, they both exhibited less hydrogen evolution of 698 μmol g−1 h−1 and 900 μmol g−1 h−1, respectively.
 | ||
| Fig. 29 Chemical structure of NP. | ||
In another report, a new nanocomposite g-C3N4–Cu-TCPP was prepared comprised of a tetra carboxyphenyl porphyrin p-TCPP and Cu as an interfacial linker.138 A pillar-like structure was formed and the material enhanced the photocatalytic hydrogen production. The nanocomposite upon the photocatalytic reaction was fairly stable and its H2 release rate was 10.3 times higher than that of pure g-C3N4 and 9.6 times higher than TCPP-g-C3N4 (Table 6, entry 21). Following the previous research of small metal linkage of porphyrins with graphitic carbon nitride, a C3N4–Cu-THPP nanohybrid was synthesized, bearing a tetrakis-(4-hydroxyphenyl)porphyrin (THPP) (Fig. 30).139 This composite showed a great photocatalytic activity that was 17.3 times higher compared to g-C3N4 and 11.6 times greater than g-C3N4/THPP. Therefore, the presence of Cu was the key factor of the enhanced hydrogen production (Table 6, entry 22).
 | ||
| Fig. 30 Chemical structure of THP, ZnPy-5, ZnPy-6. | ||
In addition, the presence of pyrimidine groups and N atoms at the peripheral of a zinc porphyrin (ZnPy) was studied.140 Asymmetric porphyrins bearing one benzoic acid and three pseudo pyrimidines (ZnPy-5 and ZnPy-6) were used as sensitizers at Pt-loaded g-C3N4 for visible-light-driven H2 evolution. The pseudo pyrimidine groups act as electron-donating groups whereas benzoic acid acts as electron-withdrawing/anchoring group. It was found that the presence of pyrimidine groups improves the photosensitization on g-C3N4 and exhibit H2 evolution production activity of 585 mmol h−1 for g-C3N4–ZnPy-6 and 418 mmol h−1 for g-C3N4–ZnPy-5 (Table 6, entries 23 and 24). It is evident that the presence and the position of N atoms in the pseudo-pyrimidines influence the activity. Therefore, both porphyrins increased the electron transfer towards g-C3N4 and ZnPy-6 with the lowest steric hindrance of N atoms showed better performance for H2 evolution compared to ZnPy-5. In all the above examples graphitic carbon nitride is coupled via noncovalent interactions with a porphyrin molecule. Another approach, that is not commonly used is the covalent linkage between the porphyrin and the g-C3N4. A tetrakis driazinyl phenyl porphyrin (TDPP) was copolymerized with urea in order to synthesize a g-CNU-TDPP material (Fig. 31).141 This material doped with porphyrin via covalent bond performed 4 times greater photocatalytic activity 7.6 mmol g−1 h−1 compared to the undoped g-CNU (Table 6, entry 25). The H2 evolution photocatalytic reaction was carried out in aqueous solution using TEOA (10%) as sacrificial agent and 2 wt% Pt as cocatalyst.
 | ||
| Fig. 31 Chemical structure of g-CNU-TDPP. | ||
The most common metal oxide that has been developed is TiO2 and upon its structural modification with a co-catalyst like Pt can become appropriate for H2 evolution.147 In order to extend the light absorption of metal oxide photocatalysts from the UV light to visible light it is essential to modify the metal oxide. One way to achieve it is the insertion of a dye-sensitized material that is able to absorb visible light. In this part of the review, we will focus on porphyrin-based molecules, that have been used as sensitizers since 2014.
Inoue and coworkers constructed a system able to produce hydrogen and useful oxygenated products from water under visible light irradiation.148 A tin(IV) tetracarboxyphenyl porphyrin Sn(IV)TCPP was used as a sensitizer and absorbed on a Pt-doped TiO2, in aqueous acetonitrile with cyclohexane. Upon irradiation with light the system 0.49 μmol of H2 and oxygenated cyclohexane products, cyclohexeneoxide, 2-cyclohexenol, 2-cyclohexenone, and cyclohexanone from the substrate, cyclohexene (Table 7, entry 1). The system did not produce any products after the photocatalytic reaction at the absence of the photosensitizer and upon addition of sulphuric acid with C > 10−4 M the porphyrin was desorbed from the TiO2 due to its protonation. The same porphyrin Sn(IV)TCPP was used as a photosensitizer in a photocatalytic hydrogen evolution reaction.149 Platinum dioxide (PtO2) was used as a heterogeneous proton-reduction catalyst and triethanolamine (TEOA) as sacrificial electron. After optimization of the system 4.8 μmol h−1 of H2 was produced upon visible light irradiation (Table 7, entry 2). When EDTA was used as SED lower amount of hydrogen was produced 0.42 μmol h−1 compared to TEOA possibly due to its lower reduction rate (Eox(TEOA) = 1.07 V and Eox(EDTA) = 1.17 V) or due to fewer amine units. It was found that hydrogen production occurred via a reductive mechanism and chlorin species were formed upon the photocatalytic reaction as the main intermediate. A series of meso substituted tin-based porphyrins (SnTPP(SO3H)4, SnTPP(COOH)4, SnTPyP, SnTMPyP(OH)2, SnTMPyP(Cl)2) were synthesized and used as photosensitizers for photocatalytic hydrogen evolution reaction under visible light irradiation.150 The photosensitizers with different functional groups were absorbed onto a Pt/TiO2 surface and various parameters were evaluated during the photochemical reaction such as the pH, Pt/TiO2 concentrations, light intensities and Pt nanoparticles with diverse surface stabilizers. After evaluation of the above parameters, the maximum hydrogen evolution was obtained for SnTPyP/TiO2/Pt in the presence of EDTA at pH = 9.0 obtaining 232 μmol of H2 (Table 7, entry 3). It was resolved that the reduced species formed after the photocatalytic reaction were long-lived in order to complete the electron transfer and that the chromophore was not essential to be bound onto TiO2 surface for H2 evolution. In an effort to exclude the use of noble metal cocatalysts onto TiO2, a graphene-like MoS2 has been added onto TiO2 as an alternative cocatalyst promoting higher H2 evolution rates upon photocatalytic reaction. In a report, Zn tetracarboxyphenyl porphyrin (Fig. 3) sensitized MoS2/TiO2 nanocomposite as a photocatalyst. The system was able to absorb visible light and produce H2 with a rate of 10.2 μmol h−1 and a TON of 261 after 12 h of irradiation (Table 7, entry 4).151 Upon comparison with ZnTCPP-Pt/TiO2 under the same photocatalytic conditions the photocatalyst with MoS2 produce H2 with 1.45 times higher rate displaying the high activity of MoS2. The same research group replaced TiO2 with ZnO and prepared ZnTPP-MoS2/ZnO nanocomposite that was able to produce H2 with a hydrogen evolution rate of 75 μmol h−1 g−1 under visible light irradiation (Table 7, entry 5).152 Cuprous oxide Cu2O can be used as a cocatalyst sensitized with free base tetracarboxyphenyl porphyrin TCPP/Cu2O.153 The composite upon visible light irradiation produced higher hydrogen compared to pure TCPP or pure Cu2O. Also, the system proved to be quite stable, since after five recycles the hydrogen production barely changed 2 mmol mg−1 (Table 7, entry 6). In addition, the same porphyrin TCPP was introduced onto an antennae-like Cu2O/NiO heterostructure in order to enhance the photocatalytic hydrogen production.154 Therefore, upon light irradiation in aqueous TEOA solution the rate of H2 evolution followed the order Cu2O/NiO/TCPP > Cu2O/NiO > pure Cu2O (Table 7, entry 7). This result indicates that NiO and Cu2O can improve the photocatalytic hydrogen activity in the presence of the porphyrin photosensitizer, due to the strong interaction between the three components and due to electric field, that ids produced at the interface of Cu2O kai NiO. In an attempt to prepare stable and highly efficient photocatalysts, TiO2 microsphere (TiO2MS) was linked via metallic Cu with a tetracarboxyphenyl porphyrin TCPP.155 The linkage between copper and the porphyrin was done with coordination and electrostatic interactions. This hybrid material was active towards H2 evolution and produced about 6 times more hydrogen compared to TiO2MS and 3.8 times higher in the absence of Cu (TiO2MS-TCPP) (Table 7, entry 8). These results indicate the importance of Cu in the hybrid material facilitating the electron transfer from TCPP to TiO2 MS. Another research group produced a photocatalyst composed of aluminum porphyrin adsorbed onto TiO2 nanoparticles with Pt clusters (Al(III)TCPP/TiO2/Pt).156 The system upon irradiation and with the absence of a sacrificial electron donor was able to produce at the same time H2 and H2O2 from water upon only one-photon (Table 7, entry 9). In addition, Coutsolelos and coworkers established an alternative methodology of H2 evolution by self-organizing porphyrin based photocatalysts onto a TiO2 scaffold.157 Among the synthesized porphyrins ZnTCP, PdTCP, PtTCP (Fig. 32) only PdTCP and PtTCP showed photocatalytic activity. When PdTCP porphyrin was adsorbed onto TiO2, H-aggregates were formed, whereas in the case of PtTCP J-aggregates were shaped. Upon photocatalytic experiments, the hydrogen evolution of PtTCP/TiO2 material was greater (146 μmol H2) compared to PdTCP/TiO2 (52 μmol H2) (Table 7, entries 10 and 11). This difference at their catalytic performance might be due to the variation of self-organization of the two porphyrins onto TiO2. In the case of adsorption of both porphyrins onto TiO2 (PdTCP/PtTCP/TiO2) the system became more efficient towards H2 evolution producing 30.2 mmol g−1 (Table 7, entry 12). This result is considered to be one of the most efficient efforts on H2 evolution of this type of systems.
| Entry | PS (C) | Catalyst (C) | SED (C) | Solvent | Light source | Irr. time (h) | TON/H2 | H2 | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Sn(IV)TCPP | TiO2/Pt (4.8 × 10−6 M) | 10% aq. acetonitrile | Water | (λ > 480 nm) | 5 | 0.49 μmol | 148 | |
| 2 | Sn(IV)TCPP | PtO2 (22 mg L−1) | TEOA, 60 mM | Aqueous phosphate buffer (0.1 M) | Visible light (λ > 400 nm) | 4.8 μmol h−1 | 149 | ||
| 3 | SnTPyP | TiO2/Pt | EDTA | Water | λ > 390–650 nm | 4 | 232 μmol | 150 | |
| 4 | ZnTCPP | SnO2/TiO2 | TEOA, 0.2 M | Water | Visible light (λ > 420 nm) | 12 | 261 | 10.2 μmol h−1 | 151 |
| 5 | ZnTCPP | SnO2/ZnO | TEOA, 0.2 M | Water | Visible light (λ > 420 nm) | 9 | 516 | 75 μmol h−1 g−1 | 152 |
| 6 | TCPP | Cu2O | TEOA | Water | Visible light (λ > 420 nm) | 8 | 2 mmol mg−1 | 153 | |
| 7 | TCPP | Cu2O/NiO | TEOA | Water | Visible light (λ > 400 nm) | 6 | 3 mmol g−1 | 154 | |
| 8 | TCPP | TiO2–Cu | TEOA | Water | 300 W Xenon lamp | 6 | 8 mmol g−1 | 155 | |
| 9 | Al(III)TCPP | TiO2/Pt | H2O/CH3CN (1/9 v/v) | Water, pH = 4.01 | 420 nm LED light | 2 | 0.3 μmol | 156 | |
| 10 | PdTCP | TiO2 | TEOA 15 vol% | CH3CN/H2O (1:1 v/v), pH = 8.0 |
White LED lamp, 40 W | 120 | 52 μmol | 157 | |
| 11 | Pt TCP | TiO2 | TEOA 15 vol% | CH3CN/H2O (1:1 v/v), pH = 8.0 |
White LED lamp, 40 W | 200 | 146 μmol | 157 | |
| 12 | PdTCP/PtTCP | TiO2 | TEOA 15 vol% | CH3CN/H2O (1:1 v/v), pH = 8.0 |
White LED lamp, 40 W | 200 | 22733 |
30.2 mmol g−1 | 157 |
| 13 | ZnP-dyad | TiO2/Pt | Ascorbic acid 0.5 M | Water, pH = 5.0 | White light, 80 mW | 120 | 12800 |
173000 μmol g−1 h−1 |
158 |
| 14 | YD2-o-C8 | TiO2/Pt | Ascorbic acid 0.5 M | Water, pH = 5.0 | White light, 80 mW | 120 | 11900 |
272000 μmol g−1 h−1 |
158 |
| 15 | LG-5 | TiO2/Pt | TEOA 20 vol% | Water | Xe lamp, 450 W | 5 | 6582 | 3291 μmol g−1 h−1 | 159 |
| 16 | LG-DtT | TiO2/Pt | TEOA 20 vol% | Water | Xe lamp, 450 W | 5 | 13282 |
6641 μmol g−1 h−1 | 159 |
| 17 | LGtT | TiO2/Pt | TEOA 20 vol% | Water | Xe lamp, 450 W | 5 | 14792 |
7396 μmol g−1 h−1 | 159 |
| 18 | TSPP | Cu2O | TEOA | Water | Xe lamp, 300 W | 6 | 0.83 mmol g−1 h−1 | 160 | |
| 19 | TSPP | ZnO NR-Cu | TEOA | Water | Xe lamp, 300 W | 6 | 0.69 mmol g−1 h−1 | 161 | |
| 20 | ZnTmMpHPP | TiO2 | CH3OH | Water | Xe lamp, 300 W | 7 | 326.3 μmol h−1 | 162 | |
| 21 | THP | TiO2/Cu | TEOA | Water | Xe lamp, 300 W | 6 | 13 mmol g−1 | 163 | |
| 22 | THPP | TiO2/Pt | TEOA 10 vol% | Water | Visible light (λ > 420 nm) | 5 | 576.8 μmol g−1 h−1 | 164 | |
| 23 | PdTHPP | TiO2/Pt | TEOA 10 vol% | Water | Visible light (λ > 420 nm) | 5 | 2025.4 μmol g−1 h−1 | 164 | |
| 24 | ZnTHPP | TiO2/Pt | TEOA 10 vol% | Water | Visible light (λ > 420 nm) | 5 | 1239.8 μmol g−1 h−1 | 164 | |
| 25 | THPP | NiO | TEOA 20 vol% | Water | Xe lamp, 300 W | 8 | 8 mmol g−1 | 165 | |
| 26 | TpTph | TiO2 | Ascorbic acid 0.25 M | Water | Xe lamp, 300 W (λ > 420 nm) | 4 | 5602 μmol h−1 g−1 | 166 | |
| 27 | BDP-Por-BDP(Im) | PtTiO2 | Ascorbic acid 1 M | Water | white LED lamp, 40 W | 72 | 18600 |
225 mmol h−1 g−1 | 167 |
 | ||
| Fig. 32 Chemical structure of ZnTCP, PdTCP, PtTCP. | ||
Push–pull porphyrins were found to have a large impact on the power conversion efficiency when used as sensitizers in dye-sensitized solar cells (DSCCs). Inspired by DSCC this type of porphyrins were investigated as photosensitizers for H2 production. The presence of a carboxylic group on the dye act as an anchoring group and is attached onto TiO2. Therefore, ZnP-dyad and YD2-o-C8 dyads were synthesized and attached onto platinized TiO2 particles and photodriven H2 evolution experiments were conducted (Fig. 33).158 All measurements were done in an aqueous solution in the presence of ascorbic acid as sacrificial electron donor. The YD2-o-C8/TiO2/Pt material produced 19.11 ml of H2, while ZnP-dyad/TiO2/Pt 15.95 ml of H2 (Table 7, entries 13 and 14).
 | ||
| Fig. 33 Chemical structure of ZnP-dyad and YD2-o-C8. | ||
Three different porphyrin-based dyes were synthesized and their photocatalytic H2 production was evaluated.159 The dyes LG-5, and LG-DtT possessed three different 4-ethynyl thiophene and a cyanoacrylic acid as an anchoring group (Fig. 34). Photocatalytic hydrogen production measurements were conducted in the presence of TEOA as sacrificial electron donor under visible light irradiation. All the photocatalytic systems composed of the dyes attached onto TiO2/Pt via the anchoring group. The LG-tT/TiO2/Pt material proved to be the most efficient system with a H2 evolution rate of 7396 μmol g−1 h−1 after 5 h of light irradiation. The activity towards H2 evolution followed the order LG-tT/TiO2/Pt > LGDtT/TiO2/Pt > LG5/TiO2/Pt (Table 7, entries 15–17).
 | ||
| Fig. 34 Chemical structure of LG-5, LG-tT and LG-DtT. | ||
In another attempt to prepare an efficient nanocatalyst for H2 generation, tetrakis sulfophenyl porphyrin (TSPP) was synthesized and assembled with cubic Cu2O (Fig. 35).160 Since porphyrins with sulfonyl groups possess stronger electron-withdrawing abilities they were used as an alternative dye. Therefore, the combination of light-harvesting TSPP porphyrin (electron donor) and Cu2O cubic phase (electron acceptor) improved the photocatalytic performance towards hydrogen production. The TSPP/Cu2O composite upon light irradiation produces 0.83 mmol h−1 g−1 of hydrogen, in the presence of TEOA as a SED (Table 7, entry 18). This hydrogen evolution was 5 times higher compared to Cu2O. The same porphyrin TSPP was used as a light harvester and was attached on a copper-implanted ZnO nanorod.161 This nanocomposite showed high photocatalytic performance upon H2 production of 0.69 mmol g−1 h−1 (Table 7, entry 19).
 | ||
| Fig. 35 Chemical structures of TSPP, ZnTmMpHPP and MTHPP (M = H2, Pd, Zn). | ||
It is well known that modification of TiO2 with porphyrin dyes can enhance the photocatalytic H2 production. A plethora of porphyrins have been used varying their anchoring groups. In this report a m-methoxy-p-hydroxylphenyl porphyrin (ZnTmMpHPP) was linked to TIO2 (Fig. 35).162 The hydrogen evolution rate of this photocatalyst was 2 times higher than pure TiO2, reaching 326.3 μmol h−1 (Table 7, entry 20). In a different example, tetrahydroxyphenyl porphyrin THP was bound to TiO2 microsphere with the presence of copper nanoparticles (CuNPs) (Fig. 35).163 The prepared nanocomposite TiO2 microsphere/CuNPs/THPP upon light irradiation showed enhanced photocatalytic activity towards H2 production by a factor of 14 compared to the pure TiO2 microsphere (Table 7, entry 21). This high activity and stability of the system might be due to the special interaction between TiO2 microsphere/CuNPs and THPP.
A series of free-base and metalated meso-carboxyphenyl porphyrins were synthesized and linked with TiO2.164 Upon addition of Pt nanoparticles, the hybrids THPP-TiO2/Pt, PdTHPP-TiO2/Pt and ZnTHPP-TiO2/Pt were formed (Fig. 35). Upon photocatalytic hydrogen evolution experiments, all materials produced hydrogen, with PdTHPP-TiO2/Pt achieving the best result 2025.4 μmol g−1 h−1. The ZnTHPP-TiO2/Pt photocatalyst with the zinc metallated porphyrin achieved a hydrogen rate of 1239.8 μmol g−1 h−1, greater than THPP-TiO2/Pt 576.8 μmol g−1 h−1, due to the effective light harvesting and electron transfer of ZnTHPP to TiO2 (Table 7, entries 22–24). The best activity of PdTHPP-TiO2/Pt might be attributed to the presence of the two metal catalysts Pd and Pt. Two-dimension nanomaterials of NiO were sensitized with tetrahydroxyphenyl porphyrins (THPP) in order to form effective photocatalysts.165 The system showed a high and stable photocatalytic activity towards hydrogen production (Table 7, entry 25).
A porphyrin-based organic polymer was synthesized (TpTph) and TiO2 was loaded onto the polymer in order to form photocatalyst TpTph-TiO2 (Fig. 36).166 The hydrogen evolution ability of the system TpTph-TiO2 was investigated and it was found that the H2 production was much greater (5602 μmol h−1 g−1) compared to TpTph (21 μmol h−1 g−1) and bulk TiO2 (39 μmol h−1 g−1) (Table 7, entry 26).
 | ||
| Fig. 36 Chemical structure of TpTph. | ||
Our research group recently reported the synthesis of BODIPY-(Zn)Porphyrin hybrids (Fig. 37) and their application in photocatalytic H2 production from water utilizing platinum-doped titanium dioxide nanoparticles (PtTiO2).167 More specifically the hybrid photosensitizers were employed in a dye-sensitized photocatalytic system (DSP) after their chemisorption onto the surface of the PtTiO2. The authors showed that the covalent attachment of the BDP moiety (BDP-Por) illustrated greater efficiency (17500 TONs) compared to the axial coordination (BDP(Im)-Por, 13700 TONs) while the triad system BDP-Por-BDP(Im) overwhelmed the other hybrids in terms of activity (18600 TONs). Noteworthy, photoelectrochemical studies of the synthesized hybrids on TiO2 films displayed the same trend in terms of photocurrent density values.
 | ||
| Fig. 37 Chemical structures of BDP-Por, BDP(Im)-Por and BDP-Por-BDP(Im). | ||
One example of DSPEC towards H2 production was provided by Moore and coworkers.168 The authors grafted Co(II) and Fe(II) metalloporphyrins to a gallium phosphide semiconductor and created a photocathode for visible light-driven H2 evolution which displayed enhanced photoelectrochemical efficiency over unmodified electrodes. The porphyrins contained a 4-vinylphenyl group (Fig. 38a) in order to be attached to the semiconductor, while the resulting hybrid material with cobalt inside the porphyrin core exhibited greater hydrogen production rate and stability in comparison to the analogous assemblies with Fe(II). The same research group further improved their system by utilizing a polypyridine coating on the surface of GaP semiconductor to provide binding sites for the coordination of the cobalt porphyrin catalysts (Fig. 38b).169 Indeed their approach enhanced the overall performance of the photocathode.
 | ||
| Fig. 38 (A) Schematic illustration of the GaP semiconductor grafted with metalloporphyrins, Reproduced from ref. 168, with permission from Royal Society of Chemistry, Copyright 2016. (B) Schematic illustration of the polypyridine modified GaP semiconductor binding Co–porphyrin catalyst, Reproduced from ref. 169, with permission from American Chemical Society, Copyright 2017. (C) Molecular structures of the free base porphyrin and the Si phthalocyanines used in the tandem cell, (D) schematic illustration of the tandem system with a TiO2–phthalocyanine DSSC, Reproduced from ref. 170, with permission from Royal Society of Chemistry, Copyright 2016. (E) Graphic representation of the DSPEC with a TiO2/ruthenium-dye photoanode and the Pt-porphyrin/TiO2 cathode. Reproduced from ref. 172, with permission from American Chemical Society, Copyright 2019. | ||
Sherman et al. prepared a tandem cell composed of two dye-sensitized photoanodes, which was able to generate H2 under visible light irradiation and in the absence of any applied electrical bias.170 The authors utilized a free-base porphyrin and a Si phthalocyanine as chromophores to harvest distinct portions of the visible spectrum (Fig. 38c). Although a DSPEC based on SnO2-porphyrin photoanode was not able to oxidize hydroquinone and use the resulting electrons to produce hydrogen at the cathode, when it was incorporated in a tandem system with a TiO2–phthalocyanine DSSC (Fig. 38d), the additional electrons provided by the DSSC enabled efficient H2 generation.
In another study Sakai,171 Ozawa and coworkers applied a platinum(II) porphyrin bearing single pyridyl anchoring group on a TiO2 cathode and showed that it catalyze H2 production electrocatalytically. As a side investigation of this work, the authors constructed DSPECs with a FTO/TiO2/ruthenium-dye photoanode and the Pt-porphyrin anchored on the FTO/TiO2 cathode (Fig. 38e) where they observed a small photo-induced electric current between the electrodes. The same research group further explored this system and altered the methyl groups on the Ru complex (Fig. 38e) with phenyl ones.172 The new DSPEC comprised of the Pt-porphyrin cathode and the TiO2/Ru-py photoanode gave substantial H2 evolution upon visible light irradiation, without applying any external bias. The electron donor in both above studies was EDTA.
Light assisted hydrogen production can be also achieved from porphyrin-modified glassy carbon electrodes.173 Ramírez and co-workers applied Co(II) and Cu(II) octaethyl-porphyrins supramolecular systems on glassy carbon electrodes as photoelectrocatalysts and showed that no photosensitizer was required for hydrogen evolution (Fig. 39a). The irradiation wavelength, the anchoring group and the supramolecular aggregation were proven significant factors related to the photoelectrochemical performance. Another work published by the same research group explored graphite paste electrodes with Co(II) and Fe(III) octaethyl-porphyrins.174 Both modified systems presented photoelectrocatalytic hydrogen production, which was enhanced when a mixture of the two metallated porphyrins was deposited on the graphite paste electrode. The authors quantified the overall performance of the later system to 70.9 μmol h−1 cm−2 of hydrogen which corresponds to a TON of 8100. Subsequently, the researchers further improved this system by incorporating N-octylpyridinium hexafluorophosphate ionic liquid as binder and using the Co-octaethylporphyrin.175
 | ||
| Fig. 39 (A) Schematic illustration of Co(II) and Cu(II) octaethyl-porphyrin supramolecular systems on glassy carbon electrodes, Reproduced from ref. 173, with permission from Elsevier, Copyright 2017. (B) Schematic illustration of tetraphenyl-manganese(III) porphyrin as photosensitizer deposited on a CuFe2O4 electrode, Reproduced with permission from ref. 176, which is an open access article distributed under the Creative Commons Attribution License (CC BY), MDPI. (C) Molecular structures of the free base porphyrin covalently connected to the cucurbit[7]uril hosts, (D) schematic illustration of the tetra-substituted porphyrins bearing thiol groups covalently attached on exfoliated MoSe2 nanosheets, Reproduced from ref. 179, with permission from Royal Society of Chemistry, Copyright 2020. (E) Graphic representation of the electron transfer root from the PMOFs to the Pt/C nanowires and Pt/C nanodots, Reproduced from ref. 181, with permission from John Wiley and Sons, Copyright 2020. (F) Energy diagram of the porphyrin-cobalt dyad on the NiO towards photoelectrochemical hydrogen production. Reproduced from ref. 182, with permission from John Wiley and Sons, Copyright 2021. | ||
Tetrasubstituted metalloporphyrins have been used also by other research groups towards the construction of DSPECs for hydrogen production. In a similar study, Li et al. used tetraphenyl-manganese(III) porphyrin (MnTPP) as photosensitizer deposited on a CuFe2O4 electrode and applied the resulting photocathode in photoelectrochemical water splitting where it was proven productive (Fig. 39b).176 Tuncel and co-workers synthesized a supramolecular assembly based on cucurbit[7]uril hosts covalently connected to a free-base porphyrin core (TTP-4CB7) (Fig. 39c) and applied this assembly in visible light driven hydrogen production.177 The authors showed that TPP-4CB7 by its own can act as a photocatalyst towards H2 evolution but photocatalytic activity was significantly improved in the presence of TiO2. The CB7 moieties were utilized as anchoring groups to coordinate with TiO2 and form a homogenous system. The final nanocomposite which contained both TiO2 and Pt co-catalyst, exhibited efficient hydrogen production 24.5 mmol h−1 g−1 at an onset potential of −10 mV and in the absence of any SED. In another report, Li et al. prepared long chain coordination complexes based on Zr(IV) meso-tetraphenyl porphyrin (ZrTPP) linked via the bridging group of a dicarboxylic acid.178 These 1-dimensional complexes displayed efficient electrocatalytic hydrogen evolution as well as photoelectrochemical performance.
Blanco et al. explored the covalent attachment of tetra-substituted porphyrins bearing thiol groups on exfoliated MoSe2 nanosheets (Fig. 39d) towards hydrogen production both electrocatalytically and photoelectrochemically.179 The resulting hybrid materials exhibited an enhancement of the hydrogen evolution onset potential under white light irradiation in comparison to the initial material. The same research group further improved their system through the conjugation of these porphyrins with MoS2/Ag2S/Ag nanocomposites deposited on a Digital Versatile Disc.180
Zhang and co-workers utilized the known porphyrin MOFS in order to confine Pt/C quantum dots (QDs) inside their pores and apply them in photoelectrocatalysis.181 More specifically the authors used PCN-222 with nanochannels and PCN-221 with nanocages to host ultrafine Pt/C nanowires and uniform Pt/C nanodots, respectively (Fig. 39e). Under visible light irradiation, these host-guest materials exhibited highly efficient electrocatalytic performance towards hydrogen evolution reaction. Moreover, it was shown that both materials had fast and shape-dependent charge transfer behavior since the charge transfer from PCN-222 to the Pt/C nanowires was faster than from PCN-221 to Pt/C nanodots. Charisiadis et al. have recently contributed in the field of photoelectrochemical hydrogen evolution utilizing a Zn–porphyrin covalently connected to a cobaloxime molecule.182 The porphyrin moiety was equipped with a carboxylic acid anchoring group to be attached on NiO semiconductor (Fig. 39f). The resulting photocathode was employed in photoelectrochemical H2 production and this was the first report of a noble metal free porphyrin-cobalt dyad aiming to this application.
3. Porphyrins for CO2 reduction
The direct conversion of solar irradiation into chemical energy through the CO2 reduction represents a very interesting and promising scientific area. The photocatalytic CO2 reduction typically involves a PS which absorbs light and generates charge separate states (electrons and holes). The excited electrons are then used to activate a catalyst where the CO2 reduction takes place. Porphyrin based materials have been extensively employed in photocatalytic CO2 reduction, mainly as catalysts. It has been demonstrated that several metallo-porphyrins with transition metals (Fe, Co, Ni, etc.) present high activity towards the electro- and the photo-catalytic CO2 reduction.183,184 Moreover, due to their exceptional optical properties and their fast charge transfer ability, porphyrin derivatives have been also used as photosensitizers in photocatalytic CO2 reduction systems.185 The porphyrin based catalytic systems that will be described is this section will be divided in homogeneous molecular materials and various heterogeneous approaches such as MOFs, COFs, porphyrin-supported systems on metal oxides, porphyrins in combination with nanomaterials and DSPECs.3.1 Homogeneous systems
Homogenous catalytic systems contain a catalyst, a PS and a SED that are all soluble in a solution. In these systems, the molecular structure of the components is usually well-defined, and this enables the determination of the corresponding catalytic mechanism. Moreover, the homogenous systems present higher selectivity and it is easier to identify the structural parameters that influence the catalytic performance.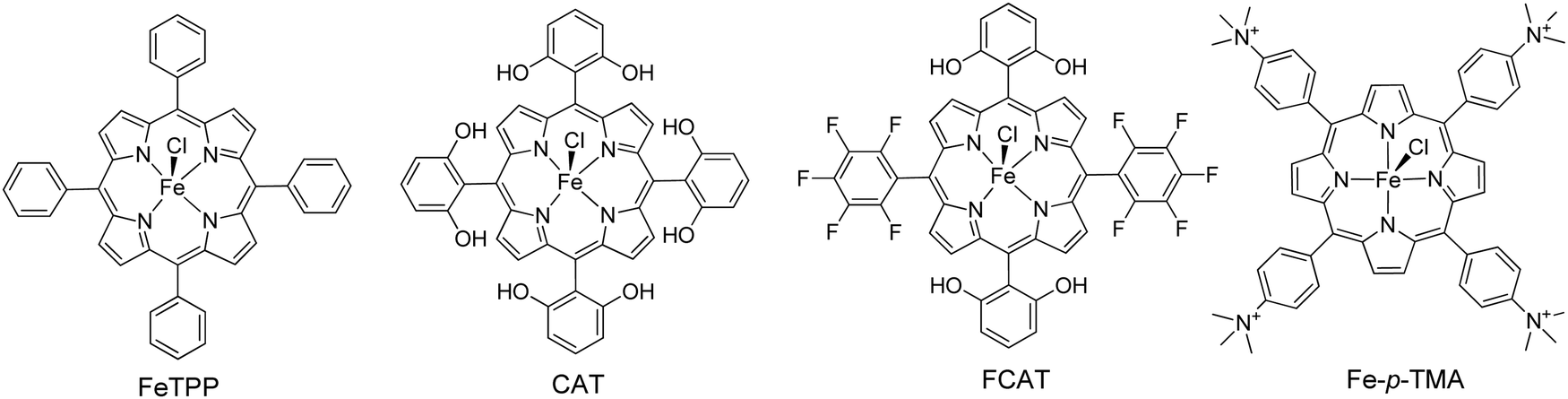 | ||
| Fig. 40 Structure of iron porphyrin molecular catalysts for CO2 reduction. | ||
| Entry | Catalyst (C) | PS (C) | SED | Solvent | Light source | Irrad. time | TON | TOF (h−1) | Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | FeTPP, 0.01 mM | — | TEA, 5% | DMF | Xenon lamp, 300 W | 170 h | 70 | — | 186 | |
| 2 | FeTPP, 0.01 mM | — | TEA, 0.36 M | CH3CN | Xenon lamp, BG40 optical filter | 10 h | 17 | — | 8 | 187 |
| 3 | CAT, 0.01 mM | — | TEA, 0.36 M | CH3CN | Xenon lamp, BG40 optical filter | 10 h | 28 | 7.7 | 93 | 187 |
| 4 | FCAT, 0.01 mM | — | TEA, 0.36 M | CH3CN | Xenon lamp, BG40 optical filter | 10 h | 23 | 6.7 | 76 | 187 |
| 5 | CAT, 0.002 mM | Ir(ppy)3, 0.2 mM | TEA, 0.36 M | CH3CN | Xenon lamp, 150 W, λ > 420 nm | 55 h | 140 | 93 | 188 | |
| 6 | CAT, 0.002 mM | 9CNA, 0.2 mM | TEA, 0.05 M | CH3CN | Xenon lamp, 150 W, λ > 400 nm | 45 h | 60 | 100 | 188 | |
| 7 | Fe-p-TMA, 0.002 mM | — | TEA, 0.05 M | CH3CN | Solar simulator, λ > 420 nm | 47 h | 33 | 100 | 189 | |
| 8 | Fe-p-TMA, 0.002 mM | — | BIH, 0.02 M | CH3CN | Solar simulator, λ > 420 nm | 47 h | 63 | 100 | 189 | |
| 9 | Fe-p-TMA, 0.002 mM | Purpurin | TEA, 0.05 M | CH3CN/H2O (1/9), pH = 6.7 | Solar simulator, λ > 420 nm | 47 h | 60 | 95% | 190 | |
| 10 | Fe-p-TMA, 0.002 mM | Ir(ppy)3, 0.2 mM | TEA, 0.05 M | CH3CN | Solar simulator, λ > 420 nm | 102 h | 367 (CO) | 78 (CO) | 191 | |
| 79 (CH4) | 17 (CH4) | |||||||||
| 11 | Fe-p-TMA, 0.002 mM | Ir(ppy)2(bpy), 0.2 mM | TEA, 0.05 M | CH3CN | Solar simulator, λ > 420 nm | 47 h | 178 (CO) | 57 (CO) | 192 | |
| 32 (CH4) | 10 (CH4) | |||||||||
| 12 | Fe-p-TMA, 0.002 mM | Ir(ppy)3, 0.2 mM | TEA, 0.05 M | CH3CN | Solar simulator, λ > 420 nm | 47 h | 198 (CO) | 78 (CO) | 191 and 192 | |
| 31 (CH4) | 12 (CH4) | |||||||||
| 13 | Fe-p-TMA, 0.002 mM | Ir(ppy)2(bpy), 0.2 mM | TEA, 0.05 M | CH3CN/H2O (3/7) | Solar simulator, λ > 420 nm | 47 h | 24 (CO) | 75 (CO) | 192 | |
| 3 (CH4) | 9 (CH4) | |||||||||
| 14 | Fe-p-TMA, 0.01 mM | Phen2, 1 mM | TEA, 0.1 M | DMF | Solar simulator, λ > 435 nm | 102 h | 140 (CO) | 75 (CO) | 193 | |
| 29 (CH4) | 15 (CH4) | |||||||||
| 15 | Fe-p-TMA, 0.006 mM | PMI-OH, 0.87 mM | SA, 0.5 M | H2O | 150 W EKE bulb | 48 h | 30 (CO) | 195 | ||
| 16 | CAT, 0.0002 mM | CuPP, 0.1 mM | BIH, 0.1 M | DMF | White LED, λ > 400 nm | 23 h | 16109 |
7650 | 95 | 196 |
| 17 | CoTPPS, 0.01 mM | [Ru(bpy)3]Cl2, 0.5 mM | AA, 0.1 M | H2O, pH = 6.7 | Xenon lamp, λ > 400 nm | 4 h | 926 | 456 | 82 | 198 |
| 18 | CoTPPS, 0.0005 mM | [Ru(bpy)3]Cl2, 0.5 mM | AA, 0.1 M | H2O, pH = 6.7 | Xenon lamp, λ > 400 nm | 4 h | 4000 | 2400 | 41 | 198 |
| 19 | CoTPPS, 0.005 mM | CuPS, 0.5 mM | AA, 0.1 M | H2O, pH = 6.7 | Xenon lamp, λ > 400 nm | 4 h | 1085 | 380 | 90 | 199 |
| 20 | CoTMPyP, 0.005 mM | CuPS, 0.5 mM | AA, 0.1 M | H2O, pH = 6.7 | Xenon lamp, λ > 400 nm | 4 h | 2680 | 1600 | 77 | 199 |
| 21 | Co(oTMPyP), 0.005 mM | CuPS, 0.5 mM | SA, 0.1 M | H2O, pH = 6.7 | Xenon lamp, λ > 400 nm | 12 h | 4000 (CO) | 1170 | 90 | 200 |
| 22 | Co(TMAP), 0.005 mM | CuPS, 0.5 mM | SA, 0.1 M | H2O, pH = 6.7 | Xenon lamp, λ > 400 nm | 8 h | 460 (CO) | 210 | 77 | 200 |
| 23 | FeCl-1-Ru, 0.05 mM | TEA, 5% | DMF | Mercury lamp, λ > 305 nm | 24 h | 11.4 | 201 | |||
| 24 | Co-1-Ru, 0.05 mM | TEA, 5% | DMF | Mercury lamp, λ > 305 nm | 24 h | 4.7 | 201 | |||
| 25 | [Dyad 1 pic]PF6, 0.05 mM | TEOA, 20% | DMF | Xenon lamp, λ > 520 nm | 6 h | 27 | 6 | 202 | ||
| 26 | [Dyad 2 pic]OTf, 0.05 mM | TEOA, 20% | DMF | Xenon lamp, λ > 520 nm | 8 h | 32 | 4 | 202 | ||
| 27 | [Dyad 3 pic]OTf, 0.05 mM | TEOA, 20% | DMF | Xenon lamp, λ > 520 nm | 7 h | 332 | 60 | 202 | ||
| 28 | Zn-1-Re, 0.05 mM | TEA, 5% | DMF | Mercury lamp, 200 W, λ > 375 nm | 24 h | 13 | 206 | |||
| 29 | Re-TXP-Zn, 0.05 mM | TEOA, 20% and BIH, 0.1 M | DMF | Xenon lamp, 200 W, λ > 450 nm | 24 h | 195 | 207 | |||
| 30 | ZnP-phen = Re, 0.05 mM | BIH, 0.05 M and PhOH 0.1 M | DMA | LED lamp, λ = 420 nm | 24 h | 900 | 99.9 | 208 | ||
| 31 | Mn(phen)(CO)3Br, 2 mM | ZnTPP, 0.5 mM | TEA, 0.1 M | CH3CN/H2O (20/1) | Xenon lamp, 500 W | 3 h | 119 (CO) | 210 | ||
| 19 (HCOOH) | ||||||||||
| 32 | Mn(HPEAB)(CO)3Cl, 0.0015 mM | ZnTPP, 0.0019 mM | TEA, 16.7% | CH3CN/H2O (95/5) | LED diode, λ = 625 nm | 100 min | 0.3 | 0.75 | 211 |
The same group, in an effort to increase the stability of the catalytic system, examined the performance of iron catalyst CAT in presence of a PS.188 When they used an iridium complex (Ir(ppy)3, Fig. 41) as PS, CO formation was observed with high efficiency (140 TON) and selectivity (93%) (Table 8, entry 5). However, Ir(ppy)3 in an expensive compound and for that reason, a cheaper organic chromophore such as 9-cyanoanthracene (9CNA, Fig. 41) was also studied. In this case the catalytic performance was reduced (60 TON), but the selectivity was even better (100%) (Table 8, entry 6). In both cases, visible light (λ > 400 nm) was employed for the excitation of the PS. Under these conditions the catalytic activity remained stable for more than 50 h of irradiation, indicating that no significant degradation of the iron catalyst CAT occurs. Rao et al. reported another approach to enhance the stability of the catalytic system.189 More specifically, they prepared a modified tetraphenyl iron porphyrin (Fe-p-TMA, Fig. 40) by introducing positively charged trimethylammonio groups at the para position of each phenyl ring. This catalyst reduces CO2 to CO with high selectivity (100%), under visible light irradiation (λ > 420 nm) in CH3CN without the assistance of a PS and remains stable for several days. In the presence of TEA as SED the CO formation reached 33 TON, while when they used 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as SED the TON increased to 63 (Table 8, entries 7 and 8). Catalyst Fe-p-TMA demonstrated increased activity compared to CAT, since the formed Fe(0)–CO2 adduct is stabilized more efficiently by through-space coulombic interactions between the positive charge of the trimethylamino substituents (+N(Me)3) and the partially negative charged oxygen atoms of the CO2. The positively charged trimethylammonio groups make catalyst Fe-p-TMA water soluble and based on this property, its catalytic performance was further examined in aqueous solution.190 Under visible light irradiation (λ > 420 nm) and with the assistance of a low-cost organic PS, such as purpurin (Fig. 41), Fe-p-TMA selectively reduce CO2 into CO with catalytic selectivity of 95% and turnover number up to 60 (Table 8, entry 9). The catalytic CO reduction was maintained for more than 48 h of irradiation and is limited mainly by the progressive degradation of the purpurin PS. Interestingly, the same group reported that Fe-p-TMA catalyst can also catalyze the eight-electron reduction of CO2 to CH4 upon visible light irradiation at ambient pressure and temperature.191 The catalytic system operates in CH3CN solution, using Ir(ppy)3 (Fig. 41) as PS and TEA as SED. After 102 h of irradiation, the gaseous products that formed were CO, CH4 and H2 with TONs of 367, 79 and 26 respectively (Table 8, entry 10). CO was the major product of the photocatalysis and CH4 production begins only after a large amount of CO has built up, indicating that CO is an intermediate in the CH4 formation process. The catalytic activity is retained for more than 4 days with no evidence for degradation of the sensitizer or catalyst, highlighting the exceptional stability of the system. Robert and Bonin and co-workers were further examined the efficiency of catalyst Fe-p-TMA in the presence of a modified iridium-based PS (Ir(ppy)2(bpy), Fig. 41).192 In CH3CN solution the main reduction product was CO (178 TON) and a considerable amount of CH4 (32 TON) was also produced (Table 8, entry 11). Under the same experimental conditions Ir(ppy)3 PS presented similar catalytic performance (Table 8, entry 12), indicating that Ir(ppy)2(bpy) is a suitable alternative PS. Three different amines (TEA, TEOA and DIPEA) were examined as SEDs and TEA demonstrated the best efficiency. Remarkably, the CO2 reduction to CO and CH4 was also observed in 70% aqueous solution (Table 8, entry 13). The TONs of both CO and CH4 were lower compared to the CH3CN solution due to the limited CO2 solubility in the catalytic solvent mixture and the instability of the PS upon prolonged irradiation. The same group reported that Fe-p-TMA catalyst retains its ability to perform the 8e−/8H+ reduction of CO2 to CH4, even after the replacement of the iridium based PSs with cheaper organic PSs.193 The phenoxazine-based organic PS (Phen2, Fig. 41) demonstrated efficient reduction of CO2 to CO and CH4 with TONs of 149 and 29, respectively (Table 8, entry 14). The catalysis was performed in DMF solution with TEA as SED and the catalytic system is stable for more than 4 days of continuous visible-light (λ > 435 nm) irradiation. Moreover, they noticed that the addition of an external acid such as 2,2,2-trifluoroethanol (TFE), increased the efficiency of the system since the production of both CO and CH4 was noticeably improved. Notably, when Phen2 was replaced by a phenazine-based PS (Phen1, Fig. 41), only CO formation was observed. This result was attributed to the lower reducing ability of Phen1, which hampers the reduction of CO2 beyond CO. The same group in a recent report evaluated six phenoxazine derivatives as visible light photosensitizers for the photo-initiated CO2 to CO reduction, using Fe-p-TMA as catalyst in CH3CN solution.194 The photcatalytic studies demonstrated that the production of CO is strongly related to the oxidation potential of the Phen sensitizer. This unexpected result indicates that photosensitizer regeneration by the SED is the rate-limiting step of this catalytic system. Stupp and co-corkers also examined the catalytic activity of Fe-p-TMA catalyst in combination with an amphiphilic hydroxyl-substituted perylene monoimide (PMI-OH, Fig. 41) chromophore.195 The water-soluble PMI-OH exhibits reversible structural, optical, and electrochemical properties in response to pH. The catalysis was performed in basic conditions using sodium ascorbate (SA) as SED and CO formation was observed with a TON of 30 (Table 8, entry 15). In a recent work, Han and co-workers reported the preparation of a highly active copper purpurin complex (CuPP, Fig. 41), which was employed as PS with an iron porphyrin (CAT, Fig. 40) as catalyst for visible-light-driven CO2 reduction.196 The coordination of the redox-active Cu center directly to the purpurin dye red-shifted the absorption maxima of purpurin to the visible region. Moreover, electrochemical studies revealed that the incorporation of Cu produces a much more reducing PS, facilitating challenging multielectron reductions. Under visible-light irradiation and in the presence of BIH as SED, this system achieves 16109 TON of CO from CO2 with 95% selectivity (Table 8, entry 16), which is among the highest reported for a homogeneous noble metal-free system.
