
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight olefin synthesis from a diversity of renewable and fossil feedstocks: state-of the-art and outlook†
Sergei A.
Chernyak
 a,
Massimo
Corda
a,
Jean-Pierre
Dath
b,
Vitaly V.
Ordomsky
*a and
Andrei Y.
Khodakov
*a
a,
Massimo
Corda
a,
Jean-Pierre
Dath
b,
Vitaly V.
Ordomsky
*a and
Andrei Y.
Khodakov
*a
aUniversity of Lille, CNRS, Centrale Lille, University of Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, Lille, France. E-mail: vitaly.ordomsky@univ-lille.fr; andrei.khodakov@univ-lille.fr
bDirection Recherche & Développement, TotalEnergies SE, TotalEnergies One Tech Belgium, Zone Industrielle Feluy C, B-7181 Seneffe, Belgium
First published on 31st August 2022
Abstract
Light olefins are important feedstocks and platform molecules for the chemical industry. Their synthesis has been a research priority in both academia and industry. There are many different approaches to the synthesis of these compounds, which differ by the choice of raw materials, catalysts and reaction conditions. The goals of this review are to highlight the most recent trends in light olefin synthesis and to perform a comparative analysis of different synthetic routes using several quantitative characteristics: selectivity, productivity, severity of operating conditions, stability, technological maturity and sustainability. Traditionally, on an industrial scale, the cracking of oil fractions has been used to produce light olefins. Methanol-to-olefins, alkane direct or oxidative dehydrogenation technologies have great potential in the short term and have already reached scientific and technological maturities. Major progress should be made in the field of methanol-mediated CO and CO2 direct hydrogenation to light olefins. The electrocatalytic reduction of CO2 to light olefins is a very attractive process in the long run due to the low reaction temperature and possible use of sustainable electricity. The application of modern concepts such as electricity-driven process intensification, looping, CO2 management and nanoscale catalyst design should lead in the near future to more environmentally friendly, energy efficient and selective large-scale technologies for light olefin synthesis.
 Sergei A. Chernyak | Dr Sergei Chernyak received his MS (2011) and PhD (2017) degrees in physical chemistry at the Chemistry Department of Lomonosov Moscow State University (MSU). From 2018 to 2021, he worked as a senior researcher at the laboratory of Catalysis and Gas Electrochemistry (MSU) and in the beginning of 2021, he joined the UCCS laboratory as a post-doc in the frame of the “Take-Off” project. His research interests involve development of new nanostructures for catalytic hydrogenation of CO and CO2 and carbon nanomaterials chemistry including synthesis and investigation of carbon nanotubes, graphene nanoflakes, carbon dots and their composites. |
 Massimo Corda | Massimo Corda obtained his master's degree at University of Cagliari (Italy) in September 2020. Afterwards, he spent a semester at Humboldt University (Berlin, Germany) working on the synthesis of metal oxide catalysts for biomass conversion. In September 2021, he joined the UCCS-CNRS laboratory (Lille, France) as a PhD Student, under the supervision of Dr Andrei Khodakov and Dr Vitaly Ordomsky. His current work focuses on the development of new catalysts for CO2 conversion. |
 Jean-Pierre Dath | Dr Jean-Pierre Dath is currently Senior Program Manager at Total S. E., where he is acting as leader of the hydrogen R&D program. Before occupying this position, he worked for more than 25 years in the field of heterogeneous catalysis applied to fine chemicals, polyolefins, refining and base chemicals. He is the co-author of nearly 40 publications and 100 patents. |
 Vitaly V. Ordomsky | Dr Vitaly V. Ordomsky received his MSc (2006) and PhD (2009) degree from Moscow State University in the field of zeolite catalysis. He was a postdoctoral researcher in the Department of Chemical Engineering at the Eindhoven University in the Netherlands, working on biomass conversion. Afterwards, he received a permanent position as a researcher in 2013 at CNRS (France) in the Energy group of UCCS laboratory in Lille University working in C1 chemistry mainly focusing on Fischer–Tropsch synthesis. From 2016 to 2019, he was a CNRS researcher in E2P2L laboratory in Shanghai. His main research field is development of new nano-materials for sustainable catalysis. |
 Andrei Y. Khodakov | Dr Andrei Y. Khodakov obtained his PhD degree in 1991 from Zelinsky Institute of Organic Chemistry of USSR Academy of Sciences and Dr Sci (Hab.) in 2002 from University of Lille. In 1992–1999, he worked as a postdoctoral researcher in academic and industrial laboratories in France, UK and USA. From 1999, he is a researcher of the National Centre for Scientific Research (CNRS) in France. Since 2017, he is a CNRS Senior Research Director in the UCCS-CNRS Laboratory in the University of Lille and Centrale Lille Institute. His research addresses catalytic synthesis of fuels and chemicals from renewable and fossil resources. |
1. Light olefins: general information, demand and feedstocks
Light olefins (LO; ethylene, propylene and butylenes) are important feedstocks and platform molecules for the chemical industry (Fig. 1). The global olefin market is set to grow at a rate of 4.5% from 2020 and is expected to reach USD 329.30 Billion by 2028.1 The current driver of LOs demand is the high penetration of polymers, plastics and fibers in electronic devices and vehicles.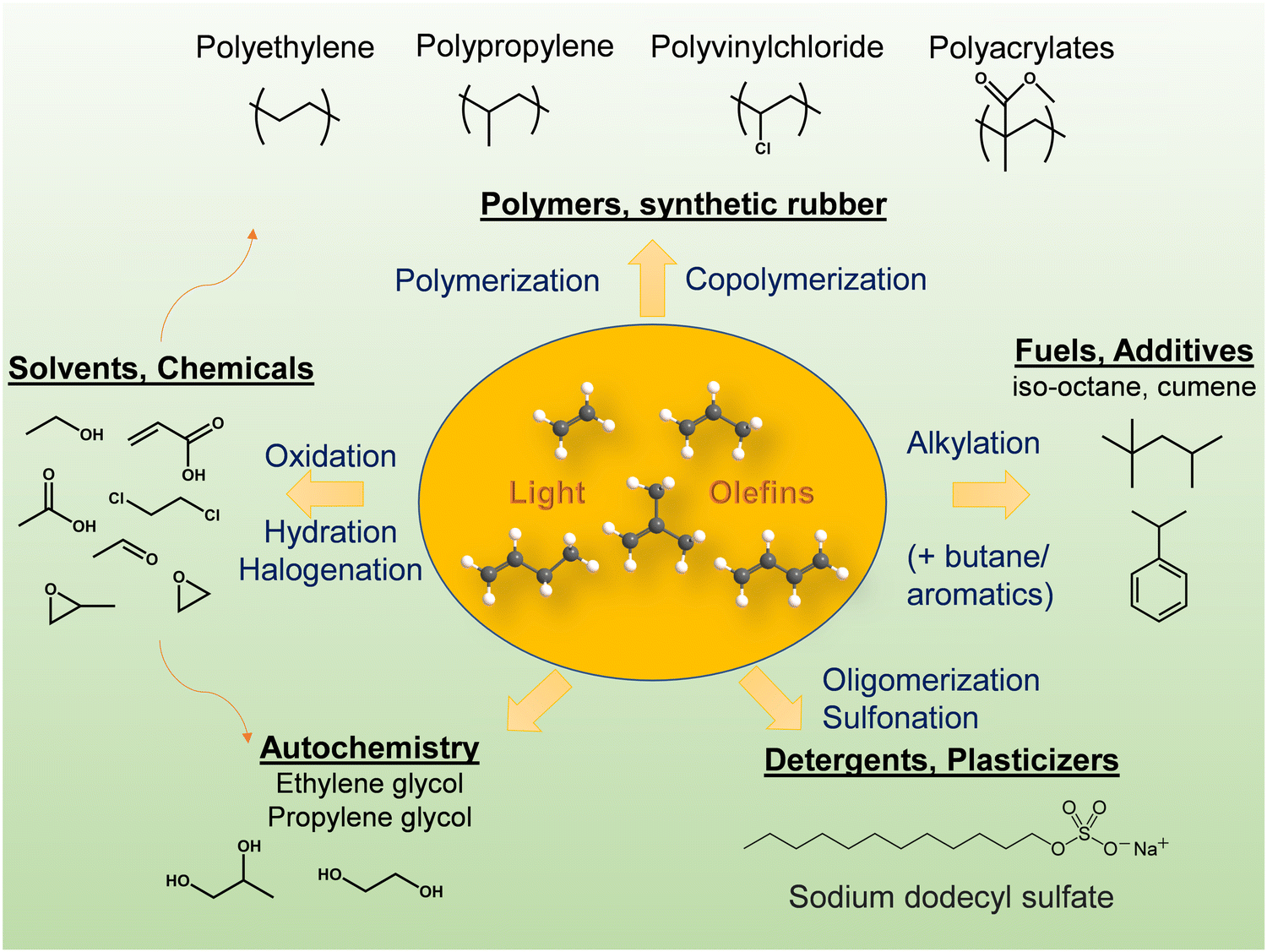 | ||
| Fig. 1 Light olefins, platform molecules for the chemical industry. | ||
Ethylene is the largest-volume chemical and has a broad range of applications and end-uses. It is often seen as a barometer of the performance of the whole chemical industry. Ethylene is employed in manufacturing polyethylene, ethylene oxide, ethylene glycols, ethanol, ethoxylates, PVC, ethylbenzene, ethanolamine, etc. The ethylene-based products are used in the following industries: packaging, transportation, electrical/electronic, textile, construction, consumer chemicals, metals, coatings, adhesives, food industry, agriculture and medicine.
The world consumption of propylene is over 100 million tons and is expected to raise 4% per year.2 Propylene is mostly consumed for manufacturing polypropylene.3 The latter is one of the most versatile polymers due to a combination of good mechanical and chemical properties. Propylene is also used to produce acrylonitrile, propylene oxide, alcohols, cumene and acrylic acid (Fig. 1). The chemical industry is experiencing a “propylene gap” – an excess of demand for propylene over its production.3
The main uses of butylene are in the production of gasoline additives and polybutadiene rubber. High-purity butylene is used as a co-monomer for different sorts of polyethylene. About 15% of butylenes are converted to specific chemicals: butadiene, butylene oxide, acetic acid, vinyl acetate, isobutylene, methyl t-butyl ether (MTBE) and solvents.
The literature shows a sharply growing interest in the synthesis of LOs, which can be produced from a diversity of renewable and fossil raw materials. In this review, we will consider the main groups of LO synthesis technologies depending on the feedstock (Fig. 2).
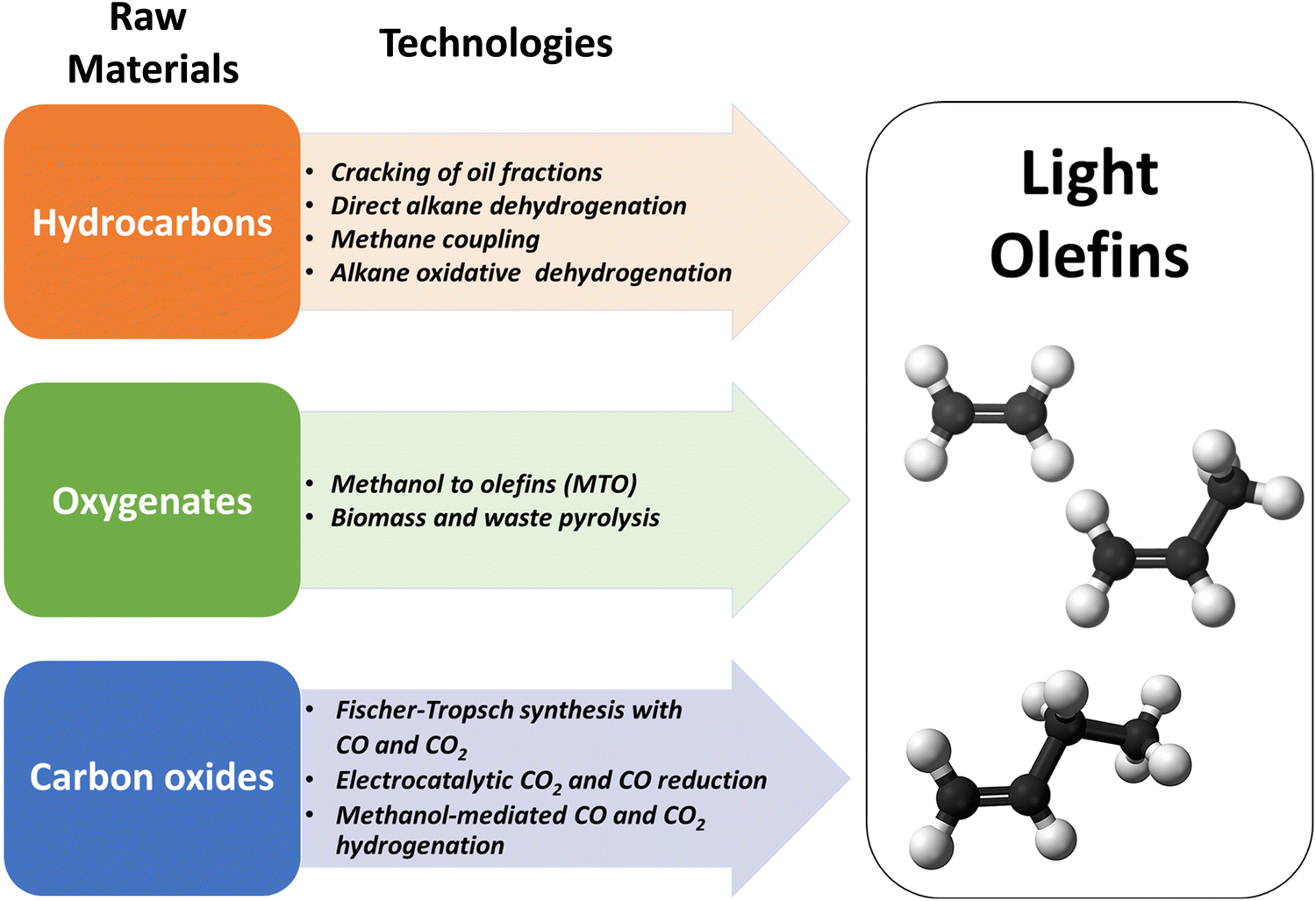 | ||
| Fig. 2 Raw materials and technologies for the synthesis of LOs. | ||
First, hydrocarbon feedstocks such as oil, natural gas or shale gas can be employed for LO synthesis. The hydrocarbon-feedstock based LO synthesis technologies involve steam-cracking of light oil and gas fractions, fluid catalytic cracking (FCC), methane coupling and direct or oxidative dehydrogenation of propane and butane. Steam cracking and FCC currently account for 81% of propylene production.4
The second type of feedstocks for LO synthesis involves oxygenates such as biomass, organic waste or methanol. The olefins are produced from these feedstocks either via pyrolysis or methanol-to-olefins (MTO) process. Finally, in recent years, CO and CO2 have emerged as important raw materials for the synthesis of fuels and chemicals. CO is a major component of syngas, which is produced by gasification of alternative fossil (heavy oil, shale gas, coal) and renewable feedstocks (biomass, organic and plastic waste). Monetization of CO2, emitted by power plants and other industrial facilities, is principally driven by environmental concerns and sustainability. The newly developed technologies for LO synthesis involve CO or CO2-based Fischer–Tropsch synthesis or methanol-mediated CO/CO2 hydrogenation. In addition to the thermochemical and thermocatalytic processes, electrochemical conversion of CO2 to LOs, and in particular to ethylene, has been attracting much attention.
Previous reviews about LO synthesis have only focused on specific routes such as Fischer–Tropsch synthesis,5–9 methanol-mediated routes,7–11 direct dehydrogenation,12–14 oxidative dehydrogenation,14–16 cracking,4,17–19 methanol-to-olefins process20,21 or on specific materials (e.g. zeolites22). To the best of our knowledge, no attempt has been made in the past to examine and compare in a single review article, a wide range of LO synthesis reactions involving diverse raw renewable and fossil materials and technologies. This review includes not only a discussion of the literature mainly published in the last 10 years but also a comparative analysis of LO synthesis routes from various fossil and renewable feedstocks using several quantitative criteria such as selectivity, productivity, stability, temperature, pressure, technological maturity and sustainability.
2. Light olefin synthesis from hydrocarbons
2.1. Cracking of hydrocarbons
Steam cracking (SC) and fluid catalytic cracking (FCC) are currently the most common routes for LO production. The first commercial plant for ethylene and propylene production has been built by Union Carbide Corporation in Clendenin in 1925. Nowadays, these two technologies produce together worldwide about 80–90% of ethylene and propylene.4,19,23–26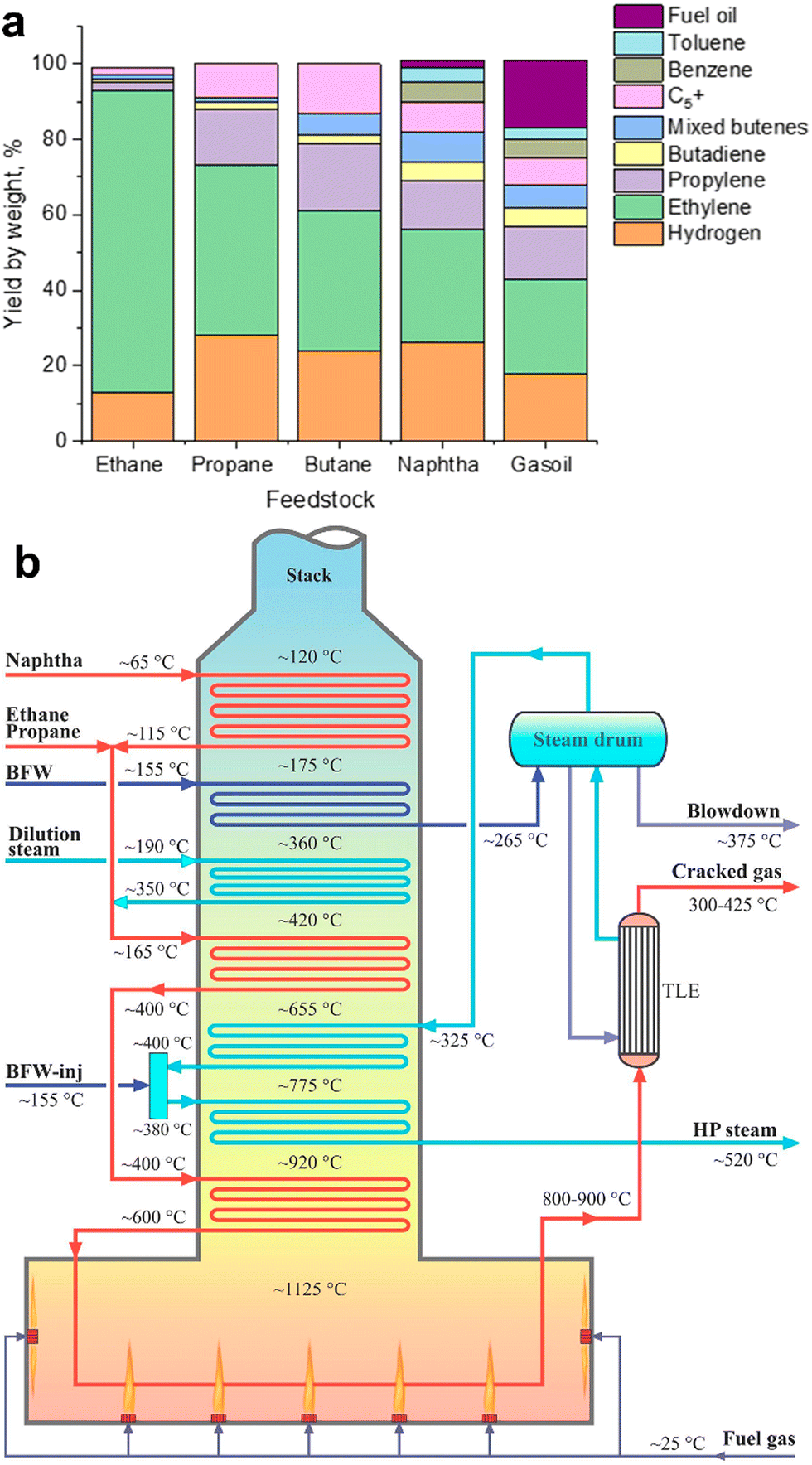 | ||
| Fig. 3 Hydrocarbon steam cracking: yields of various petrochemical fractions from a steam cracker using ethane, propane, butane, naphtha and gasoil as feedstock (a);4 schematic diagram of a typical cracking furnace in the SC plants (b). Reprinted from ref. 19 Copyright (2019), with permission from Elsevier. | ||
Propylene is a by-product of ethylene production in SC.30 The yields of ethylene and propylene vary31 between 24–80% and 1.5–18%, respectively, depending on the feedstock and operating conditions (Fig. 3a). If ethane is used as feedstock, only 0.019 t of propylene is produced for every ton of ethylene made.32 Higher amounts of propylene are produced from SC of propane, butane, naphtha and longer-chain hydrocarbons. Operating conditions also affect the selectivity of this technology. Propylene and butylene are produced at lower SC temperatures, while ethylene production requires higher temperatures. A recent trend in steam crackers has been the use of shale gas as feedstock. This leads to even higher SC temperatures, higher ethylene yields, with less propylene produced as a by-product.
The conventional naphtha-based SC plants are composed of four main sections: cracking furnaces, quench sections, compression, chilling units and separation columns. The cracking furnace is the heart of SC plants. The main parts of a cracking furnace29 are the radiation section including numerous burners on the walls and bottom of the firebox, tubular reactors, convection section, heat transfer facilities and stack (Fig. 3b). The heated feedstock stream enters the tube reactors in the furnace, where the burners provide a temperature up to 1100–1200 °C. The reactions take place in a fraction of a second (0.4–1.0 s) in the tubular reactors. The heat of flue gases is transferred to the feed/steam, high-pressure steam and boiler feed water (BFW).
The SC process is conventionally conducted without any catalyst. The possible use of catalyst in the SC process allows reducing the reaction temperature and energy costs33 by 10–20%. Catalytic SC operates at the temperature range of 750–780 °C, lower than the thermal process by about 50–100 °C. In addition, catalyst-assisted SC gave34–36 higher propylene and aromatics yields. R&D on catalytic SC of hydrocarbons to LOs started34 in the late 1960s. Three types of catalysts were tested in SC, i.e., acidic catalysts, basic catalysts and transition metal oxide catalysts. Zeolites with strong acidity and high surface area (ZSM-22, ZSM-5, MOR, ZSM-48, etc.). have been considered. Bimetallic and trimetallic oxide catalysts were also reported34 for naphtha cracking. The redox catalysts such as V–Sr–Al, Mo–Li–MgO, CaMn–NaW, SrMn–NaW, Li/Mg/Mo, Li/Mg/Bi, BaFeAl and NaW/BaFe, showed higher yields of LOs. Note that large amounts of steam and high temperature deactivate conventional cracking catalysts. That is the reason why no commercial application of catalytic SC has been reported so far.37
Ultra-stable Y (USY) zeolites with higher Si/Al ratios are typically used as catalysts in the FCC process. They are obtained by hydrothermal treatment of the parent Y zeolite.25 USY zeolites exhibit improved stability and high cracking activity due to the generation of strong acid sites.
On average, each catalyst particle goes through about 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles,40 before it is discarded. A given particle may spend 1 to 3 months in the unit.
000 cycles,40 before it is discarded. A given particle may spend 1 to 3 months in the unit.
It was uncovered41 in the 1980ies that the addition of ZSM-5 zeolite to the Y-zeolite based FCC catalysts boosted the selectivity to propylene and reduced the selectivity to naphtha olefins. The amount of ZSM-5 used in the FCC ranges from 0.2 to 3 wt% of the USY catalyst. The addition of the ZSM-5 zeolite increases the propylene yield from about 5 wt% in conventional FCC units to about 20 wt% depending on the type of feedstock.42–45 The advantage of the ZSM-5 additive relative to Y-zeolite, is its capacity to crack naphtha olefins to propylene. Besides ZSM-5 zeolite, several other high-silica zeolites (MCM-22,46 ITQ-1347 and others25) have been considered as additives for the enhancement of LO selectivity.
The LO synthesis from heavy oil fraction in FCC is an example of a bifunctional process involving two types of acid sites. First, heavy feeds (e.g., vacuum gas oil and residua) crack to naphtha olefins over large pore Y-zeolite. Then, the produced naphtha olefins diffuse into the ZSM-5 zeolite and crack into the C2–C4 olefins (Fig. 4), mostly propylene. Because of the low reactivity of the propyl carbenium ions, propylene does not convert by hydrogen transfer to propane as easily as butenes. High propylene yields in FCC result48 therefore, from naphtha over-cracking and fast diffusion of target LOs. The zeolite pore diameter plays an essential role49 in minimizing hydrogen transfer reactions. Smaller pores of the ZSM-5 zeolite hinder bimolecular reactions and, consequently, enhance cracking relative to hydrogen transfer and thus, the formation of LOs.
 | ||
| Fig. 4 Production of aromatics vs. LOs in FCC over Y and ZSM-5 zeolites (a) Effect of the incorporation of 2.5 wt% of pentasil at USY FCC Ecat on yield of paraffins and olefins at a conversion level of 60 wt%. An additive containing 10 wt% ZSM-5 zeolite was used (b).24 | ||
The FCC process requires continuous catalyst regeneration and removal of deposited coke by oxidative treatments. The regeneration often results in zeolite dealumination and in a decrease in the amount of Brønsted acid sites. The exothermic coke oxidation in the FCC catalyst regeneration provides energy for the endothermic cracking reactions.
Promotion of ZSM-5 zeolite catalyst with several elements may improve its catalytic performance. For instance, it has been reported that the addition of phosphorous compounds enhanced50 the desired stability and catalytic activity in the cracking of C4 alkenes and naphtha into propylene and ethylene. Increasing the LOs yield has been also achieved25 by incorporation of alkali earth metals in the ZSM-5 zeolite. The transition metals added to ZSM-5 may facilitate25 dehydrogenation of alkanes to alkene and their cracking to LOs.
2.2. Direct alkane dehydrogenation
Direct dehydrogenation (DDH) of light alkanes is an industrially-scaled family of reactions, which occur over platinum-based or chromium-based catalysts (Oleflex and CATOFIN processes). Propane, iso-butane and n-butane are typically used as feedstocks to obtain corresponding olefins.51 The most desirable product is propylene due to the extremely high global demand for this compound.52 This is why, most publications in this field have focused on propane DDH. The ethane dehydrogenation has not been commercialized due to the economical irrelevance, which comes from the competition with the SC process.13 Despite the commercial realization, DDH still faces several serious issues: catalyst degradation during the reaction/regeneration, high price of Pt and ecological/health risks originating from the Cr-based compounds and chlorine from the Pt precursor. These problems motivate the design of highly selective and stable systems and development of new catalysts, which may compete with the industrially-used Pt-based and Cr-based counterparts.In recent years, several detailed reviews focused on DDH have been published.13–15 Thus, in this paper, we will briefly describe the trends, focusing on the most recent publications, with a particular focus on propane DDH.
2.2.1.1. Pt-Based catalysts. Since the main challenges in propane DDH over Pt-based catalysts are long-term operation and high production rate, the majority of studies have addressed the stability and selectivity-conversion correlations. Since the catalysts based only on platinum suffer from cracking and insufficient activity, different promoters have been applied. For example, in the Oleflex process, tin is used to increase the selectivity of Pt catalysts. Recently, Motagamwala52et al. presented synthesis of a highly active and stable Pt–Sn catalyst, which involves atomic mixing of the active components. Intermetallic ultra-small (mainly 0.5–2 nm) nanoparticles supported on SiO2 exhibited thermodynamically limited propane conversion as well as high stability (Fig. 5a and b). Moreover, a propylene yield of ∼10% beyond the thermodynamic limit was achieved at 250 °C over Pt–In catalyst in the electro-assisted process.53 The intermetallic alloy-based Pt–Zn catalysts have exhibited high stability in propane DDH (Fig. 5c and d).54 The authors revealed the unique activity of single-site [PtZn4] nanoclusters and highlighted the crucial role of Zn in preventing cracking and methane formation. It was also pointed out that the SiO2 support was more preferable than alumina. A weaker interaction between silica and active metals enhances the formation of intermetallic phases and increases the catalytic activity. The Pt1Zn1 intermetallic nanoparticles on silica zeolite with MFI structure showed55 the best stability among other catalysts. The optimality of this particular composition of nanoalloy crystals was further proven by Ingale56et al. using atomic layer deposition of ZnO on the SiO2. Encapsulation of sub-nanometric Pt–Zn alloy clusters inside the silicalite allowed achieving stable operation at 550 °C for 13
000 min without hydrogen addition to the inlet.57 The stability of Pt–Zn system has been enhanced by the localization of Zn species on the surface of Pt nanoparticles, which prevented Pt aggregation.58
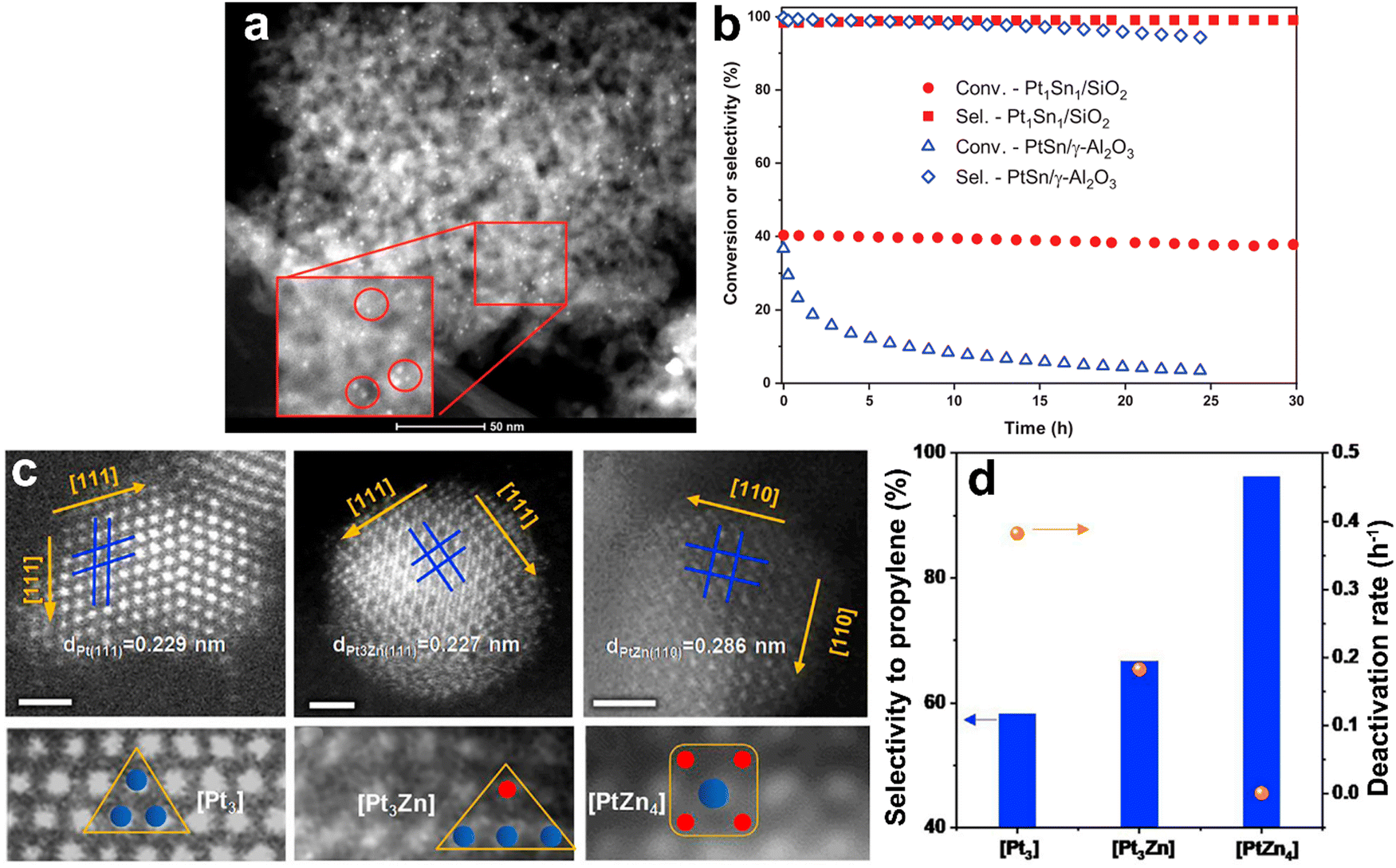 | ||
| Fig. 5 PtSn/SiO2 and PtZn/SiO2 intermetallic catalysts for propane DDH: bright-field TEM image of Pt–Sn nanoparticles formed on SiO2 (a), catalytic performance using pure propane and comparison with benchmark alumina-supported catalyst (b). From ref. 52 reprinted with permission from AAAS. AC-HAADF-STEM images of [Pt3], [Pt3Zn], and [PtZn4] ensembles of PtZn/SiO2 catalyst (c) and corresponding catalytic performance of these ensembles (d). Reprinted from ref. 54 Copyright (2021), with permission from Elsevier. | ||
An ultra-stable catalyst was developed for propane DDH using a double decoration of PtGa active sites by Pb and Ca: almost no deactivation was observed for 1 month.59 Pb blocked the Pt3 sites, which were proposed to be labile to deactivation, while Ca had an electron-donor function and afforded an electron-enriched single-atom-like Pt1 site. Combined DFT and experimental analysis of Pt–Ga system revealed the role of Ga as a structural promotor, which reduced the Pt interaction with support and increased both the Pt site isolation and mobility of surface atoms.60 Such effects led to the significant enhancement of the catalyst activity, selectivity, and stability. Gallium facilitated the activation and heterolytic dissociation of C–H bonds, while cerium suppressed the Pt sintering by increasing metal–support interactions.61 The calcium addition blocked strong acid sites and enhanced the Ga reducibility and formation of Pt–Ga alloy.62
The effect of support on the performance of Pt-based catalysts was studied and the important role of silanol “nests” for the alloying promotion of Pt catalysts promoted with rare-earth metals was observed.63,64 Covering of the Pt-based catalysts by the atomic layers of silica or alumina positively affected the stability and activity by preventing the active phase from sintering and coking.65,66 Moreover, the SiO2 thin layer allowed decreasing the Pt loading to 0.1 wt% without any loss of activity. In the case of the alumina support, the function of unsaturated Al3+ sites was relevant to the anchoring of Pt atoms by bonding them in the Al–O–Pt fragments and enhancing the sintering resistance of Pt–Sn2 clusters.67 Moreover, unsaturated alumina contained a lot of oxygen vacancies, which reduced the activation barriers.68 At the same time, the effect of strong interaction of Pt-based particles with the zeolite support was not always positive: encapsulation of Pt-clusters inside the NaX zeolite matrix strongly passivated the active phase and hindered the propane activation compared to the metal particles localized on the outer surface of the zeolite.69
2.2.1.2. Non-Pt-based catalysts. The development of new active platinum-free catalysts is another trend in propane DDH. First, other precious metals were tested. The new RhCu single-atom alloy was found active in propane DDH.70 Despite the Rh itself does not catalyse this process due to the strong coking, its atomic dispersion over copper nanoparticles resulted in the synergy effect: single-atom distribution prevented the over-dehydrogenation and bonding with carbon atoms of the Rh, while the latter greatly enhanced the stability of copper against sintering. As a result, selectivity to propene of 100% was achieved at a higher reaction rate than on the benchmark Pt/Al2O3 catalyst. Another alternative system was proposed by Chang71et al., where Ir single atom doping boosted the Ga2O3 activity via tuning the Lewis acid–base interactions on the catalyst surface. The promotion of Ru catalyst with phosphorous led to the formation of the separate Ru clusters and lower energy barrier for propane DDH: the synthesis of RuP active sites supported on silica sharply increased the catalyst stability.72
Second, there is an increasing focus on propane DDH catalysts without precious metals. The most popular systems are Mo, Ti, Cr, V, Zn, Ga, Sn or Co, which are used without or with promoters.14 Ni, Si, Sn, Na, and N-doped carbon were used as promoters to enhance the catalyst stability, activity and selectivity.73–83 Molybdenum-based species were proposed as active sites for propane DDH. For example, the molybdenum nitride single crystals with porous surfaces were considered as new perspective systems.84 Such catalysts showed a high density of Lewis acid sites on the top layer attributed to the Mo–N1/6 and Mo–N1/3 species, which effectively activated C–H bonds without the destruction of carbon-carbon bonds at 500 °C. A propane conversion of 11% and ∼95–97% propylene selectivity were observed. In another study, machine learning predicted the activity of NiMo catalyst, which was then proved experimentally.85 Titania containing oxygen vacancies were also confirmed as the active phase for DDH of propane, while their propane conversion and stability can be further improved by Sc promotion.77,86 The iron-based catalyst supported on alumina after the sulfidation of the surface87 showed high conversion up to 52% and was extremely selective to propylene (∼99%). Finally, recent studies showed the great potential of Zn-based catalysts supported on silicalite.88 The authors found that the method of support preparation was crucial for the formation of ZnOx active species and the proposed approach provided flexibility for adjusting the active site concentration and structure.
