
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne Soai reaction, two mechanisms?
Yannick
Geiger

Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: y.geiger@rug.nl
First published on 24th January 2022
Abstract
For over 25 years the chemistry community has puzzled over the mechanism of the Soai reaction, a fascinating chemical process which achieves chiral symmetry breaking by combining autocatalysis with asymmetric amplification. In 2020, the groups of Denmark and Trapp each made a proposal, based on extensive experimental work, on what is the catalytic species there: either a tetrameric product alkoxide aggregate (“SMS tetramer”) or a product-substrate dimer (“hemiacetal”). These models seemingly oppose and exclude each other; however, they might also be both valid since the studies were conducted on different substrates which are not necessarily equivalent. This is shown in this Viewpoint by an in-depth comparison of the two studies and of data from earlier reports, which opens up to a discussion on this scenario's far-reaching implications on the fundamental understanding of asymmetry-amplifying autocatalysis.
 Yannick Geiger | Yannick Geiger was born in Nantes (France) and raised in Frankfurt am Main (Germany). He studied chemistry at the University of Strasbourg (France), which ended with the obtention of a PhD in 2019 under the supervision of Dr Stéphane Bellemin-Laponnaz. His work was centered on hyperpositive non-linear effects and mechanistic studies of ephedrine-catalysed dialkylzinc additions, for which he obtained the 2020 “Henri Kagan” PhD prize of the French Chemical Society. He currently works as a post-doctoral researcher at the University of Groningen (Netherlands) on chiral selectivity and symmetry breaking in self-replicators in the group of Prof Sijbren Otto. |
For over 25 years, the famous Soai reaction has fascinated the scientific community for its unique features and performances, as well as for the mystery that surrounds its mechanism.1,2 It consists in the asymmetric addition of ZniPr2 to pyrimidyl-5-aldehydes yielding the corresponding zinc alkoxide in high enantiomeric excess (ee; Fig. 1). The reaction is autocatalytic – that is, the formed zinc alkoxide promotes its own formation (presumably through a catalytic intermediate) – and asymmetry-amplifying: a chiral initiator of low enantiomeric purity is sufficient to obtain a high ee zinc alkoxide, much higher than the ee of the initiator. There are several variants of the pyrimidyl substrate, from which the tert-butyl-alkynyl-substituted pyrimidyl aldehyde tBuPm shows superlative performances (Fig. 1): it is capable of amplifying the tiny ee generated by statistical fluctuations in the direct, non-enantioselective addition of ZniPr2 to the aldehyde, and thus produces randomly R- or S-zinc alkoxides tBuPmII in the absence of any chiral trigger.3,4 This is referred to as chiral symmetry breaking, which has been achieved otherwise only through deracemization processes.5–7 Adding a chiral initiator then merely defines the sign of the upcoming alkoxide. To do this, unlikely chiral moieties such as cryptochiral hydrocarbons, enantiomorphic crystals, enantiotopic crystal faces and chiral isotopomers are sufficient.2 Such strong chiral amplification phenomena are believed to have played a role in the emergence of biological homochirality, which is intrinsically linked to the origin of life.8,9 Much more than a “signature” of life, the spatial specificity conferred by single chiral and homochiral building blocks (i.e. sugars and amino acids) is a necessity for the tremendous efficiency of today's biomolecules in catalysis,10 electron transfer11 and information transfer.12–14 Thus, it probably opened pathways for the emergence of function in the early steps of chemical evolution.14,15 Although current opinions rather point at polymerisation models13,16 and the emergence of biopolymer function14,17 to explain homochirogenesis, it is not excluded that amplification on a small molecule-level might also have played a role.
