
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePutting precision and elegance in enzyme immobilisation with bio-orthogonal chemistry
Xiaolin
Pei†
 a,
Zhiyuan
Luo†
a,
Li
Qiao
a,
Qinjie
Xiao
a,
Pengfei
Zhang
*a,
Anming
Wang
*a and
Roger A.
Sheldon
*bc
a,
Zhiyuan
Luo†
a,
Li
Qiao
a,
Qinjie
Xiao
a,
Pengfei
Zhang
*a,
Anming
Wang
*a and
Roger A.
Sheldon
*bc
aCollege of Materials, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Key Laboratory of Organosilicon Material Technology, Hangzhou Normal University, Zhejiang Province, Hangzhou, 311121, Zhejiang, P. R. China
bMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO Wits, 2050, Johannesburg, South Africa. E-mail: roger.sheldon@wits.ac.za
cDepartment of Biotechnology, Section BOC, Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, The Netherlands
First published on 3rd August 2022
Abstract
The covalent immobilisation of enzymes generally involves the use of highly reactive crosslinkers, such as glutaraldehyde, to couple enzyme molecules to each other or to carriers through, for example, the free amino groups of lysine residues, on the enzyme surface. Unfortunately, such methods suffer from a lack of precision. Random formation of covalent linkages with reactive functional groups in the enzyme leads to disruption of the three dimensional structure and accompanying activity losses. This review focuses on recent advances in the use of bio-orthogonal chemistry in conjunction with rec-DNA to affect highly precise immobilisation of enzymes. In this way, cost-effective combination of production, purification and immobilisation of an enzyme is achieved, in a single unit operation with a high degree of precision. Various bio-orthogonal techniques for putting this precision and elegance into enzyme immobilisation are elaborated. These include, for example, fusing (grafting) peptide or protein tags to the target enzyme that enable its immobilisation in cell lysate or incorporating non-standard amino acids that enable the application of bio-orthogonal chemistry.
 Xiaolin Pei | Pei Xiaolin is an associate professor at the College of Materials, Chemistry and Chemical Engineering, Hangzhou Normal University, China. He obtained BSc (Biochemical Engineering) from Shaanxi University of Science & Technology, China, MSc (Biochemistry and Molecular Biology) from Jilin University under the supervision of Prof. Feng Yan, China and PhD (Biochemical Engineering) from Zhejiang University under the supervision of Prof. Yang Lirong, China. His current research interest is focused on biosynthesis of fine chemicals and natural products, including discovery, directed evolution and rational design of enzymes for cascade biosynthesis. |
 Zhiyuan Luo | Zhiyuan Luo received his BSc degree in 2019 and is currently a master degree candidate in the Chemical Synthetic Biology lab under supervision of Prof. Anming Wang at the College of Materials, Chemistry and Chemical Engineering, Hangzhou Normal University, China. His research focuses on the covalent immobilization of proteins through Tag/Catcher system and artificial amino acids for biocatalytic applications. |
 Li Qiao | Li Qiao earned her MS degree (2018) from Hangzhou Normal University under the supervision of Prof. Pengfei Zhang and completed her PhD degree (2022) in chemistry at Zhejiang University under the supervision of Prof. Yanguang Wang and Prof. Ping Lu. Now she is a postdoctoral researcher in the Chemical Synthetic Biology lab at Hangzhou Normal University. Her current research focuses on organic photosynthesis and building photobiocatalysis systems based on biological semiconductors. |
 Pengfei Zhang | Pengfei Zhang was born in Anhui, China. He obtained his Bachelor's degree (1986) from Huaibei Normal University, China, and completed his PhD degree (2001) at Zhejiang University under the supervision of Prof. Zhenchu Chen. Currently, he is a professor in the College of Science at Hangzhou Normal University. His current research effort focuses on developing green and efficient synthetic methodologies for coupling reaction, carbohydrate chemistry, enzymatic catalytic reaction, and chemical industry. |
 Anming Wang | Anming Wang obtained his PhD in biochemical engineering at Nanjing Tech University under the supervision of Prof. Shubao Shen and Prof. Pingkai Ouyang (An academician of the Chinese Academy of Engineering). Now, he is a professor in college of Materials, Chemistry and Chemical Engineering at Hangzhou Normal University. His research interests cover bio-orthogonal enzyme immobilization and synthetic biology. |
 Roger A. Sheldon | Roger Sheldon, widely known for developing the E factor for assessing environmental impacts of chemical processes, received the RSC Green Chemistry Award and the Biocat Lifetime Achievement Award for contributions to biocatalysis. He was elected a Fellow of the Royal Society (2015) and Honorary Fellow of the Royal Society of Chemistry (2019). He has a PhD from Leicester University (UK) and was at Shell Research Amsterdam (1969–1980), DSM-Andeno (1980–1990), Delft University of Technology (1991–2007) and CEO of CLEA Technologies (2006–2015). He is currently Distinguished Professor of Biocatalysis Engineering at the University of the Witwatersrand (South Africa). |
1. Introduction
Biocatalysis is green and sustainable1 and of fundamental importance for the envisioned transition to a climate-neutral, bio-based circular economy2,3 and achieving the UN Sustainable Development Goals.4 Thanks to spectacular advances in molecular biology it has become a mature technology with industrial applications, e.g. in pharmaceuticals manufacture.5,6 Biocatalytic processes consist of two types: (i) fermentations using growing microbial whole cells and (ii) reactions mediated by isolated enzymes. Whole cell mediated processes have the advantage that cofactors and fresh enzyme are continuously regenerated if necessary but the disadvantage that contamination by other (iso) enzymes present in the cells can lead to selectivity losses. Isolated enzymes are more expensive, owing to the added costs of isolation and purification, but don’t have the contamination problem.However, isolated enzymes are soluble in water which is an environmentally attractive solvent but it presents a problem for their efficient recovery and recycling. Consequently, isolated enzymes are generally applied on a single-use, throw-away basis which is both economically and environmentally unattractive and not conducive to a circular economy. This problem can be solved by immobilisation of the enzyme as a free-flowing solid, i.e. as a heterogeneous catalyst.7 Immobilisation also suppresses unfolding of the enzyme, resulting in increased thermal stability with the possibility of using the enzyme in continuous flow operation.8
2. Enzyme immobilisation
In this review we shall be concerned with the immobilisation of isolated enzymes. Enzyme immobilisation is divided into two categories: (i) those involving attachment to an insoluble support (carrier) (Fig. 1(A)), usually a natural or synthetic organic polymer or an inorganic solid such as silica or alumina and (ii) carrier-free self-immobilisation (Fig. 1(B)) by intermolecular cross-linking of enzyme molecules to form what is effectively an insoluble cross-linked polymer.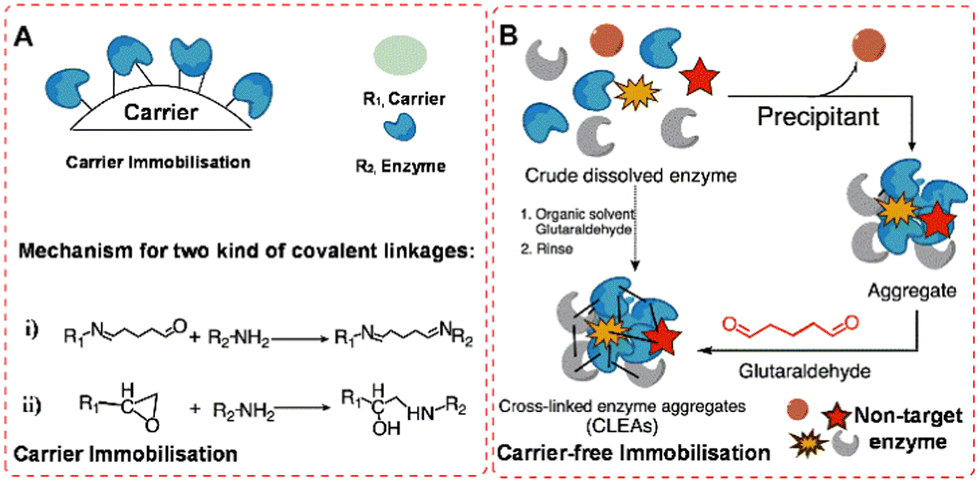 | ||
| Fig. 1 Random covalent enzyme immobilisation in traditional methods: (A) carrier immobilisation model (i), glutaraldehyde-mediated; (ii), epoxy-activation; (B), carrier-free immobilisation model. | ||
The first method can be subdivided on the basis of the nature of the bonding of the enzyme to the carrier. The simplest and least invasive is physical adsorption which generally works well in reactions in water-free organic media. The most well-known example of this technique is Novozyme 435 which consists of C. antarctica lipase (CaLB) immobilised on a macroporous resin.9 Alternatively, ionic bonding can be used by, for example, adsorbing the enzyme on an anionic or cationic ion-exchange resin, depending on the overall surface charge of the enzyme. Simple adsorption and ionic binding are widely used but they both suffer from the drawback that they are susceptible to leaching of the enzyme under aqueous conditions, depending on the pH, ionic strength and temperature, especially under the harsh conditions of many industrial processes.
Another method of binding to a carrier is affinity immobilisation which makes use of polymers containing surface chelating functionalities, such as iminodiacetic acid, preloaded with metal ions – Ni2+, Zn2+, Fe2+, Co2+ and Cu2+ – that exhibit a strong affinity for the imidazole groups in surface histidine residues of the enzyme. It has the advantage of high activity recovery through non-invasive binding.10,11 Moreover, recombinant proteins are usually produced containing a so-called His-tag – a string of six to nine histidine residues – attached to the N- or C-terminus – to enable purification through affinity binding. This creates the possibility of combining enzyme isolation, purification and immobilisation into a single operation.
Leaching of enzyme from the surface of immobilisates can be circumvented by forming stronger covalent bonds between e.g. aldehyde or epoxide functionalities on the carrier surface and free amino groups in lysine residues present on the surface of most enzymes (Fig. 1(A)). Immobilisation through covalent bonding is widely used in industry, for example, using polymethacrylates containing epoxide functionalities.12 Covalent bonding has the advantage of physically and chemically stable binding of the enzyme to the carrier but it is not feasible to regenerate the carrier from a spent catalyst.
Methods for immobilisation of enzymes on carriers have one serious shortcoming in common: a huge dilution of activity, ranging from 90% to >99%, accompanied by proportional reductions in catalyst and volumetric productivities (space-time yields). This problem is obviated by employing carrier-free methods in which enzyme molecules are covalently linked together to form microscopic solid particles. The most widely used method for carrier-free immobilisation is as cross-linked enzyme aggregates (CLEAs). The method, first described in 2000,13 involves precipitation of the enzyme from an aqueous solution as physical aggregates, without disturbing its tertiary structure. This could be by addition of, for example, a saturated solution of ammonium sulfate at optimum pH. Subsequent cross-linking by addition of a di-aldehyde (Fig. 1(B)), usually glutaraldehyde, produces insoluble aggregates with retention of their tertiary structure and activity.
CLEAs are cost-effective14 and, since precipitation is generally used to purify enzymes, they can be produced directly from fermentation broth. They are characterised by high catalyst and volumetric productivities and improved storage and operational stability to denaturation by heat, organic solvents and autolysis. They also have excellent stability towards leaching in aqueous media and can easily be recovered and recycled. They are highly porous materials and diffusional limitations are not observed with the bioconversions commonly used in organic synthesis. CLEAs have been successfully applied with many enzymes in a wide range of industrial settings.14,15
The shortcomings of CLEAs are derived from their relatively fragile and small, non-uniform particles (5–50 μm and typically below 10 μm). This is a serious drawback for operation in fixed bed reactors owing to the high pressure drop over the column. It can be counteracted by blending with non-compressible particles such as controlled-pore glass but the size and fragility of CLEA particles remains an issue for large scale applications. Alternatively, addition of a cross-linker to a suspension of an enzyme in a water-in-oil emulsion produces CLEAs as uniform enzyme microspheres-spherezymes-with a particle size of 50 μm and a size distribution of ±3%.16 However, because of the strong and rapid covalent linkage formation using di-aldehydes, random linkage is inevitable, and some non-target enzyme proteins can be immobilised in the CLEAs (Fig. 1(B)), which hinders one-step purification and immobilisation.
More recently, enzyme immobilisation has become even more important in streamlining the biocatalytic production of chemicals in bio-refineries as a result of the increasing emphasis on the development of a circular bio-based economy. In order to optimise the cost-contribution of enzymes their immobilisation has become an integral part of biocatalytic process development that facilitates the integration of production and downstream processing.17 For example, traditionally enzymes are optimised using various protein engineering techniques and when the optimum variant has been selected an immobilisation procedure is developed. However, it could well be that the optimum enzyme variant, obtained by protein engineering, does not afford the optimum immobilised biocatalyst. It makes more sense, therefore, to integrate enzyme immobilisation into the screening protocol of the protein engineering.18,19
Primarily aimed at further streamlining of biocatalytic processes, a multitude of methods for the cost-effective in vivo immobilisation of enzymes have been developed in recent years20 by combining enzyme production with in vivo immobilisation to afford a one-step, cost-effective production of an immobilised enzyme. This is achieved by genetic fusion of a self-assembly promoting protein to the target enzyme. For example, certain microorganisms produce inclusions of polyhydroxyalkanoates (PHAs) for energy storage and in vivo fusion of the central enzyme of PHA biosynthesis, polyester synthase (PhaC), to the target enzyme affords a method for one-step production, from a renewable resource, of an enzyme immobilised on a bio-based plastic.21,22
In another variation on this theme, protein engineering was employed to optimise the production of catalytic inclusion bodies (CatIBs).23 Bacterial inclusion bodies (IBs) are usually encountered as worthless catalytically inactive material formed as an undesirable consequence of heterologous over-expression of recombinant genes. Catalytically active IB formation can be achieved by fusing short peptide tags or aggregation-inducing protein domains to the target protein, affording cost-effective, carrier-free immobilised enzymes (CatIBs) in vivo.24 Furthermore, the activities of CatIBs are comparable with those of the corresponding soluble enzymes, which indicates a lack of diffusion limitations. A further improvement in the context of large scale applications is provided by the development of magnetically recoverable CatIBs.25 For example, magnetic cross-linked inclusion bodies of the lipase CaLB exhibited 114% activity recovery, with only 15% loss of activity after recycling 10 times due to excellent stability.26
Finally, in the wake of the burgeoning circular bio-based economy, waste biopolymers27,28 contained in agricultural, forestry, marine wastes and food waste, are becoming available in large amounts as inexpensive carriers for enzymes. Examples include lignocellulose derived biopolymers29 and magnetic cellulose.30 We can expect that such renewable, biodegradable polymers will find broad applications in the cost-effective and environmentally attractive immobilisation of enzymes in the future.
Regardless of which covalent approach is used, random covalent bond formation, with various amino acid residues in the target enzyme, is a problem that needs to be addressed. It can lead to loss of tertiary structure and, consequently, loss of activity. This is a long-standing question that has attracted the attention of biologists, materials scientists and chemists.31 What is clearly needed to address this problem is more precision in forming covalent bonds between the enzyme and a carrier or other enzyme molecules. If this can be successfully integrated with the isolation and purification of recombinant enzymes directly from cell lysate, this will lead to cost-effective, broadly applicable methodologies.
3. Enzyme immobilisation through genetic modification
Genetic modification of enzymes has been used to facilitate their immobilisation on carriers. For example, Zbasic2 is a silica binding module (SBM) with a size of ca. 7 kDa and a high positive charge density resulting from exposed clusters of multiple arginine residues on one side.32In vivo attachment of Zbasic2 to target enzymes, affords chimeric enzymes in which the positive charge density directs their non-covalent attachment to a negatively charged support such as silica. Thus, in vivo fusion of Zbasic2 to a D-amino acid oxidase in E. coli BL21 (DE3) afforded a chimera that was subsequently non-covalently attached in a defined orientation to a silica carrier32 The technique was also used to immobilise enzymes in the walls of microfluidic flow reactors.32,33Similarly, in vivo generation of fusion constructs of cellulose-binding module (CBM) with the ω-transaminase from Bacillus megaterium enabled immobilisation of the CBM-tagged enzyme, with ca. 95% activity retention, on macroporous cellulose scaffolds derived from delignified wood.34
3.1 Genetic modification for affinity immobilisation
The most widely used method is genetic insertion of poly(His) tags that can bind to metal ions. This forms the basis of the EziG (EnginZyme AB Sweden) technology for immobilisation of a broad range of enzymes.10,11,35–38 It involves fusion of the His6-tag to target enzymes at the N or C terminus. Following the fermentation, the His-tagged recombinant enzyme is separated from cell lysate by affinity binding of the imidazole groups in His residues to surface Ni2+, Co2+ or Fe3+ ions coordinated to surface iminodiacetic acid functions on controlled porosity glass (CPG). The immobilised enzymes are especially suitable for use in packed bed reactors (PBRs).11 In the case of purified enzymes, the maximum loading on the EziGTM carrier is 80% (Fig. 2(A)).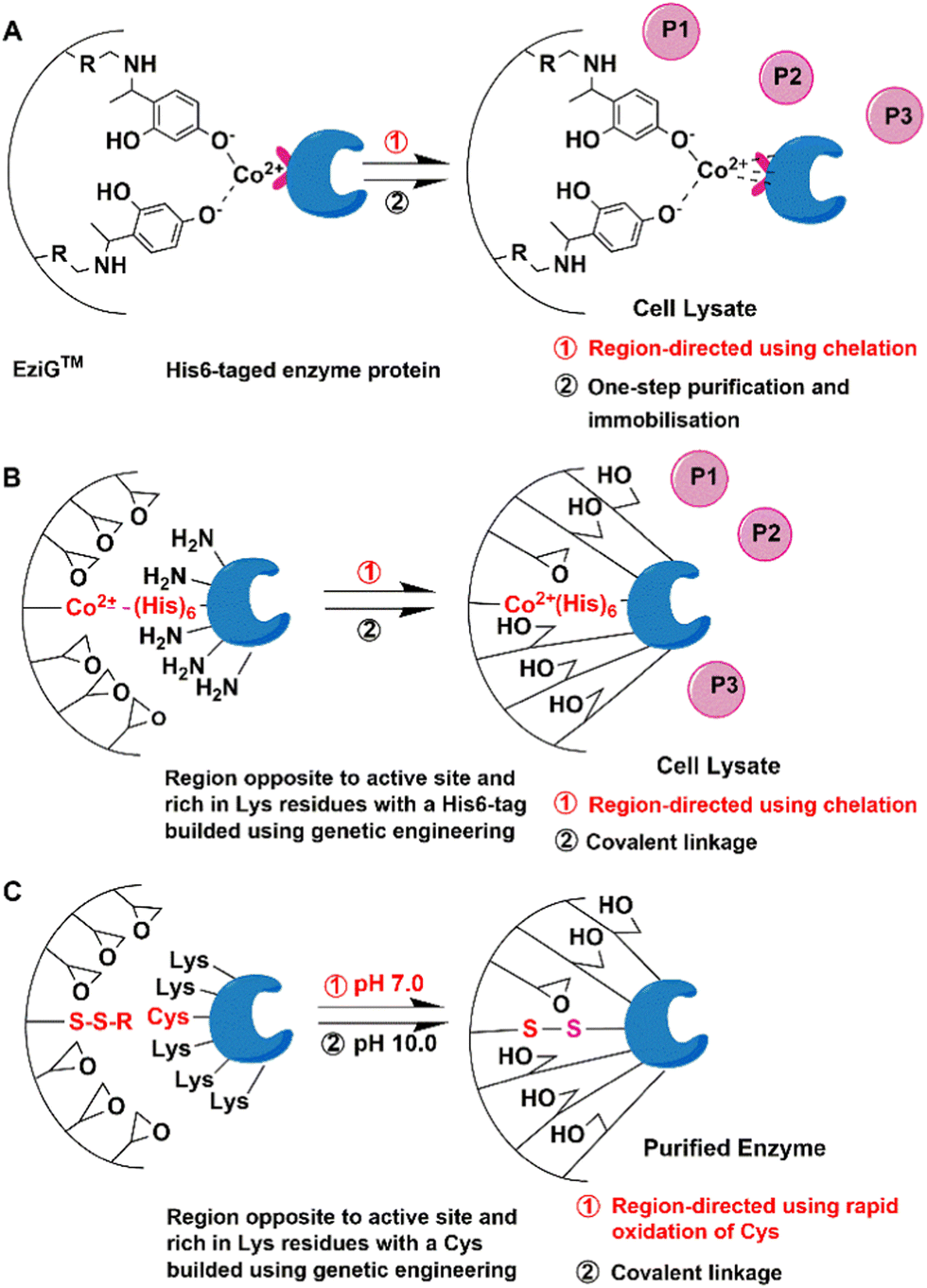 | ||
| Fig. 2 Scheme of directed affinity immobilisation and multipoint covalent immobilisation of enzymes by genetic modification. | ||
However, some enzymes such as P450 monooxygenases11 and ω-transaminase,39 exhibited reduced activities (less than 50% of the free enzymes) which may have resulted from aggregation of enzymes in the internal pores of EZiG™ and hidden active sites inaccessible for substrates.