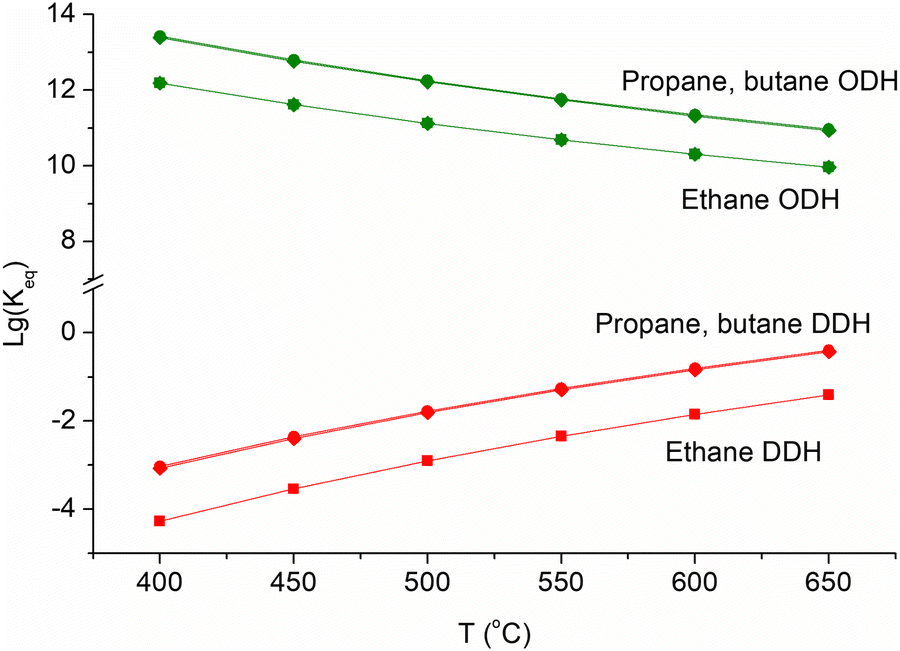 | ||
| Fig. 6 Equilibrium constants of direct (red) and oxidative (green) alkane dehydrogenation processes. | ||
The catalysts for ethane DDH are usually similar to those for propane and can be also divided into Pt or Cr-based and other systems. Several attempts for the fixation of active sites of the Pt- and Cr-based catalysts inside the zeolite structure were realized. Single-atomic Pt distribution inside the 2D-layered Ti–Si-zeolite nanosheets and embedding of nanoclusters in HZSM-5 resulted in significantly better stability, higher activity and ethylene selectivity.91,92 Application of MFI silicalite as support allowed obtaining highly active and stable Cr-based catalysts. Their catalytic performance was attributed to the CrII(O–Si![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) )2 sites identified by operando XAS.93 New catalyst formulations were proposed for ethane DDH. The performance of single crystals of vanadium oxides, nitride and cobalt-based catalysts supported on ZSM-5 and CeO294–96 was promising. The Ga-doping of alumina led to the formation of active Ga-Al sites, while DFT calculations predicted the high activity of NiCu single-atom alloys.97,98
)2 sites identified by operando XAS.93 New catalyst formulations were proposed for ethane DDH. The performance of single crystals of vanadium oxides, nitride and cobalt-based catalysts supported on ZSM-5 and CeO294–96 was promising. The Ga-doping of alumina led to the formation of active Ga-Al sites, while DFT calculations predicted the high activity of NiCu single-atom alloys.97,98
It is also important to mention the electro-assisted approach to ethane DDH, which allows the achievement of higher ethylene yields at lower temperatures. For example, an ethane conversion of 75.2% and an ethylene selectivity of 100% were observed using the proton-conductive NiCu-doped Nb–Ti–Mn–O electrodes at 700 °C.99 In another study, an ethylene yield of 26.7% was achieved at only 550 °C over the PtGa–ZSM-5 and Pr–Ba–Fe–Mo–O ceramic textile anodes.100 Theoretical calculations made by the authors showed that such an approach improved energy efficiency by ∼50% compared to conventional ethane SC.
Dehydrogenation of n- and iso-butanes to butenes is a commercial route and has been realized in the C4 and C3/C4 OLEFLEX UOP processes. Several studies focused on Pt-based catalysts have been published. Chen101et al. compared the efficiency of different types of Pt clusters supported on a hybrid non-diamond-graphene base. They demonstrated that the most active form was Pt3 clusters, which showed an ultra-high selectivity of ∼99%, conversion of ∼35% and, hence, a high reaction rate at a low synthesis temperature of 450 °C compared to single-atom and nanoparticles (Fig. 7). Application of Ti-silicalite and LaAlO3-perovskite as supports as well as promotion by MgO and CaO increased the dispersion and sintering resistance of Pt nanoparticles and suppressed side reactions and coking.102–104 Promotion with potassium was found to improve the efficiency of Cr–Al catalysts in isobutane DDH. In the activated catalysts, potassium preferably interacts with alumina and modifies the average dispersion of Cr3+Ox species. Higher conversion and selectivity were observed in the potassium promoted catalysts.105 Regarding other active systems, the Mo4+ species in the alumina-supported catalysts were identified as the most active sites for butane and isobutane DDH, while careful support design is needed for the active site stabilization.106,107
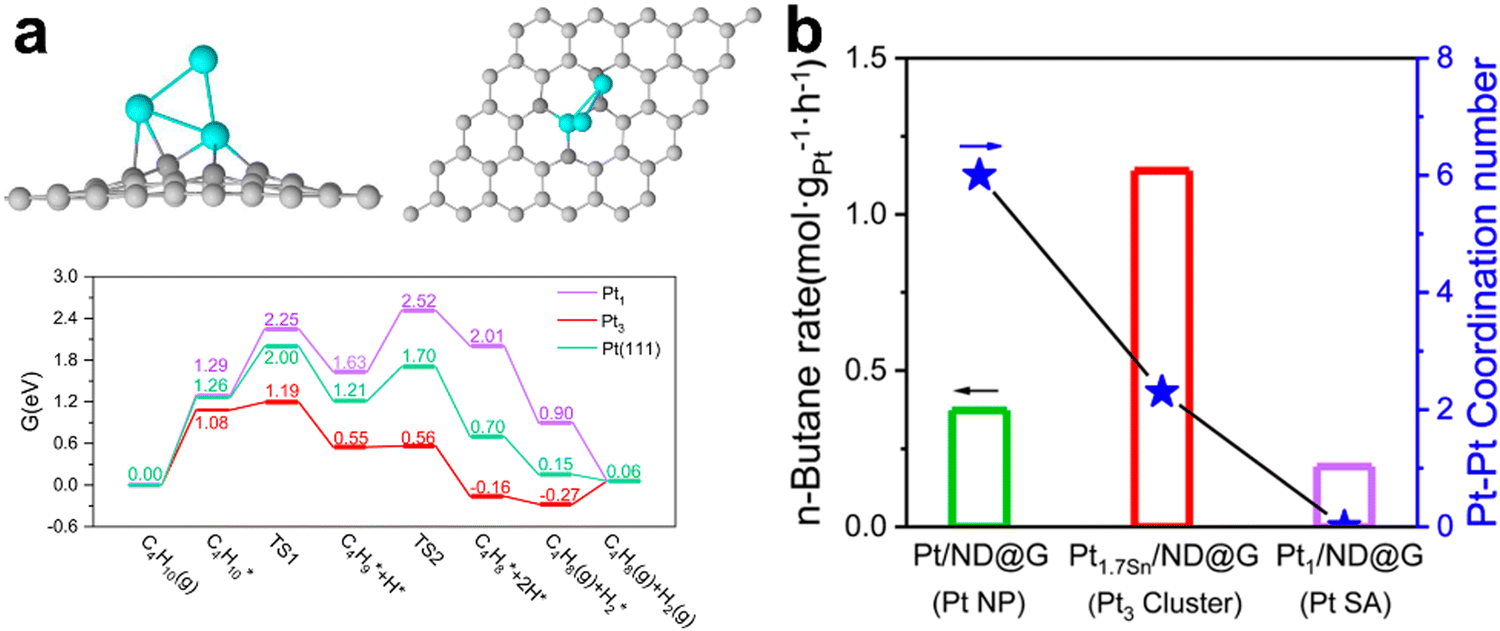 | ||
| Fig. 7 DFT calculations for the structure of Pt3/graphene catalyst and energy profile of butane dehydrogenation to 2-butene of n-butane DDH over different platinum configurations (a) and the conversion rate of n-butane over the Pt catalysts with different Pt–Pt coordination number (b).101 | ||
2.3. Alkane oxidative dehydrogenation
Oxidative dehydrogenation (ODH), which is carried out in the presence of an oxidising agent, improves the thermodynamics of light alkane dehydrogenation (Fig. 6). The catalyst stability can be also improved, since coke deposition can be slowed down in the presence of an oxidising agent. The deposited coke can be removed by oxidation. Different oxidising agents can be used. Air and oxygen are commonly used for alkane ODH. The presence of oxygen increases the conversion of alkanes, but it can lower the LO selectivity due to the abundant release of unwanted carbon dioxide and carbon monoxide.15 During the past two decades, the use of “soft” oxidants such as CO2, N2O, and even sulphur108–112 has been considered most promising, since it does not lead to hydrocarbon overoxidation, implies the rational utilization of the most ecology-risked greenhouse gases and prevents the formation of explosive mixtures.113 Somewhat lower LO selectivity of ODH relative to DDH is due to higher reactivity and higher probability of oxidation of LO compared to light alkanes in the presence of oxidizing agents.Chemical-looping ODH (CL-ODH) is one of the possible routes to overcome the overoxidation of alkane ODH intermediates and to minimize the production of carbon oxides. This strategy is based on the Mars-van Krevelen mechanism, which commonly operates in numerous reactions of partial oxidation. In the chemical looping, the reaction is divided into two parts: first, alkane ODH is occurred by lattice oxygen of reducible metal oxides and, second, the catalyst is regenerated via the oxidation by air or CO2.114,115
The CO2-assisted ODH often coincides with the unwanted side process of dry reforming, which is prominent at temperatures higher than ∼500 °C. CO2, as O2, may remove carbon from the catalyst by reverse Boudouard reaction: CO2 + C ↔ 2CO. The CO2-assisted ODH compared to DDH, promotes higher conversion at the same temperature or enables the same conversion at the lower temperature.113
The literature concerning the conventional and CO2-assisted ODH of ethane, propane, and butanes up to 2020 was considered in detail in the recently published reviews.14,15,116,117 We will focus only on the very recent publications in order to reveal the “hottest” trends in these areas. We will discuss new catalytic systems including non-metallic ones, new approaches for enhancing the LO selectivity of metal oxide catalysts for O2–ODH by nanoscale material design, chemical looping and CO2-assisted ODH process.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and bridged oxygen M–O–M groups catalyzing the conversion of alkanes to LOs, and electrophilic ones such as M–O–O–M and M–O2− responsible for deep oxidation to carbon oxides.117 Both types of sites are usually present on the surface simultaneously. Operando studies of propane O2-ODH showed112 that the rate-determining step of propane ODH was iso-propoxide formation on the surface of the V–Zr–O catalyst. The decomposition of iso-propoxide resulted either in propylene or in oxidation to acetone, followed by oxidation to acetate/formate and, finally, to COx.
O and bridged oxygen M–O–M groups catalyzing the conversion of alkanes to LOs, and electrophilic ones such as M–O–O–M and M–O2− responsible for deep oxidation to carbon oxides.117 Both types of sites are usually present on the surface simultaneously. Operando studies of propane O2-ODH showed112 that the rate-determining step of propane ODH was iso-propoxide formation on the surface of the V–Zr–O catalyst. The decomposition of iso-propoxide resulted either in propylene or in oxidation to acetone, followed by oxidation to acetate/formate and, finally, to COx.
2.3.1.1. Non-metallic catalysts. Non-metallic catalysts based on the boron nitride, isolated boron and carbon nitride catalysts were discovered a few years ago. Such systems prevent olefin oxidation and show extremely high selectivity to LO up to more than 90%.118 Application of non-metallic compounds gave nearly 80% selectivity to propylene at the 14% conversion already at 490 °C. The first studies of hexagonal boron nitride (h-BN) and BN-nanotubes have proposed a possible mechanism for propane activation. The >B–O–O–N< species were assumed to be the active sites for the reaction.118 At the initial stage, BN is oxidized by oxygen, then propane gives hydrogen to these active sites forming the nitroxyl radical on the surface and propyl radical. Then, nitroxyl stabilizes the propyl radical and prevents its further oxidation to carbon oxides, while the second hydrogen from the latter forms the >B–OH group and gives propylene and water. Recent studies debated the precise mechanism of ODH over BN. Several authors now propose the double-localized combined surface-gas phase reaction pathway, which includes activation of propane by both the catalyst surface and radicals in the gaseous phase.119–121
Several approaches have been proposed to further increase the activity and productivity of h-BN. For example, the application of fibrous silica as the support further enhances the h-BN catalytic performance. It was assumed that higher h-BN dispersion facilitated higher activity or productivity. The active sites in such a system are surface B2O3 nanoclusters. In some cases, however, higher dispersion of active phase can decrease the LO selectivity.122,123 Another approach for the optimization implies N2 plasma-assisted treatment of h-BN (Fig. 8a) resulting in three-boron active sites. Consequently, a propane conversion of 26% and a propylene selectivity of 89.4% were achieved at 520 °C.124 Crystallinity was found to be an important parameter for the h-BN performance and its higher degree enhanced O2 binding. A selectivity of 82% at a conversion of 42% was obtained.125 Moreover, use of BPO4 as a nonmetallic oxygen vacancy source for the h-BN@BPO4@h-BN sandwich-like structure resulted in O-vacancies, which increased the propane adsorption. At the same time, the confinement of BPO4 between the two h-BN layers may weaken the propylene adsorption.126 Such configuration also changes the mechanism of the process from Eley–Rideal to Langmuir–Hinshelwood.
 | ||
| Fig. 8 New approaches for the design of propane ODH catalysts: (a) plasma-assisted generation of N-defects and its evolution into “BOx” during the ODH reaction of propane over BN catalyst. From ref. 124 Reprinted with permission from John Wiley and Sons. (b) Transition state structure of –B[OH⋯O(H)–Si]2 reacting with oxygen and propane in boron-based system. From ref. 127 Reprinted with permission from AAAS. | ||
A series of recent studies optimized the unique properties of boron-based catalysts supported on silica, zeolite, or silicon matrix. Lu128et al. found high productivity of boron oxide supported on silica compared with h-BN catalyst and such a catalyst showed activity even at 405 °C. It was proposed that, analogously to nitroxyls in BN, oxidized boron species on the surface acted as stabilizing agents for the alkoxide intermediates preventing their further oxidation. Nevertheless, the structure of the active sites for B–Si-based catalysts is also debatable. The B[OH⋯O(H)–Si]2 fragments in the isolated boron clusters supported on zeolite framework (Fig. 8b) were found to be active and stable due to the prevention of hydrolysis and leaching of surface boron oxide species. The stabilization of active sites was due to the presence of the B–O–SiOx linkage127 detected using 11B NMR.129 Despite the differences in the exact active centre structure, the B–OH groups seem to be the most probable species responsible for the propane activation. This was also pointed out in the case of boron-doped silicon particles synthesized by the laser pyrolysis technique.130 A rather high propane conversion of ∼44% and a selectivity to propylene and ethylene of ∼80% were achieved on the boron/zeolite catalyst.127
Another type of metal-free system is carbon-based nanomaterials, such as graphitic carbon nitride or N-doped mesoporous carbon.131,132 The edge-localized carbonyl groups were proposed to be active centres in carbon nitride for propane activation. A combined C2 + C3 olefin selectivity of 89.6% at the conversion of 12.8% was measured on this catalyst at 500 °C.131
2.3.1.2. Metal oxide catalysts. The catalytic systems based on the metal oxides can be divided into several groups: noble metals supported on oxide matrix; reducible metal oxides of Mo, V, Co, and Ni; non-reducible oxides of the rare-earth elements; perovskites; alkali- and alkaline-earth metal oxides.14,15 As it was mentioned before, the main disadvantage of such catalysts is the formation of electrophilic active sites, which oxidize the hydrocarbons to carbon oxides. The latter fact was confirmed by recent publications, which concluded that the over-oxidation of the catalyst surface led to the formation of electrophilic centers. A less intensive oxygen treatment of the catalyst surface, achieved both by catalyst design and by reducing the oxygen concentration, promotes the dominance of nucleophilic sites.133–136 This phenomenon is independent of the catalyst structure and works for different systems.
Propane ODH can be also performed using bifunctional catalysts. A tandem In2O3–Pt/Al2O3 catalyst was proposed, where platinum nanoclusters were responsible for the DDH of propane, while indium oxide took part in the oxidation of released hydrogen.137 The authors covered Pt/alumina catalyst by indium oxide via atomic layer deposition and reached an yield of >30% by the careful material design. Overcoating led to the performance similar to the non-platinum system showing hindering of the reagents' access to the metallic surface (Fig. 9). Moreover, indium coating prevented platinum from sintering and increased the catalyst stability.
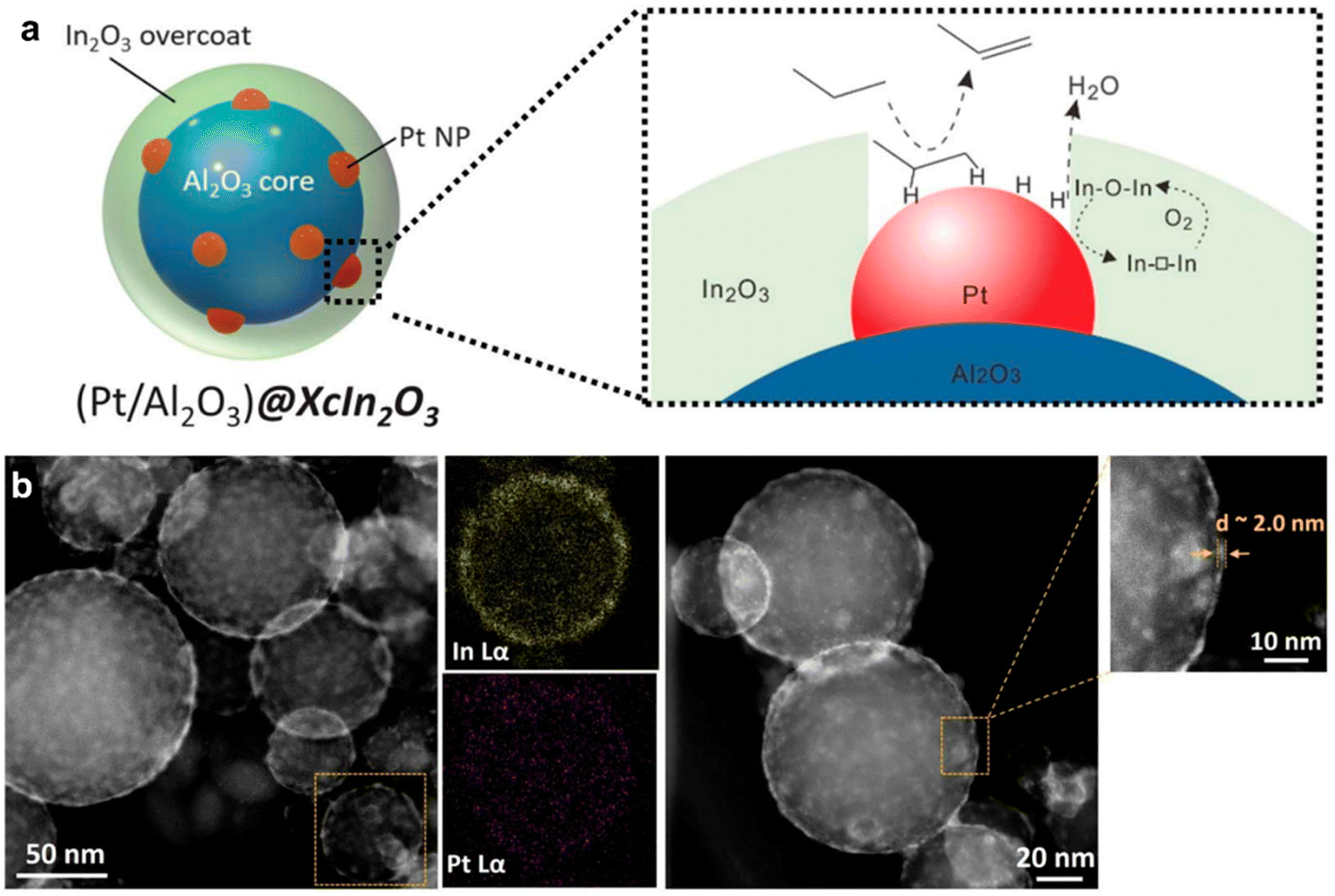 | ||
| Fig. 9 Tandem catalyst design for propane ODH process and scheme of the reaction (a); STEM-EDX analysis of Pt/alumina@In2O3 catalyst (b). From ref. 137 reprinted with permission from AAAS. | ||
Two catalytic systems have attracted scientific interest for ethane ODH: M1 orthorhombic MoVTeNbOx mixed oxide and NiO-based catalysts. Both types allow a selectivity to ethylene up to 97% and a yield up to 75%. The benefit of the M1 MoVTeNbOx structure relative to other systems is lower oxidizing olefin ability at high alkane conversion. Consequently, a high yield of ethylene up to 75% and a productivity of up to 1770 gC2H4 kgcat−1 h−1 were achieved134,138 The selectivity and productivity of such catalyst can be significantly improved by tuning the V5+ surface concentration, using the conductive supports, modification by Nd, Mn, Ge or Ga or realizing the process under supercritical conditions.138–141 The latter makes it possible to achieve an ethylene productivity up to 288 gC2H4 kgcat−1 h−1 at as low temperature as 280 °C. This is crucial for MoVTeNbOx, since the M1 structure may strongly deactivate at temperatures higher than 360 °C.142,143 The careful design and modification of the NiO-based systems by tuning the surface oxygen concentration, addition of Sn, Ti and Nb oxides, or simple variation of NiO concentration can also help to achieve a higher olefin selectivity and a higher productivity.135,136,144–147 Finally, a technico-economic analysis of the ethane ODH indicates its potential for industrial implementation, competitive with the SC process.148
The Cr-based catalysts are well-known for the propane DDH process. They were initially used in the CATOFIN process, where several parallel reactors were packed with the Cr-alumina catalyst. The studies of Cr-based systems in CO2-ODH have focused on the effects of CO2 compared to DDH and O2-ODH, effects of different supports, Cr concentration, catalyst pre-treatment, and specific operating conditions. Xie149et al. studied zirconia-based CrOx catalysts and found that carbon dioxide slightly reduced the activity in the first 30 min of the process. After 2 h, the deactivation rate in the presence of CO2 was significantly lower and led to a much higher conversion: ∼60% compared to ∼30% without CO2. The in situ Raman investigation revealed that oligomeric and unsaturated isolated Cr3+ species were the main active sites for the CO2-assisted process. Carbon dioxide prevented both the Cr3+ reduction to Cr2+ and catalyst coking. At the same time, Michorczyk150et al. showed that the CO2 addition could bring both positive and negative effects to the propane dehydrogenation over Cr-based catalysts, depending on the support: the formation rate and selectivity to propylene increased over dealuminated Si-beta zeolite due to inhibiting the cracking of propane, while the alumina-enriched support enhanced CO2 strong adsorption and poisoning of the active sites. Polymeric and isolated Cr6+ sites were suggested to be responsible for the ODH activity in the silicalite-1-supported Cr-based catalyst. Interestingly, the polymeric species were more active but less selective to propylene.151 The activity of Cr6+ species was also demonstrated for ethane CO2-ODH over the Cr/TUD-1 mesoporous silica based catalyst.152 The authors of the latter work also revealed that the addition of O2 to CO2 enhanced stability. On the one hand, it facilitated coke removal and re-oxidation of Cr2O3 or CrOx components but on the other hand, it reduced the selectivity to ethylene because of over-oxidation.
Additional inhibition of carbon deposition can be also reached by promoting the catalyst by CaO, which increases both the conversion and the selectivity of ODH.153 Al-Mamoori154et al. proposed an interesting approach for CO2-ODH involving physically mixing of the Cr/H-ZSM-5 catalyst with a K–Ca oxide–carbonate mixture. The two-step process, in which the K–Ca component catches CO2, while the Cr catalyst dehydrogenates ethane, allowed achieving a selectivity to ethylene of 88% at the 25% ethane conversion. Conducting the propane CO2-ODH in supercritical conditions provided a three times higher propylene productivity on the Cr/SiO2 catalyst compared to the tests in regular conditions. The phenomenon is explained by a highly dense reagent mass-flow and an intensification of the process.155
A study of the effect of carbon support on the performance of the Cr-based catalyst in the CO2-ODH propane process showed that the porosity and surface graphitization of the support were the main factors responsible for the best activity and selectivity. The undoped graphene nanoflakes and oxidized carbon nanotubes, which possessed a developed mesoporous structure and the highest sp2/sp3-C surface ratio, showed a propylene yield up to 20%.156 The porosity effect in the silica support was studied for ethane CO2-ODH. The pore size of 7 nm was the best for an optimal conversion of ∼26% and an ethylene selectivity of 81%.157 The carbon-supported Fe–Cr catalyst was developed after the study of the activity of carbonized stainless-steel reactor and at 700 °C high ethane selectivity was achieved, up to 82% at a conversion of about 20%.158
Along with Cr-based catalysts, other non-precious metal binary compounds such as FeOx, NiO, VOx, MoCx, MoOx, Ga2O3, GaN, and ZnO exhibited promising activity in CO2-ODH. Lawson159–161et al. obtained a set of different oxide catalysts and their mixture supported on HZSM-5 using 3D-printing. This approach increased the activity and selectivity of catalysts. The Cr–V–Zr–Ga–Ox/HZSM-5 hybrid catalyst showed the best performance. The conversion was enhanced up to 40% and selectivity up to ∼95%. Moreover, the removal of CO2 from the alkane gas feed decreased both conversion and selectivity to propylene showing the importance of carbon dioxide. The ternary Pt–Co–In/CeO2 catalyst was developed recently for this process and showed promising results at 550 °C: the conversion of propane was about 50% at the 95% propylene selectivity due to the nanoalloys of Pt with Co and In and efficient reagent activation by metallic Pt–Co–In and ceria species.162
Photocatalytic conversion of ethane to ethylene can be mentioned separately. The Pd/TiO2 catalysts showed an ethylene productivity up to ∼614 μmol gcat−1 h−1. The mechanism of the process included activation of ethane molecules over TiO2 and conversion of CO2 to CO over electron enriched Pd nanoclusters.163,164 Both CO2-assisted ODH and DDH were realized in these works.
To summarize, CO2 can play multiple roles in the CO2-ODH process. The reaction of CO2 with hydrocarbons not only makes the process thermodynamically favorable but also inhibits methane formation.165 One of the most important roles of CO2 is the prevention of coking. CO2 may remove carbon from the surface by reverse Boudouard reaction. However, compared to O2, carbon dioxide is a much softer oxidant. Recent studies showed that the rate of the reverse Boudouard process should be too low under the reaction conditions (the rate is comparable with that of the main reaction). The main function of CO2 in this reaction was to prevent carbon formation rather than eliminating it after the deposition.149 Another important function of CO2 is re-oxidation of the reduced active phase. Such phenomenon was observed in the systems with reducible oxides, for example, MoOx,166 Pd/CeZrAlOx,167 Ga2O3.168 This process occurs in the case of the one-stage “looping-like” ODH, when alkane is directly oxidized by lattice oxygen resulting in the olefin and water production, after which the reduced metal species are re-oxidized by CO2. At the same time, the oxidation of the catalyst may have a negative effect. CO2 was found to oxidize Sn to SnOx in the Pt–Sn nanoalloy system, deactivating the sample.169 The authors reported that this process could be easily inhibited by decreasing the CO2:C3H8 ratio, which noticeably raised the conversion. Finally, CO2 can also mask non-selective sites on the catalyst surface, thus increasing the selectivity and, sometimes, the conversion.170
| CnH2n+2 + MeOx → CnH2n +MeOx−1 + H2O |
| MeOx−1 + 1/2O2 → MeOx |
The vast majority of the recently published studies focus on ethane CL-ODH, since ethylene is not produced by direct dehydrogenation in the industry compared to propylene and butylenes. Despite the significant advantage of steady-state O2-ODH of ethane in terms of lower reaction temperatures compared to SC, the safety and selectivity of this process are still limiting factors for its implementation. From this point of view, CL-ODH can be seen as a solution. No explosive mixtures of hydrocarbons with oxygen are generated in CL-ODH. The selectivity of looping can be much higher, since the oxidation is carried out not by molecular but by lattice oxygen.
Different catalytic systems for O2- and CO2-based CL-ODH have been studied in recent years and promising results for olefin yield were reported over the promoted or modified perovskite-based or other mixed oxide materials. Ding172et al. studied the effect of alkali promoters on the performance of LaMnO3 perovskite and concluded that the sodium compounds like Na3PO4 or Na2WO4 enhanced the stability of the catalyst. The yield of ethylene increased up to 60% at 775 °C during 25 redox cycles. A similar effect was found when using the modification of CuMn2O4 by Na2WO4: core-shell structure, where the CuMn2O4 mixed oxide is in the core covered by tungstate. The selectivity increased up to 86.4% at 720 °C and a conversion level of 58.8%.173 The effect of Mo-doping was studied in the catalytic systems consisting of Co–Mo–Fe2O3 and Mo–V–O oxides. Molybdenum was found to be a selectivity-responsible component, which decreased the oxidation ability of CoFe2O4 and increased the V–O binding energy reducing the overoxidation of ethane and propane.174,175 The Mo-doped catalyst gave a selectivity of 87.3% to ethylene in C2H6 CL-ODH at a conversion of 56.2% and 825 °C. The propane CL-ODH was studied at 500 °C and the conversion was 36% with a selectivity to propylene of 89% during the 100 cycles. An ethylene selectivity close to 100% at ∼16% conversion of ethane at 600 °C was achieved using the Ni-based HY zeolite-supported catalyst with two simultaneously present active sites embedded into the cages of the matrix. The Ni2+ Lewis acid sites were responsible for dehydrogenation, while the NiO nanoclusters acted as selective hydrogen combustion centers.144
Modification of SrFeO3 perovskite by Ce facilitated the formation of surface oxygen vacancies during the CO2-assisted CL-ODH of ethane.176 The catalyst showed a selectivity of 82% at a conversion of 29% at 725 °C. The benefit of CO2 over O2 as an oxidant was observed due to the formation of more nucleophilic centers in the former case during the regeneration stage. Another perovskite-type structure was observed in FeOx/TiO2, where the transition between the FeTiO3 and Fe2O3 phases facilitated the ethylene formation with a selectivity and a conversion of 91% and 10% respectively, at only 500 °C.115 CO2-Assisted CL-ODH was also realized using an electrochemical approach at 600 °C: porous single-crystal CeO2 electrodes were designed and used with solid single-crystal Y2O3/ZrO2 electrolyte and an ethylene selectivity of ∼95% at a conversion of 10% was observed.177 During the process, ethylene is generated on the anode while, in parallel, the CO2 reduction to CO on the cathode gave oxygen ions transferred to the anode.
An interesting approach for the realization of both CO2 and O2 CL-ODH involves the application of the molten salts as the reaction medium with or without oxidizing catalyst. Modification of La0.8Sr0.2FeO3 by lithium carbonate shells enhanced the transport of O22− species to the reagent and blocked the non-selective sites giving at 700 °C a very high ethylene yield of 59% at a more than 90% selectivity.178 Moreover, the molten salts without any catalysts were applied for CO2-CL-ODH. This process is schematically depicted in Fig. 10a. The following reactions were proposed based on in situ DRIFTS:179
| 2OH− + CO2CO32− + H2O | (3) |
| CO32− + H22OH− + CO | (4) |
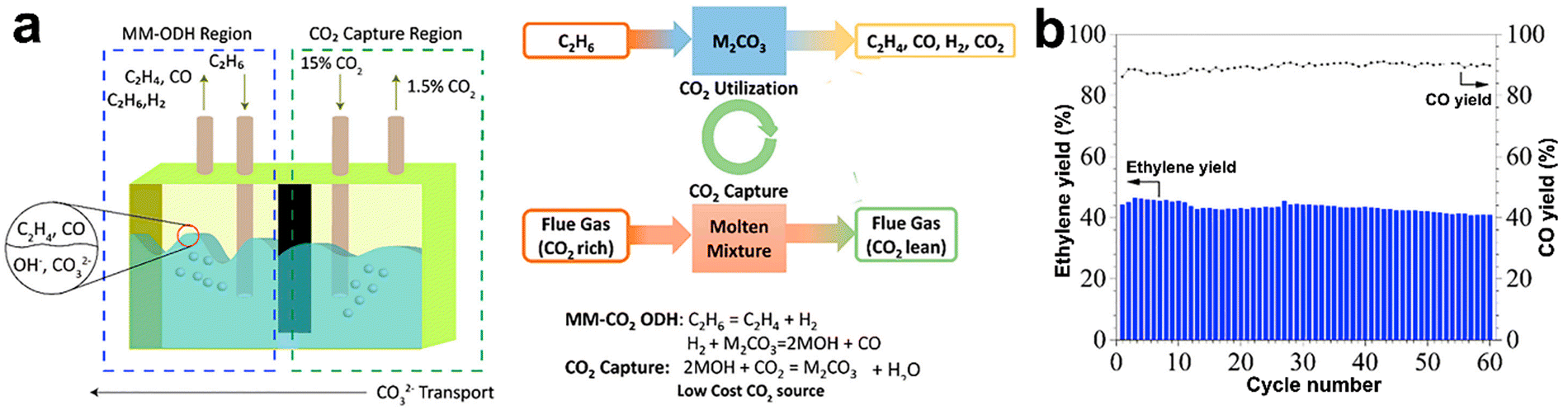 | ||
| Fig. 10 Molten-salt-mediated-CO2-ODH scheme (a) and CO2 utilization performance using this process (b). Reprinted from ref. 179 Copyright (2021), with permission from Elsevier. | ||
Significant effort was made180 for the techno-economic analysis of O2-CL-ODH of ethane. The authors compared the capital costs, energy efficiency, and ecological risks for this process with SC and concluded that noticeable benefits for energy saving, final ethylene price, and lower CO2 emission could be achieved in O2-CL-ODH due to a higher product yield, reduction of temperature used, lower safety risks, absence of air separation and exothermic character of the process. The sensitive analysis showed that the main parameter for the technology economics was the ethane price. The CO2-assisted looping in molten salts was also modeled. Up to 44.6% of energy saving was demonstrated, accompanied by highly effective “super-equilibrium” CO2 transformation to CO.179 Looping approach was considered for propane conversion. Modelling estimated a maximum yield of propylene of 73.4%, which is attractive even compared with the already realized DDH technology.181
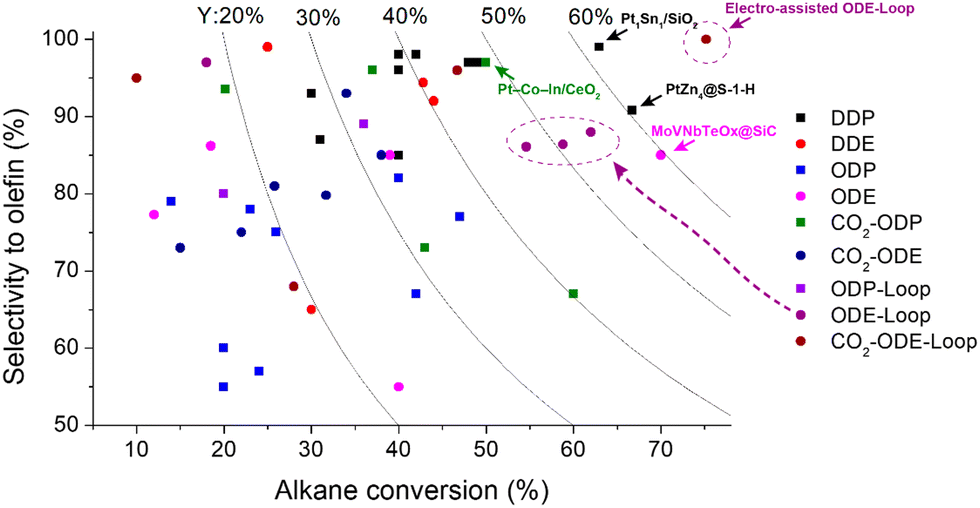 | ||
| Fig. 11 Selectivity and conversion of the catalysts in different dehydrogenation processes: DDP and DDE – direct dehydrogenation of propane and ethane; ODP and ODE – oxidative dehydrogenation of propane and ethane with oxygen; CO2-ODP and CO2-ODE – CO2-assisted oxidative dehydrogenation of propane and ethane; loop – chemical looping approach. Square and circle symbols show the propane- and ethane-based processes, respectively. Curves represent the yields of the olefins. Catalytic data from ref. 52, 54, 56, 57, 61, 63, 75, 88, 92–95, 99, 114, 118, 122–125, 127, 129, 131, 135, 137, 138, 142, 144, 146, 149, 151–153, 157, 159, 162, 166, 172, 173, 175, 176, 178, 179 and 182–187. | ||
CL-ODH can be considered the most promising technology for ethane conversion to ethylene. CL-ODH can be conducted at a much lower temperature compared to ethane SC. The chemical looping approach allows avoiding a direct reaction between an oxidizing agent and alkanes and reducing the undesired side products. Moreover, looping can be compatible with the commercial Oleflex and CATOFIN processes, where several parallel reactors operate in a cycle mode with periodic regeneration.