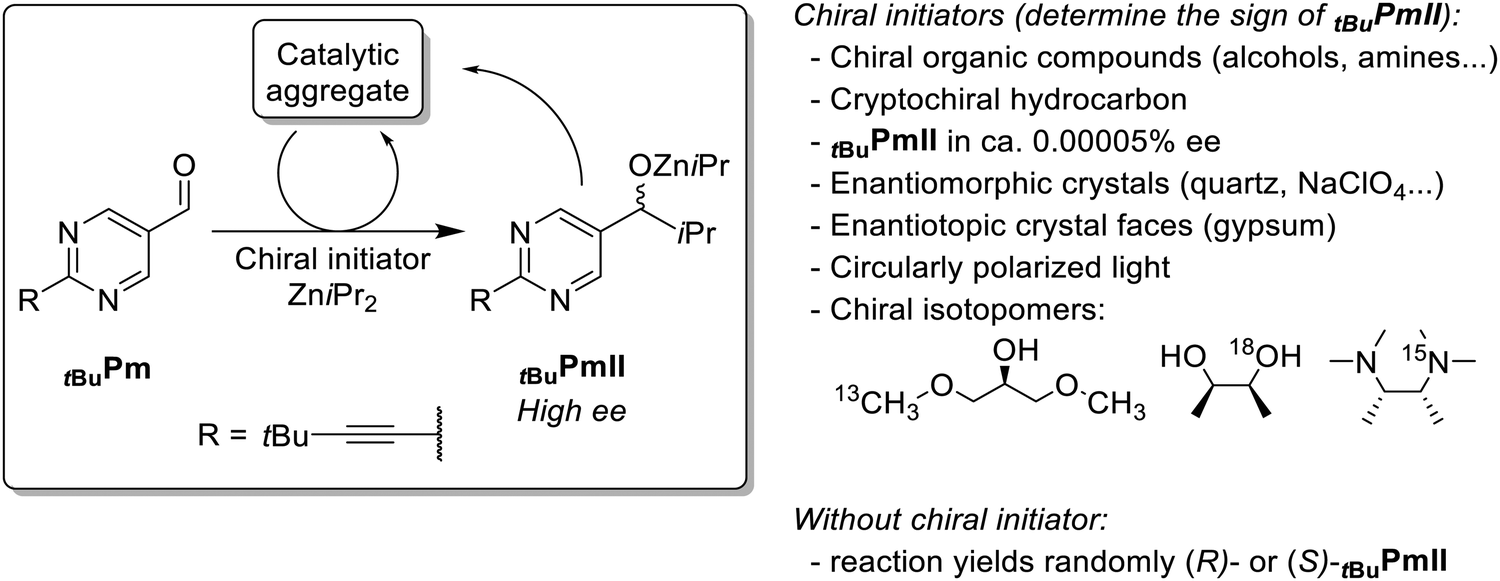 | ||
| Fig. 1 General scheme for the Soai reaction based on the tBuPm aldehyde and the tBuPmII alkylzinc alkoxide. The stereochemical outcome of the reaction (i.e. whether (R)- or (S)-tBuPmII is obtained) can be determined by various chiral initiators; otherwise, the outcome is driven by statistical fluctuations in the early stage of the reaction.2 | ||
How exactly the Soai reaction works has been a mystery and subject of fierce debate since Soai's first report in 1995.1 Following Kagan's18 and Noyori's19,20 pioneering works on non-linear effects (NLE) it was soon proposed that aggregates of the chiral zinc alkoxide are the catalytically active species.21 However, the highly fluctional nature of dialkylzinc chemistry, involving numerous interdependent aggregates in fast equilibrium, made any further study a tremendous challenge. Most of the reported investigations are of theoretical nature (kinetic modelling22–24 or quantum chemical calculations25–29). Experimental work has been performed mostly on isolated parts of the system (e.g.1H DOSY NMR30 and circular dichroism31 on the zinc alkoxide alone, 1H NMR on zinc alkoxide + ZniPr232). Notable exceptions are Blackmond's and Brown's reports of peculiar kinetic features (inverse temperature-dependence on reaction rate, induction periods, kinetic orders in reaction components)30 and of a transient hemiacetal33 during the reaction.
It is only in 2020 that the groups of Scott Denmark34,35 and Oliver Trapp36 have come each with a conclusive proposal for the mechanism behind the Soai reaction, based on extensive experimental studies supplemented by kinetic modelling or Density Functional Theory (DFT) calculations. As both studies result into seemingly contradictory conclusions – at least on first sight – the present author wishes to provide some food for thought by comparing and discussing these two proposals. A closer look reveals that both proposals might be complementary instead of contradictory, leading to far-reaching implications for the fundamental understanding of asymmetry-amplifying autocatalysis in general. This paper uses a nomenclature derived from Denmark's notation:35Pm and Py refer to pyrimidyl- and pyridylaldehydes, respectively; PmII and PyII to the respective chiral zinc alkoxides resulting from ZniPr2 addition. Lower-case prefixes indicate the substituents para to the aldehyde or alcohol moiety, e.g.tBuPy and TMSPy for the tBu-alkyne and TMS-alkyne substituted pyridine aldehydes.