3.2 Genetic amination and modification for directed and multi-point immobilisation
Genetic amination,40,41 compared to chemical amination using chemical modification in vitro, refers to techniques for the in vivo introduction of surface primary amino groups by inserting Lys in the desirable region or genetic fusion of enzymes to oligomers of amino acids containing free amino groups, e.g. His, Lys or Arg. This produces chimeric His-, Lys- or Arg-tagged enzymes. Depending on these residues, subsequent multi-point immobilisation on cation exchange resins and/or covalent bond formation with chemically reactive groups, have also been described.42,43For example, a tag consisting of three Lys alternating with three Gly residues at the end of the β chain, far from the active site, was used to efficiently control the immobilisation of penicillin G acylase from E.coli by covalent attachment to a glyoxyl agarose support.44 The substrate could freely access the well-protected active site of the enzyme and, therefore, the immobilised PGA mutant maintained its excellent catalytic properties in the synthesis of cephalosporins.
The same approach was also successfully applied to the covalent immobilisation of Geobacillus thermocatenulatus lipase.40 The active site is protected and the enzyme is directed to form multipoint attachments to the support.
In methods involving poly-His tag chelation in combination with covalent bond formation, the latter can, at least partially, be random. This can be avoided by using more precise bio-orthogonal chemistry, otherwise known as orthogonal bio-conjugation with nonstandard amino acids insertion.52–55 Bio-orthogonal chemistry is usually defined as any chemical reaction that occurs in living systems without interfering with native biochemical processes,56–58 that is chemical reactions in vivo.59–62 However, the definition of bio-orthogonal chemistry has been gradually expanded to include chemical reactions in vitro as applied in the materials science and medical science.63–65
This review focuses on biomolecule-mediated covalent ligation using a variety of so-called Tag/Catcher systems, enzyme-mediated ligation, incorporation of non-standard amino acids and tyrosine-mediated oxidation ligation for bio-orthogonal immobilisation.
4. Tag-mediated bio-orthogonal covalent immobilisation of enzymes
Protein tags are peptide sequences genetically grafted onto recombinant proteins. Grafting is usually at either the C- or N-terminus or sometimes into the coding sequence, with the specific aim of separating the recombinant protein in pure, immobilised form directly from cell lysate. A wide variety of tags are applied in biomolecule-mediated covalent immobilisation of enzymes directly from cell lysate with excellent precision and activity retention.66–704.1 Tag-mediated self-catalytic biorthogonal ligation
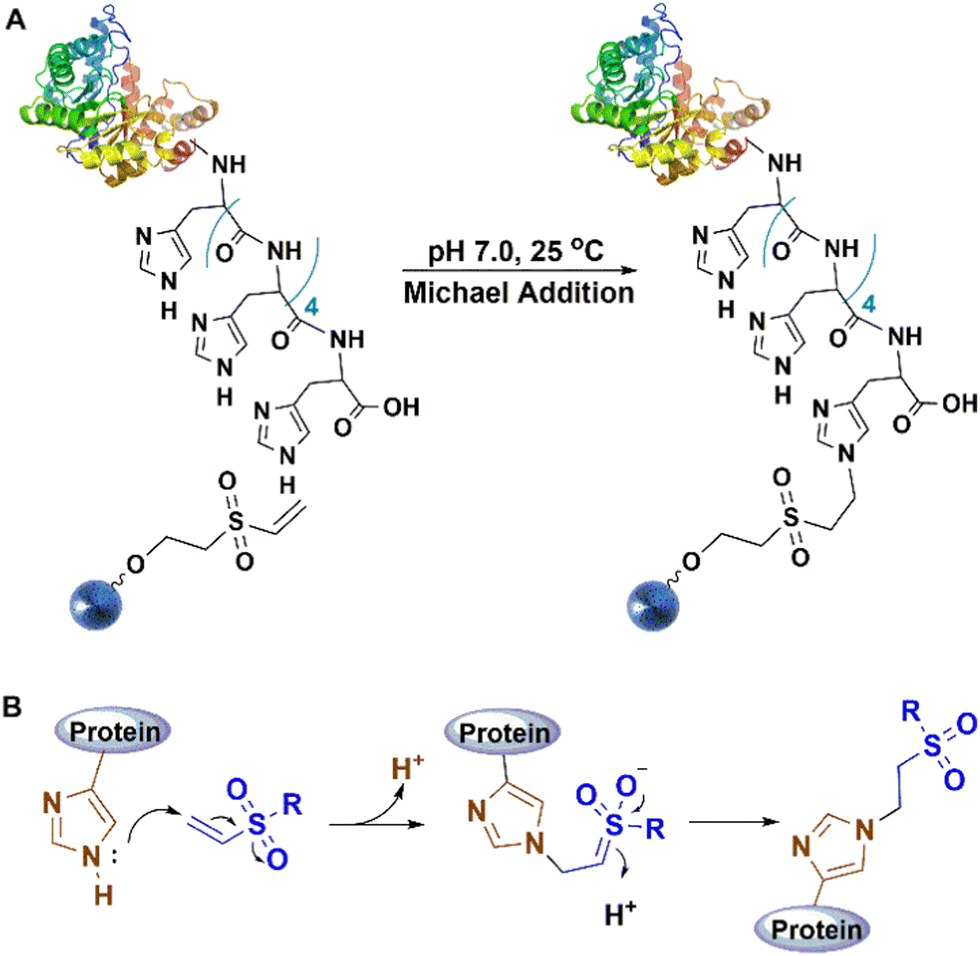 | ||
| Fig. 3 Reaction mechanism of His-tag reaction with vinyl sulfone. | ||
The method offers two significant advantages compared to existing covalent linking methods: high density and specificity. This facilitates the application of recombinant proteins in biosensor and array fabrication, and drug delivery systems, in addition to biocatalysis.17,35,72
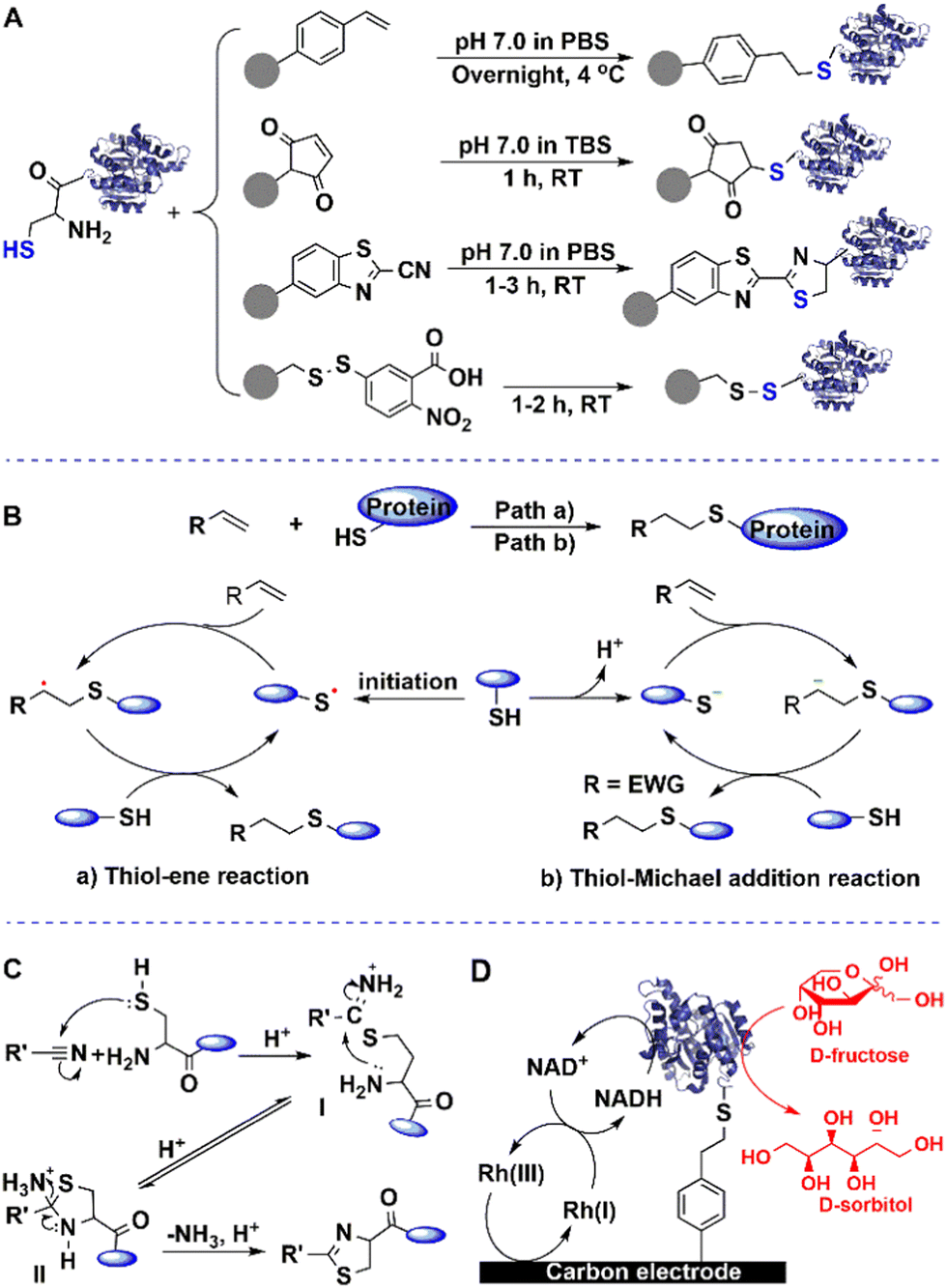 | ||
| Fig. 4 Immobilisation of enzymes through terminal cysteine-tags. | ||
The combination of cysteine-tagged proteins with a thiol−ene click bio-orthogonal reaction opens up perspectives for site-specific covalent immobilisation of enzymes with high activity retention. Moreover, positioning a cysteine tag at different locations in the polypeptide chain should allow a controllable orientation of the enzyme. The method also demonstrated good potential in screening of an epidermal growth factor receptor (EGFR)-tag and its inhibitor ibrutinib.75
When this tag is combined with streptavidin affinity technology, it can enhance the current streptavidin-based solid-phase bio-assay systems. Streptavidin (Stv-C) and streptactin present intrinsic affinity toward Strep II-tag, a synthetic peptide consisting of eight amino acids (Trp-Ser-His-Pro-Gln-Phe-Glu-Lys), and can be fused to target recombinant proteins to facilitate their isolation, purification and immobilisation on solid carriers.76 To further optimise the affinity and immobilisation, a streptavadin mutant was constructed by fusing a six-amino acid peptide (Gly-Gly-Ser-Gly-Cys-Pro) to its C-terminus (Fig. 4(A)).77 This single cysteine residue functions as a unique immobilisation site for bio-orthogonal conjugation using sulfhydryl chemistry.
When five tandem cysteine repeats (Cys-tag) have been fused at termini of enzyme proteins,78 this Cys-tag can facilitate the covalent attachment of proteins to the surface of a maleimide-modified support. In high-throughput functional proteomics as little as 5 pg of the purified Cys-tagged enhanced green fluorescent protein (EGFP) was immunodetected by this enhanced chemiluminescence system. The bio-orthogonal reaction of the sulfhydryl moiety of the Cys residue with maleimide enables the immobilisation of target enzymes on maleimide-coated surfaces (Fig. 4(B)). The novel immobilised enzyme protein mutants facilitate use in a wide variety of solid-phase diagnostic tests and biomedical assays.
Cyanobenzothiazole (CBT)-cysteine condensation can also be used to create a biorthogonal linkage with cysteine-tagged enzymes.79,80 Thiol addition to the cyano group affords a thioimidate I, which undergoes intramolecular reaction with the terminal amino group to form the 2-aminothiazolidine II. The latter then undergoes deamination to a thiazoline. The reaction takes place under very mild conditions, and is highly selective towards terminal cysteine-containing enzymes (Fig. 4(C)).
Most noteworthy is the reversible immobilisation of enzymes using S–S bond formation via this tag when the Cys residue is not essential and necessary for the target enzyme.81 On the one hand, the enzyme is covalently attached on a functionalised support and presents stronger stability than that obtained using other immobilisation methods, and on the other hand, enzymes can be removed from the support using dithiothreitol (DTT) solution when the enzyme activity has reached a low to unacceptable level. With this facile chemical treatment, the support can be regenerated and reused for the immobilisation of the next fresh batch of enzyme.
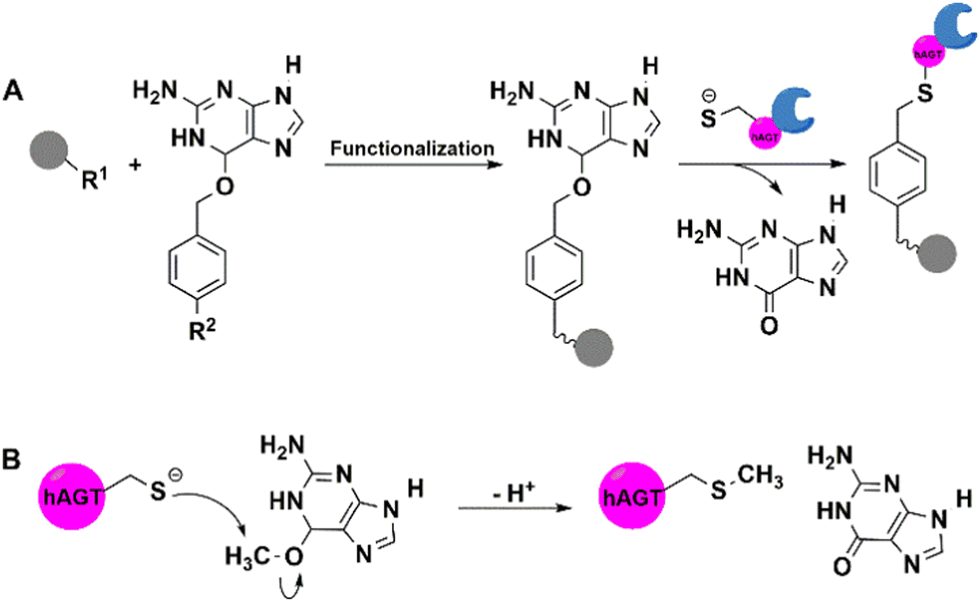 | ||
| Fig. 5 hAGT-mediated covalent immobilisation of enzyme protein on supports functionalised using O6-benzylguanine (BG) derivatives. | ||
CLIP-tag is a mutant of hAGT that was engineered87 for exclusivity with O-2-benzylcytosine (BC) derivatives. SNAP- and CLIP-tags have orthogonal substrate specificities. CLIP-tag is used in conjunction with other bio-orthogonal reactions, such as BC derivatives with azide or alkynyl groups, to achieve immobilisation.87
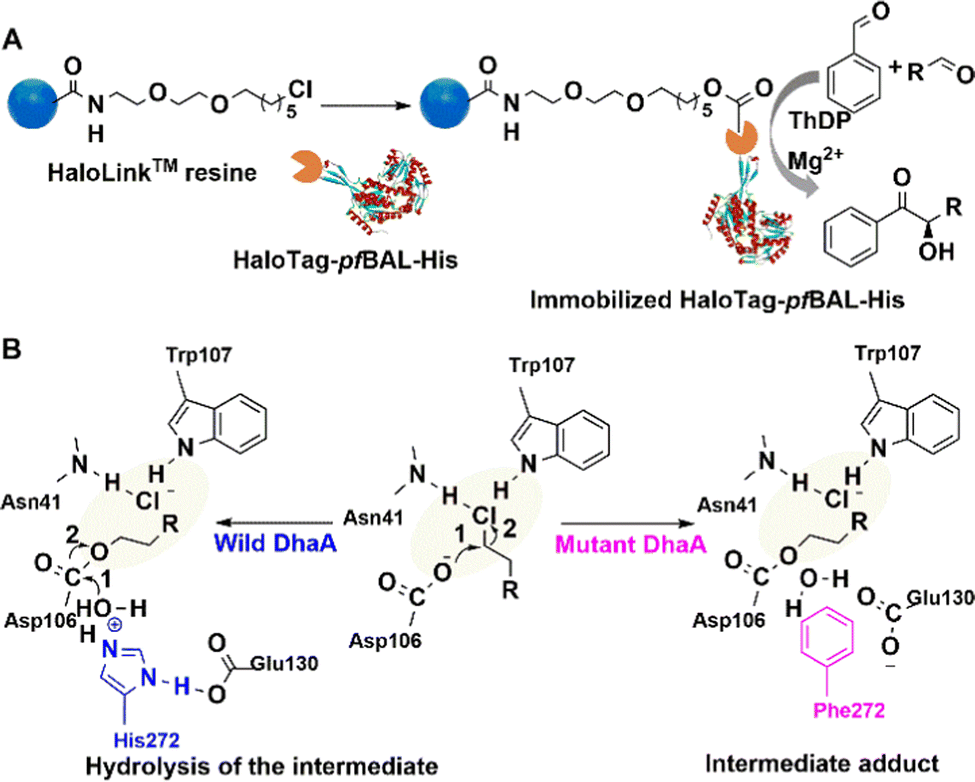 | ||
| Fig. 6 Immobilisation procedure and mechanism of HaloTag-enzyme on HaloLink Resin and the carboligation reactions. (A) Reprinted with permission from ref. 89. Copyright 2017, Elsevier. | ||
For example, Halo-tag was fused to the benzaldehyde lyase from P. fluorescence (PfBAL) without any negative effect on soluble expression and activity of the enzyme. It was subsequently immobilised on commercially available Halo-Link™ resin (Promega). The immobilisate was successfully reused in several consecutive batches in the condensation of benzaldehyde and acetaldehyde (Fig. 6(A)).89 Halo-tag is also used to study drug–protein interactions.90
4.2 Tag-mediated extra-enzymatic biorthogonal ligation
In vivo biotinylation was used, for example, to produce biotinylated bacterial magnetic particles (BacMPs),99,100 Streptavidin was introduced onto the biotinylated BacMPs by simple mixing to provide an attractive method for site-directed immobilisation on BacMPs.99 Fusion of BAP to the C-terminal of beta-glucosidase from Bacillus licheniformis, and co-expression with BirA in Escherichia coli afforded the biotinylated beta-glucosidase. The latter was simultaneously isolated from cell lysate and immobilised on BacMAPs based on the high affinity between streptavidin and biotin (Fig. 7).101 The immobilised beta-glucosidase showed excellent specific activity and thermal stability compared to the free enzyme.
