
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Main group metal polymerisation catalysts
Eszter
Fazekas
 ,
Phoebe A.
Lowy
,
Maisarah
Abdul Rahman
,
Anna
Lykkeberg
,
Yali
Zhou
,
Raju
Chambenahalli
and
Jennifer A.
Garden
*
,
Phoebe A.
Lowy
,
Maisarah
Abdul Rahman
,
Anna
Lykkeberg
,
Yali
Zhou
,
Raju
Chambenahalli
and
Jennifer A.
Garden
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: j.garden@ed.ac.uk
First published on 10th October 2022
Abstract
With sustainability at the forefront of current polymerisation research, the typically earth-abundant, inexpensive and low-toxicity main group metals are attractive candidates for catalysis. Main group metals have been exploited in a broad range of polymerisations, ranging from classical alkene polymerisation to the synthesis of new bio-derived and degradable polyesters and polycarbonates via ring-opening polymerisation and ring-opening copolymerisation. This tutorial review highlights efficient polymerisation catalysts based on Group 1, Group 2, Zn and Group 13 metals. Key mechanistic pathways and catalyst developments are discussed, including tailored ligand design, heterometallic cooperativity, bicomponent systems and careful selection of the polymerisation conditions, all of which can be used to fine-tune the metal Lewis acidity and the metal–alkyl bond polarity.
 Back (left to right): Anna Lykkeberg, Eszter Fazekas, Raju Chambenahalli, Phoebe Lowy. Front (left to right): Maisarah Abdul Rahman, Jennifer Garden, Yali Zhou | Dr Eszter Fazekas completed her undergraduate degree in Pharmaceutical Chemical Engineering at the Budapest University of Technology and Economics in Hungary. Her PhD at the University of Edinburgh focused on the design of organometallic catalysts for polymerisation and CO2 coupling, working with Dr Jennifer Garden and Prof. Michael Shaver. After a postdoctoral position at Heriot-Watt University, she returned to the Garden Group to work on the metal-catalysed synthesis of block copolymers. Phoebe Lowy graduated from Cardiff University in 2019 with an MChem in Chemistry. In her third year she spent a year with Dr Victoria Blair at Monash University in Melbourne, where she worked on the synthesis of phosphindoles and their alkali metal mediated metalation reactions. Phoebe joined the Garden group in 2020, through the SOFI2 CDT. She is developing synthetic routes towards heterotrimetallic complexes with applications in ring-opening (co)polymerisation. Maisarah Abdul Rahman graduated with a BSc in Chemistry from the University of Bristol in 2018. She joined the Garden group at the University of Edinburgh in 2019 for her MRes degree, working on the design of organometallic salen complexes as catalysts for the ROP of cyclic carbonates. She re-joined the Garden group for her PhD studies in 2021, and her research focuses on developing ProPhenol complexes for the ring-opening polymerisation of oxygenated monomers. Anna Lykkeberg studied Chemistry at the University of Edinburgh in the UK, and graduated with an MChem in 2020 after spending a year in industry. She started her PhD studies in 2020 with the SOFI2 CDT and joined Dr Jennifer Garden's group working on developing environmentally degradable hard/soft block copolymers in partnership with Croda. Yali Zhou graduated with an MSc in Medicinal and Biological Chemistry from the University of Edinburgh (2018). In the same year she started her PhD studies under the supervision of Dr Jennifer Garden. Her research focuses on the design and synthesis of novel homo- and heterometallic catalysts for the ring-opening polymerisation of cyclic esters. Dr Raju Chambenahalli obtained his MSc in Chemistry from Tumkur University. He subsequently obtained his PhD from the Indian Institute of Science Education and Research (Thiruvananthapuram, India) under the guidance of Dr Ajay Venugopal. His doctoral research focused on cationic zinc hydrides for CO2 reduction. His current research focuses on the design and synthesis of novel catalysts for block copolymers. Dr Jennifer Garden received her MSc (1st Class Hons, 2010) and PhD (2014) from the University of Strathclyde, the latter under the direction of Prof. Robert Mulvey. This was followed by two years as a postdoctoral researcher with Prof. Charlotte Williams at Imperial College London. In 2016, Jennifer joined the University of Edinburgh as the first recipient of the Christina Miller Fellowship, followed by a Ramsay Memorial Trust Fellowship, L’Oréal-UNESCO for Women in Science UK & Ireland Fellowship, and UKRI Future Leaders Fellowship. Her research combines different fields of chemistry including catalyst development and sustainable polymer synthesis. |
Key learning points(1) Main group metal catalysts have been used in a wide range of polymerisations including alkene and ring-opening (co)-polymerisations, and potentially offer a more sustainable alternative to transition metals.(2) Catalyst activity and selectivity can be optimised through choice of metal(s), ligand design and polymerisation conditions. (3) There are a wide range of accessible and effective main group catalyst systems, including combinations of mono-/multimetallic, homo-/heterometallic and mono-/bicomponent systems. (4) Metal–alkyl bond polarity is a key modulator for alkene polymerisation, with more polar metal–alkyl bonds typically showing enhanced catalytic activity. (5) Tuning the Lewis acidity of the metal is important for ring-opening (co)-polymerisation catalyst activity, as this influences the monomer coordination and nucleophilicity of the metal–oxygen bond responsible for monomer attack/insertion. |
Introduction
Main group metals have been extensively used in polymerisation reactions for over half a century. Several main group metal-based initiators have played a pivotal role in the early development of new polymeric materials, including polystyrene, polylactic acid (PLA) and polycarbonates (Scheme 1).1–3 Since these initial discoveries, main group catalysts have continued to play a key role in the production of commercial polymers. For example, tin octanoate (SnOct2) is used in the industrial production of PLA, while poly(propylene carbonate) (PPC) is produced using zinc glutarate catalyst systems.4,5 Importantly, in most polymerisations the organometallic complex is not regenerated in its original form, therefore it is sometimes referred to as an ‘initiator’ rather than a ‘catalyst’. Some metal complexes are used with an external initiator (true catalysts, as the structure of the organometallic complex is unchanged), whereas for others, part of the metal complex initiates the polymerisation (thus modifying the structure of the metal complex, and not a true catalyst). However, some organometallic complexes can follow a mixture of these different mechanisms, and so for simplicity, this review will use the term ‘main group metal catalysts’. It is also worth noting here that zinc is classed as a main group metal in spite of its position in the d-block of the periodic table, due to its 4s2 3d10 electronic configuration.6,7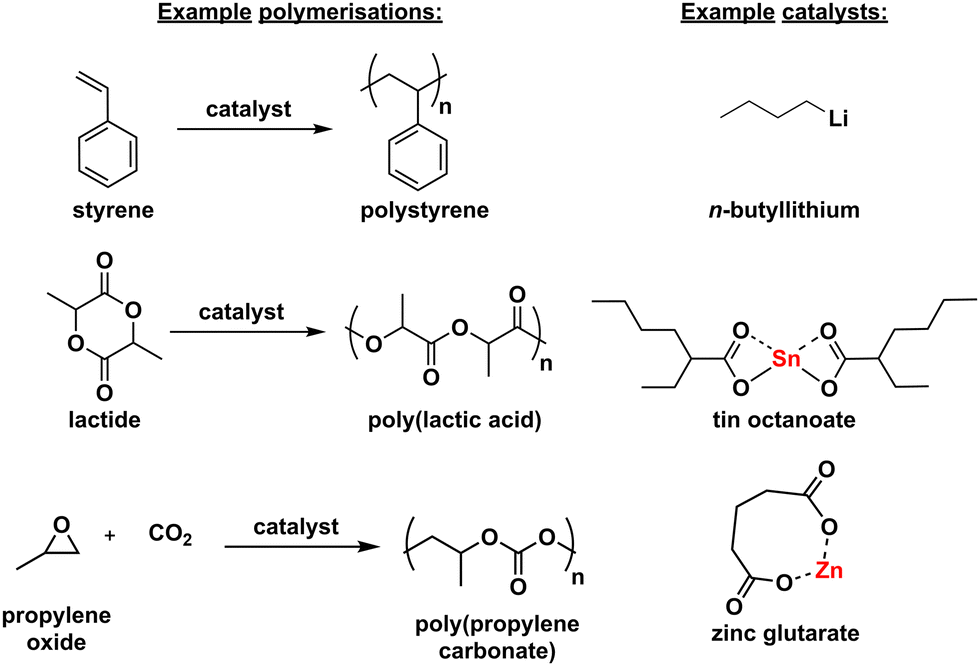 | ||
| Scheme 1 Example polymerisations using main group metal catalysts. | ||
Recent advances in mechanistic understanding and the design of new main group catalysts have delivered improvements in many different polymerisations. This review covers exciting and topical developments in polymerisation processes that are commonly catalysed by main group metal complexes (Scheme 2), with a particular focus on alkene polymerisation, the ring-opening polymerisation (ROP) of cyclic esters/cyclic carbonates, and the ring-opening copolymerisation (ROCOP) of epoxides with anhydrides or CO2.
 | ||
| Scheme 2 General polymerisation pathways catalysed by main group metal complexes. | ||
In each of these three fields, catalyst development has included both heterogeneous and homogeneous systems. Heterogeneous catalysts, which can be found in a separate phase (typically solid) from the monomers (typically liquid or gas) are often preferred in industry due to enhanced catalyst stability, as well as easier separation and reusability. Homogeneous catalysts remain in the same phase as the monomers (typically in solution), and can be stabilised on a solid carrier material to improve the catalyst stability and reusability, which can help to avoid contamination of the polymer product with traces of the catalyst. One of the key advantages of homogeneous catalysts is that they typically feature discreet, well-defined active sites, facilitating mechanistic understanding for informed catalyst design and improved performance. Thus, significant research efforts have focussed on homogeneous polymerisation catalyst development using metals from all blocks of the periodic table, with a variety of different mechanistic pathways including anionic, cationic and coordination–insertion mechanisms (see Schemes 3–6 for examples).
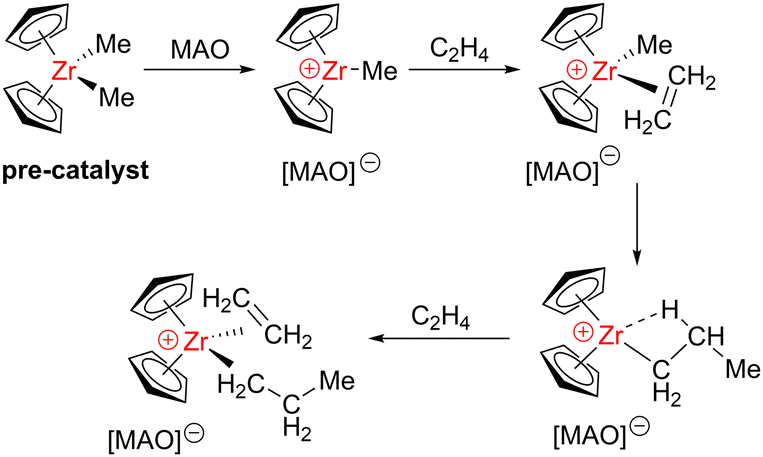 | ||
| Scheme 3 Coordination–insertion mechanism of ethylene polymerisation using a zirconocene catalyst and MAO co-catalyst. | ||
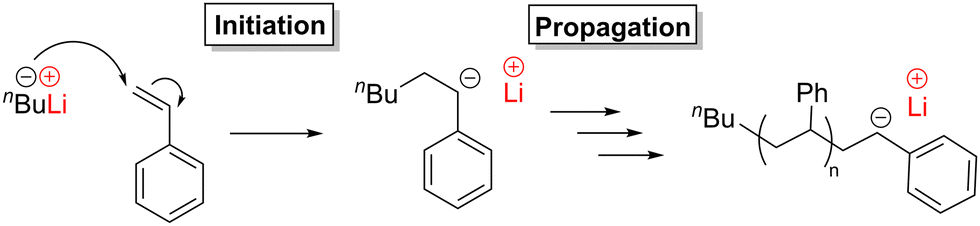 | ||
| Scheme 4 Anionic polymerisation of styrene catalysed by nBuLi. | ||
 | ||
| Scheme 5 Cationic polymerisation of isobutene initiated by (a) zinc and (b) aluminium complexes. | ||
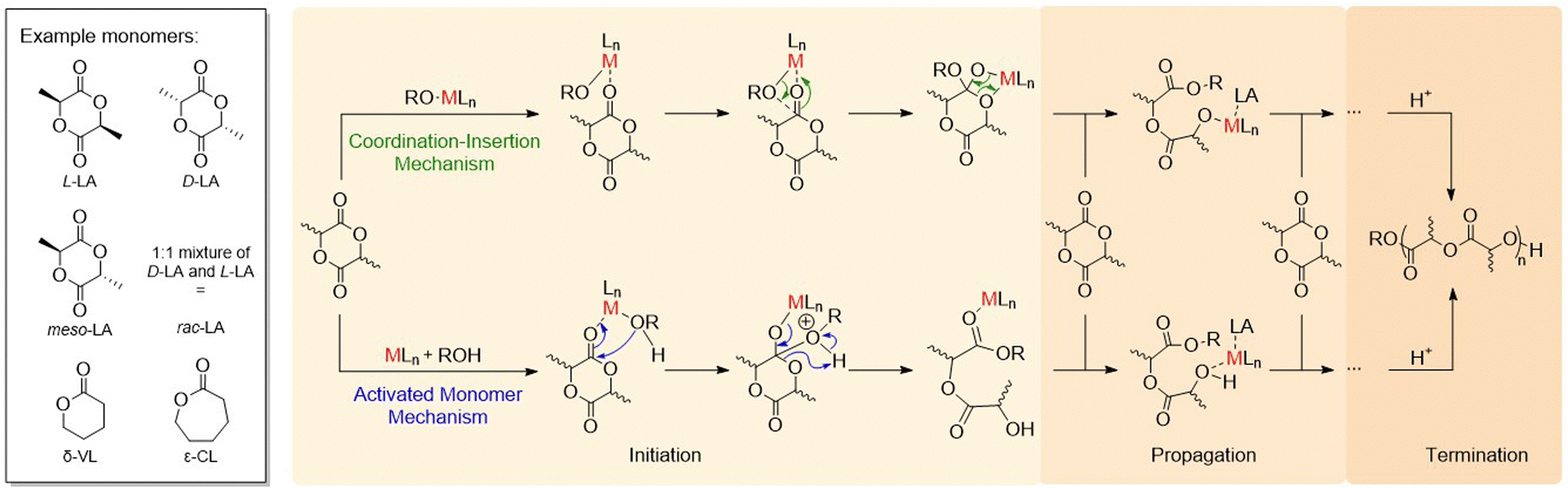 | ||
| Scheme 6 Key monomers (left) and mechanisms (right) for the ROP of lactones: coordination–insertion mechanism (top right); activated monomer mechanism (bottom right). L refers to ligand, M refers to metal and RO refers to either the initiator or the growing polymer chain. | ||
Transition metal complexes have been widely used in polymerisation processes, generally showing high reaction rates and selectivities. However, some transition metals are scarce (thus expensive) and can impose toxicity risks when embedded in the polymer products. Promising initial polymerisation studies have also been reported using f-block metals, but these catalysts can be challenging to prepare. Metal-free organocatalysts (including frustrated Lewis pairs) have been widely studied and can initiate polymerisations via the nucleophilic attack of a monomer from a lone electron pair on a heteroatom (e.g. nitrogen), yet can also suffer from toxicity issues.8 Developments in heterogeneous catalysts,9,10 transition- and f-block metal-based catalysts,11,12 frustrated Lewis pairs8 and organocatalysts13 have been extensively reviewed elsewhere. This review will therefore focus on main group metal catalysts as inexpensive, earth-abundant and low-toxicity alternatives that are economically and environmentally attractive.
Polymerisation catalyst performance can be determined using several different metrics. High activities are crucial for industrially relevant catalysts, which are typically measured using turnover numbers (TON, the number of monomer units converted by one mole of catalyst for a homopolymer), turnover frequencies (TOF, the TON achieved in a defined unit of time) or the observed reaction rate constant (kobs). Efficient catalysts can deliver control over the polymer properties, including chain length (number average molecular weight, Mn) and molecular weight distribution (dispersity, Đ). These values can be determined using size exclusion chromatography (SEC) – a technique that separates polymer chains by their hydrodynamic volume (size). Well-controlled polymerisations give more uniform chains with similar Mn values and a narrow dispersity (i.e. a value of Đ close to 1.0), while a broad dispersity indicates a larger variety in Mn that can lead to more varied polymer properties. The relative arrangement of adjacent stereocentres along the polymer chain is defined as the tacticity, which can be well-controlled (e.g. isotactic or syndiotactic) or random (atactic) (Fig. 1). The tacticity can be quantified through the relative intensity of 1H or 13C NMR resonances that are characteristic for diads, triads or tetrads (which refer to two, three or four adjacent stereocentres respectively, Fig. 1). The tacticity is described using probability values (0 < P < 1; Psyndio + Piso = 1.0), where Piso and Psyndio refer to the probability of isotactic and syndiotactic linkages, respectively.4 For example, a perfectly isotactic polymer would have a Pi value of 1.0 (and a Ps value of 0.0), whereas a perfectly syndiotactic polymer would have a Ps value of 1.0. Importantly, the tacticity can significantly influence the thermal and mechanical properties of a polymer, such as rigidity/flexibility, crystallinity, melting point and the glass transition temperature (Tg), which all determine the possible applications of the polymer material.4
 | ||
| Fig. 1 Tacticity patterns in polypropylene highlighting the stereochemistry of the chiral centres (R/S) and the relationship between adjacent stereocentres (isotactic, i or syndiotactic, s). | ||
This tutorial review focuses on developments in main group metal-catalysed polymerisations, and highlights interesting examples from across the periodic table. Due to the increasingly pressing challenge of plastic waste accumulation, examples of main group catalysts to prepare renewable and biodegradable polymers such as polyesters and polycarbonates are emphasised. These catalyst systems not only exploit the advantages of main group metals (low cost and generally low toxicity) but can also offer excellent activity and control over the polymer microstructure. Instead of a comprehensive summary, this review aims to showcase examples that feature unique activities and stereoselectivities or have delivered mechanistic understanding key to designing the next generations of main group catalysts for alkene polymerisation, ROP and ROCOP.
Main group catalysts for alkene polymerisation
Some of the earliest alkene polymerisations were achieved using main group metal catalysts but since then, metal-catalysed alkene polymerisation has been dominated by early transition metal catalysts. Transition metals are typically more efficient in activating C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, as inserting a double bond into a metal–alkyl bond is more energetically demanding for main group metal complexes.14 Challenges with the synthesis and handling of air- and moisture-sensitive main group metal–alkyl catalysts can further hinder their industrial applications. However, main group species have played a significant role as co-catalysts, when used in combination with transition metal catalysts. For example, the ubiquitous Ziegler–Natta catalysts, which are typically based on Group 4 (Ti, Zr or Hf) complexes and organoaluminium co-catalysts, have been a cornerstone of ethylene and propylene polymerisation since the 1950s.15 Trialkylaluminium (AlR3) or methylaluminoxane (MAO) compounds are used to activate these transition metal pre-catalysts towards initiation, by forming cationic transition metal–alkyl species that follow a coordination–insertion mechanism (Scheme 3). While the exact structure of MAO is still debated, a widely accepted opinion is that it consists of a mixture of aluminoxanes and methylaluminium species.16 However, it is important to note that catalysts based solely on main group metals have also been reported for alkene polymerisation, including examples from both the s- and p-block.
C double bonds, as inserting a double bond into a metal–alkyl bond is more energetically demanding for main group metal complexes.14 Challenges with the synthesis and handling of air- and moisture-sensitive main group metal–alkyl catalysts can further hinder their industrial applications. However, main group species have played a significant role as co-catalysts, when used in combination with transition metal catalysts. For example, the ubiquitous Ziegler–Natta catalysts, which are typically based on Group 4 (Ti, Zr or Hf) complexes and organoaluminium co-catalysts, have been a cornerstone of ethylene and propylene polymerisation since the 1950s.15 Trialkylaluminium (AlR3) or methylaluminoxane (MAO) compounds are used to activate these transition metal pre-catalysts towards initiation, by forming cationic transition metal–alkyl species that follow a coordination–insertion mechanism (Scheme 3). While the exact structure of MAO is still debated, a widely accepted opinion is that it consists of a mixture of aluminoxanes and methylaluminium species.16 However, it is important to note that catalysts based solely on main group metals have also been reported for alkene polymerisation, including examples from both the s- and p-block.
Reactive organoalkali metal catalysts for olefin polymerisation
Alkali metal alkyl compounds are well-known to initiate anionic polymerisations, due to the metal–alkyl bonds (e.g. R–Li) being highly polarised and thus prone to forming anionic R− species that can perform nucleophilic attack. For example, nBuLi polymerises ethylene, albeit with moderate activity and generating low molecular weight polymers. This poor reactivity was attributed to the lack of substituent groups on the monomer, as alkene substituents can stabilise the growing (propagating) polymer chain. Thus, stabilised monomers such as conjugated alkenes (e.g. styrene and butadiene) or alkenes featuring polar moieties (e.g. methyl methacrylate) can be rapidly polymerised by alkyllithium initiators.17 For example, styrene can be polymerised using nBuLi, albeit with poor control over Mn (broad dispersities) and the stereochemistry (atactic polystyrene) (Scheme 4). The polymerisation control was subsequently improved through a variety of methods,17 which included replacing nBuLi with sBuLi,18 lowering the reaction temperature (−30 °C) and using additives such as ligands, water or alkali metal salts (e.g. LiOtBu).19 Importantly, these methodologies became ‘living’, which refers to a precisely controlled polymerisation process without chain transfer and termination that can proceed until all available monomer is consumed. Consequently, these developments afforded polystyrene with fine-tuned molecular weights, dispersities and tacticities, which improved the thermomechanical properties. Nevertheless, commercial polystyrene is mainly produced through free radical polymerisation; the relatively high cost of anionic polymerisation (due to the use of solvents, monomer purification, cooling and expensive initiators) outweighs the advantages of well-controlled nBuLi-based catalyst systems.20 The reactivity of alkyllithium initiators can also be boosted by metal additives such as ZnEt2, MgBu2 or Mg(HMDS)2 (where HMDS is bis(trimethylsilyl)amide).21–23 For example, while using either nBuLi or Mg(HMDS)2 alone gave only trace conversion of methyl methacrylate, combining these two species in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio gave 90% conversion under the same reaction conditions. Deeper mechanistic studies revealed the presence of mixed-metal (heterometallic) species. While heterometallic catalysts based solely on main group metals are far rarer in alkene polymerisation than in ROP or ROCOP, these studies indicate that heterometallic main group catalysts may be an untapped area in alkene polymerisation.
:1 ratio gave 90% conversion under the same reaction conditions. Deeper mechanistic studies revealed the presence of mixed-metal (heterometallic) species. While heterometallic catalysts based solely on main group metals are far rarer in alkene polymerisation than in ROP or ROCOP, these studies indicate that heterometallic main group catalysts may be an untapped area in alkene polymerisation.
Alkaline earth metal catalysts for alkene polymerisation
Alkaline earth metal complexes can also directly initiate anionic polymerisations. Gibson and co-workers employed the first discrete and well-defined Mg complex for the living polymerisation of methyl methacrylate. The binuclear enolate-bridged MgBDI catalyst 1 (Fig. 2, where BDI is β-diiminate) showed excellent activity under mild conditions, giving up to 95% monomer conversion in 10 minutes (THF solvent, −30 °C) with well-controlled molecular weights, narrow dispersities (Đ = 1.1) and high syndiotacticity.24 The living nature of the polymerisation was supported by the linear relationship observed between Mn and monomer conversion. Solution-state NMR studies suggested that the dimeric complex dissociates into the monomeric analogue in THF solvent, which was postulated to act as the catalytically active species. | ||
| Fig. 2 Structures of MgBDI complex 1, Ca-half sandwich complex 2 and Ba complexes 3 and 4. | ||
Discrete organometallic complexes based on heavier alkaline earth metals (Ca, Sr and Ba) have also been investigated as alkene polymerisation catalysts.25 Upon descending Group 2, these complexes become increasingly nucleophilic compared to lighter alkaline earth metals such as Be and Mg, due to the enhanced polarity of the metal–alkyl bonds, which renders the activation of alkene monomers more feasible.
A series of Ca fluorenyl catalysts for the living polymerisation of styrene were reported by Harder and co-workers (2, Fig. 2).26–28 Vacant sites for monomer coordination were generated by the dissociation of THF from Ca. The ligand design was crucial in controlling the polymer tacticity: increasing the steric bulk of the fluorenyl substituents gave a significant syndiotactic bias in the polystyrene, which delivers industrially advantageous properties such as low density and a high melting point (273 °C).26 This complex combines structural motifs of catalysts used for classical anionic polymerisation (i.e. the ionic character of nBuLi) and coordination–insertion polymerisation (i.e. the cyclopentadienyl ligand of titanocenes). Group 2 metals therefore show some promise to bridge between the conventional alkene polymerisation mechanisms of anionic polymerisation (Group 1 metals) and coordination–insertion polymerisation (transition metals). In spite of the stereocontrol, the polymers displayed relatively broad dispersities (Đ = 1.4–2.9). Furthermore, butadiene polymers and styrene/isoprene block copolymers could also be synthesised using this methodology.
Homoleptic and heteroleptic barium benzyl complexes (3 and 4 respectively, Fig. 2) have shown high activity in the anionic polymerisation of styrene at 40 °C.29 Using catalyst 3 the observed Mn values (90 kg mol−1) gave good agreement with theoretical values, assuming a bivalent mechanism, where two polystyrene chains simultaneously grow on the metal centre, initiated by both triphenylpropyl groups. The relatively broad dispersity (for a living polymerisation, Đ = 1.2) of the obtained polymers was attributed to a slow initiation step, caused by the presence of four sterically demanding phenyl groups close to the Ba centre. Atactic polymer chains were detected, which was attributed to the rapid exchange of benzyl groups on the Ba centre, indicating that the growing polymer chains might have also been exchanging with each other leading to epimerisation (interconversion of the stereocentres along the polymer backbone thus a loss of control over the tacticity).
Zinc and p-block metal catalysts for alkene polymerisation
When combined with tertiary butyl halides, [Zn(C6F5)2] initiated isobutene polymerisation (which can incorporate up to 15% of an isoprene comonomer).30 These isobutene–isoprene copolymers are commonly known as butyl rubber and are widely used in the manufacturing of tubes, tyres, gloves, chewing gum and other commodities. Unlike the anionic polymerisation mechanism reported for s-block metal initiators, these reactions undergo cationic polymerisation, where the positive charge resides on the penultimate carbon of a growing polymer chain. Diarylzinc reagents can abstract the halide from alkyl halides, thereby creating a carbocation which subsequently inserts isobutene to form polyisobutene (Scheme 5a). These reactions are usually carried out at low temperatures (<−50 °C) and under inert conditions (free from air and moisture) owing to the instability of active carbocationic species.The equimolar combination of Cp2AlMe and B(C6F5)3 generates cationic [Cp2Al]+ species, which can generate high molecular weight polyisobutene (318 kg mol−1 at −30 °C) albeit with a broad dispersity (Đ = 1.8). The same compound could successfully copolymerise isobutene and isoprene with 2.7% isoprene incorporated. The authors proposed that the aluminium centre acts as a Lewis acid (an electron pair acceptor) and initiates the polymerisation, while the counter ion [MeB(C6F5)3]− assists in stabilising the positive charge created on the polymer chain (Scheme 5b).31
Equimolar mixtures of AlR3 (R = Me, Et) and co-catalysts (such as MAO, triarylboranes or salts of weakly coordinating borate anions), were reported to catalyse the homo- and copolymerisation of ethylene and propylene.32 A cationic aluminium alkyl compound, {MeC(NiPr)2}AlMe+, was used in ethylene polymerisation to give high molecular weight polyethylene (Mn = 272 kg mol−1) with a broad molecular weight distribution (Đ = 3.3).33 Mononuclear cationic aluminium complexes, based on pincer ligands (5, Fig. 3)34 or Schiff bases with a pendant arm (6, Fig. 3),35 were also shown to catalyse ethylene polymerisation with Mn values ranging from 2.4 to 7.8 kg mol−1.
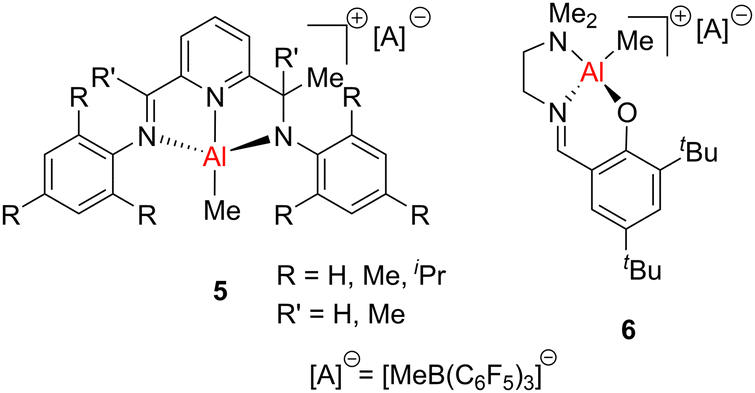 | ||
| Fig. 3 Cationic aluminium compounds for olefin polymerisation. | ||
Zinc and lighter congeners of p-block metal compounds (Al and Ga) have been studied for the synthesis of highly reactive polyisobutene (HR-PIB). HR-PIBs are low molecular weight (Mn = 0.5–5 kg mol−1) polyisobutenes featuring reactive vinylidene end groups (i.e. exo-olefin end groups, preferably >75% content), which are essential intermediates in the preparation of additives for fuel and lubricants. Ethylzinc chloride in combination with tBuCl was shown to successfully produce HR-PIBs featuring up to 90% exo-olefin groups at room temperature, with Mn ranging from 10 to 29 kg mol−1 (Đ = 1.6–2.5).36 AlCl3, GaCl3, alkyl aluminium dichlorides (RAlCl2, R = Et, iPr, iBu), and alkoxy aluminium dichlorides ((RO)AlCl2, R = Bu, Hex, iPr) combined with dialkyl ethers (R′2O; R′ = iPr, tBu) were also studied as catalyst systems to produce HR-PIBs (Mn = 0.8–2.0 kg mol−1, Đ > 3.0). These systems yield polyisobutenes that contain a high content of reactive exo-olefin end groups (80–95%).37 Here, the metal precursors act as initiators while the role of the dialkyl ether is to facilitate rapid abstraction of the β-proton of the methyl group in a growing polymer chain, thereby creating exo-olefin end groups.