The Denmark proposal: alkoxide tetramer through Zn–N coordination
The first proposal came from Denmark and co-workers,34 later complemented by a second report.35 They claim that the active catalyst of the Soai reaction is a homochiral tetramer of the product alkoxide TMSPyII (cf. note vide infra) assembled in a square-macrocycle-square (SMS) conformation (Fig. 2, middle), on the base of own and other's DFT calculations, previously reported crystal structures37 and DOSY NMR studies on tBu- and AdamPmII, respectively.30 Through an NMR study of various alkylzinc alkoxides they show that the isopropyl groups in TMSPyII are essential to drive away from the (otherwise stable and inert) Zn4O4-cubes formed by less bulky alkylzinc alkoxides19,20 (“cube-escape”), leading to the presumed N-coordinated SMS-tetramer. Mixed catalyst–substrate experiments, with TMSPyII as (non-auto)catalyst and various aromatic aldehydes, showed that at least one nitrogen atom on the substrate's aromatic ring is essential for catalysis to take place, presumably through N-coordination to the catalyst. An N,O-two-point-binding of the aldehyde substrate to the homochiral TMSPyII-tetramer (Fig. 2, right) and the transition state for the ZniPr2 addition were modelled via DFT calculations; the asymmetric amplification was explained by the racemic (meso) TMSPyII-tetramer being kinetically incompetent as well as thermodynamically favoured over the homochiral tetramer, thus being in line with Kagan's principles18 and Frank's model for autocatalytic asymmetric amplification.38 Heterochiral tetramers (in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 enantiomeric ratio) were not investigated. In the second report, further mixed catalyst–substrate-experiments, DFT studies and non-quantitative kinetic modelling address differences between TMSPyII and TMSPmII (reactivity, sensitivity to excess ZniPr2) and the role of the alkyne substituent (improvement of selectivity and solubility, prevention of unproductive aggregates).35
:1 enantiomeric ratio) were not investigated. In the second report, further mixed catalyst–substrate-experiments, DFT studies and non-quantitative kinetic modelling address differences between TMSPyII and TMSPmII (reactivity, sensitivity to excess ZniPr2) and the role of the alkyne substituent (improvement of selectivity and solubility, prevention of unproductive aggregates).35
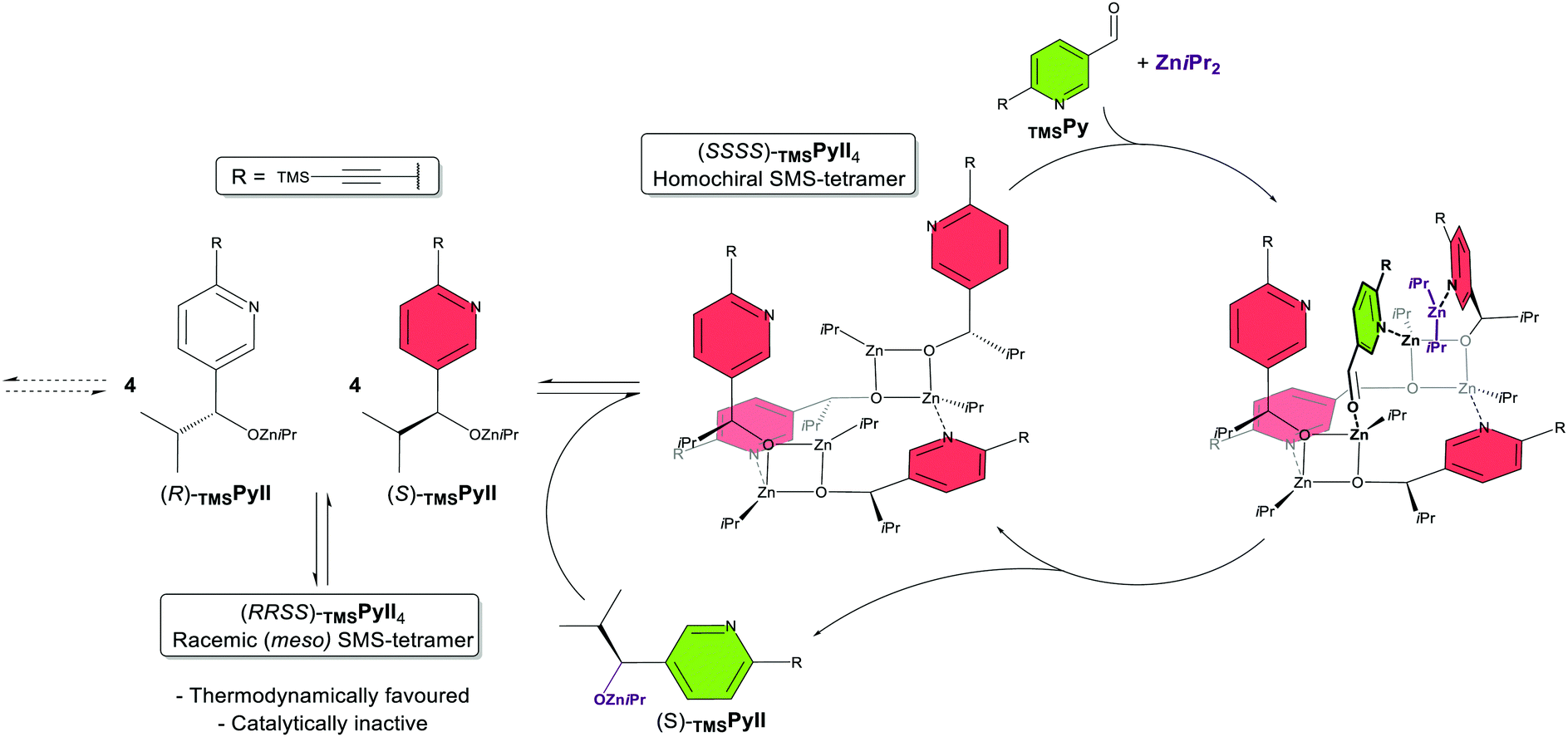 | ||