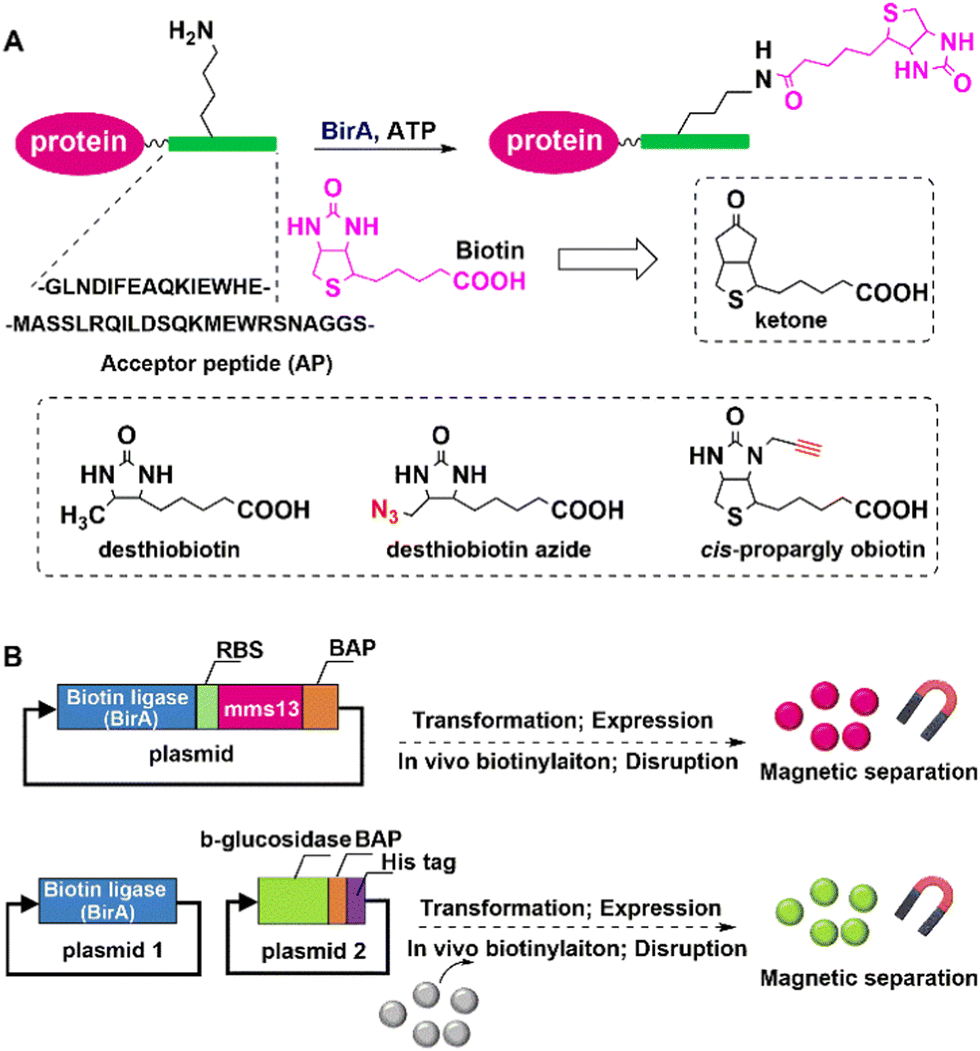 | ||
| Fig. 7 Site-specific protein labeling using biotin ligase (A) and protein immobilisation by the biotinylation system in vivo (B). | ||
SrtA acts as a sort of molecular “stapler” that mediates site-specific cross-linking of enzyme molecules with increased thermal stability. For example, Staphylococcus aureus SrtA mediated the cross-linking of (S)-ketoreductase (KRED II) from C. parapsilosis to produce a thermostable immobilised biocatalyst for efficient enantioselective reduction of ketones.107 Thus, SrtA mediated conjugation of KRED II subunits, via coupling of a native N-terminal glycine and an added GGGGSLPETGG peptide at the C-terminus of KRED II to form mainly cross-linked dimers and trimers. The resulting cross-linked KRED catalysed the smooth reduction of 2-hydroxyacetophenone to (S)-1-phenyl-1,2-ethanediol, in 99.9% yield and >99.9% enantioselectivity (Fig. 8). The scope of the method was demonstrated in efficient, highly enantioselective reductions of a series of substituted acetophenones with a further eight KREDs.107
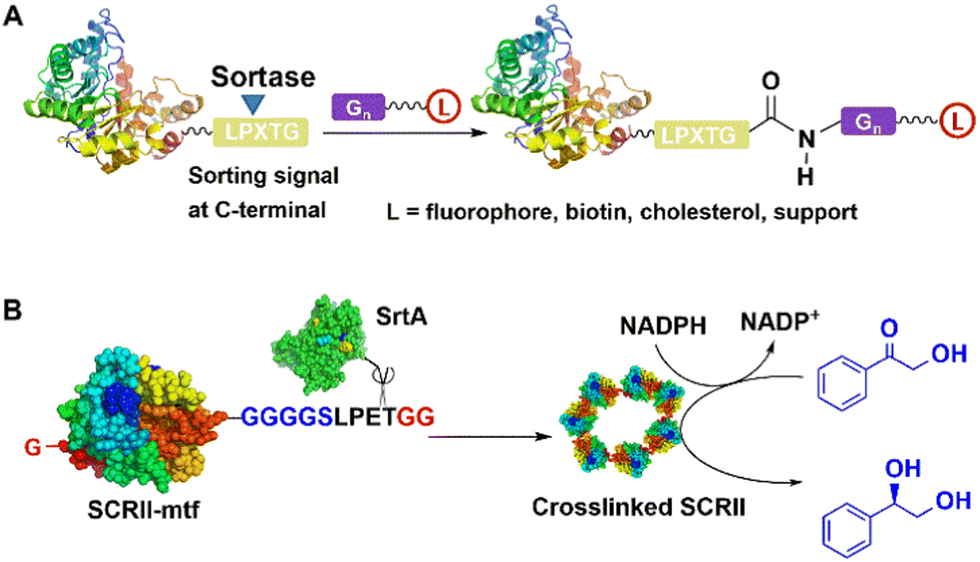 | ||
| Fig. 8 SrtA-catalysed conjugation (A) of enzyme and formation of (S)-ketoreductase (KRED II) ring (B) for enantioselective reduction of 2-hydroxyacetophenone. | ||
The S. aureus SrtA also mediated the ligation of a range of proteins to solid supports, such as polymer beads, and glass surfaces modified to display an oligoglycine motif.108–110 This transpeptidation methodology provides a general, robust, and gentle approach to the selective covalent immobilisation of proteins on solid supports. SrtA-mediated ligation was also used to attach the photosystem I (PSI) complex from Synechocystis sp. PCC 6803 to a conductive gold surface in the desired orientation for enhancing electron transfer in the conversion of light energy to electrical energy.111
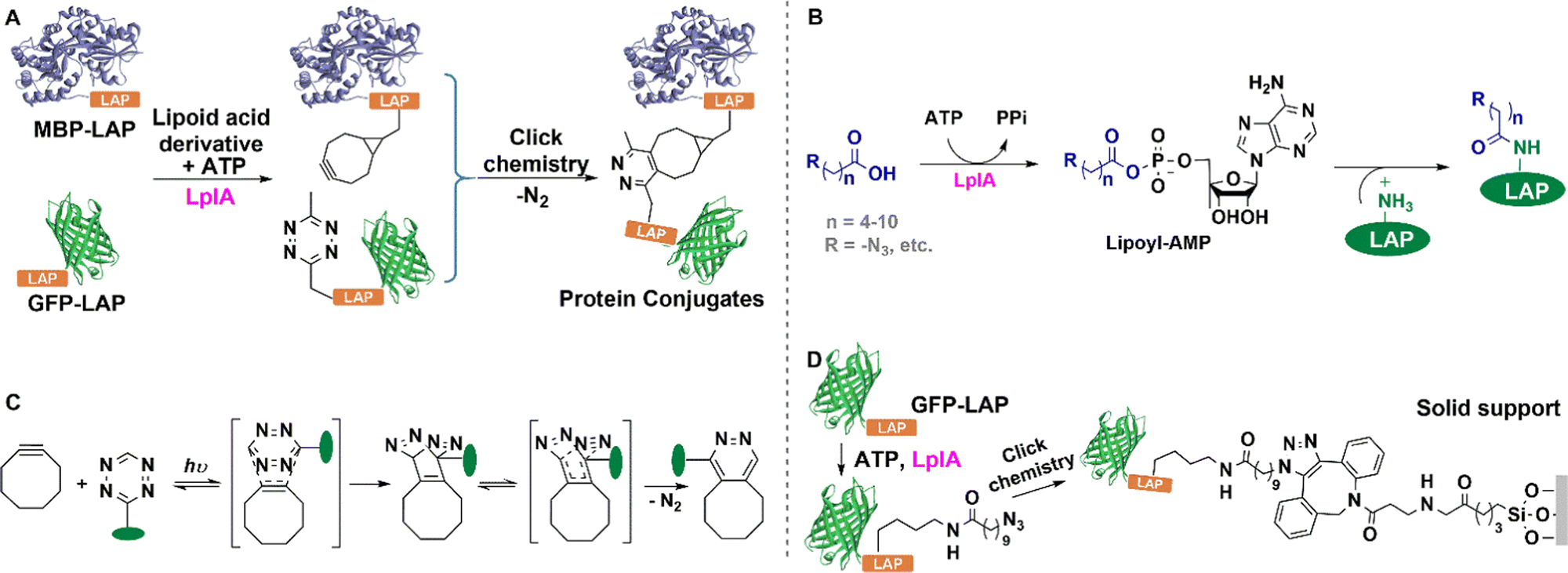 | ||
| Fig. 9 Site-specific ligation by click reaction of azide groups using lipoic acid ligase and subsequent chemical modification of GFP. | ||
5. Natural amino acid-mediated bio-orthogonal covalent immobilisation of proteins
5.1 Ligation via isopeptide bonds in tag/catcher systems
In covalent immobilisation through intermolecular cross-linking of enzymes, e.g. in CLEAs, enzyme molecules form solid state structures with increased stability and facile recovery. However, building proteins into larger, post-translational assemblies in a defined and stable way is still a challenging task. A promising approach relies on so-called tag/catcher systems (Fig. 10). Tags and Catchers usually consist of peptide/protein pairs respectively, derived from the cleavage of protein domains containing intramolecular isopeptide or ester bonds that form autocatalytically under physiological conditions. Cleavage affords a mixture of the cleaved peptide (the Tag) and the remaining polypeptide (the Catcher) which are genetically fused to recombinant proteins of interest. Intermolecular isopeptide or ester bond formation can subsequently be spontaneously reconstituted in the cell lysate. Tag/Catcher systems are widely used in numerous biotechnological and medical applications.116 Indeed, spontaneous covalent bond formation is potentially the most convenient and cost-effective method for one-step purification and immobilisation of enzymes.117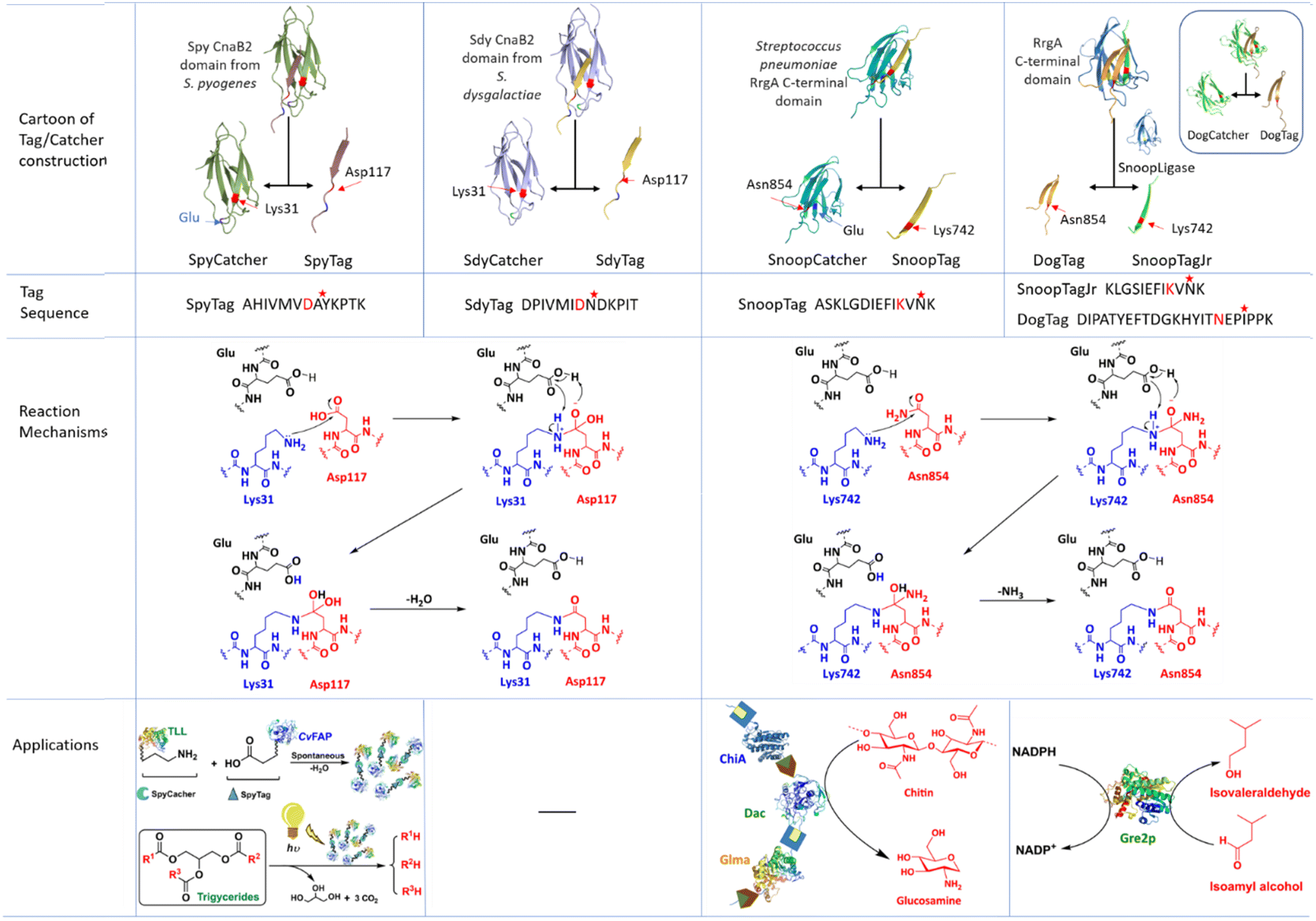 | ||
| Fig. 10 Contsruction, sequence, isopeptide bond formation mechanism and application of Tag/Catcher system used in producing self-cross-linked enzyme aggregates. | ||
The Spytag/Spycatcher technology constitutes a versatile platform for the development of novel biomaterials69,118,123–125 including synthetic multi-enzyme complexes for use in cascade biocatalysis.126–128 Thus, more efficient enzymatic cascade reactions are enabled by spatially confining enzymes using the SpyTag/SpyCatcher technology. Spatial proximity results in increased rates of mass transfer through so-called substrate channeling, e.g. as in metabolomes.69,126,127,129 Thus, fusion of Thermomyces lanuginosus lipase (TLL) with a carboxylic acid photodecarboxylase from Chlorella variabilis (CvFAP) using the Spytag/Spycatcher technique was employed in a one-pot cascade conversion of triglycerides to hydrocarbon biofuels.130
The SpyTag/SpyCatcher technique is also used to immobilise enzymes with precision on, for example, glyoxylagarose. Thus, a fusion of the SpyTag peptide with the target enzyme is conducted in vivo and the resulting cell lysate is allowed to react with the SpyCatcher which is attached to the glyoxylagarose support.68 Control of macromolecular topology with genetically encoded SpyTag–SpyCatcher chemistry69,123,131 is also used to engineer protein hydrogels from tandem modular elastomeric and enzyme proteins that can find applications in tissue engineering, controlled drug release132 and immobilisation of enzymes in multi-enzyme nanodevices.129,133–136
A single target enzyme molecule can be fused to both a SpyTag and a SpyCatcher and subsequently undergo cyclisation to form circular proteins, so-called SpyRings, through intramolecular formation of the original isopeptide bond, or formation of enzyme aggregates through intermolecular coupling.69 Circular proteins occur widely in nature, in plants animals and bacteria, and generally exhibit exceptional thermal stability and resistance to hydrolysis and proteolysis.137 For example, a SpyRing cyclised cephalosporin C acylase (SRCCA) was obtained by fusing SpyTag and SpyCatcher to the N- and C-termini of cephalosporin C acylase (CCA). The circular SRCCA exhibited higher thermal stability and tolerance to organic solvents and denaturation in general compared with the free CCA. In addition, the high number of Lys residues in the SpyRing of SRCCA facilitated its immobilisation by multipoint, oriented attachment to, for example, glyoxyl agarose.68,131
Improved variants of the SpyTag/SpyCatcher system, obtained through the application of computer-aided design and directed evolution, have also been described.138–140 However, limitations of the SpyTag/SpyCatcher system are that it occurs at termini of polypeptide chains141,142 and that it often induces the formation of active inclusion bodies in E. coli.143
The SnoopTag/SpyCatcher pair offers a robust and facile method to immobilise enzymes.122 In this method BslA, a small (16 kDa) protein present in bacterial biofilms which self-assembles to form a monolayer at a hydrophobic/hydrophilic interface, is functionalised with SpyTag or SnoopTag and deposited on a surface. A fusion protein of the target enzyme with the partner protein, SpyCatcher or SnoopCatcher, is allowed to spontaneously form a covalent isopeptide bond with the SpyTag or SnoopTag function immobilised on the surface.