2.4. Methane coupling
Methane is one of the most abundant carbon-based molecules on the earth. Natural and alternative gases (shale gas, biogas) are getting more and more demanded by the economy, due to the substitution of oil and coal as polluting energy sources.188 Methane is used for the production of chemicals by an indirect pathway through the formation of syngas (CO/H2) with subsequent FT synthesis.189 Alternatively, syngas can be converted to methanol with subsequent conversion of methanol by MTO to LOs.10 However, the multistep processes require high energy consumption for the separation and purification of the intermediates and products. The direct route for methane conversion to high-value products through oxidative (OCM) and non-oxidative routes (NOCM) would be highly desirable.The reaction mechanism should involve the abstraction of H atom from methane with the generation of hydroxyl group using surface oxygen and subsequent recombination to produce water and oxygen vacancy. The oxygen vacancies are then regenerated by the reaction with gaseous oxygen. The methyl radicals undergo coupling to produce ethane followed by dehydrogenation to ethylene (Fig. 12).16
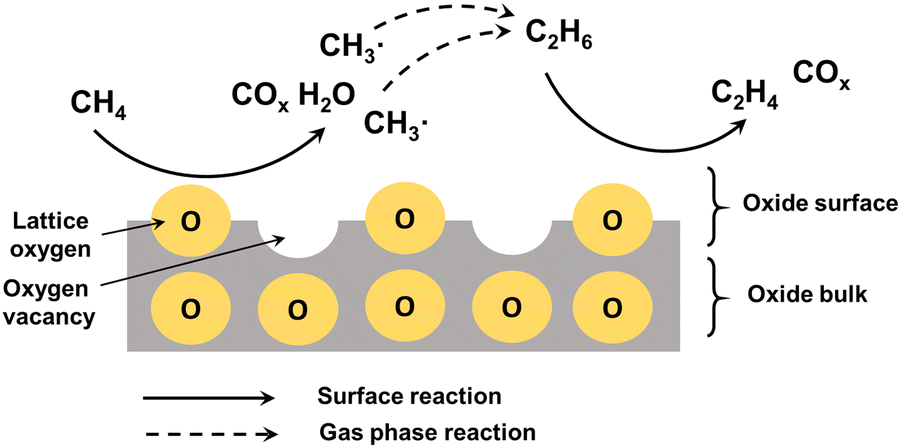 | ||
| Fig. 12 Mechanism of oxidative coupling of methane to ethylene over alkali metal catalyst. | ||
Traditionally, oxide catalysts doped by alkali metals, modified transition metal oxides, rare earth metal oxides, perovskites and pyrochlore catalysts188,190–192 have been used for the OCM reaction. The statistical analysis of about 1000 catalytic compositions (Fig. 13) indicate that rare-earth metal oxides and alkaline metal oxides demonstrate the best performance for the OCM reactions.193
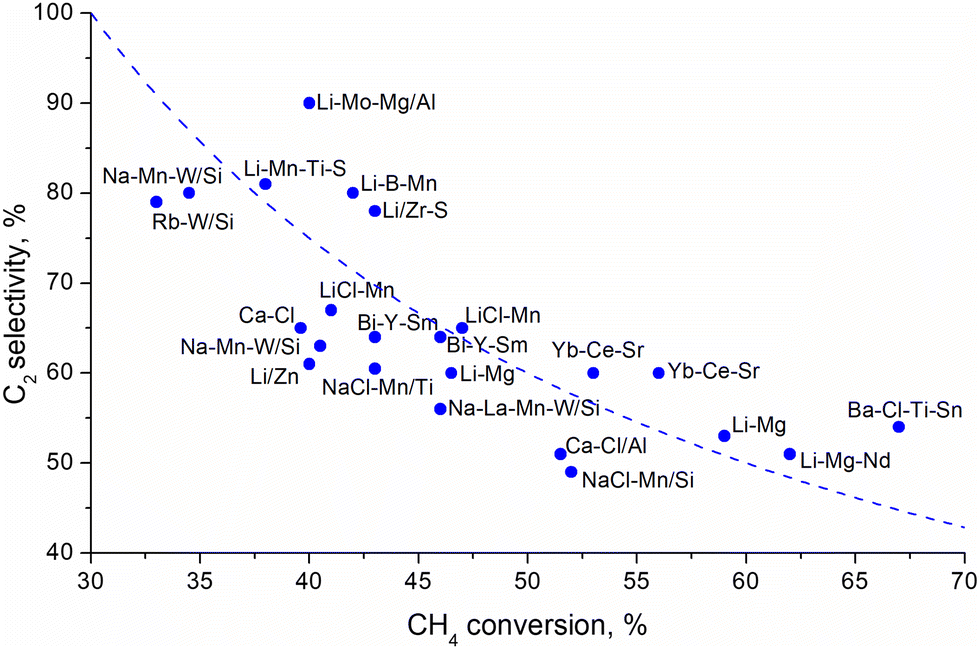 | ||
| Fig. 13 Selectivity to the C2 products as a function of methane conversion over various catalsyst.193 Dashed line indicates 30% yield. | ||
2.4.1.1. Alkali earth metal doped oxide catalysts. Conventional metal oxide catalysts exhibit relatively low C2 selectivity and fast deactivation. The presence of promoters changes the catalytic performance due to the presence of oxygen vacancies needed for methane activation.194–196 Thus, modification of CeO2 by Ca2+ with a similar ionic radius to Ce4+ provided the highest content of vacancies and basic sites with the best performance in OCM reaction.197 Among substituted titanates, SrTiO3 provided198 a selectivity of about 66% with the highest C2 yield up to 25%. Mesoporous TiO2 with supported MnxOy–Na2WO4 has demonstrated195 stable catalytic performance in OCM for 16 h. Li-Doped SnO2 catalyst exhibited196 the best performance among other dopants due to the high amount of electrophilic oxygen species in the catalyst. Mobile lattice oxygen was responsible199 for the production of C2 hydrocarbons in the lanthanum-based perovskite catalysts. The rare earth oxides (Sm2O3, TbOx, PrOy and CeO2) doped with alkali metals showed200 improved catalytic performance to C2 hydrocarbons in terms of activity, selectivity and stability due to the presence of strong basic sites.
One of the most efficient and studied systems for OCM reaction is Li-doped MgO.201,202,212,213 The reaction proceeds201via interaction of methane with polarised Li+O− centres:
| Li+O− + CH4 = Li+OH− + CH3˙ |
| 2Li+OH− = Li+O2− + Li+V0 + H2O |
| Li+O2− + Li+ V0 + 1/2O2 = 2Li+O− |
2.4.1.2. Transition metal oxides. One of the most conventional transition metal oxide catalysts for OCM reaction is Mn–Na2WO4/SiO2. This catalyst provides a C2 yield as high as 27%, which is one of the best in the literature195,206,216–218 (Table 1). For example, Wang219et al. have studied the effect of the preparation method of Na2WO4–Mn/SiO2 catalysts on their performance in the OCM reaction. The catalysts were prepared by impregnation, slurry and sol–gel. The catalyst prepared by mixture slurry demonstrated stable performance during 500 h at 800 °C with a C2 hydocarbon yield and a selectivity as high as 31% and 71%, respectively.
| Catalyst | Reaction conditions | CH4 conv., % | C2 sel., % | C2 yield, % | Ref. | ||
|---|---|---|---|---|---|---|---|
| T, °C | CH4:O2 |
GHSV, h−1 | |||||
| Alkali metals | |||||||
| Li/MgO | 720 | 1:2 |
— | 38 | 50 | 19 | 201 |
| Li/MgO-SG | 700 | 3:1 |
2400 | 39 | 66 | 26 | 202 |
| Li/MgO-IWI | 26 | 61 | 16 | ||||
| Li2CaSiO4 | 800 | 4:1 |
— | 30 | 72 | 22 | 203 |
| Li–Sm2O3/MgO | 700 | 4:1 |
2400 | 21 | 62 | 13 | 204 |
| Transition metal oxides | |||||||
| Na2WO4/Mn/SiO2 | 850 | 3.5:1 |
10000 |
32 | 45 | 14 | 205 |
| Na2WO4/Mn/SiO2 | 800 | 4:1 |
— | — | — | 24 | 206 |
| Mn–Na2WO4/SiO2 | 800 | 4:1 |
— | 28 | 73 | 18 | 207 |
| MnOx–Na2WO4/SiO2 | 770 | 4:1 |
60000 |
23 | 70 | 16 | 208 |
| A2B2O7 Pyrochlore catalysts | |||||||
| La2Sn2O7 | 727 | 2:1 |
— | 30 | 15 | 4 | 209 |
| Sm2Sn2O7 | 750 | 2:1 |
— | 40 | 49 | 20 | 210 |
| La2Ce1.5Ca0.5O7 | 800 | 4:1 |
18000 |
32 | 70 | 22 | 211 |
The catalysts containing only Na and W oxides are inactive in the OCM reaction, however, the Na/W/Mn composites demonstrated synergetic effects with a high yield of C2 products.218,220 The catalysts containing Na and Mn oxides or Mn and W are selective but have low activity. Thus, each component in the three-component systems plays important role: Mn promotes oxygen mobility between lattice oxygen and gas phase, W dissociates methane on the surface, while Na is needed to maintain Mn and W on the catalyst surface (Fig. 14).
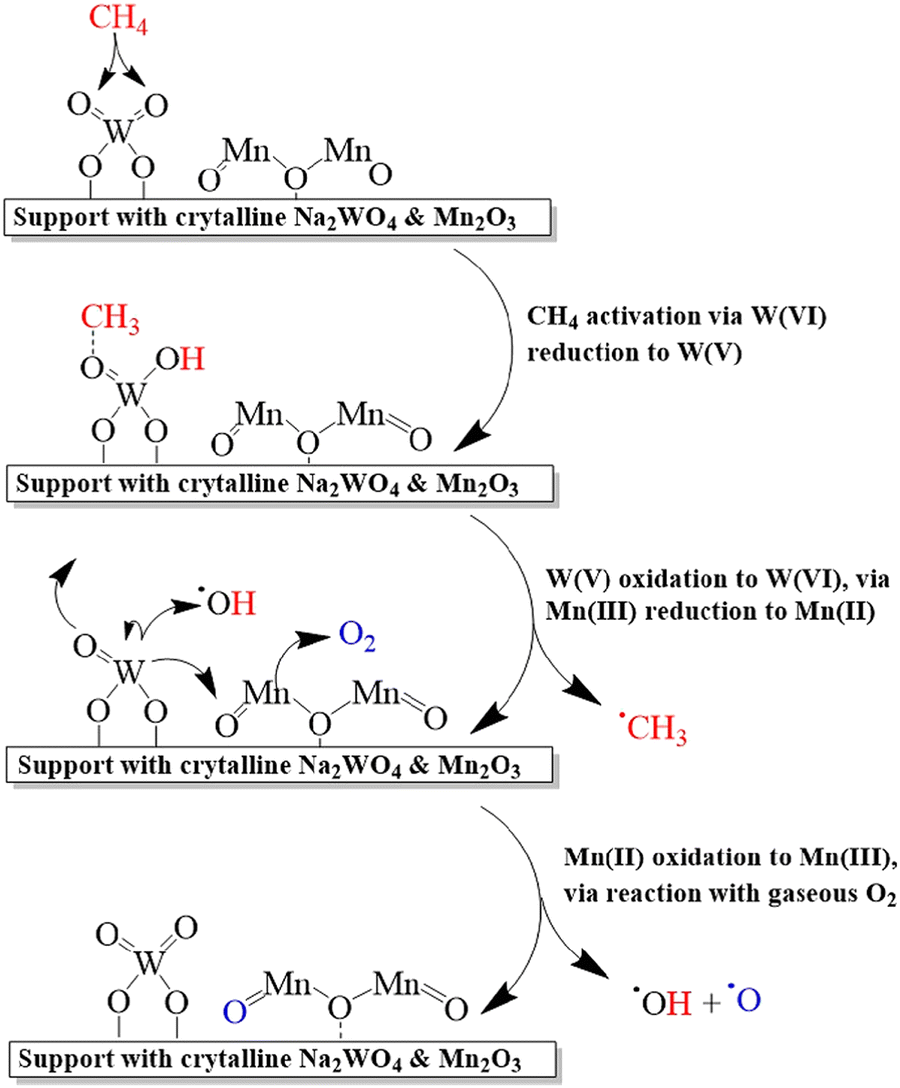 | ||
| Fig. 14 Mechanism of oxidative coupling of methane to ethylene over Mn–Na2WO4 catalyst. Reprinted with permission from ref. 217 Copyright 2019 American Chemical Society. | ||
A study of electronic properties and structure of Mn and W in the Mn–Na2WO4/SiO2 catalyst demonstrated the absence of correlation of OCM activity with the crystallinity.221 Thus, heating of catalysts leads to the transformation of W oxide to molten Na2WO4 with significant distortion of bond orders and a switch of the tetrahedral Td to octahedral Oh symmetry. The authors concluded that the Oh-W6+ species were inactive in OCM reaction, however, Td-W6+ activated methane in the presence of Mn3+ octahedral sites.
2.4.1.3. A2B2O7 Pyrochlore catalysts. The A2B2O7 pyrochlore catalysts are among the most promising for OCM due to their high stability, basicity and a high content of oxygen vacancies.222–224 The different types of pyrochlore catalysts can be prepared by variation of A to B ratio between 1.46 and 1.78.222 The pyrochlore structure contains oxygen vacancies, which increase the oxygen mobility in the catalyst.225
The catalytic activity of A2B2O7 pyrochlore (A – rare earth metals, B – Ti, Sn, Zr) increases with a decrease in the B–O strength.210 Wang222,223et al. investigated the structure, amount of oxygen sites, vacancies and alkaline sites in different types of pyrochlore catalysts (Table 1). The OCM catalytic activity changed in the order La2Ce2O7 > La2Zr2O7 > La2Ti2O7, which is consistent with the amount of surface-active oxygen.223 The optimization of La2Ce2O7 by doping with Ca led to a better yield (22%) due to higher alkalinity and higher oxygen mobility.211 Substitution of Ce4+ by Ca2+ and Sr2+ having a similar ionic radius resulted in the higher selectivity and yield of C2 hydrocarbons. It correlates with high reduction temperature and the amount of lattice oxygen according to H2-TPR. La2Ce1.5Sr0.5O7 demonstrated the best performance among all the studied catalysts.
Zeolites such as ZSM-5, ZSM-11, MCM-22 modified by transition metals such as Mo or Zn demonstrate activation of methane at 700 °C with dimerization to ethane, dehydrogenation to ethylene and rapid subsequent aromatization to benzene over Brønsted acid sites.227 Thus, the LO synthesis from methane via NOCM seems difficult over the Mo catalysts.
The Pt-based catalysts have demonstrated a significantly higher ethylene selectivity. In 1990s, Belgued228 reported the conversion of methane to ethane over Pt/SiO2 catalyst already at 250 °C, however, the yield was very low. Later, Dumesic229 demonstrated that the addition of Sn to Pt/SiO2 was beneficial for ethylene formation during methane coupling (Fig. 15). The bimetallic and stepped surfaces were found to be efficient for ethylene generation.52,229 Varma230 reported the same effect using a bimetallic Pt–Bi/ZSM-5 catalyst that could afford more than 90% selectivity to the C2 species.
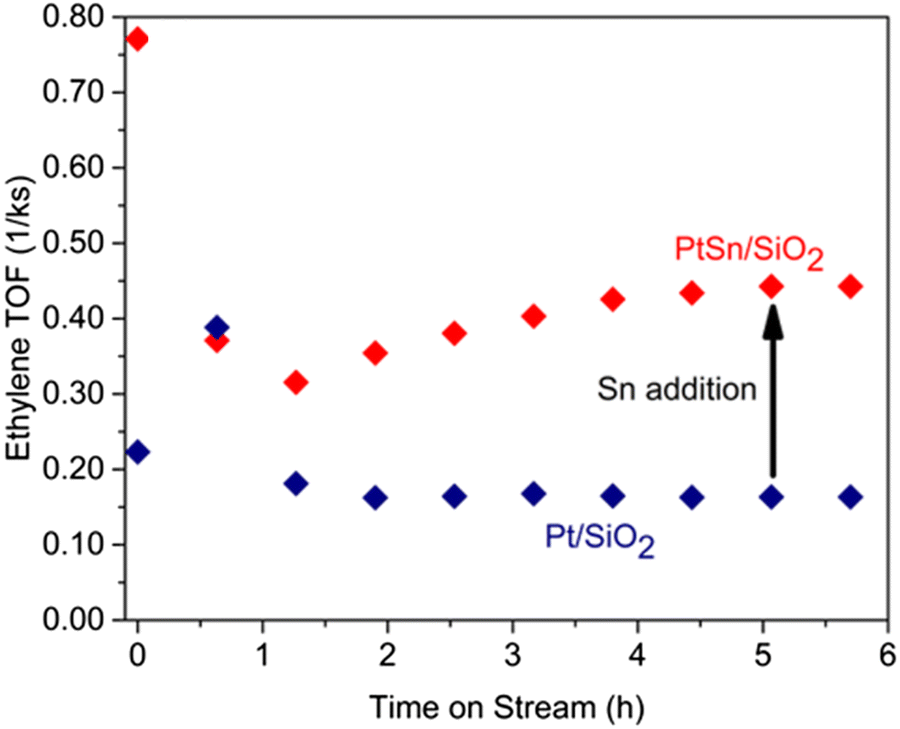 | ||
| Fig. 15 Ethylene turnover frequency values of the PtSn/SO2 and Pt/SiO2 in NOCM. | ||
The GaN catalysts have also demonstrated a high potential for the conversion of methane to ethylene. Kopyscinski231et al. used GaN/SBA-15 for direct conversion of methane to ethylene with a selectivity as high as 71% and reduced coke formation.232 Further, they studied the regeneration capability of GaN catalyst and demonstrated renitridation of Ga2O3 by ammonia.
The catalysts consisting of single iron sites embedded in a silica matrix have been proposed for NOCM. A high selectivity towards ethylene (>50%) at relatively high conversion (>30%) has been exclusively reported233 by DICP in Dalian. A mechanism has been proposed,234 where the true active site is an iron carbide, on which ethylene is formed through the methyl group formation and recombination.
NOCM can proceed with selectivity up to 90% over PtSn, PtBi and GaN catalysts, however, the yield of the product is very low (<2%) due to the thermodynamic limitations at higher temperatures.
3. Light olefin synthesis from oxygenates
3.1. Methanol to olefins (MTO)
The Methanol to Olefins process (MTO) has been studied for more than forty years. To date, MTO is the only technology used for the industrial production of LOs from non-petroleum resources. The information about the most common MTO catalysts can be found in several review articles.20,235–238 Undoubtedly, the ZSM-5 and SAPO-34 zeolites have shown the best results in terms of methanol conversion and selectivity to LOs. These two materials are the most studied and are the only ones exploited in the MTO industrial plants.21 Representative MTO data obtained with SAPO-34 and ZSM-5 catalysts are given in Table S2 (ESI†).The main strength of the MTO process is complete MeOH conversion and high selectivity to light olefins. The catalyst in MTO usually deactivates after several hours of operation. Catalyst regeneration is therefore an important part of MTO process. While the short-term stability is limited to a few hours, the long-term stability of zeolite catalysts, and in particular SAPO-34, can exceed236 450 reaction-regeneration cycles. Generally, SAPO-34 shows complete conversion and a combined ethene + propene selectivity of around 80–85%.239–242 In particular, this material allows obtaining high LO selectivities due to its mild acidity, and the presence of eight-membered ring pore openings, which limits the product molecular size and introduces shape selectivity. On the other hand, a small zeolite pore diameter causes fast catalyst deactivation. For the latter reason, industrially SAPO-34 is used as a MTO catalyst in fluidized bed reactors. The second common catalyst, ZSM-5, possesses medium size ten-membered ring pore openings. On the one hand, it shows lower ethene + propene selectivities compared to SAPO-34; on the other hand, it allows obtaining higher selectivities to propene (> 50%) and slower deactivation than SAPO-34.243–245 As a consequence, the ethene/propene ratio can be controlled by modifying the catalyst: for the ZSM-5 based catalysts, the propene/ethene ratio can be as high as 11.243
The definition of MTO as an autocatalytic reaction is widely accepted. Inside the zeolite cages, LOs produced in the initial reaction stages, react to form the so-called hydrocarbon pool species (HCPs), which in turn promote the LO production. Two types of cycles have been defined to describe the HCP model: the olefinic cycle is based on the formation of higher olefins, which are cracked to produce propylene; the aromatic cycle, which is mainly related to the ethylene production. To better explain the reaction mechanism, Zhang246 and co-workers recently presented an additional cycle, based on Methyl Cyclopentadiene (MCP) species. Inside the zeolite cages, the latter species can be produced starting from olefins and they in turn can produce aromatics. As a result, this cycle can be considered as bridging the olefinic and aromatic cycles, forming a hypercycle247 (Fig. 16).
 | ||
| Fig. 16 Scheme of the MTO reaction mechanism: initial non-autocatalytic stage; autocatalytic stage (hypercycle); coking. Reprinted with permission from ref. 247 Copyright 2021 American Chemical Society. | ||
The formation of HCP species inside the zeolite cages is also linked with the main weak point of MTO catalysts, i.e. catalyst deactivation due to carbon deposition and pore occlusion. A better understanding of the mechanisms of coke evolution is necessary in order to control this phenomenon and either to limit it or to use it for beneficial purposes. Wang240 and colleagues conducted a study on the nature of coke species and the mechanism of coke expansion inside SAPO-34 zeolite. The combination of DFT calculations, Matrix-Assisted Laser Desorption/Ionization Fourier-Transform Ion Cyclotron Resonance Mass Spectrometry (MALDI FT-ICR MS) and isotope labelling revealed a two-phase deactivation mechanism: first, polycyclic aromatic hydrocarbons (PAHs) form inside the CHA cages, with pyrene being the largest one possible due to the cage dimension, then different PAH units are cross-linked to form bigger species via cage-passing growth.
Recently, Structure Illumination Microscopy (SIM) has been used as a powerful tool to examine the spatiotemporal evolution of carbonaceous species inside molecular sieves. Using a combination of SIM, DFT calculations and molecular dynamics (MD)249 simulations, Gao248et al. and Yang249et al. explored the deactivation phenomena for the SAPO-34 catalysts. In particular, they focused on defining the coke composition and on localizing the regions of affected crystals. For nanoparticles with a diameter of around 5 nm, both groups observed an initial stage in which HCPs appear in the center of the crystal, followed by the expansion of these deposits to the rim. Finally, with the advancement of the reaction, HCPs transform into coke. Furthermore, Gao et al. performed MTO on SAPO-34 catalyst with the crystallites of 5, 13, 17 and 50 μm to evaluate the crystallite size effect and performed MD simulations to evaluate the spatiotemporal distribution of methanol, LOs and C4+ species in SAPO-34 crystals during MTO. According to their work,248 a smaller crystallite size is related to a better spatial distribution of HCPs and coke after catalyst deactivation (Fig. 17a and b), while for large crystals, these carbonaceous species form only in correspondence of the rim (Fig. 17d).
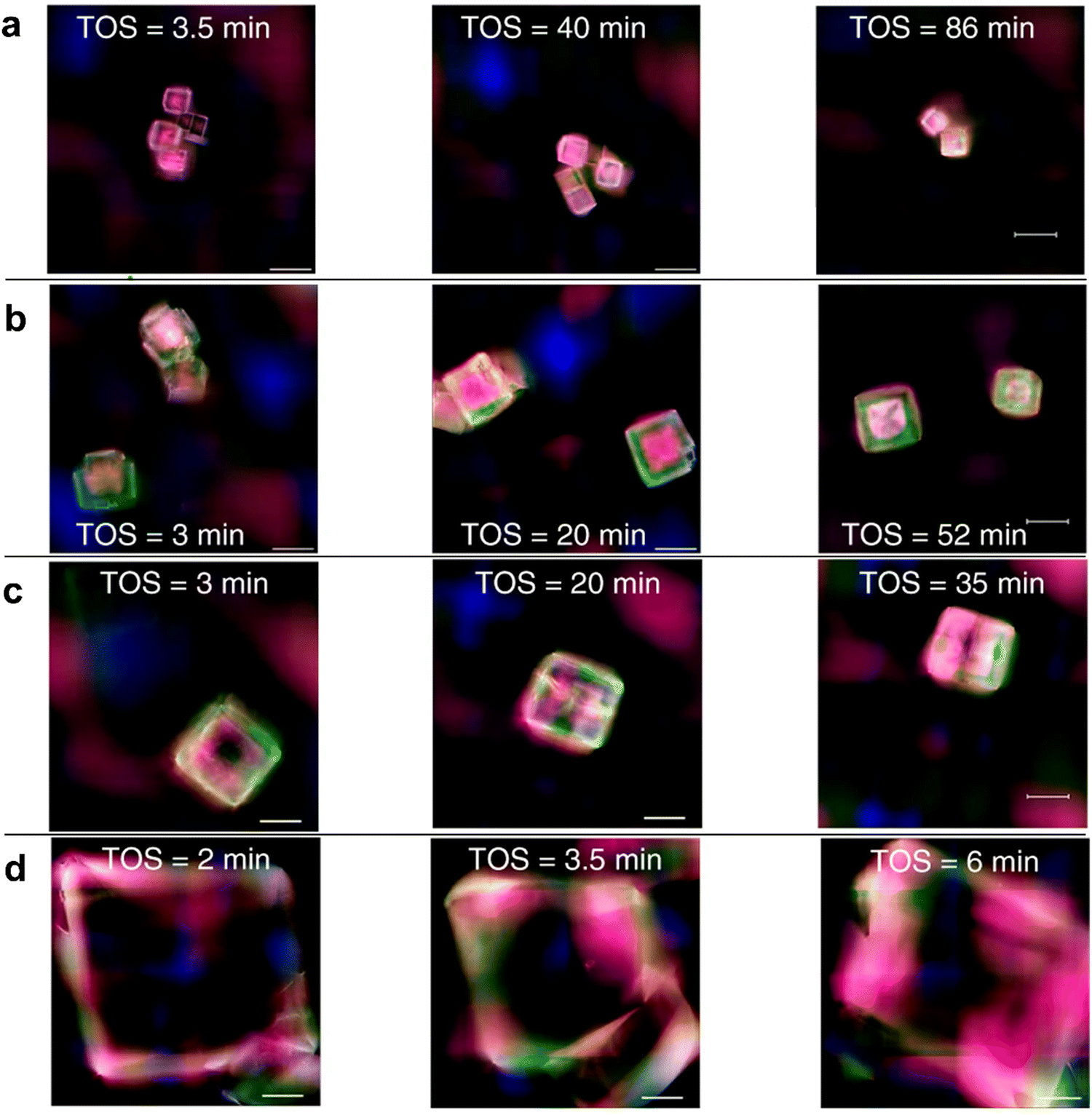 | ||
| Fig. 17 Evolution of HCP species inside SAPO-34 crystals of different sizes with time. (a) 5 nm; (b) 12 nm; (c) 17 nm; (d) 50 nm. The different colours represent different species: blue: benzenic carbocations; green: naphtalenic carbocations; red: phenantrenic carbocations; pink: pyrenic carbocations.248 | ||
Yang249et al. studied coke evolution in order to define a way to extend the SAPO-34 catalyst lifetime. They investigated the long-term effects of MTO conversion on the SAPO-34 zeolite exploiting 1D 27Al or 31P MAS NMR and 2D 31P–27Al HETCOR MAS NMR spectroscopy, as well as XRD and DFT calculations. They first analyzed the coke effect: with its formation, the SAPO-34 unit cell is subjected to a reversible expansion. In fact, the 31P MAS NMR spectra of deactivated samples showed a chemical shift to high fields for the signal related to the tetrahedrally coordinated P(IV) atoms (−30.1 ppm), with respect to the fresh sample. After the catalyst regeneration, the chemical shift turned back to the original value. For AlPO4 zeolites this parameter correlates with the mean P–O–Al bond angle. Likewise, both the results of XRD Rietveld refinements of deactivated and fresh samples and DFT calculations agree with a reversible expansion of the SAPO-34 unit cell. Water, which formed during MTO, adsorbed on the Brønsted acid site and it might compete with the coke-forming species.250 In particular, water co-feeding was evaluated in order to increase the catalyst lifetime and selectivity to LOs. Nonetheless, long exposure to moisture determined the irreversible Si–O–Al bond hydrolysis, leading to a partial collapse of the structure. In fact, after exposing SAPO-34 to moisture for 90 days, a signal related to the Si–OH group appeared at 1.8 ppm in the 1H MAS NMR spectrum. It has been pointed out that the catalyst pre-coking allows reducing this phenomenon. In fact, no relevant evidence of Si–O–Al bond hydrolysis has been detected for the pre-coked samples. Moreover, pre-coking and water co-feeding promoted an increase in the single-pass lifetime and the stability after multiple recycles. Another solution to slow the catalyst deactivation and improve the catalyst lifetime is H2 co-feeding at high partial pressures, as has been proposed by Bhan's group.251,252 They managed to increase 70 times the cumulative turnover frequency towards hydrocarbons. Moreover, they reported that an increase in the H2 partial pressure led to a decrease in the ethylene to propylene ratio, due to the suppression of the aromatic cycle. To prove the role of H2, they performed analogous experiments using He and no influence on the catalyst lifetime has been observed. Recently Akhgar et al. increased the catalyst lifetime by synthesizing hierarchical SAPO-34 with a green synthesis route.253
Recently, Zhou241et al. proposed a method to exploit the role of coke in the control of selectivity to LOs and ethene/propene ratio. The SAPO-34 zeolites deactivated by coke deposition have been treated with steam at 953 K. On the one hand, the pore volume has been recovered due to coke decomposition, on the other hand, the HCP species have been produced inside the zeolite cavities, with GC-MS showing naphthalene as the predominant coke species with molecular weight less than 200. The recovery of the pore volume allows restoring a complete MeOH conversion (for the coked material, it was around 75%). Similarly, the production of HCP species, related to this steam cracking process, determines a recovery of the LO selectivity to 88%. DFT calculations and isotopic labelling were exploited to prove that the predominance of naphthalene inside the cages was related to an increase in the ethene to propene ratio. As a result, the selectivity to ethene increases from 49% (fresh SAPO-34) to 58% (coked SAPO-34 treated with steam at 953 K for 20 min). Alternatively, different works showed that ethene selectivity could be enhanced by limiting propene diffusion inside the CHA-type zeolites. Liu's group254 observed this effect by incorporating Zn cations in SAPO-34. Zn-SAPO-34 allowed higher C2H4 and total LO selectivities compared to SAPO-34 and was able to reach the highest ethene selectivity obtained with SAPO-34 in a significantly shorter time. Not only Zn ions promote the formation of HCP species like lower methylbenzenes, which in turn facilitate ethene production, but also it has been observed a superior amount of naphthalene species in the external CHA cavities of Zn-SAPO-34, which limit propylene diffusion. Ding255et al. limited propene diffusion by the deposition of SiO2 in SAPO-34 pores. This result agrees with the work done by Han256et al. about the diffusivity of LOs over the SAPO-34 catalyst in working conditions. To avoid adsorption and to evaluate only the diffusion properties, they exploited C2–C4 alkanes as probe molecules, considering that their size was comparable to the ones of the corresponding olefins. They observed that ethane probe molecules diffused generally via an intracrystalline pathway, while the 75% of propane probe molecules diffused via an intercrystalline mode. Consequently, the ethene to propene ratio increased with coke deposition, due to a reduction of pore size, which in turn limited the propene diffusivity.256
On the other hand, reducing the zeolite acidity and limiting hydrogen transfer increase the propene selectivity. Lin243et al. proposed a new material obtained through the incorporation of Ta(V) and Al(III) in the ZSM-5 structure. The structure of the fresh material and its stability during MTO did not change after the introduction of the metal ions. On the contrary, the selectivity to olefins and the propene/ethene ratio increased compared to the results obtained with HZSM-5. This was due to the influence of Ta(V) on the acidity of the zeolite. Also, paraffins and coke production were lowered due to the drop of hydrogen transfer reactions. Interestingly, Ta was responsible for the formation of the first C–C bond in propylene. Yarulina257–259 and co-workers also evaluated the effects of different Al loadings, post-synthesis desilication and metal incorporation on the propene selectivity. It has been pointed out that propene selectivity depends on the Brønsted acid site concentration. In addition, the incorporation of extra-framework alkaline metals promotes isolation of the Brønsted acid sites. The latter property favours propylene as the production of aromatic HCPs requires a vicinal Brønsted acid site. Nonetheless, Lewis acid sites generated with the alkaline earth metal incorporation suppress aromatic growth as they destabilize cyclic carbocations that yield the aromatic cycle. Hence, it has been observed that the presence of Lewis acid sites reduces the formation of polyaromatic species, and the catalyst stability can be improved by tuning the Lewis acid site/Brønsted acid site ratio according to a volcano plot (Fig. 18).257–259
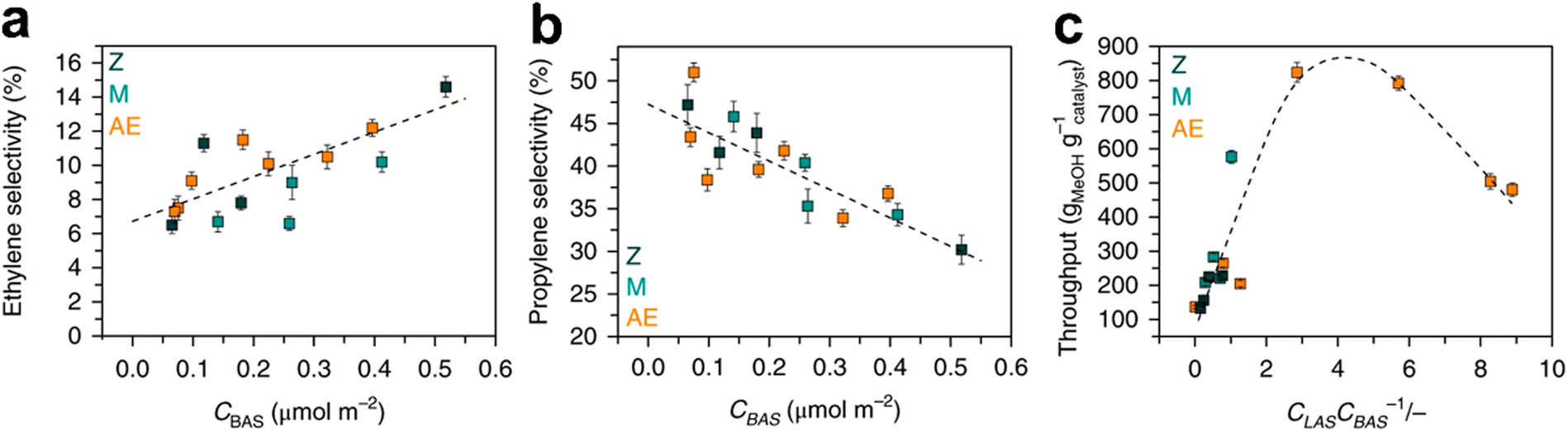 | ||
| Fig. 18 Influence of acid sites concentration on selectivity in MTO. (a) Dependence of ethylene selectivity on the Brønsted acid site concentration; (b) dependence of propylene selectivity on the Brønsted acid site concentration; (c) dependence of reaction throughput on the Lewis acid site/Brønsted acid site concentrations ratio. Z (black points): pre-synthetically modified ZSM-5 with different Si/Al ratio; M (green points): post-synthetically modified ZSM-5 by desilication or dealumination; AE (orange points): post-synthetically modified zeolites by incorporation of alkaline-earth metals.257 Reprinted by permission from Springer Nature: Springer Nature, Nature Chemistry, structure–performance descriptors and the role of Lewis acidity in the methanol-to-propylene process, Irina Yarulina et al., © 2018. | ||
3.2. Biomass and waste pyrolysis
Pyrolysis, which is thermal degradation in the absence of oxygen, allows carbon materials to be converted into valuable chemicals. This route has been exploited to obtain LOs from non-petroleum sources, like biomass or plastic waste. Unlike fossil fuels, biomass has a high oxygen content. It is a less efficient and bulky resource, but at the same time, it is abundant, carbon-neutral and low-cost. Likewise, plastic waste is an abundant, bulky and cheap feedstock, but compared to organic matter, it is a synthetic and pollutant material. Hence, exploiting waste for pyrolysis, as an alternative to incineration, is particularly interesting from an environmental point of view.Although LOs can be usually found among the components of pyrolysis products, they can be considered secondary products and still very few studies focus on the production of these species. The pyrolysis products are composed of a solid phase (char), a liquid fraction and a gaseous one: LO products are to be found in the latter fraction,260–266 although liquid bio-oils obtained from pyrolysis can be further cracked to obtain LO.267 Depending on the heating rate, pyrolysis is classified into slow (<2 °C s−1), fast (up to 200 °C s−1) and flash pyrolysis (up to 2500 °C s−1). The fast pyrolysis promotes the production of gaseous species; hence it should be preferred in case of LO production. In addition, the pyrolytic process may be performed with or without a catalyst. In the former case, the catalyst may be mixed with the raw materials (in situ) or separated from them (ex situ). In the ex situ catalytic pyrolysis (ex situ CP), the biomass is first pyrolyzed without catalyst and the resulting vapor products are transported to the catalyst bed downstream the pyrolyzer. The composition of ex situ pyrolysis products obtained with catalysts and different types of lignocellulosic biomass feedstock is presented in Table S3 (ESI†).
Wang268et al. studied the poplar wood pyrolysis using HZSM-5 as a catalyst and demonstrated that the ex situ catalytic fast pyrolysis (CFP) was a more convenient route to maximize the LO synthesis from biomass. In situ CFP yielded a higher amount of aromatics, char and coke and exhibited three times lower LO yield.268 Along with this study, other works revealed a higher LO yield due to the separation of the catalyst from biomass during the process.269–271 In the same way, Xue272 and co-workers observed an increase in LO production using an ex situ layout for polyethylene (PE), polypropylene (PP), polystyrene (PS) and polyethylene terephthalate (PET) pyrolysis. Overall, ex situ CFP usually increased the aromatics and olefin yields.260–266 In fact, an ex situ configuration allows transforming only the gaseous fraction, thus reducing undesired reactions of liquid and solid products.
Undoubtedly, the catalyst plays a key role in this process, too. The main catalyst used for biomass pyrolysis is ZSM-5: Yang273et al. compared it to CaO, SAPO-34 and MCM-41 and observed that ZSM-5 allowed the highest olefins and aromatics yields. Nonetheless, according to their results, SAPO-34 and ZSM-5 show a similar LO yield. To increase LO production from biomass, ZSM-5 has also been impregnated with other metals. For instance, Huang270et al. studied the catalytic properties of La-modified ZSM-5 in the pyrolysis of different biomass feedstocks. For the sugarcane bagasse conversion, the catalyst exhibits a 21.2 C mol% LO carbon yield. Likewise, Zhang269 and co-workers pyrolyzed corn stalk, cellulose, hemicellulose and lignin using a Fe/ZSM-5 catalyst: for cellulose pyrolysis, they observed a total LO carbon yield of 6.98 C mol% with a selectivity to ethene of 63.99 C mol%. With a composite feedstock like corn stalk, the LO yield decreased to 5.27 C mol%. Also, Shang260et al. pointed out that the combination of a hollow structure and Sn impregnation enhanced ZSM-5 catalytic activity for LO synthesis. They improved LO total carbon yield from 4.2 C mol% (ZSM-5) to 12.9 C mol% (Sn/M-ZSM-5). Similarly, for plastic waste pyrolysis, ZSM-5 is a common catalyst: Artetxe266et al. performed the high-density polyethylene (HDPE) catalytic pyrolysis using HZSM-5 catalyst, with a LO yield of 62.9 wt%. Eschenbacher265 and co-workers used it as an additive to FCC catalysts in low-density PE (LDPE) pyrolysis, obtaining a LO yield of 53 wt%. Nonetheless, other materials have been taken into consideration for plastic waste pyrolysis. For example, Cherednichenko274et al. studied the catalytic properties of MeVO3 and MeVO4 (Me = La, Gd, Lu) in the PE, PP and PET pyrolysis. Compared to the non-catalytic process, they observed an increase in LO yield, when using LaVOx and GdVOx. Witpathomwong275et al. performed waste tyre pyrolysis using Ru/MCM-48 as a catalyst, obtaining a LO yield of ca. 4.5%. Donaj et al. obtained an ethylene yield of 22.3 wt% and a propylene yield of 21.1 wt% using TiCl4/MgCl2 (Ziegler-Natta catalyst) in the catalytic pyrolysis of a mixture of polyolefins.276 Steam pre-treatment can further improve67,270 the properties of zeolite-based catalysts in biomass and waste conversion to LO.262,265 Dealumination caused by steam reduces the amount of strong acid sites in the zeolite. Moreover, water competes with the pyrolysis vapours for the adsorption on acid sites. Consequently, a minor fraction of strong acid in the zeolite promotes higher LO, which are produced on weak acid sites.
A parameter that affects the process energy consumption is the pyrolysis temperature. Thermal analysis should be considered before choosing the pyrolysis temperature. In fact, this parameter is strictly related to the feedstock. The biomass is composed mainly of hemicellulose, which decomposes at 220–315 °C, cellulose, which decomposes at 315–400 °C, and lignin, which decomposes at a broad temperature range, between 160 and 900 °C.277 Chen's group261 observed that increasing the temperature from 400 °C to 700 °C resulted in a higher fraction of the gaseous phase, while LO yield varied differently for cellulose and lignin pyrolysis: in the former case, LO weight yield increased with temperature up to 600 °C and decreased with further raising the temperature; in the latter case, it continuously increased up to 700 °C. Also for plastic waste, the decomposition temperature varies depending on the feedstock: for PE, PP and PET degradation stops between 450 °C and 490 °C.274
From another point of view, the feedstock influences the process selectivity. LOs are mainly obtained by cracking the feedstock, which produces intermediates that are finally catalytically converted into the final product. The nature of these intermediates depends on the nature of the feedstock components at the molecular level. Cellulose and hemicellulose produce alcohols and ketones, which can adsorb in the zeolitic acid sites and further react; lignin, instead, produces predominantly aromatics, which in turn cause catalyst coking.269 The results obtained from the pyrolysis of different types of feedstocks at 500–600 °C are reported in Fig. 19. For pure phase feedstocks, the total LO yield decreases following the series: cellulose > hemicellulose > lignin. This trend reflects also composite feedstocks, as the LO yield diminishes with increasing lignin content. At the same time, higher LO yields are obtained from the pyrolysis of polyolefins (PE and PP in particular), compared to other plastic waste feedstocks.274
 | ||
| Fig. 19 LO production from ex situ catalytic pyrolysis of biomass. The compositions of different lignocellulosic feedstocks are reported in the pie charts (C = cellulose; HC = hemicellulose; L = lignin). Each experiment is reported with “type of feedstock|catalyst”. Data obtained from ref. 260, 261 and 268–272. | ||
Moreover, lignocellulosic biomass and plastic waste have been used as combined feedstock. Wang278et al. studied the pyrolysis of paper biomass (C 76.5 wt%, HC 15.2 wt%, L 3.3 wt%) mixed with a blend of polyolefins (HDPE, PP and PET). They obtained a 28 wt% LO yield with a biomass/plastics ratio of 5:1 in the presence of 40%Co/30%CeO2/30%Al2O3 catalyst. Dorado et al. mixed 13C-cellulose with HDPE, LDPE, PP, PS, and PET to evaluate the origin of carbon in the products of pyrolysis over HZSM-5. They observed that with HDPE, LDPE and PP, the LO products originated mainly from the plastic source and the LO C-yield was around 25–35%, while with PS and PET it was around 10% and mainly originates from cellulose.
3.3. Summary
MTO is a mature technology, characterized by a complete methanol conversion and a high LO selectivity. SAPO-34 and ZSM-5 are the most suitable catalysts for the MTO process. The composition, acidity and diffusivity of these two types of zeolite can be modified in order to control the ethene/propene product ratio. Nevertheless, catalyst stability is still a property that needs to be improved. SAPO-34 is particularly affected by deactivation by coke formation and some interesting results in slowing down this phenomenon have been obtained either with pre-coking and water or H2 co-feeding. Because of the different stability of SAPO-34 and ZSM-5 catalyst families, two different MTO processes were developed. The MTO process developed by UOP involves SAPO-34 catalyst, which deactivates at a high rate and thus, proceeds in a fluidized bed reactor, while the Lürgi process, which takes place on a ZSM-5 zeolite, which is more resistant to carbon deposition, occurs in a fixed bed reactor.LOs are still considered secondary products of biomass and waste pyrolysis. To date, this technique is far from being considered mature for LO production and major efforts are required. Nevertheless, the available information points out that the experimental layout, the catalyst choice and the feedstock composition influence the output of the process and LO yields.
4. Light olefin synthesis from carbon oxides
4.1 Fischer–Tropsch synthesis with CO and CO2
In recent decades, there has been a growing interest in converting non-conventional carbonaceous feedstocks (biomass, organic waste, non-conventional gas, coal) into chemical products. Non-petroleum carbon feedstocks can be converted into liquid fuels or building-block chemicals via their gasification into syngas (mixture of carbon monoxide and hydrogen), followed by syngas conversion into hydrocarbons or oxygenates.279,280 Conventionally, FT synthesis has been used for the synthesis of hydrocarbon fuels such as diesel and jet fuels. Over the past 20 years, LOs have become target products of the Fischer–Tropsch to Olefins (FTO) process.Nowadays, carbon dioxide is the most important industrial pollutant and the major reason for global climate changes. The CO2 chemical conversion to LOs could contribute to lower consumption of fossil feedstocks,281 which are major sources of greenhouse gas emissions.280,282–286 The key issues in the CO2 hydrogenation reactions are the availability and sustainable production of hydrogen. To ensure the sustainability of LO synthesis from CO2, hydrogen for hydrogenation must be produced using renewable energy287 without additional CO2 emissions.288,289
The CO2 hydrogenation into olefins290 proceeds via a combination of the reverse water gas shift reaction (RWGS) and Fischer–Tropsch (FT) synthesis:
| RWGS: CO2 + H2 ↔ CO+ H2O ΔHR = +38 KJ mol−1 |
| FT synthesis: nCO + 2nH2 → CnH2n + nH2O ΔHR = −165 KJ mol−1 |
Both cobalt and iron catalysts are used in FT processes. Compared to cobalt, iron catalysts are much more flexible relative to the reaction conditions and olefin selectivity. Because of fast WGS reaction, the main side product of FT synthesis on iron catalysts is not water (as on cobalt), but carbon dioxide. Iron catalysts can be used for FT synthesis with syngas containing different H2/CO ratios. Due to their remarkable capacity to catalyse water gas shift (WGS), RWGS and FT reactions, iron is also the most common catalyst for the CO2-FT process.291,292 Dual bed and hybrid catalysts consisting of a FT catalyst for CO hydrogenation to hydrocarbons and an acid catalyst (e.g. zeolite) for the subsequent creaking of long-chain olefins to LOs are also considered.293 However, hydrogen transfer occurring within the zeolites often results in significant production of light paraffins and aromatics294–296 and thus, makes combinations of a FT catalyst and a zeolite not suitable for selective LO synthesis from CO2 or CO.
Molybdenum carbide,297–299 sulfide300 and nitride301 catalysts exhibit noticeable activity in the CO/CO2 hydrogenation. Though these catalysts usually have much lower activity compared to more conventional metallic or iron carbide catalysts, one of their important features is their stability in the presence of different impurities in syngas. The most important catalytic activity in the CO/CO2 hydrogenation was observed for molybdenum sulphide and molybdenum carbide systems. These catalysts also display high activity in WGS reaction. The unpromoted molybdenum carbide or sulphide catalysts exhibit fast methanation and fast WGS reactions with methane and CO2 being respectively the major products. The promotion of carbide and sulphide catalysts with alkali ions shifts the selectivity from methane to methanol, higher alcohols and olefins. In 2016, a group from Shanghai Advanced Research Institute reported302 remarkable catalytic performance of Mn-promoted Co2C nanoprisms (Fig. 20) with specifically exposed facets of (101) and (020) in syngas conversion to olefins under mild reaction conditions (250 °C, 1 bar). The reports of this group showed a major deviation of hydrocarbon distributions from the classical Anderson–Schulz–Flory (ASF) model. Alkali metal promoters facilitated303,304 stabilization of Co2C nanoprisms. The olefin selectivity over these catalysts was enhanced by optimizing the promotion and reaction conditions (temperature and reaction pressure). The groups of Weckhuysen and de Jong designed305 a Co/Mn/Na/S catalyst, which exhibited at 240 °C and 1 bar the C2–C4 olefin selectivity up to 54%. 10 nm Co nanoparticles of hcp Co metal phase were detected in the used samples. Na and S acted as electronic promoters on the Co surface. Interestingly, very low CO2 selectivity was observed on the Co/Mn/Na/S catalyst. Representative catalytic data for LO production using FT synthesis are shown in Table S4 (ESI†).
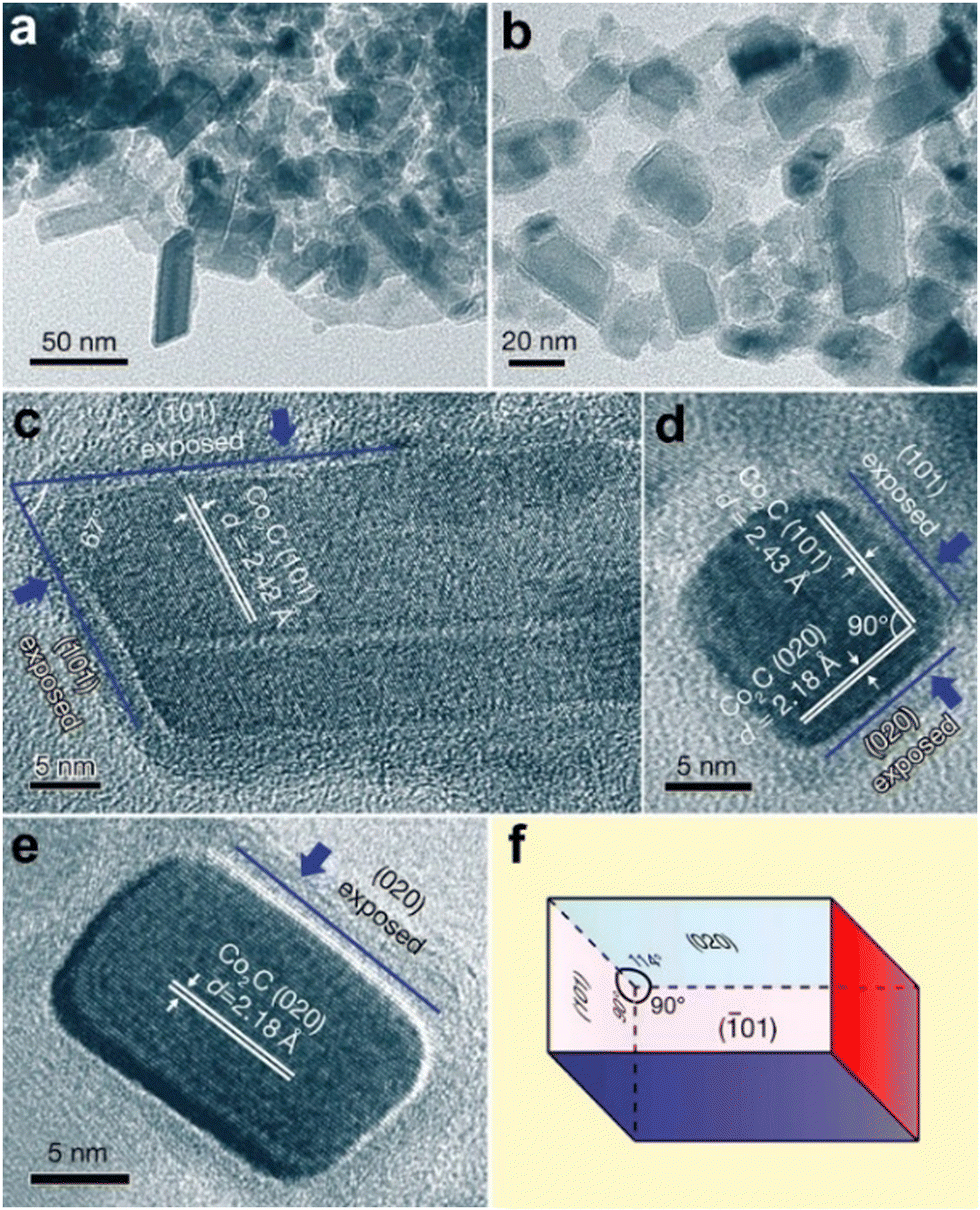 | ||
| Fig. 20 TEM images of the CoMn catalysts after reaching steady state. (a and b) Low-resolution TEM images. (c–e) High-resolution images of Co2C nanoprisms with exposed facets of (101), (101) and (020). (d) Distance (length) of the lattice fringes. (f) The Co2C nanoprism has a parallelepiped shape, with four rectangular faces and two rhomboid faces.302 Reprinted by permission from Springer Nature: Springer Nature, Nature, Cobalt carbide nanoprisms for direct production of lower olefins from syngas, Liangshu Zhong et al., © 2016. | ||
– flexibility in terms of activity, selectivity and reaction conditions;
– high selectivity to olefins within the ASF hydrocarbon distribution;
– sensitivity of reaction rate and selectivity to the promotion and support;
– activity in the WGS reaction and the possibility to use syngas having variable H2/CO ratios;
– presence of different phases potentially active in FT synthesis;
– relative stability in the presence of syngas impurities compared to cobalt catalysts;
– lower cost compared to other metal catalysts.
Iron catalysts for FT synthesis can be either fused, precipitated or supported. Preparation of iron catalysts is a complex process.311 The goal is to produce desirable chemical, physical, catalytic and mechanical properties in the final catalysts. The preparation variables and conditions such as precipitation pH and temperature, type of impregnation, washing, drying, and calcination temperatures, and reduction environment and temperature, require careful optimization. The catalyst synthesis may involve different metals, precursors, promoters and support.
Generally, iron nitrate is the most common metal precursor, because of its high solubility in water and low cost. Supported catalysts usually have higher iron dispersion compared to the fused catalysts or catalysts prepared by precipitation. The formation of barely reducible iron silicate, iron aluminate or iron titanate should be avoided, since it results in difficult iron carbidization and relatively lower FT reaction rates. In addition, the catalysts may exhibit strong deactivation. Carbon deposition is often the main cause of the loss of activity of iron-based catalysts. Interestingly, the catalyst preparation method mostly influences the catalyst activity, while the hydrocarbon selectivity is affected to a lesser extent by the catalyst preparation technique.
The activation procedure is an important step in the design of iron FT catalysts. To convert iron oxide into iron carbide or metallic iron, iron catalysts are usually activated in either hydrogen, carbon monoxide or syngas. The activation atmosphere strongly affects the phase composition and catalytic performance of iron FT catalysts. Numerous reports suggest that the activation procedure should be optimized for a given catalyst. Our previous results suggest312 that activation in CO can be more favourable for obtaining the highest conversion in FT synthesis over silica-supported iron catalysts than activation in hydrogen.
Depending on the conditions of catalyst activation in hydrogen, iron oxides are partially reduced to magnetite and wustite (FeO). Small amounts of metallic iron are being produced in hydrogen at the temperature typical of FT synthesis. In the presence of carbon monoxide or syngas, different iron carbides appear. Both metallic iron and iron carbides in the reduced samples have surface sites for CO dissociation and are active in FT synthesis. In hydrogen, iron oxides undergo stepwise reduction via the following steps:313
| α-Fe2O3 → Fe3O4 → FeO → α-Fe. |
| α-Fe2O3 → γ -Fe2O3 → Fe3O4 → Fe5C2 |
Iron carbides can be also obtained from metallic iron and wustite (FeO). However, the concentrations of metallic iron and wustite in the partially reduced catalysts are usually much lower than that of Fe3O4. It was suggested317 that during the FT synthesis, α-Fe was mostly converted into χ-Fe5C2, whereas FeO was the main source for ε-Fe2C. Several iron carbides have been described in the literature:318 FeC carbide, hexagonal ε-Fe2C, pseudo-hexagonal ε′-Fe2.2C, monoclinic Hägg χ-Fe5C2 carbide and orthorhombic θ-Fe3C cementite. Each of these carbides can be detected during FT synthesis on iron catalysts. The carbide phase composition depends on the H2/CO ratio, iron dispersion, promoters, support, the extent of iron reduction and temperature.319 Recent machine learning simulation320 indicated that Fe5C2, Fe7C3, and Fe2C were the three stable bulk phases under FT synthesis, producing olefins.
Iron carbide in the catalysts for conducting FT synthesis can be also obtained using alternative methods. Carbidisation of finely divided iron oxide in a flow of propane, butane or pentane conducted by Emmett321 and co-workers resulted in the formation of Hagg iron carbide. Selective conversion of iron into iron carbide was also observed322 during the pre-treatment of nanocrystalline iron catalysts with methane at 500–580 °C.
Activation of iron catalysts for CO2 hydrogenation has not been extensively studied in the literature. On the one hand, Visconti323et al. synthesized a high surface area K-promoted iron-based catalyst. It was found that activation treatment using CO/H2 mixtures at 350 °C yielded highly active catalysts in the CO2 hydrogenation to LOs at mild process conditions (300 °C and 5 bar). On the other hand, Wang et al.324 activated Fe/ZrO2 catalysts promoted with different alkali metals using an H2 flow rate of 50 mL min−1 at 400 °C for 5 h. They reported higher CO2 conversions (up to 43%) with higher selectivity to LOs (about 44%), while methane selectivity was limited to less than 20%. In another study, Shafer et al.325 activated iron catalysts promoted with K, Rb, and Cs using a flow of CO at 270 °C for 24 h and evaluated the performance of these catalysts for carbon dioxide hydrogenation. They reported a CO2 conversion of around 20% with a high selectivity to C2–C4 fraction (50% olefins and paraffins). However, they also reported a high selectivity to methane (40%).
Iron catalysts are very versatile with rich chemistry during their preparation, activation and reaction. They can be adapted for various operating conditions of CO/CO2 hydrogenation reactions. In addition, the structure of iron catalysts can significantly evolve as a function of the operating conditions. Schulz calls this phenomenon “catalyst self-organization”.326 In the lack of hydrogen and carbon monoxide and at higher concentrations of carbon dioxide and water, the reaction medium can become oxidizing. Some amounts of Fe3O4 magnetite are usually found in the working iron catalyst under FT reaction conditions. The presence of magnetite, however, can be favourable for the WGS and RWGS reactions, which adjust the CO/CO2/H2 ratio in the reacting feed gas. Thus, during FT synthesis iron species may be distributed among several phases: e.g. carbides, oxides and metallic iron. The iron carbides may transform from one into another and in magnetite as a function of operating conditions.
| CO + H2O ↔ CO2 + H2. |
| nH2 + 2nCO(CH2)n + nCO2 |
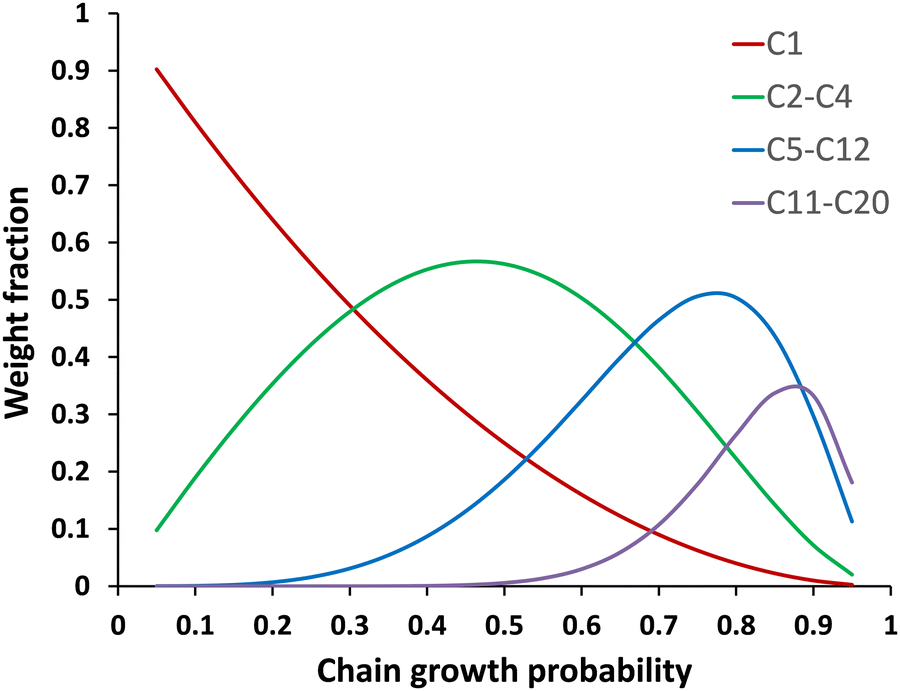 | ||
| Fig. 21 Hydrocarbon selectivity as function of the chain growth probability factor (α). | ||
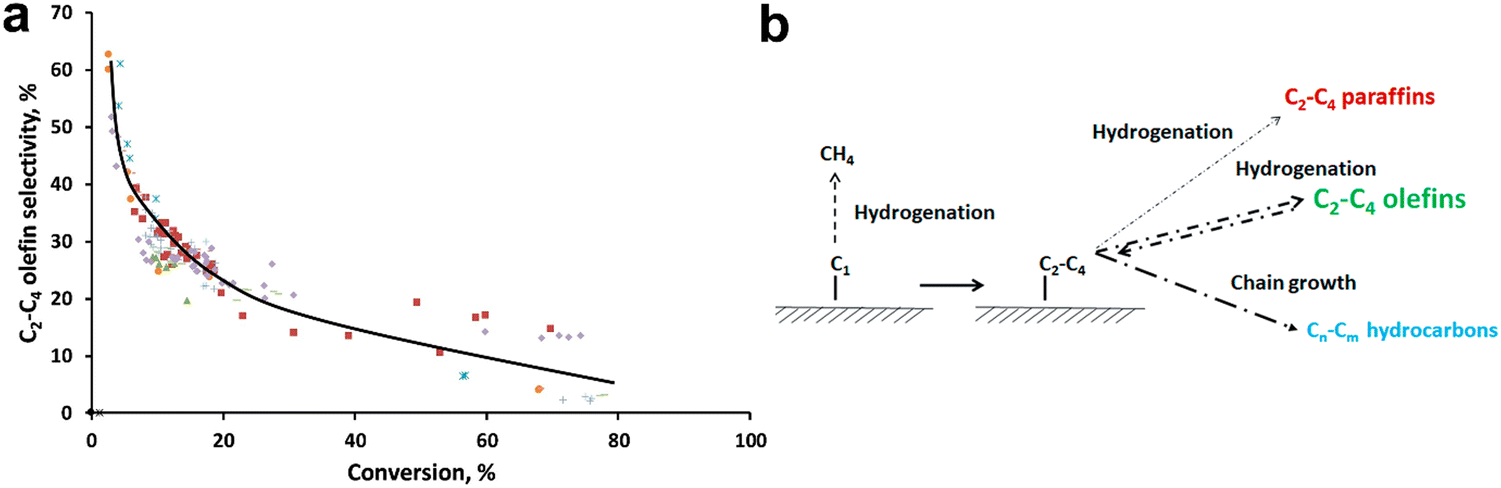 | ||
| Fig. 22 (a) LO selectivity versus carbon monoxide conversion over promoted Fe/SiO2, catalysts. Fe/Promoter = 100:2, H2/CO = 1, WHSV = 2.25–6.75 L (g h)−1, P = 10 bar. (b) Surface polymerization paths in high temperature FT synthesis over iron catalysts. Reprinted from ref. 334, Copyright (2020), with permission from Elsevier. | ||
First, the hydrogenation rate of adsorbed hydrocarbon fragments should be reduced. The high hydrogenation rate of C1 fragments would lead to major production of methane. The primary hydrogenation of the adsorbed C2–C4 hydrocarbon fragments and secondary olefin hydrogenation leading to relatively cheap light paraffins should be also slowed down. Second, the selectivity to LOs can be improved by slowing down the chain growth. It is expected that slower rates of chain growth would result in a smaller average carbon number of hydrocarbons and avoid the formation of large quantities of long chain hydrocarbons. All these strategies should increase the selectivity to LO. The olefin selectivity in FT synthesis can be also improved335 by co-feeding with carboxylic acids. The effect has been assigned to the stabilization of olefins in the presence of acids with intermediate formation of esters followed by their subsequent decomposition. Recently, Single-Event MicroKinetic modelling was extended330 to investigate the influence of catalyst properties i.e. catalyst descriptors, on the yield of C2–C4 olefins in FTO process. The three catalyst descriptors, i.e. atomic chemisorption enthalpies of hydrogen (QH), carbon (QC), and oxygen (QO) in the SEMK modelling approach have a combined effect on the conversion, whereas the selectivity to LOs is found to be less sensitive to QO.
CO2 hydrogenation to hydrocarbons (also called CO2-FT synthesis) is even a more complex multi-stage reaction9,323,336,337 than FT synthesis with CO. The first step in CO2-FT synthesis over iron catalysts is CO2 hydrogenation to CO (via RWGS) followed by the formation of C1 adsorbed monomers. Then, the adsorbed C1 monomers transform similar to those in FT synthesis with syngas. Our recent results suggest338 that the rates of hydrogenation of adsorbed C2–C4 species and rates of oligomerization could be different over the catalysts with and without alkaline promoters. Over the catalysts containing alkaline metals, the oligomerization to the C5+ hydrocarbons limits the LO selectivity, while the contribution of hydrogenation of adsorbed C2–C4 species or secondary hydrogenation of LOs to paraffins is not significant. This implies that, to boost the LO selectivity over the catalysts promoted with alkaline metals, the surface oligomerization should be hindered after the formation of the C4 surface fragments. Similar effects were previously observed in the CO hydrogenation over iron catalysts.334 Though the CO and CO2 hydrogenation have much in common, it is rare that the catalysts showing high selectivity to LOs in FT synthesis exhibit high LO selectivity in CO2 hydrogenation and vice versa.
The structural promoters (silica, alumina, or other oxides) modify the catalyst texture, enhance iron dispersion, affect iron reduction/carbidisation and improve the catalyst stability and attrition resistance. They do not affect, however, the intrinsic activity of active sites, often expressed as turnover frequency (TOF).
The electronic promoters interact directly with the active sites and affect their intrinsic activity. Consequently, the rate of adsorption/desorption and elementary reaction steps can be affected. The electronic promoters for iron FT catalyst can be divided334 into three major groups: (i) alkali metals, (ii) transition metals and (iii) sodium–sulphur promoter. Numerous studies have shown that addition of small amounts of potassium to iron catalysts used for FT synthesis affects the performance of such catalysts in a variety of ways.339–345 It is well-known that the IA group alkaline metals, especially potassium, are essential promoters in iron FT catalysts. They have a markable effect on both activity and selectivity. The role of potassium on iron-based catalysts has been previously studied; however, the effect of potassium on catalyst activity varies considerably as a function of the catalyst composition, temperature and the presence/amount of binders.
The alkali promotion affects the electronic structure of the active phase. The promotion with alkali metals varies as a function of catalytic support.347 A part of iron and alkali species can form mixed oxide compounds with the support, which do not have any activity in FT synthesis. The electronic interaction of iron species and alkali may modify the intrinsic reaction rate and selectivity. Alkali ions could enhance344 carbon monoxide dissociation, because of election-donation effect on the iron species from basic oxygen species. The promotion by alkali metals increases the olefin to paraffin ratio in the reaction products. The chain growth probability and selectivity to C5+ hydrocarbons are much higher over alkaline-promoted iron catalysts. These two simultaneous phenomena do not necessarily lead to higher selectivity to LOs.345 The WGS activity, carbon deposition and catalyst deactivation rates usually increase in the alkaline-promoted catalysts.
The second group of promoters includes transition metals and more particularly copper.316,348,349 The major copper function is to decrease the temperature required for the reduction and carbidisation of iron oxides, while the reports about the effect of copper on the selectivity are still controversial. Wachs350et al. and O’Brien351et al. did not observe any effect of copper on the product selectivity. Bukur352et al. reported an increase in the average molecular weight of hydrocarbon products over Cu-promoted iron catalysts. Coville353et al. observed a decrease in methane selectivity after the promotion with copper. The promotion with Mn usually leads to higher LO selectivity compared to non-promoted Fe catalysts.354–358 The recently proposed (Fig. 23) multifunctional hydrophobic core–shell FeMn@Si catalyst346 suppressed the CO2 and CH4 selectivity with an olefin yield of up to 36.6% at a CO conversion of 56.1%. The promotion of iron catalysts with molybdenum has shown a significant increase in the catalytic activity,359,360 a higher LO selectivity and an enhanced stability.361 The Cr-promoted precipitated Fe catalysts showed enhanced selectivity for longer chain hydrocarbons.356,362
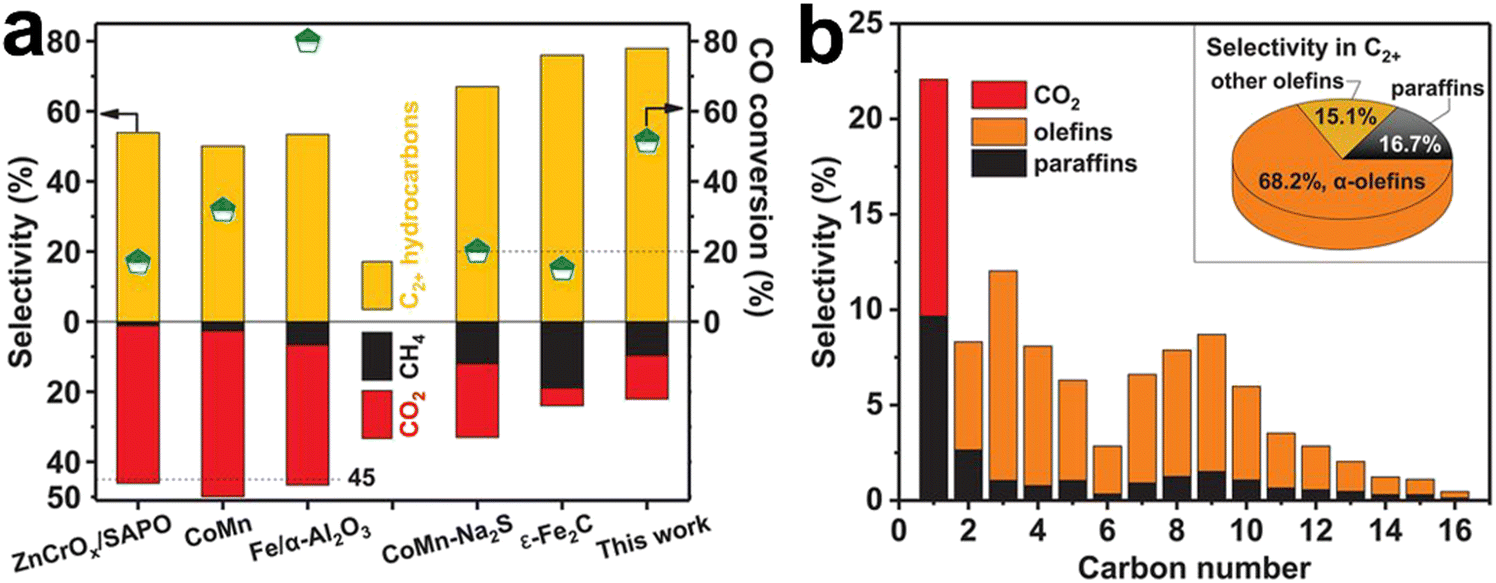 | ||
| Fig. 23 Catalytic performance for syngas to olefins. (a) Product distribution (including CO2) of FeMn@Si-c in comparison with previous works. (b) Detailed product distribution (including CO2) and the selectivity of olefins in C2+ hydrocarbons (inset) over FeMn@Si-c. From ref. 346 reprinted with permission from AAAS. | ||
The group of de Jong308,309,363et al. proposed a promotion strategy, which involves the combined addition of sodium and sulphur to the iron catalyst. Higher C2–C4 olefin selectivity and less significant methane production were reported, while the overall activity was only slightly improved.
Recently, we uncovered364,365 a new type of promotion of iron catalysts with “soldering metals”, which have low melting points such as tin, antimony, lead and bismuth. The promotion of iron catalysts with bismuth results in the formation of core–shell structures in the activated catalysts (Fig. 24). A several-fold increase in FT reaction rates was observed in the presence of small amounts of these elements. The effect of these promoters was one of the strongest ever observed in the literature.366,367 The promotion with these elements has two particular features. First, these metals are highly mobile under the conditions of high temperature FT synthesis. Their migration during catalyst activation and catalytic reaction has been observed368 by in situ TEM and NAP-XPS. Second, these metals have several oxidation states. The mechanistic study364 suggests that these elements can facilitate CO dissociation by scavenging oxygen from the surface of iron carbide by the promoters resulting in their re-oxidation. The strong interaction of iron and promoters369 leads to higher TOF. The presence of these metals can also stabilize iron carbide nanoparticles from sintering. Bi, Pb, Sn and Sb can be therefore considered as both electronic and structural promoters of iron FT catalysts.
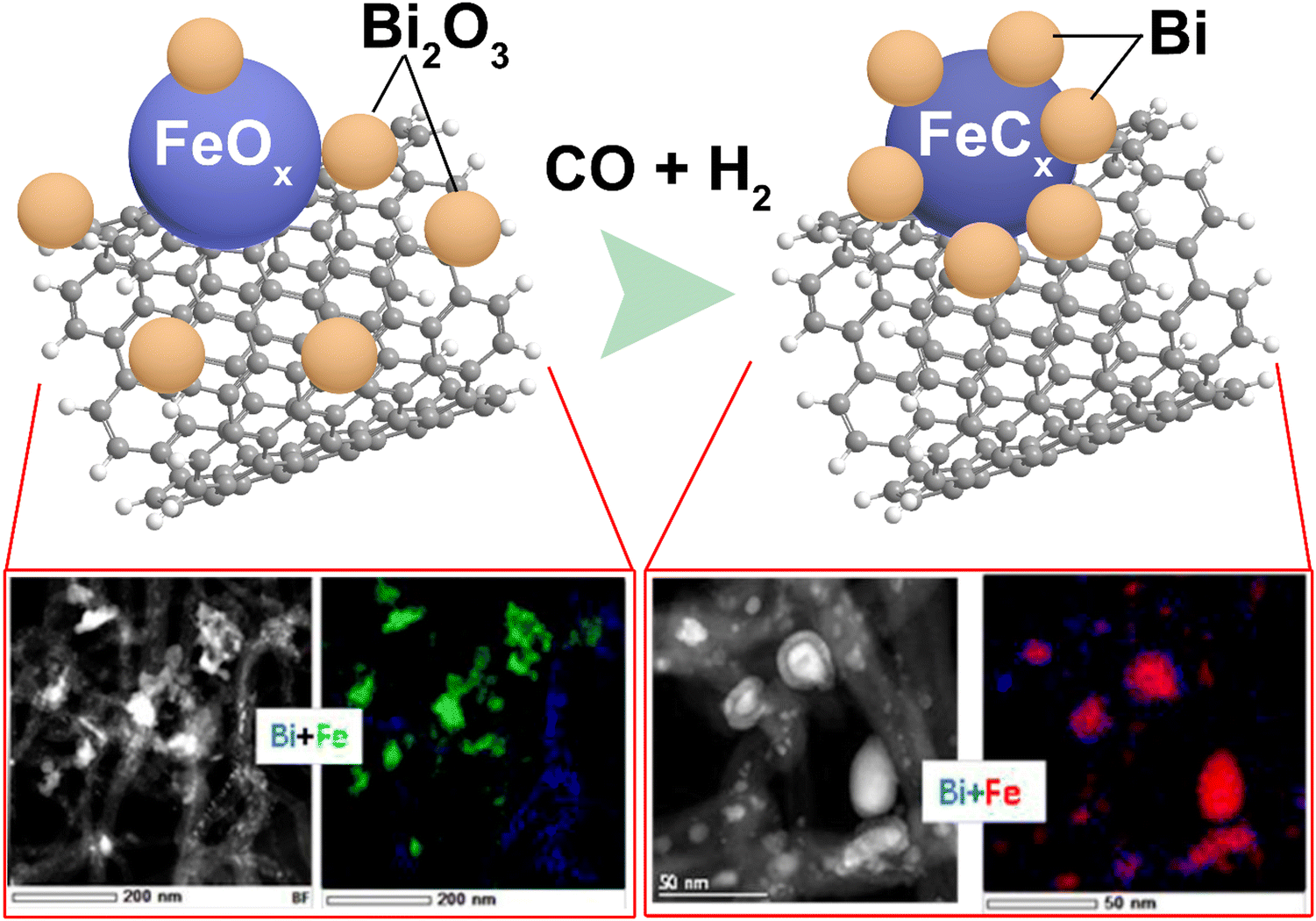 | ||
| Fig. 24 Migration of bismuth during catalysts activation and STEM-EDX images of the FeBi/CNT catalysts before (a) and after (a) carbidisation. | ||
The iron FT catalysts developed in the last decades are usually supported. Historically, mechanical strength was a major motivation for the development of supported iron catalysts, especially for application in fluidized bed or slurry reactors.370 The catalyst supports may favourably affect numerous characteristics of iron catalysts such as:
– mechanical stability and low attrition of supported iron FT catalysts;
– enhancement of iron dispersion in the supported FT catalysts;
– electronic interaction of iron species with the support;
– influence of the diffusion on the catalytic performance of the supported iron catalysts;
– the emergence of additional active sites related to the support sites, which affect the conversion of the intermediates and products of FT synthesis. The term “bifunctional catalysts” is often employed.
The preparation of supported catalysts usually involves conditioning of the support, deposition of active phase and promoters using different precursors followed by drying and calcination at high temperatures. The use of preformed support materials is a potential way to alleviate the catalyst particle breakage (attrition) problem in fluidized bed and slurry reactors. Most of the previously developed supported catalysts showed, however, lower activity and exhibited undesirable high methane selectivity.371 The inadequate activity and selectivity toward desired hydrocarbon products have been attributed372 to strong support–metal interaction, the reduction of the promotion effectiveness due to dissolution in the support matrix373 and ineffective preparation methods.374
The iron–support mixed compound can be produced during catalyst calcination, catalyst activation procedure or during the reaction. The higher temperature in the presence of water vapor can favour their formation. Possible solutions to reduce the formation of iron–support mixed compounds could be the optimization of support morphology and chemical composition and conditions of catalyst calcination and reduction. Some iron support mixed compounds can be still produced in FT synthesis at higher carbon monoxide conversion.
Another important characteristic of the catalysts, which can be affected by the support, is iron dispersion. The size of metal oxide nanoparticles on the supported catalysts is principally affected by the decomposition of metal precursors, germination and growth of metal oxide crystallites. Similar to previous results for cobalt catalysts,375,376 the support pore diameters seem to be a more important parameter compared to the surface area for controlling iron dispersion. Smaller iron particles and consequently higher iron dispersion are usually obtained in narrow pore supports. The effect of support pore size on iron dispersion has recently been studied in detail for silica supported catalysts.312 Larger iron oxide crystallites were detected in large pore supports. The larger iron particles were much easier to carbidise than smaller ones located in narrow pore supports. Y zeolites were used377 to confine iron nanoparticles within the zeolite cavities and yielded catalysts exhibiting 36.2% selectivity to LOs at a CO conversion of 91.2%.
Several publications suggest that high support surface area is not a prerequisite for better catalytic performance, as it can lead to the formation of mixed iron-support compounds and lower carburization. Promising results for CO hydrogenation to olefins were reported using low surface area supports such as α-alumina.367 The main disadvantage of catalytic support with low interaction toward the iron phase is sintering of iron carbide nanoparticles during the reaction and activation resulting in the loss of active surface area.
Carbon materials (carbon nanofibers, carbon nanotubes, carbon mesoporous materials)378 are broadly used as supports for iron FT catalysts. They have many advantages.379 First, carbon supports do not interact with iron oxides with the formation of mixed barely reducible and barley carbidised species. Second, higher dispersion and smaller particles of the active species can be obtained because of the higher surface area of carbon materials and their highly developed porosity.347 Third, because of the rich surface chemistry, the catalytic behavior of carbon-supported catalysts can be controlled via proper activation and post-treatment methods. The amounts and types of oxygen-containing functionalities in carbon materials could change surface acid hydrophilic/hydrophobic properties. Note that carbon supported catalysts generally have weak mechanical stability. In addition, handling of most the conventional catalysts requires oxidative treatments during either catalyst preparation, activation or regeneration. Possible oxidation limits the use of carbon materials as supports.
Riedel380et al. claimed that the iron phases in the non-reduced catalyst comprised mostly of α-Fe2O3 and Fe3O4. During the activation, magnetite and hematite phases are consumed and a new, most probably oxidic iron phase is generated, which seems to be responsible for the activity in the RWGS reaction. Finally, FT activity starts with the formation of Hägg carbide (χ-Fe5C2) by the reaction of iron with carbon or carbon monoxide. In order to activate the CO2 molecules, it is crucial to modify the surface basicity to enhance the adsorption ability towards CO2. The impact of impregnating with several rare earth and transition metals on the catalytic properties in CO2 hydrogenation has been widely studied in the last years. Wang et al.324 added alkali metal ions to Fe/ZrO2 catalysts. They found that these metals (except Li) considerably reduced the selectivity to methane and lower paraffins at the same time that they improved the selectivity to LOs and C5+ hydrocarbons, predominantly C5+ olefins. Additionally, they established that impregnation with Na, K, or Cs enhanced the CO2 conversion as well. Additionally, the most promising results for lower olefin synthesis were achieved over a K-modified Fe/ZrO2 catalyst. This better performance of potassium promoted catalysts was explained by assuming that K could speed up the Hägg carbide formation.
Other extensively studied promoting metals are manganese and zinc. Mn can act as both an electronic modifier and a structural promoter for iron catalysts. It is claimed that the addition of Mn increases the olefin/paraffin ratio in the CO2 hydrogenation and at the same time, restrains the formation of CH4.381,382 In addition, manganese enhances carburization, dispersion, and reduction of iron oxides, and at the same time, significantly improves the surface basicity of the catalyst.383 Nevertheless, a load excess of Mn reduces the promotional effect.381 Iron catalysts simultaneously promoted with zinc and alkaline metals were reported384–386 to exhibit high selectivity to long chain olefins in the CO2-FT synthesis. Zhang385et al. reported extremely high selectivity to LOs in the CO2 hydrogenation after the combined promotion of iron catalysts with potassium and Zn.
ZrO2 is currently the most common support9 for CO2 hydrogenation catalyst, due to its high surface basicity, which favours adsorption of CO2 at the reaction condition. Wang324et al. performed numerous experiments with iron catalysts supported by SiO2, Al2O3, TiO2, ZrO2, mesoporous carbon and carbon nanotubes (Table 2). The best LO selectivity was observed on the ZrO2 and TiO2 supported catalysts (46% and 43% respectively). Another studied support in the CO2 hydrogenation is ceria (CeO2). Torrente-Murciano387et al. prepared Fe catalysts by using ceria with different morphological properties. Among the different ceria materials used, the one presenting cubic morphology facilitated the reducibility of Fe species. This result was supported by the shift of the initial reduction temperature towards lower temperatures, giving place to the highest olefin/paraffin ratio compared to the rod-type and nanoparticle-type ceria.
| Support | CO2 conv. (%) | Carbon selectivity (C mol%) | Hydrocarbon distributiond (C mol%) | Yieldd (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CO | CnHmc | C-oxy | CH4 | C2–4 |
C2–4 | C5+ |
C5+ | C2-4 |
||
|
a Reaction conditions: W(catalyst) = 1.0 g, H2/CO2 = 3, T = 613 K, P = 2 MPa, F = 20 mL min−1, time on stream = 10 h.
b Meso-C denotes mesoporous carbon.
c CnHm denotes hydrocarbons; other products are CO and oxygenates.
d C2–4: C2–C4 olefins; C2–4: C2–C4 paraffins; C5+: C5+ olefins; C5+: C5+ paraffins.
|
||||||||||
| SiO2 | 7.1 | 91.6 | 8.1 | 0.3 | 71.8 | 17.7 | 8.6 | 0.5 | 1.3 | 0.1 |
| TiO2 | 21.0 | 54.5 | 37.6 | 7.9 | 21.8 | 42.6 | 7.2 | 17.8 | 10.7 | 4.8 |
| Meso-Cb | 32.8 | 31.0 | 57.6 | 11.4 | 31.8 | 29.8 | 25.6 | 1.9 | 10.1 | 6.0 |
| c-CNT | 34.8 | 12.0 | 73.7 | 14.3 | 25.5 | 33.8 | 10.5 | 19.4 | 11.2 | 13.7 |
| Al2O3 | 32.5 | 16.8 | 76.3 | 6.9 | 16.7 | 36.1 | 6.5 | 28.3 | 13.4 | 15.9 |
| ZrO2 | 41.7 | 15.1 | 64.4 | 20.5 | 19.8 | 45.6 | 8.2 | 17.6 | 8.6 | 17.0 |
The structure of iron catalysts depends on choosing the appropriate active phase, active phase dispersion, support, promoters, etc. Optimization of the catalyst structure could therefore result in an enhanced selectivity and yield of LO. Optimization of catalyst activation conditions (e.g. treatment with CO, hydrogen and syngas, temperature of activation) can increase the concentration of active sites in the catalysts and catalytic performance in the olefin synthesis. Finally, reaction temperature, reactant pressures, residence time and co-feeding for example with organic acids335 also affect the reaction rate and selectivity of CO and CO2 hydrogenation.
4.2 Methanol-mediated CO and CO2 hydrogenation
The main drawback of FT synthesis is the kinetic limitations concerning the LO selectivity since the product distribution is set by the ASF statistics. Only up to 60 mol% of C2–C4 olefins can be obtained among the hydrocarbon fractions in both CO2- and CO-based FT synthesis (Fig. 21). The presence of large amounts of by-products complicates the product separation. In particular, the production of large amounts of methane and light paraffins is undesirable, due to the difficulties of their removal from the LO mixture. Thus, FT synthesis is more suitable for the production of liquid fuels, which do require a high selectivity to specific hydrocarbons, while more selective routes for COx-to-LO process need to be developed.One of the possible solutions can be CO2 or CO hydrogenation to LOs through the methanol-mediated route. This approach was proposed a few years ago by the groups of Bao and Wang388,389 for the syngas-to-LO case but then was also extended for the CO2 hydrogenation to LOs. It implies a two-step process: during the first stage, CO or CO2 are hydrogenated to methanol and then the latter is converted to LOs through methanol-to-olefins (MTO) reaction (Fig. 25). In general, the methanol-mediated process can be considered as a combination of the main reactions:
| CO2 + 3H2 → CH3OH + H2O |
| CO + 2H2 → CH3OH |
| 2CH3OH → CH3OCH3 + H2O |
| CH3OH/CH3OCH3 → CnH2n + H2O |
| CO2 (or CO) + 4(or 3)H2 → CH4 + 2(or 1)H2O |
| CO2/CO + H2 → C5+ + H2O ← CH3OH/CH3OCH3 |
| CO2 + H2 ↔ CO + H2O |
| CnH2n + H2 → CnH2n+2 |
 | ||
| Fig. 25 Components of the tandem catalysts for LO synthesis from CO2: mixture of methanol synthesis and MTO catalysts. | ||
As a result, the carbon oxides-free LO selectivity up to ∼90% on a carbon basis can be achieved (Table 3). Such promising LO selectivity of the tandem process is due to the higher selectivity of MTO process (>85%). Thus, compared to FT synthesis, the absence of ASF kinetic limitations facilitates selective LO synthesis. The amount of coproduced methane in the outlet products is relatively low. Note, however, that because of higher catalyst activity in the WGS and RWGS reactions, both CO and CO2 hydrogenation processes produce respectively large amounts of CO2 and CO.390
| Catalyst (CO + H2) | P (bar) | T (°C) | WHSV NL gcat−1 h−1 | X CO (%) | S CO2 (%) | S LO (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a CO- or CO2-free selectivity to LO. b GHSV, h−1. c CO was added to the reaction mixture. | |||||||
| ZnCrOx-AlPO-18 | 40 | 390 | 1200b | 25.3 | 48 | 83 | 392 |
| ZrCeZnOx-SAPO-34 | 25 | 400 | 3.9 | 24.1 | 44.8 | 80.2 | 393 |
| ZnO-ZrO2-SAPO-34 | 30 | 400 | 3.6 | 25 | 41 | 75 | 394 |
| ZrCeZnOx-SAPO-34 | 10 | 400 | 3.9 | 12.6 | 44.6 | 82 | 395 |
| ZnAlO-SAPO-34 | 30 | 400 | 3.0 | 24 | 44 | 80.6 | 396 |
| Mn-Ga-SAPO-34 | 25 | 400 | 4.875 | 8.6 | 44.5 | 68.3 | 397 |
| CrMnGa-SAPO-34 | 30 | 400 | 3.0 | 37.6 | 46.2 | 87.4 | 398 |
| Catalyst (CO2 + H2) | P (bar) | T (°C) | WHSV (NL gcat−1 h−1) | X CO2 (%) | S CO (%) | S LO (%) | Ref. |
|---|---|---|---|---|---|---|---|
| ZnGa2O4-SAPO | 30 | 370 | 5.4 | 13 | 48 | 86 | 399 |
| In2O3-ZrO2-SAPO | 20 | 400 | 2.16 | 36.2 | 86.5 | 43 | 400 |
| 20 | 400c | 2.16c | 37.9c | 50c | 72c | ||
| ZnO/Y2O3-SAPO-34 | 40 | 390 | 1.8 | 27.6 | 85 | 83.9 | 401 |
| Cu-CeO2-SAPO-34 | 20 | 396 | 5.8 | 15.2 | 57 | 63.1 | 402 |
| NiCu/CeO2-SAPO-34 | 20 | 375 | 12 | 15 | 65 | 77 | 403 |
| ZnAl2O4-SAPO-34 | 30 | 370 | 15.9 | 15 | 49 | 87 | 404 |
| InCrOx-SAPO-34 | 35 | 350 | 1.14 | 33.6 | 55 | 75 | 405 |
| In2O3-SAPO-34 | 30 | 350 | 9 | 14.1 | 60.9 | 76.9 | 406 |
| ZnZrOx&Bio-ZSM-5 | 30 | 380 | 2.0 | 10 | 82 | 60 | 407 |
| ZnO/ZrO2-MnSAPO-34 | 20 | 380 | 4800b | 21.3 | 42.2 | 61.7 | 408 |
| Mn2O3/ZnO-SAPO-34 | 30 | 380 | 3.6 | 29.8 | 55.1 | 80.2 | 409 |
Despite the theoretically high selectivity to LO in the methanol-mediated route, it also faces several problems, which come from the same origin: methanol synthesis catalysts usually operate at lower temperatures (250–300 °C) and high pressures of 3–5 MPa, while the MTO process is realized at 400–500 °C and quasi atmospheric pressure. Thus, it is difficult to find intermediate optimal conditions, where tandem catalysts will show noticeable activity and high selectivity in both processes. Usually, the CO and CO2 hydrogenations using the methanol-mediated routes proceed at temperatures of 350–400 °C, which are too high for classic methanol synthesis, and pressures up to 5 MPa, which are too high for the MTO process. This induces several shortcomings of the tandem process, which are discussed in the following paragraphs. The LO synthesis in the methanol-mediated routes is facilitated by coupling methanol synthesis and MTO. Despite the CO2 hydrogenation to methanol is a thermodynamically unfavorable process at higher temperatures, the MTO stage is favoured by the thermodynamics and drives the overall reaction to LOs.391 The main parameters of LO synthesis via methanol-mediated route are total pressure, relative pressures of carbon oxides and hydrogen, temperature and space velocity.
In case of CO2 hydrogenation, the majority of studied catalysts showed CO-free LO selectivity in range of 60 to 95% depending on the catalyst and operating conditions.
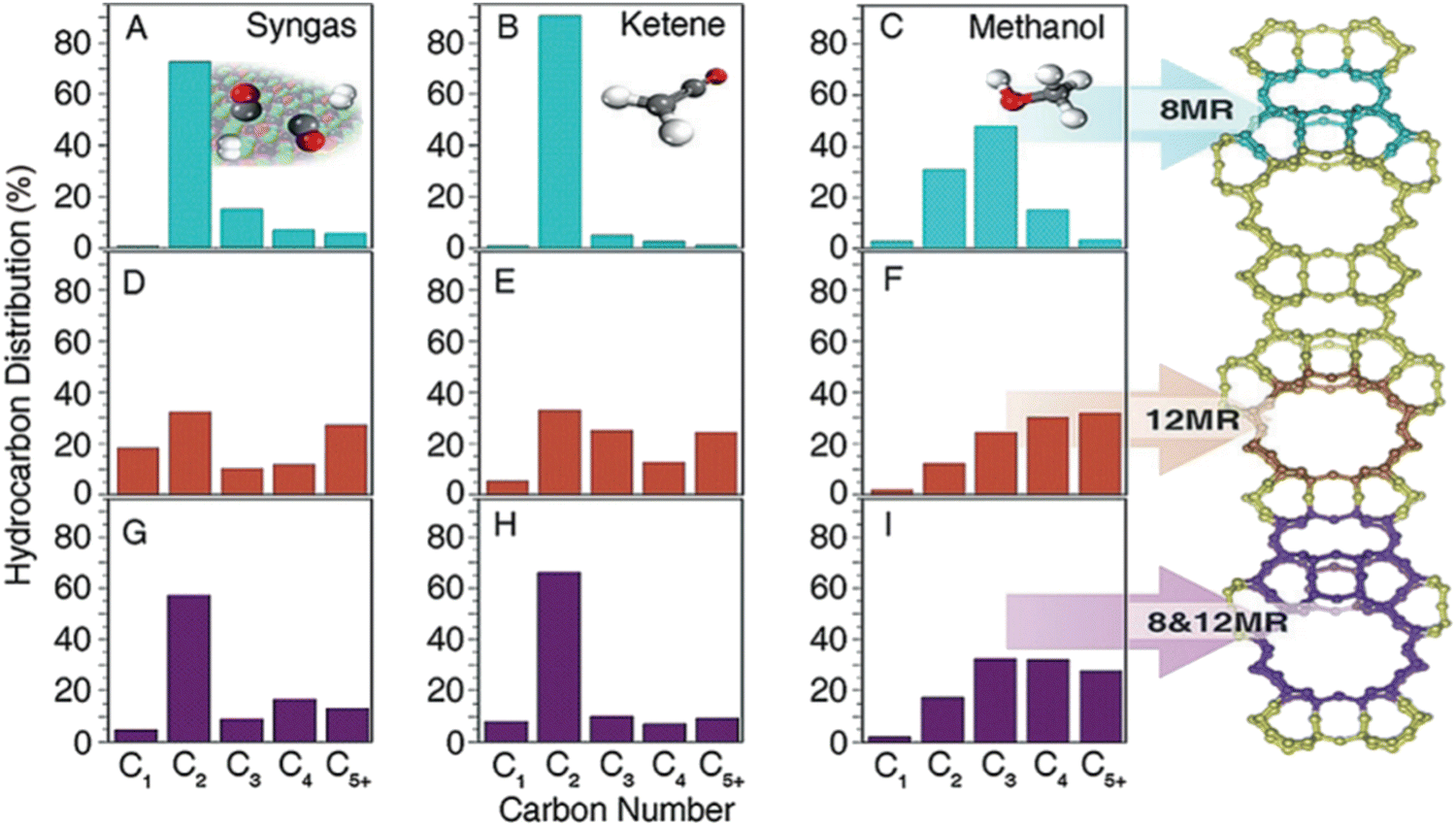 | ||
| Fig. 26 Hydrocarbon distributions in the conversion of syngas, ketene, and methanol over different sites of MOR zeolites: syngas over ZnCrOx-MOR (A, D and E); ketene over MOR (B, E and F); methanol over MOR (C, F and I). From ref. 410 reprinted with permission from John Wiley and Sons. | ||
Together with ketene-mediated route, the formaldehyde-mediated one is also proposed. Wang et al.412 confirmed by time-resolved DRIFTS and in situ XPS study that formaldehyde HCO* and H2CO* species were first produced after the CO adsorption and protonation. Then, such species are rapidly converted to HCOO* or H2COO* by the lattice oxygen, which is then hydrogenated to methoxy CH3O*. At the last step, methoxy species are protonated forming methanol, which is transferred to the zeolite. The methanation process can also occur by hydrogenation of methoxy species with C–O bond breaking. In parallel, adsorbed CO may interact with OH* produced by water dissociation forming carboxylate COOH*, which is then hydrogenated to HCOOH* and decomposes into CO2 and H2. The formate and methoxy-species were also detected by in situ DRIFTS in the work of Luo413et al. The authors proposed that oxygen vacancies activated CO molecules to produce formate intermediates with further hydrogenation of the latter. The formaldehyde-formate initiation was also proposed.412
One of the first attempts to reveal the mechanism of CO2 hydrogenation to LOs through the methanol route was published391 in 2017 for zinc-zirconia oxide mixed with SAPO-34. First, the authors proved that such a process was tandem and consisted of coupling reactions of MeOH synthesis and MTO by testing separately oxide phase and zeolite. The zinc-zirconia sample showed only methanol in the products, while SAPO-34 did not work at all. Moreover, increasing the proximity between two components led to the growth of LO selectivity and a decrease in the CO yield. This may suggest that the same intermediates are involved in the LO and CO formation, while SAPO-34 drives their transformation towards the olefins. Combined DRIFTS and MS studies during the reaction revealed the presence of both HCOO* and CH3O* species on the ZnZrO surface without SAPO-34 and almost only HCOO* with the zeolite. This suggests that the CH3O* species are moved to the zeolite to form olefins during the tandem process:
| CO2 + H2 → oxide → HCOO* → HCO* → H3CO* → H3COH → SAPO-34 → LO |
 | ||
| Fig. 27 Proposed oxygen vacancy-assisted mechanism of methanol formation on the ZnAl2O4 surface during CO2 hydrogenation to LO (a); in situ FTIR spectra of adsorbed species on ZnAl2O4: CO adsorption (dotted lines) and then switching to H2/CO flow at 533 K (b and c), CO2 adsorption (dotted lines) and then switching to H2/CO2 flow at 623 K (d and e). Reprinted with permission from ref. 404 Copyright 2020 American Chemical Society. | ||
Wang405et al. studied In-based catalysts for CO2 hydrogenation to LOs and proposed the mechanism not only for the direct process, but also for the side reactions such as methanation and RWGS. Using DFT combined with DRIFTS and MS, they showed that after the initial CO2 adsorption on the oxygen vacancies resulting in formate HCOO* and its hydrogenation to H3COO*, the latter might be protonated or hydrogenated to methane by breaking the C–O bond. The RWGS process is realized via the formation of carboxylate COOH* and COOH2* which are decomposed to CO and H2O. In this study, the authors also showed that addition of ceria or chromium oxide to indium oxide inhibited the methanation process due to the much higher free energy barriers for methane formation from CH3O* species compared to pristine In2O3. At the same time, the chromium oxide-modified indium catalyst demonstrated a higher RWGS activity due to a stronger electron interaction between its surface and COOH* species.
| Parameter | Effect |
|---|---|
| Hydrogenation component | Higher hydrogenation ability – higher selectivity to paraffins, methane (Cu) |
| Zn oxide + Cr/Ga/Al/Zr oxides = universal catalyst for both CO and CO2 hydrogenation | |
| In or Ga is suitable only for CO2 hydrogenation, because of the reduction of their oxides in CO to the metallic phase which exhibits high methanation activity | |
| Zeolite type, acidity | Hydrocarbon distribution. ZSM-5: propene + C5+ and aromatics; SAPO-34, SSZ-13 and MOR: mostly ethene, propene; RUB – for propene, butane |
| Moderate acidity – best selectivity to LO | |
| The proximity between oxide and zeolite | In the case of metals with high mobility (Zn, In) moderate proximity avoids deactivation of zeolite sites by metal ions |
| Unreducible and immobile metal oxides (Cr2O3, ZrO2): higher proximity is better for activity and selectivity | |
| Oxygen vacancies concentration | A large concentration of oxygen vacancies produces a favorable effect on the reaction but should be in combination with hydrogen activation sites |
| Temperature | Optimal: 350–400 °C; lower – weaker MTO, higher – olefin hydrogenation, intensive RWGS |
| Pressure | Optimal: 10–30 bar. Lower – poor activity in methanol production, higher – olefin hydrogenation |
| Space velocity | Higher – better LO selectivity, lower conversion; lower – higher conversion, lower LO selectivity due to hydrogenation to paraffins. Compromise at ∼3–4 NL gcat−1 h−1 |
The activity of tandem catalysts is usually assigned to the concentration of oxygen vacancies, which was the highest for the most active systems (Fig. 28a).399,404,413,416 It can be explained by the fact that oxygen vacancies facilitate chemisorption and activation of both CO and CO2. Dang416et al. studied the effect of zirconia on the In-based catalysts for CO2 hydrogenation and uncovered the presence of the In1-xZrxOy mixed oxide. Such phase contains a large number of oxygen vacancies, which sharply enhance the chemisorption of CO2 and stabilization of reaction intermediates. This fact also leads to lower RWGS activity and, hence lower CO formation rate. A linear dependence of site-time yields for both methanol and LOs on the density of oxygen vacancies was found for the ZnCeZrO/SAPO-34 catalysts prepared with different complexing agents. The authors concluded that the vacancies not only activated the CO2 molecules, but also stabilized the methanol-related intermediates. In the case of CO hydrogenation, a sharp increase in oxygen vacancy number allowed the authors to obtain LOs already at atmospheric pressure and a relatively low temperature (300 °C).412
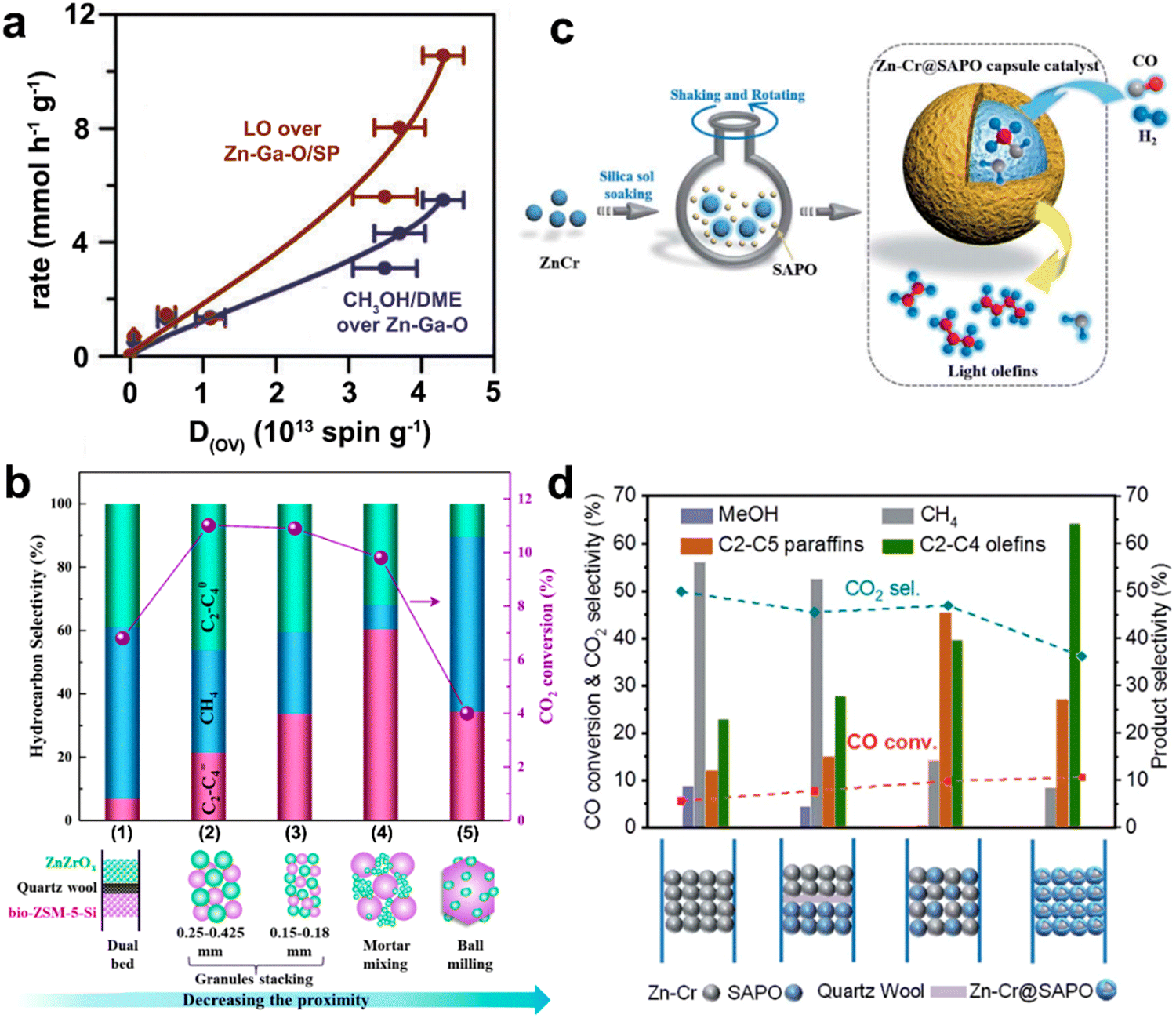 | ||
| Fig. 28 Effect of oxygen vacancies and proximity on the catalyst performance in tandem process. (a) Correlation of the formation rates of CH3OH/DME (Zn–Ga–O) and C2–C4 olefins (Zn–Ga–O/SAPO-34) with the density of oxygen vacancies D(OV).399 (b) Effect of proximity between ZnZrOx and ZSM-5 components on the catalytic performance of the bifunctional catalysts in CO2 hydrogenation: dual bed configuration (1), granules stacking with a different size (2 and 3), mixing of two powders by mortar mixing (4), and ball milling (5). Reprinted (adapted) with permission from ref. 407 Copyright 2021 American Chemical Society. (c) Preparation of the core-shell ZnCrOx@SAPO-34 catalyst. (d) Performance of this core-shell catalyst comparing with sole ZnCrOx, ZnCrOx and SAPO separated by quartz wool, and ZnCrOx with SAPO physically mixed.417 | ||
On other hand, hydrogen dissociation is found to occur on the –Zn–O–, –In–O, or –Ga–O– fragments.399,404 Note that a higher fraction of such fragments may lead to excessive hydrogenation and formation of paraffins or methane. At the same time, Zhu418et al. found that a specific catalyst such as MnO2 could provide both CO activation and further hydrogenation. To summarize, to realize the first “methanol” stage of the tandem process, the corresponding catalyst must contain two main active sites: CO or CO2 activation and H2 dissociation. This is the reason, why some systems with a larger amount of O vacancies such as Mg–Al–O showed much lower activity than those with a lower concentration of vacancies.
In the case of the second “MTO” step, the most relevant catalyst parameters are Brønsted acidity and zeolite morphology. Moderate strength is necessary, because it allows selective conversion of MeOH/DME intermediates to LOs. The strong Brønsted acid sites, instead, facilitate hydrogen transfer reactions and the formation of paraffinic hydrocarbons. In SAPO-34, NH3-TPD exhibits two main peaks: the first one, at around 200 °C, is related to medium strength acid sites; the second one, between 300–400 °C, is related to stronger sites. Luo413,416et al. have observed a reduction of the intensity of the latter peak, and a shift towards lower temperatures, after mixing SAPO-34 with oxide catalyst. The decrease in acid site density and strength has been attributed to the intimate contact between the two phases.413 Liu et al. performed CO hydrogenation to LOs using a Zn-ZrO2/SSZ-13 bifunctional catalyst. They tuned the acidity via ion exchange, by gradually increasing the H+ content from NaSSZ-13 up to HSSZ-13. They observed an improvement in the LO selectivity with increasing Brønsted acid site concentration up to 0.10 mmol g−1, followed by a reduction of LO selectivity with a further increase in the acid site amount. Along with this, they saw a continuous increase in C2–C4 paraffins selectivity with Brønsted acid site concentration. Similarly, Jiao et al. evaluated the influence of variation of the acid strength on the C2–C4 olefins/paraffins ratio. When NH3-TPD shows a peak at 395 °C, the C2–C4 olefins/paraffins ratio is 0.9.388 When the NH3-TPD peak shifts to lower temperatures with a peak at 350 °C, this ratio increases to 4.7. The strength of the acid sites can be controlled either through controlling the Si content or introducing transition metal (i.e. Mn, Zn, Zr) in the SAPO-34 structure.404,408 Addition of transition metals and decrease in Si/Al ratio promote the reduction of the strength and density of acid sites. As a consequence, an improvement in the LO selectivity can be observed.
Along with the reaction conditions, the LO selectivity can be also tuned by zeolite type and porosity. SAPO-34 is the most frequently used material for MTO stage, since it gives the highest LO yield among all the studied systems due to the unique size of cage and moderate acidity.235 The majority of papers have addressed SAPO-34 as a zeolite component for LO synthesis via the methanol-mediated route.408,419–422 Wang396et al. provided the analysis of SAPO type effect on the selectivity of CO2 hydrogenation and showed that SAPO-34 possessed the highest one towards LO fraction. At the same time, DFT calculations and catalytic experiments showed that SAPO-18 significantly enhanced the olefin-to-paraffin ratio in CO hydrogenation compared to SAPO-34 due to the suppression of hydride transfer. Consequently, higher selectivity to LOs but also to C5+ hydrocarbons was observed but at the slightly lower conversion rate.392 Hierarchical SAPO-34 porosity improved the mass-transfer inside zeolite phase. Nitric acid treatment of SAPO increased the LO selectivity due to both enhanced porosity and reduced acidity, which prevented coking.423 Stability enhancement of catalyst by the preparation of SAPO-34 hierarchical structure was also studied.394 Hierarchical zeolite structure suppresses polyaromatics formation and prevents coking, when the reaction is conducted in the presence of hydrogen.
Hierarchical porosity is also considered an important factor for the high LO selectivity in the case of ZSM-5-based ZnZrOx catalyst. Biomass-derived hierarchical ZSM-5 may provide a ∼65% CO-free selectivity to LOs and only less than 1% of aromatics.407 Bao et al. demonstrated that selective shielding of different types of MOR zeolite cages might vary the ethylene fraction in the products of syngas conversion. While the larger channel 12MR zeolites facilitate the formation of a broad range of hydrocarbons from methane to C5+, the smaller-sized “side pockets” 8MR counterparts selectively produce ethylene.410 Further studies showed that the confinement effect of the MOR cages played an important role in ketene transformations. Larger 12MR channels facilitate longer contact time of ketene and, hence, formation of a broader range of products and coking, while the 8MR confinement leads to protonation of ketene and to the formation of stable acylium ions, which further participate in catalytic reactions.424 Recently, Wang et al. showed that the application of H-RUB-13 zeolite instead of SAPO-34 provided a shift in the product distribution to propylene–butylene fraction due to the changes in hydrocarbon pool mechanism from the aromatic-mediated to alkene-based cycle.415
The last but not least important feature that may impact the activity and selectivity of the COx methanol-mediated hydrogenation to LO is the proximity of the components. The diffusion of the as-formed methanol from oxide to zeolite may play an important role in both primary and secondary reactions. The proximity of the components may regulate the local methanol concentration, which should be optimal to achieve the best LO yields.425
There are physical and chemical approaches to regulate the diffusion of reaction intermediates. The first one implies variation of the distance between hydrogenation and acid sites by a different type of mechanical mixing.388,391,407,409 Oxide and zeolite should be mixed, since their layer-by-layer packing facilitates methane production and RWGS (Fig. 28b). In general, physical mixing and grinding in the mortar, are the most optimal ways to prepare selective and active tandem catalysts. Using, for example, ball milling could be excessive and may affect negatively the performance.407 Recently, In- and Zn-species were shown to be mobile under the LO synthesis reaction conditions and can block the Brønsted acid sites and hence, destroy the catalyst bifunctionality.426 Such processes can be intensified due to the smaller distance between the components and reduce the activity and selectivity of the catalysts. Ding427et al. studied the effects of the proximity of hydrogenation and acid components in ZnCrOx-SAPO-34 and MnOx-SAPO-34. The optimal degree of proximity of ZnCrOx and SAPO-34 is required for higher LO selectivity, while in the MnOx-SAPO-34 catalyst, the activity and selectivity were higher at closer proximity.
The second chemical route for tuning the proximity consists of the formation of a core-shell structure, where the oxide component is surrounded by zeolite. Such design should increase the extent of methanol transformation to LOs and suppress the further hydrogenation of the latter products to paraffins. An interesting strategy was proposed by Tan417et al. The authors covered Zn–Cr granules with SAPO-34 crystallites using the silica sol as a binder and obtained core-shell structure, which was noticeably more active and selective to LOs than a regular mixture of Zn–Cr and SAPO-34 (Fig. 28c and d). The advantages of core-shell catalyst imply fast diffusion of methanol and water from Zn–Cr surface to SAPO-34 channels, which prevents the WGS reaction and promotes the LO formation. Synthesized LOs have less chances to be further hydrogenated on the Zn–Cr surface due to the protection of the latter by a SAPO shell. A similar effect of zeolite shell protection for secondary processes was observed in the case of CuZnZr oxide catalyst.428 Preparation of SAPO-34 in presence of the ZnZrO2 catalyst covered by alumina gave a better performance in CO2 hydrogenation than the physically mixed components and composite without alumina.429 The authors propose that Al2O3 plays a role of stabilizer for the oxide and adhesive for SAPO-34 covering of its surface: without alumina, higher selectivity to methane was observed.
Thus, two strategies should be applied for the preparation of oxide-zeolite tandem catalysts for LO synthesis from COx. For the metals exhibiting noticeable mobility, such as In and Zn, the catalyst preparation should involve building the “buffer” interface between oxide and zeolite components in order to prevent the blocking of zeolite Brønsted acid sites by the mobile metal. When working with less mobile metals such as Cr and Mn, the maximum proximity of oxide and zeolite is desirable to reach the best performance in LO synthesis.
The stability of tandem catalysts is one of the main advantages of methanol-mediated route. The long-term studies have shown high stability of the tandem catalysts. This seems to be one of the main advantages of direct LO synthesis from CO compared to the conventional MTO process, where the catalyst regeneration should be done every few hours due to the coking and, hence, pore blocking of the zeolite. The presence of hydrogen and water inside the reaction mixture over the tandem catalysts facilitates the removal of carbon species. For example, the CO2 conversion decreases from 23% to 18%, while the selectivity to LOs remained constant after 124 h of reaction over ZnGa2O4/SAPO-34 at 400 °C.399 For the In2O3–ZnZrOx/SAPO-34, the CO2 conversion remained constant as well as LO selectivity for at least 92 h.423 The similar trend was observed over other catalysts, which worked without noticeable conversion and selectivity changes for a long period of time.388,407,416,417,430
In addition, some new approaches for the tandem process should be mentioned. Hydrothermal conversion of CO2 to ethanol with further dehydration of the latter was proposed in the work of Zeng431et al. The authors used copper catalyst introduced into the structure of Ti-based MOF MIL-125-NH2 and reached the 90% selectivity to ethylene at 100 °C. They found that copper-based centres converted CO2 to ethanol, while titanium-based ones were more efficient in the EtOH dehydration to ethylene.
 | ||
| Fig. 29 Effect of reaction pressure on CO and CO2 conversion and selectivity to LO (a), selectivity to CO (b) in methanol-mediated hydrogenation. Data from ref. 391, 409 and 432. | ||
Temperature plays an important role for both selectivity and conversion of the catalysts. In general, higher temperature results in better conversion. Note that WGS or RWGS and olefin hydrogenation processes also intensify with heating (Fig. 30). The most suitable range for the best LO selectivity is 370–390 °C (Fig. 30c). Raveendra433et al. showed that an increase in temperature from 350 to 400 and then to 450 °C led to a decrease in the LO selectivity and growth of CH4 and paraffin amount. Similar results were obtained for the Zn–Cr-SAPO-34 catalyst: at a temperature higher than 400 °C, the authors also observed strong methanation and LO selectivity decrease.417 The specific complex oxide systems, such as Zn–Ce–Zr–O allowed Wang412,415et al. to achieve the high selectivity to LOs with slow WGS and methanation rate at 300 °C in case of syngas transformation and 350 °C for CO2 hydrogenation. The authors ascribed low WGS/RWGS activity to the unique structure of the catalyst: the catalyst increases the formation energy barrier of carboxylate species, which are responsible for both CO and CO2 formation in methanol synthesis.
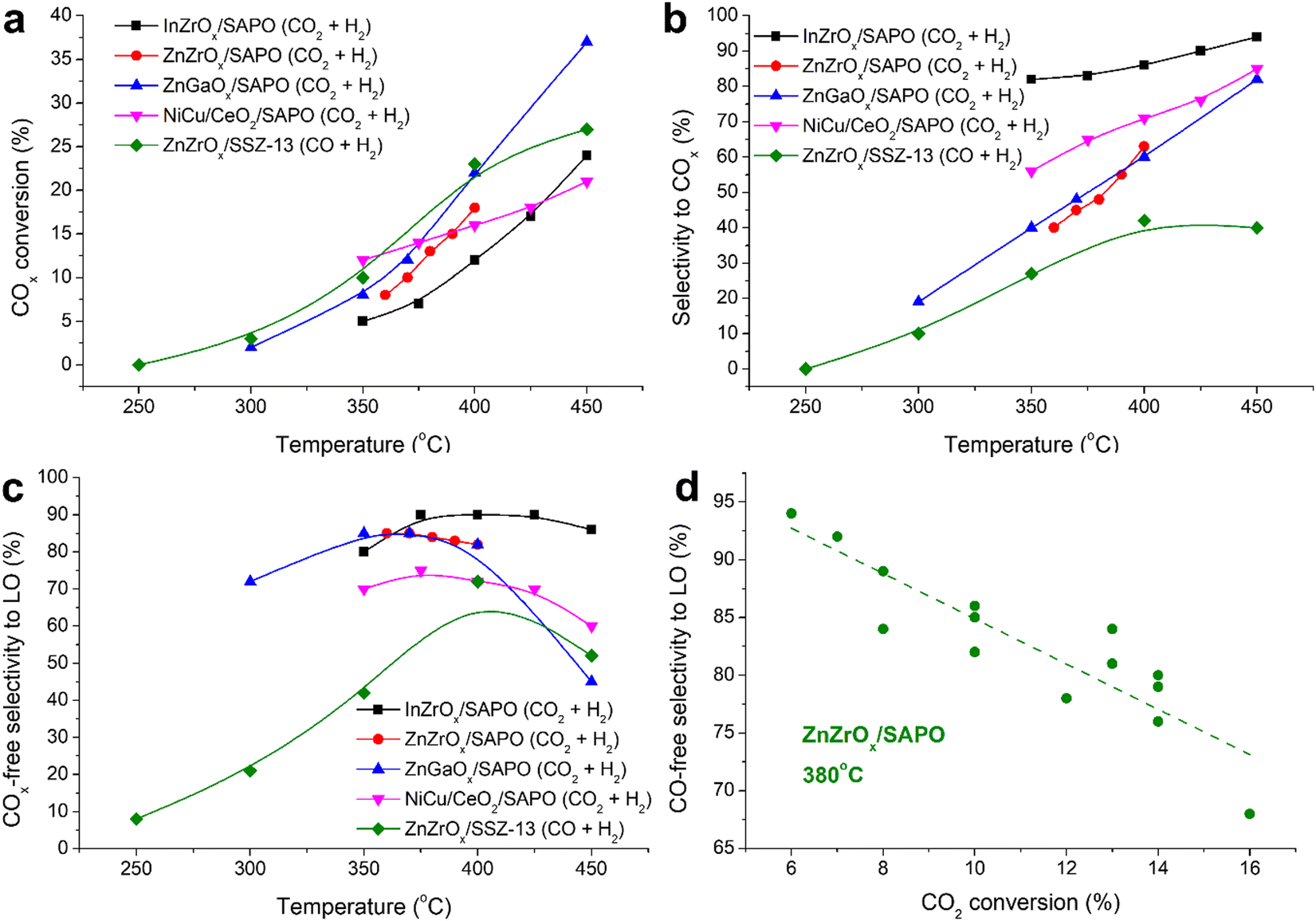 | ||
| Fig. 30 Effect of reaction temperature on the CO/CO2 conversion (a), selectivity to CO/CO2 (b) and CO/CO2-free selectivity to LO in methanol-mediated CO2 or CO hydrogenation (c). Ref. 391, 399, 403, 430 and 432 selectivity-conversion plot for CO-free selectivity to LO in the methanol-mediated CO2 hydrogenation (d) based on data from ref. 391. | ||
The space velocity (SV) affects the selectivity and product yield in different ways. First, an increase in SV leads to higher LO selectivity in both CO and CO2 hydrogenation.391,417 Higher SV significantly reduces the conversion.403 Faster flow decreases the contact time of the reagents and olefins and thus, suppresses the secondary processes such as LO hydrogenation.
Therefore, an optimum should be found for the optimal ratio between LO selectivity and conversion. For example, in the work by Numpilai434et al., optimization of reaction parameters gave the best LO yield of 7.31% in CO2 methanol-mediated hydrogenation at WHSV of only 1500 mL g−1 h−1 despite the lowest olefin-to-paraffin ratio observed at this space velocity. Sedighi and Mohammadi modelled402 the performance of CuCe/SAPO-34 catalyst and proposed different space velocities for the highest LO selectivity (17330 mL g−1 h−1) and LO yield (5400 mL g−1 h−1).
The main side processes of methanol-mediated LO synthesis routes are WGS in the case of CO as a source and reverse WGS (RWGS), when CO2 is used and the selectivity of these reactions may reach 90%. It is the most crucial in the case of CO2 hydrogenation, since the thermodynamics favors RWGS at higher temperatures in opposite to the methanol formation.391 At the same time, despite high yield of CO2 or CO, the separation of carbon oxides and their recirculation may solve it. For example, in the CO2 hydrogenation reaction, co-feeding with CO is known to significantly reduce the rate of RWGS, because of Le Chatelier's principle.393
Fig. 30d shows the selectivity-conversion dependence based on the previously published data for CO2 hydrogenation. The catalytic data used to plot this figure are also available in Table S5 (ESI†). It can be concluded that higher conversion results in lower LO selectivity, because of secondary processes: hydrogenation goes to the end and forming olefins are converted to the corresponding paraffins.
– They have comparably good performance for both CO2 and CO hydrogenation, contrary to the In and Mn-based catalysts. That is important for CO recycling in CO2 hydrogenation and CO2 recycling in CO hydrogenation.
– Zn is a much cheaper and wider-more spread element than In or Ga.
– They possess much less ecological risks than Cr-based ones which should noticeably reduce the costs of the technology.
SAPO-34 and ZSM-5 are two main possible options for the zeolite component and the choice should be done depending on the desired olefins: SAPO-34 for ethylene-propylene and ZSM-5 for propylene. Since the hydrogen presence significantly reduces the deactivation even in highly unstable in MTO SAPO-34, the choice between the zeolite type could be based only on their selectivity. Nevertheless, more detailed and long-term stability studies of tandem catalysts are necessary to clarify their real operation life span.
4.3 Electrocatalytic CO2 and CO reduction
Electrocatalytic CO2 and CO reduction reactions (CO2RR and CORR) are of great interest, because of mild reaction conditions and possible use of renewable electric power. The synthesis of LO by electrocatalysis is particularly motivating, because the existing methods require high temperature and are usually accompanied by the formation of large amounts of CO2.435 The biggest challenge for electrocatalysis is coupling of two CO2 or CO molecules,436 which is rather difficult in comparison with thermocatalytic reactions such as FT synthesis or methanol-mediated routes.10 However, the thermocatalytic routes have demonstrated a rather low selectivity to LOs due to ASF distribution in the case of FT synthesis and intensive co-production of CO2 or CO via the WGS or RWGS reactions.There are several key advantages of CO2RR and CORR, which make electrocatalysis highly attractive:437
- The use of H2O as a reducing agent is highly desirable, because it avoids additional technological process (e.g. hydrogen production), high energy consumption and generation of greenhouse gases (CO2, CH4,…).
- The reaction can be performed under very mild reaction conditions with electricity as the main energy source. Electricity can be considered renewable, if it comes from resources such as solar, geothermal, hydropower, wind energies and biomass. The cost of renewable electricity is now decreasing due to the continued substitution of fossil fuels with renewable resources.
- The coupling of C1 to olefins using electrocatalysis proceeds by a different route in comparison with the polymerization mechanism in thermocatalytic reactions and results in a higher LO selectivity (60–80%).
- The electrocatalytic CO2 reduction to LOs can be performed in both modes: direct route from CO2 to olefins by CO2RR438 and indirect route by CO2 conversion to CO with subsequent conversion of CO to LOs by CORR.439
Direct electrochemical reduction of CO2 is strongly energy demanding. The electrocatalytic reduction of CO can be performed with higher activity in comparison with CO2 due to a more positive equilibrium potential and lower consumption of protons and electrons:
| 2CO2 + 12H+ + 12e− = C2H4 + 4H2O E = 0.06 V |
| 2CO + 8H+ + 8e− = C2H4 + 2H2O E = 0.28 V |
The anode enables oxygen evolution reaction (OER) with generation of oxygen and protons:
| 2H2O = O2 + 4H+ + 4e− E = 1.23 V |
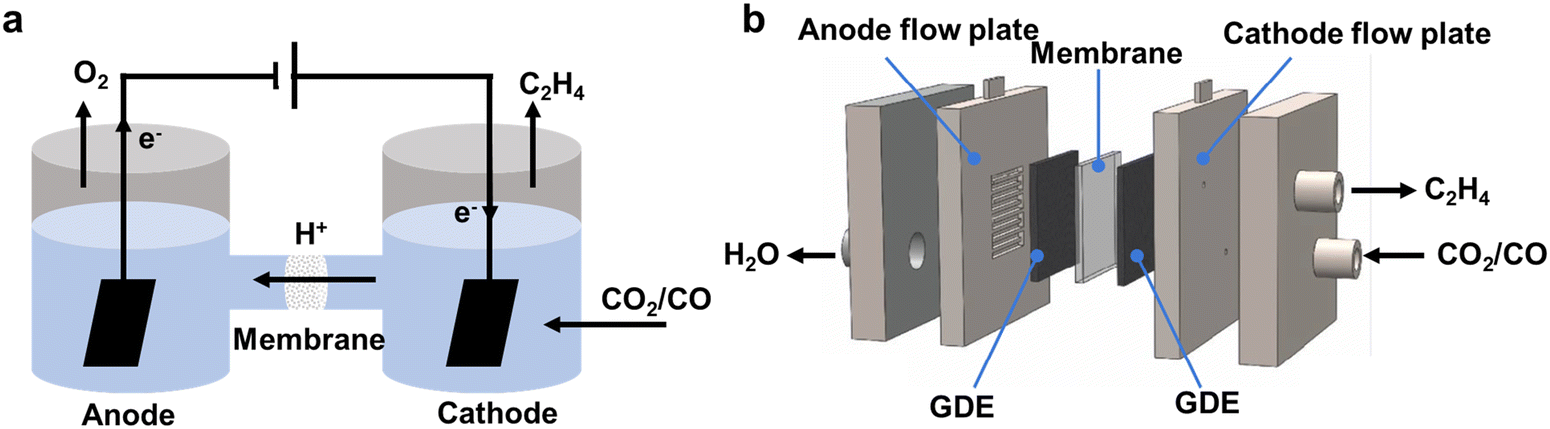 | ||
| Fig. 31 Schematic illustration of electrocatalytic reactors: H-cell (a) and MEA cell (b). Reprinted (adapted) with permission from ref. 441 Copyright 2018 American Chemical Society. | ||
The key challenge for the development of electrocatalytic reactors is low solubility of gases in aqueous solutions. Although the achieved selectivities are relatively high (up to 87%), the process is still far from industrial implementation, due to low energy conversion efficiency and high voltages. The MEA reactor is the most promising way of electrocatalytic reduction of CO2 and CO, because it does not consume aqueous electrolytes and can be scaled up.442
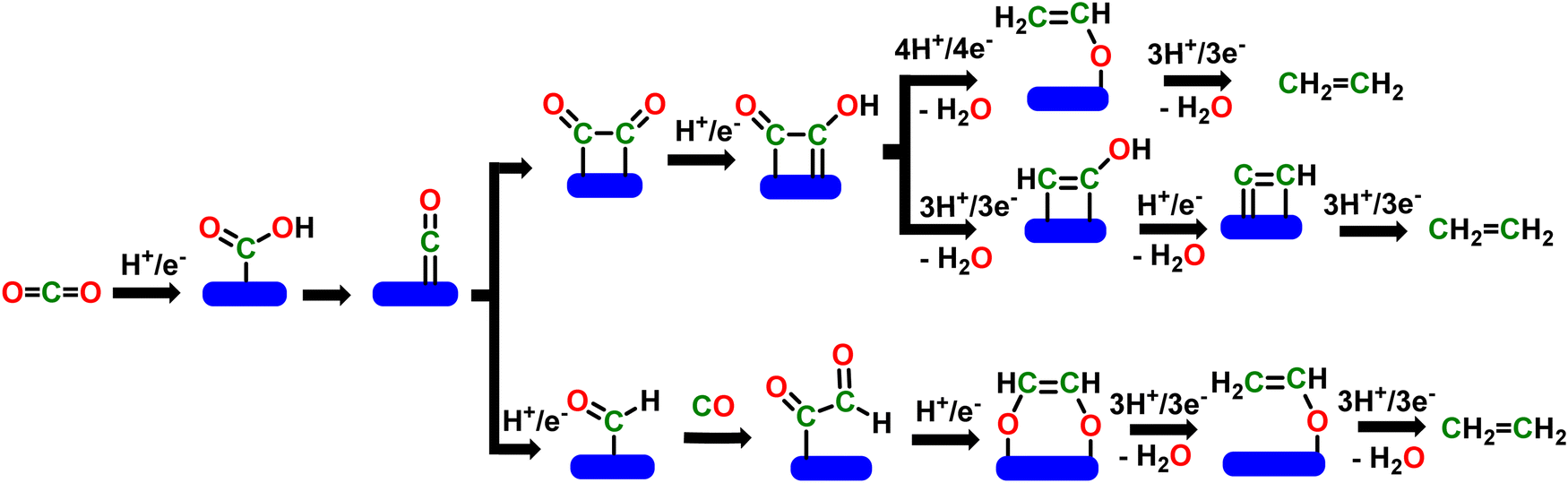 | ||
| Fig. 32 Mechanism of the electrocatalytic reduction of CO2 to ethylene. | ||
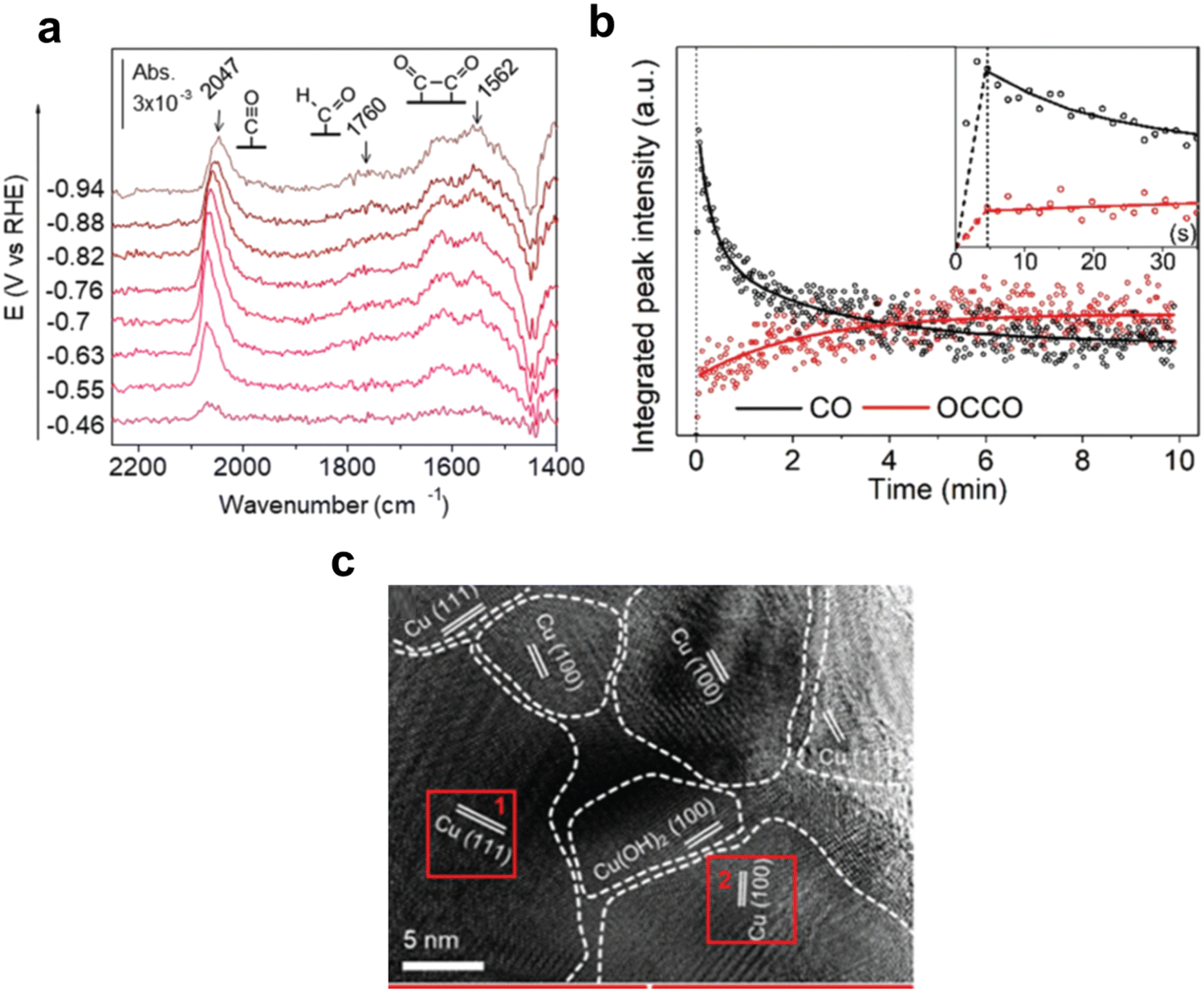 | ||
| Fig. 33 In situ ATR-SEIRAS analysis of Cu electrode (a), the integrated peak intensities (b) and HRTEM images (c). Reproduced from ref. 444. | ||
Subsequent conversion of these C–C coupled species to ethylene has been explained by H+ and electron transfer with the formation of CH2CHO* species as key intermediates with subsequent hydrogenation to C2H4.445,447 The side process is hydrogenation of CO group with the generation of C2H5OH.448 There is an alternative theory, which considers *CH*COH as intermediate species. The dehydration of these species yields adsorbed *C*CH fragments with their further hydrogenation to C2H4 (Fig. 32).449 At the same time, the hydrogenation of *CH*COH leads to ethanol.
The reaction selectivity to ethylene or alcohol is strongly affected by the surface coverage of electrode. Thus, the presence of high content of CO in the reactor in CORR results in higher ethanol selectivity and lower selectivity to ethylene in comparison with CO2RR.450
 | ||
| Fig. 34 Demonstration of Cu clustering in the process of CO2RR and HER (a) with the Faradaic efficiency at different potential (b). Reprinted by permission from Springer Nature: Springer Nature, Nature Catalysis, ref. 454 © (2019). | ||
The deposition of organic compounds has been used to modify adsorption and reactivity of reagents, intermediates and products and thus, to control the efficiency of CO2RR. For example, the deposition of polyamine on the surface of Cu improved the efficiency to C2H4 to 87% (Fig. 35).455 An increase in the degree of the methylation of amine resulted in lower selectivity to C2H4. The effect of amine NH2 group has been assigned to the higher surface pH, higher CO concentration and higher stabilization of intermediates.454 This assumption has been supported by the study of the effect of pH on the selectivity of CO2RR.455 The presence of amino groups led to the increase in the contribution of CO32− anions and to the rate of the coupling with the highest efficiency toward C2H4. In the case of CORR, the pH effect was less significant.450
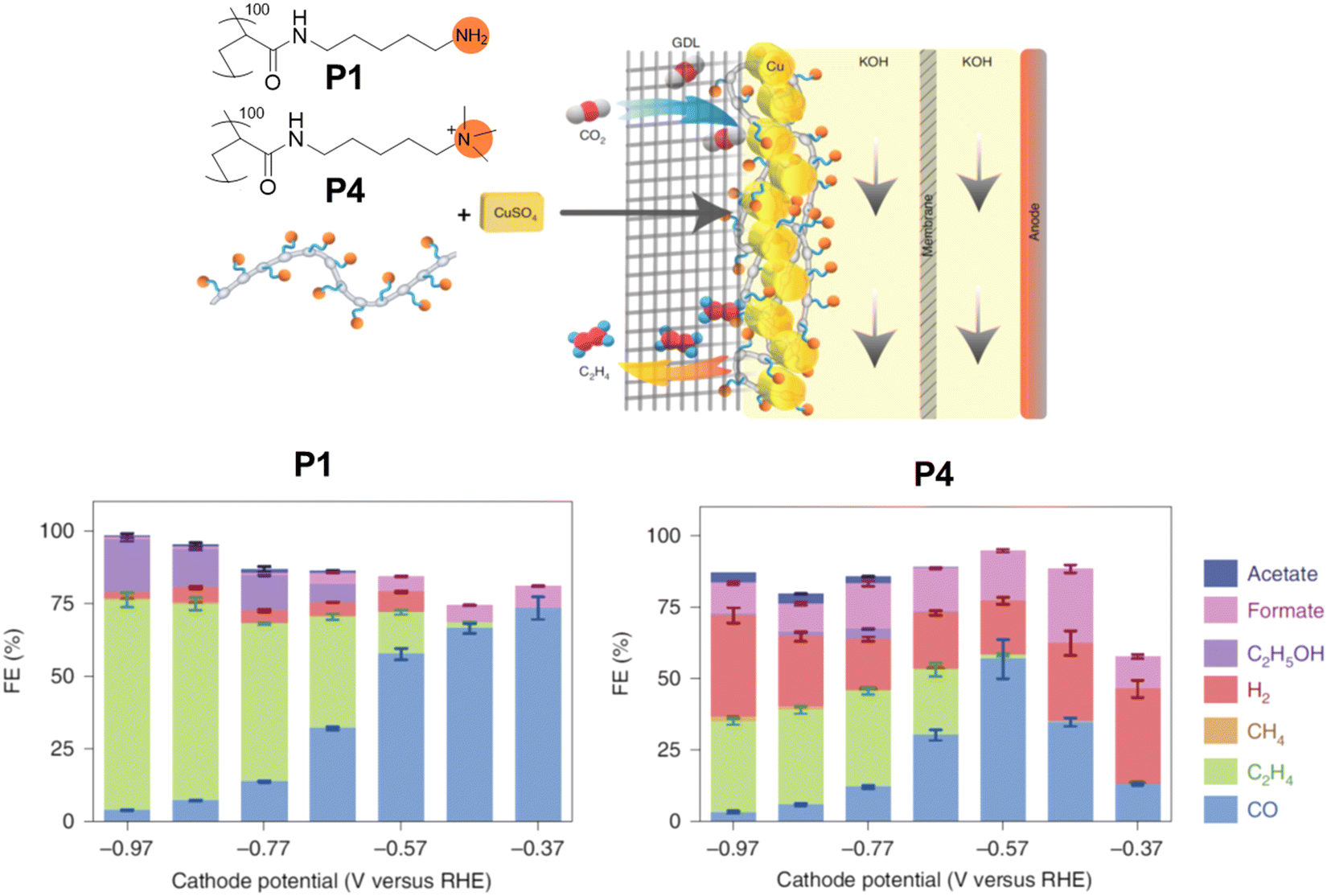 | ||
| Fig. 35 Product distributions in the electrocatalytic CO2RR over Cu catalysts with different polyamines (P1 and P4) at different cathode potentials. Reprinted by permission from Springer Nature: Springer Nature, Nature Catalysis, ref. 455 © (2020). | ||
Generally, CORR provided lower efficiency to C2H4 (50%) in comparison with CO2RR (87%). The phenomenon could be assigned to the lower solubility of CO in the polar solutions and weak interaction with the electrode surface in comparison with CO2. The increase in support hydrophobicity by PTFE resulted in an increase in Faraday efficiency (FE) to C2H4 to 52.7%.456 The variation of the concentration of CO in the reactor significantly affected FE to C2H4.450 The pure CO led to the broad distribution of the coupling products with a FE to C2H4 of about 30%. At the same time, the low CO concentration resulted in H2 generation by HER reaction with low FE to C2H4. The intermediate CO concentration provided an optimal 50% FE. DFT modelling explains this effect by promoting hydrogenation of *CH*COH to oxygenates at higher CO coverage.
The MEA cell with GDE electrodes provides an efficient solution for upscaling the electrocatalytic reduction. The key challenges are the relatively low current density and the long-term stability of the cell including the transformation of Cu catalyst. The opportunity to avoid alkaline catholyte in MEA cells is highly desirable for industrial implementation.
The energy efficiency for the conversion of CO2 and CO to C2H4 is still relatively low (30%) and requires additional efforts to improve it by the design of electrocatalytic cells and catalysts.
5. Assessment of LO synthesis routes
Various routes are explored in this review for selective LO synthesis from renewable and fossil feedstocks such as hydrocarbons, oxygenates and carbon oxides. We conducted their comparative analysis (Table 6) using several quantitative characteristics: selectivity, productivity, severity of operating conditions (temperature and pressure), stability, technological maturity and sustainability. Currently, LO synthesis faces new challenges. First, renewable raw materials should progressively replace fossil feedstocks. Second, there is a strong need for the development of new zero-waste and highly selective chemical processes. The goal is to enhance the selectivity of LO synthesis and to reach an atom efficiency close to 100%. The benign process design should avoid the cogeneration of undesirable and often toxic by-products. Third, the energy efficiency of LO synthesis technologies should be also significantly improved in order to run them at ambient temperature and pressure, whenever possible. Reduction of energy consumption via the application of more energy efficient technologies should result in zero emission of greenhouse gases, which are the major reasons for climate change.| Route | LO Selectivity | Productivity | Catalyst stability | P (bar) | T (°C) | TRL | S |
|---|---|---|---|---|---|---|---|
| a LO selectivities excluding co-produced CO or CO2. | |||||||
| SC | 30–85% | 50–120 mmol cm−3 h−1 (per reactor volume) | Catalysts are not involved in the process | 1–2 | 800–950 | 9 | C |
| FCC | 30–40% | 600–20000 mmol cm−3 h−1 (per reactor volume) |
0.1–5 s in a cycle | 1–2 | 500–700 | 9 | C |
| Long term = 3 months (15000 cycles) |
|||||||
| DDH | 85–95% | 20–300 mmol gcat−1 h−1 in reaction cycle | 10–12 min per cycle | 0.3–2 | 550–750 | 9 | B |
| Long term 3–5 years460 | |||||||
| ODH | 80–90% | 10–100 mmol gcat−1 h−1 | Predicted ∼2 years for looping cycling180 | 1–2 | 400–750 | 6-7 | B |
| MC | 50–70% | 20–200 mmol gcat−1 h−1 in the reaction cycle | 200 h | 1–2 | 700–900 | 6-7 | C |
| MTO | >85% | 30–100 mmol gcat−1 h−1 | 40 h at 50% conversion in a cycle | 1–2 | 400–500 | 9 | B |
| Long term >4–5 months236 (>450 cycles) | |||||||
| BWP | <20% | 0.5–1 mmol gcat−1 h−1 | n.a. | 1 | 400–700 | 4–5 | C |
| FT COx | ∼30% (60%a) | 5–70 mmol gcat−1 h−1 | >200 h | 20–40 | 330–350 | 5–7 | B |
| MM COx | <50% (90%a) | 2–5 mmol gcat−1 h−1 | >200 h | 20–40 | 330–400 | 4–5 | A |
| EC | >90% | 200 mmol gcat−1 h−1 | >24 h | 1 | Mostly at RT | 3 | A |
SC and FCC are historical technologies of LO synthesis. Ethylene is the main product of SC, while FCC mostly produces propylene, among the LO fraction. Synthesis of LO from renewable raw materials such as biogas-derived methane, syngas from gasification of biomass, organic or plastic waste and CO2 produced by human activities is currently of particular interest. The optimum LO selectivity, small number and quantity of by-products seem to be the most important parameters in the context of the development of efficient and zero-waste processes. Analysis of available data suggests (Table 6) that on-purpose direct dehydrogenation, MTO and oxidative dehydrogenation combined with chemical looping are currently the most selective routes for LO synthesis. Attaining high LO selectivity is a difficult issue for methane coupling, FT synthesis and biomass pyrolysis. FT synthesis and wax pyrolysis produce major amounts of cheap methane and light paraffins. In addition to methane and paraffins, methane coupling, FT synthesis, methanol-mediated route and conventional oxidative dehydrogenation generate significant amounts of CO or CO2. Separation and recycling of co-produced CO or CO2 require major energy consumption and represent a significant challenge for these technologies. Electrocatalytic reduction of CO2 to LOs is a very attractive process, due to the low reaction temperature and opportunity for use of green electricity, however, it has several challenges. Although significant progress has been achieved in this field, a high Faradaic efficiency to ethylene requires low current density, limiting the olefin yield. Another limitation of the electrocatalytic route is the production of only ethylene from COx, whereas other olefins are also required.
Productivity is another important figure of merit in any chemical process. Most of LO synthesis technologies involve a catalyst. To compare the productivity of catalytic processes (Table 6), we expressed the productivity normalized by the amount of catalyst. Note that in FCC, the catalyst is not exposed continuously to the reaction, while in SC, the cracking reactions occur without a catalyst. These are the reasons, why in SC and FCC the productivities have been normalised to the reactor volume rather than to the catalyst weight. It should be also considered, that FCC, direct dehydrogenation, oxidative dehydrogenation with chemical looping and MTO require periodic or continuous catalyst regenerations, which decrease the time-averaged process productivity. The catalyst regeneration issue is particularly crucial in the FCC process, where the catalysts with extremely high productivity can operate without regeneration only for a few fractions of a second. The duration of the reaction cycle (before the regeneration) is a few minutes in direct dehydrogenation and a few hours in MTO.
Higher LO productivity has been reported in methane oxidative coupling (∼200 mmol gcat−1 h−1) compared to many other processes. Interestingly, oxidative dehydrogenation, MTO, FT synthesis and electrocatalysis have rather similar LO productivity. The LO productivity is relatively low in the methanol-mediated CO/CO2 hydrogenation and can be considered as the most important challenge of this process. Reaching high LO productivity and selectivity remains demanding in the biomass/waste pyrolysis.
Catalyst stability is commonly considered a determining parameter for chemical processes and in particular, for the synthesis of relatively cheap commodities such as LOs. Since the catalysts in FCC, direct dehydrogenation and MTO require frequent regenerations, we should distinguish (i) the catalyst stability within a reaction cycle and (ii) long-term stability, which includes both catalytic reaction and catalyst regeneration. The amount of available information about the catalyst stability depends on the level of maturity of a specific LO synthesis process. More extensive long-term stability data are provided for the processes, which have reached industrial implementation such as FCC, direct dehydrogenation and MTO. In these processes, the catalysts can operate after consecutive regenerations for several months. For FCC, the long-term catalyst stability is usually limited by 1-3 months; in this process, the fresh catalyst is continuously added to the reactor during the operation. The catalyst long-term stability for MTO can exceed 3–4 months. The catalysts for the direct dehydrogenation or looping oxidative dehydrogenation processes may have a long-term stability of 2–3 years. For other processes, the catalyst stability has been limited by the duration of laboratory tests (about 200–300 h). Much lesser information about catalyst stability has been provided for electrocatalysis and waste/biomass pyrolysis.
Many processes for LO synthesis necessitate high temperatures and thus, require major energy consumption. SC of light hydrocarbons seems to be a particularly energy consuming process. Endothermic processes such as direct dehydrogenation, FCC and biomass pyrolysis occurring at temperatures higher than 400 °C also require high energy consumption. The LO synthesis reactions requiring medium temperature (250–400 °C) involve exothermic Fischer–Tropsch synthesis and methanol-mediated hydrogenation. At the same time, these routes require the LO extraction from the mixture of low-valued hydrocarbons in FT synthesis and carbon oxides in both FT synthesis and methanol-mediated process. The product separation can therefore, strongly impact the total energy consumption. The best energy efficiency has been reached in electrocatalytic processes occurring at ambient temperature, though the future potential of the electrocatalytic processes should be further studied. The energy for LO synthesis can be also supplied by renewable electricity: process electrification combined with process intensification becomes a priority in the petrochemical industry. If the energy for conducting a chemical process is supplied by fossil feedstocks, high energy consumption may also lead to the generation of supplementary greenhouse gas emissions.
Interestingly, most LO synthesis processes proceed at near ambient pressure with exception of FT synthesis and methanol-mediated carbon oxides hydrogenation requiring moderate pressure (20–40 bar). We can assume that pressure cannot be considered a strongly influencing parameter compared to temperature.
Cost and technological maturity usually determine scaling up these chemical reactions to the industrial level. Here, we use a Technology Readiness Level (TRL) scale from 1 to 9 for each process.461 TRL is a method for assessing the maturity of different technologies, developed at NASA in the 1970s and then adopted by the EU in 2010. A higher TRL corresponds to more mature technologies. For example, TRL of 1 means “Basic principles observed” and TRL of 9 indicates “Actual system proven in operational environment”. Note that in addition to the technological cost, the cost of chemical commodities such as LOs is strongly affected by geographic localisation and the availability of cheap feedstocks.462,463 Nevertheless, we can suggest that SC, FCC and direct dehydrogenation can currently present the cheapest options for LO synthesis, while LOs produced in MTO seem more expensive.464 The relatively low cost of olefins produced by SC and FCC is to a larger extent due to the high maturity of these technologies. In mid-term, cheap LO synthesis technologies could involve oxidative dehydrogenation in the chemical looping mode, methane coupling, methanol-mediated CO/CO2 hydrogenation. Extensive production of carbon oxides, low-value methane and light hydrocarbons seem to be a major obstacle to the implementation of CO- and CO2-based FT synthesis. Waste/biomass pyrolysis does not seem to be suitable for large scale implementation, however, it can provide an acceptable solution for small-scale LO production plants. Finally, the current state of the art makes it difficult to evaluate the future potential electrocatalytic LO synthesis from CO2. Expanding electrocatalysis from the synthesis of only ethylene to the synthesis of propylene or butylenes can be challenging and is one of the most interesting directions for future works.
The final criteria for the comparison of different routes for the synthesis of LOs is their sustainability. According to ref. 465, sustainability implies the usage of resources and energy at a rate, at which they can be replaced naturally. Also, the generation of waste cannot be faster than the rate of its renewal. From this viewpoint, SC, FCC, methane thermal coupling as well as biomass and waste pyrolysis cannot be considered sustainable approaches, because of the huge amount of side products and/or strong energy demand. We assume these processes possess the lowest grade of sustainability (rated as C).
Alkane dehydrogenation, both non-oxidative and oxidative, despite the high energy consumption and fossil sources, may give a high selectivity towards LOs and also produce recyclable hydrogen and CO. The MTO process requires lower energy and gives high selectivity but the source – methanol/DME – is now mostly produced by coal gasification with subsequent syngas conversion to LOs. Thus, the sustainability of MTO is debatable and depends on the development of “green” methanol or DME production from CO2 and electrolyze-derived hydrogen. FT synthesis gives noticeable amounts of methane along with LO and its sustainability also depends on the carbon source – CO or CO2, derived from fossil fuels, biomass, waste or captured CO2. Therefore, DH, MTO, and FT synthesis are considered as processes with medium or medium-high sustainability (rated as B). Finally, electrocatalysis and methanol-mediated COx hydrogenation, with COx from renewable feedstocks, can be assigned with the highest level of sustainability (rated as A). Here we should also point out some important stipulations for this: renewable energy costs comparable with that of fossil fuels and high efficiency of CO recycle in thermocatalytic CO2 hydrogenation.
6. Conclusions
Historically, LOs have been produced from hydrocarbons using SC and FCC. Currently, LOs can be produced using several technologies at renewable and fossil feedstocks, under a wide range of operating conditions. Among the LO synthesis routes considered in this study, the following methods stand out: alkane direct dehydrogenation, alkane oxidative dehydrogenation and MTO. These methods have great potential in the short term and have already reached technological and scientific maturities. Their advantages are high LO selectivity, low amount of by-products, high flexibility considering the desired olefins and less severe operating conditions compared to more conventional methods of LO synthesis, such as SC and FCC. Additional advancements are required for oxidative dehydrogenation. The chemical looping strategies and use of CO2 as an oxidizing agent have to be developed further in order to reach higher industrial maturity in particular for ethylene production. A combination of thermo- and electrocatalysis has also shown promising results for LO synthesis in the dehydrogenation processes. Recent publications about single-metal and nanoalloy-based catalysts with well-defined structures showed the perspectives to go beyond the deactivation limitations and to increase the catalyst lifetime and productivity.Major progress should be made in the field of methanol-mediated CO and CO2 direct hydrogenation to LOs. The efforts in this field should address reducing the production of carbon oxides and increasing the single pass LO yield. In the purely electrocatalytic approach, new selective nanostructured and molecular supported catalysts with well controlled micro-environment at the active site should be elaborated. New cells and catalysts with high Faradaic efficiency are still needed to achieve satisfactory performance in different operating regimes. More information should be provided on the stability of electrocatalysts for LO synthesis beyond a few tens of hours.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge financial support from the European Union (TAKE-OFF H2020 project).References
- Reports and Data, Olefins Market Size, Share And Industry Analysis By Type (Ethylene, Propylene, Butadiene, Butylene, Others), By Applications (Olefin Glycols, Olefin Oxides, Ethylbenzene, Polyolefins, Cumene), By Distribution Channel (Distributors, Wholesale) And Region, https://www.reportsanddata.com/report-detail/olefins-market.
- T. K. Phung, T. L. M. Pham, K. B. Vu and G. Busca, J. Environ. Chem. Eng., 2021, 9, 105673 CrossRef CAS.
- S. Najari, S. Saeidi, F. Gallucci and E. Drioli, Rev. Chem. Eng, 2021, 37, 363–406 CrossRef CAS.
- A. Akah, J. Williams and M. Ghrami, Catal. Surv., 2019, 23, 265–276 CrossRef CAS.
- H. M. Torres Galvis and K. P. De Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef CAS.
- Y. H. Choi, Y. J. Jang, H. Park, W. Y. Kim, Y. H. Lee, S. H. Choi and J. S. Lee, Appl. Catal., B, 2017, 202, 605–610 CrossRef CAS.
- M. Ronda-Lloret, G. Rothenberg and N. R. Shiju, ChemSusChem, 2019, 12, 3896–3914 CrossRef CAS PubMed.
- R.-P. P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- O. A. Ojelade and S. F. Zaman, J. CO2 Util., 2021, 47, 101506 CrossRef CAS.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- X. Pan, F. Jiao, D. Miao and X. Bao, Chem. Rev., 2021, 121, 6588–6609 CrossRef CAS PubMed.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- C. Li and G. Wang, Chem. Soc. Rev., 2021, 50, 4359–4381 RSC.
- T. Otroshchenko, G. Jiang, V. A. Kondratenko, U. Rodemerck and E. V. Kondratenko, Chem. Soc. Rev., 2021, 50, 473–527 RSC.
- S. Najari, S. Saeidi, P. Concepcion, D. D. Dionysiou, S. K. Bhargava, A. F. Lee and K. Wilson, Chem. Soc. Rev., 2021, 50, 4564–4605 RSC.
- Y. Gambo, S. Adamu, G. Tanimu, I. M. Abdullahi, R. A. Lucky, M. S. Ba-Shammakh and M. M. Hossain, Appl. Catal., A, 2021, 623, 118273 CrossRef CAS.
- V. Blay, B. Louis, R. Miravalles, T. Yokoi, K. A. Peccatiello, M. Clough and B. Yilmaz, ACS Catal., 2017, 7, 6542–6566 CrossRef CAS.
- I. Amghizar, J. N. Dedeyne, D. J. Brown, G. B. Marin and K. M. Van Geem, React. Chem. Eng., 2020, 5, 239–257 RSC.
- M. Fakhroleslam and S. M. Sadrameli, Fuel, 2019, 252, 553–566 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- M. Yang, D. Fan, Y. Wei, P. Tian and Z. Liu, Adv. Mater., 2019, 31, 1902181 CrossRef CAS PubMed.
- Q. Zhang, J. Yu and A. Corma, Adv. Mater., 2020, 32, 2002927 CrossRef PubMed.
- A. Akah and M. Al-Ghrami, Appl. Petrochem. Res., 2015, 5, 377–392 CrossRef CAS.
- A. Corma, E. Corresa, Y. Mathieu, L. Sauvanaud, S. Al-Bogami, M. S. Al-Ghrami and A. Bourane, Catal. Sci. Technol., 2017, 7, 12–46 RSC.
- S. Suganuma and N. Katada, Fuel Process. Technol., 2020, 208, 106518 CrossRef CAS.
- M. Fakhroleslam and S. M. Sadrameli, Ind. Eng. Chem. Res., 2020, 59, 12288–12303 CrossRef CAS.
- E. N. Shafei, M. Z. Albahar, M. F. Aljishi, A. N. Aljishi, A. S. Alnasir, H. H. Al-Badairy and M. A. Sanhoob, React. Chem. Eng., 2022, 7, 123–132 RSC.
- S. M. Sadrameli, Fuel, 2015, 140, 102–115 CrossRef CAS.
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS.
- S. M. Sadrameli, Fuel, 2016, 173, 285–297 CrossRef CAS.
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS.
- J. S. Plotkin, Catal. Today, 2005, 106, 10–14 CrossRef CAS.
- M. A. Bari Siddiqui, A. M. Aitani, M. R. Saeed and S. Al-Khattaf, Top. Catal., 2010, 53, 1387–1393 CrossRef CAS.
- Y. Yoshimura, N. Kijima, T. Hayakawa, K. Murata, K. Suzuki, F. Mizukami, K. Matano, T. Konishi, T. Oikawa, M. Saito, T. Shiojima, K. Shiozawa, K. Wakui, G. Sawada, K. Sato, S. Matsuo and N. Yamaoka, Catal. Surv., 2001, 4, 157–167 CrossRef.
- K. Kubo, H. Iida, S. Namba and A. Igarashi, Appl. Catal., A, 2015, 489, 272–279 CrossRef CAS.
- K. Kubo, H. Iida, S. Namba and A. Igarashi, Catal. Commun., 2012, 29, 162–165 CrossRef CAS.
- J. Wan, Y. Wei, Z. Liu, B. Li, Y. Qi, M. Li, P. Xie, S. Meng, Y. He and F. Chang, Catal. Lett., 2008, 124, 150–156 CrossRef CAS.
- A. Corma and B. W. Wojciechowski, Catal. Rev., 1985, 27, 29–150 CrossRef CAS.
- C. Wu, Y. Cheng and Y. Jin, Ind. Eng. Chem. Res., 2009, 48, 12–26 CrossRef CAS.
- J. M. Maselli and A. W. Peters, Catal. Rev., 1984, 26, 525–554 CrossRef CAS.
- E. T. Habib, X. Zhao, G. Yaluris, W. C. Cheng, L. T. Boock and J.-P. Gilson, Advances in Fluid Catalytic Cracking, Catalytic Science Series, Zeolites for Cleaner Technologies, 2002, pp. 105–130 Search PubMed.
- M. F. Alotibi, B. A. Alshammari, M. H. Alotaibi, F. M. Alotaibi, S. Alshihri, R. M. Navarro and J. L. G. Fierro, Catal. Surv., 2020, 24, 1–10 CrossRef CAS.
- S. Al-Khattaf, M. R. Saeed, A. Aitani and M. T. Klein, Energy Fuels, 2018, 32, 6189–6199 CrossRef CAS.
- A. Aitani, Catal. Today, 2000, 60, 111–117 CrossRef CAS.
- A. Usman, M. A. B. Siddiqui, A. Hussain, A. Aitani and S. Al-Khattaf, Chem. Eng. Res. Des., 2017, 120, 121–137 CrossRef CAS.
- A. Corma and J. Martinez-Triguero, J. Catal., 1997, 165, 102–120 CrossRef CAS.
- R. Castaneda, A. Corma, V. Fornes, J. Martinez-Trirguero and S. Valencia, J. Catal., 2006, 238, 79–87 CrossRef CAS.
- O. Awayssa, N. Al-Yassir, A. Aitani and S. Al-Khattaf, Appl. Catal., A, 2014, 477, 172–183 CrossRef CAS.
- J. M. Arandes, I. Torre, M. J. Azkoiti, J. Ereña, M. Olazar and J. Bilbao, Energy Fuels, 2009, 23, 4215–4223 CrossRef CAS.
- H. E. Van Der Bij and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7406–7428 RSC.
- J. G. Speight, Handbook of Petroleum Refining, CRC Press, 2016 Search PubMed.
- A. H. Motagamwala, R. Almallahi, J. Wortman, V. O. Igenegbai and S. Linic, Science, 2021, 373, 217–222 CrossRef CAS PubMed.
- J. Zhang, R. Ma, H. Ham, K. Shimizu and S. Furukawa, JACS Au, 2021, 1, 1688–1693 CrossRef CAS PubMed.
- S. Chen, Z. J. Zhao, R. Mu, X. Chang, J. Luo, S. C. Purdy, A. J. Kropf, G. Sun, C. Pei, J. T. Miller, X. Zhou, E. Vovk, Y. Yang and J. Gong, Chem, 2021, 7, 387–405 CAS.
- S. W. Han, H. Park, J. Han, J. C. Kim, J. Lee, C. Jo and R. Ryoo, ACS Catal., 2021, 11, 9233–9241 CrossRef CAS.
- P. Ingale, K. Knemeyer, P. Preikschas, M. Ye, M. Geske, R. Naumann d’Alnoncourt, A. Thomas and F. Rosowski, Catal. Sci. Technol., 2021, 11, 484–493 RSC.
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D. C. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19450–19459 CrossRef CAS PubMed.
- L. Xie, Y. Chai, L. Sun, W. Dai, G. Wu, N. Guan and L. Li, J. Energy Chem., 2021, 57, 92–98 CrossRef.
- Y. Nakaya, F. Xing, H. Ham, K. I. Shimizu and S. Furukawa, Angew. Chem., Int. Ed., 2021, 60, 19715–19719 CrossRef CAS PubMed.
- P.-A. Payard, L. Rochlitz, K. Searles, L. Foppa, B. Leuthold, O. V. Safonova, A. Comas-Vives and C. Copéret, JACS Au, 2021, 1, 1445–1458 CrossRef CAS PubMed.
- H. C. Kwon, Y. Park, J. Y. Park, R. Ryoo, H. Shin and M. Choi, ACS Catal., 2021, 11, 10767–10777 CrossRef CAS.
- S. Rimaz, L. Chen, A. Monzón, S. Kawi and A. Borgna, Chem. Eng. J., 2021, 405, 126656 CrossRef CAS.
- R. Ryoo, J. Kim, C. Jo, S. W. Han, J. C. Kim, H. Park, J. Han, H. S. Shin and J. W. Shin, Nature, 2020, 585, 221–224 CrossRef CAS PubMed.
- H. Park, H. Park, J.-C. Kim, M. Choi, J. Y. Park and R. Ryoo, J. Catal., 2021, 404, 760–770 CrossRef CAS.
- N. V. Srinath, A. Longo, H. Poelman, R. K. Ramachandran, J. Y. Feng, J. Dendooven, M. F. Reyniers and V. V. Galvita, ACS Catal., 2021, 11320–11335 CrossRef CAS.
- P. Wang, J. Yao, Q. Jiang, X. Gao, D. Lin, H. Yang, L. Wu, Y. Tang and L. Tan, Appl. Catal., B, 2022, 300, 120731 CrossRef CAS.
- X. Zhu, T. Wang, Z. Xu, Y. Yue, M. Lin and H. Zhu, J. Energy Chem., 2021, 65, 293–301 CrossRef.
- Z. Xie, Z. Li, P. Tang, Y. Song, Z. Zhao, L. Kong, X. Fan and X. Xiao, J. Catal., 2021, 397, 172–182 CrossRef CAS.
- Z. Zhang, W. Xu, X. Ye, Y. Xi, C. Qiu, L. Ding, G. Liu and Q. Xiao, Catal. Sci. Technol., 2021, 11, 5250–5259 RSC.
- R. T. Hannagan, G. Giannakakis, R. Réocreux, J. Schumann, J. Finzel, Y. Wang, A. Michaelides, P. Deshlahra, P. Christopher, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Science, 2021, 372, 1444–1447 CrossRef CAS.
- Q. Y. Chang, K. Q. Wang, Z. J. Sui, X. G. Zhou, D. Chen, W. K. Yuan and Y. A. Zhu, ACS Catal., 2021, 11, 5135–5147 CrossRef CAS.
- T. Yang, Y. Zhong, J. Li, R. Ma, H. Yan, Y. Liu, Y. He and D. Li, ACS Appl. Mater. Interfaces, 2021, 13, 33045–33055 CrossRef CAS PubMed.
- C. Huang, D. Han, L. Guan, L. Zhu, Y. Mei, D. He and Y. Zu, Fuel, 2022, 307, 121790 CrossRef CAS.
- Y. Dai, Y. Wu, H. Dai, X. Gao, S. Tian, J. Gu, X. Yi, A. Zheng and Y. Yang, J. Catal., 2021, 395, 105–116 CrossRef CAS.
- Y. Yue, J. Fu, C. Wang, P. Yuan, X. Bao, Z. Xie, J. M. Basset and H. Zhu, J. Catal., 2021, 395, 155–167 CrossRef CAS.
- Q. Wang, W. Xu, Z. Ma, F. Yu, Y. Chen, H. Liao, X. Wang and J. Zhou, ChemCatChem, 2021, 13, 1009–1022 CrossRef CAS.
- Q. H. Shang, J. N. Liu, W. Z. Lang, X. Yan, X. J. Guo and Y. J. Guo, Ind. Eng. Chem. Res., 2021, 60, 12811–12820 CrossRef CAS.
- Y. Qu, G. Li, T. Zhao, Z. Zhang, M. Douthwaite, J. Zhang and Z. Hao, ACS Sustainable Chem. Eng., 2021, 1–11 Search PubMed.
- Z. Bian, N. Dewangan, Z. Wang, S. Pati, S. Xi, A. Borgna, H. Kus and S. Kawi, ACS Appl. Nano Mater., 2021, 4, 1112–1125 CrossRef CAS.
- P. Castro-Fernández, D. Mance, C. Liu, I. B. Moroz, P. M. Abdala, E. A. Pidko, C. Copéret, A. Fedorov and C. R. Müller, ACS Catal., 2021, 11, 907–924 CrossRef.
- Y. Wang, Y. Suo, J. T. Ren, Z. Wang and Z. Y. Yuan, J. Colloid Interface Sci., 2021, 594, 113–121 CrossRef CAS PubMed.
- C. S. Praveen, A. P. Borosy, C. Copéret and A. Comas-Vives, Inorg. Chem., 2021, 60, 6865–6874 CrossRef CAS PubMed.
- Y. Yuan, C. Brady, R. F. Lobo and B. Xu, ACS Catal., 2021, 11, 10647–10659 CrossRef CAS.
- G. Lin, Y. Su, X. Duan and K. Xie, Angew. Chem., Int. Ed., 2021, 60, 9311–9315 CrossRef CAS PubMed.
- T. Wang, X. Cui, K. T. Winther, F. Abild-Pedersen, T. Bligaard and J. K. Nørskov, ACS Catal., 2021, 11, 6290–6297 CrossRef CAS.
- Z. Xie, T. Yu, W. Song, J. Li, Z. Zhao, B. Liu, Z. Gao and D. Li, ACS Catal., 2020, 10, 14678–14693 CrossRef CAS.
- L. Sharma, S. C. Purdy, K. Page, S. Rangarajan, H. Pham, A. Datye and J. Baltrusaitis, ACS Appl. Nano Mater., 2021, 4, 10055–10067 CrossRef CAS.
- D. Zhao, X. Tian, D. E. Doronkin, S. Han, V. A. Kondratenko, J.-D. Grunwaldt, A. Perechodjuk, T. H. Vuong, J. Rabeah, R. Eckelt, U. Rodemerck, D. Linke, G. Jiang, H. Jiao and E. V. Kondratenko, Nature, 2021, 599, 234–238 CrossRef CAS PubMed.
- C. R. Riley, A. De La Riva, I. L. Ibarra, A. K. Datye and S. S. Chou, Appl. Catal., A, 2021, 624, 118309 CrossRef CAS.
- B. Eryildirim, H. Arbag, N. Oktar and G. Dogu, Int. J. Hydrogen Energy, 2021, 46, 5296–5310 CrossRef CAS.
- B. Qiu, Y. Zhang, Y. Liu and Y. Zhang, Appl. Surf. Sci., 2021, 554, 149611 CrossRef CAS.
- Y. Pan, A. Bhowmick, W. Wu, Y. Zhang, Y. Diao, A. Zheng, C. Zhang, R. Xie, Z. Liu, J. Meng and D. Liu, ACS Catal., 2021, 11, 9970–9985 CrossRef CAS.
- S. De, S. Ould-Chikh, A. Aguilar, J. L. Hazemann, A. Zitolo, A. Ramirez, S. Telalovic and J. Gascon, ACS Catal., 2021, 11, 3988–3995 CrossRef CAS.
- X. Duan, L. Ye and K. Xie, Catal. Sci. Technol., 2021, 11, 6573–6578 RSC.
- H. Guo, C. Miao, W. Hua, Y. Yue and Z. Gao, Microporous Mesoporous Mater., 2021, 312, 110791 CrossRef CAS.
- Y. Hosono, H. Saito, T. Higo, K. Watanabe, K. Ito, H. Tsuneki, S. Maeda, K. Hashimoto and Y. Sekine, J. Phys. Chem. C, 2021, 125, 11411–11418 CrossRef CAS.
- S. P. Batchu, H. L. Wang, W. Chen, W. Zheng, S. Caratzoulas, R. F. Lobo and D. G. Vlachos, ACS Catal., 2021, 11, 1380–1391 CrossRef CAS.
- Y. Zhang, B. Wang, M. Fan, D. Li and R. Zhang, Fuel, 2021, 306, 121641 CrossRef CAS.
- X. Zhang, L. Ye, H. Li, F. Chen and K. Xie, ACS Catal., 2020, 10, 3505–3513 CrossRef CAS.
- W. Wu, L.-C. Wang, H. Hu, W. Bian, J. Y. Gomez, C. J. Orme, H. Ding, Y. Dong, T. He, J. Li and D. Ding, ACS Catal., 2021, 11, 12194–12202 CrossRef CAS.
- X. Chen, M. Peng, X. Cai, Y. Chen, Z. Jia, Y. Deng, B. Mei, Z. Jiang, D. Xiao, X. Wen, N. Wang, H. Liu and D. Ma, Nat. Commun., 2021, 12, 2664 CrossRef CAS PubMed.
- Y. Li, M. Ge, J. Wang, M. Guo, F. Liu, M. Han, Y. Xu and L. Zhang, Chinese J. Chem. Eng., 2021, 32, 203–211 CrossRef CAS.
- M. Shao, C. Hu, X. Xu, Y. Song and Q. Zhu, Appl. Catal., A, 2021, 621, 118194 CrossRef CAS.
- M. Chu, Y. Liu, J. Gong, C. Zhang, X. Wang, Q. Zhong, L. Wu and Y. Xu, Chem. – Eur. J., 2021, 27, 11643–11648 CrossRef CAS PubMed.
- D. A. Nazimov, O. V. Klimov, A. V. Saiko, S. N. Trukhan, T. S. Glazneva, I. P. Prosvirin, S. V. Cherepanova and A. S. Noskov, Catal. Today, 2021, 375, 401–409 CrossRef CAS.
- E. Cheng and J. Notestein, J. Catal., 2021, 397, 212–222 CrossRef CAS.
- J. Wang, Z. Song, M. Han, X. Li and L. Zhang, Mol. Catal., 2021, 511, 111746 CrossRef CAS.
- M. B. Ansari and S. E. Park, Energy Environ. Sci., 2012, 5, 9419–9437 RSC.
- G. Wu, F. Hei, N. Guan and L. Li, Catal. Sci. Technol., 2013, 3, 1333–1342 RSC.
- A. M. Arinaga, S. Liu and T. J. Marks, Catal. Sci. Technol., 2020, 10, 6840–6848 RSC.
- Q. Zhu, S. L. Wegener, C. Xie, O. Uche, M. Neurock and T. J. Marks, Nat. Chem., 2013, 5, 104–109 CrossRef CAS PubMed.
- J. J. Ternero-Hidalgo, M. Daturi, G. Clet, P. Bazin, M. A. Bañares, R. Portela, M. O. Guerrero-Pérez, J. Rodríguez-Mirasol and T. Cordero, Catal. Today, 2022, 387, 197–206 CrossRef CAS.
- E. Gomez, B. Yan, S. Kattel and J. G. Chen, Nat. Rev. Chem., 2019, 3, 638–649 CrossRef CAS.
- S. Chen, C. Pei, X. Chang, Z. J. Zhao, R. Mu, Y. Xu and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 22072–22079 CrossRef CAS PubMed.
- M. H. Jeong, J. Sun, G. Young Han, D. H. Lee and J. W. Bae, Appl. Catal., B, 2020, 270, 118887 CrossRef CAS.
- X. Jiang, L. Sharma, V. Fung, S. J. Park, C. W. Jones, B. G. Sumpter, J. Baltrusaitis and Z. Wu, ACS Catal., 2021, 11, 2182–2234 CrossRef CAS.
- J. M. Venegas, W. P. McDermott and I. Hermans, Acc. Chem. Res., 2018, 51, 2556–2564 CrossRef CAS PubMed.
- J. T. Grant, C. A. Carrero, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. E. Specht, W. P. McDermott, A. Chieregato and I. Hermans, Science, 2016, 354, 1570–1573 CrossRef CAS PubMed.
- X. Zhang, R. You, Z. Wei, X. Jiang, J. Yang, Y. Pan, P. Wu, Q. Jia, Z. Bao, L. Bai, M. Jin, B. Sumpter, V. Fung, W. Huang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 8042–8046 CrossRef CAS PubMed.
- J. M. Venegas, Z. Zhang, T. O. Agbi, W. P. McDermott, A. Alexandrova and I. Hermans, Angew. Chem., Int. Ed., 2020, 59, 16527–16535 CrossRef CAS PubMed.
- P. Kraus and R. P. Lindstedt, J. Phys. Chem. C, 2021, 125, 5623–5634 CrossRef CAS.
- D. Ding, B. Yan, Y. Wang and A. H. Lu, ChemCatChem, 2021, 13, 3312–3318 CrossRef CAS.
- R. Belgamwar, A. G. M. Rankin, A. Maity, A. K. Mishra, J. S. Gómez, J. Trébosc, C. P. Vinod and O. Lafon, ACS Sustainable Chem. Eng., 2020, 8, 16124–16135 CrossRef CAS.
- Z. Liu, B. Yan, S. Meng, R. Liu, W. D. Lu, J. Sheng, Y. Yi and A. H. Lu, Angew. Chem., Int. Ed., 2021, 60, 19691–19695 CrossRef CAS PubMed.
- T. C. Wang, J. L. Yin, X. J. Guo, Y. Chen, W. Z. Lang and Y. J. Guo, J. Catal., 2021, 393, 149–158 CrossRef CAS.
- Q. Liu, C. Chen, Q. Liu, Y. Wu, F. Xing, C. Cheng and C. Huang, Appl. Surf. Sci., 2021, 537, 147927 CrossRef CAS.
- H. Zhou, X. Yi, Y. Hui, L. Wang, W. Chen, Y. Qin, M. Wang, J. Ma, X. Chu, Y. Wang, X. Hong, Z. Chen, X. Meng, H. Wang, Q. Zhu, L. Song, A. Zheng and F. S. Xiao, Science, 2021, 372, 76–80 CrossRef CAS PubMed.
- W. D. Lu, D. Wang, Z. Zhao, W. Song, W. C. Li and A. H. Lu, ACS Catal., 2019, 9, 8263–8270 CrossRef CAS.
- H. Yan, S. Alayoglu, W. Wu, Y. Zhang, E. Weitz, P. C. Stair and J. M. Notestein, ACS Catal., 2021, 11, 9370–9376 CrossRef CAS.
- J. Chen, P. Rohani, S. G. Karakalos, M. J. Lance, T. J. Toops, M. T. Swihart and E. A. Kyriakidou, Chem. Commun., 2020, 56, 9882–9885 RSC.
- L. Cao, P. Dai, L. Zhu, L. Yan, R. Chen, D. Liu, X. Gu, L. Li, Q. Xue and X. Zhao, Appl. Catal., B, 2020, 262, 118277 CrossRef.
- W. Zhang, G. Zhao, T. Muschin and A. Bao, Surf. Interface Anal., 2021, 53, 100–107 CrossRef CAS.
- M. Huš, D. Kopač, D. Bajec and B. Likozar, ACS Catal., 2021, 11, 11233–11247 CrossRef PubMed.
- A. de Arriba, B. Solsona, A. M. Dejoz, P. Concepción, N. Homs, P. R. de la Piscina and J. M. López Nieto, J. Catal., 2022, 408, 388–400 CrossRef CAS.
- E. Moreno-Barrueta, C. Alvarado-Camacho, J. F. Durán-Pérez, A.-A. Morales-Pérez and C. O. Castillo, Catal. Today, 2022, 394–396, 161–177 CrossRef CAS.
- C. Alvarado-Camacho, J. Poissonnier, J. Thybaut and C. O. Castillo, React. Chem. Eng., 2022, 7, 619–640 RSC.
- H. Yan, K. He, I. A. Samek, D. Jing, M. G. Nanda, P. C. Stair and J. M. Notestein, Science, 2021, 371, 1257–1260 CrossRef CAS PubMed.
- P. Yan, Y. Chen and Y. Cheng, Chem. Eng. J., 2022, 427, 131813 CrossRef CAS.
- C. Xin, F. Wang and G. Q. Xu, Appl. Catal., A, 2021, 610, 117946 CrossRef CAS.
- E. V. Lazareva, V. M. Bondareva, D. A. Svintsitskiy, A. V. Ishchenko, A. S. Marchuk, E. P. Kovalev and T. Y. Kardash, Catal. Today, 2021, 361, 50–56 CrossRef CAS.
- I. I. Mishanin and V. I. Bogdan, Catal. Lett., 2021, 151, 2088–2093 CrossRef CAS.
- I. I. Mishanin, A. N. Kalenchuk, K. I. Maslakov, V. V. Lunin, A. E. Koklin, E. D. Finashina and V. I. Bogdan, Russ. J. Phys. Chem. A, 2016, 90, 1132–1136 CrossRef CAS.
- D. A. Svintsitskiy, T. Y. Kardash, E. V. Lazareva, A. A. Saraev, E. A. Derevyannikova, M. Vorokhta, B. Šmíd and V. M. Bondareva, Appl. Catal., A, 2019, 579, 141–150 CrossRef CAS.
- C. Wang, B. Yang, Q. Gu, Y. Han, M. Tian, Y. Su, X. Pan, Y. Kang, C. Huang, H. Liu, X. Liu, L. Li and X. Wang, Nat. Commun., 2021, 12, 5447 CrossRef CAS PubMed.
- X. Zhao, M. D. Susman, J. D. Rimer and P. Bollini, Catal. Sci. Technol., 2021, 11, 531–541 RSC.
- D. Delgado, B. Solsona, R. Sanchis, E. Rodríguez-Castellón and J. M. López Nieto, Catal. Today, 2021, 363, 27–35 CrossRef CAS.
- Y. Abdelbaki, A. de Arriba, B. Solsona, D. Delgado, E. García-González, R. Issaadi and J. M. López Nieto, Appl. Catal., A, 2021, 623, 1–12 CrossRef.
- A. M. Gaffney, J. W. Sims, V. J. Martin, N. V. Duprez, K. J. Louthan and K. L. Roberts, Catal. Today, 2021, 369, 203–209 CrossRef CAS.
- Z. Xie, Y. Ren, J. Li, Z. Zhao, X. Fan, B. Liu, W. Song, L. Kong, X. Xiao, J. Liu and G. Jiang, J. Catal., 2019, 372, 206–216 CrossRef CAS.
- P. Michorczyk, K. Zeńczak-Tomera, B. Michorczyk, A. Wegrzyniak, M. Basta, Y. Millot, L. Valentin and S. Dzwigaj, J. CO2 Util., 2020, 36, 54–63 CrossRef CAS.
- J. Wang, Y. H. Song, Z. T. Liu and Z. W. Liu, Appl. Catal., B, 2021, 297, 120400 CrossRef CAS.
- M. Numan, T. Kim, C. Jo and S. E. Park, J. CO2 Util., 2020, 39, 101184 CrossRef CAS.
- Y. Gao, X. Jie, C. Wang, R. M. J. Jacobs, W. Li, B. Yao, J. R. Dilworth, T. Xiao and P. P. Edwards, Ind. Eng. Chem. Res., 2020, 59, 12645–12656 CrossRef CAS.
- A. Al-Mamoori, S. Lawson, A. A. Rownaghi and F. Rezaei, Appl. Catal., B, 2020, 278, 119329 CrossRef CAS.
- M. A. Tedeeva, A. L. Kustov, P. V. Pribytkov, N. D. Evdokimenko, B. Sarkar and L. M. Kustov, Mendeleev Commun., 2020, 30, 195–197 CrossRef CAS.
- S. A. Chernyak, A. L. Kustov, D. N. Stolbov, M. A. Tedeeva, O. Y. Isaikina, K. I. Maslakov, N. V. Usol’tseva and S. V. Savilov, Appl. Surf. Sci., 2022, 578, 152099 CrossRef CAS.
- Z. Deng, X. Ge, W. Zhang, S. Luo, J. Shen, F. Jing and W. Chu, Chinese J. Chem. Eng., 2021, 34, 77–86 CrossRef CAS.
- I. I. Mishanin, T. V. Bogdan, A. E. Koklin and V. I. Bogdan, Chem. Eng. J., 2022, 446, 137184 CrossRef CAS.
- S. Lawson, A. Farsad, B. Adebayo, K. Newport, K. Schueddig, E. Lowrey, F. Polo-Garzon, F. Rezaei and A. A. Rownaghi, Adv. Sustainable Syst., 2021, 5, 1–15 Search PubMed.
- S. Lawson, K. A. Newport, A. Axtell, C. Boucher, B. Grant, M. Haas, M. Lee, F. Rezaei and A. A. Rownaghi, ACS Sustainable Chem. Eng., 2021, 9, 5716–5727 CrossRef CAS.
- A. Farsad, S. Lawson, F. Rezaei and A. A. Rownaghi, Catal. Today, 2021, 374, 173–184 CrossRef CAS.
- F. Xing, Y. Nakaya, S. Yasumura, K. Shimizu and S. Furukawa, Nat. Catal., 2022, 5, 55–65 CrossRef CAS.
- Q. Li, H. Yue, C. Liu, K. Ma, S. Zhong, B. Liang and S. Tang, Chem. Eng. J., 2020, 395, 125120 CrossRef CAS.
- R. Zhang, H. Wang, S. Tang, C. Liu, F. Dong, H. Yue and B. Liang, ACS Catal., 2018, 8, 9280–9286 CrossRef CAS.
- J. Liu, N. He, Z. Zhang, J. Yang, X. Jiang, Z. Zhang, J. Su, M. Shu, R. Si, G. Xiong, H. Bin Xie and G. Vilé, ACS Catal., 2021, 11, 2819–2830 CrossRef CAS.
- T. D. Nguyen, W. Zheng, F. E. Celik and G. Tsilomelekis, Catal. Sci. Technol., 2021, 11, 5791–5801 RSC.
- E. Nowicka, C. Reece, S. M. Althahban, K. M. H. Mohammed, S. A. Kondrat, D. J. Morgan, Q. He, D. J. Willock, S. Golunski, C. J. Kiely and G. J. Hutchings, ACS Catal., 2018, 8, 3454–3468 CrossRef CAS.
- Y. Liu, G. Zhang, J. Wang, J. Zhu, X. Zhang, J. T. Miller, C. Song and X. Guo, Chinese J. Catal., 2021, 42, 2225–2233 CrossRef CAS.
- N. Veeraraghavan Srinath, H. Poelman, L. Buelens, J. Dendooven, M.-F. Reyniers, G. B. Marin and V. V. Galvita, J. Catal., 2022, 408, 356–371 CrossRef CAS.
- M. L. Balogun, S. Adamu, M. S. Ba-Shammakh and M. M. Hossain, J. Ind. Eng. Chem., 2021, 96, 82–97 CrossRef CAS.
- L. Zeng, Z. Cheng, J. A. Fan, L. S. Fan and J. Gong, Nat. Rev. Chem., 2018, 2, 349–364 CrossRef CAS.
- W. Ding, K. Zhao, S. Jiang, Z. Zhao, Y. Cao and F. He, Appl. Catal., A, 2021, 609, 117910 CrossRef CAS.
- T. Wang, Y. Gao, Y. Liu, M. Song, J. Liu and Q. Guo, Fuel, 2021, 303, 121286 CrossRef CAS.
- X. Tian, C. Zheng, F. Li and H. Zhao, ACS Sustainable Chem. Eng., 2021, 9, 8002–8011 CrossRef CAS.
- S. Chen, L. Zeng, R. Mu, C. Xiong, Z. J. Zhao, C. Zhao, C. Pei, L. Peng, J. Luo, L. S. Fan and J. Gong, J. Am. Chem. Soc., 2019, 141, 18653–18657 CrossRef CAS PubMed.
- X. Tian, C. Zheng and H. Zhao, Appl. Catal., B, 2021, 303, 120894 CrossRef.
- L. Ye, X. Duan and K. Xie, Angew. Chem., 2021, 133, 21914–21918 CrossRef.
- Y. Gao, X. Wang, J. Liu, C. Huang, K. Zhao, Z. Zhao, X. Wang and F. Li, Sci. Adv., 2020, 6, eaaz9339 CrossRef CAS PubMed.
- J. Liu, Y. Gao, X. Wang and F. Li, Cell Rep. Phys. Sci., 2021, 2, 100503 CrossRef CAS.
- G. Luongo, F. Donat, M. Krödel, C.-C. Cormos and C. R. Müller, Renewable Sustainable Energy Rev., 2021, 149, 111403 CrossRef CAS.
- J. Liu, R. Hu, X. Liu, Q. Zhang, G. Ye, Z. Sui and X. Zhou, Chinese J. Chem. Eng., 2022, 47, 165–173 CrossRef.
- J. Wang, X. Chang, S. Chen, G. Sun, X. Zhou, E. Vovk, Y. Yang, W. Deng, Z. J. Zhao, R. Mu, C. Pei and J. Gong, ACS Catal., 2021, 11, 4401–4410 CrossRef CAS.
- K. Shimoda, S. Ishikawa, K. Matsumoto, M. Miyasawa, M. Takebe, R. Matsumoto, S. Lee and W. Ueda, ChemCatChem, 2021, 13, 3132–3139 CrossRef CAS.
- M. A. Tedeeva, A. L. Kustov, P. V. Pribytkov, G. I. Kapustin, A. V. Leonov, O. P. Tkachenko, O. B. Tursunov, N. D. Evdokimenko and L. M. Kustov, Fuel, 2022, 313, 122698 CrossRef CAS.
- X. Li, H. Chen, W. Liu, J. Shen, S. Luo and F. Jing, Mol. Catal., 2021, 509, 111658 CrossRef CAS.
- S. A. Theofanidis, C. Loizidis, E. Heracleous and A. A. Lemonidou, J. Catal., 2020, 388, 52–65 CrossRef CAS.
- M. Numan, E. Eom, A. Li, M. Mazur, H. W. Cha, H. C. Ham, C. Jo and S. E. Park, ACS Catal., 2021, 11, 9221–9232 CrossRef CAS.
- A. Galadima and O. Muraza, J. Ind. Eng. Chem., 2016, 37, 1–13 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- T. Jiang, J. Song, M. Huo, N. Yang, J. Liu, J. Zhang, Y. Sun and Y. Zhu, RSC Adv., 2016, 6, 34872–34876 RSC.
- F. Papa, P. Luminita, P. Osiceanu, R. Birjega, M. Akane and I. Balint, J. Mol. Catal. A: Chem., 2011, 346, 46–54 CrossRef CAS.
- J. S. Lee and S. T. Oyama, Catal. Rev., 1988, 30, 249–280 CrossRef CAS.
- U. Zavyalova, M. Holena, R. Schlögl and M. Baerns, ChemCatChem, 2011, 3, 1935–1947 CrossRef CAS.
- E. N. Voskresenskaya, Vi. G. Roguleva and A. G. Anshits, Catal. Rev., 1995, 37, 101–143 CrossRef CAS.
- M. Yıldız, J. Ind. Eng. Chem., 2019, 76, 488–499 CrossRef.
- L. Peng, J. Xu, X. Fang, W. Liu, X. Xu, L. Liu, Z. Li, H. Peng, R. Zheng and X. Wang, Eur. J. Inorg. Chem., 2018, 1787–1799 CrossRef CAS.
- V. J. Ferreira, P. Tavares, J. L. Figueiredo and J. L. Faria, Ind. Eng. Chem. Res., 2012, 51, 10535–10541 CrossRef CAS.
- D. V. Ivanov, L. A. Isupova, E. Y. Gerasimov, L. S. Dovlitova, T. S. Glazneva and I. P. Prosvirin, Appl. Catal., A, 2014, 485, 10–19 CrossRef CAS.
- I. Kim, G. Lee, H. Bin Na, J.-M. Ha and J. C. Jung, Mol. Catal., 2017, 435, 13–23 CrossRef CAS.
- T. W. Elkins, S. J. Roberts and H. E. Hagelin-Weaver, Appl. Catal., A, 2016, 528, 175–190 CrossRef CAS.
- T. Ito, J. Wang, C. H. Lin and J. H. Lunsford, J. Am. Chem. Soc., 1985, 107, 5062–5068 CrossRef CAS.
- A. Farsi, A. Moradi, S. Ghader and V. Shadravan, Chinese J. Chem. Phys., 2011, 24, 70–76 CrossRef CAS.
- T. Matsumoto, M. Saito, S. Ishikawa, K. Fujii, M. Yashima, W. Ueda and T. Motohashi, ChemCatChem, 2020, 12, 1968–1972 CrossRef CAS.
- T. W. Elkins, B. Neumann, M. Bäumer and H. E. Hagelin-Weaver, ACS Catal., 2014, 4, 1972–1990 CrossRef CAS.
- J. Y. Lee, W. Jeon, J.-W. Choi, Y.-W. Suh, J.-M. Ha, D. J. Suh and Y.-K. Park, Fuel, 2013, 106, 851–857 CrossRef CAS.
- R. Ghose, H. T. Hwang and A. Varma, Appl. Catal., A, 2014, 472, 39–46 CrossRef CAS.
- T. W. Elkins and H. E. Hagelin-Weaver, Appl. Catal., A, 2015, 497, 96–106 CrossRef CAS.
- N. S. Hayek, G. J. Khlief, F. Horani and O. M. Gazit, J. Catal., 2019, 376, 25–31 CrossRef CAS.
- A. T. Ashcroft, A. K. Cheetham, M. L. H. Green, C. P. Grey and P. D. F. Vernon, J. Chem. Soc., Chem. Commun., 1989, 1667 RSC.
- C. Petit, J. L. Rehspringer, A. Kaddouri, S. Libs, P. Poix and A. Kiennemann, Catal. Today, 1992, 13, 409–416 CrossRef CAS.
- J. Xu, Y. Zhang, Y. Liu, X. Fang, X. Xu, W. Liu, R. Zheng and X. Wang, Eur. J. Inorg. Chem., 2019, 183–194 CrossRef.
- S. D. Senanayake, J. A. Rodriguez and J. F. Weaver, Acc. Chem. Res., 2020, 53, 1488–1497 CrossRef CAS PubMed.
- D. Schröder and J. Roithová, Angew. Chem., Int. Ed., 2006, 45, 5705–5708 CrossRef PubMed.
- K. Qian, R. You, Y. Guan, W. Wen, Y. Tian, Y. Pan and W. Huang, ACS Catal., 2020, 10, 15142–15148 CrossRef CAS.
- L. Luo, Y. Jin, H. Pan, X. Zheng, L. Wu, R. You and W. Huang, J. Catal., 2017, 346, 57–61 CrossRef CAS.
- S. Arndt, T. Otremba, U. Simon, M. Yildiz, H. Schubert and R. Schomäcker, Appl. Catal., A, 2012, 425–426, 53–61 CrossRef CAS.
- D. Kiani, S. Sourav, J. Baltrusaitis and I. E. Wachs, ACS Catal., 2019, 9, 5912–5928 CrossRef CAS.
- A. Palermo, J. Holgadovazquez, A. Lee, M. Tikhov and R. Lambert, J. Catal., 1998, 177, 259–266 CrossRef CAS.
- J. Wang, L. Chou, B. Zhang, H. Song, J. Zhao, J. Yang and S. Li, J. Mol. Catal. A: Chem., 2006, 245, 272–277 CrossRef CAS.
- R. T. Yunarti, S. Gu, J.-W. Choi, J. Jae, D. J. Suh and J.-M. Ha, ACS Sustainable Chem. Eng., 2017, 5, 3667–3674 CrossRef CAS.
- C. A. Ortiz-Bravo, S. J. A. Figueroa, R. Portela, C. A. Chagas, M. A. Bañares and F. S. Toniolo, J. Catal., 2022, 408, 423–435 CrossRef CAS.
- J. Xu, L. Peng, X. Fang, Z. Fu, W. Liu, X. Xu, H. Peng, R. Zheng and X. Wang, Appl. Catal., A, 2018, 552, 117–128 CrossRef CAS.
- J. Xu, Y. Zhang, X. Xu, X. Fang, R. Xi, Y. Liu, R. Zheng and X. Wang, ACS Catal., 2019, 9, 4030–4045 CrossRef CAS.
- Y. Zhang, J. Xu, X. Xu, R. Xi, Y. Liu, X. Fang and X. Wang, Catal. Today, 2020, 355, 518–528 CrossRef CAS.
- J. Xu, R. Xi, X. Xu, Y. Zhang, X. Feng, X. Fang and X. Wang, J. Rare Earths, 2020, 38, 840–849 CrossRef.
- C. Karakaya and R. J. Kee, Prog. Energy Combust. Sci., 2016, 55, 60–97 CrossRef.
- D. Wang, J. H. Lunsford and M. P. Rosynek, Top. Catal., 1996, 3, 289–297 CrossRef CAS.
- M. Belgued, P. Pareja, A. Amariglio and H. Amariglio, Nature, 1991, 352, 789–790 CrossRef CAS.
- D. Gerceker, A. H. Motagamwala, K. R. Rivera-Dones, J. B. Miller, G. W. Huber, M. Mavrikakis and J. A. Dumesic, ACS Catal., 2017, 7, 2088–2100 CrossRef CAS.
- Y. Xiao and A. Varma, ACS Catal., 2018, 8, 2735–2740 CrossRef CAS.
- K. Dutta, V. Chaudhari, C.-J. Li and J. Kopyscinski, Appl. Catal., A, 2020, 595, 117430 CrossRef.
- K. Dutta, M. Shahryari and J. Kopyscinski, Ind. Eng. Chem. Res., 2020, 59, 4245–4256 CrossRef CAS.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- H. E. Toraman, K. Alexopoulos, S. C. Oh, S. Cheng, D. Liu and D. G. Vlachos, Chem. Eng. J., 2021, 420, 130493 CrossRef CAS.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- P. Barger, in Catalytic Science Series: Volume 3. Zeolites for Cleaner Technologies, ed. M. Guisnet and J.-P. Gilson, 2002, pp. 239–260 Search PubMed.
- M. Stöcker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- S. Peng, M. Gao, H. Li, M. Yang, M. Ye and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 21945–21948 CrossRef CAS PubMed.
- N. Wang, Y. Zhi, Y. Wei, W. Zhang, Z. Liu, J. Huang, T. Sun, S. Xu, S. Lin, Y. He, A. Zheng and Z. Liu, Nat. Commun., 2020, 11, 1079 CrossRef CAS PubMed.
- J. Zhou, M. Gao, J. Zhang, W. Liu, T. Zhang, H. Li, Z. Xu, M. Ye and Z. Liu, Nat. Commun., 2021, 12, 17 CrossRef CAS PubMed.
- L. Yang, C. Wang, L. Zhang, W. Dai, Y. Chu, J. Xu, G. Wu, M. Gao, W. Liu, Z. Xu, P. Wang, N. Guan, M. Dyballa, M. Ye, F. Deng, W. Fan and L. Li, Nat. Commun., 2021, 12, 4661 CrossRef CAS PubMed.
- L. Lin, M. Fan, A. M. Sheveleva, X. Han, Z. Tang, J. H. Carter, I. da Silva, C. M. A. Parlett, F. Tuna, E. J. L. McInnes, G. Sastre, S. Rudić, H. Cavaye, S. F. Parker, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, M. P. Attfield, Y. Liu, C. C. Tang, B. Han and S. Yang, Nat. Commun., 2021, 12, 822 CrossRef CAS PubMed.
- Y. Xue, J. Li, P. Wang, X. Cui, H. Zheng, Y. Niu, M. Dong, Z. Qin, J. Wang and W. Fan, Appl. Catal., B, 2021, 280, 119391 CrossRef CAS.
- S. Li, H. Yang, S. Wang, M. Dong, J. Wang and W. Fan, Microporous Mesoporous Mater., 2022, 329, 111538 CrossRef CAS.
- W. Zhang, Y. Zhi, J. Huang, X. Wu, S. Zeng, S. Xu, A. Zheng, Y. Wei and Z. Liu, ACS Catal., 2019, 9, 7373–7379 CrossRef CAS.
- S. Lin, Y. Zhi, W. Chen, H. Li, W. Zhang, C. Lou, X. Wu, S. Zeng, S. Xu, J. Xiao, A. Zheng, Y. Wei and Z. Liu, J. Am. Chem. Soc., 2021, 143, 12038–12052 CrossRef CAS PubMed.
- M. Gao, H. Li, W. Liu, Z. Xu, S. Peng, M. Yang, M. Ye and Z. Liu, Nat. Commun., 2020, 11, 3641 CrossRef CAS PubMed.
- L. Yang, C. Wang, L. Zhang, W. Dai, Y. Chu, J. Xu, G. Wu, M. Gao, W. Liu, Z. Xu, P. Wang, N. Guan, M. Dyballa, M. Ye, F. Feng, W. Fan and L. Li, Nat. Commun., 2021, 12, 4661 CrossRef CAS PubMed.
- M. Luo, Y. Fu, B. Hu, D. Wang, B. Wang and G. Mao, Appl. Catal., A, 2019, 570, 209–217 CrossRef CAS.
- S. S. Arora, D. L. S. Nieskens, A. Malek and A. Bhan, Nat. Catal., 2018, 1, 666–672 CrossRef CAS.
- S. S. Arora, Z. Shi and A. Bhan, ACS Catal., 2019, 9, 6407–6414 CrossRef CAS.
- S. Akhgar, J. Towfighi and M. Hamidzadeh, J. Sol–Gel Sci. Technol., 2020, 95, 253–264 CrossRef CAS.
- J. Zhong, J. Han, Y. Wei, S. Xu, T. Sun, X. Guo, C. Song and Z. Liu, J. Energy Chem., 2019, 32, 174–181 CrossRef.
- J. Ding, X. Shen, J. Zhou, Y. Ye, H. Liu and Z. Xie, ChemCatChem, 2020, 12, 6420–6425 CrossRef CAS.
- J. Han, Z. Liu, H. Li, J. Zhong, W. Zhang, J. Huang, A. Zheng, Y. Wei and Z. Liu, ACS Catal., 2020, 10, 8727–8735 CrossRef CAS.
- I. Yarulina, K. De Wispelaere, S. Bailleul, J. Goetze, M. Radersma, E. Abou-Hamad, I. Vollmer, M. Goesten, B. Mezari, E. J. M. Hensen, J. S. Martínez-Espín, M. Morten, S. Mitchell, J. Perez-Ramirez, U. Olsbye, B. M. Weckhuysen, V. Van Speybroeck, F. Kapteijn and J. Gascon, Nat. Chem., 2018, 10, 804–812 CrossRef CAS PubMed.
- I. Yarulina, S. Bailleul, A. Pustovarenko, J. R. Martinez, K. De Wispelaere, J. Hajek, B. M. Weckhuysen, K. Houben, M. Baldus, V. Van Speybroeck, F. Kapteijn and J. Gascon, ChemCatChem, 2016, 8, 3057–3063 CrossRef CAS.
- S. Bailleul, I. Yarulina, A. E. J. Hoffman, A. Dokania, E. Abou-Hamad, A. D. Chowdhury, G. Pieters, J. Hajek, K. De Wispelaere, M. Waroquier, J. Gascon and V. Van Speybroeck, J. Am. Chem. Soc., 2019, 141, 14823–14842 CrossRef CAS PubMed.
- J. Shang, G. Fu, Z. Cai, X. Feng, Y. Tuo, X. Zhou, H. Yan, C. Peng, X. Jin, Y. Liu, X. Chen, C. Yang and D. Chen, Bioresour. Technol., 2021, 330, 124975 CrossRef CAS PubMed.
- M. Yang, J. Shao, Z. Yang, H. Yang, X. Wang, Z. Wu and H. Chen, J. Anal. Appl. Pyrolysis, 2019, 137, 259–265 CrossRef CAS.
- C. Hu, C. Liu, Q. Liu, H. Zhang, S. Wu and R. Xiao, Fuel Process. Technol., 2020, 210, 106562 CrossRef CAS.
- H. Zhang, R. Xiao, B. Jin, D. Shen, R. Chen and G. Xiao, Bioresour. Technol., 2013, 137, 82–87 CrossRef CAS PubMed.
- C. Hu, H. Zhang and R. Xiao, Energy Convers. Manage., 2018, 177, 765–772 CrossRef CAS.
- A. Eschenbacher, R. J. Varghese, M. S. Abbas-Abadi and K. M. Van Geem, Chem. Eng. J., 2022, 428, 132087 CrossRef CAS.
- M. Artetxe, G. Lopez, M. Amutio, G. Elordi, J. Bilbao and M. Olazar, Chem. Eng. J., 2012, 207–208, 27–34 CrossRef CAS.
- C. Hong, F. Gong, M. Fan, Q. Zhai, W. Huang, T. Wang and Q. Li, J. Chem. Technol. Biotechnol., 2013, 88, 109–118 CrossRef CAS.
- K. Wang, P. A. Johnston and R. C. Brown, Bioresour. Technol., 2014, 173, 124–131 CrossRef CAS PubMed.
- S. Zhang, M. Yang, J. Shao, H. Yang, K. Zeng, Y. Chen, J. Luo, F. A. Agblevor and H. Chen, Sci. Total Environ., 2018, 628–629, 350–357 CrossRef CAS PubMed.
- W. Huang, F. Gong, M. Fan, Q. Zhai, C. Hong and Q. Li, Bioresour. Technol., 2012, 121, 248–255 CrossRef CAS PubMed.
- C. Hu, R. Xiao and H. Zhang, Bioresour. Technol., 2017, 243, 1133–1140 CrossRef CAS PubMed.
- Y. Xue, P. Johnston and X. Bai, Energy Convers. Manage., 2017, 142, 441–451 CrossRef CAS.
- M. Yang, J. Shao, H. Yang, K. Zeng, Z. Wu, Y. Chen, X. Bai and H. Chen, Bioresour. Technol., 2019, 273, 77–85 CrossRef CAS PubMed.
- A. G. Cherednichenko, E. B. Markova, T. F. Sheshko and E. A. Morozova, Catal. Today, 2021, 379, 80–86 CrossRef CAS.
- C. Witpathomwong, R. Longloilert, S. Wongkasemjit and S. Jitkarnka, Energy Procedia, 2011, 9, 245–251 CrossRef CAS.
- P. J. Donaj, W. Kaminsky, F. Buzeto and W. Yang, Waste Manage., 2012, 32, 840–846 CrossRef CAS PubMed.
- P. K. W. Likun, H. Zhang and R. Xiao, Energy Fuels, 2020, 34, 8376–8394 CrossRef CAS.
- J. Chattopadhyay, T. S. Pathak, R. Srivastava and A. C. Singh, Energy, 2016, 103, 513–521 CrossRef CAS.
- Q. Zhang, W. Deng and Y. Wang, J. Energy Chem., 2013, 22, 27–38 CrossRef CAS.
- G. Centi and S. Perathoner, Stud. Surf. Sci. Catal., 2004, 153, 1–8 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, Waste Manag., 2009, 29, 2625–2643 CrossRef CAS PubMed.
- H. Chand, P. Choudhary, A. Kumar, A. Kumar and V. Krishnan, J. CO2 Util., 2021, 51, 101646 CrossRef CAS.
- A. Kumar, P. Choudhary, A. Kumar, P. H. C. Camargo and V. Krishnan, Small, 2022, 18, 2101638 CrossRef CAS PubMed.
- R. J. O’Brien, L. Xu, R. L. Spicer and B. H. Davis, Energy Fuels, 1996, 10, 921–926 CrossRef.
- Y. Jin and A. K. Datye, J. Catal., 2000, 196, 8–17 CrossRef CAS.
- T. Riedel, G. Schaub, K. W. Jun and K. W. Lee, Ind. Eng. Chem. Res., 2001, 40, 1355–1363 CrossRef CAS.
- R. Liu, D. Leshchev, E. Stavitski, M. Juneau, J. N. Agwara and M. D. Porosoff, Appl. Catal., B, 2021, 284, 119787 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- C. Panzone, R. Philippe, A. Chappaz, P. Fongarland and A. Bengaouer, J. CO2 Util., 2020, 38, 314–347 CrossRef CAS.
- Y. Hou, J. Li, M. Qing, C.-L. Liu and W.-S. Dong, Mol. Catal., 2020, 485, 110824 CrossRef CAS.
- M. Guisnet and N. S. Gnep, Appl. Catal., A, 1996, 146, 33–64 CrossRef CAS.
- A. Martínez and C. López, Appl. Catal., A, 2005, 294, 251–259 CrossRef.
- X. Yang, R. Wang, J. Yang, W. Qian, Y. Zhang, X. Li, Y. Huang, T. Zhang and D. Chen, ACS Catal., 2020, 10, 3797–3806 CrossRef CAS.
- D.-V. N. Vo and A. A. Adesina, Appl. Catal., A, 2011, 399, 221–232 CrossRef CAS.
- T. Li, M. Virginie and A. Y. Khodakov, Appl. Catal., A, 2017, 542, 154–162 CrossRef CAS.
- D.-V. N. Vo, C. G. Cooper, T.-H. Nguyen, A. A. Adesina and D. B. Bukur, Fuel, 2012, 93, 105–116 CrossRef CAS.
- C. Liu, M. Virginie, A. Griboval-Constant and A. Y. Khodakov, Catal. Today, 2016, 261, 137–145 CrossRef CAS.
- S. F. Zaman, A. A. Alzahrani, S. Podila and Y. Al Hamed, Asia-Pac. J. Chem. Eng., 2020, 15, e2516 CAS.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–87 CrossRef CAS PubMed.
- Z. Li, L. Zhong, F. Yu, Y. An, Y. Dai, Y. Yang, T. Lin, S. Li, H. Wang, P. Gao, Y. Sun and M. He, ACS Catal., 2017, 7, 3622–3631 CrossRef CAS.
- C. Zhang, S. Li, L. Zhong and Y. Sun, J. Phys. Chem. C, 2021, 125, 6061–6072 CrossRef CAS.
- J. Xie, P. P. Paalanen, T. W. van Deelen, B. M. Weckhuysen, M. J. Louwerse and K. P. de Jong, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- J. van de Loosdrecht, F. G. Botes, I. M. Ciobica, A. Ferreira, P. Gibson, D. J. Moodley, A. M. Saib, J. L. Visagie, C. J. Weststrate and J. W. Niemantsverdriet, in Comprehensive Inorganic Chemistry II (2nd Edition), ed. J. Reedijk and K. Poeppelmeier, Elsevier, 2013, pp. 525–557 Search PubMed.
- P. Zhai, Y. Li, M. Wang, J. Liu, Z. Cao, J. Zhang, Y. Xu, X. Liu, Y.-W. Li, Q. Zhu, D. Xiao, X.-D. Wen and D. Ma, Chem, 2021, 7, 3027–3051 CAS.
- J. Xie, J. Yang, A. I. Dugulan, A. Holmen, D. Chen, K. P. de Jong and M. J. Louwerse, ACS Catal., 2016, 6, 3147–3157 CrossRef CAS.
- H. M. Torres Galvis, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, J. Am. Chem. Soc., 2012, 134, 16207–16215 CrossRef CAS PubMed.
- M. Casavola, J. Hermannsdörfer, N. de Jonge, A. I. Dugulan and K. P. de Jong, Adv. Funct. Mater., 2015, 25, 5309–5319 CrossRef CAS.
- K. M. Brunner, G. E. Harper, K. Keyvanloo, B. F. Woodfield, C. H. Bartholomew and W. C. Hecker, Energy Fuels, 2015, 29, 1972–1977 CrossRef CAS.
- K. Cheng, M. Virginie, V. V. Ordomsky, C. Cordier, P. A. Chernavskii, M. I. Ivantsov, S. Paul, Y. Wang and A. Y. Khodakov, J. Catal., 2015, 328, 139–150 CrossRef CAS.
- M. M. Moyer, C. Karakaya, R. J. Kee and B. G. Trewyn, ChemCatChem, 2017, 9, 3090–3101 CrossRef CAS.
- S. J. Liaw and B. H. Davis, Top. Catal., 2000, 10, 133–139 CrossRef CAS.
- Q. Hao, L. Bai, H. Xiang and Y. Li, Fuel Process. Technol., 2008, 89, 1358–1364 CrossRef CAS.
- P. A. Chernavskii, V. O. Kazak, G. V. Pankina, Y. D. Perfiliev, T. Li, M. Virginie and A. Y. Khodakov, Catal. Sci. Technol., 2017, 7, 2325–2334 RSC.
- R. P. Mogorosi, N. Fischer, M. Claeys and E. van Steen, J. Catal., 2012, 289, 140–150 CrossRef CAS.
- A. Wielers, J. Catal., 1989, 117, 1–18 CrossRef CAS.
- M. Luo, H. Hamdeh and B. H. Davis, Catal. Today, 2009, 140, 127–134 CrossRef CAS.
- Q.-Y. Liu, C. Shang and Z.-P. Liu, J. Am. Chem. Soc., 2021, 143, 11109–11120 CrossRef CAS PubMed.
- H. H. Podgurski, J. T. Kummer, T. W. DeWitt and P. H. Emmett, J. Am. Chem. Soc., 1950, 72, 5382–5388 CrossRef CAS.
- W. Arabczyk, W. Konicki, U. Narkiewicz, I. Jasińska and K. Kałucki, Appl. Catal., A, 2004, 266, 135–145 CrossRef CAS.
- C. G. Visconti, M. Martinelli, L. Falbo, A. Infantes-Molina, L. Lietti, P. Forzatti, G. Iaquaniello, E. Palo, B. Picutti and F. Brignoli, Appl. Catal., B, 2017, 200, 530–542 CrossRef CAS.
- J. Wang, Z. You, Q. Zhang, W. Deng and Y. Wang, Catal. Today, 2013, 215, 186–193 CrossRef CAS.
- W. D. Shafer, G. Jacobs, U. M. Graham, H. H. Hamdeh and B. H. Davis, J. Catal., 2019, 369, 239–248 CrossRef CAS.
- H. Schulz, Catal. Today, 2014, 228, 113–122 CrossRef CAS.
- H. Schulz, Catal. Today, 2013, 214, 140–151 CrossRef CAS.
- Y. Xu, X. Li and M. Ding, Chem, 2021, 7, 1977–1980 CAS.
- H. H. Anderson, R. B. Schultz, J. F. Hofer and L. J. E. Storch, US Bur. Mines Bull., 1959, 580, 1–30 Search PubMed.
- A. Chakkingal, L. Pirro, A. R. Costa da Cruz, A. J. Barrios, M. Virginie, A. Y. Khodakov and J. W. Thybaut, Chem. Eng. J., 2021, 419, 129633 CrossRef CAS.
- J. Yang, W. Ma, D. Chen, A. Holmen and B. H. Davis, Appl. Catal., A, 2014, 470, 250–260 CrossRef CAS.
- N. S. Govender, F. G. Botes, M. H. J. M. de Croon and J. C. Schouten, J. Catal., 2014, 312, 98–107 CrossRef CAS.
- T. Olewski, B. Todic, L. Nowicki, N. Nikacevic and D. B. Bukur, Chem. Eng. Res. Des., 2015, 95, 1–11 CrossRef CAS.
- A. J. Barrios, B. Gu, Y. Luo, D. V. Peron, P. A. Chernavskii, M. Virginie, R. Wojcieszak, J. W. Thybaut, V. V. Ordomsky and A. Y. Khodakov, Appl. Catal., B, 2020, 273, 119028 CrossRef CAS.
- B. Gu, A. Y. Khodakov and V. V. Ordomsky, Chem. Commun., 2018, 54, 2345–2348 RSC.
- J. Ding, L. Huang, W. Gong, M. Fan, Q. Zhong, A. G. Russell, H. Gu, H. Zhang, Y. Zhang and R. P. Ye, J. Catal., 2019, 377, 224–232 CrossRef CAS.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- A. J. Barrios, D. V. Peron, A. Chakkingal, A. I. Dugulan, S. Moldovan, K. Nakouri, J. Thuriot-Roukos, R. Wojcieszak, J. W. Thybaut, M. Virginie and A. Y. Khodakov, ACS Catal., 2022, 3211–3225 CrossRef CAS.
- T. J. Donnelly and C. N. Satterfield, Appl. Catal., 1989, 52, 93–114 CrossRef CAS.
- R. A. Dictor and A. T. Bell, J. Catal., 1986, 97, 121–136 CrossRef CAS.
- H. Arakawa and A. T. Bell, Ind. Eng. Chem. Process Des. Dev., 1983, 22, 97–103 CrossRef CAS.
- J. Li, X. Cheng, C. Zhang, J. Wang, W. Dong, Y. Yang and Y. Li, J. Chem. Technol. Biotechnol., 2017, 92, 1472–1480 CrossRef CAS.
- M. C. Ribeiro, G. Jacobs, B. H. Davis, D. C. Cronauer, A. J. Kropf and C. L. Marshall, J. Phys. Chem. C, 2010, 114, 7895–7903 CrossRef CAS.
- W. Ngantsoue-Hoc, Y. Zhang, R. J. O’Brien, M. Luo and B. H. Davis, Appl. Catal., A, 2002, 236, 77–89 CrossRef CAS.
- K. Cheng, V. V. Ordomsky, B. Legras, M. Virginie, S. Paul, Y. Wang and A. Y. Khodakov, Appl. Catal., A, 2015, 502, 204–214 CrossRef CAS.
- Y. Xu, X. Li, J. Gao, J. Wang, G. Ma, X. Wen, Y. Yang, Y. Li and M. Ding, Science, 2021, 371, 610–613 CrossRef CAS PubMed.
- K. Cheng, V. V. Ordomsky, M. Virginie, B. Legras, P. A. Chernavskii, V. O. Kazak, C. Cordier, S. Paul, Y. Wang and A. Y. Khodakov, Appl. Catal., A, 2014, 488, 66–77 CrossRef CAS.
- D. Peña, L. Jensen, A. Cognigni, R. Myrstad, T. Neumayer, W. van Beek and M. Rønning, ChemCatChem, 2018, 10, 1300–1312 CrossRef.
- N. Lohitharn, J. G. Goodwin Jr. and E. Lotero, J. Catal., 2008, 255, 104–113 CrossRef CAS.
- I. E. Wachs, D. J. Dwyer and E. Iglesia, Appl. Catal., 1984, 12, 201–217 CrossRef CAS.
- R. J. O’Brien and B. H. Davis, Catal. Lett., 2004, 94, 1–6 CrossRef.
- D. B. Bukur, D. Mukesh and S. A. Patel, Ind. Eng. Chem. Res., 1990, 29, 194–204 CrossRef CAS.
- H. Xiong, M. Moyo, M. A. M. Motchelaho, L. L. Jewell and N. J. Coville, Appl. Catal., A, 2010, 388, 168–178 CrossRef CAS.
- J. Abbott, N. J. Clark and B. G. Baker, Appl. Catal., 1986, 26, 141–153 CrossRef.
- J.-D. Xu, K.-T. Zhu, X.-F. Weng, W.-Z. Weng, C.-J. Huang and H.-L. Wan, Catal. Today, 2013, 215, 86–94 CrossRef CAS.
- H. Wang, Y. Yang, J. Xu, H. Wang, M. Ding and Y. Li, J. Mol. Catal. A Chem., 2010, 326, 29–40 CrossRef CAS.
- T. Herranz, S. Rojas, M. Ojeda, F. J. Pérez-Alonso, P. Terreros, K. Pirota and J. L. G. Fierro, Chem. Mater., 2006, 18, 2364–2375 CrossRef CAS.
- Z. Yang, Z. Zhang, Y. Liu, X. Ding, J. Zhang, J. Xu and Y. Han, Appl. Catal., B, 2021, 285, 119815 CrossRef CAS.
- S. Qin, C. Zhang, J. Xu, B. Wu, H. Xiang and Y. Li, J. Mol. Catal. A: Chem., 2009, 304, 128–134 CrossRef CAS.
- A. Alayat, E. Echeverria, D. N. Mcllroy and A. G. McDonald, Fuel Process. Technol., 2018, 177, 89–100 CrossRef CAS.
- R. M. Malek Abbaslou, J. Soltan and A. K. Dalai, Fuel, 2011, 90, 1139–1144 CrossRef CAS.
- D. J. Duvenhage and N. J. Coville, Catal. Lett., 2005, 104, 129–133 CrossRef CAS.
- H. M. Torres Galvis, A. C. J. Koeken, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, J. Catal., 2013, 303, 22–30 CrossRef CAS.
- V. V. Ordomsky, Y. Luo, B. Gu, A. Carvalho, P. A. Chernavskii, K. Cheng and A. Y. Khodakov, ACS Catal., 2017, 7, 6445–6452 CrossRef CAS.
- B. Gu, V. V. Ordomsky, M. Bahri, O. Ersen, P. A. Chernavskii, D. Filimonov and A. Y. Khodakov, Appl. Catal., B, 2018, 234, 153–166 CrossRef CAS.
- B. Gu, S. He, D. V. Peron, D. R. Strossi Pedrolo, S. Moldovan, M. C. Ribeiro, B. Lobato, P. A. Chernavskii, V. V. Ordomsky and A. Y. Khodakov, J. Catal., 2019, 376, 1–16 CrossRef CAS.
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef CAS PubMed.
- B. Gu, D. V. Peron, A. J. Barrios, M. Bahri, O. Ersen, M. Vorokhta, B. Šmíd, D. Banerjee, M. Virginie, E. Marceau, R. Wojcieszak, V. V. Ordomsky and A. Y. Khodakov, Chem. Sci., 2020, 11, 6167–6182 RSC.
- D. V. Peron, A. J. Barrios, A. Taschin, I. Dugulan, C. Marini, G. Gorni, S. Moldovan, S. Koneti, R. Wojcieszak, J. W. Thybaut, M. Virginie and A. Y. Khodakov, Appl. Catal., B, 2021, 292, 120141 CrossRef CAS.
- K. Keyvanloo, M. K. Mardkhe, T. M. Alam, C. H. Bartholomew, B. F. Woodfield and W. C. Hecker, ACS Catal., 2014, 4, 1071–1077 CrossRef CAS.
- R. J. O’Brien, L. Xu, S. Bao, A. Raje and B. H. Davis, Appl. Catal., A, 2000, 196, 173–178 CrossRef.
- R. Xu, P. S. Vengsarkar, D. Roe and C. B. Roberts, Energy & Fuels, 2017, 31, 4343–4352 CrossRef CAS.
- D. B. Bukur and C. Sivaraj, Appl. Catal., A, 2002, 231, 201–214 CrossRef CAS.
- J. Xu, C. H. Bartholomew, J. Sudweeks and D. L. Eggett, Top. Catal., 2003, 26, 55–71 CrossRef CAS.
- A. Y. Khodakov, A. Griboval-Constant, R. Bechara and V. L. Zholobenko, J. Catal., 2002, 206, 230–241 CrossRef CAS.
- A. Y. Khodakov, R. Bechara and A. Griboval-Constant, Appl. Catal., A, 2003, 254, 273–288 CrossRef CAS.
- C. C. Amoo, M. Li, A. Noreen, Y. Fu, E. Maturura, C. Du, R. Yang, X. Gao, C. Xing and N. Tsubaki, ACS Appl. Nano Mater., 2020, 3, 8096–8103 CrossRef CAS.
- Y. Chen, J. Wei, M. S. Duyar, V. V. Ordomsky, A. Y. Khodakov and J. Liu, Chem. Soc. Rev., 2021, 50, 2337–2366 RSC.
- B. Sun, K. Xu, L. Nguyen, M. Qiao and F. F. Tao, ChemCatChem, 2012, 4, 1498–1511 CrossRef CAS.
- T. Riedel, H. Schulz, G. Schaub, K. W. Jun, J. S. Hwang and K. W. Lee, Top. Catal., 2003, 26, 41–54 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884–890 RSC.
- L. Xu, Q. Wang, D. Liang, X. Wang, L. Lin, W. Cui and Y. Xu, Appl. Catal., A, 1998, 173, 19–25 CrossRef CAS.
- T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, Appl. Catal., A, 2006, 311, 66–75 CrossRef CAS.
- Z. Zhang, H. Yin, G. Yu, S. He, J. Kang, Z. Liu, K. Cheng, Q. Zhang and Y. Wang, J. Catal., 2021, 395, 350–361 CrossRef CAS.
- J. Zhang, S. Lu, X. Su, S. Fan, Q. Ma and T. Zhao, J. CO2 Util., 2015, 12, 95–100 CrossRef CAS.
- Z. Zhang, G. Huang, X. Tang, H. Yin, J. Kang, Q. Zhang and Y. Wang, Fuel, 2022, 309, 122105 CrossRef CAS.
- L. Torrente-Murciano, R. S. L. Chapman, A. Narvaez-Dinamarca, D. Mattia and M. D. Jones, Phys. Chem. Chem. Phys., 2016, 18, 15496–15500 RSC.
- F. Jiao, J. J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., 2016, 128, 4803–4806 CrossRef.
- C. Ratnasamy and J. P. Wagner, Catal. Rev., 2009, 51, 325–440 CrossRef CAS.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- J. Su, H. Zhou, S. Liu, C. Wang, W. Jiao, Y. Wang, C. Liu, Y. Ye, L. Zhang, Y. Zhao, H. Liu, D. Wang, W. Yang, Z. Xie and M. He, Nat. Commun., 2019, 10, 1297 CrossRef PubMed.
- F. Meng, X. Li, P. Zhang, L. Yang, S. Liu and Z. Li, Appl. Surf. Sci., 2021, 542, 148713 CrossRef CAS.
- M. Wang, Z. Wang, S. Liu, R. Gao, K. Cheng, L. Zhang, G. Zhang, X. Min, J. Kang, Q. Zhang and Y. Wang, J. Catal., 2021, 394, 181–192 CrossRef CAS.
- F. Meng, X. Li, P. Zhang, L. Yang, G. Yang, P. Ma and Z. Li, Catal. Today, 2021, 368, 118–125 CrossRef CAS.
- M. Wang, J. Kang, X. Xiong, F. Zhang, K. Cheng, Q. Zhang and Y. Wang, Catal. Today, 2021, 371, 85–92 CrossRef CAS.
- G. Yang, F. Meng, P. Zhang, L. Yang and Z. Li, New J. Chem., 2021, 45, 7967–7976 RSC.
- P. Zhang, F. Meng, L. Yang, G. Yang, X. Liang and Z. Li, Ind. Eng. Chem. Res., 2021, 60, 13214–13222 CrossRef CAS.
- X. Liu, M. Wang, C. Zhou, W. Zhou, K. Cheng, J. Kang, Q. Zhang, W. Deng and Y. Wang, Chem. Commun., 2018, 54, 140–143 RSC.
- L. Tan, P. Zhang, Y. Cui, Y. Suzuki, H. Li, L. Guo, G. Yang and N. Tsubaki, Fuel Process. Technol., 2019, 196, 106174 CrossRef CAS.
- J. Li, T. Yu, D. Miao, X. Pan and X. Bao, Catal. Commun., 2019, 129, 105711 CrossRef.
- M. Sedighi and M. Mohammadi, J. CO2 Util., 2020, 35, 236–244 CrossRef CAS.
- M. Ghasemi, M. Mohammadi and M. Sedighi, Microporous Mesoporous Mater., 2020, 297, 110029 CrossRef.
- X. Liu, M. Wang, H. Yin, J. Hu, K. Cheng, J. Kang, Q. Zhang and Y. Wang, ACS Catal., 2020, 10, 8303–8314 CrossRef CAS.
- S. Wang, P. Wang, Z. Qin, W. Yan, M. Dong, J. Li, J. Wang and W. Fan, J. Catal., 2020, 391, 459–470 CrossRef CAS.
- S. Lu, H. Yang, Z. Zhou, L. Zhong, S. Li, P. Gao and Y. Sun, Chin. J. Catal., 2021, 42, 2038–2048 CrossRef CAS.
- W. Li, K. Wang, G. Zhan, J. Huang and Q. Li, ACS Sustainable Chem. Eng., 2021, 9, 6446–6458 CrossRef CAS.
- M. Tong, L. Gapu Chizema, X. Chang, E. Hondo, L. Dai, Y. Zeng, C. Zeng, H. Ahmad, R. Yang and P. Lu, Microporous Mesoporous Mater., 2021, 320, 111105 CrossRef CAS.
- J. Mou, X. Fan, F. Liu, X. Wang, T. Zhao, P. Chen, Z. Li, C. Yang and J. Cao, Chem. Eng. J., 2021, 421, 129978 CrossRef CAS.
- F. Jiao, X. Pan, K. Gong, Y. Chen, G. Li and X. Bao, Angew. Chem., Int. Ed., 2018, 57, 4692–4696 CrossRef CAS PubMed.
- X. Fu, J. Li, J. Long, C. Guo and J. Xiao, ACS Catal., 2021, 11, 12264–12273 CrossRef CAS.
- S. Wang, P. Wang, D. Shi, S. He, L. Zhang, W. Yan, Z. Qin, J. Li, M. Dong, J. Wang, U. Olsbye and W. Fan, ACS Catal., 2020, 10, 2046–2059 CrossRef CAS.
- Y. Luo, S. Wang, S. Guo, K. Yuan, H. Wang, M. Dong, Z. Qin, W. Fan and J. Wang, Catal. Sci. Technol., 2021, 11, 338–348 RSC.
- Y. Peng, L. Wang, Q. Luo, Y. Cao, Y. Dai, Z. Li, H. Li, X. Zheng, W. Yan, J. Yang and J. Zeng, Chem, 2018, 4, 613–625 CAS.
- S. Wang, L. Zhang, W. Zhang, P. Wang, Z. Qin, W. Yan, M. Dong, J. Li, J. Wang, L. He, U. Olsbye and W. Fan, Chem, 2020, 6, 3344–3363 CAS.
- S. Dang, P. Gao, Z. Liu, X. Chen, C. Yang, H. Wang, L. Zhong, S. Li and Y. Sun, J. Catal., 2018, 364, 382–393 CrossRef CAS.
- L. Tan, F. Wang, P. Zhang, Y. Suzuki, Y. Wu, J. Chen, G. Yang and N. Tsubaki, Chem. Sci., 2020, 11, 4097–4105 RSC.
- Y. Zhu, X. Pan, F. Jiao, J. Li, J. Yang, M. Ding, Y. Han, Z. Liu and X. Bao, ACS Catal., 2017, 7, 2800–2804 CrossRef CAS.
- Z. Zhang, Y. Huang, H. Ma, W. Qian, H. Zhang and W. Ying, Catal. Commun., 2021, 152, 106292 CrossRef CAS.
- Y. Ni, Y. Liu, Z. Chen, M. Yang, H. Liu, Y. He, Y. Fu, W. Zhu and Z. Liu, ACS Catal., 2019, 9, 1026–1032 CrossRef CAS.
- T. Numpilai, S. Kahadit, T. Witoon, B. V. Ayodele, C. K. Cheng, N. Siri-Nguan, T. Sornchamni, C. Wattanakit, M. Chareonpanich and J. Limtrakul, Top. Catal., 2021, 64, 316–327 CrossRef CAS.
- M. Yang, B. Li, M. Gao, S. Lin, Y. Wang, S. Xu, X. Zhao, P. Guo, Y. Wei, M. Ye, P. Tian and Z. Liu, ACS Catal., 2020, 10, 3741–3749 CrossRef CAS.
- S. Dang, S. Li, C. Yang, X. Chen, X. Li, L. Zhong, P. Gao and Y. Sun, ChemSusChem, 2019, 12, 3582–3591 CrossRef CAS PubMed.
- W. Chen, G. Li, X. Yi, S. J. Day, K. A. Tarach, Z. Liu, S.-B. Liu, S. C. Edman Tsang, K. Góra-Marek and A. Zheng, J. Am. Chem. Soc., 2021, 143, 15440–15452 CrossRef CAS PubMed.
- H. Jiang, Z. Hou and Y. Luo, ACS Catal., 2020, 10, 13518–13523 CrossRef CAS.
- Y. Wang, G. Wang, L. I. Wal, K. Cheng, Q. Zhang, K. P. Jong and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 17735–17743 CrossRef CAS PubMed.
- Y. Ding, F. Jiao, X. Pan, Y. Ji, M. Li, R. Si, Y. Pan, G. Hou and X. Bao, ACS Catal., 2021, 11, 9729–9737 CrossRef CAS.
- J. Chen, X. Wang, D. Wu, J. Zhang, Q. Ma, X. Gao, X. Lai, H. Xia, S. Fan and T.-S. S. Zhao, Fuel, 2019, 239, 44–52 CrossRef CAS.
- G. Wang, Y. Wang, J. Cao, X. Wang, Y. Yi and F. Liu, Microporous Mesoporous Mater., 2020, 291, 109693 CrossRef.
- J. Gao, C. Jia and B. Liu, Catal. Sci. Technol., 2017, 7, 5602–5607 RSC.
- L. Zeng, Y. Cao, Z. Li, Y. Dai, Y. Wang, B. An, J. Zhang, H. Li, Y. Zhou, W. Lin and C. Wang, ACS Catal., 2021, 11, 11696–11705 CrossRef CAS.
- X. Liu, W. Zhou, Y. Yang, K. Cheng, J. Kang, L. Zhang, G. Zhang, X. Min, Q. Zhang and Y. Wang, Chem. Sci., 2018, 9, 4708–4718 RSC.
- G. Raveendra, C. Li, B. Liu, Y. Cheng, F. Meng and Z. Li, Catal. Sci. Technol., 2018, 8, 3527–3538 RSC.
- T. Numpilai, C. Wattanakit, M. Chareonpanich, J. Limtrakul and T. Witoon, Energy Convers. Manage., 2019, 180, 511–523 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- S. Xie, W. Ma, X. Wu, H. Zhang, Q. Zhang, Y. Wang and Y. Wang, Energy Environ. Sci., 2021, 14, 37–89 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- Y. Zhou and B. S. Yeo, J. Mater. Chem. A, 2020, 8, 23162–23186 RSC.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- W. E. Mustain, M. Chatenet, M. Page and Y. S. Kim, Energy Environ. Sci., 2020, 13, 2805–2838 RSC.
- E. Pérez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 3621–3624 CrossRef PubMed.
- Y. Kim, S. Park, S.-J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Energy Environ. Sci., 2020, 13, 4301–4311 RSC.
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C.-T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. Golam Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- R. Chen, H. Su, D. Liu, R. Huang, X. Meng, X. Cui, Z. Tian, D. H. Zhang and D. Deng, Angew. Chem., Int. Ed., 2020, 59, 154–160 CrossRef CAS PubMed.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- Y. Wang, D. Wang, C. J. Dares, S. L. Marquard, M. V. Sheridan and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 278–283 CrossRef CAS PubMed.
- H. A. Maddah, Am. Sci. Res. J. Eng. Technol. Sci, 2018, 45, 49–63 Search PubMed.
- European Commission, Horiz. 2020 – Work Program. 2014-2015 Gen. Annex. Extr. from Part 19 – Comm. Decis. C, 2014, 1.
- T. Ren, B. Daniëls, M. K. Patel and K. Blok, Resour., Conserv. Recycl., 2009, 53, 653–663 CrossRef.
- A. Boulamanti and J. A. Moya, Renewable Sustainable Energy Rev., 2017, 68, 1205–1212 CrossRef CAS.
- Z. Zhao, J. Jiang and F. Wang, J. Energy Chem., 2021, 56, 193–202 CrossRef.
- I. T. Horváth, Chem. Rev., 2018, 118, 369–371 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1cs01036k |
| This journal is © The Royal Society of Chemistry 2022 |