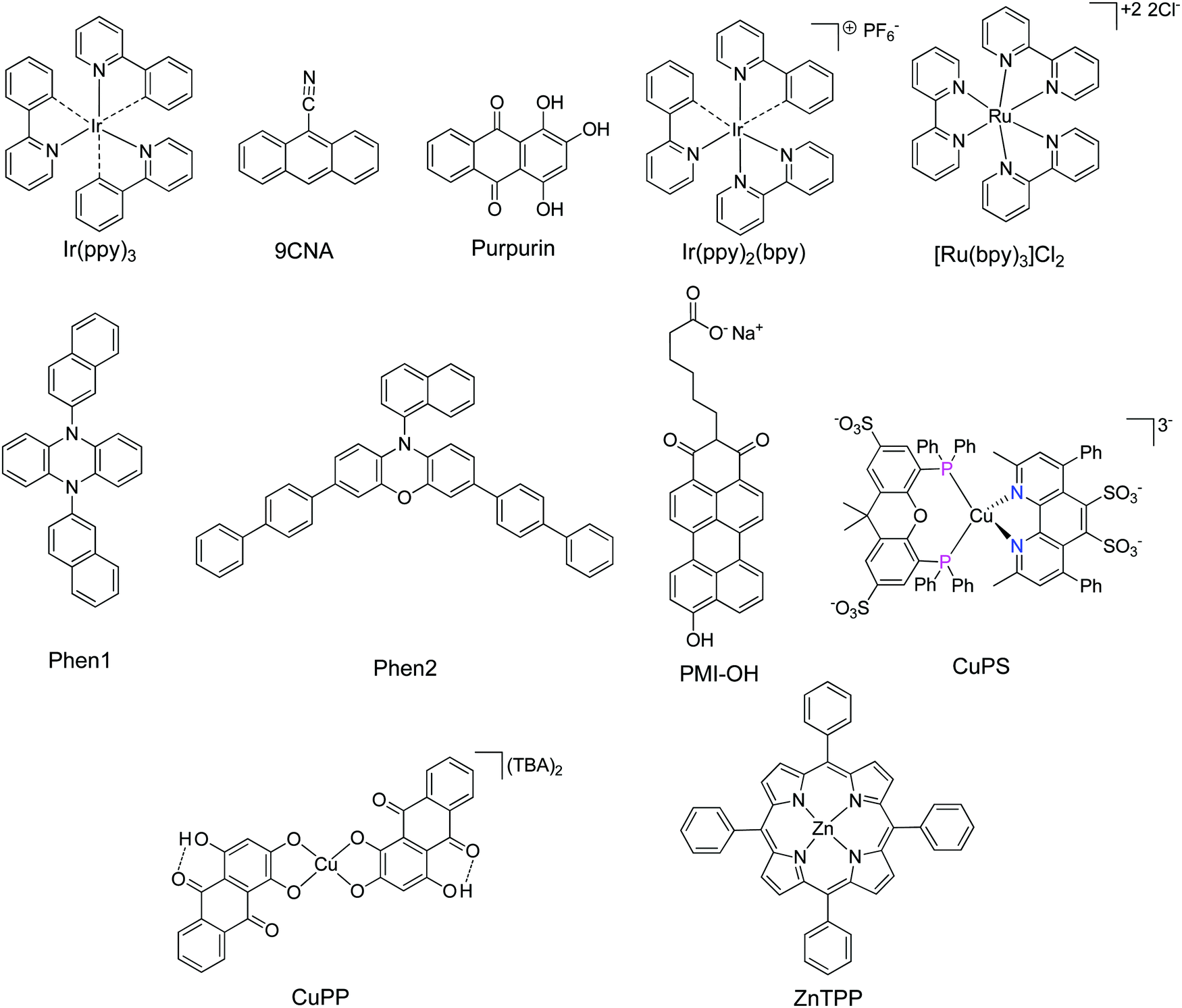 | ||
| Fig. 41 Structure of molecular derivatives employed as PS for CO2 conversion. | ||
Apart from iron porphyrins, cobalt metallated porphyrins have been used extensively as catalysts for the electo- or photo-catalytic reduction of CO2.197 Sakai and co-workers reported that the water soluble cobalt porphyrin CoTPPS (Fig. 42) achieves with high selectivity the CO2 to CO reduction in fully aqueous media.198 A ruthenium complex ([Ru(bpy)3]Cl2, Fig. 41) was utilized as PS and AA as SED. Under visible light irradiation (λ > 400 nm) CO formation was observed with high efficiency (926 TON) and selectivity (82%) (Table 8, entry 17). Interestingly, when they reduced the concentration of the catalyst from 10 μM to 0.5 μμ the TON increased to 4000, but the selectivity of the system was significantly reduced to 41% (Table 8, entry 18). The catalysis is active for 4 h and the degradation of the ruthenium PS is proposed as the major cause for the cease of photocatalysis. The same group successfully replaced the precious metal-based [Ru(bpy)3]Cl2 PS with an earth-abundant copper(I)-based water soluble PS (CuPS, Fig. 41).199 CoTPPS in the presence of CuPS in aqueous solution catalyzed the CO2 to CO reduction with high selectivity (90%), achieving 1085 TON (Table 8, entry 19). In an effort to explore the effect of the peripheral groups of the cobalt metalloporphyrin catalyst onto the CO2 photodriven reduction, they replaced the four sulfonatophenyl groups with N-methylpyridinium. Under the same experimental conditions, the new catalyst CoTMPyP (Fig. 42) demonstrated improved catalytic performance (2680 TON, Table 8, entry 20) compared to CoTPPS, but the CO selectivity over H2 formation was decreased from 90 to 77%. Electrochemical studies revealed that the multielectron chargeable character of CoTMPyP is responsible for the outstanding catalytic enhancement. Catalyst CoTMPyP can store up to 6-electrons and the final CO releasing step is assisted via intramolecular electron transfer from the reducing equivalents stored within the four methylpyridinium acceptor moieties. The same group in a later report examined two more positively charged cobalt catalysts, Co(TMAP) and Co(oTMPyP) (Fig. 42) in combination with the molecular CuPS PS.200 Catalyst Co(oTMPyP) presented significantly superior activity achieving efficient CO2 to CO reduction with a TON of 4000 after 12 h of irradiation (Table 8, entries 21 and 22).
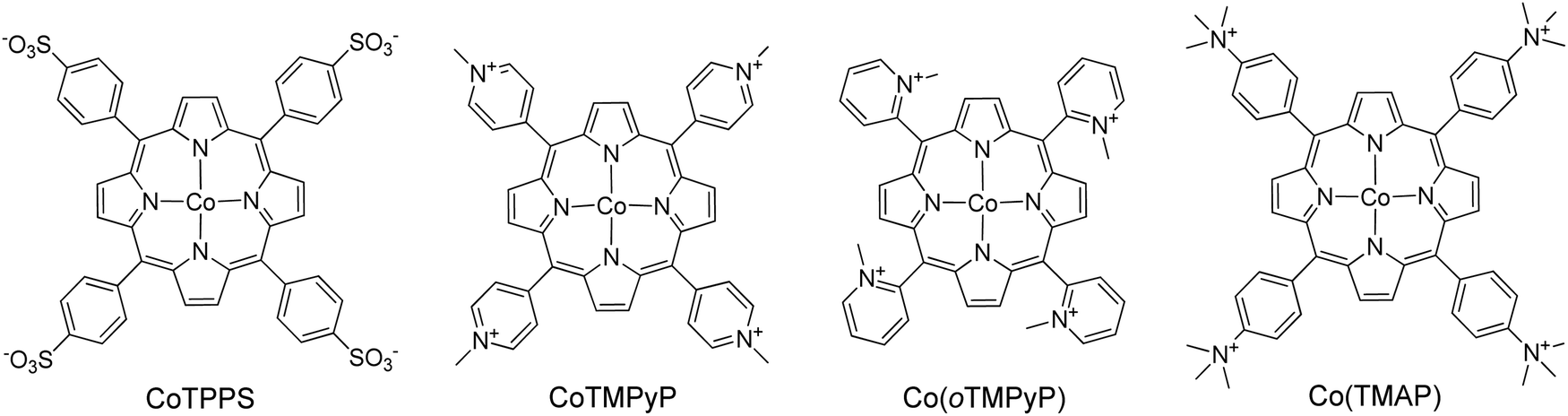 | ||
| Fig. 42 Structure of cobalt porphyrin molecular catalysts for CO2 reduction. | ||
In all the above examples the catalyst and the PS were not covalently connected and the electronic communication between the two components was usually controlled by the diffusion in the solution. Schwalbe and co-workers integrated the catalyst and the PS into a single molecular structure, in order to accelerate the electron transfer between them.201 They prepared two different dinuclear complexes (FeCl-1-Ru and Co-1-Ru, Fig. 43), where a Ru complex was covalently connected with a phenanthroline-extended porphyrin. The porphyrin ring was metallated with Fe or Co and served as catalyst, while the Ru moiety was the PS. Photocatalytic studies demonstrated that dyads FeCl-1-Ru and Co-1-Ru can reduce CO2 to CO in the presence of TEA as SED with relatively low activity (11.4 and 4.7 TON, respectively) (Table 8, entries 23 and 24). In both dyads, the covalently connected Ru complex increased the catalytic activity relative to the reference compounds where the Ru PS was absent. The Ru fragment shifted the reduction potentials of the catalysts to less negative values and thus the photochemical quenching reaction with the SED became more efficient, improving the CO2 reduction activity. Furthermore, they revealed that near UV-light (λ > 305 nm) was necessary for CO2 reduction, suggesting that in this system the Ru complex is not a “real” PS and probably through an antenna effect in the near UV region assists the photochemical reaction.
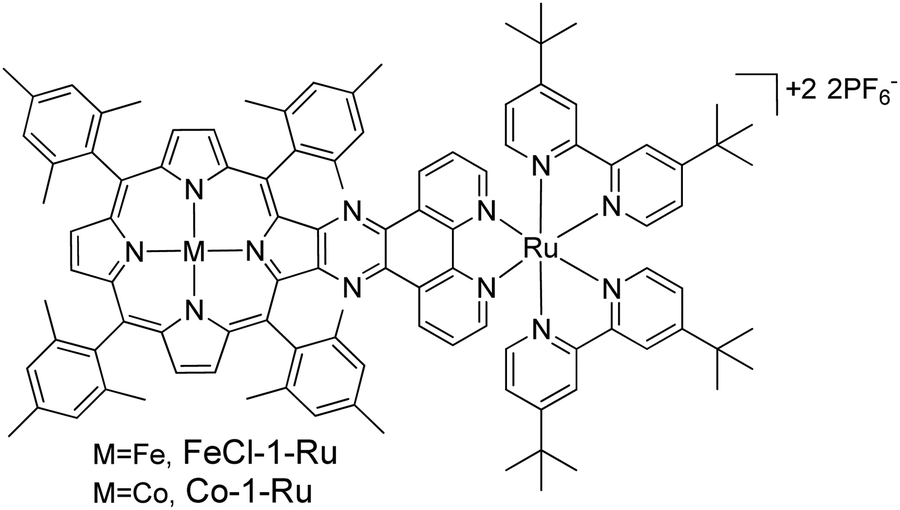 | ||
| Fig. 43 Structure of the PS-catalyst dyads FeCl-1-Ru and Co-1-Ru. | ||
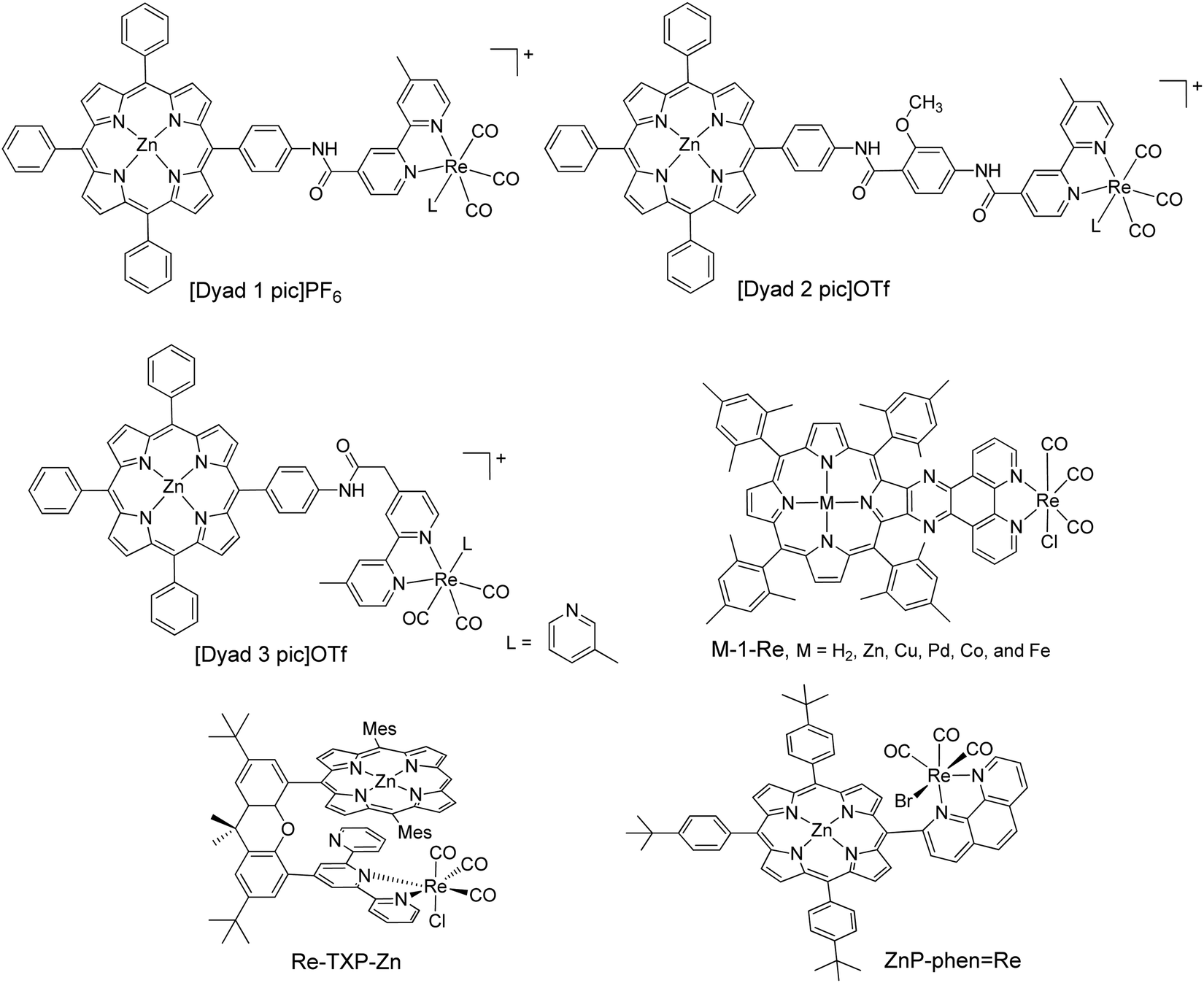 | ||
| Fig. 44 Structure of the porphyrin-Re dyads studies for CO2 reduction. | ||
Tschierlei, Schwalbe and co-workers developed a series of heterodinuclear complexes, where a porphyrin unit was connected with a rhenium phenanthroline tricarbonyl chloride catalytic center (Fig. 44), for application in photocatalytic CO2 to CO reduction.206 The linker between the two components was an extended delocalized π-system with partial electron storage ability and this was expected to enhance the catalytic performance. The porphyrin ring was metallated with various metals (H2, Zn, Cu, Pd, Co, and Fe), in an effort to examine the influence of the metal on the photochemical activity. Interestingly, the zinc derivative (Zn-1-Re, Fig. 44) was the most active compound in this series showing a TON of 13 after 24 h of illumination using a >375 nm cutoff filter (Table 8, entry 28), while all other complexes were inactive under these conditions. The superior activity of Zn-1-Re was attributed to the strong impact of the Zn metal on the nature of the excited states of the porphyrin unit, favoring the electron transfer process from the porphyrin chromophore to the Re catalyst.
The same group reported an analogue dyad (Re-TXP-Zn), consisting of a porphyrin unit as photosensitizer and a rhenium terpyridine unit as catalytically active site (Fig. 44).207 In this hetero-Pacman architecture the two moieties were connected together through a rigid xanthene backbone, which breaks the conjugation between the two components, but still can facilitate energy or electron transfer between the photosensitizer and the catalyst due to their close proximity. Photocatalytic experiments using BIH as SED revealed that the Re-TXP-Zn heterodimer catalyzes the CO2 to CO reduction, achieving a TON of 195 after 24 h of irradiation (Table 8, entry 29). The catalytic performance was reduced by changing the SED from BIH to TEA and by using the non-metallated porphyrin ring. Moreover, the intramolecular dyad presented higher catalytic performance compared to the intermolecular systems that consisted of a 1:1 mixture of the corresponding mononuclear analogues, indicating that the close proximity of the photosensitizer and catalytic active species appears to be highly advantageous.
Kuramochi et al. reported another chromophore-catalyst dyad (ZnP-phenRe), in which a rhenium phenanthroline tricarbonyl catalyst was directly connected with the meso-position of a zinc porphyrin (Fig. 44).208 The photocatalytic CO2 reduction studies were carried out in N,N-dimethylacetamide (DMA) solutions containing BIH as electron donor and phenol as proton source. The formation of CO was observed with high selectivity and activity, achieving more than 900 TONs (Table 8, entry 30). Interestingly, the catalytic activity stops after 24 h of irradiation, due to the consumption of BIH, while further addition of BIH to the reaction mixture regenerated the photocatalytic system, with the same catalytic rate. Emission spectroscopy studies revealed that porphyrin phosphorescence was efficiently quenched by BIH, suggesting that the electron transfer process proceeds via the T1 long-lived excited state of the porphyrin. The high durability of ZnP-phenRe dyad was attributed to the rapid electron transfer from the reduced zinc porphyrin to the Re complex, suppressing the undesired electron accumulation on the zinc porphyrin.
The same group continued their studies on the ZnP-phen = Re dyad and examined in more detail various basic parameters that influence the catalytic performance.209 Initially, the role of the Zn metal of the porphyrin ring was investigated. The dyad with the free base porphyrin presented significantly lower catalytic activity compared to the Zn analogue. This was assigned to the very fast decomposition of the non-metallated porphyrin, due to hydrogenation of the pyrrole double bonds. Moreover, the BIH was replaced by TEA, which is a weaker reductant and is more often used as SED in similar catalytic systems. The CO was produced with high selectivity, but the performance (TONs) was lower for more than one order of magnitude compared to the BIH. Finally, the effects of several proton sources (water, methanol, 2,2,2-trifluoroethanol (TFE), phenol, acetic acid), having different acidities, were examined. The presence of TFE and phenol enhanced significantly the catalytic activity with the trend TFE > PhOH > H2O ≈ MeOH, while acetic acid deactivated completely the catalytic system.
Replacement of Re-based catalysts with their Mn-analogues would be a huge step towards the development of more affordable and abundant catalysts. However, the Mn derivatives are photosensitive, undergoing decarbonylation after photoexcitation, and therefore the use of an additional PS is mandatory. Toward this goal, Zhang et al. reported a homogeneous catalytic system where a manganese phenanthroline tricarbonyl complex (Mn(phen)(CO)3Br, Fig. 45) was employed as catalyst and zinc tetraphenyl porphyrin (ZnTPP, Fig. 41) as photosensitizer.210 In this case the catalyst and the PS were not covalently connected and the electronic communication between the two components was controlled by the diffusion. Under visible light irradiation, CO2 could be reduced to CO (119 TON) as gas product and formic acid (19 TON) as liquid product (Table 8, entry 31).
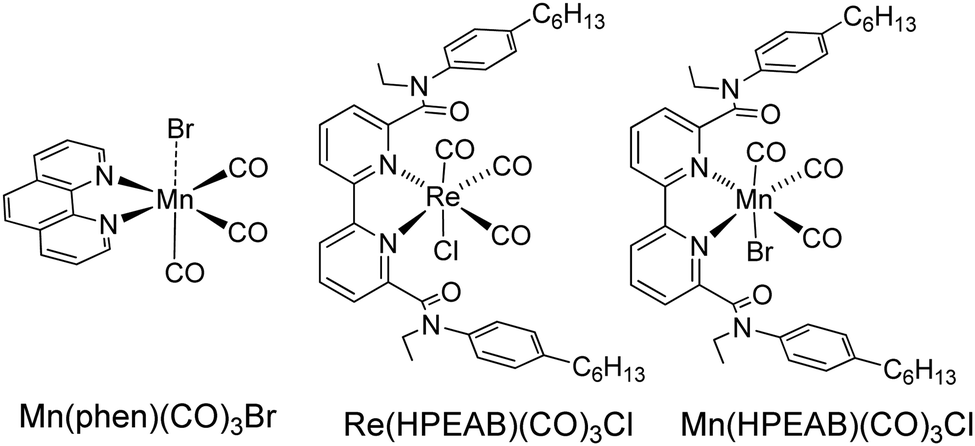 | ||
| Fig. 45 Structure of molecular Mn and Re catalysts for CO2 reduction. | ||
Shipp et al. synthesized two molecular Re and Mn tricarbonyl complexes bearing a bipyridyl ligand functionalized with sterically hindering substituents (Fig. 45), and their propensity to act as catalysts for the photochemical reduction of CO2 has been examined.211 The electron-withdrawing amide groups were selected to reduce the reduction potential of the catalysts, allowing the use of less strongly-reducing PS. No photochemical CO2 reduction was observed for the rhenium complex (Re(HPEAB)(CO)3Br) in the presence of ZnTPP (Fig. 41) as PS and TEA as SED. This was attributed to the short (3.6 ns) triplet excited state lifetime of the Re catalyst, which prevents the bimolecular diffusion-controlled electron transfer. Notably, the manganese complex (Mn(HPEAB)(CO)3Br, Fig. 45) can act as a CO2 to CO reduction catalyst after photosensitization by ZnTPP under red light irradiation (λ > 600 nm), achieving 0.3 TONs (Table 8, entry 32). The relatively low catalytic activity is likely due to inefficient reduction of the catalyst by the reduced porphyrin, since this process was only weakly thermodynamically favorable.
The problems associated with the application of molecular catalysts in the visible-light driven reduction of CO2 include, among others, low product selectivity and simultaneous hydrogen production. Biocatalysts are promising candidates to overcome these issues, due to their excellent reaction and substrate selectivity, especially for the reduction and utilization of CO2. Thus, several photocatalytic CO2 reduction systems were developed based on enzymes that served as biocatalysts.212 Amao et al. reported the efficient reduction of CO2 to formic acid with a catalytic system where formate dehydrogenase (FDH) was employed as catalyst, a water-soluble zinc porphyrin (ZnTPPS) as PS and TEOA as SED (Fig. 46a).213 In this system the presence of an extra electron carrier molecule facilitates the electronic communication between the PS and the catalyst and enhances the catalytic activity. Thus, a series of 2,2′-bipyridinium (2,2′-BPs) salt derivatives (Fig. 46b) were used as electron carriers to investigate their effect in the light-induced conversion of CO2.214 The photocatalytic studies demonstrated that redox potential and dihedral angle of 2,2′-BPs influence the activity. The highest formic acid production rate (70 μM h−1) was obtained with 1,10-ethylene-2,20-bipyridinium dibromide (DB, Fig. 46b), since it possess the lowest redox potential and the smallest dihedral angle.
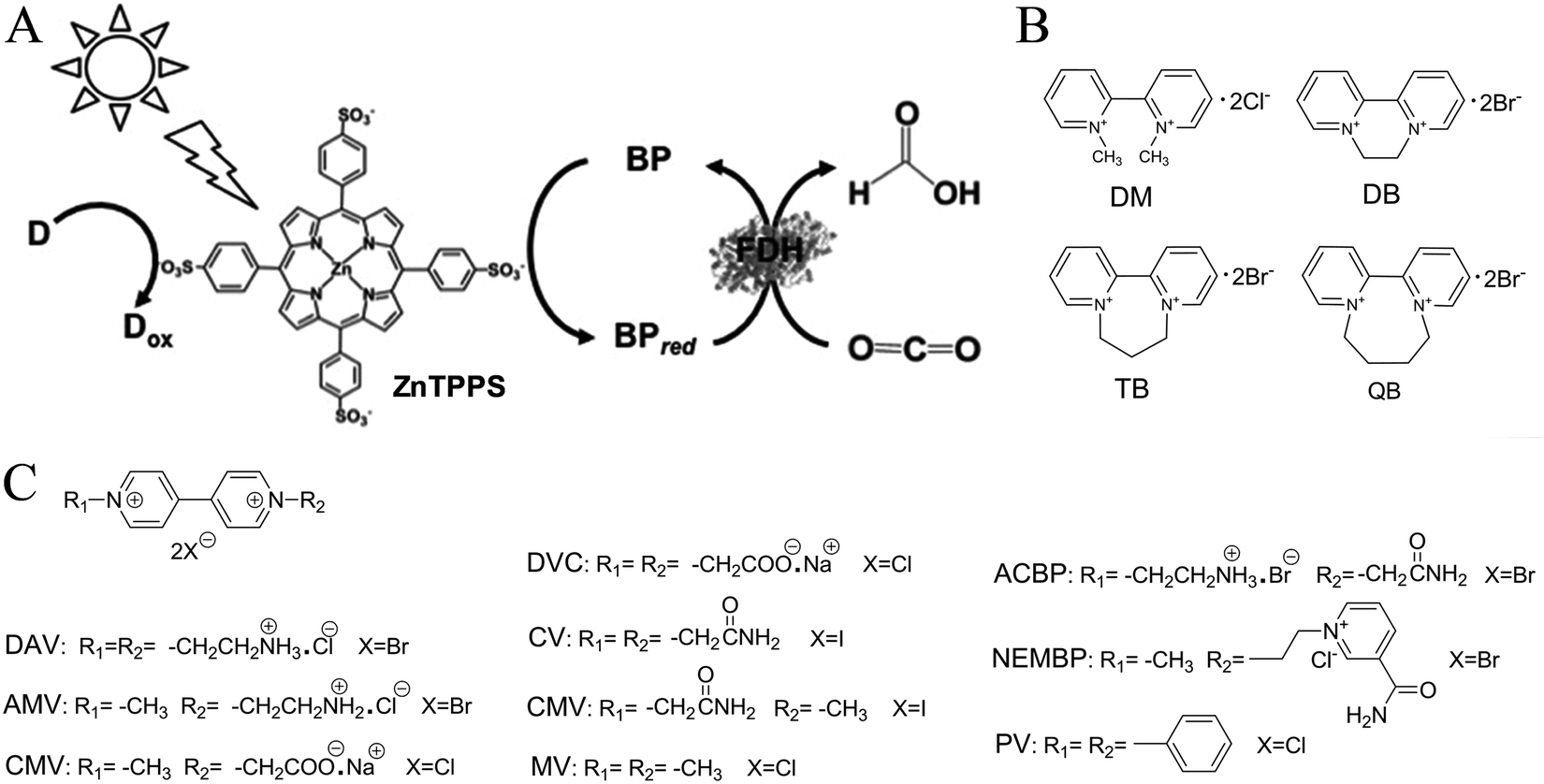 | ||
| Fig. 46 (A) Schematic representation of the visible-light driven redox system for the reduction of CO2 to formate using FDH enzyme, (B) chemical structures of various 2,2′-bipyridinium salts. (C) Various 4,4′-bipyridinium salts. | ||
Interestingly, when the electron carrier DB was replaced by 1,1′-diaminoethyl-4,4′-bipyridinium salt (DAV, Fig. 46c), the production rate of formic acid increased to 120 μM h−1.215 The improvement of CO2 photoreduction was ascribed to the higher affinity of DAV for FDH enzyme and its stronger interaction with the ZnTPPS porphyrin, due to the presence of the positively charged amino groups. The same group in an effort to improve the catalytic performance of this system examined a series of viologen-based 4,4′-bipyridinium (4,4′-BPs) salts (Fig. 46c) as electron carriers.216 The derivatives with the cationic aminoethyl-groups (DAV and AMV) presented higher formic acid production rates (120 and 100 μM h−1, respectively) compared to the 4,4′-BPs with the anionic carboxymethyl-groups (CMV and DCV, 50 and 39 μM h−1, respectively). Interestingly, this catalytic system retains its activity even in ionic liquid media.217 Two viologen derivatives with carbamoyl groups (CV and CMV, Fig. 46c) were also synthesized and applied as electron carriers to the FDA-based photoinduced CO2-formic acid conversion system.218 Both of them presented higher catalytic activity compared to the non-substituted MV (Fig. 46c). However, the compound with only one carbamoyl group (CMV) was more efficient compared to di-substituted CV, achieving formic acid production rates similar to DAV. The above results indicate that the presence of aminoethyl or carbamoyl group in 4,4′-BPs improves the catalytic performance. Thus, Miyaji and Amao prepared the ACBP (Fig. 46c) electron carrier, which possess in its structure both the amino and the carbamoyl group.219 Nevertheless, the photocatalytic formation of formic acid, in presence of ACBP was very low, even lower than that observed for MV. This was attributed to the low energy level of the singly occupied molecular orbital (SOMO) of ACBP, which influences negatively the reduction rate of CO2. The same group prepared a nicotinamide-modified viologen derivative (NEMBP, Fig. 46c) and was applied as electron carrier.220,221 The nicotinamide group was introduced in an effort to enhance the affinity of NEMBP with FDH, since the nicotinamide group is present in the structure of the natural occurring NAD+ co-enzyme. However, the NEMBP derivative presented disappointing results in all kinetic parameters for CO2 to formic acid reduction, most likely due to the unfavorable SOMO energy level. Similarly, the visible-light induced formation of malic acid from pyruvic acid and CO2 with the application of malic enzyme (ME) as biocatalyst was also reported from the same group.222,223 ME is an attractive biocatalyst for the carbon–carbon bond formation, using CO2 as a feedstock, since it catalyzes the insertion of CO2 as a “carboxy group” into pyruvic acid. In this system, ZnTPPS was employed as PS, TEOA as SED and PV (Fig. 46c) as electron mediator. In the presence of magnesium ions (co-factor for ME) the formation of malic acid was observed, while in the catalytic system without Mg2+ only the intermediate oxaloacetic acid was obtained.
The conversion of CO2 into methanol is of great importance since methanol is a fuel with high energy density and is an important precursor for the industrial synthesis of chemicals such as plastics and silicone. Amao and Kataoka reported the photocatalytic conversion of CO2 to methanol with a system consisting of three different dehydrogenases (formate dehydrogenase (FDH), aldehyde dehydrogenase (AldDH) and alcohol dehydrogenase (ADH), Fig. 47a).224 In this system a water-soluble porphyrin (H2TPPS) was used as PS, TEOA was the SED, MV was employed as electron mediator and the production of methanol proceeds via formate and formaldehyde as intermediates. Under visible-light irradiation, the formation of methanol was observed with very low selectivity since the major product of the photoreduction was formic acid. Ji et al. followed the same biocatalytic approach for the photo-activated reduction of CO2 to methanol.225 However, instead of a simple porphyrin derivative, they utilized a macromolecular structure (TCPP/EY4/Rh4, Fig. 47b) as PS. The preparation of TCPP/EY4/Rh4 hybrid involves the amidation of the tetra carboxy-porphyrin (H2TCPP) by melamine, followed by sensitization with eosin Y (EY), and finally the covalent linkage of a rhodium-based organometallic complex [Cp*RhCl2]2. The fabricated TCPP/EY4/Rh4 macromolecule was found to self-assemble into supramolecular assemblies in water through π–π stacking and hydrogen bonding, mimicking the structure and function of chlorosomes in green photosynthetic bacteria. Photocatalytic studies in the presence of TEOA, demonstrated an efficient photogeneration of NADH. The sensitization of the porphyrin core by EY improved the absorbance in the visible region, while the Rh complex served as mediator, accelerating the electron transfer efficacy, and eliminating the charge recombination. When this NADH photoregeneration process was coupled with a mixture of three dehydrogenases (FDH, AldDH and ADH), 38 μM methanol was synthesized from CO2 after 2 h of visible light irradiation.
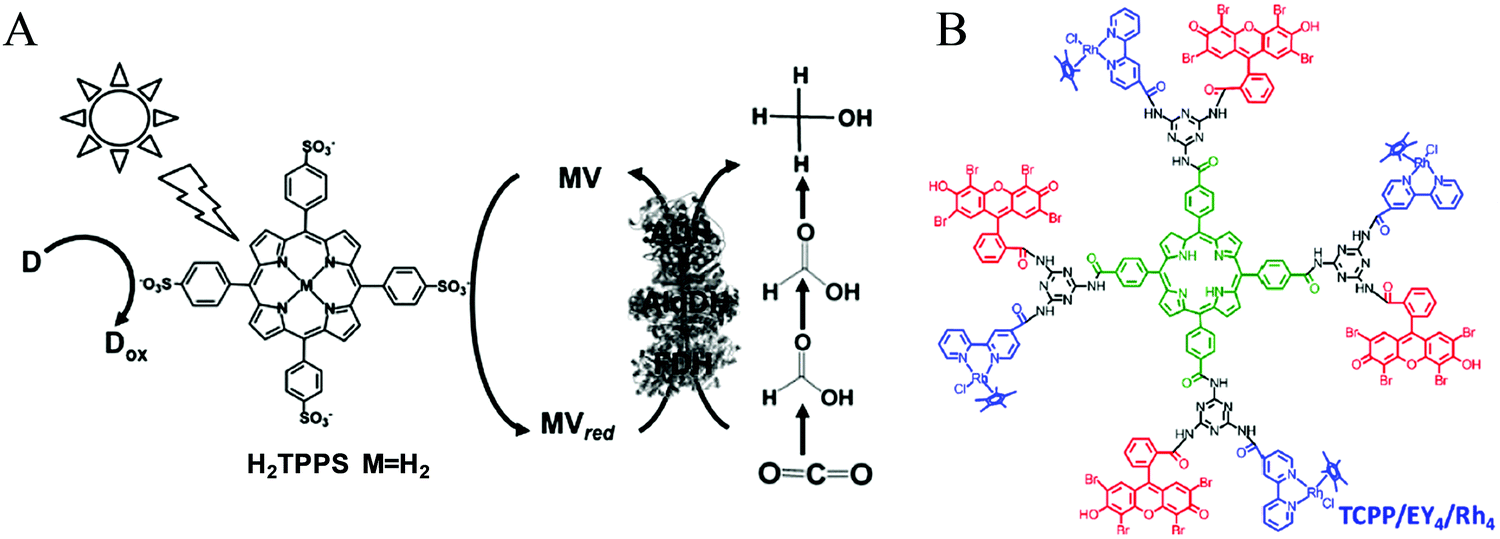 | ||
| Fig. 47 (A) Schematic representation of the visible-light driven redox system for the reduction of CO2 to methanol using three different dehydrogenases FDH, AldDH and ADH. Reproduced from ref. 224, with permission from Elsevier, Copyright 2018. (B) Chemical structure of the macromolecular hybrid TCPP/EY4/Rh4. | ||
3.2 Heterogeneous systems
Although homogeneous catalytic systems display high activity and selectivity in the photocatalytic CO2 reduction, their practical application is limited due to the difficulties in catalyst separation from the reaction mixture and recycling.226,227 One promising approach to address this issue is to anchor homogeneous molecular photocatalysts onto a solid support or to combine homogeneous catalysts with metal–organic frameworks (MOFs). The resulting catalytic systems may be considered as “heterogenized” homogeneous systems since they possess the advantages of both homogeneous and heterogeneous catalysts.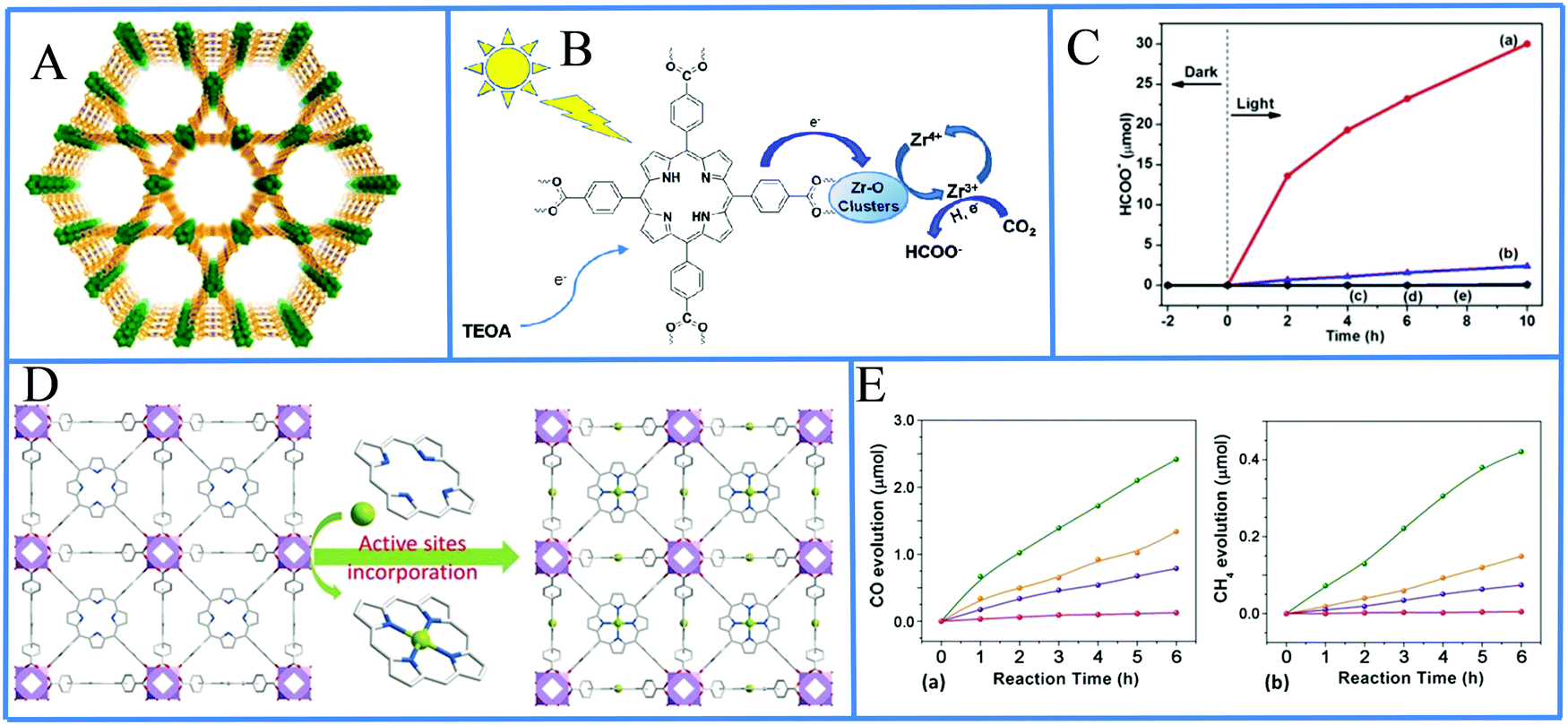 | ||
| Fig. 48 (A) View of the 3D network of PCN-222. Reproduced from ref. 228, with permission from American Chemical Society, Copyright 2015. (B) Proposed mechanism for photocatalytic CO2 reduction over PCN-222 under visible-light irradiation. Reproduced from ref. 228, with permission from American Chemical Society, Copyright 2015. (C) Amount of HCOO− produced as a function of the time of light irradiation over (a) PCN-222 and (b) H2TCPP. Reproduced from ref. 229, with permission from Royal Society of Chemistry, Copyright 2020. (D) View of the 3D network of MOF-525-Co featuring a highly porous framework and incorporated active sites. Reproduced from ref. 231, with permission from John Wiley and Sons, Copyright 2016. (E) Time time-dependent (a) CO and (b) CH4 evolution over MOF-525-Co (green), MOF-525-Zn (orange) and MOF-525 (purple) photocatalysts. Reproduced from ref. 231, with permission from John Wiley and Sons, Copyright 2016. | ||
| Entry | MOF name | Metal node | Linker | Cocatalyst or additives | SED | Solvent | Light source | Major products Rate | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | PCN-222 | Zr | H2TCPP | — | TEOA, 9% | CH3CN | Xenon lamp, 300 W, λ > 420 nm | HCOO−, 60 μmol g−1 h−1 | 228 |
| 2 | PCN-222-Zn | Zr | H2TCPP | — | Ethylene glycol, 20 mM | H2O | Xenon lamp, 300 W, λ > 420 nm | HCOO−, 120 μmol g−1 h−1 | 229 |
| 3 | PCN-222(Fe) | Zr | FeTCPP | — | TEOA, 9% | CH3CN | Xenon lamp, 500 W, λ > 420 nm | HCOO−, 347 μmol g−1 h−1 | 230 |
| 4 | PCN-222(Co) | Zr | CoTCPP | — | TEOA, 9% | CH3CN | Xenon lamp, 500 W, λ > 420 nm | HCOO−, 313 μmol g−1 h−1 | 230 |
| 5 | PCN-222(Ni) | Zr | NiTCPP | — | TEOA, 9% | CH3CN | Xenon lamp, 500 W, λ > 420 nm | HCOO−, 373 μmol g−1 h−1 | 230 |
| 6 | PCN-222(Cu) | Zr | CuTCPP | — | TEOA, 9% | CH3CN | Xenon lamp, 500 W, λ > 420 nm | HCOO−, 333 μmol g−1 h−1 | 230 |
| 7 | MOF-525-Co | Zr | CoTCPP | — | TEOA, 20% | CH3CN | Xenon lamp, 300 W, λ > 400 nm | CO, 201 μmol g−1 h−1 | 231 |
| CH4, 36.8 μmol g−1 h−1 | |||||||||
| 8 | MOF-525-Zn | Zr | ZnTCPP | — | TEOA, 20% | CH3CN | Xenon lamp, 300 W, λ > 400 nm | CO, 112 μmol g−1 h−1 | 231 |
| CH4, 11.6 μmol g−1 h−1 | |||||||||
| 9 | MOF-525 | Zr | H2TCPP | — | TEOA, 20% | CH3CN | Xenon lamp, 300 W, λ > 400 nm | CO, 64 μmol g−1 h−1 | 231 |
| CH4, 6.2 μmol g−1 h−1 | |||||||||
| 10 | Rh-PMOF-1 | Zr | Rh(TCPP)Cl | — | TEOA, 20% | CH3CN | Xenon lamp, 300 W, λ > 400 nm | HCOO−, 78 μmol g−1 h−1 | 121 |
| 11 | ZrPP-1-Co | Zr | CoTHPP | — | TEOA, 20% | CH3CN | Xenon lamp, 300 W, λ > 420 nm | CO, 14 μmol g−1 h−1 | 233 |
| CH4, 0.5 μmol g−1 h−1 | |||||||||
| 12 | PCN-138 | Zr | H2TCPP and TBTB | — | TIPA, 20% | H2O | Xenon lamp, 300 W, λ > 420 nm | HCOO−, 125 μmol g−1 h−1 | 234 |
| 13 | PCN-224(Cu) | Zr | CuTCPP | — | — | H2O | Xenon lamp, 300 W, λ > 300 nm | CO, 3.72 μmol g−1 h−1 | 235 |
| CH4, 1.36 μmol g−1 h−1 | |||||||||
| 14 | PCN-224(Cu)/TiO2 | Zr | CuTCPP | TiO2 NPs | — | H2O | Xenon lamp, 300 W, λ > 300 nm | CO, 37.21 μmol g−1 h−1 | 235 |
| CH4, 0.21 μmol g−1 h−1 | |||||||||
| 15 | CTU/TiO2 | Zr | CuTCPP | TiO2 NPs | — | H2O | Xenon lamp, 300 W, λ > 300 nm | CO, 31.32 μmol g−1 h−1 | 236 |
| CH4, 0.15 μmol g−1 h−1 | |||||||||
| 16 | PCN-221(Fe) | Zr | FeTCPP | — | H2O, 1,2% | EtOAc | Xenon lamp, 300 W, λ > 400 nm | CO, 0.52 μmol g−1 h−1 | 237 |
| CH4, 1.52 μmol g−1 h−1 | |||||||||
| 17 | MAPbI3@PCN-221(Fe) | Zr | FeTCPP | Perovskite QDs | H2O, 1.2% | EtOAc | Xenon lamp, 300 W, λ > 400 nm | CO, 14.16 μmol g−1 h−1 | 237 |
| CH4, 6.24 μmol g−1 h−1 | |||||||||
| 18 | PMOF/Re | Zr | ZnTCPP | Re(bpy) catalyst and 3% TFE | BIH | DMF | LED lamp, 60 W, λ > 500 nm | CO, 2204 TON or 535 μmol g−1 h−1 | 238 |
| 19 | Al-PMOF | Al | AlTCPP | — | TEOA, 16.7% | CH3CN | Mercury lamp, 3 × 125 W | HCOO−, 165 μmol g−1 h−1 | 240 |
| 20 | NH2-rGO/Al-PMOF | Al | AlTCPP | NH2-rGO | TEOA, 16.7% | CH3CN | Mercury lamp, 3 × 125 W | HCOO−, 685 μmol g−1 h−1 | 240 |
| 21 | Co/PMOF | Co | CoTCPP | — | TEOA, 16.7% | CH3CN | Mercury lamp, 3 × 125 W | HCOO−, 77 μmol g−1 h−1 | 241 |
| 22 | PMOF 1 | Cd | H2TCPP | — | TEA, 20% | CH3CN | Xenon lamp, 300 W, λ > 420 nm | CO, 10 μmol g−1 h−1 | 239 |
| 23 | PMOF 1 | Cd | H2TCPP | Ru(bpy)3Cl2 | TEA, 20% | CH3CN | Xenon lamp, 300 W, λ > 420 nm | CO, 56 μmol g−1 h−1 | 239 |
| 24 | In | H2TCPP | — | L-AP | EtOAc | Xenon lamp, 300 W, λ > 400 nm | CO, 144 μmol g−1 h−1 | 242 | |
| 25 | D-TiMOF | Ti | ZnTCPP and NH2BDC | — | TEOA, 20% | CH3CN | Xenon lamp, λ > 350 nm | CO, 59.55 μmol g−1 h−1 | 244 |
| 26 | Zn/PMOF | Zn | H2TCPP | — | H2O | Gas phase | Mercury lamp, 300 W | CH4, 8.7 μmol g−1 h−1 | 245 |
| 27 | PCN-601 | Ni | NiTCPP | — | H2O | Gas phase | Xenon lamp, λ > 410 nm | CO, 6 μmol g−1 h−1 | 246 |
| CH4, 10.1 μmol g−1 h−1 | |||||||||
| 28 | 2D/Zn-MOF | Zn | ZnTCPP | ZIF-67 | TEOA, 16.7% | CH3CN/MeOH (4/1) | Xenon lamp, λ > 420 nm | CO, 117.8 TON | 248 |
| 29 | 3D/Zn-MOF | Zn | ZnTCPP | ZIF-67 | TEOA, 16.7% | CH3CN/MeOH (4/1) | Xenon lamp, λ > 420 nm | CO, 63.6 TON | 248 |
| 30 | PPF-3 | Co | H2TCPP | Au NPs | EtOH, 20% | CH3CN | Xenon lamp, 300 W, λ > 400 nm | HCOO−, 42.7 μmol g−1 h−1 | 250 |
| 31 | g-CNQDs/PMOF | Co | CoTCPP | g-CNQDs | TEOA, 20% | CH3CN/H2O (3/1) | Mercury lamp, 300 W, λ > 420 nm | CO, 16.1 μmol g−1 h−1 | 252 |
| CH4, 6.86 μmol g−1 h−1 |
Ye and co-workers followed a similar approach and prepared MOF-525 using H2TCPP as linker and Zr6 oxo-clusters as metal nodes.231 Additionally, they post-metallated the porphyrin units with Co and Zn, generating the isostructural MOF-525-Co and MOF-525-Zn, respectively (Fig. 48d). The incorporation of metal centers into porphyrin rings of PMOFs is a promising approach to fabricate stable single-atom catalysts for CO2 photoreduction. In both cases the presence of the metal centers increased significantly the CO2 adsorption ability of the PMOFs. Photocatalytic CO2 reduction studies were conducted using TEOA as SED and resulted in the efficient conversion of CO2 to CO and CH4. MOF-525-Co showed the highest catalytic activity (Fig. 48e), achieving a CO evolution rate of 201 μmol g−1 h−1, and a CH4 evolution rate of 36.8 μmol g−1 h−1 (Table 9, entry 7). These values are much higher compared to MOF-525-Zn and MOF-525 (Table 9, entries 8 and 9). The MOF-525-Co composite presents enhanced catalytic performance since the CO2 reduction takes place not only in the Zr clusters but also in the Co metal centers. Additionally, MOF-525-Co exhibits excellent stability and retained its photocatalytic activity for three recycling cycles.
Liu et al. reported the preparation of Rh-PMOF-1 using Zr6 clusters as metal nodes and a tetra carboxy-porphyrin metallated with Rh (RhTCPP) as linker (Fig. 49a).232 In this case the porphyrin was already metallated before the construction of the PMOF. Under light irradiation Rh-PMOF-1 showed excellent catalytic activity towards CO2 reduction, leading to the formation of formate ion with up to 99% selectivity and a rate of 78 μmol g−1 h−1 (Table 9, entry 10). The crystalline catalyst can be isolated from the reaction mixture by centrifugation and can be reused for 3 times without loss of activity. In this system, the Rh-porphyrin has a dual role, since it acts as antenna absorbing visible light and injecting electrons to the Zr6 oxo-clusters, and also as a catalytic center carrying out the CO2 reduction.
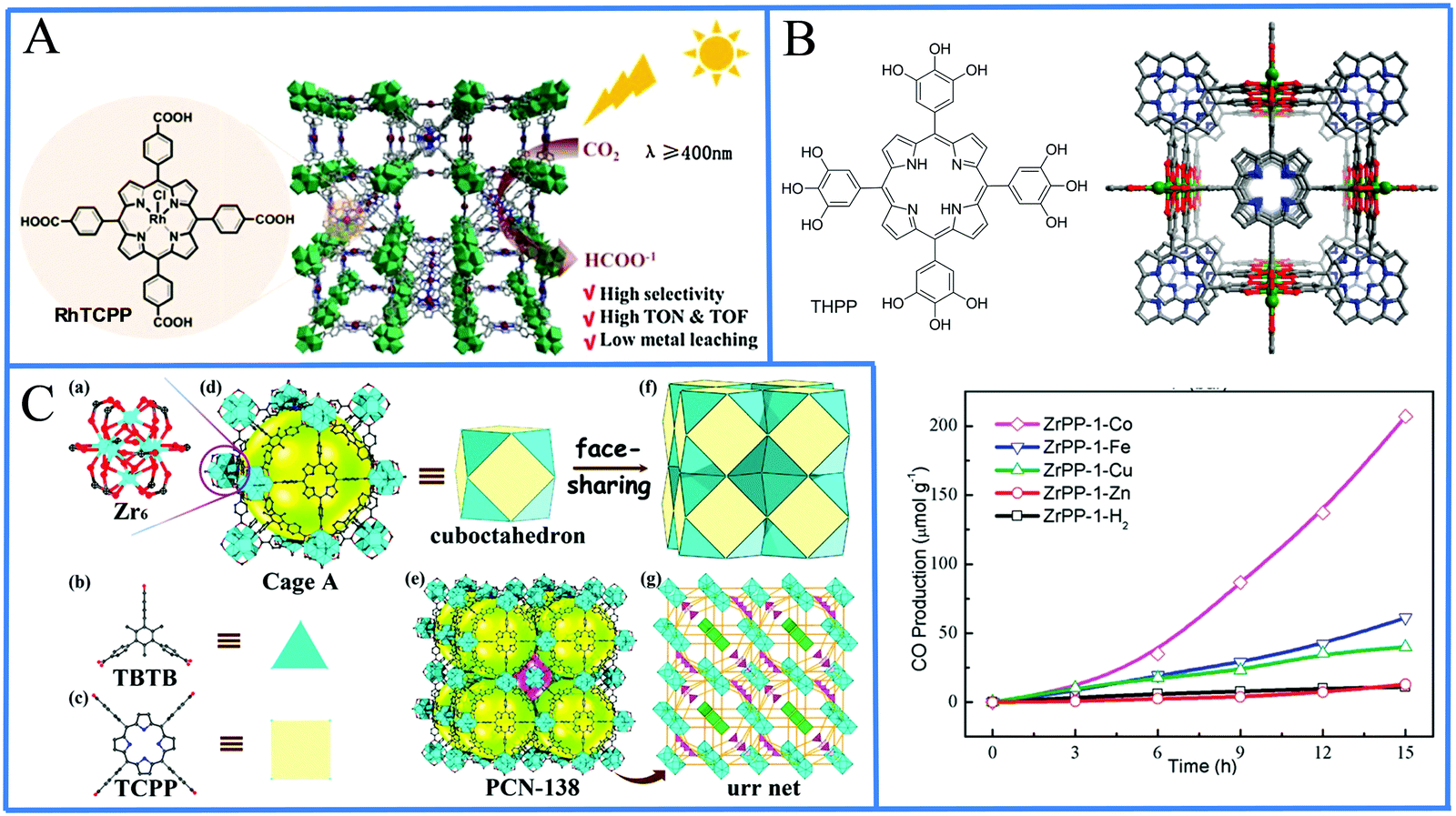 | ||
| Fig. 49 (A) Chemical structure of RhTCPP and representation of catalytic behavior of Rh-PMOF-1. Reproduced from ref. 232, with permission from Elsevier, Copyright 2018. (B) Chemical structure of THPP, 3D view ZrPP-1 PMOF and time courses of CO evolution from CO2 photoreduction with catalysts ZrPP-1-M under visible-light irradiation. Reproduced from ref. 233, with permission from John Wiley and Sons, Copyright 2018. (C) Structure of PCN-138. Reproduced from ref. 234, with permission from American Chemical Society, Copyright 2019. | ||
Chen et al. reported the preparation of ZrPP-1 PMOF where Zr6 clusters were also used as metal nodes.233 However, instead of the common tetra carboxy-porphyrins, polyphenolate-decorated porphyrin (THPP) was employed as linkers (Fig. 49b). These unusual PMOFs based on phenolic porphyrins were very robust in acidic media, and more impressively remain intact even upon immersion in saturated NaOH solution. Since photocatalysis is strongly associated with the metal inside the porphyrin core, for the construction of the PMOFs apart from the free base porphyrin a series of metallated analogues (with Zn, Cu, Fe and Co) were also employed as linkers, to study their effects on the catalytic properties. Upon light illumination, no noticeable CO evolution was observed for the non metallated (ZrPP-1-H2) and the Zn metallated (ZrPP-1-Zn) PMOF. The corresponding PMOFs metallated with Cu and Fe displayed efficient photocatalytic activity for the CO2 to CO reduction (Fig. 49b). The Co-substituted analogue (ZrPP-1-Co) exhibited the highest efficiency with a CO generation rate of 14 μmol g−1 h−1 and also a small amount of CH4 (0.5 μmol g−1 h−1), was detected in the gaseous and liquid phases (Table 9, entry 11). These catalysts exceptionally stable and can be recycled without appreciable loss of activity. Based on theoretical calculations they proposed the following catalytic mechanism: the uniformly distributed porphyrins absorb incident photons and transfer generated electrons to the Co2+ cations. Then successive reductive quenching steps, from Co2+ to Co+ and finally to Co0, occur with the aid of TEOA as SED. The Co0 species can activate CO2 (with structural transition from the linear to the bent) to form an excited adduct Co2+–CO, which dissociates upon irradiation and liberates CO.
Qiu et al. were also used Zr6 oxo-clusters as metal nodes for the preparation of the mixed-ligand MOF PCN-138 (Fig. 49c).234 In this MOF two different carboxy-derivatives were employed as linkers, more specifically the free base tetra carboxy porphyrin (H2TCPP) and the 4,4′,4′′-(2,4,6-trimethylbenzene-1,3,5-triyl)tribenzoate (TBTB). The combination of these two ligands resulted in the formation of an unusual cuboctahedron Archimedean solid, which possess large cages facilitating the CO2 adsorption. PCN-138 presented efficient photocatalytic activity toward CO2 reduction in the presence of triisopropanolamine (TIPA) as SED, producing formate as the only product with a rate of 125 μmol g−1 h−1 (Table 9, entry 12). Recycling studies suggested that PCN-138 is stable and can be reused without an obvious decrease in the activity during four continuous runs. In this system, the porphyrins act as PS absorbing the irradiation and transferring photoelectrons to the Zr–oxo clusters, while the Zr metal centers act as catalysts enabling the reduction of CO2 to HCOO−.
Wang and co-workers reported the preparation of PCN-224(Cu) with the employment of copper metallated tetra carboxy-porphyrins (CuTCPP) as linkers and Zr6 clusters as metal nodes (Fig. 50a).235 Under irradiation PCN-224(Cu) catalyzed the CO2 reduction, producing CO and CH4 with a rate of 3.72 and 1.36 μmol g−1 h−1, respectively (Table 9, entry 13). Interestingly, the in situ integration of TiO2 NPs resulted in the formation of a new material PCN-224(Cu)/TiO2 that presented significantly improved catalytic efficiency and selectivity, producing CO with a rate of 37.21 μmol g−1 h−1 (Table 9, entry 14). The enhanced photocatalytic performance in the presence of TiO2 NPs was attributed to the operation of a Z-scheme mechanism, which suppressed the unfavorable recombination of charge carriers. The same group synthesized a similar composite material CTU/TiO2.236 The only difference compared to PCN-224(Cu)/TiO2 is the use of two different carboxy-linkers (CuTCPP and terephthalic acid (BDC)) instead of only one (CuTCPP). However, this modification did not improve the catalytic performance, since the CO production rate for CTU/TiO2 was 31.32 μmol g−1 h−1 (Table 9, entry 15). Wu et al. reported the construction of PCN-221(Fe), using also Zr6 metal nodes and iron metallated carboxy-porphyins as linkers.237 PCN-221(Fe) exhibited low CO2 reduction activity producing CO and CH4 with a rate of 0.52 and 1.52 μmol g−1 h−1, respectively (Table 9, entry 16). Noteworthy, the encapsulation of low-cost CH3NH3PbI3 (MAPbI3) perovskite QDs into the pores of PCN-221(Fe) by a sequential deposition route (Fig. 50b) significantly enhanced the photocatalytic performance. The novel composite material MAPbI3@PCN-221(Fe) catalyzed the CO2 reduction to CO and CH4 with conversion rates of 14.16 and 6.24 μmol g−1 h−1, respectively (Table 9, entry 17), using water as electron source. The close contact of QDs to the Fe catalytic sites, enables the photogenerated electrons to transfer rapidly form the QDs to the Fe catalytic sites and enhance the overall photocatalytic activity. Moreover, the composite photcatalyst MAPbI3@PCN-221(Fe) displayed increased stability compared to the parent PCN-221(Fe). This was attributed to the competitive light absorption between perovskite QDs and PCN-221(Fe), which can decrease the photodegradation of PCN-221(Fe).
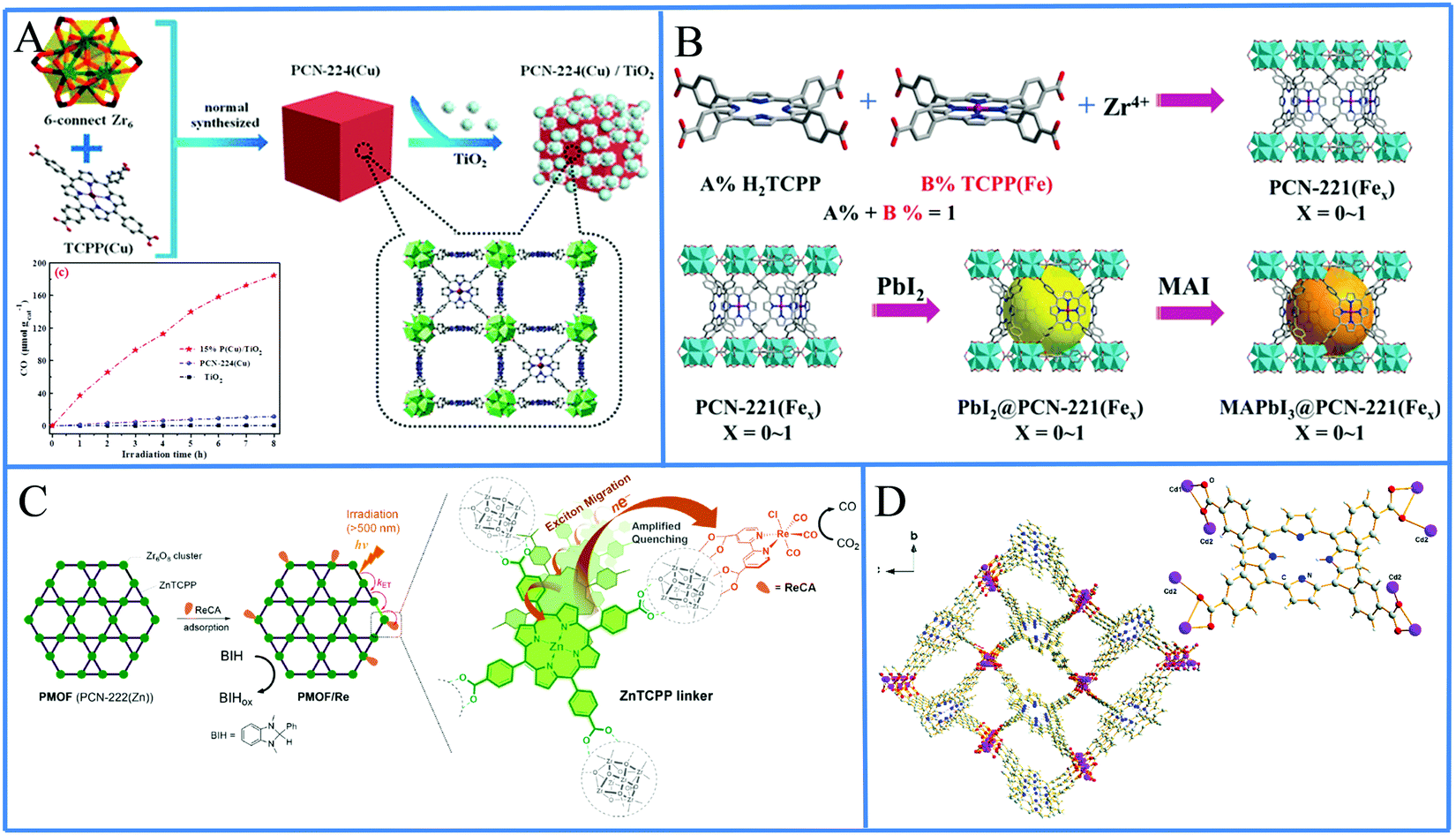 | ||
| Fig. 50 (A) View of PCN-224(Cu), schematic representation of PCN-224(Cu)/TiO2 composite material and CO evolution from these materials. Reproduced from ref. 235, with permission from American Chemical Society, Copyright 2019. (B) Preparation of composite material MAPbI3@PCN-221(Fe) after encapsulation perovskite QDs into the pores of PCN-221(Fe). Reproduced from ref. 237, with permission from John Wiley and Sons, Copyright 2019. (C) Schematic representation of the PMOF/Re hybrid system for photocatalytic CO2 reduction. Reproduced from ref. 238, with permission from American Chemical Society, Copyright 2021. (D) The coordination mode of the free base H2TCPP linker and view of the PMOF 1 mesoporous material. Reproduced from ref. 239, with permission from Royal Society of Chemistry, Copyright 2019. | ||
Choi et al. followed an alternative approach and combined the light harvesting properties of PMOFs with the catalytic performance of molecular reduction catalysts.238 More specific, a PMOF was initially prepared with Zr6 metal nodes and ZnTCPP porphyrin as linkers. Then the postmodification with a well know Re bipyridine catalyst resulted in the formation of the hybrid photocatalyst PMOF/Re (Fig. 50c). The Re complex bears carboxylate anchoring groups and it was chemically anchored to the Zr oxo-clusters. PMOF/Re demonstrated high CO2 to CO conversion activity achieving more than 2200 TONs, in the presence of BIH as SED and 3% trifluoroethanol (TFE) as additive (Table 9, entry 18). The obtained high CO selectivity (>99%) indicate that the catalysis occurs mainly at the anchored Re catalyst, rather than on the Zr6 clusters, which produce mainly formate.228 The high catalytic performance arises mainly from the efficient and rapid photoelectron transfer from the porphyrin linkers to the Re catalytic centers, mimicking the natural photosynthetic systems.
Apart from Zr other metals were also employed as metal nodes for the preparation of PMOFs. Sadeghi et al. reported the preparation of Al-PMOF with the application of Al3+ metal ions as metal nodes and the tetra carboxy-porphyrin (H2TCPP) as linker.240 During the formation process of this PMOF the porphyrin ring was also metallated with Aluminum. Al-PMOF employed as photocatalyst and CO2 to formate reduction was observed with a rate of 165 μmol g−1 h−1, in MeCN as solvent and TEOA as SED (Table 9, entry 19). Remarkably, the incorporation of amine-functionalized graphene (NH2-rGO) into the Al-PMOF resulted in the formation of a new photocatalyst NH2-rGO/Al-PMOF. The catalytic activity of this composite material was significantly improved, producing formate with a rate of 685 μmol g−1 h−1 (Table 9, entry 20). The same group reported the preparation of Co/PMOF using Co metal ions as metal nodes and tetra carboxy-porphyrin as organic linker.241 Under visible-light irradiation and in the presence of TEOA as SED, Co/PMOF presented efficient CO2 photoconversion to formate with a rate of 77 μmol g−1 h−1 (Table 9, entry 21). Co/PMOF reusability and stability were examined through recycling tests and there were no remarkable losses of photoactivity even after three cycles of photoreaction.
Li et al. used Cd2+ metal ions as metal nodes and in combination with the free base carboxy-porphyrin (H2TCPP) they contracted PMOF 1 (Fig. 50d).239 PMOF 1 in the presence of TEA as SED demonstrated a relative low CO2 to CO catalytic activity with a conversion rate of 10 μmol g−1 h−1 (Table 9, entry 22). However, after the addition of Ru(bpy)3Cl2 as co-catalyst the efficiency of CO production was increased about 5-fold, reaching 56 μmol g−1 h−1 (Table 9, entry 23). The Ru complex was found to act not only as light absorber but also as a photocatalyst, increasing the photogenerated charge separation by eliminating the holes. The catalytic system presented moderate stability, since during the second cycle a reduction on the CO production rate was observed.
Wang and et al. used In3+ ions as metal nodes, H2TCPP as linker and after the encapsulation of Fe3+ ions In–FeTCPP-MOF was obtained (Fig. 51a).242 Under light irradiation In–FeTCPP-MOF presented efficient CO2 to CO activity (144 μmol g−1 h−1), in the presence of L-ascorbgyl palmitate (L-AP) as SED (Table 9, entry 24). In this system the MOF matrix provides the electrons to the iron catalytic centers, where the CO2 reduction takes place. Moreover, In–FeTCPP-MOF can be recycled for three cycles of 24 h photocatalytic reactions without deactivation (Fig. 51a). The cobalt analogue (In–CoTCPP-MOF) was also prepared, though its catalytic performance was lower compared to the iron one.
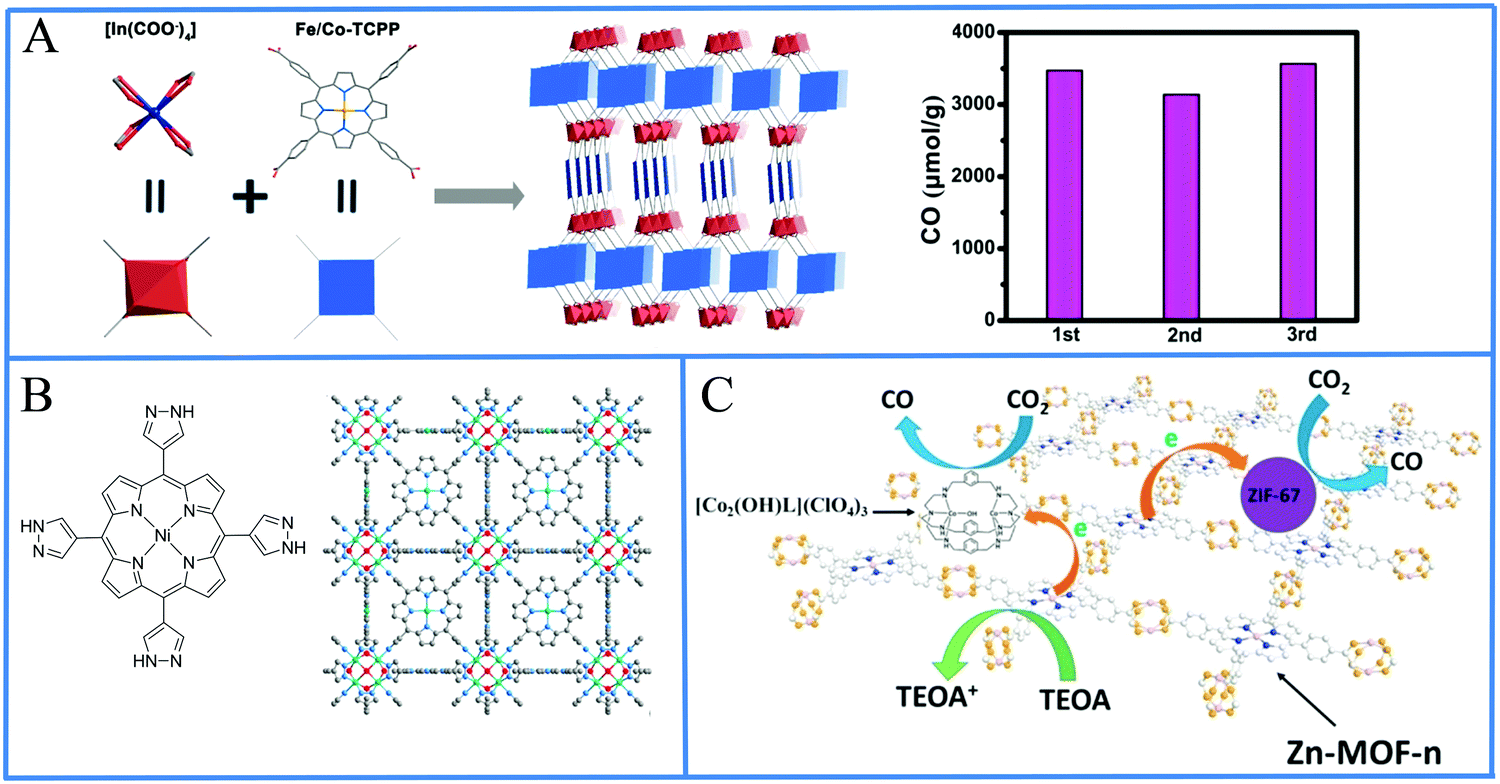 | ||
| Fig. 51 (A) Synthesis and structure of the In–Fe/CoTCPP-MOFs and recycle experiments for CO production with In–Fe–TCPP–MOF. Reproduced from ref. 242, with permission from American Chemical Society, Copyright 2020. (B) Chemical structure of pyrazolyl-substituted porphyrin and crystal structure of PCN-601. Reproduced from ref. 246, with permission from American Chemical Society, Copyright 2020. (C) Schematic representation of 2D/Zn-MOF. Reproduced from ref. 248, with permission from Elsevier, Copyright 2018. | ||
One of the first reported MOFs used for CO2 reduction is NH2-MIL-125(Ti), which is composed of titanium oxo-clusters and 2-aminoterephthalic acid (NH2BDC) as organic linker.243 However, the CO2 conversion efficiency of NH2-MIL-125(Ti) is limited due to its weak electron transfer ability. To overcome this problem Chen et al. reported the preparation of a dual ligand Ti-based MOF (D-TiMOF), where Zn carboxy-porphyrin (ZnTCPP) and NH2BDC share the coordination nodes of the Ti oxo-clusters.244 The incorporation of the porphyrin unit enhanced the optical absorption properties and significantly facilitated the electron transfer from the porphyrin to Ti clusters. D-TiMOF presented high CO2 reduction activity, producing selectively CO with a rate of 59.55 μmol g−1 h−1 (Table 9, entry 25). In addition, photocatalytic recycling studies were performed and the obtained D-TiMOF showed stable catalytic performance for three cycles.
All the above photocatalytic CO2 reductions studies with the PMOFs photocatalysts were performed in the liquid phase and require the addition of an organic compound as SED. The organic solvents are usually toxic and environmentally not friendly, and also the use of SEDs makes this approach economically unfavorable. An alternative method includes the realization of the CO2 reduction studies in the gas phase, using water vapor as SED. Moreover, since the catalysis proceeds in the gaseous phase can take full advantage of the high gas uptake of MOFs. Sharifnia and co-workers prepared Zn/PMOF by using Zn as metal nodes and H2TCPP as linkers.245 The catalytic experiments were performed in the gas phase and Zn/PMOF catalyzed the CO2 to CH4 conversion with a rate of 8.7 μmol g−1 h−1 (Table 9, entry 26).
Cao co-workers reported the construction of PCN-601 which is composed of Ni8 oxo-clusters and Ni metalloporphyrin ligands connected via pyrazolyl groups (Fig. 51b).246 In contrast to the carboxylate groups, the pyrazolyl substituents of the porphyrin possess a larger π-conjugation system and cause higher π–d orbital overlaps with the Ni oxo-nodes. These features offer stronger coordination bonds and faster ligand-to-metal electron transfer for the reduction process at the reactive Ni clusters. The photocatalytic studies were also conducted in the gas phase with H2O vapor as SED and PCN-601 demonstrated efficient and durable photocatalytic activity, converting CO2 to CO and CH4 with rates of 6 and 10.1 μmol g−1 h−1, respectively (Table 9, entry 27). Additionally, PCN-601 exhibited fairly reproducible activity for 5 cycles in 50 h. Liu and co-workers also performed photocatalytic CO2 reduction studies in gas phase using H2O vapor as SED.247 However, instead of PMOF material they examined two porphyrin precursors (CoTCPP and NiTCPP) that can be used of the preparation of PMOFs. CoTCPP showed better CO2 to CO conversion activity (0.4 μmol g−1 h−1) compared to NiTCPP (0.24 μmol g−1 h−1), due to the higher activity and adsorption ability of cobalt metal sites. This work can provide guidance for the synthesis of porphyrin-based MOF.
In general, the efficiency of bulk PMOFs is limited, mainly as a result of the low optical absorption efficiency of the photoactive units embedded in MOF matrices and the fast charge recombination of the photogenerated electron–hole pairs. In addition, adsorption of substrates and desorption of products in micropores show lower mass transport rates than on surfaces. An attractive approach to address these challenges is the development of ultrathin two dimensional (2D) MOF nanosheets. Sun and co-workers reported the preparation of ultrathin 2D PMOF nanosheets (2D/Zn-MOF) using Zn ions as metal nodes and ZnTCPP as linker (Fig. 51c).248 As control the corresponding 3D bulk MOF (3D/Zn-MOF) was also prepared. Both PMOFs were applied as PS with a zeolitic imidazolate framework (ZIF-67) as catalyst for the CO2 photoreduction. 2D/Zn-MOF presented higher photocatalytic activity and selectivity for CO2 to CO reduction (TONCO = 117.8, TONH2 = 11.6, and selectivityCO = 91.0%) compared to 3D/Zn-MOF (TONCO = 63.6, TONH2 = 7.5, and selectivityCO = 89.5%) (Table 9, entries 28 and 29). The enhanced performance of 2D/Zn-MOF arises from the improved CO2 adsorption capacity and charge transport capability, as well as the longer lifetime of the electron–hole pairs. In an effort to replace ZIF-67 with a molecular catalyst, a highly efficient dinuclear cobalt complex [Co2(OH)L](ClO4)3 (L = N[(CH2)2NHCH2(m-C6H4)CH2NH(CH2)]3N)]249 (Co2) was used as catalyst with 2D/Zn-MOF as PS (Fig. 51c).248 However, under the same experimental conditions this combination presented lower catalytic activity (TONCO = 68.7, TONH2 = 15.6, and selectivityCO = 81.5%).
Chen et al. prepared thin porphyrin paddle-wheel framework nanosheets (PPF-3) by the coordination of Co metal nodes with the free base carboxy-porphyrin (H2TCPP), following a PVP-assisted solvothermal synthetic route.250 PVP molecules were used as structural direction agents to restrain growth along the c-axis, leading to the formation of sheet-like structures. To further improve the light absorption ability of PPF-3, Au nanoparticles (Au NPs) were successfully loaded on the surface of PPF-3 nanosheets by electrostatic interactions. Photocatalytic studies showed than CO2 was reduced to formate as the sole product, with a production rate of 42.7 μmol g−1 h−1 (Table 9, entry 30). During the catalysis, Au NPs serve mainly as light-harvesting antennas, injecting photoelectrons to the Co catalytic centers (Fig. 52a). Moreover, when they used thicker nanosheets or PPF-3 without Au NPs the catalytic performance was significantly reduced. Computational studies were also performed to study the catalytic properties of 2D PMOs, using density functional theory (DFT) calculations.251 Cobalt porphyrin (CoTCPP) was selected as linker, while Co, Zn and Zr oxo-clusters were tested as metal nodes. The computational results demonstrated that the PMOF with the Co oxo-cluster (Co-PMOF) is the most promising material for the CO2 to CH4 reduction, due to the collaborative contribution of the Co oxo-cluster and the cobalt porphyrin during the catalysis.
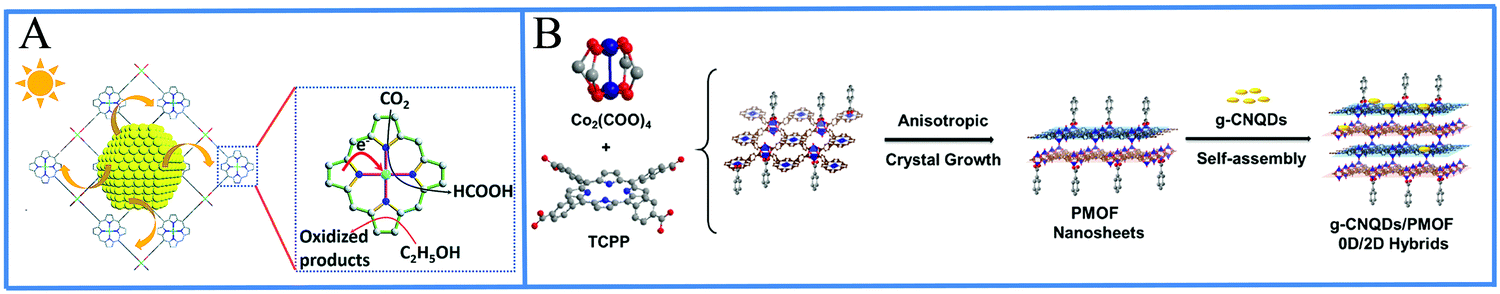 | ||
| Fig. 52 (A) Proposed mechanism for plasmon-enhanced photocatalytic activity of PPF-3 towards CO2 reduction. Reproduced from ref. 250, with permission from Royal Society of Chemistry, Copyright 2019. (B) Schematic illustration of the preparation of g-CNQDs/PMOF hybrids. Reproduced from ref. 252, with permission from American Chemical Society, Copyright 2019. | ||
Zheng et al. reported a novel hybrid material (g-CNQDs/PMOF) by coordinating zero-dimensional carbon nitride quantum dots (g-CNQDs) with 2D ultrathin porphyrin MOF.252 The thickness of the 2D-PMOF is in the range of 1.7–4.6 nm (equal to 4–10 coordination layers) and contains Co oxo-clusters connected together by CoTCPP linkers. g-CNQDs were then tightly adsorbed on the PMOF through hydrophobic and π–π stacking interactions (Fig. 52b). The hybrid g-CNQDs/PMOF presented improved catalytic activity compared to the unmodified PMOF, producing CO and CH4 with evolution rates of 16.1 and 6.86 μmol g−1 h−1, respectively (Table 9, entry 31). In this system g-CNQDs provide photo-generated electrons to the Co catalytic sites, enhancing not only the catalytic activity for CO2 reduction but also the selectivity for CH4 production.
162 μmol g−1 h−1 (accounting for a TON of 20.2), while the selectivity was 78% (Table 10, entry 1).
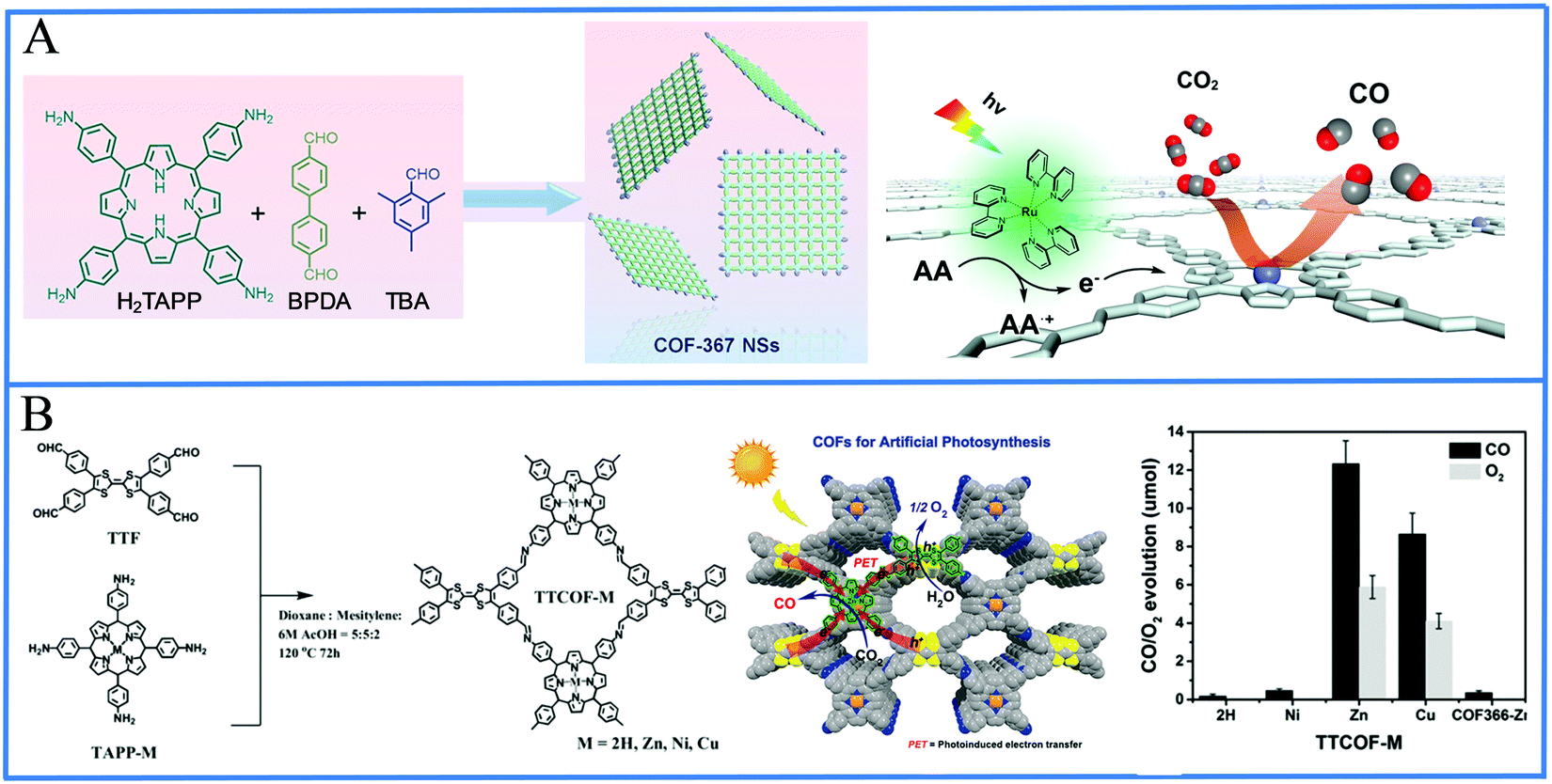 | ||
| Fig. 53 (A) Schematic illustration of synthesis of the COF-367 NSs and proposed mechanism for the photocatalytic conversion of CO2 into CO over COF-367-Co NSs under visible-light irradiation with [Ru(bpy)3]Cl2 as the PS and AA as the electron donor. Reproduced from ref. 253, with permission from American Chemical Society, Copyright 2019. (B) Schematic representation of the synthesis of TTCOF-M through the condensation of TTF and TAPP-M, structure of TTCOF-M and catalytic performance of TTCOF-M. Reproduced from ref. 254, with permission from John Wiley and Sons, Copyright 2019. | ||
| Entry | COF name | Porphyrin | Organic linker | Cocatalyst or additives | SED | Solvent | Light source | Major products rate | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | COF-367-Co | CoTAPP | BPDA | Ru(bpy)3Cl2 | AA, 0.1 M | H2O | Xenon lamp, 300 W, λ > 420 nm | CO, 10162 μmol g−1 h−1 or 20.2 TON |
253 |
| 2 | TTCOF-Zn | ZnTAPP | TTF | — | — | H2O | Xenon lamp, 300 W, λ > 420 nm | CO, 2.1 μmol g−1 h−1 | 254 |
| 3 | COF-366 | H2TAPP | TPA | — | — | H2O | Xenon lamp, λ > 430 nm | CO, 3.9 μmol g−1 h−1 | 256 |
| 4 | TAPBB-COF | H2TAPP | BDB | — | — | H2O | Xenon lamp, λ > 430 nm | CO, 12.4 μmol g−1 h−1 | 256 |
| 5 | NiP-TPE-COF | NiTAPP | TPE | Ru(bpy)3Cl2 | TEOA, 16.7% | CH3CN/H2O (3/1) | Xenon lamp, 300 W, λ > 400 nm | CO, 525 μmol g−1 h−1 | 257 |
| 6 | CoP-TPE-COF | CoTAPP | TPE | Ru(bpy)3Cl2 | TEOA, 16.7% | CH3CN/H2O (3/1) | Xenon lamp, 300 W, λ > 400 nm | CO, 2414 μmol g−1 h−1 | 257 |
| 7 | PD-COF-23-Ni | NiTAPP | DPP-CHO | — | TEOA, 9.1% | CH3CN/H2O (3/2) | Xenon lamp, 300 W | CO, 40 μmol g−1 h−1 | 258 |
| 8 | PD-COF-23 | H2TAPP | DPP-CHO | — | TEOA, 9.1% | CH3CN/H2O (3/2) | Xenon lamp, 300 W | CO, 20.9 μmol g−1 h−1 | 258 |
| 9 | COF-367-CoIII | CoIIITAPP | BPDA | — | TEA, 18% | CH3CN | Xenon lamp, 300 W, λ > 380 nm | HCOO−, 93 μmol g−1 h−1 CO, 5.5 μmol g−1 h−1 | 259 |
| CH4, 10.1 μmol g−1 h−1 | |||||||||
| 10 | COF-367-CoII | CoIITAPP | BPDA | — | TEA, 18% | CH3CN | Xenon lamp, 300 W, λ > 380 nm | HCOO−, 48.6 μmol g−1 h−1 CO, 16.5 μmol g−1 h−1 | 259 |
| CH4, 12.8 μmol g−1 h−1 | |||||||||
| 11 | Ni-PCD@TD-COF | NiTAPP | TP and DBT | Ru(bpy)3Cl2 | TEOA, 20% | CH3CN/H2O (3/1) | Xenon lamp, λ > 420 nm | CO, 478 μmol g−1 h−1 | 260 |
| 12 | α-Fe2O3@Por-CTF | H2TBPP-CHO | TPh | Ru(bpy)3Cl2 and α-Fe2O3 | TEOA, 20% | DMF | Visible light | CO, 320 μmol g−1 h−1 | 261 |
| 13 | COP-P | H2TBPP | — | — | H2O | — | Xenon lamp, 300 W | CO, 4.58 μmol g−1 h−1 | 262 |
| 14 | COP-P/TiO2 | H2TBPP | — | TiO2 NPs | H2O | Xenon lamp, 300 W | CO, 5.7 μmol g−1 h−1 | 263 | |
| CH4, 0.26 μmol g−1 h−1 |
Lan and co-workers synthesized a series of crystalline 2D rigid porphyrin-tetrathiafulvalene COFs (TTCOF-M, M = 2H, Zn, Ni, Cu) by Schiff-base condensation between the corresponding tetra amino-porphyrin (TAPP-M, M = 2H, Zn, Ni, Cu) and 2,3,6,7-tetra (4-formylphenyl)-tetrathiafulvalene (TTF) by solvothermal method (Fig. 53b).254 The covalent coupling between porphyrins and TTF within COF enables the efficient electron transfer from the TTF to the porphyrin moiety. The photocatalytic studies of all TTCOF-M were conducted in aqueous solution without additional PS and SED. Among them, TTCOF-Zn exhibited the highest CO production rate of 2.1 μmol g−1 h−1 and selectivity (ca. 100%) (Table 10, entry 2). At the same time, the formation of O2 was also detected, since the photogenerated holes in TTF are capable of oxidizing H2O, which serves as SED.
Wang et al. following an already published procedure prepared COF-366255 and examined its activity towards CO2 reduction.256 The synthesis COF-366 involves the imine-bond formation between tetra amino-porphyrin (H2TAPP) and terephthaldehyde (TPA) (Fig. 54a). COF-366 has good carrier mobility and visible light absorption capacity, but its photocatalytic performance was low with a CO2 to CO conversion rate of 3.9 μmol g−1 h−1 (Table 10, entry 3). In an effort to increase the catalytic efficiency they replaced TPA with 2,5-dibromo-terephthalaldehyde (BDB), resulting in the formation of TAPBB-COF (Fig. 54b).256 The presence of bromine functional groups adjusted the valence band of TAPBB-COF to a more suitable position, resulting in an improved catalytic performance by a factor of 3. TAPBB-COF selectively reduced CO2 with H2O as a SED, and CO was evolved with a rate of 12.4 μmol g−1 h−1 (Table 10, entry 4).
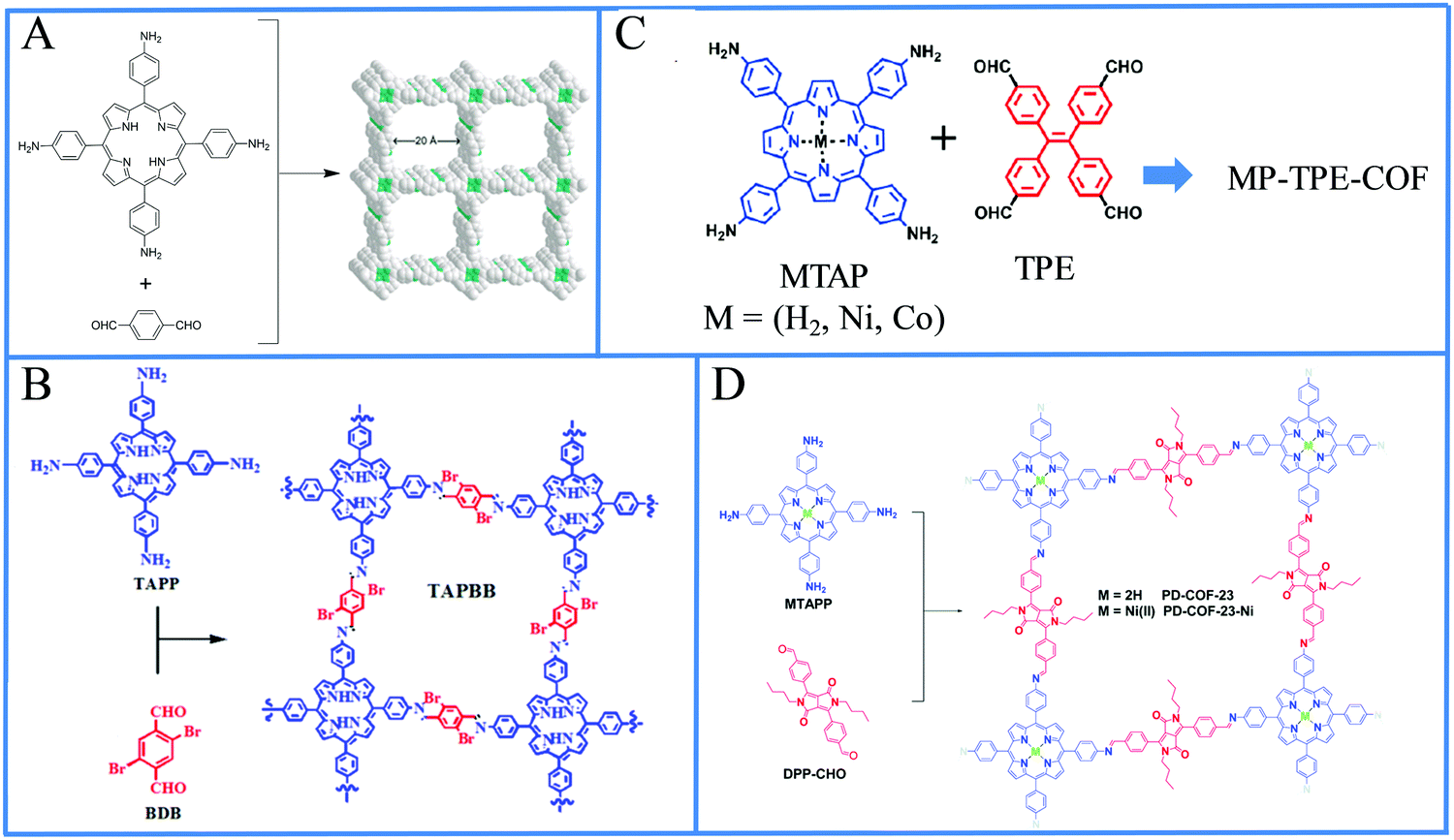 | ||
| Fig. 54 (A) Preparation of COF-366 through condensation reaction between H2TAPP and terephthaldehyde. Reproduced from ref. 256, with permission from John Wiley and Sons, Copyright 2020. (B) Synthetic procedure of TAPBB-COF. Reproduced from ref. 256, with permission from John Wiley and Sons, Copyright 2020. (C) Schematic depiction for the synthesis of MP-TPE-COF, (D) synthetic route for PD-COF-23 and PD-COF-23-Ni. Reproduced from ref. 258, with permission from Royal Society of Chemistry, Copyright 2020. | ||
Lv et al. reported the synthesis of two metalloporphyrin based COFs (NiP-TPE-COF and CoP-TPE-COF) using 4,4′,4′′,4′′′-(ethane-1,1,2,2-tetrayl) tetrabenzaldehyde (TPE) and the corresponding tetra amino-porphyrin (MTAPP, M = Ni, Co) (Fig. 54c).257 The catalytic studies were conducted in the presence of [Ru(bpy)3]Cl2 as PS and TEOA as SED. NiP-TPE-COF exhibited efficient CO evolution rate of 525 μmol g−1 h−1 (Table 10, entry 5) and 93% selectivity. On the other hand, CoP-TPE-COF provided higher catalytic activity (2414 μmol g−1 h−1, Table 10, entry 6) but with significantly lower selectivity (61%). The high catalytic activity can be attributed to the rapid transfer of photogenerated electrons, while the selectivity is directly related to the competitive reaction of the protons.
Xu et al. prepared two donor–acceptor 2D COFs, PD-COF-23 and PD-COF-23-Ni from acid-catalyzed Schiff-base reaction between the free base (H2TAPP) or the Ni metallated (NiTAPP) porphyrin and a di-formyl substituted diketopyrrolopyrrole derivative (DPP-CHO) (Fig. 54d).258 PD-COF-23-Ni showed higher CO production efficiency of 40 μmol g−1 h−1 (Table 10, entry 7) than PD-COF-23 (20.9 μmol g−1 h−1, Table 10, entry 8), in the absence of any additional PS. The selectivity of both COFs is high (99%) and only a trace amount of CH4 was detected. During the catalysis electron transfer from the porphyrin moiety to DPP unit takes place and subsequently, CO2 is reduced to CO by the two electrons accumulated near DPP. Meanwhile, when the porphyrin is metallated the Ni center also catalyzes the CO2 reduction.
Gong et al. constructed COF-367-Co from the condensation of cobalt metallated porphyrin CoTAPP with 4,4′-biphenyldicarboxaldehyde (BPDA) (Fig. 55a).259 In this COF the spin state of cobalt can be manipulated by simply changing the oxidation state of Co centered in the porphyrin ring. The photocatalytic CO2 reduction studies demonstrated that COF-367-CoIII(CoIII, S = 0) exhibits enhanced activity and remarkably higher selectivity to formate than COF-367-CoII (CoII, S = 1/2) (Table 10, entries 9 and 10). COF-367-CoIII possesses higher charge separation efficiency and lower energy barrier for the HCOO− formation than COF-367-CoII and these accounts for the improved activity of the former. This work provides a significant guideline to regulate the catalytic performance by rationally manipulating the spin state of metal centers.
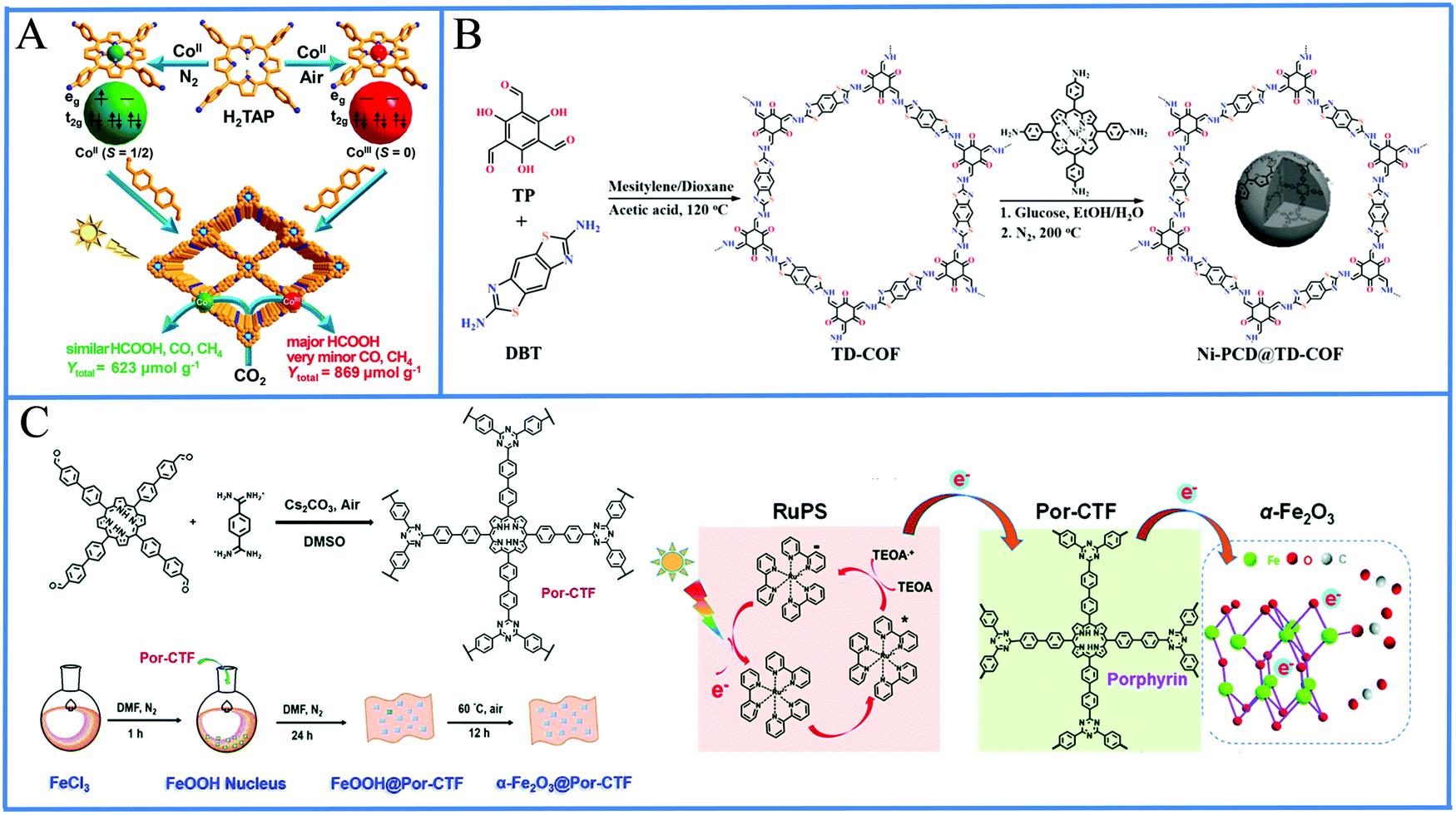 | ||
| Fig. 55 (A) Rational fabrication of COF-367-Co featuring different spin states of Co ions toward photocatalytic CO2 reduction. Reproduced from ref. 259, with permission from American Chemical Society, Copyright 2020. (B) Schematic illustration for the synthesis of TD-COF and Ni-PCD@TD-COF. Reproduced from ref. 260, with permission from John Wiley and Sons, Copyright 2020. (C) Synthetic route for the Por-CTF, fabrication of the α-Fe2O3@Por-CTF hybrid photocatalyst and proposed electron transfer mechanism for the photochemical CO2 reduction. Reproduced from ref. 261, with permission from Royal Society of Chemistry, Copyright 2019. | ||
Wang and co-workers developed a novel material (Ni-PCD@TD-COF), where inside the pores of a COF nanostructure (TD-COF) they incorporated metalloporphyrin-based carbon dots (Ni-PCD) (Fig. 55b).260 The COF scaffold prepared with the imine-bond formation between 2,4,6-triformylphloroglucinol (TP) and 2,6-diaminobenzo[1,2-d:4,5-d′]bisthiazole (DBT). Then NiTAPP porphyrin was introduced and after selective pyrolysis of glucose Ni-PCD@TD-COF was obtained. The photocatalytic activity of Ni-PCD@TD-COF was evaluated in the presence of [Ru(bpy)3]Cl2 as PS and TEOA as SED. Efficient CO evolution was observed with a rate of 478 μmol g−1 h−1 (Table 10, entry 11) and high selectivity (98%). When Ni metal was replaced with Co the catalytic activity increased, while the selectivity decreased to only 49%. Notably, the photocatalytic system is also effective for low concentration of CO2, simulating flue gas from the power plant.
Jin and co-workers fabricated a porphyrin-based covalent triazine framework (Por-CTF) decorated with α-Fe2O3 nanoparticles, denoted as α-Fe2O3@Por-CTF.261 The covalent triazine framework Por-CTF was prepared via polycondensation of terephthalamidine (TPh) with tetra formyl-substituted porphyrin derivative (H2TBP-CHO) and then the in situ growth of a-Fe2O3 NPs resulted in the formation of α-Fe2O3@Por-CTF (Fig. 55c). Under irradiation and in the presence of [Ru(bpy)3]Cl2 as PS and TEOA as SED, CO evolution was observed with a ratio of 320 μmol g−1 h−1 (Table 10, entry 12). In this ternary system, the porous framework serves as electron relay facilitator, enabling the electron transfer from the Ru PS to the Fe catalytic sites, and reducing the photocurrent carrier recombination.
Another promising method for the development of photocatalytic CO2 reduction systems is the use of microporous covalent organic polymers (COPs) as photocatalysts. Zhang et al. prepared a porphyrin based COP (COP-P) based on tetrabiphenylporphyrin (H2TBPP) through the Scholl reaction.262 The catalytic performance of COP-P was evaluated in the presence of H2O as SED, and CO was produced with a rate of 1.23 μmol g−1 h−1. Interestingly, hollowrization and post-sulfonation modulated the photocatalytic performance of COP-P, giving 4.8 times enhanced production of CO (7.54 μmol g−1 h−1, Table 10, entry 13). Post-sulfonation improved the separation and transfer ability of photogenerated carriers, while hollowrization increased the surface area of COP-P polymer and thereby enhanced CO2 uptake. The same group in an effort to increase the catalytic performance reported the preparation of a hybrid material COP-P/TiO2, where TiO2 NPs were composited with the COP-P polymer.263 The formation of COP-P/TiO2 involved the hydrothermal in situ synthesis of TiO2 in the presence of COP-P polymer template. The COP-P/TiO2 composite material presented improved photocatalytic activity (5.7 μmol g−1 h−1) relative to the initial COP-P or with the material fabricated through simple physical mixing of COP-P with TiO2 NPs. In this system CH4 was also detected but with much smaller rate (0.26 μmol g−1 h−1, Table 10, entry 14).
 | ||
| Fig. 56 Chemical structure of the porphyrin derivatives that were employed with TiO2 for CO2 photoreduction. | ||
| Entry | Metal oxide | Porphyrin | Cocatalyst or additives | SED | Solvent | Light source | Major products rate | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | TiO2 (powder) | ZnPy | — | H2O | Gas phase | Xenon lamp, 300 W | CO, 8.07 μmol g−1 h−1 | 264 |
| CH4, 1.01 μmol g−1 h−1 | ||||||||
| 2 | TiO2 (powder) | ZnPyP–RuBiPy | — | Na2SO3 | H2O | Xenon lamp, 500 W, λ > 400 nm | CH3OH, 68 μmol g−1 h−1 | 266 |
| 3 | TiO2 (P25) | CuTCPP | — | H2O | Gas phase | Xenon lamp, 300 W | CO, 13.6 μmol g−1 h−1 | 267 |
| CH4, 1.0 μmol g−1 h−1 | ||||||||
| 4 | TiO2 (P25m) | CuTCPP | — | H2O | Gas phase | Xenon lamp, 300 W | CO, 2.68 μmol g−1 h−1 | 268 |
| CH4, 19.39 μmol g−1 h−1 | ||||||||
| 5 | TiO2 (NSs) | H2TCPP | — | TEOA, 20% | CH3CN | Xenon lamp, 300 W, λ > 420 nm | CO, 141.74 μmol g−1 h−1 | 269 |
| CH4, 2.97 μmol g−1 h−1 | ||||||||
| 6 | Na(1−x)LaxTaO(3+x) | CoTPP | — | H2O | H2O | Mercury lamp, 77 W | MeOH, 36.2 μmol g−1 h−1 | 270 |
| EtOH, 21.4 μmol g−1 h−1 | ||||||||
| 7 | TiO2 | ZnPCA | Re catalyst | BIH, 0.1 M | DMF | Xenon lamp, 450 W, λ > 550 nm | CO, 1028 TON | 271 |
| 8 | TiO2 | ZnPCNPA | Re catalyst | BIH, 0.1 M | DMF | Xenon lamp, 450 W, λ > 550 nm | CO, 820 TON | 271 |
| 9 | TiO2 | ZnP | Re catalyst | BIH, 0.1 M and TEOA | DMF | LED lamp, 450 W, λ > 500 nm | CO, 2488 TON | 272 |
Mele et al. also prepared two novel composite materials by loading polycrystalline TiO2 powder with lipophilic copper phthalocyanine or porphyrin (CuPc and CuPp, Fig. 56).265 The photocatalytic studies demonstrated that the presence of the sensitizers is beneficial, and enables the reduction of CO2 in aqueous suspension, affording formic acid. CuPc presented higher catalytic activity compared to CuPp, producing formate with a rate of 26 μmol g−1 h−1.
Wang and co-workers utilized a Zn porphyrin–Ru polypyridyl complex (ZnPyP–RuBiPy, Fig. 56) to sensitize TiO2 nanotubes.266 The ZnPyP–RuBiPy derivative consists of one porphyrin unit in the center, which is surrounded by four peripheral ruthenium polypyridyl molecules. The ZnPyP–RuBiPy/TiO2 composite material demonstrated efficient catalytic activity, producing methanol as the only product with a rate of 68 μmol g−1 h−1 (Table 11, entry 2).
Wang et al. prepared a series of tetra carboxy-porphyrins (MTCPP, M = H2, Co, Ni, Cu) (Fig. 56) for the sensitization of commercial P25 TiO2.267 In all the cases the photocatalytic CO2 reduction activity of the assembled MTCPP/TiO2 nanocomposites was significantly improved compared to the bare P25. The enhanced catalytic activity of the composite material was attributed to a strengthened capability in light absorption and enhanced separation efficiency of photo-induced electrons and holes. In addition, the incorporated metal atoms inside the porphyrin ring also play an important role in the adsorption and activation of CO2 molecules. Among the various porphyrin derivatives, CuTCPP presented the highest performance producing CO and CH4 with rates of 13.6 and 1.0 μmol g−1 h−1, respectively (Table 11, entry 3). The other porphyrin complexes showed lower activity with the order NiTCPP > CoTCPP > H2TCPP.
Following the same approach, Wang and co-workers anchored CuTCPP to P25m TiO2 NPs.268 Initially, the commercial P25 TiO2 were hydrothermally treated, resulting in the formation of P25m, which possess an enlarged specific surface area and increased number of hydroxyl groups on the surface. The influence of CuTCPP content on the photocatalytic activity was investigated and the highest catalytic performance was observed when the hydrothermally treated P25m were sensitized by 0.5% CuTCPP, producing CH4 and CO with rates of 19.39 and 2.68 μmol g−1 h−1, respectively (Table 11, entry 4). On the other hand, the non-sensitized P25m TiO2 presented significantly lower performance (0.42 μmol g−1 h−1 CH4 and 1.7 μmol g−1 h−1 CO), under the same conditions. Noteworthy in the composite material the major product of the CO2 reduction was CH4.
Gao et al. utilized the free base tetra carboxy porphyrin (H2TCPP) for the sensitization of TiO2 nanosheets, following a simple self-assemble approach (Fig. 57a).269 Ultrathin TiO2 nanosheets were selected in this work, since they present higher catalytic activity compared to the corresponding TiO2 nanotubes or nanoparticles. The composite H2TCPP/TiO2 material demonstrated high catalytic activity and after visible irradiation the formation of both CO and CH4 was observed. The amount of H2TCPP in H2TCPP/TiO2 composites had significant influence on the photoreduction activity (Fig. 57a). The TiO2 nanosheets with 11.5% H2TCPP showed the best catalytic performance with a CO production rate of 141.74 μmol g−1 h−1 (Table 11, entry 5), which is more than 37 times higher compared to the pristine TiO2 nanosheets. In this system, H2TCPP works as the light-harvesting unit to generate light-induced photoelectrons, while the connection between the H2TCPP and TiO2 facilitates the electron injection to the CB of TiO2 as well as inhibiting the recombination of electron–hole pairs. Then the highly active facets of TiO2 nanosheets serve as catalytic centers and efficiently convert the CO2 into CO and CH4.
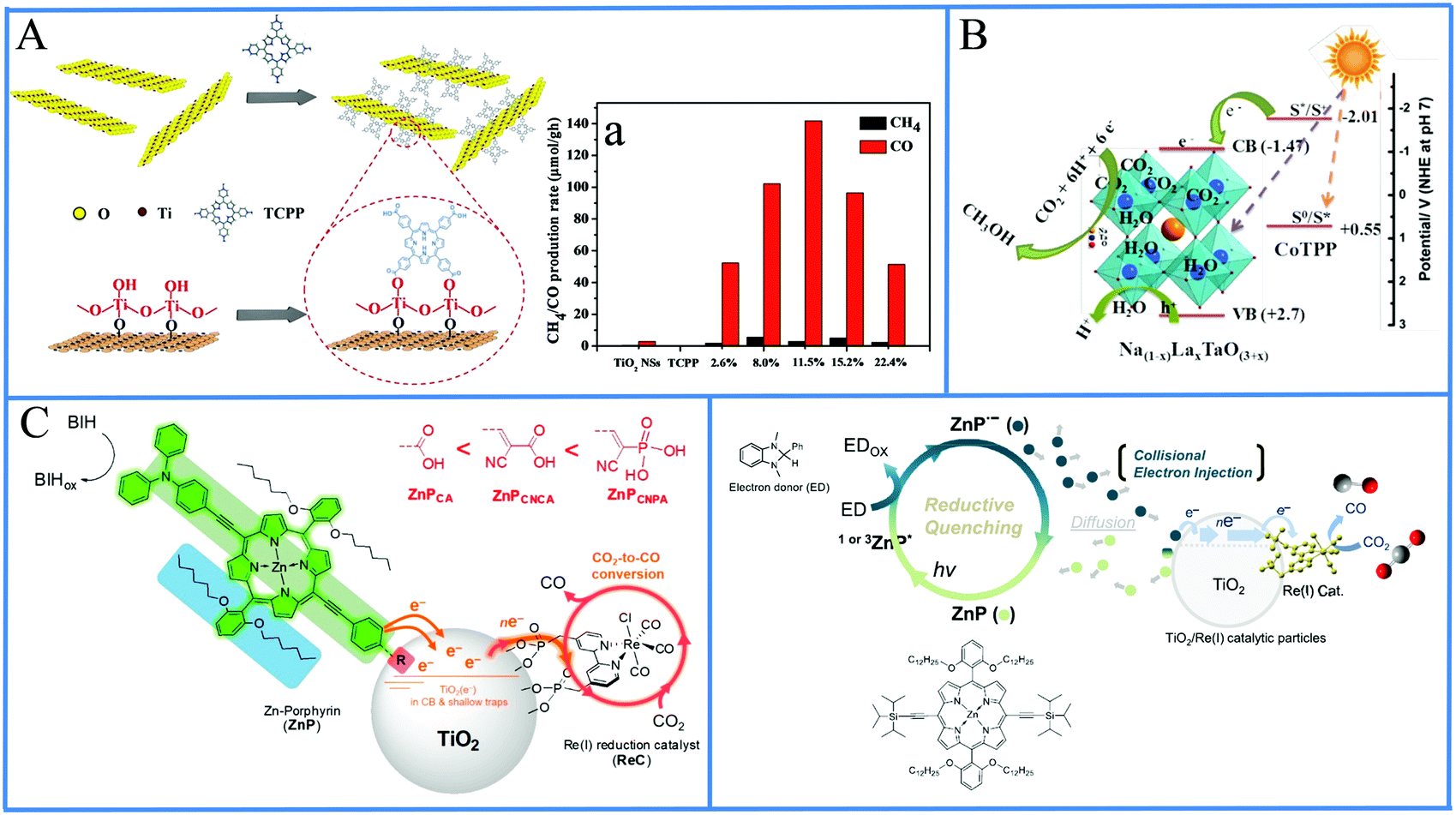 | ||
| Fig. 57 (A) Schematic illustration of the synthesis of H2TCPP/TiO2 nanosheets and photocatalytic CO2 reduction performance over different porphyrin loadings. Reproduced from ref. 269, with permission from Elsevier, Copyright 2019. (B) Photocatalytic reduction of CO2 on CoTPP sensitized Na(1−x)LaxTaO(3+x). Reproduced from ref. 270, with permission from Elsevier, Copyright 2016. (C) Sequential electron transfer process in a DSP hybrid system from zinc porphyrin PS to TiO2 (molecular immobilizer and electron mediator) and finally to Re reduction catalyst. Reproduced from ref. 271, with permission from American Chemical Society, Copyright 2018. (D) Schematic description of the overall reduction processes with ZnP porphyin. Reproduced from ref. 272, with permission from American Chemical Society, Copyright 2020. | ||
Apart from TiO2 other materials were also sensitized with porphyrin chromophores for the development of efficient CO2 photoreduction systems. Viswanathan and co-workers explored the photo catalytic activity of Lanthanum modified sodium tantalate (Na(1−x)LaxTaO(3+x)), in conjunction with Co tetra phenylporphyrin (CoTPP) as sensitizer (Fig. 57b).270 The composite material after UV-visible irradiation catalyzed the efficient CO2 reduction, producing methanol and ethanol as the main products with rates of 36.2 and 21.4 μmol g−1 h−1, respectively (Table 11, entry 6). Sensitized tantalate absorbs both visible and UV irradiation, resulting in the direct injection of photo electrons to the conduction band of active Na(1−x)LaxTaO(3+x) surface, where the catalytic CO2 conversion takes place. The presence of CoTPP besides sensitization also hinders charge carrier recombination.
Dye-sensitized photocatalysis (DSP) is another emerging approach for the development of efficient CO2 reduction systems. In DSP the PS and the catalyst are anchored onto the surface of a semiconductor such as TiO2, which serves as scaffold and electron mediator. This approach has received much attention due to the easy fabrication, facile tunability, the improvement of stability, and no limitation of solvent selection based on the immobilization of components on TiO2 particles. Won et al. prepared a series of Zn–porphyrin dyes and anchored onto a TiO2 along with the well-studied Ru bipyridine catalyst to construct a dye-sensitized photocatalytic system (DSP) (Fig. 57c).271 For the porphyrin chromophores three different anchoring groups (–COOH, –CNCOOH, –CNPO3H2) were utilized to examine their influence on the CO2 catalytic activity, while the Re complex was immobilized onto the TiO2 surface through two methyl phosphonic acid groups. The porphyrin with the –COOH anchoring group (ZnPCA) showed the best activity producing CO with a TON of 1028 after 42 h of irradiation (Table 11, entry 7). However, the porphyrin with the –CNPO3H2 group (ZnPCNPA) revealed the highest stability, showing no leveling-off tendency for >90 h giving a TON of 820 (Table 11, entry 8). The high performance of ZnPCA can be attributed to the more efficient electron injection due to the favorable electronic delocalization of the carboxylic acid. On the other hand, in the ZnPCNPA derivative the phosphonic acid group presents the stronger binding affinity and suppresses desorption from TiO2. The same group in a subsequent report utilized a Zn porphyrin (ZnP, Fig. 57d) without any anchoring groups in the analogous TiO2/Ru-catalyst system.272 ZnP possess bulky alkyl groups, which significantly lowers the possibility of an electron injection pathway through chemical or physical adsorption of the porphyrin onto the TiO2 surface. In this system, without any dye immobilization on the TiO2 surface, the dissolved porphyrin can effectively transport its excited-state electrons to the heterogeneous catalytic TiO2/Re particles via a collisional electron transfer pathway. Interestingly, this binary hybrid system exhibited more efficient photochemical CO2-to-CO conversion activity (2488 TON after 46 h, Table 11, entry 9) compared to the corresponding ternary system. The enhanced CO2-to-CO conversion efficiency was attributed to the better light-absorbing ability of the porphyrin is solution compared to the immobilized form onto the TiO2.
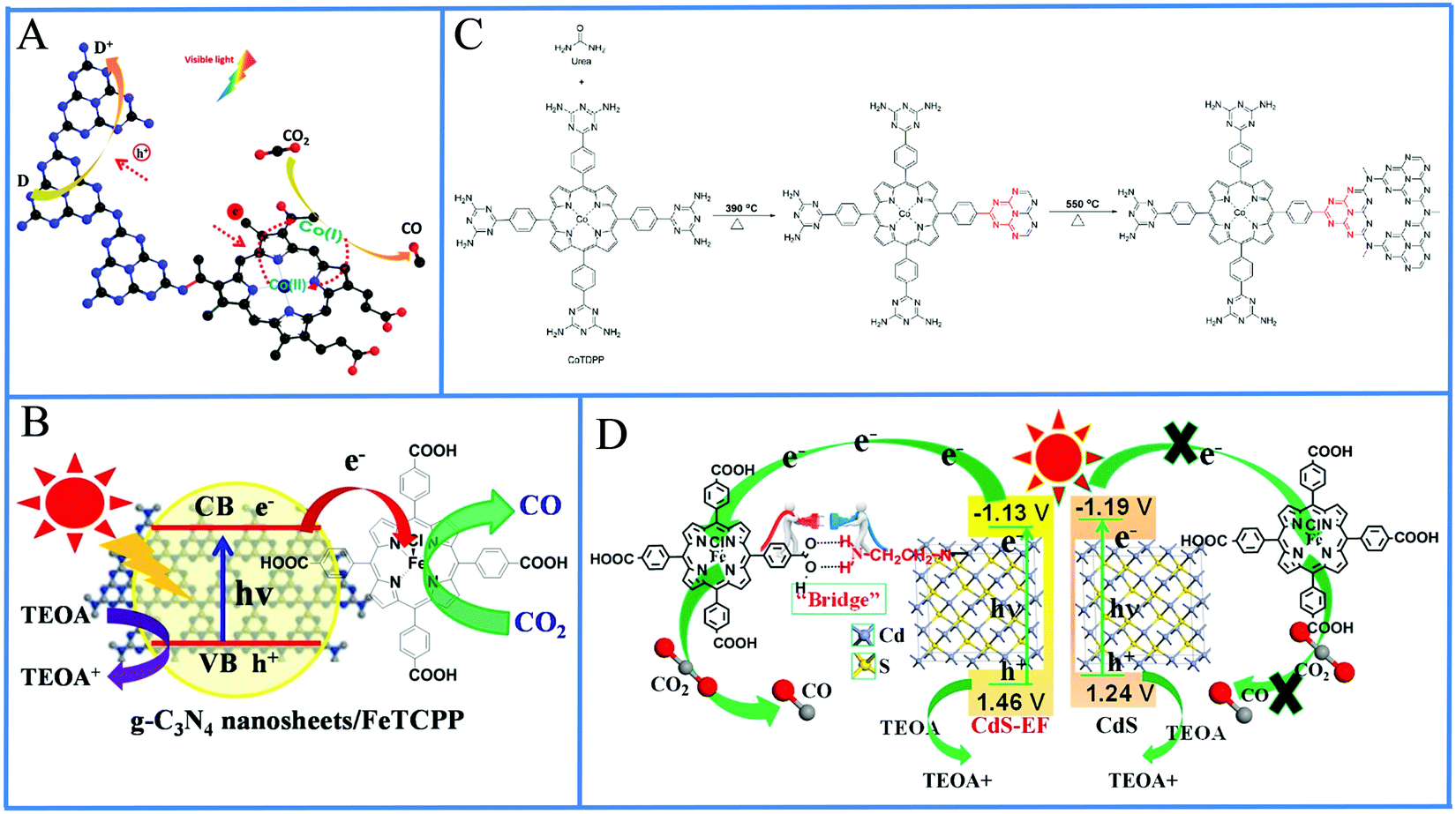 | ||
| Fig. 58 (A) CO2 reduction with a Co–porphyrin loaded C3N4 heterojunction system. Reproduced from ref. 273, with permission from Elsevier, Copyright 2017. (B) Structure of g-C3N4/FeTCPP heterogeneous catalyst system. Reproduced from ref. 274, with permission from Elsevier, Copyright 2018. (C) Schematic illustration of the formation of g-CNU-CoTDPP nanoparticle photocatalysts. (D) Schematic diagram of CO2 photoreduction over CdS-EF/FeTCPP and CdS/FeTCPP hybrid catalysts. Reproduced from ref. 276, with permission from Elsevier, Copyright 2018. | ||
| Entry | Nanomaterial | Porphyrin | Cocatalyst or additives | SED | Solvent | Light source | Major products rate | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | g-C3N4 (melon) | Co–P | — | TEOA | Gas phase | Xenon lamp, 300 W, λ > 400 nm | CO, 17 μmol g−1 h−1 | 273 |
| 2 | g-C3N4 (nanosheets) | FeTCPP | — | TEOA, 20% | CH3CN/H2O (3/1) | Xenon lamp, 300 W, λ > 420 nm | CO, 1087 μmol g−1 h−1 | 274 |
| 3 | g-C3N4 (condensed nanosheets) | CoTDPP | — | TEOA, 20% | CH3CN | White LED lamp, 5 W, λ > 400 nm | CO, 57 μmol g−1 h−1 | 275 |
| 4 | CdS | FeTCPP | — | TEOA, 20% | CH3CN/H2O (3/1) | Xenon lamp, 300 W, λ > 420 nm | CO, 7.5 μmol g−1 h−1 | 276 |
| CH4, 0.18 μmol g−1 h−1 | ||||||||
| 5 | CdS/Bi2S3 | FeTCPP | — | TEOA, 20% | CH3CN/H2O (3/1) | Xenon lamp, 300 W, λ > 420 nm | CO, 37.9 μmol g−1 h−1 | 277 |
| CH4, 0.2 μmol g−1 h−1 | ||||||||
| 6 | CuInS2/ZnS QDs | FeTPP | — | TMPD, 0.1 M | DMSO | Laser pointer, 4.5 mW, 450 nm | CO, 55 TON | 278 |
| 7 | CuInS2/ZnS QDs | FeTMA | — | TEOA, 0.015 M | H2O | Laser pointer, 4.5 mW, 450 nm | CO, 450 TON | 279 |
| 8 | CuInS2/ZnS QDs | CoTPPS | — | SA, 5 mM and TCEP, 5 mM | H2O | LED, 450 nm | CO, 84101 TON |
280 |
Lin et al. fabricated a highly efficient g-C3N4/FeTCPP heterogeneous catalyst by integrating tetra carboxy-porphyrin (FeTCPP) with g-C3N4 nanosheets via a simple self-assembly approach (Fig. 58b).274 The strong interaction between the two components originates from both the π–π stacking of tri-s-triazine units with the porphyrin aromatic ring and via the hydrogen bonding of the carboxyl groups on FeTCPP with the amino groups of g-C3N4 nanosheets. The obtained g-C3N4/FeTCPP hybrid exhibits high activity for CO2 reduction under visible-light irradiation, producing CO with a rate of 1087 μmol g−1 h−1 (Table 12, entry 2) and selectivity up to 98%. The catalytic reduction of CO2 includes photoexcitation of g-C3N4 nanosheets, charge transfer from g-C3N4 to FeTCPP, formation of Fe0TCPP and finally evolution of CO. Thus, the cost-effective g-C3N4 nanosheets act as the light-harvesting unit and iron based FeTCPP serves as the catalytic site. The presence of the carboxyl substituents in the phenyl rings improves the photocatalytic activity since it enhances the interaction between g-C3N4 nanosheets and FeTCPP, and stabilizes the CO2 adduct via intramolecular hydrogen bonding formation.
Tian et al. reported the preparation of semiconductor nanoparticles through covalent coupling and assembly of metalloporphyrin with condensed carbon nitride derived from urea (g-CNU).275 In detail, 5,10,15,20-tetrakis(4-(2,4-diaminotriazinyl)phenyl)-metalloporphyrin (MTDPP, M = Fe, Co, Ni) was employed as an organic monomer and after copolymerization in the presence of urea the g-CNU-MTDPP (M = Fe, Co, Ni) nanoparticle photocatalysts were fabricated (Fig. 58c). Among the three hybrids, the cobalt-metallated material (g-CNU-CoTDPP) presented the higher performance catalyzing the CO2 to CO reduction with a rate of 57 μmol g−1 h−1 (Table 12, entry 3) and selectivity of 79%. This nanoparticle photocatalyst showed exceptional photo stability against photo-corrosion under light irradiation, due to effective charge separation and transfer, enabling for long-term utilization.
CdS is another semiconductor material that was employed for the sensitization of iron porphyrin molecular catalysts. CdS is a traditional semiconductor with a narrow band gap (2.4 eV), excellent light harvesting capability, and sufficiently negative CB potential, which make it a promising visible-light absorber. Li et al. prepared ethylenediamine-functionalized CdS (CdS-EF) and pure CdS without functionalization (CdS) and both of them were employed as light absorbers in combination with FeTCPP catalyst for CO2 photoreduction (Fig. 58d).276 The CdS-EF/FeTCPP hybrid photocatalyst presents enhanced catalytic activity compared to CdS/FeTCPP, producing CO and CH4 with rates of 7.5 and 0.18 μmol g−1 h−1, respectively (Table 12, entry 4). The photocatalytic process mainly consists of three steps, the generation of photoelectrons in CdS-EF upon illumination, followed by the electron injection to FeTCPP, and finally CO2 reduction on FeTCPP. The superior activity of CdS-EF/FeTCPP hybrid system was attributed to the formation of hydrogen bonds between the amino groups of CdS-EF and the carboxyl groups of FeTCPP. These H-bonds provide efficient channels for interfacial electron transfer and facilitate the electronic interaction between CdS and FeTCPP.
The same group reported the preparation of CdS/Bi2S3 heterostructures which were also employed as photosensitizer with the FeTCPP molecular catalyst.277 The narrow band gap semiconductor Bi2S3 was coupled with CdS via an ion exchange reaction to improve the charge separation efficiency and enhance the visible-light harvesting ability of CdS. Interestingly, the obtained CdS/Bi2S3/FeTCPP hybrid presented enhanced activity, catalyzing the CO to CO and CH4 reduction with rate of 37.9 and 0.2 μmol g−1 h−1, respectively (Table 12, entry 5). Two major reasons may account for the enhanced performance: (i) the Bi2S3 improves charge separation efficiency and (ii) the amount of surface defects on CdS nanorods are reduced during the ion-exchange reaction.
Colloidal quantum dots (QDs) were also employed as photosensitizers for CO2 reduction. Quantum confinement of their electrons makes their absorption and electrochemical properties tunable by simply changing their diameter. Moreover, QDs have large surface area-to-volume ratios, and their surface enable the adsorption of molecular catalysts to form quasi-static complexes that mediate fast and efficient charge transfer between sensitizer and catalyst. Weiss and co-workers reported the photosensitization of an iron porphyrin catalyst (FeTPP) with colloidal, heavy metal-free CuInS2/ZnS quantum dots (QDs) (Fig. 59a).278 Upon irradiation at 450 nm, the QD donates three electrons to form the catalytically active species Fe0TPP, which binds CO2 and reduces it to CO with 84% selectivity. The catalytic system achieved a TON of 55, using N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) as SED (Table 12, entry 6). The catalyst sensitization in this system is highly efficient, due to the ultrafast electron transfer between the QD and FeTPP. This ultrafast photoreduction is attributed to the formation of a quasistatic QD/FeTPP complex, probably driven by the interaction between Fe and sulfur on the surface of the QD.
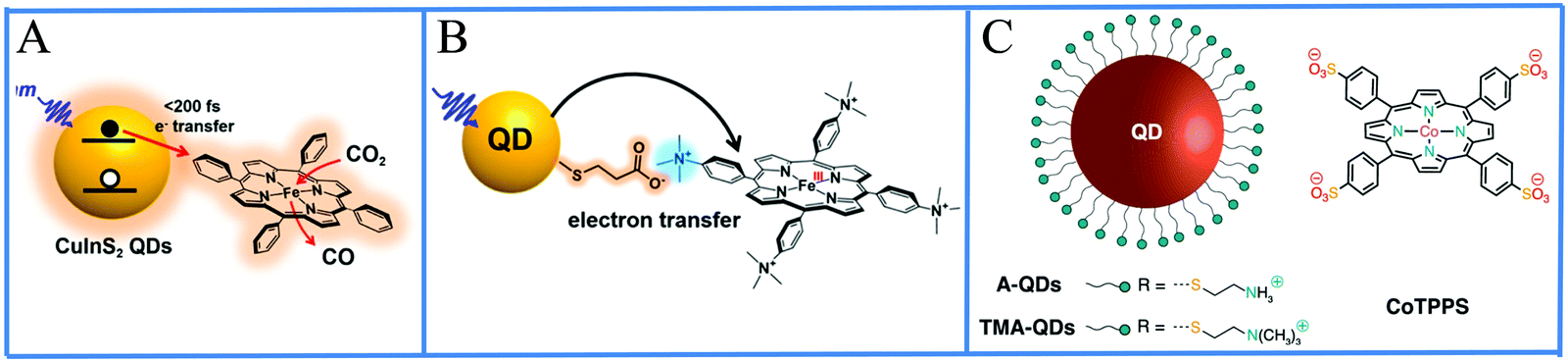 | ||
| Fig. 59 Photocatalytic CO2 reduction with CuInS2/ZnS QDs as PS and as catalyst: (A) the neutral FeTPP, Reproduced from ref. 278, with permission from American Chemical Society, Copyright 2017. (B) The positively charged FeTMA Reproduced from ref. 279, with permission from American Chemical Society, Copyright 2018 and (C) the negatively charged CoTPPS. Reproduced from ref. 280, with permission from American Chemical Society, Copyright 2021. | ||
The same group one year later extended this work by combining negatively charged colloidal CuInS2/ZnS QDs with positively charged trimethylamino-functionalized iron porphyrin catalyst (FeTMA).279 The catalytic studies were performed in H2O using TEOA as SED and a TON of 450 was obtained after 30 h of irradiation with selectivity of ∼99% (Table 12, entry 7). The catalytic performance increased significantly compared to the previous system, in which both the CuInS2 QDs and the FeTPP porphyrin were uncharged and dissolved in DMSO. This enhancement was attributed to the formation of electrostatically assembled superstructures, where one FeTMA is funneled photoelectrons from several QDs. The creation of these catalytically active superstructures is driven by the binding of negatively charged carboxylate tail groups on the QDs with the positively charged trimethylamino groups on the FeTMA (Fig. 59b). Three years later, the same team utilized positively charged CuInS2/ZnS colloidal QDs as PS and a negatively charged Co–porphyrin (CoTPPS) as catalyst (Fig. 59c).280 The synthesized QDs were coated in ligands terminated with amines/ammoniums groups (A-QDs), or with trimethylammoniums groups (TMA-QDs). The catalytic studies with the A-QDs were performed in pure water and CO formation was observed with an exceptional high TON of 84101 and selectivity 99.1% (Table 12, entry 8). The enhanced catalytic activity was attributed to the strong electrostatic attraction of the QDs with the catalyst, which promotes fast multielectron delivery and colocalization of protons, CO2, and catalyst at the source of photoelectrons. Moreover, the TMA-QDs presented lower performance (38473 TON) compared to A-QDs, under the same conditions. This was ascribed to the presence of free amines in A-QDs, which capture CO2 as carbamic acid that serves as a reservoir for CO2, effectively increasing its solubility in water, and lowers the onset potential for catalytic CO2 reduction by the Co–porphyrin. The breakthrough efficiency of this system represents a significant step in the development of reaction networks for direct solar-to-fuel conversion.
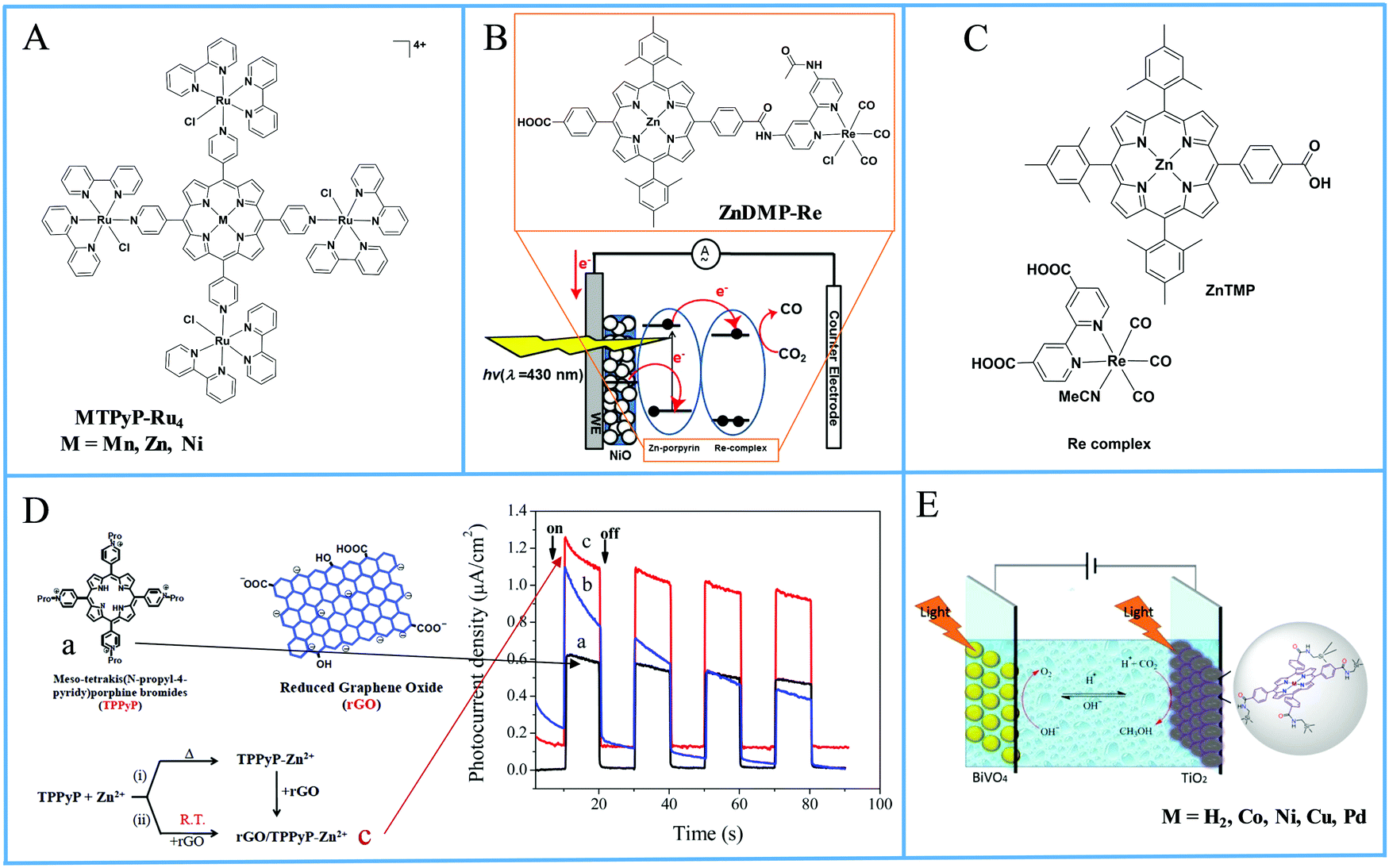 | ||
| Fig. 60 (A) Molecular structures of metallated TPyP porphyrin coordinated to four Ru-bis(pyridine)chloride moieties via the pyridyl groups, (B) schematic illustration of the photoelectrochemical scheme using ZnDMP-Re dyad, Reproduced from ref. 283, with permission from Elsevier, Copyright 2014. (C) Molecular structures of the ZnTMP photosensitizer and Re complex catalyst, (D) chemical structures and photocurrent density of TPPyP, reduced graphene oxide (rGO) and the nanohybrid photoelectrocatalyst rGO/TPPyP-Zn, Reproduced from ref. 285, with permission from Elsevier, Copyright 2018. (E) Schematic illustration of the artificial photosynthesis cell consisted of metalloporphyrin-modified TiO2/FTO electrodes photocathode and BiVO4 photoanode. Reproduced from ref. 286, with permission from Elsevier, Copyright 2019. | ||
| Entry | Porphyrin | Semiconductor | Solvent and ingredients | Products | Light source | Ref. |
|---|---|---|---|---|---|---|
| 1 | MTPyP-Ru4 | ITO | 0.1 M NaClO4(aq) | HCOOH, CO, MeOH and CH2O | 500 W Xe–Hg lamp | 281 |
| 2 | CoTPP | g-C3N4/FTO | 0.1 M KCl(aq) | HCOOH | 300 W Xe lamp | 282 |
| 3 | ZnDMP-Re | NiO/FTO | NBu4PF6 (0.1 M) in DMF | CO | 500 W Xe lamp | 283 |
| 4 | ZnTMP | Li-NiO/FTO | NBu4PF6 (0.1 M) in DMF | CO | 500 W Xe lamp | 284 |
| 5 | TPPyP-Zn | Glassy carbon | 0.1 M Na2SO3(aq) | not studied | 500 W Xe lamp λ > 420 nm | 285 |
| 6 | MTCPP | TiO2/FTO | 0.1 M NaHCO3(aq) | MeOH | 300 W Xe lamp | 286 |
| 7 | HNTM-Au-SA | Gas diffusion electrode | 0.1 M KHCO3(aq) | CO and HCOOH | 300 W Xe lamp λ > 420 nm | 287 |
| 8 | MOF-NS-Co | Gas diffusion electrode | 0.1 M KHCO3(aq) | CO and HCOOH | 300 W Xe lamp λ > 420 nm | 288 |
| 9 | CoTMPP | Perovskite/NiOx/FTO | [6,6]-Phenyl C61 butyric acid methyl ester (PCBM)/polyethylenimine/Ag | CO and H2 | Solar light simulator | 289 |
| 10 | Cu(T4H3MPP) | Ti/TiO2 nanotubes | 0.1 M Na2SO4(aq) | MeOH and EtOH | 300 W Xe lamp | 290 |
| 11 | CuTCPP | Copper foam | 0.5 M NaHCO3(aq) | EtOH | 300 W Xe lamp | 291 |
| 12 | Cu@porphyrin-COFs | Electrodeposited Copper | 0.5 M KHCO3(aq) | MeOH, EtOH, (CH3CH(OH)2) and (CH3COCH3) | 300 W Xe lamp | 292 |
| 13 | HOOC-(Zn)DMP-tpy-Ru | NiO | 0.1 M Bu4NPF6 in MeCN:H2O 9:1 |
— | White LED PAR38 lamp (17 W) | 293 |
In another report, Kou et al. synthesized a dyad (ZnDMP-Re) comprising of a zinc-porphyrin sensitizer moiety bearing a carboxylic anchoring group and a rhenium-complex catalytic moiety for CO2 reduction (Fig. 60b).283 The authors initially showed that ZnDMP-Re dyad was able to perform photoreduction of CO2 in solution with TEA as SED and then applied this system is a photoelectrochemical scheme without any sacrificial reagents. In detail, the adsorbed dyad on p-type NiO nanoparticles was deposited on FTO as working electrode and exhibited light induced reduction of CO2 to CO in DMF solution using platinum as counter electrode (Table 13, entry 3). Noteworthy the performance was improved via the co-adsorption of ZnTCPP as additional photosensitizer to the dyad. The same research group further examined this system in a recent report by doping NiO with Li ions in order to promote the hole carrier mobility within the semiconductor.284 In this system the authors co-adsorbed the ZnTMP photosensitizer and the Re complex catalyst (Fig. 60c) on the NiO semiconductor and indeed observed enhanced CO2 photoreduction under 6% of Li ions doping (Table 13, entry 4).
Zeng et al. prepared porphyrin–graphene nanocomposites in aqueous media utilizing the intramolecular electrostatic interactions as well as the π–π stacking between TPPyP and reduced graphene oxide (rGO) (Fig. 60d).285 Zn metallation was followed to afford the nanohybrid photoelectrocatalyst rGO/TPPyP-Zn which was applied in CO2 reduction under visible light irradiation. The best performing system was rGO/TPPyP-Zn nanocomposite which exhibited higher photocurrent than TPPyP and rGO/TPPyP composites (Table 13, entry 5).
Symmetrical porphyrins, as in the previous report, have attracted significant attention for photoelectrochemical CO2 reduction. In the same direction, Jing and co-workers286 prepared porphyrin-modified TiO2/FTO electrodes based on the TCPP (meso-tetra-p-carboxyphenyl-porphyrin), and the corresponding metallated with Co, Ni, Cu and Pd derivatives. In detail, the TiO2/FTO electrodes were initially functionalized with amino groups via aminopropyltriethoxysilane and subsequently covalently linked with MTCPP porphyrins yielding TiO2 modified photocathodes. The authors showed that these photocathodes can efficiently convert CO2 to MeOH photoelectrocatalytically and afterwards combined them with a BiVO4 photoanode, which was performing water oxidation, to form an artificial photosynthesis cell (Fig. 60e and Table 13, entry 6).
In another report, Wang and co-workers287 synthesized zirconium PMOF hollow nanotubes using the widespread TCPP and afterwards anchored Au, Cu, and Co as single porphyrin-metal atom catalysts to facilitate CO2 conversion (named as HNTM-Au-SA, HNTM-Cu-SA, and HNTM-Co-SA, Fig. 61a). The authors investigated the electrochemical CO2 reduction in dark as well as under visible light irradiation, where they observed high TOF performance (Table 13, entry 7). The photo-coupled electrochemical experiments showed that similar activity could be obtained at a lower overpotential with a positive shift of 20, 100, and 130 mV for the Co, Cu and Au modified PMOF, respectively. This trend was consistent with the calculated energy gaps while gas chromatography and NMR experiments verified that the reduction products were CO and HCOOH. The same research group further explored this system and prepared 2D PMOF nanosheets (MOF-NS) using again TCPP and zirconium salt (Fig. 61a).288 In this report, the authors employed Cu, Co and Fe as anchored elements to improve the catalytic activity of the material. Under visible-light irradiation, CO2 was efficiently converted to CO with a 100 mV positive shift compared to the electrocatalytic measurement under dark, for the MOF-NS-Co system (Table 13, entry 8).
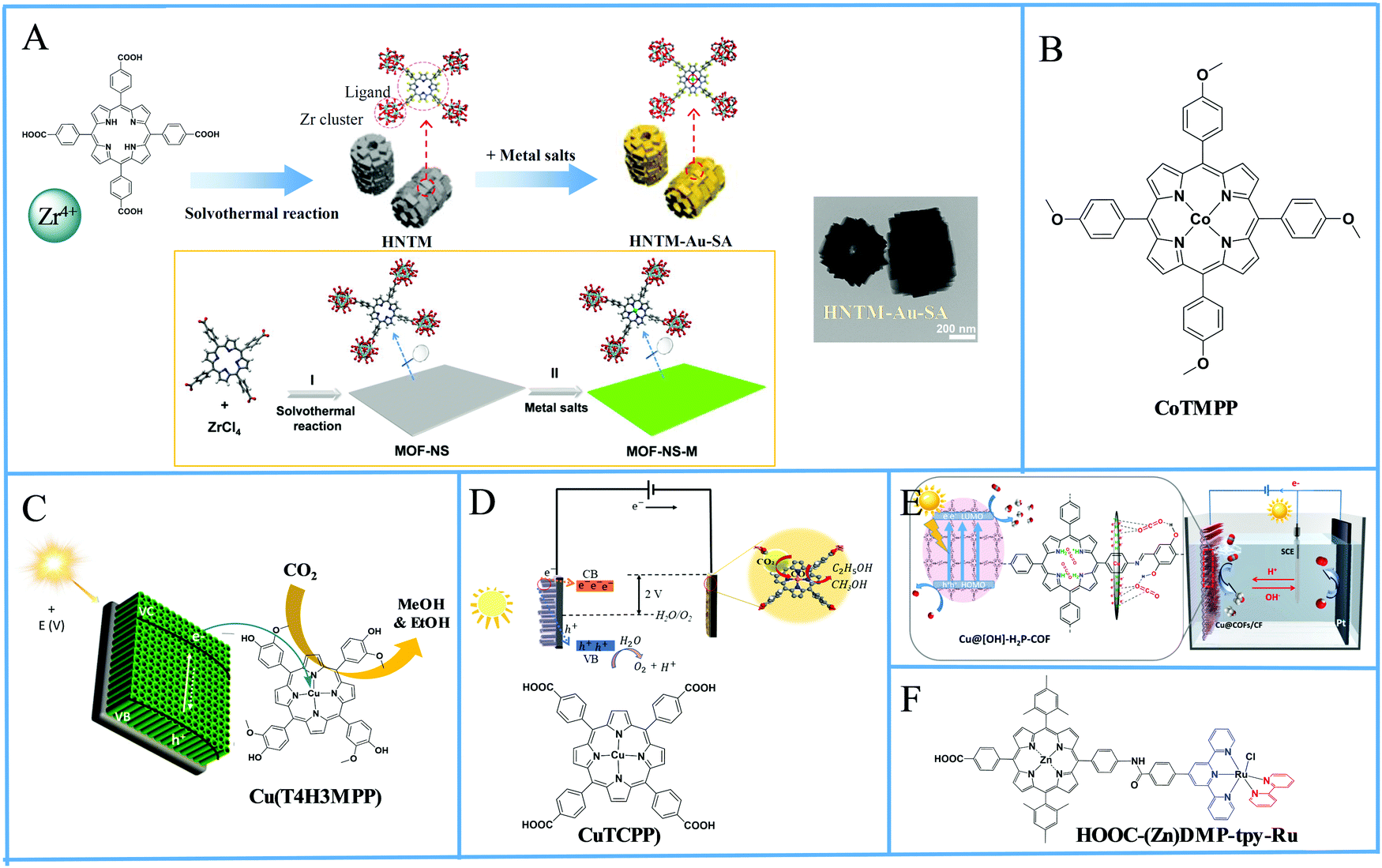 | ||
| Fig. 61 (A) Schematic illustration PMOF hollow nanotubes HNTM-Au-SA, HNTM-Cu-SA, and HNTM-Co-SA as well as the 2D PMOF nanosheets (MOF-NS) using the same TCPP porphyrin and ZrCl4 salt, Reproduced with permission from ref. 287, which is an open-access article distributed under the Creative Commons Attribution 4.0 International License, 2019 Springer Nature. (B) Molecular structure of CoTMPP porphyrin, (C) chemical structure of Cu(T4H3MPP) and schematic illustration of the resulting photoanodes, Reproduced from ref. 290, with permission from Elsevier, Copyright 2020. (D) Two-compartment photoelectrochemical cell with CuTCPP as a catalyst for CO2 reduction, Reproduced from ref. 291, with permission from Elsevier, Copyright 2020. (E) Schematic illustration of the photoelectrochemical cell consisted of Cu@porphyrin-COFs nanorods photocathode, Reproduced from ref. 292, with permission from Elsevier, Copyright 2020. (F) Chemical structure of Zn porphyrin–ruthenium complex dyad. | ||
Reisner and co-workers designed a tandem photoelectrochemical device able to convert carbon dioxide to carbon monoxide coupled with water oxidation in the absence of external bias.289 The authors utilized the noble metal free Co(II)-meso-tetrakis(4-methoxyphenyl) porphyrin (CoTMPP), as a molecular catalytic moiety immobilized via π–π intramolecular interactions onto carbon nanotubes sheets. Perovskite photocathodes were prepared using the above porphyrin–carbon nanotubes assemblies and subsequently incorporated in a tandem device with a BiVO4 photoanode bearing a known Co WOC (Fig. 61b). The above tandem call was able to convert CO2 to CO and H2 at the photocathode while water oxidation was performed at the photoanode (Table 13, entry 9). The authors revealed that light intensity is an important factor determining the selectivity of the system and showed that the highest selectivity towards CO formation was achieved at 0.1 sun light intensity.
In the following reports copper metallated porphyrins were utilized for the conversion of carbon dioxide into alcohols. Brito et al. modified the surface of Ti/TiO2 nanotube electrodes by immobilizing the Cu(T4H3MPP) metallated porphyrin (Fig. 61c) and investigated the photoelectrocatalytic performance of the resulting photoanodes for CO2 reduction.290 The authors showed that their system has selectivity towards the formation of MeOH and EtOH, while displayed enhanced activity compared to photocatalysis and electrocatalysis in the same conditions (Table 13, entry 10). Another report by Cheng and co-workers used the Cu(II)-meso-tetra-p-carboxyphenyl-porphyrin to prepare PMOFs nanosheets (NSs) on top of which CuS NPs were attached providing the catalytic moieties for CO2 reduction.291 This catalytic system was employed as electrocathode for CO2 conversion in the two-compartments photoelectrochemical cell with TiO2 nanotube photoanodes performing water oxidation (Fig. 61d) and exhibited efficient and selective light induced CO2 conversion to ethanol (Table 13, entry 11).
Wang et al.292 prepared the same porphyrin COFs we previously reported in the heterogeneous photocatalytic H2 production paragraph (Fig. 19b). In detail, the authors synthesized porphyrin COF nanorods and subsequently electrodeposited Cu on them (Fig. 61e). The resulting material Cu@porphyrin-COFs nanorods presented high photoelectrocatalytic performance for CO2 reduction, producing methanol, ethanol, ethane-1,1-diol and acetone (Table 13, entry 12). The authors showed via13C labeling experiments that the liquid products derived from CO2 and via DFT calculations that hydroxyl groups of free-base porphyrin COFs have an important role in the catalytic process. Charisiadis et al. contributed in the field of DSPECs towards CO2 reduction by applying a Zn porphyrin–ruthenium complex dyad (Fig. 61f) anchored on a NiO photocathode to fabricate a p-type DSPEC (Table 13, entry 13).293
4. Phthalocyanines for photocatalytic H2 evolution
Because of their high light-harvesting ability and outstanding physicochemical and optical features, Pc derivatives have been widely exploited as photosensitizers or catalysts in the field of photocatalytic H2 production from water. Their chemical diversity results in photophysical properties that can be modulated. Many Pc derivatives have been, therefore, prepared to date in order to investigate the relationship between molecular structure and photocatalytic performance in more depth. Recent examples of both Pc-based photosensitizers and catalysts in photocatalytic hydrogen production will be described in the following section.4.1 Metal oxides combined with phthalocyanines
The success of phthalocyanine in the field of artificial solar energy conversion devices, particularly in molecular photovoltaics,36 has inspired chemists to investigate the activity of Pc derivatives covalently and/or non-covalently linked to semiconductors as a sensitizer for photocatalytic hydrogen production. Previous reports on Pc-based DSSCs have emphasized the lack of directionality of the excited state of the Pcs and poor coupling of the LUMO with the TiO2 conduction band as bottle-neck that mainly limits the power conversion efficiencies.36,294 Later research demonstrated that the rational design of push–pull Pc derivatives with an anchoring group that provides charge transfer directionality from the Pc to the semiconductor is an effective technique for overcoming this challenge.294 The engineering of Pc derivatives for the DSSCs system have been focused on molecular structures based on unsymmetrical functionalized A3B type derivatives with an anchoring group.295 As a result, asymmetric phthalocyanine derivatives have been favoured for panchromatic sensitization of semiconductors in photocatalytic hydrogen production as well.Zhang et al. reported unsymmetrically substituted ZnPc bearing three tert-butyl groups and the carboxylic acid anchoring group (Zn-tri-PcNc) and symmetrical tetra-carboxy-zinc naphthalocyanine (Zn-tetra-Nc) derivatives (Fig. 62) as dyes to sensitized TiO2 for photocatalytic H2 production.296 Zn-tri-PcNc and Zn-tetra-Nc show strong visible/near-IR light absorption at 694 and 766 nm, respectively, and Pc sensitized/TiO2 exhibit broader absorption spectra from 550 to 900 nm compared to that of pristine TiO2. The photocatalytic performance of Zn-tri-Pc-Nc and Zn-tetra-Nc sensitized TiO2 was investigated in the presence of Pt as a co-catalyst and EDTA as a sacrificial reagent. After irradiation of 5 h, Zn-tri-PcNc/TiO2 achieved a H2 yield of 567.4 μmol with a TON of 7565, which is much higher than that of Zn-tetra-Nc/TiO2 (236.5 μmol H2 with a TON of 3153). The higher photocatalytic activity of Zn-triPcNc was attributed to a good balance between diminishing dye aggregation by introducing bulky tert-butyl groups and improving the charge transfer directionality in the excited state, which was achieved by the push–pull structure.
 | ||
| Fig. 62 Molecular structures of Zn-tri-PcNc and Zn-tetra-Nc and schematic representation of a H2 production of Zn-tri-PcNc-TiO2-AA photocatalyst. | ||
Even though the efficiency of solar energy conversion devices is governed by several factors based on photo-induced charge generation and separation, one of the most important parameters is the light-harvesting ability of the photoactive component. Thus, light-harvesting systems with broad absorption to cover large portions of the solar spectrum are very desirable. However, the preparation of a single dye to fulfil this requirement with dye sensitization systems remains a major challenge. In this respect, the combination of two or more dyes with a complementary optical property appears to be an attractive strategy to cover the entire visible region and even the near-infrared part (NIR). This tactic has been widely used in DSSCs to achieve panchromatic sensitization of TiO2 films.36,295 The related approach is also used in semiconductor-based photocatalysts, particularly TiO2, by complexing TiO2 with either small molecules such as acids or alcohols, (TEA, EDTA, ascorbic acid (AA) etc.) or D–π–A organic dyes to increase the light-harvesting ability. Many kinds of electron donor molecules have been used both as a sacrificial reagent for the formation of charge-transfer complexes on the surface of TiO2 to increase the photocatalytic activity of photocatalyst.297 In this case, it is worthy to mention that the absorption expansion of the semiconductor is based on the ligand-to-metal charge transfer (LMCT) between the absorber and the semiconductor rather than the common dye sensitization mechanism. However, LMCT complexes (TiO2-small molecule) are not sufficient for the development of the stable visible light photocatalyst.
In 2015, further developments were reported by Zhang et al. in terms of panchromatic and stable photocatalytic systems involving the addition of ascorbic acid on TiO2 and co-sensitization with ZnPc (Fig. 63).298 As mentioned, Pcs possess attractive light absorption features in the visible and near-infrared regions of the solar spectrum, exhibiting a very intense absorption at 300–400 nm (Soret band) and at 700 nm (Q band), but are transparent over a large area of the visible spectrum which makes them interesting dyes to be employed in this cocktail approach. The Zn-tri-PcNc-TiO2-AA photocatalytic system exhibited a much higher photoactivity (162.2 μmol h−1) for H2 production than that of the LMCT AA-TiO2 complex (126.2 μmol h−1), mainly due to its improved light-harvesting efficiency in the visible and near IR spectral region. Indeed, it was confirmed by DRS spectra that Zn-tri-PcNc-TiO2 absorbs in the range of 600–900 nm, but the co-sensitized Zn-tri-PcNc-TiO2-AA system exhibits a much broader absorption in the range of 400–900 nm, including the characteristic absorption bands of the surface AA-TiO2 complex and Zn-tri-PcNc molecules. This two-channel system, which collects photoelectrons in both the 400–600 nm and 600–800 nm range, resulted in higher hydrogen production than single components.
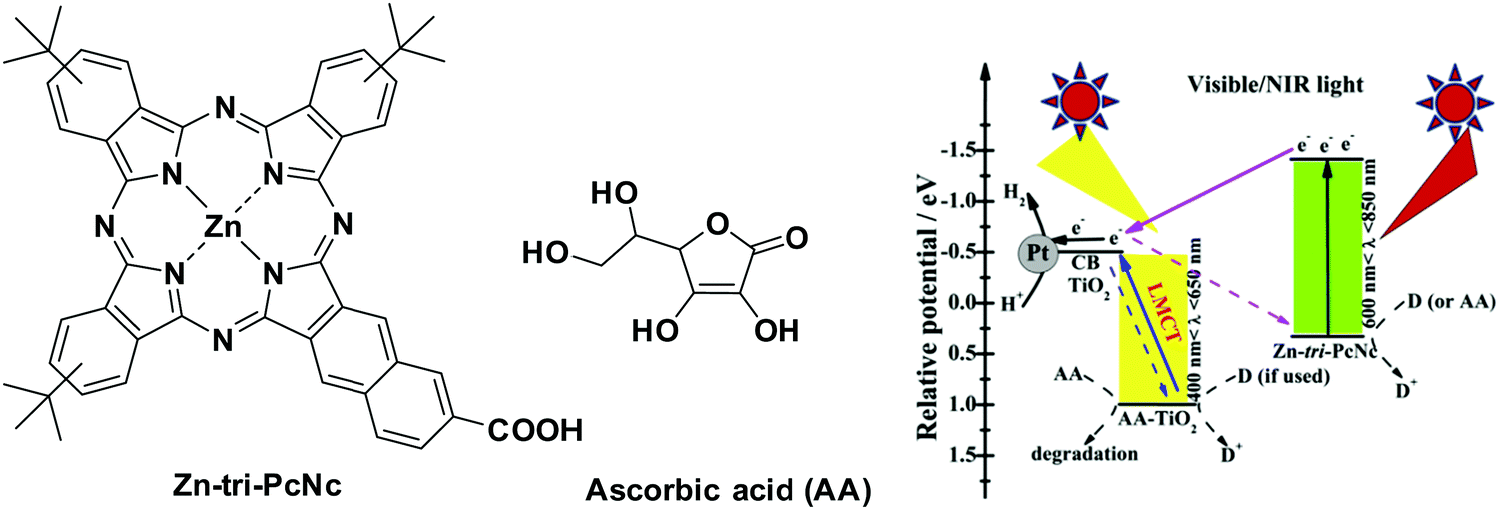 | ||
| Fig. 63 Schematic representation of H2 production of Zn-tri-PcNc-TiO2-AA photocatalyst. Reproduced from ref. 298, with permission from Elsevier, Copyright 2015. | ||
TiO2 has several considerable advantages in terms of non-toxicity and stability. The additional improvement of photocatalytic activity could be achieved by tuning of surface area, crystal structure, shape, size etc. In particular, the introduction of porosity into TiO2 in a hierarchical architecture plays an important role not only to increase the surface area and pore volume but also to improve the light-scattering and light-harvesting abilities of photocatalysts.299 Amritanjali Tiwari et al. introduced unsymmetrical ZnPc (PCH001) (Fig. 64) decorated with three tert-butyl and two carboxylic acid groups, to sensitize hierarchical porous TiO2 (HPT) particles.300 The ZnPc sensitized Pt/HPT photocatalyst showed attractive H2 producing activity (2260 μmol mmol g−1 in 5 h) with the highest TON of 18080 when compared with the reference N716 Ru(II)-bipyridyl dye. The high hydrogen production is attributed to the hierarchically porous structures of TiO2, which allow excellent transport of the reactant mass and higher light absorption capacity, as well as the efficient electron transfer arising from the push–pull structure of asymmetric ZnPc.
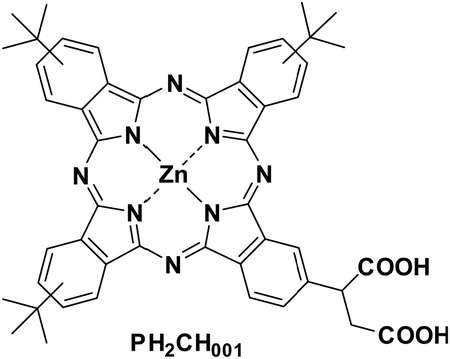 | ||
| Fig. 64 Molecular structure of PH2CH001. | ||
As mentioned previously, many photocatalytic systems require noble metal-based co-catalysts to increase hydrogen production efficiency by trapping photoexcited electrons and reducing charge recombination on the TiO2 surface. Despite the benefits of co-catalyst, both the supply and cost of these noble metals such as platinum (Pt) limit their practical applications. Recently, Moon et al. reported the use of CuPc sensitized TiO2 as the photocatalyst in photocatalytic hydrogen evolution without the aid of noble metal as a cocatalyst.301 The photocatalytic performance of CuPc/TiO2 catalysts was tested in the aqueous methanol solution in which methanol also acts as a sacrificial reagent. The photocatalyst CuPc/TiO2 produced an H2 production rate of 0.95 μmol h−1 with a high apparent quantum yield (AQY) of 6.68% at 420 nm monochromatic light irradiation. In this system, TiO2 acts as an electron mediator and CuPc serve as both a sensitizer and a charge transfer mediator. Upon photoexcitation of the photocatalyst with λ > 420 nm visible light, the excited electron–hole pair occurs only at the CuPc rather than TiO2, while the excited electron is transferred to the CB of TiO2 and used in the hydrogen evolution reaction, while the holes at HOMO of CuPc were scavenged by methanol (Fig. 65). Due to the absence of photoinduced electron and hole pair in TiO2, the internal charge recombination is avoided in the CuPc/TiO2 nanocomposite.
 | ||
| Fig. 65 Proposed mechanism of photocatalytic H2 evolution with CuPc/TiO2 nanocomposites. Reproduced from ref. 301, with permission from Elsevier, Copyright 2020. | ||
The nature of central metal atoms at the inner cavity of the Pc macrocycles also impacts on the physical, electronic, and optical properties of the Pcs and indeed the catalytic activity of Pcs is strongly related to central metal atoms. This aspect is particularly important in the case of electrocatalytic activity of the Pc derivatives since in most cases metal atoms act as the catalytic active center of the multi-electron reaction process. In order to investigate the influence of the central metal on the performance of photocatalytic hydrogen evolution, tetracarboxylic acid Pc derivatives with Zn and Co atoms as central metals (Fig. 66) have been reported by Ince and coworkers.302 The hydrogen production rate of ZnPc/TiO2 and CoPc/TiO2 photocatalyst in the presence of Pt co-catalyst was determined as the 3448 and 3328 μmol g−1 h−1, respectively. The improved photocatalytic activity of ZnPc sensitized TiO2 was attributed to superior the spectral response of Pc sensitizers, in fact, ZnPc/TiO2 revealed a significant red-shift of the Pcs Q-band absorption with a high molar extinction coefficient.
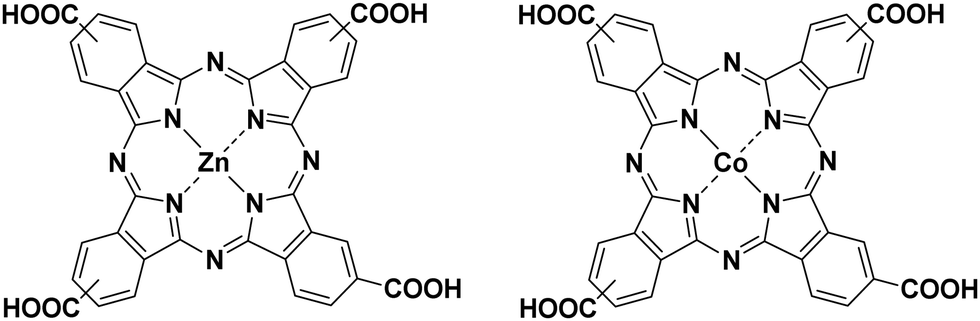 | ||
| Fig. 66 Molecular structures of tetracarboxylic acid ZnPc and CoPc. | ||
Despite the outstanding optical and photophysical properties of Pcs, the solar energy conversion efficiency of this family of sensitizers has been mainly limited by the strong tendency of Pcs to form aggregates both in solution and in the solid-state; this is more pronounced than in porphyrins, due to their more π-extended system. In the case of solar cells, significant progress in Pc-sensitized DSSCs came with the ZnPc 1 (known as TT40) bearing bulky 2,6-diphenylphenoxy peripheral substituents which completely suppresses macrocycle aggregation and impressive power conversion efficiencies over 6% were achieved (Fig. 67).303,304
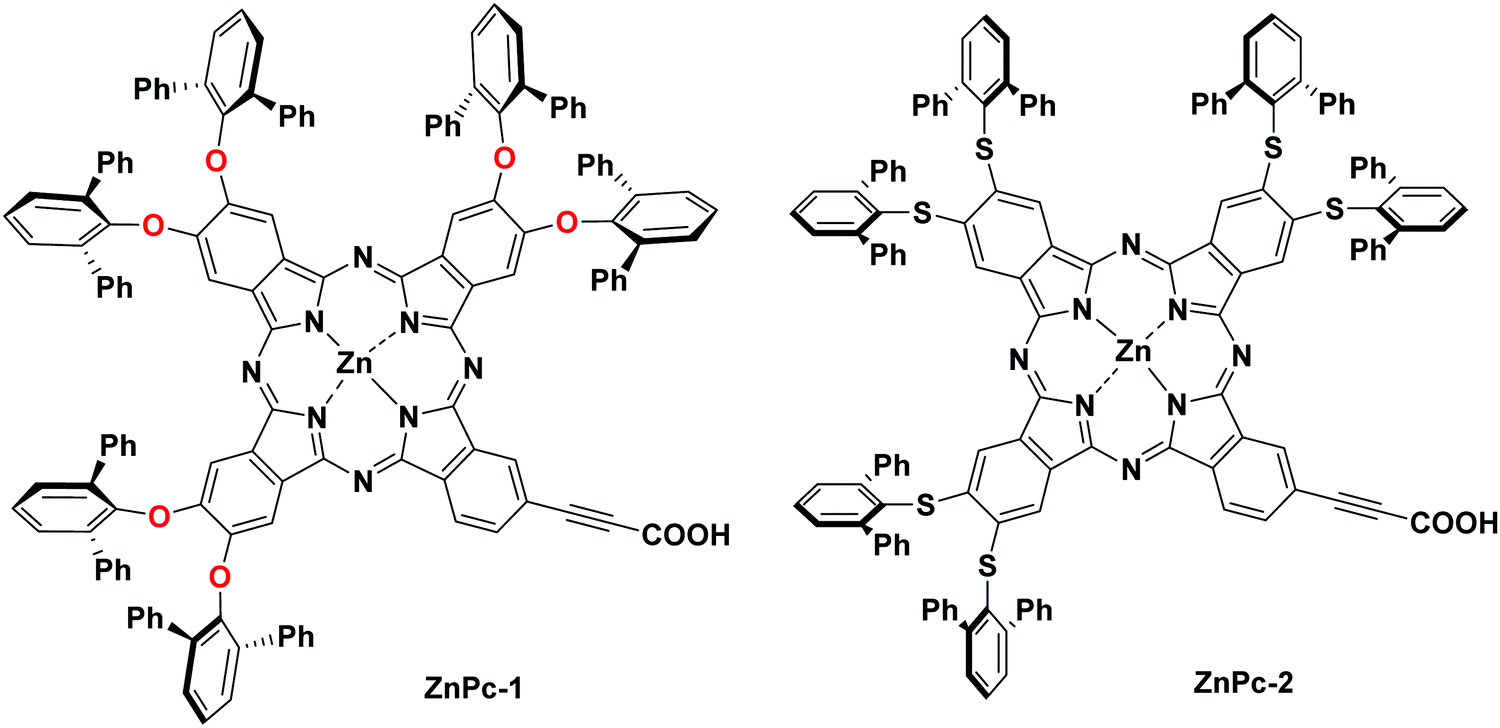 | ||
| Fig. 67 Molecular structures of phthalocyanine sensitizers ZnPc-1 and ZnPc-2. | ||
A new Pc derivative in which the oxygen atoms in the bulky phenoxy substituents were replaced with stronger electron-donating sulfur atoms (ZnPc 2) was also used as photosensitizer into the DSSC device, exhibited a relatively low photovoltaic conversion efficiency of 2.5% as compared with that of ZnPc 1.305 Ince et al. recently compared the photocatalytic activity of ZnPc-1 and ZnPc-2 sensitized TiO2 photocatalysts in the photocatalytic hydrogen evolution process in the absence and presence of co-catalyst.306 The produced hydrogen amount of ZnPcs derivatives (ZnPc-1/TiO2 and ZnPc-2/TiO2) were found as 1.221 mmol g−1 h−1 and 0.864 mmol g−1 h−1 with a corresponding solar-to-hydrogen (STH) efficiencies of 3.15% and 2.22% respectively, for the first hour. In the presence of Pt, hydrogen generation increased after 8 hours of illumination, reaching 32808 mmol g−1 and 16067 mmol g−1 for ZnPc 1 and 2, respectively. The introduction of the thioether units ZnPc-2 at the peripheral positions of the Pc results in a dramatic red shift in the absorption spectra, by decreasing the LUMO level and increasing the HOMO level of ZnPc-2 in comparison with the levels of ZnPc-1 derivative. However, the expansion of the light-harvesting range of ZnPc-2 could not positively affect on the H2 evolution performance of ZnPc-2 even in the presence of the co-catalyst. The authors suggested that lower LUMO and higher HOMO energy levels of ZnPc-2 which might be hindering both the efficient electron transfer and dye regeneration could constitute a possible explanation for low hydrogen production.
Only very recently, new Pc sensitizers ZnPc-3 and ZnPc-4 peripherally anchored through an imidazole group have been synthesized in order to investigate the effect of the anchoring group on the performance of photocatalytic hydrogen evolution, by the same authors (Fig. 68).307 The photocatalytic performance of ZnPc-3 was compared with ZnPc-5 (known as TT1) in which the carboxy group is directly attached to the macrocycle in the same conditions. The photocatalytic performance of ZnPcs sensitized TiO2 was tested in the presence of TEOA without using any co-catalyst. ZnPc-3 was capable of efficient photocatalytic H2 generation, exhibiting the highest H2 evolution rates of 0.4006 mmol g−1 h−1, whereas the composite of ZnPc-4/TiO2 and ZnPc-5/TiO2 show the relatively low photocatalytic activity of 0.3319 mmol g−1 h−1 and 0.3555 mmol g−1 h−1, respectively. After 20 h irradiation, ZnPc-3 produces a hydrogen production rate of 3.4187 mmol g−1 with a TON of 14863, higher than ZnPc-5 in the absence of co-catalyst. All ZnPcs exhibited a similar light-harvesting ability and proper HOMO–LUMO energy levels. The origin of the photocatalytic activity differences was, thus, attributed to the efficient electron injection into the semiconductor's conduction band for the ZnPc3/TiO2 photocatalyst. The reason for the high performance of ZnPc-3 is claimed to be due to the fact that the stronger electron-withdrawing character of imidazole facilitates the electron injection from excited Pc into the semiconductor's conduction band. The low performance of symmetrically substituted derivative, ZnPc-4, was explained by the extension of electron delocalization over the whole molecule limiting the directional charge transfer.
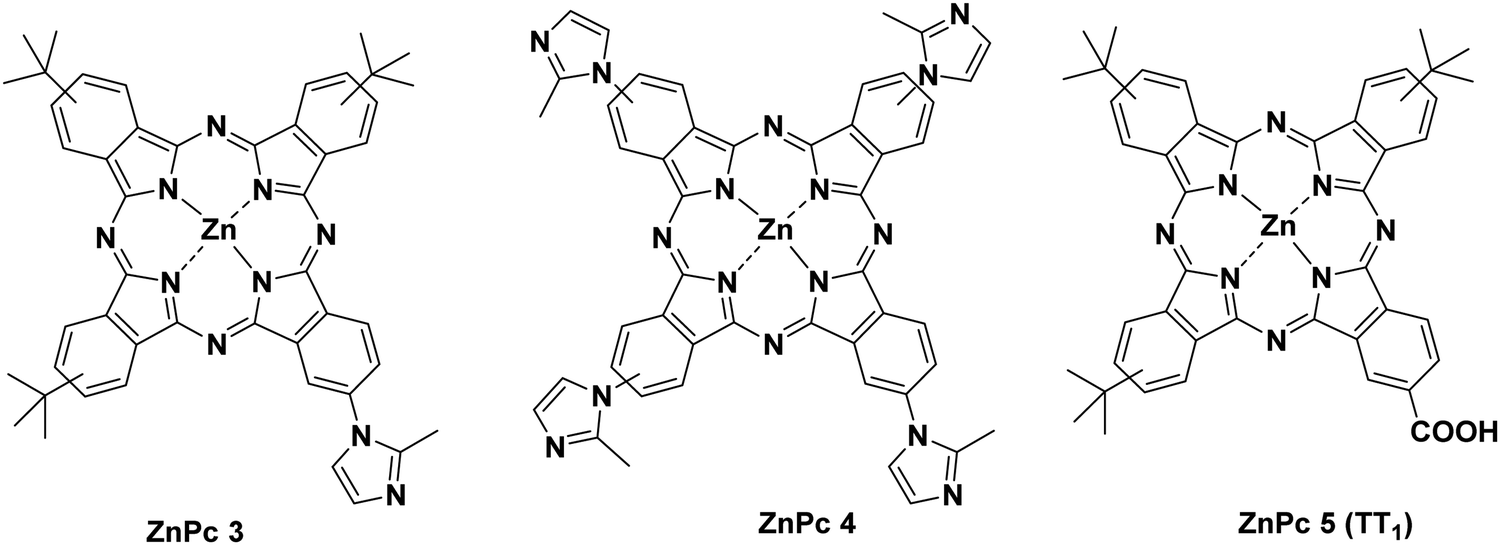 | ||
| Fig. 68 Chemical structures of ZnPc-3, ZnPc-4, and ZnPc-5. | ||
As a member of the synthetic porphrinoid family, subphthalocyanines (SubPcs) have been widely utilized as attractive building blocks in the construction of efficient artificial photosynthetic devices that mimic the natural photosynthetic systems for the conversion and storage of solar energy. SubPcs are aromatic compounds with a 14–π electron system consisting of a central boron atom, a lower homolog of Pcs, which possess a cone-shaped structure, in contrast to the wide range of planar aromatic compounds.308 Their high solubility in common organic solvents arising from a low tendency to aggregate and particularly their intense absorption in the visible region (500–700 nm), render them ideal light-harvesting material in the concept of artificial photosynthetic systems. SubPcs are synthetically versatile compounds, and their physicochemical properties can be optimized by introducing appropriate groups in the peripheral or axial positions. SubPcs have been extensively used in organic photovoltaic solar cells as both electron donor and acceptor components owing to their tunable HOMO–LUMO energy levels together with excellent charge transport properties.309 SubPc sensitizers decorated with various peripheral substituents have been incorporated into dye-sensitized solar cells, yet moderate power conversion efficiencies were obtained due to their low-lying LUMO levels.310,311
Inspired by the potential for use of SubPc in DSSCs, Ince and co-workers introduced SubPc derivatives as sensitizers for the sensitized photocatalyst. This is the very first example of SubPc as a sensitizer in the field of photocatalytic H2 production.312 To compare the influence of the anchoring group position on the photocatalytic activity of SubPcs, a series of SubPcs having a carboxylic acid anchoring group at either a peripheral (SubPc 1–2) or axial (SubPc 3) position were synthesized (Fig. 69). SubPc3/TiO2 exhibited the best photocatalytic activity with a hydrogen evolution rate of 1396 μmol h−1 which is much higher than that of SubPc 1 and 2 (771 and 658 μmol g−1, respectively). The activity of all SubPcs increases significantly after 8 h irradiation, the evolved hydrogen amounts of SubPc1–3/TiO2 reaches to 5038, 5135 and 8062 μmol g−1, respectively. Further improvement of hydrogen generation up to 22345 μmol g−1 (for SubPc 3) was achieved by using Pt as a co-catalyst. SubPc 3/TiO2 photocatalyst gave the highest hydrogen production rate, which is attributed to its higher LUMO energy level, which provides sufficient driving force for an efficient charge transfer from the LUMO orbital of the dye to the conduction band of TiO2.
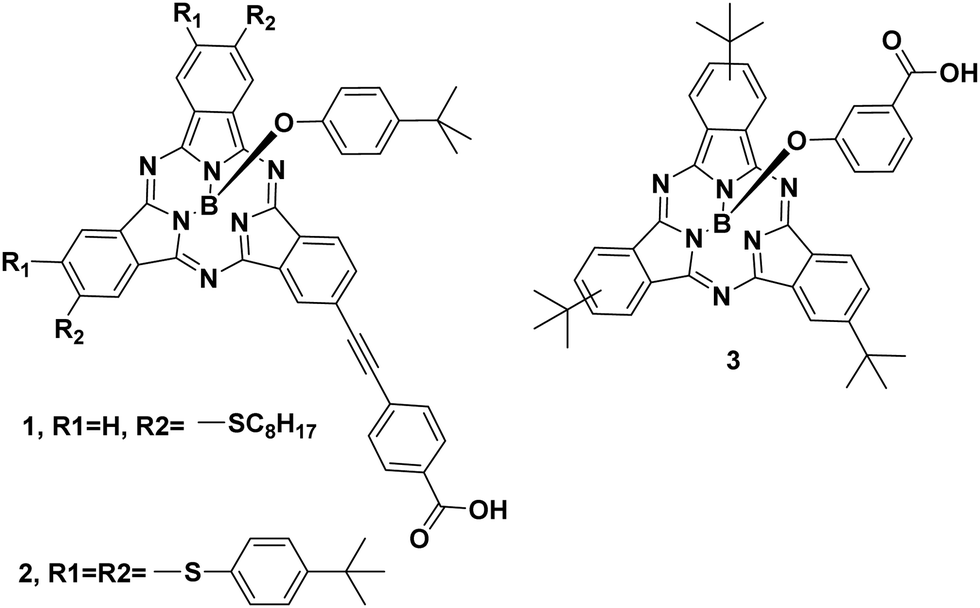 | ||
| Fig. 69 Molecular structures of SubPc 1, 2 and 3. | ||
4.2 Phthalocyanine DSPSCs for H2 production
Over the last ten years, there have been a significant advance in the design and development of dye-sensitized photoelectrochemical cells (DSPECs) for water splitting.32 Similar to DSSCs, nevertheless both devices share a common architecture, the semiconductor photoelectrodes are sensitized by molecular dyes. Porphyrins and phthalocyanines have shown great potential as sensitizers in DSSCS, so the interest in incorporating these chromophores into photoelectrodes for applications in DSPECs is not surprising. Porphyrins have received more attention than Phthalocyanines as sensitizers in DSPECs and in a previous section of this review, the most relevant articles have been summarized. As aforementioned, Sherman et al. reported a tandem DSPEC system for the light driven generation of H2 from hydroquinone using only light energy and no applied electrical bias.170 The tandem cell configuration is based on the combination of a dye-sensitized photoelectrochemical cell based on a SnO2 photoanode, which is sensitized by a free-base porphyrin, in series with dye-sensitized solar cells in which a TiO2 photoanode is sensitized by a Si phthalocyanine. There is a niche for the development of phthalocyanine-based DSPECs and we expect this research topic to accelerate in the future.A promising approach for the engineering of a photoelectrochemical water-splitting system is based on an organo-photocathode of a p–n bilayer and a semiconductor photoanode. In 2016, Abe et al. reported a water-splitting system comprising of a TiO2 photoanode and a ZnPc/C60 bilayer modified by Pt as a photocathode.313 This system demonstrated stoichiometric decomposition of water into H2 and O2 successfully occurred at bias voltages lower than the theoretical value (i.e., 1.23 V), with efficiencies ca. 0.1% at a small bias voltage of 0.25 V. Remarkably, distinct from the reference system TiO2/Pt, the system is capable of water splitting without any externally applied bias was observed. Abe and co-workers extended this work but exploring other metal oxides as photoanode. In 2017, a photoelectrochemical water-splitting system employing WO3 as a photoanode and Pt loaded ZnPc/C60 bilayer as photocathode was reported.314 Although the system led to the stoichiometric formation of H2 and O2 due to water decomposition under a low bias voltage of 0.1 V, the most efficient water-splitting occurred at 0.6 V with ca. 0.07%. In the case of the reference system WO3/Pt bias voltages more than 0.4 V were needed to be applied for water splitting. The same research group reported in 2020 a photoelectrochemical water-splitting system based on the same Pt loaded ZnPc/C60 bilayer photocathode but using a photoanode comprised with a mixed-valence cobalt(II,III) oxide (Co3O4) disperse in a Nafion membrane on a nanoporous BiVO4.315 The stoichiometric generation of H2 and O2 occurred by applying a bias voltage as low as 0.1 V, which was superior to the reference system comprising BiVO4 and Pt, ant the light-to-hydrogen conversion efficiency was ca. 0.08% at 0.6 V. The presence of a cobalt cocatalyst not only improved the water oxidation process but also the stability of the device even in an acidic medium.
4.3 Graphitic carbon nitrides combined with phthalocyanines
In addition to TiO2, another interesting photocatalyst material is graphitic carbon nitrides (g-C3N4) which has been successfully used in photocatalytic H2 evolution reactions. The effectiveness of g-C3N4 is related to its 2D planar structure, relatively narrow energy bandgap (2.7 eV), adequate energy levels, chemical and thermal stability. Graphite carbon nitride attracts particular attention as a low-cost alternative to its inorganic relatives as it contains earth-abundant elements (C, H, N). However, in order to overcome the low absorption and charge mobility of g-C3N4 (<460 nm), bandgap engineering through metal doping and dye sensitization must be employed.316Zhang et al. explored the influence of panchromatic co-sensitization on the photocatalytic activity of graphitic carbon nitride-based photocatalysts.317 They reported complementary absorption spectra over g-C3N4 for an indole-based D–π–A organic dye (LI-4) and an asymmetric ZnPc derivative (Zn-tri-PcNc) (Fig. 70). Zn-tri-PcNc exhibits absorption in the region of 650–700 nm, and LI-4 shows strong absorption in the visible region between 400–600 nm. Upon irradiation of the LI-4/g-C3N4/Zn-tri-PcNc photocatalyst, the photogenerated electrons from both sensitizers and internal excitation of g-C3N4 are transferred to the co-catalyst Pt loaded on g-C3N4 and used for H2 production. Improved light-harvesting was achieved by sensitization of g-C3N4 with both dyes, thus leading to H2 production activity of 371.4 μmol h−1 with a TON of 7428 h−1, which is higher than the one achieved with each dye separately (LI-4/g-C3N4; 233.8 μmol h−1 and Zn-tri-PcNc/g-C3N4; 132.3 μmol h−1) under visible/near-infrared (NIR) light irradiation.
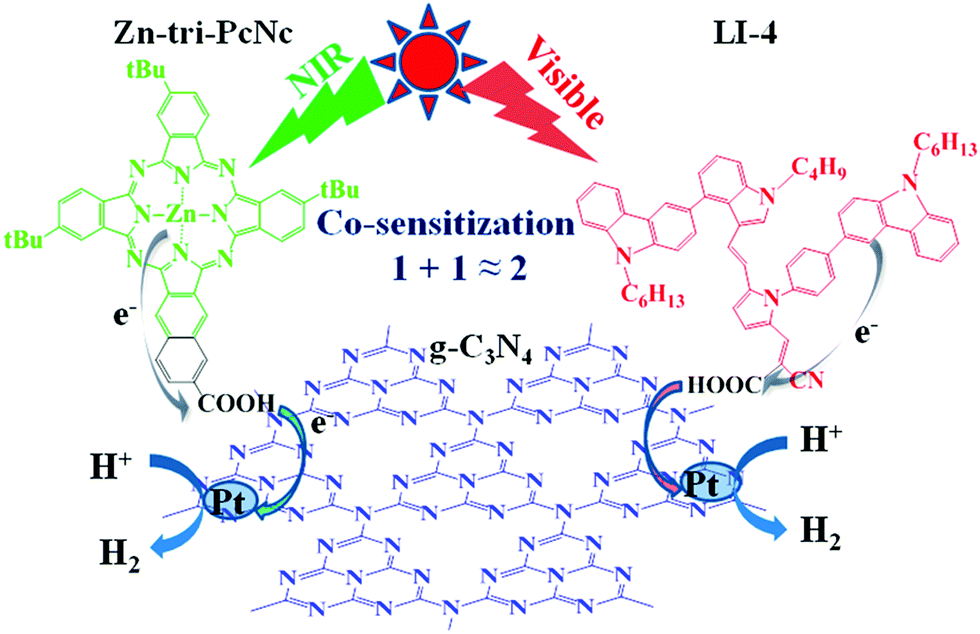 | ||
| Fig. 70 H2 production over LI-4/g-C3N4/Zn-tri-PcNc. Reproduced from ref. 317, with permission from American Chemical Society, Copyright 2015. | ||
To determine the effect of the number of carboxylic acids and nature of the substituents on the performance of Pc sensitized g-C3N4, Li and Peng reported on a series of symmetrically and unsymmetrically substituted ZnPcs with different peripheral groups (Fig. 71).318 Although all ZnPc derivatives have identical optical and electrochemical characteristics, the electron transport and charge recombination kinetics between these compounds and the semiconductor were shown to be the most important factors influencing photocatalytic activity. When placed onto g-C3N4, symmetrical PCs exhibit more efficient electron transport and reduced charge recombination kinetics than asymmetric Pcs, as evidenced by PL quenching and time-resolved fluorescence studies. Zn-tri-PcNc-1, which possesses a “push–pull” structure with three bulky tert-butyl groups and carboxylic acid anchoring group, was sensitized with g-C3N4 photocatalyst and resulted in an H2 production efficiency of 203 μmol h−1 with a maximal AQY of 0.71%. On the other hand, the symmetric ZnPcs gave a slightly lower H2 production efficiency than those of the asymmetric ZnPcs. Once again, this result confirmed that the lack of charge transfer directionality of the symmetrical ZnPc and weak coupling of LUMO with the conduction band of g-C3N4 was detrimental to the conversion efficiency of the photocatalytic system. The incorporation of two carboxylic acid functions in Zn-tri-PcNc-2 has led to the best photoactivity with the highest H2 production rate of 263 μmol h−1 and a TON of 12826 under λ ≥ 500 nm light irradiation for 10 h, which corresponds to an AQY of 0.92%. This result is mainly due to an enhancement in the electron transfer rate from the excited sensitizer to the g-C3N4 surface and the increased electron lifetime for H2 production which directly affects the photoactivity. Additionally, when Zn-tri-PcNc-2 and Zn-tri-PcNc-3, consisting of tert-butyl and n-butoxy groups respectively, were compared and Zn-tri-PcNc-2 showed a higher photoactivity due to the higher steric hindrance of the tert-butyl group and its stronger electron-donating ability. The H2 production of ZnPcs over g-C3N4 decreased in the order Zn-tri-PcNc-2 > Zn-tri-PcNc-1 > Zn-tri-PcNc-3 > Zn-tetrad-Nc-3 > Zn-tetrad-Pc-1 > Zn-tetrad-Pc-2. This result shed light on the importance of charge transfer directionality, aggregation degree of Pcs and number of anchoring groups toward photocatalytic efficiency.
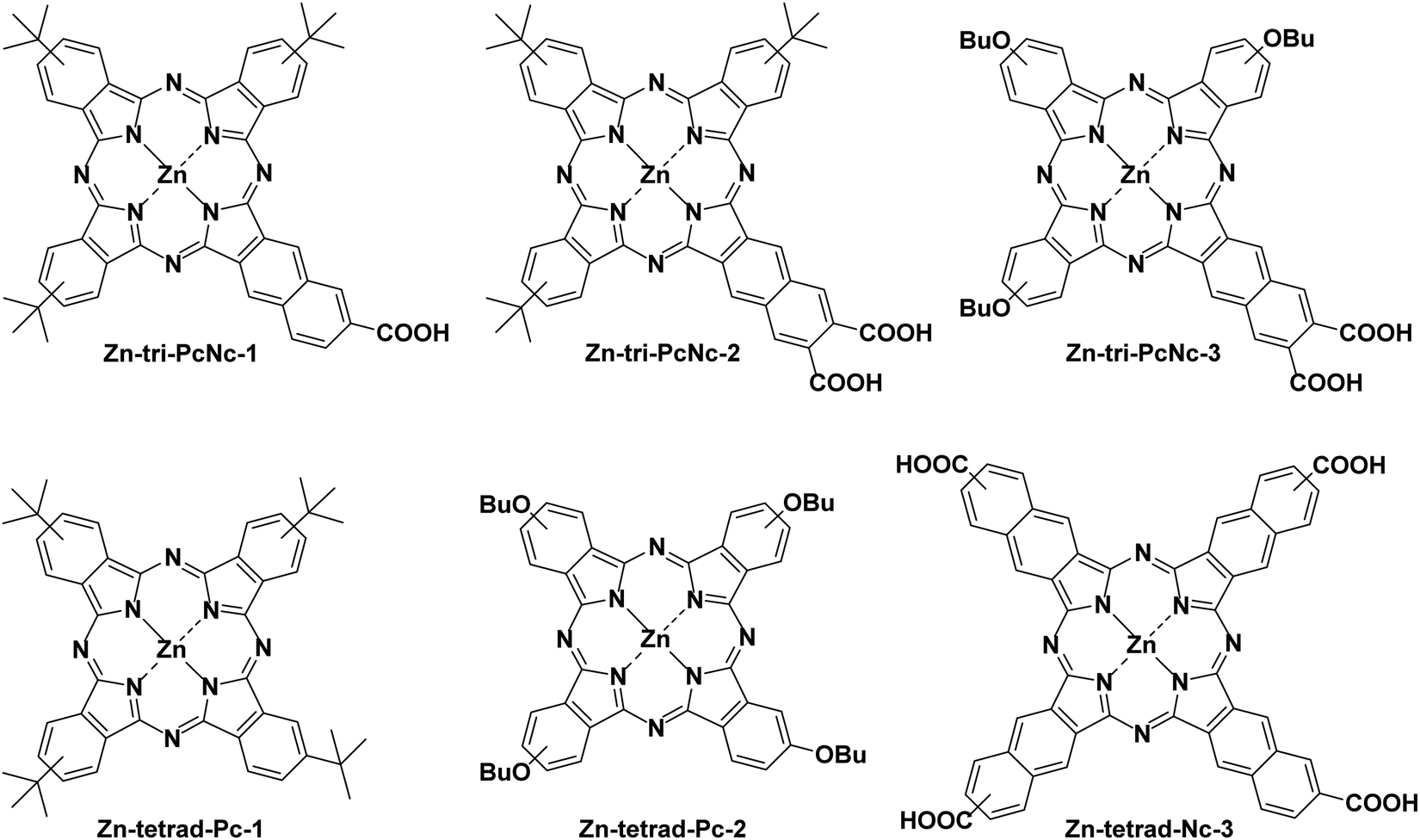 | ||
| Fig. 71 Molecular structures of ZnPc derivatives. | ||
Following a similar approach, Li and co-workers reported on a highly asymmetric A2BC type ZnPc (Zn-di-PcNcTh) bearing four bulky 2,6-diphenylphenoxy substituents (Fig. 72), a thiophene unit and a carboxyl-naphthalene unit with the goal of studying the influence of molecular structure, symmetry and carboxyl anchoring group of ZnPc derivatives on the photosensitized H2 production performance over g-C3N4.319 The symmetrical analogue containing eight bulky 2,6-diphenylphenoxy substituents (Zn-tetrad-Pc-1) was also prepared and tested for the comparison purposes.320
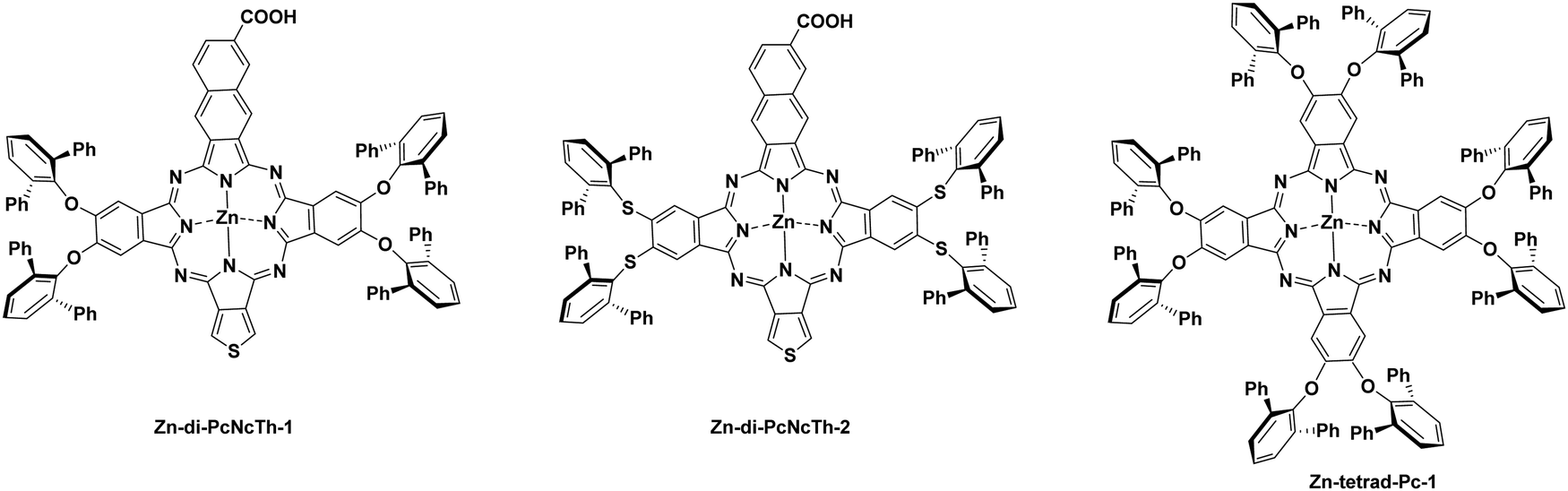 | ||
| Fig. 72 Molecular structures of A2BC type ZnPcs. | ||
The Zn-di-PcNcTh derivatives show an important redshift of about 39 nm in the absorption maximum at 739 nm relative to the symmetrical Pc analogue (Zn-tetrad-Pc-1). Zn-di-PcNcTh-1 was used to sensitize Pt-loaded g-C3N4 for H2 production, which showed an H2 production efficiency of 249 μmol h−1 with TON of 9960.8, much higher than the pristine Pt/g-C3N4. A higher AQY (3.05%) was obtained under 730 nm monochromatic light irradiation, higher than that of Zn-tetrad-Pc-1 (1.14%) without the carboxyl group. The origin of this lower H2 production activity of the symmetrical Zn-tetrad-Pc-1 was ascribed to both the lack of directionality and lower dye loading on the Pt/g-C3N4 owing to the absence of the anchoring group. Co-adsorbent was not required since the peripherally substituted bulky groups suppressed the aggregation of both Pc. In the following report, the same authors used electron-rich diphenylthiophenol derivative instead of diphenylphenoxy in order to produce a redshift in the absorption into the near–IR region.321 The novel Zn-di-PcNcTh-2 shows maximum absorbance at 744 nm, thus representing redshifts of ca. 16 nm, compared to Zn-di-PcNcTh-1. After optimizing the photocatalytic condition, the Zn-di-PcNcTh-2 sensitized Pt/g-C3N4 showed H2 evolution activity (∼232 μmol h−1) with TON of ∼23187 h−1 during the first run of 10 h visible light irradiation and much higher AQY values (4.57%, 5.53%, 4.09% and 2.35% at 730, 760, 780 and 800 nm, respectively) compared to the Zn-di-PcNcTh-1.
In 2018, Liu et al. reported a ZnPc derivative, with a carboxyl-naphthalene unit and three 15-crown-5 ether moieties at the peripheral positions (Fig. 73), as a photosensitizer of Pt/g-C3N4.322 The resulting photocatalyst exhibits an H2 evolution activity of 163 μmol h−1 with a TON of 3105 h−1. Since the crown ethers can encapsulate alkali metals, Li+, Na+, or K+ were added to the ZnPc solution before the dye sensitization process to assess the effect of alkali cation on the H2 production activity. It was determined that when alkali metals are inserted in the Pc, photocatalytic activity is enhanced. The best activity of 476 μmol h−1 with a TON of 9067 h−1 was obtained when K+ ions combine with ZnPc's 15- crown-5 ethers. This result is arising from the positive effect of increased electron injection from the excited Pc to the g-C3N4 and decreased charge recombination which was confirmed by fluorescence quenching experiments. In addition, the photocatalytic activity order of the alkali ions is Li < Na < K, which is compatible with the radii of these ions and the strength of the ion-ether interaction.
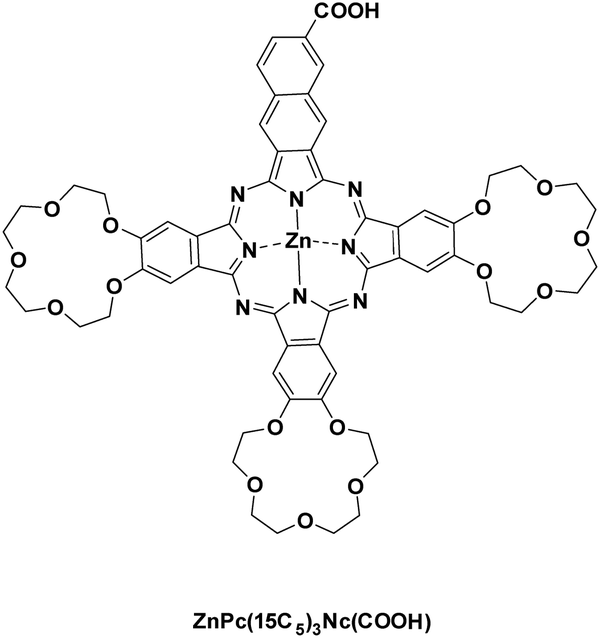 | ||
| Fig. 73 Molecular structure of unsymmetrically substituted three 15-crown-5 ethers combining with alkali metal ions (such as Li+, Na+, or K+). | ||
A nanohybrid catalyst constituted by a SiPc covalently bonded to an N-doped ultrasmall reduced graphene oxide (N-usRGO) nanosheet was reported by Yang and co-workers.323 The SiPc was grafted to N-usRGO through 1,3-dipolar cycloaddition of azomethine ylides and the resulting material exhibits a much improved photocatalytic activity compared to N-usRGO, due to the better electron transfer from photoexcited Pcs to the Pt co-catalyst in the SiPc-N-usRGO ensembles (Fig. 74). Upon photoexcitation of N-usRGO/SiPc/Pt catalyst at >400 nm, the photoexcited electrons are transferred from the SiPcs to the N-usRGO nanosheet and then to the Pt co-catalyst onto the N-usRGO nanosheets. This cascade of electron transfer processes finally realized a hydrogen production amount of 4.5 μmol g−1 in 6 h with AQY of 1.3% at 365 nm.
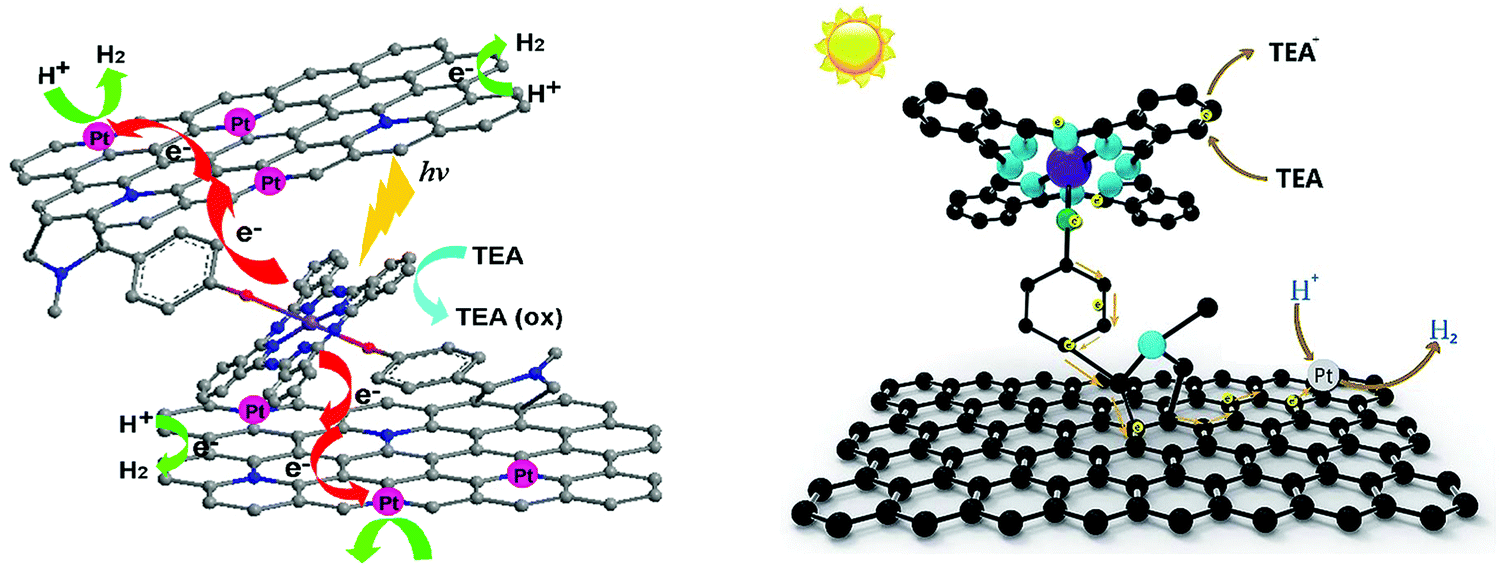 | ||
| Fig. 74 (a) Mechanism of H2 evolution in N-usRGO/SiPc/Pt photocatalyst. (b) The electron transfer in MnPcG/Pt photocatalyst. Reproduced from ref. 323, with permission from American Chemical Society, Copyright 2015. | ||
Similarly, covalent functionalization of graphene with a manganese phthalocyanine (MnPc) by a 1,3-dipolar cycloaddition reaction was demonstrated to improve the light-absorption of the nanohybrid photocatalyst into the visible-light range.324 Photophysical measurements confirmed that, in a hybrid catalyst in which graphene not only acts as an electron acceptor but also could act as an electron mediator, an efficient photo-induced electron transfer process occurs from the MnPc to the graphene. MnPcG/Pt photocatalyst exhibits much improved photocatalytic activity with an H2 production activity of 8.59 and 1.45 μmol mg−1 under 10 h of UV-vis and visible light (>400 nm) irradiation, respectively, compared to non-covalently functionalized graphene MnPcCl/G. This result was attributed to the slow rate of the recombination process and the fast electron transfer dynamics.
4.4 Phthalocyanines as catalysts in H2 production systems
Apart from the use of Pc derivatives as dye sensitizers, attention has also been drawn to the use of Pc derivatives as a water reduction catalysis. For example, Yaun et al. demonstrated that NiPc, with an iridium complex as a photosensitizer, is an efficient and stable catalyst for visible-light-driven hydrogen production in a homogeneous system.325 This system exhibited a hydrogen production amount of 112.2 mmol with TON up to 680 after irradiation for 8 h irradiation. The H2 evolution rate in NiPc–IrPS–TEOA system dramatically decreased when irradiation time was prolonged to 24 h due to the decomposition of the iridium complex rather than the NiPc catalyst. Furthermore, the stability of NiPc–IrPS–TEOA system was compared with a similar system in which NiPc is replaced by a [Ni(bpy)3]2+, in this organometallic complex the bonds between the metal and the ligand are dative. The 24 h visible light irradiation of [Ni(bpy)3]2+-IrPS-TEOA revealed that the deactivation of the H2 evolution system is due to the decomposition of [Ni(bpy)3]2+. This result was attributed to the presence of strong Ni–N Sigma bonds in the center of the Pc macrocycle which prevents the removal of Ni cation from the central cavity of the Pc and therefore leads to higher chemical stability. A possible reaction mechanism of the light-driven H2 production from NiPc–IrPS–TEOA system is illustrated in Fig. 75. Upon photoexcitation, the electron travels from IrPS to the NiPc which leads to the formation of the H-NiPc intermediate by breaking of Ni–N coordination bonds in NiPc. It is inferred that the site of initial protonation in NiPc is the pyridyl N with dechelation and that proton transfer from this ligand leads to the formation of [H–NiPc–H] complex followed by transformation into [Ni–H–H–Pc] species which is responsible for H2 generation.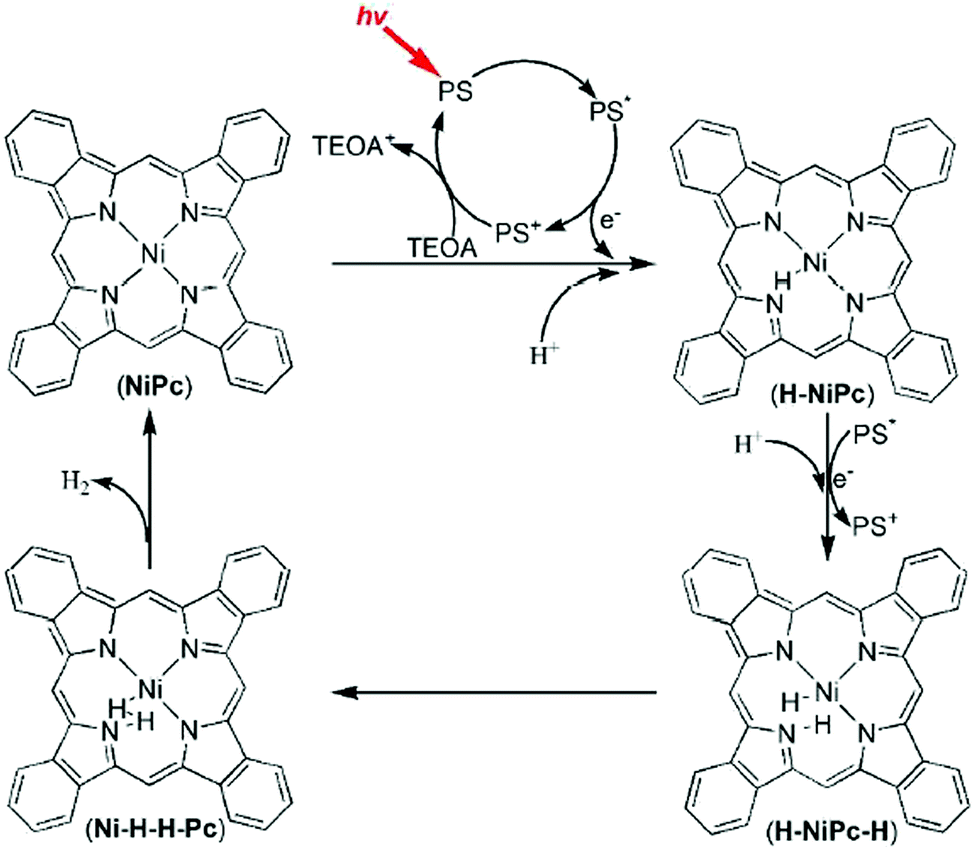 | ||
| Fig. 75 Proposed mechanism of hydrogen formation by NiPc–IrPS–TEOA system. Reproduced from ref. 325, with permission from Royal Society of Chemistry, Copyright 2016. | ||
Song and co-workers reported on a catalyst for photoinduced H2 generation based on a biomimetic model inspired on [FeFe]-hydrogenases, a family of metalloenzymes that can catalyze proton reduction to hydrogen in different microorganisms, as an active site and RuPc as a photosensitizer (Fig. 76).326 This photoactive complex catalyst was tested in a catalytic system composed of Et3N as an electron donor and H2O as a proton source under visible-light (λ > 400 nm) irradiation. However, the system exhibited low catalytic efficiency with 0.13 × 10−3 mmol of H2 after 180 min irradiation. The weak intramolecular ET from the excited RuPc macrocycle to the catalytic diiron complex as well as the low stability of the catalyst under the experimental conditions accounted for the low catalytic performance.
 | ||
| Fig. 76 Model complex for the active site of [FeFe]-hydrogenases. | ||
5. Phthalocyanines for photocatalytic CO2 reduction
Pc derivatives have been widely used in photocatalytic CO2 conversion to boost light absorption and charge separation efficiency by heterogenizing semiconductors through a variety of immobilization approaches, as previously indicated. This section will focus on heterostructured composite systems in which Pcs are used as either sensitizers or catalysts for photocatalytic CO2 reduction (Table 14).| PS | Catalyst | SED | Solvent | Light source | Irr. time | H2 production | TON | AQY | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Zn-Tri-PcNc | TiO2/Pt(0.1 wt%) | EDTA | Water | Visible light (λ > 420 nm) | 5 | 567.4 μmol | 7565 | 0.2%@700 nm | 304 |
| Zn-Tetra-Nc | TiO2/Pt(0.1 wt%) | EDTA | Water | Visible light (λ > 420 nm) | 5 | 236.5 μmol | 3153 | 304 | |
| Zn-Tri-PcNc | TiO2-AA/Pt | AA | Water | λ > 420 nm) | 5 | 162.2 μmol h−1 | 0.97%@700 nm | 306 | |
| ZnPc (PCH-001) | Pt/TiO2 (HPT) | TEOA | Water | λ > 420 nm | 5 | 2260 μmol | 18080 |
11.57% @690 | 308 |
| CuPc | TiO2 | CH3OH | Water | Visible light (λ > 420 nm) | 7 | 0.95 μmol h−1 | 6.68%@420 nm | 309 | |
| ZnPc | TiO2/Pt | TEOA | Water | Visible light (λ > 420 nm) | 8 | 3448 μmol g−1 h−1 | — | — | 310 |
| CoPc | TiO2/Pt | TEOA | Water | Visible light (λ > 420 nm) | 8 | 3328 μmol g−1 h−1 | — | — | 310 |
| ZnPc1 (TT40) | TiO2/Pt | TEOA | Water | Visible light (λ > 420 nm) | 8 | 5.387 mmol g−1 h−1 | 314 | ||
| ZnPc2 | TiO2/Pt | TEOA | Water | Visible light (λ > 420 nm) | 8 | 2.259 mmol g−1 h−1 | 314 | ||
| ZnPc 3 | TiO2 | TEOA | Water | Visible light (λ > 420 nm) | 20 | 3.4187 mmol gmmol g−1 | 14863 |
315 | |
| ZnPc4 | TiO2 | TEOA | Water | Visible light (λ > 420 nm) | 20 | 1.3078 mmol g−1 | 3441 | 315 | |
| ZnPc 5 (TT1) | TiO2 | TEOA | Water | Visible light (λ > 420 nm) | 20 | 1.3042 mmol g−1 | 3142 | 315 | |
| SubPc 1 | TiO2/Pt | TEOA | Water | Visible light (λ > 420 nm) | 8 | 20332 μmol g−1 |
— | — | 320 |
| SubPc 2 | TiO2/Pt | TEOA | Water | Visible light (λ > 420 nm) | 8 | 16179 μmol g−1 |
— | — | 320 |
| SubPc 3 | TiO2/Pt | TEOA | Water | Visible light (λ > 420 nm) | 8 | 22345 μmol g−1 |
— | — | 320 |
| LI-4/Zn-tri-PcNc | g-C3N4/Pt | AA | Water | Visible light (λ > 420 nm) | 30 | 371.4 μmol h−1 | 7428 | 1.75%@700 nm | 325 |
| Zn-Tri-PcNc-1 | g-C3N4/Pt | AA | Water | λ > 5000 nm | 10 | 203 μmol h−1 | 0.71%@700 nm | 326 | |
| Zn-Tri-PcNc-2 | g-C3N4/Pt | AA | Water | λ > 5000 nm | 10 | 263 μmol h−1 | 0.92%@700 nm | 326 | |
| Zn-Tri-PcNc-3 | g-C3N4/Pt | AA | Water | λ > 5000 nm | 10 | 101 μmol h−1 | 0.35%@700 nm | 326 | |
| Zn-Tetrad-Pc-1 | g-C3N4/Pt | AA | Water | λ > 5000 nm | 10 | 47.7 μmol h−1 | 0.17%@700 nm | 326 | |
| Zn-Tetrad-Pc-2 | g-C3N4/Pt | AA | Water | λ > 5000 nm | 10 | 40.9 μmol h−1 | 0.15%@700 nm | 326 | |
| Zn-Tetrad-Pc-3 | g-C3N4/Pt | AA | Water | λ > 5000 nm | 10 | 54.5 μmol h−1 | 0.19%@700 nm | 326 | |
| Zn-Di-PcNcTh-1 | g-C3N4/Pt | AA | Water | λ > 420 nm | 10 | 249 μmol h−1 | 9960.8 h−1 | 3.05%@730 nm | 327 |
| Zn-Tetrad-Pc-1 | g-C3N4/Pt | AA | Water | λ > 420 nm | 10 | 126.3 μmol h−1 | — | 1.14%@730 nm | 328 |
| Zn-Di-PcNcTh-2/g-C3N4 | g-C3N4/Pt | AA | Water | Visible light (λ > 420 nm) | 10 | 232 μmol h−1 | 23187 h−1 |
5.53%@760 | 329 |
| ZnPc–Pt | g-C3N4/Pt | TEOA | Water | Visible light (λ > 420 nm) | 10 | 163 μmol h−1 | 3105 h−1 | 1.88%@700 | 330 |
| ZnPc–Pt/K+ | g-C3N4/Pt | TEOA | Water | Visible light (λ > 420 nm) | 10 | 476 μmol h−1 | 9067 h−1 | 2.74%@700 | 330 |
| ZnPc–Pt/Na+ | g-C3N4/Pt | TEOA | Water | Visible light (λ > 420 nm) | 10 | 280 μmol h−1 | 2.41%@700 | 330 | |
| ZnPc–Pt/Li+ | g-C3N4/Pt | TEOA | Water | Visible light (λ > 420 nm) | 10 | 159.2 μmol h−1 | 2.06%@700 | 330 | |
| SiPc | N-usRGO/Pt | TEA | Water | Xe lamp, | 6 | 4.5 μmol g−1 | 1.3%@365 | 331 | |
| MnPc | Graphene Pt | TEA | Water | λ > 400 nm | 10 | 1.45 μmol mg_1 | 0.06%@670 | 332 | |
| IrPS | NiPc | TEOA | Water | Visible light (λ > 420 nm) | 8 | 112.2 μmol | 680 | 333 | |
| RuPc | [FeFe]-Hydrogenases | TEA | Water | 3 | 0.13 × 10−3 mmol | — | 334 |
5.1 Phthalocyanines as catalysts in heterogeneous CO2 reduction systems
Although most catalysts for solar activated CO2 reduction are mainly based on semiconductor-based materials, as already mentioned, the absorption of sunlight and the charge separation in single photocatalyst systems are limited. To address such challenges, the construction of a Z-scheme heterojunction that can strike a good compromise between charge separation, charge transfer directionality and improved light-harvesting ability appears to be a much more feasible way of improving solar energy conversion systems. Many Pc derivatives have been used in the construction of Z-scheme photocatalysts for photocatalytic CO2 conversion. The coupling of Pcs derivatives with suitable semiconductors to form Z-scheme photocatalysts provides both extended absorptions in the range of the solar emission spectrum as well as superior charge transfer and separation due to their appropriate band alignment, rich redox chemistry, and high photochemical stability.Among the metal oxide semiconductors, BiVO4 is a promising photocatalyst since it offers several advantages such as chemical stability and narrow bandgap. However, its photocatalytic performance is mainly limited by slow charge separation, poor absorption in the visible range and low reduction potential. Nevertheless, attempts to enhance the charge separation process have been made by controlling the morphology, doping of metal atoms and forming heterostructures.327
As an example, Bian et al. fabricated dimension-matched ZnPc/BiVO4 nanocomposites as an efficient photocatalyst for photocatalytic reduction of CO2 based on a novel Z-scheme charge transfer mechanism.328 ZnPc/BVNS nanocomposite was prepared by hydrogen bond interaction between the hydroxyl groups on BVNS and N atoms in the ligand of ZnPc, resulting in a stable and efficient Z scheme system (Fig. 77). As a major product, ZnPc/BiVO4 photocatalyst converts CO2 into CO, while the formation of CH4 is also detected. Notably, the photocatalytic performance of ZnPc/BiVO4 composite on CO2 reduction achieved 16-fold higher quantum efficiency than that of BiVO4 NPs. This dramatically enhanced photocatalytic activity was attributed to the established direct Z-scheme system, which promoted efficient charge separation, good match of band positions of ZnPc with BVTS and superior catalytic activity of central Zn2+ cation. In this particular case, upon photoexcitation, excited electrons in the CB of BVNS recombine with the holes in the HOMO of the ZnPc due to the compatible band positions of ZnPc and BVNS. Thus, electrons and holes with stronger reduction and oxidation abilities remain in ZnPc and BVTS, leading to increased photocatalytic activity. It should be noted that in this system, the electron transfer from the ligand to the central metal cation increases the catalytic activity of Zn for CO2 reduction.
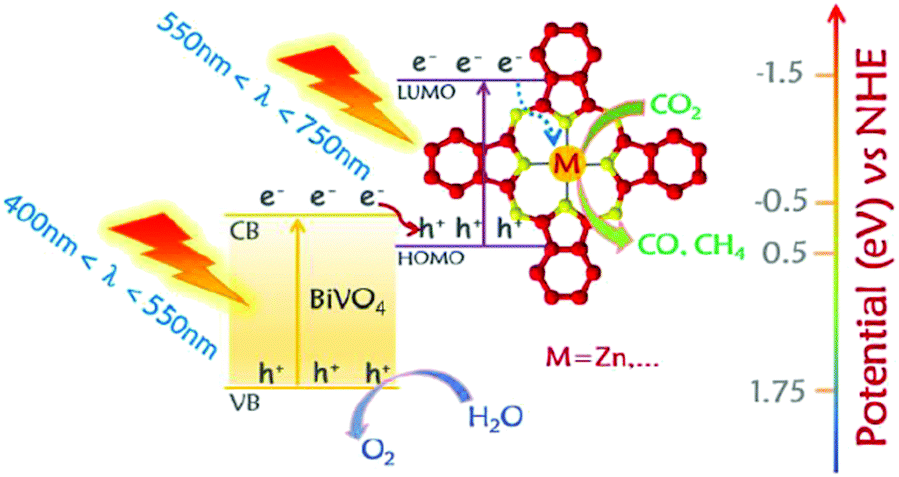 | ||
| Fig. 77 Z-Scheme mechanism of ZnPc/BVNS nanocomposite. Reproduced from ref. 328, with permission from John Wiley and Sons, Copyright 2019. | ||
Although the conversion efficiency of this Pc-based photocatalyst seems promising, the same authors stated that the limited photocatalytic activity was due to the aggregation tendency of ZnPc, which prevents the usage of the optimum amount of ZnPc required for catalytic activity.
The degree of aggregation of organic planar macrocyclic compounds like phthalocyanine has been shown to have a significant impact on light absorption, emission, active center area, and charge separation, all of which are important for photoinduced electron transfer processes.40 Therefore, macrocycle aggregation should be suppressed in such applications, specifically on a solid support surface. Considering this, in a later work, the same collaborators further improved the efficiency and stability of ZnPc/BiVO4 based Z-scheme heterojunction by using a graphene layer to minimize self-aggregation of ZnPc onto BVNS.329 Acid treatment of graphene increases the number of hydroxyl groups on the surface and enabled ZnPc to bind to both BVNS and graphene surface via hydrogen bonding. Indeed, the optimized ultrathin ZnPc/graphene/BiVO4 heterojunctions exhibited superior photocatalytic conversion of CO2 to CO, (14-fold higher) than that of bare BiVO4 nanosheet. This improvement can be attributed to two main factors: (i) graphene's two-dimensional structure and further functionalization of graphene with OH groups allows for a larger amount of highly dispersed ZnPc assembly on BVNS with the proper thickness; (ii) graphene acts as an electron bridge for optimum electronic coupling between ZnPc and BiVO4, resulting in increased charge separation in the Z scheme (Fig. 78).
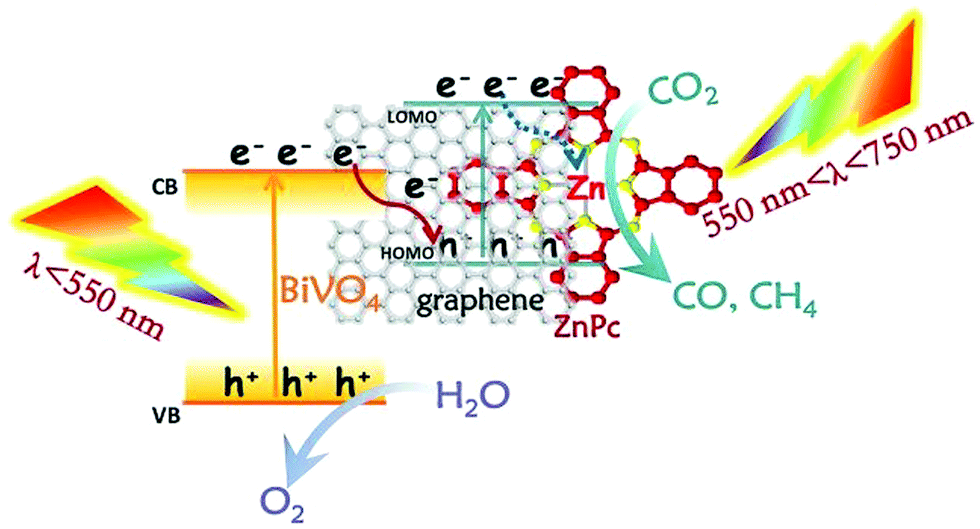 | ||
| Fig. 78 Graphene-modulated ZnPc/BiVO4 Z-scheme heterojunction. Reproduced from ref. 329, with permission from Royal Society of Chemistry, Copyright 2020. | ||
Very recently, the further improved conversion efficiency of CO2 to CO has been achieved by CuPc/Au-BVNS heterojunctions, which exhibit a 9-time and 35-time increase in photoactivity compared to pristine BVNS and the BiVO4 nanoflake, respectively.330 The authors attributed this success to the use of Au nanoparticles as an interfacial modulator, which improved electron transfer directionality in BVNS to CuPc, as well as increased CuPc loading amounts due to a superior interaction between AuNP and the N-atom of the Pc macrocycle. Indeed, as compared to the previously reported H-bond interfacial interaction, the Au nanoparticle had a better connection with the N-atom of the MPc ligand, resulting in a faster Z-scheme charge-transfer mechanism. The CuPc/Au-BVNS heterojunction system produced CO and CH4 as main reduction products by a direct Z-scheme charge carrier transfer mechanism, wherein the photogenerated electrons of the CuPc and holes of the BVNS are protected, while the photogenerated electrons of the BVNS and holes of the CuPc are recombined. The protected electrons of CuPc are efficiently utilized for the reduction of CO2 to CO (Fig. 79).
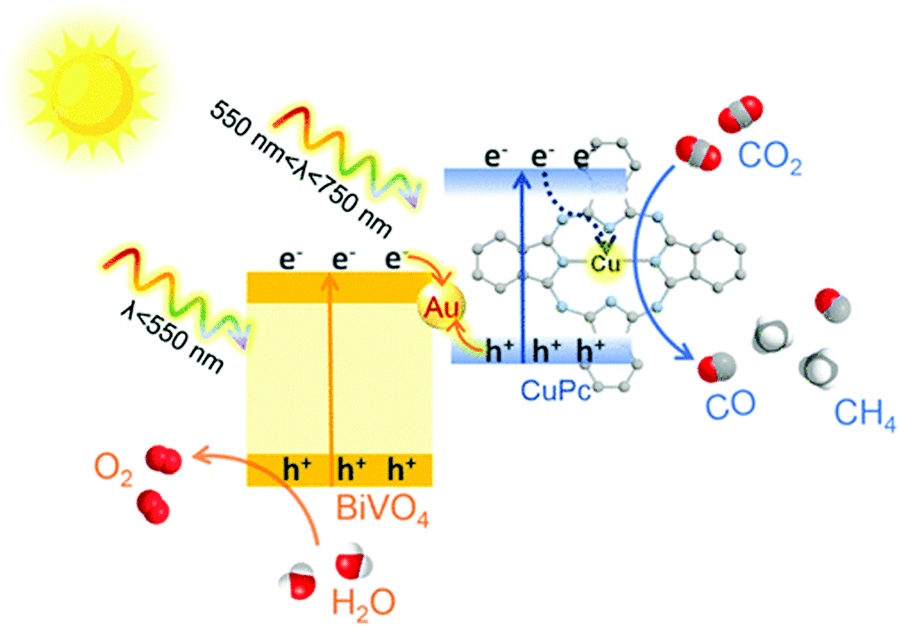 | ||
| Fig. 79 CuPc/Au-BVNS Z-scheme heterojunction. Reproduced from ref. 330, with permission from American Chemical Society, Copyright 2021. | ||
Another semiconductor photocatalyst that has been used for photocatalytic CO2 reduction is tungsten trioxide (WO3), as its narrow bandgap (2.4–2.8 V) and stable physicochemical properties make it a promising candidate for photocatalytic applications.331 On the other hand, the use of WO3 in the photocatalytic process encounters many limits such as small surface area, limited charge separation and absorption in the visible region. Since the HOMO energy band level of Pcs is close to the CB level of WO3, combining Pc and WO3 under the Z scheme system has been found to be a feasible way to increase the photocatalytic activity of WO3-based photocatalysts. Recently, Li et al. reported Z-scheme FePc/WO3 composite for the reduction of CO2 to CH4.332 Compared to the WO3 nanoplate, FePc/P-WO3 nanocomposites exhibited 8-fold high photocatalytic activity for converting CO2 to CO and CH4. This success was explained by the high surface area of nanocomposites, the better charge separation between FePc and WO3 and the catalytic activity of central metal cation in the Pc macrocycle (Fig. 80).
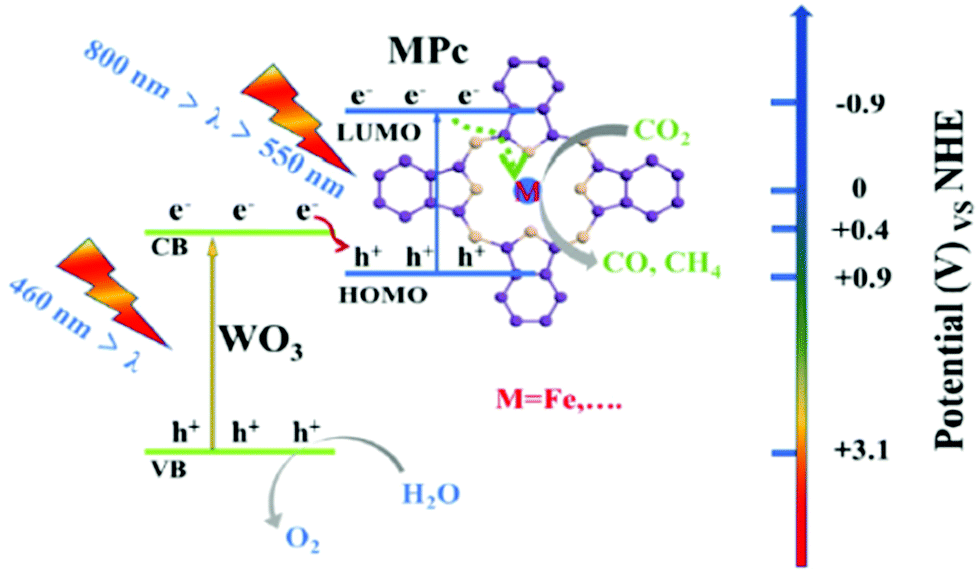 | ||
| Fig. 80 FePc/P-WO3 Z-scheme heterojunction. Reproduced from ref. 332, with permission from Elsevier, Copyright 2020. | ||
Metal-free organocatalyst such as graphitic carbon nitride (g-C3N4) has drawn a lot of attention due to its suitable band position, high photochemical stability, two-dimensional structure and low cost which make it favourable alternatives to the metal oxide-based semiconductors.333 Recent studies have shown that coupling g-C3N4 with metal oxide in heterostructure form or coupling with a similar 2D structure of organic semiconductors like Pcs has a positive effect on charge separation as well as efficient solar photon harvesting and photocatalytic activity. For example, Li et al. reported a heterostructure g-C3N4/CuPc composite for photocatalytic conversion of CO2. In this study, the ultrathin dimension-matched metal phthalocyanine/treated g-C3N4 (MPc/T-CN) was constructed through the H-bonding formation between N atom of Pc and hydroxyl groups on CN. The treatment of g-C3N4 nanolayers with HNO3 solution increases the number of hydroxyl groups on the CN surfaces, resulting in a uniform distribution of MPc on the CN and preventing Pc aggregation on the surface, improving the photocatalytic activity and stability of the MPc/CN heterojunction. When comparing the photoactivity, this hybrid photocatalyst exhibited a 10-fold higher CO production rate than pristine g-C3N4.334 The advanced photocatalytic activity was ascribed to the unexpected transfer of excited high-level-energy electrons (HLEEs) from g-C3N4 to the ligand of CuPc and then to the central metal ions (Cu2+) with potential catalytic function for CO2 reduction reactions (Fig. 81).
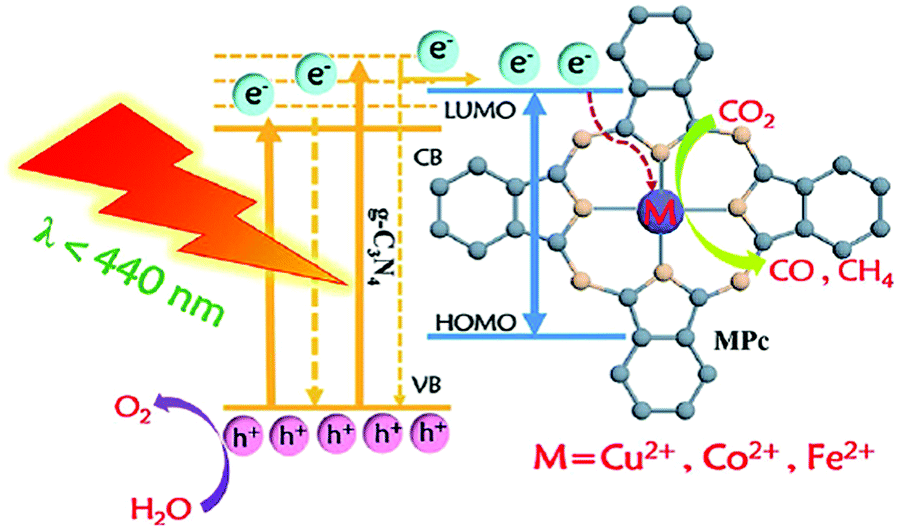 | ||
| Fig. 81 HLEEs transfer process in the MPc/T-CN heterojunction. Reproduced from ref. 334, with permission from Elsevier, Copyright 2020. | ||
Due to the excellent CO2 adsorption capacity and the suitable band edge position, which has a good match with CN for effective HLEEs transfer, the CuPc-based system exhibit better photocatalytic activity than FePc and CoPc. This study emphasized in particular that the formation of dimensional match interface by coupling with two-dimensional g-C3N4 and planar Pc ensures increased charge transfer.
Of the transition metal-containing Pc derivatives, cobalt phthalocyanine (CoPc) has been identified as one of the most promising electrocatalysts for CO2 reduction owing to its ultra-high selectivity and reasonable efficiencies for CO production.335 Immobilization of molecular catalysts onto conductive supports has proven to be a promising tactic to promote the selectivity and stability of the resulting hybrid photocatalyst.
For this purpose, a hybrid heterogeneous photocatalyst was prepared in which polymeric CoPc as a catalytic center was immobilized onto mesoporous carbon nitrite as a light harvester in order to be used in CO2 reduction under visible light irradiation.336 The deposition of CoPPc on mpg-CNx by an in situ polymerization method gives the formation of the polymer–CNx interface which allows efficient photoexcited electron transfer from CNx to the attached CoPPc catalyst. The photocatalytic activities of pristine mpg-CNx and the mpg-CNx/CoPPc hybrid catalyst were tested in CO2-saturated MeCN with TEOA as a sacrificial electron donor. While mpg-CNx/CoPPc exhibits a high conversion efficiency for the CO2 to CO, the former one generates only trace amounts of CO. Indeed, this hybrid catalyst generated 1076 μmol g−1 CO after 60 h with 85% selectivity and 90 turnovers. In the CO2 reduction process, upon photoexcitation, the electron travels from the CNx to CoPc, forming Co(I) centre on which CO2 binds followed by a second electron transfer from mpg-CNx led to the conversion of CO2 to CO (Fig. 82). The success of this hybrid catalyst has been attributed to both controlling the amount of catalyst and creating a better interface between Pc and CNx that facilitates electron transfer thanks to the in situ polymerization technique.
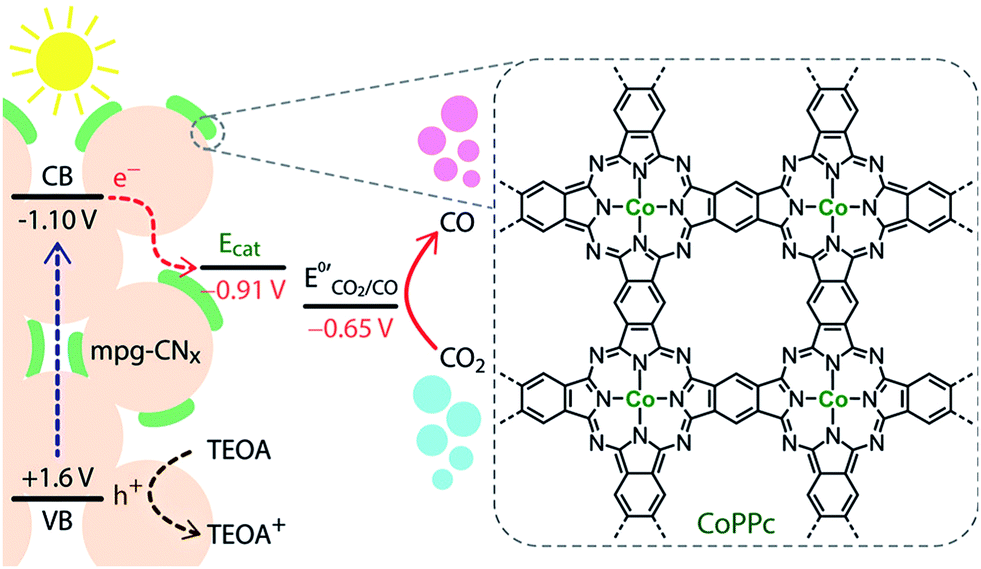 | ||
| Fig. 82 mpg-CNx/CoPPc hybrid photocatalyst. Reproduced from ref. 336, with permission from John Wiley and Sons, Copyright 2019. | ||
Liu et al. demonstrated the improved photocatalytic reduction of CO2 using the hybrid photocatalyst formed by CoPc immobilized on phosphorus-doped graphitic carbon nitride.337 It was found that the doping of phosphorous atoms into CN's framework enlarges the absorption spectra of g-C3N4 by reducing the bandgap and increasing the stability, while the incorporation of CoPc retarded charge recombination. Upon irradiation, the excited electrons at CB of PCN are transferred to the LUMO of CoPc where a CO2 reduction reaction takes place (Fig. 83). The CoPc@P–g-C3N4 hybrid photocatalyst exhibited a conversion efficiency yield of 295 μmol g−1 for CO with the stability of up to six consecutive runs.
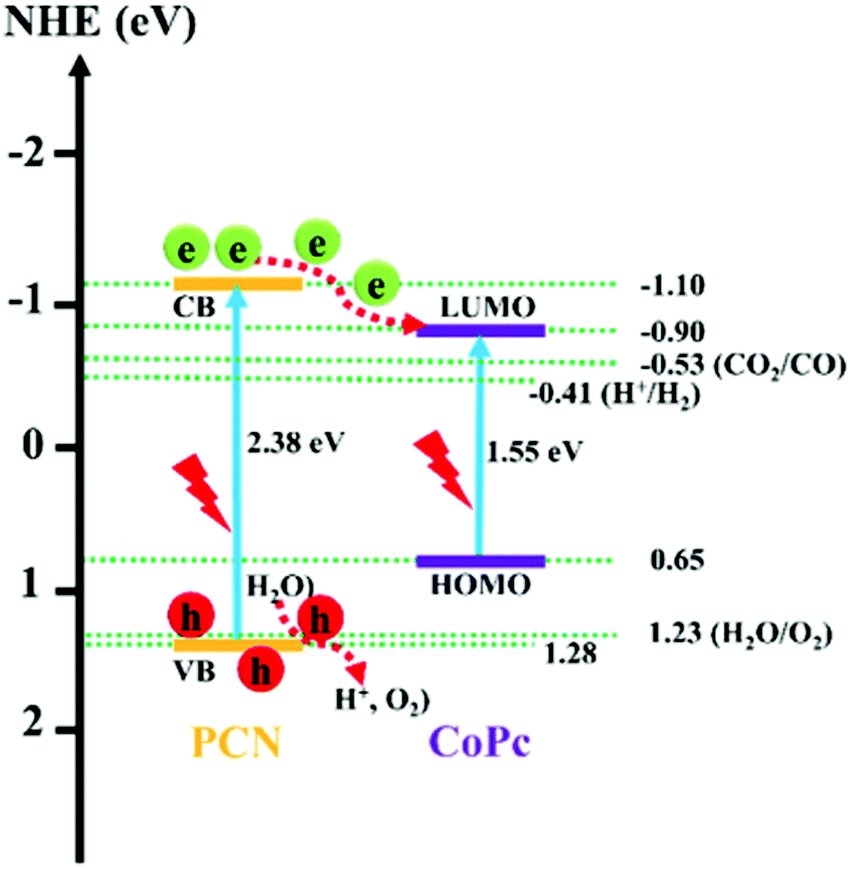 | ||
| Fig. 83 Photoreduction of CO2 with water using Co@PCN hybrid photocatalyst. Reproduced from ref. 337, with permission from Elsevier, Copyright 2021. | ||
Lin and co-workers prepared the first example of photoelectrochemical cell CO2 reduction with molecular catalyst driving both CO2 reduction and H2O oxidation using noble-metal-free materials.338 This system is very efficient in the suppression of concurrent H2 evolution and promoting CO generation. Light-driven CO2 reduction was performed in a two-electrode photoelectrochemical cell (PEC) composed of a fluorinated Co4O4 cubane complex-modified BiVO4 photoanode and a perfluorinated cobalt phthalocyanine complex-modified carbon cloth (cc) cathode. The use of hydrophobic fluorinated molecular catalyst is an efficient strategy to improve the long-term stability of the photoelectrodes in an aqueous solution. Under 1 sun AM 1.5 G illumination, simultaneous O2 and CO evolution in a ratio of about 2:1 in a CO2-saturated NaHCO3 aqueous solution were achieved with high faradaic efficiency up to 87% for CO production and a remarkable solar energy conversion efficiency up to 0.44% at a cell potential of 0.8 V.
5.2 Phthalocyanines as photosensitizers in heterogeneous CO2 reduction systems
Incorporation of Pcs as a photosensitizer onto semiconductors by means of either covalent or non-covalent interaction to form a hybrid photocatalyst is another way to improve the efficiency and selectivity of CO2 photoreduction under visible light illumination. In this photocatalytic system, Pcs play an essential role as a sensitizer in harvesting visible light and promoting charge injection into the conduction band of the catalyst.For example, a hybrid photocatalyst consisting of CoPc covalently linked to graphene oxide was reported by Kumar et al.339 The photoreduction activity of GO-CoPc was tested by using TEA as SED and water as a solvent. The methanol was obtained as a major product and the conversion rate of methanol reached 78.7893 μmol g−1 cat h−1 under visible-light irradiation for 48 h. The GO-CoPc photocatalyst exhibited superior catalytic activity in comparison with the bare GO and the physical 1:1 mixture of GO–CoPc, methanol was obtained as a major product. The authors suggested that the covalent attachment of CoPc to GO reduces the bandgap of GO, resulting in efficient electron injection from CoPc into the conduction band of GO, which greatly increases the conversion efficiency of CO2 with H2O to CH3OH (Fig. 84).
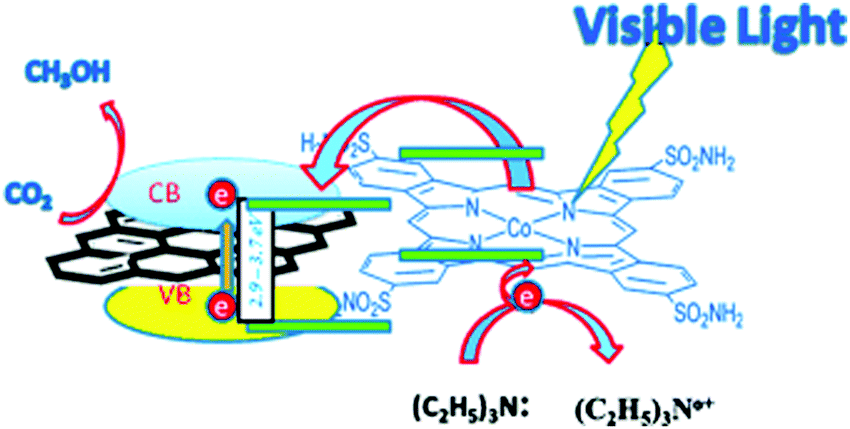 | ||
| Fig. 84 Possible mechanistic pathway of CO2 reduction by using GO-CoPc photocatalyst. Reproduced from ref. 339, with permission from John Wiley and Sons, Copyright 2014. | ||
With the same motivation, Kumar et al. prepared a hybrid photocatalyst composed of cobalt(II) phthalocyanine tetracarboxylate (CoPc–COOH) and g-C3N4 for the visible light assisted reduction of CO2 to methanol.340 The photocatalytic activity of the hybrid photocatalyst was tested in a water/DMF system in the presence of TEA as a sacrificial agent under visible light. The hybrid photocatalyst produced a higher CH3OH yield (12.9 mmol g−1) with a conversion rate of 538.75 mmol h−1 g−1cat. than its homogeneous counterparts, CoPc-COOH and g-C3N4. The better catalytic activity was found to be originated from the combination of Pc with g-C3N4 which potentially confer a synergetic effect such as superior charge transfer, large surface area and high CO2 concentration due to the high binding ability of the CoPc complex with CO2 (Fig. 85).
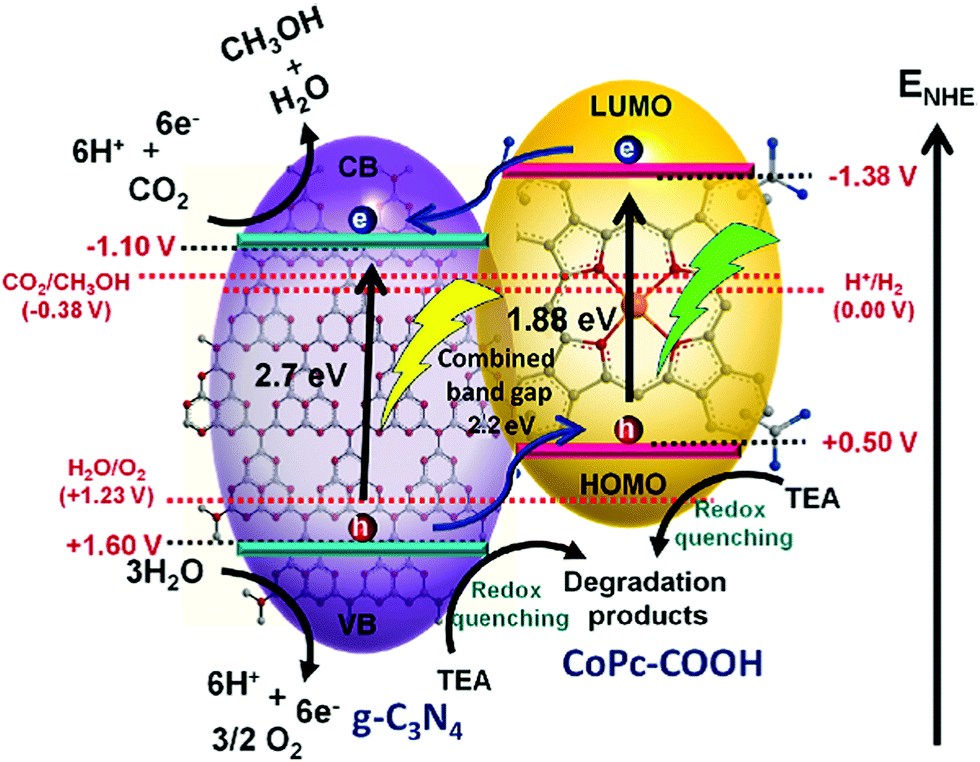 | ||
| Fig. 85 Mechanism of CO2 photoreduction by using g-C3N4/CoPc-COOH photocatalyst. Reproduced from ref. 340, with permission from Elsevier, Copyright 2019. | ||
Recently, the same authors developed a novel hybrid photocatalyst by immobilization of CoPc onto bismuth-oxybromide (BOB).341 Methanol was obtained as a main CO2 reduction product during the photoreduction experiments over CoPc/BOB hybrid photocatalyst in DMF/water solution by using TEA as a sacrificial electron donor under visible light illumination. The efficiency of methanol production of up to 3485 μmol g−1 with a formation rate of 145.2 μmol h−1 g−1, which is significantly higher than that of pristine CoPc and BOB photocatalysts. Once again, the higher photocatalytic activity was attributed to an efficient photo-stimulated electron transfer from CoPc to BOB, a higher concentration of CO2 at the surface, and obtaining the appropriate band gap for reducing CO2 to methanol by binding CoPc to the BOB surface.
Another type of composite, composed of tin phthalocyanine supported to mesoporous ceria (meso-CeO2) was prepared and used as a photocatalyst for visible light assisted photoreduction of CO2.342meso-CeO2 has been inserted at the axial position of Tin(IV) phthalocyanine dichloride via –OH groups on the surface, resulting in a recyclable and reusable photocatalyst (Fig. 86). The photocatalytic CO2 reduction performance of heterogeneous SnPc@CeO2 was tested in a DMF–water–triethylamine mixture, methanol and CO were observed as photoreduction products. Compared to the meso-CeO2 and homogeneous SnPcCl2, SnPc@CeO2 exhibits a higher product formation rate for methanol (97.5 μmol h−1 g−1 cat) and CO (35.0 μmol h−1 g−1 cat) thanks to the better charge injection from the excited state of the SnPc into the meso-CeO2 conduction band.
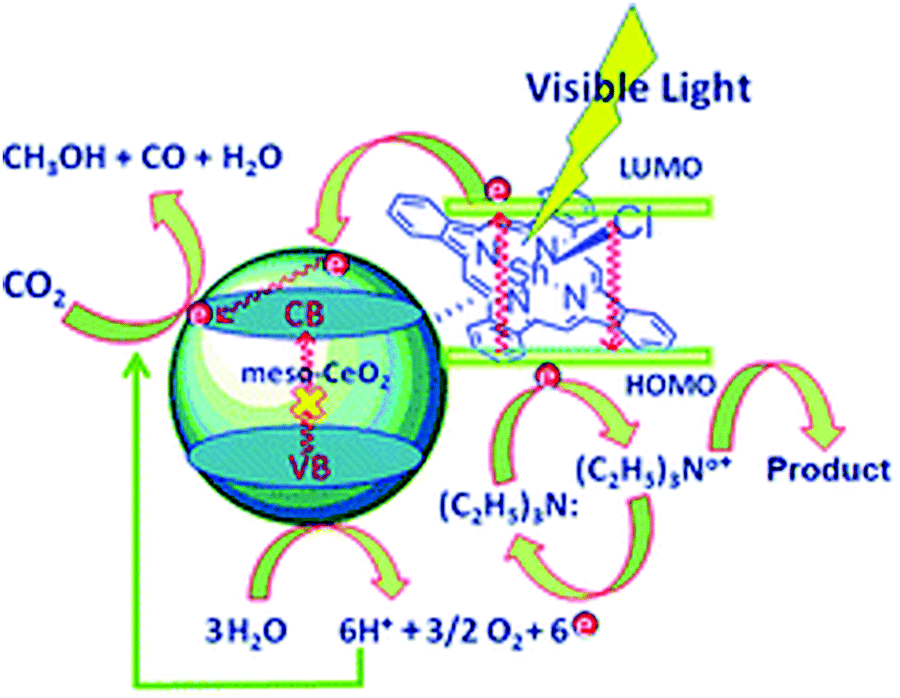 | ||
| Fig. 86 Mechanism of CO2 reduction over SnPc@CeO2 catalyst. Reproduced from ref. 342, with permission from Royal Society of Chemistry, Copyright 2015. | ||
6. Summary and outlook
As discussed in this review, plenty of porphyrins and phthalocyanines have been mostly employed as photo- and redox-active building blocks in photocatalytic H2 production and CO2 reduction schemes. The inspiration for all the above systems is derived from the natural photosynthesis, motivating scientists to prepare a vast number of artificial photosynthetic models. This review thoroughly described the role of porphyrin and phthalocyanine derivatives as photosensitizers, catalysts as well as co-catalysts and the effect of their structural modifications at the photocatalytic activity towards H2 production and CO2 reduction. Before analyzing the problems that most of the systems have, we can come to some conclusions. It is apparent that the homogeneous photocatalytic schemes can provide more insights into the catalytic mechanism whereas they are generally less stable compared to analogous heterogeneous systems. We can also point out that despite the numerous photocatalytic CO2 conversion approaches, there are not many examples generating C2+ reduction products which are considerably more valuable. Moreover, from a statistical perspective, one can see that during the last years the research in CO2 reduction is increasing more rapidly compared to the growth of H2 production studies.Scientists working in this field must deal with plenty of issues and challenges due to some inherent characteristics of the photocatalytic systems. Firstly, most of the systems described herein, have relatively low activity and therefore cannot compete with the present industrial methods to produce H2 (from natural gas, oil, coal or electrolysis). To improve the activity of these schemes it is important to understand in depth the catalytic mechanism of the photocatalytic processes and identify the key factors determining the activity and selectivity. Another challenging endeavor concerns the improvement of stability of the catalytic systems and eliminate the presence of sacrificial agents. The dye sensitization on semiconductors opened the way for the development of molecular-based DSPECs and tandem cell devices based on porphyrins and phthalocyanines. These systems are one step closer to a real-world application since there is no need for sacrificial agents and they offer more stable catalysts and photosensitizers. There is also a need for low-cost materials which can be solved by focusing on the development of noble-metal free systems.
In summary, the increasing energy demands, and environmental issues caused by the over-exploitation of fossil fuels render the need for renewable, clean, and environmentally benign energy sources undoubtedly urgent. The zero-emission energy carrier, H2 is an ideal alternative to carbon-based fuels especially when it is generated photocatalytically from water. Additionally, the photocatalytic conversion of CO2 into chemical fuels and raw materials will both provide the CO2 emission balance and have a significant environmental and economic impact. Therefore, the research on the photocatalytic systems discussed herein is an important challenge and has attracted plenty of scientific attention.
Abbreviation list
| AA | Ascorbic acid |
| AcrH2 | Dihydroacridine |
| BODIPY | Boron-dipyrromethene |
| BPY | 2,2′-Bipyridine |
| CA | Chloranilic acid |
| CB | Conduction band |
| COF | Covalent organic framework |
| CTAB | Cetyltrimethyl ammonium bromide |
| DSP | Dye-sensitized photocatalyst |
| DSPEC | Dye sensitized photoelectrochemical cell |
| DSSC | Dye sensitized solar cell |
| EDTA | Ethylenediaminetetraacetic acid |
| FTO | Fluorine doped tin oxide |
| HER | Hydrogen evolution reaction |
| HLEEs | High-level-energy electrons |
| HNTM | Hollow nanotubes |
| HOMO | Highest occupied molecular orbital |
| LBG | N,N′-Bis(octadecyl)-L-Boc-glutamic diamide |
| LED | Light-emitting diode |
| LP | Liposome |
| LUMO | Lowest unoccupied molecular orbital |
| MeCN | Acetonitrile |
| MOF | Metal organic framework |
| MOPS | 3-Morpholinopropane-1-sulfonic acid |
| MTAB | Myristyltrimethylammonium bromide |
| MV | Methyl viologen |
| NI | Naphthalimide |
| NIR | Near-infrared |
| NPs | Nanoparticles |
| ONCs | Organic nanocrystals |
| PCOF | Porphyrinic covalent organic framework |
| Pcs | Phthalocyanines |
| PMOF | Porphyrin metal organic framework |
| POMs | Polyoxometalates |
| Por | Porphyrins |
| PS | Photosensitizer |
| PVP | Polyvinylpyrrolidone |
| RGO | Reduced-graphene oxide |
| SAC | Single atom catalyst |
| SC | Semiconductor |
| SDS | Sodium dodecyl sulfate |
| SED | Sacrificial electron donor |
| TCEP | Tris-(2-carboxyethyl)phosphine |
| TEA | Triethylamine |
| TEOA | Triethanolamine |
| TFA | Trifluoroacetic acid |
| TON | Turnover number |
| TON | Turnover frequency |
| TsOH | P-Toluenesulfonic acid |
| VB | Valence band |
| WOR | Water oxidation reaction |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the General Secretariat for Research and Technology (GSRT) and Hellenic Foundation for Research and Innovation (HFRI; project code: 508). This research has been co-financed by the European Union and Greek national funds through the Regional Operational Program “Crete 2014–2020,” project code OPS: 5029187. Moreover, the European Commission's Seventh Framework Program (FP7/2007–2013) under grant agreement no. 229927 (FP7-REGPOT-2008-1, Project BIO-SOLENUTI) and the Special Research Account of the University of Crete are gratefully acknowledged for the financial support.References
- Q. Schiermeier, J. Tollefson, T. Scully, A. Witze and O. Morton, Nature, 2008, 454, 816–823 CrossRef CAS PubMed.
- O. Almora, D. Baran, G. C. Bazan, C. Berger, C. I. Cabrera, K. R. Catchpole, S. Erten-Ela, F. Guo, J. Hauch, A. W.-Y. Ho-Baillie, T. J. Jacobsson, R. A.-J. Janssen, T. Kirchartz, N. Kopidakis, Y. Li, M. A. Loi, R. R. Lunt, X. Mathew, M. D. McGehee, J. Min, D. B. Mitzi, M. K. Nazeeruddin, J. Nelson, A. F. Nogueira, U. W. Paetzold, N.-G. Park, B. P. Rand, U. Rau, H. J. Snaith, E. Unger, L. Vaillant-Roca, H.-L. Yip and C. J. Brabec, Adv. Energy Mater., 2021, 11, 2002774 CrossRef CAS.
- V. Humphrey, J. Zscheischler, P. Ciais, L. Gudmundsson, S. Sitch and S. I. Seneviratne, Nature, 2018, 560, 628–631 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, J. P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed.
- D. A.-J. Rand and R. M. Dell, Hydrogen Energy: Challenges and Prospects, Royal Society of Chemistry, 2008, pp. 1–300 Search PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- T. Keijer, T. Bouwens, J. Hessels and J. N.-H. Reek, Chem. Sci., 2020, 12, 50–70 RSC.
- R. J. Cogdell, T. H.-P. Brotosudarmo, A. T. Gardiner, P. M. Sanchez and L. Cronin, Biofuels, 2010, 1, 861–876 CrossRef CAS.
- H. Shen, T. Peppel, J. Strunk and Z. Sun, Sol. RRL, 2020, 4, 1900546 CrossRef CAS.
- W. Luc, M. Jouny, J. Rosen and F. Jiao, Energy Environ. Sci., 2018, 11, 2928–2934 RSC.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- B. Kumar, J. P. Brian, V. Atla, S. Kumari, K. A. Bertram, R. T. White and J. M. Spurgeon, Catal. Today, 2016, 270, 19–30 CrossRef CAS.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- F. Gong, H. Zhu, Y. Zhang and Y. Li, J. CO2 Util., 2018, 28, 221–227 CrossRef CAS.
- Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC.
- M. Kirch, J.-M. Lehn and J.-P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345–1384 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- W. Fan, Q. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2013, 15, 2632–2649 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- B. Cecconi, N. Manfredi, T. Montini, P. Fornasiero and A. Abbotto, Eur. J. Org. Chem., 2016, 5194–5215 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- J. F. Huang, Y. Lei, T. Luo and J. M. Liu, ChemSusChem, 2020, 13, 5863–5895 CrossRef CAS PubMed.
- M. Watanabe, Sci. Technol. Adv. Mater., 2017, 18, 705–723 CrossRef CAS.
- X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 RSC.
- M. Joseph and S. Haridas, Int. J. Hydrogen Energy, 2020, 45, 11954–11975 CrossRef CAS.
- S. N. Yun, N. Vlachopoulos, A. Qurashi, S. Ahmad and A. Hagfeldt, Chem. Soc. Rev., 2019, 48, 3705–3722 RSC.
- A. B. Munoz-Garcia, I. Benesperi, G. Boschloo, J. J. Concepcion, J. H. Delcamp, E. A. Gibson, G. J. Meyer, M. Pavone, H. Pettersson, A. Hagfeldt and M. Freitag, Chem. Soc. Rev., 2021, 50, 12450–12550 RSC.
- P. T. Xu, N. S. McCool and T. E. Mallouk, Nano Today, 2017, 14, 42–58 CrossRef CAS.
- B. B. Beyene and C.-H. Hung, Coord. Chem. Rev., 2020, 410, 213234 CrossRef CAS.
- S. C. Zhang, H. N. Ye, J. L. Hua and H. Tian, EnergyChem, 2019, 1, 100015 CrossRef.
- M. V. Martínez-Díaz, G. De La Torre and T. Torres, Chem. Commun., 2010, 46, 7090–7108 RSC.
- G. Bottari, O. Trukhina, M. Ince and T. Torres, Coord. Chem. Rev., 2012, 256, 2453–2477 CrossRef CAS.
- K. Ladomenou, M. Natali, E. Iengo, G. Charalampidis, F. Scandola and A. G. Coutsolelos, Coord. Chem. Rev., 2015, 304-305, 38–54 CrossRef CAS.
- J. Min Park, J. H. Lee and W. D. Jang, Coord. Chem. Rev., 2020, 407, 213157 CrossRef.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Coord. Chem. Rev., 2021, 428, 213605 CrossRef CAS.
- T. Lazarides, M. Delor, I. V. Sazanovich, T. M. Mc Cormick, I. Georgakaki, G. Charalambidis, J. A. Weinstein and A. G. Coutsolelos, Chem. Commun., 2014, 50, 521–523 RSC.
- A. Panagiotopoulos, K. Ladomenou, D. Sun, V. Artero and A. G. Coutsolelos, Dalton Trans., 2016, 45, 6732–6738 RSC.
- G. B. Bodedla, W.-Y. Wong and X. Zhu, J. Mater. Chem. A, 2021, 9, 20645–20652 RSC.
- G. Landrou, A. A. Panagiotopoulos, K. Ladomenou and A. G. Coutsolelos, J. Porphyrins Phthalocyanines, 2016, 20, 534–541 CrossRef CAS.
- N. Queyriaux, E. Giannoudis, C. D. Windle, S. Roy, J. Pécaut, A. G. Coutsolelos, V. Artero and M. Chavarot-Kerlidou, Sustainable Energy Fuels, 2018, 2, 553–557 RSC.
- E. Giannoudis, E. Benazzi, J. Karlsson, G. Copley, S. Panagiotakis, G. Landrou, P. Angaridis, V. Nikolaou, C. Matthaiaki, G. Charalambidis, E. A. Gibson and A. G. Coutsolelos, Inorg. Chem., 2020, 59, 1611–1621 CrossRef CAS.
- L. Mintrop, J. Windisch, C. Gotzmann, R. Alberto, B. Probst and P. Kurz, J. Phys. Chem. B, 2015, 119, 13698–13706 CrossRef CAS PubMed.
- S. Salzl, M. Ertl and G. Knör, Phys. Chem. Chem. Phys., 2017, 19, 8141–8147 RSC.
- S. S. Nurttila, R. Becker, J. Hessels, S. Woutersen and J. N.-H. Reek, Chem. – Eur. J., 2018, 24, 16395–16406 CrossRef CAS PubMed.
- P. Lang, J. Habermehl, S. I. Troyanov, S. Rau and M. Schwalbe, Chem. – Eur. J., 2018, 24, 3225–3233 CrossRef CAS PubMed.
- J. C. Manton, C. Long, J. G. Vos and M. T. Pryce, Dalton Trans., 2014, 43, 3576–3583 RSC.
- X. Liu, J. Zhou, M. Meng, G. Y. Zhu, Y. Tan, X. Chen, J. Wei, D. B. Kuang, Y. Y. Wu, S. Su, T. Cheng, Y. Zhou and C. Y. Liu, Appl. Catal., B, 2021, 286, 119836 CrossRef CAS.
- D. N. Tritton, G. B. Bodedla, G. Tang, J. Zhao, C. S. Kwan, K. C.-F. Leung, W. Y. Wong and X. Zhu, J. Mater. Chem. A, 2020, 8, 3005–3010 RSC.
- K. Zheng, G. B. Bodedla, Y. Hou, J. Zhang, R. Liang, J. Zhao, D. Lee Phillips and X. Zhu, J. Mater. Chem. A, 2022, 10, 4440–4445 RSC.
- M. Natali, A. Luisa, E. Iengo and F. Scandola, Chem. Commun., 2014, 50, 1842–1844 RSC.
- B. B. Beyene and C. H. Hung, Sustainable Energy Fuels, 2018, 2, 2036–2043 RSC.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry: Metal-Organic Frameworks and Covalent Organic Frameworks, Wiley-VCH, Weinheim, 2019 Search PubMed.
- J. Otsuki, J. Mater. Chem. A, 2018, 6, 6710–6753 RSC.
- T. Keijer, T. Bouwens, J. Hessels and J. N.-H. Reek, Chem. Sci., 2021, 12, 50–70 RSC.
- C. M. Drain, A. Varotto and I. Radivojevic, Chem. Rev., 2009, 109, 1630–1658 CrossRef CAS.
- Y. Zhong, S. Liu, J. Wang, W. Zhang, T. Tian, J. Sun and F. Bai, APL Mater., 2020, 8, 120706 CrossRef CAS.
- C. Zhang, P. Chen, H. Dong, Y. Zhen, M. Liu and W. Hu, Adv. Mater., 2015, 27, 5379–5387 CrossRef CAS PubMed.
- H. Wang, Y. Song, Z. Wang, C. J. Medforth, J. E. Miller, L. Evans, P. Li and J. A. Shelnutt, Chem. Mater., 2008, 20, 7434–7439 CrossRef CAS.
- J. Wang, Y. Zhong, L. Wang, N. Zhang, R. Cao, K. Bian, L. Alarid, R. E. Haddad, F. Bai and H. Fan, Nano Lett., 2016, 16, 6523–6528 CrossRef CAS.
- Y. Zhong, Y. Hu, J. Wang, J. Wang, X. Ren, J. Sun and F. Bai, MRS Adv., 2019, 4, 2071–2078 CrossRef CAS.
- N. Zhang, L. Wang, H. Wang, R. Cao, J. Wang, F. Bai and H. Fan, Nano Lett., 2018, 18, 560–566 CrossRef CAS PubMed.
- Y. Liu, L. Wang, H. Feng, X. Ren, J. Ji, F. Bai and H. Fan, Nano Lett., 2019, 19, 2614–2619 CrossRef CAS PubMed.
- L. J. Liu, Y. D. Lai, H. H. Li, L. T. Kang, J. J. Liu, Z. M. Cao and J. N. Yao, J. Mater. Chem. A, 2017, 5, 8029–8036 RSC.
- X. Tian, C. Lin, Z. Zhong, X. Li, X. Xu, J. Liu, L. Kang, G. Chai and J. Yao, Cryst. Growth Des., 2019, 19, 3279–3287 CrossRef CAS.
- Z. Zhang, Y. Zhu, X. Chen, H. Zhang and J. Wang, Adv. Mater., 2019, 31, 1806626 CrossRef PubMed.
- J. Jing, J. Yang, Z. Zhang and Y. Zhu, Adv. Energy Mater., 2021, 11, 2101392 CrossRef CAS.
- G. Yang, C. Lin, X. Feng, T. Wang and J. Jiang, Chem. Commun., 2020, 56, 527–530 RSC.
- Q. Zou, L. Zhang, X. Yan, A. Wang, G. Ma, J. Li, H. Möhwald and S. Mann, Angew. Chem., Int. Ed., 2014, 53, 2366–2370 CrossRef CAS PubMed.
- J. Li, A. Wang, L. Zhao, Q. Dong, M. Wang, H. Xu, X. Yan and S. Bai, ACS Appl. Mater. Interfaces, 2018, 10, 28420–28427 CrossRef CAS PubMed.
- Q. Zou, M. Abbas, L. Zhao, S. Li, G. Shen and X. Yan, J. Am. Chem. Soc., 2017, 139, 1921–1927 CrossRef CAS PubMed.
- K. Liu, R. Xing, Y. Li, Q. Zou, H. Möhwald and X. Yan, Angew. Chem., Int. Ed., 2016, 55, 12503–12507 CrossRef CAS PubMed.
- K. Liu, M. Abass, Q. Zou and X. Yan, Green Energy Environ., 2017, 2, 58–63 CrossRef.
- E. Nikoloudakis, K. Karikis, J. Han, C. Kokotidou, A. Charisiadis, F. Folias, A. M. Douvas, A. Mitraki, G. Charalambidis, X. Yan and A. G. Coutsolelos, Nanoscale, 2019, 11, 3557–3566 RSC.
- R. Chang, E. Nikoloudakis, Q. Zou, A. Mitraki, A. G. Coutsolelos and X. Yan, ACS Appl. Bio Mater., 2020, 3, 2–9 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- N. Kol, L. Adler-Abramovich, D. Barlam, R. Z. Shneck, E. Gazit and I. Rousso, Nano Lett., 2005, 5, 1343–1346 CrossRef CAS PubMed.
- K. Karikis, A. Butkiewicz, F. Folias, G. Charalambidis, C. Kokotidou, A. Charisiadis, V. Nikolaou, E. Nikoloudakis, J. Frelek, A. Mitraki and A. G. Coutsolelos, Nanoscale, 2018, 10, 1735–1741 RSC.
- G. Charalambidis, E. Kasotakis, T. Lazarides, A. Mitraki and A. G. Coutsolelos, Chem. – Eur. J., 2011, 17, 7213–7219 CrossRef CAS PubMed.
- G. Charalambidis, E. Georgilis, M. K. Panda, C. E. Anson, A. K. Powell, S. Doyle, D. Moss, T. Jochum, P. N. Horton, S. J. Coles, M. Linares, D. Beljonne, J. V. Naubron, J. Conradt, H. Kalt, A. Mitraki, A. G. Coutsolelos and T. S. Balaban, Nat. Commun., 2016, 7, 12657 CrossRef CAS PubMed.
- K. Karikis, E. Georgilis, G. Charalambidis, A. Petrou, O. Vakuliuk, T. Chatziioannou, I. Raptaki, S. Tsovola, I. Papakyriacou, A. Mitraki, D. T. Gryko and A. G. Coutsolelos, Chem. – Eur. J., 2016, 22, 11245–11252 CrossRef CAS PubMed.
- E. Nikoloudakis, K. Karikis, M. Laurans, C. Kokotidou, A. Sole-Daura, J. J. Carbo, A. Charisiadis, G. Charalambidis, G. Izzet, A. Mitraki, A. M. Douvas, J. M. Poblet, A. Proust and A. G. Coutsolelos, Dalton Trans., 2018, 47, 6304–6313 RSC.
- V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Chem. Commun., 2021, 57, 4055–4058 RSC.
- E. Nikoloudakis, M. Pigiaki, M. N. Polychronaki, A. Margaritopoulou, G. Charalambidis, E. Serpetzoglou, A. Mitraki, P. A. Loukakos and A. G. Coutsolelos, ACS Sustainable Chem. Eng., 2021, 9, 7781–7791 CrossRef CAS.
- E. Giannoudis, E. Benazzi, J. Karlsson, G. Copley, S. Panagiotakis, G. Landrou, P. Angaridis, V. Nikolaou, C. Matthaiaki, G. Charalambidis, E. A. Gibson and A. G. Coutsolelos, Inorg. Chem., 2020, 59, 1611–1621 CrossRef CAS PubMed.
- E. Nikoloudakis, G. Charalambidis, M. Vasila, E. Orfanos, P. Angaridis, G. A. Spyroulias and A. G. Coutsolelos, Polyhedron, 2021, 208, 115421 CrossRef CAS.
- G. B. Bodedla, L. Li, Y. Che, Y. Jiang, J. Huang, J. Zhao and X. Zhu, Chem. Commun., 2018, 54, 11614–11617 RSC.
- G. B. Bodedla, G. Tang, J. Zhao and X. Zhu, Sustainable Energy Fuels, 2020, 4, 2675–2679 RSC.
- G. B. Bodedla, J. Huang, W. Y. Wong and X. Zhu, ACS Appl. Nano Mater., 2020, 3, 7040–7046 CrossRef CAS.
- E. R. Clark and D. M. Kurtz, Jr., Dalton Trans., 2016, 45, 630–638 RSC.
- E. R. Clark and D. M. Kurtz, Inorg. Chem., 2017, 56, 4584–4593 CrossRef CAS PubMed.
- H. Kotani, T. Miyazaki, E. Aoki, H. Sakai, T. Hasobe and T. Kojima, ACS Appl. Energy Mater., 2020, 3, 3193–3197 CrossRef CAS.
- I. I. Alkhatib, C. Garlisi, M. Pagliaro, K. Al-Ali and G. Palmisano, Catal. Today, 2020, 340, 209–224 CrossRef CAS.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- K. Sasan, Q. Lin, C. Y. Mao and P. Feng, Chem. Commun., 2014, 50, 10390–10393 RSC.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H.-L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- F. Leng, H. Liu, M. Ding, Q. P. Lin and H. L. Jiang, ACS Catal., 2018, 8, 4583–4590 CrossRef CAS.
- T. He, S. Chen, B. Ni, Y. Gong, Z. Wu, L. Song, L. Gu, W. Hu and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 3493–3498 CrossRef CAS PubMed.
- G. Lan, Y. Y. Zhu, S. S. Veroneau, Z. Xu, D. Micheroni and W. Lin, J. Am. Chem. Soc., 2018, 140, 5326–5329 CrossRef CAS PubMed.
- S. Li, H. M. Mei, S. L. Yao, Z. Y. Chen, Y. L. Lu, L. Zhang and C. Y. Su, Chem. Sci., 2019, 10, 10577–10585 RSC.
- S. Yuan, T.-F. Liu, D. Feng, J. Tian, K. Wang, J. Qin, Q. Zhang, Y.-P. Chen, M. Bosch, L. Zou, S. J. Teat, S. J. Dalgarno and H.-C. Zhou, Chem. Sci., 2015, 6, 3926–3930 RSC.
- X. Wang, X. Zhang, W. Zhou, L. Liu, J. Ye and D. Wang, Nano Energy, 2019, 62, 250–258 CrossRef CAS.
- Q. Zuo, T. Liu, C. Chen, Y. Ji, X. Gong, Y. Mai and Y. Zhou, Angew. Chem., Int. Ed., 2019, 58, 10198–10203 CrossRef CAS PubMed.
- Y. Diao, N. Xu, M. Q. Li, X. Zhu and Z. Xu, Inorg. Chem., 2020, 59, 12643–12649 CrossRef CAS PubMed.
- H. Hu, Z. Wang, L. Cao, L. Zeng, C. Zhang, W. Lin and C. Wang, Nat. Chem., 2021, 13, 358–366 CrossRef CAS PubMed.
- H. Hu, L. Zeng, Z. Li, T. Zhu and C. Wang, Chin. J. Catal., 2021, 42, 1345–1351 CrossRef CAS.
- P. Jin, L. Wang, X. Ma, R. Lian, J. Huang, H. She, M. Zhang and Q. Wang, Appl. Catal., B, 2021, 284, 119762 CrossRef CAS.
- C. Lin, C. Han, H. Zhang, L. Gong, Y. Gao, H. Wang, Y. Bian, R. Li and J. Jiang, Inorg. Chem., 2021, 60, 3988–3995 CrossRef CAS PubMed.
- Z. Chen, J. Wang, S. Zhang, Y. Zhang, J. Zhang, R. Li and T. Peng, ACS Appl. Energy Mater., 2019, 2, 5665–5676 CrossRef CAS.
- J. Wang, L. Xu, T. Wang, S. Chen, Z. Jiang, R. Li, Y. Zhang and T. Peng, Adv. Funct. Mater., 2021, 31, 2009819 CrossRef CAS.
- Z. Fan, K. Nomura, M. Zhu, X. Li, J. Xue, T. Majima and Y. Osakada, Commun. Chem., 2019, 2, 55 CrossRef.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X. Y. Gao, L. Gao, L. Qiao, X. B. Li, L. Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed.
- K. C. Ranjeesh, L. George, V. C. Wakchaure, Goudappagouda, R. N. Devi and S. S. Babu, Chem. Commun., 2019, 55, 1627–1630 RSC.
- K. C. Ranjeesh, L. George, A. Maibam, S. Krishnamurty and S. S. Babu, ChemCatChem, 2021, 13, 1717–1721 CrossRef CAS.
- M. H.-F. Ali, G. Bengasi, K. Baba and N. D. Boscher, ACS Appl. Energy Mater., 2020, 3, 9848–9855 CrossRef.
- L. Xie, J. Tian, Y. Ouyang, X. Guo, W. Zhang, U. P. Apfel, W. Zhang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 15844–15848 CrossRef CAS PubMed.
- P. Liao, Y. Hu, Z. Liang, J. Zhang, H. Yang, L. Q. He, Y. X. Tong, J. M. Liu, L. Chen and C. Y. Su, J. Mater. Chem. A, 2018, 6, 3195–3201 RSC.
- X. Zhao, X. Zhang, Y. Liang, Z. Hu and F. Huang, Macromolecules, 2021, 54, 4902–4909 CrossRef CAS.
- R. X. Li, X. F. Liu, T. Liu, Y. B. Yin, Y. Zhou, S. K. Mei and J. Yan, Electrochim. Acta, 2017, 237, 207–216 CrossRef CAS.
- X. F. Liu, R. X. Li, X. T. Ren, Y. B. Yin, S. K. Mei, T. Liu and J. Yan, J. Catal., 2017, 348, 314–320 CrossRef CAS.
- X. Li, K. Li, D. Wang, J. Huang, C. Zhang, Y. Du and P. Yang, J. Porphyrins Phthalocyanines, 2017, 21, 179–188 CrossRef CAS.
- R. Ge, X. Li, S. Z. Kang, L. Qin and G. Li, Appl. Catal., B, 2016, 187, 67–74 CrossRef CAS.
- Q. Luo, K. Zhu, S. Z. Kang, L. Qin, S. Han, G. Li and X. Li, J. Porphyrins Phthalocyanines, 2018, 22, 877–885 CrossRef CAS.
- K. Zhu, Q. Luo, S. Z. Kang, L. Qin, G. Li and X. Li, Nanoscale, 2018, 10, 18635–18641 RSC.
- L. Zhang, L. Qin, S. Z. Kang, G. D. Li and X. Li, ACS Sustainable Chem. Eng., 2019, 7, 8358–8366 CrossRef CAS.
- Y. J. Yuan, D. Chen, J. Zhong, L. X. Yang, J. J. Wang, Z. T. Yu and Z. G. Zou, J. Phys. Chem. C, 2017, 121, 24452–24462 CrossRef CAS.
- Q. Luo, R. Ge, S. Z. Kang, L. Qin, G. Li and X. Li, Appl. Surf. Sci., 2018, 427, 15–23 CrossRef CAS.
- S. Mei, J. Gao, Y. Zhang, J. Yang, Y. Wu, X. Wang, R. Zhao, X. Zhai, C. Hao, R. Li and J. Yan, J. Colloid Interface Sci., 2017, 506, 58–65 CrossRef CAS PubMed.
- E. S. Da Silva, N. M.-M. Moura, M. G.-P. M.-S. Neves, A. Coutinho, M. Prieto, C. G. Silva and J. L. Faria, Appl. Catal., B, 2018, 221, 56–69 CrossRef CAS.
- J. Wang, Y. Zheng, T. Peng, J. Zhang and R. Li, ACS Sustainable Chem. Eng., 2017, 5, 7549–7556 CrossRef CAS.
- Y. Zheng, J. Wang, J. Zhang, T. Peng and R. Li, Dalton Trans., 2017, 46, 8219–8228 RSC.
- J. Wang, D. Liu, Q. Liu, T. Peng, R. Li and S. Zhou, Appl. Surf. Sci., 2019, 464, 255–261 CrossRef CAS.
- L. Li, G. B. Bodedla, Z. Liu and X. Zhu, Appl. Surf. Sci., 2020, 499, 143755 CrossRef CAS.
- K. Zhu, M. Zhang, X. Feng, L. Qin, S. Z. Kang and X. Li, Appl. Catal., B, 2020, 268, 118434 CrossRef CAS.
- M. Zhang, K. Zhu, L. Qin, S. Z. Kang and X. Li, Catal. Sci. Technol., 2020, 10, 1640–1649 RSC.
- P. Zeng, Y. Zheng, S. Chen, H. Liu, R. Li and T. Peng, New J. Chem., 2020, 44, 11237–11247 RSC.
- S. Tian, S. Chen, X. Ren, R. Cao, H. Hu and F. Bai, Nano Res., 2019, 12, 3109–3115 CrossRef CAS.
- G. Maduraiveeran, M. Sasidharan and W. Jin, Prog. Mater. Sci., 2019, 106, 100574 CrossRef CAS.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS PubMed.
- Y. Xue, S. Sun, Q. Wang, Z. Dong and Z. Liu, J. Mater. Chem. A, 2018, 6, 10595–10626 RSC.
- S. Martha, P. Chandra Sahoo and K. M. Parida, RSC Adv., 2015, 5, 61535–61553 RSC.
- K. Kurimoto, T. Yamazaki, Y. Suzuri, Y. Nabetani, S. Onuki, S. Takagi, T. Shimada, H. Tachibana and H. Inoue, Photochem. Photobiol. Sci., 2014, 13, 154–156 CrossRef CAS PubMed.
- A.-M. Manke, K. Geisel, A. Fetzer and P. Kurz, Phys. Chem. Chem. Phys., 2014, 16, 12029–12042 RSC.
- E. Koposova, X. Liu, A. Pendin, B. Thiele, G. Shumilova, Y. Ermolenko, A. Offenhäusser and Y. Mourzina, J. Phys. Chem. C, 2016, 120, 13873–13890 CrossRef CAS.
- Y. Yuan, H. Lu, Z. Ji, J. Zhong, M. Ding, D. Chen, Y. Li, W. Tu, D. Cao, Z. Yu and Z. Zou, Chem. Eng. J., 2015, 275, 8–16 CrossRef CAS.
- Y. J. Yuan, J. R. Tu, Z. J. Ye, H. W. Lu, Z. G. Ji, B. Hu, Y. H. Li, D. P. Cao, Z. T. Yu and Z. G. Zou, Dyes Pigm., 2015, 123, 285–292 CrossRef CAS.
- B. Zhuang, L. Xiangqing, R. Ge, S. Kang, L. Qin and G. Li, Appl. Catal., A, 2017, 533, 81–89 CrossRef CAS.
- P. Wang, M. Xi, S. Z. Kang, L. Qin, S. Han and X. Li, Int. J. Hydrogen Energy, 2020, 45, 6508–6518 CrossRef CAS.
- X. Guo, X. Li, L. Qin, S. Z. Kang and G. Li, Appl. Catal., B, 2019, 243, 1–9 CrossRef CAS.
- F. Kuttassery, S. Sagawa, S. Mathew, Y. Nabetani, A. Iwase, A. Kudo, H. Tachibana and H. Inoue, ACS Appl. Energy Mater., 2019, 2, 8045–8051 CrossRef CAS.
- V. Nikolaou, G. Charalambidis, K. Ladomenou, E. Nikoloudakis, C. Drivas, I. Vamvasakis, S. Panagiotakis, G. Landrou, E. Agapaki, C. Stangel, C. Henkel, J. Joseph, G. Armatas, M. Vasilopoulou, S. Kennou, D. M. Guldi and A. G. Coutsolelos, ChemSusChem, 2021, 14, 961–970 CrossRef CAS PubMed.
- P. Y. Ho, M. F. Mark, Y. Wang, S. C. Yiu, W. H. Yu, C. L. Ho, D. W. McCamant, R. Eisenberg and S. Huang, ChemSusChem, 2018, 11, 2517–2528 CrossRef CAS PubMed.
- P. S. Gangadhar, S. Gonuguntla, S. Madanaboina, N. Islavath, U. Pal and L. Giribabu, J. Photochem. Photobiol., A, 2020, 392, 112408 CrossRef CAS.
- P. Wang, M. Xi, L. Qin, S. Z. Kang, Y. Fang and X. Li, ACS Appl. Nano Mater., 2019, 2, 7409–7420 CrossRef CAS.
- M. Xi, P. Wang, M. Zhang, L. Qin, S. Z. Kang and X. Li, Appl. Surf. Sci., 2020, 529, 147200 CrossRef CAS.
- Q. Wang, Y. Shi, Q. Ma, D. Gao, J. Zhong, J. Li, F. Wang, Y. He and R. Wang, J. Mater. Sci.: Mater. Electron., 2017, 28, 2123–2127 CrossRef CAS.
- X. Feng, Z. Liu, L. Qin, S. Z. Kang and X. Li, Phys. Chem. Chem. Phys., 2020, 22, 13528–13535 RSC.
- L. Y. Huang, J. F. Huang, Y. Lei, S. Qin and J. M. Liu, Catalysts, 2020, 10, 656 CrossRef CAS.
- M. Xi, B. Zhuang, Y. Chen, L. Qin, S. Z. Kang and X. Li, J. Solid State Chem., 2021, 298, 122103 CrossRef CAS.
- G. Mukherjee, J. Thote, H. B. Aiyappa, S. Kandambeth, S. Banerjee, K. Vanka and R. Banerjee, Chem. Commun., 2017, 53, 4461–4464 RSC.
- V. Nikolaou, G. Charalambidis, G. Landrou, E. Nikoloudakis, A. Planchat, R. Tsalameni, K. Junghans, A. Kahnt, F. Odobel and A. G. Coutsolelos, ACS Appl. Energy Mater., 2021, 4, 10042–10049 CrossRef CAS.
- D. Khusnutdinova, A. M. Beiler, B. L. Wadsworth, S. I. Jacob and G. F. Moore, Chem. Sci., 2017, 8, 253–259 RSC.
- A. M. Beiler, D. Khusnutdinova, B. L. Wadsworth and G. F. Moore, Inorg. Chem., 2017, 56, 12178–12185 CrossRef CAS PubMed.
- B. D. Sherman, J. J. Bergkamp, C. L. Brown, A. L. Moore, D. Gust and T. A. Moore, Energy Environ. Sci., 2016, 9, 1812–1817 RSC.
- K. Morita, K. Takijiri, K. Sakai and H. Ozawa, Dalton Trans., 2017, 46, 15181–15185 RSC.
- K. Morita, K. Sakai and H. Ozawa, ACS Appl. Energy Mater., 2019, 2, 987–992 CrossRef CAS.
- C. Canales, A. F. Olea, L. Gidi, R. Arce and G. Ramírez, Electrochim. Acta, 2017, 258, 850–857 CrossRef CAS.
- L. Gidi, J. Honores, J. Ibarra, R. Arce, M. J. Aguirre and G. Ramírez, New J. Chem., 2019, 43, 12727–12733 RSC.
- L. Gidi, J. Honores, J. Ibarra, R. Arce, M. J. Aguirre and G. Ramírez, Catalysts, 2020, 10, 239 CrossRef CAS.
- X. Li, A. Liu, D. Chu, C. Zhang, Y. Du, J. Huang and P. Yang, Catalysts, 2018, 8, 108 CrossRef.
- Y. Kumar, B. Patil, A. Khaligh, S. E. Hadi, T. Uyar and D. Tuncel, ChemCatChem, 2019, 11, 2994–2999 CrossRef CAS.
- X. Li, Y. Zhang, W. Wang, J. Meng, K. Li, W. Lin, Z. Peng, J. Wan and Z. Hu, Int. J. Hydrogen Energy, 2019, 44, 18072–18082 CrossRef CAS.
- M. Blanco, M. Lunardon, M. Bortoli, D. Mosconi, L. Girardi, L. Orian, S. Agnoli and G. Granozzi, J. Mater. Chem. A, 2020, 8, 11019–11030 RSC.
- L. Girardi, M. Blanco, S. Agnoli, G. A. Rizzi and G. Granozzi, Nanomaterials, 2020, 10, 1266 CrossRef CAS PubMed.
- Y. H. Xiao, W. Tian, S. Jin, Z. G. Gu and J. Zhang, Small, 2020, 16, 2005111 CrossRef CAS PubMed.
- A. Charisiadis, E. Giannoudis, Z. Pournara, A. Kosma, V. Nikolaou, G. Charalambidis, V. Artero, M. Chavarot-Kerlidou and A. G. Coutsolelos, Eur. J. Inorg. Chem., 2021, 1122–1129 CrossRef CAS.
- P. Gotico, Z. Halime and A. Aukauloo, Dalton Trans., 2020, 49, 2381–2396 RSC.
- L. Zou, R. Sa, H. Lv, H. Zhong and R. Wang, ChemSusChem, 2020, 13, 6124–6140 CAS.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- J. Grodkowski, D. Behar, P. Neta and P. Hambright, J. Phys. Chem. A, 1997, 101, 248–254 CrossRef CAS.
- J. Bonin, M. Chaussemier, M. Robert and M. Routier, ChemCatChem, 2014, 6, 3200–3207 CrossRef CAS.
- J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed.
- H. Rao, J. Bonin and M. Robert, Chem. Commun., 2017, 53, 2830–2833 RSC.
- H. Rao, J. Bonin and M. Robert, ChemSusChem, 2017, 10, 4447–4450 CrossRef CAS PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- H. Rao, J. Bonin and M. Robert, J. Phys. Chem. C, 2018, 122, 13834–13839 CrossRef CAS.
- H. Rao, C. H. Lim, J. Bonin, G. M. Miyake and M. Robert, J. Am. Chem. Soc., 2018, 140, 17830–17834 CrossRef CAS PubMed.
- M. Kientz, G. Lowe, B. G. McCarthy, G. M. Miyake, J. Bonin and M. Robert, ChemPhotoChem, 2022 DOI:10.1002/cptc.202200009.
- A. Dannenhoffer, H. Sai, D. Huang, B. Nagasing, B. Harutyunyan, D. J. Fairfield, T. Aytun, S. M. Chin, M. J. Bedzyk, M. Olvera De La Cruz and S. I. Stupp, Chem. Sci., 2019, 10, 5779–5786 RSC.
- H. Yuan, B. Cheng, J. Lei, L. Jiang and Z. Han, Nat. Commun., 2021, 12, 1835 CrossRef CAS PubMed.
- G. F. Manbeck and E. Fujita, J. Porphyrins Phthalocyanines, 2015, 19, 45–64 CrossRef CAS.
- A. Call, M. Cibian, K. Yamamoto, T. Nakazono, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9, 4867–4874 CrossRef CAS.
- X. Zhang, M. Cibian, A. Call, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9, 11263–11273 CrossRef CAS.
- X. Zhang, K. Yamauchi and K. Sakai, ACS Catal., 2021, 11, 10436–10449 CrossRef CAS.
- C. Matlachowski and M. Schwalbe, Dalton Trans., 2015, 44, 6480–6489 RSC.
- C. D. Windle, M. W. George, R. N. Perutz, P. A. Summers, X. Z. Sun and A. C. Whitwood, Chem. Sci., 2015, 6, 6847–6864 RSC.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- E. E. Benson and C. P. Kubiak, Chem. Commun., 2012, 48, 7374–7376 RSC.
- C. D. Windle, E. Pastor, A. Reynal, A. C. Whitwood, Y. Vaynzof, J. R. Durrant, R. N. Perutz and E. Reisner, Chem. – Eur. J., 2015, 21, 3746–3754 CrossRef CAS PubMed.
- C. Matlachowski, B. Braun, S. Tschierlei and M. Schwalbe, Inorg. Chem., 2015, 54, 10351–10360 CrossRef CAS PubMed.
- P. Lang, M. Pfrunder, G. Quach, B. Braun-Cula, E. G. Moore and M. Schwalbe, Chem. – Eur. J., 2019, 25, 4509–4519 CrossRef CAS PubMed.
- Y. Kuramochi, Y. Fujisawa and A. Satake, J. Am. Chem. Soc., 2020, 142, 705–709 CrossRef CAS PubMed.
- Y. Kuramochi and A. Satake, Chem. – Eur. J., 2020, 26, 16365–16373 CrossRef CAS PubMed.
- J. X. Zhang, C. Y. Hu, W. Wang, H. Wang and Z. Y. Bian, Appl. Catal., A, 2016, 522, 145–151 CrossRef CAS.
- J. D. Shipp, H. Carson, S. J.-P. Spall, S. C. Parker, D. Chekulaev, N. Jones, M. Y. Mel'nikov, C. C. Robertson, A. J.-H. M. Meijer and J. A. Weinstein, Dalton Trans., 2020, 49, 4230–4243 RSC.
- Y. Amao, Sustainable Energy Fuels, 2018, 2, 1928–1950 RSC.
- Y. Amao, Chem. Lett., 2017, 46, 780–788 CrossRef CAS.
- Y. Amao, R. Abe and S. Shiotani, J. Photochem. Photobiol., A, 2015, 313, 149–153 CrossRef CAS.
- S. Ikeyama and Y. Amao, Sustainable Energy Fuels, 2017, 1, 1730–1733 RSC.
- S. Ikeyama and Y. Amao, Photochem. Photobiol. Sci., 2018, 17, 60–68 CrossRef CAS PubMed.
- F. Secundo and Y. Amao, RSC Adv., 2020, 10, 42354–42362 RSC.
- S. Ikeyama, T. Katagiri and Y. Amao, J. Photochem. Photobiol., A, 2018, 358, 362–367 CrossRef CAS.
- A. Miyaji and Y. Amao, New J. Chem., 2021, 45, 5780–5790 RSC.
- A. Miyaji and Y. Amao, ChemNanoMat, 2021, 7, 626–634 CrossRef CAS.
- A. Miyaji and Y. Amao, New J. Chem., 2020, 44, 18803–18812 RSC.
- Y. Amao, S. Ikeyama, T. Katagiri and K. Fujita, Faraday Discuss., 2017, 198, 73–81 RSC.
- T. Katagiri, S. Ikeyama and Y. Amao, J. Photochem. Photobiol., A, 2018, 358, 368–373 CrossRef CAS.
- Y. Amao and R. Kataoka, Catal. Today, 2018, 307, 243–247 CrossRef CAS.
- X. Ji, J. Wang, Y. Kang, L. Mei, Z. Su, S. Wang, G. Ma, J. Shi and S. Zhang, ACS Catal., 2018, 8, 10732–10745 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- H. Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- J. Jin, New J. Chem., 2020, 44, 15362–15368 RSC.
- R. Hariri and S. Dehghanpour, Appl. Organomet. Chem., 2021, 35, e6422 CrossRef CAS.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- J. Liu, Y. Z. Fan, X. Li, Z. Wei, Y. W. Xu, L. Zhang and C. Y. Su, Appl. Catal., B, 2018, 231, 173–181 CrossRef CAS.
- E. X. Chen, M. Qiu, Y. F. Zhang, Y. S. Zhu, L. Y. Liu, Y. Y. Sun, X. Bu, J. Zhang and Q. Lin, Adv. Mater., 2018, 30, 1704388 CrossRef PubMed.
- Y. C. Qiu, S. Yuan, X. X. Li, D. Y. Du, C. Wang, J. S. Qin, H. F. Drake, Y. Q. Lan, L. Jiang and H. C. Zhou, J. Am. Chem. Soc., 2019, 141, 13841–13848 CrossRef CAS PubMed.
- L. Wang, P. Jin, J. Huang, H. She and Q. Wang, ACS Sustainable Chem. Eng., 2019, 7, 15660–15670 CrossRef CAS.
- L. Wang, P. Jin, S. Duan, H. She, J. Huang and Q. Wang, Sci. Bull., 2019, 64, 926–933 CrossRef CAS.
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- S. Choi, W. J. Jung, K. Park, S. Y. Kim, J. O. Baeg, C. H. Kim, H. J. Son, C. Pac and S. O. Kang, ACS Appl. Mater. Interfaces, 2021, 13, 2710–2722 CrossRef CAS PubMed.
- Q. Li, Y. Luo, Y. Ding, Y. Wang, Y. Wang, H. Du, R. Yuan, J. Bao, M. Fang and Y. Wu, Dalton Trans., 2019, 48, 8678–8692 RSC.
- N. Sadeghi, S. Sharifnia and T. O. Do, J. Mater. Chem. A, 2018, 6, 18031–18035 RSC.
- N. Sadeghi and M. Sillanpää, Photochem. Photobiol. Sci., 2021, 20, 391–399 CrossRef CAS PubMed.
- S.-S. Wang, H.-H. Huang, M. Liu, S. Yao, S. Guo, J.-W. Wang, Z.-M. Zhang and T.-B. Lu, Inorg. Chem., 2020, 59, 6301–6307 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- S. Chen, F. Yang, H. Gao, J. Wang, X. Chen, X. Zhang, J. Li and A. Li, J. CO2 Util., 2021, 48, 101528 CrossRef CAS.
- N. Sadeghi, S. Sharifnia and M. Sheikh Arabi, J. CO2 Util., 2016, 16, 450–457 CrossRef CAS.
- Z. B. Fang, T. T. Liu, J. Liu, S. Jin, X. P. Wu, X. Q. Gong, K. Wang, Q. Yin, T. F. Liu, R. Cao and H. C. Zhou, J. Am. Chem. Soc., 2020, 142, 12515–12523 CrossRef CAS PubMed.
- J. Xu, X. Liu, Z. Zhou and M. Xu, Appl. Surf. Sci., 2020, 513, 145801 CrossRef CAS.
- L. Ye, Y. Gao, S. Cao, H. Chen, Y. Yao, J. Hou and L. Sun, Appl. Catal., B, 2018, 227, 54–60 CrossRef CAS.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- L. Chen, Y. Wang, F. Yu, X. Shen and C. Duan, J. Mater. Chem. A, 2019, 7, 11355–11361 RSC.
- C. Wang, X. M. Liu, M. Zhang, Y. Geng, L. Zhao, Y. G. Li and Z. M. Su, ACS Sustainable Chem. Eng., 2019, 7, 14102–14110 CrossRef CAS.
- C. Zheng, X. Qiu, J. Han, Y. Wu and S. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 42243–42249 CrossRef CAS PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J. L. Wang, D. Q. Yuan and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed.
- S. Wan, F. Gándara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, X. Duan, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- L. J. Wang, R. L. Wang, X. Zhang, J. L. Mu, Z. Y. Zhou and Z. M. Su, ChemSusChem, 2020, 13, 2973–2980 CrossRef CAS PubMed.
- H. Lv, R. Sa, P. Li, D. Yuan, X. Wang and R. Wang, Sci. China Chem., 2020, 63, 1289–1294 CrossRef CAS.
- N. Xu, Y. Diao, X. Qin, Z. Xu, H. Ke and X. Zhu, Dalton Trans., 2020, 49, 15587–15591 RSC.
- Y.-N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 CrossRef CAS PubMed.
- H. Zhong, R. Sa, H. Lv, S. Yang, D. Yuan, X. Wang and R. Wang, Adv. Funct. Mater., 2020, 30, 2002654 CrossRef CAS.
- S. Zhang, S. Wang, L. Guo, H. Chen, B. Tan and S. Jin, J. Mater. Chem. C, 2019, 8, 192–200 RSC.
- Y. Zhang, Y. Wang, L. C. An, J. Chen, T. Zhang and Y. H. Zhang, Mater. Chem. Front., 2020, 4, 2754–2761 RSC.
- Y. Zhang, G. L. Zhang, Y. T. Wang, Z. Ma, T. Y. Yang, T. Zhang and Y. H. Zhang, J. Colloid Interface Sci., 2021, 596, 342–351 CrossRef CAS PubMed.
- K. Li, L. Lin, T. Peng, Y. Guo, R. Li and J. Zhang, Chem. Commun., 2015, 51, 12443–12446 RSC.
- G. Mele, C. Annese, L. D'Accolti, A. De Riccardis, C. Fusco, L. Palmisano, A. Scarlino and G. Vasapollo, Molecules, 2015, 20, 396–415 CrossRef PubMed.
- Y. Song, J. Li and C. Wang, J. Mater. Res., 2018, 33, 2612–2620 CrossRef CAS.
- Z. Wang, W. Zhou, X. Wang, X. Zhang, H. Chen, H. Hu, L. Liu, J. Ye and D. Wang, Catalysts, 2020, 10, 654 CrossRef CAS.
- L. Wang, S. Duan, P. Jin, H. She, J. Huang, Z. Lei, T. Zhang and Q. Wang, Appl. Catal., B, 2018, 239, 599–608 CrossRef CAS.
- H. Gao, J. Wang, M. Jia, F. Yang, R. S. Andriamitantsoa, X. Huang, W. Dong and G. Wang, Chem. Eng. J., 2019, 374, 684–693 CrossRef CAS.
- V. Jeyalakshmi, S. Tamilmani, R. Mahalakshmy, P. Bhyrappa, K. R. Krishnamurthy and B. Viswanathan, J. Mol. Catal. A: Chem., 2016, 420, 200–207 CrossRef CAS.
- D. I. Won, J. S. Lee, Q. Ba, Y. J. Cho, H. Y. Cheong, S. Choi, C. H. Kim, H. J. Son, C. Pac and S. O. Kang, ACS Catal., 2018, 8, 1018–1030 CrossRef CAS.
- S. Choi, C. H. Kim, J. O. Baeg, H. J. Son, C. Pac and S. O. Kang, ACS Appl. Energy Mater., 2020, 3, 11581–11596 CrossRef CAS.
- G. Zhao, H. Pang, G. Liu, P. Li, H. Liu, H. Zhang, L. Shi and J. Ye, Appl. Catal., B, 2017, 200, 141–149 CrossRef CAS.
- L. Lin, C. Hou, X. Zhang, Y. Wang, Y. Chen and T. He, Appl. Catal., B, 2018, 221, 312–319 CrossRef CAS.
- S. Tian, S. Chen, X. Ren, Y. Hu, H. Hu, J. Sun and F. Bai, Nano Res., 2020, 13, 2665–2672 CrossRef CAS.
- P. Li, C. Hou, X. Zhang, Y. Chen and T. He, Appl. Surf. Sci., 2018, 459, 292–299 CrossRef CAS.
- P. Li, X. Zhang, C. Hou, Y. Chen and T. He, Appl. Catal., B, 2018, 238, 656–663 CrossRef CAS.
- S. Lian, M. S. Kodaimati, D. S. Dolzhnikov, R. Calzada and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 8931–8938 CrossRef CAS PubMed.
- S. Lian, M. S. Kodaimati and E. A. Weiss, ACS Nano, 2018, 12, 568–575 CrossRef CAS PubMed.
- F. Arcudi, L. Đorđević, B. Nagasing, S. I. Stupp and E. A. Weiss, J. Am. Chem. Soc., 2021, 143, 18131–18138 CrossRef CAS PubMed.
- M. García, M. J. Aguirre, G. Canzi, C. P. Kubiak, M. Ohlbaum and M. Isaacs, Electrochim. Acta, 2014, 115, 146–154 CrossRef.
- J. Liu, H. Shi, Q. Shen, C. Guo and G. Zhao, Green Chem., 2017, 19, 5900–5910 RSC.
- Y. Kou, S. Nakatani, G. Sunagawa, Y. Tachikawa, D. Masui, T. Shimada, S. Takagi, D. A. Tryk, Y. Nabetani, H. Tachibana and H. Inoue, J. Catal., 2014, 310, 57–66 CrossRef CAS.
- R. Nakazato, Y. Kou, D. Yamamoto, T. Shimada, T. Ishida, S. Takagi, H. Munakata, K. Kanamura, H. Tachibana and H. Inoue, Res. Chem. Intermed., 2021, 47, 269–285 CrossRef CAS.
- R. Zeng, G. Chen, C. Xiong, G. Li, Y. Zheng, J. Chen, Y. Long and S. Chen, Appl. Surf. Sci., 2018, 434, 756–762 CrossRef CAS.
- Y. Dong, R. Nie, J. Wang, X. Yu, P. Tu, J. Chen and H. Jing, Chin. J. Catal., 2019, 40, 1222–1230 CrossRef CAS.
- D. Yang, H. Yu, T. He, S. Zuo, X. Liu, H. Yang, B. Ni, H. Li, L. Gu, D. Wang and X. Wang, Nat. Commun., 2019, 10, 3844 CrossRef PubMed.
- Y. Zhou, L. Zheng, D. Yang, H. Yang, Q. Lu, Q. Zhang, L. Gu and X. Wang, Small Methods, 2021, 5, 2000991 CrossRef CAS PubMed.
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194 CrossRef CAS PubMed.
- J. F.-D. Brito, K. Irikura, C. M. Terzi, S. Nakagaki and M. V.-B. Zanoni, J. CO2 Util., 2020, 41, 101261 CrossRef CAS.
- J. Cheng, X. Yang, X. Xuan and J. Zhou, Chem. Eng. J., 2020, 392, 123799 CrossRef CAS.
- B. Wang, F. Yang, Y. Dong, Y. Cao, J. Wang, B. Yang, Y. Wei, W. Wan, J. Chen and H. Jing, Chem. Eng. J., 2020, 396, 125255 CrossRef CAS.
- A. Charisiadis, E. Glymenaki, A. Planchat, S. Margiola, A.-C. Lavergne-Bril, E. Nikoloudakis, V. Nikolaou, G. Charalambidis, A. G. Coutsolelos and F. Odobel, Dyes Pigm., 2021, 185, 108908 CrossRef CAS.
- J. J. Cid, J. H. Yum, S. R. Jang, M. K. Nazeeruddin, E. Martínez-Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358–8362 CrossRef CAS PubMed.
- M. Urbani, M. E. Ragoussi, M. K. Nazeeruddin and T. Torres, Coord. Chem. Rev., 2019, 381, 1–64 CrossRef CAS.
- X. Zhang, L. Yu, C. Zhuang, T. Peng, R. Li and X. Li, RSC Adv., 2013, 3, 14363–14370 RSC.
- G. Kim and W. Choi, Appl. Catal., B, 2010, 100, 77–83 CrossRef CAS.
- X. Zhang, B. Peng, T. Peng, L. Yu, R. Li and J. Zhang, J. Power Sources, 2015, 298, 30–37 CrossRef CAS.
- M. Fang, G. Dong, R. Wei and J. C. Ho, Adv. Energy Mater., 2017, 7, 1700559 CrossRef.
- A. Tiwari, N. V. Krishna, L. Giribabu and U. Pal, J. Phys. Chem. C, 2018, 122, 495–502 CrossRef CAS.
- H. S. Moon and K. Yong, Appl. Surf. Sci., 2020, 530, 147215 CrossRef CAS.
- E. Genc, A. C. Yüzer, G. Yanalak, E. Harputlu, E. Aslan, K. Ocakoglu, M. Ince and I. H. Patir, Renewable Energy, 2020, 162, 1340–1346 CrossRef CAS.
- M. E. Ragoussi, J. J. Cid, J. H. Yum, G. De La Torre, D. Di Censo, M. Grätzel, M. K. Nazeeruddin and T. Torres, Angew. Chem., Int. Ed., 2012, 51, 4375–4378 CrossRef CAS PubMed.
- M. E. Ragoussi, J. H. Yum, A. K. Chandiran, M. Ince, G. de la Torre, M. Grätzel, M. K. Nazeeruddin and T. Torres, ChemPhysChem, 2014, 15, 1033–1036 CrossRef CAS PubMed.
- M. Ince, R. Kuboi, T. Ince, K. Yoshimura, D. Motoyoshi, M. Sonobe, R. Kudo, S. Mori, M. Kimura and T. Torres, Sustainable Energy Fuels, 2021, 5, 584–589 RSC.
- E. Genc Acar, A. C. Yüzer, G. Kurtay, G. Yanalak, E. Harputlu, E. Aslan, K. Ocakoglu, M. Güllü, M. Ince and I. H. Patir, ACS Appl. Energy Mater., 2021, 4, 10222–10233 CrossRef CAS.
- A. C. Yüzer, E. Genc, G. Kurtay, G. Yanalak, E. Aslan, E. Harputlu, K. Ocakoglu, I. Hatay Patir and M. Ince, Chem. Commun., 2021, 57, 9196–9199 RSC.
- C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS PubMed.
- T. M. Grant, D. S. Josey, K. L. Sampson, T. Mudigonda, T. P. Bender and B. H. Lessard, Chem. Rec., 2019, 19, 1093–1112 CrossRef CAS PubMed.
- M. Ince, A. Medina, J. H. Yum, A. Yella, C. G. Claessens, M. V. Martínez-Díaz, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. – Eur. J., 2014, 20, 2016–2021 CrossRef CAS PubMed.
- M. Urbani, F. A. Sari, M. Grätzel, M. K. Nazeeruddin, T. Torres and M. Ince, Chem. – Asian J., 2016, 11, 1223–1231 CrossRef CAS PubMed.
- A. Celil Yüzer, E. Genc, E. Harputlu, G. Yanalak, E. Aslan, K. Ocakoglu, I. Hatay Patir and M. Ince, Dalton Trans., 2020, 49, 12550–12554 RSC.
- T. Abe, K. Fukui, Y. Kawai, K. Nagai and H. Kato, Chem. Commun., 2016, 52, 7735–7737 RSC.
- Y. Kawai, K. Nagai and T. Abe, RSC Adv., 2017, 7, 34694–34698 RSC.
- T. Murakami, K. Ikezoi, K. Nagai, H. Kato and T. Abe, ChemElectroChem, 2020, 7, 5029–5035 CrossRef CAS.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- X. Zhang, T. Peng, L. Yu, R. Li, Q. Li and Z. Li, ACS Catal., 2015, 5, 504–510 CrossRef CAS.
- X. Zhang, L. Yu, R. Li, T. Peng and X. Li, Catal. Sci. Technol., 2014, 4, 3251–3260 RSC.
- Y. Guo, S. Song, Y. Zheng, R. Li and T. Peng, Dalton Trans., 2016, 45, 14071–14079 RSC.
- S. Song, Y. Guo, T. Peng, J. Zhang and R. Li, RSC Adv., 2016, 6, 77366–77374 RSC.
- P. Zeng, J. Wang, Y. Guo, R. Li, G. Mei and T. Peng, Chem. Eng. J., 2019, 373, 651–659 CrossRef CAS.
- Q. Liu, J. Wang, D. Liu, R. Li and T. Peng, J. Power Sources, 2018, 396, 57–63 CrossRef CAS.
- J. Huang, Y. Wu, D. Wang, Y. Ma, Z. Yue, Y. Lu, M. Zhang, Z. Zhang and P. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 3732–3741 CrossRef CAS PubMed.
- D. Wang, J. Huang, X. Li, P. Yang, Y. Du, C. M. Goh and C. Lu, J. Mater. Chem. A, 2015, 3, 4195–4202 RSC.
- Y. J. Yuan, J. R. Tu, H. W. Lu, Z. T. Yu, X. X. Fan and Z. G. Zou, Dalton Trans., 2016, 45, 1359–1363 RSC.
- L. C. Song, F. X. Luo, B. B. Liu, Z. C. Gu and H. Tan, Organometallics, 2016, 35, 1399–1408 CrossRef CAS.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- J. Bian, J. Feng, Z. Zhang, Z. Li, Y. Zhang, Y. Liu, S. Ali, Y. Qu, L. Bai, J. Xie, D. Tang, X. Li, F. Bai, J. Tang and L. Jing, Angew. Chem., Int. Ed., 2019, 58, 10873–10878 CrossRef CAS PubMed.
- J. Bian, J. Feng, Z. Zhang, J. Sun, M. Chu, L. Sun, X. Li, D. Tang and L. Jing, Chem. Commun., 2020, 56, 4926–4929 RSC.
- J. Bian, L. Sun, Z. Zhang, Z. Li, M. Chu, X. Li, D. Tang and L. Jing, ACS Sustainable Chem. Eng., 2021, 9, 2400–2408 CrossRef CAS.
- H. Zheng, J. Z. Ou, M. S. Strano, R. B. Kaner, A. Mitchell and K. Kalantar-Zadeh, Adv. Funct. Mater., 2011, 21, 2175–2196 CrossRef CAS.
- B. Li, L. Q. Sun, J. Bian, N. Sun, J. W. Sun, L. Q. Chen, Z. J. Li and L. Q. Jing, Appl. Catal., B, 2020, 270, 118849 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. Sun, J. Bian, J. Li, Z. Zhang, Z. Li, Y. Qu, L. Bai, Z. D. Yang and L. Jing, Appl. Catal., B, 2020, 277, 119199 CrossRef CAS.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- S. Roy and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 12180–12184 CrossRef CAS PubMed.
- G. Liu, Y. Wang, Y. Zhou, J. Cao, M. Yuan and H. Lv, J. Colloid Interface Sci., 2021, 594, 658–668 CrossRef CAS PubMed.
- Y. Wang, Y. Zhu, L. C. Sun and F. Li, ACS Appl. Mater. Interfaces, 2020, 12, 41644–41648 CrossRef CAS PubMed.
- P. Kumar, A. Kumar, B. Sreedhar, B. Sain, S. S. Ray and S. L. Jain, Chem. – Eur. J., 2014, 20, 6154–6161 CrossRef CAS PubMed.
- A. Kumar, P. K. Prajapati, M. S. Aathira, A. Bansiwal, R. Boukherroub and S. L. Jain, J. Colloid Interface Sci., 2019, 543, 201–213 CrossRef CAS PubMed.
- P. K. Prajapati and S. L. Jain, Dalton Trans., 2019, 48, 4941–4948 RSC.
- P. Kumar, A. Kumar, C. Joshi, R. Singh, S. Saran and S. L. Jain, RSC Adv., 2015, 5, 42414–42421 RSC.
| This journal is © The Royal Society of Chemistry 2022 |