Enzyme cyclisation using the SpyTag/SpyCatcher or SnoopTag/SnoopCatcher systems is a simple and effective method to improve the thermal stability of enzymes. For example, construction of four cyclised variants of trehalose synthase (TreS, EC 5.4.99.16) from Thermomonospora curvata, using various Tag/Catcher pairs120 led to a much larger increase in thermostability than that achieved using site-directed mutagenesis. Similarly, fusion of a Catcher module to the C-terminus of luciferase and a Tag module to its N-terminus enabled a spontaneous reaction between the Catcher and Tag to afford a circular luciferase with increased thermal stability.121
Analogous strategies can be used to assemble multi-enzyme complexes or metabolons, which are natural assemblies of enzymes functioning in cascade biosynthesis in vivo, such as terpene biosynthesis.118 It was used, for example, in a cascade synthesis of amorpha-4,11-diene.127 An interesting elaboration of the tag/catcher theme is provided by the in vivo one-pot construction of immobilised multi-enzyme cascades on PHA microspheres to afford a versatile toolbox for enzyme immobilisation in vivo. Tag/Catcher pairs, e.g. SdyTag/SdyCatcher and SnoopTag/SnoopCatcher were genetically fused to the N- and C-termini of Ralstonia eutropha PHA synthase (PhaC)146 to enable the in vivo covalent immobilisation of a monomeric amylase and a dimeric organophosphate hydrolase as target enzymes (Fig. 10).20,145
The DogTag/Dog Catcher system was derived from cleavage of a Streptococcus pneumonia adhesin and optimised through structure-based design and evolution, to increase the reaction rate 250-fold (Table 1). DogTag mediated loop insertion in variants of isovaleraldehyde reductase Gre2p did not result in any significant loss of activity (Fig. 10).139
| Tag/catcher system | Protein domain and microorganism source | Mutations | Amino acids involved in isopeptide bond formation | Partner proteins content and reaction time | Second-order rate constant for binding reaction (M−1 s−1) | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Tag | Catcher | Terminal | Internal | |||||
| SpyTag/SpyCatcher | CnaB2, Streptococcus pyogenes | Q97D and K108E (SpyTag003) | T91E, Q97D, N103D, and K108E (SpyCatcher003) | Lys 31 of SpyCatcher and Asp117 of SpyTag | 100 nM, 2 min | (5.5 ± 0.6) × 105 (sfGFP) | 87 ± 8 (sfGFP) | 66, 147 and 148 |
| 322 ± 4 (EGFP) | ||||||||
| SdyTag/SdyCatcher | CnaB2, Streptococcus dysgalactiae | N | N | Lys 31 of SdyCatcher and Asp117 of SdyTag | 10 μM, 80 min | 75 ± 6 (EGFP) | N | 116, 144 and 146 |
| SnoopTag/SnoopCatcher | RrgA, Streptococcus pneumoniae | N | G842T, D848G | Lys742 of DogCatcher and Asn854 of DogTag | 10 μM, 15 min | N | N | 118 and 145 |
| DogTag/DogCatcher | RrgA, Streptococcus pneumoniae | G842T, N847D, and D848G | N744D, N780D, K792T, and N825D | Lys742 of DogCatcher and Asn854 of DogTag | 5 μM,5 min | 1000 ± 80 (sfGFP) | 760 ± 20 (sfGFP) | 139 and 145 |
| D737E and N746T | ||||||||
5.2 Enzyme mediated bio-orthogonal ligation for precision immobilisation
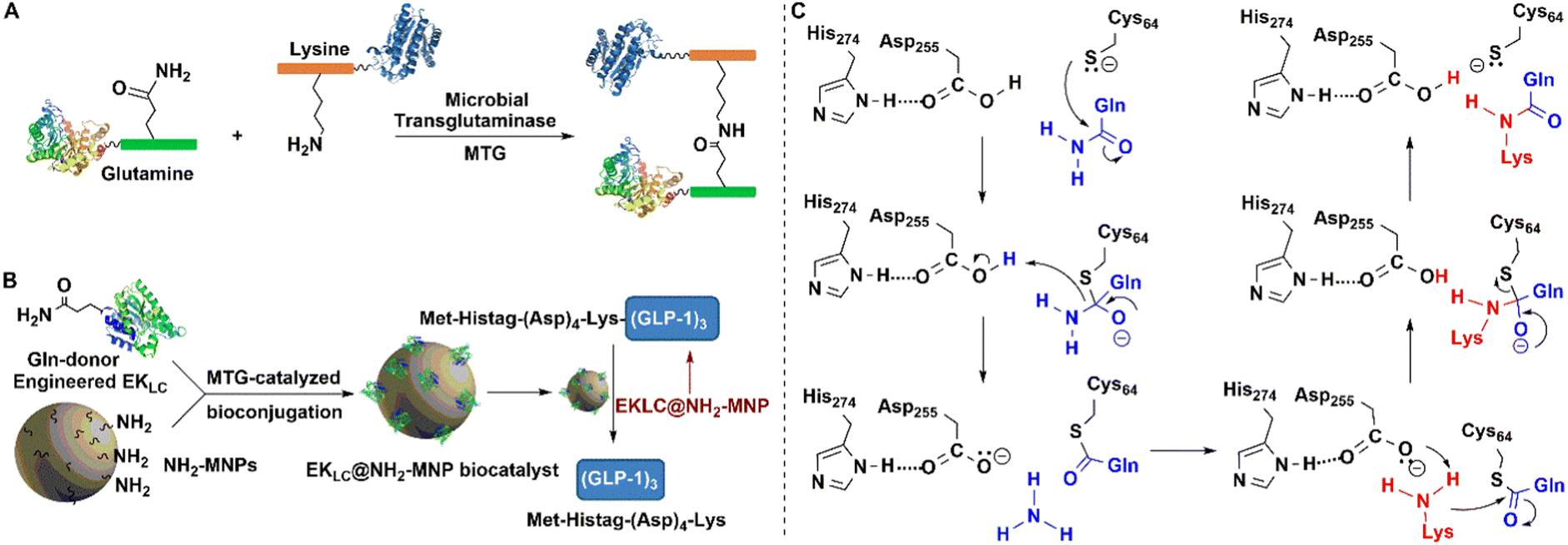 | ||
| Fig. 11 Covalent protein assembly catalysed by microbial transglutaminase depending on polyTag. (B) Reprinted with permission from ref. 154. Copyright 2019, Elsevier. | ||
MTG reacts with a broad range of amine substrates but is particularly effective with long-chain amines. In contrast with other enzymatic conjugation methods that react exclusively at protein termini, MTG catalysed transamidation also occurs at accessible glutamine residues located at internal sites.152
Building both a lysine and a glutamine residue into a single peptide tag, designated as a PolyTag, and fusing to either an N- or C-terminus, enables the MTG catalysed formation of cross-linked aggregates of target proteins and enzymes153 (Fig. 11(A)). This enzyme mediated transformation starts with a nucleophilic attack of the sulfhydryl group of cysteine on the γ-carboxamide of glutamine to afford an energy-rich thioester bond. Subsequent attack of the lysine amino group on the latter results in bioconjugation through the Lys–Gln bond (Fig. 11(C)). MTG-catalysed bioconjugation was used, for example, in the site-directed, covalent immobilisation of an engineered Enterokinase (EK) on amine-modified magnetic nanoparticles (Fig. 11(B)) to afford a robust and recyclable immobilisate.153,154
In this way a genetically encoded “aldehyde tag” was introduced into the recombinant lipase. The aldehyde tag was used to attach the target enzyme to pendant primary amine functionalities on the surface of a support via Schiff's base formation and subsequent reduction. Overall this affords a one-step purification and site-directed covalent immobilisation of the target enzyme.155 Moreover, the novel immobilised lipase was more thermally stable than a more traditional immobilised lipase (Fig. 12). The technique was also successfully applied to immobilisation of an alkaline phosphatase (AP) and methyltryptophan oxidase (MTOX).156
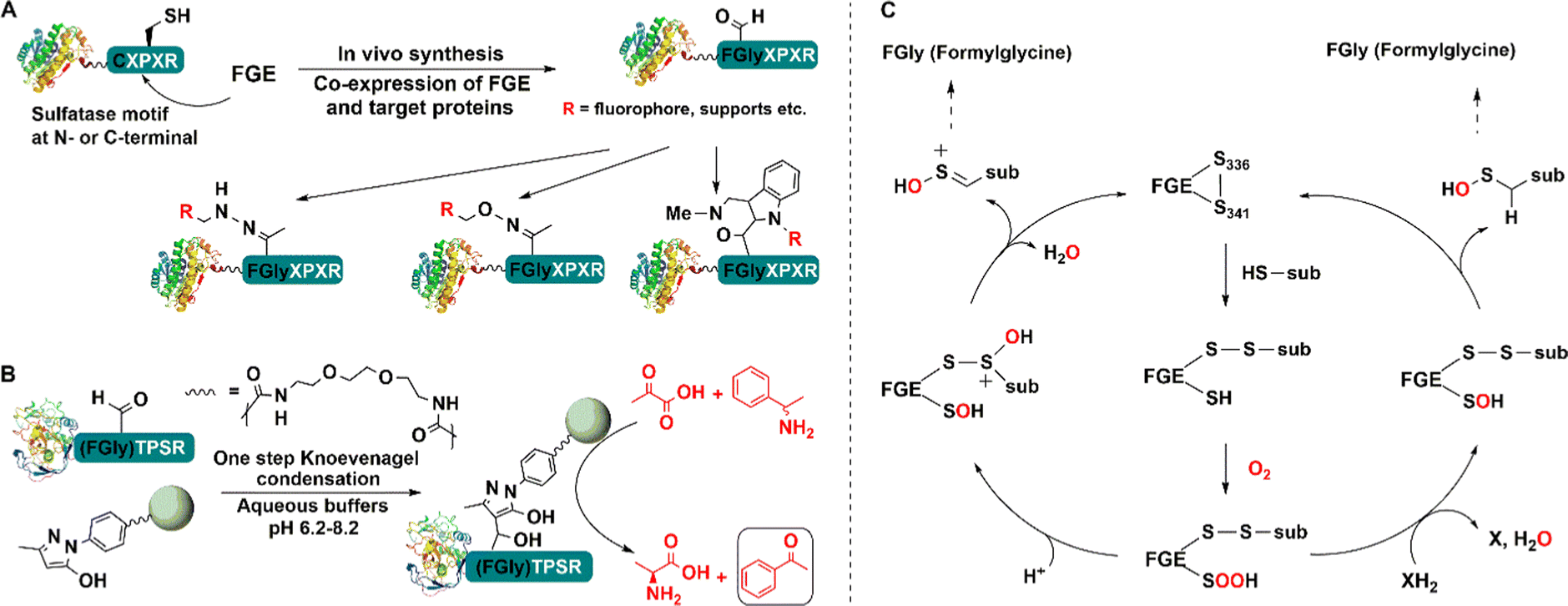 | ||
| Fig. 12 Bio-orthogonal immobilisation of aldehyde-tagged enzymes (A) on pyrazolone-functionalized support (B) and linking reaction mechanism (C). | ||
Alternatively, an agarose support modified with an aryl substituted pyrazolone can be used in conjunction with facile Knoevenagel condensation with the aldehyde tag (Fig. 12(B)).157 Immobilisation of the S-transaminase from Vibrio fluvalis (VfTA) using this method afforded an immobilisate with high activity and stability in the conversion of pyruvate and rac-1-phenylethylamine (PEA) to L-alanine and acetophenone Moreover, it was reused in multiple recycles without any leaching.157
5.2.3.1 Tyrosine ligase (TL). Tyrosine ligases (TL) such as tubulin tyrosine ligase (TTL) catalyse the connection reaction of a small tyrosine derivative, which is easy to synthesize, to any protein that carries a short peptide tag (the so-called Tub-tag, VDSVEGEGEEEGEE) at its C-terminus.158 This novel strategy for rapid chemo-selective C-terminal protein modifications on isolated proteins or cell lysates is used in biochemistry, cell biology and immobilisation (Fig. 13).158,159 The canonical amino acid tyrosine is not the only substrate of TTL.159,160o-NO2 -tyrosine,161o-F-tyrosine,162o-I-tyrosine,163o-CHO-tyrosine, o-NH2-tyrosine and o-azido-tyrosine are also substrates.164 This substrate versatility of TTL enables the facile and convenient covalent modification immobilisation, and cross-linking of enzymes.
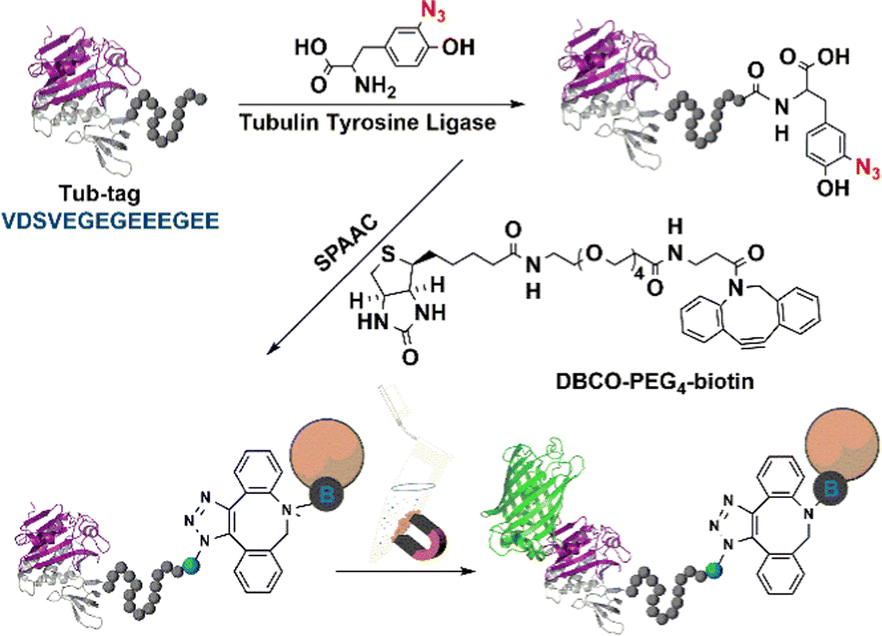 | ||
| Fig. 13 Tubulin tyrosine ligase catalytic orthogonal immobilisation of enzymes on functionalised support. Reprinted with permission from ref. 158. Copyright 2015, John Wiley and Sons. | ||
The TTL-mediated chemo-enzymatic ligation of unnatural tyrosine derivatives such as 3-azido-L-tyrosine and 3-formyl-L-tyrosine158 enabled bio-orthogonal functionalisation in a total yield of up to 99% using moderate enzyme concentrations and short reaction times. Thus, TTL-catalysed incorporation of 3-azido-L- and O-propargyl-L-tyrosine into the respective proteins enables in vitro generation of C-terminal fused proteins using Cu-catalysed click chemistry.164 However, covalent ligation occurs only at the C-termini of enzymes and is, therefore not suitable for multipoint covalent attachment to carriers.
5.2.3.2 Enzymatic oxidative ligation using tyrosinase. Introduction of an o-nitro tyrosine residue into a protein through amber stop codon suppression,165,166 followed by its reduction to an amino group enables the use of aniline coupling with Na2S2O4165,166 or NaIO4167 in phosphate buffer at pH 6.5 as a mild, selective method for protein bio-conjugation. However, chemical methods for oxidizing tyrosine and its derivatives are generally not green and sustainable167 and their scope is limited by competing oxidation of other moieties present in some proteins, e.g. cysteines and 1,2-diols found in glycans. This limitation was overcome by using the milder oxidant, potassium ferricyanide, in the coupling reaction (Fig. 14).167–171 The method can be used in antibody-drug conjugates and enzyme immobilisation.172
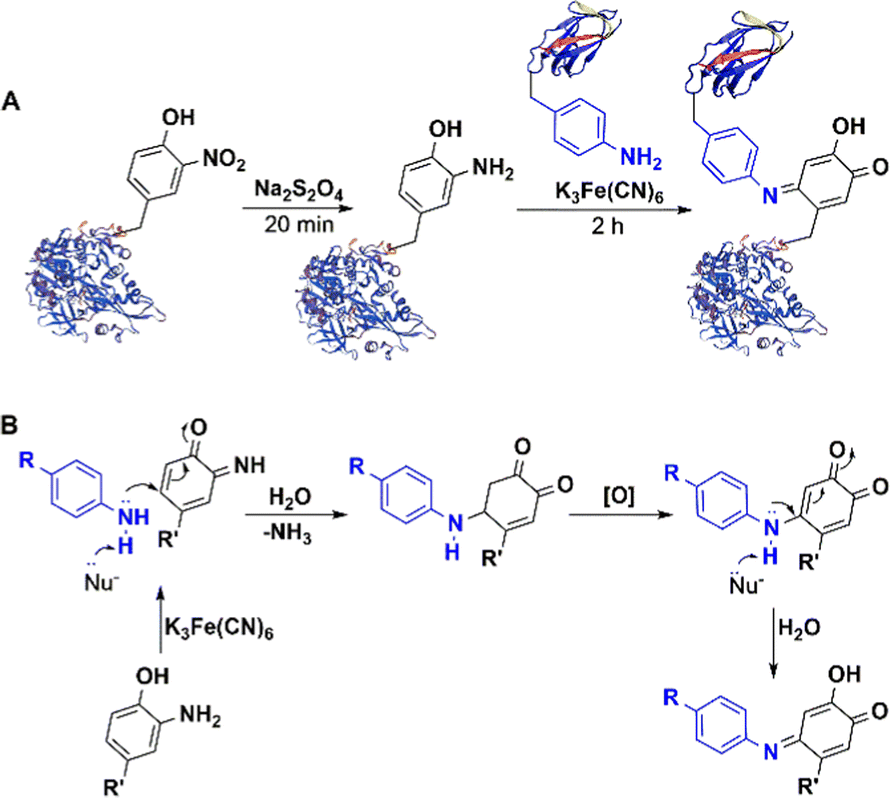 | ||
| Fig. 14 Bio-orthogonal conjugation of nitrotyrosine-containing protein and ferricyanide catalysed aniline coupling. | ||
On the other hand, some copper- and iron-dependent oxidoreductases, such as peroxidases, tyrosinase and laccase173 catalyse the mild, aerobic one-electron oxidation of tyrosine residues in proteins to achieve bio-orthogonality. For example, tyrosinase, a di-copper oxidase, catalyses the formation of o-quinone moieties (dopaquinone) via the intermediate formation of o-hydroxytyrosine.
This constitutes the first step in so-called melanogenesis, i.e. the biosynthesis of the natural pigments, melanins. Alternatively, intermolecular condensation of the o-quinone moiety with reactive functionalities, such as thiol and amino groups, in other residues, leads to cross-linking of the protein molecules. This is widely used in the processing of meat, whey and flour proteins to modify the texture of foods.174–176
Similarly, tyrosinase catalyses aerobic oxidation of exposed tyrosine residues into o-quinones that subsequently react rapidly with cysteine residues on target peptides and proteins to afford a convenient synthesis of bio-conjugates. It was used, for example, to achieve a 20-fold enhancement of the cellular delivery of CRISPR-Cas9 by attaching cationic peptides.177 Tyrosinase-mediated oxidative coupling of tyrosine tags on peptides and proteins has been applied (Fig. 15).170,178,179
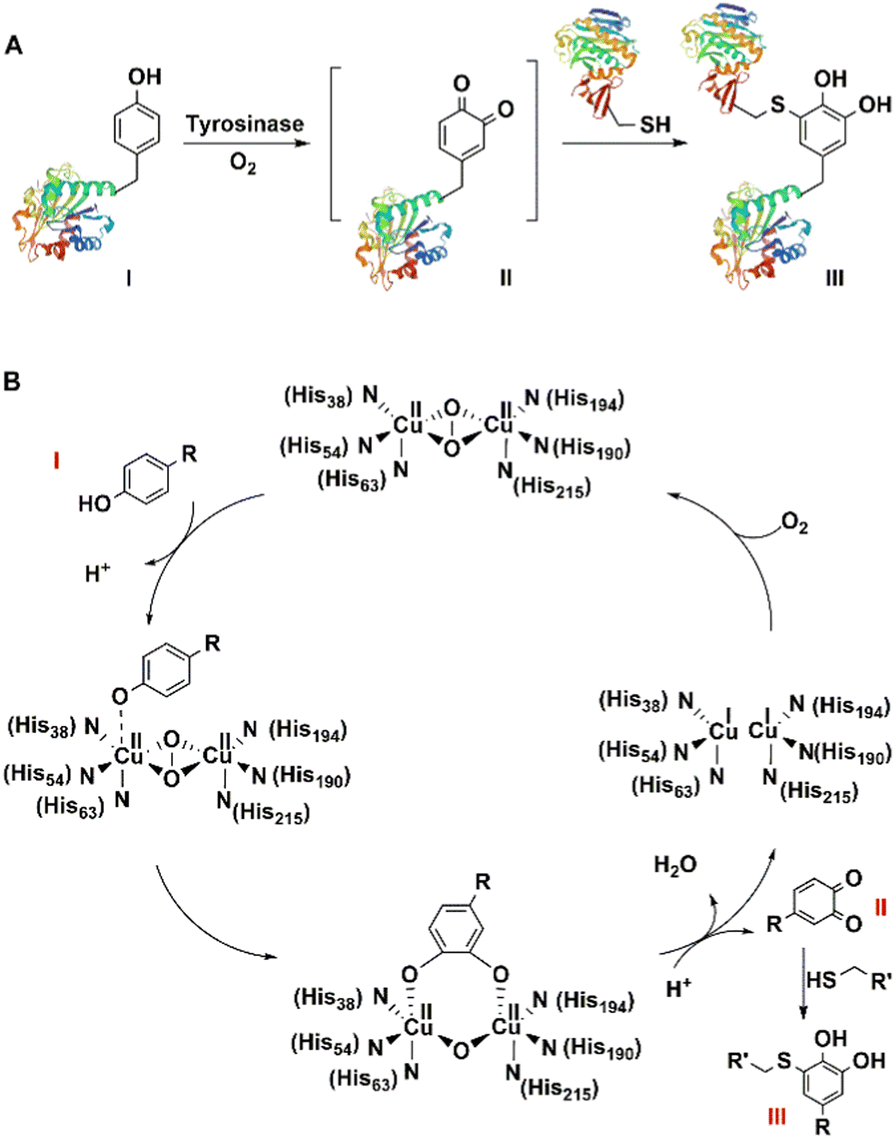 | ||
| Fig. 15 Site-Specific bioconjugation (A) and mechanism (B) through enzymatic tyrosine–cysteine bond formation. | ||
Tyrosinase from Agaricus bisporus (abTYR), catalysed the oxidation of tyrosine to o-quinone intermediates which coupled with N-terminal proline residues in high yield. This coupling strategy was successfully used for the attachment of a variety of phenols to a series of protein substrates, including self-assembled viral capsids, enzymes, and a chitin binding domain (CBD).180
Mushroom tyrosinase catalyses the formation of reactive o-quinones on unstructured, tyrosine-rich sequences such as hemaglutinin (HA) tags, YPYDVPDYA.181 The method is used to modify the C-termini of antibodies and proteins.170
5.2.3.3 Other oxidoreductases. Genetically attaching a Y-tag (GGGGY or GGYYY) peptide to the C-terminus of alkaline phosphatase (BAP)182 from Escherichia coli afforded a BAP construct (Fig. 16(A)). Subsequently, horseradish peroxidase (HRP) catalyses the hydrogen peroxide oxidation of exposed tyrosine residues on Y-tag to form phenoxy radicals, resulting in the site-specific formation of highly cross-linked conjugates via a radical coupling reaction. When used in chemical labeling, HRP-catalysed ligation also proceeded better than other oxidative coupling between N-methylluminol and tyrosine residues on protein,183 which resulted from its priority activation of N-methylluminol (Fig. 16(B)) in the presence of small amounts of H2O2.