Summary
Catalysts based solely on main group metals remain underexplored in alkene polymerisation compared to those based on transition metals. While main group metals are most commonly used as co-catalysts in Ziegler–Natta type alkene polymerisations, a handful of promising alkene polymerisation catalysts featuring only main group metals have been reported, where the mechanisms differ from the typical coordination–insertion mechanism (Schemes 3–5). In the s block, highly polar organometallic initiators based on lithium or heavier alkaline earth metals follow anionic mechanisms. While the polymerisation mechanism of some cationic organozinc (d block) and organoaluminium (p block) complexes is not completely understood, they are unlikely to follow a coordination–insertion mechanism yet it is challenging to disprove that the catalytic activity originates from trace transition metal impurities. There has been a recent explosion in the development of main group complexes capable of performing reactions that were thought to lie solely in the domain of transition metal chemistry. While alkene polymerisation catalysts based solely on main group metals remain scarce, some efficient examples have been reported. The highlighted results may serve as a base to better exploit abundant, inexpensive and low-toxicity main group metals and thus provide more sustainable alternatives to some transition metal-catalysed processes for polyolefin synthesis.Main group catalysts for ring-opening polymerisation
Main group metal catalysts from across the periodic table have been reported for the ROP of various monomers, including cyclic esters (lactones), cyclic ethers (epoxides) and amides (lactams).38 As most of this research has focused on lactone ROP, this section predominantly focuses on catalyst development for the ROP of lactones, including lactide (LA), ε-caprolactone (ε-CL) and δ-valerolactone (δ-VL) (Scheme 6, left). High-performance, degradable and biocompatible polyesters produced via lactone ROP have various applications spanning from packaging to biomedicine. To date, most research efforts in ROP have focused on lactide. Lactone ROP using main group organometallic catalysts is generally proposed to proceed via a coordination–insertion mechanism (Scheme 6, top right). The initiation step involves the Lewis basic (electron pair donor) monomer coordinating to a Lewis acidic (electron pair acceptor) metal centre, which activates the carbonyl group towards nucleophilic attack and ring-opening. The nucleophile is typically an alkoxide, alkyl, amido, or halide group bonded to the metal centre. Sequential coordination, ring-opening and insertion of subsequent monomers into the polymer chain leads to propagation and is followed by termination (typically by using a protic source such as water). Alternatively, ROP may operate through an activated monomer mechanism (Scheme 6, bottom right). Similarly to the coordination–insertion mechanism, the monomer is activated through metal coordination, yet in this case, nucleophilic attack occurs from an external protic nucleophile (commonly an alcohol or an amine). Monomer coordination and nucleophilic attack are key mechanistic steps in both pathways, and careful choice of the ligand and metal can enhance monomer coordination through increasing the metal Lewis acidity and providing accessible coordination sites.ROP initiated by organometallic catalysts can occur via “living” and/or “immortal” polymerisation (Fig. 4). “Living” ROP involves the continuous growth of polymer chains from an active site (initiator), with the chain-ends remaining active for further monomer insertion, until the reaction is terminated.39,40 Well-controlled living polymerisations can generate polymers with a narrow dispersity and targeted molecular weights. This can be especially advantageous in LA ROP, as many industrial applications require high molecular weight PLA with Mn values above 100 kg mol−1. Living ROP reactions can be instantly terminated upon the addition of protic compounds such as water or alcohol. Conversely, immortal ROP (iROP) tolerates the presence of protic species, which can act as both nucleophiles (i.e. external initiators) and chain transfer agents (CTAs). A CTA transfers the growing polymer chain to another molecule. For example, the reaction of a propagating metal–Opolymer chain (e.g. M–OP) with an exogeneous alcohol (H–OR) forms a new initiating species (M–OR) along with a dormant polymer chain (H–OP). The dormant polymer chain can be reactivated through deprotonation by another M–OP′ chain, to reform a propagating M–OP (along with dormant H–OP′). As each CTA can initiate a polymer chain, each catalyst facilitates the formation of more than one polymer chain; this can lead to enhanced catalyst productivity and typically generates polymers with narrow dispersities.39 However, it should be noted that not all metal-based initiators can tolerate the presence of large quantities of CTAs, which is the major drawback of iROP.
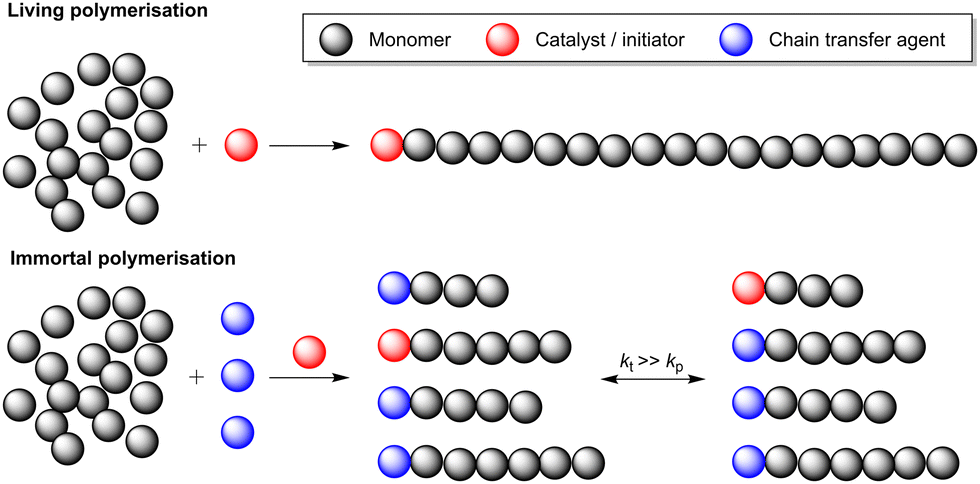 | ||
| Fig. 4 Living polymerisation and immortal polymerisation. kp and kt are rate constants of chain propagation and termination, respectively. | ||
Tin octanoate (SnOct2, Scheme 1), was amongst the earliest ROP initiators and is still the standard catalyst used industrially owing to its solubility in many lactones, stability in storage and good catalytic performance.41 Furthermore, SnOct2 can be used to produce high molecular weight PLA (Mn > 100–1000 kg mol−1) via the bulk polymerisation of LA, with the use of an alcohol co-initiator leading to activity enhancements. However, toxicity issues with SnOct2 have sparked a drive to explore alternative initiators based on less toxic metals while still maintaining good activity and control over the polymer properties.
Homometallic ROP catalysts based on s-block metals and zinc
Alkali and alkaline earth metal complexes have attracted much attention as initiators for lactone ROP. Not only are s-block metals typically earth-abundant, inexpensive and have low-toxicity but many are also highly Lewis acidic and oxophilic. These features make main group metal-based catalysts attractive candidates for the ROP of cyclic esters. Zinc has been included in this section because it is a divalent metal that bears several similarities to magnesium, including the ionic radii, coordination numbers and oxidation state. This section therefore covers some of the key developments in s-block and Zn catalysts for ROP.Alkali metal catalysts for ROP
Catalysts based on alkali metals (e.g. Li, Na, K) have shown great promise in lactone ROP to produce aliphatic polyesters. Simple alkali metal complexes such as butyllithium and lithium tert-butoxide can act as highly active initiators, yet often give poorly controlled polymerisations affording low molecular weight polymers with broad dispersities. This can result from side-reactions such as intra- or intermolecular transesterifications (Scheme 7). Simple alkali metal complexes also have a tendency to form aggregates, which can result in solubility issues. | ||
| Scheme 7 Intra- and intermolecular transesterification reactions of LA, which can lead to decreased molecular weights and broad dispersities. RO/R′O refers to either the initiator or the polymer chain. | ||
These drawbacks can be overcome using carefully tailored ligands. For example, a series of alkali metal complexes supported by both bulky ligands and crown-ethers have been exploited in cyclic ester ROP achieving high catalytic activities, high molecular weights, narrow dispersities and high isoselectivities (Fig. 5). The alkali metal is sandwiched between the phenolate ligand and the crown-ether, and the confined space may prevent the metal coordinating to an ester group of the growing polymer chain thus minimising transesterification. The coordinated crown ether disfavours aggregation, thereby accelerating the reaction. Complexes 7a–7d and 8a–8d all catalysed the highly isoselective and living ROP of racemic (rac) LA.42 Potassium-based complex 8c was the most active with complete monomer conversion achieved in 1.5 minutes (toluene, 100 equiv. LA, 1 equiv. BnOH, 25 °C). The use of electron-rich ligands was shown to enhance the catalytic activity, with identical reactivity trends observed for the K complexes (8c > 8b > 8a) and the Na complexes (7c > 7b > 7a). This was attributed to the electron-donating nature of the methoxy (7c/8c) and tert-butyl (7b/8b) groups, which afford more negatively charged and thus basic phenoxy units to activate the nucleophilic alcohol through hydrogen bonding and thus accelerate ROP. However, complexes 7d and 8d, bearing the sterically most demanding substituents (two tert-butyl groups), showed lower catalytic activities due to the highly crowded metal centres. This suggests that both the ligand electronics and availability of coordination sites can affect the polymerisation performance. It is important to note that with other main group ROP catalysts, including Al-based examples, the use of electron-donating substituents can decrease catalyst performance by increasing the electron density on the metal centre, which decreases the Lewis acidity for monomer coordination and thus slows the propagation rate (vide infra). However, Na complexes (7a–7c) followed the same trend as the K analogues albeit with slightly lower catalytic activities on account of the lesser ionic character and thus lower Lewis basicity of the Na–O bond compared to the K–O bond. Increased ROP catalyst activities are often observed upon descending a group in the periodic table. This feature has also been attributed to the larger metals featuring a greater number of available coordination sites, which enhances monomer coordination and thus the propagation rate, albeit often at the cost of reduced (stereo)control. To the best of our knowledge, the highest PLA isoselectivity reported to date using alkali metal catalysts was achieved with potassium complex 8a at −70 °C, giving Piso = 0.94. Reduced polymerisation rate and improved polymerisation control often go hand-in-hand, thus a key target is to develop catalysts that deliver excellent activities as well as exquisite control.
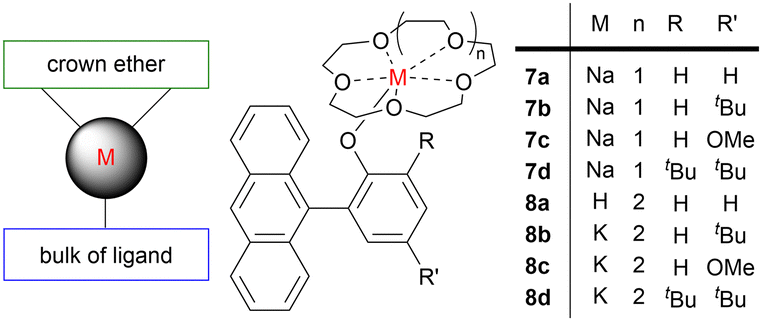 | ||
| Fig. 5 Structural representation of some alkali metal-based catalysts for cyclic ester ROP (M refers to metal: Na or K, left) and examples of alkali metal sandwich complexes (right). | ||
Alkaline earth metal and zinc catalysts for ROP
Recently, there has been an increased interest in alkaline earth metal-based ROP catalysts. The main challenge for developing these catalysts is the synthesis of stable and easy-to-handle complexes, as heteroleptic Group 2 complexes can undergo a “Schlenk equilibrium” that converts heteroleptic compounds of general formula LMX to homoleptic compounds ML2 and MX2. This reaction can become increasingly significant for the larger alkaline earth metals, thus most Group 2 catalysts to date have been based on Mg.Chiral oxazolyl aminophenoxide Mg complexes have been reported (9a–9b, Fig. 6), which display high activities and isoselectivities toward rac-LA ROP in the presence of co-initiator iPrOH (toluene, 25 °C).43 Using a racemic mixture of 9a and 9b as the catalyst system, extremely high TOF values (TOF ≤54000 h−1) were achieved along with isotactic stereoblock PLA (Piso = 0.80) of high molecular weights (Mn = 461 kg mol−1). Detailed NMR spectroscopic investigations indicated that both the ligand chirality and the dimeric structure of the active species contributed to the high isoselectivity.
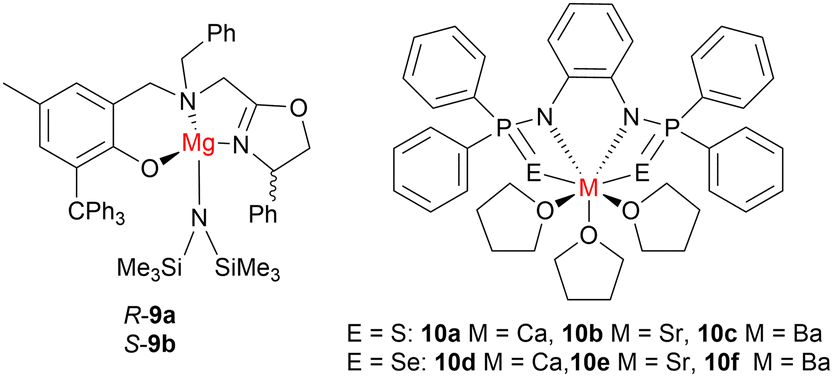 | ||
| Fig. 6 Examples of alkaline earth metal initiators for cyclic ester ROP. | ||
For larger alkaline earth metals, the use of sterically demanding monoanionic ligands is an efficient method of directing the Schlenk equilibrium towards the heteroleptic species. For example, Panda and co-workers reported alkaline earth metal complexes (10a–10f, Fig. 6) bearing bisphosphinoselenoic amine ligands [{Ph2P(Se)NH}2C6H4] for living rac-LA ROP (toluene, 25 °C).44 This was the first example of the ROP catalytic cycle being initiated via in-built phosphorous-chalcogen (S, Se) double bonds, in the absence of an external initiator. The highest activity was achieved using Ba complex 10f as an initiator, which gave complete conversion of 1000 equiv. of rac-LA in less than 10 minutes. Both the S- and Se-based complexes exhibited similar trends in terms of activity and stereoselectivity, with the larger alkaline earth metals achieving higher catalytic activities (Ba > Sr > Ca) but lower isoselectivities (Ca > Sr > Ba). Ca complex 10a provided the most stereocontrolled polymer (Piso = 0.87) while Ba complexes gave atactic PLA. Notably, the S-based ligands (10a–10c) showed higher stereoselectivity than the corresponding Se analogues (10d–10f). These observations highlight that isoselectivity can be achieved from two different modes: either by changing the active metal sites or by changing the ligand system. Additionally, all complexes (10a–10f) were extremely fast initiators for ε-CL ROP, allowing almost complete conversions of 1000 equiv. of monomer within 30 seconds.
Zinc catalysts have been extensively exploited in ROP chemistry, largely owing to the biocompatibility and excellent catalyst performance. While many active monometallic zinc catalysts have been developed, the majority of these are neutral complexes. An unusual example of a cationic zinc-lactate complex bearing a bis(phosphinimine) pincer ligand (11, Fig. 7) was reported as the first cationic complex to catalyse LA ROP at room temperature via a living coordination–insertion pathway.45 Complex 11 converted 180 equiv. rac-LA to polymer in 50 min (CD2Cl2 solvent), yielding slightly heterotactic-enriched PLA (Psyndio = 0.63) with narrow dispersities (Đ = 1.1–1.3) and targeted molecular weights. It is worth noting that in the absence of an exogeneous initiator, ROP was triggered by the reactive methyl O-lactate nucleophile of 11.
 | ||
| Fig. 7 Examples of mono- and bimetallic Zn catalysts for LA ROP. | ||
While many efficient monometallic zinc catalysts have been reported, bimetallic zinc catalysts often display significant activity enhancements in LA ROP. For example, Tolman, Hillmyer and co-workers reported a series of mono- and dinuclear zinc alkoxide complexes supported by a diiminophenolate ligand (12a–12b, Fig. 7).46 Notably, dimeric alkoxide complex 12b displayed high activity (96% conversion in 5 min), whereas the monomeric analogue 12a, without an alkoxide initiating/propagating group, was inactive towards rac-LA ROP. Complex 12b exhibited good control, producing PLA with narrow dispersities and high molecular weights (Mn < 130 kg mol−1, Đ ≈ 1.4) at low catalyst loadings (<0.1 mol% catalyst loading). Since these pioneering studies, a range of highly active bimetallic zinc catalysts have been reported for cyclic ester ROP. Of particular note are the dinuclear zinc complexes reported by Williams and co-workers (13a–13d, Fig. 7). Supported by diphenylamine-based macrocycle ligands, these complexes show exceptional activity in rac-LA ROP (TOF ≤60000 h−1, 0.1 mol% catalyst loading, 25 °C).47 Interestingly, the conformation of the ancillary macrocyclic ligand was shown to control both the Zn–Zn distance and the ROP activity. Di-zinc catalysts adopting a “folded” ligand conformation (13a) (TOF = 20 300 h−1) outperformed those with a “planar” conformation (13c) (TOF = 30 h−1) under identical conditions (THF, 0.1 mol% catalyst loading, 25 °C). The striking activity difference between these two conformations was attributed to enhanced accessibility of zinc active sites in the “folded” 13a complex, facilitating both monomer coordination and insertion simultaneously. Both Zn-alkoxide (Zn-OiPr) and Zn-bis(trimethylsilyl)amide (Zn-HMDS) initiators were investigated; higher polymerisation rates were observed using HMDS complexes 13a–13b while the alkoxide analogues 13c–13d displayed better control. Varying the Zn–Zn proximity by increasing the diamine chain length altered the activity; complex 13a was found to be 1.5 times faster than 13b, reflecting the higher efficiency of intermetallic cooperation stemming from the closer proximity of metal centres.
Homometallic ROP catalysts based on p-block metals
Across LA ROP, there is often a trade-off between high activity and high stereocontrol, and catalysts that can deliver both features are relatively rare. Significant research efforts in cyclic ester ROP have focussed on aluminium-based catalysts due to the earth-abundance, low toxicity and high Lewis acidity of this metal, and a particular focus has been placed on aluminium salen complexes. While the use of aluminium salen initiators has led to significant improvements in stereocontrol, these initiators typically require high catalyst loadings and long reaction times. Catalyst systems with heavier Group 13 metal complexes are still relatively rare, however, some examples have been reported in recent years.In 1996, Spassky et al. reported the stereoselective ROP of rac-LA, using a chiral binaphthyl Schiff base (also known as salen-type) aluminium alkoxide complex 14 (Fig. 8), to afford optically active isotactic PLA with 19% conversion (toluene, 70 °C).48 Narrow dispersities were obtained even at high conversions after an extended reaction time (90%, Đ = 1.0–1.1, >48 h), indicating minimal transesterification using the sterically hindered complex 14. Since this initial report, several Al–alkyl salen (see 15–17, Fig. 8 for representative examples) or alkoxide salen complexes have been developed for LA ROP.49,50 For example, bulky tBu ligand substituents can hinder monomer coordination and reduce the propagation rate, while a longer ligand backbone (diamine linker) can enhance the reactivity. The latter is attributed to more flexible ligands facilitating the formation of key transition states. The electronics of the ligand substituents are also important. For example, electron-withdrawing substituents (such as Cl) at the ortho-positions of the phenolate moiety can increase the Lewis acidity of the metal, enhancing monomer coordination and thus leading to enhanced propagation rates. Importantly, sterically demanding substituents such as tBuMe2Si (17) delivered high conversions as well as isoselective PLA (Piso ≤ 0.98), highlighting that there is a fine balance between activity and control when it comes to catalyst design. Furthermore, complex 17 could be used under solvent-free bulk polymerisation conditions and delivered well-controlled PLA with a high melting temperature (Tm ≤ 189 °C) and narrow dispersities (Đ = 1.0–1.1), even at high conversions and extended reaction times (98%, 1–2 h), indicating that minimal transesterification occurred.
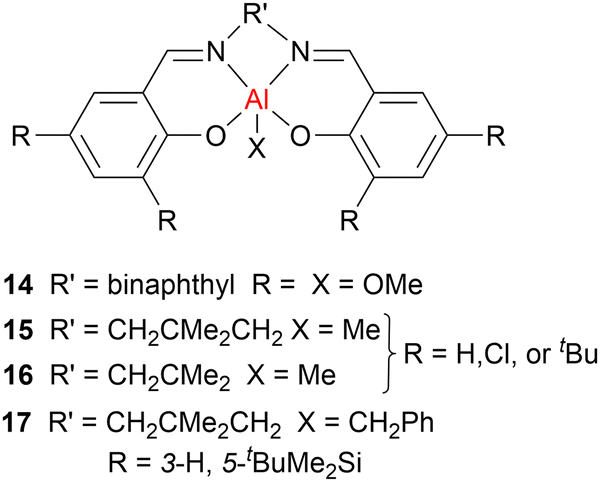 | ||
| Fig. 8 Examples of salen Al catalysts used in cyclic ester ROP. Cat+ refers to cation. | ||
Aluminium alkoxide complexes featuring trifluoromethyl substituents have also been reported for rac-LA and ε-CL ROP (for example 18, Fig. 9).51 High activities were observed in ε-CL polymerisation with TOF up to 200 h−1 (in THF, room temperature, 1 h). The experimental molecular weights matched the expected values even at a high monomer (M): initiator (I) ratio (M/I < 600). While low activity was observed for LA polymerisation in THF at room temperature (TOF ≈ 6 h−1), high activities were obtained under melt conditions (TOF ≤ 1260 h−1), yielding isotactic-enriched stereoblock PLA (Piso ≤ 0.87).
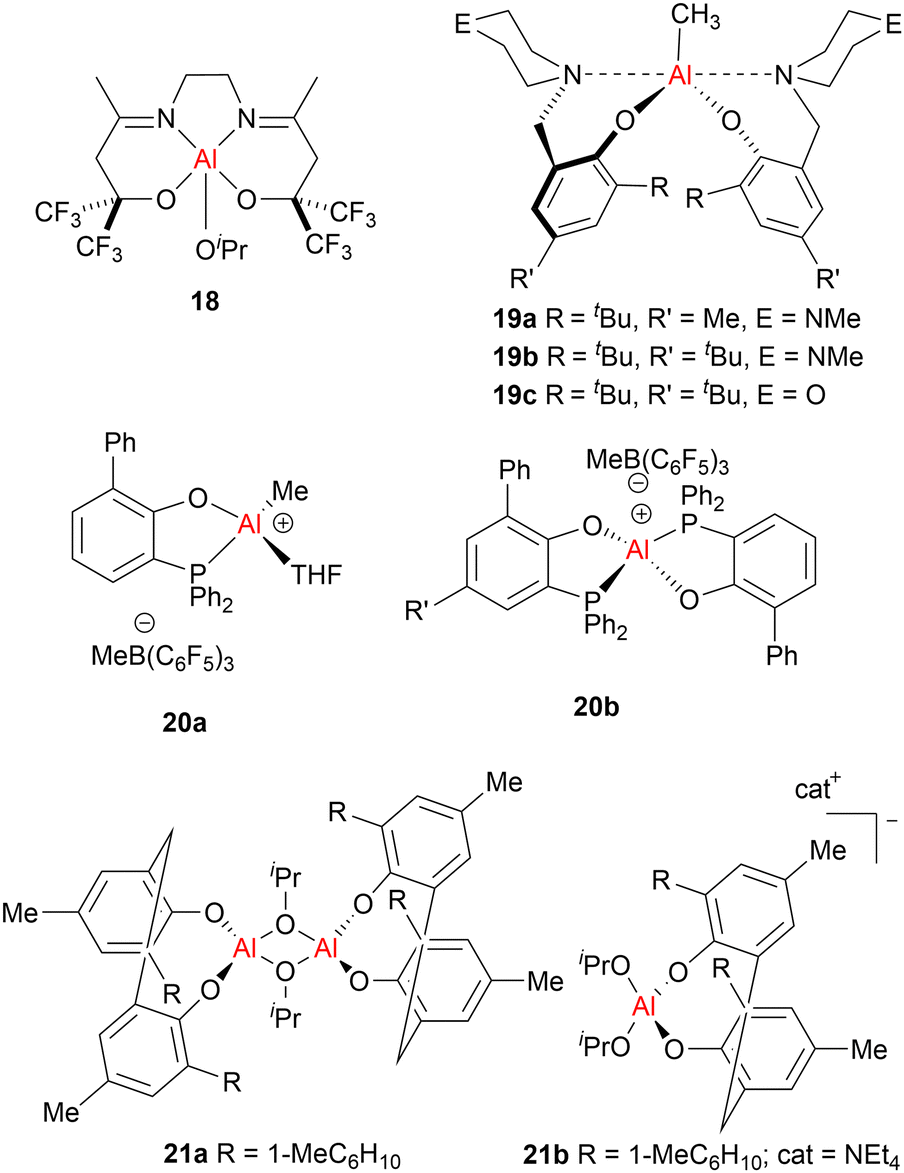 | ||
| Fig. 9 Examples of phenoxy-Al catalysts used in cyclic ester ROP and/or PO ROP. Cat+ refers to cation. | ||
A series of aluminium methyl complexes (19a–19c, Fig. 9) bearing monoanionic aminephenolate ligands, were reported by Kerton and co-workers.52 Complexes 19a–19c are efficient catalysts for ε-CL polymerisation in the presence of BnOH (TOF ≈ 1000 h−1, in toluene, 80 °C), yet were completely inactive for the ROP of rac-LA. In spite of the similarities between different cyclic esters, not all ROP catalysts are effective for multiple monomers. While this system typically gave PCL of the targeted Mn and narrow dispersities, suggesting a living polymerisation, a slight deviation from the expected Mn was observed at higher M/I ratios which suggests some termination by chain transfer. Contrary to some other reported aluminium alkoxide ROP catalysts, kinetic studies indicated the absence of an induction period which could imply that the polymerisation proceeds via an activated monomer mechanism instead of the commonly proposed coordination–insertion mechanism.
Cationic Al complexes (20a and 20b, Fig. 9) coordinated by bidentate O, P-phosphinophenolate ligands have also been developed for the ROP of ε-CL and propylene oxide (PO).53 These cationic aluminium complexes catalysed ε-CL polymerisation quantitatively in 2 h (>95%, in toluene, 75 °C). ROP initiation through ε-CL insertion into the Al–OPh bond exclusively yielded PCL capped with phosphinophenolate oxide {PPO} at the ester end of the PCL chain. This suggests that the {PPO}− moiety may play two roles: a supporting ligand and an initiating group in ε-CL ROP. PO polymerisation was catalysed readily at room temperature to afford atactic polypropylene oxide (>50% conversion, 15 min, CH2Cl2).
While the development of bimetallic aluminium ROP catalysts generally lags behind those based on zinc, Okuda and Braune demonstrated the first cooperative bimetallic aluminate complexes for the controlled ROP of propylene oxide (PO).54 In this study, neutral bis-aluminium complexes (21a, Fig. 9) and anionic aluminates (21b, featuring Al3+ surrounded by four anionic ligands to give a negatively charged [AlR4]− “ate” moiety) were unreactive towards the ROP of PO when used independently. However, combining neutral complex 21a with “ate” complex 21b resulted in good activity and control for PO polymerisation (77% conversion, CH2Cl2, 25 °C, 3 h). Supported by NMR studies, the cooperative behaviour of 21a and 21b was proposed to occur via PO activation by coordination to the neutral monomeric adduct of 21a, followed by alkoxy group transfer from the “ate” complex 21b.
While it has been well established that descending Group 1 of the periodic table can give enhanced ROP activity, the first heavier Group 13 catalysts for LA ROP were reported relatively recently. A series of Schiff base-supported gallium initiators (22a–22c, Fig. 10) were developed for LA and ε-CL ROP (toluene, 100 °C, with 4 equiv. BnOH).55 These initiators generated polymers with narrow dispersities (ĐPLA = 1.10–1.21, ĐPCL = 1.06–1.12) and PLA with enriched isotacticity (Piso ≤ 0.78). Although the tetranuclear Ga complexes displayed good control in ROP, direct comparison of activity vs. the Al complexes is not possible due to the different reaction conditions employed (temperature, catalyst loading, solvent, monomer concentration and purity).
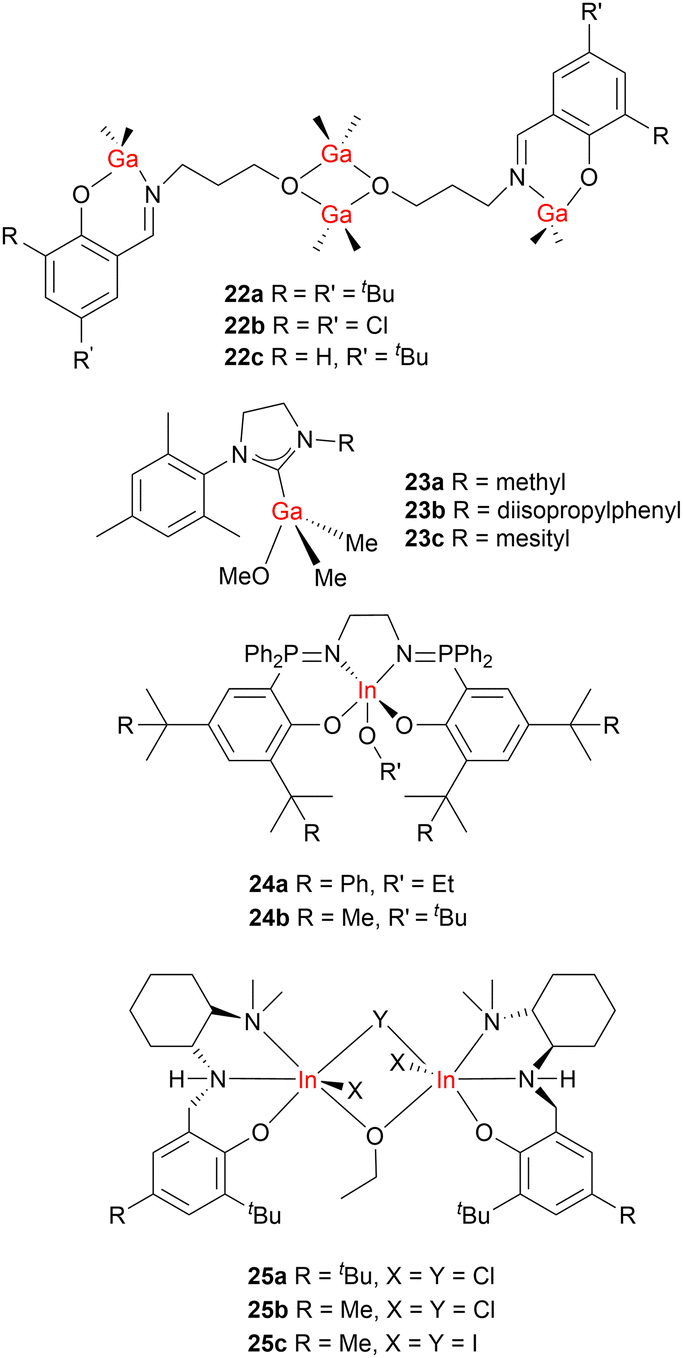 | ||
| Fig. 10 Examples of heavier Group 13 metal (Ga and In) catalysts for cyclic ester ROP. | ||
Recently, Horeglad and co-workers reported a series of alkylgallium alkoxide complexes stabilised by a range of N-heterocyclic carbenes (NHCs) including SI(Me–Mes), SI(Dipp–Mes) or SI(Mes–Mes)) (23a–23c, Fig. 10). These complexes were highly active and stereoselective in rac-LA ROP, even under mild conditions (in CH2Cl2, −20 °C).56–58 The NHC influences both the ROP activity and selectivity, as the efficient interaction of rac-LA with the Ga-methoxide unit depends on space available for monomer insertion (reactivity: SI(Me–Mes) (23a) > SI(Dipp–Mes) (23b) ≫ SI(Mes–Mes) (23c); Piso-max = 0.83, 0.78 and 0.56, respectively).
The first indium-phosphasalen complexes displaying good performance for rac-LA polymerisations were reported by Williams and co-workers (24a–24b, Fig. 10).59 In the absence of any chiral additives or ligands, 24a–24b rapidly produced isotactic-enriched PLA even with relatively low catalyst loadings (0.2 mol% catalyst loading). Complex 24a displayed higher isoselectivity than 24b (Piso = 0.92 vs. 0.75), which was attributed to the steric bulk of the ortho-cumyl substituents providing more controlled monomer orientation to the metal centre. Mehrkhodavandi and co-workers subsequently introduced a series of enantiopure and racemic dinuclear indium complexes (25a–25c, Fig. 10) that were active for LA ROP with good control (Đ = 1.04–1.26).60 In particular, complex 25a afforded high molecular weight PLA from a high M/I ratio of over 2100 (Mn < 350 kg mol−1, Đ = 1.04). Interestingly, enantiopure (R,R/R,R)-24a showed strong selectivity for L-LA, evidenced by the high polymerisation rate of L-LA (3.4 × 10−3 s−1vs. 0.25 × 10−3 s−1 for D-LA), yet generated atactic PLA from rac-LA. This indicates potential competing monomer coordination and ring-opening of the complex in rac-LA ROP as well as the presence of transesterification and epimerisation side-reactions.
Heterometallic ROP catalysts based on main group metals
While most of the metal-based ROP catalysts developed to date are homometallic (containing one type of metal), the concept of heterometallic (mixed-metal) cooperativity has emerged as an attractive method of enhancing catalyst performance. Indeed, many heterometallic ROP catalysts have outperformed their homometallic counterparts. The reasons underpinning these activity enhancements remain somewhat unclear, and further mechanistic investigations are required to uncover catalyst features that lead to improved performance. The origins of cooperativity can vary between different heterometallic catalysts: in some cases both metal centres are directly involved in the reaction, whereas in others, catalysis takes place on the primary metal while the secondary metal modulates its reactivity.61 While several heterometallic ROP catalysts have been developed, those that solely feature main group metals are still relatively rare. Examples where a main group metal has been paired with a transition or f-block metal have been reviewed elsewhere, and so this section focuses only on examples where both metals are from the main group.61,62Dagorne and co-workers developed a series of Li/Al mono- and bis-benzoxide complexes (26a–26d, Fig. 11), and tested their application in rac-LA ROP.63 While the sterically hindered mono-benzoxide (26a–26b) complexes were inactive, the bis-benzoxide analogues (26c–26d) polymerised rac-LA at 25 °C. This activity is rather uncommon for Al-based catalysts, which typically require higher temperatures of 70–110 °C.42,48,64,65 The enhanced activity compared to Al-based alkoxides was attributed to the dissociation of the complexes to a neutral Al alkoxide complex (proposed to coordinate the monomer) and LiOBn (proposed to act as the nucleophile source to initiate ROP). Notably, these monometallic components were tested individually and shown to be inactive under identical conditions, suggesting that both metals were required for ROP and thus further corroborating the Li/Al cooperativity. It is worth highlighting that heterometallic Li/Al complexes 26c and 26d appear to be significantly slower than other alkali metal catalysts such as 7a–7d and 8a–8d,42 although direct comparisons are limited by the different reaction conditions. A similar phenomenon has also been observed with heterometallic Na/Al catalysts for LA ROP based on the TrenSal ligand, which outperforms the monometallic Al complex yet is slower than the homometallic sodium analogues.66 However, these heterometallic alkali metal/aluminium catalysts can deliver improved polymerisation control compared to alkali metal catalysts.
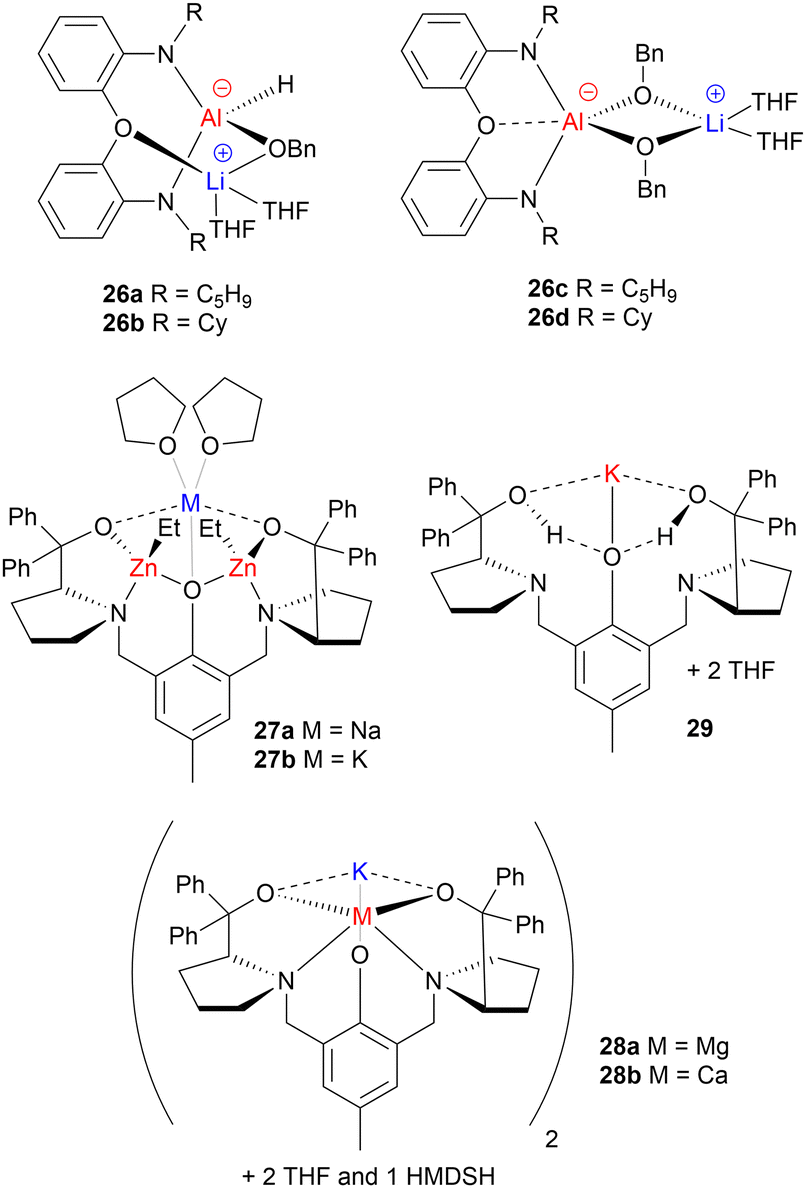 | ||
| Fig. 11 Examples of heterometallic main group catalysts for cyclic ester ROP (along with homometallic analogue 29). | ||
Exceptionally active heterotrimetallic Na/Zn2 and K/Zn2 complexes supported by a ProPhenol ligand have been reported for cyclic ester ROP (27a–27b, Fig. 11).67 K/Zn2 complex (27b) showed extremely high catalytic reactivity (kobs = 1.7 × 10−2 s−1) for rac-LA ROP in the presence of two equiv. of BnOH co-initiator, converting 60 equiv. of rac-LA in just 20 s in THF at 25 °C. Both 27a and 27b outperformed their homometallic counterparts, by combining the high activities of the Na/K complexes with the good control of the bis-Zn analogue. Experimental results suggested that the larger and more electropositive metal (Na or K) acts as the monomer coordination site, with the Zn-alkoxide unit acting as the nucleophile for monomer insertion. Switching from Na (27a) to the larger K (27b) increased the catalyst activity by a factor of 5, highlighting the key role that the alkali metal plays in monomer coordination and activation. Spectroscopic studies suggested that incorporating Na/K could labilise the Zn–Et bond through an “ate” activation (vs. the bis-Zn complex), leading to enhanced catalyst performance.67,68 The formation of alkali metal zincates is proposed to simultaneously enhance the Lewis acidity of the more electropositive metal (Na/K) and boost the nucleophilicity of the organo group on the”ate” metal (Zn) towards nucleophilic attack. These versatile catalysts also displayed excellent activities in the ROP of ε-CL and δ-VL.
Recently, a new methodology was reported to access heterometallic cooperativity in LA ROP, by combining homometallic complexes with simple salt additives in situ.69 This approach circumvents the need to isolate heterometallic complexes, which can be challenging to synthesise. The heterometallic catalyst systems were prepared via two routes: either by isolating the heterometallic complexes (28a–28b, Fig. 11) and adding 1 equiv. of BnOH as a co-initiator, or by adding 1 equiv. of an inorganic salt (Mg(OBn)2, Ca(OBn)2 or Zn(OBn)2) to an alkali metal complex such as homo-K complex 29 (Fig. 11). Reactivity studies of both routes suggested that the use of salt additives delivered similar activity enhancements to the isolated heterometallic complexes. The activity enhancements were attributed to Lewis acidic metal-based activation of the carbonyl group towards nucleophilic attack, as has previously been reported for other organic transformations. NMR spectroscopic studies revealed that the two different routes to prepare the catalyst systems each generated a similar mixture of solution-state products. This not only suggests that the in situ use of inorganic salts can generate similar catalyst systems to the isolated species but also highlights the importance of analysing isolated heterometallic catalysts in the solution state, as some catalysts that are assumed to be heterometallic may actually rearrange in solution.
Summary
ROP is currently dominated by main group chemistry. While Sn(Oct)2 is used industrially in LA ROP, there has been significant progress in developing alternative main group catalysts. Aluminium and zinc-based complexes have been the most widely researched alternatives, respectively delivering excellent stereocontrol and outstanding activity. The catalytic activities are significantly influenced by both the choice of the metal and the supporting ligands. s-Block metal complexes are generally more active than p-block complexes but often give poor polymerisation control, highlighting the importance of careful catalyst design to access both features simultaneously. Within the same group of the periodic table, larger metals often offer a greater number of monomer coordination sites, which generally contributes to higher activity in ROP. The increased M–R bond polarity upon descending a group can also influence the catalyst performance, for example by switching the mechanism from coordination–insertion to anionic. Other features may also play an important role, including the catalyst aggregation and solution-state structure. Notably, heterometallic cooperativity can be accessed either via isolated heterometallic complexes or by using salt additives to boost the activity of homometallic complexes. Both approaches display the potential to improve catalyst performance, hence providing an attractive approach for future catalyst design.Main group catalysts for ring-opening copolymerisation
Epoxide ring-opening copolymerisation (ROCOP) with cyclic anhydrides or CO2 is commonly used to prepare useful degradable materials such as aliphatic polyesters and polycarbonates, respectively (Scheme 8). ROCOP is also a useful method of preparing other polymeric materials such as poly-β-peptides and polypeptoids (N-alkylated polymers) via the copolymerisation of CO with aziridines, however, this route remains relatively underexplored. Therefore, this section predominantly focuses on main group catalysts utilised in the ROCOP of epoxides with CO2 or anhydrides.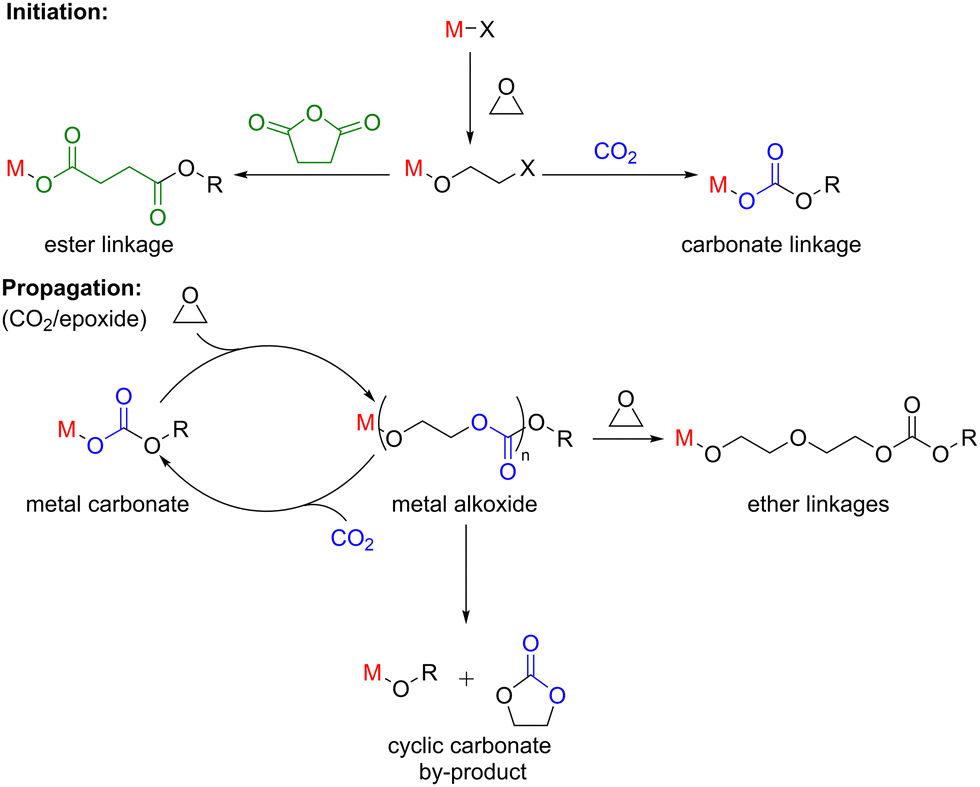 | ||
| Scheme 8 ROCOP of epoxides with CO2 or anhydrides (top); proposed catalytic cycle for epoxide ring-opening copolymerisation with CO2 (bottom). | ||
During epoxide ROCOP with CO2 or anhydrides, a series of reactions occur at the active Lewis acidic metal, which can coordinate (thus activate) monomers towards nucleophilic attack (Scheme 8). The polymerisation is typically initiated by the coordination of an epoxide monomer and subsequent ring opening via nucleophilic attack from an initiating group (e.g. a carboxylate, alkoxide or halide), resulting in the formation of a metal alkoxide intermediate. A carbonyl group (of CO2 or anhydride) is then inserted into the metal alkoxide bond during chain propagation, forming a metal–carbonate/carboxylate species, respectively. The carbonate/carboxylate group subsequently ring opens another metal-coordinated epoxide, resulting in a new metal–alkoxide species. Propagation continues as a ‘cycling’ process between metal–alkoxide and metal–carbonate/carboxylate intermediates. Five-membered ring cyclic carbonate by-products can also form as the thermodynamic product of CO2/epoxide ROCOP through depolymerisation or ‘back-biting’ reactions. The extent of cyclic carbonate formation often depends on the catalyst and the ceiling temperature for the final polymer (i.e. the temperature at which the rates of polymerisation and depolymerisation are equal). The percentage of cyclic by-products is also dependent on the relative rate of ring closure (resulting in cyclic carbonate) vs. monomer insertion (generating polymer). Additionally, bimodal molecular weight distributions are commonly observed for epoxide ROCOP with CO2 or anhydrides, which is generally attributed to impurities such as water and hydrolysed epoxides (diols), both of which can act as initiators. Owing to mechanistic similarities between ROP and the ROCOP of epoxides with CO2 or anhydrides, catalysts applied in these processes are often structurally alike, with some catalysts showing high activities in both types of polymerisations.
While ROP (vide supra) is an attractive process for the production of aliphatic polyesters and polycarbonates, there are some advantages to accessing these materials via ROCOP of epoxides with cyclic anhydrides or CO2. Using epoxide/anhydride ROCOP to generate polyesters gives access to a far broader range of material properties vs. ROP, as ROCOP uses two functionalised monomers instead of one.70 The production of aliphatic polyesters by ROP is often limited by thermodynamic constraints and a lack of commercially available monomers. In contrast, epoxide/anhydride ROCOP can employ a much broader range of monomers that are commercialised at scale, and is also more thermodynamically feasible. Notable examples include the production of polyesters containing aromatic repeat units in the polymer backbone, a feature that is challenging to access through ROP. Additionally, epoxide/anhydride ROCOP can be applied to a range of monomers derived from biomass (e.g. limonene oxide and maleic anhydride). Despite these advantages, epoxide/anhydride ROCOP catalysis remains underdeveloped compared to cyclic ester ROP and there is a need to design highly active, selective and tolerant catalysts. Using CO2 as the co-monomer instead of anhydrides brings additional sustainability benefits as CO2 has low toxicity, is renewable and can be sourced from abundant industrial waste streams.71 Furthermore, life cycle analysis for polyurethanes produced using polycarbonates made from CO2/epoxide ROCOP shows that these materials consume around 20% less raw petrochemicals and produce up to 19% fewer CO2 emissions compared to conventional petrochemicals routes.72
In 1969, Inoue et al. pioneered the copolymerisation of CO2 and epoxides by combining ZnEt2 with water, CO2 and PO to yield a small quantity of polymeric material.3 While this enabled the first synthesis of polycarbonates from CO2, the process was limited by low catalytic activities and the formation of undesired by-products such as cyclic carbonates and polymer chains containing a high content of ether linkages (Scheme 8).73 Inoue and co-workers also reported one of the first single site Al–porphyrin catalysts for the co-polymerisation of PO and phthalic anhydride (PA). This catalyst generated highly controlled polyester with alternating PO/PA monomer insertion. A range of well-defined catalysts have since been developed for the ROCOP of epoxides with CO2 or anhydrides, many of which include main group metals. While both homogeneous and heterogeneous catalysts are known for epoxide/CO2 ROCOP, most commercial catalysts are currently heterogeneous, such as zinc glutarate catalyst systems. These catalysts have been extensively studied and early mechanistic studies indicated that bimetallic polymerisation processes are important. In epoxide/CO2 ROCOP, short spatial distances between metal–metal active sites (4–5 Å) are thought to be key for significant copolymerisation activity (see below for further details). While most commercial poly(propylene carbonate) (PPC) is produced using zinc glutarate catalyst systems, the composition of heterogeneous catalysts is often ambiguous with poorly defined active sites.5 Heterogeneous catalysts can also suffer from low polymerisation rates and give low CO2 uptake, yielding poly(ether carbonates) (Scheme 8, bottom right).74 Some of these challenges can be overcome using homogeneous catalysts, including the production of highly controlled polymers with >99% carbonate linkages and no/trace cyclic carbonate by-products under moderate pressures of CO2 (usually <10 bar and sometimes as low as 1 bar).
A major breakthrough in CO2/epoxide ROCOP was the use of highly active Zn-β-diiminate (ZnBDI) catalysts (30a–30h, Fig. 12), where cyclohexene-oxide (CHO) was used as the epoxide monomer. Mechanistic studies by Coates and co-workers revealed that bimetallic epoxide enchainment was the rate-determining step, where tightly bound ZnBDI dimers and monomeric complexes that were unable to dimerise were unreactive, whereas loosely bound dimers were highly active in polymerisation. NMR spectroscopy and X-ray diffraction studies indicated that the aggregation state and metal–metal proximity were influenced by the ligand substituents (Fig. 12), and that close intermetallic distances (through the formation of loosely bound dimers) generally enhanced the catalytic activity.75 As the catalysts exist in monomer–dimer equilibria in solution, the formation of loosely bound dimers is promoted at high catalyst concentrations, i.e. the catalyst activity is concentration dependent. Therefore, the loosely bound dimeric ZnBDI catalysts perform well in neat epoxide (rather than under dilute solvent conditions). However, the use of neat epoxide limits the epoxide conversion due to viscosity issues. Asymmetric catalyst 30e (Fig. 12) was the most active of the series with a TOF of around 700 h−1 and 99% polycarbonate linkages (7 bar CO2 pressure, 50 °C). It is worth noting that 30e does not contain the most sterically demanding substituents, however the authors propose that it is sufficiently bulky to form a reactive loose dimer. Increasing the steric bulk beyond this optimal level (30a and 30f) reduced the catalyst activity. Mechanistic investigations showed rapid insertion of CO2, whereas epoxide insertion was relatively slow. A bimetallic mechanism was elucidated, involving epoxide coordination to one zinc centre, while the propagating polymer chain on the second zinc centre instigates nucleophilic attack (vide infra). This bimetallic transition state would be hard to access for monomeric species, hence the loss in activity with very sterically bulky ligands or under very dilute conditions. The highly active loosely bound dimers are also kinetically more favourable than the tightly bound dimers, making these the best performing of the study. This study inspired the development of many other dinuclear catalysts featuring proximal metal centres, including those based on dinucleating ligand scaffolds. While some highly active monometallic catalysts have been reported, there are indications that some of these also operate via a dimeric mechanism, which can lead to decreased efficiency at low catalyst loadings. There have been significant research efforts to prepare bimetallic catalysts where the two metals are held in close proximity within one ligand framework, and over time, the optimal metal–metal distance was established as 3–5 Å.61 The use of such bimetallic catalysts allows access to lower catalyst loadings and therefore higher molecular weight polymers, unlike more concentration-sensitive monometallic catalysts. This opens up the possibility of using solvents (instead of neat epoxide), overcoming the aforementioned viscosity issues and enabling higher monomer conversions.
 | ||
| Fig. 12 β-Diiminate zinc catalysts for the alternating copolymerisation of CHO and CO2. Loosely bound dimers are highly reactive catalysts (bottom right). | ||
Homometallic alkali metal catalysts for ROCOP
While the highly polar nature of organoalkali metal reagents typically requires stringent air- and moisture-free conditions, alkali metal carboxylates are easier to handle and have also shown success in epoxide/anhydride ROCOP.76 Simple metal acetate catalysts (MOAc, where M = Li, Na, K, Cs; 31, Fig. 13) were all active for the ROCOP of PA and CHO. These studies showed that the catalytic activity depended on the strength of interaction between the metal and acetate; loosely bound ion pairs showed activity whereas closely bound contact pairs did not, which was attributed to a lack of monomer coordination sites. Correspondingly, CsOAc was the most active, however, the study focussed on KOAc as it is commercially available, has low toxicity and displayed a reasonable TOF value of 99 h−1 at 150 °C. The activity of KOAc could be boosted by increasing the ion pair separation by using a more stable anion. For example, CF3COOK features an electron-withdrawing CF3 group and gave double the activity of KOAc (approximate respective TOF values of 48 h−1 and 23 h−1; 110 °C). Based on DFT calculations, the authors posited that fully separated ion pairs such as CsOAc operate through an anionic polymerisation route via the acetate anion, whereas loose ion pairs like KOAc proceed through a coordination–insertion mechanism with K+ activating the monomer for nucleophilic attack. While alkali metal catalysts have shown success in ROCOP, including the use of LiOtBu to perform ROCOP of epoxides with CS2, far more catalyst development has focussed on di- and tri-valent metal complexes.77 | ||
| Fig. 13 Examples of Group 1, Group 2 and zinc catalysts for ROCOP of epoxides with anhydrides or CO2. | ||
Alkaline earth metal and zinc catalysts for ROCOP
While early pioneering studies highlighted the ability of Zn systems to catalyse CO2/epoxide ROCOP, subsequent catalyst development has expanded to divalent alkaline earth metals as well as sophisticated Zn catalysts. Good catalyst performance has been achieved using simple metal alkoxide salts. For example, Groysman, Mazzeo and co-workers reported a mononuclear Mg alkoxide catalyst (32, Fig. 13) with a BnOH or bis(triphenylphosphine)iminium chloride (PPNCl) co-catalyst, which was active for PA/CHO ROCOP.78 Onium salts such as PPNCl are common co-catalysts in both ROP and ROCOP, and can boost the catalyst activities via the chloride anion coordinating to a metal centre and thereby increasing the nucleophilicity of a metal–X bond (where X is an initiating group or a growing polymer chain).79–81 Notably, the choice of co-catalyst can produce vastly different results. While BnOH gave full conversion in 24 h with >99% ester selectivity, PPNCl gave 83% conversion and 92% ester linkages (albeit with lower catalyst loading). Solvent effects are also impactful, as while excess CHO improved conversion (>99% in 24 h for a 1:100:800 ratio of catalyst:anhydride:epoxide, vs. 61% in 96 h for a 1:100:100 ratio, both in 1 mL toluene), moving to neat polymerisations in CHO saw a marked drop in selectivity (25% ester linkages) along with significant polyether formation.
As early studies in CO2/epoxide ROCOP suggested that intermetal proximity is a key feature, this set a precedent for targeting bimetallic complexes. Williams and co-workers synthesised di-Mg and di-Zn complexes with a macrocyclic ligand (33a–d, Fig. 13) for CHO/CO2 and CHO/PA ROCOP.82 While similarities exist between epoxide/CO2 and epoxide/anhydride ROCOP, catalysts do not necessarily translate seamlessly between the two. This is demonstrated by the di-Mg complex with a bromide initiator (33a) being active for CHO/CO2 (TOF = 15 h−1, 1 bar CO2) where the di-Zn analogue (33b) is inactive. In contrast, for CHO/PA Mg-based 33a is less active (TOF = 9 h−1) than the di-Zn analogue 33b (TOF = 17 h−1). The initiating X group also modulates activity; swapping the bromide co-ligand for acetate gave increased activity in CHO/CO2 ROCOP for di-Mg (33c, TOF = 35 h−1, 1 bar CO2) and activated the previously inactive di-Zn complex (33d, TOF = 18 h−1, 1 bar CO2).83 The higher activity of the di-Mg catalyst was attributed to the more nucleophilic metal–carbonate bond in di-Mg vs. di-Zn due to the lower electronegativity of Mg (giving a more polarised metal–carbonate bond) and the lower Lewis acidity of Mg (decreasing the strength of the metal–Ocarbonate bond); both of these features enhance the rate-determining nucleophilic attack upon an epoxide and thereby improve the catalyst activity. Better suppression of “back-biting” was also seen for the di-Mg system, with 4–6% cyclic carbonate yield observed with di-Zn and none with di-Mg.
Ding and co-workers prepared a bimetallic di-Zn catalyst based on the Trost ligand (34a, Fig. 13) which gave higher TOF values (142 h−1, 20 bar CO2) than the earliest CHO/CO2 ROCOP catalysts and remained active at 1 bar CO2 (97% conversion at 1 bar vs. >99% at 20 bar).84 Complex 34a also gave reduced selectivity when polymerisations were carried out in neat epoxide (94% carbonate linkages) vs. in toluene (>99%). Changes within ligand substituents can also have important implications, as when the aryl groups are changed from phenyl (34a) to electron-withdrawing 3,5-(CF3)2Ph groups (34b), the carbonate selectivity dropped to only 22%.
More recently, flexible di-Zn catalysts with exceptionally high activity for CO2/epoxide ROCOP were reported by Rieger and co-workers (35a, Fig. 13).85 This substantial activity is demonstrated by a TOF value around 9100 h−1 at 40 bar CO2 pressure and 100 °C, which was a marked increase over previous ROCOP catalysts. Even at lower CO2 pressures catalyst 35a was still highly active, achieving TOF around 3600 h−1 at 5 bar CO2 pressure. The importance of the flexible tether between the two Zn centres was supported by comparison against a control complex with a more rigid flexible ligand scaffold, which displayed low activity. Notably, 35a was the first Zn-based catalyst for epoxide/CO2 ROCOP where the rate-determining step (typically the epoxide ring opening) was shown to switch between epoxide ring opening and CO2 insertion depending on the CO2 pressure, which indicates the adaptability and tuneability of the tethered di-Zn species. Further improvement of the ligand to incorporate electron-withdrawing CF3 groups (35b, Fig. 13) led to unprecedented reactivities (TOF up to 155000 h−1).86
Although ROCOP of epoxides with either CO2 or anhydrides already offers a potentially broad monomer and polymer scope, catalysts capable of controlled terpolymerisation are important tools to further expand the range of available polymer structures and thus material properties. Zn-based catalysts have been exploited in the production of terpolymers via controlled ROCOP, including the use of ZnBDI catalysts (30h, Fig. 12) that enabled the controlled one-pot terpolymerisation of CHO, CO2 and diglycolic anhydride (DGA) to selectively produce di-block poly(ester-block-carbonate) with minimal tapering between the two blocks (Scheme 9, 50 °C, 7 bar CO2 pressure).87 IR spectroscopic studies revealed that the polyester block is formed first, in spite of the fact that polyester formation is slower than polycarbonate formation. While the rate-determining step is the epoxide insertion, these studies showed there was a product-determining step that precedes epoxide insertion (Scheme 9). Firstly, the zinc alkoxide species reacts irreversibly with DGA, so only when the DGA is almost fully consumed does CO2 incorporation become competitive. Polycarbonate formation is quicker than polyester formation because CHO insertion into zinc-carbonate (polycarbonate, Scheme 9, right-hand cycle) is faster than into zinc-carboxylate (polyester, Scheme 9, left-hand cycle). This highlights the exciting potential to extend the scope of polyesters produced via epoxide/anhydride ROCOP to an even broader variety of poly(carbonate-block-ester) materials with wide-ranging properties, and to obtain terpolymers with well-controlled structures through a sophisticated one-pot route.
 | ||
| Scheme 9 Terpolymerisation of CHO with DGA and CO2 respectively by ZnBDI(OAc) to give poly(ester-block-carbonate). P′ and P′′ are growing polyester and polycarbonate chains respectively. | ||
Homometallic ROCOP catalysts containing p-block metals
While the most notable ROCOP catalysts based on p-block metals generally involve Al supported by salen ligands, examples using other ligands and other Group 13 metals such as indium and gallium have been reported, including complexes that can also catalyse cyclic ester ROP (e.g.22 and 24b, Fig. 10).55,88 Binary salen-based catalyst/co-catalyst systems are well-established for cyclic ester ROP and have also been extensively explored for ROCOP. Binary systems typically involve a metal-salen catalyst paired with an onium salt co-catalyst that enhances the activity in polymerisation (see catalyst 36, Fig. 14 as a representative example). These binary systems typically proceed via a monometallic but bicomponent mechanism, where the catalyst and co-catalyst are both involved in the propagation step (Scheme 10). The epoxide coordinates to Al and is ring-opened by the associated co-catalyst carboxylate species, which is formed from the ring-opened anhydride. Subsequent anhydride insertion occurs to generate an ester linkage. The catalyst exists in an equilibrium between the active and dormant states, depending on the coordination number of the aluminium centre. In its five-coordinate state, a vacant site is available for monomer coordination and the catalyst is active. However, if aluminium is six-coordinate, no further monomer can coordinate, and the catalyst is dormant. | ||
| Fig. 14 Examples of p-block metal-based catalysts for ROCOP of epoxides with CO2 or anhydrides. | ||
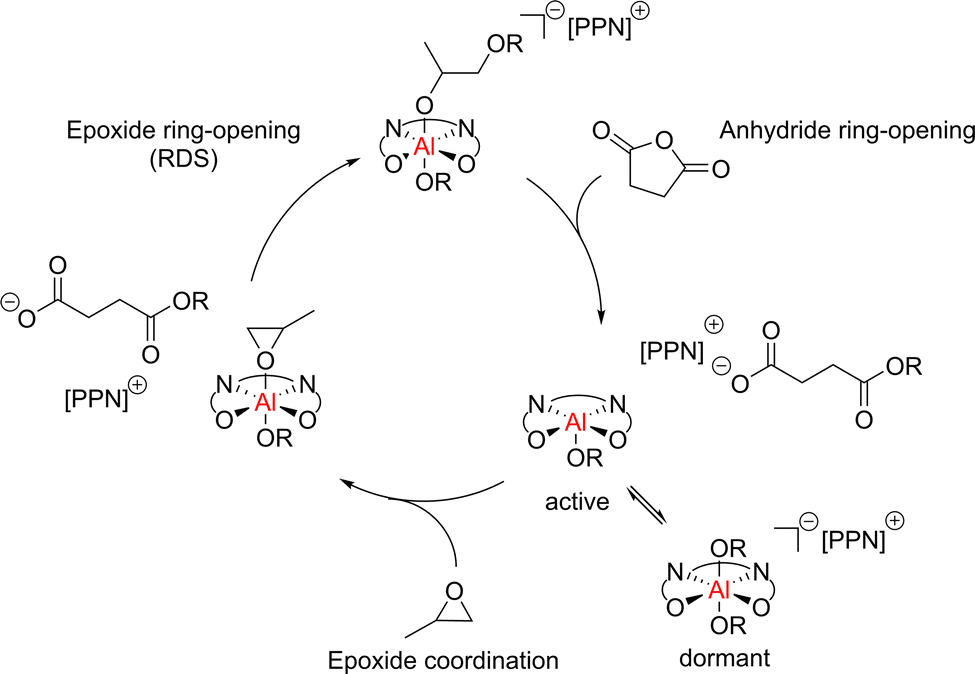 | ||
| Scheme 10 General aluminium salen catalysed ROCOP mechanism for propylene oxide and succinic anhydride with PPNCl co-catalyst. OR refers to the growing polymer chain. | ||
The efficiency of binary catalyst systems for epoxide/CO2 and epoxide/anhydride ROCOP is often reduced at high dilutions, as both components are required for polymerisation. Therefore, ROCOP catalysts have been reported where the co-catalyst was covalently attached to the salen backbone, such as the bifunctional Al(salen) system reported by Coates and co-workers (37, Fig. 14).89 At higher catalyst loadings, the binary system of 36/PPNCl was faster (36 TOF = 112 h−1, 37 TOF = 93 h−1; catalyst:anhydride:epoxide ratio of 1:400:2000) whereas at lower loadings the bifunctional catalyst 37 showed superior activity (35 TOF = 22 h−1, 37 TOF = 87 h−1; 1:2000:10000) owing to the catalyst components being in optimal proximity even at high dilution. The bifunctional Al(salen) catalyst system 37 provided well-controlled polymerisation, suggesting a lack of transesterification and with limited polyether linkages from epoxide homopolymerisation (unlike the faster transition metal analogues featuring Co and Cr).
Bimetallic Al phenoxy-imine complexes have also been reported for epoxide/anhydride ROCOP (Fig. 14).90 As with many salen catalysts, a nucleophilic co-catalyst was used; 4-dimethylaminopyridine (DMAP) improved the ester selectivity (63% to >99%) for succinic anhydride/CHO ROCOP. Notably bimetallic catalysts with a short Al–Al distance (38a) showed comparable activity to the monometallic analogue (39). This observation suggests that even though 38a is bimetallic, only one of the aluminium metals is involved in the propagation (although chain transfer between the two aluminium centres may occur). In contrast, 38b was twice as active as 38a and 39, which was attributed to the greater intermetallic distance and reduced steric hindrance at Al enabling simultaneous propagation of polymer chains on both Al centres. This example highlights that intermetallic proximity can modulate the catalyst activity, and that not all multimetallic catalysts display activity enhancements in ROCOP.
While indium- and gallium-based catalysts remain scarce, recent studies have established that both metals have potential in ROCOP. Williams and co-workers reported an active In(phosphasalen) complex for CO2/epoxide ROCOP (24b, Fig. 10).88 The use of co-catalysts was evaluated yet was not advantageous, and either decreased the activity (DMAP) or directed the selectivity towards undesired cyclic carbonate formation (PPNCl). The In(phosphasalen) complex was more active than the In(salen) analogue. This was attributed to the phosphasalen ligand scaffold reducing the Lewis acidity of the metal, thus increasing the reactivity of the intermediate indium carbonate species towards nucleophilic attack and ring-opening of the epoxide. A combination of kinetic, spectroscopic and structural studies of isolated intermediates suggested a mononuclear mechanism. Although catalysts in this field typically operate through bimetallic mechanisms, Group 13-based catalysts tend to deviate from this, as seen here and for Al(salen) catalysts including 36 (Fig. 14).89 The mononuclear mechanism for the indium-based catalyst (24b) is notable, even compared to Al(salen) 36, which although monometallic, still operates through a bicomponent catalytic system. The monometallic mechanism for 24b was attributed to the relatively large ionic radius of indium facilitating monomer (and polymer) coordination. Although not the most active catalyst, the In(phosphasalen) complex 24b showed good stereocontrol giving isotactic polycarbonate (TOF = 15 h−1, Piso = 0.86) and is also active at 1 bar CO2 pressure. Recently, Schulz and co-workers have produced a series of tetranuclear gallium-based catalysts that are active for epoxide/anhydride ROCOP (22a, Fig. 10).55 With BnOH as the initiator, 95–99% conversion was reached in 12 hours for CHO with succinic or maleic anhydride ROCOP at 100 °C. Good selectivity towards polyester was achieved, with no evidence of polyether formation.
Heterometallic main group catalysts for ROCOP
Heterometallic cooperativity has been well-exploited in numerous chemical transformations and is an attractive means to enhance catalyst reactivity and selectivity compared to the homometallic counterparts, yet the concept is still gathering momentum in polymerisation processes. The first homogeneous, heterometallic catalyst for epoxide/CO2 ROCOP to be isolated was based on main group metals (Mg and Zn) and showed significant activity enhancements compared to the homometallic analogues.82 Since then, a range of heterometallic catalysts have been developed, many of which feature a main group metal paired with a transition metal or f-block metal. As these complexes have been comprehensively reviewed elsewhere, this review will solely focus on heterometallic complexes where both metals are from the main group.61Heterometallic ROCOP catalysts were targeted as mechanistic studies had shown that some bimetallic catalysts could outperform their monometallic analogues.75 For example, experimental and computational mechanistic studies for a homobimetallic zinc complex (40, Fig. 15) showed that a “chain shuttling” mechanism can occur (Scheme 11).91,92 One metal was proposed to coordinate and activate the epoxide (M2, Scheme 11), whilst the other (M1) was proposed to provide the source of the (metal–carbonate) nucleophile. Kinetic studies showed that the epoxide coordination and ring opening was the rate-determining step, whereas the reaction mechanism was zero-order in CO2. These observations suggested that each metal played a distinct role in the propagation step and that heterometallic catalysts may therefore offer enhanced catalyst performance by tailoring the two different metals to these two different roles. Enhancing the epoxide coordination would require a Lewis acidic metal (M2), whereas increasing the rate of nucleophilic attack and epoxide ring-opening would require a labile metal–carbonate (M1–O) bond.
 | ||
| Fig. 15 Examples of homo- and heterobimetallic complexes employed in epoxide/CO2 ROCOP. | ||
 | ||
| Scheme 11 Proposed chain-shuttling mechanism for CHO/CO2 ROCOP. | ||
In 2015, a heterometallic Zn/Mg analogue of 40 was reported for CO2/CHO ROCOP (41a, Fig. 15),82 which significantly outperformed both of the homobimetallic analogues (41a, TOF = 34 h−1vs. di-Mg, TOF = 15 h−1; di-Zn, inactive).82 When compared to a 50:50 mixture of the homobimetallic complexes, heterobimetallic Zn/Mg complex 41a showed five times greater activity for CO2/CHO ROCOP (0.1 mol% catalyst loading, neat epoxide, 1 bar CO2, 80 °C), suggesting a synergic relationship between the metals. Additionally, 41a displayed relatively good polymerisation control, generating >99% carbonate linkages with only trace cyclic carbonate. Catalyst 41a also displayed excellent activities in anhydride/epoxide ROCOP showing 40 times greater activity than a 50:50 mixture of the homobimetallic complexes under identical conditions. Exchanging the bromide for carboxylate co-ligands gave significant activity improvements in CHO/CO2 ROCOP. The best catalyst at high pressure (41e), featuring p-CF3-substituted benzoate co-ligands, displayed TOF values of up to 8800 h−1 (0.01 mol% catalyst loading, neat epoxide, 20 bar CO2, 120 °C).91 All of the catalysts 41a–41j were also highly active at low pressure (1 bar CO2).91 For example, the heterometallic p-nitro-benzoate complex (41f) (TOF = 124 h−1) was significantly more active than either the di-Zn or di-Mg analogues (TOF = 18 h−1 and 30 h−1 respectively) under identical conditions (0.1 mol% catalyst loading, neat epoxide, 80 °C and at 1 bar CO2). The enhanced activities of the heterometallic complexes was attributed to Lewis acidic Mg enhancing epoxide coordination and the labile Zn-carbonate bond accelerating the nucleophilic attack on the basis of DFT and kinetic studies.
While heterometallic cooperativity can lead to significantly improved catalyst performance, it is important to highlight that not all heterometallic complexes are cooperative.93 Understanding the features that lead to heterometallic cooperativity is key for designing the next generation of catalysts for epoxide/CO2 ROCOP.93 Factors that are emerging as important for epoxide ROCOP with CO2 or anhydrides include the Lewis acidity of the metal and nucleophilicity of the M–O(polymer) bond, the metal size and coordination number, and M–M′ proximity.
Only a few heterometallic complexes have been employed for epoxide/anhydride ROCOP, yet these have displayed promising activities. Recently, a series of heterobimetallic complexes containing Al and Group 1 metals (Na, K, Rb, Cs) were reported to show excellent catalytic performance for PA/CHO ROCOP (42a–42d, Fig. 16).94 Kinetic studies and DFT calculations suggested that these complexes operate via a similar chain shuttling mechanism to that proposed for epoxide/CO2 ROCOP. Compared to the other alkali metal/Al complexes, the combination of Al/K (42b) displayed the highest catalytic activity (with TOF of approximately 1100 h−1, 0.25 mol% catalyst loading, T = 100 °C) as well as excellent selectivity and polymerisation control (Đ = 1.06). These studies highlighted that the internuclear separation (metal–metal distance), which increases in the expected order of Al–Na < Al–K < Al–Rb < Al–Cs, is important for reducing the relative transition state energies of key intermediates. Additionally, the Lewis acidity of each alkali metal (Cs > Rb > K > Na) was considered, as more Lewis acidic M form stronger M–Ocarboxylate bonds, which need to be broken in the propagation step. Lewis acidity trends of the Group 1 metal ions and intermetallic distance trends suggest that the most active complexes would be Al/Cs or Al/Na respectively. The excellent activity of the Al/K complex highlights the importance of an appropriate balance of these two demands.94
 | ||
| Fig. 16 Heterobimetallic catalysts containing aluminium and Group 1 metals employed in epoxide/anhydride ROCOP. | ||
Highly active heterometallic (Mg/Zn) catalysts (41g and 41i, Fig. 15) have been reported that are capable of switching between lactone ROP and CO2/epoxide ROCOP to produce a series of poly(ester-block-carbonates) through a one-pot process (Scheme 12).72 Potential sustainability benefits of this method include that both blocks are degradable and have the potential to be made from renewable resources. For example, a series of poly(cyclohexene carbonate-block-decalactone-block-cyclohexene carbonate) [PCHC-PDL-PCHC] materials was prepared using ε-decalactone (which can be derived from biomass), CHO and CO2. The catalyst 41i displayed high selectivities (CO2 selectivity >99%), high monomer conversions (>90%) and yielded block copolymers with predictable compositions. Additionally, the ABA-block polymers incorporated 6–23 wt% CO2 and showed superior properties compared to poly(cyclohexene carbonate) (PCHC), specifically improved thermal stability and high toughness. This is a key finding, as high molecular weight polycarbonates often suffer from poor physical properties such as brittleness and high glass transition temperatures. The selectivity towards lactone ROP or CO2/epoxide ROCOP depended on the nature of the M–O bond at the growing polymer chain-end. Specifically, a M-alkoxide bond inserts lactone whereas a M–carbonate unit inserts epoxide. The presence of either a M–carbonate or M–alkoxide bond and thus the insertion of an epoxide or lactone monomer is dependent on the presence or absence of a CO2 atmosphere, respectively.
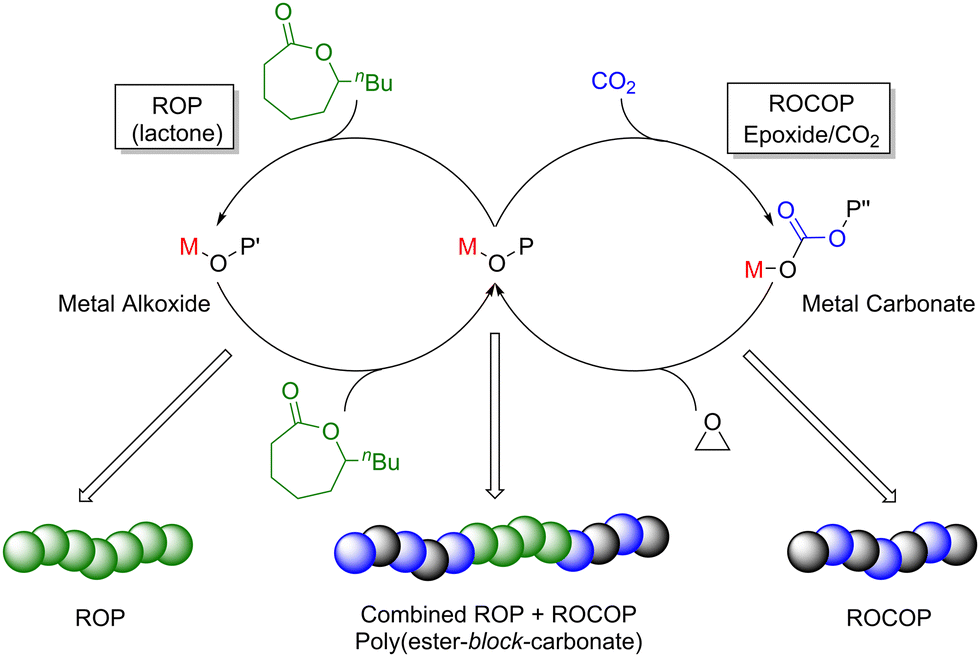 | ||
| Scheme 12 Catalytic cycles accessed by a single, switchable catalyst for the production of poly(ester-block-carbonates). | ||
Summary
Main group element-based catalysts are strong contenders for the ROCOP of epoxides with CO2 or anhydrides. Though many do not yet match the high activity seen with transition metal catalysts, main group metals still offer good activity and control paired with low toxicity and high abundance, which makes them attractive targets for ROCOP catalysis in terms of balancing sustainability with catalyst performance. While this research area has so far been dominated by Zn and Al, recent studies have shown that a broader range of metals can be used including Group 1 and 2 metals. Epoxide ROCOP with CO2 or anhydrides has been proposed to operate via a bimetallic mechanism. The development of bimetallic epoxide/CO2 ROCOP catalysts has led to highly active di-Zn catalysts, including systems that maintain high activity at atmospheric pressure, which is an exciting and industrially relevant milestone. Although lesser explored, interest in epoxide/anhydride ROCOP has also been increasing, with advanced catalyst design delivering improved catalyst activities, enhanced control over the polymer microstructure and efficient catalysis at low catalyst loadings. Emerging reports on heterometallic cooperativity indicate that key features of cooperative heterocombinations include metal size, M–OR bond strength and M–M′ proximity. Further studies will be key to fully understand, predict and optimise useful heterocombinations to lead to even more significant advances in ROCOP catalysis.Conclusions
Main group polymerisation catalysts have helped to pioneer the development of many new polymer materials that are now widely used, including polystyrene, polylactic acid and polypropylene carbonate. While these initial catalyst systems were typically simple metal salts or metal–alkyl reagents, the introduction of ancillary ligands has led to improved mechanistic understanding and enhanced catalyst performance, with main group catalysts displaying high activities across alkene polymerisation, ROP and ROCOP. In all three fields, most of the reported main group catalysts feature metals from Group 1, Group 2, Group 13 (especially Al) or Zn. Within olefin polymerisation, the metal–alkyl bond polarity is key, with more polarised metal–alkyl bonds typically leading to enhanced catalyst activities. Within ROP and ROCOP, the metal Lewis acidity is particularly important, as Lewis basic monomer coordination (thus activation) is typically a key mechanistic step that facilitates the nucleophilic attack and ring opening of lactones, lactams and epoxides. The source of the nucleophile is also important, and can determine the RO(CO)P mechanism. For example, in ROP, metal-based catalysts featuring very strong metal–alkoxide bonds in the presence of an exogeneous alcohol typically favour an activated monomer mechanism, whereas those featuring a weaker metal-alkoxide bond typically follows a coordination–insertion mechanism. While alkene polymerisations and ring-opening (co)polymerisations progress mainly via coordination–insertion or anionic mechanisms, the ideal catalyst features differ for each class of polymerisation and for different types of monomer.Highlighting the importance of main group metal Lewis acidity in ROP and ROCOP, there is a general trend towards enhanced catalyst activities upon descending the s-block. This has been attributed to metals with larger ionic radii possessing a greater number of monomer coordination sites leading to faster chain propagation. However, tuning the Lewis acidity of the metal is a balancing act. Descending the s-block increases the polarity of the metal–R bond (e.g. where R is alkoxide, carboxylate or carbonate), which can boost the nucleophilicity thus enhancing attack and insertion of monomers. However, increasing the ionic character of the M–R bond can also result in the metal binding the reactive polymer chain end too tightly, disfavouring nucleophilic attack. In some cases, increased polarisation leads to charge separation of the metal and initiating group, which can shift the polymerisation mechanism from coordination–insertion to anionic. Carefully fine-tuning both the metal Lewis acidity and the metal–R bond character is thus key to accessing optimum catalyst activity. This has been achieved through the use of carefully designed ligands to improve both the activity and polymerisation control of main group metal catalysts. The careful selection of an appropriate polymerisation solvent is also important, as Lewis basic solvents may alter the catalyst structure (typically by decreasing the catalyst aggregation state and/or charge separation of the metal and co-ligand) and thus modulating the catalyst performance.
More recently, the incorporation of a heterometal has emerged as an alternative method of modifying the metal Lewis acidity as well as the M–R bond polarity. Overall, heterometallic catalysts applied in ROP and ROCOP have often displayed enhanced reactivity and overall catalytic performance compared to the homometallic analogues. However, due to the highly extensive research into homometallic catalysts, it is not surprising that current heterometallic catalyst performance often falls short of the homometallic frontrunners. Recently, systematic studies have started to uncover which heterometallic catalyst features can lead to enhanced catalyst performance. In some cases, heterometallic combinations are starting to compete with the best-performing homometallic catalysts. Heterometallic cooperativity is therefore an interesting area to explore for future advancements in RO(CO)P catalyst design. However, further studies are required to be able to predict whether heterometallic catalysts will be “cooperative” or “uncooperative”, as not all heterocombinations display activity enhancements. The heterometallic polymerisation catalysts reported to date are just the tip of the iceberg in terms of the vast number of heterocombinations available, both within the main group and beyond.
Since their advent, main group polymerisation catalysts have opened up access to new polymer structures, and their use in developing pioneering new materials continues to this day. Main group catalysts can deliver careful control over the polymer structure. Changing the coordination environment of the metal centre mid-polymerisation, for example from a metal-alkoxide to a metal–carboxylate by using CO2 as a chemical “switch”, has led to the selective production of terpolymers (and higher multi-block polymers) in one-pot reactions from mixtures of monomers. With the current drive to use earth-abundant metals in catalysis, combined with the growing demand for biodegradable polymer materials from renewable sources, the use of main group metals in polymerisation catalysis will surely continue as an exciting and impactful area of research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Prof. Manfred Bochmann for helpful discussions, as well as the UKRI Future Leaders Fellowship (J. A. G. and E. F. MR\T042710\1), British Ramsay Memorial Trust (J. A. G.), L’Oréal-UNESCO For Women in Science UK & Ireland Fellowship (J. A. G.), the University of Edinburgh Principal's Career Development PhD Scholarship (Y. Z.), Croda (A. L.), MARA Malaysia (M. A. R.) and the EPSRC (R. C. EP/V049003/1) and EPSRC SOFI2 Centre for Doctoral Training (A. L and P. L. EP/S023631/1) for funding.Notes and references
- N. D. Scott, J. F. Walker and V. L. Hansley, J. Am. Chem. Soc., 1936, 58, 2442–2444 CrossRef.
- W. H. Carothers, G. L. Dorough and F. J. van Natta, J. Am. Chem. Soc., 1932, 54, 761–772 CrossRef.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B Polym. Lett., 1969, 7, 287–292 CrossRef.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef PubMed.
- C. Lichtenberg and J. Okuda, Angew. Chem., Int. Ed., 2013, 52, 5228–5246 CrossRef CAS PubMed.
- M. Hong, J. Chen and E. Y.-X. Chen, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS.
- G. G. Hlatky, Chem. Rev., 2000, 100, 1347–1376 CrossRef PubMed.
- Z. B. Shifrina, V. G. Matveeva and L. M. Bronstein, Chem. Rev., 2020, 120, 1350–1396 CrossRef PubMed.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef PubMed.
- Z. Hou and Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1–22 CrossRef.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef.
- E. S. Tabatabaie, S. Dehghanpour, E. Mosaddegh, R. Babaahmadi, A. Chipman, B. F. Yates and A. Ariafard, Dalton Trans., 2019, 48, 6997–7005 RSC.
- K. Soga and T. Shiono, Prog. Polym. Sci., 1997, 22, 1503–1546 CrossRef.
- M. E. Z. Velthoen, A. Muñoz-Murillo, A. Bouhmadi, M. Cecius, S. Diefenbach and B. M. Weckhuysen, Macromolecules, 2018, 51, 343–355 CrossRef CAS PubMed.
- M. Van Beylen, S. Bywater, G. Smets, M. Szwarc and D. J. Worsfold, Polysiloxane Copolymers/Anionic Polymerization, Springer, Berlin, Heidelberg, 1988, pp. 87–143 Search PubMed.
- H. L. Hsieh and I. W. Wang, Macromolecules, 1986, 19, 299–304 CrossRef CAS.
- L. Cazzaniga and R. E. Cohen, Macromolecules, 1989, 22, 4125–4128 CrossRef CAS.
- K. Matyjaszewski, T. E. Patten and J. Xia, J. Am. Chem. Soc., 1997, 119, 674–680 CrossRef CAS.
- H. Fouilloux and C. M. Thomas, ChemCatChem, 2022, 14, e202101673 CrossRef CAS.
- M.-L. Hsueh, B.-T. Ko, T. Athar, C.-C. Lin, T.-M. Wu and S.-F. Hsu, Organometallics, 2006, 25, 4144–4149 CrossRef CAS.
- H. L. Hsieh, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 379–386 CrossRef.
- A. P. Dove, V. C. Gibson, E. L. Marshall, A. J. P. White and D. J. Williams, Chem. Commun., 2002, 1208–1209 RSC.
- P. Dabringhaus, M. Schorpp, H. Scherer and I. Krossing, Angew. Chem., Int. Ed., 2020, 59, 22023–22027 CrossRef PubMed.
- S. Harder, F. Feil and K. Knoll, Angew. Chem., Int. Ed., 2001, 40, 4261–4264 CrossRef PubMed.
- S. Harder, F. Feil and A. Weeber, Organometallics, 2001, 20, 1044–1046 CrossRef.
- D. F.-J. Piesik, K. Häbe and S. Harder, Eur. J. Inorg. Chem., 2007, 5652–5661 CrossRef.
- A. Weeber, S. Harder, H. H. Brintzinger and K. Knoll, Organometallics, 2000, 19, 1325–1332 CrossRef.
- S. Garratt, A. Guerrero, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2004, 43, 2166–2169 CrossRef CAS PubMed.
- M. Bochmann and D. M. Dawson, Angew. Chem., Int. Ed. Engl., 1996, 35, 2226–2228 CrossRef CAS.
- J. S. Kim, L. M. Wojcinski, S. Liu, J. C. Sworen and A. Sen, J. Am. Chem. Soc., 2000, 122, 5668–5669 CrossRef CAS.
- M. P. Coles and R. F. Jordan, J. Am. Chem. Soc., 1997, 119, 8125–8126 CrossRef CAS.
- M. Bruce, V. C. Gibson, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 2523–2524 RSC.
- P. A. Cameron, V. C. Gibson, C. Redshaw, J. A. Segal, M. D. Bruce, A. J. P. White and D. J. Williams, Chem. Commun., 1999, 1883–1884 RSC.
- A. Guerrero, K. Kulbaba and M. Bochmann, Macromolecules, 2007, 40, 4124–4126 CrossRef CAS.
- I. V. Vasilenko and S. V. Kostjuk, J. Macromol. Sci., Part A: Pure Appl. Chem., 2021, 58, 725–735 CrossRef CAS.
- O. Nuyken and S. D. Pask, Polymers, 2013, 5, 361–403 CrossRef.
- S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861–2871 CrossRef.
- R. B. Grubbs and R. H. Grubbs, Macromolecules, 2017, 50, 6979–6997 CrossRef.
- A. J. Nijenhuis, D. W. Grijpma and A. J. Pennings, Macromolecules, 1992, 25, 6419–6424 CrossRef.
- Y. Sun, J. Xiong, Z. Dai, X. Pan, N. Tang and J. Wu, Inorg. Chem., 2016, 55, 136–143 CrossRef.
- J. Hu, C. Kan, H. Wang and H. Ma, Macromolecules, 2018, 51, 5304–5312 CrossRef.
- J. Bhattacharjee, A. Harinath, H. P. Nayek, A. Sarkar and T. K. Panda, Chem. – Eur. J., 2017, 23, 9319–9331 CrossRef PubMed.
- C. A. Wheaton and P. G. Hayes, Chem. Commun., 2010, 46, 8404–8406 RSC.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS PubMed.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433–4451 CrossRef.
- M. Bouyahyi, E. Grunova, N. Marquet, E. Kirillov, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Organometallics, 2008, 27, 5815–5825 CrossRef.
- N. Ikpo, S. M. Barbon, M. W. Drover, L. N. Dawe and F. M. Kerton, Organometallics, 2012, 31, 8145–8158 CrossRef.
- M. Haddad, M. Laghzaoui, R. Welter and S. Dagorne, Organometallics, 2009, 28, 4584–4592 CrossRef CAS.
- W. Braune and J. Okuda, Angew. Chem., Int. Ed., 2003, 42, 64–68 CrossRef CAS PubMed.
- S. Ghosh, E. Glöckler, C. Wölper, A. Tjaberings, A. H. Gröschel and S. Schulz, Dalton Trans., 2020, 49, 13475–13486 RSC.
- A. M. Dąbrowska, A. Hurko, K. Durka, M. Dranka and P. Horeglad, Organometallics, 2021, 40, 1221–1234 CrossRef.
- P. Horeglad, G. Szczepaniak, M. Dranka and J. Zachara, Chem. Commun., 2012, 48, 1171–1173 RSC.
- P. Horeglad, M. Cybularczyk, B. Trzaskowski, G. Z. Zukowska, M. Dranka and J. Zachara, Organometallics, 2015, 34, 3480–3496 CrossRef CAS.
- D. Myers, A. J. P. White, C. M. Forsyth, M. Bown and C. K. Williams, Angew. Chem., Int. Ed., 2017, 56, 5277–5282 CrossRef CAS PubMed.
- I. Yu, A. Acosta-Ramírez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758–12773 CrossRef CAS.
- W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252 CrossRef CAS PubMed.
- Z. Cai, D. Xiao and L. H. Do, Comments Inorg. Chem., 2019, 39, 27–50 CrossRef CAS.
- F. Hild, P. Haquette, L. Brelot and S. Dagorne, Dalton Trans., 2010, 39, 533–540 RSC.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072–4073 CrossRef CAS.
- X. Pang, H. Du, X. Chen, X. Wang and X. Jing, Chem. – Eur. J., 2008, 14, 3126–3136 CrossRef CAS PubMed.
- Y. Zhou, G. S. Nichol and J. A. Garden, Eur. J. Inorg. Chem., 2022, e202200134 CAS.
- W. Gruszka, A. Lykkeberg, G. S. Nichol, M. P. Shaver, A. Buchard and J. A. Garden, Chem. Sci., 2020, 11, 11785–11790 RSC.
- W. Gruszka, H. Sha, A. Buchard and J. A. Garden, Catal. Sci. Technol., 2022, 12, 1070–1079 RSC.
- W. Gruszka and J. A. Garden, Chem. Commun., 2022, 58, 1609–1612 RSC.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef.
- N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Peña Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef PubMed.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1985, 107, 1358–1364 CrossRef.
- K. Soga, E. Imai and I. Hattori, Polym. J., 1981, 13, 407–410 CrossRef.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef.
- C.-M. Chen, X. Xu, H.-Y. Ji, B. Wang, L. Pan, Y. Luo and Y.-S. Li, Macromolecules, 2021, 54, 713–724 CrossRef.
- J. Diebler, H. Komber, L. Häußler, A. Lederer and T. Werner, Macromolecules, 2016, 49, 4723–4731 CrossRef CAS.
- D. Wannipurage, T. S. Hollingsworth, F. Santulli, M. Cozzolino, M. Lamberti, S. Groysman and M. Mazzeo, Dalton Trans., 2020, 49, 2715–2723 RSC.
- A. J. Gaston, Z. Greindl, C. A. Morrison and J. A. Garden, Inorg. Chem., 2021, 60, 2294–2303 CrossRef CAS.
- A. Rae, A. J. Gaston, Z. Greindl and J. A. Garden, Eur. Polym. J., 2020, 138, 109917 CrossRef CAS.
- G. Trott, P. K. Saini and C. K. Williams, Phil. Trans. R. Soc. A, 2016, 374, 20150085 CrossRef.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef.
- Y. Xiao, Z. Wang and K. Ding, Chem. – Eur. J., 2005, 11, 3668–3678 CrossRef.
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef PubMed.
- S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579–4582 RSC.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef PubMed.
- A. Thevenon, A. Cyriac, D. Myers, A. J. P. White, C. B. Durr and C. K. Williams, J. Am. Chem. Soc., 2018, 140, 6893–6903 CrossRef CAS PubMed.
- B. A. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef CAS PubMed.
- F. Isnard, M. Lamberti, C. Pellecchia and M. Mazzeo, ChemCatChem, 2017, 9, 2972–2979 CrossRef CAS.
- G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC.
- A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS.
- A. C. Deacy, C. B. Durr and C. K. Williams, Dalton Trans., 2020, 49, 223–231 RSC.
- W. T. Diment, G. L. Gregory, R. W. F. Kerr, A. Phanopoulos, A. Buchard and C. K. Williams, ACS Catal., 2021, 11, 12532–12542 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |