
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The mechanochemical synthesis of polymers
Annika
Krusenbaum
 a,
Sven
Grätz
a,
Getinet Tamiru
Tigineh
bc,
Lars
Borchardt
*a and
Jeung Gon
Kim
*c
a,
Sven
Grätz
a,
Getinet Tamiru
Tigineh
bc,
Lars
Borchardt
*a and
Jeung Gon
Kim
*c
aAnorganische Chemie I, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: lars.borchardt@rub.de
bDepartment of Chemistry, Bahir Dar University, Peda Street 07, PO Box 79, Bahir Dar, Amhara, Ethiopia
cDepartment of Chemistry and Research Institute of Physics and Chemistry, Jeonbuk National University, Jeon-Ju, Jeollabuk-do, 54896, Republic of Korea. E-mail: jeunggonkim@jbnu.ac.kr
First published on 18th March 2022
Abstract
Mechanochemistry – the utilization of mechanical forces to induce chemical reactions – is a rarely considered tool for polymer synthesis. It offers numerous advantages such as reduced solvent consumption, accessibility of novel structures, and the avoidance of problems posed by low monomer solubility and fast precipitation. Consequently, the development of new high-performance materials based on mechanochemically synthesised polymers has drawn much interest, particularly from the perspective of green chemistry. This review covers the constructive mechanochemical synthesis of polymers, starting from early examples and progressing to the current state of the art while emphasising linear and porous polymers as well as post-polymerisation modifications.
 Annika Krusenbaum | Annika Krusenbaum studied chemistry at Ruhr-University Bochum. In 2019 she joined the group of Prof. Borchardt as PhD candidate working on the mechanochemical synthesis of porous polymers. |
 Sven Grätz | S. Grätz studied chemistry and chemical engineering in Dresden (Germany) and Strasbourg (France). He went on to obtained his PhD in 2018 from the Dresden University of Technology in the group of Prof. Borchardt where he worked on different mechanochemical synthesis. He is currently a senior scientist in the same group working on mechanochemical polymerization and in situ monitoring of mechanochemical reactions. |
 Getinet Tamiru Tigineh | Getinet Tamiru Tigineh is a full-time associate professor of chemistry at BDU, Ethiopia. He received his PhD in applied chemistry from NTUST, Taiwan in 2015 under the supervision of Prof. Ling Kang Liu. In 2020, he joined prof. Jeung Gon Kim's group as a visiting professor at JBNU, South Korea. His research interest covers the design and mechanochemical synthesis of molecules. |
 Lars Borchardt | L. Borchardt received his PhD 2013 in inorganic chemistry from Technische Universität Dresden, Germany. After a postdoctoral stay at ETH Zürich, Switzerland he got awarded a junior research group in 2015 and started working on mechanochemistry. Since 2019 he is professor for inorganic chemistry at Ruhr-University of Bochum. |
 Jeung Gon Kim | J. G. Kim is an associate professor in Chemistry at Jeonbuk National University, which he joined in the fall of 2015. He earned his PhD degree at University of Pennsylvania under the supervision of Professor Patrick J. Walsh in 2005. After postdoctoral work with Professor Geoffrey W. Coates (Cornell) and Professor Sukbok Chang (KAIST/IBS) and industry career at LG Chem. and Samsung Cheil Industries, he now independently leads a team, focusing on the development of new methods for making, breaking, and decorating polymers. |
Introduction
Mechanochemistry is a fast-growing area in chemistry. The efficient energy dispersion and mass transportation due to the use of mechanical forces has facilitated the elimination of solvents and warranted cleaner, faster, and more straightforward chemical syntheses than conventional solvent-based reactions.1–4 With many reported examples, mechanochemistry has become a reliable tool in synthesis. In particular, the application of mechanochemistry to the synthesis of molecules difficult or impossible to obtain by conventional methods has advanced many areas of chemistry,5,6 and mechanochemical activation has now been established as a promising technique alongside thermal-, photo-, and electroactivation.Polymer mechanochemistry has a rich history. From the dawn of polymer science, chemical events related to mechanical actions such as the grinding, shearing, and stretching of polymer chains have been a topic of interest. In the 1930s, Staudinger and coworkers reported that the action of an external force on polymer chains induces their scission and thus reduces molecular weight, viscosity, and strength,7–9 defining polymer mechanochemistry as the science and engineering of chain breakage caused by the failure, fatigue, or abrasion of polymeric materials.10 Although the increasing understanding of destructive polymer mechanochemistry has inspired its numerous and diverse ‘smart’ applications,11–13 the development and applications of constructive monomer-to-polymer mechanochemistry lag behind.
The 100 year history of modern polymer science has witnessed the development and commercialisation of many new polymers,14 with the development and refinement of novel polymerisation techniques resulting in a previously unimaginable level of variety and precision. However, the underlying synthetic process has remained unchanged, i.e., most polymerisations are still carried out in a fluidic phase using either a large excess of solvent or the melting of solid monomers upon excessive heating. This approach features the drawbacks of significant initial investment, high energy consumption, and problematic waste disposal/safety control15,16 and is barely applicable to the synthesis of polymers with limited solubility or miscibility.
Recently, mechanochemical polymer synthesis has drawn much attention in view of the inherent advantages of mechanochemistry such as greenness and access to novel reactivity.15 Additionally, solid-state mechanochemical polymer synthesis allows one to avoid solubilising group usage and low polymerisation degrees (DPs) caused by fast precipitation, i.e., is well suited for the production of structures that cannot be accessed by solution processes.
The first mechanochemical polymerisation was demonstrated in 1959 by the Kargin group, who used vibrational ball milling to polymerise vinyl monomers.17 Several sporadic studies followed, including those of vinyl monomer polymerisation by Oprea in the 1980s18 and by Kuzuya in the 1990s.19 As mechanochemistry was not well established at that time, these studies did not receive deserved attention. In addition to the development of green and sustainable processes, mechanochemistry drew much interest at the beginning of the 21st century. The mechanochemical Gilch polymerisation of poly(phenylene vinylene) reported by Ravnsbæk and Swager in 2014 highlighted the potential of constructive polymer mechanochemistry.20 Since then, many research teams confirmed the suitability of mechanochemistry for the construction of one- to three-dimensional polymers via step-and chain-growth polymerisations.
The re-emergence of mechanochemistry within the last decade has inspired several reviews focusing on the broad topic of mechanochemistry and several subdisciplines.21–26
While the general discipline of mechanochemistry relies on the generation of reactive sites by either shearing, grinding, or stretching, the subdiscipline polymer mechanochemistry is nowadays divided into two different communities.27,28 Based on Staudinger's work on molecular weight reduction of polystyrene, the broad field of sonication and force spectroscopy emerged as a very important aspect of polymer mechanochemistry.29 The field expanded rapidly around the careful design and activation of mechanophores and has already been covered in several reviews.29–35
In contrast to this, we herein focus on the other part of polymer mechanochemistry, which comprises of mechanochemical polymerisation reactions induced by shearing and grinding during mortar-and-pestle synthesis, high-speed ball milling and reactive extrusion. To the best of our knowledge, this aspect of polymer mechanochemistry has not been addressed in a review by now, although it is recently gaining huge attention by the chemical community.
This review provides insights into the constructive mechanochemical synthesis of polymers, starting with the basics of mechanochemistry and then describing the syntheses of linear and porous polymers in separate chapters. We believe that this structure is convenient, as the related articles are very different in scope and originate from different communities. Nevertheless, we aim to bring them together within this review, showcasing the underlying similarities.
Mechanochemistry
It was Matthew Carey Lea in 1892 who postulated that mechanochemical reactions can be fundamentally different to thermochemical ones and it was Wilhelm Ostwald who coined the term “mechanochemistry” in 1907 as distinct branch of chemistry alongside electrochemistry, photochemistry, and thermochemistry.36,37 Since these days, the field of mechanochemistry has constantly expanded, from Staudinger's pioneering work on polymer mechanochemistry in the 1930s8 over different reports of supramolecular mechanochemistry in the 1980s38,39 and solid–solid organic synthesis strategies in the 1990s.40–42Today, the IUPAC defines a mechanochemical reaction as a chemical reaction induced by the direct absorption of mechanical energy.27 Despite the rapid expansion of shearing- and grinding-induced mechanochemistry and the routine publication of new synthetic procedures for various products, the mechanism at microscopic and molecular levels, however, remains unclear. High-speed ball mills and reactive extruders are often described as ‘black boxes’, as the chemical processes therein are shielded by enclosures, which are generally made of metals or ceramics.43 This leads to one of the major obstacles of mechanochemistry, which is the lack of a valid theory describing the ongoing reactions entirely, while being ubiquitously applicable towards various synthetic protocols. As of now, three different theories, namely the hot-spot theory,44,45 the magma-plasma model,46,47 and the pseudo-fluid model,48 are known for mechanochemistry, each capable to describe certain phenomena of various subdisciplines.49,50 Unfortunately, they all fail to explain mechanochemical polymerization reactions thoroughly. Any theory, developed in the future, should take the following important observations into consideration:
• Milling achieves the intimate mixing of substrates on the sub-micrometre scale, constantly creates fresh reactive surface, and thus decreases diffusion limitations.51
• Mechanical stress generates heat52,53 and minor temperature increases can lead to huge increases of the mechanochemical reaction rate.54–57
• The milling parameters play a major role in enabling and controlling mechanochemical reactions.56,58–60
• The collision inside the vessel can lead to the generation of radicals.61,62
Mechanochemical reactors
The simplest mechanochemical method has been (and still is) the utilisation of the mortar and pestle to induce chemical reactions between solid materials. Although this protocol is still used for the generation of polymers,63,64 the method itself has certain limitations. Owing to the size of the mortar, the typical synthesis batch is in the milligram to lower gram range and cannot be upscaled to meet the needs of industrial applications.65 Additionally, the synthesis time usually does not exceed several hours and is mainly limited by the human operator, while grinding itself is utterly slow. The most important drawback is the inhomogeneous energy input. In particular, with longer grinding times, the energy input continuously decreases, which makes the entire process barely reproducible.65 Likewise, reproducibility depends on the experimenter, as Stolle and co-workers have shown.66These drawbacks have been addressed by the invention of automated grinding devices. An important step in this direction was accomplished by the development of a motorised mortar-and-pestle setup by Retsch in 1923.67 Automated ball mills designed for size reduction, such as tumbler mills (invented in 1870) and vibrational ball mills (invented in 1930), are also used for mechanochemical syntheses.68
A tumbler mill is a horizontally mounted grinding drum operated with the aid of gravity, as the milling bodies (either balls or rods) drop from a certain height onto the vessel wall and the reaction mixture to induce a reaction between the particles. Tumbler mills have not been developed for chemical reactions in the laboratory, as laboratory-scale applications require the mill and, hence, the dropping height and impact energy, to be much smaller. To counteract this, in 1961, Fritsch placed four vertically mounted tumblers onto a counter-rotating sun disc to develop the first planetary ball mill (a lab-scale state-of-the-art planetary ball mill is shown in Fig. 1a).67
 | ||
| Fig. 1 Upper part: Examples of (a) a planetary ball mill (Fritsch planetary ball mill P7) and (b) a vibrational ball mill (Retsch mixer mill MM400). Lower part: Procedure of milling jar filling for (left) a planetary ball mill and (right) a vibrational ball mill. (I) The empty milling jars are equipped with a seal and milling balls. (II) The mill charge is added to the jar. (III) Milling jars are properly closed. (IV) Implementation of the milling jars in the respective ball mill. | ||
Because of this setup, the grinding media are accelerated by rotation, which leads to friction and collisions between the milling balls, the jar, and the reactants,69,70 and the kinetic energy is transferred to the reaction particles to induce bond breakage, bond formation, and size reduction. Furthermore, the kinetic energy is transformed into thermal energy, which results in a continuous temperature rise inside the milling jar.22,71,72
Another ball mill ubiquitously used for lab-scale mechanochemical synthesis is the vibrational ball mill, which is usually equipped with two to six milling jars (Fig. 1b). During the milling process, the jar follows a horizontal, vertical, or elliptical course at high frequencies.73 Owing to the inertia of the milling material and the mill charge, the fast changes of direction during vibration result in collisions and friction of the interior with itself and the vessel walls. The milling frequency and the horizontal/vertical deflection are directly linked to impact energy.43 In contrast to planetary ball milling, which is dominated by shear forces, vibrational ball milling is dominated by impact forces, as in the latter case, the sample is exposed to short pulses of mechanical energy.65
The design of high-speed ball mills has significantly evolved during the development of mechanochemistry. As mentioned previously, the mechanical energy is partially converted into thermal energy to heat up the milling jar. Consequently, energy management lacks the controllability required for upscaling to industrial batch sizes of kilograms or even tons.69 Retsch GmbH invented a cryomill, i.e., a vibrational ball mill equipped with an internal cooling system. Although this mill was originally developed for polymer milling, chemists have adapted the underlying principle to conduct reactions at constant temperatures. In this case, impact energy can be considered to be independent of thermal energy, which delivers new insights into the underlying mechanochemical mechanism.74,75 Concurrently, a mill setup with electric cuffs enables the heating of steel milling jars during synthesis, which is especially useful for temperature-dependent reactions such as the mechanochemical generation of microporous polymers53 and metal–organic frameworks57 or for obtaining fundamental insights into the temperature dependence of mechanochemical reactions.76,77 These latest developments make temperature-regulating milling equipment more accessible for mechanochemists around the globe.
The invention of transparent milling vessels made of quartz, sapphire, poly(perfluoroalkoxy alkane)s or poly(methyl methacrylate) allows the simultaneous mechano- and photochemical acceleration in a photoreactor78–81 as well as in situ (e.g., Raman spectroscopic) measurements, thus providing access to non-isolatable reactive intermediates.82–84 Likewise, in situ powder X-ray diffraction enables the real-time detection of crystalline materials and their transformations during milling.78,85–87 Recently, Emmerling and Van Wüllen reported the first monitoring of a mechanochemical reaction by in situ solid-state NMR spectroscopy.88 Specifically, the 31P spectrum with and without proton decoupling was monitored in a self-made miniaturised ball mill on an NMR sample head. Another important in situ technique is the simultaneous measurement of temperature and pressure using a gas pressure and temperature measurement system. In particular, for the synthesis of porous polymers, the recording of pressure build-up can assist mechanism elucidation.53,89
Although planetary and vibrational ball mills are highly efficient for polymer synthesis,90,91 their low material throughput is a definite drawback. Typically, milling jars have a volume of 1–500 mL, which limits the batch sizes to several grams, as jars are usually filled to one-third with milling balls, one-third with reactants, and one-third with void volume.
Common mistakes to be avoided when operating ball mills are as follows.
• The use of too few or too many milling balls, e.g., the erroneous use of only one 10 mm milling ball inside a 50 mL vessel.
• The use of milling balls with the wrong size, e.g., the use of overly large (>20 mm) or small (<5 mm) milling balls in vibrational ball mills.
• The use of incompatible balls and vessels, e.g., zirconium oxide balls in steel vessels.
• Underestimation of pressure build-up during the reaction, for example, risk of explosion during work with permanganates, carbides, etc.
Unlike ball milling, reactive extrusion facilitates continuous and scalable synthesis.92 An extruder comprises a feed port, one (single-screw extruder; SSE) or two (twin screw extruder; TSE) screws placed inside a heatable barrel, and an exit port.93 In an SSE, the screw diameter gradually increases in the direction from the feed port towards the exit port, which increases the compressive force acting on the material as it passes the barrel. The two screws of a TSE can rotate in the same or opposite directions, applying shear to the sample.94 Additionally, several different setups can be realised by exchanging screw segments for reverse segments, toothed segments, or kneading blocks to allow different mechanical actions.92 In polymer manufacturing, SSEs and TSEs are widely used for the mixing of molten polymers and additives.
Mechanochemical reaction parameters
While classical synthetic procedures are mainly dependent on the nature and content (concentration) of the employed solvent, these parameters play a subordinate role in mechanochemistry. Although standard reaction parameters such as stoichiometry, temperature, and reaction time also apply, there are other important parameters that allow the sophisticated design of mechanochemical processes. For mechanochemical synthesis inside a high-speed ball mill, the nature of the milling material is a crucial variable along with the type of the ball mill itself.95 The density of the milling material is directly related to its moment of inertia, and therefore, the kinetic energy transferred to the sample.22 The most frequently utilised milling materials are agate (2.7 g cm−3), silicon nitride (3.3 g cm−3), corundum (3.8 g cm−3), zirconium dioxide (5.7 g cm−3), stainless steel (7.8 g cm−3) and tungsten carbide (14.3 g cm−3).43,96 Although heavier milling materials afford increased yields in several cases,97,98 this behaviour is not always directly transferable to mechanochemical polymer synthesis. The use of higher-density milling materials might not only enhance polymerisation, but also favour the destruction of the synthesised polymers.20,53,99,100 In addition to material density, chemical resistance also plays a significant role; hence, tungsten carbide and zirconium dioxide are primarily selected for lab-scale processing. For example, stainless steel corrodes upon contact with strong acids, which can lead to the contamination and rapid degradation of the milling material.26 Nevertheless, as it is significantly cheaper, stainless steel is the material of choice for large-scale industrial applications.The size and number of milling balls are just as important as the nature of the milling material. For lab-scale applications, milling ball size usually ranges between 0.1 and 30 mm.43 To adjust the milling ball size, one has to keep the overall mass constant owing to the coherence between mass and energy.101 Furthermore, one can also mention the ball-to-powder mass ratio, which is defined as the total mass of milling balls divided by the total mill charge mass and can be adapted for process fine-tuning.26 In this regard, to ensure the adequate trajectories of milling media, one should maintain a uniform filling degree, which is ideally one-third of the grinding bowl volume.69 To achieve this optimal filling degree, one can change the reaction scale or the jar size or add a grinding auxiliary, often called a bulking agent,3,95 which should be chemically inert towards the reaction mixture.74 In terms of polymer mechanochemistry, one should ensure that the bulking agent can be removed from the usually solid product. For example, one can use salts (e.g., NaCl) that are easily removable by water, which is beneficial in ecological terms.102 In contrast, silica is inappropriate for insoluble polymers, as it has to be removed with hydrofluoric acid, which may destroy the synthesised polymer. In particular, when one or more liquid starting materials are used, a bulking agent is of high importance, as it can function as an adsorbent and therefore allows one to circumvent the formation of sticky mud-like reaction mixtures, which hinders sufficient energy transfer.26
In a neat milling environment, only solids are involved, which creates a homogeneous sample distribution without the possibility of lump formation. In many cases, however, catalytic amounts of liquids can benefit mechanochemical syntheses.25,103–105 This liquid-assisted grinding (LAG) developed from the poorly quantified addition of a certain amount of solvent droplets to a mechanochemical reaction and was formerly referred to as solvent-drop or solvent-assisted grinding.3,23,106,107 Recently, the methodology has been defined by the LAG parameter η, which is the ratio between the amount of liquid (μL) and the total mass of solid components (mg) and usually lies between 0 (no liquid) and 1 (slurry or even homogenous solution).3 LAG can significantly enhance the yield or speed of a reaction, even if the starting materials are poorly soluble in the applied liquid.108 Although the latter phenomenon is scarcely understood, it is assumed that a small amount of liquid is permanently saturated with reactants or that a mobile surface layer is formed.3,109 Furthermore, larger amounts of liquids can act as porogenes and stabilise pore formation during the synthesis of porous polymers.53,89 For example, for the mechanochemical synthesis of poly(lactic acid) (PLA), LAG limited polymer chain degradation while speeding up propagation to afford a high-molecular-weight product.99 In addition, polymers can also be used as grinding auxiliaries (polymer-assisted grinding) during mechanochemical processing.110–112
Another very important parameter is the frequency (for vibrational mills) or rotation rate (for planetary mills) of ball milling.113,114 In either case, higher agitation frequency accelerates the movement of milling balls and thus increases their kinetic energy.24,115 Although the milling frequency is easy to adjust, the accompanying temperature change is difficult to regulate and must be taken into consideration (particularly considering the exothermal character of most polymerizations to compensate a negative entropy). In the context of polymer mechanochemistry, we again notice that a higher frequency might also result in the destruction of the synthesised polymers,116,117 as has been observed for different reaction systems and for both rotational and vibrational ball mills. This usually leads to the trend, that the chain length of a given polymer is growing with increasing reaction time for the first minutes of a reaction and consequently either plateaus if the longer polymer chains are degraded, or even decreases if the speed of degradation exceeds the reaction rate.19
This, however, is not surprising, as the degradation of polymers under mechanical force is systematically studied since the middle of the last century. Especially the detailed studies by Kausch on polymer degradation kinetics enabled novel insights into the mechanochemical degradation of polymers and led to the establishment of a variety of different systematic reports nowadays.118–124 Choi and Peterson have initiated systematic studies on mechanical force-induced polymer degradation by ball milling,117 revealing that milling frequency has the greatest impact on the degradation rate of polystyrene. For future studies, the balance between reaction and degradation rates must be considered to produce high-molecular-weight polymers.
Mechanochemical synthesis of linear polymers
The solvent-free mechanochemical synthesis of polymers was pioneered by the groups of Kargin, Oprea, and Kuzuya,19,125,126 who investigated the polymerisation of vinyl monomers and showed that mechanical action can indeed deliver sufficient energy and mixing efficiency for polymer synthesis. Further research broadened the scope of mechanochemical polymer synthesis and revealed many features distinguishing it from conventional techniques.20,90,127–129 Unfortunately, the knowledge gained from studies of conventional solution-based systems is not always valid for solid-state mechanochemical polymerisation, as in the latter case, concurrent chain degradation, the heterogeneous nature of solid-state conditions, and unexpected reaction pathways result in previously unseen architectures.20,99,125,130,131 This chapter focuses on mechanochemical (chain-growth and step-growth) polymerisations affording linear polymers as well as on post-polymerisation modification and provides a summary of the current issues.Chain-growth polymerisation
Before discussing the essential aspects and details of mechanochemical chain-growth polymerisation, we outline a brief history of its development.
In 1959 Kargin and coworkers disclosed the first mechanochemical polymerisation of methyl methacrylate and styrene in a vibrational ball mill.17 Shortly thereafter, the same group expanded the reaction scope to crystalline sodium acrylate and acrylamide.125,138 Continuing studies by Oprea in the 1980s and by Kuzuya in the 1990s covered many of the vinyl monomers known to undergo polymerisation (Scheme 1).18 The adopted strategy was used to convert (meth)acrylates,19,130,139–145 (meth)acrylamides,125,126,141,146 styrenes,128,141,147–150 and other structurally diverse vinyl monomers149–152 into the corresponding polymers under solvent-free ball-milling conditions. Moreover, the copolymerisation of these monomers has also been investigated,125,130,140,149,153–155 as is discussed below.
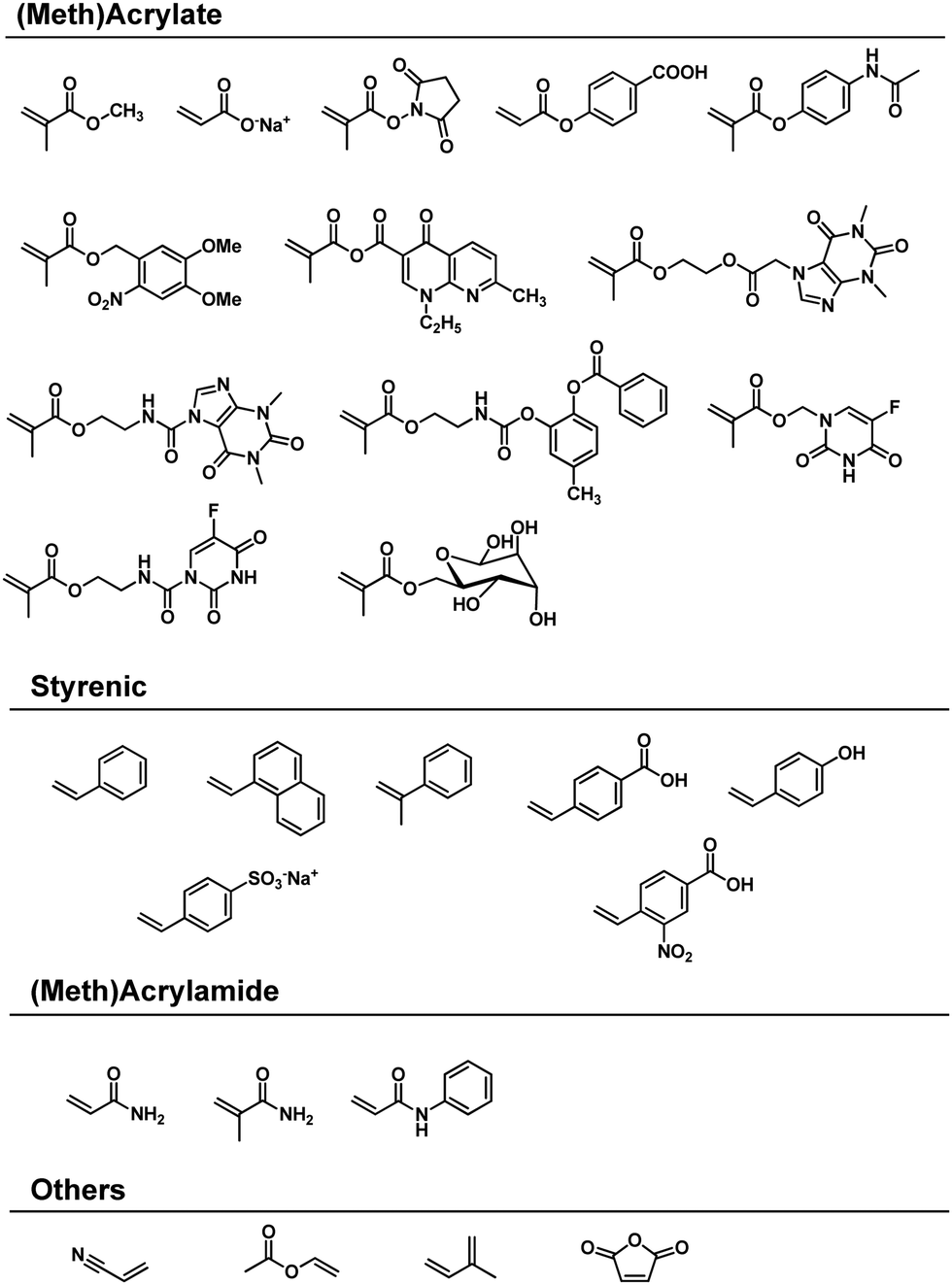 | ||
| Scheme 1 Reactive vinyl monomers used for solvent-free mechanochemical polymerisations. | ||
Conventional chain-growth polymerisation is usually initiated using compounds such as azobisisobutyronitrile (AIBN), benzoyl peroxide, or alkyl lithiums. However, mechanochemical ball milling can produce polymers from the corresponding vinyl compounds without any of these initiators, which makes the origin of initiation a topic of considerable debate.
Kargin and coworkers reported the mechanochemical polymerisation of MMA and styrene and claimed the importance of using NaCl and SiO2 as additives. According to them, the surface activation of these inorganic solids due to mechanical forces forms active radical or ionic sites, consequently causing initiation.17,139 This hypothesis was supported by several studies on active radicals and ionic sites formed by mechanical forces acting on the surface of inorganic solids.156,157
Later, Hasegawa continued related works using alumina, clay minerals, quartz, marble, and limestone activators. However, no direct observation of the propagating species was made.158–164
The hypothesis of ionic site formation at the surface of inorganic solid additives is not necessarily wrong, but its necessity is debatable, e.g., the Oprea group reported that the ball-milling of acrylonitrile produces polyacrylonitrile even without additives.151,165 All monomers presented in Scheme 1 do not require external initiators for ball-milling polymerisation. Both Oprea and Kuzuya proposed that the initiation step involves electron transfer from the milling material to the substrate.18,19,126,141 Strong collisions between ball and vessel supply energy greater than the milling materials' work function, i.e. the threshold energy for electron emission. Electron transfer from the milling material generates a radical anion that triggers the observed polymerisation (Fig. 2). In agreement with this hypothesis, the addition of radical inhibitors such as oxygen, phenol, and hydroquinone slows down the reaction progress.126 Later, Kuzuya's group observed the propagating radical species using electron spin resonance (ESR) spectroscopy146 and reasoned that the milling jar material is critical for polymerisation.19
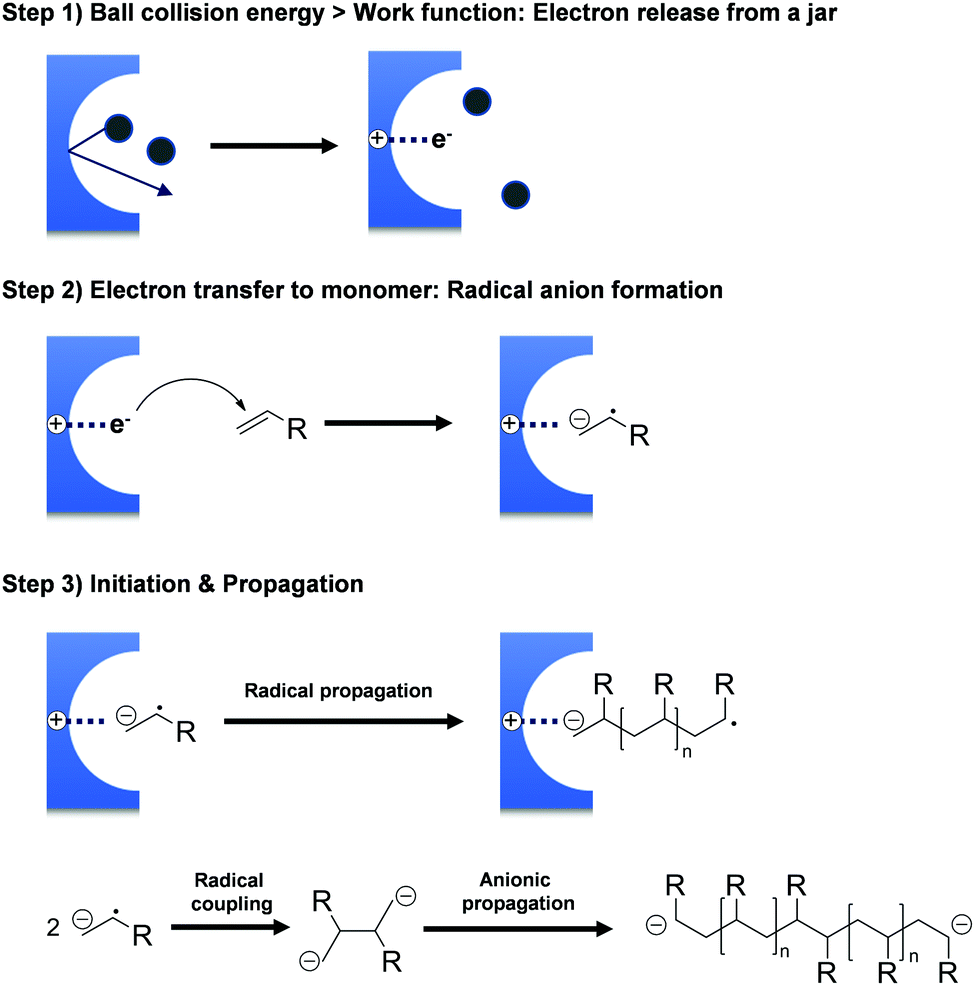 | ||
| Fig. 2 Mechanism of initiator-free mechanochemical polymerisation. | ||
Highly efficient initiation was observed in a stainless-steel reactor, whereas no polymerisation was observed in a Teflon jar. Indeed, metal-based containers might more efficiently deliver electrons and thus facilitate initiation; however, monomer self-initiation cannot be excluded completely.166,167
To elucidate this topic further and understand why some monomers do not show any reactivity during ball milling, Kuzuya and coworkers conducted theoretical studies on the relationship between monomer structure and reactivity.19,132 They found that the structure of the lowest unoccupied molecular orbital (LUMO) of the monomer was directly correlated to initiation efficiency (Fig. 3). The shape of the LUMO was further proposed to be a more pertinent factor than its energy. Only monomers with LUMOs localised on the vinyl unit underwent solid-state polymerisation (Fig. 3a and b). When the LUMO extended to substituent orbitals, radical anion formation was sluggish (Fig. 3c).
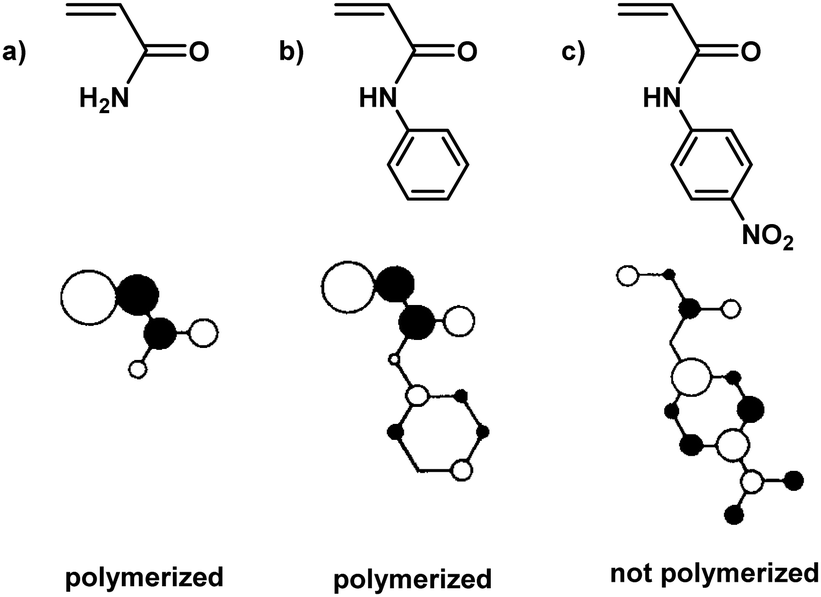 | ||
| Fig. 3 LUMO localisation of selected acrylamide monomers. | ||
Kuzuya and coworkers further monitored conversion, radical concentration, and molecular weight changes during the reaction of a methyl acrylate monomer.19,146 At the beginning (<5 min), only a small amount of the active radical species was generated. High monomer concentration resulted in fast chain propagation, and chains with the highest molecular weight were produced at a monomer conversion of 10%. In continuation of the reaction a fast monomer consumption and a decrease in molecular weight was observed. This is because after reaching the threshold molecular weight, the polymer chains undergo degradation by ball milling and the homolytic bond dissociation produces short polymers with an active radical chain-end (macroradical) resulting in an increase in radical concentration. This phenomenon had two implementations, i.e., fast polymerisation within 5–20 min and a maximum molecular weight limited by chain degradation. After 20 min, the polymer chains experienced degradation until the minimal molecular weight was reached. Fig. 4 summarises this reaction mechanism, showing that in the early state, propagation is dominant (stage 1) and once high-molecular-weight chains appear, mechanical force-induced chain degradation becomes dominant (stage 2), which results in molecular weight reduction.
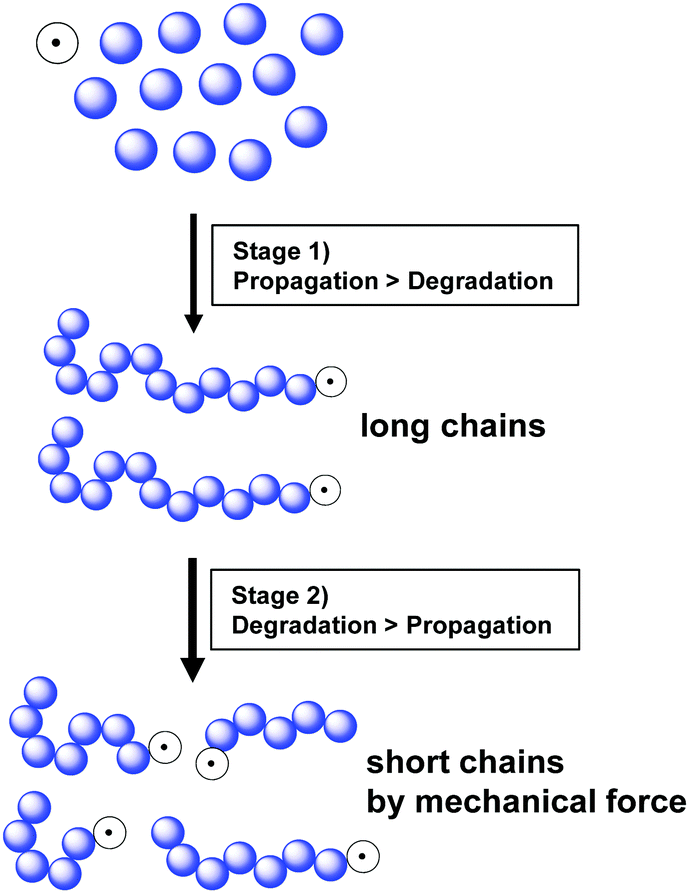 | ||
| Fig. 4 Schematic of mechanochemical polymerisation and subsequent chain degradation. | ||
In their studies, Kuzuya and coworkers obtained mechanochemically prepared polymers with narrow molecular weight distributions, ascribing this to acute polymerisation induced by mechanical forces.145,168–170 However, it is important to understand that this behaviour was mainly caused by mechanical degradation and not by controlled polymerisation kinetics. Hence, full control over chain length, as in the cases of living or controlled polymerisation, has scarcely been realised mechanochemically.
Today, chain length can be adjusted using reversible deactivation radical polymerisation.171,172 Although mechanochemical radical polymerisation has been studied for over 60 years, the related controlled polymerisation has not been realised until Cho and Bielawski's recent report.128 These researchers developed a mechanochemical atom radical transfer polymerisation (ATRP) of solid 2-vinylnaphthalene. The polymerisation system comprised Cu powder, CuBr, tris(2-pyridylmethyl)amine, and 1-bromo-1-phenylethane and allowed for high monomer conversion, linear chain growth proportional to the monomer consumption rate, and narrow dispersity. The realisation of control over chain length using different monomer-to-initiator ratios confirmed the reversible activation/deactivation mechanism previously observed for the solution state. The retardation of polymerisation in the presence of TEMPO and the isolation of phenylethyl-TEMPO also supported a radical mechanism. Despite this great success, it needs to be emphasised that chain degradation still remained uncontrolled, limiting the polymer molecular weight (Mn).
The differences between mechanochemical and solution-phase radical polymerisation are particularly obvious for copolymerisations.125,130 As an illustrative example, Fig. 5 shows the 13C NMR spectra of four different polymers based on polyacrylamide and poly(sodium acrylate), namely (A) a physical blend, (B) a copolymer prepared by solution-phase radical polymerisation, (C) a copolymer prepared by mechanochemical solid-state polymerisation, and (D) a diblock copolymer.130 The carbonyl environments of (A) and (D) are similar because of the lack of an acrylamide–sodium acrylate sequence in the polymer chains. The spectra of products obtained by solution-phase (B) and solid-state (C) polymerisation, however, showed signals at δ = 183.5 and 180.5 ppm, which corresponded to heterojunctions. Whereas the completely random copolymer (B) exclusively showed heterojunction peaks, polymer (C) showed the signals of both homojunctions (major) and heterojunctions (minor). Consequently, the formation of a multiblock-type polymer is anticipated if a mechanochemical pathway is involved. These results support those of the early study of Kargin and coworkers on the copolymerisation of acrylamide and methyl methacrylate (Table 1).125
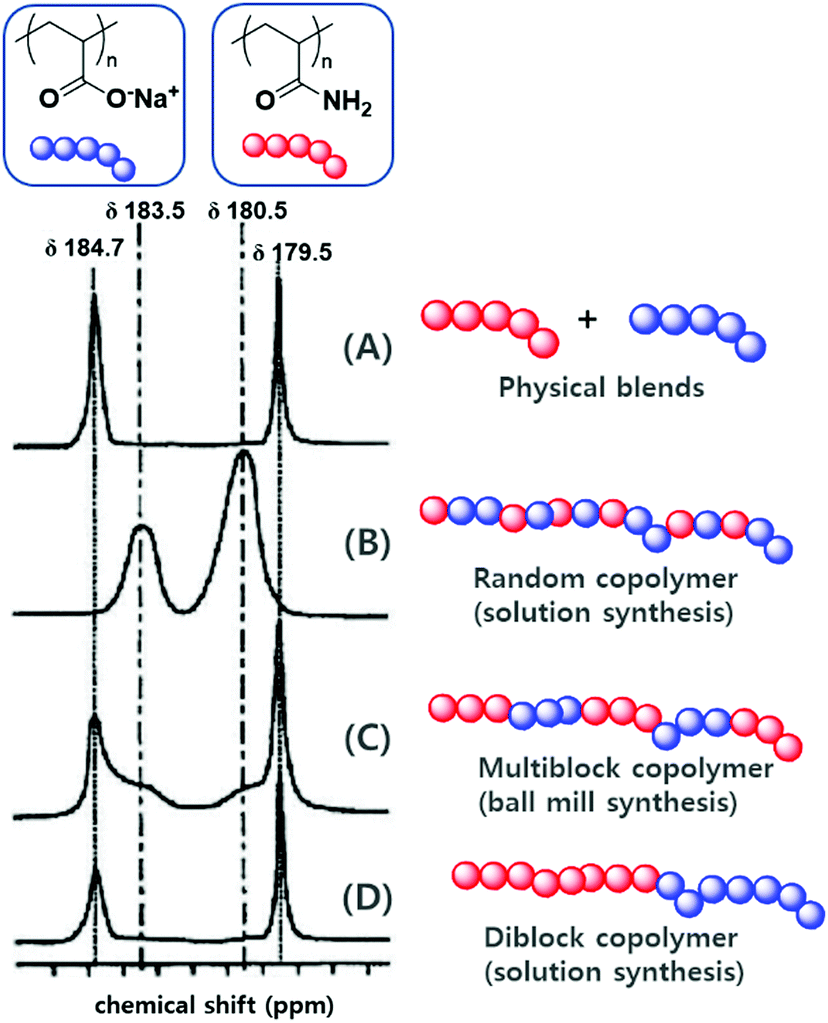 | ||
| Fig. 5 13C NMR signals of the carbonyl groups of (A) a physical blend of polyacrylamide and poly(sodium acrylate), (B) a random copolymer of polyacrylamide and poly(sodium acrylate) prepared by solution-phase radical polymerisation, (C) a multiblock copolymer of polyacrylamide and poly(sodium acrylate) prepared by ball milling, (D) a diblock copolymer of polyacrylamide and poly(sodium acrylate) prepared by ball milling. Reproduced with permission from ref. 130. Copyright (2000). The Pharmaceutical Society of Japan. | ||
| Monomers | Temp. (°C) | Condition | Relative reactivity ratios | ||
|---|---|---|---|---|---|
| M1 | M2 | γ 1 | γ 2 | ||
| AA | MMA | 70 | Benzene, 0.15% AIBN | 4.2 | 0.12 |
| −65 | No solvent, 1% NaCl | 36.6 | 4.2 | ||
Under the conditions of conventional solution polymerisation (70 °C in benzene, AIBN), the addition of acrylamide was dominant regardless of the active chain-end structure because of the higher reactivity of acrylamide compared to that of methyl methacrylate. In the initial stage, polyacrylamide was primarily produced. However, solid-state polymerisation at −65 °C afforded a strikingly different product. In this case, the heterogeneous distribution of solid monomers caused each monomer particle to become a domain in which one monomer exists excessively. Thus, the concentration factor overrode end-group reactivity, especially the heteropropagation tendency of methyl methacrylate end radicals. Each chain end preferentially underwent homopropagation before entering another monomer particle, which afforded a multiblock-like copolymer structure. In addition, the copolymerization of orthogonally soluble monomers, i.e., when monomers lack solubility in a common solvent, can profit from a mechanochemical pathway. Bielawski and Cho presented the copolymerization of hydrophobic 2-vinyl naphthalene and ionic sodium styrene sulfonate in their above mentioned ATRP study.128
The mechanical fracture of polymers and the resulting physical changes have been known for a long time, and the underlying mechanism has been of interest since the dawn of polymer science.7–9 In 1952, Watson and coworkers showed that mechanically cleaved rubber produced polymer fragments with a radical end, known as macroradical.173,174 That these species indeed initiate polymerization reactions was then reported by Goto and Fujiwara in 1963.175
The species obtained by the mechanical breakdown of poly(vinyl acetate) (PVA) in a solution of vinyl acetate (VA) were found to consume VA. The fact that this consumption was quenched in the presence of hydroquinone supported radical propagation. This experiment has been extended to the synthesis of various block copolymers, especially those not accessible by conventional methods (Fig. 6).
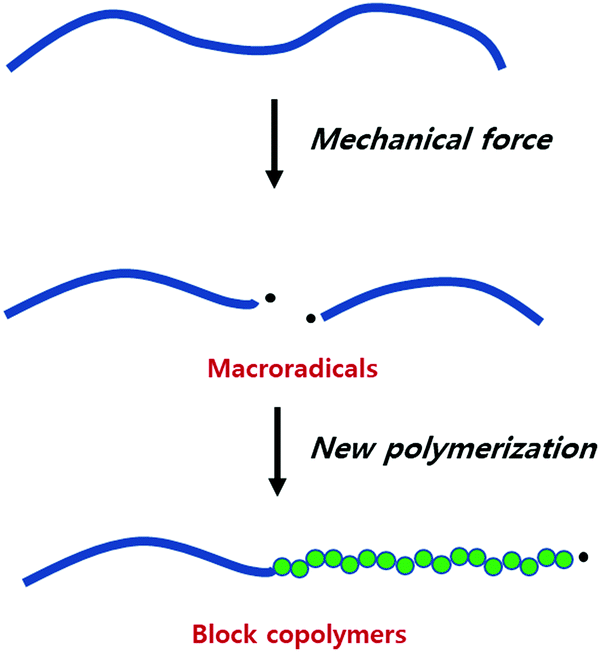 | ||
| Fig. 6 Illustration of macroradical formation-induced block copolymerisation. | ||
The involvement of mechanically produced macroradicals was also reported by Sohma and coworkers in their study on the mechanochemical synthesis of polytetrafluoroethylene (PTFE) block copolymers.131 ESR spectroscopy showed that the mechanical fracture of solid PTFE produced PTFE macroradicals, which, in view of their low mobility, were not terminated under anaerobic conditions and initiated chain propagation upon the introduction of monomers such as MMA, VA, and ethylene. This method allowed the synthesis of block copolymers (e.g., PTFE-b-PMMA, PTFE-b-PVA, and PTFE-b-PE) difficult to obtain under common polymerisation conditions. Kuzuya and coworkers also used the macroradical formation and concurrent polymerisation of solid-state monomers to produce amphiphilic polymers. Specifically, the ball milling of polymethacrylate,142,144 poly(4-vinylpyridine),170,176 or cellulose168 produced the corresponding macroradicals that initiated the radical polymerisation of solid-state monomers to afford block copolymers of various compositions.
The capping of active macroradicals with functional small molecules is another way to obtain end-functionalised polymers. Ito's group realised this concept through ball milling.177 Polymeric macroradicals formed in situ under ball milling conditions underwent radical coupling reaction with a prefluorescent nitroxide based reagent. A series of luminescent polymeric materials were obtained from polystyrene, polyethylene, poly(methyl methacrylate), poly(phenylene sulfide), and polysulfone, showing off its synthetic versatility.
Initially, the Kim group showed that conventional organic superbase catalysts such as 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) and triazabicyclodecene can polymerise solid lactide and trimethylene carbonate without a solvent and that ball milling enables efficient initiator-catalyst mixing even at very low concentrations. The number and mass of the milling balls significantly influenced mechanochemical conversion.99 At a constant total mass of moving parts, a comparison of one 12 mm ball with five 7 mm balls showed that faster conversion was achieved in the latter case (89% vs. 49%, respectively). A similar trend was observed for poly(trimethylene carbonate) synthesis,91 and it was concluded that the magnitude of the impact energy (single event) was more influential than the total energy input.
While neat grinding resulted in the anticipated chain degradation after a DP of 100 (Fig. 7, solid line), LAG selectively retarded chain degradation and enhanced the polymerisation rate (Fig. 7, dotted line).99 Among the tested liquid additives, toluene (η = 0.2 mL mg−1) exhibited the best effect, i.e., no signature of chain degradation was observed up to 1 × 105 Da. The exact mechanism of LAG-mediated chain protection is not clear, particularly considering that this effect was not observed for poly(trimethylene carbonate) synthesis. We hypothesise that the protective effect of LAG is due to the combined influence of the absorption of impact energy, material lubrication, and mass transport enhancement.
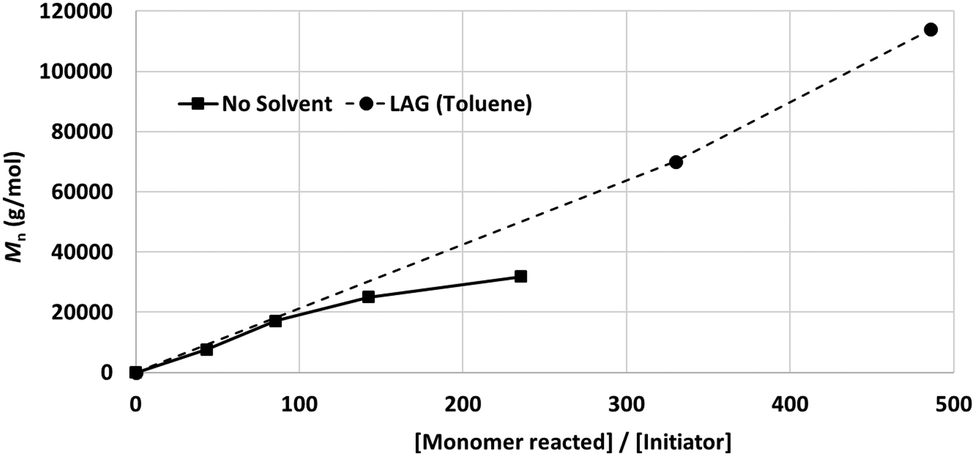 | ||
| Fig. 7 Effect of LAG on the production of high-molecular weight PLA. | ||
Mechanochemical ROP can be a powerful technique for obtaining block copolymers.181 For instance, macroinitiators with terminal hydroxy groups, such as poly(ethylene oxide) (PEO), can be extended by lactide polymerisation. As PEO is hydrophilic and is not miscible with lactide and the organic catalyst (DBU), initial trials using high-molecular-weight PEO were unsuccessful because of poor mixing. This problem was overcome by increasing milling energy through the use of larger milling balls and LAG (THF, η = 0.2 mL mg−1), which resulted in the successful synthesis of triblock copolymers.
A warrantable concern for mechanochemical polymerisation reactions is whether they indeed proceed in the solid state. Therefore, the authors of the latter study performed instant temperature measurements at the end of polymerisation, revealing only a slight increase to 44 °C, which is below the melting temperature of PLA (110 °C), PEO (60 °C), and lactide (95 °C). As the resulting mixture was a fine powder, the formation of a eutectic mixture occasionally seen in mechanochemical synthesis was ruled out.
Step-growth polymerization
Step-growth polymerisation refers to the successive reaction of multifunctional monomers to afford dimers, trimers, oligomers, and long-chain polymers. According to Carothers equation, the formation of the desired polymer chain is warranted by high reaction efficiency and low side-reaction probability.183 The fact that mechanochemical reactions can proceed at maximal monomer concentration (i.e., in the absence of a solvent) results in increased reaction rates and efficiencies, which makes mechanochemical techniques particularly suitable for step-growth polymerisation. At the same time, the lack of the solubility limitation allows the preparation of polymers hardly accessible in solution, such as unsubstituted polyphenylenes.Early attempts relied on the simple manual grinding of reactive monomers using mortar and pestle. In 2002, Qu and coworkers described the solid-state synthesis of doped polyaniline (PANI)184–187 based on the 30 min hand-grinding of frozen aniline at −20 °C with the inorganic acid H4SiW12O40 (Fig. 8A). This technique has drawn much attention in the polyaniline community because of its synthetic simplicity, greenness, and unique properties but exhibited all the above mentioned drawbacks of manual grinding concerning reproducibility and scalability.188,189 To meet these challenges, Kaner and coworkers applied ball milling to the mechanochemical synthesis of PANI.190,191 The authors used anilinium salts such as anilinium chloride and oxidants such as ammonium peroxysulphate, studying the effect of dopants and other derivatised anilines. Posudievsky and coworkers showed that the molecular weight of PANI prepared by ball milling was similar to that of PANI prepared using a solution-based method. Here, one should note that PANI was produced by a facile polycondensation reaction that does not require high mechanical energy. For this reason, syntheses could be performed even with simple mortar-and-pestle grinding. An in-depth study of the influence of grinding parameters was, unfortunately, not considered necessary at that time.
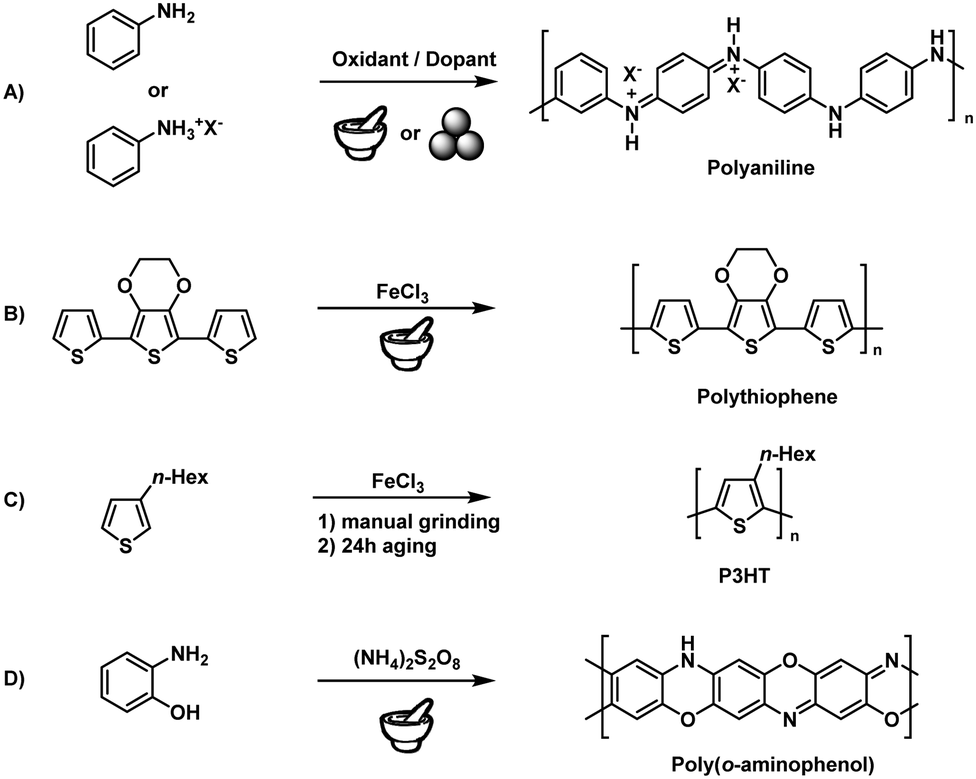 | ||
| Fig. 8 Solid-state conducting polymer synthesis: early examples. | ||
Solid-state syntheses of other conducting polymers have also been reported. Nurulla and coworkers synthesised poly(thiophene) derivatives by the hand-grinding of terthiophene and FeCl3 (Fig. 8B), while Takahara and coworkers used 3-hexylthiophene to synthesise poly(3-hexylthiophene) (P3HT) by grinding and subsequent aging for 24 h (Fig. 8C).192–194 Notably, aging seemed to be an important factor. Whereas room-temperature aging only yielded low-molecular-weight products (Mn = 3000 Da), Kumar's group showed that aging at elevated temperatures (100 °C) produces high-molecular-weight polymers in good yields, using poly(3,4-propylenedioxythiophene) as an example.195 Zoromba synthesised poly(o-aminophenol) using an automated mortar-and-pestle grinder (Retsch RM200) (Fig. 8D).196
Similar to chain-growth polymerisation, in the 2010s, mechanochemical step-growth polymerisations advanced their focus from a purely chemical perspective to a more mechanochemical point of view that involves the study of milling parameters. In 2014, Swager and coworkers studied mechanochemically promoted solid-state Gilch polymerisation with an emphasis on these parameters.20 High-speed ball milling of bis(chloromethyl)benzene (monomer) and tBuOK (base) produced poly(phenylene vinylene) (PPV) with a molecular weight of up to 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da in less than 1 h with high reproducibility, whereas the solution process often required more than 24 h and afforded a large deviation in molecular weights between each synthesis. Notably, no PPV was formed when slow vibration or small balls were used, which suggests that a certain mechanical energy threshold needs to be overcome for efficient synthesis. The progressing chain degradation with increasing reaction time limited the molecular weight to ∼40 kDa.
000 Da in less than 1 h with high reproducibility, whereas the solution process often required more than 24 h and afforded a large deviation in molecular weights between each synthesis. Notably, no PPV was formed when slow vibration or small balls were used, which suggests that a certain mechanical energy threshold needs to be overcome for efficient synthesis. The progressing chain degradation with increasing reaction time limited the molecular weight to ∼40 kDa.
Borchardt and coworkers continued this focus on the impact of milling parameters on step-growth polymerisation by investigating a series of mechanochemical polycondensations of poly(azomethine), polyphenylene, and polyimide.90,197–199 The first investigation probed Schiff base formation from p-benzenedicarbaldehyde and p-phenylenediamine (Fig. 9),197 revealing that polymerisation efficiency was proportional to the density of milling materials and ball size. This result again demonstrates that the introduced mechanical energy promotes polycondensation. Mechanically prepared poly(azomethine) exhibited a longer chain length than that prepared by solution-phase methods, which was ascribed to the fast precipitation of polymers during classical synthesis.
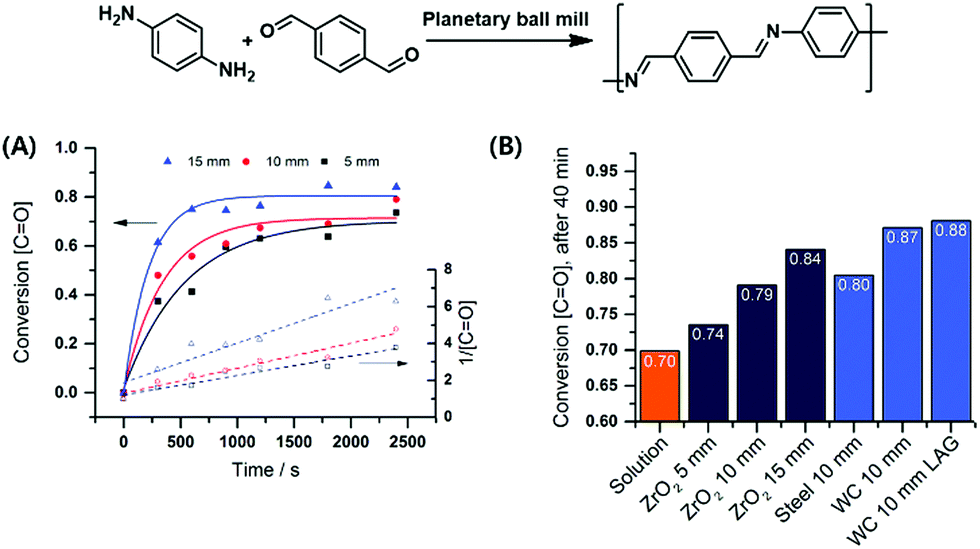 | ||
| Fig. 9 Polycondensation via highly efficient mechanochemical Schiff base formation: effects of (A) ball size and (B) milling materials. Reproduced with permission from ref. 197. Copyright (2016). The Royal Society of Chemistry. | ||
However, this trend holds only to a certain extent, as demonstrated by the same group for the Pd-catalysed Suzuki polycondensation of 1,4-dibromobenzene and 1,4-phenyldiboronic acid.90,198 In the presence of Pd(OAc)2 as the catalyst and K2CO3 as the base, 30 min milling in a planetary ball mill yielded p-polyphenylene (PPP) with a high DP of up to 164. Notably, heavy milling balls made from tungsten carbide afforded a lower DP than lighter milling balls such as those made from silicon nitride, as the higher impact energy in the former case resulted in chain degradation. This hypothesis was substantiated by post-polymer milling experiments in which the milling of freshly synthesised PPP decreased DP when the impact energy was sufficiently high. This type of mechanochemical polycondensation is comparatively robust to the change of substrate substitution pattern. In addition to the mentioned A2B2-type reaction affording linear polymers, AB-type homopolymerisations or A2B-type polymerisations affording hyperbranched polymers were also possible.
A major breakthrough in the field of step-growth polymerisation was the development of direct mechanocatalysis, in which case the reaction is catalysed by milling balls and not by intentionally added soluble or solid-state catalysts, as in the abovementioned polymerisations. Vogt et al. used Pd milling balls to promote the conversion of 4-bromophenylboronic acid to PPP within a ball mill,198 achieving high DPs of up to 199 in the absence of ligands or salts. This study substantiated the strong influence of impact energy and the need to adjust it by tuning milling parameters. Indeed, a certain critical impact energy is necessary, as milling at <200 rpm did not induce any conversion; however, excessively high impact energies led to chain degradation.
The impact energy can be adjusted by tuning the hardness of the milling vessel. If Pd milling balls are used in soft Teflon vessels and not in zirconia or steel vessels, chain degradation is hindered because of the decrease in the power of impact events (elastic vs. inelastic collision). Moreover, this change also results in significantly reduced abrasion.
Song and coworkers made similar observations during their mechanochemical synthesis of polyurethane100 based on the vibrational ball milling-induced reaction of diisocyanates with a series of biomass-derived diols (e.g., 2,5 bis(hydroxymethyl)furan) at ambient temperature (Fig. 10A). Whereas chain degradation occurred during fast (and hence, energetic) vibrational milling at 30 Hz, it was strongly hindered during slower milling at 20 Hz (Fig. 10B). Notably, the reaction was complete within 1 h, i.e., was faster than the corresponding solution-phase reaction.
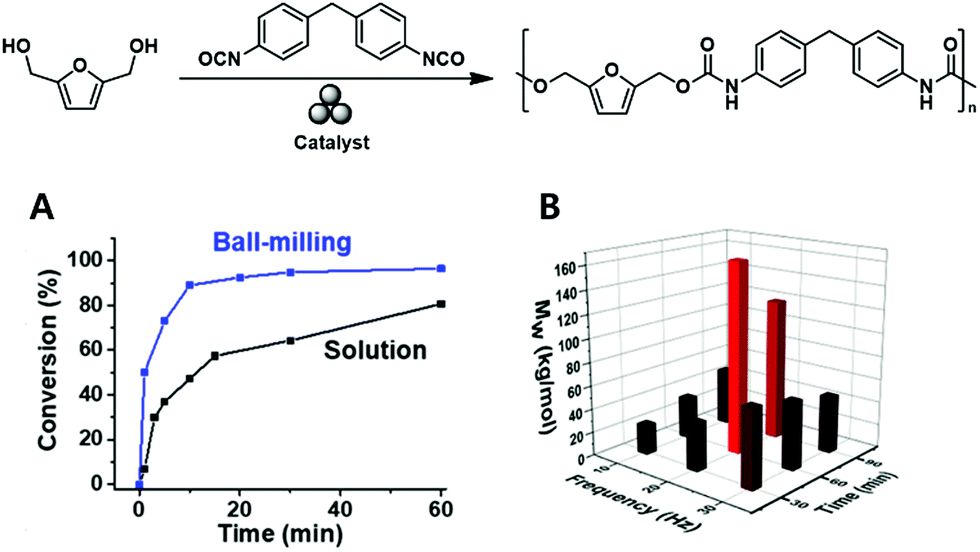 | ||
| Fig. 10 Mechanochemical solid-state polyurethane synthesis from a furan-diol: (A) milling optimisation and (B) comparison of ball-milling and solution-phase polymerisations in terms of reaction rate. Reproduced with permission from ref. 100. Copyright (2020). The American Chemical Society. | ||
The synthesis of polyimides requires excess heating and long reaction times in solution synthesis. The ‘beat and heat’ process consists of ball milling of dianhydride and diamine, rapidly forming polyamic acid, and subsequent heating at 200 °C, shortening the process time to 1 h. Furthermore, the process enabled the synthesis of polymers inaccessible through solution-based processes.199
Such an economy of time was also observed by Shi and coworkers who synthesised polyamides from bis-benzoxazine and bis-isocyanate.64 The manual grinding of monomer mixtures in an agate mortar for 1 h produced a polyamide in good yield and with a high molecular weight comparable to that obtained in solution-phase reactions after 6 h.
Post-polymerization modification
Post-polymerization modification (PPM), the chemical operation on an existing polymeric chain or side-functional group, provides new possibilities in polymer science, as it can be utilized to introduce chemical units that are not compatible with conventional polymerization methods.200,201 In addition, chemical modifications beginning from a parental polymer produce a library of functional polymers with identical chain-lengths and topologies, but with different functional groups, allowing the facile and acute study of structure–property relationships. However, a distinct disadvantage of post-polymerization modifications is the often limited solubility of the polymers, which narrows the opportunities for syntheses by conventional solvent-based methods. Recently, examples for solvent-free solid-state post-polymerization modifications have been reported, enabling to overcome the given limitation of solubility.These procedures mainly focus on two different approaches, as it is either possible to change the functional groups of a polymer, or to modify the polymeric chain itself.
One distinct example for the latter was presented by the Moores group, reporting the transformation of chitin, the second most abundant biopolymer, into chitosan (Fig. 11).202 During the first step, ball milling of neat chitin softened the polymer to an amorphous disordered state, which enabled the mixing with NaOH during a second ball milling step. The mixture of chitin and the base underwent a final aging step under high humidity to facilitate deacetylation, giving high-molecular weight chitosan. Similar to aforementioned polymerization methods, the milling media density was found to be highly influential during the polymer chain modification. By the use of stainless steel as milling material, a higher deacetylation efficiency was exhibited, however, the viscosity decreased in comparison to the application of zirconia as milling material, attributed to the increase of chain degradations due to the higher energy impact of the heavier milling material.
 | ||
| Fig. 11 Schematic mechanochemical transformation of chitin to chitosan. | ||
The mechanochemical amorphization of well-known polymers was additionally examined by Hirotsu and coworkers by ball milling of crystalline cellulose.203 Since the strong hydrogen bonds between the cellulose chains limit the processability in solution, the group utilized planetary ball milling to turn crystalline cellulose into an amorphous state by breaking the hydrogen bond network between the hydroxy groups. As a result, the free hydroxy groups can participate in chemical reactions more efficiently (Fig. 12).
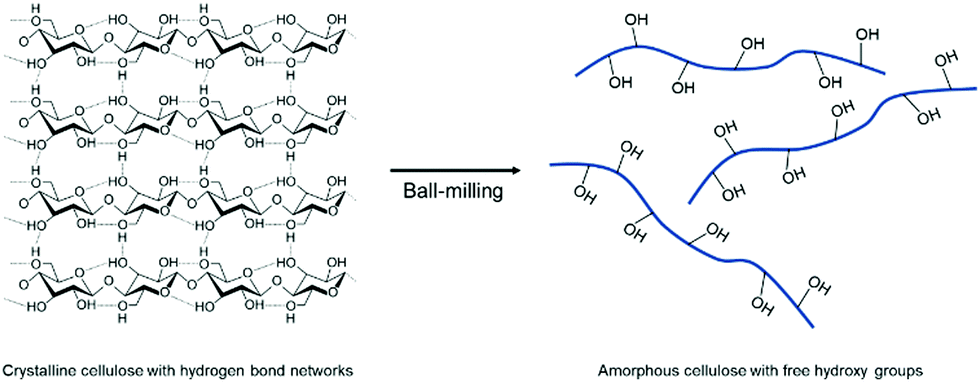 | ||
| Fig. 12 Phase transition and free hydroxy group generation by ball milling of crystalline cellulose. | ||
Based on this approach the modification of the polymers’ functional groups was possible in a further step. Moores and Friščić attempted the phosphorylation of milling induced amorphous cellulose by a mechanochemical functionalization with phosphorus pentoxide.204 To highlight the versatility of this mechanochemical post-polymerization modification, various polymers were brought to reaction with P2O5 by ball milling, including cellulose nanocrystals, poly(ethylene oxide), poly(vinyl alcohol), poly(vinyl chloride), and lignin. Although the approach enabled the circumvention of the conventional energy-intensive solution method, corrosive phosphoric pentoxide was needed to be applied for the mechanochemical process, which is a distinct drawback for a green synthesis procedure.
Another drawback of the mechanochemical modification of polymer functionalities is a lack of process control, as presented by Hirotsu and Qiu.205,206 By milling polypropylene, maleic anhydride, and initiator together, polypropylene macroradicals were generated. This was achieved by either direct mechanical chain degradation or by initiator-induced chain disproportionation, whereupon a reaction with maleic anhydride occurred, yielding a maleic anhydride grafted polypropylene (MAPP). Since the radical process is uncontrollable under ball milling conditions, the resulting functional polymer exhibited an undesirable high melt flow index, attributed to chain crosslinking reactions. Interestingly, the mechanochemically prepared MAPP showed a lower flow ability than those obtained in melt, thus less chain scission happened during ball milling.
Since uncontrollable conditions are undesirable, the Kim group reported the first regulated mechanochemical post-polymerization modification of the polymer main chain functionalities.127 Thereby, a quick condensation reaction between a model aldehyde-polymer, poly(4-vinyl benzaldehyde), and assorted amines or solid amine surrogates was promoted by ball milling (Fig. 13), resulting in a complete aldehyde conversion towards the imine within 30 minutes. This functionality modification showed no sign of uncontrolled side reactions and the molecular weight of the polymer increased gradually, while the dispersity remained unchanged. Interestingly, the reaction with an ionic substrate, ammonium carbamate, did not hamper the reaction rate regardless of the immiscibility of the reactants. In a further approach Kim and Kim additionally verified that the immiscibility is indeed not a limiting factor for mechanochemical post-polymerization modifications, as they achieved the conjugation of hydrophobic molecules and an ionic polymer, poorly soluble in organic solvents, by ball milling.207
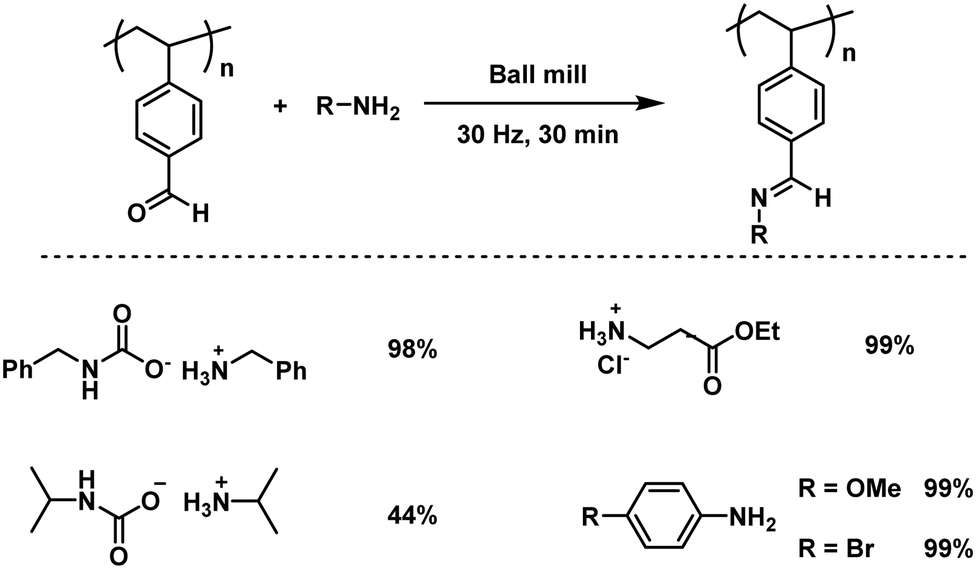 | ||
| Fig. 13 PPM of an aldehyde-polymer with amines induced by high-speed ball milling. | ||
In addition to the modification of the main chain functionalities, it is furthermore possible to modify the chain end functionalities of a polymer by mechanochemistry, even in spite of a low-end group concentration. Moores and Friščić realized the solid-state mechanochemical ω-functionalization of poly(ethylene glycol), by the activation of the terminal hydroxy group with a base (Fig. 14).208 Thereby, a series of end-functionalized PEGs was obtained with moderate to good yields without any solvent. The report used low molecular weight PEG (Mn = 750 and 2000 kDa), in which no degradation was anticipated.
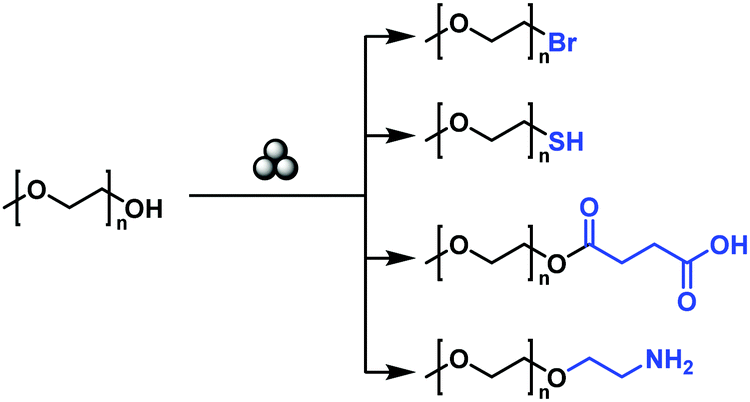 | ||
| Fig. 14 Mechanochemical ω-functionalisation of poly(ethylene glycol). | ||
Challenges and outlook
Carothers equation, describing the influence of monomer concentration on the polymerization efficiency, showcases why the solvent-free and thus highly concentrated environment of a ball mill is in principle well-suited for step growth polymerizations. Likewise, the possibility to form radicals during the mechanical impact event of milling showcases that this technique is also well-suited for initiator-free chain-growth polymerizations. Nevertheless, mechanochemical polymerization is still in its infancy and there are many aspects urging for improvement. For example, polymerization kinetics established for classical homogeneous systems are not comparable to what is observed in solid-state. The fast initiation over propagation is a criterion for narrow molar mass distribution, but the physical blending of initiator and monomer brings an inherent delay and unevenness.Additionally, a tailored polymer synthesis is urgently necessary, as in many examples chain growth competes with chain degradation. This results in a dilemma between the utilization of high impact conditions for higher yields and faster reaction kinetics and of low impact conditions for suppressing chain degradation. The lack of understanding of the mechanism of chain degradation led to a trial and error-based research. In this context, the impact of LAG, temperature, and post-synthesis aging for instance is not understood so far.
Moreover, the scalability is a challenging issue for future commercialization. For industrial applications, continuous processes are preferred to enable the synthesis of kilograms or even tons per hour. However, the tool that was mostly presented herein, ball milling, generally could not meet this criterion. Therefore, the engineering of ball mills or the transfer to other mechanochemical methods, such as to extrusion, should be advanced further.
Another drawback is the reproducibility of mechanochemical reaction protocols, as it is often depending on the use of the same or similar equipment. Problems usually arise if rather exotic devices or home-built vessels are used. Lately, we have observed a trend that many mechanochemical laboratories use the same mills and thus this issue should soon be behind us.
Although, there are still distinct deficiencies of mechanochemical polymerization reactions that need to be circumvented in the future, the advantages of solvent-free synthesis protocols, such as the avoidance of toxic solvents and the circumvention of solubility-based limitations, render the whole area as highly important for future applications. We are certain that other common polymerization techniques, such as polyaddition reactions, will soon be transferred into solvent-free mechanochemical protocols. One interesting example would be the establishment of controlled/living polymerizations, which is highly required as it allows the block copolymer synthesis by sequential monomer addition.
Highlighting this, there is plenty of room for new reactions to be transformed into a mechanochemical process, but even more desirable is a deeper understanding of the underlying principles of mechanochemical polymerization. Moreover, it is highly necessary to monitor and follow mechanochemical polymerization reactions in situ. Modern in situ techniques based on Raman spectroscopy or XRD, which are coupled directly into the vibrating mill will help to elucidate this missing information in the future.
Mechanochemical synthesis of porous polymers
Porous polymers have diverse applications in fields of industrial relevance209,210 such as catalysis,211–214 molecular separation,215,216 gas storage,217–222 and energy storage in batteries and supercapacitors.223,224 These materials feature large pore volumes and high specific surface areas and can be classified as macroporous (pore diameter > 50 nm), mesoporous (pore diameter = 2–50 nm), or microporous (pore diameter < 2 nm).225,226 Categorisation by material classes opens up a broad range of various polymer groups such as porous coordination polymers (PCPs)227–229 or porous organic polymers (POPs).230,231While many classical solvent-based syntheses require expensive starting materials or catalysts,232,233 mechanochemistry obviates the need for solubilising groups and allows one to avoid low DPs due to the rapid precipitation of products.234 The need to reduce solvent waste has inspired several studies on the general mechanochemical synthesis of porous materials235,236 such as porous carbons,237–248 porous coordination polymers249,250 (e.g., metal–organic frameworks (MOFs)251–255), and purely organic polymers.53,102,256–260
Up to now, porous polymers have been exclusively synthesised by step-growth polymerisation, while particularly using polycondensation reactions. Although this allows the following chapter to be integrated into the previous one, the available reports are much more numerous than in the case of linear polymers and often have a different focus because of the difference in the relevant material properties. For instance, the specific surface area, pore size, and pore volume are more important properties than DP or dispersity degree. Moreover, as long as the respective polymers are insoluble in common organic solvents, even a lower degree of polymerization might be favourable in order to ensure a better pore accessibility. Herein the focus is therefore shifted from the degree of polymerization to the specific surface areas of the porous polymers. As coordination polymers such as MOFs have been discussed in detail in previous reviews, we exclusively focus on porous polymers built from covalently bonded monomers and identify three major techniques to mechanochemically build up these polymers, namely Scholl polymerisation, Friedel–Crafts polymerisation, and imine/enamine/amide formation.
Scholl polymerisation
The Scholl polymerization is a step-growth polymerization, in which aromatic compounds are coupled in the presence of (catalytic) amounts of a Lewis acid and an oxidant. Thereby, the para position of the respective monomers is favoured, firstly leading to the formation of smaller oligomers that react with each other to form an interconnected network. During this oxidative coupling reaction, the Lewis acid enables the elimination of hydrogen that is released as HCl. While this process takes long reaction times of up to 48 h and the necessity of often hazardous solvents in the classical solution based synthesis,261–264 mechanochemistry surpasses these drawbacks, rendering the approach extremely versatile to cross-link various monomers within very short reaction times of down to 5 minutes.The first proposal in this regard was presented by the group of Dai, adapting the mechanochemical oxidative coupling procedure towards conjugated nanoporous polymers (CNPs).256 In the given case various carbazole-containing building blocks were polymerized in a vibrational ball mill within 30 min and the building block geometry had a direct effect onto the SSABET of the polymers (Fig. 15). The most important detail, however, is the utilisation of anhydrous FeCl3 instead of FeCl3·6H2O as both a Lewis acid and an oxidant, which significantly simplified the synthetic procedure. The number of FeCl3 equivalents strongly influenced the reaction outcome. For instance, the polymer yield could be increased from 54% to 94% by increasing the amount of Lewis acid from 1.25 eq. to 5 eq. A similar result was obtained when the same group advanced this protocol to soluble nanoporous networks,265 starting from a ladder-like monomer structure (3,3,3′,3′-tetramethyl-2,2′,3,3′-tetrahydro-1,1′-spirobi[indene]-6,6′-diol) (Fig. 16). The synthesis was carried out with 1–4 mass equivalents of FeCl3 in a vibrational ball mill, and the increase in FeCl3 quantity was accompanied by an increased yield and a decreased solubility of the generated polymers in common organic solvents. Independently, Borchardt and coworkers came to the same conclusion when working on mechanically produced microporous thiophene polymers (Fig. 17).102 Despite facilitating high yields, large FeCl3 loadings strongly affected the SSABET of the polymer, with specific surface areas of almost 1900 m2 g−1 achieved for high-speed ball milling with 12 eq. of FeCl3. This is particularly surprising, as the reference polymer prepared using solution-phase chemistry featured a significantly lower SSABET. The authors hypothesised that the instant precipitation of the product from the reaction mixture, which occurs in solution but not during mechanochemical processing, is responsible for this significant difference.266 Subsequent publications point towards the same, i.e., confirm the important effect of FeCl3 loading on the outcome of mechanochemical Scholl reactions. A more detailed study of reaction parameters using qualitative and quantitative tools such as the design of experiments (Fig. 17) helped to identify crucial factors of influence. The investigation of the relations and cross-relations of reaction parameters such as milling time, ball-to-powder ratio, ball size, and milling frequency has shown that only FeCl3 loading and milling frequency have a significant impact on the reaction. Specifically, increased milling frequency induced pore collapse to decrease SSABET, which was verified in a subsequent study on the Scholl polymerisation of 1,3,5-triphenylbenzene by the same group.53 In this work, a 5 min reaction afforded a polymer with a surface area of >1000 m2 g−1 in >99% yield. The insufficiently high impact energy observed at low milling frequencies, short milling times, and small milling material densities or ball sizes results in poor yields and modest specific surface areas. Despite providing quantitative yields, the opposite parameter combination was found to be similarly undesirable, as excessive energy input devastated internal porosity. Although the origin of the profound impact of the oxidant remains unclear, two factors might play a role. First, FeCl3 is very hygroscopic, and as many experiments were conducted under ambient conditions, moisture was rapidly drawn from the air. If only a small excess of the oxidant is used in the reaction, this might lead to deactivation below the required stoichiometry or to a pseudo-LAG experiment with an excessively high η.
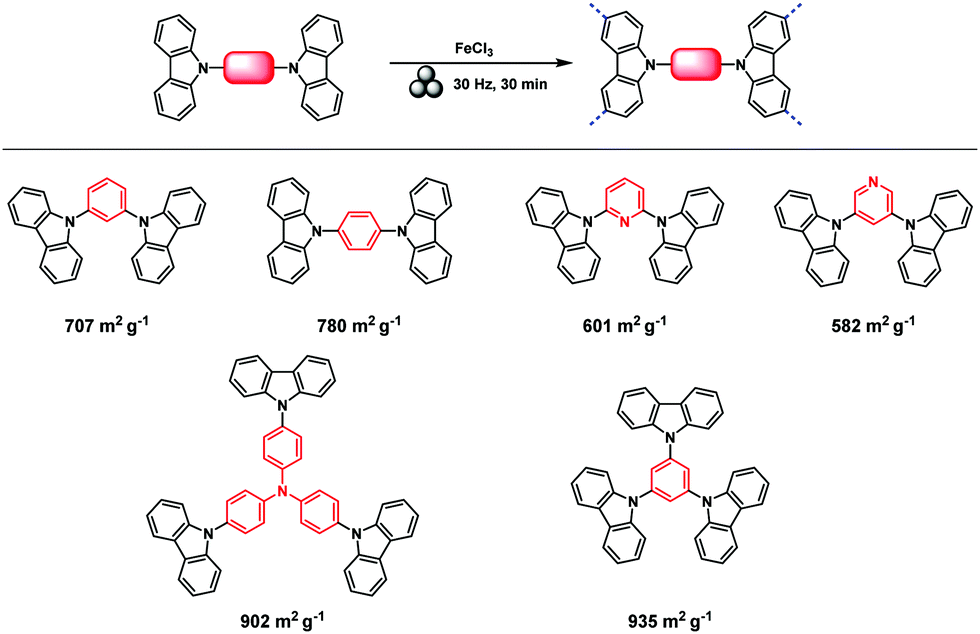 | ||
| Fig. 15 Top: Scholl model reaction for the generation of nanoporous conjugated polycarbazoles by ball milling. Bottom: Different building blocks and SSABET values of the corresponding polymers. Adapted with permission from ref. 256. Copyright (2017). The American Chemical Society. | ||
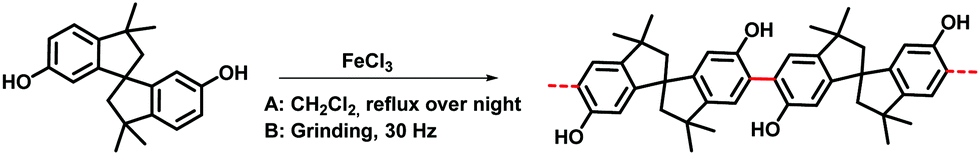 | ||
| Fig. 16 Classical (A) and mechanochemical (B) oxidative coupling polymerisation of 3,3,3′,3′-tetramethyl-2,2′,3,3′-tetrahydro-1,1′-spirobi[indene]-6,6′-diol. Reprinted with permission from ref. 265. | ||
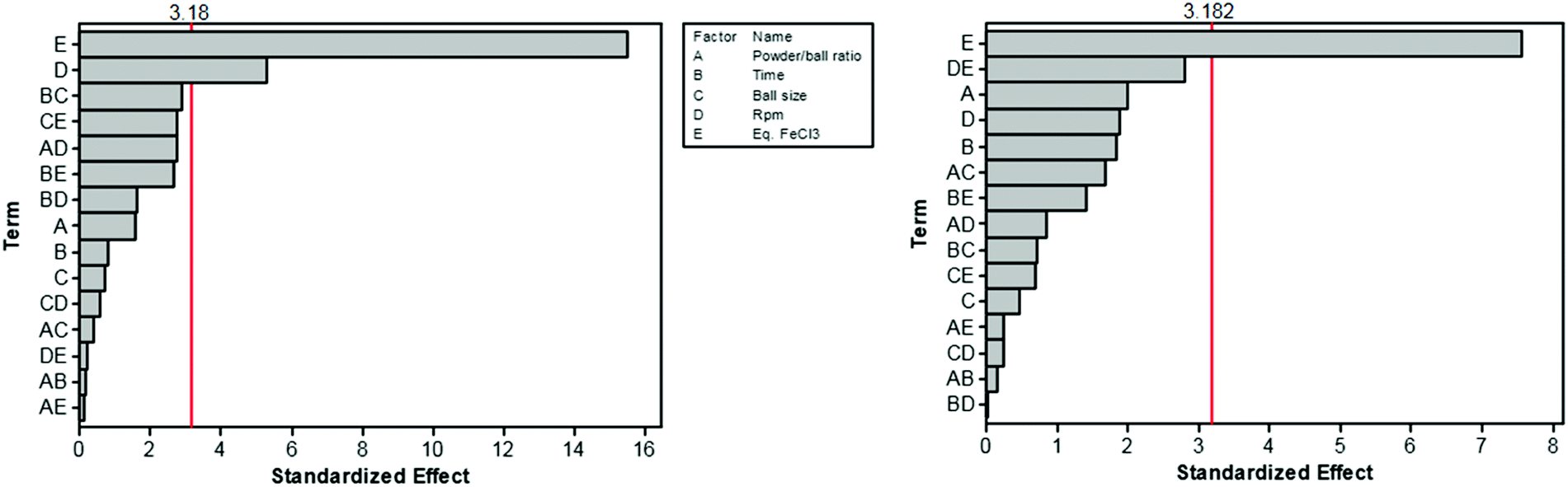 | ||
| Fig. 17 Design of experiment of the mechanochemical Scholl reaction yielding a microporous thiophene polymer. The most important influence onto the surface area (left) and the yield (right) are the eq. FeCl3 (E) and the frequency (D). Adapted from ref. 102 with permission from The Royal Society of Chemistry. | ||
Second, FeCl3 is often simultaneously used as a bulking material and thus influences the rheology of the milling process.
One of the key questions relates to the origin of porosity in the mechanochemical Scholl reaction producing porous polymers. While conventional solution-based syntheses often claim the monomer molecular structure, size, and geometry to be responsible for porosity (molecular design concept), we believe that this is rather unlikely for mechanochemical approaches, particularly for the Scholl reaction. Typically, monomers yielding highly porous polymers in solution do not do so when used in mechanochemical synthesis, and vice versa, the porosity of mechanochemically produced polymers can exceed that of the same polymers synthesised in solution. We believe that the reaction kinetics, cross-linking, LAG, and the atmosphere within the milling vessel significantly influence polymerisation and, hence, the porosity of the produced polymer. As mechanochemical reactions are conducted in sealed milling vessels, reactions producing gaseous by-products, e.g., HCl for the Scholl reaction, build up gas pressure in the vessel. Borchardt and coworkers measured a pressure increase of 12 bar using gas pressure and temperature sensors (GTM system) integrated into the vessel cap and found that the emerging HCl is capable of inflating the polymer and is therefore responsible for high SSABET (Fig. 18).53 In this context, LAG, that is the addition of small quantities of a liquid, impacts the mechanochemical Scholl polymerisation in three ways. First, if the liquid is volatile, it can contribute to the build-up of internal pressure; second, if the liquid is halogenated, it can react with FeCl3 to form highly reactive intermediates and thus accelerate HCl formation; and third, halogenated liquids can serve as cross-linkers in Friedel–Crafts-type reactions. In any case, the amount of added liquid is very crucial, as revealed by the simultaneous studies of Horike and coworkers and Borchardt and coworkers, who probed the polymerisation of 1,3,5-triphenylbenzene. Both groups synthesised highly porous polymers in as little as 5 min by neat grinding; however, the addition of MeOH led to pore collapse in one case and to increased porosity in the other case,53,267 which was ascribed to differences in the amount of used liquid. Whereas the Borchardt group conducted LAG experiments with 1 mL of MeOH (η = 0.25 μL mg−1), Horike and coworkers determined the optimal amount of MeOH as 0.1–0.3 mL (η = 0.03–0.08 μL mg−1). This substantiates the enormous importance and sensitivity of LAG in mechanochemical approaches and shows that the liquid additives should not be treated as conventional solvents, but rather as catalysts that exert strong influence in the smallest quantities.
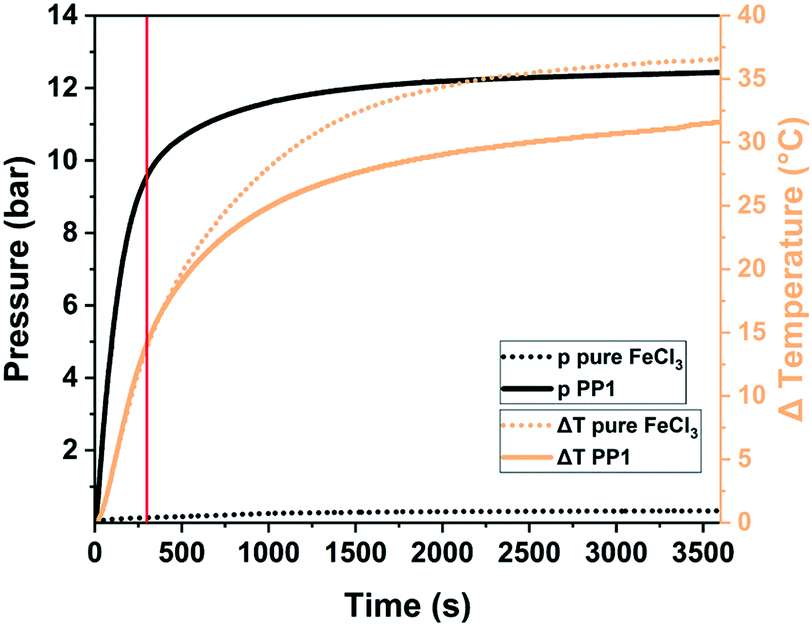 | ||
| Fig. 18 Pressure (solid black) and temperature (solid orange) evolution during the mechanochemical Scholl reaction of 1,3,5-triphenylbenzene (PP1). The pressure and temperature changes for the milling of pure FeCl3 are shown as dotted lines. The usual synthesis time of 5 min is marked by a solid red line. Reproduced with permission from ref. 53. Copyright (2020). The Royal Society of Chemistry. | ||
In summary, the mechanochemical Scholl reaction can be carried out over a period as small as 5 min in the absence of large amounts of toxic solvents. The yield and specific surface area of the synthesised polymers are strongly dependent on the amount of Lewis acid, milling frequency, and amount of liquid used for LAG experiments. Special scrutiny, however, has to be paid to the evaluation of the reaction parameters, as even the smallest changes can make the difference between low and high yield and no or high porosity.
Friedel–Crafts polymerisation
The Friedel–Crafts polymerization is a step-growth polymerization that is driven by a non-directed electrophilic aromatic substitution. Similar to the Scholl polymerization, a catalytic amount of Lewis acid, but in contrast no additional oxidant, is required for the C–C coupling. During the polymerization, the Lewis acid creates an electrophilic reagent (either an external cross-linker or an intramolecular group) that is capable to break the aromaticity of the targeted ring. After the release of a proton, often as HCl, the rearomatization takes place, whereby oligomers are formed that interconnect towards a porous network. Conventional Friedel–Crafts alkylation of porous organic polymers usually requires long heating times of up to 36 h,268 gel formation,269 or prolonged stirring (up to 48 h) in hazardous solvents such as dichloromethane, chloroform, or 1,2-dichloroethane.270 These deficiencies can be circumvented using a mechanochemical approach drastically decreasing the synthesis time down to 35 minutes. Despite the similarities between both reactions, the mechanochemical Friedel–Crafts reaction yields porous polymers with higher surface areas than Scholl coupling under similar conditions.53,267,271 While the mechanochemical Scholl reaction of various monomers leads to the formation of conjugated porous polymers, the additional crosslinker induced by the Friedel–Crafts polymerization breaks the conjugation and functions as a spacer between the monomers, allowing a higher flexibility during network formation, accompanied by an enhanced SSABET of the resulting polymer. Recently, the Borchardt group has compared the Scholl and Friedel–Crafts polymerisations of 1,3,5-triphenylbenzene. While the neat-grinding Scholl reaction yielded a polymer with an SSABET of 658 m2 g−1,53 the introduction of a cross-linker allowed the synthesis of highly flexible polymers with SSABET values of up to 1670 m2 g−1.271 Nevertheless, the surface area of polymers generated by the mechanochemical Friedel–Crafts reaction was highly dependent on the monomer building blocks used, as investigated by the group of Horike (Fig. 19).267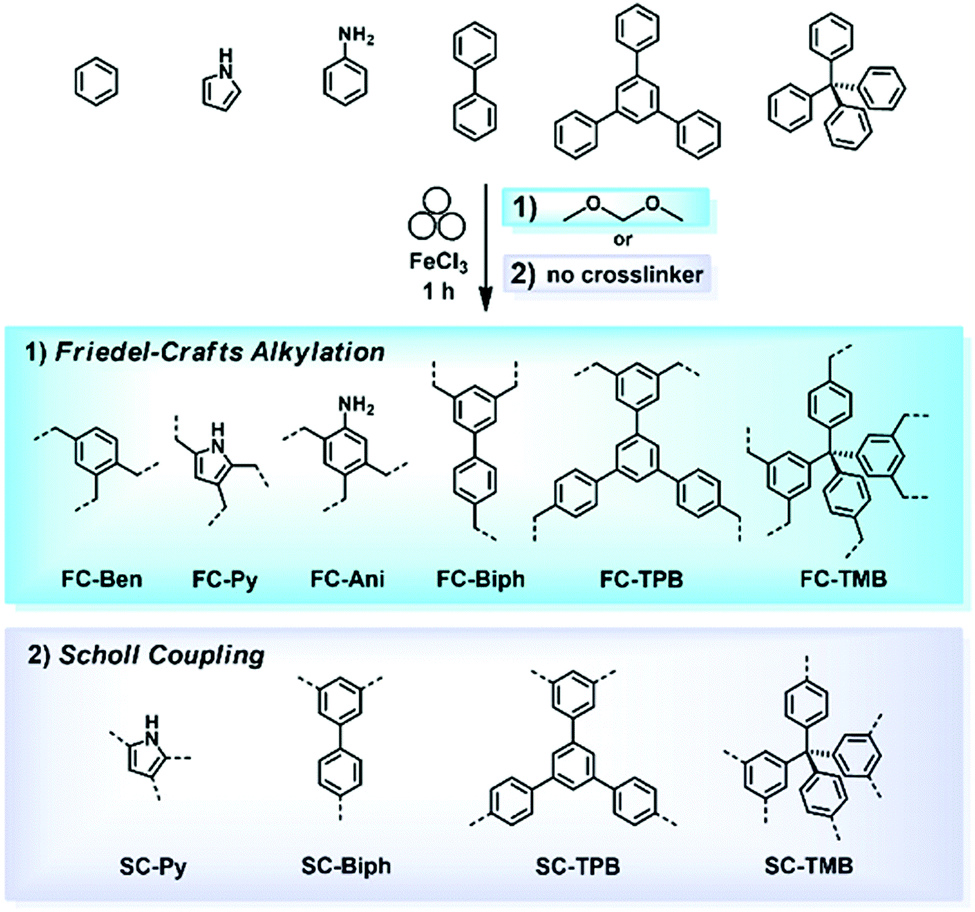 | ||
| Fig. 19 Mechanochemical generation of hyper-cross-linked polymers by (1) Friedel–Crafts alkylation and (2) the Scholl reaction. All products were obtained by ball milling for 1 h in the presence of FeCl3 as a Lewis acid. Reprinted with permission from ref. 267. Copyright (2020). The American Chemical Society. | ||
Friedel–Crafts alkylations are not limited to the utilisation of external cross-linkers, and intramolecular groups can also function as cross-coupling reagents, as presented by Borchardt and coworkers.89 In this case, 4,4′-bis(chloromethyl)-1,1′-biphenyl (BCMCP) was milled with anhydrous FeCl3 in a planetary ball mill to yield a microporous polymer after 35 min. Upon evaluating the milling parameters, the group identified the differences between mechanochemical Scholl and Friedel–Crafts reactions. Whereas the SSABET of polymers produced by the Scholl reaction mainly depended on the amount of Lewis acid equivalents, this parameter was of minor importance for the Friedel–Crafts reaction. Similarly, the reaction time hardly influenced polymer porosity. The strongest effect was exerted by grinding frequency.
Analogous to the Scholl reaction, excessively low frequencies led to an incomplete reaction and thus to the generation of non-porous polymers or oligomers, while excessively high frequencies caused porosity degradation. Furthermore, SSABET could be further increased using LAG, as already observed for the Scholl reaction. Initially, the boiling point of the added liquid seemed to be correlated with the obtained specific surface area, which suggested that the liquid might serve as a porogene. Although recent investigations show that this simple correlation does not hold,53 the advanced synthesis approach allowed SSABET to be increased to 1720 m2 g−1, which exceeded the value of 1450 m2 g−1 obtained for the solution-based reference (Fig. 20).89
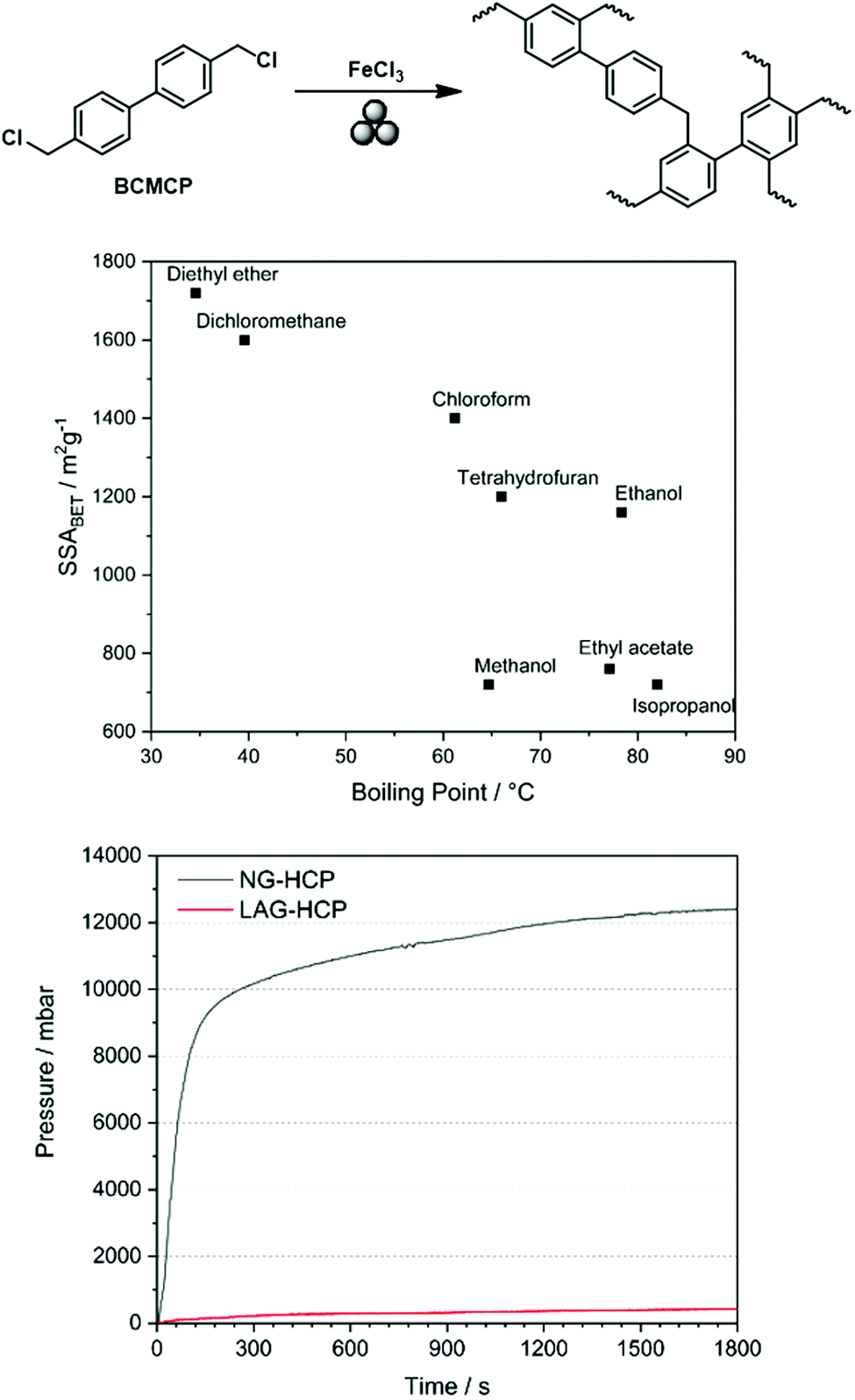 | ||
| Fig. 20 Top: Scheme of mechanochemical Friedel–Crafts alkylation yielding a hyper-cross-linked polymer. Middle: Correlation between the boiling point of the liquid added to the milling jar for LAG and the SSABET of the generated polymer. Bottom: Pressure development during neat grinding-based (black) and LAG-based (red) syntheses of hyper-cross-linked polymers. Reprinted with permission from ref. 89. Copyright (2019). Beilstein J. Org. Chem. | ||
In 2019, the group of Zhou proved the versatility of mechanochemical Friedel–Crafts alkylation by generating a hyper-cross-linked polymer from 1,1,2,2-tetraphenylethene as the starting material.272 As described above,89 anhydrous FeCl3 was added to the milling jar to promote cross-coupling, and milling was performed in a planetary ball mill at 500 rpm for 35 min.
The mechanochemical Friedel–Crafts polymerisation can furthermore be used to synthesise covalent triazine frameworks, a class of porous polymers typically produced using complex and time-consuming pathways relying on ionothermal trimerisation273 or the utilisation of trifluoromethanesulfonic acid as a catalyst.274,275 Mechanochemical synthesis allows many of these issues to be circumvented (Fig. 21).276 Borchardt and coworkers linked various monomers such as carbazole, benzene, naphthalene, anthracene, and 1,3,5-triphenylbenzene with a triazine node (cyanuric chloride) in the presence of AlCl3 and ZnCl2. A yield of >90% was achieved after 1 h of milling for the carbazole system, and surface areas as high as 590 m2 g−1 were realised.
 | ||
| Fig. 21 Mechanochemical Friedel–Crafts linkage of various aromatic monomers and a triazine node to yield porous covalent triazine frameworks. Reprinted with permission from ref. 276. Copyright (2017). Wiley-VCH. | ||
The characterisation of mechanochemically derived porous polymers is challenging in two ways. First, the reaction is conducted in dense milling vessels rotating or vibrating at frequencies of several hundred times per minute, which precludes the application of most established in situ analytical approaches. Fortunately, several groups have developed advanced in situ analytics, particularly for milling processes. In principle, these methods are based on in situ Raman spectroscopy, in situ XRD, and in situ temperature/pressure sensing. The latter is a helpful tool to track mechanochemical Friedel–Crafts reactions (Fig. 20 bottom). For the abovementioned reaction, a steep increase in pressure is related to the evolution of HCl gas, and thus, the initiation of the polymerisation reaction. In view of the exothermic nature of polymerisation, a temperature peak at exactly the same time indicates reaction initiation. The second challenge of porous polymer characterisation relates to the insolubility and amorphous structure of these polymers, which precludes characterisation by solution-phase NMR spectroscopy and XRD. Borchardt and coworkers therefore established a comprehensive characterisation method based on dynamic nuclear polarisation (DNP) NMR spectroscopy, which relies on NMR signal amplification by several orders of magnitude via the transfer of the electron spin of a polarising agent in the pores of the polymer to the nuclear spin of the polymer backbone atoms.277 Thus, DNP-NMR spectroscopy allows one to elucidate the monomer connectivity, possible cross-linkages, and the atomic environment of mechanically produced polymers. Although the whole process is rather expensive and requires a detailed radical screening to be performed prior to analysis, DNP-NMR spectroscopy is a valuable characterisation method for amorphous and insoluble porous polymers that, compared to classical solid-state NMR spectroscopy, is much faster, enables access to multiple heteroatoms, and features a better signal-to-noise ratio.
In summary, although the mechanochemical Friedel–Crafts reaction exhibits disadvantages, such as the formation of HCl, which can corrode the grinding jars and the mill, this solvent-free approach can outperform its solution-based analogue in terms of synthesis time and generated surface area. Furthermore, the above reaction is not as vulnerable to grinding parameter changes as the Scholl reaction and is therefore considerably more reliable for the synthesis of porous polymers. Interestingly, the Friedel–Crafts reaction is suitable not only for the production of hyper-cross-linked polymers, but also for the formation of covalent triazine frameworks.
Imine, enamine, and amide formation
The formation of imines, enamines, and amides plays a key role in the generation of porous polymers and therefore forms a separate area of mechanochemical polycondensation reactions. Typically, these step-growth polymerisations involve monomers with nitrogen-containing groups, such as amines, and monomers with carbonyl functionalities, such as aldehydes, ketones, or carbonyl chlorides. In contrast to the polymerisation reactions mentioned previously, HCl and water can also be released during condensation. Imine, enamine, and amide formations are commonly used for the synthesis of porous polymers known as covalent organic frameworks (COFs).278 These frameworks can either feature 2D (first reported in 2005)279 or 3D (first reported in 2007)280 topologies and usually contain light elements such as boron, carbon, nitrogen, and oxygen.280 In contrast to the aforementioned porous polymer types, COFs exhibit crystalline and well-ordered structures, which require the synthesis under harsh reaction conditions in many cases.281To circumvent these issues and develop a fast and simple synthetic procedure, the group of Banerjee showcased the mechanochemical COF formation in their pioneering study in 2013 (Fig. 22).282 In this study, three isoreticular COFs were constructed by manual grinding in a mortar-and-pestle setup under ambient conditions. The advantages of this approach are the avoidance of solvents and a drastic decrease in synthesis time from days to 45 min. However, one disadvantage is the need for a long and constant energy input by manual grinding. Although several imines, enamines, and amides have been prepared in a mortar-and-pestle setup,64,283,284 a more elegant approach is ball milling,285,286 as it guarantees a certain reproducibility and does not rely on the constant energy input of the human operator.
 | ||
| Fig. 22 Mechanochemical Schiff-base reaction induced by manual grinding and signified by a colour change from orange over yellow (oligomer) towards red (polymer). Reprinted with permission from ref. 282. Copyright (2013). The American Chemical Society. | ||
One example is the generation of a bipyridine-functionalised COF in a high-speed ball mill.287 For direct comparison, a solvothermal procedure was also established, requiring harsh conditions such as ultrasonication, freeze–pumping, and heating for 72 h in a vacuum-sealed tube. In contrast, the mechanochemical synthesis was accomplished in a rapid (90 min, 30 Hz) and environmentally benign fashion under ambient conditions, circumventing the generation of solvent waste and the use of long heating times.287
A certain disadvantage of ball milling procedures is the limited batch size, which results in low throughput. The need for solvent-free scalability motivated Banerjee to transfer the well-established COF synthesis282 to a twin-screw extruder (Fig. 23).288
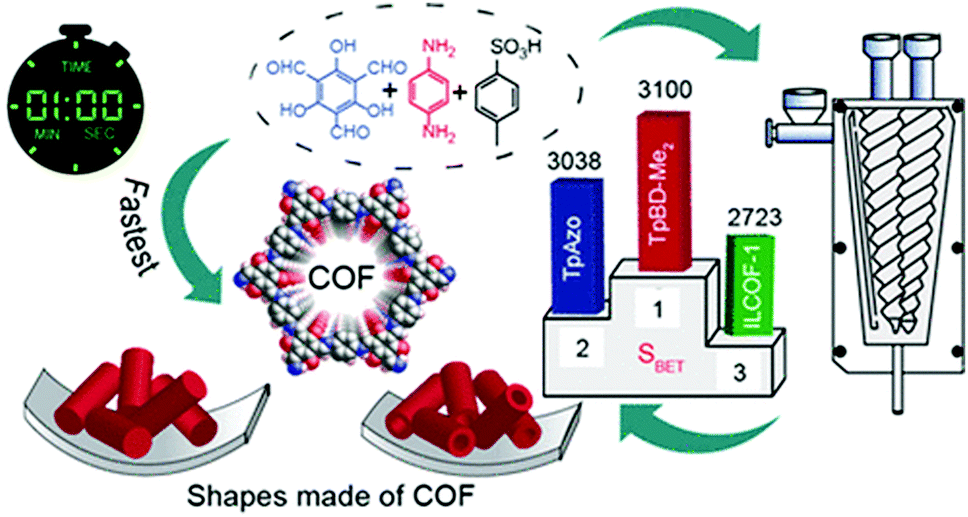 | ||
| Fig. 23 Molecular organisation concept based on the utilisation of PTSA during the mechanochemical reaction. This concept enabled the synthesis of various COFs with high specific surface areas (middle) as well as continuous synthesis (right). Reprinted with permission from ref. 288. Copyright (2017). The American Chemical Society. | ||
Although this approach might seem questionable, as the COF synthesis relies on reversible covalent bond formation and, hence, on prolonged synthesis times, the introduction of p-toluenesulfonic acid (PTSA) enabled molecular organisation and drastically reduced the synthesis time. This concept was used to realise the large-scale (several kg h−1) formation of COFs via extrusion for 5–10 min followed by heating at 170 °C for 60 s.288
The utilization of PTSA during the COF synthesis is not solely important to reduce the long reaction times. The acid furthermore acts as a template to achieve the formation of highly crystalline frameworks. Owing to the random displacement of the mechanochemically exfoliated layers, 2D COFs often exhibit a graphene-like layered structure with low-range crystallinity.289 Although this mechanochemical exfoliation is required in certain cases, e.g., for the mechanochemical delamination-based production of covalent organic nanosheets,290,291 it generally poses a threat to the synthesis of highly crystalline materials. However, in the presence of PTSA, the protonation of primary amines is followed by the formation of PTSA-amine salts, which act as a template to minimise network defects.
In addition to the template method, the addition of small quantities of liquid was found to have a positive impact on the crystallinity of mechanochemically synthesised COFs.292 The mechanochemical reactions of 1,3,5-triformylphloroglucinol and melamine was performed under various conditions, e.g., using neat grinding, LAG, and PTSA-assisted grinding. While neat grinding and ball milling with 1 mL of solvent resulted in the formation of COFs with poor crystallinity, the addition of PTSA or 3 mL of solvent generated COFs exhibiting high crystallinity and a well-defined thread- or ribbon-like morphology, respectively.292
Furthermore, the LAG approach facilitated COF formation even in a mortar-and-pestle setup.284 The related study identified enol-to-keto tautomerism as a key factor responsible for high stability, which was underlined by the fact that the prolonged water or acid treatment of the β-ketoenamine COF did not affect its crystallinity, while the imine COF was unstable even in a humid environment.284
In addition to having low crystallinity, mechanochemically generated COFs often exhibit lower porosity than their classically synthesised counterparts.287,293
Although the use of LAG increased the crystallinity of the synthesised polymers, this effect was not observed for porosity. The liquid-assisted manual grinding of imine, β-ketoimine, and imine-linked COFs (Fig. 24) resulted in the formation of polymers with low BET surface areas and reversible type II isotherms.284 The authors suggested that the mechanochemical synthesis procedure leads to thin-layered structures, hindering a long range pore formation.
 | ||
| Fig. 24 Schematic manual grinding-based LAG synthesis of (A) hexagonal and (B) tetragonal COFs as well as (C) products of the Schiff-base reaction and their respective bonding motifs (blue circles). Reproduced/adapted from ref. 284. Copyright (2014). The Royal Society of Chemistry. | ||
Despite this, the addition of PTSA not only drastically increased the crystallinity of the generated COFs, but also had a similar effect on polymer porosity.288 The template method enabled a 2–3-fold increase in BET surface area in comparison to the solvothermal methods, which was attributed to long-range porosity and ordered pore channels obtained in the former case.288
In addition to this, the group of Dai showed that the structure of the monomer is an important factor to achieve a decent porosity of the resulting polymer by the utilization of pillar[5]quinone, a monomer exhibiting an intrinsic cavity.294
Recently, the Borchardt group investigated the mechanochemical synthesis of PI-COFs with a special focus on the “shaking vs. baking” question.199 Within the two-step condensation reaction, the first step (amic acid formation) was found to proceed readily under ball milling conditions, attributed to the five-ring opening reaction exhibiting a negative enthalpy. However, the second step (imide formation) could solely be achieved by a 30 minutes’ heat treatment, although the Gibbs free energy was found to be negative for the ball milling reaction. The group calculated that the condensation is entropically unfavored, since the generated water is not capable to leave the sealed milling vessel. By a thermal treatment in an open vessel, the water can leave the reaction mixture, shifting the equilibrium towards the imide formation. This result is of high importance as it enables a deeper understanding of the question why some mechanochemical reactions proceed readily, while others require an additional heating step.
In summary, mechanochemical imine, enamine, and amide formations are widely used to generate COFs but are also suitable for the formation of other porous polymers. One of the biggest problems faced by the synthesis of various COFs is their lower crystallinity and porosity compared to those of their counterparts prepared by solution-based synthesis. Furthermore, especially for the synthesis of 2D COFs, special care has to be taken in the parameter evaluation, as already manual grinding might lead to a mechanochemically exfoliation of the layers. Nevertheless, molecular organisation concepts such as PTSA-assisted grinding allow one to minimise network defects and counteract low crystallinities and porosities. Furthermore, these concepts allow reaction upscaling, which renders the entire process highly interesting for the industry. In general, all three mechanochemical polymerisation reactions are sustainable alternatives to classical solvent-based syntheses, allowing the rapid and inexpensive generation of various porous polymers.
Other polycondensation reactions
In addition to the three main synthetic strategies mentioned above, several other polycondensation-based pathways have been reported.In 2015, Dai and coworkers295 reported a base-catalysed polymerisation reaction between tetrahydroxy compounds and tetrafluoro compounds induced by high-speed ball milling and affording polymers with oxygen bridges. The obtained polymers of intrinsic microporosity (PIM) showed specific surface areas of >500 m2 g−1 and featured higher molecular weights and narrower molecular weight distributions than their counterparts prepared in solution, which was attributed to the fast precipitation of the latter in the classical synthesis. The same group transferred this concept of hydroxyl-fluoro-group coupling to charged porous polymers (CPPs) (Fig. 25)296 and realised cationic or anionic (depending on the nature of the fluorine-activated ionic monomer) CPPs. It is important to mention that depending on the backbone of the respective CPP, this mechanochemical concept drastically reduces the reaction time (60–90 min) as compared to the solvent-based process (dozens of hours). The cationic CPP polymer was further modified by a simple ion-exchange method to generate CPPs with phenolic or proline counter-anions for capturing and recovering SO2 from the atmosphere.
 | ||
| Fig. 25 Mechanochemical polycondensation affording charged porous polymers and enabling the synthesis of anionic (bottom) and cationic (top) polymers. Reprinted with permission from ref. 296. Copyright (2015). Wiley-VCH. | ||
Another concept of porous polymer synthesis relies on cross-coupling reactions. Borchardt and coworkers synthesised hyperbranched polyphenylenes90via the Suzuki polymerisation of 3,5-dibromophenylboronic acid. The Pd-catalysed reactions of arylboronic acids and aryl halides have been proven to afford linear polyphenylenes. The introduction of branching points (Fig. 26) via an A2B1-type monomer provided access to a mesoporous hyperbranched polymer, which was obtained in up to 84% yield and featured a branching degree of 0.74, which is close to the literature value of 0.7.297
 | ||
| Fig. 26 (A) Mechanochemical Suzuki polycondensation affording hyperbranched polyphenylene. (B) Infrared spectra of the monomer (9) and the polymer (10) confirming the polymerisation. (C) Thermal analysis of polymers generated by milling for 30 min (green) and 120 min (orange). Reproduced from ref. 90. Copyright (2017). The Royal Society of Chemistry. | ||
A recent example is the mechanochemical Ullmann cross-coupling of electron-deficient aromatic monomers containing C–X bonds.298 This approach involves the dehalogenation of aryl halides with Mg followed by the formation of a Grignard-type intermediate and thus circumvents the need for electron-rich aromatic building blocks and potential methylene cross-linking, which otherwise limits the aromaticity and conductivity of the porous polymers. As a result, the aromatic ring-knitting reaction enables the solvent-free and straightforward synthesis of prospective electrode materials and illustrates the possibility of using various reaction types for the mechanochemical synthesis of porous polymers.298
Challenges and outlook
Although the mechanochemical generation of porous polymers is a versatile and sustainable alternative to the classical solvent-based approach, it exhibits certain drawbacks. One of the most prominent disadvantages is the degradation of porosity due to prolonged milling times or very high milling frequencies. Additionally, the milling material density and ball size can also have a significant impact in this regard. In the case of excessive energy input due to any of these parameters, the as-synthesised polymers may collapse because of mechanical degradation, which leads to a deterioration of the polymer network, the formation of oligomers, and the associated SSABET decline. Moreover, these parameters also determine the abrasion degree of the milling material. During mechanochemical synthesis, debris produced by excessive abrasion might not only contaminate the polymer, but also clog its pores to invalidate the results of surface area measurements. Especially for Scholl and Friedel–Crafts polymerisations, one needs to consider the formation of HCl as a by-product, as it may corrode not only the beakers, but also the mill itself. Furthermore, the formation of HCl leads to a large pressure increase inside the milling vessel, which results in the need for appropriately sealed milling beakers and limits the choice of milling devices as well as upscaling possibilities.Although some of these disadvantages require a longer evaluation of the experimental parameters to be optimised or cannot be avoided at all, the advantages of the mechanochemical synthesis of porous polymers over the classical solvent-based approach generally overweigh the related disadvantages. This is not only due to the significantly shorter reaction times, the avoidance of expensive catalysts and starting materials, and the circumvention of the premature precipitation of polymers from the reaction solution, but also due to the avoidance of large quantities of toxic solvent waste, which drastically enhances the green metrics of the porous polymers (see subsequent section for details).
Until now, only step-growth polymerisation has been used in the synthesis of porous polymers. In general, these approaches are merely a copy of the protocols used in solution. In the future, we believe that hyperbranched polymers will be easily accessed by chain-growth polymerisation. As it has been shown that order and crystallinity are not a prerequisite for the successful application of such materials, the indirect growth of such systems is not a problem. This is especially true because highly ordered structures are difficult to obtain without the reversibility and long reaction times provided by solution syntheses.
However, this is a problem for the characterisation of materials prepared by mechanochemistry, which are generally insoluble, have low crystallinities, and can only be characterised by solid-state methods. However, the latter methods have improved over the years, and access to DNP-NMR spectroscopy or XPS is becoming easier for scientists in all disciplines.
Furthermore, linear and porous polymer communities often use different terminologies and adhere to different standards. As mechanochemical polymerisation seems to readily produce insoluble linear polymers, these communities should come together and establish a common language.
Green metrics discussion
In the previous chapters, we described the differences between classical and mechanochemical syntheses based on reaction times, on the hazardous potential of the solvents or on the need for a high energy demand due to heating. However, these concepts are relatively vague and a detailed analysis of the underlying principles requires certain standardized parameters. Based on the twelve principles of green chemistry,299 a green metrics discussion is a versatile tool for identifying the sustainability of a chemical process,16,300 enabling the standardised quantification of the ecological impact of a reaction pathway and providing comparability for a variety of processes according to predefined categories. In the early 1990s, the environmental factor (E-factor) was introduced by Sheldon in response to the growing awareness of waste generation in chemical manufacturing throughout the 1980s.301,302 The E-factor is defined as the total waste mass of a process divided by the product mass and therefore accounts for auxiliary chemicals and waste generated by workup.Therefore, a high E-factor indicates an environmentally unfriendly process. However, this factor is strongly dependent on the definition of waste.302 Previously, waste was defined as everything not being a product, with the exception of water, so as not to distort the factor. Nevertheless, the disposal or recycling of water might require additional processing steps and should therefore be included in the calculation. As the E-factor can change by orders of magnitude upon water inclusion, even for otherwise harmless processes, calculations are often performed both with and without water.
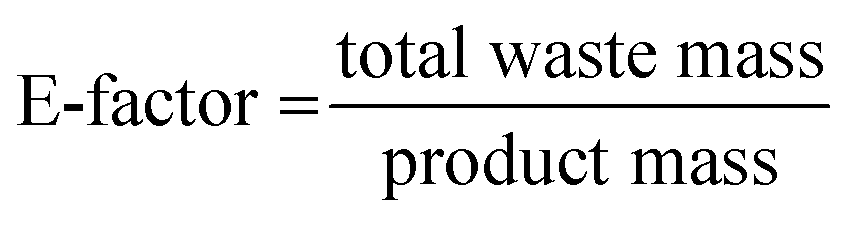
The second concept, which has been well known since the early 1990s, is the atom economy (AE), which is defined as the molecular weight of the desired product divided by the sum of the molecular weight of all reactants in percent.303
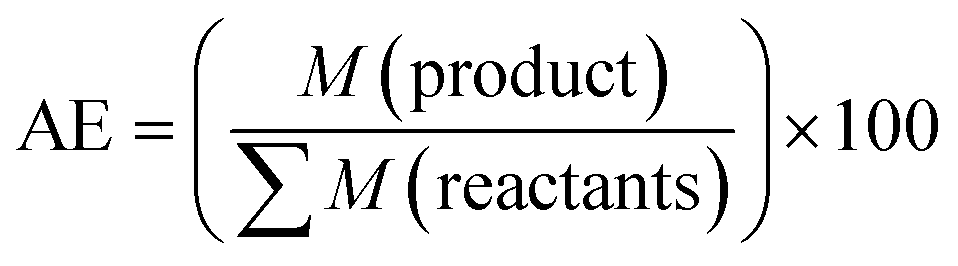
Among the more specific parameters related to AE, one should mention carbon efficiency (CE), which is calculated as the amount of carbon in the product divided by the amount of carbon in the reactants in percent.304,305 Similarly, the reaction mass efficiency (RME) is also based on the AE, expressing the mass of reactants remaining in the product.304 Although both concepts are sufficient for the comparison of various routes towards one product, they do not account for waste and are therefore inadequate for the improvement of process conditions.
To achieve this, the mass intensity (MI) can be determined, which is another very important green chemistry metrics and is defined as the total mass included in the process divided by the mass of the product.306
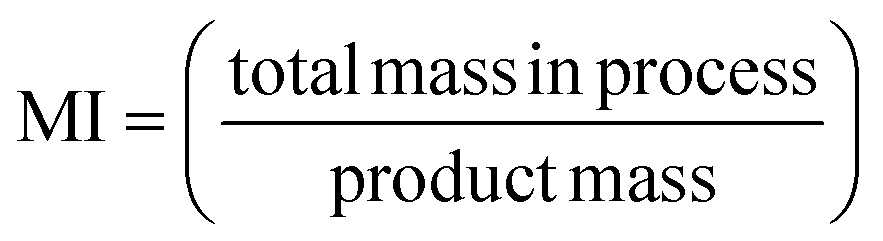
A distinct example for the green metrics discussion of a mechanochemical polymer synthesis was provided in 2017 by Grätz et al.,90 who discussed the sustainability of a mechanochemical Suzuki polymerisation and directly compared it to that of a solvent-based approach in terms of AE, MI, and MP. For the calculation, the washing procedure was excluded for both processes, as it was assumed to involve equivalent amounts of solvent. Although this assumption allows one to simplify the system, an in-depth analysis for comparison with other processes still requires to consider the mass of all involved chemicals, including those utilised for washing.
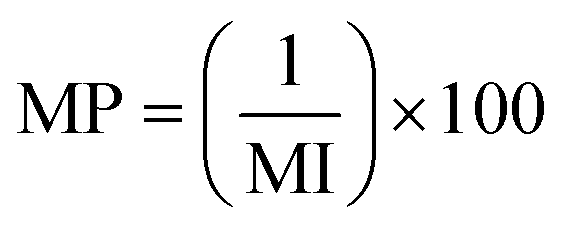
During the investigation, the authors calculated the AE to be 38% for each process, as both reaction pathways exhibited the same stoichiometry. Nevertheless, distinct differences were found in the case of MI, which was calculated as 71.4 for the solvent-based approach and as 22.0 for the mechanochemical reaction. By avoiding the use of the bulking material during the mechanochemical reaction, one could decrease the above value to 9.4, which is fairly close to the ideal value of unity and corresponds to a MP of 10.6% that exceeds the solution-based MP by an order of magnitude. In addition, the reaction time was compared, as a shorter process can generally be declared as more favourable. In contrast to the 6 h reaction in solution, mechanochemical Suzuki polymerisation was accomplished within 30 min.
Recently, the same group calculated the green metrics of a mechanochemical Friedel–Crafts polymerisation.271 In this study, 1,3,5-triphenylbenzene was cross-linked with either DCM or CHCl3 in the presence of the Lewis acid AlCl3, which served as the bulk material and the catalyst. The authors provided a detailed calculation of AE, MI, MP, and the E-factor for the solvent-based synthesis, for the mechanochemical synthesis, and for the mechanochemical synthesis without additional bulk material. In the case of no additional bulk material, 6 eq. AlCl3 was used to catalyse the reaction. While the solvent-based DCM cross-linking synthesis utilised 20 mL of DCM, the mechanochemical approach only required 0.63 mL to cross-link the monomer. Therefore, the mass productivity increased from 3.18% to 23.47%, which corresponded to E-factors of 30.34 and 2.11 for the solvent-based and for the mechanochemical reaction without bulk material, respectively. For the CHCl3-based cross-linking, the reduction in solvent volume from 20 mL to 0.78 mL led to a decrease in the E-factor from 35.76 (solvent-based synthesis) to 1.87 (mechanochemical synthesis without bulk material). As the E-factor equals zero in the ideal case, the mechanochemical approach offers an opportunity to drastically decrease the environmental impact of the reaction and provides further important benefits. Whereas the solvent-based synthesis required a time of 48 h and was followed by a 24 h Soxhlet extraction in 100 mL MeOH and 100 mL CHCl3, the mechanochemical reaction was accomplished within 30 min and was followed by a short rinse of the polymer in 100 mL H2O and 100 mL acetone. Although the mechanochemical synthesis drastically reduced the environmental impact as well as the synthesis/workup time and the toxicity of the liquids required for workup, the resulting porous polymer outperformed its solvent-based equivalent in terms of SSABET and yield.271
Exemplary for other mechanochemical polymerization reactions, the Suzuki and the Friedel–Crafts polymerization can therefore be considered as extremely advantageous in comparison to the solvent-based approach, since the avoidance of solvent waste drastically increases the overall sustainability and the time reduction further promotes energy efficiency as well as cost reduction.90,271
Although mechanochemical polymerisations can generally be declared as more environmentally benign than their solution-based counterparts, other synthetic methods may even exceed the sustainability of mechanochemical procedures. Examples of these are reaction pathways that do not require solvents or bulking agents, e.g., polymerisation from the melt.307 Nevertheless, such reactions often demand prolonged heating times, which has a negative impact on the overall energy consumption and must be taken into consideration.
To quantify the energy consumption of ball milling procedures and compare it to that of other prominent methodologies, Stolle et al. determined the dependence of molar energies on batch size and line power consumption (Fig. 27).22,308 Using various process data, the authors concluded that the energy efficiency of a vibrational ball mill exceeds that of a planetary ball mill, whereas ultrasound and microwave methods are the least efficient.22 These results render ball milling procedures well suited for the efficient processing of polymers to enhance the overall sustainability.
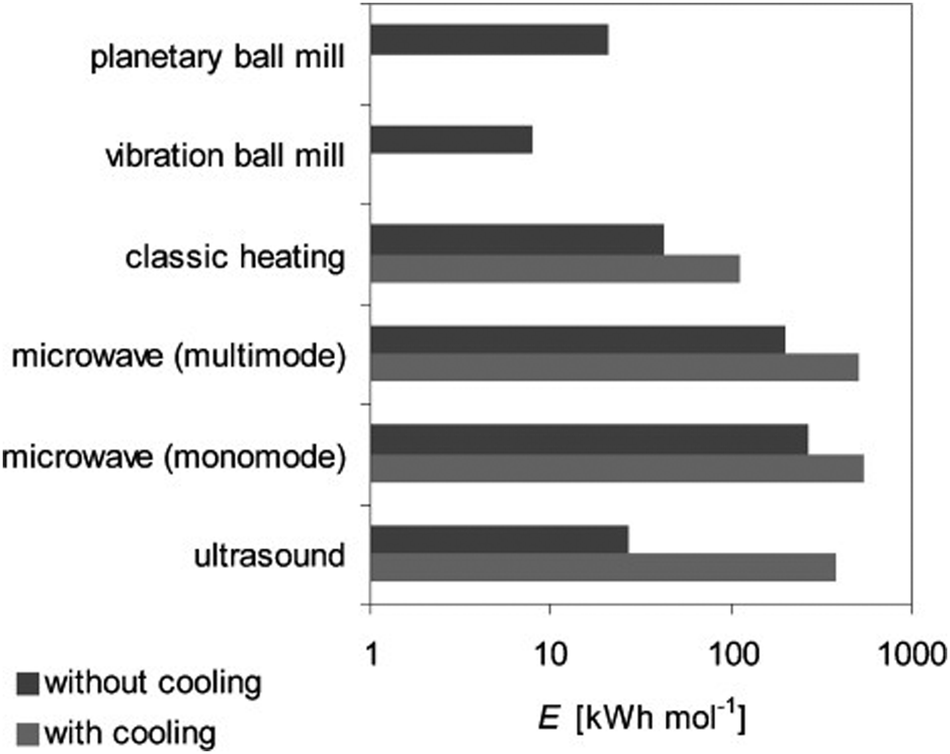 | ||
| Fig. 27 Comparison of the energy consumptions of different processes without cooling (dark grey) and with cooling (light grey). Reprinted from ref. 308. Copyright (2010). Wiley-VCH. | ||
Although mechanochemical polymerisations can generally be declared as sustainable and sufficient alternatives to solution-based analogues, which is attributed to the avoidance of solvent waste and the circumvention of mass transport limitations and insufficient energy transfer, it is essential to consider the characteristics of the generated material. Most polymers exhibit excellent corrosion resistance with an attractive cost-to-performance ratio,309 which results in large-scale production and environmental pollution due to polymer persistence. In addition to the sustainable generation of polymers, the use of renewable resources and effective waste recycling are important factors for a green and sustainable future.
Conclusion
The International Union of Pure and Applied Chemistry (IUPAC) announced ‘mechanochemistry’ as one of 10 chemical innovations expected to change the world. While classical syntheses rely on the addition of solvents, mechanochemistry induces reactions through mechanical forces, enabling sustainable syntheses by avoiding large amounts of waste. In particular, this approach greatly benefits polymer synthesis, allowing one to bypass the need for solubilising groups, to avoid low polymerisation degrees due to rapid precipitation, and to access new innovative structures. Nevertheless, mechanochemistry is still widely unknown to chemists in general and, although extremely powerful, is very rarely considered as a tool for polymer synthesis. This review provides insights into and an understanding of constructive mechanochemical polymer synthesis by summarising examples of various mechanochemical polymerisations with higher yields, shorter reaction times, and improved green chemistry metrics compared to those achieved in classical synthetic routes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. B., A. K., and S. G. gratefully acknowledge the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) for support of the Mechanocarb project (award number 03SF0498) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (“Mechanocat”, grant agreement no. 948521) as well as the Deutsche Forschungsgemeinschaft for funding the Project 469290370. The part of J. G. K. and G. T. is supported by the Samsung Research Funding & Incubation Center of Samsung Electronics under Project Number SRFC-MA1902-05.References
- S. L. James and T. Friščić, Mechanochemistry, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC
.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Mechanochemistry: opportunities for new and cleaner synthesis, Chem. Soc. Rev., 2012, 41, 413–447 RSC
- J.-L. Do and T. Friščić, Mechanochemistry: A Force of Synthesis, ACS. Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed
- J. G. Hernández, Mechanochemistry, Beilstein J. Org. Chem., 2017, 13, 2372–2373 CrossRef PubMed
- C. Bolm and J. G. Hernández, Mechanochemistry of Gaseous Reactants, Angew. Chem., Int. Ed., 2019, 58, 3285–3299 CrossRef CAS PubMed
- T. Friščić, C. Mottillo and H. M. Titi, Mechanochemistry for Synthesis, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed
- H. Staudinger and E. O. Leupold, Über Isopren und Kautschuk, 18. Mitteil.: Viscositäts-Untersuchungen an Balata, Ber. Dtsch. Chem. Ges. A/B, 1930, 63, 730–733 CrossRef
- H. Staudinger and H. F. Bondy, Über Isopren und Kautschuk, 19. Mitteil.: Über die Molekülgröße des Kautschuks und der Balata, Ber. Dtsch. Chem. Ges. A/B, 1930, 63, 734–736 CrossRef
- H. Staudinger and W. Heuer, Über hochpolymere Verbindungen, 93. Mitteil.: Über das Zerreißen der Faden-Moleküle des Poly-styrols, Ber. Dtsch. Chem. Ges. A/B, 1934, 67, 1159–1164 CrossRef
- P. A. May and J. S. Moore, Polymer mechanochemistry: techniques to generate molecular force via elongational flows, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC
- J. Li, C. Nagamani and J. S. Moore, Polymer mechanochemistry: from destructive to productive, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed
- N. Willis-Fox, E. Rognin, T. A. Aljohani and R. Daly, Polymer Mechanochemistry: Manufacturing Is Now a Force to Be Reckoned With, Chem, 2018, 4, 2499–2537 CAS
- G. de Bo, Polymer Mechanochemistry and the Emergence of the Mechanophore Concept, Macromolecules, 2020, 53, 7615–7617 CrossRef
- A. S. Abd-El-Aziz, M. Antonietti, C. Barner-Kowollik, W. H. Binder, A. Böker, C. Boyer, M. R. Buchmeiser, S. Z. D. Cheng, F. D’Agosto, G. Floudas, H. Frey, G. Galli, J. Genzer, L. Hartmann, R. Hoogenboom, T. Ishizone, D. L. Kaplan, M. Leclerc, A. Lendlein, B. Liu, T. E. Long, S. Ludwigs, J.-F. Lutz, K. Matyjaszewski, M. A. R. Meier, K. Müllen, M. Müllner, B. Rieger, T. P. Russell, D. A. Savin, A. D. Schlüter, U. S. Schubert, S. Seiffert, K. Severing, J. B. P. Soares, M. Staffilani, B. S. Sumerlin, Y. Sun, B. Z. Tang, C. Tang, P. Théato, N. Tirelli, O. K. C. Tsui, M. M. Unterlass, P. Vana, B. Voit, S. Vyazovkin, C. Weder, U. Wiesner, W.-Y. Wong, C. Wu, Y. Yagci, J. Yuan and G. Zhang, The Next 100 Years of Polymer Science, Macromol. Chem. Phys., 2020, 221, 2000216 CrossRef CAS
-
P. T. Anastas and J. C. Warner, Green chemistry. Theory and practice, Oxford Univ. Press, Oxford, 1st edn, 2000 Search PubMed
- P. Anastas and N. Eghbali, Green chemistry: principles and practice, Chem. Soc. Rev., 2010, 39, 301–312 RSC
- V. A. Kargin and N. A. Plate, Chemical grafting on crystal surfaces, Vysokomol. Soed, 1959, 1, 330 CAS
- C. V. Oprea and M. Popa, Mechanochemically Synthesized Polymers with Special Properties, Polym.-Plast. Technol. Eng., 1989, 28, 1025–1058 CrossRef CAS
- M. Kuzuya, S. Kondo and A. Noguchi, A new development of mechanochemical solid-state polymerization of vinyl monomers: prodrug syntheses and its detailed mechanistic study, Macromolecules, 1991, 24, 4047–4053 CrossRef CAS
- J. B. Ravnsbæk and T. M. Swager, Mechanochemical Synthesis of Poly(phenylene vinylenes), ACS Macro Lett., 2014, 3, 305–309 CrossRef
- G.-W. Wang, Mechanochemical organic synthesis, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC
- A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Ball milling in organic synthesis: solutions and challenges, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC
- E. Boldyreva, Mechanochemistry of inorganic and organic systems: what is similar, what is different?, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC
- N. R. Rightmire and T. P. Hanusa, Advances in organometallic synthesis with mechanochemical methods, Dalton Trans., 2016, 45, 2352–2362 RSC
- D. Tan, L. Loots and T. Friščić, Towards medicinal mechanochemistry: evolution of milling from pharmaceutical solid form screening to the synthesis of active pharmaceutical ingredients (APIs), Chem. Commun., 2016, 52, 7760–7781 RSC
- D. Tan and F. García, Main group mechanochemistry: from curiosity to established protocols, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC
-
IUPAC Compendium of Chemical Terminology, ed. M. Nič, J. Jirát, B. Košata, A. Jenkins and A. McNaught, IUPAC, Research Triagle Park, NC, 2009 Search PubMed
- R. T. O’Neill and R. Boulatov, The many flavours of mechanochemistry and its plausible conceptual underpinnings, Nat. Rev. Chem., 2021, 5, 148–167 CrossRef
- M. A. Ghanem, A. Basu, R. Behrou, N. Boechler, A. J. Boydston, S. L. Craig, Y. Lin, B. E. Lynde, A. Nelson, H. Shen and D. W. Storti, The role of polymer mechanochemistry in responsive materials and additive manufacturing, Nat. Rev. Mater., 2020, 67, 1159 Search PubMed
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Mechanochemical tools for polymer materials, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC
- J. Ayarza, Z. Wang, J. Wang, C.-W. Huang and A. P. Esser-Kahn, 100th Anniversary of Macromolecular Science Viewpoint: Piezoelectrically Mediated Mechanochemical Reactions for Adaptive Materials, ACS Macro Lett., 2020, 9, 1237–1248 CrossRef CAS
- Z. Wang, X. Pan, L. Li, M. Fantin, J. Yan, Z. Wang, Z. Wang, H. Xia and K. Matyjaszewski, Enhancing Mechanically Induced ATRP by Promoting Interfacial Electron Transfer from Piezoelectric Nanoparticles to Cu Catalysts, Macromolecules, 2017, 50, 7940–7948 CrossRef CAS
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Mechanically controlled radical polymerization initiated by ultrasound, Nat. Chem., 2017, 9, 135–139 CrossRef CAS
- S. He, M. Stratigaki, S. P. Centeno, A. Dreuw and R. Göstl, Tailoring the Properties of Optical Force Probes for Polymer Mechanochemistry, Chemistry, 2021, 27, 15889–15897 CrossRef CAS PubMed
- S. Akbulatov and R. Boulatov, Experimental Polymer Mechanochemistry and its Interpretational Frameworks, ChemPhysChem, 2017, 18, 1422–1450 CrossRef CAS PubMed
- M. C. Lea, IV, Disruption of the silver haloid molecule by mechanical force, London, Edinburgh Dublin Philos. Mag. J. Sci., 1892, 34, 46–50 CrossRef
-
W. Ostwald, Prinzipien der Chemie: eine Einleitung in alle chemischen Lehrbücher, Akademische Verlagsgesellschaft m. b. H., Leipzig, 1907 Search PubMed
- F. Toda, K. Tanaka and A. Sekikawa, Host–guest complex formation by a solid–solid reaction, J. Chem. Soc., Chem. Commun., 1987, 279–280 RSC
- A. O. Patil, D. Y. Curtin and I. C. Paul, Solid-state formation of quinhydrones from their components. Use of solid-solid reactions to prepare compounds not accessible from solution, J. Am. Chem. Soc., 1984, 106, 348–353 CrossRef CAS
- J. Schmeyers, F. Toda, J. Boy and G. Kaupp, Quantitative solid–solid synthesis of azomethines, J. Chem. Soc., Perkin Trans. 2, 1998, 989–994 RSC
- F. Toda, H. Takumi and M. Akehi, Efficient solid-state reactions of alcohols: dehydration, rearrangement, and substitution, J. Chem. Soc., Chem. Commun., 1990, 1270 RSC
- M. O. Rasmussen, O. Axelsson and D. Tanner, A Practical Procedure for the Solid-Phase Synthesis of Racemic 2,2′-Dihydroxy-1,1′-binaphthyl, Synth. Commun., 1997, 27, 4027–4030 CrossRef CAS
-
L. Borchardt and S. Grätz, in Handbuch der Geodäsie, ed. W. Freeden and R. Rummel, Springer Berlin Heidelberg, Berlin, Heidelberg, 2019, pp. 1–28 Search PubMed
- F. P. Bowden and A. D. Yoffe, Initiation and Growth of Explosion in Liquids and Solids, Am. J. Phys., 1952, 20, 250–251 CrossRef
-
F. P. Bowden and A. D. Yoffe, Fast Reactions in Solids, Academic Press, New York, 1958 Search PubMed
-
P. A. Thiessen, K. Meyer and G. Heinicke, Grundlagen der Tribochemie, Akademie-Verlag, Berlin, 1967 Search PubMed
-
P. Bálaz, in Extractive metallurgy of activated minerals, ed. P. Baláž, Elsevier, Amsterdam, Oxford, 2000, pp. 1–14 Search PubMed
- X. Ma, W. Yuan, S. E. J. Bell and S. L. James, Better understanding of mechanochemical reactions: Raman monitoring reveals surprisingly simple ‘pseudo-fluid’ model for a ball milling reaction, Chem. Commun., 2014, 50, 1585–1587 RSC
- R. Weichert and K. Schönert, On the temperature rise at the tip of a fast running crack, J. Mech. Phys. Solids, 1974, 22, 127–133 CrossRef
- P. G. Fox, Mechanically initiated chemical reactions in solids, J. Mater. Sci., 1975, 10, 340–360 CrossRef CAS
- M. Carta, S. L. James and F. Delogu, Phenomenological Inferences on the Kinetics of a Mechanically Activated Knoevenagel Condensation: Understanding the “Snowball” Kinetic Effect in Ball Milling, Molecules, 2019, 24(19), 3600 CrossRef CAS PubMed
- H. Kulla, M. Wilke, F. Fischer, M. Röllig, C. Maierhofer and F. Emmerling, Warming up for mechanosynthesis – temperature development in ball mills during synthesis, Chem. Commun., 2017, 53, 1664–1667 RSC
- A. Krusenbaum, S. Grätz, S. Bimmermann, S. Hutsch and L. Borchardt, The mechanochemical Scholl reaction as a versatile synthesis tool for the solvent-free generation of microporous polymers, RSC Adv., 2020, 10, 25509–25516 RSC
- K. Užarević, V. Štrukil, C. Mottillo, P. A. Julien, A. Puškarić, T. Friščić and I. Halasz, Exploring the Effect of Temperature on a Mechanochemical Reaction by in Situ Synchrotron Powder X-ray Diffraction, Cryst. Growth Des., 2016, 16, 2342–2347 CrossRef
- T. Seo, N. Toyoshima, K. Kubota and H. Ito, Tackling Solubility Issues in Organic Synthesis: Solid-State Cross-Coupling of Insoluble Aryl Halides, J. Am. Chem. Soc., 2021, 143, 6165–6175 CrossRef CAS PubMed
- F. Fischer, K.-J. Wenzel, K. Rademann and F. Emmerling, Quantitative determination of activation energies in mechanochemical reactions, Phys. Chem. Chem. Phys., 2016, 18, 23320–23325 RSC
- N. Cindro, M. Tireli, B. Karadeniz, T. Mrla and K. Užarević, Investigations of Thermally Controlled Mechanochemical Milling Reactions, ACS Sustainable Chem. Eng., 2019, 7, 16301–16309 CrossRef CAS
- F. Delogu and G. Cocco, Kinetics of amorphization processes by mechanical alloying: A modeling approach, J. Alloys Compd., 2007, 436, 233–240 CrossRef CAS
- E. Gil-González, M. R. Del Rodríguez-Laguna, P. E. Sánchez-Jiménez, A. Perejón and L. A. Pérez-Maqueda, Unveiling mechanochemistry: Kinematic-kinetic approach for the prediction of mechanically induced reactions, J. Alloys Compd., 2021, 866, 158925 CrossRef
- M. Danielis, S. Colussi, C. de Leitenburg, L. Soler, J. Llorca and A. Trovarelli, The effect of milling parameters on the mechanochemical synthesis of Pd–CeO2 methane oxidation catalysts, Catal. Sci. Technol., 2019, 9, 4232–4238 RSC
- T. Yamamoto, S. Kato, D. Aoki and H. Otsuka, A Diarylacetonitrile as a Molecular Probe for the Detection of Polymeric Mechanoradicals in the Bulk State through a Radical Chain-Transfer Mechanism, Angew. Chem., Int. Ed., 2021, 60, 2680–2683 CrossRef CAS PubMed
- K. Gobindlal, Z. Zujovic, P. Yadav, J. Sperry and C. C. Weber, The Mechanism of Surface-Radical Generation and Amorphization of Crystalline Quartz Sand upon Mechanochemical Grinding, J. Phys. Chem. C, 2021, 125, 20877–20886 CrossRef CAS
- M. J. Mancheño, S. Royuela, A. de La Peña, M. Ramos, F. Zamora and J. L. Segura, Introduction to Covalent Organic Frameworks: An Advanced Organic Chemistry Experiment, J. Chem. Educ., 2019, 96, 1745–1751 CrossRef
- X. Sun, W. Shi, X.-Y. Zhou and S. Ding, Facile Mechanochemical Preparation of Polyamide-derivatives via Solid-state Benzoxazine-isocyanide Chemistry, Chin. J. Polym. Sci., 2020, 42, 7668 Search PubMed
- D. Hasa and W. Jones, Screening for new pharmaceutical solid forms using mechanochemistry: a practical guide, Adv. Drug Delivery Rev., 2017, 117, 147–161 CrossRef CAS PubMed
- F. Schneider, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Energetic assessment of the Suzuki–Miyaura reaction: a curtate life cycle assessment as an easily understandable and applicable tool for reaction optimization, Green Chem., 2009, 11, 1894 RSC
- L. Takacs, The historical development of mechanochemistry, Chem. Soc. Rev., 2013, 42, 7649–7659 RSC
-
A. J. Lynch and C. A. Rowland, The History of Grinding, Society of Mining, Metallurgy and Exploration, Littleton, Colorado, 2005 Search PubMed
- A. Stolle, R. Schmidt and K. Jacob, Scale-up of organic reactions in ball mills: process intensification with regard to energy efficiency and economy of scale, Faraday Discuss., 2014, 170, 267–286 RSC
- C. F. Burmeister and A. Kwade, Process engineering with planetary ball mills, Chem. Soc. Rev., 2013, 42, 7660–7667 RSC
- B. Rodríguez, A. Bruckmann, T. Rantanen and C. Bolm, Solvent-Free Carbon-Carbon Bond Formations in Ball Mills, Adv. Synth. Catal., 2007, 349, 2213–2233 CrossRef
- C. Burmeister, L. Titscher, S. Breitung-Faes and A. Kwade, Dry grinding in planetary ball mills: Evaluation of a stressing model, Adv. Powder Technol., 2018, 29, 191–201 CrossRef CAS
- W. Pickhardt, S. Grätz and L. Borchardt, Direct Mechanocatalysis: Using Milling Balls as Catalysts, Chem. – Eur. J., 2020, 26, 12903–12911 CrossRef CAS PubMed
- B. P. Hutchings, D. E. Crawford, L. Gao, P. Hu and S. L. James, Feedback Kinetics in Mechanochemistry: The Importance of Cohesive States, Angew. Chem., Int. Ed., 2017, 56, 15252–15256 CrossRef CAS PubMed
- N. Kumar and K. Biswas, Fabrication of novel cryomill for synthesis of high purity metallic nanoparticles, Rev. Sci. Instrum., 2015, 86, 83903 CrossRef PubMed
- J. Andersen and J. Mack, Insights into Mechanochemical Reactions at Targetable and Stable, Sub-ambient Temperatures, Angew. Chem., Int. Ed., 2018, 57, 13062–13065 CrossRef CAS PubMed
- J. Andersen, J. Brunemann and J. Mack, Exploring stable, sub-ambient temperatures in mechanochemistry via a diverse set of enantioselective reactions, React. Chem. Eng., 2019, 4, 1229–1236 RSC
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Real-time and in situ monitoring of mechanochemical milling reactions, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed
- J. G. Hernández, Mechanochemical borylation of aryldiazonium salts; merging light and ball milling, Beilstein J. Org. Chem., 2017, 13, 1463–1469 CrossRef PubMed
- T. Friščić, C. Mottillo and H. M. Titi, Mechanochemistry for Synthesis, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed
- V. Štrukil and I. Sajko, Mechanochemically-assisted solid-state photocatalysis (MASSPC), Chem. Commun., 2017, 53, 9101–9104 RSC
- V. Štrukil, D. Gracin, O. V. Magdysyuk, R. E. Dinnebier and T. Friščić, Trapping Reactive Intermediates by Mechanochemistry: Elusive Aryl N-Thiocarbamoylbenzotriazoles as Bench-Stable Reagents, Angew. Chem., Int. Ed., 2015, 54, 8440–8443 CrossRef PubMed
- D. Gracin, V. Štrukil, T. Friščić, I. Halasz and K. Užarević, Laboratory real-time and in situ monitoring of mechanochemical milling reactions by Raman spectroscopy, Angew. Chem., Int. Ed., 2014, 53, 6193–6197 CrossRef CAS PubMed
- M. Tireli, M. Juribašić Kulcsár, N. Cindro, D. Gracin, N. Biliškov, M. Borovina, M. Ćurić, I. Halasz and K. Užarević, Mechanochemical reactions studied by in situ Raman spectroscopy: base catalysis in liquid-assisted grinding, Chem. Commun., 2015, 51, 8058–8061 RSC
- I. Halasz, A. Puškarić, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. E. Dinnebier, B. Patel, W. Jones, V. Strukil and T. Friščić, Real-time in situ powder X-ray diffraction monitoring of mechanochemical synthesis of pharmaceutical cocrystals, Angew. Chem., Int. Ed., 2013, 52, 11538–11541 CrossRef CAS PubMed
- I. Halasz, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. C. Nightingale, R. E. Dinnebier and T. Friščić, In situ and real-time monitoring of mechanochemical milling reactions using synchrotron X-ray diffraction, Nat. Protoc., 2013, 8, 1718–1729 CrossRef PubMed
- L. Batzdorf, F. Fischer, M. Wilke, K.-J. Wenzel and F. Emmerling, Direct in situ investigation of milling reactions using combined X-ray diffraction and Raman spectroscopy, Angew. Chem., Int. Ed., 2015, 54, 1799–1802 CrossRef CAS PubMed
- J. G. Schiffmann, F. Emmerling, I. C. B. Martins and L. van Wüllen, In-situ reaction monitoring of a mechanochemical ball mill reaction with solid state NMR, Solid State Nucl. Magn. Reson., 2020, 109, 101687 CrossRef CAS PubMed
- S. Grätz, S. Zink, H. Kraffczyk, M. Rose and L. Borchardt, Mechanochemical synthesis of hyper-crosslinked polymers: influences on their pore structure and adsorption behaviour for organic vapors, Beilstein J. Org. Chem., 2019, 15, 1154–1161 CrossRef PubMed
- S. Grätz, B. Wolfrum and L. Borchardt, Mechanochemical Suzuki polycondensation – from linear to hyperbranched polyphenylenes, Green Chem., 2017, 19, 2973–2979 RSC
- S. Park and J. G. Kim, Mechanochemical synthesis of poly(trimethylene carbonate)s: an example of rate acceleration, Beilstein J. Org. Chem., 2019, 15, 963–970 CrossRef CAS PubMed
- D. E. Crawford and J. Casaban, Recent Developments in Mechanochemical Materials Synthesis by Extrusion, Adv. Mater., 2016, 28, 5747–5754 CrossRef CAS PubMed
-
C. Rauwendaal, Polymer extrusion, Hanser Publication, Munich, Cincinnati, 5th edn, 2014 Search PubMed
-
J. R. Wagner, E. M. Mount and H. F. Giles, Extrusion. The definitive processing guide and handbook, Andrew Elsevier, Oxford, 2nd edn, 2014 Search PubMed
- J. L. Howard, Q. Cao and D. L. Browne, Mechanochemistry as an emerging tool for molecular synthesis: what can it offer?, Chem. Sci., 2018, 9, 3080–3094 RSC
- J. G. Hernández and C. Bolm, Altering Product Selectivity by Mechanochemistry, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed
- S. Grätz, M. Oltermann, C. G. Vogt and L. Borchardt, Mechanochemical Cyclodehydrogenation with Elemental Copper: An Alternative Pathway toward Nanographenes, ACS Sustainable Chem. Eng., 2020, 8, 7569–7573 CrossRef
- R. Thorwirth, A. Stolle and B. Ondruschka, Fast copper-, ligand- and solvent-free Sonogashira coupling in a ball mill, Green Chem., 2010, 12, 985 RSC
- N. Ohn, J. Shin, S. S. Kim and J. G. Kim, Mechanochemical Ring-Opening Polymerization of Lactide: Liquid-Assisted Grinding for the Green Synthesis of Poly(lactic acid) with High Molecular Weight, ChemSusChem, 2017, 10, 3529–3533 CrossRef CAS PubMed
- C. Oh, E. H. Choi, E. J. Choi, T. Premkumar and C. Song, Facile Solid-State Mechanochemical Synthesis of Eco-Friendly Thermoplastic Polyurethanes and Copolymers Using a Biomass-Derived Furan Diol, ACS Sustainable Chem. Eng., 2020, 8, 4400–4406 CrossRef CAS
- T. Szuppa, A. Stolle, B. Ondruschka and W. Hopfe, Solvent-free dehydrogenation of γ-terpinene in a ball mill: investigation of reaction parameters, Green Chem., 2010, 12, 1288 RSC
- S. Grätz, M. Oltermann, E. Troschke, S. Paasch, S. Krause, E. Brunner and L. Borchardt, Solvent-free synthesis of a porous thiophene polymer by mechanochemical oxidative polymerization, J. Mater. Chem. A, 2018, 6, 21901–21905 RSC
- T. Friščić and W. Jones, Recent Advances in Understanding the Mechanism of Cocrystal Formation via Grinding, Cryst. Growth Des., 2009, 9, 1621–1637 CrossRef
- V. Sládková, J. Cibulková, V. Eigner, A. Šturc, B. Kratochvíl and J. Rohlíček, Application and Comparison of Cocrystallization Techniques on Trospium Chloride Cocrystals, Cryst. Growth Des., 2014, 14, 2931–2936 CrossRef
- T. Friscić, A. V. Trask, W. Jones and W. D. S. Motherwell, Screening for inclusion compounds and systematic construction of three-component solids by liquid-assisted grinding, Angew. Chem., Int. Ed., 2006, 45, 7546–7550 CrossRef PubMed
- N. Shan, F. Toda and W. Jones, Mechanochemistry and co-crystal formation: effect of solvent on reaction kinetics, Chem. Commun., 2002, 2372–2373 RSC
- G. A. Bowmaker, Solvent-assisted mechanochemistry, Chem. Commun., 2013, 49, 334–348 RSC
- J.-L. Do, C. Mottillo, D. Tan, V. Štrukil and T. Friščić, Mechanochemical ruthenium-catalyzed olefin metathesis, J. Am. Chem. Soc., 2015, 137, 2476–2479 CrossRef CAS PubMed
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, The role of solvent in mechanochemical and sonochemical cocrystal formation: a solubility-based approach for predicting cocrystallisation outcome, CrystEngComm, 2009, 11, 418–426 RSC
- V. Declerck, E. Colacino, X. Bantreil, J. Martinez and F. Lamaty, Poly(ethylene glycol) as reaction medium for mild Mizoroki-Heck reaction in a ball-mill, Chem. Commun., 2012, 48, 11778–11780 RSC
- L. Konnert, M. Dimassi, L. Gonnet, F. Lamaty, J. Martinez and E. Colacino, Poly(ethylene) glycols and mechanochemistry for the preparation of bioactive 3,5-disubstituted hydantoins, RSC Adv., 2016, 6, 36978–36986 RSC
- D. Hasa, G. S. Rauber, D. Voinovich and W. Jones, Cocrystal Formation through Mechanochemistry: from Neat and Liquid-Assisted Grinding to Polymer-Assisted Grinding, Angew. Chem., Int. Ed., 2015, 54, 7371–7375 CrossRef CAS PubMed
- A. A. Gečiauskaitė and F. García, Main group mechanochemistry, Beilstein J. Org. Chem., 2017, 13, 2068–2077 CrossRef PubMed
- J. Andersen and J. Mack, Mechanochemistry and organic synthesis: from mystical to practical, Green Chem., 2018, 20, 1435–1443 RSC
- J. M. Andersen and J. Mack, Decoupling the Arrhenius equation via mechanochemistry, Chem. Sci., 2017, 8, 5447–5453 RSC
- G. Janke and G. Schmidt-Naake, Synthesis of Block and Graft Copolymers in a Vibratory Mill, Chem. Eng. Technol., 2001, 24, 711–715 CrossRef CAS
- G. I. Peterson, W. Ko, Y.-J. Hwang and T.-L. Choi, Mechanochemical Degradation of Amorphous Polymers with Ball-Mill Grinding: Influence of the Glass Transition Temperature, Macromolecules, 2020, 53, 7795–7802 CrossRef CAS
- T. Q. Nguyen and H.-H. Kausch, Kinetics of polymer degradation in transient elongational flow, Makromol. Chem., 1989, 190, 1389–1406 CrossRef CAS
- T. Q. Nguyen and H.-H. Kausch, GPC Data Interpretation in Mechanochemical Polymer Degradation, Int. J. Polym. Anal. Charact., 1998, 4, 447–470 CrossRef CAS
- T. Q. Nguyen, Q. Z. Liang and H.-H. Kausch, Kinetics of ultrasonic and transient elongational flow degradation: a comparative study, Polymer, 1997, 38, 3783–3793 CrossRef CAS
-
T. Q. Nguyen and H.-H. Kausch, in Macromolecules, ed. K. A. Armitstead, Springer, Berlin, Heidelberg, 1992, pp. 73–182 Search PubMed
- V. Štrukil, Highly Efficient Solid-State Hydrolysis of Waste Polyethylene Terephthalate by Mechanochemical Milling and Vapor-Assisted Aging, ChemSusChem, 2020, 14(1), 330 CrossRef PubMed
- V. P. Balema, I. Z. Hlova, S. L. Carnahan, M. Seyedi, O. Dolotko, A. J. Rossini and I. Luzinov, Depolymerization of polystyrene under ambient conditions, New J. Chem., 2021, 45, 2935–2938 RSC
- Y. Li, J. Li, S. Guo and H. Li, Mechanochemical degradation kinetics of high-density polyethylene melt and its mechanism in the presence of ultrasonic irradiation, Ultrason. Sonochem., 2005, 12, 183–189 CrossRef CAS PubMed
- K. A. Plate and V. A. Kargin, Mechanochemical reactions of polymerization and degradation at low temperatures, J. Polym. Sci., Part C: Polym. Symp., 1963, 4, 1027–1041 CrossRef
- C. Simionescu, C. Vasiliu Oprea and J. Nicoleanu, Mechanochemically initiated polymerizations—5. Polymerization by vibratory milling of acrylamide and methacrylamide, Eur. Polym. J., 1983, 19, 525–528 CrossRef CAS
- N. Ohn and J. G. Kim, Mechanochemical Post-Polymerization Modification: Solvent-Free Solid-State Synthesis of Functional Polymers, ACS Macro Lett., 2018, 7, 561–565 CrossRef CAS
- H. Y. Cho and C. W. Bielawski, Atom Transfer Radical Polymerization in the Solid-State, Angew. Chem., Int. Ed., 2020, 59, 13929–13935 CrossRef CAS PubMed
- J. Sohma, Mechanochemistry of polymers, Prog. Polym. Sci., 1989, 14, 451–596 CrossRef CAS
- S.-I. Kondo, I. Hatakeyama, S. Hosaka and M. Kuzuya, Mechanochemical Solid-State Polymerization (X): The Influence of Copolymer Structure in Copolymeric Prodrugs on the Nature of Drug Release, Chem. Pharm. Bull., 2000, 48, 1882–1885 CrossRef CAS PubMed
- M. Sakaguchi and J. Sohma, Copolymerizations initiated by mechano-radicals on particle surfaces of poly(tetrafluoroethylene), J. Appl. Polym. Sci., 1978, 22, 2915–2924 CrossRef CAS
- S. Chatani, C. J. Kloxin and C. N. Bowman, The power of light in polymer science: photochemical processes to manipulate polymer formation, structure, and properties, Polym. Chem., 2014, 5, 2187–2201 RSC
- S. Yamago and Y. Nakamura, Recent progress in the use of photoirradiation in living radical polymerization, Polymer, 2013, 54, 981–994 CrossRef CAS
- R. N. Carmean, M. B. Sims, C. A. Figg, P. J. Hurst, J. P. Patterson and B. S. Sumerlin, Ultrahigh Molecular Weight Hydrophobic Acrylic and Styrenic Polymers through Organic-Phase Photoiniferter-Mediated Polymerization, ACS Macro Lett., 2020, 9, 613–618 CrossRef CAS
- M. Fantin, S. Park, Y. Wang and K. Matyjaszewski, Electrochemical Atom Transfer Radical Polymerization in Miniemulsion with a Dual Catalytic System, Macromolecules, 2016, 49, 8838–8847 CrossRef CAS PubMed
- B. M. Peterson, S. Lin and B. P. Fors, Electrochemically Controlled Cationic Polymerization of Vinyl Ethers, J. Am. Chem. Soc., 2018, 140, 2076–2079 CrossRef CAS PubMed
- M. Zhao, H. Zhang, C. Gu and Y. Ma, Electrochemical polymerization: an emerging approach for fabricating high-quality luminescent films and super-resolution OLEDs, J. Mater. Chem. C, 2020, 8, 5310–5320 RSC
- V. A. Kargin, V. A. Kabanov and N. Rapaport-Molodtsova, The mechano-chemical initiation of polymerization of crystalline salts of acrylic acid, Polym. Sci. USSR, 1962, 3, 657–664 CrossRef
- V. A. Kargin and N. A. Platé, Polymerization and grafting processes on freshly formed surfaces, J. Polym. Sci., 1961, 52, 155–158 CrossRef CAS
- S.-I. Kondo, D. Shichijyou, Y. Sasai, Y. Yamauchi and M. Kuzuya, Synthesis of DNA Conjugate by Mechanochemical Solid-State Polymerization and Its Affinity Separation of Oligonucleotides Having Single-Base Difference by Capillary Electrophoresis, Chem. Pharm. Bull., 2005, 53, 863–865 CrossRef CAS PubMed
- S.-I. Kondo, K. Murase and M. Kuzuya, Mechanochemical Solid-State Polymerization. VI. Quantum Chemical Considerations for Structural Criteria of Mechanically Polymerizable Vinyl Monomers, Chem. Pharm. Bull., 1994, 42, 768–773 CrossRef CAS
- N. Doi, Y. Yamauchi, R. Ikegami, M. Kuzuya, Y. Sasai and S.-I. Kondo, Photo-responsive polymer micelles from o-nitrobenzyl ester-based amphiphilic block copolymers synthesized by mechanochemical solid-state copolymerization, Polym. J., 2020, 52, 1375–1385 CrossRef CAS
- Y. Yamauchi, N. Doi, S.-I. Kondo, Y. Sasai and M. Kuzuya, Characterization of a novel polymeric prodrug of an antibacterial agent synthesized by mechanochemical solid-state polymerization, Drug Devlivery Res., 2020, 81, 867–874 CrossRef CAS PubMed
- S.-I. Kondo, H. Mori, Y. Sasai and M. Kuzuya, Conventional Synthesis of Amphiphilic Block Copolymer Utilized for Polymeric Micelle by Mechanochemical Solid-State Polymerization, Chem. Pharm. Bull., 2007, 55, 389–392 CrossRef CAS PubMed
- S.-I. Kondo, Y. Sasai, M. Kuzuya and S. Furukawa, Synthesis of Water-Soluble Polymeric Prodrugs Possessing 4-Methylcatechol Derivatives by Mechanochemical Solid-State Copolymerization and Nature of Drug Release, Chem. Pharm. Bull., 2002, 50, 1434–1438 CrossRef CAS PubMed
- M. Kuzuya, S.-I. Kondo, A. Noguchi and N. Noda, Mechanistic study on mechanochemical polymerization of acrylamide, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 489–494 CrossRef CAS
- C. V. Oprea and F. Weiner, Mechanochemisch eingeleitete Polymerisationsreaktionen. 7. Einfluß der Radikalinitiatoren und -akzeptoren auf die durch Schwingmahlung des Styrols mechanochemisch eingeleitete Polymerisation, Angew. Makromol. Chem., 1984, 126, 89–105 CrossRef CAS
- C. Simionescu, C. V. Oprea and C. Negulianu, Copolymerizations Mechanochemically Initiated–I, Eur. Polym. J., 1979, 15, 1037–1042 CrossRef CAS
- C. V. Oprea and M. Popa, Mechanochemically initiated copolymerizations. 5. Characterization of some binary and ternary copolymers obtained mechanochemically by vibratory milling, Acta Polym., 1984, 35, 261–264 CrossRef CAS
- C. V. Oprea, I. Avram and R. Avram, Mechanochemisch ausgelöste Polymerisationsreaktionen I. Mechanochemische Homopolymerisation durch Schwingmahlung von Styrol und Acrylnitril, Angew. Makromol. Chem., 1978, 68, 1–15 CrossRef CAS
- C. V. Oprea and M. Popa, Mechanochemically initiated polymerizations. Characterization of poly(acrylonitrile) mechanochemically
synthesized by vibratory grinding, Angew. Makromol. Chem., 1980, 92, 73–88 CrossRef CAS
- C. V. Oprea and M. Popa, Mechanochemically initiated copolymerizations. 8. Copolymerization of acetonitrile with styrene by vibratory milling, Acta Polym., 1986, 37, 177–179 CrossRef CAS
- G. V. Oprea and M. Popa, Mechanochemically initiated copolymerizations. 4. Mechanochemical synthesis of some binary and ternary copolymers by vibratory milling, Acta Polym., 1983, 34, 612–615 CrossRef
- S.-I. Kondo, K. Murase and M. Kuzuya, Mechanochemical Solid-State Polymerization. VII. The Nature of Hydrolysis of Novel Polymeric Prodrugs Prepared by Mechanochemical Copolymerization, Chem. Pharm. Bull., 1994, 42, 2412–2417 CrossRef CAS
- C. V. Oprea, M. Popa and S. Dumitriu, Mechanochemically initiated copolymerizations, 10. Kinetic data on the copolymerization of the acrylonitrile/vinyl acetate/α-methyl styrene ternary system by vibratory milling, Angew. Makromol. Chem., 1988, 157, 93–103 CrossRef CAS
- R. E. Benson and J. E. Castle, Reactions of Freshly Formed Surfaces of Silica, J. Phys. Chem., 1958, 62, 840–843 CrossRef CAS
- Y. Momose, K. Yamada and K. Nagayama, Mechanochemical polymerization on disintegrated sand surface, J. Polym. Sci., Part B: Polym. Lett. Ed., 1974, 12, 623–628 CAS
- M. Hasegawa, Y. Akiho and Y. Kanda, Mechanochemical polymerization of methyl methacrylate initiated by the grinding of inorganic compounds, J. Appl. Polym. Sci., 1995, 55, 297–304 CrossRef CAS
- M. Hasegawa, M. Kimata and S.-I. Kobayashi, Mechanochemical polymerization of styrene initiated by the grinding of quartz, J. Appl. Polym. Sci., 2001, 82, 2849–2855 CrossRef CAS
- M. Hasegawa, M. Kimata and S.-I. Kobayashi, Mechanochemical copolymerization of methyl methacrylate and styrene initiated by the grinding of quartz, J. Appl. Polym. Sci., 2002, 84, 2011–2017 CrossRef CAS
- M. Hasegawa, M. Kimata and I. Takahashi, Mechanochemical polymerization of styrene initiated by the grinding of layered clay minerals, Adv. Powder Technol., 2007, 18, 541–554 CrossRef CAS
- M. Kimata, M. Hasegawa and N. Kotake, Mechanochemical polymerization of methyl methacrylate initiated by the grinding of quartz in an n-heptane solvent, Powder Technol., 2013, 235, 336–340 CrossRef CAS
- M. Kimata, T. Sato and M. Hasegawa, Mechanochemical Polymerization of Styrene by Grinding of the Minerals at Bead Mill, J. Soc. Powder Technol., Jpn., 2014, 51, 424–429 CrossRef CAS
- M. Kimata, S. Suzuki, N. Kotake and M. Hasegawa, Preparation of Composite Materials of Waste Clay and Polymethyl Methacrylate Using Mechanochemial Polymerization in an Organic Solvent, J. Soc. Powder Technol., Jpn., 2014, 51, 10–15 CrossRef CAS
- C. V. Oprea and M. Popa, Mechanochemically initiated polymerizations II. Influence of some factors on the mechanochemical homopolymerization of acrylonitrile by vibratory milling, Angew. Makromol. Chem., 1980, 90, 13–22 CrossRef CAS
- H. Riazi, A. A. Shamsabadi, M. C. Grady, A. M. Rappe and M. Soroush, Experimental and Theoretical Study of the Self-Initiation Reaction of Methyl Acrylate in Free-Radical Polymerization, Ind. Eng. Chem. Res., 2018, 57, 532–539 CrossRef CAS
- K. S. Khuong, W. H. Jones, W. A. Pryor and K. N. Houk, The mechanism of the self-initiated thermal polymerization of styrene. Theoretical solution of a classic problem, J. Am. Chem. Soc., 2005, 127, 1265–1277 CrossRef CAS PubMed
- N. Doi, Y. Sasai, Y. Yamauchi, T. Adachi, M. Kuzuya and S.-I. Kondo, Development of Novel Polymeric Prodrugs Synthesized by Mechanochemical Solid-State Copolymerization of Hydroxyethylcellulose and Vinyl Monomers, Chem. Pharm. Bull., 2015, 992–997 CrossRef CAS PubMed
- S.-I. Kondo, S. Hosaka and M. Kuzuya, Mechanochemical Solid-State Polymerization. VIII. Novel Composite Polymeric Prodrugs Prepared by Mechanochemical Polymerization in the Presence of Pharmaceutical Aids, Chem. Pharm. Bull., 1998, 46, 669–673 CrossRef CAS PubMed
- S.-I. Kondo, K. Yamamoto, Y. Sawama, Y. Sasai, Y. Yamauchi and M. Kuzuya, Characterization of Novel pH-Sensitive Polymeric Micelles Prepared by the Self-Assembly
of Amphiphilic Block Copolymer with Poly-4-vinylpyridine Block Synthesized by Mechanochemical Solid-State Polymerization, Chem. Pharm. Bull., 2011, 59, 1200–1202 CrossRef CAS PubMed
- M. Destarac, Industrial development of reversible-deactivation radical polymerization: is the induction period over?, Polym. Chem., 2018, 9, 4947–4967 RSC
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Reversible-deactivation radical polymerization (Controlled/living radical polymerization): from discovery to materials design and applications, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS
- M. Pike and W. F. Watson, Mastication of rubber, I. Mechanism of plasticizing by cold mastication, J. Polym. Sci., 1952, 9, 229–251 CrossRef CAS
- M. Kuzuya, S.-I. Kondo, A. Noguchi and N. Noda, Nature of mechanoradical formation and reactivity with oxygen in methacrylic vinyl polymers, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 97–103 CrossRef CAS
- K. Goto and H. Fujiwara, Mechano-chemical polymerization of vinyl acetate, J. Polym. Sci., Part B: Polym. Lett., 1963, 1, 505–508 CrossRef CAS
- S.-I. Kondo, Y. Asano, N. Koizumi, K. Tatematsu, Y. Sawama, Y. Sasai, Y. Yamauchi, M. Kuzuya and S. Kurosawa, Novel pH-Responsive Polymeric Micelles Prepared through Self-assembly of Amphiphilic Block Copolymer with Poly-4-vinylpyridine Block Synthesized by Mechanochemical Solid-State Polymerization, Chem. Pharm. Bull., 2015, 63, 489–494 CrossRef PubMed
- K. Kubota, N. Toyoshima, D. Miura, J. Jiang, S. Maeda, M. Jin and H. Ito, Introduction of a Luminophore into Generic Polymers via Mechanoradical Coupling with a Prefluorescent Reagent, Angew. Chem., Int. Ed., 2021, 60, 16003–16008 CrossRef CAS PubMed
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Organocatalytic ring-opening polymerization, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed
-
J. E. McGrath, in Sustainability & Green Polymer Chemistry: Biocatalysis and Biobased Polymers, ed. H. N. Cheng and R. A. Gross, American Chemical Society, Washington, DC, 2020, vol. 2, pp. 1–22 Search PubMed
- C. Jérôme and P. Lecomte, Recent advances in the synthesis of aliphatic polyesters by ring-opening polymerization, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef PubMed
- G. S. Lee, B. R. Moon, H. Jeong, J. Shin and J. G. Kim, Mechanochemical synthesis of poly(lactic acid) block copolymers: overcoming the miscibility of the macroinitiator, monomer and catalyst under solvent-free conditions, Polym. Chem., 2019, 10, 539–545 RSC
- T. F. Burton, J. Pinaud, N. Pétry, F. Lamaty and O. Giani, Simple and Rapid Mechanochemical Synthesis of Lactide and 3S-(Isobutyl)morpholine-2,5-dione-Based Random Copolymers Using DBU and Thiourea, ACS Macro Lett., 2021, 10, 1454–1459 CrossRef CAS
- W. H. Carothers, Polymers and polyfunctionality, Trans. Faraday Soc., 1936, 32, 39 RSC
- T. Abdiryim, R. Jamal and I. Nurulla, Doping effect of organic sulphonic acids on the solid-state synthesized polyaniline, J. Appl. Polym. Sci., 2007, 105, 576–584 CrossRef CAS
- J. Gong, X.-J. Cui, Z.-W. Xie, S.-G. Wang and L.-Y. Qu, The solid-state synthesis of polyaniline/H4SiW12O40 materials, Synth. Met., 2002, 129, 187–192 CrossRef CAS
- T. Abdiryim, Z. Xiao-Gang and R. Jamal, Comparative studies of solid-state synthesized polyaniline doped with inorganic acids, Mater. Chem. Phys., 2005, 90, 367–372 CrossRef CAS
- X.-S. Du, C.-F. Zhou, G.-T. Wang and Y.-W. Mai, Novel Solid-State and Template-Free Synthesis of Branched Polyaniline Nanofibers, Chem. Mater., 2008, 20, 3806–3808 CrossRef CAS
- I. Bekri-Abbes and E. Srasra, Investigation of structure and conductivity properties of polyaniline synthesized by solid–solid reaction, J. Polym. Res., 2011, 18, 659–665 CrossRef CAS
- S. Bhadra, N. H. Kim, K. Y. Rhee and J. H. Lee, Preparation of nanosize polyaniline by solid-state polymerization and determination of crystal structure, Polym. Int., 2009, 58, 1173–1180 CrossRef CAS
- J. Huang, J. A. Moore, J. H. Acquaye and R. B. Kaner, Mechanochemical Route to the Conducting Polymer Polyaniline, Macromolecules, 2005, 38, 317–321 CrossRef CAS
- O. Y. Posudievsky, O. A. Goncharuk, R. Barillé and V. D. Pokhodenko, Structure–property relationship in mechanochemically prepared polyaniline, Synth. Met., 2010, 160, 462–467 CrossRef CAS
- T. Abdiryim, R. Jamal, C. Zhao, T. Awut and I. Nurulla, Structure and properties of solid-state synthesized poly(3′,4′-ethylenedioxy-2,2′:5′,2′′-terthiophene), Synth. Met., 2010, 160, 325–332 CrossRef CAS
- X. Mamtimin, R. Matsidik, A. Obulda, S. Sidik and I. Nurulla, Solid-state polymerization of EDOT derivatives containing acenaphthenequinoxaline ring, Fibers Polym., 2013, 14, 1066–1072 CrossRef CAS
- T. Hirai, Y. Nagae, K. L. White, K. Kamitani, M. Kido, T. Uchiyama, M. Nishibori, Y. Konishi, K. Yokomachi, R. Sugimoto, K. Saigo, T. Ohishi, Y. Higaki, K. Kojio and A. Takahara, Solvent free oxidative coupling polymerization of 3-hexylthiophene (3HT) in the presence of FeCl3 particles, RSC Adv., 2016, 6, 111993–111996 RSC
- A. Kumar, R. Singh, S. P. Gopinathan and A. Kumar, Solvent free chemical oxidative polymerization as a universal method for the synthesis of ultra high molecular weight conjugated polymers based on 3,4-propylenedioxythiophenes, Chem. Commun., 2012, 48, 4905–4907 RSC
- M. S. Zoromba and M. H. Abdel-Aziz, Ecofriendly method to synthesize poly(o-aminophenol) based on solid state polymerization and fabrication of nanostructured semiconductor thin film, Polymer, 2017, 120, 20–29 CrossRef CAS
- S. Grätz and L. Borchardt, Mechanochemical polymerization – controlling a polycondensation reaction between a diamine and a dialdehyde in a ball mill, RSC Adv., 2016, 6, 64799–64802 RSC
- C. G. Vogt, S. Grätz, S. Lukin, I. Halasz, M. Etter, J. D. Evans and L. Borchardt, Direct Mechanocatalysis: Palladium as Milling Media and Catalyst in the Mechanochemical Suzuki Polymerization, Angew. Chem., Int. Ed., 2019, 58, 18942–18947 CrossRef CAS PubMed
- T. Rensch, S. Fabig, S. Graetz and L. Borchardt, Mechanochemically-assisted Synthesis of Polyimides, ChemSusChem, 2022, 15(1), e202101975, DOI:10.1002/cssc.202101975
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Synthesis of functional polymers by post-polymerization modification, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, 50th Anniversary Perspective: Polymer Functionalization, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS
- T. Di Nardo and A. Moores, Mechanochemical amorphization of chitin: impact of apparatus material on performance and contamination, Beilstein J. Org. Chem., 2019, 15, 1217–1225 CrossRef CAS PubMed
- W. Qiu, F. Zhang, T. Endo and T. Hirotsu, Milling-induced esterification between cellulose and maleated polypropylene, J. Appl. Polym. Sci., 2004, 91, 1703–1709 CrossRef CAS
- B. G. Fiss, L. Hatherly, R. S. Stein, T. Friščić and A. Moores, Mechanochemical Phosphorylation of Polymers and Synthesis of Flame-Retardant Cellulose Nanocrystals, ACS Sustainable Chem. Eng., 2019, 7, 7951–7959 CrossRef CAS
- W. Qiu and T. Hirotsu, A New Method to Prepare Maleic Anhydride Grafted Poly(propylene), Macromol. Chem. Phys., 2005, 206, 2470–2482 CrossRef CAS
- W. Qiu, T. Endo and T. Hirotsu, A novel technique for preparing of maleic anhydride grafted polyolefins, Eur. Polym. J., 2005, 41, 1979–1984 CrossRef CAS
- J. W. Lee, J. Park, J. Lee, S. Park, J. G. Kim and B.-S. Kim, Solvent-Free Mechanochemical Post-Polymerization Modification of Ionic Polymers, ChemSusChem, 2021, 14, 3801–3805 CrossRef CAS PubMed
- M. Y. Malca, P.-O. Ferko, T. Friščić and A. Moores, Solid-state mechanochemical ω-functionalization of poly(ethylene glycol), Beilstein J. Org. Chem., 2017, 13, 1963–1968 CrossRef CAS PubMed
- S. Kitagawa, R. Kitaura and S. Noro, Functional porous coordination polymers, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed
- P. Kaur, J. T. Hupp and S. T. Nguyen, Porous Organic Polymers in Catalysis: Opportunities and Challenges, ACS Catal., 2011, 1, 819–835 CrossRef CAS
- Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Porous polymer catalysts with hierarchical structures, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC
- N. B. McKeown and P. M. Budd, Polymers of intrinsic microporosity (PIMs): organic materials for membrane separations, heterogeneous catalysis and hydrogen storage, Chem. Soc. Rev., 2006, 35, 675–683 RSC
- K. Dong, Q. Sun, X. Meng and F.-S. Xiao, Strategies for the design of porous polymers as efficient heterogeneous catalysts: from co-polymerization to self-polymerization, Catal. Sci. Technol., 2017, 7, 1028–1039 RSC
- S. Kim, B. Kim, N. A. Dogan and C. T. Yavuz, Sustainable Porous Polymer Catalyst for Size-Selective Cross-Coupling Reactions, ACS Sustainable Chem. Eng., 2019, 7, 10865–10872 CrossRef CAS
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Conjugated microporous poly(aryleneethynylene) networks, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed
- O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Synthetic Strategies, Structure Patterns, and Emerging Properties in the Chemistry of Modular Porous Solids, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS
- A. N. Li, R.-F. Lu, Y. Wang, X. Wang, K.-L. Han and W.-Q. Deng, Lithium-doped conjugated microporous polymers for reversible hydrogen storage, Angew. Chem., Int. Ed., 2010, 49, 3330–3333 CrossRef CAS PubMed
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Porous Polymer Networks: Synthesis, Porosity, and Applications in Gas Storage/Separation, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS
- K. Cousins and R. Zhang, Highly Porous Organic Polymers for Hydrogen Fuel Storage, Polymers, 2019, 11(4), 690 CrossRef CAS PubMed
- L. Borchardt, M. E. Casco and J. Silvestre-Albero, Methane Hydrate in Confined Spaces: An Alternative Storage System, ChemPhysChem, 2018, 19, 1298–1314 CrossRef CAS PubMed
- M. E. Casco, F. Rey, J. L. Jordá, S. Rudić, F. Fauth, M. Martínez-Escandell, F. Rodríguez-Reinoso, E. V. Ramos-Fernández and J. Silvestre-Albero, Paving the way for methane hydrate formation on metal-organic frameworks (MOFs), Chem. Sci., 2016, 7, 3658–3666 RSC
- V. Rozyyev, D. Thirion, R. Ullah, J. Lee, M. Jung, H. Oh, M. Atilhan and C. T. Yavuz, High-capacity methane storage in flexible alkane-linked porous aromatic network polymers, Nat. Energy, 2019, 4, 604–611 CrossRef CAS
- L. Wang, J. Ding, S. Sun, B. Zhang, X. Tian, J. Zhu, S. Song, B. Liu, X. Zhuang and Y. Chen, Viologen-Hypercrosslinked Ionic Porous Polymer Films as Active Layers for Electronic and Energy Storage Devices, Adv. Mater. Interfaces, 2018, 5, 1701679 CrossRef
- W. Li, J. Liu and D. Zhao, Mesoporous materials for energy conversion and storage devices, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS
- D. Xu, J. Guo and F. Yan, Porous ionic polymers: Design, synthesis, and applications, Prog. Polym. Sci., 2018, 79, 121–143 CrossRef CAS
- K. Ariga, A. Vinu, Y. Yamauchi, Q. Ji and J. P. Hill, Nanoarchitectonics for Mesoporous Materials, Bull. Chem. Soc. Jpn., 2012, 85, 1–32 CrossRef CAS
-
A. M. P. Peedikakkal and N. N. Adarsh, in Cellulose-Based Superabsorbent Hydrogels, ed. M. I. H. Mondal, Springer International Publishing; Imprint, Springer, Cham, 2020, pp. 1–44 Search PubMed
- S. Noro, J. Mizutani, Y. Hijikata, R. Matsuda, H. Sato, S. Kitagawa, K. Sugimoto, Y. Inubushi, K. Kubo and T. Nakamura, Porous coordination polymers with ubiquitous and biocompatible metals and a neutral bridging ligand, Nat. Commun., 2015, 6, 5851 CrossRef CAS PubMed
- J.-S. M. Lee, K. Otake and S. Kitagawa, Transport properties in porous coordination polymers, Coord. Chem. Rev., 2020, 421, 213447 CrossRef CAS
- T. Zhang, G. Xing, W. Chen and L. Chen, Porous organic polymers: a promising platform for efficient photocatalysis, Mater. Chem. Front., 2020, 4, 332–353 RSC
- T.-X. Wang, H.-P. Liang, D. A. Anito, X. Ding and B.-H. Han, Emerging applications of porous organic polymers in visible-light photocatalysis, J. Mater. Chem. A, 2020, 8, 7003–7034 RSC
- M.-Y. Wang, Q.-J. Zhang, Q.-Q. Shen, Q.-Y. Li and S.-J. Ren, Truxene-based Conjugated Microporous Polymers via Different Synthetic Methods, Chin. J. Polym. Sci., 2020, 38, 151–157 CrossRef CAS
- X.-J. Zhang, N. Bian, L.-J. Mao, Q. Chen, L. Fang, A.-D. Qi and B.-H. Han, Porous Polybenzimidazoles via Template-Free Suzuki Coupling Polymerization: Preparation, Porosity, and Heterogeneous Catalytic Activity in Knoevenagel Condensation Reactions, Macromol. Chem. Phys., 2012, 213, 1575–1581 CrossRef CAS
- L. Tan and B. Tan, Hypercrosslinked porous polymer materials: design, synthesis, and applications, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC
- C.-A. Tao and J.-F. Wang, Synthesis of Metal Organic Frameworks by Ball-Milling, Crystals, 2021, 11, 15 CrossRef CAS
- Z. Wang, Z. Li, M. Ng and P. J. Milner, Rapid mechanochemical synthesis of metal-organic frameworks using exogenous organic base, Dalton Trans., 2020, 49, 16238–16244 RSC
- M. E. Casco, E. Zhang, S. Grätz, S. Krause, V. Bon, D. Wallacher, N. Grimm, D. M. Többens, T. Hauß and L. Borchardt, Experimental Evidence of Confined Methane Hydrate in Hydrophilic and Hydrophobic Model Carbons, J. Phys. Chem. C, 2019, 123, 24071–24079 CrossRef CAS
- D. Leistenschneider, N. Jäckel, F. Hippauf, V. Presser and L. Borchardt, Mechanochemistry-assisted synthesis of hierarchical porous carbons applied as supercapacitors, Beilstein, J. Org. Chem., 2017, 13, 1332–1341 CAS
- D. Leistenschneider, K. Wegner, C. Eßbach, M. Sander, C. Schneidermann and L. Borchardt, Tailoring the porosity of a mesoporous carbon by a solvent-free mechanochemical approach, Carbon, 2019, 147, 43–50 CrossRef CAS
- D. Leistenschneider, K. Zürbes, C. Schneidermann, S. Grätz, S. Oswald, K. Wegner, B. Klemmed, L. Giebeler, A. Eychmüller and L. Borchardt, Mechanochemical Functionalization of Carbon Black at Room Temperature, C, 2018, 4, 14 Search PubMed
- C. Schneidermann, C. Kensy, P. Otto, S. Oswald, L. Giebeler, D. Leistenschneider, S. Grätz, S. Dörfler, S. Kaskel and L. Borchardt, Nitrogen-Doped Biomass-Derived Carbon Formed by Mechanochemical Synthesis for Lithium-Sulfur Batteries, ChemSusChem, 2019, 12, 310–319 CrossRef CAS PubMed
- C. Schneidermann, P. Otto, D. Leistenschneider, S. Grätz, C. Eßbach and L. Borchardt, Upcycling of polyurethane waste by mechanochemistry: synthesis of N-doped porous carbon materials for supercapacitor applications, Beilstein J. Nanotechnol., 2019, 10, 1618–1627 CrossRef PubMed
- E. Zhang, G.-P. Hao, M. E. Casco, V. Bon, S. Grätz and L. Borchardt, Nanocasting in ball mills – combining ultra-hydrophilicity and ordered mesoporosity in carbon materials, J. Mater. Chem. A, 2018, 6, 859–865 RSC
- J. Zhu, J. Yang, R. Miao, Z. Yao, X. Zhuang and X. Feng, Nitrogen-enriched, ordered mesoporous carbons for potential electrochemical energy storage, J. Mater. Chem. A, 2016, 4, 2286–2292 RSC
- M. E. Casco, F. Badaczewski, S. Grätz, A. Tolosa, V. Presser, B. M. Smarsly and L. Borchardt, Mechanochemical synthesis of porous carbon at room temperature with a highly ordered sp2 microstructure, Carbon, 2018, 139, 325–333 CrossRef CAS
- M. E. Casco, S. Kirchhoff, D. Leistenschneider, M. Rauche, E. Brunner and L. Borchardt, Mechanochemical synthesis of N-doped porous carbon at room temperature, Nanoscale, 2019, 11, 4712–4718 RSC
- Z. Zhang, C. Zhu, N. Sun, H. Wang, Z. Tang, W. Wei and Y. Sun, One-Pot Solvent-Free Synthesis of Nitrogen and Magnesium Codoped Mesoporous Carbon Composites for CO2 Capture, J. Phys. Chem. C, 2015, 119, 9302–9310 CrossRef CAS
- Q. Wang, Y. Mu, W. Zhang, L. Zhong, Y. Meng and Y. Sun, A facile solvent-free route to synthesize ordered mesoporous carbons, RSC Adv., 2014, 4, 32113–32116 RSC
- R. Chan-Navarro, D. Corpus-Coronado, B. M. Muñoz-Flores, M. Loredo-Cancino, N. Waksman, R. Ramírez and V. M. Jiménez-Pérez, Luminescent Sensing of Volatile Organic Compounds Using a Zn-based Coordination Polymer with Tunable Morphology, J. Inorg. Organomet. Polym. Mater., 2017, 27, 467–473 CrossRef CAS
- P. Zhang, H. Li, G. M. Veith and S. Dai, Soluble porous coordination polymers by mechanochemistry: from metal-containing films/membranes to active catalysts for aerobic oxidation, Adv. Mater., 2015, 27, 234–239 CrossRef CAS PubMed
- M. Klimakow, P. Klobes, A. F. Thünemann, K. Rademann and F. Emmerling, Mechanochemical Synthesis of Metal−Organic Frameworks: A Fast and Facile Approach toward Quantitative Yields and High Specific Surface Areas, Chem. Mater., 2010, 22, 5216–5221 CrossRef CAS
-
T. Friščić, in Encyclopedia of inorganic and bioinorganic chemistry, ed. R. A. Scott, John Wiley and Sons, Inc, Hoboken, NJ, 2011, pp. 1–19 Search PubMed
- L. Tröbs, M. Wilke, W. Szczerba, U. Reinholz and F. Emmerling, Mechanochemical synthesis and characterisation of two new bismuth metal organic frameworks, CrystEngComm, 2014, 16, 5560–5565 RSC
- T.-H. Wei, S.-H. Wu, Y.-D. Huang, W.-S. Lo, B. P. Williams, S.-Y. Chen, H.-C. Yang, Y.-S. Hsu, Z.-Y. Lin, X.-H. Chen, P.-E. Kuo, L.-Y. Chou, C.-K. Tsung and F.-K. Shieh, Rapid mechanochemical encapsulation of biocatalysts into robust metal-organic frameworks, Nat. Commun., 2019, 10, 5002 CrossRef PubMed
- A. D. Katsenis, A. Puškarić, V. Štrukil, C. Mottillo, P. A. Julien, K. Užarević, M.-H. Pham, T.-O. Do, S. A. J. Kimber, P. Lazić, O. Magdysyuk, R. E. Dinnebier, I. Halasz and T. Friščić, In situ X-ray diffraction monitoring of a mechanochemical reaction reveals a unique topology metal-organic framework, Nat. Commun., 2015, 6, 6662 CrossRef CAS PubMed
- X. Zhu, C. Tian, T. Jin, K. L. Browning, R. L. Sacci, G. M. Veith and S. Dai, Solid-State Synthesis of Conjugated Nanoporous Polycarbazoles, ACS Macro Lett., 2017, 6, 1056–1059 CrossRef CAS
- C. Schneidermann, N. Jäckel, S. Oswald, L. Giebeler, V. Presser and L. Borchardt, Solvent-Free Mechanochemical Synthesis of Nitrogen-Doped Nanoporous Carbon for Electrochemical Energy Storage, ChemSusChem, 2017, 10, 2416–2424 CrossRef CAS PubMed
- Y. G. Kryazhev, I. V. Anikeeva, M. V. Trenikhin, A. B. Arbuzov, T. I. Gulyaeva and V. A. Drozdov, The Use of Mechanical Activation of Polyvinyl Chloride in the Presence of Alkalis for Synthesis of Porous Carbon Materials, Prot. Met. Phys. Chem. Surf., 2020, 56, 712–715 CrossRef CAS
- D. Leistenschneider, L. H. Heß, A. Balducci and L. Borchardt, Solid-state transformation of aqueous to organic electrolyte – Enhancing the operating voltage window of ‘ in situ electrolyte’ supercapacitors, Sustainable Energy Fuels, 2020, 4, 2438–2447 RSC
- D. Leistenschneider, C. Schneidermann, F. Hippauf, S. Grätz and L. Borchardt, The “In Situ Electrolyte” Concept: Using Activation Chemicals as Electrolytes for Carbon-Based Supercapacitors, Adv. Sustainable Syst., 2018, 2, 1800087 CrossRef
- X. Xie, J. Wang, J. Zheng, J. Huang, C. Ni, J. Cheng, Z. Hao and G. Ouyang, Low-cost Scholl-coupling microporous polymer as an efficient solid-phase microextraction coating for the detection of light aromatic compounds, Anal. Chim. Acta, 2018, 1029, 30–36 CrossRef CAS PubMed
- T. Jin, Y. Xiong, X. Zhu, Z. Tian, D.-J. Tao, J. Hu, D. Jiang, H. Wang, H. Liu and S. Dai, Rational design and synthesis of a porous, task-specific polycarbazole for efficient CO2 capture, Chem. Commun., 2016, 52, 4454–4457 RSC
- F. Jiang, T. Jin, X. Zhu, Z. Tian, C.-L. Do-Thanh, J. Hu, D. Jiang, H. Wang, H. Liu and S. Dai, Substitution Effect Guided Synthesis of Task-Specific Nanoporous Polycarbazoles with Enhanced Carbon Capture, Macromolecules, 2016, 49, 5325–5330 CrossRef CAS
- Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, Microporous Polycarbazole with High Specific Surface Area for Gas Storage and Separation, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS PubMed
- X. Zhu, Y. Hua, C. Tian, C. W. Abney, P. Zhang, T. Jin, G. Liu, K. L. Browning, R. L. Sacci, G. M. Veith, H.-C. Zhou, W. Jin and S. Dai, Accelerating Membrane-based CO2 Separation by Soluble Nanoporous Polymer Networks Produced by Mechanochemical Oxidative Coupling, Angew. Chem., Int. Ed., 2018, 57, 2816–2821 CrossRef CAS PubMed
- J. Schmidt, J. Weber, J. D. Epping, M. Antonietti and A. Thomas, Microporous Conjugated Poly(thienylene arylene) Networks, Adv. Mater., 2009, 21, 702–705 CrossRef CAS
- J.-S. M. Lee, T. Kurihara and S. Horike, Five-Minute Mechanosynthesis of Hypercrosslinked Microporous Polymers, Chem. Mater., 2020, 32, 7694–7702 CrossRef CAS
- H. Zhou, Q. Chen, X. Tong and H. Liu, Hypercrosslinked mesoporous polymers: Synthesis, characterization and its application for catalytic reduction of 4-nitrophenol, Colloids Interface Sci. Commun., 2020, 37, 100285 CrossRef CAS
- K. Schute and M. Rose, Metal-free and Scalable Synthesis of Porous Hyper-cross-linked Polymers: Towards Applications in Liquid-Phase Adsorption, ChemSusChem, 2015, 8, 3419–3423 CrossRef CAS PubMed
- V. Rozyyev, Y. Hong, M. S. Yavuz, D. Thirion and C. T. Yavuz, Extensive Screening of Solvent-Linked Porous Polymers through Friedel-Crafts Reaction for Gas Adsorption, Adv. Energy Sustainable Res., 2021, 2100064 CrossRef
- A. Krusenbaum, J. Geisler, F. J. L. Kraus, S. Grätz, M. V. Höfler, T. Gutmann and L. Borchardt, The mechanochemical Friedel-Crafts polymerization as a solvent-free cross-linking approach toward microporous polymers, J. Polym. Sci., 2022, 60(1), 62 CrossRef CAS
- Q. Pan, Z. Xu, S. Deng, F. Zhang, H. Li, Y. Cheng, L. Wei, J. Wang and B. Zhou, A mechanochemically synthesized porous organic polymer derived CQD/chitosan–graphene composite film electrode for electrochemiluminescence determination of dopamine, RSC Adv., 2019, 9, 39332–39337 RSC
- P. Kuhn, M. Antonietti and A. Thomas, Porous, covalent triazine-based frameworks prepared by ionothermal synthesis, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Porous, fluorescent, covalent triazine-based frameworks via room-temperature and microwave-assisted synthesis, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS PubMed
- X. Zhu, C. Tian, S. M. Mahurin, S.-H. Chai, C. Wang, S. Brown, G. M. Veith, H. Luo, H. Liu and S. Dai, A superacid-catalyzed synthesis of porous membranes based on triazine frameworks for CO2 separation, J. Am. Chem. Soc., 2012, 134, 10478–10484 CrossRef CAS PubMed
- E. Troschke, S. Grätz, T. Lübken and L. Borchardt, Mechanochemical Friedel-Crafts Alkylation-A Sustainable Pathway Towards Porous Organic Polymers, Angew. Chem., Int. Ed., 2017, 56, 6859–6863 CrossRef CAS PubMed
- S. Grätz, M. de Olivera Junior, T. Gutmann and L. Borchardt, A comprehensive approach for the characterization of porous polymers using 13C and 15N dynamic nuclear polarization NMR spectroscopy, Phys. Chem. Chem. Phys., 2020, 22, 23307–23314 RSC
- C. S. Diercks and O. M. Yaghi, The atom, the molecule, and the covalent organic framework, Science, 2017, 355(6328), eaal1585 CrossRef PubMed
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Porous, crystalline, covalent organic frameworks, Science, 2005, 310, 1166–1170 CrossRef PubMed
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Designed synthesis of 3D covalent organic frameworks, Science, 2007, 316, 268–272 CrossRef CAS PubMed
- P. Zhang and S. Dai, Mechanochemical synthesis of porous organic materials, J. Mater. Chem. A, 2017, 5, 16118–16127 RSC
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, Mechanochemical synthesis of chemically stable isoreticular covalent organic frameworks, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed
- Y. Peng, G. Xu, Z. Hu, Y. Cheng, C. Chi, D. Yuan, H. Cheng and D. Zhao, Mechanoassisted Synthesis of Sulfonated Covalent Organic Frameworks with High Intrinsic Proton Conductivity, ACS Appl. Mater. Interfaces, 2016, 8, 18505–18512 CrossRef CAS PubMed
- G. Das, D. Balaji Shinde, S. Kandambeth, B. P. Biswal and R. Banerjee, Mechanosynthesis of imine, β-ketoenamine, and hydrogen-bonded imine-linked covalent organic frameworks using liquid-assisted grinding, Chem. Commun., 2014, 50, 12615–12618 RSC
- Y. Yang, F. Bu, J. Liu, I. Shakir and Y. Xu, Mechanochemical synthesis of two-dimensional aromatic polyamides, Chem. Commun., 2017, 53, 7481–7484 RSC
- G. Li, J. Ye, Q. Fang and F. Liu, Amide-based covalent organic frameworks materials for efficient and recyclable removal of heavy metal lead(II), Chem. Eng. J., 2019, 370, 822–830 CrossRef CAS
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, A mechanochemically synthesized covalent organic framework as a proton-conducting solid electrolyte, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, Constructing Ultraporous Covalent Organic Frameworks in Seconds via an Organic Terracotta Process, J. Am. Chem. Soc., 2017, 139, 1856–1862 CrossRef CAS PubMed
- K. Preet, G. Gupta, M. Kotal, S. K. Kansal, D. B. Salunke, H. K. Sharma, S. Chandra Sahoo, P. van der Voort and S. Roy, Mechanochemical Synthesis of a New Triptycene-Based Imine-Linked Covalent Organic Polymer for Degradation of Organic Dye, Cryst. Growth Des., 2019, 19, 2525–2530 CrossRef CAS
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, Chemically stable multilayered covalent organic nanosheets from covalent organic frameworks via mechanical delamination, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed
- C. Zou, Q. Li, Y. Hua, B. Zhou, J. Duan and W. Jin, Mechanical Synthesis of COF Nanosheet Cluster and Its Mixed Matrix Membrane for Efficient CO2 Removal, ACS Appl. Mater. Interfaces, 2017, 9, 29093–29100 CrossRef CAS PubMed
- H. Lv, X. Zhao, H. Niu, S. He, Z. Tang, F. Wu and J. P. Giesy, Ball milling synthesis of covalent organic framework as a highly active photocatalyst for degradation of organic contaminants, J. Hazard. Mater., 2019, 369, 494–502 CrossRef CAS PubMed
- L. Rajput and R. Banerjee, Mechanochemical Synthesis of Amide Functionalized Porous Organic Polymers, Cryst. Growth Des., 2014, 14, 2729–2732 CrossRef CAS
- K. Jie, Y. Zhou, Q. Sun, B. Li, R. Zhao, D. Jiang, W. Guo, H. Chen, Z. Yang, F. Huang and S. Dai, Mechanochemical synthesis of pillar5quinone derived multi-microporous organic polymers for radioactive organic iodide capture and storage, Nat. Commun., 2020, 11, 1086 CrossRef CAS PubMed
- P. Zhang, X. Jiang, S. Wan and S. Dai, Advancing polymers of intrinsic microporosity by mechanochemistry, J. Mater. Chem. A, 2015, 3, 6739–6741 RSC
- P. Zhang, X. Jiang, S. Wan and S. Dai, Charged Porous Polymers using a Solid C–O Cross-Coupling Reaction, Chem. – Eur. J., 2015, 21, 12866–12870 CrossRef CAS PubMed
- Y. H. Kim and O. W. Webster, Hyperbranched polyphenylenes, Macromolecules, 1992, 5561–5572 CrossRef CAS
- H. Chen, J. Fan, Y. Fu, C.-L. Do-Thanh, X. Suo, T. Wang, I. Popovs, D. Jiang, Y. Yuan, Z. Yang and S. Dai, Benzene Ring Knitting Achieved by Ambient-Temperature Dehalogenation via Mechanochemical Ullmann-Type Reductive Coupling, Adv. Mater., 2021, 33, e2008685 CrossRef PubMed
-
P. T. Anastas and J. C. Warner, Green chemistry. Theory and practice, Oxford Univ. Press, Oxford, 1998 Search PubMed
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, The Green ChemisTREE: 20 years after taking root with the 12 principles, Green Chem., 2018, 20, 1929–1961 RSC
- R. A. Sheldon, Organic synthesis-past, present and future., Chem. Ind., 1992, 903–906 CAS
- R. A. Sheldon, Metrics of Green Chemistry and Sustainability: Past, Present, and Future, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS
- B. M. Trost, The atom economy–a search for synthetic efficiency, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Metrics to ‘green’ chemistry—which are the best?, Green Chem., 2002, 4, 521–527 RSC
- M. Tobiszewski, M. Marć, A. Gałuszka and J. Namieśnik, Green Chemistry Metrics with Special Reference to Green Analytical Chemistry, Molecules, 2015, 20, 10928–10946 CrossRef CAS PubMed
- A. D. Curzons, D. N. Mortimer, D. J. C. Constable and V. L. Cunningham, So you think your process is green, how do you know?—Using principles of sustainability to determine what is green – a corporate perspective, Green Chem., 2001, 3, 1–6 RSC
- M. Rose, N. Klein, I. Senkovska, C. Schrage, P. Wollmann, W. Böhlmann, B. Böhringer, S. Fichtner and S. Kaskel, A new route to porous monolithic organic frameworks via cyclotrimerization, J. Mater. Chem., 2011, 21, 711–716 RSC
- R. Thorwirth, F. Bernhardt, A. Stolle, B. Ondruschka and J. Asghari, Switchable selectivity during oxidation of anilines in a ball mill, Chem. – Eur. J., 2010, 16, 13236–13242 CrossRef CAS PubMed
- R. Mülhaupt, Green Polymer Chemistry and Bio-based Plastics: Dreams and Reality, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef
| This journal is © The Royal Society of Chemistry 2022 |