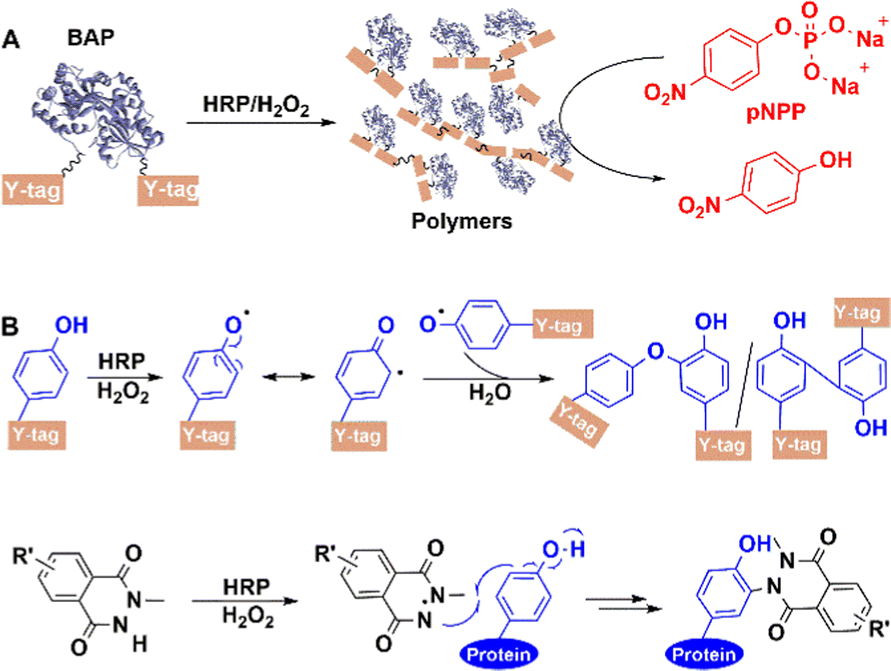 | ||
| Fig. 16 HRP-catalysed conjugation of enzyme proteins with Y-tag at the N-termini and C-termini. | ||
Similarly, laccases catalyse the aerobic one-electron oxidation of tyrosine, tryptophan, and cysteine residues to form radicals which subsequently form mixtures of oligomers and polymers.184 This can be used to perform site-specific cross-linking of enzyme proteins with peptide tags.185 For example, both BAP and antibody-binding proteins (pG2PAs)186 contain a tyrosine residue (Y-tag) at the N- or C-termini (Fig. 17), which is oxidised by laccase to afford protein polymers. Co-polymers of BAP and pG2PA were also prepared by mixing aqueous solutions of small Y-tagged substrates.
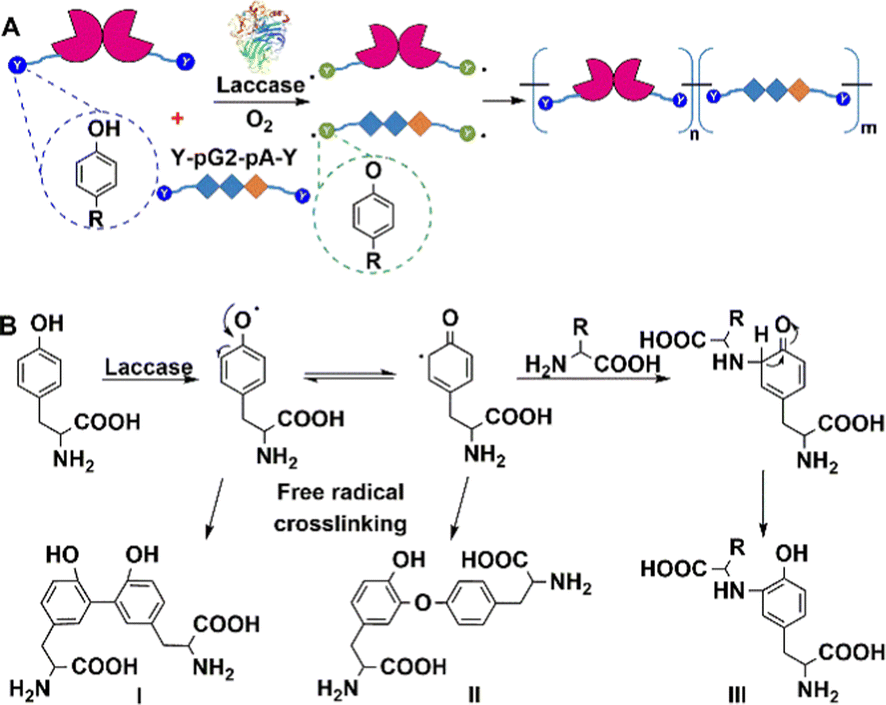 | ||
| Fig. 17 Laccase-catalysed site-specific enzyme protein cross-linking reaction. (A) Reprinted with permission from ref. 186. Copyright 2018, Elsevier. | ||
6. Bio-orthogonal immobilisation mediated by non-standard amino acids (NSAAs)
Traditional covalent immobilisation methods often lead to loss of activity and accompanying reduced catalyst and volumetric productivities, through the random formation of covalent bonds with the enzyme.51 It is especially important, therefore, to achieve more precision in covalent bond formation to fully retain or even enhance activity. Genetic encoding of NSAAs is one way to achieve this. Random and unwanted covalent linkages are circumvented by incorporating NSAAs with reactive groups into the enzyme to enable mild and precise bio-orthogonal coupling of the enzyme to a carrier or to other molecules of the enzyme in cell lysate.Genetic code expansion is used to insert non-standard amino acids into a single site52,187 or multiple sites54,188 of proteins. This enables the site-specific or multi-point covalent attachment of enzymes using bio-orthogonal chemistry. A variety of covalent ligation reactions, such as copper-catalysed azide–alkyne cycloadditions (CuAAC),189–191 strain-promoted azide–alkyne cycloadditions (SPAAC),192 and inverse-electron demand Diels–Alder reactions (IEDDA), are used depending on the nature of the inserted NSAA. The natural genetic code only allows for 20 standard amino acids in protein translation but recently developed biosynthetic approaches enable the insertion of non-standard amino acids (NSAAs). Thus far, nearly 200 distinct NSAAs were incorporated into proteins to endow them with novel physical, chemical, and biological properties.193
6.1 Copper-catalysed azide–alkyne cycloadditions (CuAAC)
The copper-catalysed azide-alkyne coupling (CuAAC), the classic example of Click chemistry, was independently reported by Sharpless and Meldal in 2001.194 It has been widely used in biology, materials science and biomedicine.190,195 It is a versatile reaction based on its exquisite selectivity and diversity of building blocks that can be synthesized and the fact that the triazole products are extremely stable under physiological conditions. The method is suitable for protein labelling,191,196 biomolecular ligation197–199 and enzyme immobilisation.200–203The utilisation of NSAAs has proven to be an effective method for site-specific immobilisation of proteins.203 Every protein molecule is covalently attached to a support at the same site. For example, the non-standard p-azido-L-phenylalanine (p-AzF) was inserted into a GFP reporter system for site-specific covalent immobilisation on solid-carriers.202 Recently, we incorporated another p-propargyloxy-L-phenylalanine (p-PaF) into a KRED, and then induced the formation of cross-linked enzymes (CLEs) by adding bis-PEG3-azide and CuI to cell lysate in a microwave reactor.190 The robust CLEs were combined with TiO2 and Cp*Rh(bpy) to develop a biohybrid photocatalytic hydrogenation system, which was used in the enantio-selective synthesis of R-1-[3,5-bis(trifluoromethyl)-phenyl]ethanol, an important intermediate in the synthesis of the anticancer drug Crezotinib (Fig. 18).
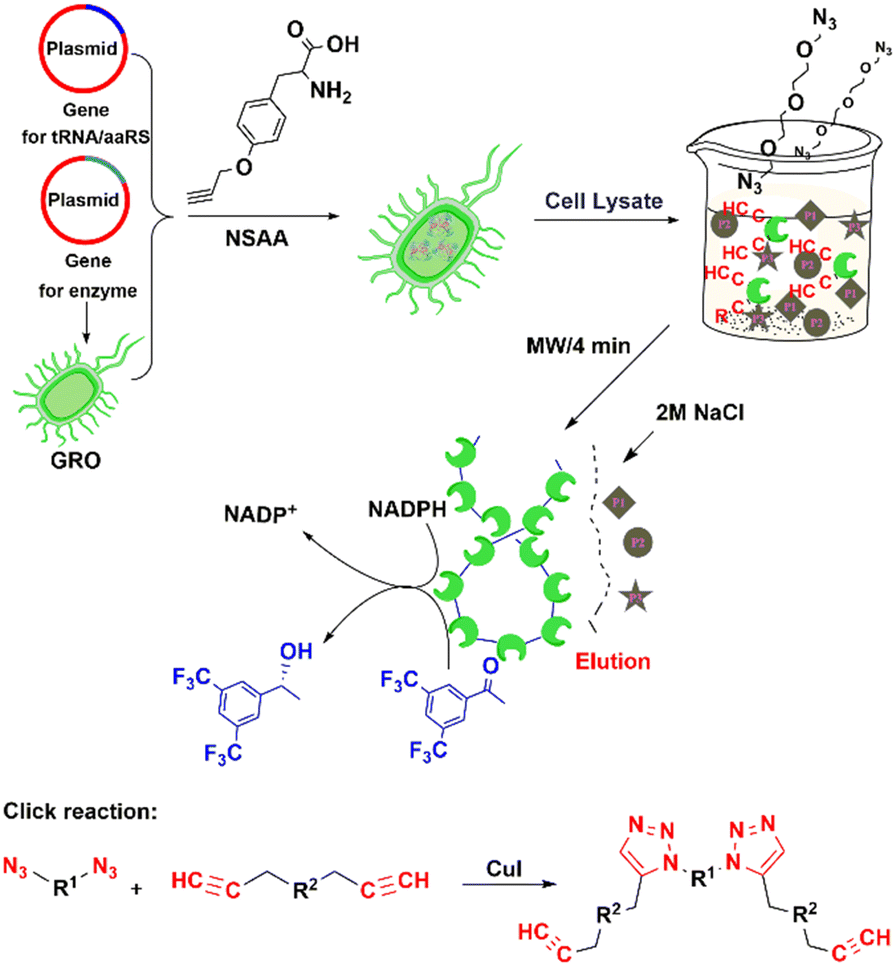 | ||
| Fig. 18 Process for preparing alcohol dehydrogenase cross-linked enzymes (CLEs) from cell lysate. Reprinted with permission from ref. 190. Copyright 2021, American Chemical Society. | ||
The copper catalyst is usually prepared with an appropriate chelating ligand to maintain the Cu(I) oxidation state.204 The cycloaddition reaction between azide and alkyne is usually performed in aqueous buffer to produce a stable and inert 1,4-triazole.205
6.2 Strain-promoted azide–alkyne cycloadditions (SPAAC)
Cyclooctyne is the smallest stable cyclic alkyne and is distorted by 17° from the preferred linear alkyne geometry. This results in sufficient ring strain to enable metal-free [3+2] cycloaddition of azides. Hence, the reaction's name: strain-promoted azide–alkyne cycloaddition (SPAAC).206 Biological applications of this reaction have generally been limited by its low reaction rate but the latter can be accelerated using microwave irradiation of low power at low temperatures.207Amber codon suppression was used to replace five tyrosine residues (Tyr50, 137, 243, 274, and 355) with azido phenylalanine in a recombinant lipase. The mutants were coupled to a solid support using the SPAAC reaction and the obtained AzPhe-Lip243 and AzPhe-Lip274 showed higher catalytic activity compared with the natural enzyme. This clearly demonstrates the importance of NSAA insertion for directing covalent immobilisation.202 The method has broad scope in one-step enzyme purification and immobilisation of enzymes. It was used, for example, in the multiple site insertion of p-azido-L-Phe (p-AzF) in an aldehyde ketone reductase (AKR) followed by immobilisation as cross-linked enzymes (CLEs) directly from cell lysate supernatant under microwave irradiation.192 The specific activities of the AKR-CLEs were 1–2 times that of the free enzyme (Fig. 19). The method has the potential to generate a range of bio-orthogonally cross-linked enzymes, for use in industrial bocatalysis, directly from cell lysate.
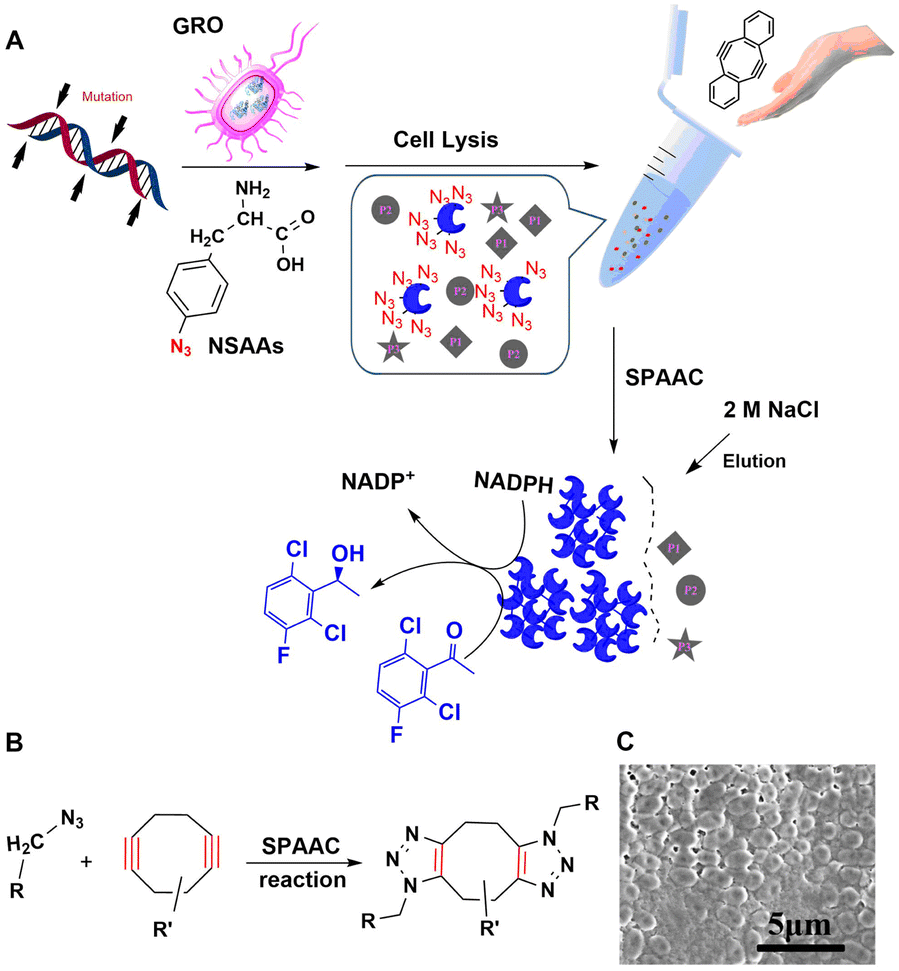 | ||
| Fig. 19 Preparation, reaction mechanism and morphology of rapidly and precisely cross-linked enzyme (RP-CLEs) of aldehyde ketone reductase. Reprinted with permission from ref. 192. Copyright 2020, American Chemical Society. | ||
López-Gallego and coworkers208 have developed a one-pot cell-free protein synthesis (CFPS) and immobilisation to enable on-demand fabrication of protein-based biomaterials. It involved the incorporation of azide-substituted amino acids into nascent proteins followed by selective immobilisation on a variety of solid supports using SPAAC. Here, if the insertion sites of NSAAs are rationally designed in space (Fig. 19(C)), the protein rings can be obtained209 and further used as protein-based biomaterials for drug delivery or directly as clinical protein.
6.3 Inverse-electron demand Diels–Alder reactions (IED-DA)
The inverse-electron-demand Diels–Alder (IEDDA) reaction involves the cycloaddition of an electron-rich dienophile with an electron-poor diene.210 For example, cycloaddition of tetrazines with strained trans-cyclooctenes has attracted increasing attention based on its high rate, biocompatibility and scope as a site-directed bio-orthogonal technique.211–213 The reaction tolerates a range of buffered conditions appropriate for protein handling, requires no catalyst, and boasts tunable rate constants of up to 106 M−1 s−1, orders of magnitude faster than 3+2 cycloadditions or oxime formation.213–215As shown in Fig. 20(A) a tetrazine containing NSAA (Tet2.0) was genetically inserted in human carbonic anhydrase II (tsCA) to enable its IEDDA immobilisation onto trans-cyclooctene (TCO) modified surfaces.216 By genetically encoding the Tet2.0 the precise site of incorporation, and thereby the orientation of the protein after immobilisation, can be easily controlled, as shown in Fig. 20(B). This affords a rapid, gentle and general method for bio-orthogonal immobilisation using IEDDA (Fig. 20(C)).
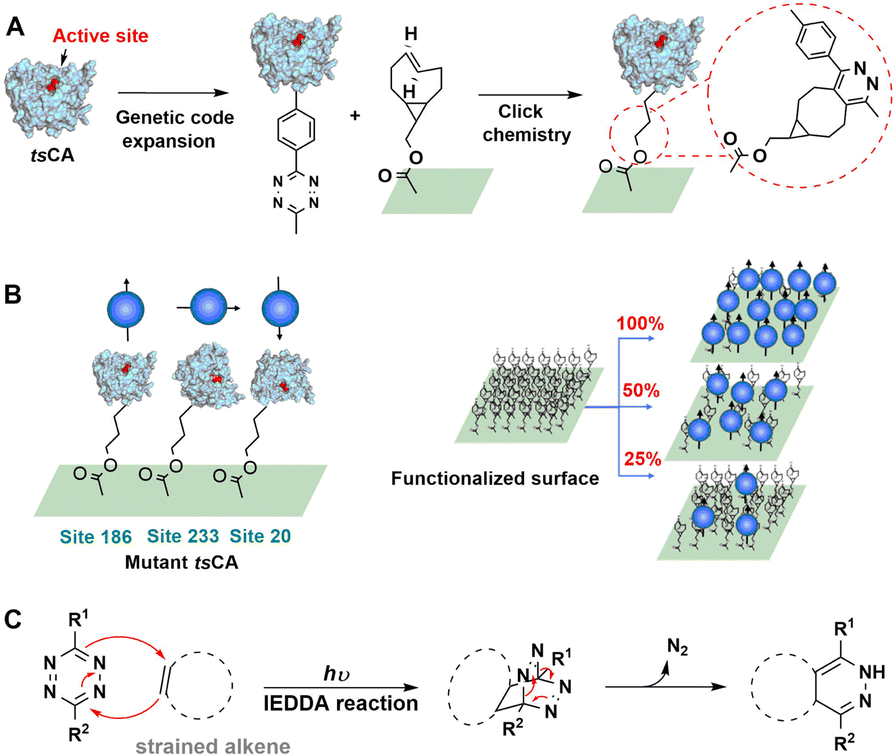 | ||
| Fig. 20 Bio-orthogonal immobilisation of NSAA-inserted human carbonic anhydrase II (tsCA) on TCO-functionalised surfaces. Reprinted with permission from ref. 216. Copyright 2019, American Chemical Society. | ||
7. Challenges for the future
Rapid and precise covalent immobilisation of enzymes in combination with the use of continuous processing will continue to play an important role in the design of the next generation of biotechnological processes for application in a bio-based circular economy.217–2197.1 Design of an improved biological tag for covalent enzyme ligation
Genetically encoded peptide–protein coupling systems, such as the SpyTag/SpyCatcher and related two-component systems (see Section 5.1), have expanded the protein bioconjugation toolbox. More recently, three-component systems with added advantages such as SpyTag/KTag/SpyLigase,220 SnoopTagJr/DogTag/Snoop Ligase117 and SpyTag/BTag/Spy Stapler120,221 have emerged. Three-component ligation systems, such as SpyTag/KTag/SpyLigase, are expected to address the problem of formation of inclusion bodies when the 116-residue SpyCatcher is fused to target enzymes.66 For example, in the SpyTag/KTag/SpyLigase system the original Catcher from CnaB2 is re-split into two parts, K tag and one small protein domain termed SpyLigase which catalyses the bio-orthogonal ligation of the two tags.220Such systems possess most of the advantages of the two-component systems but require only two short peptide tags for side-chain ligation through genetic fusion. This enables the construction of large enzyme protein conjugates for covalent immobilisation directly with minimum loss of function, from expressed recombinant proteins and involving an entirely new mode of bioconjugation, that is via spontaneous intermolecular isopeptide bond222 and possibly mechanical bonding.221
Additional benefits to the bioconjugates, such as improved activity and stability, can also be obtained by solving the problem of the limitation of binding sites to the C- and N- termini of the target enzyme. This would result in significant improvements in covalent immobilisation via isopeptide formation which forms the basis for the generation of insoluble circular enzymes as elegant examples of carrier-free enzyme immobilisation that are more in line with a circular economy.
7.2 Rapid and controllable chemical linkage reaction for precise enzyme immobilisation
Traditional covalent immobilisation methods, such as using epoxide cross-linkers is time-consuming, often taking several days because of the slow linking reaction.223,224 Moreover, enzyme tertiary structures are often destroyed and active sites are buried. Proteolysis can also occur, resulting in substantial activity losses and decreases in volumetric productivities. All in all this results in a decrease in cost-effectiveness of the procedure.51 Thus, it is especially important from the cost-effectiveness viewpoint to achieve precise covalent cross-linking to expose fully the active sites and retain high enzyme activities, perhaps even higher than that observed in vivo.225,226NSAA insertion is an efficient tool for enabling covalent bond formation between the enzyme and the carrier. Thus, depending on the enzyme structure analysis, various NSAAs can be incorporated into the protein to accurately guide and control the covalent linkage to a carrier or to other enzyme molecules to form cross-linked enzyme (CLEs). This could afford a green method to generate metal-free bio-orthogonally cross-linked enzymes192 and allow their facile separation from cell lysates for industrial biocatalysis. Moreover, rapid covalent protein ligation, in several minutes168 or even seconds227via photo-controlled click chemistry, is ideal for protecting the enzyme from proteolysis. More than 170 NSAAs193 have been inserted in proteins in living organisms, indicating that more and more bio-orthogonal reaction pairs can be used to cross-link enzyme proteins. This will eventually enable the construction of sequentially cross-linked multi-enzymes for in vitro enzymatic cascade reactions and metabolic pathways in cells thereby advancing the development of in vitro synthetic biology.228,229
7.3 The effect of the carrier in enzyme immobilisation
This review is concerned with the use of bio-orthogonal chemistry to affect the modification of recombinant enzymes such that enzyme production and purification are combined in vitro. The immobilised enzyme can then be produced directly from cell lysate. The basic premise of this approach is that it will afford a cost-effective streamlining of enzyme production, purification and immobilisation. However, this assumes that the immobilisation from cell lysate, either on a carrier or carrier-free, can be accomplished simply and cost-effectively.The last step in this sequence is the immobilisation, which in many of the articles on the use of bio-orthogonal chemistry seems to be an underexposed part of the overall procedure. However, if this part of the procedure is not cost-effective then the procedure as a whole is not cost-effective and will not be used. It is self-evident, therefore, that methods for performing this last step in the sequence cost-effectively is a conditio sine qua non for the success of the overall procedure. As we noted in Section 2 developments in carriers for enzyme immobilisation tend to follow developments in chemistry as a whole, i.e. there is a transition from synthetic, hydrocarbon-based polymers towards bio-based polymers, preferably derived from waste biopolymers in a circular bio-based economy. In short, a major challenge for future developments in this field is to develop carriers that are of benign origin and are readily recycled in order to produce an absolute minimum of waste. This is in stark contrast with hydrocarbon-derived polymers which tend to end their life as difficult to recycle waste. Moreover, conversion of hydrocarbon polymers to functional enzyme carriers generally requires some surface modification, such as the introduction of epoxide groups that react with the enzyme. However, such ‘epoxy carriers’ are expensive, readily deactivated and end their life as non-recyclable waste. Hence, such carriers are gradually being phased out and already have limited commercial availability.
Hence, an important challenge for the future is the development of carriers that are produced from waste biopolymers, are readily available and recyclable and are more consistent with the general trend towards more sustainable biocatalytic processes.217,230,231 Carriers for enzyme immobilisation must be recyclable by design and the development of a bio-orthogonal methodology for enzyme immobilisation must involve the design of a readily available, inexpensive and eminently recyclable carrier as an integral part of the project, from the beginning, not as an afterthought.
The cost, commercial availability and recyclability of the carrier plays an important role in determining the commercial viability of the method as a whole.232–234 Turner and co-workers11 provided guidelines for the immobilisation of enzymes for use in the pharmaceutical industry. The immobilised enzyme should have a wt loading of >10%, an activity recovery of >50%, have >20 recycles, a cost contribution of <5%, exhibit no leaching, tolerance to organic solvents, be mechanically stable in batch and in flow and good substrate mass transport.
Recently, much attention has been devoted to the use of magnetically recoverable immobilised biocatalysts, e.g. based on magnetic cellulose30 as a technique that is used on a very large scale. Similarly, magnetic CLEAs (mCLEAs) are easily recycled using commercially available equipment.14
In the many examples discussed in Sections 4 and 5 a wide variety of carriers have been used. At the end-of-use of the enzyme, it is important that the carrier can be recycled and re-used. In the cases of enzymes that are covalently attached, drastic conditions may be necessary to separate the carrier that may be detrimental for a polymeric carrier.235 However, if enzymes are covalently attached to the carrier by forming disulfide (S–S) bonds through non-essential thiol (SH) groups in the enzyme and reactive thiol groups on the carrier. the bound protein can be subsequently released by treatment with an excess of dithiothreitol.81,236 Alternatively, concentrated aqueous HCl can be used to remove the enzymes from the carrier.235 Nevertheless, after several recycles the carrier will usually be abandoned and end its life as solid waste.
Interestingly, some polymers are particularly amenable to recycling.237–240 With a judicious choice of monomer and polymer structures it is possible to create recyclable polymers. For example, a synthetic polymer derived from γ-butyrolactone can be easily and selectively depolymerised and the monomer used to produce pristine polymer.236,239 Similarly, polycarbonates derived from epoxides and carbon dioxide can be readily recycled back to the monomer and re-polymerised under mild conditions237 and recovered polydiketoenamine monomers can be re-manufactured into the same polymer formulation, without any loss of performance.240
7.4 Machine learning-guided rationally spatial multi-enzyme immobilisation
Machine learning has been considered as a potential strategy in the pursuit of rational immobilisation of enzymes. The CapiPy program for predicting the immobilisation properties of enzymes is based on a python algorithm.241 The algorithm is composed of four modules, including the sequence blast and structure modelling, active site identification, surface and residue clustering, and immobilisation literature retrieval. CapiPy can conveniently help researchers to make a selection on the immobilisation approach of target protein, and has been tested using three separate random sets of 150 protein sequences with more than 70% overall success rate. In addition, enzyme-linked immunosorbent assay (ELISA) has been used to identify antibodies, antigens, proteins and glycoproteins. Polystyrene binding peptides (PSBPs) can be used to immobilise bioactive peptides, enzymes and antigens in water at room temperature. PSBP-SWM, a machine learning-based computational identifier, was developed to predict PSBPs for correct recognition in ELISA. The machine-based identification model contains four steps, including feature extraction, feature selection, model training and optimisation.242In the last decade, metal–organic frameworks (MOFs) have proven to be a good platform for the immobilisation of enzymes. To save experimental time and cost, several machine learning algorithms (random forest and Gaussian process regression) were used to predict enzyme-MOF properties, including enzyme loading, activity retention, immobilisation yield and reusability.243 Recently, Raman hyperspectral imaging with principal component analysis has been developed to discern the spatial and chemical distribution of immobilised enzymes.244 Furthermore, chemical imaging has been combined with machine learning to elucidate the immobilisation of enzymes for biocatalysis. The machine learning was based on multivariate curve resolution-alternating least squares (MCR-ALS), which was used to explain pure response profiles of the chemical constituents within a complex.245 Machine learning can be used to understand and improve enzyme immobilisation for biocatalytic processes, and is likely to become an active research area in the near future.
In striking contrast with the elegant orchestration of multiple biocatalytic steps in metabolic pathways in living cells, classical multi-step organic syntheses in vitro are performed in a step-by-step fashion involving isolation and often purification of intermediates and/or a solvent switch after each individual step. This affords low volumetric productivities combined with copious waste formation. The number and amounts of solvent used and the amounts of waste generated are substantially reduced by telescoping such multi-step syntheses into one-pot procedures that have higher productivities, involve fewer unit operations, smaller reactor volumes and shorter cycle times.246
One problem of creating chemocatalytic cascade processes is that the individual steps often involve vastly different conditions of temperature, pressure and solvent. In contrast, the vast majority of enzymes perform admirably in water as the solvent at ambient temperature and pressure at a defined pH. Furthermore, because of the exquisite specificity of enzymes functional group activation and protection and deprotection steps, necessary in many chemocatalytic reactions, become redundant in biocatalytic equivalents. Moreover, coupling biocatalytic steps offers the possibility of driving equilibria towards product formation by product removal.
As is also the case with individual biocatalytic reactions, immobilisation of the individual enzymes involved in a biocatalytic cascade process offers obvious processing advantages. A further advantage is that it is possible to co-immobilise enzymes that are incompatible in homogeneous solution. An excellent example of this is the co-immobilisation of a protease and a lipase in a combi-CLEA.247
In order to facilitate recycling, multiple enzymes can be co-immobilised on a carrier, e.g. for the manufacture of islatravir,248 or by cross-linking. Co-immobilisation offers the additional benefit of enzyme proximity that enables substrate channeling in which the intermediate produced by one enzyme is transferred to the next enzyme without intermediary mixing in the homogeneous phase, leading to reductions in diffusion times, higher overall activities and less accumulation of inhibitory intermediates.249,250 This phenomenon occurs widely in nature, for example in multi-enzyme cellulosomes consisting mainly of carbohydrases that catalyse the hydrolysis of cellulose and hemicellulose.251 An interesting example of such a co-immobilisation is the His-tagged ketoreductase (KRED) and His-tagged GDH attached to functionalised magnetic beads for enantioselective reduction of prochiral ketones.252
Compartmentalisation is fundamental to the functioning and protection from cellular degradation mechanisms of multiple enzymes in metabolic pathways. Much research is devoted, therefore, to designing compartmentalised cascade processes with isolated enzymes in vitro that emulate this exquisite orchestration of multi-enzyme metabolic pathways in vivo.253 The use of enzyme data bases such as EnzymeML and SABIO-RK254 in combination with machine-assisted learning255 and the use of microfluidics and robotics will play an important role in designing the next generation of biotechnological processes involving enzyme cascades in compartments.256 We expect that the design of efficient bio-orthogonal methods for precision co-immobilisation of multiple enzymes will play an indispensible role in these developments.
8. Conclusions and prospects
Enzyme immobilisation continues to be a subject of immense interest, in both industry and academia. The commercial viability of industrial biotransformations stands or falls with the cost contribution of the enzyme. Immobilisation is an enabling technology that, in addition to providing an active and stable biocatalyst, should be a relatively simple operation not requiring a highly pure enzyme preparation or an expensive support that may not be commercially available. This review is concerned with the progress of covalent immobilisation of enzymes directly from cell lysates with emphasis on the use of elegant bio-orthogonal chemistry to achieve precision in immobilisation. Bio-orthogonal chemistry has three main advantages: (i) no disruption in the structure and functioning of the enzyme, (ii) the key chemistry is integrated in the production of the recombinant enzyme and immobilisation occurs directly from cell lysate to afford a cost-efficient, green and sustainable methodology and (iii) the method is applicable to the immobilisation of enzymes either on carriers or as cross-linked enzyme aggregates.Bio-orthogonal immobilisation through, for example, genetic encoding of NSAAs and subsequent covalent bond formation allows for control of the precise position, number and manner of the covalent bond formation. Random covalent bond formation, leading to loss of activity and function that is often observed with traditional covalent immobilisation, is avoided. We expect that further integration of protein engineering with bio-orthogonal immobilisation directly from cell lysate and supported by machine learning will put precision and elegance into enzyme immobilisation to enable cost effective biocatalysis in a sustainable circular bioeconomy.
Which method shows the most promise? It is difficult to say but based on the enormous advances made in the incorporation of NSAAs in protein sequences it seems likely that significant advances will be made in the use of NSAAs for guiding the efficient immobilisation of enzymes directly from cell lysate. One way to avoid the complications associated with finding a suitable carrier is to avoid it altogether by using carrier-free immobilised enzymes. In this context the formation of circular proteins via isopeptide bonds is of particular interest.
In any case, we predict that the combination of innovative bio-orthogonal chemistry with enzyme immobilisation techniques will stimulate the further development and reduction to commercial practice of continuous-flow biocatalysis.8,257 This could be used, for example, to drive in vitro multi-enzyme cascade processes, in emulation of the exquisite orchestration of metabolic pathways in vivo, and provide truly sustainable biocatalytic methods for the industrial production of chemicals.257–261
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (22078079), the “Ten-thousand Talents Plan” of Zhejiang Province (2019R51012), the Natural Science Foundation of Zhejiang Province (LY18B060009), the “Star and light” Project for Talent Students in Hangzhou Normal University (2019) and Research Plan for Sprout Talents in University in Zhejiang Province (2020R427071).Notes and references
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed
.
- C. Raffaele and C. Daniele, Recycling, 2018, 3, 22 CrossRef
- P. Intasian, K. Prakinee, A. Phintha, D. Trisrivirat, N. Weeranoppanant, T. Wongnate and P. Chaiyen, Chem. Rev., 2021, 121, 10367–10451 CrossRef CAS PubMed
- D. Firoiu, G. H. Ionescu, A. Bndoi, N. M. Florea and E. Jianu, Sustainability, 2019, 11, 2156 CrossRef
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef
- H. H. Sun, H. F. Zhang, E. L. Ang and H. M. Zhao, Bioorg. Med. Chem., 2018, 26, 1275–1284 CrossRef CAS PubMed
- R. A. Sheldon, A. Basso and D. Brady, Chem. Soc. Rev., 2021, 50, 5850–5862 RSC
- P. De Santis, L.-E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184 RSC
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, Á. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC
- K. E. Cassimjee, M. Kadow, Y. Wikmark, M. S. Humble, M. L. Rothstein, D. M. Rothstein and J. E. Backvall, Chem. Commun., 2014, 50, 9134–9137 RSC
- M. P. Thompson, S. R. Derrington, R. S. Heath, J. L. Porter, J. Mangas-Sanchez, P. N. Devine, M. D. Truppo and N. J. Turner, Tetrahedron, 2019, 75, 327–334 CrossRef CAS
- B. Chen, J. Hu, E. M. Miller, W. Xie, M. Cai and R. A. Gross, Biomacromolecules, 2008, 9, 463–471 CrossRef CAS PubMed
- L. Q. Cao, F. van Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361–1364 CrossRef CAS PubMed
- R. A. Sheldon, Catalysts, 2019, 9, 261–291 CrossRef CAS
- H. Yamaguchi, Y. Kiyota and M. Miyazaki, Catalysts, 2018, 8, 174–190 CrossRef
- M. B. Mbanjwa, K. J. Land, T. Windvoel, P. M. Papala, L. Fourie, J. G. Korvink, D. Visser and D. Brady, Process Biochem., 2018, 69, 75–81 CrossRef CAS
- R. A. Sheldon and D. Brady, ACS Sustain. Chem. Eng., 2021, 9, 8032–8052 CrossRef CAS
- C. Bernal, K. Rodríguez and R. Martínez, Biotechnol. Adv., 2018, 36, 1470–1480 CrossRef CAS PubMed
- M. Bilal and H. M. N. Iqbal, Catal. Lett., 2019, 149, 2204–2217 CrossRef CAS
- G. Ölçücü, O. Klaus, K.-E. Jaeger, T. Drepper and U. Krauss, ACS Sustain. Chem. Eng., 2021, 9, 8919–8945 CrossRef
- F. B. H. Rehm, S. Chen and B. H. A. Rehm, Bioengineered, 2018, 9, 6–11 CrossRef PubMed
- F. B. H. Rehm, S. Chen and B. H. A. Rehm, Molecules, 2016, 21, 1370–1388 CrossRef PubMed
- V. D. Jaeger, R. Lamm, K. Kuesters, G. Oelcuecue, M. Oldiges, K.-E. Jaeger, J. Buechs and U. Krauss, Appl. Microbiol. Biotechnol., 2020, 104, 7313–7329 CrossRef CAS PubMed
- J. M. Sanchez, H. Lopez-Laguna, N. Serna, U. Unzueta, P. D. Clop, A. Villaverde and E. Vazquez, ACS Sustain. Chem. Eng., 2021, 9, 2552–2558 CrossRef CAS
- R. Koszagova, T. Krajcovic, K. Palencarova-Talafova, V. Patoprsty, A. Vikartovska, K. Pospiskova, I. Safarik and J. Nahalka, Microb. Cell. Fact., 2018, 17, 139 CrossRef PubMed
- Y. Han, X. Zhang and L. Zheng, Mol. Catal., 2021, 504, 111467 CrossRef CAS
- M. Bilal and H. M. N. Iqbal, Int. J. Biol. Macromol., 2019, 130, 462–482 CrossRef CAS PubMed
- S. Aggarwal, A. Chakravarty and S. Ikram, Int. J. Biol. Macromol., 2021, 167, 962–986 CrossRef CAS PubMed
- Y. A. Rodriguez-Restrepo and C. E. Orrego, Environ. Chem. Lett., 2020, 18, 787–806 CrossRef CAS
- A. Gennari, A. J. Fuhr, G. Volpato and C. F. V. de Souza, Carbohydr. Polym., 2020, 246, 116646 CrossRef CAS PubMed
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS
- D. Valikhani, J. M. Bolivar and B. Nidetzky, Methods Mol. Biol., 2020, 2100, 243–257 CrossRef CAS PubMed
- D. Valikhani, J. M. Bolivar, M. Viefhues, D. N. McIlroy, E. X. Vrouwe and B. Nidetzky, ACS Appl. Mater. Interfaces, 2017, 9, 34641–34649 CrossRef CAS PubMed
- A. D. Roberts, K. A. P. Payne, S. Cosgrove, V. Tilakaratna, I. Penafiel, W. Finnigan, N. J. Turner and N. S. Scrutton, Green Chem., 2021, 23, 4716–4732 RSC
- J. Coloma, T. Lugtenburg, M. Afendi, M. Lazzarotto, P. Bracco, P.-L. Hagedoorn, L. Gardossi and U. Hanefeld, Catalysts, 2020, 10, 899–913 CrossRef CAS
- W. Bohmer, A. Volkov, K. Engelmark Cassimjee and F. G. Mutti, Adv. Synth. Catal., 2020, 362, 1858–1867 CrossRef PubMed
- K. E. Cassimjee, P. Hendil-Forssell, A. Volkov, A. Krog, J. Malmo, T. E. V. Aune, W. Knecht, I. R. Miskelly, T. S. Moody and M. S. Humble, ACS Omega, 2017, 2, 8674–8677 CrossRef CAS PubMed
- M. Gand, C. Thoele, H. Mueller, H. Brundiek, G. Bashiri and M. Hoehne, J. Biotechnol., 2016, 230, 11–18 CrossRef CAS PubMed
- W. Bhmer, L. Koenekoop, T. Simon and F. G. Mutti, Molecules, 2020, 25, 2140–2156 CrossRef PubMed
- C. A. Godoy, B. de las Rivas and J. M. Guisan, Process Biochem., 2014, 49, 1324–1331 CrossRef CAS
- R. C. Rodrigues, Á. Berenguer-Murcia, D. Carballares, R. Morellon-Sterling and R. Fernandez-Lafuente, Biotechnol. Adv., 2021, 52, 107821 CrossRef CAS PubMed
- C. Ladavière, T. Delair, A. Domard, A. Novelli-Rousseau, B. Mandrand and F. Mallet, Bioconjugate Chem., 1998, 9, 655–661 CrossRef PubMed
- C. Ladavière, C. Lorenzo, A. Elaïssari, B. Mandrand and T. Delair, Bioconjugate Chem., 2000, 11, 146–152 CrossRef PubMed
- I. Serra, D. A. Cecchini, D. Ubiali, E. M. Manazza, A. M. Albertini and M. Terreni, Eur. J. Org. Chem., 2009, 1384–1389 CrossRef CAS
- S. Tural, B. Tural and A. S. Demir, Chirality, 2015, 27, 635–642 CrossRef CAS PubMed
- B. C. C. Pessela, C. Mateo, A. V. Carrascosa, A. Vian, J. L. Garcia, G. Rivas, C. Alfonso, J. M. Guisan and R. Fernandez-Lafuente, Biomacromolecules, 2003, 4, 107–113 CrossRef CAS PubMed
- C. Mateo, G. Fernandez-Lorente, E. Cortes, J. L. Garcia, R. Fernandez-Lafuente and J. M. Guisan, Biotechnol. Bioeng., 2001, 76, 269–276 CrossRef CAS PubMed
- A. A. Ruediger, W. Bremser and O. I. Strube, J. Coat. Technol. Res., 2016, 13, 597–611 CrossRef CAS
- V. Grazú, F. López-Gallego, T. Montes, O. Abian, R. González, J. A. Hermoso, J. L. García, C. Mateo and J. M. Guisán, Process Biochem., 2010, 45, 390–398 CrossRef
- V. Grazu, F. López-Gallego and J. M. Guisán, Process Biochem., 2012, 47, 2538–2541 CrossRef CAS
- L. Cao, L. v Langen and R. A. Sheldon, Curr. Opin. Biotechnol., 2003, 14, 387–394 CrossRef CAS PubMed
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS PubMed
- F. J. Isaacs, P. A. Carr, H. H. Wang, M. J. Lajoie, B. Sterling, L. Kraal, A. C. Tolonen, T. A. Gianoulis, D. B. Goodman, N. B. Reppas, C. J. Emig, D. Bang, S. J. Hwang, M. C. Jewett, J. M. Jacobson and G. M. Church, Science, 2011, 333, 348–353 CrossRef CAS PubMed
- M. J. Lajoie, A. J. Rovner, D. B. Goodman, H.-R. Aerni, A. D. Haimovich, G. Kuznetsov, J. A. Mercer, H. H. Wang, P. A. Carr and J. A. Mosberg, Science, 2013, 342, 357–360 CrossRef CAS PubMed
- M. Amiram, A. D. Haimovich, C. Fan, Y.-S. Wang, H.-R. Aerni, I. Ntai, D. W. Moonan, N. J. Ma, A. J. Rovner, S. H. Hong, N. L. Kelleher, A. L. Goodman, M. C. Jewett, D. Soell, J. Rinehart and F. J. Isaacs, Nat. Biotechnol., 2015, 33, 1272–1279 CrossRef CAS PubMed
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed
- Y. Wang, W. Song, W. J. Hu and Q. Lin, Angew. Chem., Int. Ed., 2009, 48, 5330–5333 CrossRef CAS PubMed
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed
- K. K. Palaniappan and C. R. Bertozzi, Chem. Rev., 2016, 116, 14277–14306 CrossRef CAS PubMed
- D. M. Patterson and J. A. Prescher, Curr. Opin. Chem. Biol., 2015, 28, 141–149 CrossRef CAS PubMed
- M. King and A. Wagner, Bioconjugate
Chem., 2014, 25, 825–839 CrossRef CAS PubMed
- M. F. Debets, J. C. M. van Hest and F. Rutjes, Org. Biomol. Chem., 2013, 11, 6439–6455 RSC
- H. Zhan, S. Jiang, A. M. Jonker, I. A. B. Pijpers and D. W. P. M. Löwik, J. Mater. Chem. B, 2020, 8, 5912–5920 RSC
- N. Contessi Negrini, A. Angelova Volponi, P. T. Sharpe and A. D. Celiz, ACS Biomater. Sci. Eng., 2021, 7, 4330–4346 CrossRef CAS PubMed
- A. Battigelli, B. Almeida and A. Shukla, Bioconjugate Chem., 2022, 33, 263–271 CrossRef CAS PubMed
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed
- S. C. Reddington and M. Howarth, Curr. Opin. Chem. Biol., 2015, 29, 94–99 CrossRef CAS PubMed
- Y. Wang, Y. Chang, R. Jia, H. Sun, J. Tian, H. Luo, H. Yu and Z. Shen, Process Biochem., 2020, 95, 260–268 CrossRef CAS
- W.-B. Zhang, F. Sun, D. A. Tirrell and F. H. Arnold, J. Am. Chem. Soc., 2013, 135, 13988–13997 CrossRef CAS PubMed
- A. I. Freitas, L. Domingues and T. Q. Aguiar, J. Adv. Res., 2022, 36, 249–264 CrossRef CAS PubMed
- M. Li, F. Cheng, H. Li, W. Jin, C. Chen, W. He, G. Cheng and Q. Wang, Langmuir, 2019, 35, 16466–16475 CrossRef CAS PubMed
- H. Zheng, J. Chen, L. Su, Y. Zhao, Y. Yang, H. Zeng, G. Xu, S. Yang and W. Jiang, Enzyme Microb. Technol., 2007, 41, 474–479 CrossRef CAS
- L. Zhang, N. Vila, T. Klein, G.-W. Kohring, I. Mazurenko, A. Walcarius and M. Etienne, ACS Appl. Mater. Interfaces, 2016, 8, 17591–17598 CrossRef CAS PubMed
- D. Weinrich, P.-C. Lin, P. Jonkheijm, U. T. T. Nguyen, H. Schroeder, C. M. Niemeyer, K. Alexandrov, R. Goody and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 1252–1257 CrossRef CAS PubMed
- X. Zhao, Y. Jin, X. Yuan, Z. Hou, Z. Chen, X. Fu, Q. Li, J. Wang and Y. Zhang, Anal. Chem., 2020, 92, 13750–13758 CrossRef CAS PubMed
- J. Xiong, J. He, W. P. Xie, E. Hinojosa, C. S. R. Ambati, N. Putluri, H.-E. Kim, M. X. Zhu and G. Du, J. Cell Sci., 2019, 132, jcs235390 CrossRef CAS PubMed
- G. O. Reznik, S. Vajda, C. R. Cantor and T. Sano, Bioconjugate Chem., 2001, 12, 1000–1004 CrossRef CAS PubMed
- T. Ichihara, J. K. Akada, S. Kamei, S. Ohshiro, D. Sato, M. Fujimoto, Y. Kuramitsu and K. Nakamura, J. Proteome Res., 2006, 5, 2144–2151 CrossRef CAS PubMed
- P. Wang, C. J. Zhang, G. C. Chen, Z. K. Na, S. Q. Yao and H. Y. Sun, Chem. Commun., 2013, 49, 8644–8646 RSC
- H. C. Wang, C. C. Yu, C. F. Liang, L. D. Huang, J. R. Hwu and C. C. Lin, ChemBioChem, 2014, 15, 829–835 CrossRef CAS PubMed
- A. I. Benítez-Mateos and F. Paradisi, ChemSusChem, 2022, 15, e202102030 CrossRef PubMed
- M. Kindermann, N. George, N. Johnsson and K. Johnsson, J. Am. Chem. Soc., 2003, 125, 7810–7811 CrossRef CAS PubMed
- R. Meyer and C. M. Niemeyer, Small, 2011, 7, 3211–3218 CrossRef CAS PubMed
- B. Sacca, R. Meyer, M. Erkelenz, K. Kiko, A. Arndt, H. Schroeder, K. S. Rabe and C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 9378–9383 CrossRef CAS PubMed
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed
- J. Wang, Y. Wang, J. Liu, Q. Li, G. Yin, Y. Zhang, C. Xiao, T. Fan, X. Zhao and X. Zheng, Anal. Chem., 2019, 91, 7385–7393 CrossRef CAS PubMed
- M. Macias-Contreras, H. He, K. N. Little, J. P. Lee, R. P. Campbell, M. Royzen and L. Zhu, Bioconjugate Chem., 2020, 31, 1370–1381 CrossRef CAS PubMed
- L. P. Encell, R. Friedman Ohana, K. Zimmerman, P. Otto, G. Vidugiris, M. G. Wood, G. V. Los, M. G. McDougall, C. Zimprich, N. Karassina, R. D. Learish, R. Hurst, J. Hartnett, S. Wheeler, P. Stecha, J. English, K. Zhao, J. Mendez, H. A. Benink, N. Murphy, D. L. Daniels, M. R. Slater, M. Urh, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, Curr. Chem. Genomics, 2012, 6, 55–71 CrossRef PubMed
- J. Döbber and M. Pohl, J. Biotechnol., 2017, 241, 170–174 CrossRef PubMed
- X. Fu, L. Li, X. Wen, R. Xu, Y. Xue, H. Zuo, Q. Liang, G. Feng, J. Wang and X. Zhao, J. Chromatogr. A, 2021, 1640, 461946 CrossRef CAS PubMed
- S. A. Slavoff, I. Chen, Y.-A. Choi and A. Y. Ting, J. Am. Chem. Soc., 2008, 130, 1160–1162 CrossRef CAS PubMed
- I. Chen, M. Howarth, W. Lin and A. Y. Ting, Nat. Methods, 2005, 2, 99–104 CrossRef CAS PubMed
- M. Howarth, K. Takao, Y. Hayashi and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7583–7588 CrossRef CAS PubMed
- E. de Boer, P. Rodriguez, E. Bonte, J. Krijgsveld, E. Katsantoni, A. Heck, F. Grosveld and J. Strouboulis, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7480–7485 CrossRef CAS PubMed
- J. Tykvart, P. Sacha, C. Barinka, T. Knedlik, J. Starkova, J. Lubkowski and J. Konvalinka, Protein Expr. Purif., 2012, 82, 106–115 CrossRef CAS PubMed
- S. L. Khatwani, J. S. Kang, D. G. Mullen, M. A. Hast, L. S. Beese, M. D. Distefano and T. A. Taton, Bioorg. Med. Chem., 2012, 20, 4532–4539 CrossRef CAS PubMed
- H. Miyao, Y. Ikeda, A. Shiraishi, Y. Kawakami and S. Sueda, Anal. Biochem., 2015, 484, 113–121 CrossRef CAS PubMed
- K. R. Davenport, C. A. Smiths, H. Hofstetter, J. R. Horn and O. Hofstetter, J. Chromatogr. B, 2016, 1021, 114–121 CrossRef CAS PubMed
- Y. Maeda, T. Yoshino, M. Takahashi, H. Ginya, J. Asahina, H. Tajima and T. Matsunaga, Appl. Environ. Microbiol., 2008, 74, 5139–5145 CrossRef CAS PubMed
- Y. Maeda, T. Yoshino and T. Matsunaga, Appl. Environ. Microbiol., 2010, 76, 5785–5790 CrossRef CAS PubMed
- J. Alftren, K. E. Ottow and T. J. Hobley, J. Mol. Catal. B-Enzym., 2013, 94, 29–35 CrossRef CAS
- T. Matsumoto, Y. Isogawa, T. Tanaka and A. Kondo, Biosens. Bioelectron., 2018, 99, 56–61 CrossRef CAS PubMed
- S. Garg, G. S. Singaraju, S. Yengkhom and S. Rakshit, Bioconjugate Chem., 2018, 29, 1714–1719 CrossRef CAS PubMed
- J. Fauser, S. Savitskiy, M. Fottner, V. Trauschke and B. Gulen, Bioconjugate Chem., 2020, 31, 1883–1892 CrossRef CAS PubMed
- S. M. Qafari, G. Ahmadian and M. Mohammadi, RSC Adv., 2017, 7, 56006–56015 RSC
- M. Raeeszadeh-Sarmazdeh, R. Parthasarathy and E. T. Boder, Colloids Surf., B, 2015, 128, 457–463 CrossRef CAS PubMed
- K. P. Li, R. Z. Zhang, Y. Xu, Z. M. Wu, J. Li, X. T. Zhou, J. W. Jiang, H. Y. Liu and R. Xiao, Sci. Rep., 2017, 7, 3081 CrossRef PubMed
- L. Chan, H. F. Cross, J. K. She, G. Cavalli, H. F. Martins and C. Neylon, PLoS One, 2007, 2, e1164 CrossRef PubMed
- F. Clow, J. D. Fraser and T. Proft, Biotechnol. Lett., 2008, 30, 1603–1607 CrossRef CAS PubMed
- J. E. Glasgow, M. L. Salit and J. R. Cochran, J. Am. Chem. Soc., 2016, 138, 7496–7499 CrossRef CAS PubMed
- R. K. Le, M. Raeeszadeh-Sarmazdeh, E. T. Boder and P. D. Frymier, Langmuir, 2015, 31, 1180–1188 CrossRef CAS PubMed
- M. Best, A. Degen, M. Baalmann, T. T. Schmidt and R. Wombacher, ChemBioChem, 2015, 16, 1158–1162 CrossRef CAS PubMed
- J. G. Plaks, R. Falatach, M. Kastantin, J. A. Berberich and J. L. Kaar, Bioconjugate Chem., 2015, 26, 1104–1112 CrossRef CAS PubMed
- M. Fernandez-Suarez, H. Baruah, L. Martinez-Hernandez, K. T. Xie, J. M. Baskin, C. R. Bertozzi and A. Y. Ting, Nat. Biotechnol., 2007, 25, 1483–1487 CrossRef CAS PubMed
- J. Z. Yao, C. Uttamapinant, A. Poloukhtine, J. M. Baskin, J. A. Codelli, E. M. Sletten, C. R. Bertozzi, V. V. Popik and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 3720–3728 CrossRef CAS PubMed
- M. Proeschel, M. E. Kraner, A. H. C. Horn, L. Schaefer, U. Sonnewald and H. Sticht, PLoS One, 2017, 12, e0179740 CrossRef PubMed
- C. M. Buldun, J. X. Jean, M. R. Bedford and M. Howarth, J. Am. Chem. Soc., 2018, 140, 3008–3018 CrossRef CAS PubMed
- G. Veggiani, T. Nakamura, M. D. Brenner, R. V. Gayet, J. Yan, C. V. Robinson and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1202–1207 CrossRef CAS PubMed
- S.-C. Wu, C. Wang, J. Chin and S.-L. Wong, Sci. Rep., 2019, 9, 3359 CrossRef PubMed
- Y. Chen, Y. Zhao, X. Zhou, N. Liu, D. Ming, L. Zhu and L. Jiang, Int. J. Biol. Macromol., 2021, 168, 13–21 CrossRef CAS PubMed
- Q. Xu, D. Ming, C. Shi, D. Lu, S. Tang, L. Jiang and H. Huang, Process Biochem., 2019, 80, 64–71 CrossRef CAS
- D. M. Williams, G. Kaufman, H. Izadi, A. E. Gahm, S. M. Prophet, K. T. Vanderlick, C. O. Osuji and L. Regan, ACS Appl. Nano Mater., 2018, 1, 2483–2488 CrossRef CAS
- X. Yang, J. Wei, Y. Wang, C. Yang, S. Zhao, C. Li, Y. Dong, K. Bai, Y. Li, H. Teng, D. Wang, N. Lyu, J. Li, X. Chang, X. Ning, Q. Ouyang, Y. Zhang and L. Qian, ACS Synth. Biol., 2018, 7, 2331–2339 CrossRef CAS PubMed
- W.-H. Wu, J. Wei and W.-B. Zhang, ACS Macro Lett., 2018, 7, 1388–1393 CrossRef CAS PubMed
- J. Fang and W.-b Zhang, Acta Polym. Sin., 2018, 429–444, DOI:10.11777/j.issn1000-3304.2018.18034
- J. L. Qu, S. Cao, Q. X. Wei, H. W. Zhang, R. Wang, W. Kang, T. Ma, L. Zhang, T. G. Liu, S. W. N. Au, F. Sun and J. Xia, ACS Nano, 2019, 13, 9895–9906 CrossRef CAS PubMed
- Q. Wei, S. He, J. Qu and J. Xia, Bioconjugate Chem., 2020, 31, 2413–2420 CrossRef CAS PubMed
- Y. Arfi, M. Shamshoum, I. Rogachev, Y. Peleg and E. A. Bayer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9109–9114 CrossRef CAS PubMed
- F. Peng, Q.-S. Chen, M.-H. Zong and W.-Y. Lou, Mol. Catal., 2021, 510, 111673 CrossRef CAS
- X. Zhong, Y. Ma, X. Zhang, J. Zhang, B. Wu, F. Hollmann and Y. Wang, Mol. Catal., 2022, 521, 112188 CrossRef CAS
- C. Schoene, J. O. Fierer, S. P. Bennett and M. Howarth, Angew. Chem., Int. Ed., 2014, 53, 6101–6104 CrossRef CAS PubMed
- X. Gao, J. Fang, B. Xue, L. Fu and H. Li, Biomacromolecules, 2016, 17, 2812–2819 CrossRef CAS PubMed
- M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef CAS PubMed
- A. Chatterjee, C. Mahato and D. Das, Angew. Chem., Int. Ed., 2021, 60, 202–207 CrossRef CAS PubMed
- I. W. Hamley, Biomacromolecules, 2021, 22, 1835–1855 CrossRef CAS PubMed
- L. Cai, Y. Chu, X. Liu, Y. Qiu, Z. Ge and G. Zhang, Microb. Cell. Fact., 2021, 20, 37 CrossRef CAS PubMed
- J. M. Antos, M. W.-L. Popp, R. Ernst, G.-L. Chew, E. Spooner and H. L. Ploegh, J. Biol. Chem., 2009, 284, 16028–16036 CrossRef CAS PubMed
- A. H. Keeble, A. Banerjee, M. P. Ferla, S. C. Reddington, I. N. A. K. Anuar and M. Howarth, Angew. Chem., Int. Ed., 2017, 56, 16521–16525 CrossRef CAS PubMed
- A. H. Keeble, V. K. Yadav, M. P. Ferla, C. C. Bauer, E. Chuntharpursat-Bon, J. Huang, R. S. Bon and M. Howarth, Cell Chem. Biol., 2022, 29(2), 339–350.e10 CrossRef CAS PubMed
-
A. H. Keeble and M. Howarth, in Metabolons and Supramolecular Enzyme Assemblies, ed. C. SchmidtDannert and M. B. Quin, 2019, vol. 617, pp. 443–461 Search PubMed
- F. Manea, V. G. Garda, B. Rad and C. M. Ajo-Franklin, Biotechnol. Bioeng., 2020, 117, 912–923 CrossRef CAS PubMed
- J. Tian, R. Jia, D. Wenge, H. Sun, Y. Wang, Y. Chang and H. Luo, Biotechnol. Lett., 2021, 43, 1075–1087 CrossRef CAS PubMed
- W. Dong, H. Sun, Q. Chen, L. Hou, Y. Chang and H. Luo, Int. J. Biol. Macromol., 2022, 199, 358–371 CrossRef CAS PubMed
- L. L. Tan, S. S. Hoon and F. T. Wong, PLoS One, 2016, 11, e0165074 CrossRef PubMed
- J. X. Wong, M. Gonzalez-Miro, A. J. Sutherland-Smith and B. H. A. Rehm, Front. Bioeng. Biotechnol., 2020, 8, 44 CrossRef PubMed
- J. X. Wong and B. H. A. Rehm, Biomacromolecules, 2018, 19, 4098–4112 CrossRef CAS PubMed
- P. Bitterwolf, S. Gallus, T. Peschke, E. Mittmann, C. Oelschlaeger, N. Willenbacher, K. S. Rabe and C. M. Niemeyer, Chem. Sci., 2019, 10, 9752–9757 RSC
- A. H. Keeble, P. Turkki, S. Stokes, I. N. A. K. Anuar, R. Rahikainen, V. P. Hytönen and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26523–26533 CrossRef CAS PubMed
- N. M. Rachel and J. N. Pelletier, Chem. Commun., 2016, 52, 2541–2544 RSC
- S. K. Oteng-Pabi, C. Pardin, M. Stoica and J. W. Keillor, Chem. Commun., 2014, 50, 6604–6606 RSC
- N. M. Rachel, J. L. Toulouse and J. N. Pelletier, Bioconjugate Chem., 2017, 28, 2518–2523 CrossRef CAS PubMed
- N. M. Rachel, D. Quaglia, E. Levesque, A. B. Charette and J. N. Pelletier, Protein Sci., 2017, 26, 2268–2279 CrossRef CAS PubMed
- R. Sato, K. Minamihata, R. Wakabayashi, M. Goto and N. Kamiya, Anal. Biochem., 2020, 600, 113700 CrossRef CAS PubMed
- J. H. Wang, M. Z. Tang, X. T. Yu, C. M. Xu, H. M. Yang and J. B. Tang, Colloids Surf., B, 2019, 177, 506–511 CrossRef CAS PubMed
- A. M. Wang, F. C. Du, F. Wang, Y. Q. Shen, W. F. Gao and P. F. Zhang, Biochem. Eng. J., 2013, 73, 86–92 CrossRef CAS
- H. Cho and J. Jaworski, Colloids Surf., B, 2014, 122, 846–850 CrossRef CAS PubMed
- N. Janson, T. Heinks, T. Beuel, S. Alam, M. Hohne, U. T. Bornscheuer, G. F. von Mollard and N. Sewald, ChemCatChem, 2022, 14, e202101485 CrossRef CAS
- D. Schumacher, J. Helma, F. A. Mann, G. Pichler, F. Natale, E. Krause, M. C. Cardoso, C. P. R. Hackenberger and H. Leonhardt, Angew. Chem., Int. Ed., 2015, 54, 13787–13791 CrossRef CAS PubMed
- C. Erck, R. Frank and J. Wehland, Neurochem. Res., 2000, 25, 5–10 CrossRef CAS PubMed
- C. A. Arce, J. A. Rodriguez, H. S. Barra and R. Caputo, Eur. J. Bbiochem., 1975, 59, 145–149 CrossRef CAS PubMed
- H. M. Kalisz, C. Erck, U. Plessmann and J. Wehland, Biochim. Biophys. Acta, 2000, 1481, 131–138 CrossRef CAS
- O. Monasterio, E. Nova, A. Lopez-Brauet and R. Lagos, FEBS Lett., 1995, 374, 165–168 CrossRef CAS PubMed
- M. Joniau, K. Coudijzer and M. De Cuyper, Anal. Biochem., 1990, 184, 325–329 CrossRef CAS PubMed
- A. Stengl, M. Gerlach, M. A. Kasper, C. P. R. Hackenberger, H. Leonhardt, D. Schumacher and J. Helma, Org. Biomol. Chem., 2019, 17, 4964–4969 RSC
- A. C. Obermeyer, J. B. Jarman and M. B. Francis, J. Am. Chem. Soc., 2014, 136, 9572–9579 CrossRef CAS PubMed
- R. Sangsuwan, A. C. Obermeyer, P. Tachachartvanich, K. K. Palaniappan and M. B. Francis, Chem. Commun., 2016, 52, 10036–10039 RSC
- J. M. Hooker, A. P. Esser-Kahn and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 15558–15559 CrossRef CAS PubMed
- C. R. Behrens, J. M. Hooker, A. C. Obermeyer, D. W. Romanini, E. M. Katz and M. B. Francis, J. Am. Chem. Soc., 2011, 133, 16398–16401 CrossRef CAS PubMed
- A. C. Obermeyer, J. B. Jarman, C. Netirojjanakul, K. El Muslemany and M. B. Francis, Angew. Chem., Int. Ed., 2014, 53, 1057–1061 CrossRef CAS PubMed
- A. M. Marmelstein, M. J. Lobba, C. S. Mogilevsky, J. C. Maza, D. D. Brauer and M. B. Francis, J. Am. Chem. Soc., 2020, 142, 5078–5086 CrossRef CAS PubMed
- S. L. Capehart, A. M. ElSohly, A. C. Obermeyer and M. B. Francis, Bioconjugate Chem., 2014, 25, 1888–1892 CrossRef CAS PubMed
- D. Alvarez Dorta, D. Deniaud, M. Mevel and S. G. Gouin, Chemistry, 2020, 26, 14257–14269 CrossRef CAS PubMed
- S. Sato and H. Nakamura, Molecules, 2019, 24, 3980–3996 CrossRef CAS PubMed
- S. Isaschar-Ovdat and A. Fishman, Food Chem., 2017, 232, 587–594 CrossRef CAS PubMed
- R. Lantto, E. Puolanne, K. Kruus, J. Buchert and K. Autio, J. Agric. Food Chem., 2007, 55, 1248–1255 CrossRef CAS PubMed
- M. Goldfeder, M. Kanteev, S. Isaschar-Ovdat, N. Adir and A. Fishman, Nat. Commun., 2014, 5, 4505 CrossRef CAS PubMed
- M. J. Lobba, C. Fellmann, A. M. Marmelstein, J. C. Maza, E. N. Kissman, S. A. Robinson, B. T. Staahl, C. Urnes, R. J. Lew, C. S. Mogilevsky, J. A. Doudna and M. B. Francis, ACS Central Sci., 2020, 6, 1564–1571 CrossRef CAS PubMed
- A. V. Ramsey, A. J. Bischoff and M. B. Francis, J. Am. Chem. Soc., 2021, 143, 7342–7350 CrossRef CAS PubMed
- C. S. Mogilevsky, M. J. Lobba, D. D. Brauer, A. M. Marmelstein, J. C. Maza, J. M. Gleason, J. A. Doudna and M. B. Francis, J. Am. Chem. Soc., 2021, 143, 13538–13547 CrossRef CAS PubMed
- J. C. Maza, D. L. V. Bader, L. F. Xiao, A. M. Marmelstein, D. D. Brauer, A. M. ElSohly, M. J. Smith, S. W. Krska, C. A. Parish and M. B. Francis, J. Am. Chem. Soc., 2019, 141, 3885–3892 CrossRef CAS PubMed
- M. J. Long and L. Hedstrom, ChemBioChem, 2012, 13, 1818–1825 CrossRef CAS PubMed
- K. Minamihata, M. Goto and N. Kamiya, Bioconjugate Chem., 2011, 22, 74–81 CrossRef CAS PubMed
- S. Sato, K. Nakamura and H. Nakamura, ChemBioChem, 2017, 18, 475–478 CrossRef CAS PubMed
- M. L. Mattinen, M. Hellman, P. Permi, K. Autio, N. Kalkkinen and J. Buchert, J. Agric. Food Chem., 2006, 54, 8883–8890 CrossRef CAS PubMed
- M. L. Mattinen, K. Kruus, J. Buchert, J. H. Nielsen, H. J. Andersen and C. L. Steffensen, FEBS J., 2005, 272, 3640–3650 CrossRef CAS PubMed
- D. Permana, K. Minamihata, M. Goto and N. Kamiya, J. Biosci. Bioeng., 2018, 126, 559–566 CrossRef CAS PubMed
- J. W. Chin, Nature, 2017, 550, 53–60 CrossRef CAS PubMed
- D. B. F. Johnson, J. F. Xu, Z. X. Shen, J. K. Takimoto, M. D. Schultz, R. J. Schmitz, Z. Xiang, J. R. Ecker, S. P. Briggs and L. Wang, Nat. Chem. Biol., 2011, 7, 779–786 CrossRef CAS PubMed
- A. Z. Wang and R. Kluger, Biochemistry, 2014, 53, 6793–6799 CrossRef CAS PubMed
- Y. Yin, R. Wang, J. Zhang, Z. Luo, Q. Xiao, T. Xie, X. Pei, P. Gao and A. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 41454–41463 CrossRef CAS PubMed
- J. F. Chen, J. Wang, Y. G. Bai, K. Li, E. S. Garcia, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2018, 140, 13695–13702 CrossRef CAS PubMed
- H. Li, R. Wang, A. Wang, J. Zhang, Y. Yin, X. Pei and P. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 6466–6478 CrossRef CAS
- A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Chem. Sci., 2015, 6, 50–69 RSC
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed
- R. L. Weller and S. R. Rajski, Org. Lett., 2005, 7, 2141–2144 CrossRef CAS PubMed
- Z. Lin, Y. Amako, F. Kabir, H. A. Flaxman, B. Budnik and C. M. Woo, J. Am. Chem. Soc., 2022, 144, 606–614 CrossRef CAS PubMed
- K. Liao, Y. Gong, R. Y. Zhu, C. Wang, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2021, 60, 8488–8493 CrossRef CAS PubMed
- D. Astruc, L. Y. Liang, A. Rapakousiou and J. Ruiz, Acc. Chem. Res., 2012, 45, 630–640 CrossRef CAS PubMed
- N. Hauptstein, P. Pouyan, J. Kehrein, M. Dirauf, M. D. Driessen, M. Raschig, K. Licha, M. Gottschaldt, U. S. Schubert, R. Haag, L. Meinel, C. Sotriffer and T. Luehmann, Biomacromolecules, 2021, 22, 4521–4534 CrossRef CAS PubMed
- T. Vranken, E. S. Redeker, A. Miszta, B. Billen, W. Hermens, B. de Laat, P. Adriaensens, W. Guedens and T. J. Cleij, Sens. Actuators, B, 2017, 238, 992–1000 CrossRef CAS
- J. F. Chen, K. Li, S. E. Bonson and S. C. Zimmerman, J. Am. Chem. Soc., 2020, 142, 13966–13973 CrossRef CAS PubMed
- B. K. Raliski, C. A. Howard and D. D. Young, Bioconjugate Chem., 2014, 25, 1916–1920 CrossRef CAS PubMed
- M.-H. Seo, J. Han, Z. Jin, D.-W. Lee, H.-S. Park and H.-S. Kim, Anal. Chem., 2011, 83, 2841–2845 CrossRef CAS PubMed
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed
- C. Besanceney-Webler, H. Jiang, T. Zheng, L. Feng, D. Soriano del Amo, W. Wang, L. M. Klivansky, F. L. Marlow, Y. Liu and P. Wu, Angew. Chem., Int. Ed., 2011, 50, 8051–8056 CrossRef CAS PubMed
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed
- A. M. Wang, F. C. Du, X. L. Pei, C. Y. Chen, S. G. Wu and Y. G. Zheng, J. Mol. Catal. B-Enzym., 2016, 132, 54–60 CrossRef CAS
- A. I. Benitez-Mateos, I. Llarena, A. Sanchez-Iglesias and F. Lopez-Gallego, ACS Synth. Biol., 2018, 7, 875–884 CrossRef CAS PubMed
- J. Zhang, R. Wang, Z. Luo, D. Jia, H. Chen, Q. Xiao, P. Zhang, X. Pei and A. Wang, Mater. Chem. Front., 2022, 6, 182–193 RSC
- Z. M. Png, H. Zeng, Q. Ye and J. Xu, Chem. – Asian J., 2017, 12, 2142–2159 CrossRef CAS PubMed
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed
- K. Lang, L. Davis, J. Torres-Kolbus, C. J. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298–304 CrossRef CAS PubMed
- C. H. Kim, J. Y. Axup and P. G. Schultz, Curr. Opin. Chem. Biol., 2013, 17, 412–419 CrossRef CAS PubMed
- V. F. C. Ferreira, B. L. Oliveira, A. D'Onofrio, C. M. Farinha, L. Gano, A. Paulo, G. J. L. Bernardes and F. Mendes, Bioconjugate Chem., 2021, 32, 121–132 CrossRef CAS PubMed
- R. M. Bednar, T. W. Golbek, K. M. Kean, W. J. Brown, S. Jana, J. E. Baio, P. A. Karplus and R. A. Mehl, ACS Appl. Mater. Interfaces, 2019, 11, 36391–36398 CrossRef CAS PubMed
- S. Axon and D. James, Curr. Opin. Green Sustainable Chem., 2018, 13, 140–145 CrossRef
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y. S. Ko, W. D. Jang and Y. S. Jang, Nat. Catal., 2019, 2, 18–33 CrossRef CAS
- R. A. Sheldon, Philos. Trans. R. Soc., A, 2020, 378, 20190274 CrossRef CAS PubMed
- J. O. Fierer, G. Veggiani and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1176–E1181 CrossRef CAS PubMed
- X.-D. Da, X.-L. Wu, Y. Liu and W.-B. Zhang, Curr. Protoc., 2021, 1, e99–e99 CAS
- B. Zakeri and M. Howarth, J. Am. Chem. Soc., 2010, 132, 4526–4527 CrossRef CAS PubMed
- L. Langen, M. Janssen, N. Oosthoek, S. Pereira, V. K. Vedas, F. V. Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2010, 79, 224–228 CrossRef PubMed
- C. Mateo, V. Grazu, J. M. Palomo, F. Lopez-Gallego, R. Fernandez-Lafuente and J. M. Guisan, Nat. Protoc., 2007, 2, 1022–1033 CrossRef CAS PubMed
- J. Camunas-Soler, A. Alemany and F. Ritort, Science, 2017, 355, 412–415 CrossRef CAS PubMed
- D. Balchin, M. Hayer-Hartl and F. U. Hartl, Science, 2016, 353(6294), aac4354 CrossRef PubMed
- Z. Yu and Q. Lin, J. Am. Chem. Soc., 2014, 136, 4153–4156 CrossRef CAS PubMed
- A. C. Forster and G. M. Church, Genome Res., 2007, 17, 1–6 CrossRef CAS PubMed
- H. W. H. van Roekel, B. J. H. M. Rosier, L. H. H. Meijer, P. A. J. Hilbers, A. J. Markvoort, W. T. S. Huck and T. F. A. de Greef, Chem. Soc. Rev., 2015, 44, 7465–7483 RSC
- U. Nations, Transforming our World: The 2030 Agenda for Sustainable Development: Sustainable Development Knowledge Platform, 2020, Available online: https://sustainabledevelopment.un.org/post2015/transformingourworld.
- C. Smaniotto, C. Battistella, L. Brunelli, E. Ruscio, A. Agodi, F. Auxilia, V. Baccolini, U. Gelatti, A. Odone, R. Prato, S. Tardivo, G. Voglino, F. Valent, S. Brusaferro, F. Balzarini, M. Barchitta, A. Carli, F. Castelli, C. Coppola, G. Iannelli, M. Milazzo, B. Rosina, C. Salerno, R. Siliquini and S. Sisi, Int. J. Environ. Res. Public Health, 2020, 17, 8968 CrossRef PubMed
- J. Yu, C. Wang, A. Wang, N. Li, X. Chen, X. Pei, P. Zhang and S. G. Wu, RSC Adv., 2018, 8, 16088–16094 RSC
- P. Gonzalez, F. Batista-Viera and B. M. Brena, Int. J. Biotechnol., 2004, 6, 338–345 CrossRef
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS
- T. Kumaraguru, T. Harini and S. Basetty, Tetrahedron: Asymmetry, 2017, 28, 1612–1617 CrossRef CAS
-
F. Batista-Viera, K. Ovsejevi and C. Manta, in Immobilization of Enzymes and Cells, ed. J. M. Guisan, Humana Press, Totowa, NJ, 2006, pp. 185–204 DOI:10.1007/978-1-59745-053-9_17
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, Science, 2018, 360, 398–403 CrossRef CAS PubMed
- Y. Liu, H. Zhou, J. Z. Guo, W. M. Ren and X. B. Lu, Angew. Chem., Int. Ed., 2017, 56, 4862–4866 CrossRef CAS PubMed
- M. Hong and E. Y. X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4188–4193 CrossRef CAS PubMed
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed
- D. Roura Padrosa, V. Marchini and F. Paradisi, Bioinformatics, 2021, 37, 2761–2762 CrossRef PubMed
- C. Meng, Y. Hu, Y. Zhang and F. Guo, Front. Bioeng. Biotechnol., 2020, 8, 245 CrossRef PubMed
- M. Chai, S. Moradi, E. Erfani, M. Asadnia, V. Chen and A. Razmjou, Chem. Mater., 2021, 33, 8666–8676 CrossRef CAS
- J. P. Smith, M. Liu, M. L. Lauro, M. Balasubramanian, J. H. Forstater, S. T. Grosser, Z. E. X. Dance, T. A. Rhodes, X. Bu and K. S. Booksh, Analyst, 2020, 145, 7571–7581 RSC
- N. M. Ralbovsky and J. P. Smith, Anal. Chem., 2021, 93, 11973–11981 CrossRef CAS PubMed
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed
- S. S. Mahmod, F. Yusof, M. S. Jami, S. Khanahmadi and H. Shah, Process Biochem., 2015, 50, 2144–2157 CrossRef CAS
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed
- H. O. Spivey and J. Ovadi, Methods, 1999, 19, 306–321 CrossRef CAS PubMed
- E. W. Miles, S. Rhee and D. R. Davies, J. Biol. Chem., 1999, 274, 12193–12196 CrossRef CAS PubMed
- C. You and Y. H. P. Zhang, ACS Synth. Biol., 2013, 2, 102–110 CrossRef CAS PubMed
- T. Peschke, P. Bitterwolf, K. S. Rabe and C. M. Niemeyer, Chem. Eng. Technol., 2019, 42, 2009–2017 CrossRef CAS
- S. Tsitkov and H. Hess, ACS Catal., 2019, 9, 2432–2439 CrossRef CAS
- J. Range, C. Halupczok, J. Lohmann, N. Swainston, C. Kettner, F. T. Bergmann, A. Weidemann, U. Wittig, S. Schnell and J. Pleiss, FEBS J., 2021 DOI:10.1111/febs.16318
-
C. Zeymer and D. Hilvert, in Annual Review of Biochemistry, ed. R. D. Kornberg, 2018, vol. 87, pp. 131–157 Search PubMed
- K. S. Rabe, J. Mueller, M. Skoupi and C. M. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 13574–13589 CrossRef CAS PubMed
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC
- A. I. Benitez-Mateos, D. R. Padrosa and F. Paradisi, Nat. Chem., 2022, 14, 489–499 CrossRef CAS PubMed
- C. J. Hartley, C. C. Williams, J. A. Scoble, Q. I. Churches, A. North, N. G. French, T. Nebl, G. Coia, A. C. Warden, G. Simpson, A. R. Frazer, C. N. Jensen, N. J. Turner and C. Scott, Nat. Catal., 2019, 2, 1006–1015 CrossRef CAS
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef CAS
- A. Al-Shameri, M. C. Petrich, K. J. Puring, U. P. Apfel, B. M. Nestl and L. Lauterbach, Angew. Chem., Int. Ed., 2020, 59, 10929–10933 CrossRef CAS PubMed
Footnote |
| † Zhiyuan Luo and Xiaolin Pei contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |