
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Repurposing of F-gases: challenges and opportunities in fluorine chemistry
Daniel J.
Sheldon
 and
Mark R.
Crimmin
*
and
Mark R.
Crimmin
*
Department of Chemistry, Molecular Sciences Research Hub, Imperial College London, 82 Wood Lane, Shepherds Bush, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 26th May 2022
Abstract
Fluorinated gases (F-gases) are routinely employed as refrigerants, blowing agents, and electrical insulators. These volatile compounds are potent greenhouse gases and consequently their release to the environment creates a significant contribution to global warming. This review article seeks to summarise: (i) the current applications of F-gases, (ii) the environmental issues caused by F-gases, (iii) current methods of destruction of F-gases and (iv) recent work in the field towards the chemical repurposing of F-gases. There is a great opportunity to tackle the environmental and sustainability issues created by F-gases by developing reactions that repurpose these molecules.
 Daniel J. Sheldon | Daniel Sheldon is a third year PhD student at Imperial College London. His research focuses on repurposing waste fluorinated gases using highly reactive main group compounds. He has reported new chemical reactions to upgrade F-gases including CF3H and SF6. He obtained his Master's degree from the University of Oxford in 2019. |
 Mark R. Crimmin | Mark Crimmin: Graduated from Imperial College London in 2004 and completed a MSc by research in organic synthesis at Bristol University under the supervision of Prof. Aggarwal. He received his PhD in main group chemistry and catalysis from Imperial College London in 2008 supervised by Prof. Mike Hill and Prof. Tony Barrett. In the same year, he was awarded a Royal Commission for the Exhibition of 1851 research fellowship which he took to UC Berkeley to study with Prof. Bob Bergman and Prof. Dean Toste. In 2011, he returned to London as a Royal Society University Research Fellow, initially at UCL and now back at Imperial. He was appointed as a lecturer in 2011, Senior Lecturer in 2016, Reader in Organometallic Chemistry in 2019, and full Professor in 2021. His group develop new methods for the recycling and chemical repurposing of F-gases. |
1. Introduction
Fluorinated compounds have greatly improved the quality of our life over the last century. They have found wide ranging uses in the refrigeration, pharmaceutical and agrochemical industries, and as propellants, surfactants, polymers, and fire suppressants. Due to its lack of polarizability fluorine does not engage in many intermolecular interactions, and so fluorinated compounds often have low surface energies and consequently low boiling points compared to their non-fluorinated analogues. This is true of F-gases. F-gases include chlorofluorocarbons (CFCs), hydrochlorofluorocarbons (HCFCs), hydrofluorocarbons (HFCs), perfluorocarbons (PFCs), and sulfur hexafluoride (SF6). They are a class of volatile molecules that are defined by the inclusion of at least one fluorine atom and their low boiling point, arguably hydrofluoroolefins (HFOs) can also be included in this compound class (Fig. 1).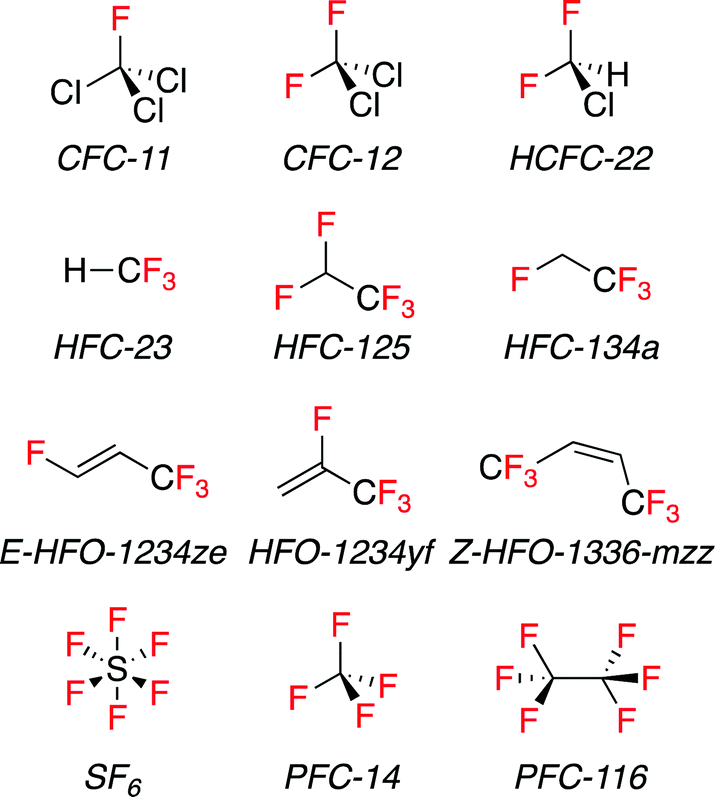 | ||
| Fig. 1 Line drawings of common F-gases. | ||
Despite their widespread usage, there are almost no naturally occurring fluorocarbons. The majority of fluorinated chemicals, including F-gases, are made from inorganic fluoride (i.e. CaF2). Reaction of CaF2 with H2SO4 produces HF; a widely used chemical intermediate.1 HF can be converted into fluorocarbons predominantly through the Swarts process and the Balz–Schiemann process.2,3 HF can also be used to make elemental fluorine, F2, which can then be used to synthesise perfluorocarbons in the Fowler process.4,5 Other major uses of F2 include the manufacture of UF6 from UF4 and in the production of SF6.6 In 2007, it was estimated there is a 100 year supply of CaF2 if the current rate of usage continues.5 While it is likely that new sources – or alternatives such as cryolite (Na3AlF6) or fluorapatite (Ca5(PO4)3F) – will be found,6 the rate of consumption is predicted only to increase as living standards increase.7
Fluorocarbons are often treated as ‘single-use’. One of the largest mechanisms of loss to the environment is through F-gas emissions. This has created a major environmental problem as many F-gases are potent greenhouse gases. Fluorine containing molecules are also discarded as a mixture of solid waste from metal manufacturing and fertiliser.6 Negligible amounts of fluorine used in industry is recycled or reclaimed, creating a major issue of sustainability. We foresee a huge demand for repurposing the fluorine in current waste streams, particularly from emitted F-gases. In this review article, we set out the environmental impact of F-gases and discuss recent work in the field for the chemical repurposing of these compounds. Recent reviews can be found on the history of fluorocarbon refrigerants and their syntheses,8–11 and one containing a discussion of the developments towards reducing F-Gas emissions.12 A further review details the advances made in using fluorinated gases in continuous flow processes.13
2. Applications of F-gases
2.1 CFCs and HCFCs
The first wide industrial application of F-gases (Table 1) was realised with CFCs in the early 1930s.14 These gases were used as refrigerants due to their low toxicity, low flammability, high volatility and high chemical and thermal stability.1,15 CFCs also later became the main components of aerosol propellants and blowing agents. Chlorotrifluoromethane (CFC-11) and dichlorodifluoromethane (CFC-12) were the primary refrigerants alongside hydrochlorofluorocarbons (HCFCs) until the 1980s, with these gases being produced on a scale of one million tonnes per annum at the peak of their production.CFCs have very long atmospheric lifetimes and diffuse into the stratosphere where they are decomposed by UV radiation.6 UV radiation homolytically cleaves the C–Cl bonds of CFCs, creating chlorine radicals. These react with ozone to create oxygen and chlorooxide radicals, which then regenerate chlorine radicals and therefore propagate the cycle.1,16 Ozone depletion causes a major environmental problem as the ozone layer protects the earth's surface from harmful UV radiation.6 The Montreal Protocol agreed a phase out of CFCs from developed countries by 1996 and from developing countries by 2010.17 It is considered one of the most successful responses to a global environmental issue. HCFCs were seen as a temporary replacement for CFCs as they have a shorter atmospheric lifetime and contribute less to ozone depletion. Chlorodifluoromethane (HCFC-22) has an ozone depleting potential (ODP) about 20 times less than CFC-12, and so it became a popular replacement.17 However, HCFCs are still ozone-depleting substances and are also being phased out. By 2030, all HCFC consumption should be halted in most developed countries, and by 2040 for developing countries.18,19 However, production of HCFCs (such as HCFC-22) as a feedstock chemical (e.g. for fluoropolymer synthesis) is permitted to continue, and as such HCFC-22 is still being produced in large amounts as a chemical intermediate by fluoropolymer and polyfluoroalkyl substance (PFAS) manufacturing plants.20 A recent report from the US environmental protection agency (EPA) suggests the release of this gas to the environment may still be a serious issue.21
2.2 HFCs
The Montreal Protocol accelerated the need to find direct replacements for CFCs and HCFCs. Hydrofluorocarbons (HFCs) were identified because they do not deplete ozone, while they have similar chemical and thermal properties to CFCs.16,22 Nowadays, the majority of HFC consumption is for refrigeration purposes, with the most common being 1,1,1,2-tetrafluoroethane (HFC-134a).17,23 While the use of HFCs was seen to ‘fix’ the issue of ozone depletion, they brought a new issue to light in that they are greenhouse gases, absorbing infrared radiation in the region 1000–1400 cm−1. Global warming potential (GWP) is a metric that considers the amount of energy 1 ton of an emitted gas will absorb over a given period relative to 1 ton of emitted CO2 (often considered over 100 years, denoted GWP100). Most HFCs have a GWP100 over 1000 and are therefore considered highly potent greenhouse gases. The Kyoto Protocol, signed in 1997, committed signatories to reduce greenhouse gas emissions with a focus on the reduction of F-gases including HFCs, PFCs and SF6.24,25 However, it faced criticism over its narrow time period and the fact it focused mainly on developed countries.24 In order to reduce greenhouse gas emissions, further stringent restrictions on the production and use of HFCs were set, although only many years later. In 2016, the Kigali Amendment to the Montreal Protocol was signed which committed signatories to ‘phase-down’ HFCs, i.e. reduce the production and consumption of HFCs.26–29 Other legislation included the EU F-gas regulations, which further sought to limit fluorinated gas emissions by banning fluorochemical refrigerants with a GWP100 exceeding 150 in automobile air conditioning systems, effecting a ban on HFC-134a in its largest and most emissive application.19,29 It also banned the marketing of refrigeration and air conditioning equipment using these refrigerants, and required periodic inspection of stationary systems using HFCs.30,31Alternative uses for HFCs are in applications such as blowing agents for producing foam plastics applied to insulation and packaging, aerosols in metered dose inhalers, fire suppressants and in the dry etching of dielectric films and fabrication of thin-film devices.22,32
2.3 HFOs
The next generation of refrigerants set to replace HFCs include hydrofluoroolefins (HFOs). HFOs are considered promising alternatives due to their low GWP and suitable chemical and physical properties.33 For example, E-1,3,3,3-tetrafluoropropene (E-HFO-1234ze) and 2,3,3,3-tetrafluoropropene (HFO-1234yf) both have a GWP100 of less than 1.19 The inclusion of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond within the F-gas leads to a more reactive compound, thus enabling a lower atmospheric lifetime and GWP100. The counterpoint is that this substitution also leads to lower stability and higher toxicity.33 It is likely that these chemicals will decompose close to the source of emission, which can lead to local pollution, along with concerns that the decomposition products may themselves possess a high GWP100. There are also some thermophysical related issues, such as the low evaporating pressure and small vaporisation enthalpy of E-HFO-1234ze resulting in a lower coefficient of performance and a lower volumetric refrigeration capacity than HFC-134a.34 In order for industry to create a refrigerant that possesses the ideal thermophysical properties and a low GWP100, refrigerant blends comprising various HFOs and HFCs have been marketed These are often designed to offer a compromise between flammability and GWP.35 For example R-449a (Opteon XP40 by Chemours) has a 1
C double bond within the F-gas leads to a more reactive compound, thus enabling a lower atmospheric lifetime and GWP100. The counterpoint is that this substitution also leads to lower stability and higher toxicity.33 It is likely that these chemicals will decompose close to the source of emission, which can lead to local pollution, along with concerns that the decomposition products may themselves possess a high GWP100. There are also some thermophysical related issues, such as the low evaporating pressure and small vaporisation enthalpy of E-HFO-1234ze resulting in a lower coefficient of performance and a lower volumetric refrigeration capacity than HFC-134a.34 In order for industry to create a refrigerant that possesses the ideal thermophysical properties and a low GWP100, refrigerant blends comprising various HFOs and HFCs have been marketed These are often designed to offer a compromise between flammability and GWP.35 For example R-449a (Opteon XP40 by Chemours) has a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 composition by % wt of HFC-32:HFC-125:HFO-1234yf:HFC134a.34,36,37 Refrigerant blends can be designed to have particular properties, perhaps superior to other pure refrigerants, but each element of added complexity increases cost, the refrigerant charge, the potential for leaks, while also decreasing reliability.14,19
:1:1:1 composition by % wt of HFC-32:HFC-125:HFO-1234yf:HFC134a.34,36,37 Refrigerant blends can be designed to have particular properties, perhaps superior to other pure refrigerants, but each element of added complexity increases cost, the refrigerant charge, the potential for leaks, while also decreasing reliability.14,19
2.4 Perfluorinated F-gases
SF6 is widely used as an electrical insulating gas in circuit breakers, transmission lines, transformers, and substations. 80% of SF6 produced between 1996–2003 was consumed by electric utilities and equipment manufacturers for electric power systems.38,39 Some of its other uses include semiconductor processing, a blanket gas for magnesium refining, thermal and sound insulation.40 It is a synthetic gas whose unique chemical and physical inertness (non-flammable, non-toxic, non-explosive) and excellent thermal conductivity mean it is ubiquitous in electrical insulation and largely irreplaceable. It is one of the most potent greenhouse gases and as such its emission creates a significant contribution to climate change, as will be discussed in further detail below. It was included in the Kyoto Protocol and EU F-gas regulations for limiting the emissions of fluorinated greenhouse gases.25,30 There are no restrictions on using SF6 in switchgear under these regulations, but there are requirements to recover SF6 where possible.30,38 The EPA established an agreement in 1995 with electric power systems to reduce emissions and in 2016 it was reported that the industrial partners who signed up had reduced their emissions by 76%.38,39 Furthermore, very recently there has been research to provide alternative gases to SF6 such as perfluoroketones, perfluoronitriles and CF3I.41–44 Newer wind-turbine technology has resulted in some SF6-free equipment or even vacuum insulated switchgear.38Perfluorocarbons find wide-ranging applications as specialist solvents, blood substitutes, textile finishers and fire suppressants.45,46 They are also used widely in the electronic industry, with applications ranging from semiconductor manufacturing, integrated circuit components and in the assembly of power electronics.25 They have low surface energies, high dielectric strength and are compressible. They are also potent greenhouse gases, with the fully saturated C1 to C6 perfluorocarbons included in the Kyoto protocol of 1997.47 Efforts to limit emissions are being carried out by several developed countries.6 In 1999, the World Semiconductor Council committed to the reduction of PFC emissions by 10% or greater by 2010 from the baseline levels of 1995.48 This included exploring alternative chemicals such as NF3, improving capture/recycle systems and the optimisation of existing processes and destruction methods.49
3. Production and environmental impact of F-gases
In general, atmospheric concentrations of F-gases are low (relative to CO2), parts per trillion, but their impact is significant. Since 1978, the Advanced Global Atmospheric Gases Experiment (AGAGE) have measured the atmospheric concentrations of synthetic greenhouse gases from remote stations.50,51 These remote stations observe air masses using gas chromatography-electron capture detection and gas chromatography-mass spectrometry techniques.50,52,53 Many of the data discussed below are reported from these measurements.18,38,47 Top-down global emission estimates are derived from atmospheric concentration measurements from AGAGE, using the two-dimensional AGAGE 12-box model.18,54,55 From 1990 onwards, developed countries have been required to report emissions of HFCs, PFCs and SF6 as part of the United Nations Framework Convention on Climate Change (UNFCCC). However, by these reports half of the world's HFC emissions were unaccounted for when compared to global emission estimates derived from AGAGE atmospheric measurements.18,29,56 The implication is that much of the world's F-gas emissions are unreported by UNFCCC. This is likely due, in part, to emissions from developing countries.57,58| F-Gas | Main uses | Atmospheric lifetime (years) | GWP100 |
|---|---|---|---|
| CFC-11, CFCl3 | Refrigerant | 52 | 5560 |
| CFC-12, CF2Cl2 | Refrigerant | 102 | 11200 |
| HCFC-22, CF2ClH | Refrigerant | 11.9 | 1960 |
| HFC-23, CF3H | By-product | 228 | 14600 |
| HFC-32, CF2H2 | Refrigerant blend component | 5.4 | 771 |
| HFC-125, C2F5H | Refrigerant blend component, fire suppressant | 30 | 3740 |
| HFC-134a, CFH2CF3 | Refrigerant, foam blowing agent, fire suppressant and propellant | 14 | 1530 |
| HFC-143a, CF3CH3 | Refrigerant blend component | 51 | 5810 |
| HFC-152a, CF2HCH3 | Refrigerant blend component, foam blowing agent and aerosol propellant | 1.6 | 164 |
| HFO-1234yf, H2CCFCF3 |
Refrigerant | 12 days | <1 |
| HFO-1234ze, (E)-HFCCHCF3 |
Refrigerant | 10 days | <1 |
| HFO-1336-mzz, (Z)-F3CCHCHCF3 |
High temperature refrigerant | 27 days | 2 |
| SF6 | Insulation gas in electric power industry | 3200 | 25200 |
| PFC-14, CF4 | Semiconductor manufacture, aluminium production | 50000 |
7380 |
| PFC-116, C2F6 | Semiconductor manufacture | 10000 |
12400 |
| PFC-218, C3F8 | Semiconductor manufacture | 2600 | 9290 |
3.1 HCFCs and HFCs
Trifluoromethane, CF3H (HFC-23), is produced on a vast scale (ca. 20 Gg year−1) largely as a by-product in the synthesis of CF2ClH (HCFC-22) by the over-fluorination of chloroform (CCl3H).60 HFC-23 is a potent greenhouse gas, with a GWP100 of 14600 and an atmospheric lifetime of 228 years.59 It has very little application itself and is either stored, destroyed or released to the environment. Its atmospheric release therefore presents a significant issue for climate change. Although commercial applications of HCFC-22 are being phased out under the Montreal Protocol, HCFC-22 is still permitted for ‘non-dispersive uses’, such as a chemical intermediate in the manufacture of fluoropolymers.20 Total HCFC-22 production has increased dramatically, from 65 Gg year−1 in 1990 to 947 Gg year−1 in 2017.61 In more recent years, this production has been dominated by developing countries, but there are still small increases in total production from developed countries despite a complete phase-out of production and consumption scheduled for 2030.61 As a consequence of increased production of HCFC-22, the atmospheric concentration of HFC-23 has been steadily increasing since at least 1978.62,63 Using data collected from AGAGE measurements, global emissions of HFC-23 were estimated as ∼15.9 Gg year−1 in 2018, the highest than at any point in history,61 an increase from the estimates of ∼12.3 Gg year−1 in 2016, ∼12.5 Gg year−1 in 2012, ∼8.9 Gg year−1 in 1995 and ∼4.3 Gg year−1 in 1978.18,64 It is noted that there was brief mitigation of global HFC-23 emissions during 2006–2009, due to initial success of abatement technologies through the UNFCCC Clean Development Mechanism (CDM), with emissions reaching a global minimum of ∼9.6 Gg year−1 in 2009.18,20 It is suggested that abatement measures have since been inadequate to offset the increasing production of HCFC-22 for non-dispersive uses, hence the return to an upwards trend in global emissions of HFC-23.20 Estimates of global emissions of HCFC-22 have remained relatively constant at ∼370 Gg year−1 during the period 2012–2016.56 Before this period of stability they increased drastically to ∼368.8 Gg year−1 in 2012, from ∼232.2 Gg year−1 in 1995 and ∼101.8 Gg year−1 in 1978.18
Production and emissions of 1,1,1,2-tetrafluoroethane (HFC-134a) begun in the 1990s as CFCs were being phased-out. HFC-134a was seen as a direct replacement for CFC-12 in motor vehicle air conditioning systems.17 HFC-134a is manufactured predominantly by fluorination of trichloroethane using hydrogen fluoride in the gas phase, with a heterogenous catalyst. The trichloroethane is itself synthesised from ethylene.23 HFC-134a has a GWP100 of 1530 and an atmospheric lifetime of 14 years.59 It is the most abundant HFC in the global atmosphere.65 Estimates suggest that more than 56% of cumulative HFC-134a produced since 1990 has been released to the environment.19 Global emission estimates derived from HFC-134a atmospheric concentration data collected from AGAGE measurements suggest a dramatic increase to ∼223 Gg year−1 in 2016, from ∼176.5 Gg year−1 in 2012, ∼109.6 Gg year−1 in 2003 and ∼10.2 Gg year−1 in 1994.18,64
1,1,1-Trifluoroethane (HFC-143a) is used in a variety of refrigerant blends, mainly R-404A.29,34,36,66,67 It was avoided as a pure refrigerant due to its high GWP100 of 5810 and because it is also considered mildly flammable.22,34,59,68 Recent research has looked at the critical properties of a binary mixture of HFC-143a and E-HFO-1234ze as an alternative refrigerant and supercritical solvent, due to the low critical temperature and high chemical stability of HFC-143a.34 The global emission estimates of HFC-143a have shown an increase from ∼1 Gg year−1 in the early 1990s to ∼23 Gg year−1 in 2012 and to ∼28 Gg year−1 in 2016.18,64
1,1-Difluoroethane (HFC-152a) has a relatively low GWP100 of 164 and was initially considered as a replacement refrigerant for CFC-12.59,69–74 However, unlike HFC-134a it is flammable, and as a result it is limited to usage as a component of refrigerant blends and as a foam blowing agent or propellant in aerosol sprays and gas dusters.75 The global emission estimates of HFC-152a have shown an increase from ∼2 Gg year−1 around 1990, to ∼53 Gg year−1 around 2012 and remained at ∼53 Gg year−1 in 2016.18,64
Difluoromethane (HFC-32) has a relatively low GWP100 of 771 and good thermodynamic properties as a refrigerant. Its usage as a pure refrigerant was limited due to the fact that it is marginally flammable.19,59,76 Similarly, pentafluoroethane (HFC-125) was considered another viable candidate for refrigeration, but it has a relatively high GWP100 of 3740.59,68,77,78 These gases are often used in refrigerant blends, such as HFC-410A (1:1, HFC-32:HFC-125).19 This blend is a primary replacement for HCFC-22 and thus HFC-410A is now widely used in residential and commercial air conditioning. The blend has practically zero ozone-depleting potential (ODP), in this sense superior to HCFC-22, however the GWP100 of HFC-410A is a 16% increase from HCFC-22. This provides an example of an environmental trade-off between ozone depletion and greenhouse gas emissions. HFC-125 is also used as fire suppression agents, owing to its electrical non-conductivity, ready vapourisation, low toxicity and non-flammability.22 The global emission estimates of HFC-125 have grown from ∼1 Gg year−1 in the early 1990s to ∼40 Gg year−1 in 2012 and to ∼62 Gg year−1 in 2016, while HFC-32 global emissions have increased from ∼1 Gg year−1 in the early 2000s to ∼20 Gg year−1 in 2012 and to ∼35 Gg year−1 in 2016.18,64
3.2 Perfluorinated F-gases
SF6 is considered one of the most potent greenhouse gases, with a GWP100 of 25200 and an atmospheric lifetime of 3200 years.38,59,79–81 SF6 is largely immune to chemical and photolytic degradation, evidenced from vertical profiles of SF6 indicating very little loss of SF6 due to photochemistry in the troposphere and lower atmosphere,38,82 meaning that its contribution to global warming is cumulative and effectively permanent.40,83
The world production of SF6 has steadily increased since the 1970s. The large amount of SF6 in electrical equipment provides a substantial source of emissions through maintenance, replacement, and leakage. It is estimated that about 12% of SF6 consumed in the manufacture and commissioning of electrical equipment is directly emitted.38 In the UK it is estimated that SF6 losses from the electrical power industry is an average of 1% per year from 2010–2016.38,84 The global atmospheric emission rate of SF6 was estimated as ∼9.0 Gg year−1 in 2018, an increase from ∼7.3 Gg year−1 in 2008, and from 2.5 Gg year−1 in 1978, using atmospheric concentration data collected at the AGAGE monitoring sites.38 The overall increase is consistent with the increase in globally installed electrical capacity.38 More recent increases are attributed to the rapid expansion of the electric power industry particularly in Asia.38 For example, electrical capacity installed in China relative to the rest of the world increased from ∼3% in 1980 to ∼43% in 2018.38,85 The adoption of renewable energy technologies has been particularly important.85 It has been assumed that renewables have a wider distribution of power (compared to localised gas or oil power stations), which consequently requires more connections in the electricity grid and therefore more gas-insulated switches, circuit breakers and transformers.38 As a result, despite an observed decrease in SF6 emissions from developed countries in recent years thanks to efforts made to replace SF6 as a blanketing gas along with technological improvements to reduce emissions from electrical equipment, this has been largely overwhelmed by major increases in emissions elsewhere in the world.38