| Fig. 2 Scheme of the autocatalytic model of the Denmark proposal.34,35 For simplicity, the autocatalytic cycle of the (R)-enantiomer has been omitted. The product alkoxide TMSPyII aggregates to a square-macrocycle-square (SMS) tetramer which can accommodate a molecule of both ZniPr2 and TMSPy aldehyde. Upon reaction of both, a molecule of TMSPyII is released which can further aggregate. Only the homochiral tetramer is catalytically active; the inactive (and more stable) meso tetramer acts as a minor enantiomer trap and thus drives asymmetric amplification. | ||
It should be noted that the study was not based on the pyrimidine system tBuPm/tBuPmII but its pyridine analogue TMSPy/TMSPyII with a TMS-alkyne substituent. The authors found it (and also its tBu-alkyne counterpart tBuPyII) to be autocatalytic and asymmetry-amplifying, much like the pyrimidine-based Soai system. This is remarkable since the unsubstituted pyridine aldehyde PyII was reported to be non-autocatalytic39 and earlier attempts on tBuPy/tBuPyII showed chiral erosion instead of amplification.40 Therefore, they used TMSPy/TMSPyII as a substrate more convenient to handle with and took this as a base for the assumption that the 2nd nitrogen on Soai's pyrimidine substrates plays only a minor role.35
The Trapp proposal: slow-forming hemiacetals
Trapp and co-workers published their study on the Soai reaction later in the same year, with a proposal quite different from Denmark's.36 Instead of an alkoxide tetramer they propose an aldehyde-alkoxide mixed dimer – a hemiacetal whose induced stereogenic centre is determined by the alkoxide's chiral configuration – which binds to a 2nd tBuPm aldehyde and ZniPr2 to form a new molecule of tBuPmII bound to the hemiacetal catalyst (Fig. 3). Incorporation of a further tBuPm moiety gives rise to a double hemiacetal, which then splits into two independent units. Thus, the catalyst-binding and subsequent reaction of aldehyde and Zn(iPr)2 resembles much the Noyori model for chiral zinc-dimethylaminoisoborneol (DAIB) complexes.19,20 However, the way how it achieves asymmetric amplification is strikingly different: the core of the model is that the initial hemiacetal formation is slow and even unfavourable, but once it has formed the autocatalytic reaction kicks in and proceeds at a much faster rate. Thus, it is based on a kinetic-controlled nonequilibrium stationary state (NESS)5 instead of Kagan's thermodynamics-based principles (racemate elimination through heterochiral aggregation with no or low catalytic activity);18 it therefore also doesn’t follow Frank's often cited model which relies on “specific mutual antagonism” of enantiomers as in the Kagan models.38 Here, the aggregate is not needed for minor enantiomer entrapment but for providing a kinetic barrier between the pool of chiral tBuPmII and the autocatalytic cycle; the off-cycle dimers (tBuPmII)2 also don’t provide any chiral entrapment (which is impossible with the dimer association constants KHetero/KHomo = 2,41 contrary to what was stated in the proposal36). Thus, the relevant factor for the catalytic outcome is which tBuPmII enantiomer is the first to generate a hemiacetal – that event is so rare that any following hemiacetal of opposite chirality will be outcompeted or won’t even appear – and that is governed purely by statistics.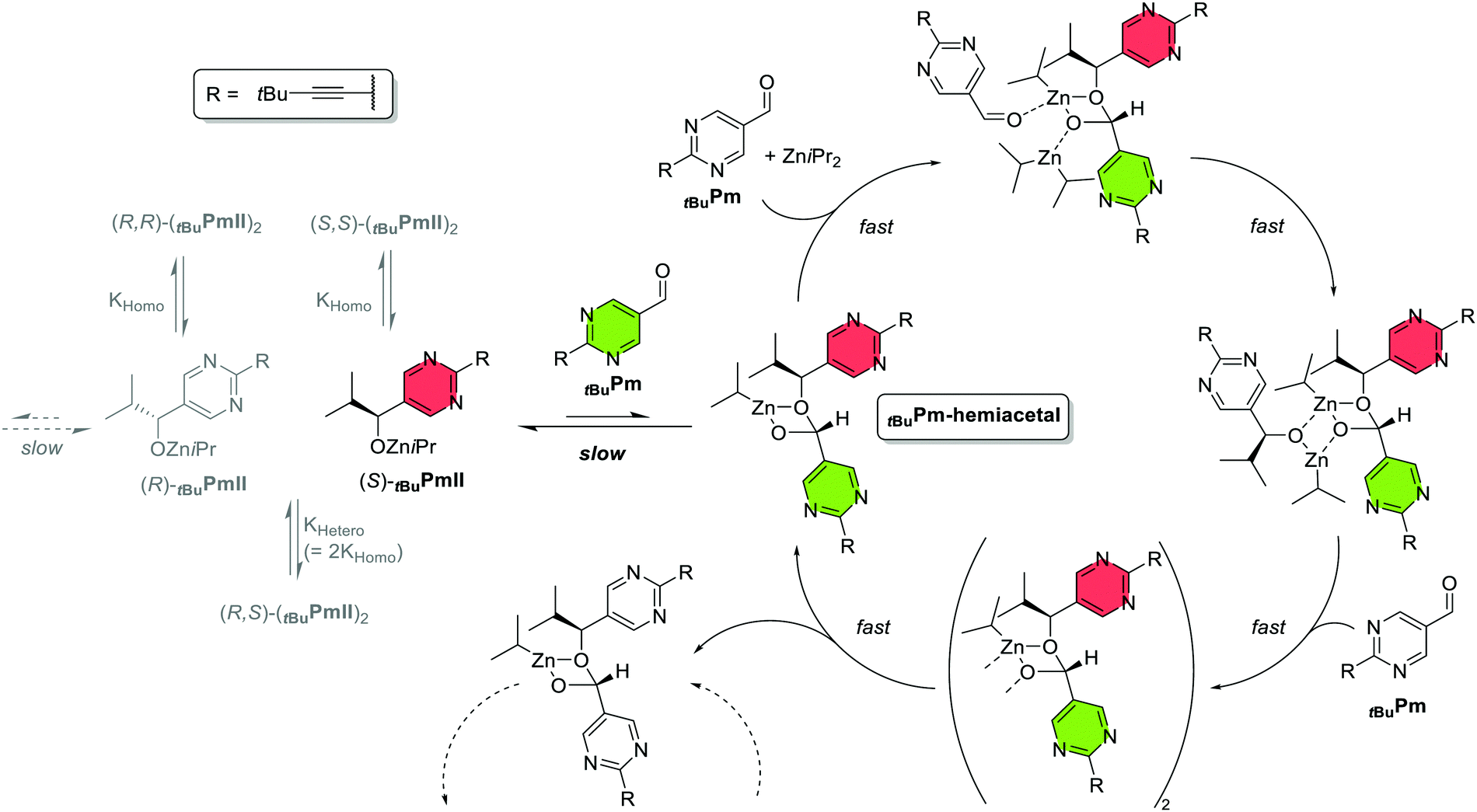 | ||
| Fig. 3 Scheme of the kinetics-based autocatalytic model of the Trapp proposal.36 For simplicity, the autocatalytic cycle of the (R)-enantiomer has been omitted. The tBuPmII alkoxide first reacts (slowly) with the tBuPm aldehyde to form a hemiacetal, which catalyses the reaction between tBuPm + ZniPr2 and the formation of a 2nd hemiacetal moiety at a much higher rate. The first tBuPmII-enantiomer to initiate this cycle determines the sign of the overall produced product. | ||
Evidence for the tBuPm aldehyde being more than just a substrate is its kinetic order of 1.9, which confirms previous reports.30,42,43 Another observation is an induction period before the actual autocatalysis starts (also previously observed with the adamantyl-alkyne and methyl derivatives AdamPm/AdamPmII and MePm/MePmII30). That period can be eliminated by adding, right after the reaction start, an aliquot of another Soai reaction which already has reached the autocatalytic phase (“doping” or “seeding” experiment). Seeding with a completed Soai reaction (i.e. where all aldehyde substrate had been consumed) or with a dissolved crystalline SMS tetramer of tBuPmII didn’t influence the induction period. They interpreted this as the autocatalyst being a transient species: it exists neither at the beginning nor at the end of the reaction but forms (and then depletes) over time, much like the hemiacetal which was monitored via in situ mass spectrometry; its maximum abundance coincides with the peak of the reaction rate. The slow rate of the hemiacetal's association/dissociation was probed by dynamic HPLC on tBuPmII in presence of isopropanol and its temperature-dependence was found to be consistent with that of the overall Soai reaction30 and of the hemiacetal alone in an earlier report (in which it was considered only as an off-loop species by the authors).331H NMR experiments further proved the strong propensity of pyrimidylaldehydes to form hemiacetals.44 Finally, extensive kinetic modelling allowed prediction of very strong chiral amplification and of the time of the inflection point as a function of initiator ee.
Two sides of the same coin?
At first glance, the two proposals seem to be quite antagonistic as they describe two radically different systems – alkoxide tetramers vs. hemiacetals as the active species, Frank-type model vs. kinetic-controlled NESS – which also exclude each other: Trapp's doping experiments with a completed Soai reaction show that tBuPmII alone cannot be the catalyst, whereas Denmark and co-workers exclude aldehyde participation based on their mixed catalyst–substrate experiments. However, this contradiction applies only if both pyrimidine- and pyridine-based systems are assumed to be identical in their mode of action. This need not be true: the newly found autocatalytic system TMSPy/TMSPyII might act following a different mechanism than the classic tBuPm/tBuPmII-system. In other words: both proposals may be valid, each for the respective system the study was based on. Several aspects of the present data, which we will show in the following, indicate that this might be well the case.– The TMSPy/TMSPyII-system was claimed to be similar to tBuPm/tBuPmII because it is also autocatalytic, asymmetry-amplifying and with an inverse temperature effect on reaction rate,34 and the SMS-tetramer model being applicable on tBuPm/tBuPmII because of a corresponding crystal structure37 and DOSY data30 of tBuPmII and AdamPmII, respectively. In principle, the inverse temperature effect can occur with any catalytic aggregate (higher-order species are usually disfavoured at high temperature) and data on alkoxides alone – without substrate and/or reactant – is insufficient to conclude on the catalyst's aggregation level during the reaction, especially in dialkylzinc chemistry. In solution, alkylzinc complexes of DAIB19,20 and N-benzylephedrine (NBE)41 alone form inactive di- and trimers, respectively, which upon addition of both aldehyde and dialkylzinc dissociate to the monomeric (with NBE also dimeric45–47) catalysts. Therefore, there is a lack of evidence for the pyridine and pyrimidine systems following necessarily the same mechanism; discrepancies in the kinetic order in aldehyde (0 with mixed system Py/TMSPyII,34 >1 with Adam- and tBuPm/PmII30,36,42,43) even tend to render that option unlikely.
– Upon discovery of the TMSPy/TMSPyII-system, Denmark and co-workers concluded that the 2nd nitrogen atom on pyrimidine is not necessary for autocatalysis to take place. However, the pyridine and pyrimidine moieties may be no comparable entities: the latter is much more electron-poor and a weaker ligand.35 This does not only render an N-coordination-based aggregate & catalytic mechanism less likely for tBuPm/tBuPmII, it should also improve the aldehyde's ability to form hemiacetals: this is favoured the more the aromatic is electron-poor. Therefore, the pyridine-based system might well catalyse via a SMS-tetramer and the pyrimidine-based system via a hemiacetal.
– Within the SMS-tetramer model, the pyrimidine's lower N-Lewis basicity (together with the presence of a 2nd nitrogen atom) was taken to explain TMSPm/TMSPmII's insensitivity towards excess ZniPr2, whereas TMSPy/TMSPyII exhibits reduced reaction rate and enantioselectivity – presumably due to ZniPr2 blocking the pyridine's nitrogen atom.35 However, TMSPm/TMSPmII following the hemiacetal-mechanism would also explain this discrepancy since no N-coordinated complexes intervene there. Furthermore, that would give a more straightforward explanation for the observed differences in reaction rate, as TMSPm/TMSPmII reacts much faster than TMSPy/TMSPyII.35 Within the SMS-tetramer model, the higher electrophilicity of the TMSPm aldehyde would have to overcompensate the weaker coordination ability of TMSPmII (needed to form the SMS-tetramer as well as to bind to the ZniPr2 reagent) to explain the exceptionally high reaction rate of TMSPm/TMSPmII, whereas between different mechanistic systems rate differences are no surprise.
From that perspective, it would be interesting to compare both models for their propensity to perform chiral symmetry breaking (when no chiral initiator is used), which has not been done with the TMSPy/TMSPyII-system yet. At a very early stage of the reaction, when only few alkoxide molecules have formed through the direct aldehyde-ZniPr2 addition, it should make a difference for the reaction dynamics whether the alkoxide must react with the abundant aldehyde to form a hemiacetal or 3 times with itself to form the SMS-tetramer. Also, with tBuPm/tBuPmII chiral symmetry breaking works best in a 1:4 toluene/Et2O mixture.3,4 Since Et2O is a coordinating solvent to some extent, one would expect this to be better compatible with a hemiacetal-mechanism since the ether solvent should disfavour higher-order aggregates; in THF TMSPmII forms exclusively dimers.32
Finally, one should consider the possibility that both mechanisms may be operating simultaneously on the same system, to different extents and depending on the reaction conditions. Soai's crystal structure37 and Blackmond's DOSY measurement30 show that tBu- and AdamPmII are, at least in principle, capable of forming SMS-tetramers. Even though these should be less favoured in pyrimidine systems (vide supra) they may appear if no other equilibria pull material away from them. Likewise, even though hemiacetals are less likely in the pyridine system, they may not be fully excluded depending on the reaction conditions: Amedjkouh and co-workers found indirect evidence for a transient hemiacetal with tBuPy/tBuPyII under heterogeneous conditions.48 It is also worth looking at the initial reaction conditions in both proposals, which favour each the respective defended model: Denmark added the aldehyde to an alcohol initiator/ZniPr2-mixture, in which SMS-tetramers (but no hemiacetals) can already have formed; in the Trapp procedure ZniPr2 is added to an aldehyde/alcohol initiator mixture, which may have already generated non-metalated hemiacetals but no alkoxide tetramers. Trapp's mixed dialkylzinc-experiments show that the starting conditions indeed have a strong influence on the final product ee.36 Thus, it is conceivable that the catalytic activity of TMSPmII with several aldehydes in Denmark's mixed catalyst–substrate experiments originates in an initially formed SMS-tetramer, even if tBuPmII operates via a hemiacetal mechanism in Trapp's conditions and with tBuPm as aldehyde. Amedjkouh's mixed autocatalyst-experiments might also shed some light on that possibility.40,49
Conclusion
The fluctional nature of asymmetric dialkylzinc additions, with its numerous aggregates of different degrees involving catalyst, product and substrate, has not only complicated its studying but has also always been good for surprises.1,19,45 Thus, it might be somehow expectable if the Soai reaction was even more complex than initially thought, with not one but several mechanisms operating. This is where the true achievement of the Denmark group lies: while Trapp's group probably cracked the mechanistic puzzle of the “historical”, pyrimidine-based Soai autocatalysis, Denmark most likely found something completely different – despite the seemingly similar substrates and behaviours in terms of autocatalysis and chiral amplification (which is a coincidence… or not). If this gets confirmed it would be most exciting: knowing not one but several of such systems might help to understand the very fundamental principles which govern asymmetry-amplifying autocatalysis, which features and processes are required. From this perspective, the insights given by the Trapp proposal are remarkable: apart from breaking with the racemate entrapment dogma, they also point to (hemi-)acetals – often disregarded in organic chemistry as uninteresting intermediates or mere protecting groups – as key species. This opens up further discussion e.g. in prebiotic sugar synthesis, where hemiacetals are likely to intervene. Furthermore, it highlights reversible covalent bonding as an alternative to coordination or hydrogen bonds to generate aggregates (an overlooked point in a field issued from metal-complex catalysis) and that a thermodynamically unfavoured reaction can play a central role in chiral amplification.In any case, further experimental work is needed (and most probably will be done) to strengthen the validity of the Denmark or Trapp proposal – or of both. The last word is not spoken in the rich story of the Soai reaction and asymmetric dialkylzinc additions and, who knows, maybe there are even more surprises to come.
Conflicts of interest
The author declares no conflict of interest.Acknowledgements
The author would like to thank Stéphane Bellemin-Laponnaz and Sijbren Otto for helpful comments on the manuscript and Damián Padín Santos for a discussion that initiated the writing of this article. This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 847675 (oLife post-doctoral fellowship programme).References
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767–768 CrossRef CAS.
- K. Soai, T. Kawasaki and A. Matsumoto, Tetrahedron, 2018, 74, 1973–1990 CrossRef CAS.
- K. Soai, I. Sato, T. Shibata, S. Komiya, M. Hayashi, Y. Matsueda, H. Imamura, T. Hayase, H. Morioka, H. Tabira, J. Yamamoto and Y. Kowata, Tetrahedron: Asymmetry, 2003, 14, 185–188 CrossRef CAS.
- D. A. Singleton and L. K. Vo, Org. Lett., 2003, 5, 4337–4339 CrossRef CAS PubMed.
- T. Buhse, J.-M. Cruz, M. E. Noble-Terán, D. Hochberg, J. M. Ribó, J. Crusats and J.-C. Micheau, Chem. Rev., 2021, 121, 2147–2229 CrossRef CAS PubMed.
- C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef PubMed.
- W. L. Noorduin, T. Izumi, A. Millemaggi, M. Leeman, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg, B. Kaptein, E. Vlieg and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 1158–1159 CrossRef CAS PubMed.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 114, 285–366 CrossRef CAS PubMed.
- D. G. Blackmond, Cold Spring Harbor Perspect. Biol., 2019, 11, a032540 CrossRef CAS PubMed.
- M. M. Green and V. Jain, Orig. Life Evol. Biosph., 2009, 40, 111 CrossRef PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS.
- J. D. Carroll, Chirality, 2009, 21, 354–358 CrossRef CAS PubMed.
- P. Cintas, Chirality in Supramolecular Assemblies: causes and consequences, John Wiley & Sons, Ltd, 1st edn, 2016, pp. 44–64 Search PubMed.
- H. I. Cruz-Rosas, F. Riquelme, A. Ramírez-Padrón, T. Buhse, G. Cocho and P. Miramontes, J. Theor. Biol., 2020, 499, 110316 CrossRef CAS PubMed.
- J. Skolnick, H. Zhou and M. Gao, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26571–26579 CrossRef CAS PubMed.
- A. Brewer and A. P. Davis, Nat. Chem., 2014, 6, 569–574 CrossRef CAS PubMed.
- M. Wu, S. I. Walker and P. G. Higgs, Astrobiology, 2012, 12, 818–829 CrossRef CAS PubMed.
- D. Guillaneux, S.-H. Zhao, O. Samuel, D. Rainford and H. B. Kagan, J. Am. Chem. Soc., 1994, 116, 9430–9439 CrossRef CAS.
- M. Kitamura, S. Okada, S. Suga and R. Noyori, J. Am. Chem. Soc., 1989, 111, 4028–4036 CrossRef CAS.
- M. Kitamura, S. Suga, H. Oka and R. Noyori, J. Am. Chem. Soc., 1998, 120, 9800–9809 CrossRef CAS.
- D. G. Blackmond, C. R. McMillan, S. Ramdeehul, A. Schorm and J. M. Brown, J. Am. Chem. Soc., 2001, 123, 10103–10104 CrossRef CAS PubMed.
- J.-C. Micheau, C. Coudret, J.-M. Cruz and T. Buhse, Phys. Chem. Chem. Phys., 2012, 14, 13239 RSC.
- M. E. Noble-Terán, J.-M. Cruz, J.-C. Micheau and T. Buhse, ChemCatChem, 2018, 10, 642–648 CrossRef.
- D. G. Blackmond, Chem. Rev., 2020, 120, 4831–4847 CrossRef CAS PubMed.
- J. Klankermayer, I. D. Gridnev and J. M. Brown, Chem. Commun., 2007, 3151–3153 RSC.
- L. Schiaffino and G. Ercolani, Angew. Chem., Int. Ed., 2008, 47, 6832–6835 CrossRef CAS PubMed.
- L. Schiaffino and G. Ercolani, ChemPhysChem, 2009, 10, 2508–2515 CrossRef CAS PubMed.
- L. Schiaffino and G. Ercolani, Chem. – Eur. J., 2010, 16, 3147–3156 CrossRef CAS PubMed.
- I. D. Gridnev and A. K. Vorobiev, ACS Catal., 2012, 2, 2137–2149 CrossRef CAS.
- M. Quaranta, T. Gehring, B. Odell, J. M. Brown and D. G. Blackmond, J. Am. Chem. Soc., 2010, 132, 15104–15107 CrossRef CAS PubMed.
- A. Matsumoto, A. Tanaka, Y. Kaimori, N. Hara, Y. Mikata and K. Soai, Chem. Commun., 2021, 57, 11209–11212 RSC.
- I. D. Gridnev, J. M. Serafimov and J. M. Brown, Angew. Chem., Int. Ed., 2004, 43, 4884–4887 CrossRef CAS PubMed.
- T. Gehring, M. Quaranta, B. Odell, D. G. Blackmond and J. M. Brown, Angew. Chem., Int. Ed., 2012, 51, 9539–9542 CrossRef CAS PubMed.
- S. V. Athavale, A. Simon, K. N. Houk and S. E. Denmark, Nat. Chem., 2020, 12, 412–423 CrossRef CAS PubMed.
- S. V. Athavale, A. Simon, K. N. Houk and S. E. Denmark, J. Am. Chem. Soc., 2020, 142, 18387–18406 CrossRef CAS PubMed.
- O. Trapp, S. Lamour, F. Maier, A. F. Siegle, K. Zawatzky and B. F. Straub, Chem. – Eur. J., 2020, 26, 15871–15880 CrossRef CAS PubMed.
- A. Matsumoto, T. Abe, A. Hara, T. Tobita, T. Sasagawa, T. Kawasaki and K. Soai, Angew. Chem., Int. Ed., 2015, 54, 15218–15221 CrossRef CAS PubMed.
- F. C. Frank, Biochim. Biophys. Acta, 1953, 11, 459–463 CrossRef CAS.
- K. Soai, S. Niwa and H. Hori, J. Chem. Soc., Chem. Commun., 1990, 982–983 RSC.
- C. Romagnoli, B. Sieng and M. Amedjkouh, Eur. J. Org. Chem., 2015, 4087–4092 CrossRef CAS.
- Y. Geiger, T. Achard, A. Maisse-François and S. Bellemin-Laponnaz, Eur. J. Org. Chem., 2021, 2916–2922 CrossRef CAS.
- F. G. Buono and D. G. Blackmond, J. Am. Chem. Soc., 2003, 125, 8978–8979 CrossRef CAS PubMed.
- D. G. Blackmond, Tetrahedron: Asymmetry, 2006, 17, 584–589 CrossRef CAS.
- O. Trapp, Front. Chem., 2020, 8, 615800 CrossRef CAS PubMed.
- Y. Geiger, T. Achard, A. Maisse-François and S. Bellemin-Laponnaz, Nat. Catal., 2020, 3, 422–426 CrossRef CAS.
- Y. Geiger, T. Achard, A. Maisse-François and S. Bellemin-Laponnaz, Chirality, 2020, 32, 1250–1256 CrossRef CAS PubMed.
- Y. Geiger, T. Achard, A. Maisse-François and S. Bellemin-Laponnaz, Chem. Sci., 2020, 11, 12453–12463 RSC.
- G. Rotunno, D. Petersen and M. Amedjkouh, ChemSystChem, 2020, 2, e1900060 CAS.
- C. Romagnoli, B. Sieng and M. Amedjkouh, Chirality, 2020, 32, 1143–1151 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |