
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePossible chemical and physical scenarios towards biological homochirality†
Quentin
Sallembien
 *a,
Laurent
Bouteiller
a,
Jeanne
Crassous
*b and
Matthieu
Raynal
*a
*a,
Laurent
Bouteiller
a,
Jeanne
Crassous
*b and
Matthieu
Raynal
*a
aSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, Equipe Chimie des Polymères, 4 Place Jussieu, 75005 Paris, France. E-mail: quentin.sallembien.2017@enscbp.fr; matthieu.raynal@sorbonne-universite.fr
bUniv Rennes, CNRS, Institut des Sciences Chimiques de Rennes, ISCR-UMR 6226, F-35000 Rennes, France. E-mail: jeanne.crassous@univ-rennes1.fr
First published on 4th April 2022
Abstract
The single chirality of biological molecules in terrestrial biology raises more questions than certitudes about its origin. The emergence of biological homochirality (BH) and its connection with the appearance of life have elicited a large number of theories related to the generation, amplification and preservation of a chiral bias in molecules of life under prebiotically relevant conditions. However, a global scenario is still lacking. Here, the possibility of inducing a significant chiral bias “from scratch”, i.e. in the absence of pre-existing enantiomerically-enriched chemical species, will be considered first. It includes phenomena that are inherent to the nature of matter itself, such as the infinitesimal energy difference between enantiomers as a result of violation of parity in certain fundamental interactions, and physicochemical processes related to interactions between chiral organic molecules and physical fields, polarized particles, polarized spins and chiral surfaces. The spontaneous emergence of chirality in the absence of detectable chiral physical and chemical sources has recently undergone significant advances thanks to the deracemization of conglomerates through Viedma ripening and asymmetric auto-catalysis with the Soai reaction. All these phenomena are commonly discussed as plausible sources of asymmetry under prebiotic conditions and are potentially accountable for the primeval chiral bias in molecules of life. Then, several scenarios will be discussed that are aimed to reflect the different debates about the emergence of BH: extra-terrestrial or terrestrial origin (where?), nature of the mechanisms leading to the propagation and enhancement of the primeval chiral bias (how?) and temporal sequence between chemical homochirality, BH and life emergence (when?). Intense and ongoing theories regarding the emergence of optically pure molecules at different moments of the evolution process towards life, i.e. at the levels of building blocks of Life, of the instructed or functional polymers, or even later at the stage of more elaborated chemical systems, will be critically discussed. The underlying principles and the experimental evidence will be commented for each scenario with particular attention on those leading to the induction and enhancement of enantiomeric excesses in proteinogenic amino acids, natural sugars, and their intermediates or derivatives. The aim of this review is to propose an updated and timely synopsis in order to stimulate new efforts in this interdisciplinary field.
 Quentin Sallembien | Quentin Sallembien was born in 1993 in Strasbourg, France. He got an engineering diploma from ENSCBP – Bordeaux INP (2017) and obtained in 2021 a doctoral degree under the supervision of Dr Matthieu Raynal (Sorbonne Université). His PhD research focused on controlling the handedness of supramolecular helical polymers by means of circularly polarized light and chiral additives. He is currently a postdoctoral researcher in the group of Prof. Renaud Nicolaÿ at ESPCI Paris, dealing with polyolefin vitrimers. |
 Laurent Bouteiller | Laurent Bouteiller is the CNRS Research Director and head of the Polymer Chemistry lab at Sorbonne Université in Paris. His main research interests are focused on the interface between polymer science and supramolecular chemistry, which involves using non-covalent interactions to assemble molecules. The low energies involved make it possible to control the formation of complex architectures and to obtain materials with reversible properties of interest in various fields (e.g. rheology, catalysis, adhesion, and coatings). |
 Jeanne Crassous | Dr Jeanne Crassous (born Costante) received her PhD in 1996 under the supervision of Prof. André collet (ENS, Lyon, France), working on the absolute configuration of bromochlorofluoromethane. After a one-year postdoctoral period studying the chirality of fullerenes in Prof. François Diederich's group (ETH Zurich, Switzerland), she was appointed a CNRS researcher at the ENS Lyon in 1998. In 2005, she joined the “Institut des Sciences Chimiques de Rennes” (University of Rennes, France) and was appointed a CNRS Director of Research in 2010. Her group works on many fields related to chirality (organometallic and heteroatomic helicenes, fundamental aspects of chirality such as parity violation effects, chiroptical activity such as electronic and vibrational circular dichroism and circularly polarized luminescence). In 2020, she received the National Prize of the Organic Chemistry Division of the French Chemical Society (DCO-SCF). |
 Matthieu Raynal | Matthieu Raynal got his PhD degree under the supervision of Dr P. Braunstein in 2009 (Strasbourg). He conducted postdoctoral studies at UPMC with L. Bouteiller (Paris) and in the group of Prof. P. W. N. M. van Leeuwen at ICIQ (Tarragona, Spain). In 2012, he was appointed as a CNRS researcher at Sorbonne Université, Paris. He is fascinated by how non-covalent interactions can be designed to control the outcome of a catalytic reaction, i.e. supramolecular catalysis. His group is currently developing supramolecular helical catalysts with particular efforts devoted to improving their chirality amplification and switchable properties. His research activities also concern the design of functional chiral assemblies and the structure–property relationship of supramolecular polymers. He recently co-edited with P. W. N. M. van Leeuwen the book “Supramolecular Catalysis: New Directions and Developments” (2022 Wiley-VCH GmbH). |
1. Introduction
In 1884, Lord Kelvin used the word chirality—derived from the Proto-Indo-European *ǵhesr through the Ancient Greek χειρ (kheír), which both mean ‘hand’—and gave the following definition: “an object is chiral if and only if it is not superimposable on its mirror image”.1 Additionally, chirality can be described based on symmetry aspects: an object is chiral if it possesses no symmetry elements of the second kind (i.e. if it is devoid of any improper axis of rotation).2 Whilst the manifestation of chirality at the macroscopic scale sparked human's curiosity from antiquity, its observation at the molecular and sub-atomic levels is relatively recent. In the 19th-century, advances made in optics,3 crystallography4 and chemistry5 paved the way to the scientific study of molecular chirality (named ‘molecular dissymmetry’ by Louis Pasteur)6 which soon after manifested itself in a variety of studied phenomena. The term “chirality” took additionally almost 80 years to be introduced in chemistry by Kurt Mislow (1962).7Chirality is found at all scales in matter, from elementary particles to cucumber tendrils,8 from screws to spiral galaxies, in living and inert systems.9 It is also an everyday concern in industry (e.g. pharma, agribusiness, and cosmetics)10–14 as well as in fundamental research (visible in countless conferences encompassing not only chemistry, physics, and biology but also economy and arts).15
Homochirality of life refers to the fact that Nature has chosen a specific handedness. Homochirality is a fascinating aspect of terrestrial biology: all living systems are composed of L-amino acids and D-sugars‡ to such an elevated extent that the occurrence of the molecules of life with different configurations (e.g.D-amino acids) is seen as a curiosity.16 Clearly, the perfect level of selectivity reached by evolution and preserved along billion years, is out of reach for currently developed artificial systems. Homochirality and life are so closely related that homochirality in Nature is considered as a stereochemical imperative.17 For example, D-sugars are building blocks of helically shaped DNA and RNA macromolecules, which store genetic information and encode the synthesis of proteins through the ligation of their constituting amino acids. Glucose monomers in glycogen, starch and cellulose also have a D configuration. This suggests that the chirality, structure, and functions of these biomacromolecules are intimately related.18
In 1857, Louis Pasteur revealed the dramatic difference in the fermentation rate of two tartaric acid enantiomers with a yeast microorganism, thus uncovering biological enantioselectivity.19–21 Pasteur was convinced that chirality was a manifestation of life, and unsuccessfully looked for the link between physical forces ruling out the Cosmos and the molecular dissymmetry observed in natural products. In 1886, an Italian chemist Arnaldo Piutti22 succeeded in isolating (R)-asparagine, mirror-image of the tasteless amino-acid (S)-asparagine, and found that it was intensely sweet.23 These discoveries refer to the link between the handedness of chiral substances and their biological properties but do not explain the origin of biological homochirality (BH).
Despite the extensive literature, the emergence of BH remains a conundrum.24–44 The key points of this intricate topic can be summarized as: how, when and where did single chirality appear and eventually lead to the emergence of life (Fig. 1).45–48 Along this line, the question of the creation of the original chiral bias appears critical (box “how?” in Fig. 1). Huge efforts have been dedicated to decipher which processes may lead to the generation of a chiral bias without the action of pre-existing enantiomerically enriched chemical species, that is without using the commonly employed routes in stereoselective synthesis. The creation of a chiral bias “from scratch”, often referred to as absolute asymmetric synthesis30,31,49,50 and spontaneous deracemization,41,51,52 actually encompasses a large variety of phenomena. Here, a distinction can be made between chiral biases that: (i) are inherent to the nature of matter itself, (ii) originate from the interaction of molecules with physical fields, particles, spins or surfaces, or (iii) emerge from the mutual interaction between molecules (Fig. 1). The first category (i) corresponds to the fact that a racemate deviates infinitesimally from its ideal equimolar composition deterministically, i.e. in direction of the same enantiomer for a given racemate, as a result of parity violation in certain interactions within nuclei.53,54 The second category (ii) refers to natural physical fields (gravitational, magnetic, and electric), light and their combinations, which under certain conditions constitute truly chiral fields,30 but also to a range of inherently chiral sources such as chiral light and polarized particles (mostly electrons), polarized electron spins, vortices, or surfaces.44 The third category (iii) encompasses processes that lead to the spontaneous emergence of chirality in the absence of detectable chiral physical and chemical sources, upon destabilization of the racemic state and stabilization of a scalemic or homochiral state. Such spontaneous mirror symmetry breaking (SMSB) phenomena40 involve interactions between molecules through auto-catalytic processes which under far-from-equilibrium conditions may lead to the emergence of enantiopure molecules. The topic has recently undergone significant progress thanks to numerous theoretical models and experimental validations, namely the deracemization of conglomerates through Viedma ripening55 and the asymmetric auto-catalysis with the Soai reaction.56 Importantly, the plausibility of the aforementioned chirality induction processes in the context of BH will depend on several parameters such as: the extent of asymmetric induction they may provide, their mode of action, i.e. if they are unidirectional (deterministic towards a single enantiomer) or bidirectional (leading to either type of enantiomers), their relevance according to prebiotic conditions present on earth 4 billion years ago, the scope of molecules it could be applied to, and their validation by experimental evidence. The first three parts of this review will provide an updated version of phenomena i–iii that are commonly discussed as plausible sources of asymmetry under prebiotic conditions and can thus be potentially accountable for the primeval chiral bias in molecules of life.
 | ||
| Fig. 1 Schematic representation of the questions and potential answers which are fundamentally related to the conundrum of the origin of the homochirality of life. This review is divided into 4 parts as indicated in the scheme. PVED: parity-violating energy difference. SMSB: spontaneous mirror symmetry breaking. | ||
However, uncovering plausible mechanisms towards the emergence of a chiral bias is not enough per se for elucidating the origin of BH. Additional fundamental challenges such as the extra-terrestrial or terrestrial origin of molecule of life precursors (box “where?” in Fig. 1), the mechanism(s) for the propagation and enhancement of the original chiral bias (box “how 2?” in Fig. 1) and the chemical/biological pathways leading to functional bio-relevant molecules are key aspects to propose a credible scenario. The detection of amino acids and sugars with preferred L and D configurations, respectively, on carbonaceous meteorites57 instigated further research for determining plausible mechanisms for the production of chiral molecules in an interstellar environment and their subsequent enantiomeric enrichment.58,59 Alternatively, hydrothermal vents in primeval oceans constitute an example of reaction domains often evoked for prebiotic chemistry which may also include potential sources of asymmetry such as high-speed microvortices.60 Some mechanisms are known for increasing an existing e.e., such as the self-disproportionation of enantiomers (SDE),61 non-linear effects in asymmetric catalysis,62,63 and stereoselective polymerization.64 Noteworthy in the present context, these processes may be applied to increase the optical purity of prebiotically relevant molecules. However, a general amplification scheme which is valid for all molecules of life is lacking.
The temporal sequence between chemical homochirality, BH and life emergence is another intricate point (box “when?” in Fig. 1). Tentative explanations try to build-up either abiotic theories considering that single chirality is created before the living systems or biotic theories suggesting that life preceded homochirality.44 Purely abiotic theories refer to reactions or physicochemical processes involving low-molecular weight organic molecules presumably present in the prebiotic soup.38,65 From a different angle, polymerization of activated building blocks is also discussed as a possible stage for the induction/enhancement of chirality,64 even though prebiotic mechanisms towards these essential-to-life macromolecules remain highly elusive.45–48 In the fifth part of this review, we will propose an update of the most plausible chemical and physical scenarios towards BH, with emphasis on the underlying principles and the experimental evidence, showing merits and limitations of each mechanism. Notably, relevant experimental investigations conducted with building blocks of life: proteinogenic amino acids, natural sugars, and their intermediates or derivatives, will be commented in regards of the different scenarios.
Ultimately, the aim of this literature review is to familiarize the novice with research dealing with BH, and to propose to the expert an updated and timely synopsis of this interdisciplinary field.
2. Parity violation (PV) and parity-violating energy difference (PVED)
“Videmus nunc per speculum in aenigmate,” (Holy Bible, I Cor. XIII, 12) which can be translated into “At present, we see indistinctly, as in a mirror” refers to the intuition that a mirror reflection is a distorted representation of the reality. The perception of a different nature of mirror-image objects is also found in the modern literature. In his famous novel “Through the Looking-Glass” by Lewis Caroll Alice raises important questions: ‘How would you like to live in Looking-glass House, Kitty? I wonder if they’d give you milk in there? Perhaps Looking-glass milk isn’t good to drink…”The Universe is constituted of elementary particles which interact through fundamental forces, namely the electromagnetic, strong, weak, and gravitational forces. Until the mid-20th century, fundamental interactions were thought to equally operate in a physical system and its image built through space inversion. Indeed, these laws were assumed by physicists to be conserved under the parity operator P (which transforms the spatial coordinates x, y, z into −x, −y, −z), i.e. parity-even. However, in 1956, Lee and Yang highlighted that parity was only conserved for strong and electromagnetic forces, and proposed experiments to test it for weak interactions.66 A few months later, Wu experimentally demonstrated that the parity symmetry is indeed broken in weak forces (which are hereby parity-odd),67 by showing that the transformation of unstable 60Co nuclei into 60Ni, through the β−-decay of a neutron into a proton, emits electrons of only left-handedness. In fact, solely left-handed electrons were emitted since W+ and W− bosons (abbreviated as W± bosons), which mediate the weak charged-current interactions, only couple with left-handed particles. Right-handed particles are not affected by weak interactions carried out by W± bosons and consequently, neutrinos, that are only generated by processes mediated by W± bosons, are all left-handed in the universe.68
The weak neutral current interactions, mediated by the Z0 boson (sometimes called Z forces), are without charge exchange and, just like the charged ones, violate the parity symmetry.69–73 Thus, all weak interactions, carried out by W± or Z0 bosons, break the fundamental parity symmetry.
Parity violation has been observed in nuclear67 and atomic Physics.74–77 In consequence, the contribution of the Z force between the nuclei and electrons produces an energy shift between the two enantiomers of a chiral molecule. The lower-energy enantiomer would thus be present in slight excess in an equilibrium mixture; this imbalance may provide a clue to the origin of biomolecular homochirality, i.e. why chiral molecules usually occur in a single enantiomeric form in nature. Such a tiny parity violation energy difference (a PVED of about 10−17 kT at 300 K) should be measurable by any absorption spectroscopy provided that ultra-high resolution can be reached.78–81 Over the past decades, various experiments have been proposed to observe parity violation in chiral molecules, including measurements of PV frequency shifts in NMR spectroscopy,82 measurements of time dependence of optical activity,83 and direct measurement of the absolute PV energy shift of the electronic ground state.79–81,84
However, it has never been unequivocally observed at the molecular level to date. Note that symmetry violation of time reversal (T) and of charge parity (CP) is actually recovered in the CPT symmetry, i.e., in the “space-inverted anti-world made of antimatter”.85 Quantitative calculations of this parity-violating energy difference between enantiomers have been improved during the last four decades,86–90 to give for example about 10−12 J mol−1 for CHFClBr.91,92 Although groups of Crassous/Darquié in France78,93–98 and Quack in Switzerland99–103 have been pursuing an experimental effort to measure PVED, thanks to approaches based on spectroscopic techniques and/or tunneling processes, no observation has unambiguously confirmed it yet. However, thanks to the combination of the contribution from the weak interaction Hamiltonian (Z3) and from the spin orbit coupling (Z2), the parity violating energy difference strongly increases with increasing nuclear charge with a commonly accepted Z5 scaling law, thus chiral heavy metal complexes might be favourable candidates for future observation of PV effects in chiral molecules.94,96 Other types of experiments have been proposed to measure PV effects, such as nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR), microwave (MW) or Mössbauer spectroscopy.79 Note that other phenomena have been taken into consideration to measure PVED such as in Bose–Einstein condensation, but those were not conclusive.104,105
The tempting idea that PVED could be the source of the tiny enantiomeric excess amplified to the asymmetry of life was put forward by Ulbricht in 1959106,107 and by Yamagata in 1966.108 With this in mind, Mason, Tranter and MacDermott109–122 defended in the eighties and early nineties that (S)-amino acids, D-sugars, α-helix or β-sheet secondary structures, and other natural products and secondary structures of biological importance are more stable than their enantiomorph due to PVED.54 However, Quack89,123 and Schwerdtfeger124,125 independently refuted these results on the strength of finer calculations, and Lente126,127 asserted that a PVED of around 10−13 J mol−1 causes an excess of only 6 × 106 molecules in one mole (against 1.9 × 1011 for the standard deviation). In reply, MacDermott claimed, by means of a new generation of PVED computations, that the enantiomeric excess of four gaseous amino acids found in the Murchison meteorite (in the solid state) could originate from their PVED.128,129 Whether PVED could have provided a sufficient bias for the emergence of BH likely depends on the related amplification mechanism, a point that will be discussed in more detail in part 4.
3. Chiral fields
Physical fields, polarized particles, polarized spins and surfaces are commonly discussed as potential chiral inducers of enantiomeric excesses in organic molecules. The aim of this part is to present selected chiral fields along with experimental observations which are relevant in the context of elucidating BH.3.1 Physical fields
 | ||
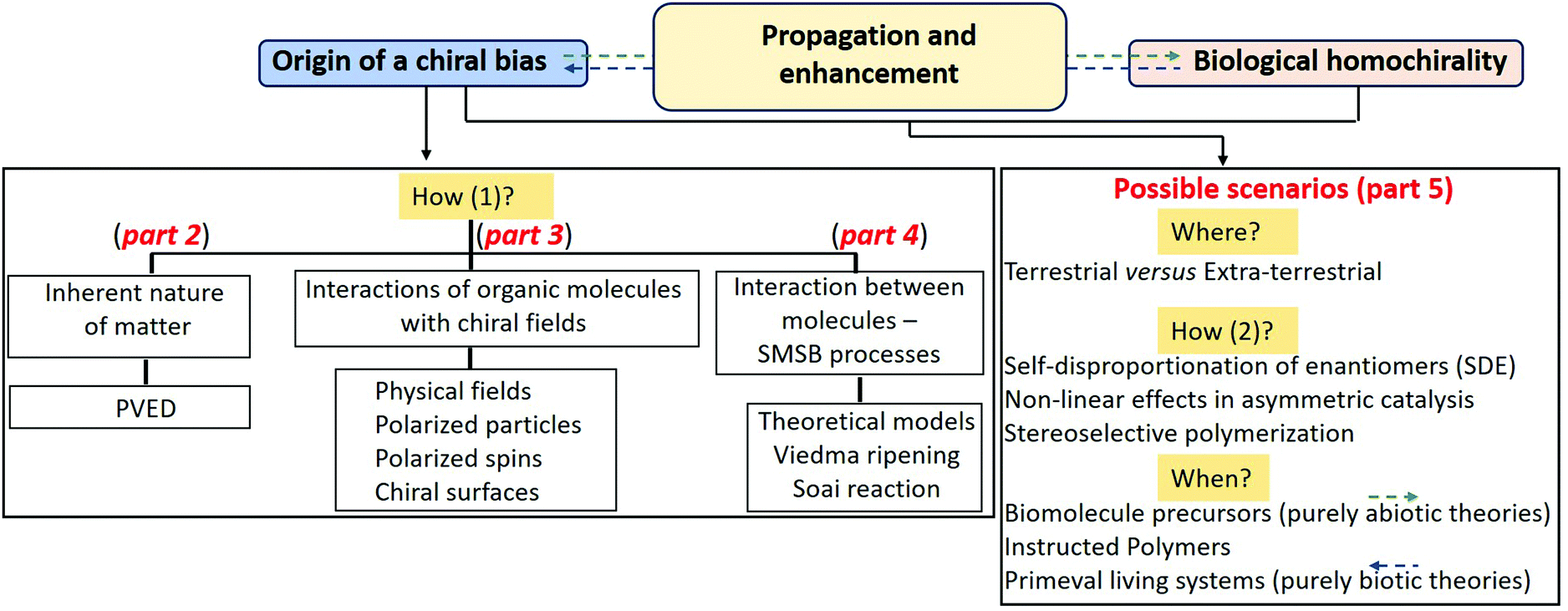
| Fig. 2 Distinction between “true” and “false” chirality30,130 by considering the effect of parity (P) and time (T) reversal on spinning cones (a) and aligned magnetic and electric fields (b). | ||
Importantly, only when interacting with a truly chiral system the energy of enantiomeric probes can be different (corresponding to diastereomeric situations), while no loss of degeneration in energy levels can happen in a falsely chiral system; however, asymmetry could be obtained for processes out of thermodynamic equilibrium.30,31 Based on these definitions, truly chiral forces may lift the degeneracy of enantiomers and induce enantioselection in a reaction system reaching its stationary state, while an influence of false chirality is only possible for kinetically controlled reaction outputs, since in this case the enantiomers remain strictly degenerate and only the breakdown of the reaction path microreversibility occurs.41 Furthermore, the extent of chiral induction that can be achieved by a chiral physical field is intimately related to the nature of its interaction with matter, i.e. with prebiotically relevant organic molecules in the context of BH. A few examples of physical fields for absolute asymmetric synthesis are mentioned in the next paragraphs.
 | ||
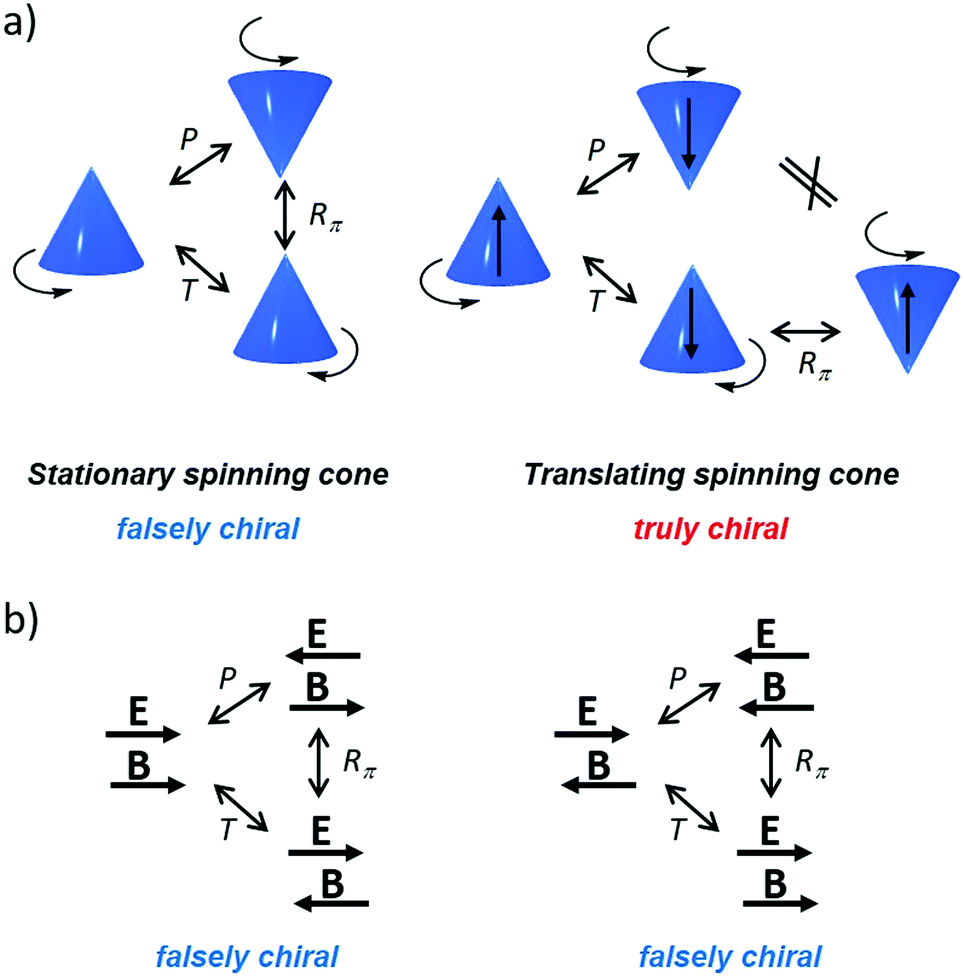
| Fig. 3 (a) Schematic representation of MChD for a racemate of a metal complex: the unpolarised light is preferentially absorbed by Λ versus Δ enantiomers. Reprinted from ref. 136 with permission from Wiley-VCH, copyright 2020. (b) Photoresolution of the chromium(III) tris(oxalato) complex. Plot of the e.e. after irradiation with unpolarised light for 25 min at λ = 695.5 nm, as a function of the magnetic field, with an irradiation direction k, either parallel or perpendicular to the magnetic field.137 | ||
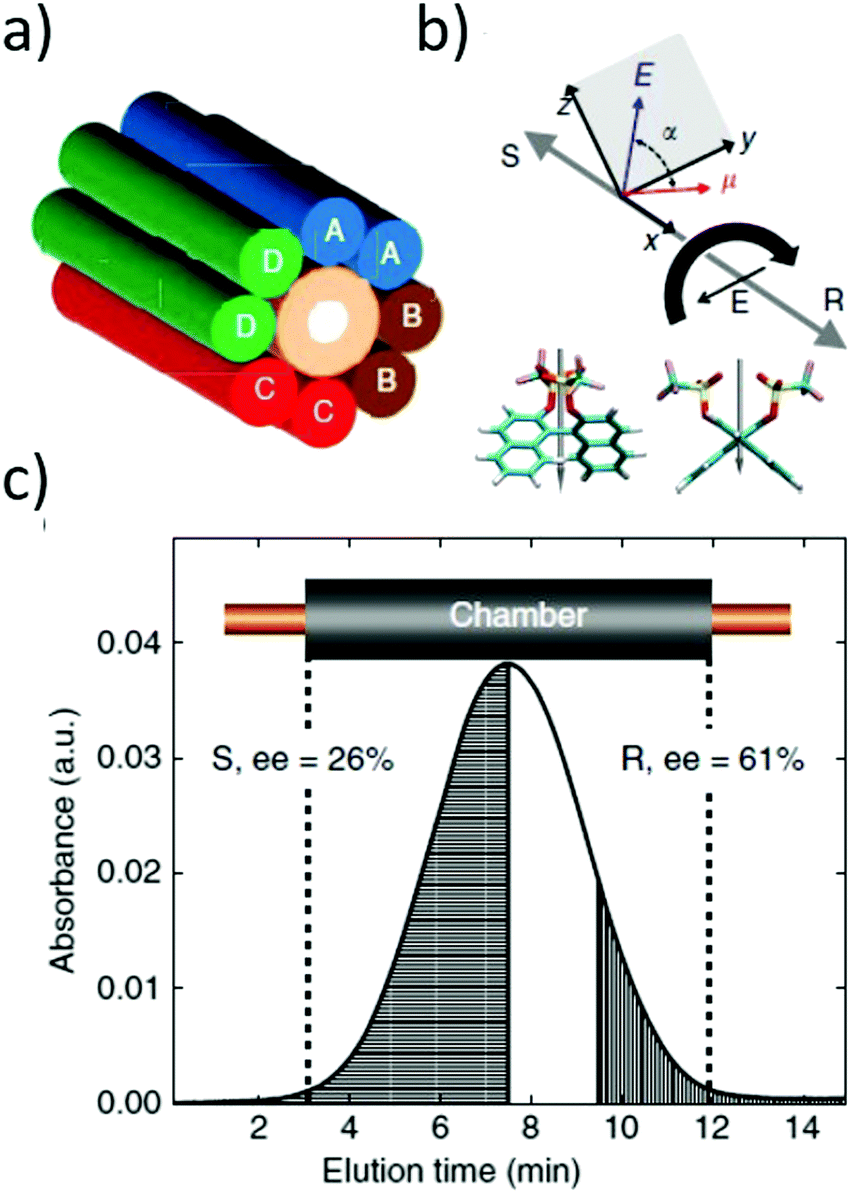 | ||
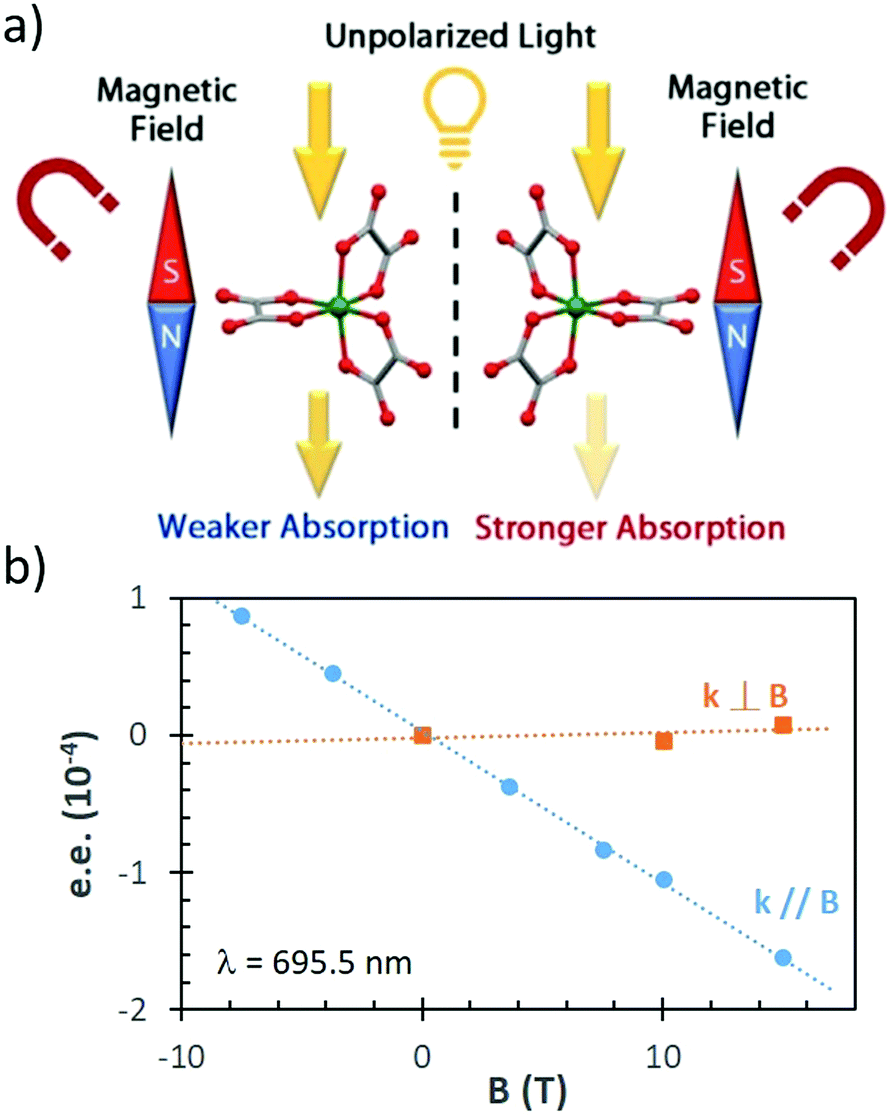
| Fig. 4 (a) Schematic representation of the experimental set-up for the separation of chiral molecules placed in a microfluidic capillary surrounded by rotating electric fields (A–D electrodes). (b) Expected directions of motion of the enantiomers of 1,1′-bi-2-naphthol bis(trifluoromethanesulfonate) for the indicated direction of rotation of REF (curved black arrow). α is the relative angle between the electric dipole moment and electric field. The grey arrows show the opposite directions of motion of the enantiomers. (c) Absorbance chromatogram from the in-line detector of a slug of (rac)-1,1′-bi-2-naphthol bis(trifluoromethanesulfonate) after exposure to clockwise REF for 45 h. The sample collected from the shaded left side of the chromatogram had an e.e. of 26% in favour of the (S) enantiomer, while the right shaded section of the chromatogram had an e.e. of 61% for the (R) enantiomer. Reprinted ref. 138. Copyright 2015. Springer Nature under Creative Commons Attribution 4.0 International License https://creativecommons.org/licenses/by/4.0/. | ||
 | ||
| Fig. 5 Control of the handedness of TPPS3 helical assemblies by the relative orientation of the angular momentum of rotation (L) and the effective gravity (Geff). TPPS3: tris-(4-sulfonatophenyl)phenyl porphyrin. Reprinted from ref. 161 with permission from Nature publishing group, copyright 2012. | ||
3.2 Polarized radiations and spins
Circular dichroism is a phenomenon, corresponding to the differential absorption of l-CPL and r-CPL at a given wavelength in the absorption region of an optically active material, as well the spectroscopic method that measures it.173,174 Enantiomers absorbing CPL of one handedness constitute non-degenerated diastereoisomeric systems, based on the interaction between two distinct chiral influences, one chemical and the other physical. Thus, one state of this system is energetically favoured, and one enantiomer preferentially absorbs CPL of one polarization state (l- or r-CPL).
The dimensionless Kuhn anisotropy (or dissymmetry) factor g allows the quantitative description of the chiroptical response of enantiomers (eqn (1)). The Kuhn anisotropy factor is expressed by the ratio between the difference in molar extinction coefficients of l-CPL and r-CPL (Δε), and the global molar extinction coefficient (ε), where εL and εR are the molar extinction coefficients for left- and right-handed CPL, respectively.175 It ranges from −2 to +2, for a total absorption of right- and left-handed CPL, respectively, and is wavelength dependent. Enantiomers have equal but opposite g values, corresponding to their preferential absorption of one CPL handedness.
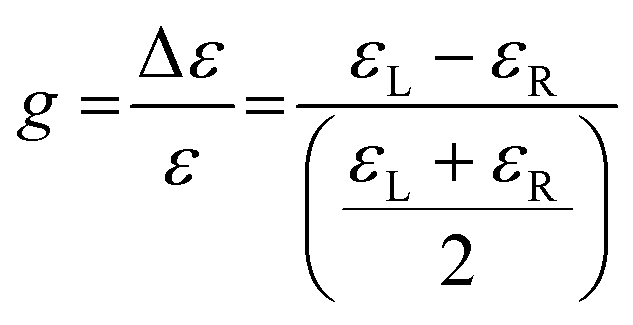 | (1) |
 | ||
Fig. 6 Simplified kinetic schemes for asymmetric (a) photolysis, (b) photoresolution and (c) photosynthesis with CPL. SR and SS are substrate enantiomers and 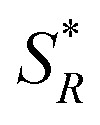 and and 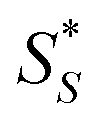 are their photoexcited states. PR and PS are the products generated from the respective photoexcited states. The thick line represents the preferential absorption of CPL by one of the enantiomers. [SS] > [SR] for asymmetric photolysis and photoresolution processes whilst [PR] > [PS] for asymmetric photosynthesis. are their photoexcited states. PR and PS are the products generated from the respective photoexcited states. The thick line represents the preferential absorption of CPL by one of the enantiomers. [SS] > [SR] for asymmetric photolysis and photoresolution processes whilst [PR] > [PS] for asymmetric photosynthesis. | ||
The first CPL-induced asymmetric partial resolution dates back to 1968 thanks to Stevenson and Verdieck, who worked with octahedral oxalato complexes of chromium(III).179 Asymmetric photoresolution was further investigated for small organic molecules,180,181 macromolecules182 and supramolecular assemblies.183 A number of functional groups such as overcrowded alkene, azobenzene, diarylethene, α,β-unsaturated ketone or fulgide were specifically-designed to enhance the efficiency of the photoresolution process.58
Kagan et al. pioneered the field of asymmetric photosynthesis with CPL in 1971, through examining hexahelicene photocyclization in the presence of iodine.184 The following year, Calvin et al. reported an e.e. of up to 2% for an octahelicene produced under similar conditions.185 Enantioenrichment by photoresolution and photosynthesis with CPL is limited in scope, since it requires molecules with high g values to be detected, and in intensity, since it is limited to g/2.
Since its discovery by Kuhn et al. ninety years ago,186,187 through the enantioselective decomposition of ethyl-α-bromopropionate and N,N-dimethyl-α-azidopropionamide, the asymmetric photolysis of racemates has attracted a lot of interest. In the common case of two competitive pseudo-first order photolytic reactions with unequal rate constants, kS and kR, for the (S) and (R) enantiomers, respectively, and if the anisotropies are close to zero, the enantiomeric excess induced by asymmetric photolysis can be approximated as eqn (2):188
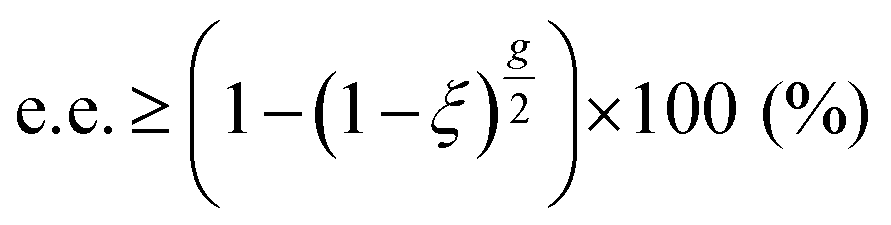 | (2) |
In 1974, the asymmetric photodecomposition of racemic camphor reported by Kagan et al. reached 20% e.e. at 99% completion, a long-lasting record in this domain.189 Three years later, Norden190 and Bonner et al.191 independently showed that enantioselective photolysis by UV-CPL was a viable source of symmetry-breaking for amino acids, by inducing an e.e. of up to 2% in aqueous solutions of alanine and glutamic acid,191 or 0.2% with leucine.190 Leucine was then intensively studied thanks to a relatively high anisotropy factor in the UV region.192 The e.e. was increased up to 1.3% in 2001 (ξ = 0.55) by Inoue et al. by exploiting the pH-dependence of the g value.193,194 In the early 2000s, Meierhenrich et al. got closer to astrophysically relevant conditions by irradiating samples in the solid state with synchrotron vacuum ultraviolet (VUV)-CPL (below 200 nm). This made it possible to avoid water absorption in the VUV, and allowed electronic transitions having higher anisotropy factors to be reached (Fig. 7).195 In 2005, a solid racemate of leucine was reported to reach 2.6% of e.e. after illumination with r-CPL at 182 nm (ξ not reported).196 More recently, the same team improved the selectivity of the photolysis process, thanks to amorphous samples of finely-tuned thickness, providing e.e. values of 5.2 ± 0.5% and 4.2 ± 0.2% for leucine,197 and alanine,198,199 respectively. A similar enantioenrichment was reached in 2014 with gaseous photoionized alanine,200 which constitutes an appealing result taking into account the detection of interstellar gases such as propylene oxide201 and glycine202 in star-forming regions.
 | ||
| Fig. 7 Anisotropy spectra (thick lines, left ordinate) of isotropic amorphous (R)-alanine (red) and (S)-alanine (blue), in the VUV and UV spectral regions. Dashed lines represent the enantiomeric excess (right ordinate) that can be induced by photolysis of rac-alanine with either left- (in red) or right- (in blue) circularly polarized light at ξ = 0.9999. Positive e.e. values correspond to scalemic mixture biased in favour of (S)-alanine. Note that enantiomeric excesses are calculated from eqn (2). Reprinted from ref. 192 with permission from Wiley-VCH, copyright 2017. | ||
Important studies in the context of BH reported the direct formation of enantio-enriched amino acids generated from simple chemical precursors, when illuminated with CPL. Takano et al. showed in 2007 that eleven amino acids could be generated upon CPL irradiation of macromolecular compounds, originating from proton-irradiated gaseous mixtures of CO, NH3 and H2O.203 Small e.e. values of +0.44 ± 0.31% and −0.65 ± 0.23% were detected for alanine upon irradiation with r- and l-CPL, respectively. Nuevo et al. irradiated interstellar ice analogues composed of H2O, 13CH3OH and NH3 at 80 K with CPL centred at 187 nm, which led to the formation of alanine with an e.e. of 1.34 ± 0.40%.204 The same team also studied the effect of CPL on regular ice analogues or organic residues coming from their irradiation in order to mimic the different stages of asymmetric induction in interstellar ices.205 Sixteen amino acids were identified, and five of them (including alanine and valine) were analysed by enantioselective two-dimensional gas chromatography, GC × GC,206 coupled to TOF mass spectrometry, to show enantioenrichments of up to 2.54 ± 0.28% e.e. Optical activities likely originated from the asymmetric photolysis of the amino acids initially formed as racemates. Advantageously, all five amino acids exhibited e.e. values of identical sign for a given polarization and wavelength, suggesting that irradiation by CPL could constitute a general route towards amino acids with a single chirality. Even though the chiral biases generated upon CPL irradiation are modest, these values can be significantly amplified through different physicochemical processes, notably those including auto-catalytic pathways (see Parts 4 and 5).
Of the leptons, electrons are one of the most universally present particles in ordinary materials. Spin-polarized electrons in nature are emitted with β− decay from radioactive nuclear particles derived from PV involving the weak nuclear interaction and spin-polarized positrons (the anti-particle of electrons) from β+ decay. In β−/β+-decay, with weak interaction, the spin angular momentum vectors of electrons/positrons are perfectly polarized as antiparallel/parallel to the vector direction of the kinetic momentum. In this meaning, spin-polarized electrons/positrons are “chiral radiation”, as well as are muons and neutrinos, which will be mentioned below. It is expected that spin-polarized leptons will induce reactions different from those triggered by CPL. For example, β− decay from 60Co is accompanied by circularly polarized gamma-rays.207 Similarly, spin-polarized muon irradiation has the potential to induce novel types of optical activities different from those of polarized photon and spin-polarized electron irradiation.
Single-handed polarized particles produced by supernovae explosions may thus interact with molecules in the proto-solar clouds.35,207–210 Left-handed electrons generated by β−-decay impinge on matter to form a polarized electromagnetic radiation through bremsstrahlung. At the end of fifties, Vester and Ulbricht suggested that these circularly-polarized “Bremsstrahlen” photons can induce and direct asymmetric processes towards a single direction upon interaction with organic molecules.107,211 From the sixties to the eighties,212–220 many experimental attempts to show the validity of the “V–U hypothesis”, generally by photolysis of amino acids in the presence of a number of β-emitting radionuclides or through self-irradiation of 14C-labeled amino acids, only led to poorly conclusive results.44,221,222 During the same period, the direct effect of high-energy spin-polarized particles (electrons, protons, positrons and muons) has been probed for the selective destruction of one amino acid enantiomer in a racemate, but without further success as reviewed by Bonner.44,54 More recent investigations by the international collaboration RAMBAS (RAdiation Mechanism of Biomolecular ASymmetry) claimed minute e.e. values (up to 0.005%) upon irradiation of various amino acid racemates with (natural) left-handed electrons.223,224
Other fundamental particles have been proposed to play a key role in the emergence of BH.207,209,225 Amongst them, electron antineutrinos have received particular attention, through the Supernova Neutrino Amino Acid Processing (SNAAP) model.226–228 Electron antineutrinos are emitted after a supernova explosion, to cool the nascent neutron star, and, by a similar reasoning to that applied with neutrinos, they are all right-handed. According to the SNAAP scenario, right-handed electron antineutrinos generated in the vicinity of neutron stars with strong magnetic and electric fields were presumed to selectively transform 14N into 14C, and this process depended on whether the spin of 14N was aligned or anti-aligned with that of the antineutrinos. Calculations predicted enantiomeric excesses for amino acids from 0.02% to a few percent, and a preferential enrichment in (S)-amino acids.
Despite important efforts, no experimental evidence has been reported to date in favour of a deterministic scenario for the generation of a chiral bias in prebiotic molecules.
In 2008, Rosenberg et al.231 irradiated adsorbed molecules of (R)-2-butanol or (S)-2-butanol on a magnetized iron substrate with low-energy SEs (10–15% of spin polarization) and measured a difference of about ten percent in the rate of CO bond cleavage of the enantiomers. Extrapolations of the experimental results suggested that an e.e. of 25% would be obtained after photolysis of the racemate at 98.6% of conversion. Importantly, the different rates in the photolysis of the 2-butanol enantiomers depend on the spin polarization of SEs, showing the first example of CISS.232–234 Later, SEs with a higher degree of spin polarization (60%) were found to dissociate Cl from epichlorohydrin (Epi) with a quantum yield 16% greater for the S form.229 To achieve this, electrons are produced by X-ray irradiation of a gold substrate and spin-filtered by a self-assembled overlayer of DNA before they reach the adlayer of Epi (Fig. 8).
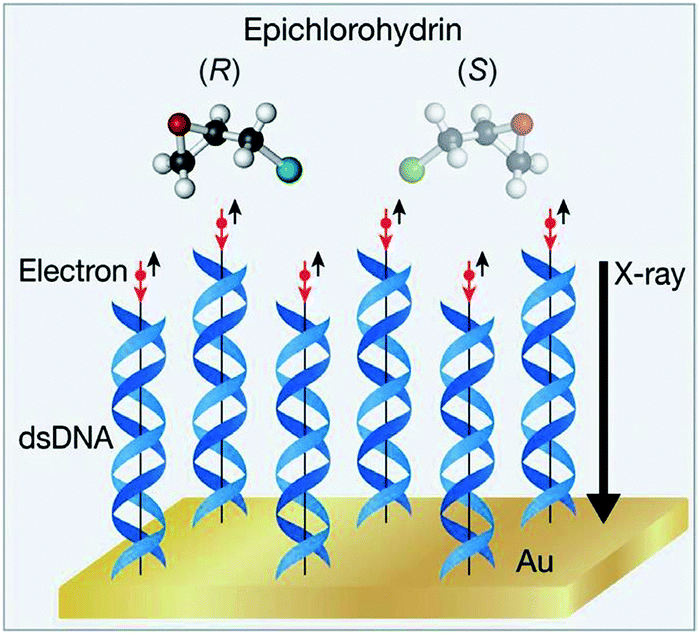 | ||
| Fig. 8 Enantioselective dissociation of epichlorohydrin by spin polarized SEs. Red (black) arrows indicate the electron's spin (motion direction, respectively). Reprinted from ref. 229 with permission from Wiley-VCH, copyright 2015. | ||
In 2018, Banerjee-Ghosh et al. showed that a magnetic field perpendicular to a ferromagnetic (FM) substrate can generate enantioselective adsorption of polyalanine, ds-DNA and cysteine.235 One enantiomer was found to be more rapidly adsorbed on the surface depending on the magnetization direction (Fig. 9). The effect is not attributed to the magnetic field per se but to the exchange interaction between the adsorbed molecules and surface electron spins, i.e. CISS.
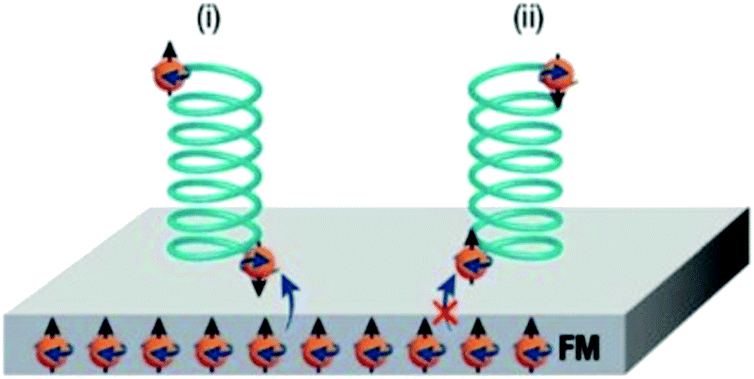 | ||
| Fig. 9 Suggested mechanism for the enantiospecific interaction triggered by chiral-induced spin selectivity. Enantiomers are sketched as opposite green helices and electrons as orange spheres with straight arrows indicating their spin orientation, which can be reversed for surface electrons by changing the magnetization direction. In contact with the perpendicularly magnetized FM surface, molecular electrons are redistributed to form a dipole, and the spin orientation at each pole depends on the chiral potentials of enantiomers. The interaction between the FM substrate and the adsorbed molecule (blue arrows) is favoured when the two spins are antiparallel leading to the preferential adsorption of one enantiomer over the other. Reprinted from ref. 230 with permission from the Royal Society of Chemistry, copyright 2019, under Creative Commons Attribution 3.0 Unported License https://creativecommons.org/licenses/by/3.0/. | ||
Enantioselective crystallization of initially racemic mixtures of asparagine, glutamic acid, and threonine, known to crystallize as conglomerates, was also observed on a ferromagnetic substrate surface (Ni(120 nm)/Au(10 nm)).230 The racemic mixtures were crystallized from aqueous solution on the ferromagnetic surfaces in the presence of two magnets, one pointing north and the other south, located at different sites of the surface. A clear enantioselective effect was observed in the formation of an excess of D- or L-crystals, depending on the direction of the magnetization orientation.
In 2020, the CISS effect was successfully applied to several asymmetric chemical processes, SEs acting as chiral reagents.236 Spin-polarized electrons, produced by a magnetized Ni/Au substrate coated with an achiral self-assembled monolayer (SAM) of carboxyl-terminated alkanethiols [HS–(CH2)x−1–COO−], caused an enantiospecific association of 1-amino-2-propanol enantiomers, leading to an e.e. of 20% in the reactive medium. The enantioselective electro-reduction of (1R/1S)-10-camphorsulfonic acid (CSA) into isoborneol was also governed by the spin orientation of SEs, injected through an electrode, with an e.e. of about 11.5% after the electrolysis of 80% of the initial amount of CSA.
Electrochirogenesis links the CISS process to biological homochirality through several theories, all based on an initial bias stemming from spin polarized electrons.232,237 Strong fields and radiations of neutron stars could align ferrous magnetic domains in interstellar dust particles, and produce spin-polarized electrons, able to create an enantiomeric excess into adsorbed chiral molecules. One enantiomer from a racemate in a cosmic cloud would merely accrete on a magnetized domain in an enantioselective manner as well. Alternatively, magnetic minerals of the prebiotic world, like pyrite (FeS2) or greigite (Fe3S4), might serve as an electrode in the asymmetric electrosynthesis of amino acids or purines, or as a spin filter in the presence of an external magnetic field, e.g. in hydrothermal vents.
3.3 Chiral surfaces
Selective adsorption is generally the consequence of reversible and preferential diastereomeric interactions between the chiral surface and one of the enantiomers,239 commonly described by the simple three-point model. But this model, assuming that only one enantiomer can present three groups that match three active positions of the chiral surface,243 fails to fully explain chiral recognition which are the fruit of more subtle interactions.244 In the second part of the XXth century, a large number of studies have focused on demonstrating chiral interactions between biological molecules and inorganic mineral surfaces.
Quartz is the only common mineral which is composed of enantiomorphic crystals. Right-handed (D-quartz) and left-handed (L-quartz) can be separated (similarly to the tartaric acid salts of the famous Pasteur experiment) and investigated independently in adsorption studies of organic molecules. The process of separation is made somewhat difficult by the presence of “Brazilian twins” (also called chiral or optical twins),242 which might bias the interpretation of the experiments. Bonner et al. in 1974245,246 measured the differential adsorption of alanine derivatives defined as % adsorbed on D-quartz – % adsorbed on L-quartz. These authors reported on the small but significant 1.4 ± 0.4% preferential adsorption of (R)-alanine over D-quartz and (S)-alanine over L-quartz, respectively. A more precise evaluation of the selectivity with radiolabelled (RS)-alanine hydrochloride led to higher levels of differential adsorption between L-quartz and D-quartz (up to 20%).247 The hydrochloride salt of alanine isopropyl ester was also found to be adsorbed enantiospecifically from its chloroform solution leading to chiral enrichment varying between 1.5 and 12.4%.248 Furuyama and co-workers also found preferential adsorption of (S)-alanine and (S)-alanine hydrochloride over L-quartz from their ethanol solutions.249,250 Anhydrous conditions are required to get sufficient adsorption of the organic molecules onto α-quartz crystals which, according to Bonner, discards α-quartz as a suitable mineral for the deracemization of building blocks of life.251 According to Hazen and Scholl,239 the fact that these studies have been conducted on powdered quartz crystals (i.e. polycrystalline quartz) has hampered a precise determination of the mechanism and magnitude of adsorption on specific surfaces of α-quartz. Some of the faces of quartz crystals likely display opposite chiral preferences which may have reduced the experimentally-reported chiral selectivity. Moreover, chiral indices of the commonest crystal growth surfaces of quartz, as established by Downs and Hazen, are relatively low (or zero), suggesting that the potential of enantiodiscrimination of organic molecules by quartz is weak in overall.252 Quantum-mechanical studies using density functional theories (DFT) have also been performed to probe the enantiospecific adsorption of various amino acids on hydroxylated quartz surfaces.253–256 In short, the computed differences in the adsorption energies of the enantiomers are modest (on the order of 2 kcal mol−1 at best) but strongly depend on the nature of amino acids and quartz surfaces. A final argument against the implication of quartz as a deterministic source of chiral discrimination of the molecules of life comes from the fact that D-quartz and L-quartz are equally distributed on earth.257,258
Calcite (CaCO3), as the most abundant marine mineral in the Archaean era, has potentially played an important role in the formation of prebiotic molecules relevant to life. The trigonal scalenohedral crystal form of calcite displays chiral faces which can yield chiral selectivity. In 2001, Hazen et al.261 reported that (S)-aspartic acid adsorbs preferentially on the (3![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 1) face of calcite, whereas (R)-aspartic acid adsorbs preferentially on the (21
1) face of calcite, whereas (R)-aspartic acid adsorbs preferentially on the (21![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 1) face. An e.e. value in the order of 0.5% on average was measured for the adsorbed aspartic acid molecules. No selectivity was observed on a centric surface that served as control. The experiments were conducted with aqueous solutions of (rac)-aspartic acid and selectivity was greater on crystals with terraced surface textures, presumably because enantiomers concentrated along step-like linear growth features. The calculated chiral indices of the (214) scalenohedral face of calcite was found to be the highest amongst 14 surfaces selected from various minerals (calcite, diopside, quartz, and orthoclase) and face-centred cubic (FCC) metals.252 In contrast, DFT studies revealed negligible difference in adsorption energies of enantiomers (<1 kcal mol−1) of alanine on the (211) face of calcite because alanine cannot establish three points of contact on the surface.262 Conversely, it is well established that amino acids modify the crystal growth of calcite crystals in a selective manner leading to asymmetric morphologies, e.g. upon crystallization263,264 or electrodeposition (Fig. 10a).259 Vaterite helicoids, produced by crystallization of CaCO3 in the presence of non-racemic mixtures of aspartic acid, were found to be single-handed (Fig. 10b).260 Enantiomeric ratio are identical in the helicoids and in solution, i.e. incorporation of aspartic acid in valerite displays no chiral amplification effect. Asymmetric growth was also observed for various organic substances with gypsum, another mineral with a centrosymmetric crystal structure.265 As expected, asymmetric morphologies produced from amino acid enantiomers are mirror image (Fig. 10).
1) face. An e.e. value in the order of 0.5% on average was measured for the adsorbed aspartic acid molecules. No selectivity was observed on a centric surface that served as control. The experiments were conducted with aqueous solutions of (rac)-aspartic acid and selectivity was greater on crystals with terraced surface textures, presumably because enantiomers concentrated along step-like linear growth features. The calculated chiral indices of the (214) scalenohedral face of calcite was found to be the highest amongst 14 surfaces selected from various minerals (calcite, diopside, quartz, and orthoclase) and face-centred cubic (FCC) metals.252 In contrast, DFT studies revealed negligible difference in adsorption energies of enantiomers (<1 kcal mol−1) of alanine on the (211) face of calcite because alanine cannot establish three points of contact on the surface.262 Conversely, it is well established that amino acids modify the crystal growth of calcite crystals in a selective manner leading to asymmetric morphologies, e.g. upon crystallization263,264 or electrodeposition (Fig. 10a).259 Vaterite helicoids, produced by crystallization of CaCO3 in the presence of non-racemic mixtures of aspartic acid, were found to be single-handed (Fig. 10b).260 Enantiomeric ratio are identical in the helicoids and in solution, i.e. incorporation of aspartic acid in valerite displays no chiral amplification effect. Asymmetric growth was also observed for various organic substances with gypsum, another mineral with a centrosymmetric crystal structure.265 As expected, asymmetric morphologies produced from amino acid enantiomers are mirror image (Fig. 10).
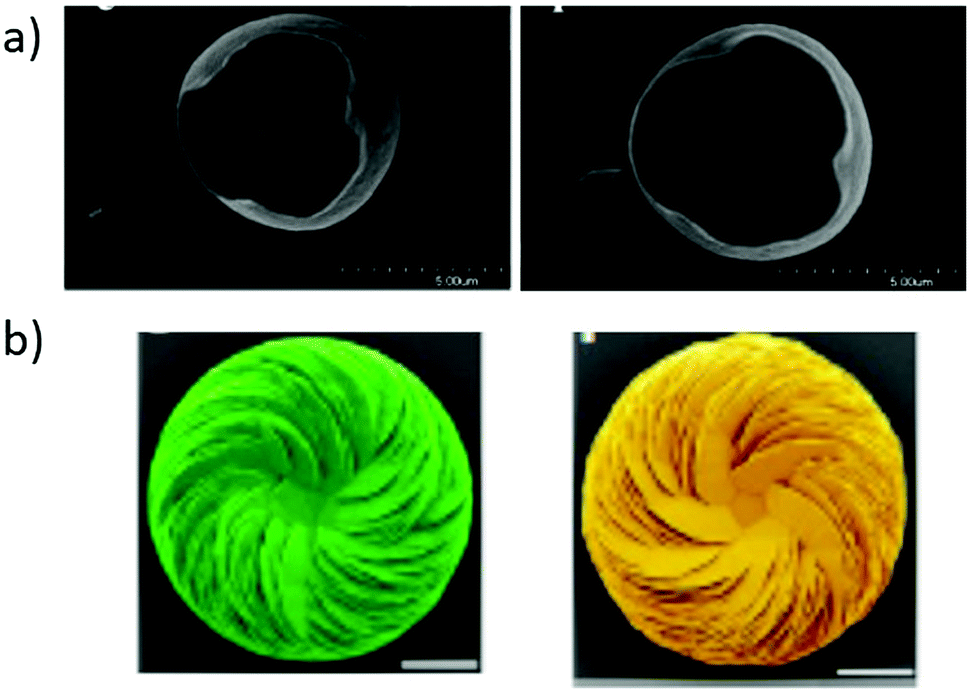 | ||
| Fig. 10 Asymmetric morphologies of CaCO3-based crystals induced by enantiopure amino acids. (a) Scanning electron micrographs (SEM) of calcite crystals obtained by electrodeposition from calcium bicarbonate in the presence of magnesium and (S)-aspartic acid (left), and (R)-aspartic acid (right). Reproduced with permission from ref. 259. Copyright 2007. American Chemical Society. (b) SEM images of vaterite helicoids obtained by crystallization in the presence of non-racemic solutions (40% e.e.) biased in favour of (S)-aspartic acid (left) and (R)-aspartic acid (right). Reprinted from ref. 260. Copyright 2019. Springer Nature under Creative Commons Attribution 4.0 International License https://creativecommons.org/licenses/by/4.0/. | ||
Clay minerals, of which some of them display high specific surface area, and adsorption and catalytic properties, are often invoked as potential promoters of the transformation of prebiotic molecules. Amongst the large variety of clays, serpentine and montmorillonite were likely the dominant ones on earth prior to life's origin.241 Clay minerals can exhibit non-centrosymmetric structures, such as the A and B forms of kaolinite, which correspond to the enantiomeric arrangement of the interlayer space. These chiral organizations are, however, not individually separable. All experimental studies claiming asymmetric inductions by clay minerals reported in the literature have raised suspicion about their validity, with no exception.242 This is because these studies employed either a racemic clay or clays which have no established chiral arrangement, i.e. presumably achiral clay minerals. Asymmetric adsorption and polymerization of amino acids reported with kaolinite266–270 and bentonite271–273 in the 1970s–1980s actually originated from experimental errors or contaminations. Supposedly enantiospecific adsorptions of amino acids with allophane,274 hydrotalcite-like compound,275 montmorillonite,276 and vermiculite277,278 also likely belong to this category.
Experiments aimed at demonstrating deracemization of amino acids in the absence of any chiral inducers or during phase transition under equilibrium conditions have to be interpreted cautiously (see the Chapter 4.2 of the book written by Meierhenrich for a more comprehensive discussion on this topic).24 Deracemization is possible under far-from-equilibrium conditions but a set of repeated experiments must then reveal a distribution of the chiral biases (see Part 4). The claimed specific adsorptions for racemic mixtures of amino acids likely originated from the different purities between (S)- and (R)-amino acids; or contaminants of biological origin such as microbial spores.279 Such issues are not old-fashioned and despite great improvement in analytical and purification techniques, the difference in enantiomer purities is most likely at the origin of the different behaviour of amino acid enantiomers observed in the crystallization of wulfingite (ε-Zn(OH)2)280 and CaCO3281,282 in two recent reports.
Very impressive levels of selectivity (on the range of 10% e.e.) were recently reported for the adsorption of aspartic acid on brushite, a mineral composed of achiral crystals of CaHPO4·2H2O.283 In this case, selective adsorption was observed under supersaturation and undersaturation conditions (i.e. non-equilibrium states) but not at saturation (equilibrium state). Likewise, opposite selectivity was observed for the two non-equilibrium states. It was postulated that mirror symmetry breaking of the crystal facets occurred during the dynamic events of crystal growth and dissolution. Spontaneous mirror symmetry breaking is not impossible under far-from-equilibrium conditions but again a distribution of the selectivity outcome is expected upon repeating the experiments under strictly achiral conditions (Part 4).
Ribó and co-workers proposed that chiral surfaces could have been involved in the chiral enrichment of prebiotic molecules on carbonaceous chondrites present on meteorites.284 In their scenario, mirror symmetry breaking during the formation of planetesimal bodies and comets may have led to a bias in the distribution of chiral fractures, screw distortions or step-kink chiral centres on the surfaces of these inorganic matrices. This in turn would have led to a bias in the adsorption of organic compounds. Their study was motivated by the fact that the enantiomeric excesses measured for organic molecules vary according to their location on the meteorite surface.285 Their measurement of the optical activity of three meteorite samples by circular birefringence (CB) indeed revealed a slight bias towards negative CB values for the Murchinson meteorite. The optically active areas are attributed to serpentines and other poorly identified phyllosilicate phases, whose formation may have occurred concomitantly to organic matter.
The implication of inorganic minerals in biasing the chirality of prebiotic molecules remains uncertain given that no strong asymmetric adsorption values have been reported to date and that certain minerals were even found to promote the racemization of amino acids286 and secondary alcohols.287 However, evidence exists that minerals could have served as hosts and catalysts for prebiotic reactions, including the polymerization of nucleotides.288 In addition, minute chiral biases provided by inorganic minerals could have driven SMSB processes into a deterministic outcome (Part 4).
α-Glycine crystallizes from water into a centrosymmetric form. In the 1980s, Lahav, Leiserowitch and co-workers demonstrated that amino acids were occluded to the basal faces (010 and 00) of glycine crystals with exquisite selectivity.289–291 For example, when a racemic mixture of leucine (1–2% wt/wt of glycine) was crystallized with glycine at an air/water interface, (R)-Leu was incorporated only into those floating glycine crystals whose (010) faces were exposed to the water solution, while (S)-Leu was incorporated only into the crystals with exposed (00) faces. This results in the nearly perfect resolution (97–98% e.e.) of Leu enantiomers. In the presence of a small amount of an enantiopure amino-acid (e.g. (S)-Leu), all crystals of Gly exposed the same face to the water solution leading to one enantiomer of a racemate being occluded in glycine crystals while the other remains in solution. These striking observations led the same authors to propose a scenario in which the crystallization of supersaturated solutions of glycine in the presence of amino-acid racemates would have led to the spontaneous resolution of all amino acids (Fig. 11).
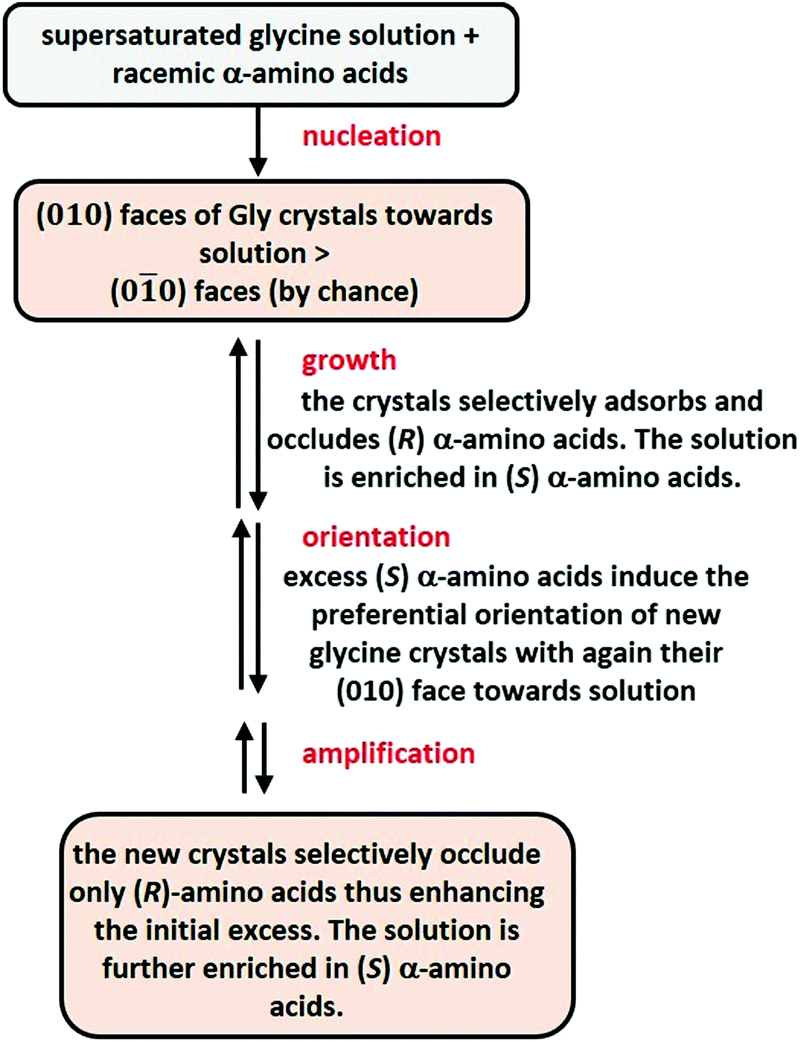 | ||
| Fig. 11 Resolution of amino acid enantiomers following a “by chance” mechanism including enantioselective occlusion into achiral crystals of glycine.289,290 | ||
This can be considered as a “by chance” mechanism in which one of the enantiotopic face (010) would have been exposed preferentially to the solution in the absence of any chiral bias. From then, the solution, enriched into (S)-amino acids, enforces all glycine crystals to expose their (010) faces to water, eventually leading to all (R)-amino acids being occluded in glycine crystals.
A somewhat related strategy was disclosed in 2010 by Soai and co-workers.292 Dehydration of centrosymmetric crystals of cytosine monohydrate yielded enantio-enriched anhydrous cytosine chiral crystals when only one of two enantiotopic faces of the crystal was put into direct contact with the hot plate. Enrichment was also observed if the dehydration was performed under reduced pressure but with a surprising inversion of the configuration of the chiral crystal relative to thermal dehydration.293 Rearrangement of the hydrogen bond network during transition between the centrosymmetric and chiral forms of the cytosine crystals is likely at the origin of this selective process. Reactions occurring at the enantiotopic surface of an organic crystal also yields enantio-enriched compounds whose optical purity can be enhanced by SMSB processes.294,295
4. Spontaneous mirror-symmetry breaking (SMSB)
4.1 Definition, models and the Soai reaction
Spontaneous mirror-symmetry breaking (SMSB) phenomenon is the process that leads to the preferential formation of one chiral state over its enantiomeric form in the absence of a detectable chiral bias or enantiomeric imbalance. As defined by Ribó and co-workers, SMSB concerns the transformation of “metastable racemic non-equilibrium stationary states (NESS) into one of two degenerate but stable enantiomeric NESSs”.301 Although this definition is somewhat in contradiction with the textbook statement that enantiomers need the presence of a chiral bias to be distinguished, it was recognized a long time ago that SMSB can emerge from reactions involving asymmetric self-replication or auto-catalysis. The connection between SMSB and BH is appealing,25,40,51,301–308 since SMSB is the unique physicochemical process that allows for the emergence and retention of enantiopurity from scratch. It is also intriguing to note that the competitive chiral reaction networks that might give rise to SMSB could exhibit replication, dissipation and compartmentalization,301,309i.e. fundamental functions of living systems.Systems able to lead to SMSB consist of enantioselective autocatalytic reaction networks, described through models dealing with either the transformation of achiral to chiral compounds, or the deracemization of racemic mixtures.301 As early as 1953, Frank described a theoretical model dealing with the former case. According to Frank's model, SMSB emerges from a system involving homochiral self-replication (one enantiomer of the chiral product accelerates its own formation) and heterochiral inhibition (the replication of the other product enantiomer is prevented).303 It is now well-recognized that the Soai reaction,56 an auto-catalytic asymmetric process (Fig. 12a), disclosed 42 years later,310 is an experimental validation of the Frank model. The reaction between pyrimidine-5-carbaldehyde and diisopropyl zinc (two achiral reagents) is strongly accelerated by their zinc alkoxy product, which is found to be enantiopure (>99% e.e.) after a few cycles of reaction/addition of reagents (Fig. 12b and c).310–312 Kinetic models based on the stochastic formation of homochiral and heterochiral dimers313–315 of the zinc alkoxy product provide good fits of the kinetic profile even though the involvement of higher species has gained more evidence recently.316–324 In this model, homochiral dimers serve as auto-catalysts for the formation of the same enantiomer of the product whilst heterochiral dimers are inactive and sequester the minor enantiomer, a Frank model-like inhibition mechanism. A hallmark of the Soai reaction is that the direction of auto-catalysis is dictated by extremely weak chiral perturbations: quartz, cryptochiral molecules, circularly polarized light, and chiral isotopomers amongst others (Fig. 12b).312 In addition, the apparent outcome of the Soai reaction performed in the absence of detectable chiral species is stochastic as expected for a truly SMSB process (Fig. 12c).325–333 On the one hand, the Soai reaction offers a credible mechanistic scenario from which homochiral biomolecules at the origin of life would have been created on a deterministic manner through a SMSB process coupled to an infinitesimal chiral bias (vide infra). This bias would have survived from a larger one despite significant erosion through racemization processes. On the other hand, the Soai reaction is more an exception than a rule in the chemical space explored to date.334–342 The exergonic and irreversible nature of the organozinc addition reaction are key for pushing the system far-from-equilibrium and for the generation and preservation of the homochiral state. On the contrary, it is assumed that prebiotic chemical reactions would have been only weakly exergonic, i.e. their products would have been more prone to racemization or to side reactions occurring in solution.37,46,301
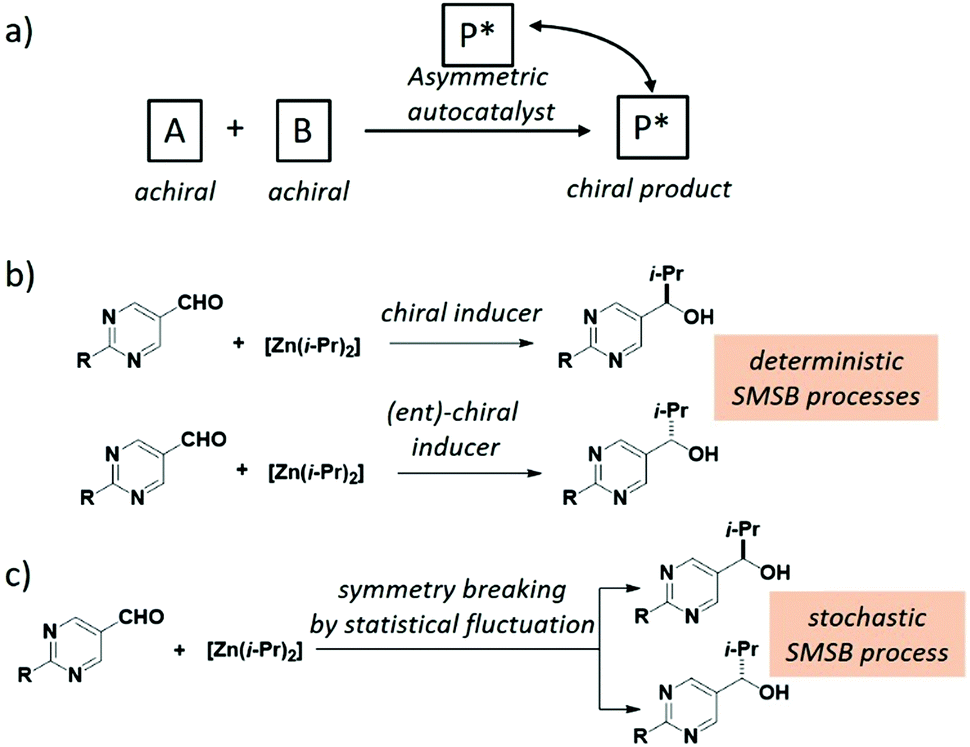 | ||
| Fig. 12 (a) General scheme for an auto-catalysed asymmetric reaction. (b) The Soai reaction performed in the presence of detected chirality, leading to highly enantio-enriched alcohol with the same configuration in successive experiments (deterministic SMSB). (c) Soai reaction performed in the absence of detected chirality, leading to highly enantio-enriched alcohol with a bimodal distribution of the configurations in successive experiments (stochastic SMSB). | ||
Many other models of spontaneous emergence of homochirality in far-from-equilibrium systems have been proposed in the literature.343–345 Most of them are derived from the Frank model but do not include any mutual inhibition reaction. The limited enantioselective (LES) model306,346 assumes that both the asymmetric auto-catalysis (similar to the homochiral self-replication in the Frank model) and the non-enantioselective auto-catalysis (the accelerated formation of both enantiomers of the product) can co-exist. SMSB emerges if these two autocatalytic processes are (i) individually compartmentalized within regions experiencing different temperatures,347,348 or (ii) driven by a constant concentration of external reagents.349 Required conditions for SMSB through the LES model could have been present in deep ocean hydrothermal plumes. Likewise, a chemical scenario has been proposed for LES based on coupled Strecker-type reactions for amino acid synthesis and degradation which have been postulated to be accelerated by a heterogeneous catalytic support such as phyllosilicates.349 However, the LES model has found no experimental evidence to date. Models for enantioselective hypercyclic replicators were recently disclosed in which the inhibition reaction in the Frank model has been replaced by mutual cross-catalytic processes occurring between families of coupled replicators.350,351 These models support a scenario in which the combination of SMSB, formation of the first (coupled) self-replicators and the emergence of their functions would have led to BH.301 This intriguing concept may foster experimental investigations of SMSB processes in polymerization/depolymerization reactions.
Imposed boundary conditions for SMSB involve “either systems open to matter exchange, or closed systems unable to equilibrate energy with their surroundings”.301 In the absence of any chiral influence, the obtained metastable NESSs are exposed to statistical fluctuations, and evolve towards scalemic or homochiral NESSs, as long as the systems are far-from-equilibrium. It is important to note that in the absence of these boundary conditions, systems will be able to equilibrate with their surrounding and the deviation from the racemic state will be lost, e.g. racemization would occur under classically employed reaction workups operated in solution.41,352 This is probably the main reason why a single SMSB process has been identified to date for a reaction performed in solution (the Soai reaction). On the contrary, SMSB processes have been observed more frequently in crystals (vide infra) or in supramolecular assemblies,353i.e. processes involving phase transition. Asymmetric reactions performed with catalytic single-handed supramolecular assemblies obtained through a SMSB process were found to yield enantio-enriched products whose configuration is left to chance.157,354 SMSB processes leading to homochiral crystals as the final state appear particularly relevant in the context of BH and will thus be discussed separately in the following section.
4.2 Homochirality by crystallization
Havinga postulated that just one enantiomorph can be obtained upon a gentle cooling of a racemate solution (i) when the crystal nucleation is rare and the growth is rapid and (ii) when fast inversion of configuration occurs in solution (i.e. racemization). Under these circumstances, only monomers with matching chirality to the primary nuclei crystallize leading to SMSB.55,355 Havinga reported in 1954 a set of experiments aimed at demonstrating his hypothesis with N,N,N-allylethylmethylanilinium iodide – an organic molecule which crystallizes as a conglomerate from chloroform (Fig. 13).355 Fourteen supersaturated solutions were gently heated in sealed tubes, then stored at 0 °C to give crystals which were in 12 cases inexplicably more dextrorotatory (measurement of optical activity by dissolution in water, where racemization is not observed). Seven other supersaturated solutions were carefully filtered before cooling to 0 °C, but no crystallization occurred after one year. Crystallization occurred upon further cooling: three crystalline products with no optical activity were obtained, while the other four showed a small optical activity ([α]D = +0.2°; +0.7°; −0.5°; −3.0°). More successful examples of preferential crystallization of one enantiomer appeared in the literature notably with tri-o-thymotide,356 and 1,1′-binaphthyl.357,358 In the latter case, the distribution of specific rotations recorded for several independent experiments is centred to zero.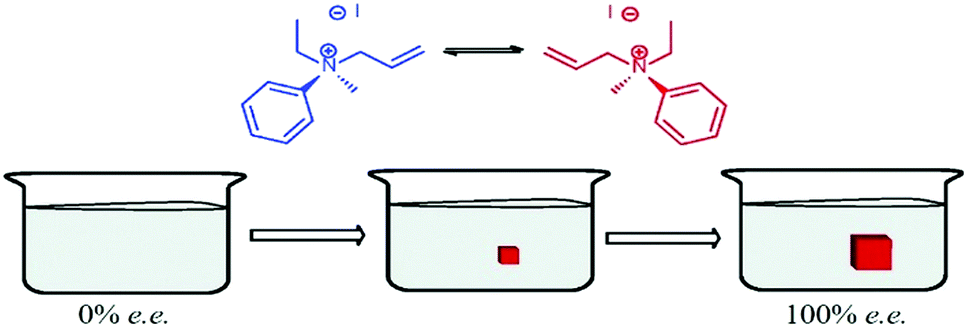 | ||
| Fig. 13 Enantiomeric preferential crystallization of N,N,N-allylethylmethylanilinium iodide as described by Havinga. Fast racemization in solution supplies the growing crystal with the appropriate enantiomer. Adapted from ref. 55 with permission from the Royal Society of Chemistry, copyright 2015. | ||
Sodium chlorate (NaClO3) crystallizes by evaporation of water into a conglomerate (P213 space group).359–361 Preferential crystallization of one of the crystal enantiomorph over the other was already reported by Kipping and Pope in 1898.362,363 From static (i.e. non-stirred) solution, NaClO3 crystallization seems to undergo an uncertain resolution, similar to Havinga's findings with the aforementioned quaternary ammonium salt. However, a statistically significant bias in favour of D-crystals was invariably observed, likely due to the presence of bio-contaminants.364 Interestingly, Kondepudi et al. showed in 1990 that magnetic stirring, during the crystallization of sodium chlorate, randomly oriented the crystallization to only one enantiomorph, with a virtually perfect bimodal distribution over several samples (±1).365 Further studies366–369 revealed that the maximum degree of supersaturation is solely reached once, when the first primary nucleation occurs. At this stage, the magnetic stirring bar breaks up the first nucleated crystal into small fragments that have the same chirality than the ‘Eve crystal’, and act as secondary nucleation centres whence crystals grow (Fig. 14). This constitutes a SMSB process coupling homochiral self-replication plus inhibition through the supersaturation drop during secondary nucleation, precluding new primary nucleation and the formation of crystals of the mirror-image form.307 This deracemization strategy was also successfully applied to 4,4′-dimethyl-chalcone,370 and 1,1′-binaphthyl (from its melt).371
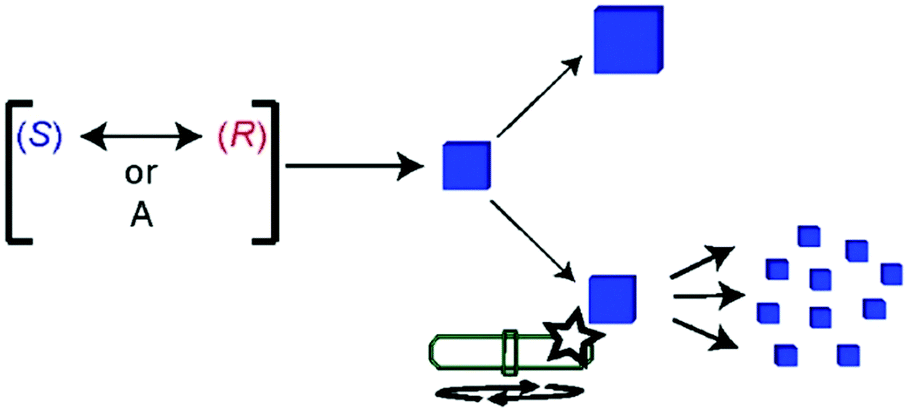 | ||
| Fig. 14 Primary nucleation of an enantiopure ‘Eve crystal’ of random chirality, slightly amplified by growing under static conditions (top, Havinga-like), or strongly amplified by secondary nucleation thanks to magnetic stirring (bottom, Kondepudi-like) from rapidly racemizing chiral molecules, (S) and (R), or achiral molecules, A. Reprinted from ref. 55 with permission from the Royal Society of Chemistry, copyright 2015. | ||
In 2005, Viedma reported that solid-to-solid deracemization of NaClO3 proceeded from its saturated solution by abrasive grinding with glass beads.373 Complete homochirality with bimodal distribution is reached after several hours or days.374 The process can also be triggered by replacing grinding with ultrasound,375 turbulent flow,376 or temperature variations.376,377 Although this deracemization process is easy to implement, the mechanism by which SMSB emerges is an ongoing highly topical question that falls outside the scope of this review.40,41,378–381
Viedma ripening was exploited for deracemization of conglomerate-forming achiral or chiral compounds (Fig. 15).55,382 The latter can be formed in situ by a reaction involving a prochiral substrate. For example, Vlieg et al. coupled an attrition-enhanced deracemization process with a reversible organic reaction (an aza-Michael reaction) between prochiral substrates under achiral conditions to produce an enantiopure amine.383 In a recent review, Buhse and co-workers identified a range of conglomerate-forming molecules that can be potentially deracemized by Viedma ripening.41 Viedma ripening also proves to be successful with molecules crystallizing as racemic compounds under the condition that the conglomerate form is energetically accessible.384 Furthermore, a promising mechanochemical method to transform racemic compounds of amino acids into their corresponding conglomerates has been recently found.385 When valine, leucine and isoleucine were milled one hour in the solid state, in a Teflon jar with a zirconium ball and in the decisive presence of zinc oxide, their corresponding conglomerates eventually formed.
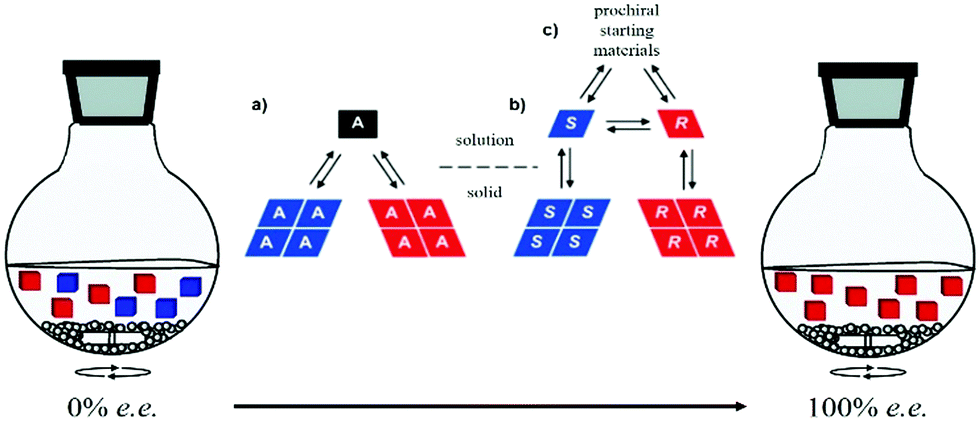 | ||
| Fig. 15 Schematic representation of Viedma ripening and solution–solid equilibria of an intrinsically achiral molecule (a) and a chiral molecule undergoing solution-phase racemization (b). The racemic mixture can result from chemical reaction involving prochiral starting materials (c). Adapted from ref. 55 with permission from the Royal Society of Chemistry, copyright 2015 and from ref. 372. Copyright 2008. American Chemical Society. | ||
Shortly after the discovery of Viedma, aspartic acid386 and glutamic acid387,388 were deracemized up to the homochiral state starting from biased racemic mixtures. The chiral γ-polymorph of glycine389 was obtained with a preferred handedness by Ostwald ripening, albeit with a stochastic distribution of the optical activities.390 Salts or imine derivatives of alanine,391,392 phenylglycine372,384 and phenylalanine391,393 were desymmetrized by Viedma ripening with DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) as the racemization catalyst. Successful deracemization was also achieved with amino acid precursors such as α-aminonitriles,394–396 α-iminonitriles,397N-succinopyridine398 and thiohydantoins.399 The first three classes of compounds could be obtained directly from prochiral precursors by coupling synthetic reactions and Viedma ripening. In the preceding examples, the direction of the SMSB process is selected by biasing the initial racemic mixtures in favour of one enantiomer or by seeding the crystallization with chiral chemical additives. In the next sections, we will consider the possibility to drive the SMSB process towards a deterministic outcome by means of PVED, physical fields, polarized particles, and chiral surfaces, i.e. the sources of asymmetry depicted in Parts 2 and 3 of this review.
4.3 Deterministic SMSB processes
The possibility to bias crystallization processes with chiral particles emitted by radionuclides was probed by several groups as summarized in the reviews of Bonner.44,54 Kondepudi-like crystallization of NaClO3 in the presence of β particles from a 39Sr90 source notably yielded a distribution of (+) and (−)-NaClO3 crystals, largely biased in favour of (+) crystals.405 It was presumed that spin polarized electrons produced chiral nucleating sites, albeit chiral contaminants cannot be excluded.
The effect of chiral additives on crystallization processes, in which the additive inhibits one of the enantiomer growth thereby enriching the solid phase with the opposite enantiomer is well established as “the rule of reversal”.412,413 In the realm of the Viedma ripening, Noorduin et al. discovered in 2020 a way of propagating homochirality between α-iminonitriles, possible intermediates in the Strecker synthesis of α-amino acids.414 These authors demonstrated that an enantiopure additive (1–20 mol%) induces an initial enantio-imbalance, which is then amplified by Viedma ripening up to a complete mirror-symmetry breaking. In contrast to the “rule of reversal”, the additive favours the formation of the product with identical configuration. The additive is actually incorporated in a thermodynamically controlled way into the bulk crystal lattice of the crystallized product of the same configuration, i.e. a solid solution is formed enantiospecifically.
CPL was successfully used in the realm of the Soai reaction to direct its outcome, either by using a chiroptical switchable additive, or by asymmetric photolysis of a racemic substrate. In 2004, Soai et al. illuminated for 48 h a photoresolvable chiral olefin with l- or r-CPL, and mixed it with the reactants of the Soai reaction to afford (S)- or (R)-5-pyrimidyl alkanol, respectively, in e.e. higher than 90%.417 In 2005, the photolyzate of a pyrimidyl alkanol racemate acted as an asymmetric catalyst for its own formation reaching e.e. greater than 99.5%.418 The enantiomeric excess of the photolyzate was below the detection level of chiral HPLC instrument but was amplified thanks to the SMSB process.
In 2009, Vlieg et al. coupled CPL with Viedma ripening to achieve complete and deterministic mirror-symmetry breaking.419 Previous investigation revealed that the deracemization by attrition of the Schiff base of phenylglycine amide (rac-1, Fig. 16a) always occurred in the same direction, the (R)-enantiomer, as a probable result of minute levels of chiral impurities.372 CPL was envisaged as a potent chiral physical field to overcome this chiral interference. Irradiation of solid–liquid mixtures of rac-1 indeed led to complete deracemization, the direction of which was directly correlated to the circular polarization of light. Control experiments indicated that the direction of the SMSB process is controlled by a non-identified chiral photoproduct generated upon irradiation of (rac)-1 by CPL. This photoproduct (S* or R* in Fig. 16b) then serves as an enantioselective crystal-growth inhibitor which mediates the deracemization process towards the other enantiomer (Fig. 16b). In the context of BH, this work highlights that asymmetric photosynthesis by CPL is a potent mechanism that can be exploited to direct deracemization processes when coupled to an amplification phenomenon.
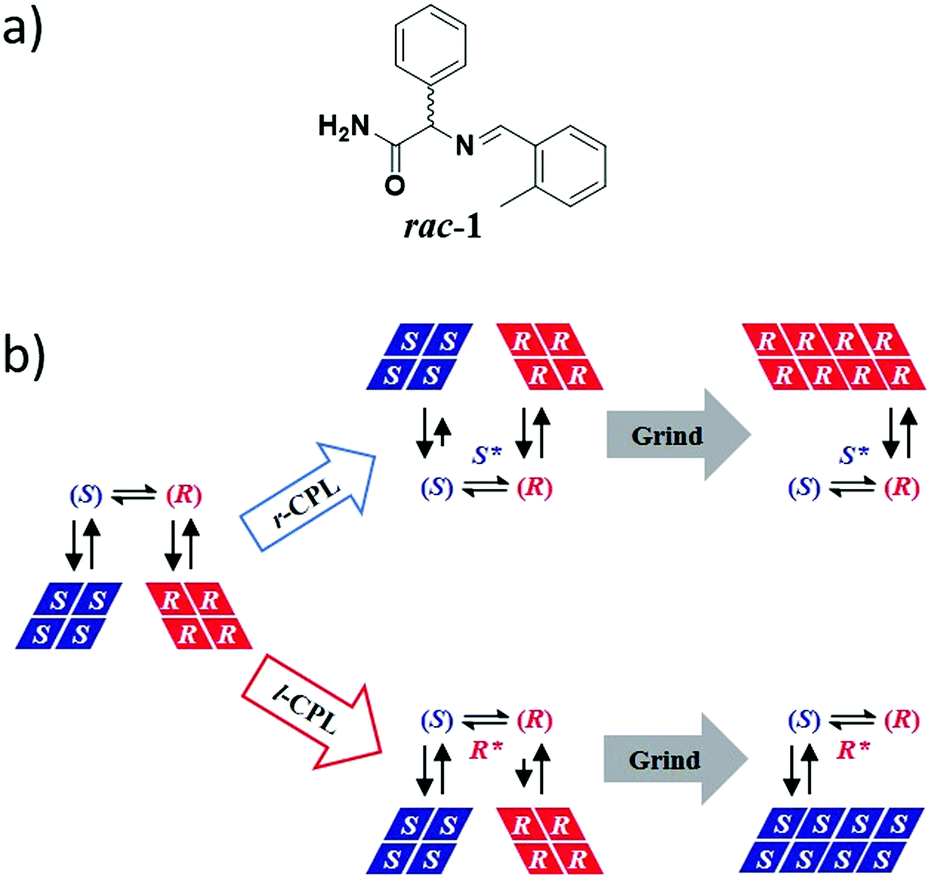 | ||
| Fig. 16 (a) Molecular structure of rac-1. (b) CPL-controlled complete attrition-enhanced deracemization of rac-1. (S) and (R) are the enantiomers of rac-1 and S* and R* are chiral photoproducts formed upon CPL irradiation of rac-1.419 | ||
5. Theories for the emergence of BH
Physical fields, CPL, polarized particles, polarized spins, chiral surfaces and SMSB processes have been presented as potential candidates for the emergence of chiral biases in prebiotic molecules. Their main properties are summarized in Table 1. The plausibility of the occurrence of these biases under the conditions of the primordial universe has also been evoked for certain physical fields (such as CPL or CISS). However, it is important to provide a more global overview of the current theories that tentatively explain the following puzzling questions: where, when and how did the molecules of life reach a homochiral state? At which point of this undoubtedly intricate process did life emerge?| Type | Truly/falsely chiral | Direction | Extent of induction | Scope | Relevance to BH | Selected references |
|---|---|---|---|---|---|---|
| a PVED ≈ 10−12–10−15 kJ mol−1.53 b However, experimental results are not conclusive (see Part 3.2(b)). c e.e.MChD = gMChD/2 with gMChD ≈ (gNCD × gMCD)/2. NCD: natural circular dichroism. MCD: magnetic circular dichroism. For the resolution of Cr complexes,137 e.e. = k × B with k = 10−5 T−1 at λ = 695.5 nm. d The minute chiral induction is amplified upon aggregation leading to homochiral helical assemblies.423 e For photolysis, e.e. depends both on g and the extent of reaction (see eqn (2) and the text in part 3.2(a)). Up to a few e.e. percent have been observed experimentally.197–199 f Recently, spin-polarized SE through the CISS effect have been implemented as chiral reagents with relatively high e.e. values (up to a ten percent) reached for a set of reactions.236 g The standard deviation for 1 mole of chiral molecules is of 1.9 × 1011.126na: not applicable. | ||||||
| PV | Truly | Unidirectional, deterministic, (+) or (−) for a given molecule | Minutea | Any chiral molecules | PVED: theo. calculations (natural) polarized particles: asymmetric destruction of racematesb | 44, 53 and 54 |
| MChD | Truly | Bidirectional, (+) or (−) depending on the relative orientation of light and magnetic field | Minutec | Chiral molecules with high gNCD and gMCD values | Proceed with unpolarised light | 137 |
| Aligned magnetic field, gravity and rotation | Falsely | Bidirectional, (+) or (−) depending on the relative orientation of angular momentum and effective gravity | Minuted | Large supramolecular aggregates | Ubiquitous natural physical fields | 161 |
| Vortices | Truly | Bidirectional, (+) or (−) depending on the direction of the vortices | Minuted | Large objects or aggregates | Ubiquitous natural physical field (pot, present in hydrothermal vents) | 151 and 160 |
| CPL | Truly | Bidirectional, (+) or (−) depending on the direction of CPL | Low to Moderatee | Chiral molecules with high gNCD values | Asymmetric destruction of racemates | 58 |
| Spin-polarized electrons (CISS effect) | Truly | Bidirectional, (+) or (−) depending on polarization | Low to highf | Any chiral molecules | Enantioselective adsorption/crystallization of racemate, asymmetric synthesis | 233 |
| Chiral surfaces | Truly | Bidirectional, (+) or (−) depending on surface chirality | Low to excellent | Any adsorbed chiral molecules | Enantioselective adsorption of racemates | 238 and 244 |
| SMSB (crystallization) | na | Bidirectional, stochastic distribution of (+) or (−) for repeated processes | Low to excellent | Conglomerate-forming molecules | Resolution of racemates | 55 and 382 |
| SMSB (asymmetric auto-catalysis) | na | Bidirectional, stochastic distribution of (+) or (−) for repeated processes | Low to excellent | Soai reaction | To be demonstrated | 312 |
| Chance mechanisms | na | Bidirectional, stochastic distribution of (+) or (−) for repeated processes | Minuteg | Any chiral molecules | To be demonstrated | 17 and 420–422 |
5.1 Terrestrial or extra-terrestrial origin of BH?
The enigma of the emergence of BH might potentially be solved by finding the location of the initial chiral bias, might it be on earth or elsewhere in the universe. The ‘panspermia’ hypothesis,424 according to which living organisms were transplanted to earth from another solar system, sparked interest on the extra-terrestrial origin of BH, but the fact that such a high level of chemical and biological evolution was present on celestial objects has not been supported by any scientific evidence.44 Accordingly, terrestrial and extra-terrestrial scenarios for the original chiral bias in prebiotic molecules will be considered in the following.Alternatively, theories suggesting that BH emerged from scratch, i.e. without any involvement of the chiral discriminating sources mentioned in Part 2–3 and SMSB processes (Part 4), have been mentioned in the literature for a long time,420 and variant versions appeared sporadically. Herein, these mechanisms are named “random” or “by chance” and are based on probabilistic grounds only (Table 1). The prevalent form comes from the fact that a racemate is very unlikely made of exactly equal amounts of enantiomers, due to natural fluctuations described statistically like coin tossing.126,432 One mole of chiral molecules actually exhibits a standard deviation of 1.9 × 1011. Putting into relation this statistical variation and putative strong chiral amplification mechanisms and evolutionary pressures, Siegel suggested that homochirality is an imperative of molecular evolution.17 However, the probability to get both homochirality and life emerging from statistical fluctuations at the molecular scale appears very unlikely.35,59,433 SMSB phenomena may amplify statistical fluctuations up to the homochiral state, yet the direction of process for multiple occurrences will be left to chance in the absence of a chiral inducer (Part 4). Other theories suggested that homochirality emerges during the formation of biopolymers “by chance”, as a consequence of the limited number of sequences that can be possibly contained in a reasonable amount of macromolecules (see Part 5.3).17,421,422 Finally, kinetic processes have also been mentioned in which a given chemical event would have occurred to a larger extent for one enantiomer over the other under achiral conditions (see one possible physicochemical scenario in Fig. 11). Hazen notably argued that nucleation processes governing auto-catalytic events occurring at the surface of crystals are rare and thus a kinetic bias can emerge from an initially unbiased set of prebiotic racemic molecules.239 Random and by chance scenarios towards BH might be attractive on a conceptual view but lack experimental evidence.
The 100 kg Murchison's meteorite that fell at Australia in 1969 is generally considered as the standard reference for extra-terrestrial organic matter (Fig. 17a).435 In fifty years, the analyses of its composition revealed more than ninety α, β, γ and δ-isomers of C2 to C9 amino acids, diamino acids, and dicarboxylic acids as well as numerous polyols including sugars (ribose,436 a building block of RNA), sugar acids and alcohols, but also α-hydroxycarboxylic acids437 and deoxy acids.434 Unequal amounts of enantiomers were also found with a quasi-exclusive predominance for (S)-amino acids57,285,438–440 ranging from 0 to 26.3 ± 0.8% e.e. values (highest e.e. being measured for non-proteinogenic α-methyl amino acids),441 and, when they are not racemates, only D-sugar acids with an e.e. of up to 82% for xylonic acid have been detected.442 These measurements are relatively scarce for sugars and in general need to be repeated, notably to definitely exclude their potential contamination by terrestrial environment. Future space missions to asteroids, comets and Mars, coupled with more advanced analytical techniques,443 will indubitably lead to a better determination of the composition of extra-terrestrial organic matter. The fact that major enantiomers of extra-terrestrial amino acids and sugar derivatives have the same configuration as the building blocks of life constitutes a promising set of results.
 | ||
| Fig. 17 (a) A fragment of a meteorite landed in Murchison, Australia, in 1969, exhibited at the National Museum of Natural History (Washington). (b) Scheme of the preparation of interstellar ice analogues. A mixture of primitive gas molecules is deposited and irradiated under vacuum on a cooled window. Composition and thickness are monitored by infrared spectroscopy. Reprinted from ref. 434 with permission from MDPI. Copyright 2019. Licensee MDPI under Creative Commons Attribution 4.0 International License hhttp://ttps://creativecommons.org/licenses/by/4.0/. | ||
To complete these analyses of the difficult-to-access outer space, laboratory experiments have been conducted by reproducing the plausible physicochemical conditions present on astrophysical ices (Fig. 17b).444 Natural ones are formed in interstellar clouds,445,446 on the surface of dust grains from which condensates a gaseous mixture of carbon, nitrogen and oxygen-based molecules (e.g. H2O, CH3OH, CH4, NH3, and CO2),447 under the influence of very low temperature (5–15 K)448 and pressure. Subsequent photochemical processes in this mantle of frost are assumed to lead to complex molecules.449 Since collapsing clouds gave birth to our solar system through the aggregation of dust grains,450 studies of their composition receive a keen interest to broaden our knowledge about the prebiotic environment. Experiments on simulated interstellar ices support the formation of many proteinaceous amino acids,451,452 and building blocks of RNA and DNA such as sugars,453 like ribose454 and deoxyribose,455 as well as nucleobases (adenine, cytosine, uracil and thymine for example).456 These molecules were obtained with no significant bias from the ideal racemic composition which supports their abiotic origin.444 However, when similar experiments are conducted under CPL irradiation, amino acids are generated with significant biases towards one enantiomer as described in Part 3.2(a).204,205
The occurrence of CPL-driven photochirogenesis on interstellar dust grains was supported by the detection of near-infrared light with significant circular polarization degrees (up to 22%),457 in parsec-sized star-forming regions, such as massive molecular clouds.458–464 Cosmic circularly polarized photons arise from synchrotron radiations emitted by neutron stars, remnants of supernovae explosions,35 through bremsstrahlung, dichroic scattering and/or light extinction (along lined up grains).465 In addition, although it was not directly observed due to dust shielding, models predicted the generation of vacuum ultraviolet (VUV) and UV-CPL under these conditions,459i.e. spectral regions of light absorbed by amino acids and sugars. Photolysis by broad band and optically impure CPL is expected to yield lower enantioenrichments than those obtained experimentally by monochromatic and quasiperfect circularly polarized synchrotron radiation (see Part 3.2a).198 However, a broad band CPL is still capable of inducing chiral bias by photolysis of an initially abiotic racemic mixture of aliphatic α-amino acids as previously debated.466,467 Likewise, CPL in the UV range will produce a wide range of amino acids with a bias towards the (S) enantiomer,195 including α,α-dialkyl amino acids.468
l- and r-CPL produced by a neutron star are equally emitted in vast conical domains in the space above and below its equator.35 However, appealing hypotheses were formulated against the apparent contradiction that amino acids have always been found as predominantly (S) on several celestial bodies,59 and the fact that CPL is expected to be portioned into left- and right-handed contributions in equal abundance within the outer space. In the 1980s, Bonner and Rubenstein proposed a detailed scenario in which the solar system, revolving around the centre of our galaxy, had repeatedly traversed a molecular cloud and accumulated enantio-enriched incoming grains.430,469 The same authors assumed that this enantioenrichment would come from asymmetric photolysis induced by synchrotron CPL emitted by a neutron star at the stage of planet formation. Later, Meierhenrich remarked in addition that, in molecular clouds, regions of homogeneous CPL polarization can exceed the expected size of a protostellar disk – or of our solar system,458,470 allowing a unidirectional enantioenrichment within our solar system, including comets.24 A solid scenario towards BH thus involves CPL as a source of chiral induction for biorelevant candidates, through photochemical processes on the surface of dust grains, and delivery of the enantio-enriched compounds on primitive earth by direct grain accretion or by impact471 of larger objects (Fig. 18).472–474
 | ||
| Fig. 18 CPL-based scenario for the emergence of BH following the seeding of the early earth with extra-terrestrial enantio-enriched organic molecules. Adapted from ref. 474 with permission from Wiley-VCH, copyright 2015. | ||
The high enantiomeric excesses detected for (S)-isovaline in certain stones of the Murchison's meteorite (up to 15.2 ± 0.2%) suggested that CPL alone cannot be at the origin of this enantioenrichment.285 The broad distribution of e.e. values (0–15.2%) and the abundance ratios of isovaline relatively to other amino acids also point to (S)-isovaline (and probably other amino acids) being formed through multiple synthetic processes that occurred during the chemical evolution of the meteorite.440 Finally, based on the anisotropic spectra,188 it is highly plausible that other physiochemical processes, e.g. racemization coupled to phase transitions or coupled non-equilibrium/equilibrium processes,378,475 have led to a change in the ratio of enantiomers initially generated by UV-CPL.59 In addition, a serious limitation of the CPL-based scenario shown in Fig. 18 is that significant enantiomeric excesses can only be reached at high conversion, i.e. by decomposition of most of the organic matter (see eqn (2) in Part 3.2(a)). Even though there is a solid foundation for CPL being involved as an initial inducer of chiral bias in extra-terrestrial organic molecules, chiral influences other than CPL cannot be excluded. Induction and enhancement of optical purities by physicochemical processes occurring at the surface of meteorites and potentially involving water and the lithic environment have been evoked but have not been assessed experimentally.285
Asymmetric photoreactions431 induced by MChD can also be envisaged notably in a neutron star environment, of tremendous magnetic fields (108–1012 T) and synchrotron radiations.35,476 Spin-polarized electrons (SPEs), another potential source of asymmetry, can potentially be produced upon ionizing irradiation of ferrous magnetic domains present in interstellar dust particles, aligned by enormous magnetic fields produced by a neutron star. One enantiomer from a racemate in a cosmic cloud could adsorb enantiospecifically on the magnetized dust particle. In addition, meteorites contain magnetic metallic centres that can act as asymmetric reaction sites upon generation of SPEs. Finally, polarized particles such as antineutrinos (the SNAAP model226–228) have been proposed as a deterministic source of asymmetry, at work in the outer space. Radioracemization must potentially be considered as a jeopardizing factor in that specific context.44,477,478 Further experiments are needed to probe whether these chiral influences have played a role in the generation of the enantiomeric imbalances detected in celestial bodies.
5.2 Purely abiotic scenarios
Emergence of life and biomolecular homochirality must be tightly linked,46,479,480 but in such a way that needs to be cleared up. As recalled recently by Glavin, homochirality by itself cannot be considered as a biosignature.59 Non proteinogenic amino acids are predominantly (S) and abiotic physicochemical processes can lead to enantio-enriched molecules. However, it has been widely substantiated that polymers of life (proteins, DNA, and RNA) as well as lipids need to be enantiopure to be functional. Considering the NASA definition of life, “a self-sustaining chemical system capable of Darwinian evolution”,481 and the “widespread presence of ribonucleic acid (RNA) cofactors and catalysts in today's terran biosphere”,482 a strong hypothesis for the origin of Darwinian evolution and life is “the abiotic formation of long-chained RNA polymers” with the ability to self-replicate.309 Current theories differ by placing the emergence of homochirality at different times of the chemical and biological evolutions leading to life. Regarding on whether homochirality happens before or after the appearance of life discriminates between purely abiotic and biotic theories, respectively (Fig. 19). In between these two extreme cases, homochirality could have emerged during the formation of primordial polymers and/or their evolution towards more elaborated macromolecules. | ||
| Fig. 19 Possible connections between the emergences of life and homochirality at the different stages of the chemical and biological evolutions. Possible mechanisms leading to homochirality are indicated below each of the three main scenarios. Some of these mechanisms imply an initial chiral bias which can be of terrestrial or extra-terrestrial origins as discussed in Part 5.1. LUCA = Last Universal Cellular Ancestor. | ||
Finally, the oligomerization of activated racemic guanosine was also inhibited on DNA and PNA templates.487 The latter being achiral, it suggests that enantiomeric cross-inhibition is intrinsic to the templated oligomerization process involving complementary nucleobases.
Several scenarios have considered that initial enantiomeric imbalances have probably been decreased by racemization but not eliminated. Abiotic theories thus rely on processes that would be able to amplify tiny enantiomeric excesses (likely ≪ 1% e.e.) up to the homochiral state. Intermolecular interactions cause enantiomer and racemate to have different physicochemical properties and this can be exploited to enrich a scalemic material into one enantiomer under strictly achiral conditions. This phenomenon of self-disproportionation of the enantiomers (SDE) is not rare for organic molecules and may occur through a wide range of physicochemical processes.61 SDE with molecules of life such as amino acids and sugars is often discussed in the framework of the emergence of BH. SDE often occurs during crystallization as a consequence of the difference in solubility between racemic and enantiopure crystals, and its implementation to amino acids was exemplified by Morowitz as early as 1969.495 It was confirmed later that a number of amino acids display high eutectic e.e. values which allows very high e.e. values to be present in solution, even from moderately biased enantiomeric mixtures.496 Serine is the most striking example since a virtually enantiopure solution (>99% e.e.) is obtained at 25 °C under solid–liquid equilibrium conditions starting from a 1% e.e. mixture only.497 Enantioenrichment was also reported for various amino acids after consecutive evaporations of their aqueous solutions498 or preferential kinetic dissolution of their enantiopure crystals.499 Interestingly, the eutectic e.e. values can be increased for certain amino acids by the addition of simple achiral molecules such as carboxylic acids.500DL-Cytidine, DL-adenosine and DL-uridine also form racemic crystals and their scalemic mixture can thus be enriched towards the D enantiomer in the same way provided t that the solution is saturated in both D and DL sugars.501 SDE of amino acids does not occur solely during crystallization;502e.g. sublimation of near-racemic samples of serine yields a sublimate which is highly enriched in the major enantiomer.503 Amplification of e.e. by sublimation has also been reported for other scalemic mixtures of amino acids,504–506 or for a racemate mixed with a non-volatile optically pure amino acid.507 Alternatively, amino acids were enantio-enriched by simple dissolution/precipitation of their phosphorylated derivatives in water.508
It is likely that prebiotic chemistry has linked amino acids, sugars and lipids in a way that remains to be determined. Merging the organocatalytic properties of amino acids with the aforementioned SDE phenomenon offers a pathway towards enantiopure sugars.509 The aldol reaction between 2-chlorobenzaldehyde and acetone was found to exhibit a strongly positive non-linear effect, i.e. the e.e. in the aldol product is drastically higher than that expected from the optical purity of the engaged amino acid catalyst.497 Again, the effect was particularly strong with serine since nearly racemic serine (1% e.e.) and enantiopure serine provided the aldol product with the same enantioselectivity (ca. 43% e.e., Fig. 20, (1)). Enamine catalysis in water was employed to prepare glyceraldehyde, the probable synthon towards ribose and other sugars, by reacting glycolaldehyde and formaldehyde in the presence of various enantiopure amino acids. It was found that all (S)-amino acids, except (S)-proline, provided glyceraldehyde with a predominant R configuration (up to 20% e.e. with (S)-glutamic acid, Fig. 20, (2)).65,510 This result coupled to SDE furnished a small fraction of glyceraldehyde with 84% e.e. Enantio-enriched tetrose and pentose sugars are also produced by means of aldol reactions catalysed by amino acids and peptides in aqueous buffer solutions, albeit in modest yields.511–513
 | ||
| Fig. 20 Selected catalytic reactions involving amino acids and sugars, and leading to the enantioenrichment of prebiotically relevant molecules. | ||
The influence of α-amino acids on the synthesis of RNA precursors was also probed. Along this line, Blackmond and co-workers reported that ribo- and arabino-amino oxazolines were enantio-enriched towards the expected D configuration when 2-aminooxazole and (RS)-glyceraldehyde were reacted in the presence of (S)-proline (Fig. 20, (3)).514 When coupled with the SDE of the reacting proline (1% e.e.) and of the enantio-enriched product (20–80% e.e.), the reaction yielded enantiopure crystals of ribo-amino-oxazoline. (S)-Proline does not act as a mere catalyst in this reaction but rather traps the (S)-enantiomer of glyceraldehyde, thus accomplishing a formal resolution of the racemic starting material. The latter reaction can also be exploited in the opposite way to resolve a racemic mixture of proline in the presence of enantiopure glyceraldehyde (Fig. 20, (4)). This dual substrate/reactant behaviour motivated the same group to test the possibility of synthetizing enantio-enriched amino acids with D-sugars. The hydrolysis of 2-benzyl α-amino nitrile yielded the corresponding α-amino amide (precursor of phenylalanine) with various e.e. values and configurations depending on the nature of the sugars.515 Notably, D-ribose provided the product with 70% e.e. biased in favour of unnatural (R)-configuration (Fig. 20, (5)). This result, which is apparently contradictory with such process being involved in the primordial synthesis of amino acids, was solved by finding that the mixture of four D-pentoses actually favoured the natural (S) amino acid precursor. This result suggests an unanticipated role of prebiotically relevant pentoses such as D-lyxose in mediating the emergence of amino acid mixtures with a biased (S) configuration.
How the building blocks of proteins, nucleic acids and lipids would have interacted between each other before the emergence of life is a subject of intense debate. The aforementioned examples in which prebiotic amino acids, sugars, and nucleotides would have mutually triggered their formation are actually not the privileged scenario of ‘origin of life’ practitioners. Most theories infer relationship at a more advanced stage of the chemical evolution. In the “RNA world”,516 a primordial RNA replicator catalysed the formation of the first peptides and proteins. Alternative hypotheses are that proteins (“metabolism first” theory) or lipids517 originated first518 or that RNA, DNA and proteins emerged simultaneously by continuous and reciprocal interactions, i.e. mutualism.519,520 It is commonly considered that homochirality would have arisen through stereoselective interactions between the different types of biomolecules, i.e. chirally matched combinations would have conducted to potent living systems whilst the chirally mismatched combinations would have declined. Such theory has notably been proposed recently to explain the splitting of lipids into opposite configurations in archaea and bacteria (known as the ‘lipide divide’)521 and their persistence.522 However, these theories do not address the fundamental question of the initial chiral bias and its enhancement.
SDE appears as a potent way to increase the optical purity of some building blocks of life but its limited scope, efficiency (an initial bias ≥1% e.e. is required) and productivity (high optical purity is reached at the cost of the mass of the material) appear detrimental for explaining the emergence of chemical homochirality. An additional drawback of SDE is that the enantioenrichment is only local, i.e. the overall material remains unenriched. SMSB processes as those mentioned in Part 4 are consequently considered as more probable alternatives towards homochiral prebiotic molecules. They disclose two major advantages: (i) a tiny fluctuation around the racemic state might be amplified up to the homochiral state in a deterministic manner, (ii) the amount of prebiotic molecules generated throughout these processes is potentially very high (e.g. in Viedma-type ripening experiments).383 Even though experimental reports of SMSB processes have appeared in the literature for the last 25 years, none of them display conditions that appear relevant to prebiotic chemistry. The quest for small-molecule reactions, exhibiting asymmetric replication and persisting high selectivity, compatible with primeval conditions has recently been suggested as a key challenge for organic chemists.523 Studying complex networks of organic chemical reactions524 instead of single auto-catalytic events might shed light on cooperative systems from which homochirality might emerge.302 In this context, open systems with a continuous supply of reactants are better suited to reach homochiral NESS and it is expected that current progresses made in studying the self-assembly process under dissipative conditions525 will be extended to chiral reaction networks.41
5.3 Homochirality through polymerization
Purely abiotic theory is based on the argument that enantiomeric cross-inhibition will ineluctably impede the formation of potent replicators. However, the fact that chemical processes may follow dramatically different mechanisms depending on the conditions has been overlooked. Likewise, stereoselective and non-selective polymerization reactions which allow regular and random arrangements of the monomer enantiomers along the polymer backbone, respectively, are ubiquitous in polymer science, and cross-inhibition is likely to be the exception rather than the norm.526,527![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the D and L enantiomorphs was found to improve the stereoselectivity of the polymerization process thanks to the selective adsorption of the more regular homochiral peptides on the quartz surface.532 The combination of (α)-quartz and a reaction mixture biased in favour of one of the amino-acid enantiomer (20% e.e.) was necessary to get homochiral sequences as the major component of the peptide stereoisomers.533 The length of peptides reached under these conditions remains limited (n < 10) which lets the question of how long and well-structured homochiral peptides sequences emerged from the prebiotic soup unanswered. One possibility is that their formation was triggered by a ribozyme, i.e. that the construction of functional and catalytic RNAs preceded the generation of peptides and proteins.516
:1 mixture of the D and L enantiomorphs was found to improve the stereoselectivity of the polymerization process thanks to the selective adsorption of the more regular homochiral peptides on the quartz surface.532 The combination of (α)-quartz and a reaction mixture biased in favour of one of the amino-acid enantiomer (20% e.e.) was necessary to get homochiral sequences as the major component of the peptide stereoisomers.533 The length of peptides reached under these conditions remains limited (n < 10) which lets the question of how long and well-structured homochiral peptides sequences emerged from the prebiotic soup unanswered. One possibility is that their formation was triggered by a ribozyme, i.e. that the construction of functional and catalytic RNAs preceded the generation of peptides and proteins.516
Synthetic chemistry aimed at mimicking prebiotic conditions for the synthesis of RNA oligomers has provided some support along this direction. Oligomers of up to 55 nucleotides can be synthetized by successive elongation of a decanucleotide with enantiopure nucleotides on Na+-montmorillonite.288 Subsequent experiments have then been conducted directly from racemic mixtures of activated mononucleotides in order to probe the possibility of generating homochiral RNA oligomers, again with Na+-montmorillonite. Activated racemic adenosine oligomerized with comparable efficiency to enantiopure D-monomers discarding significant enantiomeric cross-inhibition.534 The distribution of oligomer stereoisomers (up to 8 units under these conditions) appeared to be biased in favour of homochiral sequences. Deeper investigation of these reactions confirmed important and modest chiral selection in the oligomerization of activated adenosine535–537 and uridine, respectively.537 The co-oligomerization reaction of activated adenosine and uridine exhibited greater efficiency (up to 74% homochiral selectivity for the trimers) compared with the separate reactions of enantiomeric activated monomers.538 Again, the length of oligomers detected in these experiments is far below the estimated number of nucleotides necessary to instigate chemical evolution.540 This questions the plausibility of RNA as the primeval informational polymer. Joyce and co-workers evoked the possibility of a more flexible chiral polymer based on acyclic nucleoside analogues as an ancestor of the more rigid furanose-based replicators but this hypothesis has not been probed experimentally.541
Replication provided an advantage for achieving stereoselectivity provided that reactivity of chirally mismatched combinations are disfavoured relative to homochiral ones. A 32-residue peptide replicator was designed to probe the relationship between homochirality and self-replication.539 Electrophilic and nucleophilic 16-residue peptide fragments of the same handedness were preferentially ligated, even in the presence of their enantiomers (ca. 70% of diastereomeric excess was reached when peptide fragments EL, ED, NE, and ND were engaged, Fig. 21). The replicator entails a stereoselective autocatalytic cycle, for which all bimolecular steps are faster for matched versus unmatched pairs of substrate enantiomers, thanks to self-recognition driven by hydrophobic interactions.542 The process is very sensitive to the optical purity of the substrates, fragments embedding a single (S)/(R) amino acid substitution lacked significant auto-catalytic properties. On the contrary, stereochemical mismatches were tolerated in the replicator; single mutated templates were able to couple homochiral fragments, a process referred to as “dynamic stereochemical editing”.
 | ||
| Fig. 21 Top: Schematic representation of the stereoselective replication of peptide residues with the same handedness. Below: Diastereomeric excess (de) as a function of time. de (%)= [(TLL + TDD) − (TLD + TDL)]/Ttotal.539 | ||
Templating also appeared to be crucial for promoting the oligomerization of nucleotides in a stereoselective way. The complementarity between nucleobase pairs was exploited to achieve homochiral sequences of pyranosyl-RNA.421 Activated homochiral tetramers containing hemi self-complementary base sequences (pr(GCCG)-2′3′cyclophosphate, pr = pyranosyl-ribo) yielded relatively long oligomers (a ten of units) under mild conditions. Heterochiral tetramers (e.g. DDDL, DDLD, and DLDD stereoisomers) were found to be poorly reactive under the same conditions. Importantly, the oligomerization of the homochiral tetramer was only slightly affected when conducted in the presence of heterochiral tetramers. These results raised the possibility that a similar experiment performed with the whole set of stereoisomers would have generated “predominantly homochiral” (L) and (D) sequence libraries of relatively long p-RNA oligomers. The studies with replicating peptides or auto-oligomerizing pyranosyl tetramers undoubtedly yield peptides and RNA oligomers that are both longer and optically purer than in the aforementioned reactions (Part 5.2) involving activated monomers. Further work is needed to delineate whether these elaborated molecular frameworks could have emerged from the prebiotic soup.
Replication in the aforementioned systems stems from the stereoselective non-covalent interactions established between products and substrates. Stereoselectivity in the aggregation of non-enantiopure chemical species is a key mechanism for the emergence of homochirality in the various states of matter.543 The formation of homochiral versus heterochiral aggregates with different macroscopic properties led to enantioenrichment of scalemic mixtures through SDE as discussed in Part 5.2. Alternatively, homochiral aggregates might serve as templates at the nanoscale. In this context, the ability of serine (Ser) to preferentially form octamers when ionized from its enantiopure form is intriguing.544 Moreover, (S)-Ser in these octamers can be substituted enantiospecifically by prebiotic molecules (notably D-sugars)545 suggesting an important role of this amino acid in prebiotic chemistry. However, the preference for homochiral clusters is strong but not absolute and other clusters form when the ionization is conducted from racemic Ser,546,547 making the implication of serine clusters in the emergence of homochiral polymers or aggregates doubtful.
Lahav and co-workers investigated in detail the correlation between aggregation and reactivity of amphiphilic activated racemic α-amino acids.548 These authors found that the stereoselectivity of the oligomerization reaction is strongly enhanced under conditions for which β-sheet aggregates are initially present549 or emerge during the reaction process.550–552 These supramolecular aggregates serve as templates in the propagation step of chain elongation leading to long peptides and co-peptides with a significant bias towards homochirality. Large enhancement of the homochiral content was detected, notably for the oligomerization of rac-Val NCA in the presence of 5% of an initiator (Fig. 22).551 Racemic mixtures of isotactic peptides are desymmetrized by adding chiral initiators551 or by biasing the initial enantiomer composition.553,554 The interplay between aggregation and reactivity might have played a key role for the emergence of primeval replicators.
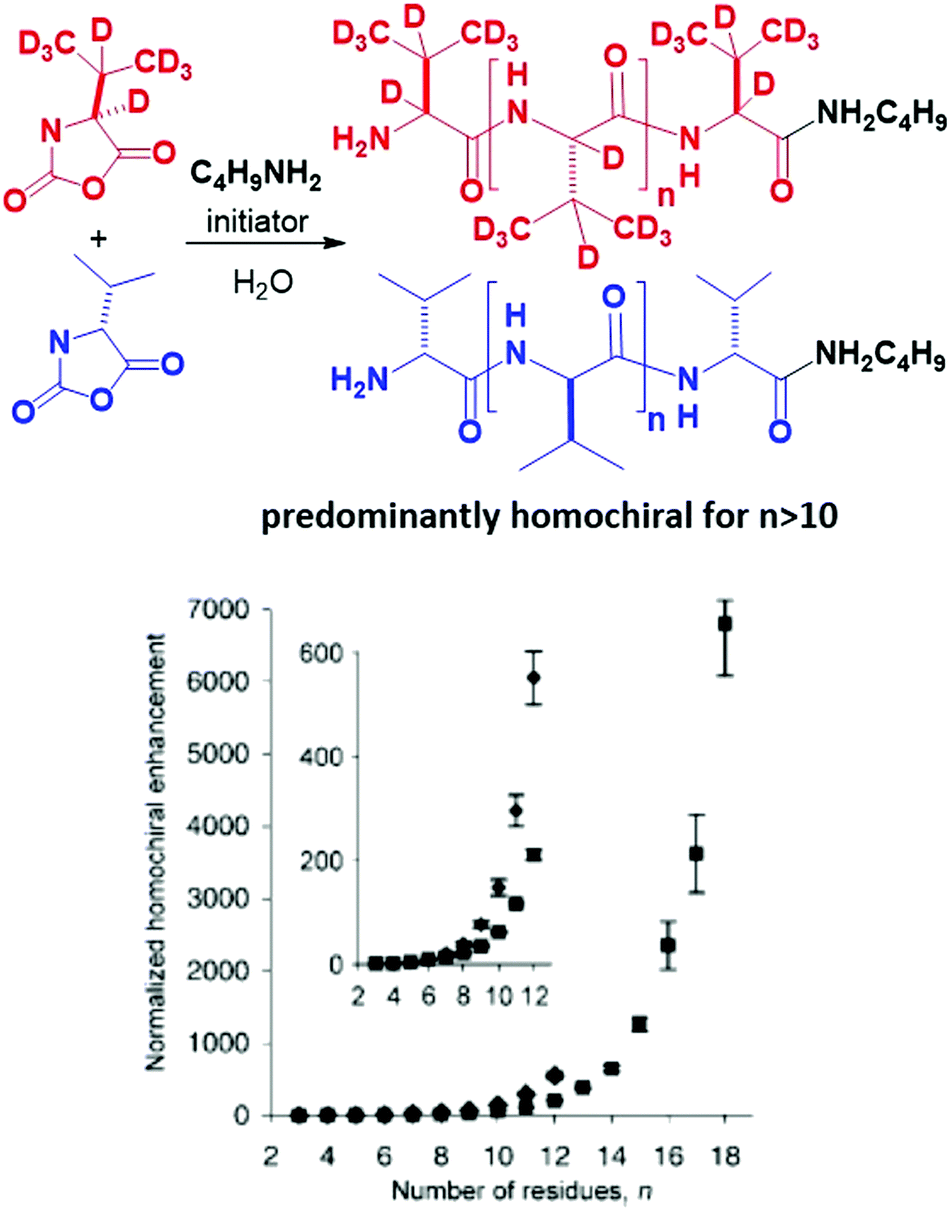 | ||
| Fig. 22 Stereoselective polymerization of rac-Val N-carboxyanhydride in the presence of 5 mol% (square) or 25 mol% (diamond) of n-butylamine as the initiator. Homochiral enhancement is calculated relative to the binomial distribution of the stereoisomers. Reprinted from ref. 551 with permission from Wiley-VCH, copyright 2008. | ||
:1 ratio of (R) and (S) α-amino acids, even though less stable than their homochiral analogues, exhibit structural requirements (folding, substrate binding and active sites) suitable for promoting early metabolism (e.g. t-RNA and DNA ligase activities).557 Likewise, several racemic membranes, i.e. composed of lipid antipodes, were found to be of comparable stability to homochiral ones.521
Several scenarios towards BH involve non-homochiral polymers as possible intermediates towards potent replicators. Joyce proposed a three-phase process towards the formation of genetic materials assuming the formation of flexible polymers, constructed from achiral or prochiral acyclic nucleoside analogues, as intermediates towards RNA and finally DNA.541 It was presumed that ribose-free monomers would be more easily accessed from the prebiotic soup than ribose ones and that the conformational flexibility of these polymers would work against enantiomeric cross-inhibition. Other simplified structures, relative to RNA, have been proposed by others.558 However, the molecular structures of the proposed building blocks are still complex relative to what is expected to be readily generated from the prebiotic soup. Brewer and Davis hypothesized a set of more realistic polymers that could have emerged from very simple building blocks such as formaldehyde, α-substituted ketones, aldehydes, alkenes, amino-acids or α-hydroxy acids.422 Polymers with random arrangement of (R) and (S) stereogenic centres are expected to be replicated through recognition of their chiral sequence. Such chiral encoding559 might allow the emergence of replicators with specific catalytic properties. If one considers that the large number of possible sequences exceeds the number of molecules present in a reasonably sized sample of these chiral informational polymers, then their mixture will not constitute a perfect racemate since certain heterochiral polymers will lack their enantiomers. This argument of the emergence of homochirality or of a chiral bias “by chance” mechanism through the polymerization of a racemic mixture was also put forward previously by Eschenmoser421 and Siegel.17 This concept has been sporadically probed notably through the template-controlled copolymerization of the racemic mixtures of two different activated amino acids.560–562 However, in the absence of any chiral bias, it is more likely that this mixture will yield informational polymers with pseudo enantiomeric like structures rather than the idealized chirally uniform polymers (see Part 5.4). Finally, the same authors also considered that pairing and replication between heterochiral polymers could operate through interaction between their helical structures, rather than on their individual stereogenic centres (Fig. 23).422 On this specific point, it should be emphasized that the helical conformation adopted by the main chain of certain types of polymers can be “amplified”, i.e. that single handed fragments may form even if composed of non-enantiopure building blocks.563 For example, synthetic polymers embedding a modestly biased racemic mixture of enantiomers adopt a single-handed helical conformation thanks to the so-called “majority-rules” effect.564–566 This phenomenon might have helped to enhance the helicity of the primeval heterochiral polymers relatively to the optical purity of their feeding monomers.
 | ||
| Fig. 23 Principle of chiral encoding in the case of a template consisting of regions of alternating helicity. The initially formed heterochiral polymer replicates by recognition and ligation of its constituting helical fragments.422 | ||
The Sandars model was modified in different ways by several groups570–574 to integrate more realistic parameters such as the possibility for polymers of all lengths to act catalytically in the breakdown of the achiral substrate into chiral monomers (instead of solely polymers of length N as in the model of Sandars).64,575 Hochberg considered in addition a closed chemical system (i.e. the total mass of matter is kept constant) which allows polymers to grow towards a finite length (see reaction scheme in Fig. 24).64 Starting from an infinitesimal e.e. bias (e.e.0 = 5 × 10−8%), the model shows the emergence of homochiral polymers in an absolute but temporary manner. The reversibility of this SMSB process was expected for a closed system. Ma and co-workers recently published a probabilistic approach which is presumed to better reproduce the emergence of the primeval RNA replicators and ribozymes in the RNA World.576 The D-nucleotide and L-nucleotide precursors are set to racemize to account for the behaviour of glyceraldehyde under prebiotic conditions; and the polynucleotide synthesis is surface- or template-mediated. The emergence of RNA polymers with RNA replicase or nucleotide synthase properties during the course of the simulation led to amplification of the initial chiral bias. Finally, several models show that cross-inhibition is not a necessary condition for the emergence of homochirality in polymerization processes. Higgs and co-workers considered all polymerization steps to be random (i.e. occurring with the same rate constant) regardless of the nature of condensed monomers and that a fraction of homochiral polymers catalyzes the formation of the monomer enantiomers in an enantiospecific manner.577 The simulation yielded homochiral polymers (of both antipodes) even from a pure racemate under conditions which favour the catalyzed over non-catalyzed synthesis of the monomers. These polymers are referred to as “chiral living polymers” as the result of their auto-catalytic properties. Hochberg modified its previous kinetic reaction scheme drastically by suppressing cross-inhibition (polymerization operates through a stereoselective and cooperative mechanism only), and by allowing fragmentation and fusion of the homochiral polymer chains.578 The process of fragmentation is irreversible for the longest chains, mimicking a mechanical breakage. This breakage represents an external energy input to the system. This binary chain fusion mechanism is necessary to achieve SMSB in this simulation from infinitesimal chiral bias (e.e.0 = 5 × 10−11%). Finally, even though not specifically designed for a polymerization process, a recent model by Ribó and Hochberg shows how homochiral replicators could emerge from two or more catalytically coupled asymmetric replicators, again without the need for the inclusion of a heterochiral inhibition reaction.350 Six homochiral replicators emerge from their simulation by means of an open flow reactor incorporating six achiral precursors and replicators in low initial concentrations and minute chiral biases (e.e.0 = 5 × 10−18%). These models should stimulate the quest of polymerization pathways which include stereoselective ligation, enantioselective synthesis of the monomers, replication and cross-replication, i.e. hallmarks of an ideal stereoselective polymerization process.
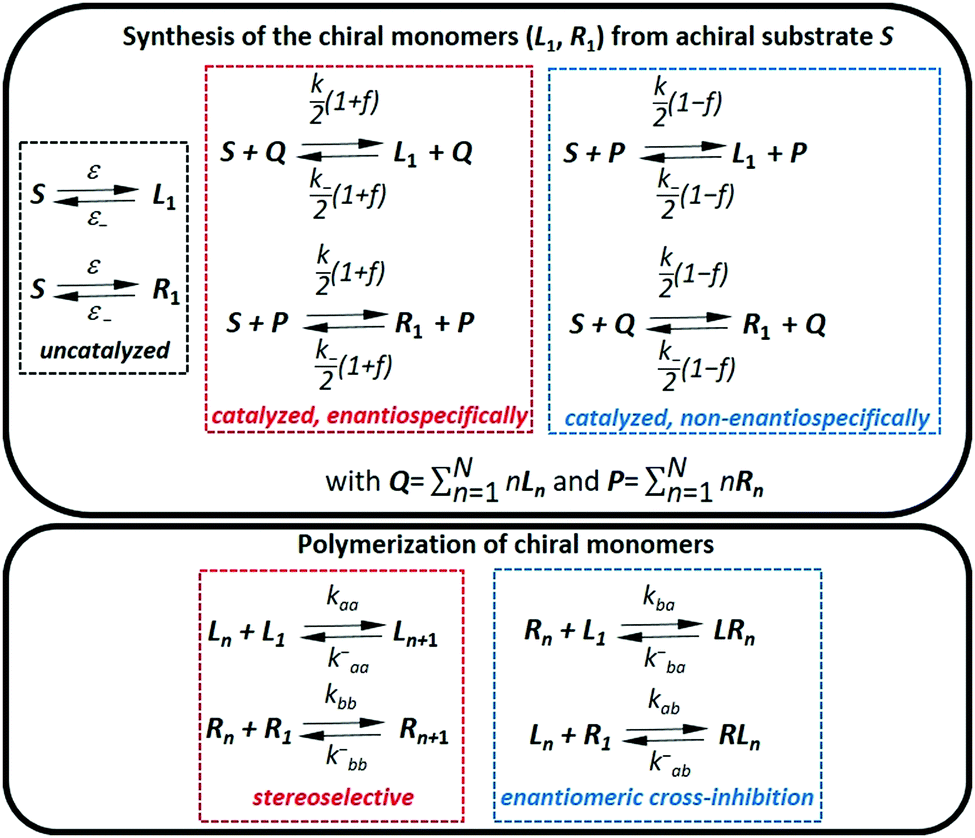 | ||
| Fig. 24 The Hochberg model for chiral polymerization in closed systems. N = maximum chain length of the polymer, f = fidelity of the feedback mechanism, Q and P are the total concentrations of left-handed and right-handed polymers, respectively. ε(ε−), k(k−), kaa(kaa−), kbb(kbb−), kba(kba−), kab(kab−) denote the forward (reverse) reaction rate constants.64 | ||
5.4 Purely biotic scenarios
In the previous two sections, the emergence of BH was dated at the level of prebiotic building blocks of life (for purely abiotic theories) or at the stage of the primeval replicators, i.e. at the early or advanced stages of the chemical evolution, respectively. In most theories, an initial chiral bias was amplified yielding either prebiotic molecules or replicators as single enantiomers. Others hypothesized that homochiral replicators and then life emerged from unbiased racemic mixtures by chance, basing their rationale on probabilistic grounds.17,421,559,577 In 1957, Fox,579 Rush580 and Wald581 held a different view and independently emitted the hypothesis that BH is an inevitable consequence of the evolution of the living matter.44 Wald notably reasoned that, since polymers made of homochiral monomers likely propagate faster, are longer and have stronger secondary structures (e.g. helices), they must have provided sufficient criteria to the chiral selection of amino acids thanks to the formation of their polymers under ad hoc conditions. This statement was supported by experiments showing that stereoselective polymerization is enhanced when oligomers adopt a α-helix conformation.485,528 However, Wald went a step further by supposing that homochiral polymers of both handedness would have been generated under the supposedly symmetric external forces present on prebiotic earth and that primordial life originated under the form of two populations of organisms, enantiomers of each other. From then, the natural forces of evolution led certain organisms to be superior to their enantiomorphic neighbours leading to life in a single form, as we know it today. The purely biotic theory of emergence of BH thanks to biological evolution, instead of chemical evolution for abiotic theories, was accompanied by large scepticism in the literature even though the arguments of Wald were developed later on by others44 and notably by Ageno (sexual reproduction naturally resolves enantiomeric populations),582 and Kuhn (the stronger enantiomeric form of life survived in the “struggle”).583 More recently, Green and Jain summarized the Wald theory into the catchy formula “Two Runners, One Tripped”,584 and called for deeper investigation on routes towards racemic mixtures of biologically relevant polymers.The Wald theory by its essence has been difficult to assess experimentally. On the one side, (R)-amino acids when found in mammals are often related to destructive and toxic effects suggesting a lack of complementary with the current biological machinery in which (S)-amino acids are ultra-predominating. On the other side, (R)-amino acids have been detected in the cell wall peptidoglycan layer of bacteria585 and in various peptides of bacteria, archaea, and eukaryotes.16 (R)-Amino acids in these various living systems have an unknown origin. Certain proponents of the purely biotic theories suggest that the small but general occurrence of (R)-amino acids in nowadays living organisms can be a relic of a time in which mirror-image living systems were “struggling”. Likewise, to rationalize the aforementioned “lipid divide”, it has been proposed that the LUCA of bacteria and archaea could have embedded a heterochiral lipid membrane, i.e. a membrane containing two sorts of lipid with opposite configurations.521
Several studies also probed the possibility to prepare a biological system containing the enantiomers of the molecules of life as we know it today. L-Polynucleotides and (R)-polypeptides were synthesized; and expectedly they exhibited chiral substrate specificity and biochemical properties that mirrored those of their natural counterparts.586–588 In a recent example, Liu, Zhu and co-workers showed that a synthesized 174-residue (R)-polypeptide catalyzes the template-directed polymerization of L-DNA and its transcription into L-RNA.587 It was also demonstrated that the synthesized and natural DNA polymerase systems operate without any cross-inhibition when mixed together in the presence of a racemic mixture of the constituents required for the reaction (D- and L primers, D- and L-templates and D- and L-dNTPs). From these impressive results, it is easy to imagine how mirror-image ribozymes would have worked independently in the early evolution times of primeval living systems.
One puzzling question concerns the feasibility for a biopolymer to synthesize its mirror-image. This has been addressed elegantly by the group of Joyce who demonstrated very recently the possibility for a RNA polymerase ribozyme to catalyze the templated synthesis of RNA oligomers of the opposite configuration.589 The D-RNA ribozyme was selected, through 16 rounds of selective amplification away from a random sequence, for its ability to catalyze the ligation of two L-RNA substrates on a L-RNA template. The 16.12t D-RNA ribozyme was eventually discovered which exhibited sufficient activity to generate full-length copies of its enantiomer through the template-assisted ligation of 11 oligonucleotides. Variants of this cross-chiral enzyme demonstrated stronger ability to polymerize nucleotide triphosphates (NTP) and trinucleotides.590 Importantly, these designed ribozymes (such as the NTP polymerase shown in Fig. 25) remain operative in the presence of racemic substrates and templates. In the hypothesis of a RNA world, it is intriguing to consider the possibility of a primordial ribozyme with cross-catalytic polymerization activities. In such a case, one can consider the possibility that enantiomeric ribozymes would have existed concomitantly and that evolutionary innovation would have favoured the systems based on D-RNA and (S)-polypeptides leading to the exclusive form of BH as present on earth nowadays. Finally, a strongly convincing evidence for the standpoint of the purely biotic theories would be the discovery in sediments of primitive forms of life based on a molecular machinery entirely composed of (R)-amino acids and L-nucleic acids.
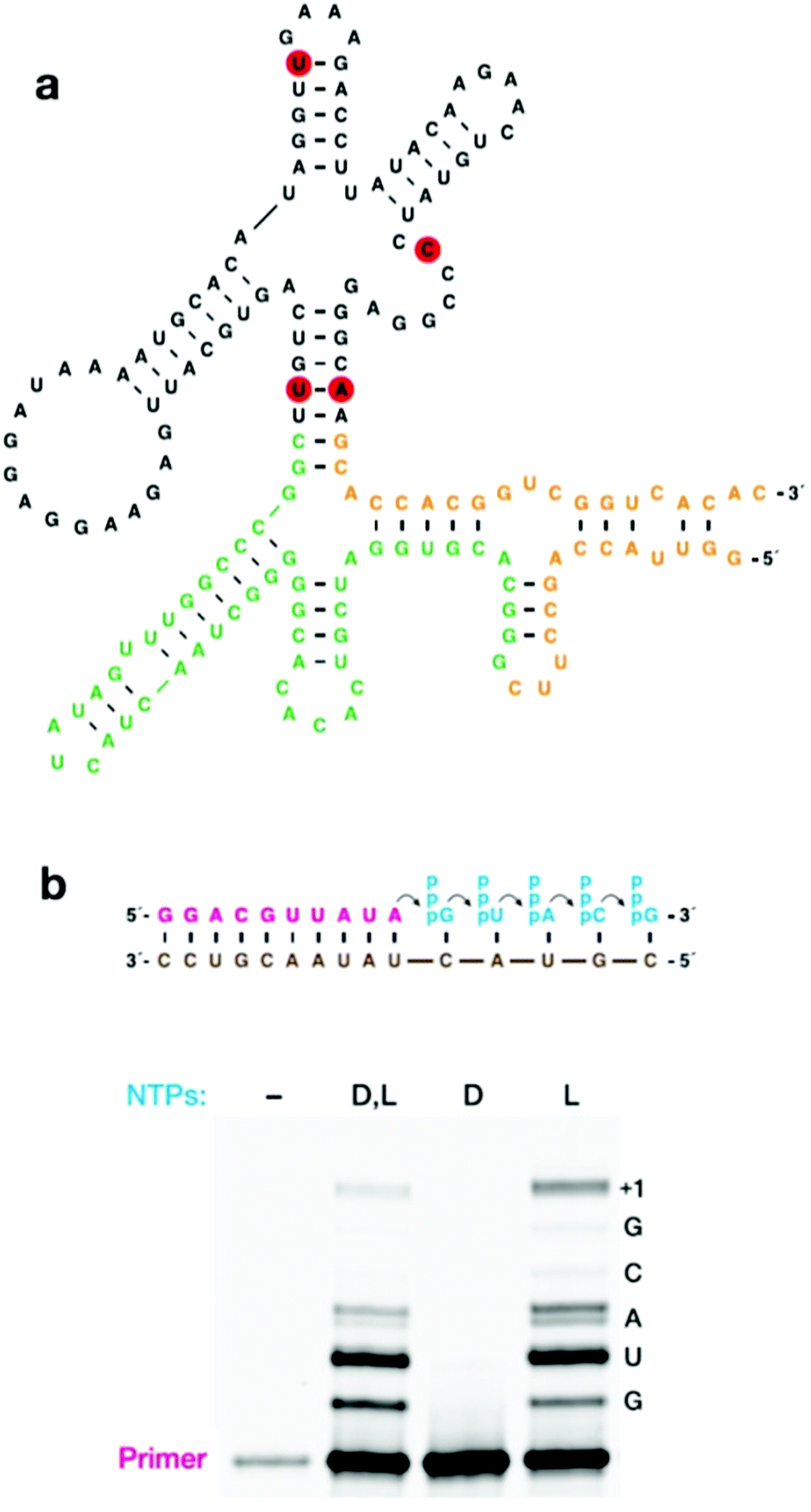 | ||
| Fig. 25 Cross-chiral ribozyme. (a) Sequence and secondary structure of the 42.9t ribozyme. Structural evolutions from the starting 16.12t ribozyme are indicated as follow: core of the 16.12t ribozyme (black), optimized nucleotide sequence (green) and primer binding sites (orange). Red circles indicate mutations relative to the core of the starting 16.12t ribozyme. (b) RNA-templated polymerization, demonstrating incorporation of all four L-NTPs, but not D-NTPs. The experiments are conducted with a L-RNA primer connected to the D-ribozyme, in presence of a separate L-RNA template, in order to direct the synthesis of a product having the sequence 5′-GUACG-3′. Reproduced with permission from ref. 590. Copyright 2020. American Chemical Society. | ||
6. Conclusions and perspectives of biological homochirality studies
Questions accumulated while considering all the possible origins of the initial enantiomeric imbalance that have ultimately led to biological homochirality. When some hypothesize a reason behind its emergence (such as for informational entropic reasons, resulting in evolutionary advantages towards more specific and complex functionalities),25,350 others wonder whether it is reasonable to reconstruct a chronology 3.5 billion years later.37 Many are circumspect in front of the pile-up of scenarios and assert that the solution is likely not expected in a near future (due to the difficulty to do all required control experiments, and fully understand the theoretical background of the putative selection mechanism).53 In parallel, the existence, and the extent, of a putative link between the different configurations of biologically relevant amino acids and sugars also remains unsolved,591 and only Goldanskii and Kuz’min studied the effects of a hypothetical global loss of optical purity in the future.429Nevertheless, great progress has been made recently for a better perception of this long-standing enigma. The scenario involving circularly polarized light as a chiral bias inducer is more and more convincing thanks to operational and analytical improvements. Increasingly accurate computational studies supply precious information, notably about SMSB processes, chiral surfaces, and other truly chiral influences. Asymmetric autocatalytic systems and deracemization processes have also undoubtedly grown in interest (notably thanks to the discoveries of the Soai reaction and the Viedma ripening). Space missions are also an opportunity: to study the in situ organic matter, its conditions of transformations, and possible associated enantio-enrichment; to elucidate the solar system origin and its history; and maybe, to find traces of chemicals with “unnatural” configurations in celestial bodies, which could indicate that the chiral selection of terrestrial BH could be a mere coincidence.
The current state of the art indicates that further experimental investigations of the possible effect of other sources of asymmetry are needed. Photochirogenesis is attractive in many respects: CPL has been detected in space, e.e. values have been measured for several prebiotic molecules found on meteorites or generated in laboratory-reproduced interstellar ices. However, this detailed postulated scenario still faces pitfalls related to the variable sources of extra-terrestrial CPL, the requirement of finely-tuned illumination conditions (almost full extent of reaction at the right place and moment of the evolutionary stages), and the unknown mechanism leading to the amplification of the original chiral biases. Strong calls to organic chemists are thus necessary to discover new asymmetric autocatalytic reactions, maybe through the investigation of complex and large chemical systems,592 that can meet the criteria of primordial conditions.40,41,302,312
Anyway, the quest for the biological homochirality origin is fruitful in many aspects. The first concerns one consequence of the asymmetry of life: the contemporary challenge of synthesizing enantiopure bioactive molecules. Indeed, many synthetic efforts are directed towards the generation of optically-pure molecules, to avoid potential side effects of racemic mixtures due to the enantioselectivity of biological receptors. These endeavors can undoubtedly draw inspiration from a range of deracemization and chirality induction processes conducted in connection with biological homochirality. One example is the Viedma ripening, which allows the preparation of enantiopure molecules displaying potent therapeutic activities.55,593 Other efforts are devoted to the building-up of sophisticated experiments and pushing their measurement limits to be able to detect tiny enantiomeric excesses, thus strongly contributing to important improvements in scientific instrumentation and acquiring fundamental knowledge at the interface between chemistry, physics, and biology. Overall, this joint endeavor at the frontier of many fields is also beneficial to materials science notably for the elaboration of biomimetic materials and emerging chiral materials.594,595
Abbreviations
| BH | Biological homochirality |
| CISS | Chiral-induced spin selectivity |
| CD | Circular dichroism |
| de | Diastereomeric excess |
| DFT | Density functional theories |
| DNA | Deoxyribonucleic acid |
| dNTPs | Deoxynucleotide triphosphates |
| e.e(s). | Enantiomeric excess(es) |
| EPR | Electron paramagnetic resonance |
| Epi | Epichlorohydrin |
| FCC | Face-centred cubic |
| GC | Gas chromatography |
| LES | Limited enantioselective |
| LUCA | Last universal cellular ancestor |
| MCD | Magnetic circular dichroism |
| MChD | Magneto-chiral dichroism |
| MS | Mass spectrometry |
| MW | Microwave |
| NESS | Non-equilibrium stationary states |
| NCA | N-Carboxy-anhydride |
| NTPs | Nucleotide triphosphates |
| NMR | Nuclear magnetic resonance |
| OEEF | Oriented-external electric fields |
| Pr | Pyranosyl-ribo |
| PV | Parity violation |
| PVED | Parity-violating energy difference |
| REF | Rotating electric fields |
| RNA | Ribonucleic acid |
| SDE | Self-disproportionation of the enantiomers |
| SEs | Secondary electrons |
| SMSB | Spontaneous mirror symmetry breaking |
| SNAAP | Supernova neutrino amino acid processing |
| SPEs | Spin-polarized electrons |
| VUV | Vacuum ultraviolet |
Author contributions
QS selected the scope of the review, made the first critical analysis of the literature and wrote the first draft of the review. JC modified Parts 1, 2 and 3.1 according to her expertise in the domains of chiral physical fields and parity violation. MR re-organized the review into its current form and extended Parts 3–5. All authors were involved in the revision and proof-checking of the successive versions of the review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The French Agence Nationale de la Recherche is acknowledged for funding the project AbsoluCat (ANR-17-CE07-0002) to MR. The GDR 3712 Chirafun from Centre National de la recherche Scientifique (CNRS) is acknowledged for allowing a collaborative network between partners involved in this review. J. C. warmly thanks Dr Benoît Darquié from the Laboratoire de Physique des Lasers (Université Sorbonne Paris Nord) for fruitful discussions and precious advice.Notes and references
- W. Thomson, Baltimore Lectures on Molecular Dynamics and the Wave Theory of Light, Cambridge Univ. Press, Warehouse, 1894, Edition of 1904, p. 619 Search PubMed.
- K. Mislow, in Topics in Stereochemistry, ed. S. E. Denmark, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007, pp. 1–82 Search PubMed.
- B. Kahr, Chirality, 2018, 30, 351–368 CrossRef CAS PubMed.
- S. H. Mauskopf, Trans. Am. Philos. Soc, 1976, 66, 1–82 CrossRef.
- J. Gal, Helv. Chim. Acta, 2013, 96, 1617–1657 CrossRef CAS.
- L. Pasteur, C. R. Hebd. Seances Acad. Sci., 1848, 26, 535–538 Search PubMed.
- C. Djerassi, R. Records, E. Bunnenberg, K. Mislow and A. Moscowitz, J. Am. Chem. Soc., 1962, 870–872 CrossRef CAS.
- S. J. Gerbode, J. R. Puzey, A. G. McCormick and L. Mahadevan, Science, 2012, 337, 1087–1091 CrossRef CAS PubMed.
- G. H. Wagnière, On Chirality and the Universal Asymmetry. Reflections on Image and Mirror Image, VHCA, Verlag Helvetica Chimica Acta, Zürich (Switzerland), 2007 Search PubMed.
- H.-U. Blaser, Rendiconti Lincei, 2007, 18, 281–304 CrossRef.
- H.-U. Blaser, Rendiconti Lincei, 2013, 24, 213–216 CrossRef.
- A. Rouf and S. C. Taneja, Chirality, 2014, 26, 63–78 CrossRef CAS PubMed.
- H. Leek and S. Andersson, Molecules, 2017, 22, 158 CrossRef PubMed.
- D. Rossi, M. Tarantino, G. Rossino, M. Rui, M. Juza and S. Collina, Expert Opin. Drug Discovery, 2017, 12, 1253–1269 CrossRef CAS PubMed.
- Editorial: Asymmetry symposium unites economists, physicists and artists, Nature, 2018, 555, 414 Search PubMed.
- Y. Nagata, T. Fujiwara, K. Kawaguchi-Nagata, Y. Fukumori and T. Yamanaka, Biochim. Biophys. Acta, Gen. Subj., 1998, 1379, 76–82 CrossRef CAS.
- J. S. Siegel, Chirality, 1998, 10, 24–27 CrossRef CAS.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737 CrossRef CAS PubMed.
- L. Pasteur, C. R. Hebd. Seances Acad. Sci., 1857, 45, 1032–1036 Search PubMed.
- L. Pasteur, C. R. Hebd. Seances Acad. Sci., 1858, 46, 615–618 Search PubMed.
- J. Gal, Chirality, 2008, 20, 5–19 CrossRef CAS PubMed.
- J. Gal, Chirality, 2012, 24, 959–976 CrossRef CAS PubMed.
- A. Piutti, C. R. Hebd. Seances Acad. Sci., 1886, 103, 134–138 Search PubMed.
- U. Meierhenrich, Amino acids and the asymmetry of life: caught in the act of formation, Springer, Berlin, 2008 Search PubMed.
- L. Morozov, Orig. Life, 1979, 9, 187–217 CrossRef CAS PubMed.
- S. F. Mason, Nature, 1984, 311, 19–23 CrossRef CAS PubMed.
- S. Mason, Chem. Soc. Rev., 1988, 17, 347–359 RSC.
- W. A. Bonner, in Topics in Stereochemistry, ed. E. L. Eliel and S. H. Wilen, John Wiley & Sons, Ltd, 1988, vol. 18, pp. 1–96 Search PubMed.
- L. Keszthelyi, Q. Rev. Biophys., 1995, 28, 473–507 CrossRef CAS PubMed.
- M. Avalos, R. Babiano, P. Cintas, J. L. Jiménez, J. C. Palacios and L. D. Barron, Chem. Rev., 1998, 98, 2391–2404 CrossRef CAS PubMed.
- B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418–3438 CrossRef PubMed.
- J. Podlech, Cell. Mol. Life Sci. CMLS, 2001, 58, 44–60 CrossRef CAS PubMed.
- D. B. Cline, Eur. Rev, 2005, 13, 49–59 CrossRef.
- A. Guijarro and M. Yus, The Origin of Chirality in the Molecules of Life: A Revision from Awareness to the Current Theories and Perspectives of this Unsolved Problem, RSC Publishing, 2008 Search PubMed.
- V. A. Tsarev, Phys. Part. Nucl., 2009, 40, 998–1029 CrossRef CAS.
- D. G. Blackmond, Cold Spring Harb. Perspect. Biol., 2010, 2, a002147–a002147 Search PubMed.
- M. Ávalos, R. Babiano, P. Cintas, J. L. Jiménez and J. C. Palacios, Tetrahedron Asymmetry, 2010, 21, 1030–1040 CrossRef.
- J. E. Hein and D. G. Blackmond, Acc. Chem. Res., 2012, 45, 2045–2054 CrossRef CAS PubMed.
- P. Cintas and C. Viedma, Chirality, 2012, 24, 894–908 CrossRef CAS PubMed.
- J. M. Ribó, D. Hochberg, J. Crusats, Z. El-Hachemi and A. Moyano, J. R. Soc., Interface, 2017, 14, 20170699 CrossRef PubMed.
- T. Buhse, J.-M. Cruz, M. E. Noble-Terán, D. Hochberg, J. M. Ribó, J. Crusats and J.-C. Micheau, Chem. Rev., 2021, 121, 2147–2229 CrossRef CAS PubMed.
- G. Palyi, Biological Chirality, Elsevier, 1st edn, 2019 Search PubMed.
- M. Mauksch and S. B. Tsogoeva, Biomimetic Organic Synthesis, John Wiley & Sons, Ltd, 2011, pp. 823–845 Search PubMed.
- W. A. Bonner, Orig. Life Evol. Biosph., 1991, 21, 59–111 CrossRef CAS PubMed.
- G. Zubay, Origins of Life on the Earth and in the Cosmos, Elsevier, 2000 Search PubMed.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 114, 285–366 CrossRef CAS PubMed.
- M. Yadav, R. Kumar and R. Krishnamurthy, Chem. Rev., 2020, 120, 4766–4805 CrossRef CAS PubMed.
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS PubMed.
- L. D. Barron, Science, 1994, 266, 1491–1492 CrossRef CAS PubMed.
- J. Crusats and A. Moyano, Synlett, 2021, 2013–2035 CrossRef CAS.
- V. I. Gol’danskiĭ and V. V. Kuz’min, Sov. Phys. Usp., 1989, 32, 1–29 CrossRef.
- D. B. Amabilino and R. M. Kellogg, Isr. J. Chem, 2011, 51, 1034–1040 CrossRef CAS.
- M. Quack, Angew. Chem., Int. Ed., 2002, 41, 4618–4630 CrossRef CAS PubMed.
- W. A. Bonner, Chirality, 2000, 12, 114–126 CrossRef CAS PubMed.
- L.-C. Sögütoglu, R. R. E. Steendam, H. Meekes, E. Vlieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2015, 44, 6723–6732 RSC.
- K. Soai, T. Kawasaki and A. Matsumoto, Acc. Chem. Res., 2014, 47, 3643–3654 CrossRef CAS PubMed.
- J. R. Cronin and S. Pizzarello, Science, 1997, 275, 951–955 CrossRef CAS PubMed.
- A. C. Evans, C. Meinert, C. Giri, F. Goesmann and U. J. Meierhenrich, Chem. Soc. Rev., 2012, 41, 5447 RSC.
- D. P. Glavin, A. S. Burton, J. E. Elsila, J. C. Aponte and J. P. Dworkin, Chem. Rev., 2020, 120, 4660–4689 CrossRef CAS PubMed.
- J. Sun, Y. Li, F. Yan, C. Liu, Y. Sang, F. Tian, Q. Feng, P. Duan, L. Zhang, X. Shi, B. Ding and M. Liu, Nat. Commun., 2018, 9, 2599 CrossRef PubMed.
- J. Han, O. Kitagawa, A. Wzorek, K. D. Klika and V. A. Soloshonok, Chem. Sci., 2018, 9, 1718–1739 RSC.
- T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456–494 CrossRef CAS PubMed.
- K. P. Bryliakov, ACS Catal, 2019, 9, 5418–5438 CrossRef CAS.
- C. Blanco and D. Hochberg, Phys. Chem. Chem. Phys., 2010, 13, 839–849 RSC.
- R. Breslow, Tetrahedron Lett, 2011, 52, 2028–2032 CrossRef CAS.
- T. D. Lee and C. N. Yang, Phys. Rev, 1956, 104, 254–258 CrossRef CAS.
- C. S. Wu, E. Ambler, R. W. Hayward, D. D. Hoppes and R. P. Hudson, Phys. Rev., 1957, 105, 1413–1415 CrossRef CAS.
- M. Drewes, Int. J. Mod. Phys. E, 2013, 22, 1330019 CrossRef.
- F. J. Hasert, et al. , Phys. Lett. B, 1973, 46, 121–124 CrossRef CAS.
- F. J. Hasert, et al. , Phys. Lett. B, 1973, 46, 138–140 CrossRef CAS.
- C. Y. Prescott, et al. , Phys. Lett. B, 1978, 77, 347–352 CrossRef.
- C. Y. Prescott, et al. , Phys. Lett. B, 1979, 84, 524–528 CrossRef.
- L. Di Lella and C. Rubbia, 60 Years of CERN Experiments and Discoveries, World Scientific, 2014, vol. 23, pp. 137–163 Search PubMed.
- M. A. Bouchiat and C. C. Bouchiat, Phys. Lett. B, 1974, 48, 111–114 CrossRef CAS.
- M.-A. Bouchiat and C. Bouchiat, Rep. Prog. Phys., 1997, 60, 1351–1396 CrossRef CAS.
- L. M. Barkov and M. S. Zolotorev, JETP, 1980, 52, 360–376 Search PubMed.
- C. S. Wood, S. C. Bennett, D. Cho, B. P. Masterson, J. L. Roberts, C. E. Tanner and C. E. Wieman, Science, 1997, 275, 1759–1763 CrossRef CAS PubMed.
- J. Crassous, C. Chardonnet, T. Saue and P. Schwerdtfeger, Org. Biomol. Chem., 2005, 3, 2218 RSC.
- R. Berger and J. Stohner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, 25 Search PubMed.
- R. Berger, in Theoretical and Computational Chemistry, ed. P. Schwerdtfeger, Elsevier, 2004, vol. 14, pp. 188–288 Search PubMed.
- P. Schwerdtfeger, The Search for Parity Violation in Chiral Molecules, in Computational Spectroscopy, ed. J. Grunenberg, John Wiley & Sons, Ltd, 2010, pp. 201–221 Search PubMed.
- J. Eills, J. W. Blanchard, L. Bougas, M. G. Kozlov, A. Pines and D. Budker, Phys. Rev. A, 2017, 96, 042119 CrossRef.
- R. A. Harris and L. Stodolsky, Phys. Lett. B, 1978, 78, 313–317 CrossRef.
- M. Quack, Chem. Phys. Lett., 1986, 132, 147–153 CrossRef CAS.
- L. D. Barron, Magnetochemistry, 2020, 6, 5 CrossRef CAS.
- D. W. Rein, R. A. Hegstrom and P. G. H. Sandars, Phys. Lett. A, 1979, 71, 499–502 CrossRef.
- R. A. Hegstrom, D. W. Rein and P. G. H. Sandars, J. Chem. Phys., 1980, 73, 2329–2341 CrossRef CAS.
- M. Quack, Angew. Chem., Int. Ed. Engl., 1989, 28, 571–586 CrossRef.
- A. Bakasov, T.-K. Ha and M. Quack, J. Chem. Phys., 1998, 109, 7263–7285 CrossRef CAS.
- M. Quack, in Quantum Systems in Chemistry and Physics, ed. K. Nishikawa, J. Maruani, E. J. Brändas, G. Delgado-Barrio and P. Piecuch, Springer Netherlands, Dordrecht, 2012, vol. 26, pp. 47–76 Search PubMed.
- M. Quack and J. Stohner, Phys. Rev. Lett., 2000, 84, 3807–3810 CrossRef CAS PubMed.
- G. Rauhut and P. Schwerdtfeger, Phys. Rev. A, 2021, 103, 042819 CrossRef CAS.
- C. Stoeffler, B. Darquié, A. Shelkovnikov, C. Daussy, A. Amy-Klein, C. Chardonnet, L. Guy, J. Crassous, T. R. Huet, P. Soulard and P. Asselin, Phys. Chem. Chem. Phys., 2011, 13, 854–863 RSC.
- N. Saleh, S. Zrig, T. Roisnel, L. Guy, R. Bast, T. Saue, B. Darquié and J. Crassous, Phys. Chem. Chem. Phys., 2013, 15, 10952 RSC.
- S. K. Tokunaga, C. Stoeffler, F. Auguste, A. Shelkovnikov, C. Daussy, A. Amy-Klein, C. Chardonnet and B. Darquié, Mol. Phys., 2013, 111, 2363–2373 CrossRef CAS.
- N. Saleh, R. Bast, N. Vanthuyne, C. Roussel, T. Saue, B. Darquié and J. Crassous, Chirality, 2018, 30, 147–156 CrossRef CAS PubMed.
- B. Darquié, C. Stoeffler, A. Shelkovnikov, C. Daussy, A. Amy-Klein, C. Chardonnet, S. Zrig, L. Guy, J. Crassous, P. Soulard, P. Asselin, T. R. Huet, P. Schwerdtfeger, R. Bast and T. Saue, Chirality, 2010, 22, 870–884 CrossRef PubMed.
- A. Cournol, M. Manceau, M. Pierens, L. Lecordier, D. B. A. Tran, R. Santagata, B. Argence, A. Goncharov, O. Lopez, M. Abgrall, Y. L. Coq, R. L. Targat, H. Á. Martinez, W. K. Lee, D. Xu, P.-E. Pottie, R. J. Hendricks, T. E. Wall, J. M. Bieniewska, B. E. Sauer, M. R. Tarbutt, A. Amy-Klein, S. K. Tokunaga and B. Darquié, Quantum Electron., 2019, 49, 288 CrossRef CAS.
- C. Fábri, Ľ. Horný and M. Quack, ChemPhysChem, 2015, 16, 3584–3589 CrossRef PubMed.
- P. Dietiker, E. Miloglyadov, M. Quack, A. Schneider and G. Seyfang, J. Chem. Phys., 2015, 143, 244305 CrossRef CAS PubMed.
- S. Albert, I. Bolotova, Z. Chen, C. Fábri, L. Horný, M. Quack, G. Seyfang and D. Zindel, Phys. Chem. Chem. Phys., 2016, 18, 21976–21993 RSC.
- S. Albert, F. Arn, I. Bolotova, Z. Chen, C. Fábri, G. Grassi, P. Lerch, M. Quack, G. Seyfang, A. Wokaun and D. Zindel, J. Phys. Chem. Lett., 2016, 7, 3847–3853 CrossRef CAS PubMed.
- S. Albert, I. Bolotova, Z. Chen, C. Fábri, M. Quack, G. Seyfang and D. Zindel, Phys. Chem. Chem. Phys., 2017, 19, 11738–11743 RSC.
- A. Salam, J. Mol. Evol., 1991, 33, 105–113 CrossRef CAS.
- A. Salam, Phys. Lett. B, 1992, 288, 153–160 CrossRef CAS.
- T. L. V. Ulbricht, Orig. Life, 1975, 6, 303–315 CrossRef CAS PubMed.
- F. Vester, T. L. V. Ulbricht and H. Krauch, Naturwissenschaften, 1959, 46, 68 CrossRef CAS.
- Y. Yamagata, J. Theor. Biol., 1966, 11, 495–498 CrossRef CAS PubMed.
- S. F. Mason and G. E. Tranter, Chem. Phys. Lett., 1983, 94, 34–37 CrossRef CAS.
- S. F. Mason and G. E. Tranter, J. Chem. Soc. Chem. Commun., 1983, 117–119 RSC.
- S. F. Mason and G. E. Tranter, Proc. R. Soc. Lond. Math. Phys. Sci., 1985, 397, 45–65 CAS.
- G. E. Tranter, Chem. Phys. Lett., 1985, 115, 286–290 CrossRef CAS.
- G. E. Tranter, Mol. Phys., 1985, 56, 825–838 CrossRef CAS.
- G. E. Tranter, Nature, 1985, 318, 172–173 CrossRef CAS.
- G. E. Tranter, J. Theor. Biol., 1986, 119, 467–479 CrossRef CAS.
- G. E. Tranter, J. Chem. Soc. Chem. Commun., 1986, 60–61 RSC.
- G. E. Tranter, A. J. MacDermott, R. E. Overill and P. J. Speers, Proc. Math. Phys. Sci., 1992, 436, 603–615 CAS.
- A. J. MacDermott, G. E. Tranter and S. J. Trainor, Chem. Phys. Lett., 1992, 194, 152–156 CrossRef CAS.
- G. E. Tranter, Chem. Phys. Lett., 1985, 120, 93–96 CrossRef CAS.
- G. E. Tranter, Chem. Phys. Lett., 1987, 135, 279–282 CrossRef CAS.
- A. J. MacDermott and G. E. Tranter, Chem. Phys. Lett., 1989, 163, 1–4 CrossRef CAS.
- S. F. Mason and G. E. Tranter, Mol. Phys., 1984, 53, 1091–1111 CrossRef CAS.
- R. Berger and M. Quack, ChemPhysChem, 2000, 1, 57–60 CrossRef CAS PubMed.
- R. Wesendrup, J. K. Laerdahl, R. N. Compton and P. Schwerdtfeger, J. Phys. Chem. A, 2003, 107, 6668–6673 CrossRef CAS.
- J. K. Laerdahl, R. Wesendrup and P. Schwerdtfeger, ChemPhysChem, 2000, 1, 60–62 CrossRef CAS PubMed.
- G. Lente, J. Phys. Chem. A, 2006, 110, 12711–12713 CrossRef CAS PubMed.
- G. Lente, Symmetry, 2010, 2, 767–798 CrossRef CAS.
- A. J. MacDermott, T. Fu, R. Nakatsuka, A. P. Coleman and G. O. Hyde, Orig. Life Evol. Biosph., 2009, 39, 459–478 CrossRef CAS PubMed.
- A. J. MacDermott, Chirality, 2012, 24, 764–769 CrossRef CAS PubMed.
- L. D. Barron, J. Am. Chem. Soc., 1986, 108, 5539–5542 CrossRef CAS.
- L. D. Barron, Nat. Chem, 2012, 4, 150–152 CrossRef CAS PubMed.
- K. Ishii, S. Hattori and Y. Kitagawa, Photochem. Photobiol. Sci., 2020, 19, 8–19 CrossRef CAS PubMed.
- G. L. J. A. Rikken and E. Raupach, Nature, 1997, 390, 493–494 CrossRef CAS.
- G. L. J. A. Rikken, E. Raupach and T. Roth, Phys. B, 2001, 294–295, 1–4 CrossRef.
- Y. Xu, G. Yang, H. Xia, G. Zou, Q. Zhang and J. Gao, Nat. Commun., 2014, 5, 5050 CrossRef CAS PubMed.
- M. Atzori, G. L. J. A. Rikken and C. Train, Chem. – Eur. J, 2020, 26, 9784–9791 CrossRef CAS PubMed.
- G. L. J. A. Rikken and E. Raupach, Nature, 2000, 405, 932–935 CrossRef CAS PubMed.
- J. B. Clemens, O. Kibar and M. Chachisvilis, Nat. Commun., 2015, 6, 7868 CrossRef CAS PubMed.
- V. Marichez, A. Tassoni, R. P. Cameron, S. M. Barnett, R. Eichhorn, C. Genet and T. M. Hermans, Soft Matter, 2019, 15, 4593–4608 RSC.
- B. A. Grzybowski and G. M. Whitesides, Science, 2002, 296, 718–721 CrossRef PubMed.
- P. Chen and C.-H. Chao, Phys. Fluids, 2007, 19, 017108 CrossRef.
- M. Makino, L. Arai and M. Doi, J. Phys. Soc. Jpn., 2008, 77, 064404 CrossRef.
- Marcos, H. C. Fu, T. R. Powers and R. Stocker, Phys. Rev. Lett., 2009, 102, 158103 CrossRef CAS PubMed.
- M. Aristov, R. Eichhorn and C. Bechinger, Soft Matter, 2013, 9, 2525–2530 RSC.
- J. M. Ribó, J. Crusats, F. Sagués, J. Claret and R. Rubires, Science, 2001, 292, 2063–2066 CrossRef PubMed.
- Z. El-Hachemi, O. Arteaga, A. Canillas, J. Crusats, C. Escudero, R. Kuroda, T. Harada, M. Rosa and J. M. Ribó, Chem. – Eur. J., 2008, 14, 6438–6443 CrossRef CAS PubMed.
- A. Tsuda, M. A. Alam, T. Harada, T. Yamaguchi, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 8198–8202 CrossRef CAS PubMed.
- M. Wolffs, S. J. George, Ž. Tomović, S. C. J. Meskers, A. P. H. J. Schenning and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8203–8205 CrossRef CAS PubMed.
- O. Arteaga, A. Canillas, R. Purrello and J. M. Ribó, Opt. Lett., 2009, 34, 2177 CrossRef PubMed.
- A. D’Urso, R. Randazzo, L. Lo Faro and R. Purrello, Angew. Chem., Int. Ed., 2010, 49, 108–112 CrossRef PubMed.
- J. Crusats, Z. El-Hachemi and J. M. Ribó, Chem. Soc. Rev., 2010, 39, 569–577 RSC.
- O. Arteaga, A. Canillas, J. Crusats, Z. El-Hachemi, J. Llorens, E. Sacristan and J. M. Ribo, ChemPhysChem, 2010, 11, 3511–3516 CrossRef CAS PubMed.
- O. Arteaga, A. Canillas, J. Crusats, Z. El-Hachemi, J. Llorens, A. Sorrenti and J. M. Ribo, Isr. J. Chem, 2011, 51, 1007–1016 CrossRef CAS.
- P. Mineo, V. Villari, E. Scamporrino and N. Micali, Soft Matter, 2014, 10, 44–47 RSC.
- P. Mineo, V. Villari, E. Scamporrino and N. Micali, J. Phys. Chem. B, 2015, 119, 12345–12353 CrossRef CAS PubMed.
- Y. Sang, D. Yang, P. Duan and M. Liu, Chem. Sci, 2019, 10, 2718–2724 RSC.
- Z. Shen, Y. Sang, T. Wang, J. Jiang, Y. Meng, Y. Jiang, K. Okuro, T. Aida and M. Liu, Nat. Commun., 2019, 10, 1–8 Search PubMed.
- M. Kuroha, S. Nambu, S. Hattori, Y. Kitagawa, K. Niimura, Y. Mizuno, F. Hamba and K. Ishii, Angew. Chem., Int. Ed., 2019, 58, 18454–18459 CrossRef CAS PubMed.
- Y. Li, C. Liu, X. Bai, F. Tian, G. Hu and J. Sun, Angew. Chem., Int. Ed., 2020, 59, 3486–3490 CrossRef CAS PubMed.
- T. M. Hermans, K. J. M. Bishop, P. S. Stewart, S. H. Davis and B. A. Grzybowski, Nat. Commun., 2015, 6, 5640 CrossRef CAS PubMed.
- N. Micali, H. Engelkamp, P. G. van Rhee, P. C. M. Christianen, L. M. Scolaro and J. C. Maan, Nat. Chem, 2012, 4, 201–207 CrossRef CAS PubMed.
- Z. Martins, M. C. Price, N. Goldman, M. A. Sephton and M. J. Burchell, Nat. Geosci., 2013, 6, 1045–1049 CrossRef CAS.
- Y. Furukawa, H. Nakazawa, T. Sekine, T. Kobayashi and T. Kakegawa, Earth Planet. Sci. Lett., 2015, 429, 216–222 CrossRef CAS.
- Y. Furukawa, A. Takase, T. Sekine, T. Kakegawa and T. Kobayashi, Orig. Life Evol. Biosph., 2018, 48, 131–139 CrossRef CAS PubMed.
- G. G. Managadze, M. H. Engel, S. Getty, P. Wurz, W. B. Brinckerhoff, A. G. Shokolov, G. V. Sholin, S. A. Terent’ev, A. E. Chumikov, A. S. Skalkin, V. D. Blank, V. M. Prokhorov, N. G. Managadze and K. A. Luchnikov, Planet. Space Sci., 2016, 131, 70–78 CrossRef CAS PubMed.
- G. Managadze, Planet. Space Sci., 2007, 55, 134–140 CrossRef CAS.
- J. H. van’t Hoff, Arch. Néerl. Sci. Exactes Nat., 1874, 9, 445–454 Search PubMed.
- J. H. van’t Hoff, Voorstel tot uitbreiding der tegenwoordig in de scheikunde gebruikte structuur-formules in de ruimte: benevens een daarmee samenhangende opmerking omtrent het verband tusschen optisch actief vermogen en chemische constitutie van organische verbindingen, J. Greven, Utrecht, 1874 Search PubMed.
- J. H. van’t Hoff, Die Lagerung der Atome im Raume, Friedrich Vieweg und Sohn, Braunschweig, 2nd edn, 1894 Search PubMed.
- A. A. Cotton, C. R. Hebd. Seances Acad. Sci., 1895, 120, 989–991 Search PubMed.
- A. A. Cotton, C. R. Hebd. Seances Acad. Sci., 1895, 120, 1044–1046 Search PubMed.
- A. A. Cotton, Ann. Chim. Phys, 1896, 7, 347–432 Search PubMed.
- N. Berova, P. Polavarapu, K. Nakanishi and R. W. Woody, Comprehensive Chiroptical Spectroscopy: Instrumentation, Methodologies, and Theoretical Simulations, Wiley, Hoboken, NJ, USA, 2012, vol. 1 Search PubMed.
- In Comprehensive Chiroptical Spectroscopy: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, and Biomolecules, ed. N. Berova, P. Polavarapu, K. Nakanishi and R. W. Woody, Wiley, Hoboken, NJ, USA, 2012, vol. 2 Search PubMed.
- W. Kuhn, Trans. Faraday Soc., 1930, 26, 293–308 RSC.
- H. Rau, Chem. Rev., 1983, 83, 535–547 CrossRef CAS.
- Y. Inoue, Chem. Rev., 1992, 92, 741–770 CrossRef CAS.
- P. K. Hashim and N. Tamaoki, ChemPhotoChem, 2019, 3, 347–355 CrossRef CAS.
- K. L. Stevenson and J. F. Verdieck, J. Am. Chem. Soc., 1968, 90, 2974–2975 CrossRef CAS.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed.
- W. R. Browne and B. L. Feringa, in Molecular Switches, ed. W. R. Browne and B. L. Feringa, John Wiley & Sons, Ltd, 2nd edn, 2011, vol. 1, pp. 121–179 Search PubMed.
- G. Yang, S. Zhang, J. Hu, M. Fujiki and G. Zou, Symmetry, 2019, 11, 474–493 CrossRef CAS.
- J. Kim, J. Lee, W. Y. Kim, H. Kim, S. Lee, H. C. Lee, Y. S. Lee, M. Seo and S. Y. Kim, Nat. Commun., 2015, 6, 6959 CrossRef CAS PubMed.
- H. Kagan, A. Moradpour, J. F. Nicoud, G. Balavoine and G. Tsoucaris, J. Am. Chem. Soc., 1971, 93, 2353–2354 CrossRef CAS.
- W. J. Bernstein, M. Calvin and O. Buchardt, J. Am. Chem. Soc., 1972, 94, 494–498 CrossRef CAS.
- W. Kuhn and E. Braun, Naturwissenschaften, 1929, 17, 227–228 CrossRef.
- W. Kuhn and E. Knopf, Naturwissenschaften, 1930, 18, 183 CrossRef CAS.
- C. Meinert, J. H. Bredehöft, J.-J. Filippi, Y. Baraud, L. Nahon, F. Wien, N. C. Jones, S. V. Hoffmann and U. J. Meierhenrich, Angew. Chem., Int. Ed., 2012, 51, 4484–4487 CrossRef CAS PubMed.
- G. Balavoine, A. Moradpour and H. B. Kagan, J. Am. Chem. Soc., 1974, 96, 5152–5158 CrossRef CAS.
- B. Norden, Nature, 1977, 266, 567–568 CrossRef CAS PubMed.
- J. J. Flores, W. A. Bonner and G. A. Massey, J. Am. Chem. Soc., 1977, 99, 3622–3625 CrossRef CAS PubMed.
- I. Myrgorodska, C. Meinert, S. V. Hoffmann, N. C. Jones, L. Nahon and U. J. Meierhenrich, ChemPlusChem, 2017, 82, 74–87 CrossRef CAS PubMed.
- H. Nishino, A. Kosaka, G. A. Hembury, H. Shitomi, H. Onuki and Y. Inoue, Org. Lett., 2001, 3, 921–924 CrossRef CAS PubMed.
- H. Nishino, A. Kosaka, G. A. Hembury, F. Aoki, K. Miyauchi, H. Shitomi, H. Onuki and Y. Inoue, J. Am. Chem. Soc., 2002, 124, 11618–11627 CrossRef CAS PubMed.
- U. J. Meierhenrich, J.-J. Filippi, C. Meinert, J. H. Bredehöft, J. Takahashi, L. Nahon, N. C. Jones and S. V. Hoffmann, Angew. Chem., Int. Ed., 2010, 49, 7799–7802 CrossRef CAS PubMed.
- U. J. Meierhenrich, L. Nahon, C. Alcaraz, J. H. Bredehöft, S. V. Hoffmann, B. Barbier and A. Brack, Angew. Chem., Int. Ed., 2005, 44, 5630–5634 CrossRef CAS PubMed.
- U. J. Meierhenrich, J.-J. Filippi, C. Meinert, S. V. Hoffmann, J. H. Bredehöft and L. Nahon, Chem. Biodivers., 2010, 7, 1651–1659 CrossRef CAS PubMed.
- C. Meinert, S. V. Hoffmann, P. Cassam-Chenaï, A. C. Evans, C. Giri, L. Nahon and U. J. Meierhenrich, Angew. Chem., Int. Ed., 2014, 53, 210–214 CrossRef CAS PubMed.
- C. Meinert, P. Cassam-Chenaï, N. C. Jones, L. Nahon, S. V. Hoffmann and U. J. Meierhenrich, Orig. Life Evol. Biosph., 2015, 45, 149–161 CrossRef CAS PubMed.
- M. Tia, B. Cunha de Miranda, S. Daly, F. Gaie-Levrel, G. A. Garcia, L. Nahon and I. Powis, J. Phys. Chem. A, 2014, 118, 2765–2779 CrossRef CAS PubMed.
- B. A. McGuire, P. B. Carroll, R. A. Loomis, I. A. Finneran, P. R. Jewell, A. J. Remijan and G. A. Blake, Science, 2016, 352, 1449–1452 CrossRef CAS PubMed.
- Y.-J. Kuan, S. B. Charnley, H.-C. Huang, W.-L. Tseng and Z. Kisiel, Astrophys. J., 2003, 593, 848 CrossRef CAS.
- Y. Takano, J. Takahashi, T. Kaneko, K. Marumo and K. Kobayashi, Earth Planet. Sci. Lett., 2007, 254, 106–114 CrossRef CAS.
- P. de Marcellus, C. Meinert, M. Nuevo, J.-J. Filippi, G. Danger, D. Deboffle, L. Nahon, L. Le Sergeant d’Hendecourt and U. J. Meierhenrich, Astrophys. J., 2011, 727, L27 CrossRef.
- P. Modica, C. Meinert, P. de Marcellus, L. Nahon, U. J. Meierhenrich and L. L. S. d’Hendecourt, Astrophys. J., 2014, 788, 79 CrossRef.
- C. Meinert and U. J. Meierhenrich, Angew. Chem., Int. Ed., 2012, 51, 10460–10470 CrossRef CAS PubMed.
- J. Takahashi and K. Kobayashi, Symmetry, 2019, 11, 919 CrossRef CAS.
- A. G. W. Cameron and J. W. Truran, Icarus, 1977, 30, 447–461 CrossRef CAS.
- P. Bargueño and R. Pérez de Tudela, Orig. Life Evol. Biosph., 2007, 37, 253–257 CrossRef PubMed.
- P. Banerjee, Y.-Z. Qian, A. Heger and W. C. Haxton, Nat. Commun., 2016, 7, 13639 CrossRef CAS PubMed.
- T. L. V. Ulbricht, Q. Rev. Chem. Soc., 1959, 13, 48–60 RSC.
- T. L. V. Ulbricht and F. Vester, Tetrahedron, 1962, 18, 629–637 CrossRef CAS.
- A. S. Garay, Nature, 1968, 219, 338–340 CrossRef CAS PubMed.
- A. S. Garay, L. Keszthelyi, I. Demeter and P. Hrasko, Nature, 1974, 250, 332–333 CrossRef CAS PubMed.
- W. A. Bonner, M. A. V. Dort and M. R. Yearian, Nature, 1975, 258, 419–421 CrossRef CAS PubMed.
- W. A. Bonner, M. A. van Dort, M. R. Yearian, H. D. Zeman and G. C. Li, Isr. J. Chem., 1976, 15, 89–95 CrossRef.
- L. Keszthelyi, Nature, 1976, 264, 197 CrossRef CAS PubMed.
- L. A. Hodge, F. B. Dunning, G. K. Walters, R. H. White and G. J. Schroepfer, Nature, 1979, 280, 250–252 CrossRef CAS.
- M. Akaboshi, M. Noda, K. Kawai, H. Maki and K. Kawamoto, Orig. Life, 1979, 9, 181–186 CrossRef CAS PubMed.
- R. M. Lemmon, H. E. Conzett and W. A. Bonner, Orig. Life, 1981, 11, 337–341 CrossRef CAS PubMed.
- T. L. V. Ulbricht, Nature, 1975, 258, 383–384 CrossRef.
- W. A. Bonner, Orig. Life, 1984, 14, 383–390 CrossRef CAS PubMed.
- V. I. Burkov, L. A. Goncharova, G. A. Gusev, K. Kobayashi, E. V. Moiseenko, N. G. Poluhina, T. Saito, V. A. Tsarev, J. Xu and G. Zhang, Orig. Life Evol. Biosph., 2008, 38, 155–163 CrossRef CAS PubMed.
- G. A. Gusev, K. Kobayashi, E. V. Moiseenko, N. G. Poluhina, T. Saito, T. Ye, V. A. Tsarev, J. Xu, Y. Huang and G. Zhang, Orig. Life Evol. Biosph., 2008, 38, 509–515 CrossRef CAS PubMed.
- A. Dorta-Urra and P. Bargueño, Symmetry, 2019, 11, 661 CrossRef CAS.
- M. A. Famiano, R. N. Boyd, T. Kajino and T. Onaka, Astrobiology, 2017, 18, 190–206 CrossRef PubMed.
- R. N. Boyd, M. A. Famiano, T. Onaka and T. Kajino, Astrophys. J., 2018, 856, 26 CrossRef.
- M. A. Famiano, R. N. Boyd, T. Kajino, T. Onaka and Y. Mo, Sci. Rep, 2018, 8, 8833 CrossRef PubMed.
- R. A. Rosenberg, D. Mishra and R. Naaman, Angew. Chem., Int. Ed., 2015, 54, 7295–7298 CrossRef CAS PubMed.
- F. Tassinari, J. Steidel, S. Paltiel, C. Fontanesi, M. Lahav, Y. Paltiel and R. Naaman, Chem. Sci, 2019, 10, 5246–5250 RSC.
- R. A. Rosenberg, M. Abu Haija and P. J. Ryan, Phys. Rev. Lett., 2008, 101, 178301 CrossRef CAS PubMed.
- K. Michaeli, N. Kantor-Uriel, R. Naaman and D. H. Waldeck, Chem. Soc. Rev., 2016, 45, 6478–6487 RSC.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Acc. Chem. Res., 2020, 53, 2659–2667 CrossRef CAS PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem, 2019, 3, 250–260 CrossRef CAS.
- K. Banerjee-Ghosh, O. Ben Dor, F. Tassinari, E. Capua, S. Yochelis, A. Capua, S.-H. Yang, S. S. P. Parkin, S. Sarkar, L. Kronik, L. T. Baczewski, R. Naaman and Y. Paltiel, Science, 2018, 360, 1331–1334 CrossRef CAS PubMed.
- T. S. Metzger, S. Mishra, B. P. Bloom, N. Goren, A. Neubauer, G. Shmul, J. Wei, S. Yochelis, F. Tassinari, C. Fontanesi, D. H. Waldeck, Y. Paltiel and R. Naaman, Angew. Chem., Int. Ed., 2020, 59, 1653–1658 CrossRef CAS PubMed.
- R. A. Rosenberg, Symmetry, 2019, 11, 528 CrossRef CAS.
- A. Guijarro and M. Yus, The Origin of Chirality in the Molecules of Life, 2008, RSC Publishing, pp. 125–137 Search PubMed.
- R. M. Hazen and D. S. Sholl, Nat. Mater., 2003, 2, 367–374 CrossRef CAS PubMed.
- I. Weissbuch and M. Lahav, Chem. Rev., 2011, 111, 3236–3267 CrossRef CAS PubMed.
- H. J. C. Ii, A. M. Scott, F. C. Hill, J. Leszczynski, N. Sahai and R. Hazen, Chem. Soc. Rev., 2012, 41, 5502–5525 RSC.
- E. I. Klabunovskii, G. V. Smith and A. Zsigmond, Heterogeneous Enantioselective Hydrogenation – Theory and Practice, Springer, 2006 Search PubMed.
- V. Davankov, Chirality, 1998, 9, 99–102 CrossRef.
- F. Zaera, Chem. Soc. Rev., 2017, 46, 7374–7398 RSC.
- W. A. Bonner, P. R. Kavasmaneck, F. S. Martin and J. J. Flores, Science, 1974, 186, 143–144 CrossRef CAS PubMed.
- W. A. Bonner, P. R. Kavasmaneck, F. S. Martin and J. J. Flores, Orig. Life, 1975, 6, 367–376 CrossRef CAS PubMed.
- W. A. Bonner and P. R. Kavasmaneck, J. Org. Chem., 1976, 41, 2225–2226 CrossRef CAS PubMed.
- P. R. Kavasmaneck and W. A. Bonner, J. Am. Chem. Soc., 1977, 99, 44–50 CrossRef CAS PubMed.
- S. Furuyama, H. Kimura, M. Sawada and T. Morimoto, Chem. Lett., 1978, 381–382 CrossRef CAS.
- S. Furuyama, M. Sawada, K. Hachiya and T. Morimoto, Bull. Chem. Soc. Jpn., 1982, 55, 3394–3397 CrossRef CAS.
- W. A. Bonner, Orig. Life Evol. Biosph., 1995, 25, 175–190 CrossRef CAS PubMed.
- R. T. Downs and R. M. Hazen, J. Mol. Catal. Chem, 2004, 216, 273–285 CrossRef CAS.
- J. W. Han and D. S. Sholl, Langmuir, 2009, 25, 10737–10745 CrossRef CAS PubMed.
- J. W. Han and D. S. Sholl, Phys. Chem. Chem. Phys., 2010, 12, 8024–8032 RSC.
- B. Kahr, B. Chittenden and A. Rohl, Chirality, 2006, 18, 127–133 CrossRef CAS PubMed.
- A. J. Price and E. R. Johnson, Phys. Chem. Chem. Phys., 2020, 22, 16571–16578 RSC.
- K. Evgenii and T. Wolfram, Orig. Life Evol. Biosph., 2000, 30, 431–434 CrossRef CAS PubMed.
- E. I. Klabunovskii, Astrobiology, 2001, 1, 127–131 CrossRef CAS PubMed.
- E. A. Kulp and J. A. Switzer, J. Am. Chem. Soc., 2007, 129, 15120–15121 CrossRef CAS PubMed.
- W. Jiang, D. Athanasiadou, S. Zhang, R. Demichelis, K. B. Koziara, P. Raiteri, V. Nelea, W. Mi, J.-A. Ma, J. D. Gale and M. D. McKee, Nat. Commun., 2019, 10, 2318 CrossRef PubMed.
- R. M. Hazen, T. R. Filley and G. A. Goodfriend, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5487–5490 CrossRef CAS PubMed.
- A. Asthagiri and R. M. Hazen, Mol. Simul, 2007, 33, 343–351 CrossRef CAS.
- C. A. Orme, A. Noy, A. Wierzbicki, M. T. McBride, M. Grantham, H. H. Teng, P. M. Dove and J. J. DeYoreo, Nature, 2001, 411, 775–779 CrossRef CAS PubMed.
- M. Maruyama, K. Tsukamoto, G. Sazaki, Y. Nishimura and P. G. Vekilov, Cryst. Growth Des., 2009, 9, 127–135 CrossRef CAS.
- A. M. Cody and R. D. Cody, J. Cryst. Growth, 1991, 113, 508–519 CrossRef CAS.
- E. T. Degens, J. Matheja and T. A. Jackson, Nature, 1970, 227, 492–493 CrossRef CAS PubMed.
- T. A. Jackson, Experientia, 1971, 27, 242–243 CrossRef CAS PubMed.
- J. J. Flores and W. A. Bonner, J. Mol. Evol., 1974, 3, 49–56 CrossRef CAS PubMed.
- W. A. Bonner and J. Flores, Biosystems, 1973, 5, 103–113 CrossRef CAS.
- J. J. McCullough and R. M. Lemmon, J. Mol. Evol., 1974, 3, 57–61 CrossRef CAS PubMed.
- S. C. Bondy and M. E. Harrington, Science, 1979, 203, 1243–1244 CrossRef CAS PubMed.
- J. B. Youatt and R. D. Brown, Science, 1981, 212, 1145–1146 CrossRef CAS PubMed.
- E. Friebele, A. Shimoyama, P. E. Hare and C. Ponnamperuma, Orig. Life, 1981, 11, 173–184 CrossRef CAS PubMed.
- H. Hashizume, B. K. G. Theng and A. Yamagishi, Clay Miner., 2002, 37, 551–557 CrossRef CAS.
- T. Ikeda, H. Amoh and T. Yasunaga, J. Am. Chem. Soc., 1984, 106, 5772–5775 CrossRef CAS.
- B. Siffert and A. Naidja, Clay Miner., 1992, 27, 109–118 CrossRef CAS.
- D. G. Fraser, D. Fitz, T. Jakschitz and B. M. Rode, Phys. Chem. Chem. Phys., 2010, 13, 831–838 RSC.
- D. G. Fraser, H. C. Greenwell, N. T. Skipper, M. V. Smalley, M. A. Wilkinson, B. Demé and R. K. Heenan, Phys. Chem. Chem. Phys., 2010, 13, 825–830 RSC.
- S. I. Goldberg, Orig. Life Evol. Biosph., 2008, 38, 149–153 CrossRef CAS PubMed.
- G. Otis, M. Nassir, M. Zutta, A. Saady, S. Ruthstein and Y. Mastai, Angew. Chem., Int. Ed., 2020, 59, 20924–20929 CrossRef CAS PubMed.
- S. E. Wolf, N. Loges, B. Mathiasch, M. Panthöfer, I. Mey, A. Janshoff and W. Tremel, Angew. Chem., Int. Ed., 2007, 46, 5618–5623 CrossRef CAS PubMed.
- M. Lahav and L. Leiserowitz, Angew. Chem., Int. Ed., 2008, 47, 3680–3682 CrossRef CAS PubMed.
- W. Jiang, H. Pan, Z. Zhang, S. R. Qiu, J. D. Kim, X. Xu and R. Tang, J. Am. Chem. Soc., 2017, 139, 8562–8569 CrossRef CAS PubMed.
- O. Arteaga, A. Canillas, J. Crusats, Z. El-Hachemi, G. E. Jellison, J. Llorca and J. M. Ribó, Orig. Life Evol. Biosph., 2010, 40, 27–40 CrossRef CAS PubMed.
- S. Pizzarello, M. Zolensky and K. A. Turk, Geochim. Cosmochim. Acta, 2003, 67, 1589–1595 CrossRef CAS.
- M. Frenkel and L. Heller-Kallai, Chem. Geol., 1977, 19, 161–166 CrossRef CAS.
- Y. Keheyan and C. Montesano, J. Anal. Appl. Pyrolysis, 2010, 89, 286–293 CrossRef CAS.
- J. P. Ferris, A. R. Hill, R. Liu and L. E. Orgel, Nature, 1996, 381, 59–61 CrossRef CAS PubMed.
- I. Weissbuch, L. Addadi, Z. Berkovitch-Yellin, E. Gati, M. Lahav and L. Leiserowitz, Nature, 1984, 310, 161–164 CrossRef CAS.
- I. Weissbuch, L. Addadi, L. Leiserowitz and M. Lahav, J. Am. Chem. Soc., 1988, 110, 561–567 CrossRef CAS.
- E. M. Landau, S. G. Wolf, M. Levanon, L. Leiserowitz, M. Lahav and J. Sagiv, J. Am. Chem. Soc., 1989, 111, 1436–1445 CrossRef CAS.
- T. Kawasaki, Y. Hakoda, H. Mineki, K. Suzuki and K. Soai, J. Am. Chem. Soc., 2010, 132, 2874–2875 CrossRef CAS PubMed.
- H. Mineki, Y. Kaimori, T. Kawasaki, A. Matsumoto and K. Soai, Tetrahedron Asymmetry, 2013, 24, 1365–1367 CrossRef CAS.
- T. Kawasaki, S. Kamimura, A. Amihara, K. Suzuki and K. Soai, Angew. Chem., Int. Ed., 2011, 50, 6796–6798 CrossRef CAS PubMed.
- S. Miyagawa, K. Yoshimura, Y. Yamazaki, N. Takamatsu, T. Kuraishi, S. Aiba, Y. Tokunaga and T. Kawasaki, Angew. Chem., Int. Ed., 2017, 56, 1055–1058 CrossRef CAS PubMed.
- M. Forster and R. Raval, Chem Commun, 2016, 52, 14075–14084 RSC.
- C. Chen, S. Yang, G. Su, Q. Ji, M. Fuentes-Cabrera, S. Li and W. Liu, J. Phys. Chem. C, 2020, 124, 742–748 CrossRef CAS.
- A. J. Gellman, Y. Huang, X. Feng, V. V. Pushkarev, B. Holsclaw and B. S. Mhatre, J. Am. Chem. Soc., 2013, 135, 19208–19214 CrossRef CAS PubMed.
- Y. Yun and A. J. Gellman, Nat. Chem, 2015, 7, 520–525 CrossRef CAS PubMed.
- A. J. Gellman and K.-H. Ernst, Catal. Lett., 2018, 148, 1610–1621 CrossRef CAS.
- J. M. Ribó and D. Hochberg, Symmetry, 2019, 11, 814 CrossRef.
- D. G. Blackmond, Chem. Rev., 2020, 120, 4831–4847 CrossRef CAS PubMed.
- F. C. Frank, Biochim. Biophys. Acta, 1953, 11, 459–463 CrossRef CAS.
- D. K. Kondepudi and G. W. Nelson, Phys. Rev. Lett., 1983, 50, 1023–1026 CrossRef CAS.
- L. L. Morozov, V. V. Kuz Min and V. I. Goldanskii, Orig. Life, 1983, 13, 119–138 CrossRef CAS.
- V. Avetisov and V. Goldanskii, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11435–11442 CrossRef CAS PubMed.
- D. K. Kondepudi and K. Asakura, Acc. Chem. Res., 2001, 34, 946–954 CrossRef CAS PubMed.
- J. M. Ribó and D. Hochberg, Phys. Chem. Chem. Phys., 2020, 22, 14013–14025 RSC.
- S. Bartlett and M. L. Wong, Life, 2020, 10, 42 CrossRef PubMed.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767–768 CrossRef CAS.
- T. Gehring, M. Busch, M. Schlageter and D. Weingand, Chirality, 2010, 22, E173–E182 CrossRef CAS PubMed.
- K. Soai, T. Kawasaki and A. Matsumoto, Symmetry, 2019, 11, 694 CrossRef CAS.
- T. Buhse, Tetrahedron Asymmetry, 2003, 14, 1055–1061 CrossRef CAS.
- J. R. Islas, D. Lavabre, J.-M. Grevy, R. H. Lamoneda, H. R. Cabrera, J.-C. Micheau and T. Buhse, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13743–13748 CrossRef CAS PubMed.
- O. Trapp, S. Lamour, F. Maier, A. F. Siegle, K. Zawatzky and B. F. Straub, Chem. – Eur. J, 2020, 26, 15871–15880 CrossRef CAS PubMed.
- I. D. Gridnev, J. M. Serafimov and J. M. Brown, Angew. Chem., Int. Ed., 2004, 43, 4884–4887 CrossRef CAS PubMed.
- I. D. Gridnev and A. Kh. Vorobiev, ACS Catal., 2012, 2, 2137–2149 CrossRef CAS.
- A. Matsumoto, T. Abe, A. Hara, T. Tobita, T. Sasagawa, T. Kawasaki and K. Soai, Angew. Chem., Int. Ed., 2015, 54, 15218–15221 CrossRef CAS PubMed.
- I. D. Gridnev and A. Kh. Vorobiev, Bull. Chem. Soc. Jpn., 2015, 88, 333–340 CrossRef.
- A. Matsumoto, S. Fujiwara, T. Abe, A. Hara, T. Tobita, T. Sasagawa, T. Kawasaki and K. Soai, Bull. Chem. Soc. Jpn., 2016, 89, 1170–1177 CrossRef CAS.
- M. E. Noble-Terán, J.-M. Cruz, J.-C. Micheau and T. Buhse, ChemCatChem, 2018, 10, 642–648 CrossRef.
- S. V. Athavale, A. Simon, K. N. Houk and S. E. Denmark, Nat. Chem, 2020, 12, 412–423 CrossRef CAS PubMed.
- A. Matsumoto, A. Tanaka, Y. Kaimori, N. Hara, Y. Mikata and K. Soai, Chem. Commun., 2021, 57, 11209–11212 RSC.
- Y. Geiger, Chem. Soc. Rev., 2022, 51, 1206–1211 RSC.
- K. Soai, I. Sato, T. Shibata, S. Komiya, M. Hayashi, Y. Matsueda, H. Imamura, T. Hayase, H. Morioka, H. Tabira, J. Yamamoto and Y. Kowata, Tetrahedron Asymmetry, 2003, 14, 185–188 CrossRef CAS.
- D. A. Singleton and L. K. Vo, Org. Lett., 2003, 5, 4337–4339 CrossRef CAS PubMed.
- I. D. Gridnev, J. M. Serafimov, H. Quiney and J. M. Brown, Org. Biomol. Chem., 2003, 1, 3811–3819 RSC.
- T. Kawasaki, K. Suzuki, M. Shimizu, K. Ishikawa and K. Soai, Chirality, 2006, 18, 479–482 CrossRef CAS PubMed.
- B. Barabas, L. Caglioti, C. Zucchi, M. Maioli, E. Gál, K. Micskei and G. Pályi, J. Phys. Chem. B, 2007, 111, 11506–11510 CrossRef CAS PubMed.
- B. Barabás, C. Zucchi, M. Maioli, K. Micskei and G. Pályi, J. Mol. Model., 2015, 21, 33 CrossRef PubMed.
- Y. Kaimori, Y. Hiyoshi, T. Kawasaki, A. Matsumoto and K. Soai, Chem. Commun., 2019, 55, 5223–5226 RSC.
- A biased distribution of the product enantiomers has been observed for Soai (ref. 332) and Soai related (ref. 333) reactions as a probable consequence of the presence of cryptochiral species: D. A. Singleton and L. K. Vo, J. Am. Chem. Soc., 2002, 124, 10010–10011 CrossRef CAS PubMed.
- G. Rotunno, D. Petersen and M. Amedjkouh, ChemSystChem, 2020, 2, e1900060 CAS.
- M. Mauksch, S. B. Tsogoeva, I. M. Martynova and S. Wei, Angew. Chem., Int. Ed., 2007, 46, 393–396 CrossRef CAS PubMed.
- M. Amedjkouh and M. Brandberg, Chem. Commun., 2008, 3043 RSC.
- M. Mauksch, S. B. Tsogoeva, S. Wei and I. M. Martynova, Chirality, 2007, 19, 816–825 CrossRef CAS PubMed.
- M. P. Romero-Fernández, R. Babiano and P. Cintas, Chirality, 2018, 30, 445–456 CrossRef PubMed.
- S. B. Tsogoeva, S. Wei, M. Freund and M. Mauksch, Angew. Chem., Int. Ed., 2009, 48, 590–594 CrossRef CAS PubMed.
- S. B. Tsogoeva, Chem. Commun., 2010, 46, 7662–7669 RSC.
- X. Wang, Y. Zhang, H. Tan, Y. Wang, P. Han and D. Z. Wang, J. Org. Chem., 2010, 75, 2403–2406 CrossRef CAS PubMed.
- M. Mauksch, S. Wei, M. Freund, A. Zamfir and S. B. Tsogoeva, Orig. Life Evol. Biosph., 2009, 40, 79–91 CrossRef PubMed.
- G. Valero, J. M. Ribó and A. Moyano, Chem. – Eur. J, 2014, 20, 17395–17408 CrossRef CAS PubMed.
- R. Plasson, H. Bersini and A. Commeyras, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16733–16738 CrossRef CAS PubMed.
- Y. Saito and H. Hyuga, J. Phys. Soc. Jpn., 2004, 73, 33–35 CrossRef CAS.
- F. Jafarpour, T. Biancalani and N. Goldenfeld, Phys. Rev. Lett., 2015, 115, 158101 CrossRef PubMed.
- K. Iwamoto, Phys. Chem. Chem. Phys., 2002, 4, 3975–3979 RSC.
- J. M. Ribó, J. Crusats, Z. El-Hachemi, A. Moyano, C. Blanco and D. Hochberg, Astrobiology, 2013, 13, 132–142 CrossRef PubMed.
- C. Blanco, J. M. Ribó, J. Crusats, Z. El-Hachemi, A. Moyano and D. Hochberg, Phys. Chem. Chem. Phys., 2013, 15, 1546–1556 RSC.
- C. Blanco, J. Crusats, Z. El-Hachemi, A. Moyano, D. Hochberg and J. M. Ribó, ChemPhysChem, 2013, 14, 2432–2440 CrossRef CAS PubMed.
- J. M. Ribó, J. Crusats, Z. El-Hachemi, A. Moyano and D. Hochberg, Chem. Sci, 2017, 8, 763–769 RSC.
- M. Eigen and P. Schuster, The Hypercycle: A Principle of Natural Self-Organization, Springer-Verlag, Berlin Heidelberg, 1979 Search PubMed.
- F. Ricci, F. H. Stillinger and P. G. Debenedetti, J. Phys. Chem. B, 2013, 117, 602–614 CrossRef CAS PubMed.
- Y. Sang and M. Liu, Symmetry, 2019, 11, 950–969 CrossRef CAS.
- A. Arlegui, B. Soler, A. Galindo, O. Arteaga, A. Canillas, J. M. Ribó, Z. El-Hachemi, J. Crusats and A. Moyano, Chem. Commun., 2019, 55, 12219–12222 RSC.
- E. Havinga, Biochim. Biophys. Acta, 1954, 13, 171–174 CrossRef CAS.
- A. C. D. Newman and H. M. Powell, J. Chem. Soc. Resumed, 1952, 3747–3751 RSC.
- R. E. Pincock and K. R. Wilson, J. Am. Chem. Soc., 1971, 93, 1291–1292 CrossRef CAS.
- R. E. Pincock, R. R. Perkins, A. S. Ma and K. R. Wilson, Science, 1971, 174, 1018–1020 CrossRef CAS PubMed.
- W. H. Zachariasen, Z. Krist. – Cryst. Mater., 1929, 71, 517–529 CrossRef CAS.
- G. N. Ramachandran and K. S. Chandrasekaran, Acta Crystallogr., 1957, 10, 671–675 CrossRef CAS.
- S. C. Abrahams and J. L. Bernstein, Acta Crystallogr. B, 1977, 33, 3601–3604 CrossRef.
- F. S. Kipping and W. J. Pope, J. Chem. Soc., Trans., 1898, 73, 606–617 RSC.
- F. S. Kipping and W. J. Pope, Nature, 1898, 59, 53 CrossRef.
- J.-M. Cruz, K. Hernández-Lechuga, I. Domínguez-Valle, A. Fuentes-Beltrán, J. U. Sánchez-Morales, J. L. Ocampo-Espindola, C. Polanco, J.-C. Micheau and T. Buhse, Chirality, 2020, 32, 120–134 CrossRef CAS PubMed.
- D. K. Kondepudi, R. J. Kaufman and N. Singh, Science, 1990, 250, 975–976 CrossRef CAS PubMed.
- J. M. McBride and R. L. Carter, Angew. Chem., Int. Ed. Engl., 1991, 30, 293–295 CrossRef.
- D. K. Kondepudi, K. L. Bullock, J. A. Digits, J. K. Hall and J. M. Miller, J. Am. Chem. Soc., 1993, 115, 10211–10216 CrossRef CAS.
- B. Martin, A. Tharrington and X. Wu, Phys. Rev. Lett., 1996, 77, 2826–2829 CrossRef CAS PubMed.
- Z. El-Hachemi, J. Crusats, J. M. Ribó and S. Veintemillas-Verdaguer, Cryst. Growth Des., 2009, 9, 4802–4806 CrossRef CAS.
- D. J. Durand, D. K. Kondepudi, P. F. Moreira Jr. and F. H. Quina, Chirality, 2002, 14, 284–287 CrossRef CAS PubMed.
- D. K. Kondepudi, J. Laudadio and K. Asakura, J. Am. Chem. Soc., 1999, 121, 1448–1451 CrossRef CAS.
- W. L. Noorduin, T. Izumi, A. Millemaggi, M. Leeman, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg, B. Kaptein, E. Vlieg and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 1158–1159 CrossRef CAS PubMed.
- C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef PubMed.
- C. Viedma, Cryst. Growth Des., 2007, 7, 553–556 CrossRef CAS.
- C. Xiouras, J. Van Aeken, J. Panis, J. H. Ter Horst, T. Van Gerven and G. D. Stefanidis, Cryst. Growth Des., 2015, 15, 5476–5484 CrossRef CAS.
- J. Ahn, D. H. Kim, G. Coquerel and W.-S. Kim, Cryst. Growth Des., 2018, 18, 297–306 CrossRef CAS.
- C. Viedma and P. Cintas, Chem. Commun., 2011, 47, 12786–12788 RSC.
- W. L. Noorduin, W. J. P. van Enckevort, H. Meekes, B. Kaptein, R. M. Kellogg, J. C. Tully, J. M. McBride and E. Vlieg, Angew. Chem., Int. Ed., 2010, 49, 8435–8438 CrossRef CAS PubMed.
- R. R. E. Steendam, T. J. B. van Benthem, E. M. E. Huijs, H. Meekes, W. J. P. van Enckevort, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Cryst. Growth Des., 2015, 15, 3917–3921 CrossRef CAS.
- C. Blanco, J. Crusats, Z. El-Hachemi, A. Moyano, S. Veintemillas-Verdaguer, D. Hochberg and J. M. Ribó, ChemPhysChem, 2013, 14, 3982–3993 CrossRef CAS PubMed.
- C. Blanco, J. M. Ribó and D. Hochberg, Phys. Rev. E, 2015, 91, 022801 CrossRef PubMed.
- G. An, P. Yan, J. Sun, Y. Li, X. Yao and G. Li, CrystEngComm, 2015, 17, 4421–4433 RSC.
- R. R. E. Steendam, J. M. M. Verkade, T. J. B. van Benthem, H. Meekes, W. J. P. van Enckevort, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Nat. Commun., 2014, 5, 5543 CrossRef CAS PubMed.
- A. H. Engwerda, H. Meekes, B. Kaptein, F. Rutjes and E. Vlieg, Chem. Commun., 2016, 52, 12048–12051 RSC.
- C. Viedma, C. Lennox, L. A. Cuccia, P. Cintas and J. E. Ortiz, Chem. Commun., 2020, 56, 4547–4550 RSC.
- C. Viedma, J. E. Ortiz, T. de Torres, T. Izumi and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 15274–15275 CrossRef CAS PubMed.
- L. Spix, H. Meekes, R. H. Blaauw, W. J. P. van Enckevort and E. Vlieg, Cryst. Growth Des., 2012, 12, 5796–5799 CrossRef.
- F. Cameli, C. Xiouras and G. D. Stefanidis, CrystEngComm, 2018, 20, 2897–2901 RSC.
- K. Ishikawa, M. Tanaka, T. Suzuki, A. Sekine, T. Kawasaki, K. Soai, M. Shiro, M. Lahav and T. Asahi, Chem. Commun., 2012, 48, 6031–6033 RSC.
- A. V. Tarasevych, A. E. Sorochinsky, V. P. Kukhar, L. Toupet, J. Crassous and J.-C. Guillemin, CrystEngComm, 2015, 17, 1513–1517 RSC.
- L. Spix, A. Alfring, H. Meekes, W. J. P. van Enckevort and E. Vlieg, Cryst. Growth Des., 2014, 14, 1744–1748 CrossRef CAS.
- L. Spix, A. H. J. Engwerda, H. Meekes, W. J. P. van Enckevort and E. Vlieg, Cryst. Growth Des., 2016, 16, 4752–4758 CrossRef CAS.
- B. Kaptein, W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2008, 47, 7226–7229 CrossRef CAS PubMed.
- T. Kawasaki, N. Takamatsu, S. Aiba and Y. Tokunaga, Chem. Commun., 2015, 51, 14377–14380 RSC.
- S. Aiba, N. Takamatsu, T. Sasai, Y. Tokunaga and T. Kawasaki, Chem. Commun., 2016, 52, 10834–10837 RSC.
- S. Miyagawa, S. Aiba, H. Kawamoto, Y. Tokunaga and T. Kawasaki, Org. Biomol. Chem., 2019, 17, 1238–1244 RSC.
- I. Baglai, M. Leeman, K. Wurst, B. Kaptein, R. M. Kellogg and W. L. Noorduin, Chem. Commun., 2018, 54, 10832–10834 RSC.
- N. Uemura, K. Sano, A. Matsumoto, Y. Yoshida, T. Mino and M. Sakamoto, Chem. – Asian J., 2019, 14, 4150–4153 CrossRef CAS PubMed.
- N. Uemura, M. Hosaka, A. Washio, Y. Yoshida, T. Mino and M. Sakamoto, Cryst. Growth Des., 2020, 20, 4898–4903 CrossRef CAS.
- D. K. Kondepudi and G. W. Nelson, Phys. Lett. A, 1984, 106, 203–206 CrossRef.
- D. K. Kondepudi and G. W. Nelson, Phys. Stat. Mech. Appl., 1984, 125, 465–496 CrossRef.
- D. K. Kondepudi and G. W. Nelson, Nature, 1985, 314, 438–441 CrossRef CAS.
- N. A. Hawbaker and D. G. Blackmond, Nat. Chem., 2019, 11, 957–962 CrossRef CAS PubMed.
- J. I. Murray, J. N. Sanders, P. F. Richardson, K. N. Houk and D. G. Blackmond, J. Am. Chem. Soc., 2020, 142, 3873–3879 CrossRef CAS PubMed.
- S. Mahurin, M. McGinnis, J. S. Bogard, L. D. Hulett, R. M. Pagni and R. N. Compton, Chirality, 2001, 13, 636–640 CrossRef CAS PubMed.
- K. Soai, S. Osanai, K. Kadowaki, S. Yonekubo, T. Shibata and I. Sato, J. Am. Chem. Soc., 1999, 121, 11235–11236 CrossRef CAS.
- A. Matsumoto, H. Ozaki, S. Tsuchiya, T. Asahi, M. Lahav, T. Kawasaki and K. Soai, Org. Biomol. Chem., 2019, 17, 4200–4203 RSC.
- D. J. Carter, A. L. Rohl, A. Shtukenberg, S. Bian, C. Hu, L. Baylon, B. Kahr, H. Mineki, K. Abe, T. Kawasaki and K. Soai, Cryst. Growth Des., 2012, 12, 2138–2145 CrossRef CAS.
- H. Shindo, Y. Shirota, K. Niki, T. Kawasaki, K. Suzuki, Y. Araki, A. Matsumoto and K. Soai, Angew. Chem., Int. Ed., 2013, 52, 9135–9138 CrossRef CAS PubMed.
- T. Kawasaki, Y. Kaimori, S. Shimada, N. Hara, S. Sato, K. Suzuki, T. Asahi, A. Matsumoto and K. Soai, Chem. Commun., 2021, 57, 5999–6002 RSC.
- A. Matsumoto, Y. Kaimori, M. Uchida, H. Omori, T. Kawasaki and K. Soai, Angew. Chem., Int. Ed., 2017, 56, 545–548 CrossRef CAS PubMed.
- L. Addadi, Z. Berkovitch-Yellin, N. Domb, E. Gati, M. Lahav and L. Leiserowitz, Nature, 1982, 296, 21–26 CrossRef CAS.
- L. Addadi, S. Weinstein, E. Gati, I. Weissbuch and M. Lahav, J. Am. Chem. Soc., 1982, 104, 4610–4617 CrossRef CAS.
- I. Baglai, M. Leeman, K. Wurst, R. M. Kellogg and W. L. Noorduin, Angew. Chem., Int. Ed., 2020, 59, 20885–20889 CrossRef CAS PubMed.
- J. Royes, V. Polo, S. Uriel, L. Oriol, M. Piñol and R. M. Tejedor, Phys. Chem. Chem. Phys., 2017, 19, 13622–13628 RSC.
- E. E. Greciano, R. Rodríguez, K. Maeda and L. Sánchez, Chem. Commun., 2020, 56, 2244–2247 RSC.
- I. Sato, R. Sugie, Y. Matsueda, Y. Furumura and K. Soai, Angew. Chem., Int. Ed., 2004, 43, 4490–4492 CrossRef CAS PubMed.
- T. Kawasaki, M. Sato, S. Ishiguro, T. Saito, Y. Morishita, I. Sato, H. Nishino, Y. Inoue and K. Soai, J. Am. Chem. Soc., 2005, 127, 3274–3275 CrossRef CAS PubMed.
- W. L. Noorduin, A. A. C. Bode, M. van der Meijden, H. Meekes, A. F. van Etteger, W. J. P. van Enckevort, P. C. M. Christianen, B. Kaptein, R. M. Kellogg, T. Rasing and E. Vlieg, Nat. Chem, 2009, 1, 729 CrossRef CAS PubMed.
- W. H. Mills, J. Soc. Chem. Ind., 1932, 51, 750–759 CrossRef CAS.
- M. Bolli, R. Micura and A. Eschenmoser, Chem. Biol., 1997, 4, 309–320 CrossRef CAS PubMed.
- A. Brewer and A. P. Davis, Nat. Chem, 2014, 6, 569–574 CrossRef CAS PubMed.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- F. H. C. Crick and L. E. Orgel, Icarus, 1973, 19, 341–346 CrossRef.
- A. J. MacDermott and G. E. Tranter, Croat. Chem. Acta, 1989, 62, 165–187 CAS.
- A. Julg, THEOCHEM, 1989, 184, 131–142 CrossRef.
- W. Martin, J. Baross, D. Kelley and M. J. Russell, Nat. Rev. Microbiol., 2008, 6, 805–814 CrossRef CAS PubMed.
- W. Wang, arXiv:2001.03532.
- V. I. Goldanskii and V. V. Kuz’min, AIP Conf. Proc., 1988, 180, 163–228 CrossRef CAS.
- W. A. Bonner and E. Rubenstein, Biosystems, 1987, 20, 99–111 CrossRef CAS PubMed.
- A. Jorissen and C. Cerf, Orig. Life Evol. Biosph., 2002, 32, 129–142 CrossRef CAS PubMed.
- K. Mislow, Collect. Czechoslov. Chem. Commun., 2003, 68, 849–864 CrossRef CAS.
- A. Burton and E. Berger, Life, 2018, 8, 14 CrossRef PubMed.
- A. Garcia, C. Meinert, H. Sugahara, N. Jones, S. Hoffmann and U. Meierhenrich, Life, 2019, 9, 29 CrossRef CAS PubMed.
- G. Cooper, N. Kimmich, W. Belisle, J. Sarinana, K. Brabham and L. Garrel, Nature, 2001, 414, 879–883 CrossRef CAS PubMed.
- Y. Furukawa, Y. Chikaraishi, N. Ohkouchi, N. O. Ogawa, D. P. Glavin, J. P. Dworkin, C. Abe and T. Nakamura, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24440–24445 CrossRef CAS PubMed.
- J. Bocková, N. C. Jones, U. J. Meierhenrich, S. V. Hoffmann and C. Meinert, Commun. Chem, 2021, 4, 86 CrossRef.
- J. R. Cronin and S. Pizzarello, Adv. Space Res., 1999, 23, 293–299 CrossRef CAS.
- S. Pizzarello and J. R. Cronin, Geochim. Cosmochim. Acta, 2000, 64, 329–338 CrossRef CAS PubMed.
- D. P. Glavin and J. P. Dworkin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5487–5492 CrossRef CAS PubMed.
- I. Myrgorodska, C. Meinert, Z. Martins, L. Le Sergeant d’Hendecourt and U. J. Meierhenrich, J. Chromatogr. A, 2016, 1433, 131–136 CrossRef CAS PubMed.
- G. Cooper and A. C. Rios, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3322–E3331 CrossRef CAS PubMed.
- A. Furusho, T. Akita, M. Mita, H. Naraoka and K. Hamase, J. Chromatogr. A, 2020, 1625, 461255 CrossRef CAS PubMed.
- M. P. Bernstein, J. P. Dworkin, S. A. Sandford, G. W. Cooper and L. J. Allamandola, Nature, 2002, 416, 401–403 CrossRef CAS PubMed.
- K. M. Ferrière, Rev. Mod. Phys., 2001, 73, 1031–1066 CrossRef.
- Y. Fukui and A. Kawamura, Annu. Rev. Astron. Astrophys., 2010, 48, 547–580 CrossRef CAS.
- E. L. Gibb, D. C. B. Whittet, A. C. A. Boogert and A. G. G. M. Tielens, Astrophys. J. Suppl. Ser., 2004, 151, 35–73 CrossRef CAS.
- J. Mayo Greenberg, Surf. Sci., 2002, 500, 793–822 CrossRef CAS.
- S. A. Sandford, M. Nuevo, P. P. Bera and T. J. Lee, Chem. Rev., 2020, 120, 4616–4659 CrossRef CAS PubMed.
- J. J. Hester, S. J. Desch, K. R. Healy and L. A. Leshin, Science, 2004, 304, 1116–1117 CrossRef PubMed.
- G. M. Muñoz Caro, U. J. Meierhenrich, W. A. Schutte, B. Barbier, A. Arcones Segovia, H. Rosenbauer, W. H.-P. Thiemann, A. Brack and J. M. Greenberg, Nature, 2002, 416, 403–406 CrossRef PubMed.
- M. Nuevo, G. Auger, D. Blanot and L. d’Hendecourt, Orig. Life Evol. Biosph., 2008, 38, 37–56 CrossRef CAS PubMed.
- C. Zhu, A. M. Turner, C. Meinert and R. I. Kaiser, Astrophys. J., 2020, 889, 134 CrossRef CAS.
- C. Meinert, I. Myrgorodska, P. de Marcellus, T. Buhse, L. Nahon, S. V. Hoffmann, L. L. S. d’Hendecourt and U. J. Meierhenrich, Science, 2016, 352, 208–212 CrossRef CAS PubMed.
- M. Nuevo, G. Cooper and S. A. Sandford, Nat. Commun., 2018, 9, 5276 CrossRef CAS PubMed.
- Y. Oba, Y. Takano, H. Naraoka, N. Watanabe and A. Kouchi, Nat. Commun., 2019, 10, 4413 CrossRef PubMed.
- J. Kwon, M. Tamura, P. W. Lucas, J. Hashimoto, N. Kusakabe, R. Kandori, Y. Nakajima, T. Nagayama, T. Nagata and J. H. Hough, Astrophys. J., 2013, 765, L6 CrossRef.
- J. Bailey, Orig. Life Evol. Biosph., 2001, 31, 167–183 CrossRef CAS PubMed.
- J. Bailey, A. Chrysostomou, J. H. Hough, T. M. Gledhill, A. McCall, S. Clark, F. Ménard and M. Tamura, Science, 1998, 281, 672–674 CrossRef CAS PubMed.
- T. Fukue, M. Tamura, R. Kandori, N. Kusakabe, J. H. Hough, J. Bailey, D. C. B. Whittet, P. W. Lucas, Y. Nakajima and J. Hashimoto, Orig. Life Evol. Biosph., 2010, 40, 335–346 CrossRef CAS PubMed.
- J. Kwon, M. Tamura, J. H. Hough, N. Kusakabe, T. Nagata, Y. Nakajima, P. W. Lucas, T. Nagayama and R. Kandori, Astrophys. J., 2014, 795, L16 CrossRef.
- J. Kwon, M. Tamura, J. H. Hough, T. Nagata, N. Kusakabe and H. Saito, Astrophys. J., 2016, 824, 95 CrossRef.
- J. Kwon, M. Tamura, J. H. Hough, T. Nagata and N. Kusakabe, Astron. J., 2016, 152, 67 CrossRef.
- J. Kwon, T. Nakagawa, M. Tamura, J. H. Hough, R. Kandori, M. Choi, M. Kang, J. Cho, Y. Nakajima and T. Nagata, Astron. J., 2018, 156, 1 CrossRef.
- K. Wood, Astrophys. J., 1997, 477, L25–L28 CrossRef.
- S. F. Mason, Nature, 1997, 389, 804 CrossRef CAS PubMed.
- W. A. Bonner, E. Rubenstein and G. S. Brown, Orig. Life Evol. Biosph., 1999, 29, 329–332 CrossRef CAS PubMed.
- J. H. Bredehöft, N. C. Jones, C. Meinert, A. C. Evans, S. V. Hoffmann and U. J. Meierhenrich, Chirality, 2014, 26, 373–378 CrossRef PubMed.
- E. Rubenstein, W. A. Bonner, H. P. Noyes and G. S. Brown, Nature, 1983, 306, 118 CrossRef.
- M. Buschermohle, D. C. B. Whittet, A. Chrysostomou, J. H. Hough, P. W. Lucas, A. J. Adamson, B. A. Whitney and M. J. Wolff, Astrophys. J., 2005, 624, 821–826 CrossRef.
- J. Oró, T. Mills and A. Lazcano, Orig. Life Evol. Biosph., 1991, 21, 267–277 CrossRef PubMed.
- A. G. Griesbeck and U. J. Meierhenrich, Angew. Chem., Int. Ed., 2002, 41, 3147–3154 CrossRef PubMed.
- C. Meinert, J.-J. Filippi, L. Nahon, S. V. Hoffmann, L. D’Hendecourt, P. De Marcellus, J. H. Bredehöft, W. H.-P. Thiemann and U. J. Meierhenrich, Symmetry, 2010, 2, 1055–1080 CrossRef CAS.
- I. Myrgorodska, C. Meinert, Z. Martins, L. L. S. d’Hendecourt and U. J. Meierhenrich, Angew. Chem., Int. Ed., 2015, 54, 1402–1412 CrossRef CAS PubMed.
- I. Baglai, M. Leeman, B. Kaptein, R. M. Kellogg and W. L. Noorduin, Chem. Commun., 2019, 55, 6910–6913 RSC.
- A. G. Lyne, Nature, 1984, 308, 605–606 CrossRef.
- W. A. Bonner and R. M. Lemmon, J. Mol. Evol., 1978, 11, 95–99 CrossRef CAS PubMed.
- W. A. Bonner and R. M. Lemmon, Bioorganic Chem, 1978, 7, 175–187 CrossRef CAS.
- M. Preiner, S. Asche, S. Becker, H. C. Betts, A. Boniface, E. Camprubi, K. Chandru, V. Erastova, S. G. Garg, N. Khawaja, G. Kostyrka, R. Machné, G. Moggioli, K. B. Muchowska, S. Neukirchen, B. Peter, E. Pichlhöfer, Á. Radványi, D. Rossetto, A. Salditt, N. M. Schmelling, F. L. Sousa, F. D. K. Tria, D. Vörös and J. C. Xavier, Life, 2020, 10, 20 CrossRef PubMed.
- K. Michaelian, Life, 2018, 8, 21 CrossRef PubMed.
- NASA Astrobiology, https://astrobiology.nasa.gov/research/life-detection/about/ (accessed Decembre 22th, 2021).
- S. A. Benner, E. A. Bell, E. Biondi, R. Brasser, T. Carell, H.-J. Kim, S. J. Mojzsis, A. Omran, M. A. Pasek and D. Trail, ChemSystChem, 2020, 2, e1900035 CAS.
- M. Idelson and E. R. Blout, J. Am. Chem. Soc., 1958, 80, 2387–2393 CrossRef CAS.
- G. F. Joyce, G. M. Visser, C. A. A. van Boeckel, J. H. van Boom, L. E. Orgel and J. van Westrenen, Nature, 1984, 310, 602–604 CrossRef CAS PubMed.
- R. D. Lundberg and P. Doty, J. Am. Chem. Soc., 1957, 79, 3961–3972 CrossRef CAS.
- E. R. Blout, P. Doty and J. T. Yang, J. Am. Chem. Soc., 1957, 79, 749–750 CrossRef CAS.
- J. G. Schmidt, P. E. Nielsen and L. E. Orgel, J. Am. Chem. Soc., 1997, 119, 1494–1495 CrossRef CAS PubMed.
- M. M. Waldrop, Science, 1990, 250, 1078–1080 CrossRef CAS PubMed.
- E. G. Nisbet and N. H. Sleep, Nature, 2001, 409, 1083–1091 CrossRef CAS PubMed.
- V. R. Oberbeck and G. Fogleman, Orig. Life Evol. Biosph., 1989, 19, 549–560 CrossRef CAS PubMed.
- A. Lazcano and S. L. Miller, J. Mol. Evol., 1994, 39, 546–554 CrossRef CAS PubMed.
- J. L. Bada, in Chemistry and Biochemistry of the Amino Acids, ed. G. C. Barrett, Springer, Netherlands, Dordrecht, 1985, pp. 399–414 Search PubMed.
- S. Kempe and J. Kazmierczak, Astrobiology, 2002, 2, 123–130 CrossRef CAS PubMed.
- J. L. Bada, Chem. Soc. Rev., 2013, 42, 2186–2196 RSC.
- H. J. Morowitz, J. Theor. Biol., 1969, 25, 491–494 CrossRef CAS PubMed.
- M. Klussmann, A. J. P. White, A. Armstrong and D. G. Blackmond, Angew. Chem., Int. Ed., 2006, 45, 7985–7989 CrossRef PubMed.
- M. Klussmann, H. Iwamura, S. P. Mathew, D. H. Wells, U. Pandya, A. Armstrong and D. G. Blackmond, Nature, 2006, 441, 621–623 CrossRef CAS PubMed.
- R. Breslow and M. S. Levine, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12979–12980 CrossRef CAS PubMed.
- M. Levine, C. S. Kenesky, D. Mazori and R. Breslow, Org. Lett., 2008, 10, 2433–2436 CrossRef CAS PubMed.
- M. Klussmann, T. Izumi, A. J. P. White, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2007, 129, 7657–7660 CrossRef CAS PubMed.
- R. Breslow and Z.-L. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9144–9146 CrossRef CAS PubMed.
- J. Han, A. Wzorek, M. Kwiatkowska, V. A. Soloshonok and K. D. Klika, Amino Acids, 2019, 51, 865–889 CrossRef CAS PubMed.
- R. H. Perry, C. Wu, M. Nefliu and R. Graham Cooks, Chem. Commun., 2007, 1071–1073 RSC.
- S. P. Fletcher, R. B. C. Jagt and B. L. Feringa, Chem. Commun., 2007, 2578–2580 RSC.
- A. Bellec and J.-C. Guillemin, Chem. Commun., 2010, 46, 1482–1484 RSC.
- A. V. Tarasevych, A. E. Sorochinsky, V. P. Kukhar, A. Chollet, R. Daniellou and J.-C. Guillemin, J. Org. Chem., 2013, 78, 10530–10533 CrossRef CAS PubMed.
- A. V. Tarasevych, A. E. Sorochinsky, V. P. Kukhar and J.-C. Guillemin, Orig. Life Evol. Biosph., 2013, 43, 129–135 CrossRef CAS PubMed.
- V. Dašková, J. Buter, A. K. Schoonen, M. Lutz, F. de Vries and B. L. Feringa, Angew. Chem., Int. Ed., 2021, 60, 11120–11126 CrossRef PubMed.
- A. Córdova, M. Engqvist, I. Ibrahem, J. Casas and H. Sundén, Chem. Commun., 2005, 2047–2049 RSC.
- R. Breslow and Z.-L. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5723–5725 CrossRef CAS PubMed.
- S. Pizzarello and A. L. Weber, Science, 2004, 303, 1151 CrossRef CAS PubMed.
- A. L. Weber and S. Pizzarello, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12713–12717 CrossRef CAS PubMed.
- S. Pizzarello and A. L. Weber, Orig. Life Evol. Biosph., 2009, 40, 3–10 CrossRef PubMed.
- J. E. Hein, E. Tse and D. G. Blackmond, Nat. Chem, 2011, 3, 704–706 CrossRef CAS PubMed.
- A. J. Wagner, D. Yu Zubarev, A. Aspuru-Guzik and D. G. Blackmond, ACS Cent. Sci, 2017, 3, 322–328 CrossRef CAS PubMed.
- M. P. Robertson and G. F. Joyce, Cold Spring Harb. Perspect. Biol, 2012, 4, a003608 Search PubMed.
- A. Kahana, P. Schmitt-Kopplin and D. Lancet, Astrobiology, 2019, 19, 1263–1278 CrossRef CAS PubMed.
- F. J. Dyson, J. Mol. Evol., 1982, 18, 344–350 CrossRef CAS PubMed.
- R. Root-Bernstein, BioEssays, 2007, 29, 689–698 CrossRef CAS PubMed.
- K. A. Lanier, A. S. Petrov and L. D. Williams, J. Mol. Evol., 2017, 85, 8–13 CrossRef CAS PubMed.
- H. S. Martin, K. A. Podolsky and N. K. Devaraj, ChemBioChem, 2021, 22, 3148–3157 CrossRef CAS PubMed.
- V. Sojo, BioEssays, 2019, 41, 1800251 CrossRef PubMed.
- A. Eschenmoser, Tetrahedron, 2007, 63, 12821–12844 CrossRef CAS.
- S. N. Semenov, L. J. Kraft, A. Ainla, M. Zhao, M. Baghbanzadeh, V. E. Campbell, K. Kang, J. M. Fox and G. M. Whitesides, Nature, 2016, 537, 656–660 CrossRef CAS PubMed.
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120–5156 CrossRef CAS PubMed.
- C. M. Thomas, Chem. Soc. Rev., 2009, 39, 165–173 RSC.
- A. Brack and G. Spach, Nat. Phys. Sci, 1971, 229, 124–125 CrossRef CAS.
- S. I. Goldberg, J. M. Crosby, N. D. Iusem and U. E. Younes, J. Am. Chem. Soc., 1987, 109, 823–830 CrossRef CAS.
- T. H. Hitz and P. L. Luisi, Orig. Life Evol. Biosph., 2004, 34, 93–110 CrossRef CAS PubMed.
- T. Hitz, M. Blocher, P. Walde and P. L. Luisi, Macromolecules, 2001, 34, 2443–2449 CrossRef CAS.
- T. Hitz and P. L. Luisi, Helv. Chim. Acta, 2002, 85, 3975–3983 CrossRef CAS.
- T. Hitz and P. L. Luisi, Helv. Chim. Acta, 2003, 86, 1423–1434 CrossRef CAS.
- H. Urata, C. Aono, N. Ohmoto, Y. Shimamoto, Y. Kobayashi and M. Akagi, Chem. Lett., 2001, 324–325 CrossRef CAS.
- K. Osawa, H. Urata and H. Sawai, Orig. Life Evol. Biosph., 2005, 35, 213–223 CrossRef CAS PubMed.
- P. C. Joshi, S. Pitsch and J. P. Ferris, Chem. Commun., 2000, 2497–2498 RSC.
- P. C. Joshi, S. Pitsch and J. P. Ferris, Orig. Life Evol. Biosph., 2007, 37, 3–26 CrossRef CAS PubMed.
- P. C. Joshi, M. F. Aldersley and J. P. Ferris, Orig. Life Evol. Biosph., 2011, 41, 213–236 CrossRef CAS PubMed.
- A. Saghatelian, Y. Yokobayashi, K. Soltani and M. R. Ghadiri, Nature, 2001, 409, 797–801 CrossRef CAS PubMed.
- J. E. Šponer, A. Mládek and J. Šponer, Phys. Chem. Chem. Phys., 2013, 15, 6235–6242 RSC.
- G. F. Joyce, A. W. Schwartz, S. L. Miller and L. E. Orgel, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 4398–4402 CrossRef CAS PubMed.
- J. Rivera Islas, V. Pimienta, J.-C. Micheau and T. Buhse, Biophys. Chem., 2003, 103, 201–211 CrossRef CAS.
- M. Liu, L. Zhang and T. Wang, Chem Rev, 2015, 115, 7304–7397 CrossRef CAS PubMed.
- S. C. Nanita and R. G. Cooks, Angew. Chem., Int. Ed., 2006, 45, 554–569 CrossRef CAS PubMed.
- Z. Takats, S. C. Nanita and R. G. Cooks, Angew. Chem., Int. Ed., 2003, 42, 3521–3523 CrossRef CAS PubMed.
- R. R. Julian, S. Myung and D. E. Clemmer, J. Am. Chem. Soc., 2004, 126, 4110–4111 CrossRef CAS PubMed.
- R. R. Julian, S. Myung and D. E. Clemmer, J. Phys. Chem. B, 2005, 109, 440–444 CrossRef CAS PubMed.
- I. Weissbuch, R. A. Illos, G. Bolbach and M. Lahav, Acc. Chem. Res., 2009, 42, 1128–1140 CrossRef CAS PubMed.
- J. G. Nery, R. Eliash, G. Bolbach, I. Weissbuch and M. Lahav, Chirality, 2007, 19, 612–624 CrossRef CAS PubMed.
- I. Rubinstein, R. Eliash, G. Bolbach, I. Weissbuch and M. Lahav, Angew. Chem., Int. Ed., 2007, 46, 3710–3713 CrossRef CAS PubMed.
- I. Rubinstein, G. Clodic, G. Bolbach, I. Weissbuch and M. Lahav, Chem. – Eur. J, 2008, 14, 10999–11009 CrossRef CAS PubMed.
- R. A. Illos, F. R. Bisogno, G. Clodic, G. Bolbach, I. Weissbuch and M. Lahav, J. Am. Chem. Soc., 2008, 130, 8651–8659 CrossRef CAS PubMed.
- I. Weissbuch, H. Zepik, G. Bolbach, E. Shavit, M. Tang, T. R. Jensen, K. Kjaer, L. Leiserowitz and M. Lahav, Chem. – Eur. J., 2003, 9, 1782–1794 CrossRef CAS PubMed.
- C. Blanco and D. Hochberg, Phys. Chem. Chem. Phys., 2012, 14, 2301–2311 RSC.
- J. Shen, Amino Acids, 2021, 53, 265–280 CrossRef CAS PubMed.
- A. T. Borchers, P. A. Davis and M. E. Gershwin, Exp. Biol. Med., 2004, 229, 21–32 CrossRef CAS PubMed.
- J. Skolnick, H. Zhou and M. Gao, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26571–26579 CrossRef CAS PubMed.
- C. Böhler, P. E. Nielsen and L. E. Orgel, Nature, 1995, 376, 578–581 CrossRef PubMed.
- A. L. Weber, Orig. Life Evol. Biosph., 1987, 17, 107–119 CrossRef CAS PubMed.
- J. G. Nery, G. Bolbach, I. Weissbuch and M. Lahav, Chem. – Eur. J, 2005, 11, 3039–3048 CrossRef CAS PubMed.
- C. Blanco and D. Hochberg, Chem. Commun., 2012, 48, 3659–3661 RSC.
- C. Blanco and D. Hochberg, J. Phys. Chem. B, 2012, 116, 13953–13967 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- H. Cao, X. Zhu and M. Liu, Angew. Chem., Int. Ed., 2013, 52, 4122–4126 CrossRef CAS PubMed.
- S. C. Karunakaran, B. J. Cafferty, A. Weigert-Muñoz, G. B. Schuster and N. V. Hud, Angew. Chem., Int. Ed., 2019, 58, 1453–1457 CrossRef CAS PubMed.
- Y. Li, A. Hammoud, L. Bouteiller and M. Raynal, J. Am. Chem. Soc., 2020, 142, 5676–5688 CrossRef CAS PubMed.
- W. A. Bonner, Orig. Life Evol. Biosph., 1999, 29, 615–624 CrossRef CAS PubMed.
- Y. Yamagata, H. Sakihama and K. Nakano, Orig. Life, 1980, 10, 349–355 CrossRef CAS PubMed.
- P. G. H. Sandars, Orig. Life Evol. Biosph., 2003, 33, 575–587 CrossRef CAS PubMed.
- A. Brandenburg, A. C. Andersen, S. Höfner and M. Nilsson, Orig. Life Evol. Biosph., 2005, 35, 225–241 CrossRef CAS PubMed.
- M. Gleiser, Orig. Life Evol. Biosph., 2007, 37, 235–251 CrossRef PubMed.
- M. Gleiser and J. Thorarinson, Orig. Life Evol. Biosph., 2006, 36, 501–505 CrossRef CAS PubMed.
- M. Gleiser and S. I. Walker, Orig. Life Evol. Biosph., 2008, 38, 293 CrossRef CAS PubMed.
- Y. Saito and H. Hyuga, J. Phys. Soc. Jpn., 2005, 74, 1629–1635 CrossRef CAS.
- J. A. D. Wattis and P. V. Coveney, Orig. Life Evol. Biosph., 2005, 35, 243–273 CrossRef CAS PubMed.
- Y. Chen and W. Ma, PLOS Comput. Biol., 2020, 16, e1007592 CrossRef CAS PubMed.
- M. Wu, S. I. Walker and P. G. Higgs, Astrobiology, 2012, 12, 818–829 CrossRef CAS PubMed.
- C. Blanco, M. Stich and D. Hochberg, J. Phys. Chem. B, 2017, 121, 942–955 CrossRef CAS PubMed.
- S. W. Fox, J. Chem. Educ., 1957, 34, 472 CrossRef CAS.
- J. H. Rush, The dawn of life, Hanover House, Signet Science Edition, 1st edn, 1957 Search PubMed.
- G. Wald, Ann. N. Y. Acad. Sci, 1957, 69, 352–368 CrossRef CAS PubMed.
- M. Ageno, J. Theor. Biol., 1972, 37, 187–192 CrossRef CAS PubMed.
- H. Kuhn, Curr. Opin. Colloid Interface Sci., 2008, 13, 3–11 CrossRef CAS.
- M. M. Green and V. Jain, Orig. Life Evol. Biosph., 2010, 40, 111–118 CrossRef PubMed.
- F. Cava, H. Lam, M. A. de Pedro and M. K. Waldor, Cell. Mol. Life Sci., 2011, 68, 817–831 CrossRef CAS PubMed.
- B. L. Pentelute, Z. P. Gates, J. L. Dashnau, J. M. Vanderkooi and S. B. H. Kent, J. Am. Chem. Soc., 2008, 130, 9702–9707 CrossRef CAS PubMed.
- Z. Wang, W. Xu, L. Liu and T. F. Zhu, Nat. Chem, 2016, 8, 698–704 CrossRef CAS PubMed.
- A. A. Vinogradov, E. D. Evans and B. L. Pentelute, Chem. Sci, 2015, 6, 2997–3002 RSC.
- J. T. Sczepanski and G. F. Joyce, Nature, 2014, 515, 440–442 CrossRef CAS PubMed.
- K. F. Tjhung, J. T. Sczepanski, E. R. Murtfeldt and G. F. Joyce, J. Am. Chem. Soc., 2020, 142, 15331–15339 CrossRef CAS PubMed.
- F. Jafarpour, T. Biancalani and N. Goldenfeld, Phys. Rev. E, 2017, 95, 032407 CrossRef PubMed.
- G. Laurent, D. Lacoste and P. Gaspard, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2012741118 CrossRef CAS PubMed.
- M. W. van der Meijden, M. Leeman, E. Gelens, W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, B. Kaptein, E. Vlieg and R. M. Kellogg, Org. Process Res. Dev., 2009, 13, 1195–1198 CrossRef CAS.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem, 2017, 1, 1–12 CrossRef.
- H. Kuang, C. Xu and Z. Tang, Adv. Mater., 2020, 32, 2005110 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Sandra Pizzarello (1933–2021). |
| ‡ Proteinogenic amino acids and natural sugars are usually mentioned as L-amino acids and D-sugars according to the descriptors introduced by Emil Fischer. It is worth noting that natural L-cysteine (R) uses the Cahn–Ingold–Prelog system, due to the sulfur atom in the side chain which changes the priority sequence. In the present review, (R)/(S) and D/L descriptors will be used for amino acids and sugars, respectively, as commonly employed in the literature dealing with BH. |
| This journal is © The Royal Society of Chemistry 2022 |