Perfluorocarbons such as CF4 (PFC-14), C2F6 (PFC-116) and C3F8 (PFC-218) are amongst the most highly potent greenhouse gases regulated under the Kyoto Protocol.47 CF4 has a GWP100 of 7380 and an atmospheric lifetime of 50000 years, C2F6 has a GWP100 of 12400 and an atmospheric lifetime of 10000 years, and C3F8 has a GWP100 of 9290 and an atmospheric lifetime of 2600 years.59 These gases are vital to the semiconductor industry for numerous low-pressure operations such as chemical etching. The gaseous waste streams from these processes are often emitted to the atmosphere due to the difficulty of thermal incineration processes. Significant CF4 releases also occur during the electrolytic production of aluminium.47 CF4 and C2F6 were found to be ubiquitous in the troposphere from the 1970s, with the less abundant C3F8 being found in the atmosphere more recently.47 It is noted that there is evidence for a natural source of CF4 in the atmosphere, likely from a lithospheric source, although there is some uncertainty in its contribution to emissions.47 Reports using atmospheric concentration data collected by AGAGE estimated the global CF4 emissions to be ∼13.9 Gg year−1 in 2019, ∼12.6 Gg year−1 in 2016 and ∼10.5 Gg year−1 during the period 2005 to 2008.47,55,56 This is slightly down from earlier emission rates, for example ∼17.7 Gg year−1 from 1980–1984.47 The reduction is attributed to increasing efficiency and reduction measures taken up by the aluminium industry. The estimated global emissions of C2F6 showed an increase from ∼2.0 Gg year−1 for 1980–1984 to ∼2.9 Gg year−1 during 2000–2004, before decreasing slightly to ∼2.3 Gg year−1 in 2005–2008. Global emissions of C2F6 were then estimated as ∼2.0 Gg year−1 in 2016 and ∼2.2 Gg year−1 in 2019.47,55,56 This trend follows the introduction of C2F6 as a fluorine source in the semiconductor industry in the early 1990s, before its gradual replacement with more efficient fluorine sources such as NF3 in more recent years.47 C3F8 global emissions increased sharply to ∼1.1 Gg year−1 in 2000–2004 from ∼0.2 Gg year−1 in 1980–1984, before declining to ∼0.8 Gg year−1 for 2005–2008 and ∼0.5 Gg year−1 for 2016–2019.47,55,56 This trend represents the increased applications for C3F8 in more recent years, such as an inert reaction medium, a dielectric and as a propellant, before attempts to gradually replace it.47 While the overall emissions of these gases are lower compared to CFCs, HCFCs and HFCs discussed above, their incredibly long atmospheric lifetimes can lead to permanent alteration of the radiative budget of the atmosphere and so even small atmospheric concentrations are to be taken seriously.47
Fig. 2a and b display the global mean atmospheric concentrations of the above synthetic greenhouse gases, showing their variation against time. Data was obtained from AGAGE GC-MS “Medusa” measurements, including the most recent data published (2020) at the time of writing.51
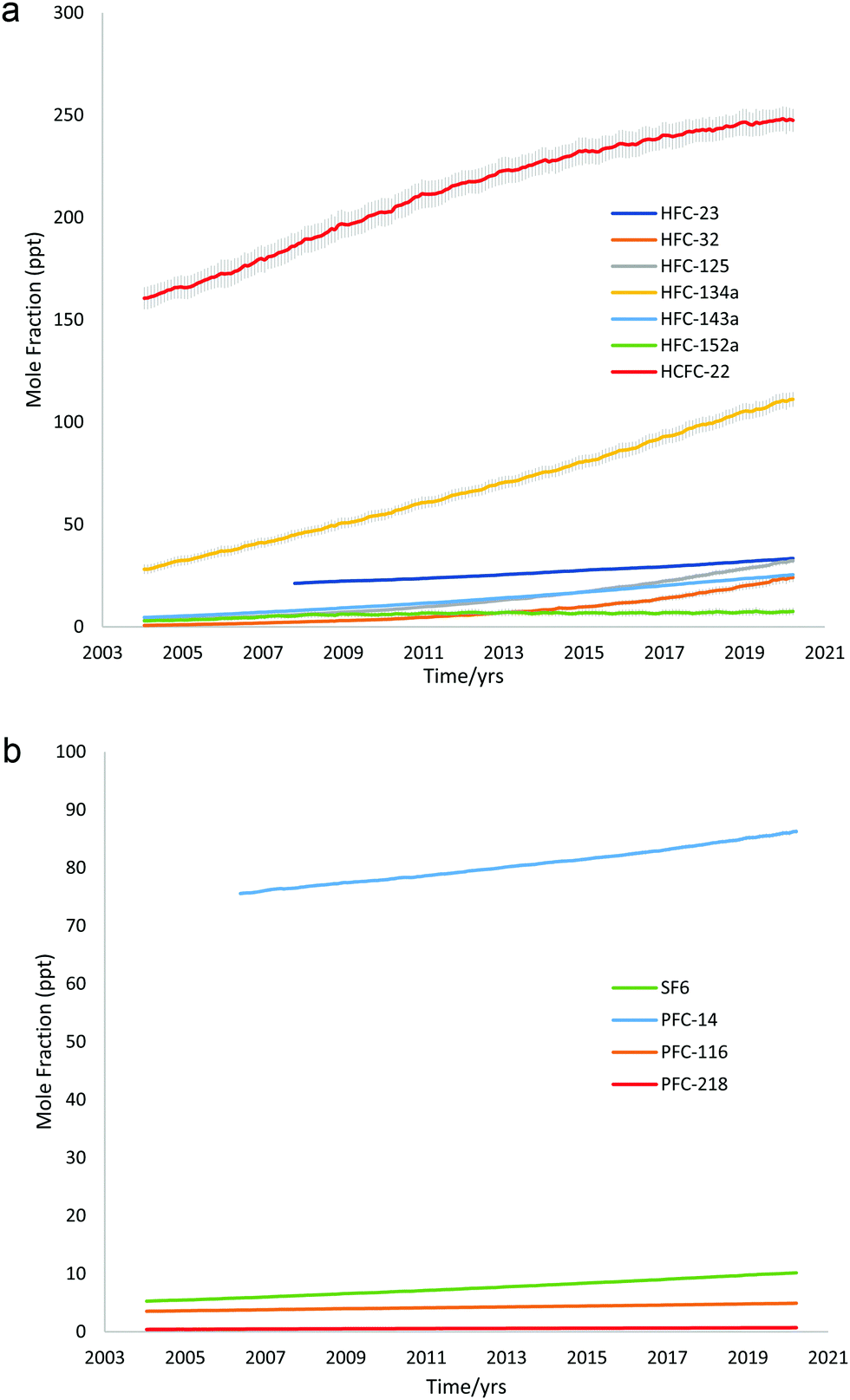 | ||
| Fig. 2 (a) Global mean atmospheric concentrations of HFCs and HCFC-22 vs. time, data from AGAGE GC-MS Medusa measurements. (b) Global mean atmospheric concentrations of PFCs and SF6vs. time, data from AGAGE GC-MS Medusa measurements. | ||
4. Destruction of F-gases
One of the ways to prevent the release of environmentally persistent F-gases into the atmosphere is through collection and immediate destruction of the emitted gases. Destruction methods for F-gases require extreme conditions due to their chemical and physical inertness. They are as a result often highly energy intensive, with methods including thermal oxidation, catalytic hydrolysis and plasma destruction.624.1 Thermal oxidation
One of the most common destruction methods is thermal oxidation. The thermal oxidation of HFC-23 (which often contains traces of HCFC-22) generally requires temperatures higher than 1200 °C and leads to the formation of CO2, HF and HCl. The HF can be recovered and sent to storage for future use, often as an alkali fluoride salt, and as such this destruction technique can allow for some fluorine material recovery. A major issue of this technique is the high operational costs associated with sustaining these extremely high temperatures and finding suitable materials compatible with both the temperature and the strongly corrosive HF. Moreover, there is the potential to form other hazardous materials during the cooling procedure of reaction flows that may pose a great environmental threat, such as dioxin.62 Under the UNFCCC Clean Development Mechanism (CDM) (2003–2014), developing countries were able to register waste treatment facilities for thermal oxidation of synthetic greenhouse gases (not including CO2).61 There were 19 HCFC-22 production plants in five developing countries approved for the destruction of waste HFC-23.20,62 As noted in Chapter 3, global emissions of HFC-23 declined between 2006–2009, and this is largely attributed to CDM projects. Since the CDM period ended, HFC-23 emissions have grown once more.20,61A few studies available in the open literature have focused specifically on HFC-134a destruction.86–91 One study carried out experiments using a tubular-type furnace to explore the important features of HFC-134a thermal decomposition. They concluded that with a temperature above 800 °C, sufficient O2 and an auxiliary fuel (H2O or CH4), HFC-134a reacts to produce HF, H2O and CO2.86 Without an auxiliary fuel capable of providing a hydrogen atom, HFC-134a reacts with O2 to produce F2 instead of HF. F2 is highly toxic and far more difficult to remove than HF.86 With insufficient H2O and O2, carbon products such as CO can be formed.87 A fine control over other factors such as steam flow rate are crucial for keeping the NOx concentration down. It appears paramount that the conditions are optimum for a destruction methodology to be both environmentally and economically viable. There are also a few examples of patents filed for the thermal oxidation of other F-Gases, such as SF6 and other PFCs.92,93 For SF6, temperatures in excess of 1100 °C are required for its combustion, and even at these temperatures combustion is often incomplete with NOx released, along with the same problems of cost and sourcing of materials.81
4.2 Catalytic hydrolysis
Catalytic hydrolysis has been thoroughly researched as an alternative, low-cost method for F-gas destruction. For this approach to be viable, catalytic materials must be both highly reactive and be able to function for long periods of time in a highly corrosive acid gas environment.94 Multiple reports, along with a number of patents,95–97 have demonstrated successful catalyst systems for the disposal of HFC-23 alongside other HCFCs, CFCs and PFCs, but only at very low concentrations. For example, a Pt/ZrO2–SO4 catalyst was shown to destroy HFC-23 by catalytic hydrolysis to form CO2 and HF, at 500–550 °C.94 In a similar manner, a nickel pyrophosphate catalyst was shown to decompose HFC-23 at 500 °C to CO2 and HF.98 Another report detailed a AlPO4–Al2O3 system that was shown to hydrolytically decompose CF4 into CO2 and HF, where the phosphate helped stabilise the catalyst from deactivation.99 Zhang and co-workers reported a platinum promoted TiO2–ZrO2 catalyst that served to convert HCFC-22 to CO2 with 95% selectivity at 340 °C.100 A major downside to these methods is that at high concentrations (>5000 ppm), the efficiency and stability of the catalysts are highly vulnerable.62 Also, the inclusion of an expensive transition metal is often required for the catalyst to convert CO to CO2 during hydrolysis and also allows for a lower operating temperature.62,99 The production of HF and HCl pose a huge challenge to any catalyst, as these compounds will react with almost all materials leading to catalyst deactivation. Catalytic technologies for SF6 destruction suffer in a similar way as the toxic products of decomposition poison the catalyst.1014.3 Plasma destruction
Plasma technology has been commercially applied for the destruction of F-gases in a plasma arc at temperatures typically between 10000 and 30000 °C.62,102–106 The PLASCON process technology, developed by the Australian Commonwealth Scientific and Research Organisation and SRL Plasma Ltd, was adapted for CFC and halon destruction in 1992, and for HFC-23 in 2007. The fluorinated products of HFC-23 destruction are HF and F2. These molecules create significant issues at the high temperatures required. In the late 1990s, a few reports detailed the abatement of HFC-23 and PFCs using surface wave plasma with very high efficiency, but poisonous COF2 was detected in the effluent gas alongside other products including CO, CO2 and HF.62,107,108 Methods to avoid the formation of other dangerous fluorochemicals have been explored, such as by using steam instead of oxygen as the oxidising gas.104 Nonthermal plasma technologies have been thoroughly investigated as a way of controlling PFCs, although these also often led to the formation of toxic byproducts.107–112 Combining plasma with a catalyst was explored in the abatement of C2F6, where it was found to produce mainly CO2 and trace CO, as the catalyst surfaces were able to adsorb and dissociate radicals that usually lead to the formation of toxic products such as F2, COF2 and CF4.113 In general, the potential for hazardous fluorinated by-products along with the operational costs associated with the exceptionally high temperatures required restrict the wide application of thermal plasma technology for F-gas destruction.62
4.4 Electric discharge
SF6 can undergo decomposition in an electrical discharge (arc, spark or corona).48,113–122 However, the products from this type of decomposition can be highly toxic, while some are potent greenhouse gases themselves. Decomposition products can include S2OF10, CF4, COF2, F2, HF, H2S, NF3, F2O, SiF4, SO2, S2F10, SF4, SO2F2, SOF2, SOF4, S2O2F10.48,114,118,123–1255. Chemical upgrading of F-gases
Much progress has been made in the field of chemical upgrading fluorocarbons into reactive building blocks, but F-gases are some of the most challenging and least reactive molecules to be studied.126–129 This is largely due to the strength of the sp3 C–F bond; the C–F bond dissociation energy of CH3F is 115 ± 4 kcal mol−1.130 In addition, alkyl fluorides are poor substrates for nucleophilic substitution or oxidative addition to metals due to a lack of charge stabilisation in the transition state for bond-breaking.130–132 Despite this, recent work has shown a range of methods for C–F activation in fluoroalkanes.133–136 We describe below the recent efforts in targeting new reactions to upgrade F-gases.5.1 HFC-23
Fluorination in the pharmaceutical and agrochemical industries is increasingly important, with both –CF3 and –CF2H moieties common in drug design. Therein lies an opportunity for a small component of waste CF3H (HFC-23) to serve as a feedstock for the synthesis of trifluoromethyl and difluoromethyl building blocks. CF3H has a low boiling point (−83 °C) and a relatively acidic C–H bond (pKA ∼ 25 in H2O).60,137 The CF3− anion generated from a deprotonation reaction has been shown to decompose under certain reaction conditions to form difluorocarbene, CF2, and a fluoride anion, F−.60,138–142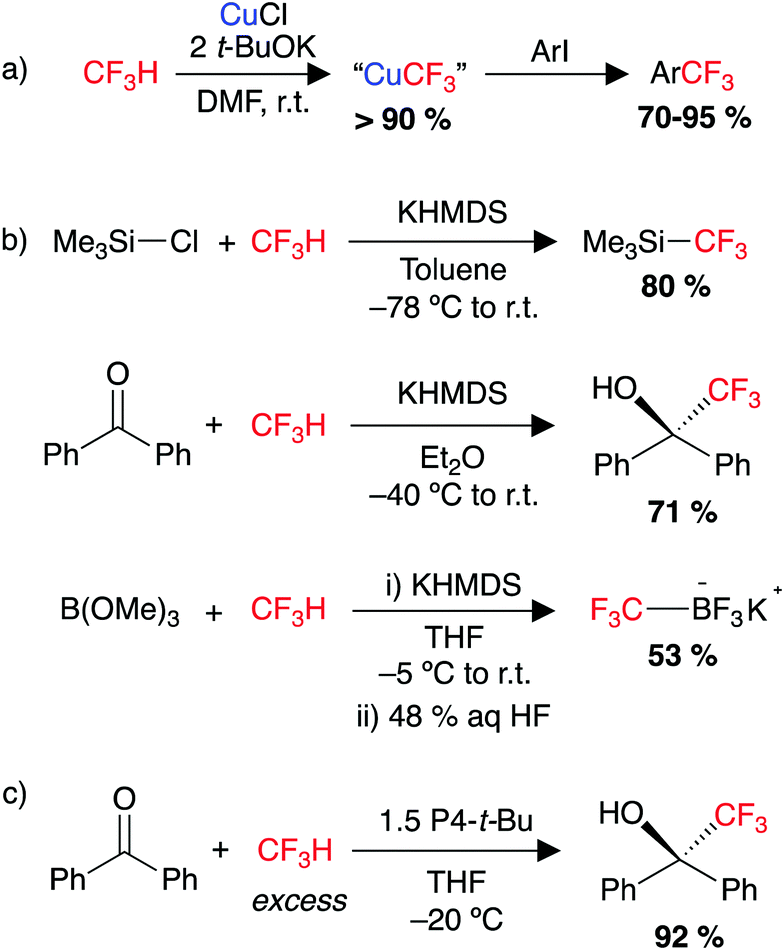 | ||
| Scheme 1 (a) Cupration of CF3H and trifluoromethylation of aryl iodides, (b) nucleophilic trifluoromethylation of a range of electrophiles using CF3H as a feedstock. HMDS = hexamethyldisilazane, (c) trifluoromethylation of carbonyl substrates (benzophenone shown) using HCF3 and P4-t-Bu superbase. P4-t-Bu = ((NMe2)3PN)3PN(t-Bu). | ||
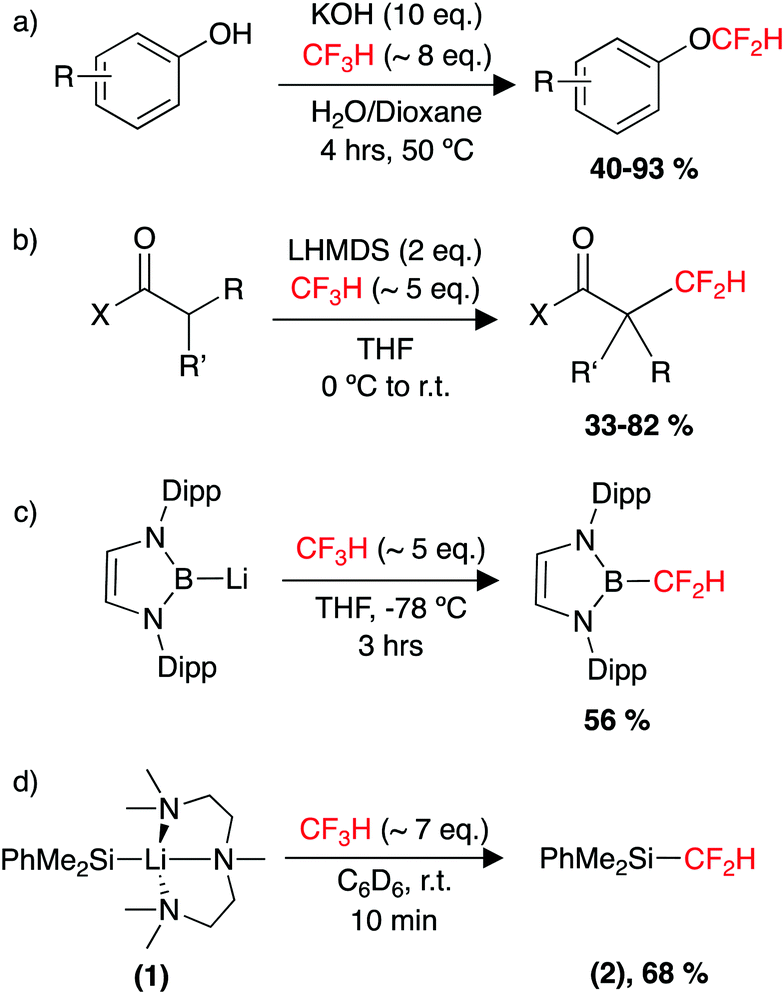 | ||
| Scheme 2 Utility of HCF3 as a feedstock, (a) in the synthesis of aryldifluoromethylethers, (b) in the α-difluoromethylation of carbonyls, (c) defluoroborylation to form an organoboron building block and (d) defluorosilylation to form an organosilicon building block. | ||
More recently in 2017, the groups of Ito and Mikami reported the defluoroborylation of CF3H using a highly nucleophilic boryl lithium reagent, to form a –CF2H containing organoboron building block (Scheme 2c).178 While a mechanistic study was not carried out for this system, the related computational study by Mikami on the α-difluoromethylation of lithium enolates was utilised to propose a pathway for the defluoroborylation.174 The authors suggest initial deprotonation of CF3H occurs to form the unstable intermediate [CF3Li], before C–F cleavage then proceeds via an SN2-type attack by the nucleophilic boryl lithium at [CF3Li], in a bimetallic transition state. While an important discovery, application of the defluoroborylation methodology was limited by issues regarding scalability. The boryl lithium reagent is extremely difficult to synthesise and is highly susceptible to degradation. The difluoromethyl organoboron building block was reported as bench stable but its utility is unknown.178
In 2020, our group reported the rapid, room-temperature defluorosilylation of CF3H with the silyl lithium reagent 1 (Scheme 2d).179 The reaction forms a valuable difluoromethyl organosilicon building block, 2, which is closely related to Me3Si–CF2H, both are known difluoromethyl transfer agents.180,181 The reaction was successfully scaled up to synthesise 2 on a gram-scale in a 68% isolated yield. An extensive DFT study was carried out to explore the mechanism of the reaction. The mechanism is proposed to involve an initial deprotonation of CF3H by 1 to form PhMe2SiH and [CF3Li·PMDETA] (PMDETA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine), a second equivalent of 1 then attacks [CF3Li·PMDETA] at the carbenoid carbon in an SN2-like fashion, resulting in the cleavage of a C–F bond and formation of a C–Si bond. Reprotonation by a further equivalent of CF3H leads to the product 2 and regenerates an equivalent of [CF3Li·PMDETA], suggesting the reaction is catalytic in [CF3Li·PMDETA]. Key intermediates and a transition state of the calculated pathway are represented in Fig. 3.
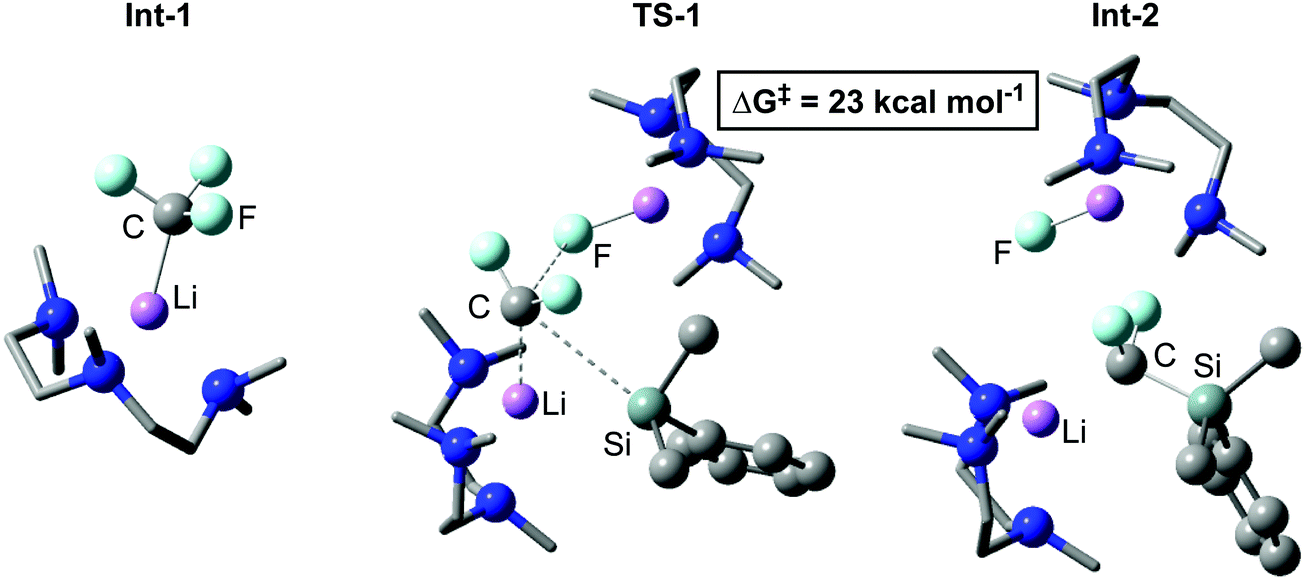 | ||
| Fig. 3 Key stationary points calculated for the C–F cleavage of an equivalent of [CF3Li·PMDETA] in the defluorosilylation of trifluoromethane. | ||
TS-1 is unusual as [CF3Li] may have been considered an unlikely electrophile. A Natural Bond Orbital (NBO) analysis was therefore carried out. The resulting Natural Population Analysis (NPA) charges revealed a positive charge at the carbenoid carbon of [CF3Li·PMDETA] as a result of the highly electron withdrawing effect of three electronegative fluorine atoms, a result noted in previous calculations on [CF3Li].182 This explains the electrophilic nature of [CF3Li·PMDETA] and why it is attacked by 1 in TS-1. The geometry of TS-1 is somewhat similar to that calculated by Mikami for the attack of a THF-stabilised lithium enolate on [CF3Li].174TS-1 is bimetallic, where one lithium cation stabilises the fluoride leaving group, and the other acts as an anchor in C–Si bond formation. It has been suggested in other work that Li⋯F interactions are crucial in stabilising similar transition states.171,174
The organosilicon building block 2 is an easy-to-use difluoromethyl transfer agent for carbonyl substrates (Scheme 3).180,181 The use of 2 has been somewhat scarce compared to Me3SiCF2H, but it is suggested that this could be due to the difficulty and cost of its synthesis.183 Greater access to easy-to-use fluorinated transfer agents often leads to an increased use of those fluorinated moieties in pharmaceutical design.179
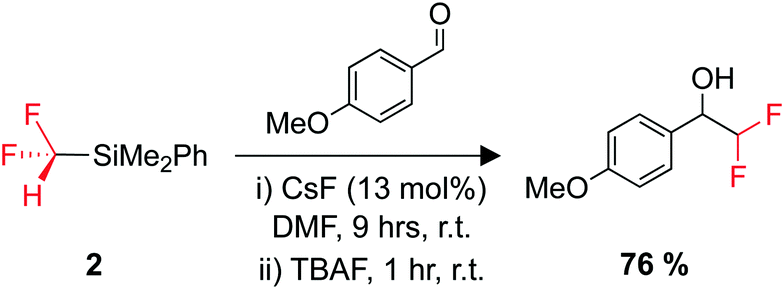 | ||
| Scheme 3 Utility of 2 as a difluoromethylating agent. TBAF = tetrabutylammonium fluoride. | ||
5.2 HFC-134a
The chemistry of HFC-134a (1,1,1,2-tetrafluoroethane) is largely limited to reactions with a base that involve a formal elimination of an equivalent of HF and subsequent deprotonation to form a nucleophilic source of the trifluorovinyl moiety.190,191 For example, the reaction of HFC-134a with 2 equiv. n-BuLi at −78 °C formed trifluorovinyllithium, reported to be stable in solution at −78 °C (Scheme 4a). Warming the solution to room temperature led to decomposition to an uncharacterised black material. The trifluorovinyllithium moiety formed at −78 °C can react with a wide range of electrophiles including metal halides, main group halides, CO2, aldehydes and epoxides.190–208 Using this method with transition metal halides resulted in the formation of transition metal complexes with a trifluorovinyl ligand.191,193 Other reports include the transfer of the trifluorovinyl group onto zinc chlorides for application in palladium-catalysed Negishi cross-coupling reactions (Scheme 4b).201,209–213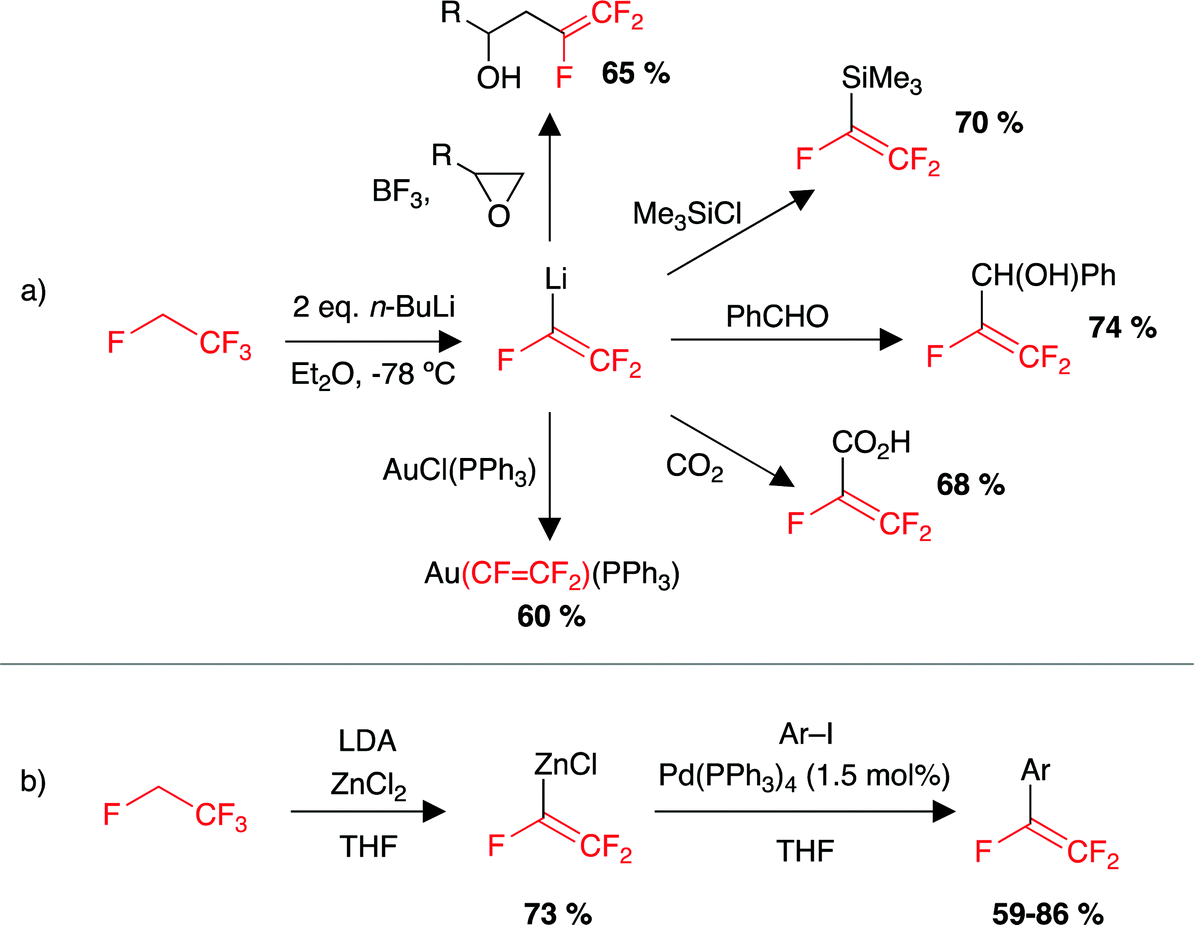 | ||
| Scheme 4 Examples of HFC-134a as a source of the trifluorovinyl moiety and utility in (a) reactions with a range of electrophiles, (b) palladium catalysed cross-coupling reactions. LDA = lithium diisopropylamide. | ||
There is a distinct lack of sp3 C–F functionalisation chemistry of HFC-134a. This is likely due to the high bond strength of the sp3 C–F bond, and as the literature suggests, deprotonation and elimination to form trifluorovinyl species appears to be far more facile.
5.3 HFC-143a, HFC-152a, HFC-32 and HFC-125
A highly fluorophilic main group cation with a carborane counterion has been reported to abstract fluoride from CF3CH3 (HFC-143a).214 The putative CH3CF2+ carbocation generated from this reaction can undergo a Friedel–Crafts addition to the fluorobenzene solvent, to form (p-F–C6H4)(CH3)CF+, which was isolated as a salt with the carborane anion (Scheme 5). The encapsulation of halocarbons (including CF3CH3) in caged supramolecular systems has also been reported.215 In general, there is a lack of functionalisation chemistry for CF3CH3, likely due to the very strong C–F bonds possessed by a terminal CF3 group. There is a similar lack of activation chemistry found in the literature of CHF2CH3 (HFC-152a).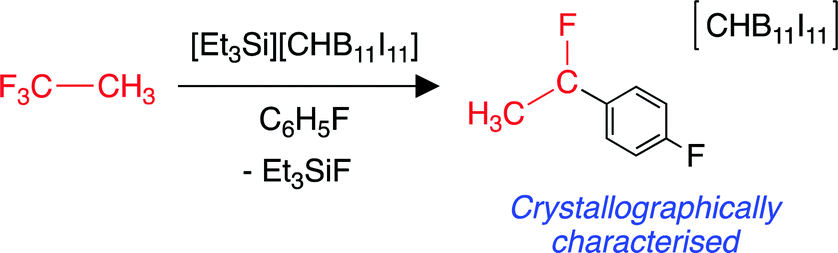 | ||
| Scheme 5 Fluoride abstraction from CF3CH3 by a main group cationic species. | ||
The use of CF2H2 (HFC-32) a nucleophilic source of the difluoromethyl group would be highly attractive, but has been largely limited by the weak acidity of CF2H2 (pKA = 35–41, gas phase proton affinity: 389 kcal mol−1) and low stability of the conjugate base to α-fluoride elimination.216,217 An elegant approach published in 2019 demonstrated the first strategy to repurpose difluoromethane as a {CF2H}− building block, through the use of a Lewis acid/base pair to deprotonate CF2H2 and capture CF2H− as a borane adduct. This adduct can then serve as a nucleophilic source of CF2H−, capable of transmetallation to Pd(II) and subsequent reductive elimination to form a C–C coupled product (Scheme 6).218
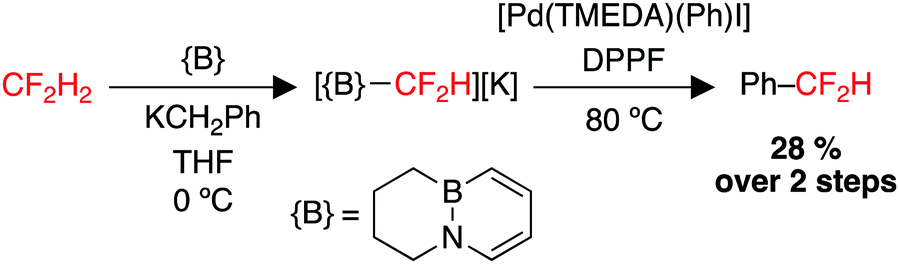 | ||
| Scheme 6 Utility of H2CF2 as a {CF2H}− building block. DPPF = 1,1′-bis(diphenylphosphino)ferrocene. TMEDA = N,N,N′,N′-tetramethylethylenediamine. | ||
Hydrodefluorination of CF2H2 has also been reported. In 2001, Jones and co-workers reported the reaction of a range of fluorocarbons with [Cp*2ZrH2] (Cp* = pentamethylcyclopentadienyl). The hydrodefluorination of CF2H2 required heating to 120 °C for more than 10 days, giving methane and [Cp*2ZrHF]. The authors proposed a radical chain mechanism.219 Andersen and coworkers showed that CF2H2 reacted with a cerium hydride compound to form the analogous cerium fluoride and CH3F.220 The oxidative addition of the C–H bond of CF2H2 to a rhodium compound under photolytic conditions has been reported, where the product was then shown to reductively eliminate CF2H2 in C6D6 at 100 °C.221 In 2006, a paper disclosed the gas-phase C–F activation of CF2H2 by laser-ablated Ti atoms to form the methylidene complex [CH2TiF2],222 and later with other transition metals.223–227 More recently, C–F activation of difluoromethane was reported using heterogeneous nanoscopic aluminium chlorofluoride in the presence of Et3SiH, forming Et3SiF amongst other products.228 A surface-bound silylium-like ion was proposed to be a crucial intermediate for C–F cleavage.
While there remains a lack of C–F functionalisation chemistry for C2F5H (HFC-125), there are quite a few examples of using pentafluoroethane as a source for installing the {C2F5} moiety onto a range of electrophiles, through deprotonation and trapping of [C2F5]−.229–234 In one example, Shibata and coworkers used a potassium base in combination with triglyme to encapsulate the K cation. The pentafluoroethyl moiety could then be transferred to a wide range of carbonyl substrates (Scheme 7a).234 A recent report demonstrated flow-conditions for the pentafluoroethylation of a wide range of ketones, aldehydes and chalcones.235 There has also been work on the cupration of C2F5H and the use of “CuCF2CF3” as a pentafluoroethylation reagent.236,237 Tsui and co-workers reported “CuCF2CF3” as a source of the ˙CF2CF3 radical for pentafluoroethylation of unactivated alkenes.238 Perfluoroalkyl groups can be used to increase the Lewis acidity of main group compounds, and thus C2F5H as a source of LiCF2CF3 has been employed in the synthesis of new main group species such as Bi(C2F5)3 (Scheme 7b).239–243 It is also utilised in ligand design for transition metal complexes.231
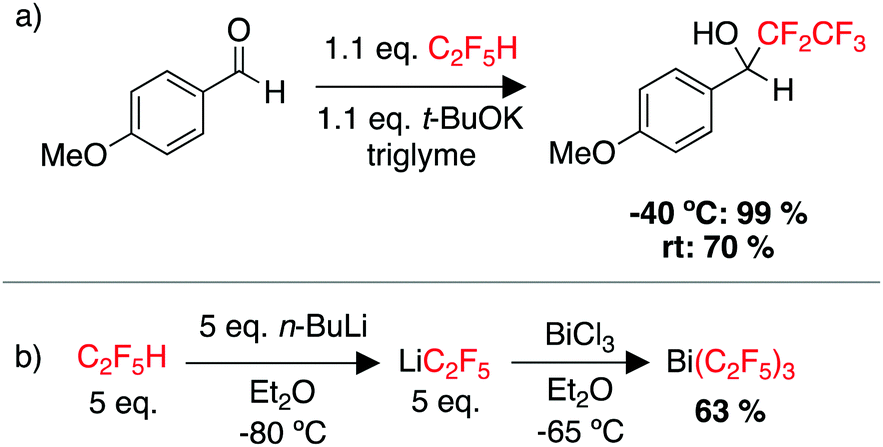 | ||
| Scheme 7 Examples of using C2F5H as a feedstock for (a) pentafluoroethylation of a carbonyl substrate, (b) the synthesis of a main group complex with C2F5 ligands. | ||
5.4 SF6
Early work in the field of SF6 degradation described the chemical breakdown of SF6 using strong reducing agents such as alkali metals.244–247 More recently, the field has been reinvigorated by multiple reports of SF6 activation using transition metals.248–255 Stoichiometric reactions of low valent, early transition-metal compounds of Ti, Zr, Cr and V with SF6 produced a range of transition metal fluorides.248,249 A β-diketiminate nickel complex was shown to activate SF6, forming nickel(II) fluoride and nickel(II) sulfide products. Braun and co-workers reported the selective decomposition of SF6 using rhodium complexes in both stoichiometric and catalytic transformations. These reactions include a range of fluorine and sulfur scavengers to give a range of fluoride and sulfide decomposition products.251,252 The same group later reported the activation of SF6 by a xantphos ligated rhodium complex to form well defined rhodium fluoride and hydrosulfide products.254 They also reported the activation of SF6 using a platinum complex, [Pt(PR3)2] (R = Cy, iPr), to generate trans-[Pt(F)(SF3)(PR3)2] (Scheme 8a). This complex was utilised in deoxyfluorination reactions of ketones, demonstrating the indirect use of SF6 as a fluorinating agent.253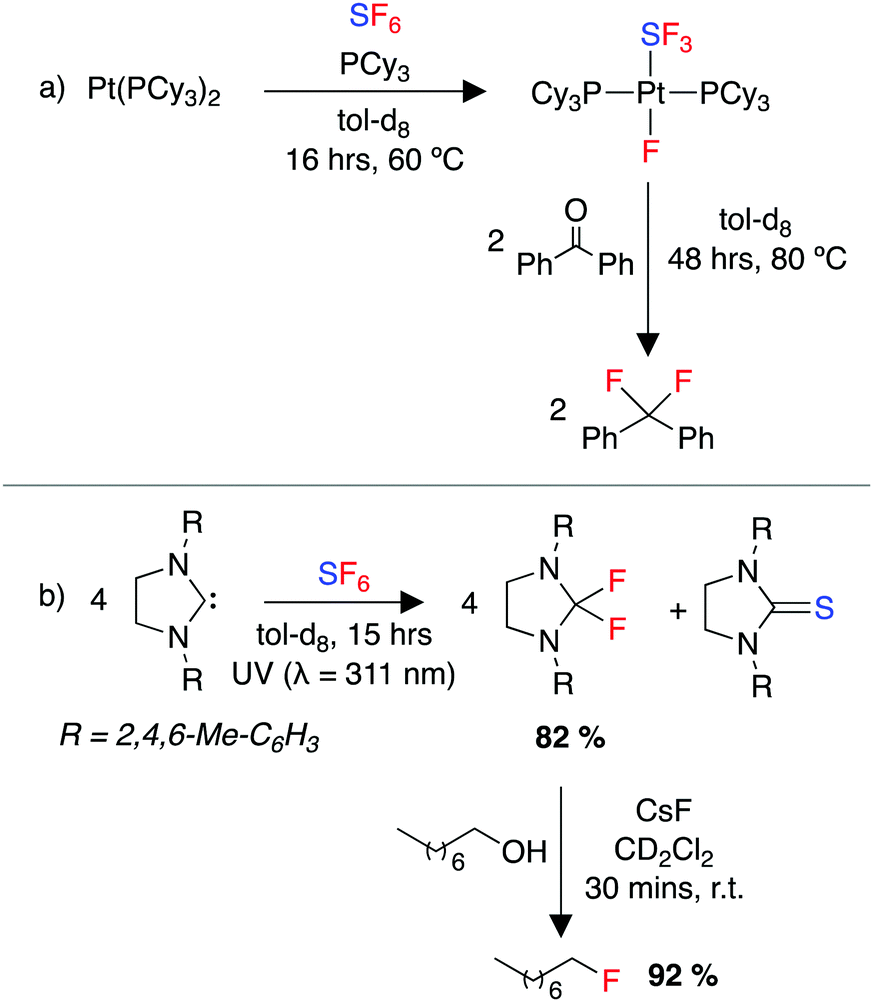 | ||
| Scheme 8 (a) Activation of SF6 by a Pt(0) complex and use of the product in a subsequent deoxyfluorination of a ketone, (b) photolytic activation of SF6 using an NHC, and subsequent deoxyfluorination of an alcohol. | ||
Recent work has utilised organic derivatives and main group compounds for the selective degradation of SF6.256,257 Rueping and co-workers developed a method for the reduction of SF6 using bipyridine based organic reductants, to form ion pair products containing a donor dication and F− and SF5− anions. These salts were isolated and applied as fluorinating agents in the deoxyfluorination of alcohols, aldehydes and carboxylic acids.257 Dielmann and co-workers demonstrated the metal-free activation of SF6 by using a highly nucleophilic phosphine.256 By varying the phosphine ligands, SF6 was degraded to non-volatile phosphine sulfide and phosphine fluoride products, or in one case a bench-stable, crystalline SF5− salt was isolated.256 These reactions were calculated to proceed via nucleophilic attack at the fluorine atom by the phosphine, in contrast to the previous SF6 degradation reports mentioned above involving alkali metals, organic reductants and transition-metal mediated transformations which were proposed to proceed by a single-electron transfer step to SF6.
A number of elegant photoredox strategies have been developed also.258–262 Notably, Braun and co-workers developed a system using an N-heterocyclic carbene (NHC) to reduce SF6 under photoredox conditions, forming a difluoroimidazolidine. This compound was then applied in the deoxyfluorination of a range of alcohols (Scheme 8b).259
In 2021, our group sought to extend our previous work in the activation of environmentally persistent fluorocarbons to SF6. We developed a transition metal free reaction that rapidly reduces SF6 under mild conditions using a highly nucleophilic aluminium(I) complex, 3.263 The reaction produces two known, well-defined aluminium(III) fluoride and sulfide complexes, 4 and 5 (Scheme 9). These two species are separable by virtue of their differing solubilities in the reaction solvent, with the sulfide species 5 forming a colourless precipitate from benzene or toluene.
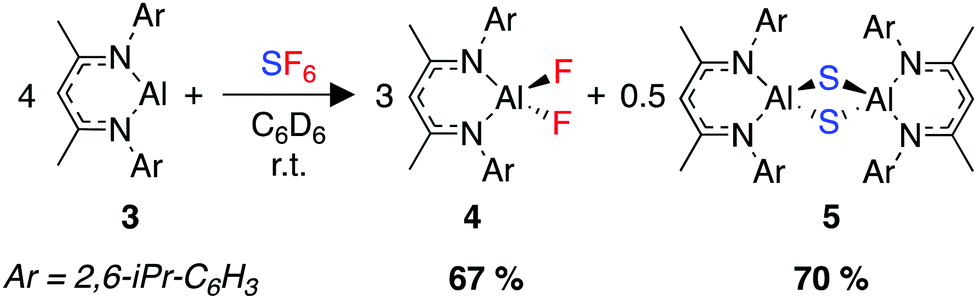 | ||
| Scheme 9 Room-temperature activation of SF6 using the aluminium(I) complex 3. | ||
Due to the limited mechanistic understanding of previous SF6 activation in the literature, we conducted an extensive computational study. The mechanism calculated suggests a series of nucleophilic steps involving the attack of 3 at the fluorine atom of an S–F bond, degrading SF6via SF4 and SF2 to the experimentally observed reaction products 4 and 5 (Fig. 4). Experimentally, no reaction intermediates were observed, and the reaction reaches completion within 15 minutes at room temperature, consistent with the small activation barriers calculated for each elementary step.
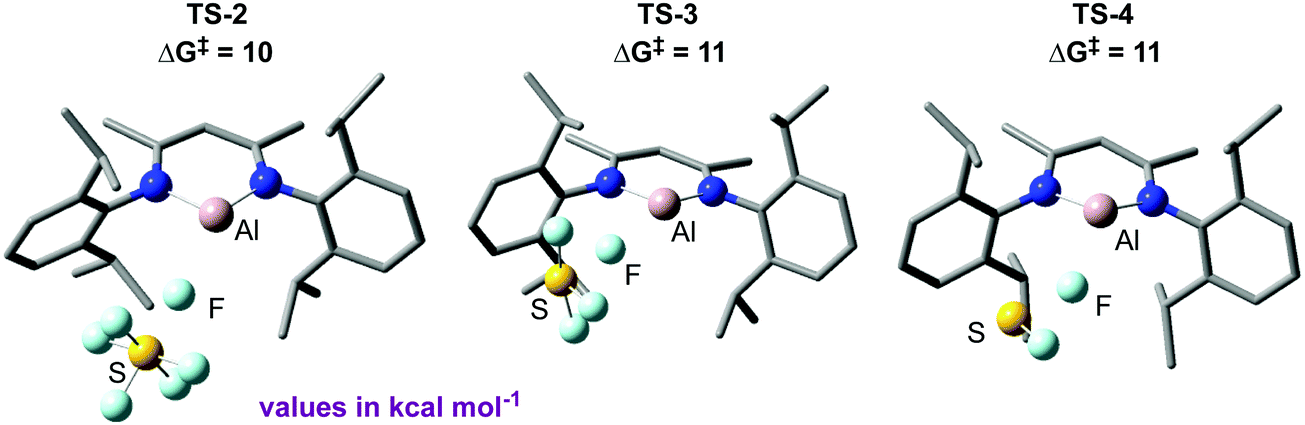 | ||
| Fig. 4 Key transition states calculated for the deconstruction of SF6 by 3. | ||
NBO analysis provided further evidence for a nucleophilic attack mechanism. Notably, an increasingly negative NPA charge at the sulfur atom was calculated as the transition state is traversed and conversely an increasingly positive NPA charge at aluminium. This suggests a transfer of electron density from aluminium to the sulfur atom. These calculations distinguished the proposed nucleophilic attack mechanism from a fluoride abstraction mechanism, where it would be expected that electron density would flow in the opposite direction. Extended Transition State-Natural Orbitals for Chemical Valence (ETS-NOCV) analysis was also carried out,264 and revealed the largest contribution to the orbital interaction between 1 and SF6 in TS-2 to be donation from the aluminium lone pair to σ*(S–F), with overlap occurring at the fluorine end of the bond.
We were able to demonstrate the utility of the products 4 and 5 as fluorinating and sulfinating agents (Scheme 10). Despite the high thermodynamic stability of the Al–F bond, a set of reactions were developed allowing transfer of the fluoride onto carbon, silicon, and boron electrophiles. The reaction with acid anhydrides allowed the formation of acyl fluorides. These compounds are of increasing importance as fluorinating agents in transition metal catalysed reactions, due to their unique balance of stability and reactivity.265–267 Overall, this process represents the repurposing of a waste greenhouse gas in the synthesis of compounds of value.
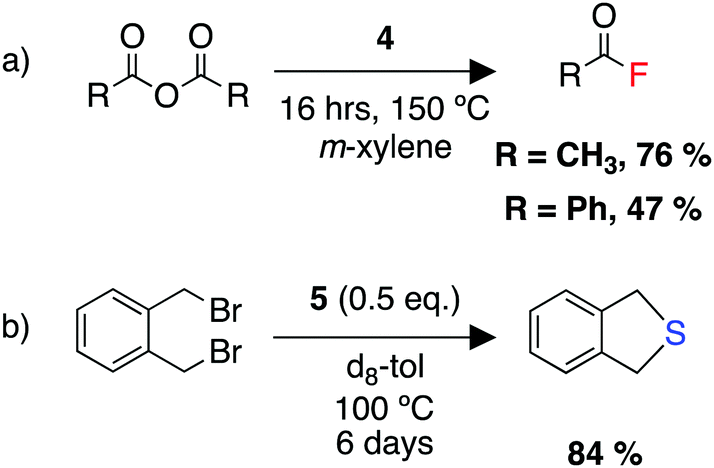 | ||
| Scheme 10 (a) Utility of 4 as a fluorinating agent for a range of electrophiles, (b) utility of 5 as a sulfinating agent. | ||
6. Conclusions and outlook
F-Gases have diverse uses across several important industries. Despite strong legislation that now controls many of these gases, data concerning emissions and atmospheric concentrations suggest that release of F-gases into the environment continues to be a problem. While several potential routes for the destruction of F-gases exist (thermal oxidation, catalytic hydrolysis, plasma treatment, electric discharge), most of these techniques are energy and cost intensive due to the extremely high temperatures required. They are often operationally difficult and carry the threat of producing hazardous decomposition products if conditions are not optimal. There is a further concern that some of these decomposition products may themselves contribute to environmental damage.It is worthwhile considering that the C–F bonds within these waste fluorinated species are valuable, having been created by the fluorination of organic molecules likely using HF derived from inorganic sources. The repurposing of waste F-gases as a chemical feedstock to produce other useful fluorinated chemicals is a highly attractive alternative to destruction. The opportunity lies within chemistry to explore and develop methods for activating and functionalising the C–H and C–F bonds in these highly inert molecules. Although research is advancing, the development of chemical methods to upgrade inert substrates such as HFCs, PFCs and SF6 is difficult. In the presence of strong nucleophiles HFCs tend to react by pathways involving an initial deprotonation, while fluoride abstraction becomes more favourable in the presence of highly electrophilic Lewis acids. There is far less chemistry known for upgrading PFCs, but methods for the photochemical and thermal activation of SF6 are emerging.
The challenge in this field remains controlling the selectivity of these reactions to produce useful chemical building blocks for onward use. While several methods are emerging to use HFC-23 as a source of both –CF3 and –CF2H groups, HFC-134a and SF6 remain underexplored synthons for the creating –CH2CF3 and –SF5 groups. Significant technical obstacles remain for scaling-up and implementing these methods for real-life F-gas remediation. Flow methods are emerging as a powerful approach in this regard.13 The ultimate aim should be to employ chemical recycling methods using recovered F-gases. In the long-term it will be necessary to consider both life-cycle analysis and technoeconomic analysis of any technologies developed – as is already standard in CO2 remediation methods.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to ERC for generous funding (Fluorofix:677367) and Imperial College London and the EPSRC for DTP studentship funding (Daniel Sheldon). AGAGE is supported principally by NASA (USA) grants to MIT and SIO, and also by: BEIS (UK) and NOAA (USA) grants to Bristol University; CSIRO and BoM (Australia): FOEN grants to Empa (Switzerland); NILU (Norway); SNU (Korea); CMA (China); NIES (Japan); and Urbino University (Italy). We are thankful to Prof. Matthew Rigby (Bristol) for scientific discussion.References
- P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley Blackwell, 2005 Search PubMed.
- G. Balz and G. Schiemann, Berichte der Dtsch. Chem. Gesellschaft A B Ser., 1927, 60, 1186–1190 CrossRef.
- J. H. Simons, J. Electrochem. Soc., 1949, 95, 47 CrossRef CAS.
- R. Fowler, W. Buford III, J. Hamilton, Jr., R. Sweet, C. Weber, J. Kasper and I. Litant, Ind. Eng. Chem., 1947, 39, 292–298 CrossRef CAS.
- R. Benner, A. Benning, F. Downing, C. Irwin, K. Johnson, A. Linch, H. Parmelee and W. Wirth, Ind. Eng. Chem., 1947, 39, 329–333 CrossRef CAS.
- G. Villalba, R. U. Ayres and H. Schroder, J. Ind. Ecol., 2007, 11, 85–101 CrossRef CAS.
- A. Harsanyi and G. Sandford, Green Chem., 2015, 17, 2081–2086 RSC.
- A. J. Sicard and R. T. Baker, Chem. Rev., 2020, 120, 9164–9303 CrossRef CAS.
- M. O. McLinden and M. L. Huber, J. Chem. Eng. Data, 2020, 65, 4176–4193 CrossRef CAS PubMed.
- R. M. Bellabarba, J. Fluorine Chem., 2021, 244, 109741 CrossRef CAS.
- M. Nappa, S. Peng and X. Sun, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds: Progress in Fluorine Science, ed. H. Groult, F. R. Leroux and A. B. T. Tressaud, Elsevier, 2017, pp. 27–69, Industrial Syntheses of Hydrohaloolefins and Related Products Search PubMed.
- B. K. Sovacool, S. Griffiths, J. Kim and M. Bazilian, Renewable Sustainable Energy Rev., 2021, 141, 110759 CrossRef CAS.
- W. C. Fu, P. M. MacQueen and T. F. Jamison, Chem. Soc. Rev., 2021, 50, 7378–7394 RSC.
- J. M. Calm and D. A. Didion, Int. J. Refrig., 1998, 21, 308–321 CrossRef CAS.
- A. J. Elliot, in Organofluorine Chemistry Principles and Applications, ed. R. E. Banks, B. E. Smart and J. C. Tatlow, Plenum Press, New York, 1994, pp. 145–157, Chlorofluorocarbons Search PubMed.
- L. E. Manzer, Science, 1990, 249, 31–35 CrossRef CAS PubMed.
- B. Xiang, P. K. Patra, S. A. Montzka, S. M. Miller, J. W. Elkins, F. L. Moore, E. L. Atlas, B. R. Miller, R. F. Weiss, R. G. Prinn and S. C. Wofsy, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17379–17384 CrossRef CAS PubMed.
- M. Rigby, R. G. Prinn, S. O’Doherty, B. R. Miller, D. Ivy, J. Mühle, C. M. Harth, P. K. Salameh, T. Arnold, R. F. Weiss, P. B. Krummel, L. P. Steele, P. J. Fraser, D. Young and P. G. Simmonds, Geophys. Res. Lett., 2014, 41, 2623–2630 CrossRef CAS.
- J. M. Calm, Int. J. Refrig., 2008, 31, 1123–1133 CrossRef CAS.
- P. G. Simmonds, M. Rigby, A. McCulloch, M. K. Vollmer, S. Henne, J. Mühle, S. O’Doherty, A. J. Manning, P. B. Krummel, P. J. Fraser, D. Young, R. F. Weiss, P. K. Salameh, C. M. Harth, S. Reimann, C. M. Trudinger, L. P. Steele, R. H.-J. Wang, D. J. Ivy, R. G. Prinn, B. Mitrevski and D. M. Etheridge, Atmos. Chem. Phys., 2018, 18, 4153–4169 CrossRef CAS.
- Releases: Facility Report|TRI Explorer|US EPA, https://enviro.epa.gov/triexplorer/release_fac?p_view=USFA&trilib=TRIQ1&sort=RE_TOLBY&sort_fmt=2&state=All+states&county=All+counties&chemical=0000075456&industry=ALL&year=2019&tab_rpt=1&fld=TRIID&fld=RELLBY&fld=TSFDSP, (accessed 1 October 2021).
- W.-T. Tsai, Chemosphere, 2005, 61, 1539–1547 CrossRef CAS PubMed.
- A. McCulloch and A. A. Lindley, Int. J. Refrig., 2003, 26, 865–872 CrossRef CAS.
- G. J.-M. Velders, A. R. Ravishankara, M. K. Miller, M. J. Molina, J. Alcamo, J. S. Daniel, D. W. Fahey, S. A. Montzka and S. Reimann, Science, 2012, 335, 922–923 CrossRef CAS PubMed.
- A. A. Lindley and A. McCulloch, J. Fluorine Chem., 2005, 126, 1457–1462 CrossRef CAS.
- EU ratifies Kigali Amendment to the Montreal Protocol | Climate Action, https://ec.europa.eu/clima/news/eu-ratifies-kigali-amendment-montreal-protocol_en, (accessed 6 July 2021).
- F. Polonara, L. J. Kuijpers and R. Peixoto, Int. J. Heat Technol., 2017, 35, S1–S8 CrossRef.
- The Kigali Amendment to the Montreal protocol: another global commitment to stop climate change, https://www.unep.org/news-and-stories/news/kigali-amendment-montreal-protocol-another-global-commitment-stop-climate, (accessed 6 August 2021).
- A. J. Manning, A. L. Redington, D. Say, S. O’Doherty, D. Young, P. G. Simmonds, M. K. Vollmer, J. Mühle, J. Arduini, G. Spain, A. Wisher, M. Maione, T. J. Schuck, K. Stanley, S. Reimann, A. Engel, P. B. Krummel, P. J. Fraser, C. M. Harth, P. K. Salameh, R. F. Weiss, R. Gluckman, P. N. Brown, J. D. Watterson and T. Arnold, Atmos. Chem. Phys., 2021, 21, 12739–12755 CrossRef CAS.
- Regulation (EU) No. 517/2014 of the European Parliament and of the Council of 16 April 2014 on fluorinated greenhouse gases and repealing Regulation (EC) No. 842/2006—European Environment Agency, https://www.eea.europa.eu/policy-documents/regulation-eu-no-517-2014, (accessed 6 August 2021).
- Y. Heredia-Aricapa, J. M. Belman-Flores, A. Mota-Babiloni, J. Serrano-Arellano and J. J. García-Pabón, Int. J. Refrig., 2020, 111, 113–123 CrossRef CAS.
- C. C. Allgood, Adv. Mater., 1998, 10, 1239–1242 CrossRef CAS.
- A. McCulloch, P. M. Midgley and P. Ashford, Atmos. Environ., 2003, 37, 889–902 CrossRef CAS.
- N. Zhang, P. Hu, L. Chen, M. Liu and Q. Chen, J. Chem. Eng. Data, 2021, 66, 2717–2722 CrossRef CAS.
- M. O. McLinden, J. S. Brown, R. Brignoli, A. F. Kazakov and P. A. Domanski, Nat. Commun., 2017, 8, 14476 CrossRef CAS PubMed.
- A. P. Fröba, S. Will and A. Leipertz, Int. J. Refrig., 2001, 24, 734–743 CrossRef.
- R. Dodangodage, P. F. Bernath, C. D. Boone, J. Crouse and J. J. Harrison, J. Quant. Spectrosc. Radiat. Transf., 2021, 272, 107804 CrossRef CAS.
- P. G. Simmonds, M. Rigby, A. J. Manning, S. Park, K. M. Stanley, A. McCulloch, S. Henne, F. Graziosi, M. Maione, J. Arduini, S. Reimann, M. K. Vollmer, J. Mühle, S. O’Doherty, D. Young, P. B. Krummel, P. J. Fraser, R. F. Weiss, P. K. Salameh, C. M. Harth, M.-K. Park, H. Park, T. Arnold, C. Rennick, L. P. Steele, B. Mitrevski, R. H.-J. Wang and R. G. Prinn, Atmos. Chem. Phys., 2020, 20, 7271–7290 CrossRef CAS.
- Overview of SF6 Emissions Sources and Reduction Options in Electric Power Systems|US EPA, https://www.epa.gov/eps-partnership/overview-sf6-emissions-sources-and-reduction-options-electric-power-systems, accessed 24 August 2021.
- L. G. Christophorou, J. K. Olthoff and R. J. Van Brunt, IEEE Electr. Insul. Mag., 1997, 13, 20–24 Search PubMed.
- H. Okubo and A. Beroual, IEEE Electr. Insul. Mag., 2011, 27, 34–42 Search PubMed.
- X. Li, H. Zhao and A. B. Murphy, J. Phys. D: Appl. Phys., 2018, 51, 153001 CrossRef.
- S. Xiao, X. Zhang, J. Tang and S. Liu, Energy Rep., 2018, 4, 486–496 CrossRef.
- M. Rabie and C. M. Franck, Environ. Sci. Technol., 2018, 52, 369–380 CrossRef CAS PubMed.
- R. K. Spence, E. D. Norcross, J. Costabile, S. McCoy, A. C. Cernaianu, J. B. Alexander, M. J. Pello, U. Atabek and R. C. Camishion, Artif. Cells, Blood Substitutes, Biotechnol., 1994, 22, 955–963 CrossRef CAS.
- D. P. Curran and J. A. Gladysz, Handbook of Fluorous Chemistry, Wiley-VCH, 2004 Search PubMed.
- J. Mühle, A. L. Ganesan, B. R. Miller, P. K. Salameh, C. M. Harth, B. R. Greally, M. Rigby, L. W. Porter, L. P. Steele, C. M. Trudinger, P. B. Krummel, S. O’Doherty, P. J. Fraser, P. G. Simmonds, R. G. Prinn and R. F. Weiss, Atmos. Chem. Phys., 2010, 10, 5145–5164 CrossRef.
- H. M. Lee, M. B. Chang and K. Y. Wu, J. Air Waste Manage. Assoc., 2004, 54, 960–970 CrossRef CAS PubMed.
- S. H. Han, H. W. Park, T. H. Kim and D. W. Park, Clean Technol., 2011, 17, 250–258 Search PubMed.
- R. G. Prinn, R. F. Weiss, P. J. Fraser, P. G. Simmonds, D. M. Cunnold, F. N. Alyea, S. O’Doherty, P. Salameh, B. R. Miller, J. Huang, R. H.-J. Wang, D. E. Hartley, C. Harth, L. P. Steele, G. Sturrock, P. M. Midgley and A. McCulloch, J. Geophys. Res.: Atmos., 2000, 105, 17751–17792 CrossRef CAS.
- R. G. Prinn, R. F. Weiss, J. Arduini, T. Arnold, H. L. DeWitt, P. J. Fraser, A. L. Ganesan, J. Gasore, C. M. Harth, O. Hermansen, J. Kim, P. B. Krummel, S. Li, Z. M. Loh, C. R. Lunder, M. Maione, A. J. Manning, B. R. Miller, B. Mitrevski, J. Mühle, S. O’Doherty, S. Park, S. Reimann, M. Rigby, T. Saito, P. K. Salameh, R. Schmidt, P. G. Simmonds, L. P. Steele, M. K. Vollmer, R. H. Wang, B. Yao, Y. Yokouchi, D. Young and L. Zhou, Earth Syst. Sci. Data, 2018, 10, 985–1018 CrossRef.
- P. G. Simmonds, S. O’Doherty, G. Nickless, G. A. Sturrock, R. Swaby, P. Knight, J. Ricketts, G. Woffendin and R. Smith, Anal. Chem., 1995, 67, 717–723 CrossRef CAS.
- R. G. Prinn, J. Huang, R. F. Weiss, D. M. Cunnold, P. J. Fraser, P. G. Simmonds, A. McCulloch, C. Harth, S. Reimann, P. Salameh, S. O’Doherty, R. H.-J. Wang, L. W. Porter, B. R. Miller and P. B. Krummel, Geophys. Res. Lett., 2005 DOI:10.1029/2004GL022228.
- M. Rigby, R. G. Prinn, S. O’Doherty, S. A. Montzka, A. McCulloch, C. M. Harth, J. Mühle, P. K. Salameh, R. F. Weiss, D. Young, P. G. Simmonds, B. D. Hall, G. S. Dutton, D. Nance, D. J. Mondeel, J. W. Elkins, P. B. Krummel, L. P. Steele and P. J. Fraser, Atmos. Chem. Phys., 2013, 13, 2691–2702 CrossRef.
- D. Say, A. J. Manning, L. M. Western, D. Young, A. Wisher, M. Rigby, S. Reimann, M. K. Vollmer, M. Maione, J. Arduini, P. B. Krummel, J. Mühle, C. M. Harth, B. Evans, R. F. Weiss, R. G. Prinn and S. O’Doherty, Atmos. Chem. Phys., 2021, 21, 2149–2164 CrossRef CAS.
- A. Engel, M. Rigby, J. B. Burkholder, R. P. Fernandez, L. Froidevaux, B. D. Hall, R. Hossaini, T. Saito, M. K. Vollmer and B. Yao, Scientific Assessment of Ozone Depletion: 2018, Global Ozone Research and Monitoring Project - Report No. 58, World Meteorological Organization, Geneva, Switzerland, 2018, Update on Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol Search PubMed.
- J. Kim, S. Li, K.-R. Kim, A. Stohl, J. Mühle, S.-K. Kim, M.-K. Park, D.-J. Kang, G. Lee, C. M. Harth, P. K. Salameh and R. F. Weiss, Geophys. Res. Lett., 2010 DOI:10.1029/2010GL043263.
- B. Yao, M. K. Vollmer, L. X. Zhou, S. Henne, S. Reimann, P. C. Li, A. Wenger and M. Hill, Atmos. Chem. Phys., 2012, 12, 10181–10193 CrossRef CAS.
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B.-R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Oxford, UK, 2021 Search PubMed.
- G. K.-S. Prakash, P. V. Jog, P. T.-D. Batamack and G. A. Olah, Science, 2012, 338, 1324–1327 CrossRef CAS PubMed.
- K. M. Stanley, D. Say, J. Mühle, C. M. Harth, P. B. Krummel, D. Young, S. J. O’Doherty, P. K. Salameh, P. G. Simmonds, R. F. Weiss, R. G. Prinn, P. J. Fraser and M. Rigby, Nat. Commun., 2020, 11, 397 CrossRef CAS PubMed.
- W. Han, Y. Li, H. Tang and H. Liu, J. Fluorine Chem., 2012, 140, 7–16 CrossRef CAS.
- D. E. Oram, W. T. Sturges, S. A. Penkett, A. McCulloch and P. J. Fraser, Geophys. Res. Lett., 1998, 25, 35–38 CrossRef CAS.
- S. A. Montzka, G. J.-M. Velders, P. B. Krummel, J. Mühle, V. L. Orkin, S. Park, H. Shah and H. Walter-Terrinoni, Scientific Assessment of Ozone Depletion: 2018, Global Ozone Research and Monitoring Project - Report No. 58, World Meteorological Organization, Geneva, Switzerland, 2018, Hydrofluorocarbons (HFCs) Search PubMed.
- D. Say, A. J. Manning, S. O’Doherty, M. Rigby, D. Young and A. Grant, Environ. Sci. Technol., 2016, 50, 11129–11136 CrossRef CAS PubMed.
- K. Srinivasan and L. R. Oellrich, Int. J. Refrig., 1997, 20, 332–338 CrossRef CAS.
- H. M.-N. T. Avelino, J. M.-N. A. Fareleira and C. M.-B. P. Oliveira, J. Chem. Eng. Data, 2006, 51, 1672–1677 CrossRef CAS.
- R. Talukdar, A. Mellouki, T. Gierczak, J. B. Burkholder, S. A. McKeen and A. R. Ravishankara, J. Phys. Chem., 1991, 95, 5815–5821 CrossRef CAS.
- S. Tomassetti, Fluid Phase Equilib., 2021, 546, 113173 CrossRef CAS.
- T. Tamatsu, T. Sato, H. Sato and K. Watanabe, Int. J. Thermophys., 1992, 13, 985–997 CrossRef CAS.
- R. Tillner-Roth, Int. J. Thermophys., 1995, 16, 91–100 CrossRef CAS.
- A. van Pelt and J. V. Sengers, J. Supercrit. Fluids, 1995, 8, 81–99 CrossRef CAS.
- T. Gierczak, R. Talukdar, G. L. Vaghjiani, E. R. Lovejoy and A. R. Ravishankara, J. Geophys. Res.: Atmos., 1991, 96, 5001–5011 CrossRef CAS.
- J. J. Orlando, J. B. Burkholder, S. A. McKeen and A. R. Ravishankara, J. Geophys. Res.: Atmos., 1991, 96, 5013–5023 CrossRef CAS.
- R. Huet and G. Johanson, Pharmaceutics, 2020, 12, 997 CrossRef CAS PubMed.
- Z. Lv, Z. Yang, H. Ma, Y. Chen and Y. Zhang, J. Fluorine Chem., 2021, 248, 109832 CrossRef CAS.
- J. W. Schmidt and M. R. Moldover, J. Chem. Eng. Data, 1994, 39, 39–44 CrossRef CAS.
- Y. Higashi, Int. J. Refrig., 1994, 17, 524–531 CrossRef CAS.
- M. Rigby, J. Mühle, B. R. Miller, R. G. Prinn, P. B. Krummel, L. P. Steele, P. J. Fraser, P. K. Salameh, C. M. Harth, R. F. Weiss, B. R. Greally, S. O’Doherty, P. G. Simmonds, M. K. Vollmer, S. Reimann, J. Kim, K.-R. Kim, H. J. Wang, J. G.-J. Olivier, E. J. Dlugokencky, G. S. Dutton, B. D. Hall and J. W. Elkins, Atmos. Chem. Phys., 2010, 10, 10305–10320 CrossRef CAS.
- G. P. Stiller, T. von Clarmann, M. Höpfner, N. Glatthor, U. Grabowski, S. Kellmann, A. Kleinert, A. Linden, M. Milz, T. Reddmann, T. Steck, H. Fischer, B. Funke, M. López-Puertas and A. Engel, Atmos. Chem. Phys., 2008, 8, 677–695 CrossRef CAS.
- L. Huang, Y. Shen, W. Dong, R. Zhang, J. Zhang and H. Hou, J. Hazard. Mater., 2008, 151, 323–330 CrossRef CAS PubMed.
- P. K. Patra, S. Lal, B. H. Subbaraya, C. H. Jackman and P. Rajaratnam, J. Geophys. Res.: Atmos., 1997, 102, 8855–8859 CrossRef CAS.
- A. R. Ravishankara, S. Solomon, A. A. Turnipseed and R. F. Warren, Science, 1993, 259, 194–199 CrossRef CAS PubMed.
- P. Widger and A. Haddad, Energies, 2018, 11, 2037 CrossRef.
- X. Fang, X. Hu, G. Janssens-Maenhout, J. Wu, J. Han, S. Su, J. Zhang and J. Hu, Environ. Sci. Technol., 2013, 47, 3848–3855 CrossRef CAS PubMed.
- M. Shin, D. Jang and J. Ha, J. Mater. Cycles Waste Manag., 2016, 18, 399–406 CrossRef CAS.
- S. A. Roh, W. H. Kim, D. S. Jung and B. K. Hong, J. Energy Inst., 2019, 92, 1842–1851 CrossRef CAS.
- T. Mi, J. Han, X. He and L. Qin, Environ. Prot. Eng., 2015, 41, 143–150 Search PubMed.
- M. Jasiński, M. Dors and J. Mizeraczyk, Plasma Chem. Plasma Process., 2009, 29, 363–372 CrossRef.
- K.-T. Kim, H. S. Kang, D. H. Lee and S. J. Lee, J. Korean Soc. Atmos. Environ., 2013, 29, 27–37 CrossRef.
- S.-S. Choi, D.-W. Park and T. Watanabe, Nucl. Eng. Technol., 2012, 44, 21–32 CrossRef CAS.
- Y. Shinji, JP Pat., JP2006122790A, 2006 Search PubMed.
- Y. Mori, T. Kyotani and T. Shinohara, TW Pat., TW510819B, 2002 Search PubMed.
- W. B. Feaver and J. A. Rossin, Catal. Today, 1999, 54, 13–22 CrossRef CAS.
- L. C. Draper and J. D. Scott, WO Pat., WO2002058824A1, 2002 Search PubMed.
- S. Shuichi, A. Toshiaki, I. Shinzo, Y. Takeshi, Y. Hisao, A. Shigeru and T. Shin, JP Pat., JP3977887B2, 1998 Search PubMed.
- J. A. Rossin and W. B. Feaver, US Pat., US6673326B1, 2004 Search PubMed.
- H. Onoda, T. Ohta, J. Tamaki and K. Kojima, Appl. Catal., A, 2005, 288, 98–103 CrossRef CAS.
- J. Y. Jeon, X.-F. Xu, M. H. Choi, H. Y. Kim and Y.-K. Park, Chem. Commun., 2003, 1244–1245 RSC.
- H. Zhang, C. F. Ng and S. Y. Lai, Appl. Catal., B, 2005, 55, 301–307 CrossRef CAS.
- R. S. Brown, J. A. Rossin and K. B.-T.-S. S.-T. Aitchison, Solid State Technol., 2001, 44, 189 CAS.
- H. Sekiguchi, T. Honda and A. Kanzawa, Plasma Chem. Plasma Process., 1993, 13, 463–478 CrossRef CAS.
- A. Gal’, A. Ogata, S. Futamura and K. Mizuno, J. Phys. Chem. A, 2003, 107, 8859–8866 CrossRef.
- A. B. Murphy, A. J.-D. Farmer, E. C. Horrigan and T. McAllister, Plasma Chem. Plasma Process., 2002, 22, 371–385 CrossRef CAS.
- T. Yamamoto and B. W. Jang, IEEE Trans. Ind. Appl., 1999, 35, 736–742 CrossRef CAS.
- A. B. Murphy and T. McAllister, Phys. Plasmas, 2001, 8, 2565–2571 CrossRef CAS.
- B. A. Wofford, M. W. Jackson, C. Hartz and J. W. Bevan, Environ. Sci. Technol., 1999, 33, 1892–1897 CrossRef CAS.
- C. L. Hartz, J. W. Bevan, M. W. Jackson and B. A. Wofford, Environ. Sci. Technol., 1998, 32, 682–687 CrossRef CAS.
- J.-S. Chang, K. G. Kostov, K. Urashima, T. Yamamoto, Y. Okayasu, T. Kato, T. Iwaizumi and K. Yoshimura, IEEE Trans. Ind. Appl., 2000, 36, 1251–1259 CrossRef CAS.
- M. B. Chang and S. J. Yu, Environ. Sci. Technol., 2001, 35, 1587–1592 CrossRef CAS PubMed.
- S. J. Yu and M. B. Chang, Plasma Chem. Plasma Process., 2001, 21, 311–327 CrossRef CAS.
- D. T. Chen, M. M. David, G. V.-D. Tiers and J. N. Schroepfer, Environ. Sci. Technol., 1998, 32, 3237–3240 CrossRef CAS.
- M. B. Chang and H. M. Lee, Catal. Today, 2004, 89, 109–115 CrossRef CAS.
- M. Shih, W.-J. Lee, C.-H. Tsai, P.-J. Tsai and C.-Y. Chen, J. Air Waste Manage. Assoc., 2002, 52, 1274–1280 CrossRef CAS PubMed.
- R. J. Van Brunt and J. T. Herron, IEEE Trans. Electr. Insul., 1990, 25, 75–94 CrossRef CAS.
- A. Derdouri, J. Casanovas, R. Grob and J. Mathieu, IEEE Trans. Electr. Insul., 1989, 24, 1147–1157 CrossRef CAS.
- I. Coll, A. M. Casanovas, L. Vial, A. Gleizes and J. Casanovas, J. Phys. D: Appl. Phys., 2000, 33, 221–229 CrossRef CAS.
- W.-T. Tsai, J. Fluorine Chem., 2007, 128, 1345–1352 CrossRef CAS.
- L. Vial, A. M. Casanovas, J. Diaz, I. Coll and J. Casanovas, J. Phys. D: Appl. Phys., 2001, 34, 2037–2051 CrossRef CAS.
- C.-H. Tsai and J.-M. Shao, J. Hazard. Mater., 2008, 157, 201–206 CrossRef CAS PubMed.
- J. Diaz, A. M. Casanovas and J. Casanovas, J. Phys. D: Appl. Phys., 2003, 36, 1558–1564 CrossRef CAS.
- Y. Kabouzi, M. Moisan, J. C. Rostaing, C. Trassy, D. Guérin, D. Kéroack and Z. Zakrzewski, J. Appl. Phys., 2003, 93, 9483–9496 CrossRef CAS.
- G. Griffin, I. Sauers, L. Christophorou, C. Easterly and P. Walsh, IEEE Trans. Electr. Insul., 1983, EI-18, 551–552 CAS.
- F. Y. Chu, IEEE Trans. Electr. Insul., 1986, EI-21, 693–725 CAS.
- I. Sauers, Plasma Chem. Plasma Process., 1988, 8, 247–262 CrossRef CAS.
- G. Coates, F. Rekhroukh and M. R. Crimmin, Synlett, 2019, 2233–2246 CAS.
- W. Chen, C. Bakewell and M. Crimmin, Synthesis, 2016, 810–821 Search PubMed.
- T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 CrossRef PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- J. Burdeniuc, B. Jedicka and R. H. Crabtree, Chem. Ber., 1997, 130, 145–154 CrossRef CAS.
- J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373–431 CrossRef CAS.
- M. R. Crimmin, M. J. Butler and A. J.-P. White, Chem. Commun., 2015, 51, 15994–15996 RSC.
- G. Coates, B. J. Ward, C. Bakewell, A. J.-P. White and M. R. Crimmin, Chem. – Eur. J., 2018, 24, 16282–16286 CrossRef CAS PubMed.
- B. Cui, S. Jia, E. Tokunaga and N. Shibata, Nat. Commun., 2018, 9, 4393 CrossRef PubMed.
- X.-W. Liu, C. Zarate and R. Martin, Angew. Chem., Int. Ed., 2019, 58, 2064–2068 CrossRef CAS PubMed.
- E. A. Symons and M. J. Clermont, J. Am. Chem. Soc., 1981, 103, 3127–3130 CrossRef CAS.
- S. Kyasa, Synlett, 2015, 1911–1912 CAS.
- C. P. Johnston, T. H. West, R. E. Dooley, M. Reid, A. B. Jones, E. J. King, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2018, 140, 11112–11124 CrossRef CAS PubMed.
- A. García-Domínguez, T. H. West, J. J. Primozic, K. M. Grant, C. P. Johnston, G. G. Cumming, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2020, 142, 14649–14663 CrossRef PubMed.
- G. K.-S. Prakash, F. Wang, Z. Zhang, R. Haiges, M. Rahm, K. O. Christe, T. Mathew and G. A. Olah, Angew. Chem., Int. Ed., 2014, 53, 11575–11578 CrossRef CAS PubMed.
- A. Lishchynskyi, F. M. Miloserdov, E. Martin, J. Benet-Buchholz, E. C. Escudero-Adán, A. I. Konovalov and V. V. Grushin, Angew. Chem., Int. Ed., 2015, 54, 15289–15293 CrossRef CAS PubMed.
- B. Folléas, I. Marek, J.-F. Normant and L. Saint Jalmes, Tetrahedron Lett., 1998, 39, 2973–2976 CrossRef.
- B. Folléas, I. Marek, J.-F. Normant and L. Saint-Jalmes, Tetrahedron, 2000, 56, 275–283 CrossRef.
- B. R. Langlois and T. Billard, Synthesis, 2003, 185–194 CrossRef CAS.
- B. R. Langlois and T. Billard, Fluorine-Containing Synthons, American Chemical Society, 2005, vol. 911, pp. 3–57, Fluoroform, Fluoral, Trifluoroacetic, and Trifluoromethanesulfinic Acids Derivatives as New Reagents for the Introduction of Polyfluorinated Moieties Search PubMed.
- T. Shono, M. Ishifune, T. Okada and S. Kashimura, J. Org. Chem., 1991, 56, 2–4 CrossRef CAS.
- R. Barhdadi, M. Troupel and J. Périchon, Chem. Commun., 1998, 1251–1252 RSC.
- A. Zanardi, M. A. Novikov, E. Martin, J. Benet-Buchholz and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901–20913 CrossRef CAS PubMed.
- P. Novák, A. Lishchynskyi and V. V. Grushin, Angew. Chem., Int. Ed., 2012, 51, 7767–7770 CrossRef PubMed.
- P. Novák, A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2012, 134, 16167–16170 CrossRef PubMed.
- G. K.-S. Prakash and M. Mandal, J. Fluorine Chem., 2001, 112, 123–131 CrossRef CAS.
- G. K.-S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757–786 CrossRef CAS PubMed.
- Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034–9037 CrossRef CAS PubMed.
- Y. Ye, S. H. Lee and M. S. Sanford, Org. Lett., 2011, 13, 5464–5467 CrossRef CAS PubMed.
- N. D. Ball, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 2878–2879 CrossRef CAS PubMed.
- E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679–1681 CrossRef CAS PubMed.
- G. G. Dubinina, H. Furutachi and D. A. Vicic, J. Am. Chem. Soc., 2008, 130, 8600–8601 CrossRef CAS PubMed.
- H. Morimoto, T. Tsubogo, N. D. Litvinas and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 3793–3798 CrossRef CAS PubMed.
- F. Wang, T. Luo, J. Hu, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. K.-S. Prakash and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 7153–7157 CrossRef CAS PubMed.
- H. Kawai, Z. Yuan, E. Tokunaga and N. Shibata, Org. Biomol. Chem., 2013, 11, 1446–1450 RSC.
- S. Okusu, K. Hirano, E. Tokunaga and N. Shibata, ChemistryOpen, 2015, 4, 581–585 CrossRef CAS PubMed.
- N. Punna, T. Saito, M. Kosobokov, E. Tokunaga, Y. Sumii and N. Shibata, Chem. Commun., 2018, 54, 4294–4297 RSC.
- K. Hirano, T. Saito, Y. Fujihira, D. M. Sedgwick, S. Fustero and N. Shibata, J. Org. Chem., 2020, 85, 7976–7985 CrossRef CAS PubMed.
- T. Saito, J. Wang, E. Tokunaga, S. Tsuzuki and N. Shibata, Sci. Rep., 2018, 8, 11501 CrossRef PubMed.
- Y. Fujihira, Y. Liang, M. Ono, K. Hirano, T. Kagawa and N. Shibata, Beilstein J. Org. Chem., 2021, 17, 431–438 CrossRef CAS PubMed.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 9811–9814 CrossRef CAS PubMed.
- J. B. Geri, M. M. Wade Wolfe and N. K. Szymczak, Angew. Chem., Int. Ed., 2018, 57, 1381–1385 CrossRef CAS PubMed.
- J. Choi, D. Y. Wang, S. Kundu, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Science, 2011, 332, 1545–1548 CrossRef CAS PubMed.
- C. S. Thomoson and W. R. Dolbier, J. Org. Chem., 2013, 78, 8904–8908 CrossRef CAS PubMed.
- T. Iida, R. Hashimoto, K. Aikawa, S. Ito and K. Mikami, Angew. Chem., Int. Ed., 2012, 51, 9535–9538 CrossRef CAS PubMed.
- B. E. Smart, The Chemistry of Halides, Psuedo-Halides and Azides: Supplement D: Part 1, Wiley, 1983, vol. 1, pp. 603–655, Fluorocarbons Search PubMed.
- T. Hiyama and H. Yamamoto, Organofluorine Compounds: Chemistry and Applications, Springer Berlin Heidelberg, 2000 Search PubMed.
- K. Honda, T. V. Harris, M. Hatanaka, K. Morokuma and K. Mikami, Chem. – Eur. J., 2016, 22, 8796–8800 CrossRef CAS PubMed.
- K. Aikawa, K. Maruyama, K. Honda and K. Mikami, Org. Lett., 2015, 17, 4882–4885 CrossRef CAS PubMed.
- K. Aikawa, K. Maruyama, J. Nitta, R. Hashimoto and K. Mikami, Org. Lett., 2016, 18, 3354–3357 CrossRef CAS PubMed.
- S. Okusu, E. Tokunaga and N. Shibata, Org. Lett., 2015, 17, 3802–3805 CrossRef CAS PubMed.
- S. Ito, N. Kato and K. Mikami, Chem. Commun., 2017, 53, 5546–5548 RSC.
- D. J. Sheldon, G. Coates and M. R. Crimmin, Chem. Commun., 2020, 56, 12929–12932 RSC.
- Y. Zhao, W. Huang, J. Zheng and J. Hu, Org. Lett., 2011, 13, 5342–5345 CrossRef CAS PubMed.
- T. Hagiwara and T. Fuchikami, Synlett, 1995, 717–718 CrossRef CAS.
- J. Kvíčala, J. Štambaský, S. Böhm and O. Paleta, J. Fluorine Chem., 2002, 113, 147–154 CrossRef.
- T. Fuchikami and I. Ojima, J. Organomet. Chem., 1981, 212, 145–153 CrossRef CAS.
- K. Hirano, S. Gondo, N. Punna, E. Tokunaga and N. Shibata, ChemistryOpen, 2019, 8, 406–410 CrossRef CAS PubMed.
- B. Musio, E. Gala and S. V. Ley, ACS Sustain. Chem. Eng., 2018, 6, 1489–1495 CrossRef CAS.
- Z. Mazloomi, A. Bansode, P. Benavente, A. Lishchynskyi, A. Urakawa and V. V. Grushin, Org. Process Res. Dev., 2014, 18, 1020–1026 CrossRef CAS.
- B. Gutmann, M. Köckinger, G. Glotz, T. Ciaglia, E. Slama, M. Zadravec, S. Pfanner, M. C. Maier, H. Gruber-Wölfler and C. Oliver Kappe, React. Chem. Eng., 2017, 2, 919–927 RSC.
- M. Köckinger, T. Ciaglia, M. Bersier, P. Hanselmann, B. Gutmann and C. O. Kappe, Green Chem., 2018, 20, 108–112 RSC.
- M. Köckinger, C. A. Hone, B. Gutmann, P. Hanselmann, M. Bersier, A. Torvisco and C. O. Kappe, Org. Process Res. Dev., 2018, 22, 1553–1563 CrossRef.
- J. Burdon, P. L. Coe, I. B. Haslock and R. L. Powell, Chem. Commun., 1996, 49–50 RSC.
- K. K. Banger, A. K. Brisdon and A. Gupta, Chem. Commun., 1997, 139–140 RSC.
- S. Duric, B. M. Schmidt, N. M. Ninnemann, D. Lentz and C. C. Tzschucke, Chem. – Eur. J., 2012, 18, 437–441 CrossRef CAS PubMed.
- K. K. Banger and A. K. Brisdon, J. Organomet. Chem., 1999, 582, 301–309 CrossRef CAS.
- A. K. Brisdon, I. R. Crossley, R. G. Pritchard and G. Sadiq, J. Organomet. Chem., 2007, 692, 2125–2130 CrossRef CAS.
- N. A. Barnes, A. K. Brisdon, W. I. Cross, J. G. Fay, J. A. Greenall, R. G. Pritchard and J. Sherrington, J. Organomet. Chem., 2000, 616, 96–105 CrossRef CAS.
- P. L. Coe, J. Burdon and I. B. Haslock, J. Fluorine Chem., 2000, 102, 43–50 CrossRef CAS.
- J. Burdon, P. L. Coe, I. B. Haslock and R. L. Powell, J. Fluorine Chem., 1997, 85, 151–153 CrossRef CAS.
- K. K. Banger, R. P. Banham, A. K. Brisdon, W. I. Cross, G. Damant, S. Parsons, R. G. Pritchard and A. Sousa-Pedrares, J. Chem. Soc., Dalton Trans., 1999, 427–434 RSC.
- J. E. Phelps, S. B. Frawley and R. G. Peters, Heteroat. Chem., 2009, 20, 393–397 CrossRef CAS.
- K. Wu and Q.-Y. Chen, Tetrahedron, 2002, 58, 4077–4084 CrossRef CAS.
- J. Burdon, P. L. Coe, I. B. Haslock and R. L. Powell, J. Fluorine Chem., 1999, 99, 127–131 CrossRef CAS.
- K. Nickisch, W. Elger, J. Cessac, N. Kesavaram, B. Das, R. Garfield, S.-Q. Shi, O. Amelkina and R. Meister, Steroids, 2013, 78, 255–267 CrossRef CAS PubMed.
- N. A. Barnes, A. K. Brisdon, F. R.-W. Brown, W. I. Cross, I. R. Crossley, C. Fish, J. V. Morey, R. G. Pritchard and L. Sekhri, New J. Chem., 2004, 28, 828–837 RSC.
- K. K. Banger, A. K. Brisdon, C. J. Herbert, H. A. Ghaba and I. S. Tidmarsh, J. Fluorine Chem., 2009, 130, 1117–1129 CrossRef CAS.
- G. DiMartino, M. B. Hursthouse, M. E. Light, J. M. Percy, N. S. Spencer and M. Tolley, Org. Biomol. Chem., 2003, 1, 4423–4434 RSC.
- D. J. Brauer and G. Pawelke, J. Organomet. Chem., 2000, 604, 43–51 CrossRef CAS.
- K. Wu and Q.-Y. Chen, J. Fluorine Chem., 2003, 122, 171–174 CrossRef CAS.
- T. Okano, M. Chokai, M. Hiraishi, M. Yoshizawa, T. Kusukawa and M. Fujita, Tetrahedron, 2004, 60, 4031–4035 CrossRef CAS.
- A. Raghavanpillai and D. J. Burton, J. Org. Chem., 2004, 69, 7083–7091 CrossRef CAS PubMed.
- K. Fuchibe, K. Shigeno, N. Zhao, H. Aihara, R. Akisaka, T. Morikawa, T. Fujita, K. Yamakawa, T. Shimada and J. Ichikawa, J. Fluorine Chem., 2017, 203, 173–184 CrossRef CAS.
- R. Anilkumar and D. J. Burton, Tetrahedron Lett., 2002, 43, 2731–2733 CrossRef CAS.
- H. Miyauchi, N. Kondo and N. Nagasaki, US Pat., US2020017429, 2020 Search PubMed.
- P. V. Ramachandran and G. V. Reddy, J. Fluorine Chem., 2008, 129, 443–446 CrossRef CAS.
- C. Douvris, E. S. Stoyanov, F. S. Tham and C. A. Reed, Chem. Commun., 2007, 1145–1147 RSC.
- D. Yang, J. Zhao, Y. Zhao, Y. Lei, L. Cao, X.-J. Yang, M. Davi, N. de Sousa Amadeu, C. Janiak, Z. Zhang, Y.-Y. Wang and B. Wu, Angew. Chem., Int. Ed., 2015, 54, 8658–8661 CrossRef CAS PubMed.
- E. P.-F. Lee, J. M. Dyke and C. A. Mayhew, J. Phys. Chem. A, 1998, 102, 8349–8354 CrossRef CAS.
- D. Chen, C. Ni, Y. Zhao, X. Cai, X. Li, P. Xiao and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 12632–12636 CrossRef CAS PubMed.
- J. B. Geri, E. Y. Aguilera and N. K. Szymczak, Chem. Commun., 2019, 55, 5119–5122 RSC.
- B. M. Kraft, R. J. Lachicotte and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 10973–10979 CrossRef CAS PubMed.
- L. Maron, E. L. Werkema, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 279–292 CrossRef CAS PubMed.
- Y. Jiao, M. E. Evans, J. Morris, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2013, 135, 6994–7004 CrossRef CAS PubMed.
- J. T. Lyon and L. Andrews, Organometallics, 2006, 25, 1341–1343 CrossRef CAS.
- J. T. Lyon and L. Andrews, Inorg. Chem., 2007, 46, 4799–4808 CrossRef CAS PubMed.
- H.-G. Cho and L. Andrews, Inorg. Chem., 2008, 47, 1653–1662 CrossRef CAS PubMed.
- H.-G. Cho and L. Andrews, Organometallics, 2009, 28, 1358–1368 CrossRef CAS.
- H.-G. Cho and L. Andrews, Organometallics, 2010, 29, 2211–2222 CrossRef CAS.
- H.-G. Cho and L. Andrews, Organometallics, 2011, 30, 477–486 CrossRef CAS.
- M. Ahrens, G. Scholz, T. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2013, 52, 5328–5332 CrossRef CAS PubMed.
- A. A. Kolomeitsev, A. A. Kadyrov, J. Szczepkowska-Sztolcman, M. Milewska, H. Koroniak, G. Bissky, J. A. Barten and G.-V. Röschenthaler, Tetrahedron Lett., 2003, 44, 8273–8277 CrossRef CAS.
- N. E. Shevchenko, V. G. Nenajdenko and G.-V. Röschenthaler, J. Fluorine Chem., 2008, 129, 390–396 CrossRef CAS.
- J. L. Butikofer, J. M. Hoerter, R. G. Peters and D. M. Roddick, Organometallics, 2004, 23, 400–408 CrossRef CAS.
- Z. Luo, X. Yang and G. C. Tsui, Org. Lett., 2020, 22, 6155–6159 CrossRef CAS PubMed.
- M. Keßler, S. Porath, H.-G. Stammler and B. Hoge, Eur. J. Inorg. Chem., 2021, 907–913 CrossRef.
- Y. Fujihira, K. Hirano, M. Ono, H. Mimura, T. Kagawa, D. M. Sedgwick, S. Fustero and N. Shibata, J. Org. Chem., 2021, 86, 5883–5893 CrossRef CAS PubMed.
- M. Ono, Y. Sumii, Y. Fujihira, T. Kagawa, H. Mimura and N. Shibata, J. Org. Chem., 2021, 86, 14044–14053 CrossRef CAS PubMed.
- A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2013, 135, 12584–12587 CrossRef CAS PubMed.
- I. Popov, S. Lindeman and O. Daugulis, J. Am. Chem. Soc., 2011, 133, 9286–9289 CrossRef CAS PubMed.
- X. Yang and G. C. Tsui, Org. Lett., 2020, 22, 4562–4567 CrossRef CAS PubMed.
- M. Niemann, B. Neumann, H.-G. Stammler and B. Hoge, Angew. Chem., Int. Ed., 2019, 58, 8938–8942 CrossRef CAS PubMed.
- S. Solyntjes, J. Bader, B. Neumann, H.-G. Stammler, N. Ignat’ev and B. Hoge, Chem. – Eur. J., 2017, 23, 1557–1567 CrossRef CAS PubMed.
- S. Pelzer, B. Neumann, H.-G. Stammler, N. Ignat’ev and B. Hoge, Chem. – Eur. J., 2016, 22, 4758–4763 CrossRef CAS PubMed.
- M. Wiesemann, J. Klösener, M. Niemann, B. Neumann, H.-G. Stammler and B. Hoge, Chem. – Eur. J., 2017, 23, 14476–14484 CrossRef CAS PubMed.
- L. A. Bischoff, J. Riefer, R. Wirthensohn, T. Bischof, R. Bertermann, N. V. Ignat’ev and M. Finze, Chem. – Eur. J., 2020, 26, 13615–13620 CrossRef CAS PubMed.
- J. R. Case and F. Nyman, Nature, 1962, 193, 473 CrossRef CAS.
- H. C. Cowen, F. Riding, E. Warhurst, A. D. Buckingham, R. J.-W. Le Fèvre, G. D. Meakins, R. N. Haszeldine, J. Jander, G. R. Clemo, B. W. Fox, R. Raper, J. F.-W. McOmie, I. M. White, O. L. Brady, P. E. Halstead, A. G. Sharpe, G. W. Gray, B. Jones, J. Cast, T. S. Stevens, F. Bell, J. W. Cook, L. Hunter, R. M. Barrer, N. Mackenzie, D. MacLeod, C. C. Barker, F. D. Casson, G. F. Laws, W. Carruthers, E. S. Lane and C. Williams, J. Chem. Soc., 1953, 4168–4188 RSC.
- G. C. Demitras and A. G. MacDiarmid, Inorg. Chem., 1964, 3, 1198–1199 CrossRef CAS.
- H. Deubner and F. Kraus, Inorganics, 2017, 5, 68 CrossRef.
- R. Basta, B. G. Harvey, A. M. Arif and R. D. Ernst, J. Am. Chem. Soc., 2005, 127, 11924–11925 CrossRef CAS PubMed.
- B. G. Harvey, A. M. Arif, A. Glöckner and R. D. Ernst, Organometallics, 2007, 26, 2872–2879 CrossRef CAS.
- P. Holze, B. Horn, C. Limberg, C. Matlachowski and S. Mebs, Angew. Chem., Int. Ed., 2014, 53, 2750–2753 CrossRef CAS PubMed.
- L. Zámostná, T. Braun and B. Braun, Angew. Chem., Int. Ed., 2014, 53, 2745–2749 CrossRef PubMed.
- L. Zámostná and T. Braun, Angew. Chem., Int. Ed., 2015, 54, 10652–10656 CrossRef PubMed.
- C. Berg, T. Braun, M. Ahrens, P. Wittwer and R. Herrmann, Angew. Chem., Int. Ed., 2017, 56, 4300–4304 CrossRef CAS PubMed.
- M. Wozniak, T. Braun, M. Ahrens, B. Braun-Cula, P. Wittwer, R. Herrmann and R. Laubenstein, Organometallics, 2018, 37, 821–828 CrossRef CAS.
- D. Dirican, N. Pfister, M. Wozniak and T. Braun, Chem. – Eur. J., 2020, 26, 6945–6963 CrossRef CAS PubMed.
- F. Buß, C. Mück-Lichtenfeld, P. Mehlmann and F. Dielmann, Angew. Chem., Int. Ed., 2018, 57, 4951–4955 CrossRef PubMed.
- M. Rueping, P. Nikolaienko, Y. Lebedev and A. Adams, Green Chem., 2017, 19, 2571–2575 RSC.
- T. A. McTeague and T. F. Jamison, Angew. Chem., Int. Ed., 2016, 55, 15072–15075 CrossRef CAS PubMed.
- P. Tomar, T. Braun and E. Kemnitz, Chem. Commun., 2018, 54, 9753–9756 RSC.
- D. Rombach and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2020, 59, 300–303 CrossRef CAS PubMed.
- D. Rombach and H.-A. Wagenknecht, ChemCatChem, 2018, 10, 2955–2961 CrossRef CAS.
- S. Kim, Y. Khomutnyk, A. Bannykh and P. Nagorny, Org. Lett., 2021, 23, 190–194 CrossRef CAS PubMed.
- D. J. Sheldon and M. R. Crimmin, Chem. Commun., 2021, 57, 7096–7099 RSC.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- Y. Ogiwara, S. Hosaka and N. Sakai, Organometallics, 2020, 39, 856–861 CrossRef CAS.
- Y. Ogiwara and N. Sakai, Angew. Chem., Int. Ed., 2020, 59, 574–594 CrossRef CAS PubMed.
- J. A. Kalow and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 3268–3269 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |