
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring contraction in synthesis of functionalized carbocycles
Chunngai
Hui
ab,
Luke
Craggs
c and
Andrey P.
Antonchick
 *abc
*abc
aMax Planck Institute of Molecular Physiology, Department of Chemical Biology, Otto-Hahn-Strasse 11, 44227 Dortmund, Germany. E-mail: andrey.antonchick@ntu.ac.uk
bTechnical University Dortmund, Faculty of Chemistry and Chemical Biology, Otto-Hahn-Strasse 6, 44221 Dortmund, Germany
cNottingham Trent University, School of Science and Technology, Department of Chemistry and Forensics, Clifton Lane, NG11 8NS Nottingham, UK
First published on 29th September 2022
Abstract
Carbocycles are a key and widely present structural motif in organic compounds. The construction of structurally intriguing carbocycles, such as highly-strained fused rings, spirocycles or highly-functionalized carbocycles with congested stereocenters, remains challenging in organic chemistry. Cyclopropanes, cyclobutanes and cyclopentanes within such carbocycles can be synthesized through ring contraction. These ring contractions involve re-arrangement of and/or small molecule extrusion from a parental ring, which is either a carbocycle or a heterocycle of larger size. This review provides an overview of synthetic methods for ring contractions to form cyclopropanes, cyclobutanes and cyclopentanes en route to structurally intriguing carbocycles.
 Chunngai Hui | Chunngai Hui obtained his BSc in Chemical Technology from the Hong Kong Polytechnic University (China) and his MSc in biotechnology from Hong Kong University of Science and Technology (China). He completed his PhD at Max Planck Institute of Molecular Physiology under the supervision of Prof. A. P. Antonchick. His research interests include the synthesis of bioactive natural products and their related biological investigations. |
 Luke Craggs | Luke E. Craggs obtained his BSc and MRes degrees at Nottingham Trent University (UK) working under Prof. Andrey P. Antonchick. He is currently undertaking his PhD at the University of Nottingham (UK) working under Dr Mattia Silvi. He has particular interests in sustainable organic synthesis. |
 Andrey P. Antonchick | Andrey P. Antonchick studied at Belarusian State University. He received a PhD at the Institute of Bioorganic Chemistry of the National Academy of Sciences of Belarus and the Max Planck Institute for Chemical Ecology (Germany). After a postdoctoral appointment with Prof. M. Rueping at Frankfurt University, he joined Prof H. Waldmann at the Max Planck Institute of Molecular Physiology (Germany). In 2011, he was appointed group leader at the Max Planck Institute of Molecular Physiology and Technical University of Dortmund. Since August 2020, he holds the position of an Associate Professor at Nottingham Trent University (UK). |
1. Introduction
Carbocycles are omnipresent in chemical pharmaceuticals, biologically active natural products, and organic functional materials. The construction of structurally intriguing carbocycles, such as highly-strained fused rings,1,2 spirocycles,3 and highly-functionalized carbocycles with congested stereocenters,4,5 remains a challenging task in organic chemistry. Conventional transformations, such as cycloadditions,6–10 cyclizations,11–18 cascade reactions,19–25 ring expansion, and ring contraction, are readily accessible to synthesize carbocycles, which are subjected to further chemical transformations to afford the desired compounds. In particular, ring contraction involves the synthesis of cyclic compounds (e.g., carbocycles, heterocycles, metallocycles, etc.) from a parental compound with a larger ring size. Compared with medium and large carbocycles, for which a variety of methods can be resorted to in most cases, the construction of small carbocycles (i.e. three to five-membered) has fewer synthetic methods available. Especially when small carbocycles are highly substituted in nature and/or have a number of stereocenters, their synthesis is very challenging. More synthetic steps may be required to prepare these systems using alternative approaches such as direct ring closure chemistry. Moreover, the synthesis of small carbocycles containing two or more contiguous quaternary carbon centers is not trivial because few effective methods are available for achieving such sterically hindered structural motifs. The development of novel synthetic methods and/or synthetic strategies for small carbocycles containing two or more contiguous quaternary carbon centers continues to be a research hot spot for synthetic scientists.It should be noted that contractive synthesis of carbocycles have been used extensively in natural product synthesis to afford highly substituted cyclic compound possessing an array of stereocenters and/or sterically hindered quaternary carbon center(s) (Fig. 1). One remarkable example is the protecting group free, eight-step synthesis of (+)-welwitindolinone A (1) featured an oxidative, pinacol-type rearrangement to construct a cyclobutane from the cyclopentane moiety of 12-epi-fischerindole I (39)26 showing good atom-economy and good chemoselectivity (Scheme 1(A)). In contrast, the first total synthesis of rac-1 required 22 steps,27–29 involving the early construction of cyclobutanone 43 and required sequences of functional group transformations. The scarcity of direct synthetic method to forge the spirocyclobutane motif in the early stage poses difficulties for the synthetic route design that may cause more steps to be required to build up the molecule skeleton and embellish the necessary functional groups. Alternatively, the late-stage ring contraction of fused indole 39 affording spiro-cyclobutane 1 significantly improves the synthetic efficiency. Another representative example of ring contractive synthesis is the four-step synthesis of prostratin (28) from crotophorbolone30 (Scheme 1(B)). The late-stage, highly-chemoselective dinitrogen extrusion on pyrazoline 46 with UV irradiation producing the cyclopropane. Importantly, the dinitrogen extrusion process required no prior protection of reactive hydroxy groups and is redox neutral. The excellent chemoselectivity of contractive synthesis of cyclopropane from pyrazoline was adopted by recently reported 20-step total synthesis of prostratin (28)31 and other cyclopropane natural products. Up-to-date, no alternative synthetic protocol en route to prostratin (28) was reported. Noteworthy, the remarkable synthetic efficiency of Baran's synthesis of (+)-welwitindolinone A (8 steps, versus 22 steps of non-contractive approach) and Wender's semi-synthesis of prostratin (28) (4 steps) could be attributed to the chemoselective ring contraction of advanced and/or late-stage intermediates.
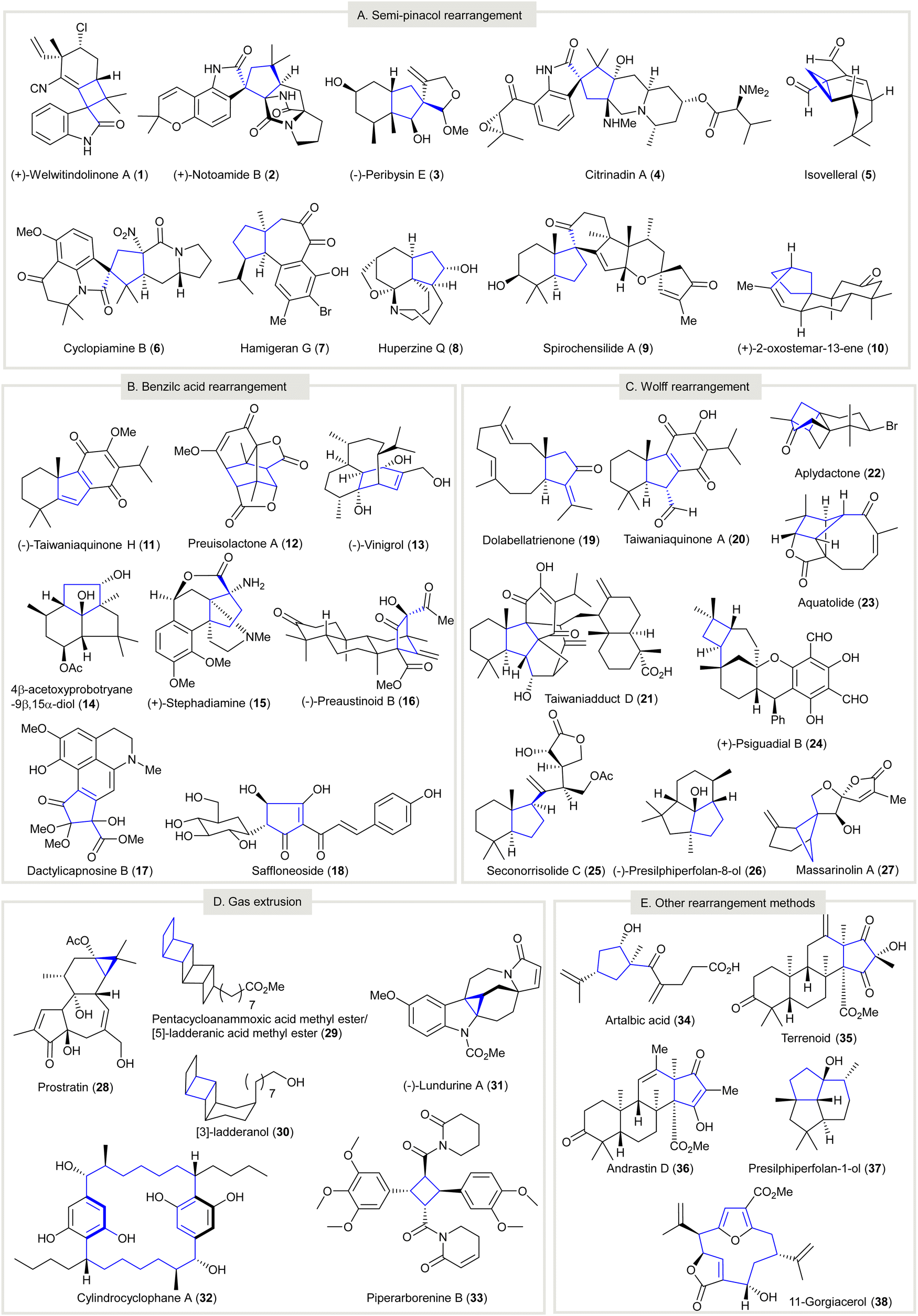 | ||
| Fig. 1 Selected natural products created by the contractive synthesis of carbocycles. (A) Semi-pinacol rearrangement. (B) Benzilic acid type rearrangement. (C) Wolff rearrangement. (D) Gas-extrusion. (E) Other rearrangement methods. | ||
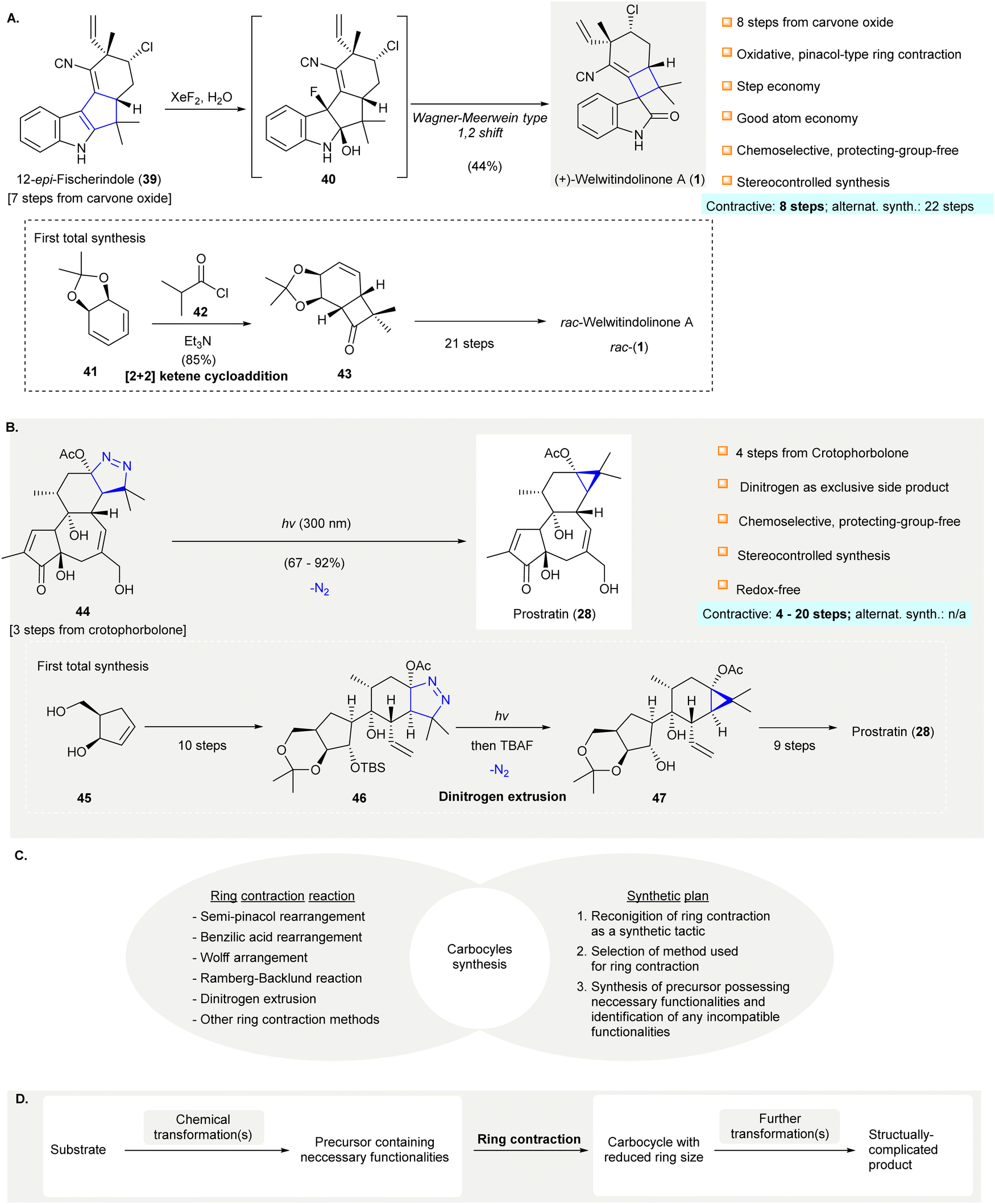 | ||
| Scheme 1 Examples of contractive carbocycle synthesis that comply with elements of efficient synthesis. (A) An oxidative pinacol-type rearrangement converted 12-epi-fisherindole (39) to oxindole (+)-welwitindolinone A (1)26 (inset, the non-contractive synthesis of rac-welwitindolinone A rac-(1)).27–29 (B) Photoinduced dinitrogen extrusion-ring contraction of pyrazoline 44 en route to prostatin (28).30 Dinitrogen extrusion in the 20-step total synthesis of prostratin (28).31 (C) Ring contraction methods and rational synthetic plan direct to successful synthesis of carbocycle. (D) A general strategy of applying ring contraction as a maneuver to prepare structurally complicated product. XeF2, xenon fluoride; TBAF, tetra-n-butylammonium fluoride. | ||
Historically, low synthetic efficiency made these practically useful but synthetically challenging small carbocycles32 inaccessible. Although various synthetic strategies have been proposed to improve the efficiency of synthetic design,33–41 and contractive synthesis of carbocycles has seen wide application in organic chemistry, they are still often overlooked (Scheme 1(C)). In general, 1,2-rearrangment methods (e.g. semi-pinacol rearrangement, benzilic acid rearrangement, Wolff rearrangement, etc.) and gas extrusion reactions (e.g. Ramberg–Backlund reaction, dinitrogen extrusion, etc.), which are well-documented, have mainly contributed to ring contraction methods. However, the use of such reactions often requires intricate synthetic design in which the precursor of ring contraction usually possesses skeletal framework structurally distinct to the final product, for instance, 39 and (+)-welwitindolinone A (1) (see Scheme 1(A)). Early recognition of ring contraction as a synthetic tactic for the target compound is essential so that ring contraction methods can be identified and a precursor carrying the necessary functional features for such transformation can be elaborated. After successfully preparing the desired carbocycle via ring contraction, the target product could be accomplished by further transformations. (Scheme 1(D)). As a promising strategy to improve synthetic efficiency, a systematic review of the contractive synthesis of carbocycles in organic synthesis could reveal significant success factors and provide insights for scientists in future research.
In this review, the synthetic applications of ring contraction enabling the synthesis of carbocyclic natural products (1–38, Fig. 1) from 2011 to 2021 were discussed. Selected examples reported before 2011 are introduced, providing a brief glance at the history of ring contraction in the synthesis of complex natural products. The representative synthesis of complex natural products using ring contraction as key strategy to fabricate skeletal carbocycles are illustrated. Methods used for the ring contraction in natural product synthesis are catalogized into three groups: 1,2-carbon-migrations, gas-extrusions, and miscellaneous rearrangements. Important information including the reaction scheme of ring contraction, the possible reaction intermediate(s) involved that hints at the reaction mechanism, and the resultant natural products are depicted. Although the contractive synthesis of carbocycles in this Review are categorized according to what we deem to be the key contributing factors, it is of note that contractive synthesis of carbocycles could be organized by other ways, for instance, ring contraction based on the changes in ring size. As such, the number of steps of either the first synthesis or, if applicable, the first asymmetric or enantioselective synthesis using an alternative approach (i.e. non-contractive) is denoted along with that of its ring contraction synthesis counterpart for comparison. Some elegant synthetic methods enabled contractive synthesis of carbocycles, which have not been applied to natural product synthesis, are also demonstrated. The transformations of each method are outlined, and how each contractive synthesis was successfully applied is discussed. Our motivation to compose this Review is to arouse the attention from the synthetic communities for ring contraction as an efficient approach to making carbocycles, especially the highly functionalized and small carbocycles. Ring contraction not included in this review are intramolecular cyclizations and cycloadditions, reductive elimination of metals from cyclic organometallic complexes, and intramolecular rearrangements resulting in simultaneous ring contractions and expansions in fused carbocycles. Finally, future method developments and applications of contractive synthesis in carbocycles were considered.
2. Selected early transformations
A 1,2-carbon migration between two vicinal atoms can create structural complexity. This is the principal mechanism of numerous, classical, named rearrangement reactions. One notable reaction is the semi-pinacol rearrangement; an organic transformation involving a 1,2-bond migration (C–C or C–H) centered on oxygen-containing carbons which migrate to vicinal electrophilic carbons generating carbonyl groups.42 The semi-pinacol reaction allows the contractive synthesis of smaller carbocycles and is applied widely in organic synthesis (Scheme 2).43–45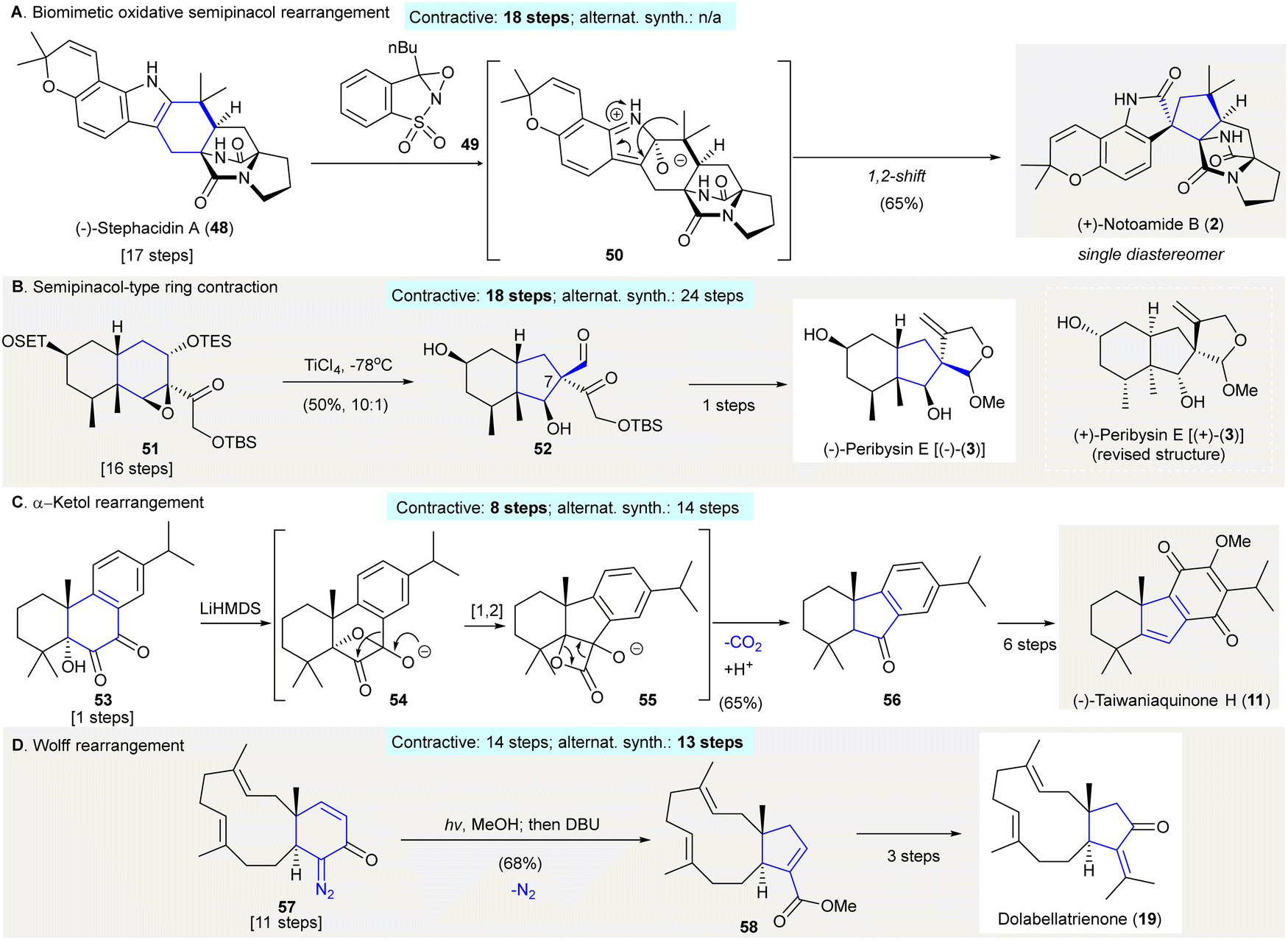 | ||
| Scheme 2 Contractive synthesis of carbocycles via 1,2-carbon migrations in organic synthesis. (A) Biomimetic, oxidative semi-pinacol rearrangement facilitated the conversion of (−)-stephacidin A (48) to (+)-notoamide B (2).47,48,61 (B) The synthesis of (−)-peribysin E [(−)-(3)] involved a semi-pinacol-type reaction to build the [6,5] fused ring in 52 from compound 51 with a [6,6] fused ring.53 (C) Benzilic acid rearrangement en route to (−)-taiwaniaquinone H (11).57 (D) Wolff arrangement featured as a key reaction in the synthesis of dolabellatrienone (19).59 | ||
The synthesis of oxindole (+)-notoamide B (2) was accomplished via a biomimetic, oxidative semi-pinacol rearrangement using Davis’ oxaziridine46 (49)47,48 (Scheme 2(A)). The regioselective epoxidation of (−)-stephacidin A (48) at the less sterically hindered α-face and subsequent epoxide opening gave intermediate 50. Ring contraction at the α-face of 50via a 1,2-shift successfully synthesized (+)-notoamide B (2) with 65% yield as a single diastereomer (contractive synthesis: 18 steps;47,48 alternative synthesis: not available) The biomimetic conversion of an indole structure to its corresponding oxindole using Davis’ oxaziridine was reported thereafter.49–52
The synthesis of (−)-peribysine E [(−)-(3)] used a semi-pinacol-type rearrangement to facilitate the contractive synthesis of a C7 quaternary center, on fused cyclopentane 5253,54 (Scheme 2(B)). Treatment of epoxide 51 with titanium chloride afforded cyclopentane 52 in 50% yield, and was converted to (−)-peribysine E [(−)-(3)] in one step55 (contractive synthesis: 18 steps;53,54 alternative synthesis: 24 steps56). The revised structure proved to be (+)-perbysine E [(+)-(3)] but was misassigned as (−)-peribysine E [(−)-(3)].
The synthesis of (−)-taiwaniaquinone H (11) featured a benzilic acid rearrangement forming the 6-5-6 tricyclic core of 5657 (Scheme 2(C)). Exposure of 1,2-diketone 53 to LiHMDS as a base resulted in the formation of oxetane intermediate 54, which underwent a 1,2-carbon migration giving intermediate lactone 55. Successive decarboxylation and protonation of 55 afforded the desired tricyclic ketone 56, a precursor to (−)-taiwaniaquinone H (11) (contractive synthesis: 8 steps;57 alternative synthesis: 14 steps58).
The synthesis of dolabellatrienone (19) relied on a Wolff rearrangement producing chiral dolabellane derivative 58 in 68% yield59 (Scheme 2(D)). Photoirradiation of the α,β-unsaturated diazoketone 57 followed by heating in neat DBU formed the ring-contracted ester 58, synthesized through an α-keto carbene intermediate (contractive synthesis: 14 steps;59 alternative synthesis: 13 steps60). The Wolff rearrangement is often used to construct 4 and 5 membered carbocycles via ring contraction in natural product synthesis.
TBS, tert-butyldimethylsilyl; TES, triethylsilyl; TiCl4, titanium tetrachloride; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; LiHMDS, lithium bis(trimethylsilyl)amide.
The synthesis of pentacycloanammoxic acid/[5]-ladderanoic acid methyl ester (29) featured a photo-induced dinitrogen extrusion giving pentacyclic ladderane ketone 6062 (Scheme 3(A)). After ketal protection of the bridged azo ketone 59, a photoinduced dinitrogen extrusion followed by deprotection affording fused cyclobutane 60 in 6% yield. It was theorized that the low yield of the dinitrogen extrusion was a result of fragmentation. The formation of unidentified oligomeric materials and problems involving polymerization were also reported in other bridged azo compounds.63–65 Later, the synthesis of [5]-ladderanoic acid methyl ester (29) was accomplished through a modified Ramberg–Bäcklund olefination66 (see Scheme 9(C)).
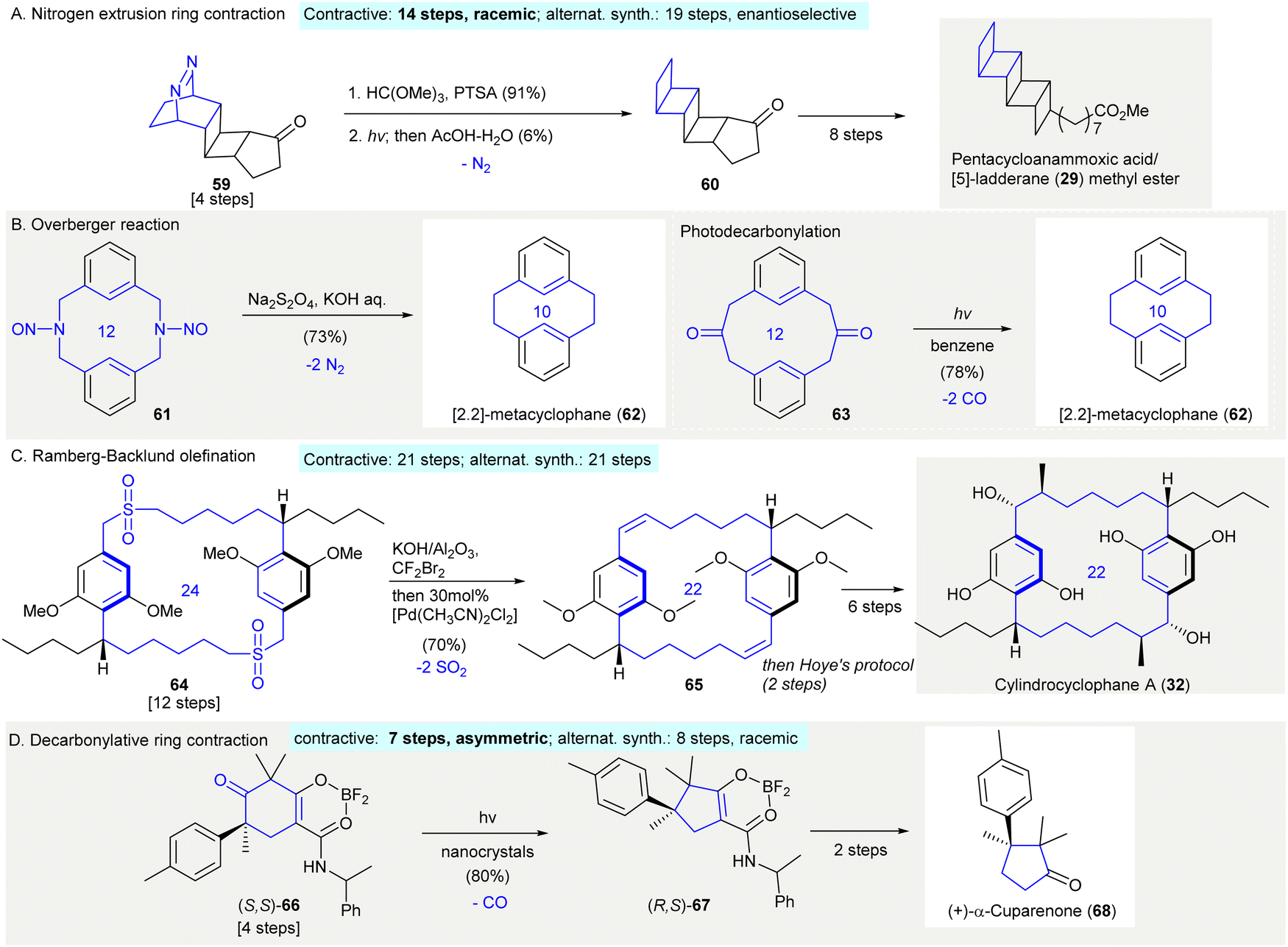 | ||
| Scheme 3 Contractive synthesis of carbocycles via gas extrusion in organic synthesis. (A) Dinitrogen extrusion-ring contraction of bridged azo ketone 59 to fused-cyclobutanes 60 in the synthesis of pentacycloanammoxic acid (29) methyl ester.62 (B) Synthesis of cyclophane 62 relied on the Overberger reaction.68 (inset: a photoinduced decarbonylation)69 (C) Double Ramberg–Bäcklund olefination resulted in macrocyclic diene 65 in the synthesis of cylindrocyclophane A (32).72 (D) Synthesis of (+)-α-cuparenone (68) relying on a solid-state photodecarbonylation.78 | ||
The Overberger reaction67 produced the 10-membered carbocycle [2,2]-metacyclophane (62) via reduction/dinitrogen extrusion68 (Scheme 3(B)). As described by Overberger,67 this reaction involved the reduction of a N-nitroso group using sodium dithionite under alkaline condition. In this reaction, dinitrogen extrusion took place giving a ring-contracted carbocycle. The treatment of N-nitroso compound 61 under standard conditions afforded [2,2] metacyclophane (62) in 72% yield, which was also synthesized by the photodecarbonylation of diketone 63 (Scheme 3(B)).69
The synthesis of cylindrocyclophane A (32) relied on a Ramberg–Bäcklund olefination70,71 to give bis(olefin) 6572 (Scheme 3(C)). The treatment of bis(sulfone) 64 with alumina-supported KOH–CBr2F273 by removing sulfur dioxide gave a diolefin. This compound was then isomerized by 30 mol% of [Pd(CH3CN)2Cl2] to give exclusively E,E-65 in 70% yield. (contractive synthesis: 21 steps;72 alternative synthesis: 21 steps74) The Ramberg–Bäcklund olefination is often employed in the synthesis of cyclophane,75 and is also used when synthesizing smaller cycloalkenes76 and fused rings77 which are not discussed in this review.
The synthesis of (+)-α-cuparenone (68) used solid-state photodecarbonylation generating (R,S)-6778 (Scheme 3(D)). The photoirradiation of (S,S)-66 as a nanocrystalline suspension in water using cetrimonium bromide afforded decarbonylated product (R,S)-67 in 80% yield, in which all vicinal carbons possessed quaternary centers. The ketone substrate had to be crystalline (i.e. (S,S)-66) and contain at least one radical-stabilizing functional group in order to stabilize the possible biradical intermediate (contractive synthesis: 7 steps,78 asymmetric; alternative synthesis: 8 steps,79 racemic). This method was also applied in the synthesis of other natural products.80
PTSA, p-toluenesulfonic acid; CTAB, Cetrimonium bromide.
3. The contractive synthesis of carbocycles via 1,2-rearrangements in organic synthesis
Ring contractions via 1,2-rearrangements have seen wide application in natural product synthesis. In this section, the use of semi-pinacol rearrangements, benzilic acid rearrangements (including variant α-ketol rearrangements), Wolff rearrangements and miscellaneous 1,2-rearrangement reactions in natural product synthesis are discussed. Additionally, novel 1,2-rearrangement methods capable of contractively synthesizing carbocycles are introduced.3.1 Semi-pinacol rearrangements in natural product synthesis
The enantioselective synthesis of (−)-citrinadin A (4) featured a substrate-controlled, oxidative semi-pinacol rearrangement of indole 69 to oxindole 71 using William's approach48–50(Scheme 4(A)). Exposure of indole 69 to an excessive amount of Davis’ oxaziridine 49 afforded epoxide (70). This compound was then subjected to a semi-pinacol rearrangement with acetic acid forming 71 in 47% yield. A variety of oxidants, besides Davis’ oxaziridine 49, such as tert-BuOCl, OsO4, and NBS, failed to give oxindole 7181 (contractive synthesis: 20 steps;49,50 non-contractive synthesis: N/A).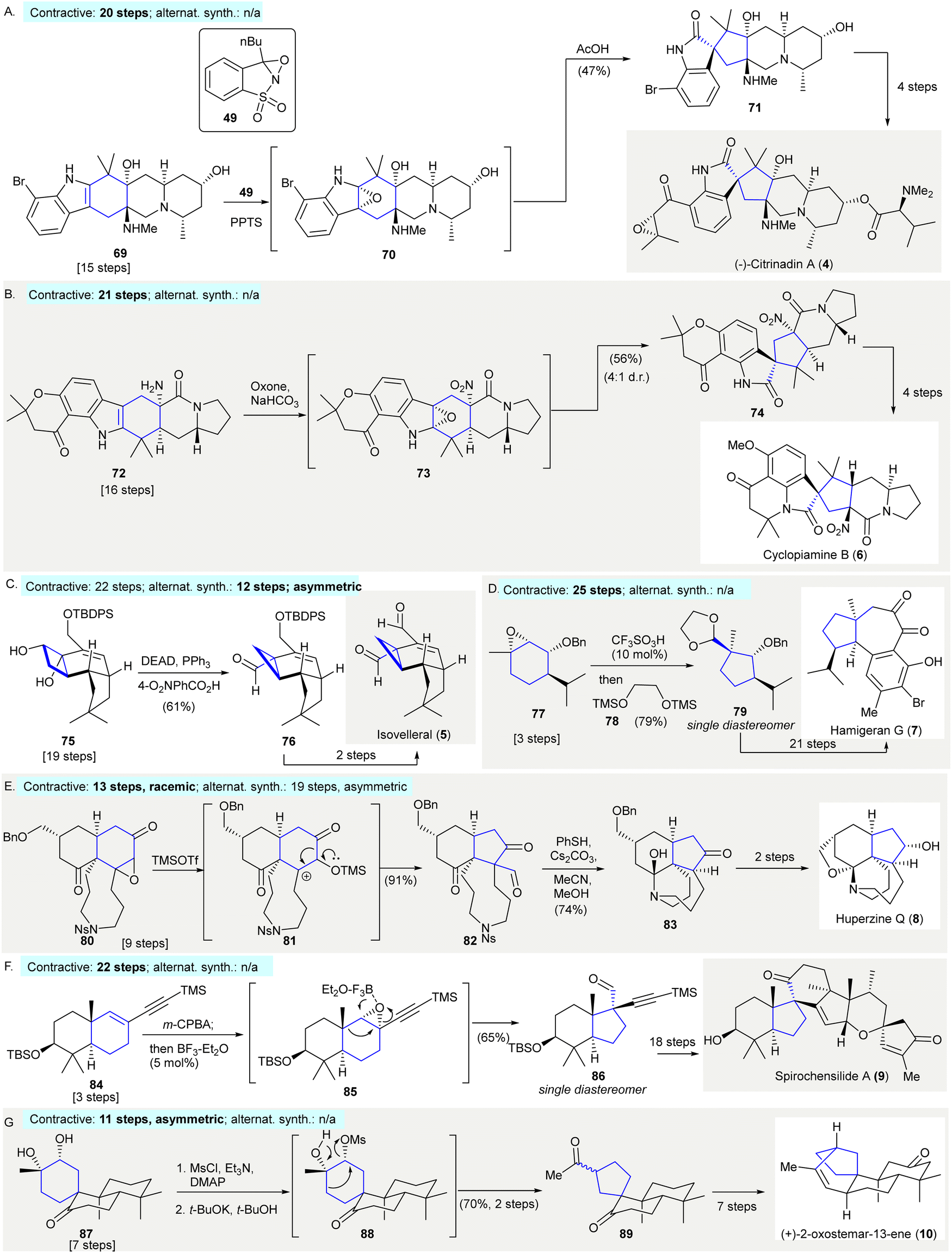 | ||
| Scheme 4 Ring contractions enabled by semi-pinacol rearrangements in natural product synthesis. A. The diastereoselective epoxidation with Davis’ oxaziridine 49 and subsequent acid-catalysed rearrangement, resulting in the formation of oxindole precursor 71 in the enantioselective synthesis of (−)-citrinadin A (4).49 (B) The synthesis of cyclopiamine B (6) featured the reaction of dimethyldioxirane via a one-pot rearrangement and tertiary amine formation to give spiroxoindole 74.82 (C) An unexpected semi-pinacol-type reaction occurred under Mitsunobu's condition in the preparation of isovelleral (5).84 (D) Synthesis of hamigeran G (7) utilized a semi-pinacol rearrangement followed by hemiketal protection yielding highly functionalized cyclopentane 79.88 (E) Selective cleavage of epoxide via a 1,2-carbon shift in the synthesis of hyperzine Q (8).89 (F) A semi-pinacol rearrangement converted [6,6]-bicycle 80 to [6,5]-bicycle 82 in the synthesis of spirochensilide A (9).91 (G) Conversion of a diol 87 to a cyclopentane 89 achieved through the selective mesylation via a 1,2-carbon migration in the synthesis of (+)-2-oxostemar-13-ene (10).92 | ||
The synthesis of cyclopiamine B (6) utilized a dimethyldioxirane-promoted, one-pot semi-pinacol rearrangement/amine oxidation and synthesized cyclopiamine B precursor 7482 (Scheme 4(B)). The treatment of indole 72 with excess dimethyldioxirane (generated in situ from acetone and oxone) resulted in the sequential stereoselective epoxidation of the indole C2–C3 bond, and amine oxidation forming a nitro group to give intermediate 73. This resulted in a semi-pinacol rearrangement giving oxindole 74 in 56% yield with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio.82 The primary amine group of 72 may have served as a hydrogen-bond donor, facilitating stereoselective epoxidation of the indole C2–C3 bond. Then, the primary amine was oxidized to a nitro group, making hydrogen bonding formation to stabilize the pseudoindoxyl side product (not depicted) no longer possible. Lastly, intermediate 7283 was used in a semi-pinacol rearrangement to give product 74. It was suggested that the chromanone moiety minimized the participation of the indole nitrogen's lone pair through intramolecular H-bonding producing oxindole 74 (contractive synthesis: 21 steps;82 non-contractive synthesis: N/A).
:1 diastereomeric ratio.82 The primary amine group of 72 may have served as a hydrogen-bond donor, facilitating stereoselective epoxidation of the indole C2–C3 bond. Then, the primary amine was oxidized to a nitro group, making hydrogen bonding formation to stabilize the pseudoindoxyl side product (not depicted) no longer possible. Lastly, intermediate 7283 was used in a semi-pinacol rearrangement to give product 74. It was suggested that the chromanone moiety minimized the participation of the indole nitrogen's lone pair through intramolecular H-bonding producing oxindole 74 (contractive synthesis: 21 steps;82 non-contractive synthesis: N/A).
The synthesis of sesquiterpenoid isovelleral (5)84 detailed the preparation of cyclopropane 76 through a semi-pinacol type rearrangement85 (Scheme 4(C)). When cyclobutanediol 75 was subjected to Mitsunobu's condition (DEAD, PPh3 and 4-O2NPhCO2H), an unexpected semi-pinacol rearrangement occurred to give cyclopropane 76 in 61% yield, without observing the expected stereo-inverted product (contractive synthesis: 22 steps,84 enantioselective; non-contractive synthesis: 12 steps,86 asymmetric). The ring contraction of cyclobutanediol formed a quaternary carbon-containing cyclopropane which could be used as an efficient strategy to prepare fused cyclopropanes.87
The synthesis of hamigeran G (7) started via the construction of cyclopentane 79 with three adjacent stereocenters and occurred through an acid-catalyzed semi-pinacol rearrangement and subsequent ketal protection88 (Scheme 4(D)). The treatment of epoxide 77 with trifluoromethanesulfonic acid and silylated diol 78 gave ketal 79 in 79% yield as single diastereomer. The stereochemistry of the C9-quaternary center of 79 matched that of hamigeran G (7) (contractive synthesis: 25 steps,88 enantioselective; non-contractive synthesis: N/A). This showed that the stereocongested compound 79 was accessible via a semi-pinacol rearrangement of epoxide 77, which was synthesized from the chiral pool chemical (R)-piperitone in three steps.2
The synthesis of huperzine Q (8) featured selective epoxide cleavage via a 1,2-carbon shift in the preparation of product 8289 (Scheme 4(E)). Exposure of epoxide 80 to TMSOTf enabled the selective opening of the epoxide, resulting in 1,2-carbon migration giving ketoaldehyde 82 in 91% yield. The cleavage of the tosyl group in 82 in the presence of thiophenol and cesium carbonate resulted in the formation of a hemiketal (not depicted), in which deformylation occurred simultaneously through the addition of methanol giving 83 in 74% yield. The synthesis of huperzine Q (8) was achieved via an additional two-step synthesis (contractive synthesis: 13 steps, racemic;89 non-contractive synthesis: 19 steps, asymmetric90).
The synthesis of (−)-spirochensilide A (9) started with the cleavage of an epoxide via a semi-pinacol rearrangement cascade of enyne 84 to generate the bicycle 8691 (Scheme 4(F)). The treatment of enyne 84 with meta-chloroperoxybenzoic acid produced epoxide 85. Subsequent addition of a substoichiometric amount of BF3·OEt2 resulted in a semi-pinacol rearrangement to give product 86 in 65% yield as a single diastereomer. Aldehyde 86 was converted to (−)-spirochensilide A (9) in further reactions (contractive synthesis: 22 steps;91 non-contractive synthesis: N/A).
The synthesis of (+)-2-oxostemar-13-ene (10) utilized a semi-pinacol rearrangement to prepare spirocyclopentane 8992 (Scheme 4(G)). The selective mesylation of the C12 hydroxy group of 87, followed by treatment with potassium tert-butoxide enabled a semi-pinacol rearrangement giving diketone 89 in 70% yield over two steps with a 1:1 epimeric ratio (contractive synthesis: 11 steps,92 asymmetric; non-contractive synthesis: N/A).
TBDPS, tert-butyldiphenylsilyl; DEAD, diethyl azodicarboxylate; TMS, trimethylsilyl; PPTS, pyridinium p-toluenesulfonate; Bn, benzyl; oxone, potassium peroxymonosulfate; Ph, phenyl; DMAP, 4-(dimethylamino)pyridine.
3.2 Benzilic acid rearrangements in natural product synthesis
A ring contractive synthesis was conducted using a benzilic acid rearrangement to synthesize preuisolactone A (12)93 (Scheme 5(A)). Koser's reagent 91 oxidized enol 90, which is a desirable intermediate formed under the effect of alkaline followed by acidification. It was proposed that lactone 93 formed via the activation of the enolized 1,2-diketone with assistance from the hypervalent iodine(III) species 92. The obtained product 93 was converted to oxetane 94 followed by 1,2-carbon migration upon workup with aqueous phosphate buffer (pH = 8) to give preuisolactone A (12) in 57% yield (contractive synthesis: 4 steps;93 alternative synthesis: N/A).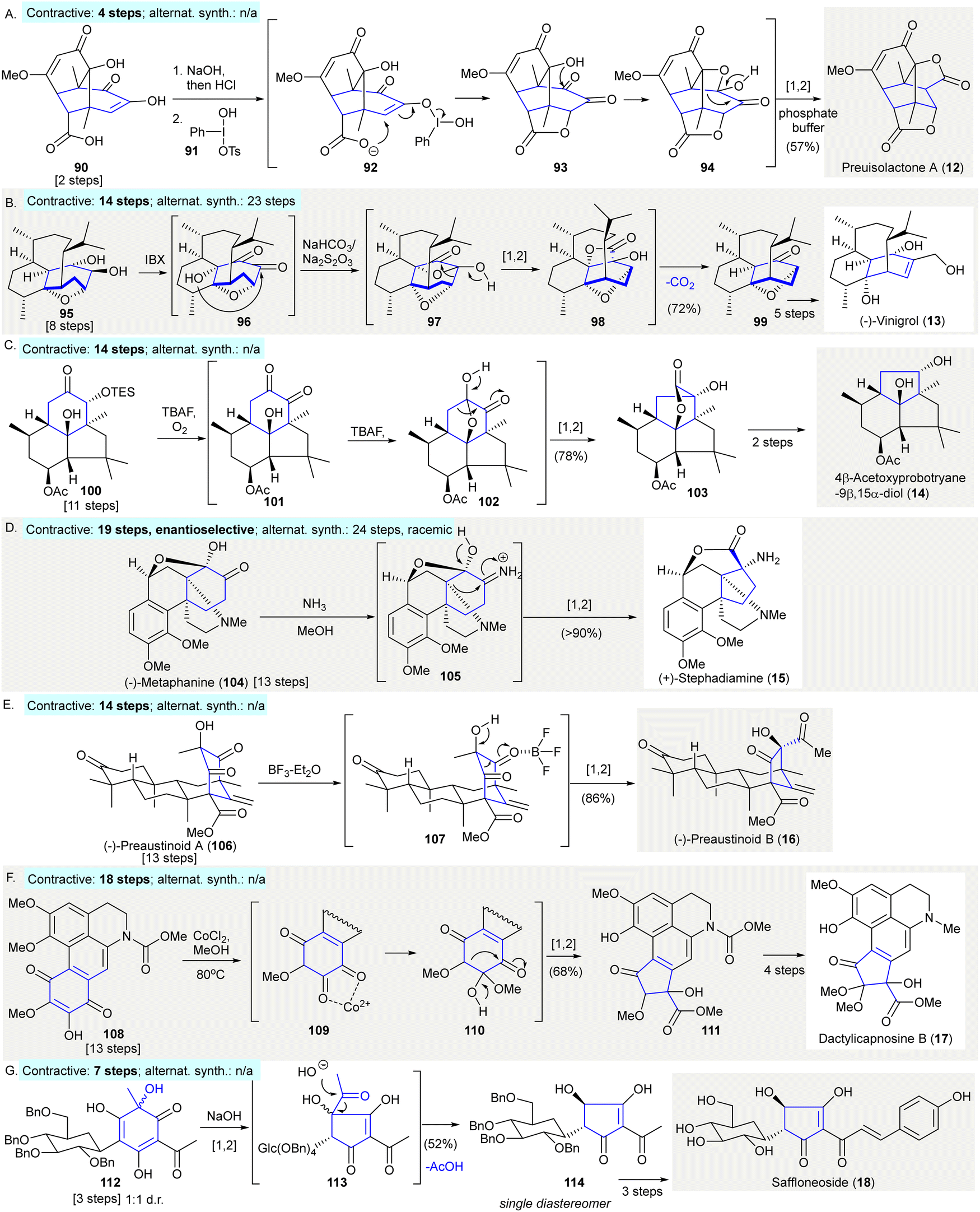 | ||
| Scheme 5 Ring contractions via benzilic acid rearrangements in natural product synthesis. (A) A late-stage benzilic acid rearrangement en route to preuisolacone A (12).93 (B) Synthesis of (−)-vinigrol (13) featured an unexpected decarboxylative ring contraction to give precursor 99.94 (C) A benzilic acid rearrangement of silyl ether 100 produced a highly-strained trans-fused structure 103 in the synthesis of 4β-Acetoxyprobotryane-9β,15α-diol (14).96 (D) An aza-benzilic acid-type rearrangement of (−)-metaphanine (104) en route to (+)-stephadiamine (15).97 (E) Conversion of (−)-preaustinoid A (106) to (−)-preaustinoid B (16) achieved via late-stage α-ketol rearrangement.99 (F) Ring contraction of p-quinone 108via cobalt-mediated benzilic acid rearrangement gave cyclopentanone 112 in the synthesis of dactylicapnosine B (17).101 (G) Base-mediated stereospecific acyloin ring contraction in the synthesis of saffloneoside (18).102 | ||
The asymmetric synthesis of (−)-vinigrol (13) featured an unexpected decarboxylative ring contraction, yielding the 1,5-butanodecahydronaphthalene core 99 as a synthetic precursor for other reactions94 (Scheme 5(B)). The oxidation of diol 95 using IBX gave 1,2-diketone 96, this reaction proceeded via the α-hydroxy group attacking the least hindered ketone to give oxetanone 97. This step was followed by a 1,2-carbon migration to give the unstable β-lactone 98. The β-lactone 98 was isolated and characterized and underwent spontaneous decarboxylation and epimerization to give desired product 99 in 72% yield (contractive synthesis: 14 steps;94,95 alternative synthesis: 23 steps95).
The synthesis of 4β-acetoxyprobotryane-9β,15α-diol (14) used a benzilic acid-type rearrangement to construct the trans-fused bicyclo[3.3.0]octane skeleton of 10396 (Scheme 5(C)). The treatment of silyl ether 100 with TBAF in the presence of oxygen resulted in desilylation, followed by rearrangement generating the 1,2-diketone 101, which underwent hemiketalization to give 101. The rearrangement in ketal 101 proceeded spontaneously, producing lactone 103 in 78% yield which contained a trans-fused bicyclo[3.3.0]octane scaffold. The further synthesis of 4β-acetoxyprobotryane-9β,15α-diol (14) was completed in two additional steps (contractive synthesis: 14 steps;96 alternative synthesis: N/A).
(+)-Stephadiamine (15) was prepared from (−)-metaphanine (104) through an aza-benzilic acid-type rearrangement97 (Scheme 5(D)). The exposure of (−)-metaphanine (104) to ammonia in methanol in situ generated imine 105, which underwent another aza-benzilic acid-type rearrangement giving (+)-stephadiamine (15) in over 90% yield. The author mentioned that stephadiamine (15) was not stable upon purification using silica gel under acidic, or basic condition (contractive: 19 steps,97 enantioselective; alternative synthesis: 24 steps,98 racemic).
The conversion of (−)-preaustinoid A (106) to (−)-preaustinoid B (16) was achieved via late-stage α-ketol rearrangement99 (Scheme 5(G)). The treatment of (−)-preaustinoid A (106) with BF3·Et2O led to an α-ketol rearrangement affording (−)-preaustinoid B (16), presumably via intermediate 107. However, attempts to use acidic, basic, or thermal conditions failed to give the desired rearrangement product (contractive: 14 steps;99 alternative synthesis: N/A).
The synthesis of dactylicapnosine B (17) made use of a cobalt-mediated benzilic acid rearrangement100 preparing tetracyclic precursor 111101 (Scheme 5(F)). p-Quinone 108 was treated with cobalt chloride in methanol providing 1,2-diketone 109. The addition of methanol to the diketone 109 gave hemiacetal 110, which underwent a benzilic acid rearrangement to give cyclopentenone 111 in 68% yield. The synthesis of dactylicapnosine B (17) was completed in seven steps (contractive synthesis: 18 steps;101 alternative synthesis: N/A).
The synthesis of saffloneoside (18) relied on a stereospecific acyloin ring contraction yielding cyclopentanone 114102 (Scheme 5(G)). The treatment of the cyclohexadienone 112 with aqueous sodium hydroxide gave the cyclopentenone-containing intermediate 113via α-ketol rearrangement. After the cleavage of the acyl moiety of 113 under basic conditions, cyclopentenone 114 was produced in 52% yield as a single diastereomer. The cyclopentenone 114 was converted to saffloneoside (18) in three steps. (Contractive synthesis: 7 steps;102 alternative synthesis: N/A)
Ph, phenyl; Ts, p-toluenesulfonyl; IBX, 2-iodoxybenzoic acid; TBAF, tetra-n-butylammonium fluoride.
3.3 Wolff rearrangements in natural product synthesis
The synthesis of taiwaniaquinone A (20)103 and taiwaniadduct D (21)104 used a Wolff rearrangement to prepare the 6-5-6 tricyclic core 116a or 116b from the 6-6-6 tricyclic diazo compound 115 (Scheme 6(A)). Irradiation of diazo compound 115 with a mercury lamp in methanol gave 116a in 30% yield as a single diastereomer. Due to decreased efficiency upon scale-up under photoirradiation, thermal conditions (BnOH, 2,4,6-collidine, 160 °C) were tested and the desired benzyl ester 116b was isolated in 56% yield as a single diastereomer (taiwaniaquinone A (20): contractive synthesis: 11 steps,103 racemic; alternative synthesis: 14 steps,105 asymmetric).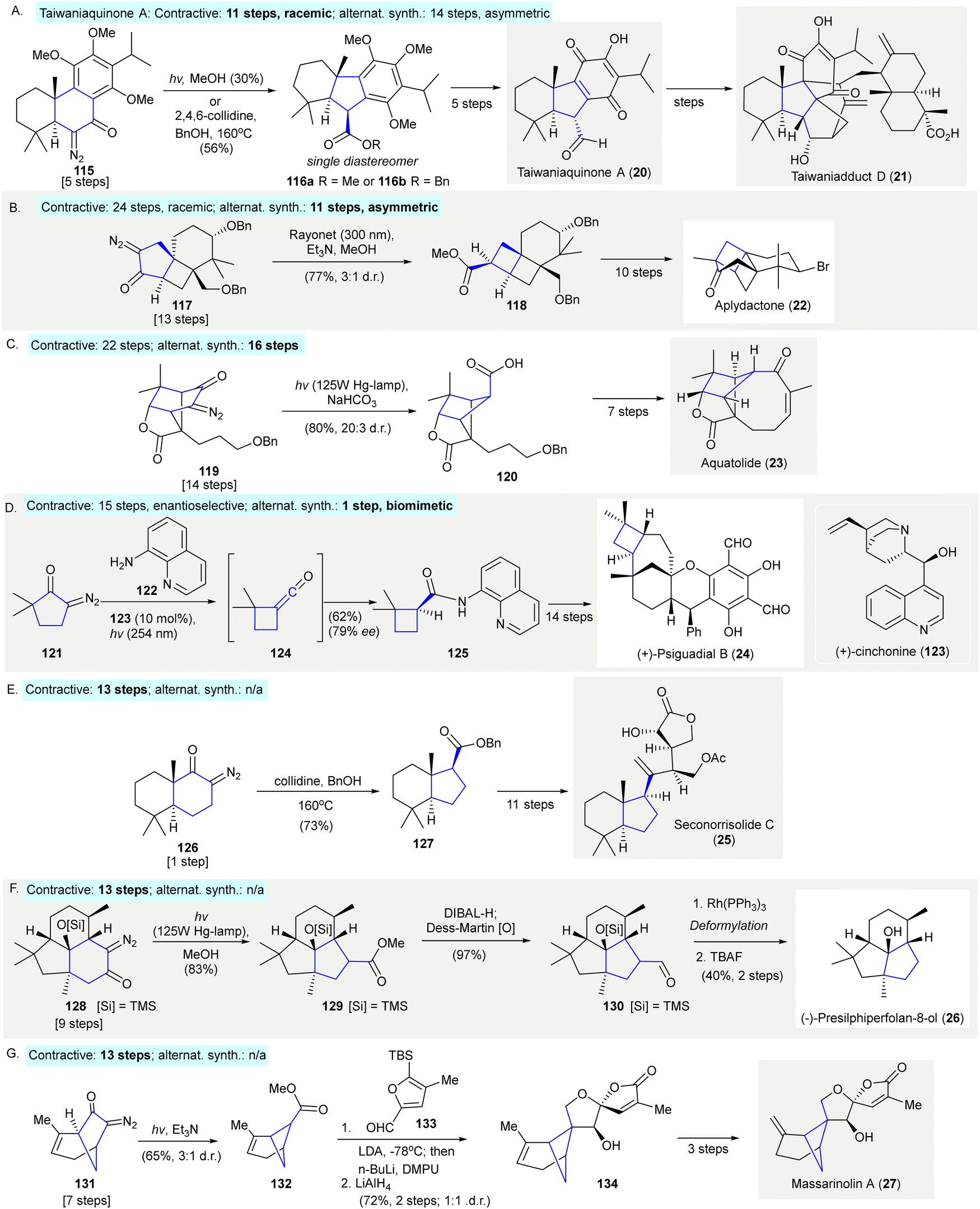 | ||
| Scheme 6 Ring contractions via Wolff rearrangements in natural product synthesis. (A) The synthesis of taiwaniaquinone A (20) and taiwaniadduct D (21) revealed a conversion of [6.6.6]-tricyclic fused ring 115 to [6.5.6] tricyclic fused ring 116a or 116b.103,104 (B) The synthesis of aplydactone (22) demonstrated the preparation of [4.4.6] tricyclc fused ring 118.106,107 (C) A Wolff rearrangement enabled the ring contraction of bridged [2.2.1] bicycle 119 to bridged [2.1.1] bicycle 120 in the synthesis of aquatolide (23).109 (D) The enantioselective synthesis of precursor 125 of (−)-psiguadial B (24) through tandem Wolff rearrangement/asymmetric ketene addition.111 (E) Synthesis of seconorrisolide C (25) used a Wolff rearrangement to prepare the [6.5] bicycle 127.115 (F) Wolff rearrangement used to prepare tricycle 129 in the enantiospecific synthesis of (−)-presilphiperfolan-8-ol (26).116 A flow Wolff rearrangement of 131 to give 132 as an key intermediate toward the preparation of massarinolin A (27).117 | ||
Trauner's synthesis of aplydactone (22) featured a Wolff rearrangement to synthesize fused cyclobutane 118106 (Scheme 6(B)). Upon photoirradiation of the α-diazo cyclopentanone 117, the ladderane 118 was formed in 77% yield with a 3:1 diastereomeric ratio. The same strategy was adopted to prepare a fused cyclobutane by Zhang107 in the total synthesis of aplydactone (22) (contractive synthesis: 24 steps,106 racemic; alternative synthesis: 11 steps,108 enantioselective).
The synthesis of aquatolide (23) featured a Wolff rearrangement to create the bicyclo[2.1.1]hexane structure of 120109 (Scheme 6(C)). Irradiation of the diazo compound 119 with a high-pressure mercury lamp in the presence of NaHCO3 gave bridged carboxylic acid 120 in 80% yield with a 20:3 diastereomeric ratio (contractive synthesis: 22 steps,109 alternative synthesis: 16 steps110).
The enantioselective synthesis (+)-psiguadial B (24) began with a tandem Wolff rearrangement/catalytic asymmetric ketene addition to afford the cyclobutane on 125111 (Scheme 6(D)). This reaction was conducted in the presence of 8-aminoquinoline (122) using (+)-cinchonine (123) as chiral organocatalyst, resulting in asymmetric nucleophilic addition to ketene 124112,113via photolysis of the α-diazo ketone, 121 giving product 125 in 66% yield and 81% ee. Further recrystallization of 125 provided enantiomerically pure material used in the enantioselective synthesis of (+)-psiguadial B (24) (contractive synthesis: steps, 15 steps;111 alternative synthesis: 1 steps114).
The synthesis of seconorrisolide C (25) used a Wolff rearrangement to prepare the [6,5]-bicycle 127115 (Scheme 6(E)). Similar to Li's approach in the synthesis of taiwaniaquinone A (20)103 and taiwaniadduct D (21)104 (see Scheme 6(A)), the treatment of diazoketone 125 with collidine and benzyl alcohol at 160 °C produced ester 127 in 73% yield, a synthetic precursor of seconorrisolide C (25) (contractive synthesis: steps, 13 steps;115 alternative synthesis: N/A).
The enantiospecific synthesis of (−)-presilphiperfolan-8-ol (26) used a Wolff rearrangement to prepare the tricyclic core of 128116 (Scheme 6(F)). Photoirradiation of diazoketone 128 gave ring contraction product 129 in 83% yield. After a two-step redox manipulation intermediate 129 was formed and when deformylated and desilylated afforded (−)-presilphiperfolan-8-ol (26) (contractive synthesis: 13 steps;116 alternative synthesis: N/A).
Very recently, Dai and co-workers reported the first total syntheses of complex bergamotane sesquiterpenes massarinolin A (27) and its congeners featuring a scalable flow photochemical Wolff rearrangement as a key reaction117 (Scheme 6(G)) (contractive synthesis: steps, 13 steps;117 alternative synthesis: N/A).
2,4,6-Collidine, 2,4,6-trimethylpyridine; Bn, benzyl.
3.4 Other 1,2-carbon migration methods in natural product synthesis
The synthesis of artalbic acid (34) utilized a photoinduced carboborative ring contraction/oxidation to give the highly-functionalized cyclopentane 139118 (Scheme 7(A)). The exposure of (S,S)-carveol-derived TBS ether 135 to UV-irradiation resulted in an isomerization.119 This was followed by boration with triethylborane 136 to generate a carbocation intermediate 137, which underwent 1,2-carbon migration yielding product 138. The successive oxidation of borane 138 with trimethylamine N-oxide and Dess–Martin periodinane afforded cyclopentane 139 in 69% yield (contractive synthesis: 5 steps;118 alternative synthesis: 12 steps120).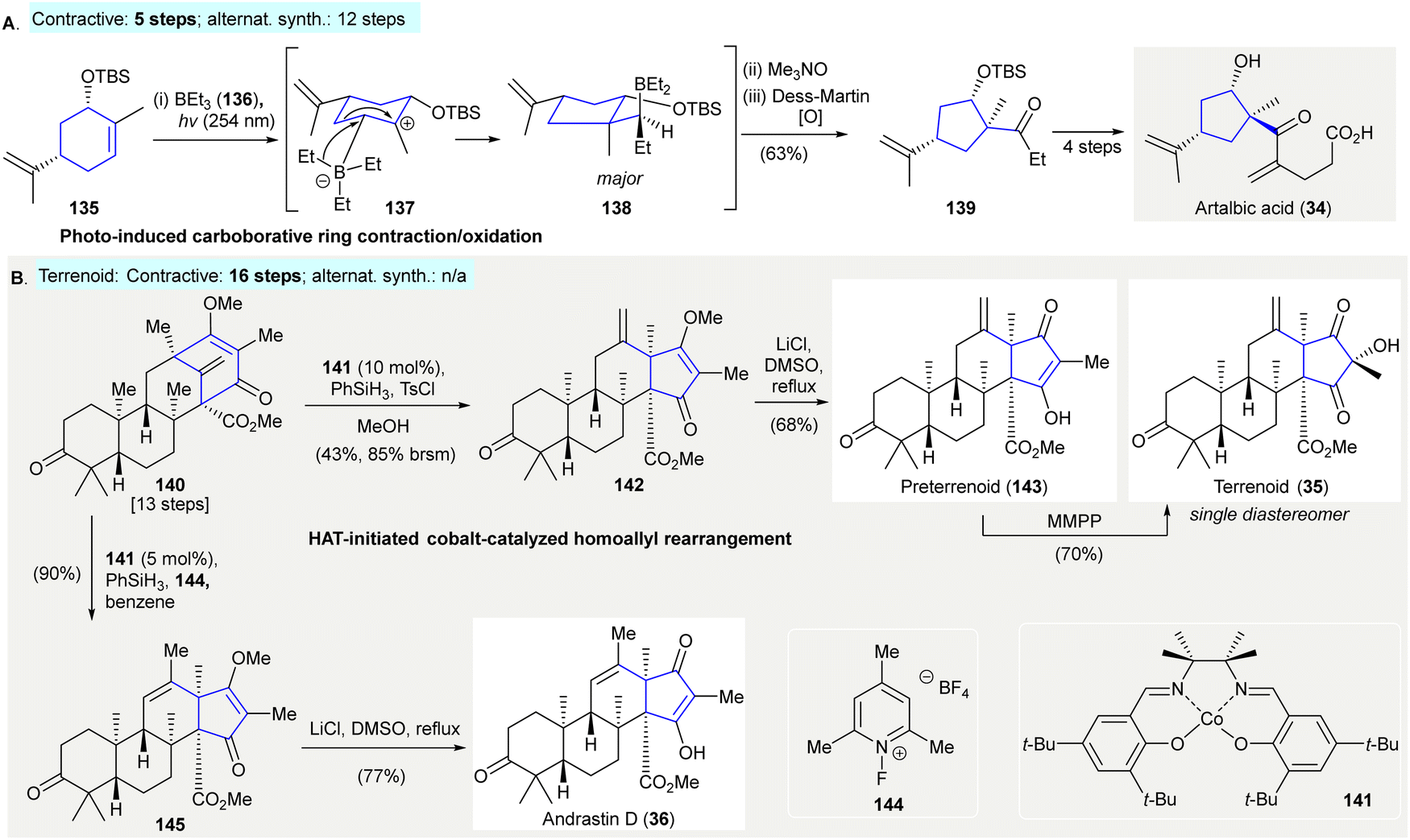 | ||
| Scheme 7 Other 1,2-carbon migration methods in natural product synthesis. (A) Photo-induced carboborative ring contraction/oxidation in the synthesis of artalbic acid (34).118 (B) HAT-initiated cobalt-catalyzed homoallyl rearrangement en route to terrenoid (35) and andrastin D (36).121 | ||
The synthesis of andrastin (36) and terretonin meroterpenes (e.g. terrennoid (35)) utilised HAT-initiated homoallyl rearrangements121 (Scheme 7(B)). The treatment of 140 with 10 mol% of cobalt(II) catalyst 141 and phenylsilane in the presence of tosyl chloride122 afforded 142 in 43% yield. The demethylation of 142 with lithium chloride under reflux yielded preterrenoid (143), which was subjected to stereoselective oxidation with magnesium monoperoxyphthalate giving terrenoid (35). Alternatively, the treatment of 140 with 5 mol% of cobalt(II) catalyst 141 in the presence of phenylsilane and 144 as an oxidant (i.e. modified Shigehisa conditions123) avoided the use of the alcohol as a trapping agent, producing the rearrangement product 145 in 90% yield. Demethylation of 145 afforded andrastin (36) in 77% yield. (Terrenoid: contractive synthesis: steps, 16 steps;121 alternative synthesis: N/A).
HAT, hydrogen atom transfer; DMSO, dimethyl sulfoxide; Ph, phenyl; MMPP, magnesium monoperoxyphthalate.
3.5 Novel 1,2-carbon migration synthetic methods in organic synthesis
The chiral substituted cyclohexene 149 was prepared from a tandem desymmetrization124/dyotropic rearrangement from meso-3,7-dibromocycloheptene 146125 (Scheme 8(A)). Upon treatment of 146 with substoichiometric amount of CuBr·SMe2/147 and n-hexyllithium, the bromocycloheptene 148 (formed by asymmetric allylic substitution (AAS)) was exposed to silica giving 149via stereospecific ring contraction. This dyotropic rearrangement occurred via a double 1,2-alkene migration followed by a 1,2-bromide migration.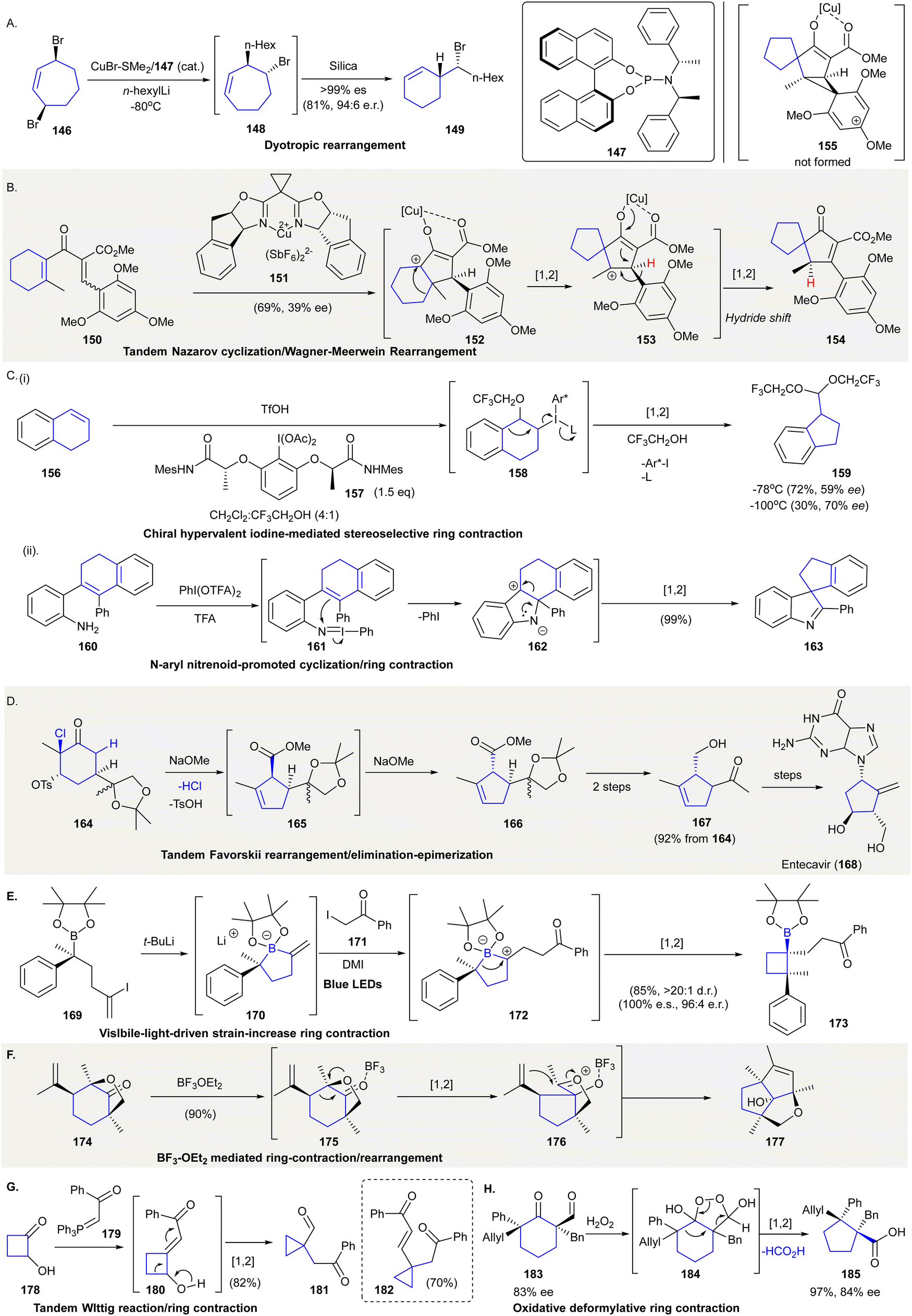 | ||
| Scheme 8 Novel 1,2-carbon migration synthetic methods in organic synthesis. (A) Desymmetrization and stereospecific ring contraction through dyotropic rearrangement.125 (B) Tandem Nazarov cyclization/Wagner–Meerwein rearrangement.138 (C) (i) Chiral hypervalent iodine(III) reagent 157 mediated stereoselective ring contraction.128 (ii) N-aryl nitrenoid-enabled cyclization/ring contraction.129 (D) A tandem Favorskii rearrangement/tosylate elimination-epimerization was used in the synthesis of Entecavir (168).139 (E) Visible light-driven ring-contraction triggered by a 1,2-metalate rearrangement.130 (F) A BF3·OEt3-mediated tandem ring contraction/rearrangement produced diquinane 177.133 (G) Tandem Wittig reaction/ring contraction.134 (H) Oxidative and deformylative ring contraction gave cyclopentane 185 containing vicinal quaternary carbon centers.135 | ||
A copper-mediated Nazarov cyclization followed by a double Wagner–Meerwein migration converted divinyl ketone 150 into spirocycle 154 in 69% yield with 39% ee126 (Scheme 8(B)). The treatment of divinyl ketone 150 with stochiometric amount of copper complex 151 resulted in a Nazarov cyclization and produced intermediate 152. The carbocation of 152 initiated a 1,2-carbon migration, generating the cationic spirocycle 153, in which a 1,2-hydride migration took place to give spirocycle 154 as the final product. It is reported that the 1,2-hydride migration was favorable due to the electron-poor aromatic substitutions present while 1,2-carbon migration was favored with electron-rich aromatic substitutions. Unexpectedly, 1,2-hydride migration took place when electron-rich 2,4,6-trimethoxyphenyl group used as substituent (i.e.153 to 154). Authors rationalized that phenyl migration is also affected by steric factors that hinder the formation of the bridged cation intermediate 153, resulting in the formation of 1,2-hydride shift product 154.
Hypervalent iodine(III) reagents have been used extensively in rearrangement reactions.127 The ring contraction of tetralone 156 relied on stoichiometric amounts of chiral hypervalent iodine(III) reagent 157 using TfOH as a Lewis acid. The reaction was theorized to proceed through phenyliodinated Intermediate 158 to afford cyclopropane 159 in 30% yield with 70% ee at −100 °C128 (Scheme 8C(i)). Another example involved the in situ generation of electrophilic iodonitrene to facilitate tandem C–N bond formation and 1,2-carbon migration to give spirocyclopentane 163129 (Scheme 8(C)(ii)). A reaction between 2-substituted anilines 160 and bis(trifluoroacetoxy)iodobenzene generated the electrophilic N-aryl nitrenoid intermediate 161, which enabled an intramolecular reaction to give tetracycle intermediate 162. Intermediate 162 then underwent a 1,2-carbon migration to afford spirocycle 163 in 99% yield. Trifluoroacetic acid was added in stoichiometric amounts, improving the reaction yield by enabling the substitution of TFA on bis(trifluoroacetoxy)iodobenzene with aniline generating the iodonitrene reactant.
The synthesis of Entecavir (168), which is an approved drug for the treatment of hepatitis B (HBV), applied a tandem Favorskii rearrangement/elimination/epimerization to prepare the cyclopentene fragment of 166 (Scheme 8(D)). Treatment of α-chlorohexanone 164 with sodium methoxide initially produced the cis-substituted Favorskii rearrangement product 165, which upon isomerization gave the more thermodynamically stable cyclopentanecarboxylate 166. Sequential transformations, including reduction, hydrolysis of the ketal, and oxidative cleavage afforded 167 in 92% yield over three steps.
The synthesis of cycloboronic ester 173 was achieved via a visible-light-driven ring contraction of five-membered alkenyl boronate complex 170130 (Scheme 8(E)). Reactions between vinyl iodide 169 and tert-butyllithium produced cyclic alkenyl boronate 170. Further reactions with iodido-compound 171 were followed by single electron oxidation to give the zwitterionic intermediate 172. Ring-contractive 1,2-metalate rearrangement131,132 of freshly prepared 172 afforded cyclobutyl boronic ester 173 in 85% yield with >20:1 diastereomeric ratio. The synthetic versatility of boronic esters enabled transformations to produce other functionalities, including the construction of adjacent quaternary stereocenters.
The Lewis acid-catalyzed 1,2-carbon migration/cyclization of substituted 1-methyl-4-isopropenyl-6-oxabicylo[3.2.1]octan-8-ones 174 produced bridged diquinane 177133 (Scheme 8(F)). The treatment of the cyclohexanone 174 with BF3·OEt2 facilitated 1,2-carbon migration to give an oxocarbenium ion 176, which underwent an ene-type reaction yielding bridged diquinane 177 in 90% yield.
The ring contraction of α-hydroxycyclobutanone 178 to cyclopropanecarbaldehyde 181 was achieved via simultaneous Wittig reaction and 1,2-carbon migration134 (Scheme 8(G)). The reaction between cyclobutanone 178 and the phosphonium ylide 179 gave olefination product 180, which underwent a 1,2-carbon migration to give cyclopropanecarbaldehyde 181 in 82% yield. The ring contraction product 182 was formed in 70% yield using two equivalents of phosphonium ylide 179.
A stereospecific oxidative ring contraction converted α-formyl cyclic ketone 183 to 185via the elimination of formic acid135 (Scheme 8(H)). The treatment of α-formyl cyclic ketone 183 with hydrogen peroxide resulted in an intramolecular cyclization. The reaction was theorized to give 1,2-dioxolane 184 as an intermediate,136,137 undergoing subsequent 1,2-carbon migration affording 185 in 97% yield. The relative configuration of both stereocenters were completely preserved, the chirality of the two stereocenters could then be transferred to other products.
TfOH, trifluoromethanesulfonic acid; Mes, mesityl; Ac, acetyl; Ph, phenyl; TFA, trifluoroacetic acid, t-Bu, tert-butyl; DMI, 1,3-dimethyl-2-imidazolidinone; Ph, phenyl; Bn, benzyl.
4. The contractive synthesis of carbocycles via gas extrusion in organic synthesis
The synthesis of (−)-lundurine A ((−)-31) featured indole cyclopropanation, involving a Lewis acid-mediated formal [3+2]-cycloaddition of the indole C2–C3 bond with tosyl hydrazine. Subsequent dinitrogen extrusion gave cyclopropane (−)-188140 (Scheme 9(A)). Exposure of tosyl hydrazine 186 to BF3·OEt2141 gave an isolatable pyrazoline intermediate 187, which underwent dinitrogen extrusion producing cyclopropane (−)-188 in 80% yield (contractive synthesis: 13 steps,140 enantioselective; alternative synthesis: 31 steps, racemic142). Later, the same research group reported a homolytic photochemical decarboxylation of γ-lactone 189 producing cyclopropane 190 in 23% yield, providing a possible biosynthetic pathway to produce Kopsia pyrroloazocine indole alkaloids, including lundurine A (31)143 (Scheme 9(B)).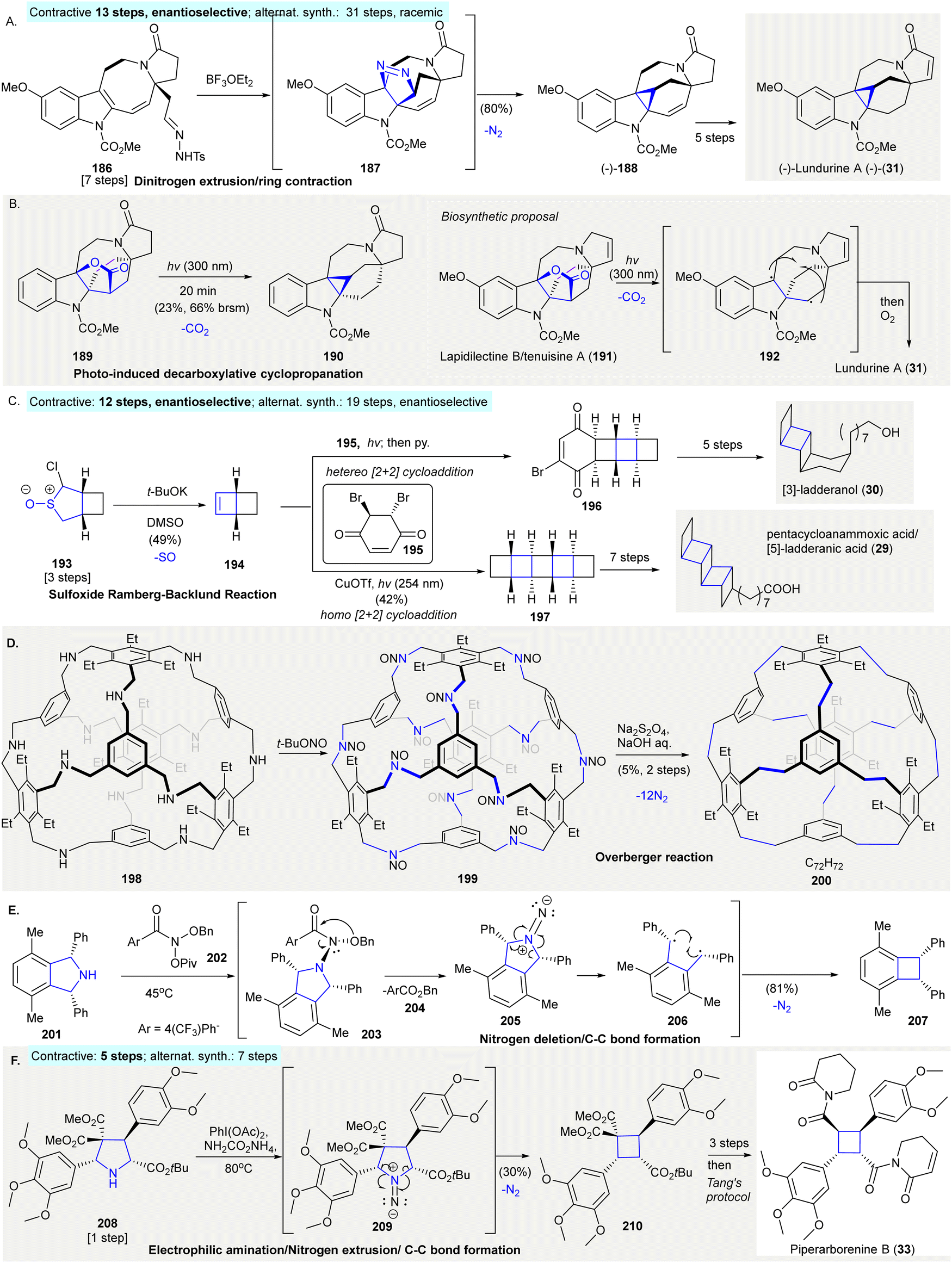 | ||
| Scheme 9 Ring contractions via gas extrusion reactions in organic synthesis. (A) The preparation of (−)-lundurine A (31) via dinitrogen extrusion.140 (B) Photoinduced decarboxylative cyclopropanation produced cyclopropane 190, structurally similar to (−)-lundurine A (31)143 (inset, a new biosynthetic proposal). (C) An atypical sulfoxide Ramberg–Bäcklund olefination in the synthesis of [3]-ladderanol (30) and pentacycloanammoxic acid (29).66 (D) Synthesis of cage structures facilitated by an Overberger reaction.146 (E) Dinitrogen deletion from secondary amine 201 using N-pivaloyloxy-N-alkoxyamide 202 produced cylcobutane 207 through 1,4-diradical C–C bond formation.149 (F) Stereoselective contraction of pyrrolidine 208 afforded cyclobutanes and was applied in the formal synthesis of piperarborenine B (33).150 | ||
The synthesis of petacycloanammoxic acid/[5]-ladderanic acid (29) and [3]-ladderanol (30) relied on an atypical sulfoxide Ramberg–Bäcklund olefination144 to produce bicyclohex[2.2.0]ene 19466 (Scheme 9(C)). α-Chlorosulfoxide 193 was treated with excess potassium tert-butoxide in DMSO to afford cyclobutene 194 in 49% yield, which then underwent either a hetero [2+2] cycloaddition with 195, followed by selective elimination of a proton with pyridine to give vinyl bromide 196. Or, the homodimerization of 194 gave [5]-ladderane pentacycle 197. Pentacycle 197 and vinyl bromide 196 which was converted to pentacycloanammoxic acid/[5]-ladderanic acid (29) and [3]-ladderanol (30) in further steps, respectively. Compared to typical Ramberg–Bäcklund olefinations using sulfone as precursors, this modified sulfoxide approach resulted in a high yield for olefin product 194 (contractive synthesis: 12 steps,66 enantioselective; alternative synthesis: 19 steps,145 enantioselective).
Overberger reaction facilitated the conversion of imine cages to hydrocarbon cages146 (Scheme 9(D)). Nitrosoamine 199, was prepared by the treatment of truncated tetrahedral [4+4] imine cages 198147 with tBuONO. 199 underwent reduction/dinitrogen extrusion under Overberger's conditions67 to give “cubic” cage derivative 200 in 5% yield over two steps. This application of Overberger's reaction enabled the synthesis of less symmetric compounds from multiple building blocks. This provided access to a large variety of structures, despite the fact that alkyne metathesis can provide higher yields when synthesizing carbon cages.148
The nitrogen deletion of secondary amines using N-pivaloyloxy-N-alkoxyamide 202 led to C–C bond formation and was applied to the ring contraction of cyclic secondary amine to carbocycles149 (Scheme 9(E)). The treatment of pyrrolidine 201 with 202 provided N–N bond containing compound 203, which underwent 1,2-rearrangement to give 1,1-diazene 205via the elimination of ester 204. Additionally, the dinitrogen extrusion of 205 produced 1,4-diradical 206 as a hypothetical intermediate. The intramolecular radical coupling of the 1,4-diradical in 206 produced 207 in 81% yield.
The iodonitrene-induced ring contraction of pyrrolidine resulted in the stereoselective synthesis of cyclobutene 210, this method was then adopted in the formal synthesis of piperarborenine B (33)150 (Scheme 9(F)). The electrophilic amination of pyrrolidine 208 was achieved via the generation of iodonitrene in situ from phenyliododiacetate and ammonium carbamate in 2,2,2-trifluoroethanol. This formed 209 as a suggested intermediate, in which dinitrogen extrusion from 209 afforded cylcobutane 210 in 30% yield. A further three-step synthesis from 210 formed a synthetic precursor, which was converted to piperarborenine B (33) in one step using Tang's protocol151 (contractive synthesis: 6 steps;150 alternative synthesis: 7 steps152)
Ts, p-toluenesulfonyl; t-Bu, tert-butyl; DMSO, dimethyl sulfoxide; Bn, benzyl; Ph, phenyl; Ac, acetyl; Piv, pivaloyl.
5. The ring contraction via miscellaneous rearrangement reactions in organic synthesis
The synthesis of the cyclocitrinol core 215 involved a tandem Ireland Claisen/strain-accelerated Cope rearrangement153 (Scheme 10(A)). An Ireland-Claisen rearrangement154 converted macrolactone 211 to the strained 10-membered ring intermediate 212. The resulting strain then drove the Cope rearrangement under unusually mild thermal conditions to give the tricyclic core 213. Successive hydrolysis of the silyl ester of 213, followed by methylation afforded methyl ester 214 in 38% yield over three steps.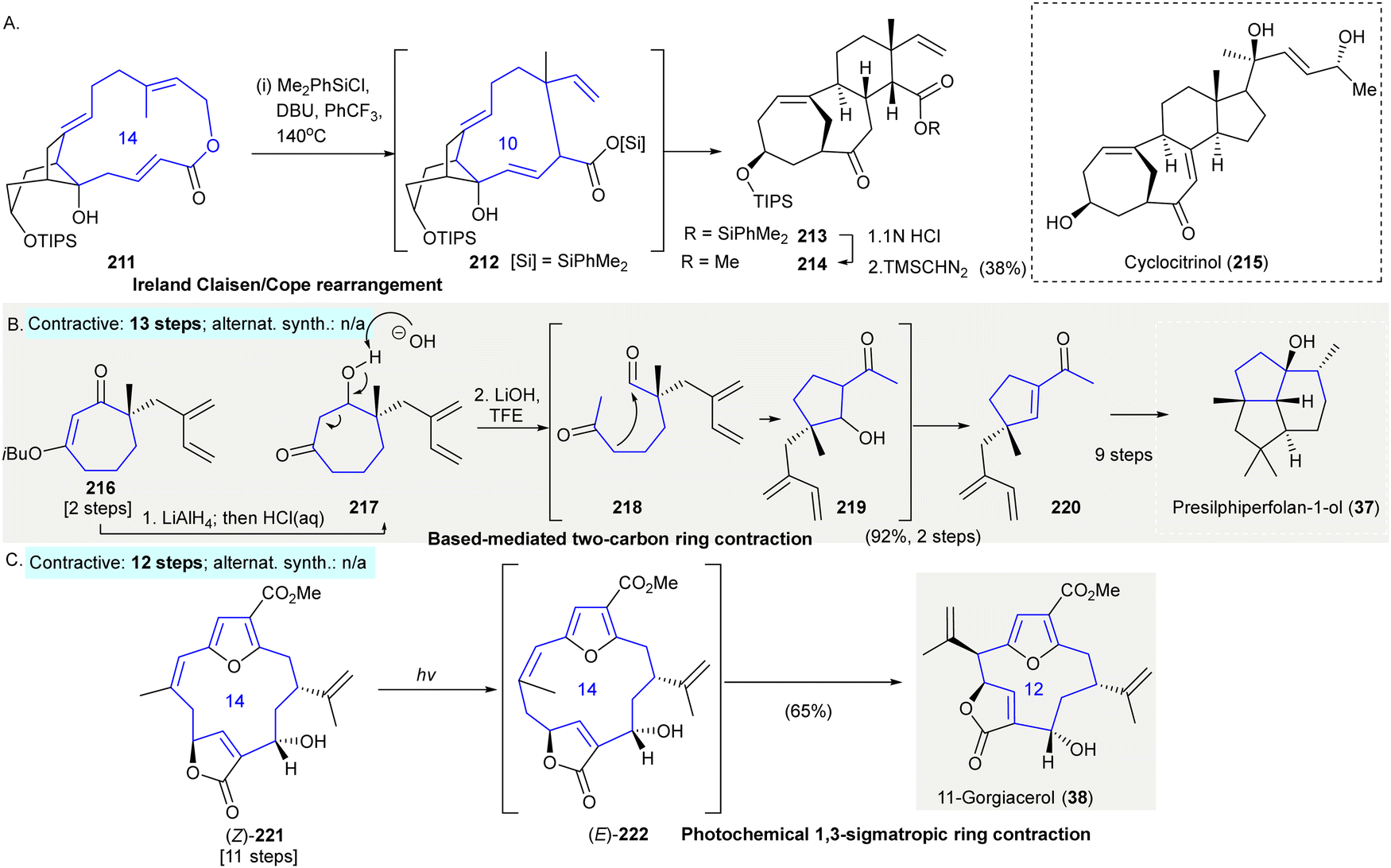 | ||
| Scheme 10 Ring contractions enabled by miscellaneous rearrangement reactions in organic synthesis. (A) A tandem ring contractive Ireland Claisen/strain-accelerated Cope rearrangement produced cyclocitrinol core 213.153 (B) A base-mediated two-carbon ring contraction provided access to acylcyclopentene 214 as a key intermediate in the synthesis of presilphiperfolan-1-ol (37).155,156 (C) A photochemical 1,3-sigmatropic ring contraction en route to 11-gorgiacerol (38).159 TIPS, triisopropylsilyl; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; Ph, phenyl; iBu, iso-butyl; TFE, 2,2,2-trifluoroethanol; TBS, tert-butyldimethylsilyl. | ||
The synthesis of presilphiperfolan-1-ol (37) used a base-mediated two-carbon ring contraction to give acylcyclopentene 220155,156 (Scheme 10(B)). The reduction of compound 216 by LiAlH4 gave intermediate β-hydroxyketone 217. When 217 was exposed to lithium hydroxide in 2,2,2-trifluoroethanol gave highly functionalized chiral acylcyclopentene 220 in 92% yield over two steps. The synthesis of presilphiperfolan-1-ol (37) was achieved from 220 in 9 additional steps (contractive synthesis: 13 steps,155,156 enantioselective; alternative synthesis: N/A)
The synthesis of 11-gorgiacerol (38) featured a photochemical 1,3-sigmatropic ring contraction (Rodriguez–Pattenden rearrangement157,158) as a key synthetic step159 (Scheme 10(C)). Photoirradiation of 221 led to olefin Z-to-E isomerization followed by a [1,3]-sigmatropic shift giving 11-gorgiacerol (38) in 65% yield (contractive synthesis: 12 steps;159 alternative synthesis: N/A)
6. Summary and outlook
In this review, the contractive synthesis of carbocycles and their respective applications were discussed. Ring contractions have played important roles in organic synthesis, enabling the creation of small carbocycles with highly condensed functionalities and stereocenters (Scheme 11). Providing the efficient synthesis of complex carbocycles26 which otherwise required multi-step, time-consuming syntheses when following conventional methods.27–29 The synthesis of carbocycles through ring contraction using well-planned methods is very practical, as they encompass one to two concepts of efficient synthesis. Including; step economy,36 chemoselectivity,39 protecting group-free synthesis,40 atom economy34 and/or redox economy.38 Methods such as semi-pinacol rearrangements and dinitrogen extrusions demonstrate late-stage modifications of complex structures, provide the possibility of late-stage diversification,160 and enable access to a diverse array of structural analogues which could be useful for compound libraries. | ||
| Scheme 11 Strategic applications of contractive synthesis in carbocycles. | ||
This review summarizes the contractive synthesis of carbocycles, highlighting synthetic methods in organic synthesis including natural product synthesis. The example that we discussed in this Review, ring contraction appear to be either a more efficient approach in term of synthetic steps or an exclusive strategy towards complex natural product synthesis. The comparison of step-efficiency of the complex natural product synthesis using or without using ring contraction approach is tabulated (Table 1). In most cases, the ring contraction involves the preparation of small carbocycles of three to five membered (i.e. cyclopropane, cyclobutane and cyclopentane). The behind rationale might be a result of two reasons: first, ring contraction would be a more efficient approach to prepare highly functionalized small carbocycles while more available methods and/or strategies are eligible to prepare the carbocycles of larger ring size, for instance, cycloaddition and ring-closing metathesis. Second, the synthesis of the ring contraction precursor, for example, a seven-membered ring which undergoes ring contraction to a six-membered carbocycles, would be more challenging that the product itself because of the higher kinetic and thermodynamic barriers associated the synthesis of medium-sized ring compared to other rings sizes. More importantly, chemoselective ring contraction of advanced and/or late-stage intermediates might provide a concise synthetic approach compared to the synthesis of the target natural products adopting alternative approaches, such as direct ring closure.
| Natural products | Reaction | Ring contraction | Steps of total synthesis (alter. syn) | Scheme |
|---|---|---|---|---|
| # denotes heterocycles. | ||||
| (+)-Welwitindolinone A | Oxidative pinacol-type rearrangement | 5 → 4 | 8 (22) | 1a |
| (−)-Peribysin E | Semi-pinacol-type reaction | 6 → 5 | 18 (24) | 2b |
| (−)-Taiwaniaquinone H | Benzilic acid rearrangement | 6 → 5 | 9 (14) | 2c |
| Dolabellatrienone | Wolff arrangement | 6 → 5 | 14 (13) | 2d |
| Pentacycloanammoxic acid/[5]-ladderane methyl ester | Dinitrogen extrusion of 1,2-diazene | 6# → 4 (fused bicyclo-hexane) | 14 (19) | 3a |
| Cylindrocyclophane A | Double Ramberg-Backlund olefination | 24 → 22 | 21 (21) | 3c |
| (+)-α-Cuparenone | Decarbonylative ring contraction | 6 → 5 | 7, asymmetric; (8, racemic) | 3d |
| Huperzine Q | Semi-pinacol rearrangement | 6 → 5 | 13, racemic (19, asymmetric) | 4e |
| (+)-2-Oxostemar-13-ene | 6 → 5 | 11, asymmetric; (n/a) | 4g | |
| (−)-Vinigrol | Benzilic acid rearrangement | 7 → 6 | 15 (23) | 5b |
| (+)-Stephadiamine | aza-Benzilic acid-type rearrangement | 6 → 5 | 19, enantioselective (24, racemic) | 5d |
| Taiwaniaquinone A | Wolff rearrangement | 6 → 5 | 11, racemic (14, asymmetric) | 6a |
| Aplydactone | 5 → 4 | 25, racemic; (11, enantioselective) | 6b | |
| Aquatolide | 6 → 5 | 22 (16) | 6c | |
| (+)-Psiguadial B | 5 → 4 | 15 (1) | 6d | |
| Artalbic acid | Photo-induced carboborative ring contraction | 6 → 5 | 5 (12) | 7a |
| (−)-Lundurine A | Dinitrogen extrusion of 1,2-diazene | 5# → 3 | 13, enantioselective; (31, racemic) | 9a |
| [5]-Ladderane acid | Sulfoxide Ramberg–Backlund reaction | 5# → 4 | 12, enantioselective (19, enantioselective) | 9c |
| Piperarborenine B | Dinitrogen extrusion of 1,1-diazene | 5# → 4 | 5 (7) | 9f |
Ring contractive synthesis often required stoichiometric amounts of reagents to facilitate rearrangements, suggesting the possibility of developing catalytic versions. This could enable the simultaneous addition of functionalities during ring contraction, which could be useful for further method development. Ring contractions using gas extrusion required the prior introduction of heteroatom(s), however, the valuable transformations provided can offset this limitation.30,31,66,140,143,146 Transformations with an emphasis on remodeling carbon skeletons were discussed, aiming to inspire innovative method development and method replacement in the synthesis of important, but synthetically elusive organic molecules. We anticipate ring contractions to function alongside conventional synthetic methods assisting in the further advancement of organic synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P. A. acknowledges the support of the DFG (AN 1064/4-1), the Boehringer Ingelheim Foundation (Plus 3) and Nottingham Trent University. C. H. acknowledges the International Max Planck Research School for Living Matter (Dortmund, Germany). The authors would like to thank the anonymous reviewers for their thought-provoking comments and apologize to colleagues whose work was not cited owing to selected coverage.References
- C. N. Ungarean, E. H. Southgate and D. Sarlah, Enantioselective polyene cyclizations, Org. Biomol. Chem., 2016, 14, 5454–5467, 10.1039/C6OB00375C.
- Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Navigating the chiral pool in the total synthesis of complex terpene natural products, Chem. Rev., 2017, 117, 11753–11795, DOI:10.1021/acs.chemrev.6b00834.
- V. A. D’yakonov, O. g A. Trapeznikova, A. de Meijere and U. M. Dzhemilev, Metal complex catalysis in the synthesis of spirocarbocycles, Chem. Rev., 2014, 114, 5775–5814, DOI:10.1021/cr400291c.
- R. Long, J. Huang, J. X. Gong and Z. Yang, Direct construction of vicinal all-carbon quaternary stereocenters in natural product synthesis, Nat. Prod. Rep., 2015, 32, 1584–1601, 10.1039/c5np00046g.
- M. Buschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Synthetic strategies toward natural products containing contiguous stereogenic quaternary carbon atoms, Angew. Chem., Int. Ed., 2016, 55, 4156–4186, DOI:10.1002/anie.201507549.
- T. Bach and J. P. Hehn, Photochemical reactions as key steps in natural product synthesis, Angew. Chem., Int. Ed., 2011, 50, 1000–1045, DOI:10.1002/anie.201002845.
- K. E. O. Ylijoki and J. M. Stryker, 5+2 Cycloaddition reactions in organic and natural product synthesis, Chem. Rev., 2013, 113, 2244–2266, DOI:10.1021/cr300087g.
- S. Poplata, A. Troster, Y. Q. Zou and T. Bach, Recent advances in the synthesis of cyclobutanes by Olefin 2+2 photocycloaddition reactions, Chem. Rev., 2016, 116, 9748–9815, DOI:10.1021/acs.chemrev.5b00723.
- X. Y. Liu and Y. Qin, Enabling syntheses of diterpenoid alkaloids and related diterpenes by an oxidative dearomatization/Diels–Alder cycloaddition strategy, Nat. Prod. Rep., 2017, 34, 1044–1050, 10.1039/c7np00033b.
- Z. S. Yin, Y. He and P. Chiu, Application of (4+3) cycloaddition strategies in the synthesis of natural products, Chem. Soc. Rev., 2018, 47, 8881–8924, 10.1039/c8cs00532j.
- D. J. Edmonds, D. Johnston and D. J. Procter, Samarium(II)-iodide-mediated cyclizations in natural product synthesis, Chem. Rev., 2004, 104, 3371–3403, DOI:10.1021/cr030017a.
- K. Gilmore and I. V. Alabugin, Cyclizations of alkynes: Revisiting Baldwin's rules for ring closure, Chem. Rev., 2011, 111, 6513–6556, DOI:10.1021/cr200164y.
- N. Krause and C. Winter, Gold-catalyzed nucleophilic cyclization of functionalized allenes: A powerful access to carbo- and heterocycles, Chem. Rev., 2011, 111, 1994–2009, DOI:10.1021/cr1004088.
- L. Li, Z. Chen, X. W. Zhang and Y. X. Jia, Divergent strategy in natural product total synthesis, Chem. Rev., 2018, 118, 3752–3832, DOI:10.1021/acs.chemrev.7b00653.
- K. Q. Ma, B. S. Martin, X. L. Yin and M. J. Dai, Natural product syntheses via carbonylative cyclizations, Nat. Prod. Rep., 2019, 36, 174–219, 10.1039/c8np00033f.
- A. Furstner, Alkyne metathesis on the rise, Angew. Chem., Int. Ed., 2013, 52, 2794–2819, DOI:10.1002/anie.201204513.
- M. R. Becker, R. B. Watson and C. S. Schindler, Beyond olefins: New metathesis directions for synthesis, Chem. Soc. Rev., 2018, 47, 7867–7881, 10.1039/c8cs00391b.
- C. Lecourt, S. Dhambri, L. Allievi, Y. Sanogo, N. Zeghbib, R. Ben Othman, M. I. Lannou, G. Sorin and J. Ardisson, Natural products and ring-closing metathesis: Synthesis of sterically congested olefins, Nat. Prod. Rep., 2018, 35, 105–124, 10.1039/c7np00048k.
- R. A. Yoder and J. N. Johnston, A case study in biomimetic total synthesis: Polyolefin carbocyclizations to terpenes and steroids, Chem. Rev., 2005, 105, 4730–4756, DOI:10.1021/cr040623l.
- K. C. Nicolaou and J. S. Chen, The art of total synthesis through cascade reactions, Chem. Soc. Rev., 2009, 38, 2993–3009, 10.1039/b903290h.
- J. Poulin, C. M. Grise-Bard and L. Barriault, Pericyclic domino reactions: Concise approaches to natural carbocyclic frameworks, Chem. Soc. Rev., 2009, 38, 3092–3101, 10.1039/b819798a.
- C. Grondal, M. Jeanty and D. Enders, Organocatalytic cascade reactions as a new tool in total synthesis, Nat. Chem., 2010, 2, 167–178, DOI:10.1038/nchem.539.
- A. C. Jones, J. A. May, R. Sarpong and B. M. Stoltz, Toward a symphony of reactivity: Cascades involving catalysis and sigmatropic rearrangements, Angew. Chem., Int. Ed., 2014, 53, 2556–2591, DOI:10.1002/anie.201302572.
- M. P. Plesniak, H. M. Huang and D. J. Procter, Radical cascade reactions triggered by single electron transfer, Nat. Rev. Chem., 2017, 1, 0077, DOI:10.1038/s41570-017-0077.
- K. Hung, X. R. Hu and T. J. Maimone, Total synthesis of complex terpenoids employing radical cascade processes, Nat. Prod. Rep., 2018, 35, 174–202, 10.1039/c7np00065k.
- P. S. Baran, T. J. Maimone and J. M. Richter, Total synthesis of marine natural products without using protecting groups, Nature, 2007, 446, 404–408, DOI:10.1038/nature05569.
- J. M. Ready, S. E. Reisman, M. Hirata, M. M. Weiss, K. Tamaki, T. V. Ovaska and J. L. Wood, A mild and efficient synthesis of oxindoles: Progress towards the synthesis of welwitindolinone a isonitrile, Angew. Chem., Int. Ed., 2004, 43, 1270–1272, DOI:10.1002/anie.200353282.
- S. E. Reisman, J. M. Ready, A. Hasuoka, C. J. Smith and J. L. Wood, Total synthesis of (+/−)-welwitindolinone A isonitrile, J. Am. Chem. Soc., 2006, 128, 1448–1449, DOI:10.1021/ja057640s.
- S. E. Reisman, J. M. Ready, M. M. Weiss, A. Hasuoka, M. Hirata, K. Tamaki, T. V. Ovaska, C. J. Smith and J. L. Wood, Evolution of a synthetic strategy: Total synthesis of (+/−)-welwitindolinone A isonitrile, J. Am. Chem. Soc., 2008, 130, 2087–2100, DOI:10.1021/ja076663z.
- P. A. Wender, J.-M. Kee and J. M. Warrington, Practical synthesis of prostratin, DPP, and their analogs, adjuvant leads against latent HIV, Science, 2008, 320, 649, DOI:10.1126/science.1154690.
- G. Tong, Z. Liu and P. Li, Total synthesis of (±)-prostratin, Chem, 2018, 4, 2944–2954, DOI:10.1016/j.chempr.2018.10.002.
- P. Huang, Principles for Synthetic Efficiency and Expansion of the Field, in Efficiency in Natural Product Total Synthesis, ed. P. Huang, Z. Yao and R. P. Hsung, Wiley, Hoboken, NJ, 2018, ch. 1, pp. 27−66 DOI:10.1002/9781118940228.ch1.
- B. M. Trost, Selectivity – A key to synthetic efficiency, Science, 1983, 219, 245–250, DOI:10.1126/science.219.4582.245.
- B. M. Trost, The atom economy – a search for synthetic efficiency, Science, 1991, 254, 1471–1477, DOI:10.1126/science.1962206.
- R. W. Hoffmann, Protecting-group-free synthesis, Synthesis, 2006, 3531–3541, DOI:10.1055/s-2006-950311.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Function-oriented synthesis, step economy, and drug design, Acc. Chem. Res., 2008, 41, 40–49, DOI:10.1021/ar700155p.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, The economies of synthesis, Chem. Soc. Rev., 2009, 38, 3010–3021, 10.1039/b821200g.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Redox economy in organic synthesis, Angew. Chem., Int. Ed., 2009, 48, 2854–2867, DOI:10.1002/anie.200806086.
- R. A. Shenvi, D. P. O’Malley and P. S. Baran, Chemoselectivity: The mother of invention in total synthesis, Acc. Chem. Res., 2009, 42, 530–541, DOI:10.1021/ar800182r.
- I. S. Young and P. S. Baran, Protecting-group-free synthesis as an opportunity for invention, Nat. Chem., 2009, 1, 193–205, DOI:10.1038/nchem.216.
- T. Gaich and P. S. Baran, Aiming for the ideal synthesis, J. Org. Chem., 2010, 75, 4657–4673, DOI:10.1021/jo1006812.
- M. Tiffeneau and J. Levy, C. R., 1923, 176, 312 CAS.
- Z. L. Song, C. A. Fan and Y. Q. Tu, Semipinacol rearrangement in natural product synthesis, Chem. Rev., 2011, 111, 7523–7556, DOI:10.1021/cr200055g.
- B. M. Wang and Y. Q. Tu, Stereoselective construction of quaternary carbon stereocenters via a semipinacol rearrangement strategy, Acc. Chem. Res., 2011, 44, 1207–1222, DOI:10.1021/ar200082p.
- T. J. Snape, Recent advances in the semi-pinacol rearrangement of α-hydroxy epoxides and related compounds, Chem. Soc. Rev., 2007, 36, 1823–1842, 10.1039/B709634H.
- F. A. Davis, J. C. Towson, D. B. Vashi, R. ThimmaReddy, J. P. McCauley, M. E. Harakal and D. J. Gosciniak, Chemistry of oxaziridines. 13. Synthesis, reactions, and properties of 3-substituted 1,2-benzisothiazole 1,1-dioxide oxides, J. Org. Chem., 1990, 55, 1254–1261, DOI:10.1021/jo00291a028.
- G. D. Artman, A. W. Grubbs and R. M. Williams, Concise, asymmetric, stereocontrolled total synthesis of stephacidins A, B and notoamide B, J. Am. Chem. Soc., 2007, 129, 6336–6342, DOI:10.1021/ja070259i.
- T. J. Greshock, A. W. Grubbs, S. Tsukamoto and R. M. Williams, A concise, biomimetic total synthesis of stephacidin A and notoamide B, Angew. Chem., Int. Ed., 2007, 46, 2262–2265, DOI:10.1002/anie.200604378.
- Z. G. Bian, C. C. Marvin and S. F. Martin, Enantioselective total synthesis of (−)-CItrinadin A and revision of its stereochemical structure, J. Am. Chem. Soc., 2013, 135, 10886–10889, DOI:10.1021/ja405547f.
- Z. G. Bian, C. C. Marvin, M. Pettersson and S. F. Martin, Enantioselective total syntheses of citrinadins A and B. stereochemical revision of their assigned structures, J. Am. Chem. Soc., 2014, 136, 14184–14192, DOI:10.1021/ja5074646.
- E. V. Mercado-Marin and R. Sarpong, Unified approach to prenylated indole alkaloids: Total syntheses of (−)-17-hydroxy-citrinalin B, (+)-stephacidin A, and (+)-notoamide I, Chem. Sci., 2015, 6, 5048–5052, 10.1039/c5sc01977j.
- B. X. Zhang, W. F. Zheng, X. Q. Wang, D. Q. Sun and C. Z. Li, Total synthesis of notoamides F, I, and R and sclerotiamide, Angew. Chem., Int. Ed., 2016, 55, 10435–10438, DOI:10.1002/anie.201604754.
- A. R. Angeles, D. C. Dorn, C. A. Kou, M. A. Moore and S. J. Danishefsky, Total synthesis of peribysin E necessitates revision of the assignment of its absolute configuration, Angew. Chem., Int. Ed., 2007, 46, 1451–1454, DOI:10.1002/anie.200604308.
- A. R. Angeles, S. P. Waters and S. J. Danishefsky, Total syntheses of (+)- and (−)-peribysin E, J. Am. Chem. Soc., 2008, 130, 13765–13770, DOI:10.1021/ja8048207.
- K. Maruoka, M. Hasegawa, H. Yamamoto, K. Suzuki, M. Shimazaki and G. Tsuchihashi, Epoxy silyl ether rearrangements: A new, stereoselective approach to the synthesis of.beta.-hydroxy carbonyl compounds, J. Am. Chem. Soc., 1986, 108, 3827–3829, DOI:10.1021/ja00273a046.
- H.-Y. Lee and C.-K. Sha, Stereoselective total synthesis of (±)-peribysin E, J. Org. Chem., 2012, 77, 598–605, DOI:10.1021/jo2021604.
- C. K. Jana, R. Scopelliti and K. Gademann, A Synthetic entry into the taiwaniaquinoids based on a biogenetic hypothesis: Total Synthesis of (−)-taiwaniaquinone H, Chem. – Eur. J., 2010, 16, 7692–7695, DOI:10.1002/chem.201001085.
- E. Alvarez-Manzaneda, R. Chahboun, E. Cabrera, E. Alvarez, A. Haidour, J. M. Ramos, R. Alvarez-Manzaneda, Y. Charrah and H. Es-Samti, An enantiospecific route towards taiwaniaquinoids. First synthesis of (−)-taiwaniaquinone H and (−)-dichroanone, Org. Biomol. Chem., 2009, 7, 5146–5155, 10.1039/B916209G.
- S. A. Snyder and E. J. Corey, Concise total syntheses of palominol, dolabellatrienone, beta-araneosene, and isoedunol via an enantioselective Diels–Alder macrobicyclization, J. Am. Chem. Soc., 2006, 128, 740–742, DOI:10.1021/ja0576379.
- E. J. Corey and R. S. Kania, Concise total synthesis of (±)-palominol and (±)-dolabellatrienone via a dianion-accelerated oxy-cope rearrangement, Tetrahedron Lett., 1998, 39, 741–744, DOI:10.1016/S0040-4039(97)10614-1.
- J. D. Sunderhaus, T. J. McAfoos, J. M. Finefield, H. Kato, S. Y. Li, S. Tsukamoto, D. H. Sherman and R. M. Williams, Synthesis and bioconversions of notoamide T: A biosynthetic precursor to stephacidin A and notoamide B, Org. Lett., 2013, 15, 22–25, DOI:10.1021/ol302901p.
- V. Mascitti and E. J. Corey, Total synthesis of (±)-pentacycloanammoxic acid, J. Am. Chem. Soc., 2004, 126, 15664–15665, DOI:10.1021/ja044089a.
- N. J. Turro, J.-M. Liu, H.-D. Martin and M. Kunze, Photoelimination of nitrogen from cyclic azo alkanes. An exceptionally labile and an exceptionally reluctant diazabicyclo[2.2.2]octene, Tetrahedron Lett., 1980, 21, 1299–1302, DOI:10.1016/S0040-4039(00)74560-6.
- M. A. Anderson and C. B. Grissom, Photolysis of 2,3-diazabicyclo[2.2.2]oct-2-ene: Electronic spin determines the distribution of Products, J. Am. Chem. Soc., 1995, 117, 5041–5048, DOI:10.1021/ja00123a005.
- J. A. Jenkins, R. E. Doehner and L. A. Paquette, Synthesis and absolute configuration of 4,5-diazatwist-4-ene, J. Am. Chem. Soc., 1980, 102, 2131–2133, DOI:10.1021/ja00526a082.
- J. A. M. Mercer, C. M. Cohen, S. R. Shuken, A. M. Wagner, M. W. Smith, F. R. Moss, M. D. Smith, R. Vahala, A. Gonzalez-Martinez, S. G. Boxer and N. Z. Burns, Chemical synthesis and self-assembly of a ladderane phospholipid, J. Am. Chem. Soc., 2016, 138, 15845–15848, DOI:10.1021/jacs.6b10706.
- C. G. Overberger, J. G. Lombardino and R. G. Hiskey, Novel reductions of N-nitrosodibenzylamines—A new reaction1, J. Am. Chem. Soc., 1958, 80, 3009–3012, DOI:10.1021/ja01545a028.
- H. Takemura, T. Shinmyozu and T. Inazu, A new synthetic method of [2.2]cyclophanes, Tetrahedron Lett., 1988, 29, 1031–1032, DOI:10.1016/0040-4039(88)85327-9.
- H. Isaji, K. Sako, H. Takemura, H. Tatemitsu and T. Shinmyozu, A new synthetic method of [2.2]cyclophanes from [3.3]cyclophane-2, 11-diones via photodecarbonylation, Tetrahedron Lett., 1998, 39, 4303–4304, DOI:10.1016/S0040-4039(98)00716-3.
- L. A. Paquette and L. S. Wittenbrook, .alpha.-Halo sulfones. VI. Evidence for the intermediacy of thiirene dioxides in the base-promoted rearrangement of.alpha.,.alpha.-dichloro sulfones, J. Am. Chem. Soc., 1967, 89, 4483–4487, DOI:10.1021/ja00993a042.
- C. Y. Meyers, A. M. Malte and W. S. Matthews, Ionic reactions of carbon tetrachloride. Survey of reactions with ketones, alcohols, and sulfones, J. Am. Chem. Soc., 1969, 91, 7510–7512, DOI:10.1021/ja01054a049.
- K. C. Nicolaou, Y. P. Sun, H. Korman and D. Sarlah, Asymmetric total synthesis of cylindrocyclophanes A and F through cyclodimerization and a Ramberg–Backlund reaction, Angew. Chem., Int. Ed., 2010, 49, 5875–5878, DOI:10.1002/anie.201003500.
- T.-L. Chan, S. Fong, Y. Li, T.-O. Man and C.-D. Poon, A new one-flask Ramberg–Bäcklund reaction, J. Chem. Soc., Chem. Commun., 1994, 1771–1772, 10.1039/C39940001771.
- T. R. Hoye, P. E. Humpal and B. Moon, Total synthesis of (−)-cylindrocyclophane A via a double Horner–Emmons macrocyclic dimerization event, J. Am. Chem. Soc., 2000, 122, 4982–4983, DOI:10.1021/ja000429q.
- G. J. Bodwell and P. R. Nandaluru, Olefination reactions in the synthesis of cyclophanes, Isr. J. Chem., 2012, 52, 105–138, DOI:10.1002/ijch.201200003.
- R. J. K. Taylor, Recent developments in Ramberg–Bäcklund and episulfone chemistry, Chem. Commun., 1999, 217–227, 10.1039/A806615I.
- J. H. Rigby, N. C. Warshakoon and A. J. Payen, Studies on chromium(0)-promoted higher-order cycloaddition-based benzannulation. Total synthesis of (+)-estradiol, J. Am. Chem. Soc., 1999, 121, 8237–8245, DOI:10.1021/ja991016w.
- A. Natarajan, D. Ng, Z. Yang and M. A. Garcia-Garibay, Parallel Syntheses of (+)- and (−)-α-cuparenone by radical combination in crystalline solids, Angew. Chem., Int. Ed., 2007, 46, 6485–6487, DOI:10.1002/anie.200700679.
- E. Wenkert, B. L. Buckwalter, A. A. Craveiro, E. L. Sanchez and S. S. Sathe, Oxycyclopropanes in organochemical synthesis. Total syntheses of (.+ −.)-.alpha.-cuparenone and (.+ −.)-.beta.-vetivone, J. Am. Chem. Soc., 1978, 100, 1267–1273, DOI:10.1021/ja00472a039.
- J. J. Dotson, S. Perez-Estrada and M. A. Garcia-Garibay, Taming radical pairs in nanocrystalline ketones: Photochemical synthesis of compounds with vicinal stereogenic all-carbon quaternary centers, J. Am. Chem. Soc., 2018, 140, 8359–8371, DOI:10.1021/jacs.8b03988.
- C. Marti and E. M. Carreira, Construction of spiro[pyrrolidine-3,3′-oxindoles] − recent applications to the synthesis of oxindole alkaloids, Eur. J. Org. Chem., 2003, 2209–2219, DOI:10.1002/ejoc.200300050.
- E. V. Mercado-Marin, P. Garcia-Reynaga, S. Romminger, E. F. Pimenta, D. K. Romney, M. W. Lodewyk, D. E. Williams, R. J. Andersen, S. J. Miller, D. J. Tantillo, R. G. S. Berlinck and R. Sarpong, Total synthesis and isolation of citrinalin and cyclopiamine congeners, Nature, 2014, 509, 318–324, DOI:10.1038/nature13273.
- X. Zhang and C. S. Foote, Dimethyldioxirane oxidation of indole derivatives. Formation of novel indole-2,3-epoxides and a versatile synthetic route to indolinones and indolines, J. Am. Chem. Soc., 1993, 115, 8867–8868, DOI:10.1021/ja00072a061.
- M. T. Hovey, D. T. Cohen, D. M. Walden, P. H. Cheong and K. A. Scheidt, A carbene catalysis strategy for the synthesis of protoilludane natural products, Angew. Chem., Int. Ed., 2017, 56, 9864–9867, DOI:10.1002/anie.201705308.
- J. M. Conia and J. P. Barnier, Cyclobutanediols-1,2.-Nouvelles syntheses de cyclobutanones et de cetones et aldehydes cyclopropaniques, Tetrahedron Lett., 1971, 12, 4981–4984, DOI:10.1016/S0040-4039(01)97605-1.
- R. Bergman, T. Hansson, O. Sterner and B. Wickberg, A total synthesis of (+)-isovelleral. The absolute configuration of the russulaceae sesquiterpenes, J. Chem. Soc., Chem. Commun., 1990, 865–867, 10.1039/C39900000865.
- C. Ebner and E. M. Carreira, Cyclopropanation strategies in recent total syntheses, Chem. Rev., 2017, 117, 11651–11679, DOI:10.1021/acs.chemrev.6b00798.
- X. Li, D. Xue, C. Wang and S. Gao, Total synthesis of the hamigerans, Angew. Chem., Int. Ed., 2016, 55, 9942–9946, DOI:10.1002/anie.201604070.
- S. Tanimura, S. Yokoshima and T. Fukuyama, Total synthesis of huperzine Q, Org. Lett., 2017, 19, 3684–3686, DOI:10.1021/acs.orglett.7b01633.
- A. Nakayama, N. Kogure, M. Kitajima and H. Takayama, Asymmetric total synthesis of a pentacyclic lycopodium alkaloid: Huperzine-Q, Angew. Chem., Int. Ed., 2011, 50, 8025–8028, DOI:10.1002/anie.201103550.
- X.-T. Liang, J.-H. Chen and Z. Yang, Asymmetric total synthesis of (−)-spirochensilide A, J. Am. Chem. Soc., 2020, 142, 8116–8121, DOI:10.1021/jacs.0c02522.
- R. Chen, F. Zhang, Y. Hua, D. Shi, X. Lei, H. Xiao, Y. Wang, S. Ding, Y. Shen and Y. Zhang, Total synthesis of stemarene and betaerene diterpenoids: Divergent ring-formation strategy and late-stage C–H functionalization, CCS Chem., 2021, 4, 987–995, DOI:10.31635/ccschem.021.202100821.
- A. J. E. Novak, C. E. Grigglestone and D. Trauner, A Biomimetic synthesis elucidates the origin of preuisolactone A, J. Am. Chem. Soc., 2019, 141, 15515–15518, DOI:10.1021/jacs.9b08892.
- L. Min, X. H. Lin and C. C. Li, Asymmetric total synthesis of (−)-vinigrol, J. Am. Chem. Soc., 2019, 141, 15773–15778, DOI:10.1021/jacs.9b08983.
- T. J. Maimone, J. Shi, S. Ashida and P. S. Baran, Total synthesis of vinigrol, J. Am. Chem. Soc., 2009, 131, 17066–17067, DOI:10.1021/ja908194b.
- W. Zhang, Z.-X. Zhou, X.-J. Zhu, Z.-H. Sun, W.-M. Dai and C.-C. Li, Asymmetric total synthesis of the highly strained 4β-acetoxyprobotryane-9β,15α-diol, J. Am. Chem. Soc., 2020, 142, 19868–19873, DOI:10.1021/jacs.0c10116.
- M. Odagi, T. Matoba, K. Hosoya and K. Nagasawa, Enantioselective total synthesis of (+)-stephadiamine through bioinspired aza-benzilic acid type rearrangement, J. Am. Chem. Soc., 2021, 143, 2699–2704, DOI:10.1021/jacs.1c00047.
- N. Hartrampf, N. Winter, G. Pupo, B. M. Stoltz and D. Trauner, Total synthesis of the norhasubanan alkaloid stephadiamine, J. Am. Chem. Soc., 2018, 140, 8675–8680, DOI:10.1021/jacs.8b01918.
- H. Li, Y. Zhang, Y. Ji, I. Franzoni, C. Guo, H. Jia and B. Hong, Enantioselective total synthesis of Berkeleyone A and Preaustinoids, Angew. Chem., Int. Ed., 2021, 60, 14869–14874, DOI:10.1002/anie.202104014.
- X. Wang, Y. Zhang, L. V. Ponomareva, Q. Qiu, R. Woodcock, S. I. Elshahawi, X. Chen, Z. Zhou, B. E. Hatcher, J. C. Hower, C.-G. Zhan, S. Parkin, M. K. Kharel, S. R. Voss, K. A. Shaaban and J. S. Thorson, Mccrearamycins A–D, geldanamycin-derived cyclopentenone macrolactams from an eastern kentucky abandoned coal mine microbe, Angew. Chem., Int. Ed., 2017, 56, 2994–2998, DOI:10.1002/anie.201612447.
- Y. Zhao, Y. Li, B. Wang, J. Zhao, L. Li, X.-D. Luo and H. Zhang, Total synthesis of dactylicapnosines A and B, J. Org. Chem., 2020, 85, 13772–13778, DOI:10.1021/acs.joc.0c01900.
- W. Gao, J. S. Jiang, Z. Chen, Y. N. Yang, Z. M. Feng, X. Zhang, X. Yuan and P. C. Zhang, Stereospecific acyloin ring contraction controlled by glucose and concise total synthesis of saffloneoside, Org. Chem. Front., 2019, 6, 1858–1862, 10.1039/c9qo00279k.
- J. Deng, R. Li, Y. Luo, J. Li, S. Zhou, Y. Li, J. Hu and A. Li, Divergent total synthesis of taiwaniaquinones A and F and taiwaniaquinols B and D, Org. Lett., 2013, 15, 2022–2025, DOI:10.1021/ol400717h.
- J. Deng, S. Zhou, W. Zhang, J. Li, R. Li and A. Li, Total synthesis of taiwaniadducts B, C, and D, J. Am. Chem. Soc., 2014, 136, 8185–8188, DOI:10.1021/ja503972p.
- E. Alvarez-Manzaneda, R. Chahboun, E. Alvarez, R. Tapia and R. Alvarez-Manzaneda, Enantioselective total synthesis of cytotoxic taiwaniaquinones A and F, Chem. Commun., 2010, 46, 9244–9246, 10.1039/C0CC03763J.
- R. Meier and D. Trauner, A Synthesis of (+/−)-Aplydactone, Angew. Chem., Int. Ed., 2016, 55, 11251–11255, DOI:10.1002/anie.201604102.
- C. Liu, R. Chen, Y. Shen, Z. Liang, Y. Hua and Y. Zhang, Total Synthesis of Aplydactone by a Conformationally Controlled C-H Functionalization, Angew. Chem., Int. Ed., 2017, 56, 8187–8190, DOI:10.1002/anie.201703803.
- A. J. Burckle, V. H. Vasilev and N. Z. Burns, A unified approach for the enantioselective synthesis of the brominated chamigrene sesquiterpenes, Angew. Chem., Int. Ed., 2016, 55, 11476–11479, DOI:10.1002/anie.201605722.
- B. Wang, Y. Z. Xie, Q. Yang, G. Z. Zhang and Z. H. Gu, Total synthesis of aquatolide: Wolff ring contraction and late-stage Nozaki–Hiyama–Kishi medium-ring formation, Org. Lett., 2016, 18, 5388–5391, DOI:10.1021/acs.orglett.6b02767.
- J. M. Saya, K. Vos, R. A. Kleinnijenhuis, J. H. van Maarseveen, S. Ingemann and H. Hiemstra, Total synthesis of aquatolide, Org. Lett., 2015, 17, 3892–3894, DOI:10.1021/acs.orglett.5b01888.
- L. M. Chapman, J. C. Beck, L. Wu and S. E. Reisman, Enantioselective total synthesis of (+)-psiguadial B, J. Am. Chem. Soc., 2016, 138, 9803–9806, DOI:10.1021/jacs.6b07229.
- B. L. Hodous and G. C. Fu, Enantioselective addition of amines to ketenes catalyzed by a planar-chiral derivative of PPY: Possible intervention of chiral Brønsted-acid catalysis, J. Am. Chem. Soc., 2002, 124, 10006–10007, DOI:10.1021/ja027466x.
- S. L. Wiskur and G. C. Fu, Catalytic asymmetric synthesis of esters from ketenes, J. Am. Chem. Soc., 2005, 127, 6176–6177, DOI:10.1021/ja0506152.
- C. G. Newton, D. N. Tran, M. D. Wodrich and N. Cramer, One-step multigram-scale biomimetic synthesis of psiguadial B, Angew. Chem., Int. Ed., 2017, 56, 13776–13780, DOI:10.1002/anie.201708333.
- T. Qiao, Y. Wang, S. Zheng, H. Kang and G. Liang, Total syntheses of norrisolide-type spongian diterpenes cheloviolene C, seconorrisolide B, and seconorrisolide C, Angew. Chem., Int. Ed., 2020, 59, 14111–14114, DOI:10.1002/anie.202005600.
- P. Hu and S. A. Snyder, Enantiospecific total synthesis of the highly strained (−)-Presilphiperfolan-8-ol via a Pd-catalyzed tandem cyclization, J. Am. Chem. Soc., 2017, 139, 5007–5010, DOI:10.1021/jacs.7b01454.
- Y.-C. Wang, C. Cui and M. Dai, Flow Chemistry-enabled divergent and enantioselective total syntheses of massarinolin A, purpurolides B, D, E, 2,3-deoxypurpurolide C, and structural revision of massarinolin A, Angew. Chem., Int. Ed., 2021, 60, 24828–24832, DOI:10.1002/anie.202109625.
- S. F. Jin, V. T. Nguyen, H. T. Dang, D. P. Nguyen, H. D. Arman and O. V. Larionov, Photoinduced carboborative ring contraction enables regio- and stereoselective synthesis of multiply substituted five-membered carbocycles and heterocycles, J. Am. Chem. Soc., 2017, 139, 11365–11368, DOI:10.1021/jacs.7b07128.
- J. A. Marshall, Photochemically induced ionic reactions of cycloalkenes, Science, 1970, 170, 137, DOI:10.1126/science.170.3954.137.
- T. Kobayashi, R. Shioi, A. Ushie, H. Abe and H. Ito, Catalytic asymmetric total synthesis of (+)-artalbic acid, Chem. Commun., 2016, 52, 9391–9393, 10.1039/C6CC04828E.
- G. Xu, M. Elkin, D. J. Tantillo, T. R. Newhouse and T. J. Maimone, Traversing biosynthetic carbocation landscapes in the total synthesis of andrastin and terretonin meroterpenes, Angew. Chem., Int. Ed., 2017, 56, 12498–12502, DOI:10.1002/anie.201705654.
- B. Gaspar and E. M. Carreira, Catalytic hydrochlorination of unactivated olefins with para-toluenesulfonyl chloride, Angew. Chem., Int. Ed., 2008, 47, 5758–5760, DOI:10.1002/anie.200801760.
- H. Shigehisa, T. Aoki, S. Yamaguchi, N. Shimizu and K. Hiroya, Hydroalkoxylation of unactivated olefins with carbon radicals and carbocation species as key intermediates, J. Am. Chem. Soc., 2013, 135, 10306–10309, DOI:10.1021/ja405219f.
- S. S. Goh, S. Guduguntla, T. Kikuchi, M. Lutz, E. Otten, M. Fujita and B. L. Feringa, Desymmetrization of meso-dibromocycloalkenes through copper(I)-catalyzed asymmetric allylic substitution with organolithium reagents, J. Am. Chem. Soc., 2018, 140, 7052–7055, DOI:10.1021/jacs.8b02992.
- S. S. Goh, P. A. Champagne, S. Guduguntla, T. Kikuchi, M. Fujita, K. N. Houk and B. L. Feringa, Stereospecific ring contraction of bromocycloheptenes through dyotropic rearrangements via nonclassical carbocation–anion pairs, J. Am. Chem. Soc., 2018, 140, 4986–4990, DOI:10.1021/jacs.8b00821.
- J. Huang, D. Lebœuf and A. J. Frontier, Understanding the fate of the oxyallyl cation following Nazarov electrocyclization: Sequential Wagner–Meerwein migrations and the synthesis of spirocyclic cyclopentenones, J. Am. Chem. Soc., 2011, 133, 6307–6317, DOI:10.1021/ja111504w.
- A. Yoshimura and V. V. Zhdankin, Advances in synthetic applications of hypervalent iodine compounds, Chem. Rev., 2016, 116, 3328–3435, DOI:10.1021/acs.chemrev.5b00547.
- U. Farid, F. Malmedy, R. Claveau, L. Albers and T. Wirth, Stereoselective rearrangements with chiral hypervalent iodine reagents, Angew. Chem., Int. Ed., 2013, 52, 7018–7022, DOI:10.1002/anie.201302358.
- T. Deng, W. Mazumdar, R. L. Ford, N. Jana, R. Izar, D. J. Wink and T. G. Driver, Oxidation of nonactivated anilines to generate N-aryl nitrenoids, J. Am. Chem. Soc., 2020, 142, 4456–4463, DOI:10.1021/jacs.9b13599.
- R. Davenport, M. Silvi, A. Noble, Z. Hosni, N. Fey and V. K. Aggarwal, Visible-light-driven strain-increase ring contraction allows the synthesis of cyclobutyl boronic esters, Angew. Chem., Int. Ed., 2020, 59, 6525–6528, DOI:10.1002/anie.201915409.
- M. Silvi, C. Sandford and V. K. Aggarwal, Merging photoredox with 1,2-metallate rearrangements: The photochemical alkylation of vinyl boronate complexes, J. Am. Chem. Soc., 2017, 139, 5736–5739, DOI:10.1021/jacs.7b02569.
- M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld and A. Studer, Radical-polar crossover reactions of vinylboron ate complexes, Science, 2017, 355, 936, DOI:10.1126/science.aal3803.
- C. Nagaraju and K. R. Prasad, An unusual ring-contraction/rearrangement sequence for making functionalized di- and triquinanes, Angew. Chem., Int. Ed., 2014, 53, 10997–11000, DOI:10.1002/anie.201407680.
- F. Cuccu, L. Serusi, A. Luridiana, F. Secci, P. Caboni, D. J. Aitken and A. Frongia, Tandem Wittig reaction-ring contraction of cyclobutanes: A route to functionalized cyclopropanecarbaldehydes, Org. Lett., 2019, 21, 7755–7758, DOI:10.1021/acs.orglett.9b02690.
- X. Yu, J. Hu, Z. Shen, H. Zhang, J. M. Gao and W. Xie, Stereospecific construction of contiguous quaternary all-carbon centers by oxidative ring contraction, Angew. Chem., Int. Ed., 2017, 56, 350–353, DOI:10.1002/anie.201609975.
- G. B. Payne, Reactions of hydrogen peroxide. X. Oxidative rearrangements with certain β-Diketones, J. Org. Chem., 1961, 26, 4793–4797, DOI:10.1021/jo01070a003.
- X. Yu, Z. Liu, Z. Xia, Z. Shen, X. Pan, H. Zhang and W. Xie, Oxidative rearrangement of malondialdehyde: Substrate scope and mechanistic insights, RSC Adv., 2014, 4, 53397–53401, 10.1039/C4RA11237G.
- J. Huang, D. Leboeuf and A. J. Frontier, Understanding the fate of the oxyallyl cation following Nazarov electrocyclization: Sequential Wagner-Meerwein migrations and the synthesis of spirocyclic cyclopentenones, J. Am. Chem. Soc., 2011, 133, 6307–6317, DOI:10.1021/ja111504w.
- H. Xu, F. Wang, W. C. Xue, Y. J. Zheng, Q. Wang, F. Y. G. Qiu and Y. H. Jing, Total synthesis of entecavir: A robust route for pilot production, Org. Process Res. Dev., 2018, 22, 377–384, DOI:10.1021/acs.oprd.8b00007.
- M. S. Kirillova, M. E. Muratore, R. Dorel and A. M. Echavarren, Concise total synthesis of lundurines A–C enabled by gold catalysis and a homodienyl retro-ene/ene isomerization, J. Am. Chem. Soc., 2016, 138, 3671–3674, DOI:10.1021/jacs.6b01428.
- É. Frank, Z. Mucsi, I. Zupkó, B. Réthy, G. Falkay, G. Schneider and J. Wölfling, Efficient approach to androstene-fused arylpyrazolines as potent antiproliferative agents. experimental and theoretical studies of substituent effects on BF3-catalyzed intramolecular [3+2] cycloadditions of olefinic phenylhydrazones, J. Am. Chem. Soc., 2009, 131, 3894–3904, DOI:10.1021/ja808636e.
- S. Arai, M. Nakajima and A. Nishida, A concise and versatile synthesis of alkaloids from kopsia tenuis: Total synthesis of (±)-lundurine A and B, Angew. Chem., Int. Ed., 2014, 53, 5569–5572, DOI:10.1002/anie.201400464.
- F. M. Miloserdov, M. S. Kirillova, M. E. Muratore and A. M. Echavarren, Unified total synthesis of pyrroloazocine indole alkaloids sheds light on their biosynthetic relationship, J. Am. Chem. Soc., 2018, 140, 5393–5400, DOI:10.1021/jacs.7b13484.
- K. Weinges, W. Sipos, J. Klein, J. Deuter and H. Irngartinger, Kondensierte ringsysteme, XVIII zur synthese von cyclobuten-derivaten aus den entsprechenden thiolanen–Kristall- und Molekülstruktur von 2,5-o-benzeno-3,4-benzo[4.2.2]propella-3,7,9-trien, Chem. Ber., 1987, 120, 5–9, DOI:10.1002/cber.19871200103.
- E. N. Hancock, E. L. Kuker, D. J. Tantillo and M. K. Brown, Lessons in strain and stability: Enantioselective synthesis of (+)-[5]-ladderanoic acid, Angew. Chem., Int. Ed., 2020, 59, 436–441, DOI:10.1002/anie.201910901.
- T. H. G. Schick, J. C. Lauer, F. Rominger and M. Mastalerz, Transformation of imine cages into hydrocarbon cages, Angew. Chem., Int. Ed., 2019, 58, 1768–1773, DOI:10.1002/anie.201814243.
- J. C. Lauer, W.-S. Zhang, F. Rominger, R. R. Schröder and M. Mastalerz, Shape-persistent [4+4] imine cages with a truncated tetrahedral geometry, Chem. – Eur. J., 2018, 24, 1816–1820, DOI:10.1002/chem.201705713.
- S. Lee, A. Yang, T. P. Moneypenny and J. S. Moore, Kinetically trapped tetrahedral cages via alkyne metathesis, J. Am. Chem. Soc., 2016, 138, 2182–2185, DOI:10.1021/jacs.6b00468.
- S. H. Kennedy, B. D. Dherange, K. J. Berger and M. D. Levin, Skeletal editing through direct nitrogen deletion of secondary amines, Nature, 2021, 593, 223–227, DOI:10.1038/s41586-021-03448-9.
- C. Hui, L. Brieger, C. Strohmann and A. P. Antonchick, Stereoselective synthesis of cyclobutanes by contraction of pyrrolidines, J. Am. Chem. Soc., 2021, 143, 18864–18870, DOI:10.1021/jacs.1c10175.
- J.-L. Hu, L.-W. Feng, L. Wang, Z. Xie, Y. Tang and X. Li, Enantioselective construction of cyclobutanes: A new and concise approach to the total synthesis of (+)-piperarborenine B, J. Am. Chem. Soc., 2016, 138, 13151–13154, DOI:10.1021/jacs.6b08279.
- W. R. Gutekunst and P. S. Baran, Total synthesis and structural revision of the piperarborenines via sequential cyclobutane C–H arylation, J. Am. Chem. Soc., 2011, 133, 19076–19079, DOI:10.1021/ja209205x.
- C. W. Plummer, C. S. Wei, C. E. Yozwiak, A. Soheili, S. O. Smithback and J. L. Leighton, Design, development, mechanistic elucidation, and rational optimization of a tandem ireland Claisen/cope rearrangement reaction for rapid access to the (iso)cyclocitrinol core, J. Am. Chem. Soc., 2014, 136, 9878–9881, DOI:10.1021/ja505131v.
- R. L. Funk, T. A. Olmstead and M. Parvez, A solution to the in,out-bicyclo[4.4.1]undecan-7-one problem inherent in ingenane total synthesis, J. Am. Chem. Soc., 1988, 110, 3298–3300, DOI:10.1021/ja00218a049.
- A. Y. Hong, M. R. Krout, T. Jensen, N. B. Bennett, A. M. Harned and B. M. Stoltz, Ring-contraction strategy for the practical, scalable, catalytic asymmetric synthesis of versatile gamma-quaternary acylcyclopentenes, Angew. Chem., Int. Ed., 2011, 50, 2756–2760, DOI:10.1002/anie.201007814.
- A. Y. Hong and B. M. Stoltz, Enantioselective total synthesis of the reported structures of (−)-9-epi-presilphiperfolan-1-ol and (−)-presilphiperfolan-1-ol: Structural confirmation and reassignment and biosynthetic insights, Angew. Chem., Int. Ed., 2012, 51, 9674–9678, DOI:10.1002/anie.201205276.
- A. D. Rodríguez, J.-G. Shi and S. D. Huang, Pinnatins A–E: Marine diterpenes of the rare gersolane class derived from a photochemically induced rearrangement of a conjugated 2,5-bridged furanocembrane precursor, J. Org. Chem., 1998, 63, 4425–4432, DOI:10.1021/jo980256b.
- Z. Yang, Y. Li and G. Pattenden, Synthesis of E-deoxypukalide, and its biomimetic conversion into deoxypseudopterolide by photochemical ring contraction involving a 1,3-allylic shift, Tetrahedron, 2010, 66, 6546–6549, DOI:10.1016/j.tet.2010.04.001.
- H. Weinstabl, T. Gaich and J. Mulzer, Application of the Rodriguez–Pattenden photo-ring contraction: Total synthesis and configurational reassignment of 11-gorgiacerol and 11-epigorgiacerol, Org. Lett., 2012, 14, 2834–2837, DOI:10.1021/ol301068h.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, The medicinal chemist's toolbox for late stage functionalization of drug-like molecules, Chem. Soc. Rev., 2016, 45, 546–576, 10.1039/C5CS00628G.
| This journal is © The Royal Society of Chemistry 2022 |