
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissecting the role of protein phosphorylation: a chemical biology toolbox
Tim
Bilbrough
 ,
Emanuele
Piemontese
and
Oliver
Seitz
*
,
Emanuele
Piemontese
and
Oliver
Seitz
*
Department of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: oliver.seitz@chemie.hu-berlin.de
First published on 21st June 2022
Abstract
Protein phosphorylation is a crucial regulator of protein and cellular function, yet, despite identifying an enormous number of phosphorylation sites, the role of most is still unclear. Each phosphoform, the particular combination of phosphorylations, of a protein has distinct and diverse biological consequences. Aberrant phosphorylation is implicated in the development of many diseases. To investigate their function, access to defined protein phosphoforms is essential. Materials obtained from cells often are complex mixtures. Recombinant methods can provide access to defined phosphoforms if site-specifically acting kinases are known, but the methods fail to provide homogenous material when several amino acid side chains compete for phosphorylation. Chemical and chemoenzymatic synthesis has provided an invaluable toolbox to enable access to previously unreachable phosphoforms of proteins. In this review, we selected important tools that enable access to homogeneously phosphorylated protein and discuss examples that demonstrate how they can be applied. Firstly, we discuss the synthesis of phosphopeptides and proteins through chemical and enzymatic means and their advantages and limitations. Secondly, we showcase illustrative examples that applied these tools to answer biological questions pertaining to proteins involved in signal transduction, control of transcription, neurodegenerative diseases and aggregation, apoptosis and autophagy, and transmembrane proteins. We discuss the opportunities and challenges in the field.
 Tim Bilbrough | Tim Bilbrough received both his Bachelor of Biomedical Sciences and Master of Drug Discovery and Development from Victoria University of Wellington, New Zealand. There he focused on the synthesis of novel vaccine adjuvants under the supervision of Prof. Gavin Painter. He is currently undertaking his PhD in Chemistry under the guidance of Prof. Dr. Oliver Seitz at the Humboldt University Berlin. There, his thesis focuses on native chemical ligation auxiliaries and the synthesis of phosphorylated protein domains. |
 Emanuele Piemontese | Emanuele Piemontese studied Pharmaceutical Chemistry and Technology at the Università di Firenze, Italy. After an exchange at the University of Eastern Finland, he developed an interest in peptide chemistry in the PeptLab group in Florence. He then joined the laboratory of Prof. Dr. Oliver Seitz at the Humboldt-Universität zu Berlin to pursue his PhD in bio-organic chemistry. His interests are directed towards the synthesis of post-translationally modified peptides, in particular heavily phosphorylated peptides, and the development of biological assays involving the peptides to investigate the mechanism of transcription. |
 Oliver Seitz | Oliver Seitz obtained his PhD from the University of Mainz in 1995. After postdoctoral research at the Scripps Research Institute in La Jolla, he moved to the University of Karlsruhe, Germany. In 2000, he became group leader at the Max-Planck Institute of Molecular Physiology in Dortmund, Germany, and in 2003 he was appointed Full Professor at the Humboldt-Universität zu Berlin. Oliver Seitz has a keen interest in developing chemistry that allows the interrogation and perturbation of cellular and biochemical processes. Recently, he is focusing on the synthesis of posttranslationally modified proteins, peptide- and nucleic acid-templated chemistry and RNA and protein imaging. He is a recipient of an ERC Advanced Grant and the Max Bergmann Medal 2018. |
1 Introduction
Phosphorylation is the most abundant post-translational modification (PTM) of proteins. Its significance is reflected in the space allocated in the genome for kinases. Genes for over 500 kinases have been identified in humans, representing 1.7% of the entire genome.1 Phosphosite, a database of reported PTMs, lists more than 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 phosphorylation sites in the proteome.2 Phosphorylation occurs most commonly on serine, followed by threonine and tyrosine, at a relative frequency of 11.2:2.5:1.3 However, phosphorylation is not limited to these sites. Though with reduced frequency, kinases also act on the side chains of cysteine, lysine, histidine, arginine, aspartic and glutamic acid.4
000 phosphorylation sites in the proteome.2 Phosphorylation occurs most commonly on serine, followed by threonine and tyrosine, at a relative frequency of 11.2:2.5:1.3 However, phosphorylation is not limited to these sites. Though with reduced frequency, kinases also act on the side chains of cysteine, lysine, histidine, arginine, aspartic and glutamic acid.4
The reversible introduction of a phosphate group has a significant effect on the protein. The large, dianionic group can change the structure of the protein as well as the local environment. Specifically, a phosphate offers a new site to form hydrogen bonds or salt bridges. This can change the activity of the protein or create a new binding site. For example, phosphorylation regulates signalling cascades such as in the mitogen-activated protein kinase (MAPK) pathway where a chain of kinases propagate a phosphorylation signal to eventually activate transcription.5 Exemplifying the creation of a new binding site, the Src Homology 2 (SH2) domain recognises phosphotyrosine-containing motifs and enables protein-protein interactions.6 Furthermore, aberrant phosphorylation is implicated in disease, including cancer. The constitutively active Bcr-Abl kinase in chronic myeloid leukaemia, for example, causes the misregulation of cell cycle signalling and leads to oncogenesis.7 These examples highlight the range of roles phosphorylation plays in diverse areas of the cell and also possible therapeutic targets.
Each phosphoform of a protein, the protein state defined by the specific combination of phosphorylated residues, is chemically and biologically distinct and results in unique outcomes. For example, different patterns of phosphorylations on a G-protein coupled receptor (GPCR) can lead to a range of different signalling outcomes (Fig. 1).8 Although proteomics has revealed an enormous array of phosphorylated proteins, the function of most phosphorylation sites remains unknown.9 It is, therefore, important to be able to understand the role of distinct phosphoforms – the function each individual phosphorylation site plays in a protein. Access to highly pure, site-specifically phosphorylated material in the quantities required for assays is, therefore, necessary to understand the role of each site. Investigations on heterogeneously phosphorylated material – either a mixture of phosphorylated and unphosphorylated material or multiple undesired sites of phosphorylation – cannot accurately dissect the role of each phosphorylation, just as a drug assay would not use a mixture of compounds. Furthermore, regulations for therapeutics and diagnostics require defined, highly pure and homogeneous material for their application.
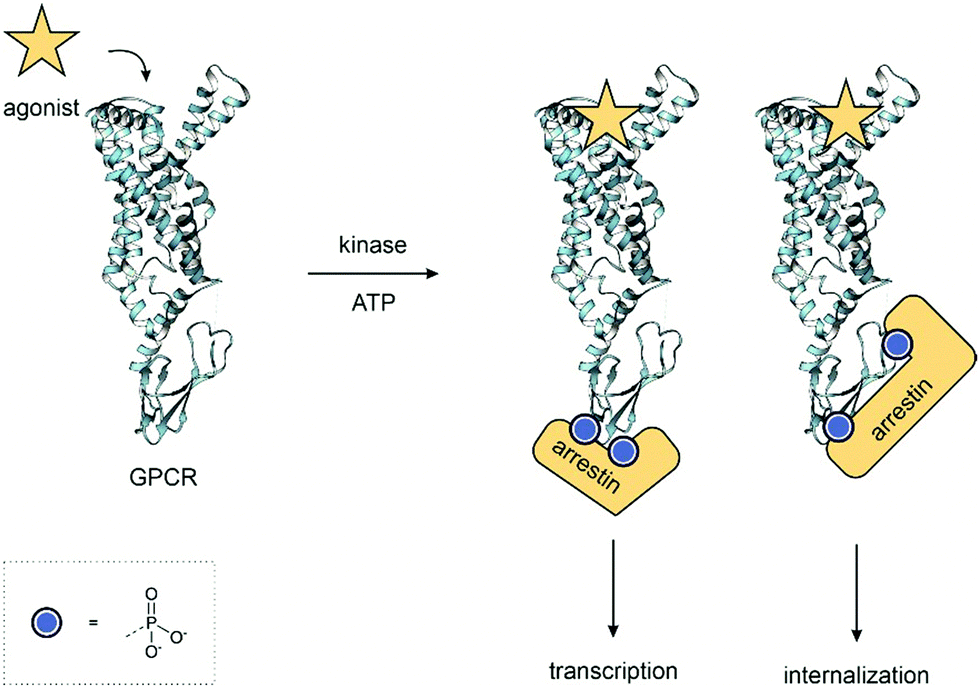 | ||
| Fig. 1 GPCR Phosphoforms – the combination of phosphorylated residues on a protein, the phosphoform, acts as a barcode. Each is unique and leads to different outcomes. In this example, the pattern of phosphorylation determines the conformation of the arrestin and leads to diverse signalling outcomes. PDB: 7LCK.10 | ||
In this review, we showcase the range of tools available to obtain homogeneous, site-specifically modified protein samples for interrogating individual phosphorylation events. Firstly, we will overview the chemical methods available to synthesise phosphopeptides (Fig. 2A) and then, secondly, discuss the methods allowing the convergent synthesis of phosphoproteins from phosphopeptide fragments (Fig. 2B). We particularly draw attention to examples of total chemical synthesis, given our lab's own interest in this area of research. Thirdly, we highlight the application of phosphoproteins and peptides to examine biological systems and reveal the function of specific phosphorylation sites (Fig. 2C).
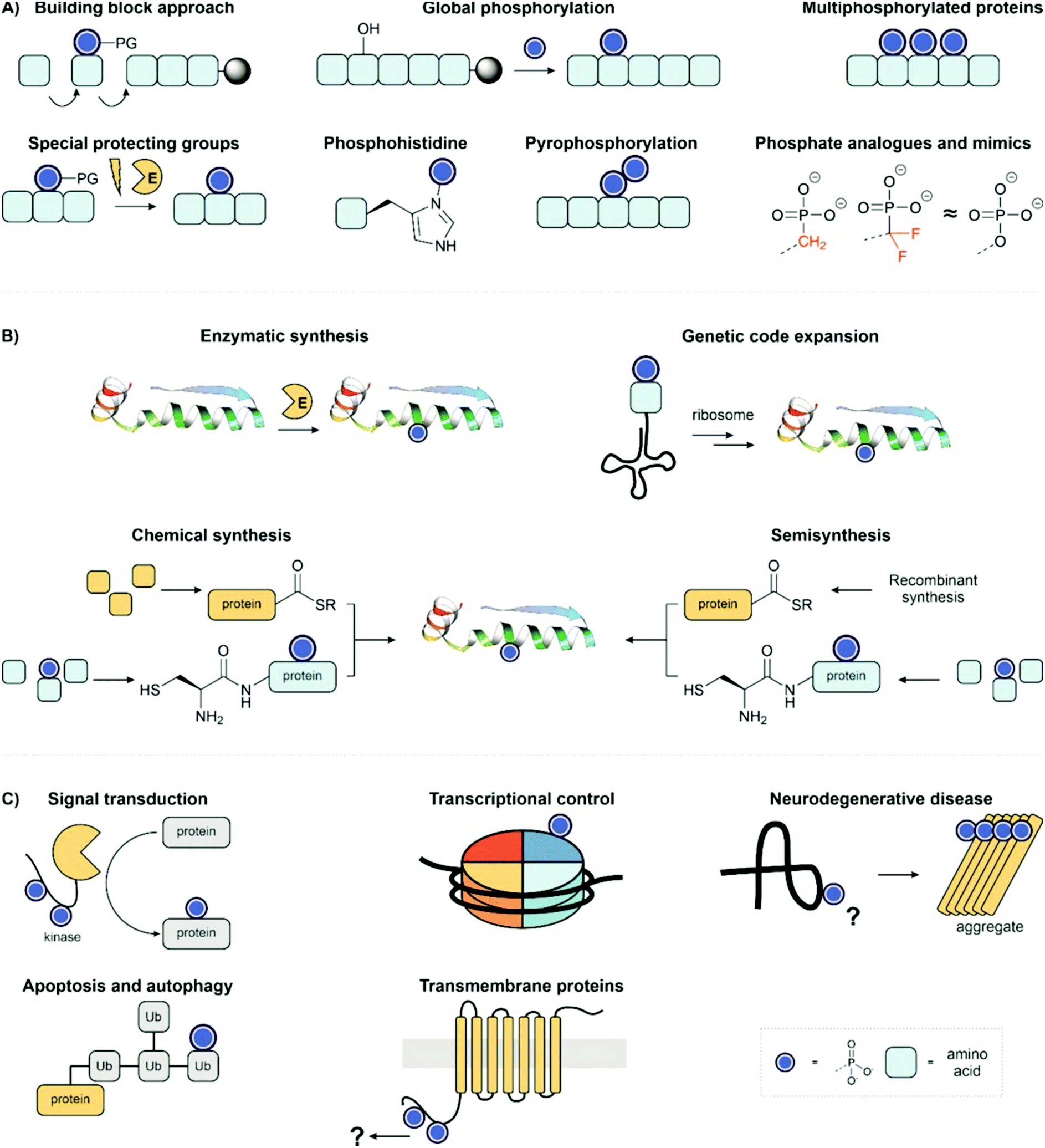 | ||
| Fig. 2 Review overview; (A) methods for synthesis of phosphopeptides; (B) strategies for preparing homogenous phosphoproteins; (C) synthetic targets and applications. PDB: 2WTT.11 | ||
2 Synthesis of phosphopeptides
Access to phosphopeptides is key for studies on the function of protein phosphorylation. They can be used alone as fragments for investigation or as invaluable building blocks employed in protein synthesis. Since the introduction of solid-phase peptide synthesis12 (SPPS), several methods to obtain phosphopeptides have been developed for both Boc13 and Fmoc14 strategies. The versatility and safety made the latter the strategy of choice for SPPS and peptide chemists focused on creating methods compatible with the Fmoc strategy.The synthetic methods for the introduction of a phosphate group fall into two categories: the building block approach and the global phosphorylation approach. In this section of the review, we will discuss the most important methods for synthesising phosphorylated peptides and underline the advantages and disadvantages of the two main approaches.
Currently, the synthesis of single or double phosphorylated peptides is routine, which allows for simple biological assays but is not sufficient to study higher phosphorylated proteins and their biological role. The bulkiness of the phosphate group and its negative charges make the chemical synthesis of phosphopeptides complicated,15 leading to many side products, like truncation or deletion sequences, due to incomplete coupling of the building blocks. Moreover, the purification and analysis of phosphopeptides synthesized with standard SPPS methods is usually challenging due to the high polarity of the products.16,17 For a detailed account of the chemical synthesis of multiphosphorylated peptides, we recommend a recent review from Samarasimhareddy et al.18
2.1 Building block approach
In the building block (or synthon) approach, phosphorylated amino acids are incorporated during the elongation of the peptide chain. This strategy is suitable for the introduction of phosphoserine, phosphothreonine and phosphotyrosine, which are usually incorporated as N-α-amino-protected amino acids with protection also on the phosphorylation.| Phospho group | Comments | Ref. | |
|---|---|---|---|
| 1 | Dibenzyl protection 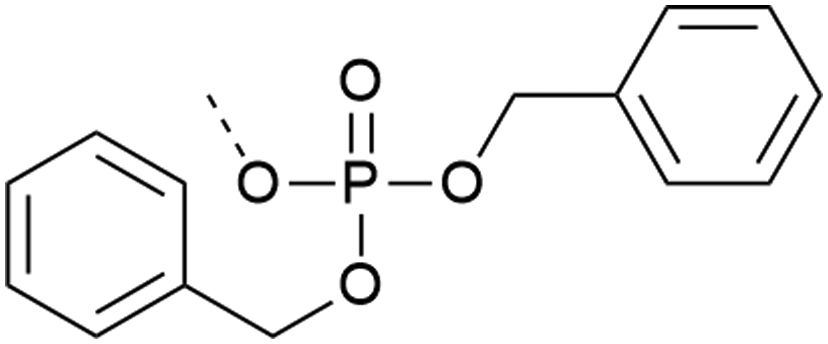 |
– High β-elimination for S and T | 19–21 |
| – Cleavage requires strong scavengers | |||
| – Best coupled with iminium reagents | |||
| – Bulky | |||
| – Can be introduced as a building block or with post-synthetic phosphorylation strategies | |||
| 2 | Monobenzyl protection 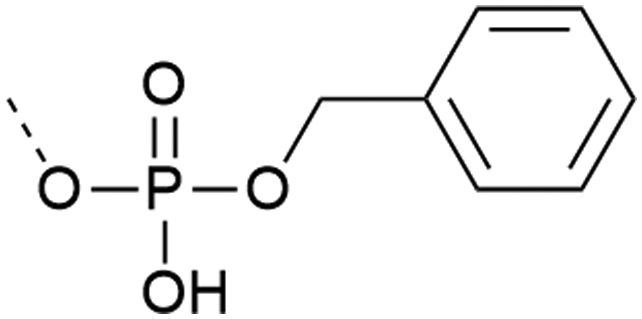 |
– Low β-elimination, but precautions necessary with MW | 20 and 22–24 |
| – Free acid can react with the activator or form piperidinium adduct during elongation | |||
| – Shelf-stable | |||
| – Commercially available | |||
| – Cleavage requires strong scavengers | |||
| – Mono-benzyl protected pY more stable upon storage compared to the correspondent of entries 1 and 3 | |||
| – Best coupled with iminium reagents | |||
| – Can be introduced as a building block or with post-synthetic phosphorylation strategies | |||
| 3 | Unprotected phosphate 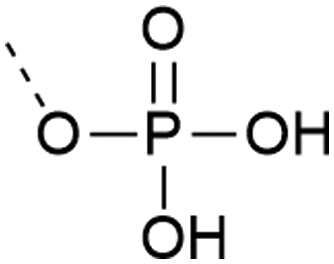 |
– Can form pyrophosphates with adjacent phosphorylated residues | 25 and 26 |
| – Free acid can react with the activator or form piperidinium adduct during elongation | |||
| – Introduced as a building block | |||
| – Low solubility of the building block in organic solvents | |||
| 4 | Di-n-propyl-phosphodiamidates 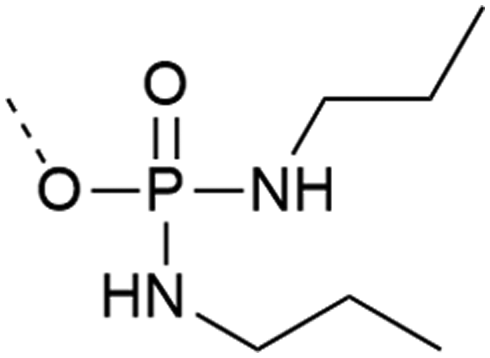 |
– Does not require an extra step after cleavage | 27 and 28 |
| – Does not react with activator or form salts | |||
| – Introduced as a building block | |||
| – Commercially available | |||
| – Used on Y | |||
| 5 | Tetramethyl phosphodiamidates  |
– Requires extra hydrolysis step after cleavage | 29 and 30 |
| – Does not react with activator or forms salts | |||
| – Cleavage conditions can form depsipeptide at S and T | |||
| – Introduced as a building block | |||
| – Commercially available for tyrosine | |||
| 6 | 1-(2-nitrophenyl)ethyl protection  |
– Light controlled deprotection at 365 nm | 31–33 |
| – Spatio-temporal control of deprotection | |||
| – In vivo application | |||
| – Can be introduced with post-synthetic phosphorylation strategies | |||
| 7 | Bhc protection 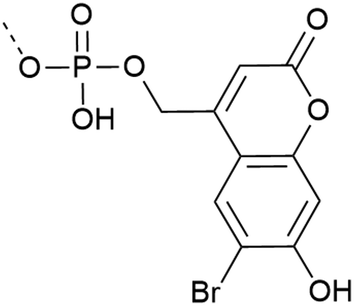 |
– Photo-deprotection at higher wavelengths is less cytotoxic (two-photon irradiation at 749 nm) | 34 |
| – Spatio-temporal control of deprotection | |||
| – In vivo application | |||
| – Used on Y | |||
| – Introduced as a building block | |||
| 8 | POM protection  |
– Esterase cleavable | 35 |
| – In vivo application – deprotection inside the cell | |||
| – Used on S and T | |||
| – Introduced as a building block. Subsequent POMylation is necessary for in vivo application | |||
| 9 | SATE protection 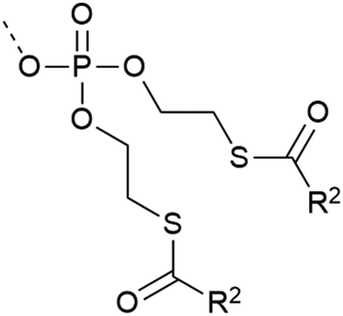 |
– Esterase cleavable | 36 and 37 |
| – Unstable in acidic solution | |||
| – In vivo application – deprotection inside the cell | |||
| – Used on Y | |||
| 10 | Phosphonate 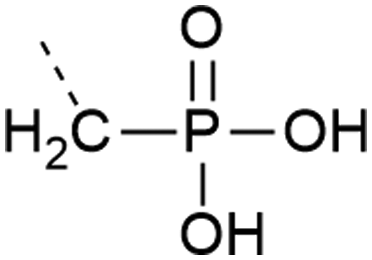 |
– Stable against phosphatases | 38 |
| – Only Y is commercially available | |||
| – May not accurately represent a phosphorylated residue | |||
| – Introduced as a building block | |||
| 11 | Difluorophosphonate 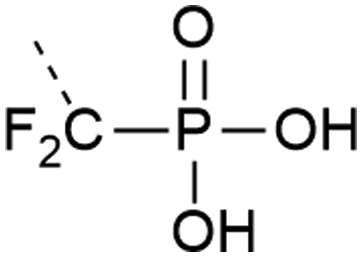 |
– Stable against phosphatases | 39 and 40 |
| – Only Y is commercially available | |||
| – pKa close to phosphorylated residue | |||
| – Introduced as a building block | |||
The use of serine and threonine phosphotriesters is problematic because the phosphate may be lost in a β-elimination reaction that occurs under the basic conditions applied during Fmoc removal. This leads to the formation of dehydroalanyl and dehydroamino-2-butyryl residues (M-80).47 The resulting double bond is a weak electrophile and can, therefore, be attacked by piperidine to form 3-(1-piperidinyl)alanine (M-13) (Fig. 3).48 Both products of these side reactions are difficult to separate from the desired peptide.
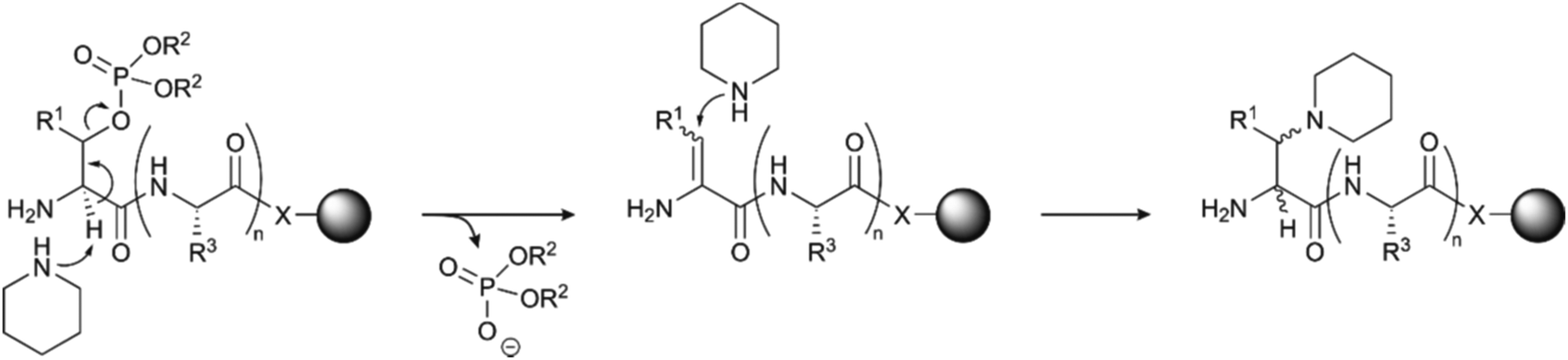 | ||
| Fig. 3 Mechanism of piperidine mediated β-elimination of the double protected phosphate group from the side chains of serine (R1 = H) or threonine (R1 = CH3) and formation of the piperidinyl-adduct. R2 is the protecting group on the phosphate (see Table 1)and R3 represents the generic side chain of an amino acid. The nature of X depends on the functionalisation of the resin. | ||
The advent of monobenzyl-protection, introduced in 1994 by Wakamiya et al.22 was a game-changer.49,50 Compared to solid-phase syntheses with phosphotriesters, the rate of β-elimination is significantly lower with phosphodiesters, which therefore provide crude materials of higher purity. However, it needs to be taken into consideration that piperidine-mediated removal of the Fmoc group leads to deprotonation of the phosphodiester. The phosphate binds a piperidinium group as a countercation.51 This piperidinium salt is not washed away and can react, as a secondary amine, with the activated amino acid in the following coupling step.52 This consumes one equivalent of activated amino acid per phosphorylation, incrementally decreasing incorporation yield as the number of phosphorylated residues increases. The problem can be overcome by increasing the number of equivalents of the amino acids and the coupling reagents or exchanging the counterion of the phosphate with a tertiary amine (usually DIEA) after the deprotection steps.51
Although the β-elimination is not completely suppressed,52 in particular with pSer53 and in microwave-assisted reactions,23 the monobenzyl protected Ser and Thr (Table 1, entry 2) have become the building block of choice for the synthesis of phosphorylated peptides.54 Mono-benzyl phosphodiester derivatives are chemically stable over long storage times53 and are commercially available. Perich et al.20 compared an array of coupling methods and proved the superiority of mixtures containing uronium based activators, HOBt (or HOAt) and DIEA, suggesting also extended reaction time for the coupling of the building blocks, in particular of Fmoc-Thr(PO(OBzl)OH)-OH. They also described low coupling yields with other activators such as PyBOP, BOP and DIC in combination with HOBt or HOAt. The authors suggested that the high reactivity of these coupling reagents allowed reactions at the phosphodiester, which reduced the amount of coupling reagent available for activation of the carboxylic acid group.
Cleavage of benzyl phosphates succeeds by treating the resin-bound phosphopeptides with commonly used cleavage cocktails comprised of trifluoroacetic acid (TFA), triisopropylsilane (TIS) and water (TFA:TIS:H2O 95:2.5:2.5). The formed benzyl cation can alkylate nucleophilic side chains of Tyr, Cys, Met and Trp,55 in particular during microwave-assisted cleavages. As a remedy, powerful scavengers such as EDT, phenol or thioanisole are added to the cleavage mixtures and heating should be avoided.31 In our laboratory, we experienced alkylation of Tyr (M + 90), and we solved the problem using the cleavage cocktail K (TFA:H2O:phenol:thioanisole:EDT 82.5:5:5:5:2.5).56,57
The synthesis of multiphosphorylated peptides remains challenging. In such cases, extended coupling times and double coupling may be required. While manually synthesizing the Phosphoryn Repeat Motif bearing six phosphorylations, O’Brien-Simpson et al.15 noticed that the most effective coupling strategy for a stretch of neighbouring phosphorylated amino acids was performing double couplings in every cycle with HBTU as an activator in the first coupling and the stronger activator58 HATU in the second. Moreover, they reported that using a 2Cl-Trt linker helped the removal of all the Bzl protecting groups (which may proceed faster in solution than on resin-bound phosphopeptides) in the cleavage step, compared with the previously used PAL-PEG based resin.
Microwave heating is widely used in peptide synthesis to improve coupling yields and decrease synthesis time.59 Jensen and co-workers used the Fmoc-Ser(PO(OBzl)OH)-OH building block in the assembly of a monophosphorylated 15-mer.60 The yields obtained in the microwave-assisted SPPS were twice as high as yields provided by “conventional” SPPS. Attard et al.53 also experienced β-elimination of the phosphate group with mono-benzyl protected building blocks (although at lower rates), in particular in microwave-assisted synthesis. While investigating alternative Fmoc cleavage conditions, they observed high purity crude material when cyclohexylamine in DCM (1:1) was used for Fmoc removal directly after the introduction of the phosphoserine building block. They recommended switching to 20% piperidine in DMF for subsequent Fmoc removal steps because β-elimination occurred preferentially at N-terminal phosphoserine. Furthermore, β-elimination has been reported to be slow with the bulky base DBU, though in this case, it may prove necessary to include scavengers for the dibenzofulvene formed upon deprotection. Caution is required when DBU is applied in the synthesis of Asp/Asn-containing sequences, which are prone to form aspartimides.61 For the synthesis of the Phosphoryn Repeat Motif DBU (2.5% v/v in DMF) was complemented by 2.5% piperidine as scavenger.15
Building blocks protected with a phosphorodiamidate such as Fmoc-Tyr[P(O)(NHR)2]-OH (R = nPr and iPr),27,28 (Table 1, entry 4) and in particular Fmoc-Tyr[P(O)(NMe2)2]-OH (Table 1, entry 5) introduced by Chao et al.29 are useful for the introduction of phosphotyrosine in a sequence. The tetramethyl phosphorodiamidate is extensively used for Fmoc synthesis of phosphotyrosine-containing peptides since it is stable in a basic environment and the phosphate group is fully protected. Deprotection of phosphorodiamidates involves two steps. First, treatment with 95% TFA for four hours and, secondly, acid hydrolysis with 10% water in TFA overnight, after the normal cleavage with scavengers. Di-n-propyl phosphodiamidates can be deprotected with a 4 hour long cleavage with 95%, without further steps.27 Phosphorodiamidates cannot react with the activators used in the coupling step and, therefore, provide more options for the coupling procedure than possible with mono-protected or unprotected phosphates.20 However, there is evidence that an N → O acyl shift at Thr (or Ser) can occur during the prolonged acid cleavage of bisdimethylamino-masked pTyr containing peptides. This side reaction was most prominent when Thr was in the +2-position to a phosphorylated tyrosine. The formation of the depsipeptide by-product (Fig. 4) can be avoided by using mono- and dibenzyl protected pTyr, which can be deprotected more quickly.30
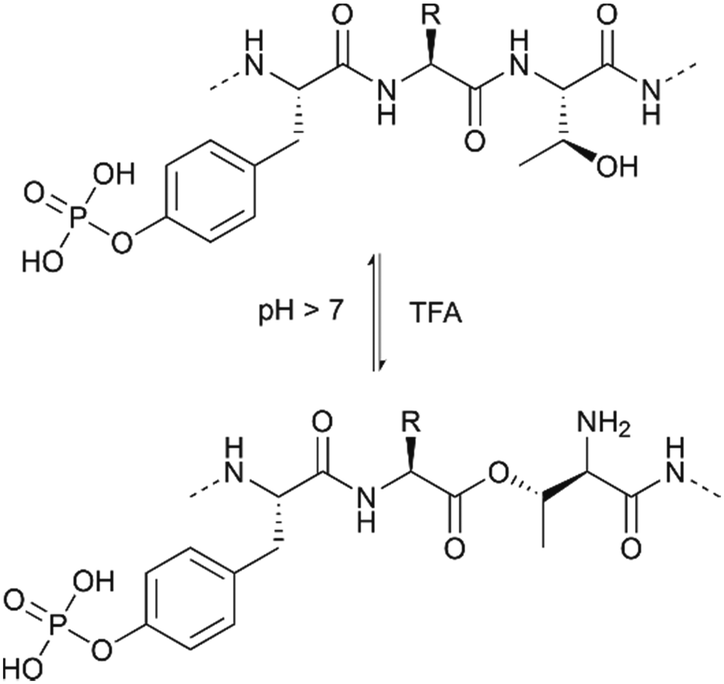 | ||
| Fig. 4 Reversible N → O acyl shift occurring at a threonine close to a pTyr residue upon prolonged TFA cleavage. | ||
2.2 Global and on-line phosphorylation
With the global phosphorylation approach (or post-synthetic phosphorylation), free hydroxyl groups are phosphorylated after the peptide bonds have been established. Phosphorylation can be carried out in solution or on solid-phase.65 The reagents for the phosphorylation are the same used for the synthesis of the phosphorylated building blocks, and usually, they are based on phosphorus(III)-based phosphoramidites. While the alkyl chains on the nitrogen are generally methyl or isopropyl groups, a wide array of protecting groups for the two oxygen atoms have been developed, but the di-O-benzyl and di-O-tert-butyl N,N-dialkyl phosphoramidites (Fig. 1, 2 and 5), introduced by Perich and Johns, are the most common reagents for the global phosphorylation of Ser, Thr and Tyr.66 These reagents are shelf-stable, highly reactive under mildly acidic conditions and easy to prepare. Typically, tetrazole is added for activation of the phosphoramidites by nucleophilic catalysis. The phosphite triester formed is oxidized to P(V), most commonly by using mCPBA,13tert-butyl-hydroperoxide67 or aqueous iodine (Fig. 6).65 Phosphate deprotection proceeds with concentrated TFA (protocol compatible with Fmoc strategy), although the Bzl is more acid-stable. If desired, the acid-stability of the phosphate protection can be increased by using the 4-chlorobenzyl group (Fig. 3 and 5).68,69 Prior to phosphorylation, the hydroxyl groups must be accessible while all other functional groups must remain protected. This can be achieved by introducing amino acids with an unprotected side chain14 or with Trt protecting groups that can be cleaved under very mild conditions (2% TFA in DCM).70 As an alternative, the t-butyldimethylsilyl (TBDMS) group offers orthogonal protection that can be selectively removed with a fluoride source.71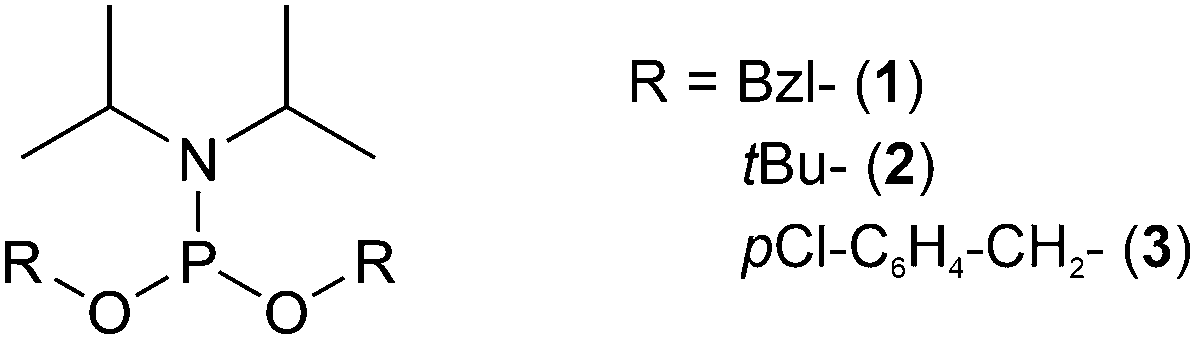 | ||
| Fig. 5 Examples of phosphoramidites used for global phosphorylation. The most commonly used reagents are di-O-benzyl (1), di-O-tert-butyl (2) and di-O-p-chlorobenzyl (3) N,N-diisopropyl phosphoramidites. | ||
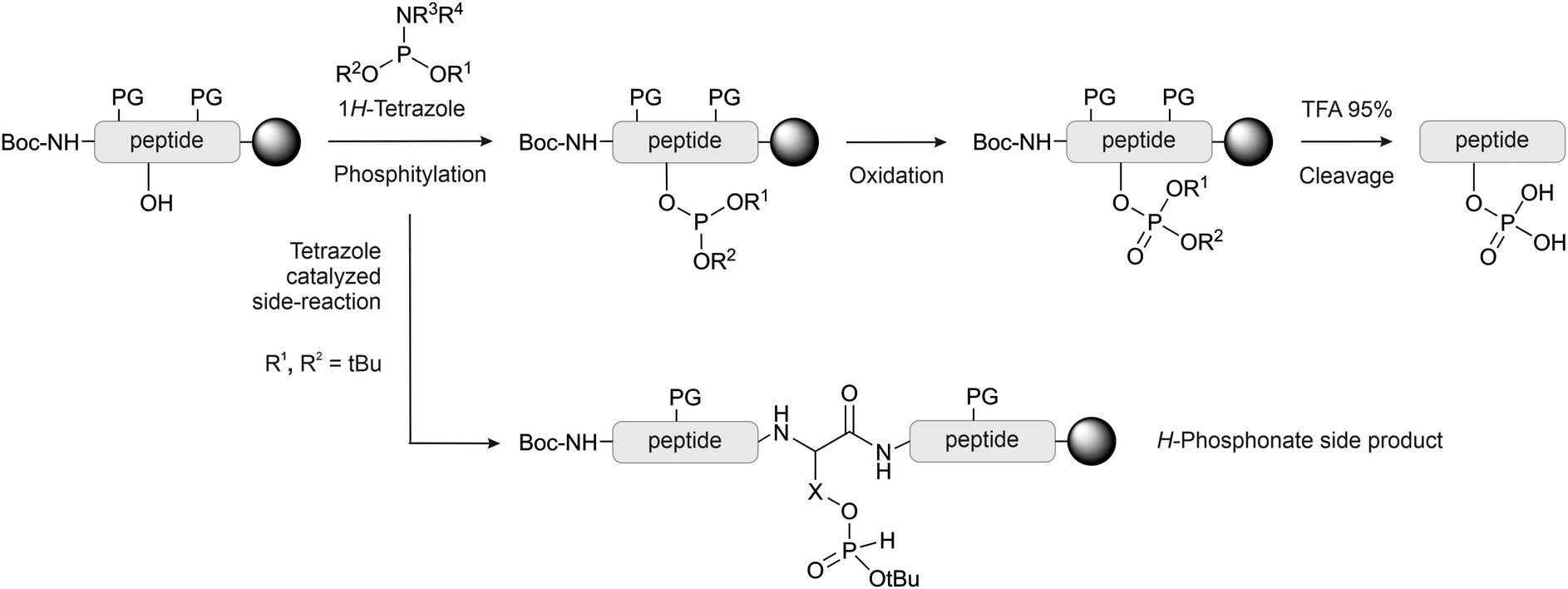 | ||
| Fig. 6 Global phosphorylation by phosphitylation and oxidation. The most common reagents for phosphitylation and their substituent are listed. Formation of H-phosphonate is the most common side reaction occurring during global phosphorylation with O-tBu-protected phosphoramidites. The side reaction can occur, albeit at a lower rate, also with the other protecting groups. X = –CH2– (pSer), –CH(CH3)– (pThr), –CH2–C6H4– (pTyr). PG: protecting group. | ||
The oxidation step can be detrimental to Cys, Met and Trp.65 Bannwarth and coworkers72 showed that 1M iodine in a mixture of 2,6-lutidine/THF/ water 40:10:172,73 allowed for smooth oxidation with no significant side reactions. Andrews et al. stated that anhydrous tBuOOH is the preferred choice in the case of Met containing peptides.67
In global phosphorylation, one of the most common side reactions is the formation of H-phosphonates (M-16) during the phosphitylation step (Fig. 6).14 The tert-butyl protecting group is particularly acid-sensitive and, therefore, H-phosphonate formation occurs more readily with tBu-protected than with Bzl-protected phosphites. Perich suggested using less concentrated 1H-tetrazole and aqueous iodine/pyridine for the oxidation step, which is known to convert H-phosphonates to phosphates. A solution of tBuOOH in anhydrous DMF was used for reactions in the presence of oxidation-sensitive amino acids.74 The oxidant should be added as quickly as possible once the phosphitylation is complete. It has been observed that waiting longer than 10 minutes can significantly increase the H-phosphonate formation.75 Daus et al. successfully used Perich's protocol to obtain a multi-phosphorylated peptide in high yield (see Section 2.3).76
On-line phosphorylation describes a method of phosphorylation that is performed directly after coupling of the hydroxyl group-containing amino acid (Fig. 7A). This strategy eases the problems deriving from steric hindrance caused by protecting groups of adjacent amino acids or by the full-length peptidic structure itself. Perich introduced the strategy for the synthesis of a tyrosine-phosphorylated Fcγ receptor peptide.77 For phosphorylation at serine, Toth and colleagues used the O-cyanoethyl-O-tBu-protected phosphoramidite. The cyanoethyl group can be selectively removed, leaving a phosphodiester moiety, which avoids the β-elimination problem during the elongation of the rest of the chain (Fig. 7B). After oxidation, treatment with DBU or piperidine removed both the cyanoethyl and the Fmoc group. The authors reported that, owing to the high rate of cyanoethyl deprotection, the reaction proceeded without β-elimination.78 The same group also used H-phosphonates as an alternative to phosphorylation with P(III) reagents (Fig. 7C).79
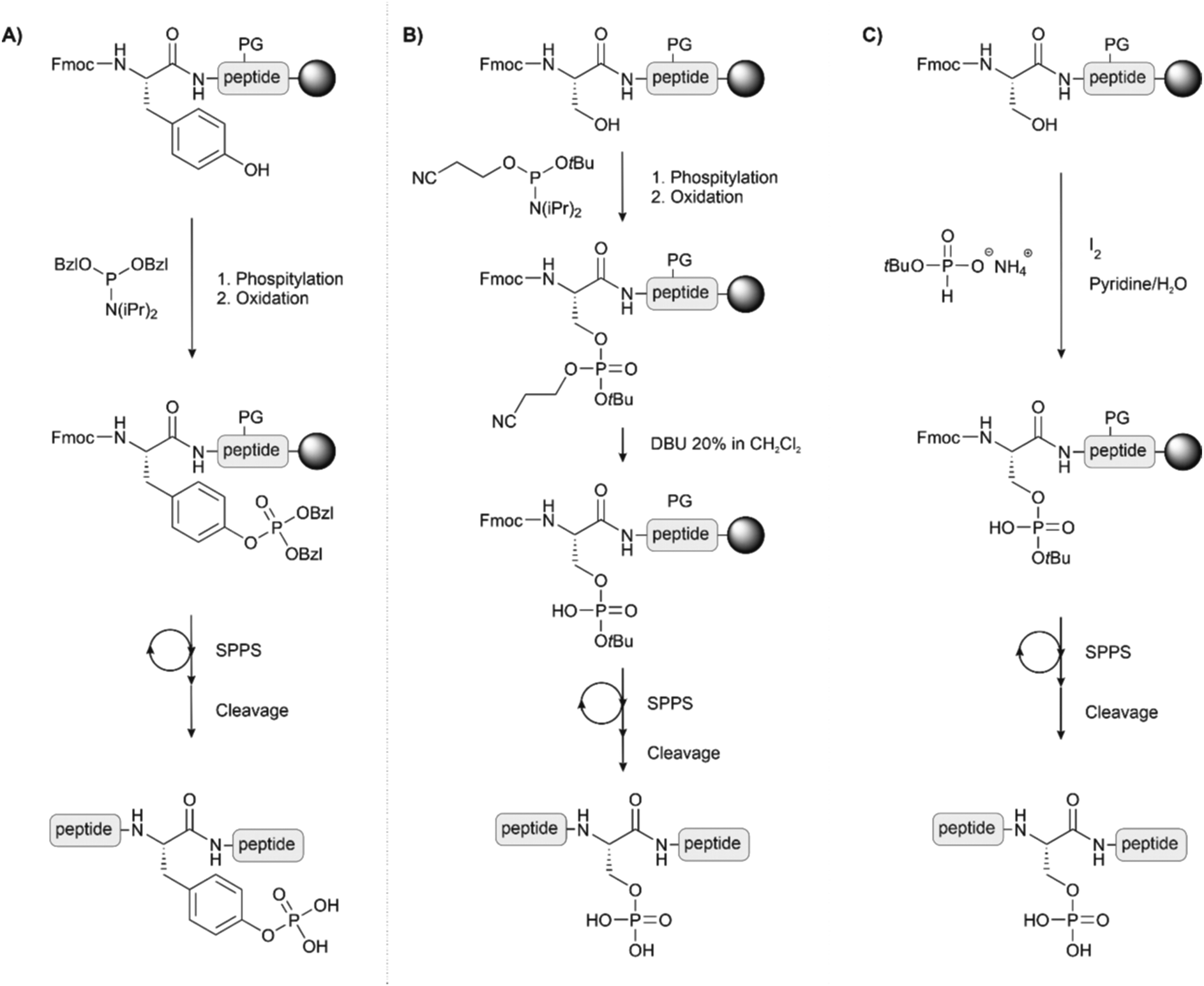 | ||
| Fig. 7 Synthetic schemes for on-line phosphorylation (A) of tyrosine, (B) of Ser or Thr by using the cyanoethyl protected phosphoramidite as phosphitylating agent and (C) of Ser, Thr or Tyr by the H-phosphonate method. PG: protecting group. | ||
Some peptides or peptide segments tend to form inter- or intramolecular aggregates, which form in the protected form on the solid support and in solution. Such difficult sequences are poorly solvated and access to functional groups is hindered. Partial remedy is provided by substituting the backbone amide protons to perturb the H-bond networks that drive aggregation. Johnson et al.80 successfully used the N-(2-hydroxy-4-methoxybenzyl) (Hmb) group as a backbone amide-protecting group in phosphopeptide synthesis (Fig. 8A). The implementation of Hmb protection comes at the price of additional steps required for blocking the Hmb hydroxyl group (with Alloc or Ac) prior to phosphorylation and deprotection before peptide cleavage. Additional steps are not necessary with backbone protection by the N-2,4,6-trimethoxybenzyl (Tmob) group (Fig. 8B).81 The two groups are removed from the backbone of the peptide during the standard TFA cleavage.
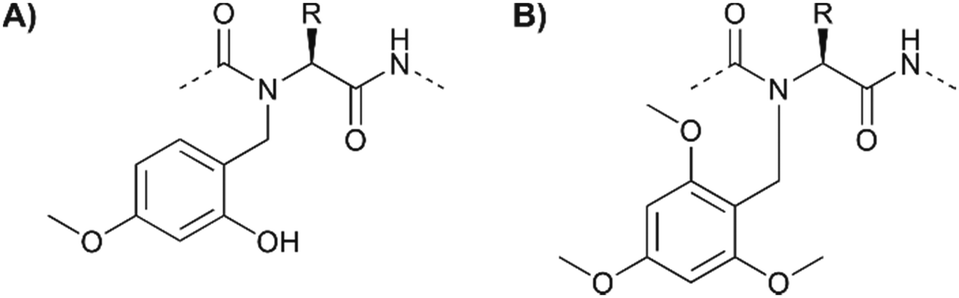 | ||
| Fig. 8 The (A) Hmb and (B) Tmob groups have been used as backbone protecting groups to reduce the aggregation of peptides and to increase the availability of serine for on-resin phosphorylation. | ||
2.3 Multiphosphorylated peptides
While the synthesis of peptides bearing only a few phosphorylation sites has become almost routine, the synthesis of multiphosphorylated peptides still presents a formidable challenge as failed couplings and side reactions accumulate with the increasing number of phosphorylated side chains.Samarasimhareddy et al.82 applied the building blocks Fmoc-Ser(HPO3Bzl)-OH and Fmoc-Thr(HPO3Bzl)-OH in the synthesis of multiphosphorylated peptide 18-mers derived from the C-terminal domain of rhodopsin. To improve coupling yields, microwave heating up to 75 °C was applied during coupling while Fmoc deprotection was performed at room temperature to minimize β-elimination (Fig. 3). The authors carefully analyzed yields after each coupling step. They found that the introduction of the first three pSer or pThr residues proceeded smoothly by using HATU as the activator in the presence of DIEA. Double couplings and extended reaction times were required for the third and fourth pSer/pThr. To achieve the introduction of the fifth and sixth phosphorylated building blocks, a higher excess of the phosphorylated building block was required in addition to double couplings. Recently, the same group used a glycan synthesiser for the automated synthesis of heavily phosphorylated peptides. More efficient control of the temperature, in particular in the Fmoc-deprotection step, allowed the synthesis of peptides bearing up to nine clustered phosphorylations in reasonable yield and purity.83 Works from Becker and Geyer demonstrated that global phosphorylation could provide access to multiphosphorylated peptides. In their synthesis of silaffin peptides, which play an important role in biomineralization, Lechner and Becker masked serine phosphorylation sites by means of Trt protection.84 Detritylation was accomplished with 1% TFA and 1% TIS in DCM prior to phosphitylation with iPr2NP(OBzl)2. The approach afforded 20mer peptides containing up to 7 pSer residues, though the yield of material purified via HPLC and ion-exchange chromatography was low. Geyer and colleagues76 relied on the use of Fmoc-Ser(TBDMS)-OH, and treatment with Bu4NF in THF was used to liberate sites subsequently targeted by phosphitylation with iPr2NP(OBzl)2. Mono-, tri- and hepta-phosphorylated silaffin peptides were used in crude form in biomineralization tests.
2.4 Special application phosphate protecting groups
For some applications in cell biological experiments, the standard phosphate protecting groups are not suitable. Photocleavable and enzyme-labile phosphate protecting groups provide opportunities for time-controlled release and cellular delivery, respectively, of phosphopeptides in biological assays.The Imperiali group developed a method to protect the phosphate on Ser, Thr and Tyr with the 1-(2-nitrophenyl)ethyl cage, which enabled light-controlled release of the phosphate group (Table 1, entry 6). The synthesis of the caged phosphopeptides succeeded by using on-line phosphorylation or pre-synthesised phosphotriester building blocks featuring a cyanoethyl protecting group, which is cleaved upon Fmoc removal.33 The protecting group was removed with UV light (365 nm) once the phosphorylated peptide was delivered into cells. The ligand peptide was attached to a cell internalization sequence from the third helix of the Antennapedia homeodomain, a well-known cell-penetrating peptide,31via a disulfide bridge that cleaves intracellular to release the ligand. Caging of phosphopeptides with the nitrophenethyl group was also used by Muir and co-workers. They employed global phosphorylation with O-1-(2-nitrophenyl)ethyl-O′-β-cyanoethyl-N,N-diisopropyl-phosphoramidite to prepare a pentapeptide containing two caged phosphoserines.32 The peptide was used for the semisynthesis of Smad2 by expressed protein ligation and subsequent biological studies (see Section 4.1). Irradiation with UV light can be toxic to cells. To enable uncaging at higher wavelengths, Nagamune and co-workers applied a coumarinylmethyl cage (6-bromo-7-hydroxycoumarin-4-ylmethyl derivative, Bhc (Table 1, entry 7).34 A phosphotriester tyrosine building block allowed the solid-phase synthesis of nonapeptides, which can bind the SH2 domain of phosphatidylinositol 3-kinase (PI3K). After microinjection into cells, uncaging was performed by one-photon UV or two-photon IR excitation.
To improve cellular delivery of phosphopeptides the negative charges of the phosphate have been masked with enzyme-labile protecting groups. Burke and coworkers35 identified the pivaloyloxymethyl (POM) moiety as an esterase cleavable protecting group (Table 1, entry 8) that can be used to protect the phosphate group of pSer and pThr. Fully phospho-protected peptides were synthesized by coupling the building block Fmoc-Thr[PO(OH)(OPOM)]-OH and, prior to cleavage, “POMylation” of the free phosphoric acid group with iodomethylpivalate (POMI) and DIEA was performed in order to mask the negative charge. Adopting methods developed for mononucleotide prodrugs,85 Imbach and colleagues installed S-acyl-2-thioethyl (SATE) groups on phosphotyrosine.36 The bis(S-pivaloyl-2-thioethyl)-protected (bis(tBuSATE)) phosphotyrosine was used in the solution-phase synthesis of a Leu-enkephalinamide derivative with increased stability to cleavage by leucine aminopeptidase (Table 1, entry 9).36 Garbay and co-workers employed the building block approach to include S-acetyl-2-thioethyl (MeSATE) protection in the synthesis of membrane permeability-improved peptides containing phosphotyrosine or phosphotyrosine mimics (see Section 2.7) targeting the SH2 domain of the adapter protein Grb2.37 It was discussed that esterases remove the tBuSATE acyl group in vivo.86 The major drawback is the instability in solutions containing more than 50% of TFA and mono dealkylation of the phosphate has been observed during standard Fmoc-deprotection procedure. The use of 2% DBU in DCM solved the latter problem.37
2.5 Phosphohistidine
Phosphohistidine presents unique challenges as a phosphorylated residue. Two isomers exist (Fig. 9A) and the P–N bond is vulnerable to acids.87 Typical SPPS conditions and biochemical isolation techniques are, therefore, incompatible with phosphohistidine. Given the difficulties in isolating proteins phosphorylated at histidine, chemical methods have enabled investigation through synthetic phosphorylation and stable analogues. Potassium phosphoramidate has been applied for the chemical phosphorylation of histidine residues (Fig. 9B). Medzihradszky et al.88 prepared histidine-phosphorylated peptides by treatment of unprotected peptides spanning a sequence of a human tyrosine phosphatase. Attwood et al.89 globally phosphorylated two histidines on two peptides deriving from histone H4. The reagent is mild enough to phosphorylate histidine even in the presence of unprotected Ser, Thr and Tyr. Hohenester et al.90 used the same method to successfully modify the histidines of myoglobin (containing 11 His residues) and other proteins (Fig. 9B). However, this technique is not specific and risks phosphorylating other nucleophilic residues like lysine. Furthermore, phosphorylation occurs preferentially on the 3-position, and the undesired di-phosphorylated product is also formed in low amounts. The phosphorylation on the 3-position is also more stable.91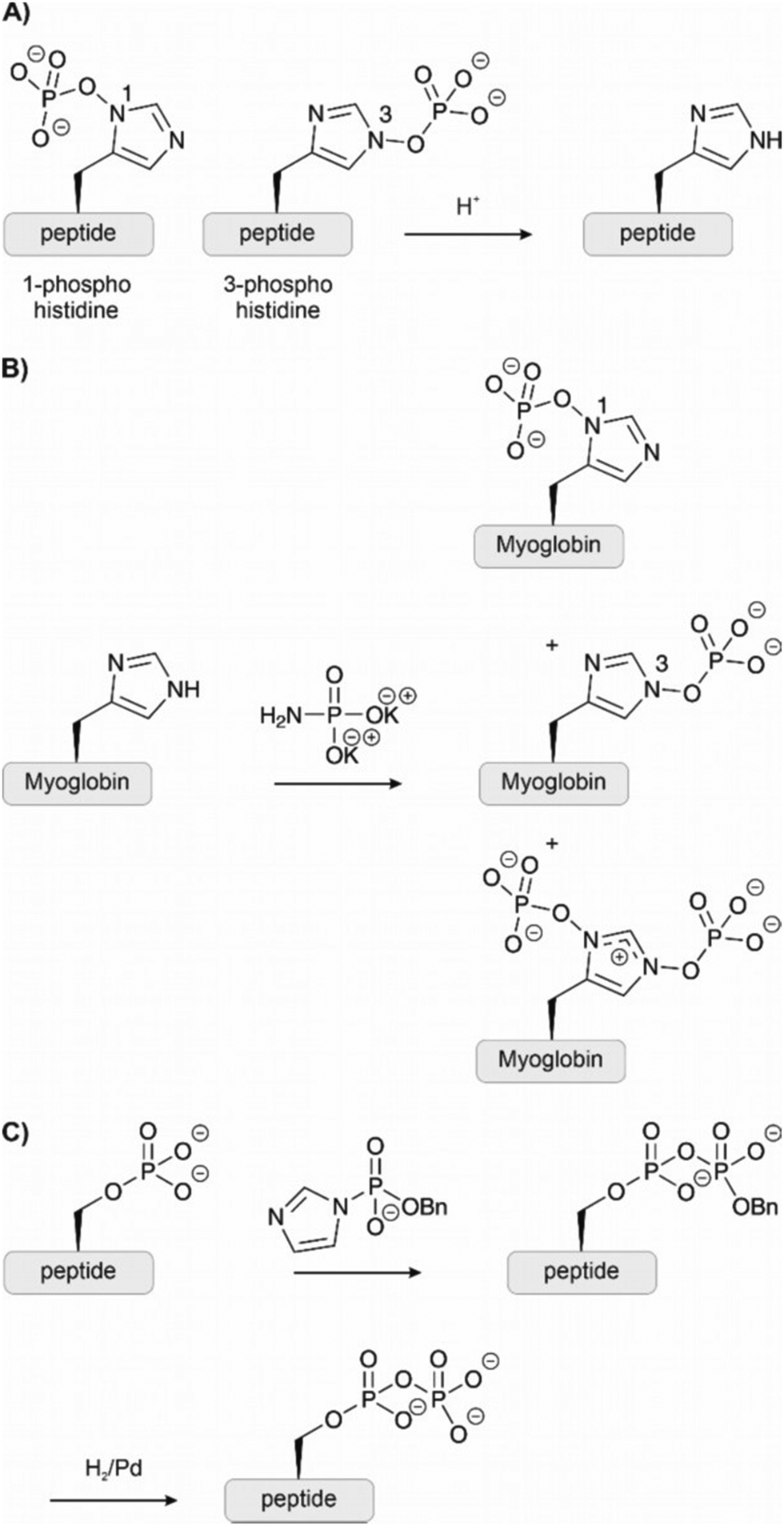 | ||
| Fig. 9 (A) The amino acid histidine can be phosphorylated on both nitrogens of the imidazole ring, forming two different isomers also known as π-phosphohistidine and τ-phosphohistidine respectively. Under acidic conditions, they are prone to hydrolysis. (B) Potassium phosphoroamidate has been used to phosphorylate histidine residues on proteins chemically. However, the reaction leads to multiple products and is not selective. (C) Using the benzyl-protected phosphorimidazolide, phosphorylated residues could be selectively converted to pyrophosphates. | ||
2.6 Pyrophosphorylation
Knowledge about pyrophosphorylation has been limited due to a lack of suitable tools and methodologies to isolate or produce pyrophosphorylated proteins. The consequences of pyrophosphorylation on a protein are, therefore, still unclear. Fiedler and co-workers92–94 have developed new reagents to access pyrophosphorylated peptides and proteins. Monobenzyl-protected phosphoric acid imidazolides react specifically with phosphorylated residues (Fig. 9C). The reaction has been performed on unprotected peptides in dimethylacetamide or water and provided access to pyrophospho-ubiquitin and myoglobin. Although pyrophosphorylation is most common on serine, the method has been applied also to peptides containing pThr and pTyr.2.7 Phosphate analogues and mimicking
Analogues of phosphates can be essential to obtain information about the impact of phosphorylation when it is difficult to obtain an authentic phosphorylated sample by other means. Often phosphate mimicking groups are employed in order to overcome dephosphorylation by phosphatases. Phosphonates, where the phosphate–alcohol bridgehead oxygen is replaced with a methylene bridge, are commonly used analogues of phosphoamino acids (see Table 1, entries 10 and 11). The C–P bond is not vulnerable to phosphatases. Phosphono derivatives have been described for serine, threonine, and tyrosine, but only the latter is commercially available. Despite the structural similarity to an authentic phosphate, they do not always accurately represent a phosphorylated residue. For example, the phosphonomethyl phenylalanine (Pmp, Table 1, entry 10) analogue has a pKa 2 of 7.72 compared to the pKa 2 of 6.22 for phosphotyrosine, which results in a different charge at neutral pH. The difluorophosphonomethyl phenylalanine (F2Pmp, Table 1, entry 11) analogue is more similar because the phosphonic acid has a pKa 2 of 5.71 and the fluorine atoms are available for hydrogen bonding.39,40 These analogues have been used in Fmoc SPPS and genetic code expansion. For example, Lu et al. applied the Fmoc-protected Pmp building block to investigate the phosphorylation of a phosphatase.38 Rogerson et al. used 2-amino-4-phosphonobutyric acid, a phosphoserine analogue, in amber codon suppression to generate a constitutively active version of the kinase Nek7.95 Mann et al.96 synthesized a dibenzyl protected serine phosphonate building block, which they used in the synthesis of a stable phosphoubiquitin probe. This building block overcame the typical problems of monobenzyl-protected building blocks (see Section 2.1.1) and had a higher similarity to a phosphoserine residue than commonly used glutamate or aspartate mimics. The phosphoubiquitin probe was used to measure ubiquitin conjugation in mitophagy (see Section 4.4).p-Carboxymethyl-L-phenylalanine was chosen as a phosphatase resistant phosphotyrosine analogue.97 The residue was incorporated into a fragment of the DNA binding domain of STAT1 through genetic code expansion. Using this substitution, a constitutively active mutant of STAT1 was created, which dimerized and bound DNA in the same way as pY701.
Methods that allow the introduction of phosphate-analogous structures on fully assembled peptides are particularly useful. Many of the methods hinge on the use of dehydroalanine (DHA), which has a unique reactivity as an electrophile that is not found among the proteinogenic amino acids. DHA can be generated chemically from cysteine or phosphoserine. Amongst the many methods available for converting cysteine to DHA,98 a two-step protocol involving a bisalkylation-elimination sequence showed high chemoselectivity (Fig. 10A). In the first step, the Cys side chain is monoalkylated upon treatment with a bis-electrophile such as 2,5-dibromohexanediacetamide (DBHDA).99 Under optimal pH, other potentially nucleophilic amino acids are either protected by protonation or not reactive enough to compete with Cys. Gentle heating to 37 °C triggers the second step, which involves an intramolecular attack of the remaining electrophilic function followed by elimination from the formed sulfonium ion. Bernandes et al.100 applied oxidative elimination using O-mesitylenesulfonylhydroxylamine (MSH) to the same end (Fig. 10A). An alternative route to DHA is provided by β-elimination of phosphoserine.101 As discussed earlier (see Section 2.1.1), this common side reaction during SPPS is here exploited to obtain a unique site for modification. The phosphoserine residue was introduced through genetic code expansion and treated with a mild base to form DHA. However, cysteine is a more suitable DHA precursor because it is easier to introduce a point mutation in recombinant expression than incorporating a phosphoserine or DHA with genetic code expansion. Once installed, a DHA unit can serve diverse modification reactions. In an impressive feat, carbon free-radical chemistry was applied on proteins under biocompatible conditions to form carbon-carbon bonds upon treatment of DHA-containing proteins with iodomethylphosphonic acid derivatives (Fig. 10A, middle right).102 The α-C radical formed upon radical addition onto DHA was quenched with NaBH4. The reactions were performed in a glove box. The method enabled the synthesis of the histone protein H3 carrying phosphonic acid modifications on serine 10.
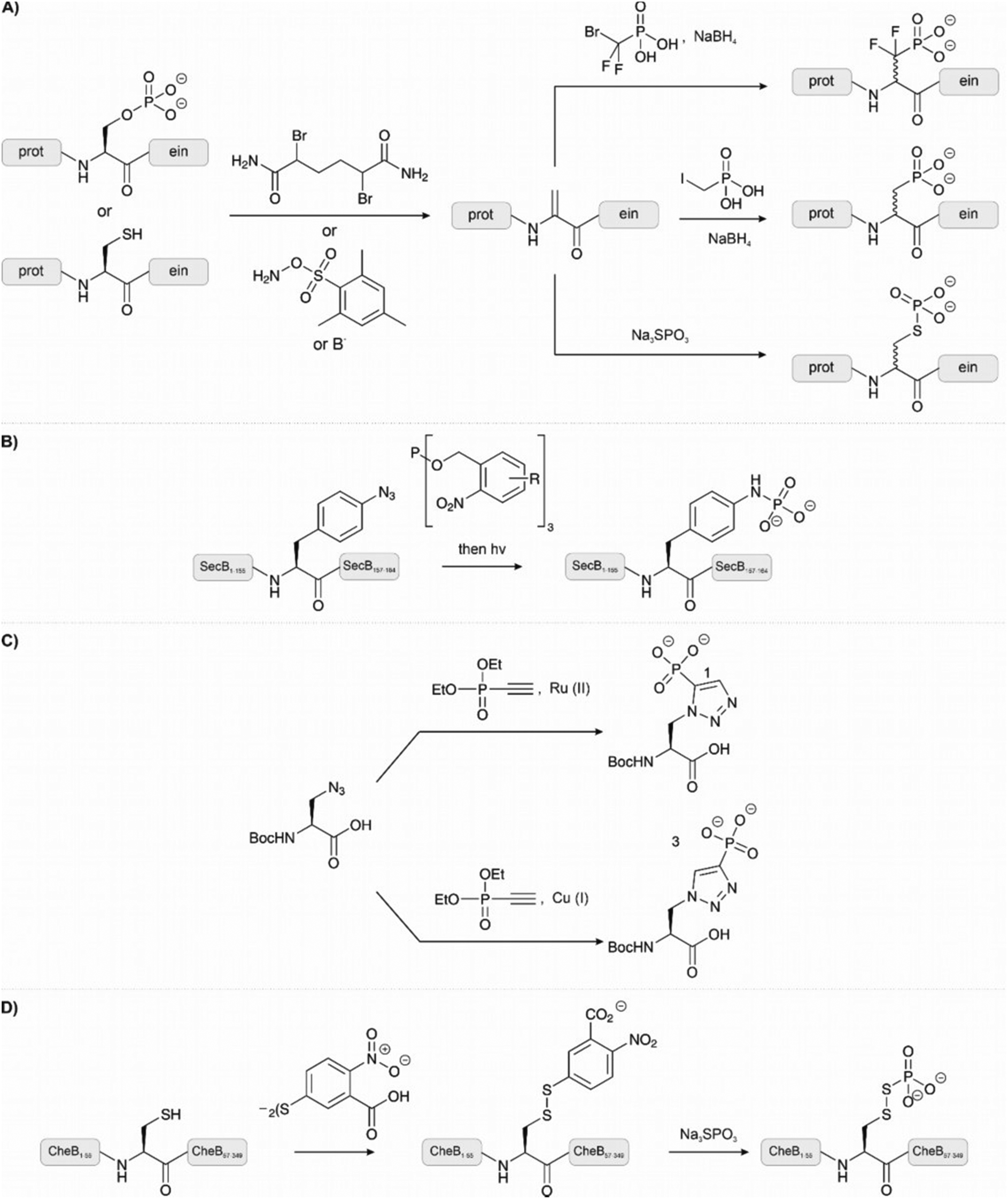 | ||
| Fig. 10 Methods for chemically modifying unprotected peptides and proteins to introduce phosphate groups and their analogues. (A) Dehydroalanine can be formed via elimination. Cysteine can be converted to dehydroalanine through alkylation and elimination or oxidative elimination. Phosphoserine undergoes elimination by treatment with a base to form dehydroalanine. From dehydroalanine, a carbon-carbon bond was formed on a protein to afford the phosphonate and difluorophosphonate homologues of serine/threonine. Alternatively, phosphothioates can be formed by the treatment of dehydroalanine with sodium thiophosphate. (B) The Staudinger-phosphite reaction enabled selective reaction at azido-phenylalanine to introduce a phosphotyrosine mimic. (C) A stable phosphohistidine mimic was formed by reacting an alkyne-phosphonate with azidolysine. (D) To introduce a stable phosphoaspartate mimic, a cysteine residue is treated with Ellman's reagent followed by incubation of the formed disulphide with sodium thiophosphate. | ||
The reactivity of DHA as a Michael acceptor can be exploited to introduce thiol modifications. A common reagent to introduce a phosphate analogue is sodium thiophosphate (Fig. 10A, lower right). To exemplify the reaction, a DHA residue was installed at the serine protease mutant subtilisin Bacillus lentus S156C and treated with sodium thiophosphate to generate the phosphorylated protein.100 Notably, the method was orthogonal in the presence of methionine and also reversible through a second elimination. The same reagent was used to generate a phosphothreonine mimic in the activation loop of protein kinase p38α.99 One downside of this method, though, is that it forms diastereomers.
To install a phosphatase-stable phosphoramidate analogue of phosphotyrosine Serwa et al.103 used a Staudinger-phosphite reaction (Fig. 10B). In this reaction, an azide reacts with a phosphite to form a phosphorimidate, which is hydrolysed to a phosphoramidate. As a proof of concept, a synthetic peptide bearing an N-terminal p-azido-phenylalanine was treated with the water-soluble phosphite and deprotected with light to afford the phosphoramidate. The reaction proceeded in aqueous buffers at physiological pH and did not require the exclusion of air. As an example, the 17 kDa protein SecB was synthesized. The unnatural p-azido-phenylalanine residue was introduced through genetic code expansion and treated with the phosphite to afford the same phosphorylation mimic. Anti-phosphotyrosine antibodies recognised the analogue. The same reaction was also applied to form phospholysine from ε-azido lysine.104 Here, the reaction was demonstrated on peptides synthesized with the Fmoc-ε-azido lysine building block. These examples highlight a straightforward reaction that can be used to generate site-specifically phosphorylated peptides and proteins, however, the use of genetic code expansion may limit its widespread adoption. The use of these building blocks in the context of SPPS, as in the ε-azido lysine example, is a more accessible technique.
Reactions on azides have also been used for the synthesis of phosphohistidine mimetics.105 An azide-alkyne click reaction between azidoalanine and an alkyne-phosphonate affords a non-hydrolysable and non-isomerising phosphohistidine analogue (Fig. 10C). The use of copper or ruthenium salts in the click reaction can change the regioselectivity of the reaction and preferentially form analogues of either 1-pHis or 3-pHis. A short tail of histone H4 was synthesized using the Boc-protected building block in SPPS. Subsequent native chemical ligation chemistry furnished a full-length H4 protein carrying the phosphohistidine analogue at position 18.
Phosphoaspartate is a particularly unstable modification. In a study by Saxl et al.,106 a modified cysteine residue was used to emulate phosphoaspartate in phosphorylated bacterial methylesterase CheB (Fig. 10D). The cysteinyl-thiophosphate was introduced through oxidation to a disulfide with Ellman's reagent, followed by thiol exchange with sodium thiophosphate. The phosphorylated disulfide should mimic the distance, flexibility, and charge of phosphoaspartate while enabling reversible modification.
3 Synthesis of phosphoproteins
3.1 Enzymatic synthesis
Historically, phosphorylated peptides and proteins have been prepared by the in vitro treatment of isolated protein with a purified kinase. For example, phosphorylation sites in the T-cell protein LAT were investigated using the purified kinases Zap-70, Lck, and Syk.107 Peptide sequencing revealed that Zap-70 phosphorylated LAT at five different tyrosine residues. Subsequently, the phosphoproteins were used to measure the interaction and activation of downstream proteins. This work revealed the importance of specific LAT tyrosine phosphorylations in recruiting signalling proteins for T-cell activation and PLCγ1 and Ras signalling pathway control.However, this method is limited in applicability. Often the target is not a substrate of a known kinase, nor is a kinase available to target the desired site. Besides, even when a kinase has been identified, its phosphorylation activity may be promiscuous, or the reaction does not go to completion.
When an authentic phosphorylated sample cannot be obtained, biologists often resort to phosphate mimicking. Point mutations are introduced at the site of interest by means of site-directed mutagenesis, for example, to replace phosphoserine or phosphothreonine with glutamate or aspartate. However, glutamate/aspartate residues are poor mimics of authentic phosphorylation: neither the size nor charge of a phosphate group is accurately emulated. Nonetheless, phosphate mimicking is still commonly employed. However, because mimicking is applied in situations where authentic samples cannot be obtained, the mimic cannot be compared with authentic phosphorylation. It seems that this lack of validation experiments has contributed to the persistence of the approach. Where comparisons have been made, the mimic often failed to replicate the properties of a real phosphate group. In one example, the biophysical properties of α-synuclein-pS129 were compared with α-synuclein(S129E/D) due to conflicting results in the literature.108 The authentic phosphorylated material was obtained from kinase phosphorylation. Importantly, α-synuclein(S129E/D) did not emulate the structural nor aggregation properties of authentically phosphorylated α-synuclein.
Another example compared the non-hydrolysable phosphonomethylenealanine (Pma, see Table 1) with a phosphothreonine to glutamate substitution in semisynthetic serotonin N-acetyltransferase.109 The interaction with a 14-3-3 protein, which recognizes a specific phosphorylated motif, was measured and no difference was found between the unphosphorylated and glutamate substituted proteins, whereas a high binding was observed for the Pma isostere. Additionally, in the synthesis of phospho-Akt1, the substitution of pT-308 with acidic residues, E or D, did not activate the kinase to the same extent as an authentic phosphate, nor did an alanine substitution sufficiently mimic unphosphorylated threonine.110
Nevertheless, there are cases where mimicking has been successfully applied. For example, Pasapera et al.111 used phosphate mimicking to examine the role of phosphorylated paxillin in focal adhesions. They expressed a phosphorylated Y31E, Y118E mutant and a Y31, Y118F mutant to represent phosphorylated and unphosphorylated paxillin respectively. The psuedophosphorylated protein was able to induce the recruitment of vinculin.
It is important to be aware of these cases when interpreting results obtained through phosphomimicking, especially when no comparison with an authentic phosphate, or isostere, is provided.
3.2 Genetic code expansion
Genetic code expansion enables the use of unnatural building blocks for ribosomal protein synthesis. In brief, expanding the genetic code requires three components: an unassigned codon, a tRNA that recognises the codon, and an aminoacyl-tRNA synthetase that recognises the amino acid and is orthogonal to other synthetase/tRNA pairs in the host. The amber stop codon, UAG, is the most frequently used unassigned codon because in E. coli it occurs rarely and is often ignored. Cell-free, bacterial and animal cell systems have been developed. The approach potentially provides a powerful tool for the in vivo investigation of phosphorylated proteins. Systems have been developed for the incorporation of phosphoserine,112 phosphothreonine,113 phosphotyrosine114 and their analogues. The field has been well-reviewed, including for incorporation of post-translational modifications.115,116 A key area absent from the literature is genetic code expansion methods for non-canonical phosphorylated amino acids. This is likely due to the lability of the N-, S- and acyl phosphate groups. Despite the power of genetic code expansion methods, there are inherent limitations. Firstly, reading of the amber codon is intrinsically inefficient. As the stop codon is usually a termination signal, tRNAs compete with release factors for the binding site, leading to poor yields. Secondly, natural amino acids may be incorporated at the site due to incorrect loading of tRNAs because of the promiscuity of aminoacyl-tRNA synthetases. Finally, given the limited number of unassigned codons, there are few opportunities to incorporate more than one unnatural amino acid into a protein.3.3 Chemical synthesis of phosphoproteins
Chemical synthesis can access virtually any protein phosphoform in arbitrary combinations with modifications that are not available by biosynthetic methods. Total chemical peptide synthesis has enabled the production of high purity compounds in amounts sufficient for research and clinical use.117 However, despite the development of new coupling reagents, backbone protection methods118,119 and microwave120 and flow121 syntheses, the efficiency of solid-phase peptide synthesis is rarely sufficient to provide pure proteins of a size that allows folding into a specific tertiary structure. Modern ligation techniques, however, can bypass the length restrictions of solid-phase peptide synthesis.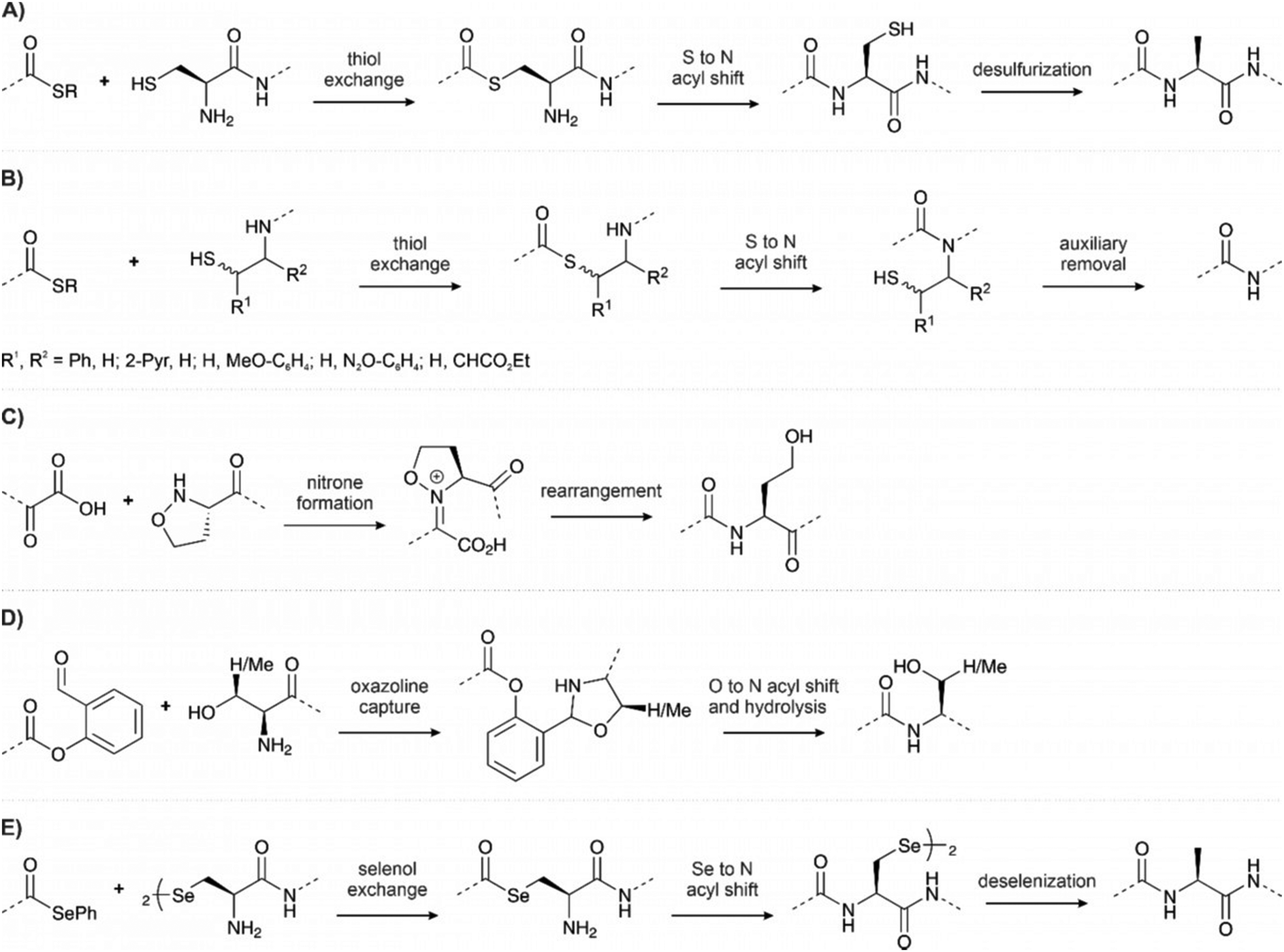 | ||
| Fig. 11 Ligation methods used in chemical protein synthesis (A) native chemical ligation, with the option of desulfurization. (B) Auxiliary-mediated ligation, followed by auxiliary removal. (C) KAHA ligation with a generic α-hydroxyacid and 5-oxaproline. (D) Serine-threonine ligation. (E) Diselenide selenoester ligation, followed by deselenization. | ||
The peptide thioester (or selenoester) is a key component of an NCL reaction. It is important to consider the chemical lability of phosphate-bearing thioesters. On the one hand, thioesters typically do not withstand the conditions applied for Fmoc removal, while, on the other hand, PTMs like phosphorylation or glycosylation are sensitive to the strong acids, like HF, used for detachment and global deprotection.125–127 With the mainstream use of Fmoc-based solid-phase peptide synthesis, researchers developed alternative strategies for the generation of thioesters.128,129
Kenner's “safety catch” resin, an N-acylsulfonamide linker, found wide use early on due to its stability under Fmoc SPPS conditions130–132 (Fig. 12A). After completing solid-phase assembly, alkylation with (trimethylsilyl)diazomethane, iodoacetonitrile or β-mercaptotriisopropylsilylethanol activates the acyl sulfonamide to enable the release of fully protected peptide thioesters upon nucleophilic attack with a thiol. Final treatment with TFA affords unprotected peptide thioesters. The Muir lab used this method for the semisynthesis of hyperphosphorylated TβR-I to create a tetra-phosphorylated peptide thioester.133 Alkylation was performed using iodoacetonitrile instead of the commonly used (trimethylsilyl)diazomethane to avoid O-methylation of the mono-benzyl protected pSer and pThr. They noticed a major side product, which was identified as cyanomethylated homocysteine formed upon the reaction of methionine with ICH2CN.134 In this case, the problem was solved by substituting the methionine with the isostere, norleucine. Mende et al. developed an improved sulfonamide “safety catch” linker, which enabled the selective detachment of full-length peptides while truncation products remained on the solid support.135 Later, this “self-purification” method was applied in our laboratory to synthesise single and multi tyrosine-phosphorylated forms of the SH3 domain (see Section 4.1).136
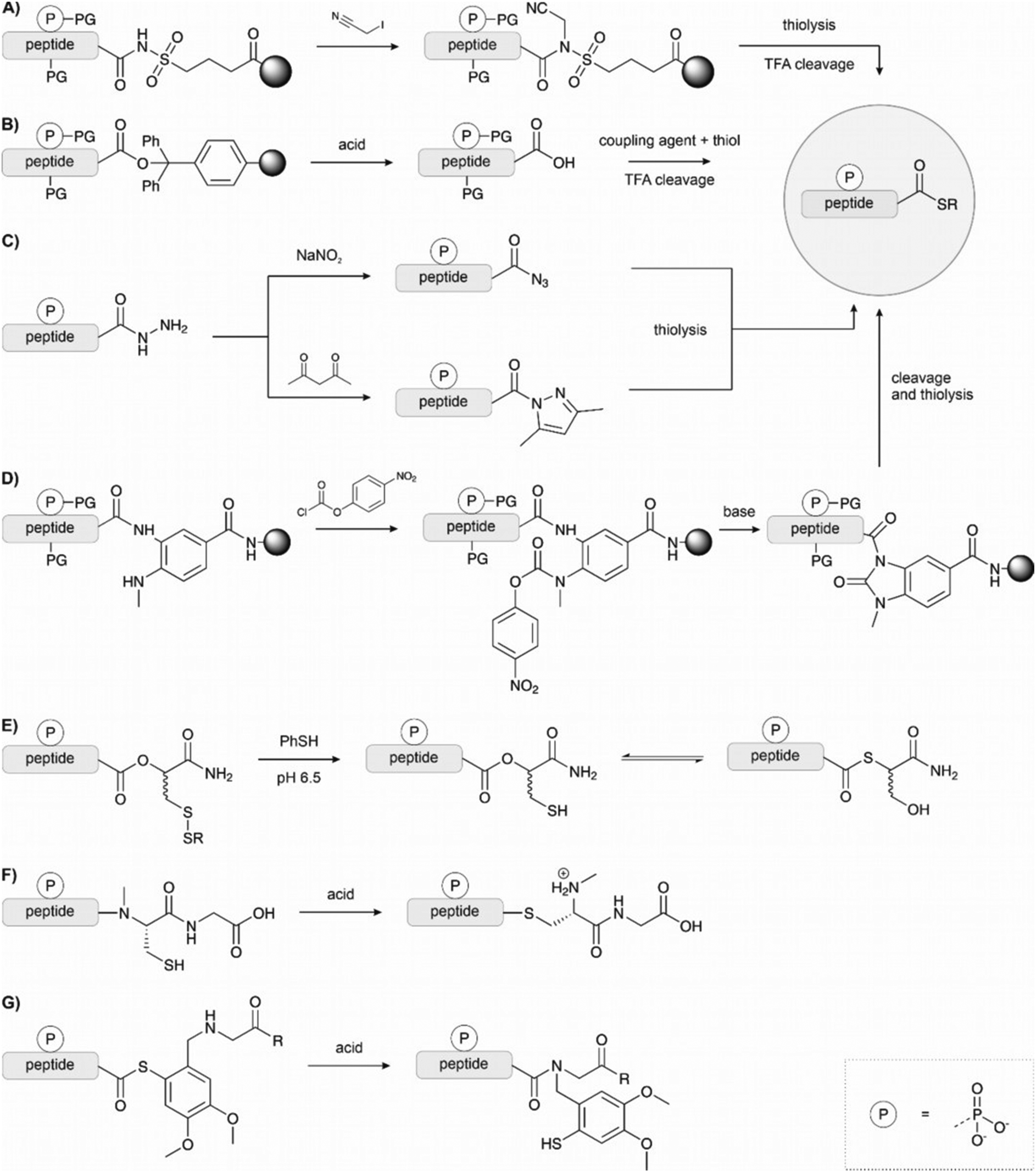 | ||
| Fig. 12 Strategies for the synthesis of peptide thioesters via Fmoc SPPS. (A) Kenner's safety catch linker relies on activation by alkylation of the N-acylsulfonamide. (B) Peptide synthesis on an acid-labile resin and thioesterification in solution. The protected peptide is cleaved from the resin with dilute TFA or a weak acid. (C) Hydrazides as thioester surrogates. The hydrazide is activated either by oxidation with sodium nitrite or the formation of the Knorr pyrazole. (D) Dawson's (Me)Dbz linker or N-acyl urea method; (E) O → S acyl shift using Botti's linker. F, (G) N → S acyl shift exploiting the reactivity of an alkylated amide, either methylated or bearing methoxybenzyl auxiliary. | ||
Solid-phase synthesis on hyper acid-labile linkers such as Trt, 2-Cl-Trt and HMPB allows cleavage of the fully protected peptide from the resin with a mild acid and the generation of peptide thioesters via activation in solution (Fig. 12B).125,137,138 However, the low solubility of protected peptides and epimerization of the C-terminal amino acid are significant drawbacks of this method.
Thioester surrogates are particularly valuable because they can be activated on-demand to form the thioester, are often easier to handle, and do not suffer from epimerization or solubility issues. Liu and coworkers introduced the peptide hydrazide method (Fig. 12C).139–141 Peptide hydrazides do not participate in NCL chemistry. However, treatment with sodium nitrite in acidic solution leads to peptidyl azides that react with mercaptans to form peptide thioesters. Liu's method was used to synthesise site-specifically phosphorylated PDZ domain of PSD-95 by Stromgard and colleagues.142 Recently, Dawson established a new hydrazide-to-thioester conversion.144 Acetoacetone induces the formation of an N-acylpyrazole, which undergoes thiolysis upon treatment with thiols. This method has also been applied to generate selenoesters.145
Blanco-Canosa and Dawson developed a diaminobenzoic acid-based linker (Dbz),146 and later on the improved, methylated version MeDbz147 (Fig. 12D). Synthesis proceeds on the more reactive amine group. After completion of the peptide chain assembly, treatment with p-nitrophenyl chloroformate affords a p-nitrophenyl urethane that is cyclized under basic conditions. Following cleavage from the resin,the newly formed C-terminal N-acyl urea then undergoes thiolysis to form a thioester. Very reactive amino acids such as glycine can react with the p-amino group of the Dbz linker, preventing cyclization later. The MeDbz linker was developed to overcome this limitation. Dawson's chemistry (also called the Nbz/MeNbz method) has found wide usage and has been used, for example, by Zhan for the synthesis of a phosphorylated fragment of MDM2,148 and by Muir for the semisynthesis of modified histone H2B.149 The same linker can also be activated with sodium nitrite to form the N-acyl benzotriazole, which acts as a good leaving group and can be thiolysed to form a thioester. This can be utilised in a similar manner to hydrazides, as a latent thioester that can be activated on demand.150 Cistrone et al.143 summarized the protocols for the native chemical ligation with the Nbz and hydrazide methods.
Several methods rely on N- or O-to-S acyl shift reactions to form a thioester.151 These methods make use of the spontaneous rearrangement initiated by the attack of a nearby thiol at the C-terminal ester or alkylated amide. Botti et al. developed a latent thioester linker that exploits the O → S shift to form a reactive thioester (Fig. 12E).152 There, the peptide is built on an ester linkage with a thiol in the β-position protected as a disulfide. Under the reducing, slightly acidic conditions of the ligation mixture, the thiol is freed, undergoes an acyl shift to form the thioester and then participates in ligation. Muir also applied this linker in the synthesis of phosphorylated H2B.153
Even though the equilibrium typically favours the N-acyl form, the balance can be shifted toward the product when an excess of a more reactive thiol intercepts the thioester intermediate. Typically, the thiol remains protected throughout the synthesis, and its deprotection triggers the rearrangement, which occurs at acidic pH. The N → S acyl shift method has been frequently used in protein synthesis, but applications in the synthesis of phosphopeptides are rare (Fig. 12F and G). For example, Aimoto and coworkers used a thiol-bearing auxiliary at the C terminus to form pSer containing thioester peptide derived from histone H3.154 Already in 2002, Vorherr and coworkers observed that acid treatment of peptides containing an amide-linked dimethoxy-mercaptobenzyl group allows the formation of thioesters.155 Aimoto et al. attached the dimethoxy-mercaptobenzyl group to an alanine residue by reductive alkylation. Then the first amino acid of the target sequence was coupled to this building block. After completion of the chain assembly, the peptide was cleaved from the resin upon treatment with aqueous TFA and TCEP, which triggered the N to S acyl shift and furnished the S-linked peptide, which was intercepted with an alkyl mercaptan. In a more recent example, Jbara et al. prepared the N-terminal fragment of histone H2A using a photocaged N-methyl cysteine linker.156 The 2-nitrobenzyl protected N-methyl cysteine was loaded onto the resin, and SPPS was performed as normal. Following TFA cleavage, UV irradiation and treatment with 3-mercaptopropionic acid under reducing conditions formed the N-terminal thioester. This fragment was ligated to the C terminal fragment to form the full H2A (see Section 4.2).
Melnyk and colleagues157,158 have developed a bis(2-sulfanylethyl)amido linker to exploit the N-to-S acyl shift for the synthesis of thioesters. The peptide is synthesized on a resin functionalized with the linker by Fmoc SPPS. Following cleavage from the resin, treatment with TCEP under mildly acidic conditions leads to the formation of the β-amino-thiol thioester. This intermediate can undergo transthioesterification with another thiol to form a reactive thioester. Furthermore, control of the oxidation state of the linker, between the thiol and disulfide forms, acts as an on/off switch and allows use as a latent thioester. This method has not yet been applied with phosphopeptides.
Aimoto and colleagues159–161 pioneered an alternative method in which fragments are joined through direct aminolysis of thioesters in the presence of silver. In this method, nucleophilic side chains must remain protected. Teruya et al.162 successfully applied this method in this synthesis of phosphorylated p53 (see Section 4.4).
Despite its’ broad applicability, NCL is, nevertheless, limited to cysteine-containing junctions. To overcome this limitation, methods have been established that temporarily introduce a surrogate thiol for ligation, such as thiolated amino acids in combination with desulfurization or auxiliary-mediated ligations (Fig. 11A and B).163 In the first technique, a thiol-bearing amino acid is introduced at the N-terminus, which is subsequently desulfurized to afford a native amino acid at the ligation junction.164 This method has been widely applied in phosphoprotein synthesis, primarily with cysteine. The cysteine is temporarily installed for ligation and then desulfurized to afford an alanine at the junction. Compared to cysteine, alanine is more common in protein sequences and allows ligation at more desirable junctions. Initially, desulfurization was accomplished by treatment with metals such as RANEY® nickel.164 The advent of radical-induced desulfurization paved the way to an extension of the methodology to other amino acids.165 A variety of thiolated building blocks have been developed to serve as precursors of amino acids. Some excellent reviews describe the state of the art.163,166–168 For example, in the solid-supported synthesis of phospho-SH3 domains, Zitterbart et al.136 adopted a method introduced by Haase et al.169 and used an unnatural penicillamine residue to afford a valine at the ligation junction (see Section 4.1).
Ligation auxiliaries are thiol-bearing scaffolds that are attached to the N-terminus of the C-terminal ligation fragment.170 A ligation auxiliary can, in theory, enable a ligation at any junction, though in practice, most auxiliaries are limited by the sterics of hindered junctions. As a result, ligations are typically performed at glycine. The majority of ligation auxiliaries developed in the first two decades after the pioneering work from Dawson171 and Kent170 focused on the benzyl type, with substituents enabling removal by photolysis or under acidic conditions. Recent developments in our lab widened the scope of ligation junctions accessible by ligation auxiliaries. The 2-mercapto-2-phenylethyl (MPE) scaffold is not limited to glycine-containing sites.172 Furthermore, cleavage proceeds through a radical-induced fragmentation reaction, which is induced under slightly basic conditions by TCEP in the presence of oxygen.173 Most recently, we introduced the 2-mercapto-2-(pyridin-2-yl)ethyl (MPyE) group, which is the first auxiliary designed to aid ligation by intramolecular base catalysis.174 The MPyE auxiliary enables ligation at sterically hindered junctions, including proline or β-branched amino acids. At the time this review was drafted, auxiliaries have found little use so far in phosphoprotein synthesis. Xu et al. used a dimethoxybenzyl auxiliary to synthesize different phosphoforms of the p62 UBA domain (see Section 4.4) by ligation at a glycine-glycine junction.175 Liu and co-workers used a glycyl-cysteamine auxiliary in the synthesis of phosphorylated di-ubiquitins (see Section 4.4).176
This technique has been successfully applied to the synthesis of phosphoproteins.178 In a notable example, the synthesis of phosphorylated interferon-induced transmembrane protein 3 (IFITM3) from influenza A revealed the compatibility of the phosphate with both the oxidation of the cyanosulfurylide and the acidic ligation conditions. This method also tolerates the presence of high levels of organic solvents, which can be useful for the synthesis of proteins prone to aggregation such as membrane proteins.
:1 AcOH:pyridine) are comparatively harsher than NCL, no side reactions were noted.
Selenoesters can be prepared from protected peptide acids, either in solution or on resin, by treatment with PBu3 and diphenyl diselenide. The SEA linker has also been applied for the synthesis of selenoesters.182 The diselenide fragment can be prepared with PMB-protected selenocysteine or another selenylated amino acid using Fmoc SPPS, which can be deselenized to the native amino acid following ligation.
Currently, native chemical ligation is the most popular ligation tool. There is a wide range of strategies available to prepare thioesters, for the ligation of fragments in a C to N or N to C direction and many extensions of the concept. Nevertheless, the alternatives to native chemical ligation offer a wider choice of possible retrosynthetic disconnections and adaptations for more difficult proteins. Furthermore, the more reactive options such as DSL can produce full proteins much faster and under dilution conditions when solubility is not an issue. However, the application of the methods may be limited since the required building blocks are not yet widely available and sometimes require difficult handling and preparation – though this may change as they gain popularity.
3.4 Semisynthesis
Semisynthesis is one of the most prevalent methods to investigate protein phosphorylation. The ligation of a synthetic fragment with a recombinant segment combines the flexibility of chemical synthesis with the high efficiency of protein biosynthesis.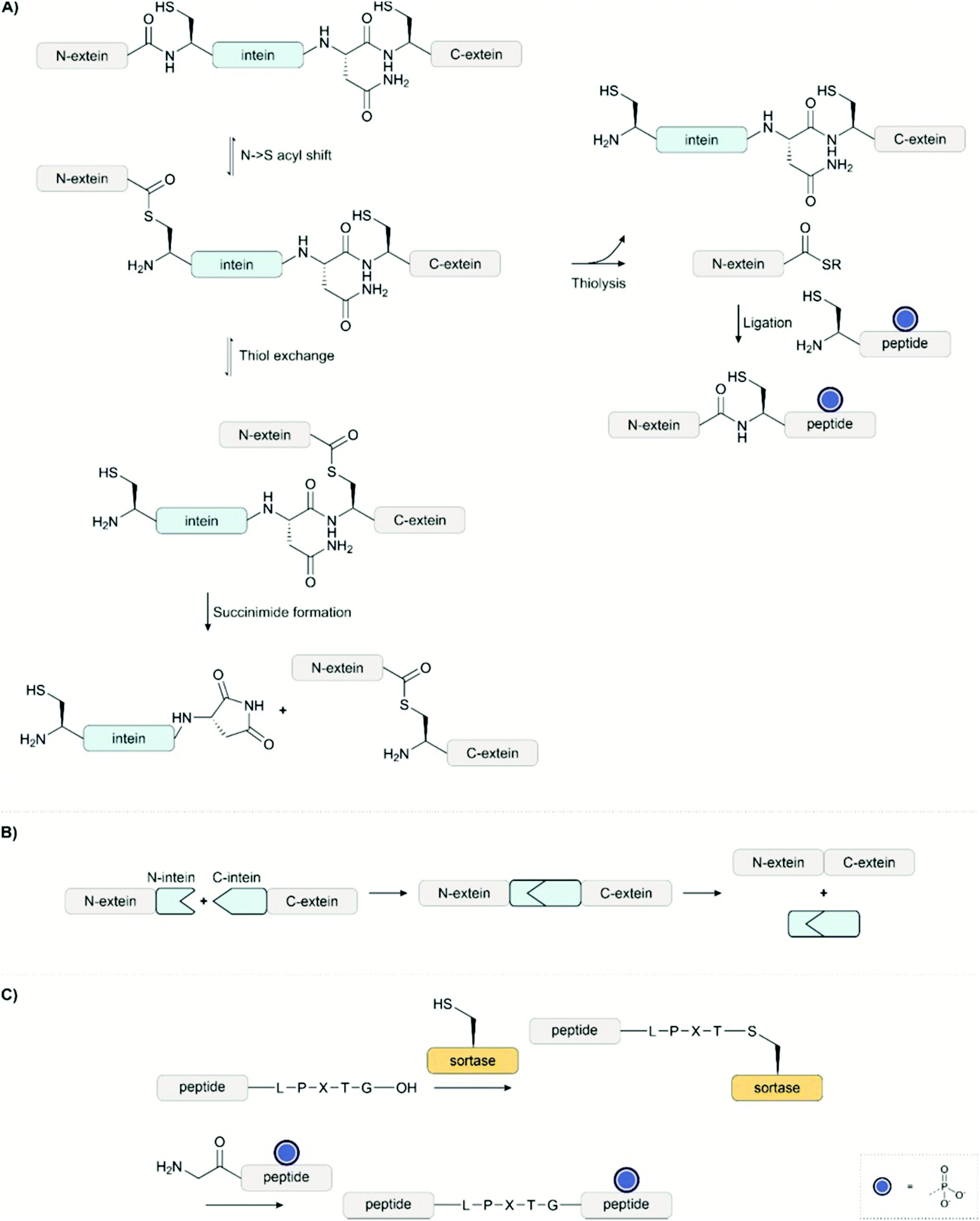 | ||
| Fig. 13 (A) An intein can autocatalytically self-excise itself from a protein. Semisynthetic technologies have taken advantage of this function. Expressed protein ligation, for example, intercepts the thioester in the second step with a reactive thiol in order to ligate a synthetic peptide. (B) In protein trans-splicing, a split intein brings two fragments together. The two fragments fuse and the intein self-excises to afford the exteins joined together by a native amide bond. (C) Sortase enables ligation at the LPXTG tag with an N-terminal glycine-bearing peptide. | ||
A synthetic fragment can also be introduced between two recombinant fragments with multiple ligation steps, though this can be more technically challenging. Muir has recently reviewed the chemoenzymatic synthesis of proteins in detail, including those bearing post-translational modifications.184
Shiraishi et al.186 applied protein trans-splicing to investigate the formation of the phosphorylated β2 adrenoreceptor (β2AR) – β-arrestin 1 complex. Khoo et al.187 used tandem trans-splicing to determine the effect of phosphorylation on the voltage-gated sodium channel, NaV1.5 (see Section 4.5).
The combination of recombinant expression with synthetic peptide synthesis has enabled access to larger and more difficult targets. For example, EPL has enabled the synthesis of STAT6,191 a 668 residue protein – over 200 residues longer than the largest totally synthetic protein. These tools have also enabled synthesis in living cells, which would otherwise not be possible. Expressed protein ligation is extremely flexible and can be performed under a wide range of conditions, while PTS and a sortase ligation require conditions closer to physiological conditions.
4 Synthetic targets and applications
In this section, we demonstrate how the methods described above have facilitated answering biological questions. Given the pivotal role of phosphorylation in cell signalling and disease processes, it is unsurprising that these areas are key targets of investigation. The wide range of targets presented emphasizes the importance of phosphorylation in diverse processes. We discuss recent examples that exemplify the techniques described in chapter 3 and highlight important developments in the field.4.1 Kinases and phosphatases in signal transduction
Signal transduction is tightly controlled by the action of kinases and phosphatases. Abnormal phosphorylation is often a cause of disease. This is exemplified in the use of kinase inhibitors such as Imatinib,192 a frontline cancer treatment. Notably, the enzymes that control phosphorylation are, themselves, controlled by phosphorylation. Key mechanisms for kinase regulation are through phosphorylation in the conserved activation loop193 and the intramolecular binding of a phosphate-recognising domain to a phosphorylated motif to bring the protein into an active or inactive state.194,195The regulation of proto-oncogene Akt1/protein kinase B depends on phosphorylation within the activation loop. However, it was unknown to what extent each phosphorylation site contributes to the kinases’ activity due to the previous inability to prepare site-specifically phosphorylated protein. Three phosphoforms of Akt, either mono- or di-phosphorylated variants, were synthesized, with phosphoserine introduced at position 473 through genetic code expansion and threonine 308 phosphorylated with the kinase PDK1. The activities of the phosphorylated kinases were measured in an in vitro kinase assay and live-cell imaging. This work revealed the key role of pThr308 in the activation of Akt – other sites contributed to the increase in activity, but this site was required for activation. The authors suggest that this could be an important outcome for diagnostic purposes, which previously used pSer473 as a biomarker.110
Muir and co-workers have applied EPL to investigate the role of phosphorylation in signalling pathways.133,134,149,183,184,196 In the first example of EPL, the C-terminal Src kinase (Csk) was engineered to bear an unnatural regulatory C-terminal tail (Fig. 14A). A conserved mechanism of regulation among the Src kinase family is autoinhibition through the intramolecular binding of an SH2 domain to a phosphorylated tyrosine residue on the C-terminal tail. This interaction brings the protein into an inactive conformation. However, despite the high similarity of Csk to other Src kinases, it does not usually bear this C-terminal tail. Fmoc solid-phase synthesis was used to prepare an 11 amino acid long C-terminal tail bearing phosphotyrosine, introduced as the Fmoc building block with no phosphate protection. The synthetic C-terminal tail was ligated with a recombinant thioester to form both the unphosphorylated and phosphorylated variants, as well as bearing a C-terminal fluorescein tag (Fig. 14B).183 The activity of the synthetic kinases was measured using a radioactive ATP phosphorylation assay on the poly(Glu, Tyr) Csk substrate (Fig. 14C). Surprisingly, it was found that the addition of the tail instead led to an increase in phosphorylation activity.
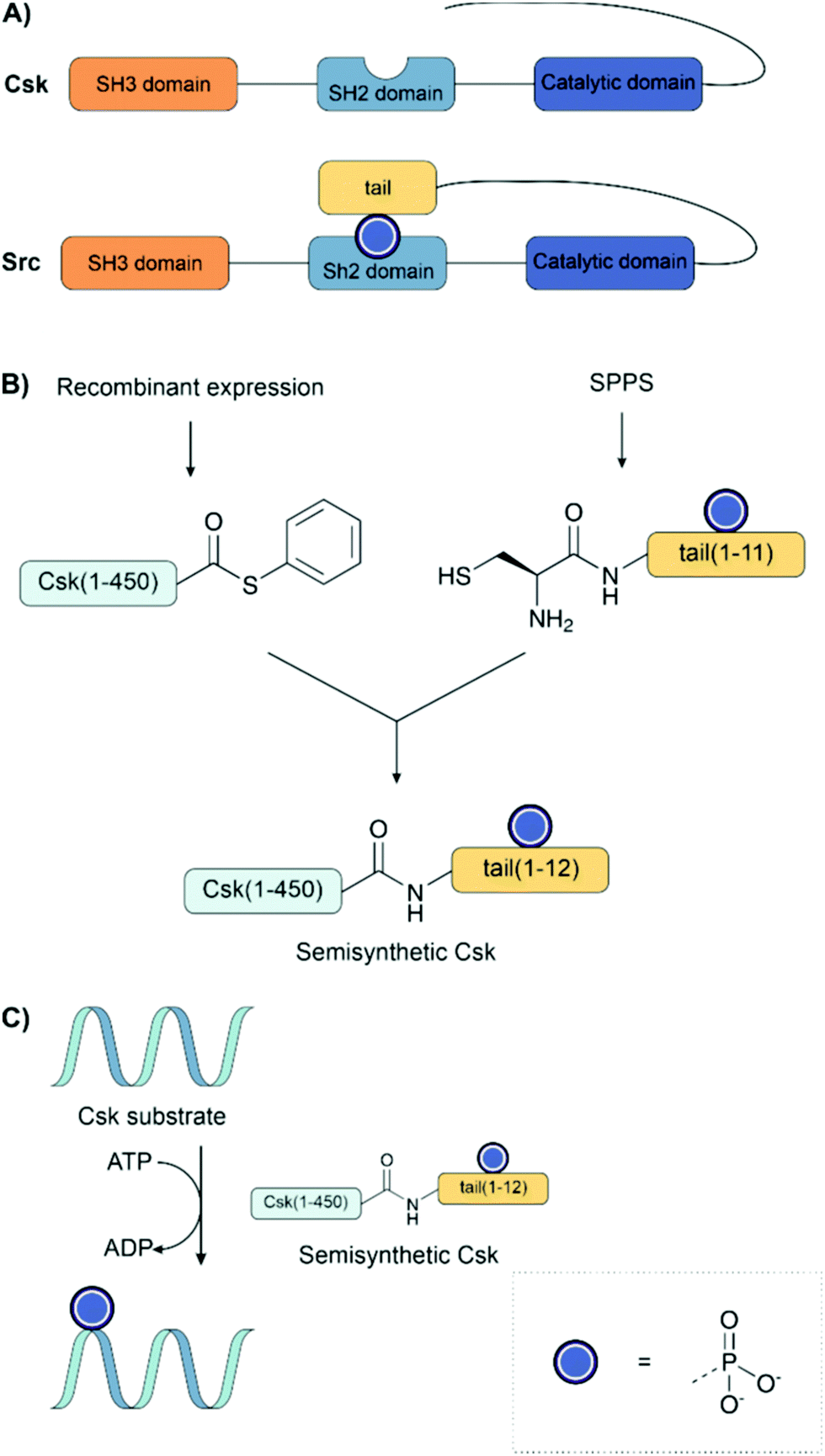 | ||
| Fig. 14 (A) Comparison of the structures of Src and Csk kinase. Src kinase naturally carries a tail with a tyrosine residue that can be phosphorylated, whereas Csk does not. (B) Expressed protein ligation to introduce an unnatural, phosphorylated tail to the kinase Csk. A phosphorylated tail was synthesized by SPPS and ligated to a recombinantly produced thioester of Csk. (C) The phosphorylation activity of the semisynthetic protein was measured with a radioactive ATP assay. | ||
Cole and co-workers have applied EPL to investigate the regulation of phosphatases, SHP-1 and SHP-2, through phosphorylation.38,197,198 Non-hydrolyzable phosphonates were required for the investigation given the inherent phosphatase activity of the target. These were installed using Fmoc-SPPS with benzyl-protected Pmp and F2Pmp (see Section 2.7) building blocks and ligated to the expressed protein thioester. EPL enabled the synthesis of a range of proteins bearing point mutations or truncated domains, which helped to dissect the protein's function (Fig. 15A). Similarly to the previous example, intramolecular binding between a phosphorylated tail and an SH2 domain was key to the regulation of enzyme activity (Fig. 15B). Here, however, it was found that each SH2 domain binds a different phosphotyrosine residue. For example, in SHP-1, it was found that the replacement of Y536 with Pmp or F2Pmp increased the catalytic activity, likely by intramolecular binding of the N-SH2 domain, thereby relieving autoinhibition. Phosphorylation at Y564 activated the phosphatase activity of SHP-1 to a much smaller extent, which was explained by intramolecular engagement of the C-SH2 domain. However, this was not sufficient to release the N-SH2 domain from the PTPase domain.198 An analogous mechanism was also found in SHP-2.38
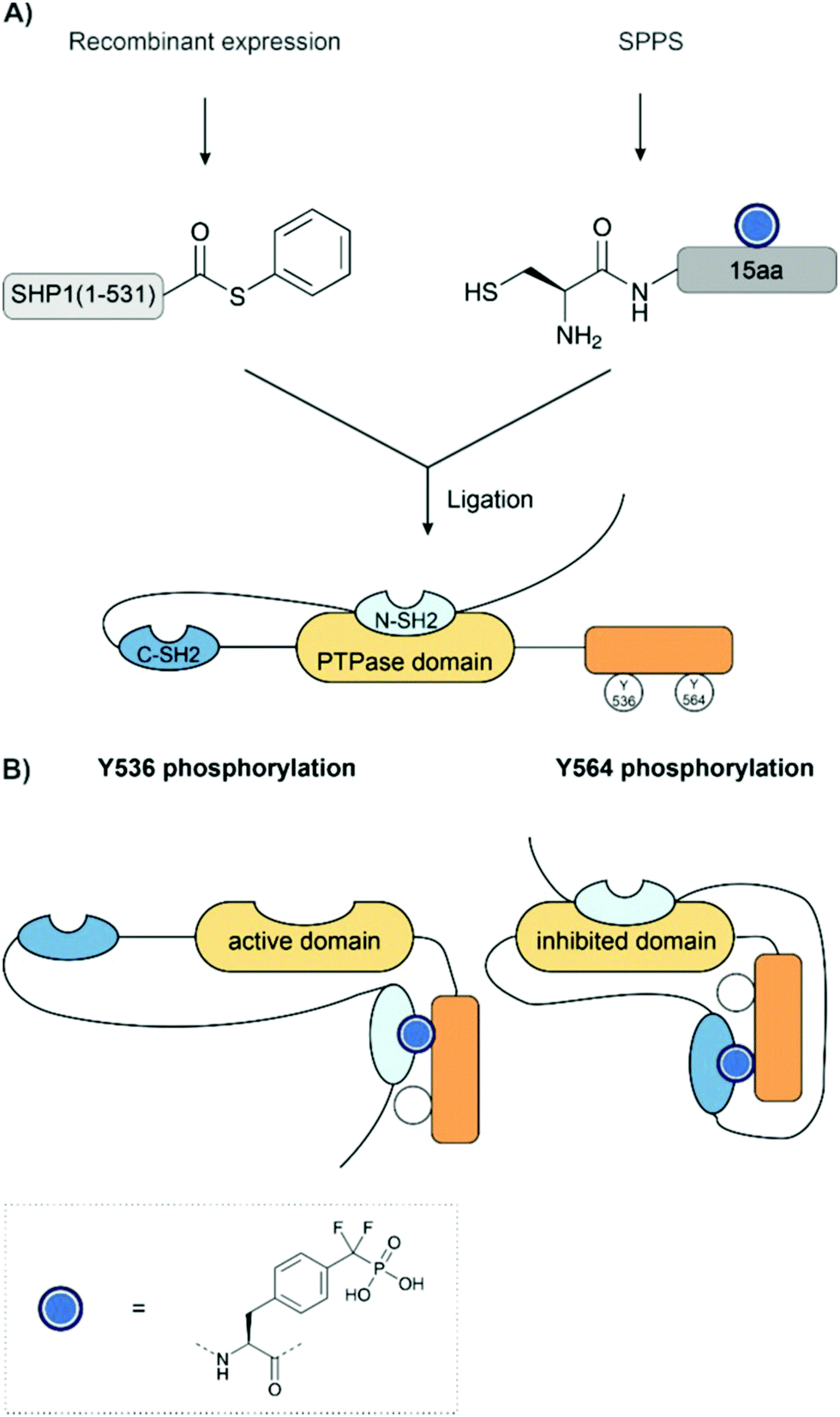 | ||
| Fig. 15 (A) Synthesis of SHP-1. SHP1 was prepared from a recombinant N-terminal thioester and a synthetic C-terminal fragment bearing a difluorophosphonotyrosine residue. (B) Binding of SHP1 SH2 domains. In the unphosphorylated state, the N-SH2 domain binds the phosphatase (PTPase) domain and inhibits activity. The phosphatase activity is enabled when the N SH2 domain binds phosphorylated Y536. The C-SH2 domain binds phosphotyrosine 564 and indirectly increases activity despite the N-SH2 domain remaining bound to the phosphatase domain. | ||
Phosphorylation within a recognition domain can modulate its affinity to a ligand. In work from our group, an array of 16 different Abl and Arg SH3 domains was synthesized on the surface of a 96-well plate (Fig. 16).136 To enable rapid analysis of all possible phosphotyrosine forms, we attached the C-terminal segments, obtained as peptide hydrazides by SPPS, in crude form to the aldehyde-functionalized plate through a hydrazone linkage. In the next step, only the full-length peptide can ligate with the N-terminal segment, a peptide thioester prepared by applying the self-purification approach described in Section 3.3.1. Binding assays performed after on-plate desulfurization and in situ folding revealed that phosphorylation within the SH3 domain of Abl kinase can both positively and negatively modulate the affinity for proline-rich ligands. Of note, monophosphorylation at every tyrosine abolished the affinity for a proline-rich peptide derived from the interdomain between the Abl SH2 and kinase domain. This interaction is known to stabilize a closed state, in which the Abl kinase has low activity. Phosphorylation could therefore facilitate the opening of the Abl kinase. On the other hand, phosphorylation at Y7, Y30 or Y52 can increase the affinity for other proline-rich peptides, whereas phosphorylation at all tyrosine residues induces unfolding. Apparently, tyrosine phosphorylation acts as a switch to fine-tune the recognition repertoire of SH3 domains.
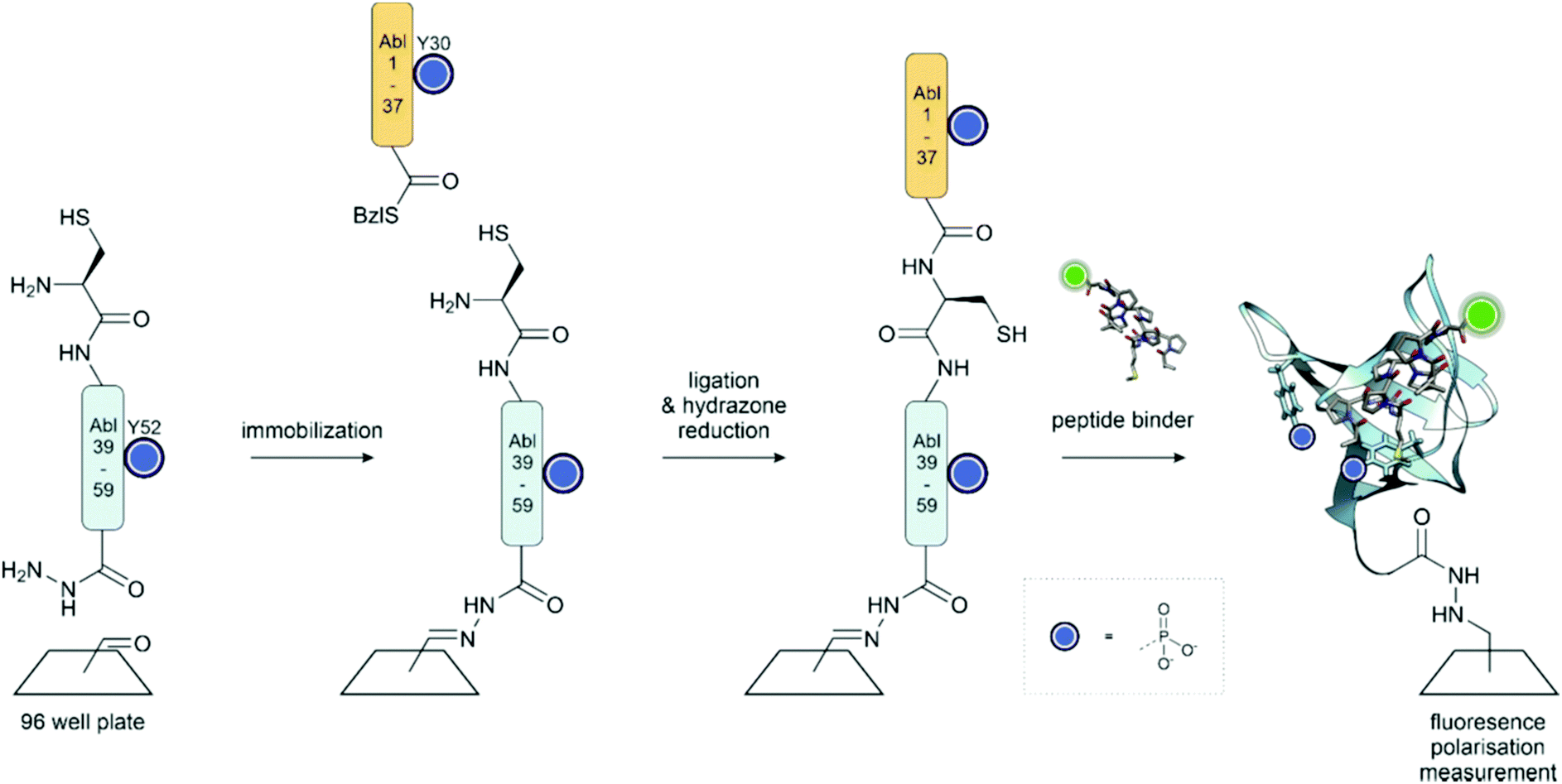 | ||
| Fig. 16 On surface synthesis of the Abl SH3 domain. The C-terminal peptide hydrazide is attached to the aldehyde functionalized plate through a hydrazone linkage. Following ligation with the thioester fragment, the hydrazone is reduced, and the cysteine for ligation is desulfurized to the native Ala residue. Surface saturation binding analysis with fluorescently labelled peptides showed that phosphorylation provides a means to fine-tune the recognition repertoire of the Abl-SH3 domain. PDB: 4JJC199 | ||
Transforming growth factor-beta (TGF-β) signalling is an important pathway controlling cellular proliferation, differentiation and migration. TGF-β signalling exerts its effect on gene expression through the action of SMADs, which are activated through phosphorylation. The TGF-β receptors are homodimeric serine/threonine kinases formed from one type I and one type II monomer. Upon ligand binding, the type II subunit phosphorylates the type I subunit and forms an activated tetrameric complex composed of one type I and one type II dimer, which phosphorylates the SMAD. The activated SMAD dimerises and then translocates to the nucleus.200
Muir and co-workers showed that tetraphosphorylation of the GS region, a regulatory segment containing a 185TTSGSGSG192 sequence, increased the catalytic activity of a type I TGF-β receptor construct in a SMAD peptide phosphorylation assay. The 20 aa tetraphosphorylated peptide thioester was synthesized via Fmoc SPPS using an alkylsulfonamide resin. This was ligated to a recombinant N-terminal cysteine fragment to form the type I TGF-β receptor construct (Fig. 17A). The semisynthetic route provided material phosphorylated at four defined sites, which was not accessible by phosphorylation with a kinase.133,134 The synthetic protein was examined in a kinase assay using the C-terminal domain of Smad2 as the substrate. Here, the tetraphosphorylated variant showed a 40-fold increase in phosphorylation activity compared to the non-phosphorylated protein. Later, the model of TGF-β activation was expanded to show that phosphorylation increased the affinity for its substrate, Smad2 and concurrently prevented binding of the inhibitory protein FKBP12 (Fig. 17B).201 The mechanism was unexpected because it does not increase the kinase activity but instead generates a site for Smad binding.
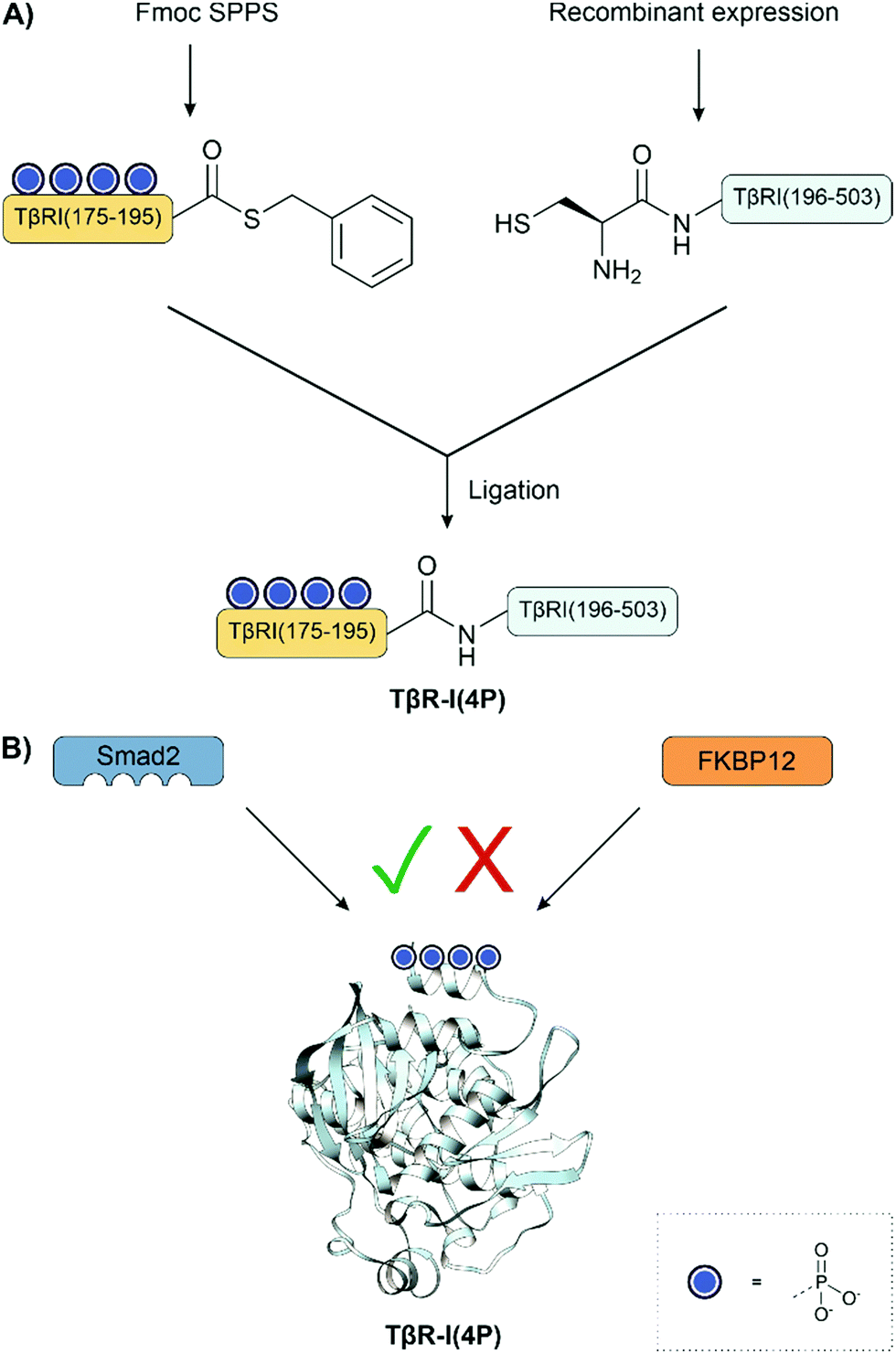 | ||
| Fig. 17 (A) Semisynthesis of type I TGF-β receptor fragments. (B) Updated binding model shows that phosphorylation increases the affinity for its substrate, Smad2 while simultaneously preventing the binding of FKBP12. PDB: 1B6C.202 | ||
The phosphorylation of Smad2 was also investigated.196 The possible combinations of the two conserved activating serine phosphorylations, S465 and S467, were synthesized. This material was not accessible by enzymatic synthesis because the known kinase phosphorylated both sites. The pS465-Smad2 was phosphorylated significantly faster at the second residue, S467, compared to unphosphorylated, and the opposite effect was not observed. Additionally, phosphorylation at S465 was key to promoting trimerization of Smad2, but both phosphorylations were required for a stable homotrimer. The combination of these two results is interesting because it shows that phosphorylation is cooperative – the first phosphorylation primes the protein for the subsequent phosphorylation to ensure the formation of a stable complex.
4.2 Control of transcription
The control of transcription is a critical cellular process that enables a cell to alter gene expression in response to signals. Controlling access to chromatin through its structure is the primary mechanism of regulation and is complemented by a vast network of enzymes that tightly control transcription at various stages.Histones are the essential structural proteins of chromatin and are, therefore, prominent targets of investigation. Genomic DNA wraps around an octameric complex made up of two of each of the four core histones, H2A, H2B, H3, and H4 to form a nucleosome.203 Histones are highly post-translationally modified on the N-terminal tails and in the core, providing a code for chromatin regulation through a variety of reader, writer, and eraser enzymes. This network of modifications finely regulates transcription. Abnormal modification patterns have been implicated in autoimmune diseases and cancer. Another interesting feature is their modularity which allows combinations of histones from different sources to form entire artificial nucleosomes. Given their size, ranging between 100 and 250 residues, they are within reach for total chemical synthesis.
The Spt-Ada-Gcn5 acetyltransferase (SAGA) complex is an important multifunction histone-regulating enzyme with both acetyltransferase and deubiquitinase activity. Brik and coworkers applied total chemical synthesis to unravel how phosphorylation of histone H2A regulates deubiquitination of ubiquitinylated histone H2BK120Ub by the SAGA complex.156 H2A, a 130 amino acid long protein, was synthesized from three segments: an N-terminal thioester, a thiazolidine-protected phosphate-bearing middle thioester, and a C-terminal fragment bearing an N-terminal cysteine (Fig. 18A). The phosphate-bearing middle fragment was prepared on the Dbz linker, and the phosphorylation at Tyr54 was installed by coupling the monobenzyl-protected phosphotyrosine during Fmoc-SPPS. The thioester of the N-terminal fragment was instead prepared by the N-methylcysteine method (see Section 3.3.1) because, with glycine as the C-terminal amino acid, using the Dbz linker would require extra protection and deprotection steps. The ligation was performed in a one-pot fashion: first, the middle fragment was ligated to the C-terminal fragment, followed by deprotection of the thiazolidine and ligation with the N-terminal fragment. The cysteine residues were desulfurized to afford the native alanine residues following ligation. After total synthesis, nucleosomes were assembled by com bining pY57-H2A or unmodified recombinant H2A with H2B120Ub, H3 and H4 in the presence of DNA (Fig. 18B). Treatment of the nucleosomes with the SAGA complex showed that this phosphorylation reduced the rate of deubiquitination.
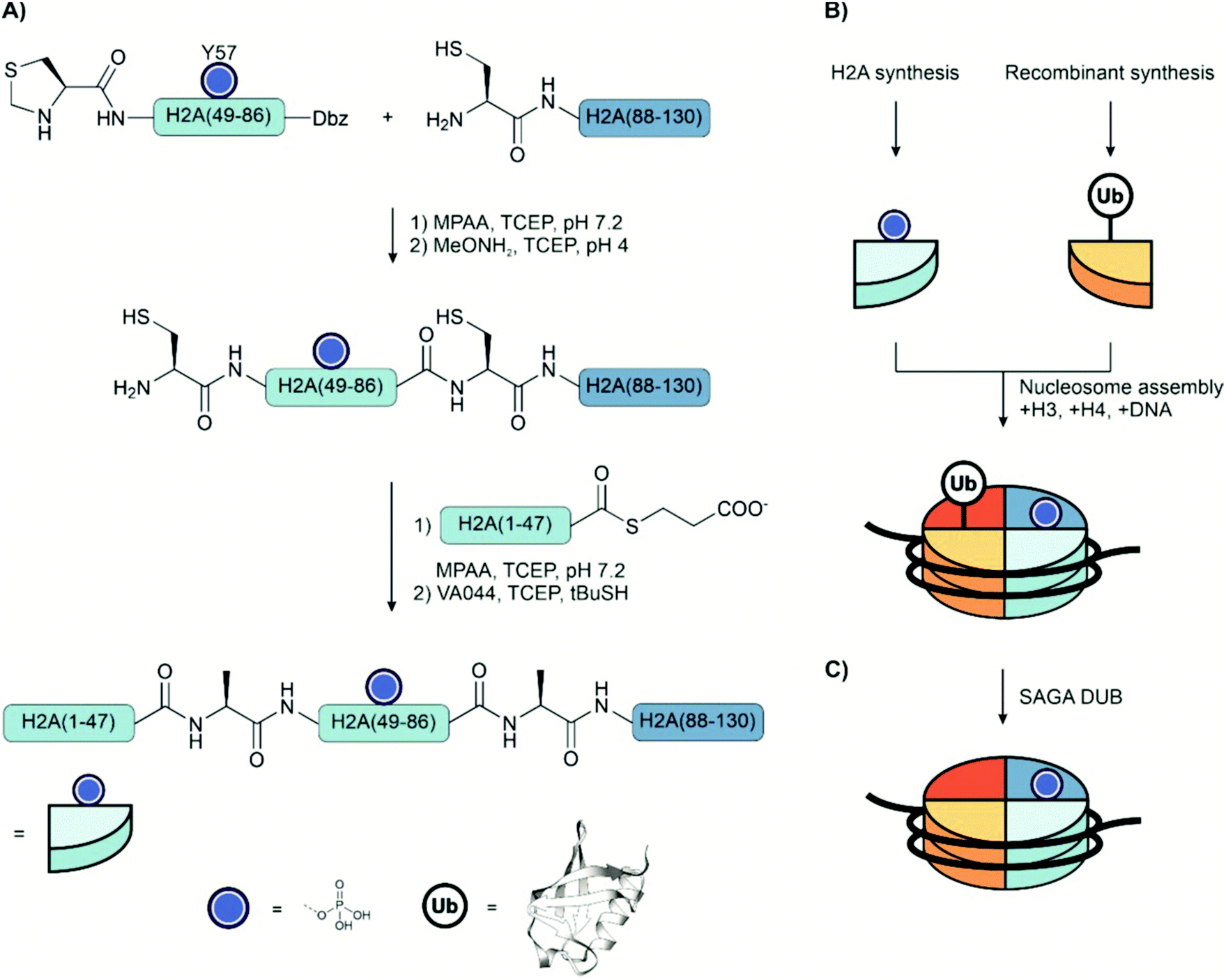 | ||
| Fig. 18 (A) Strategy for the total chemical synthesis of Tyr57 phosphorylated histone H2A. (B) Assembly of synthetic phospho-H2A, semisynthetic ubiquitinated H2B and recombinant H3 and H4 into a nucleosome. (C) Nucleosomes were incubated with the SAGA DUB module, and deubiquitination was measured by gel electrophoresis. PDB: 1UBQ204,205 | ||
In a subsequent synthetic study, Brik compared four synthetic routes to H3-pSer57; sequential, one-pot, semi-one-pot, and convergent methods. The convergent approach provided unphosphorylated and phosphorylated H3 in the highest yields.126 Using this strategy, the protein was divided into four fragments. Two pairs of fragments were synthesized that were first ligated together before being ligated to each other to form the whole protein. The product of the first ligation prepared the C-terminal fragment, which carried a thiazolidine-protected cysteine at the N-terminus, for the subsequent ligation. The ligation of the N-terminal pair resulted in a peptide hydrazide that was activated prior to the final ligation. Following all ligations, the cysteines were subjected to radical desulfurization to afford the native alanines.
The role of phosphorylation at serine 10 of H3 is disputed in the literature. There are conflicting reports as to whether it promotes acetylation of lysine residues in the H3 N-terminal domain, and different approaches have been used to investigate this site. In an early report, it was shown that phosphorylation of this site increased the rate of acetylation of Lys14 by the SAGA complex. Short peptide segments containing phosphoserine were used as the substrate for this assay.206 Later, Shogren-Knaak et al.207 used semisynthesis to investigate the same phosphorylation site and found that in nucleosomal assays phosphorylation did not affect acetylation. However, more recently, the site has been investigated again. Amber codon suppression was applied to incorporate phosphoserine into H3, and histone octamers were assembled. When the acetylation activity was measured here, where no DNA is present, there was no difference between unphosphorylated and phosphorylated material. However, in nucleosomal assays, H3-pSer10 stimulated SAGA-mediated acetylation of N-terminal domain lysine residues by up to three times.208 The reason for the difference in results between the two later investigations is not clear.
The use of highly pure, site-specifically modified material is useful for the validation of antibodies. Chiang et al.149 explored the effect of neighbouring modifications on the recognition of an antibody against a serine phosphorylation site. Expressed protein ligation in combination with desulfurization was used to synthesize histone H2B-pSer14. The N-terminal 16-mer phosphopeptide fragment was obtained as a thioester by Fmoc-SPPS with monobenzyl-protected pSer either on the Dbz linker from Dawson or the 2-hydroxy-3-mercaptopropionic acid linker from Botti (see Section 3.3.1) and ligated with the expressed C-terminal fragment. Following ligation, the cysteine residue was desulfurized to the native alanine with RANEY® nickel. The pure semisynthetic protein was used to validate a set of commercially available antibodies and examine the influence of other post-translational modifications. Notably, it was found that an antibody raised against H2B-pSer14 did not recognise this site when an acetylated lysine was nearby. The implications of this result may be concerning for studies that used these antibodies. False negatives may have been observed when attempting to identify a phosphate at this site, especially when in vivo or extracted material was being investigated.
Many transcription factors contain two or more Cys2His2 zinc finger domains. The linker regions that connect two adjacent zinc finger domains contain a threonine residue which is subject to phosphorylation. Studies of the Ikaros protein suggested that phosphorylation excluded this transcription factor from mitotic chromosomes.209 Jantz and Berg210 synthesized the 86 amino acid protein in 4 different forms: non-phosphorylated, fully phosphorylated and phosphorylated at one of the two linker domains connecting the three zinc fingers. The peptide was divided into three fragments: the C-terminal fragment presenting an N-terminal cysteine, the middle fragment with a thioester and bearing the phosphorylated residue and with the N-terminal cysteine protected by Fmoc, and the N-terminal thioester fragment. The thioesters were prepared by Fmoc-SPPS on Kenner's safety catch resin (see Section 3.3.1), and phosphothreonine was introduced with the monobenzyl protected building block. N-terminal Fmoc-protection was used to prevent self-ligation of the middle fragment. In DNA binding assays, Jantz and Berg observed an approximately 40-fold reduction in the DNA affinity with a single phosphorylation, whereas phosphorylation of both linkers reduced affinity by 130-fold. These experiments supported the hypothesis that a kinase/phosphatase pair regulates this set of proteins during the cell cycle.
Chen et al.211 applied genetic code expansion to investigate the effect of tyrosine phosphorylation on IκB-α, which inhibits NF-κB, an important transcription factor for responding to stress and in immune response regulation. IκB-α-pY42 was synthesized through genetic code expansion using phosphotyrosine bearing photolabile o-nitrobenzyl protecting groups. The unphosphorylated IκB-α caused dissociation of the NF-κB-DNA complex as expected. However, in contrast to previous reports that phosphorylation at tyrosine 42 reduced affinity for NF-κB, it was found that this phosphorylation had a minor effect on the affinity, and in fact, mediated the exchange of exogenous DNA into the NF-κB-DNA complex.
The Conibear lab used semisynthesis to investigate post-translational modifications in the nuclear protein HMGN1.212 This protein is intrinsically disordered. Modifications were investigated at both the C-terminus and N-terminus using the ligation of synthetic peptides with recombinant peptides. The fragments were synthesized with either both acetyl-lysine and phosphoserine or each modification alone. To investigate modifications at the N-terminus of the protein, a short synthetic N-terminal fragment was ligated with a recombinant C-terminal fragment. The N-terminal 10-mer fragment was synthesized as a peptide hydrazide using microwave synthesis. The phosphoserine residue was incorporated using the benzyl protected phosphoserine building block. The hydrazide was converted to the azide with sodium nitrite at pH 3, thiolysed to the methyl thioglycolate thioester and ligated with the recombinant fragment. Following ligation, the cysteine at the ligation junction was desulfurized to the native alanine. To investigate modifications at the C-terminus of the protein, the strategy was inverted – a short synthetic C-terminal fragment was ligated with a recombinant N-terminal fragment. The C-terminal 33-mer fragment was produced by SPPS carrying up to three serine phosphorylations and an N-terminal cysteine. The N-terminal thioester was produced by intein cleavage and subsequently ligated with the phosphorylated C-terminal fragment then desulfurized. The recombinant fragments were also produced enriched with 13C or 15N labels to enable NMR experiments to characterize these proteins. It was observed that the phosphorylation affected the chemical shift and conformations even of distant upstream residues. Additionally, the binding of the different variants with mononucleosomes was compared. However, no significant differences were found. There was only a slight increase in binding with multiple phosphorylations at the C-terminus.
4.3 Neurodegenerative diseases and aggregation
Aggregation of misfolded protein is implicated in the development of several neurodegenerative diseases, including α-synuclein in Parkinson's disease, tau in Alzheimer's disease, and huntingtin in Huntington's disease.213,214 Post-translational modifications may affect aggregation, though the historical difficulty in preparing site-specifically phosphorylated proteins has made further investigation challenging. Furthermore, antibodies typically used to identify phosphorylation sites are raised against extracted protein which may not be homogeneous and potentially leads to cross-reactivity.Research using site-specifically modified material in this area has revealed two common factors. Firstly, that often phosphorylation does not behave as previously expected, namely that in some cases, it had no effect, or in fact, the opposite effect. Secondly, it has emphasized the role of kinases in pathogenesis, which can be targeted therapeutically. For example, Lashuel and co-workers applied total chemical synthesis to generate mono-, di-, and tri-phosphorylated variants of microtubule-binding repeat domain K18 of tau.215 Three fragments were prepared by Fmoc-SPPS using the monobenzyl-protected pSer building block. The thioester fragments were prepared on Dawson's MeDbz resin (see Section 3.3.1). The ligations were performed in the C → N direction by using the native cysteine residues and in situ deprotection of the middle fragment's N-terminal thiazolidine. Contrary to the prevailing theory,216 their experiments revealed that hyperphosphorylation inhibits rather than promotes aggregation of K18 in vitro, with more phosphorylations having a more potent inhibitory effect. With three phosphorylations, fibril formation was completely inhibited. Full length phosphorylated tau has also been prepared.217
Tyrosine 39 of α-synuclein is a known target of the kinase c-Abl and is implicated in disease progression. Studies using defined material are of particular interest because conflicting results have been published, with differences between in vivo and in vitro studies.218,219 In one study by Dikiy et al.,219 a 156 amino acid long α-synuclein-pY39 construct was synthesized through ligations of three fragments. The longest C-terminal segment was obtained by recombinant synthesis and featured an N-terminal cysteine residue as a precursor of the natural alanine. The middle fragment was prepared as a thioester on the Dawson linker (see Section 3.3.1), had a thiazolidine-protected N-terminal cysteine and featured the phosphotyrosine, which was introduced using Fmoc-Tyr(PO3HBzl)-OH. After NCL, the thiazolidine protection was removed upon treatment with methoxyamine. A subsequent NCL with the synthetic N-terminal segment was followed by desulfurization affording the desired protein. Functional studies showed that phosphorylation of Tyr39 slowed the aggregation of the free protein but enhanced membrane association which could lead to aggregation in vivo. Of note, the authors observed similar effects with recombinant protein bearing the ‘phosphomimetic’ Y39E mutation. In a more recent study, c-Abl was used to phosphorylate Tyr39 within the N-terminal 55 amino acid fragment prepared recombinantly as a thioester-linked intein fusion.220 This fragment also contained a fluorescent label, which was attached to a cysteine residue. The 84 amino acid C-terminal segment was prepared by recombinant synthesis and incorporating O-propargyl tyrosine as an unnatural amino acid to allow click labelling with a second fluorophore. NCL provided phosphorylated and doubly fluorescently labelled α-synuclein. The aggregation of mixtures of unphosphorylated and α-synuclein-pY39 was measured using FRET. These experiments showed that the rate of aggregation was dependent on the proportion of phosphorylated protein. In agreement with the earlier report, 100% α-synuclein-pY39 slowed the aggregation. However, at lower concentrations (1–5%), aggregation was accelerated.
The C-terminus of α-synuclein contains tyrosine 125, which is subject to phosphorylation by the kinases BARK1, PLK2, CK2 and GRK5. The Lashuel group employed semisynthesis to elucidate the role of pY125.221 An N-terminal 106 amino acid segment was expressed in E. coli as an intein fusion. Splicing of the purified construct in the presence of mercaptoethanesulfonic acid (MESNa) afforded the peptide thioester, which was allowed to react with a 34-mer phosphopeptide prepared by Fmoc-SPPS. After NCL, Cys was converted to Ala. The purified protein was delivered into primary hippocampal neurons by microinjection. With the help of antibodies, localization and phosphorylation status were examined at different times after microinjection. The studies showed that the phosphate group at Tyr125 is rapidly removed. Antibodies generated with the phosphoform-defined semisynthetic α-synuclein revealed its cytoplasmatic localization, with a small proportion in the membrane. Additionally, kinase assays demonstrated that the pY125 modification did not affect phosphorylation at S129 or S87.
The Lashuel group also investigated the role of phosphorylation within the first 17 residues of exon 1 of Huntingtin (Htt), which harbours a polyQ repeat expansion and shows aggregation-related toxicity related to Huntington's disease. The Htt exon 1 segment is also highly post-translationally modified. To examine the role of phosphorylation in this domain, Lashuel and co-workers developed the semisynthesis of 89 amino acid long Htt exon 1 segment containing a phosphorylated Thr3.222 In this strategy, a synthetic phosphopeptide was ligated with an expressed fragment. The N-terminal phosphooctapeptide thioester was prepared on Dawson's Dbz linker (see Section 3.3.1) and ligated through the N-terminal cysteine of the expressed C-terminal segment. Following ligation, desulfurization yielded the native alanine at the ligation junction. AFM and TEM experiments showed that phosphorylation at Thr3 significantly reduced the rate of oligomerization and fibril formation.
4.4 Apoptosis and autophagy
Like many other processes, apoptosis and autophagy are controlled, amongst others, by protein phosphorylation. For example, p53 is an essential cellular protein involved in the response to DNA damage and with control over the cell cycle and apoptosis. Phosphorylation of p53 itself and its inhibitory protein, MDM2, both mediate its activity. Mutation of p53, or misregulation, is a hallmark of cancer. Teruya et al.162 synthesized the C-terminal domain that stretches over residues 303 – 393 of p53. The aim was to study the role of phosphorylations at Ser315 and Ser378 and acetylation at Lys320 on DNA recognition. Three fragments were prepared by Fmoc-SPPS. All segment couplings were performed by using the thioester condensation method (see Section 3.3). Both the N-terminal and the middle peptide thioesters were constructed on Kenner's safety catch linker. The middle and C-terminal segments were coupled first. Subsequently, the light-sensitive 6-nitroveratroyloxycarbonyl (Nvoc) protecting group at the N-terminus of the former middle segment was removed before the following thioester condensation and final removal of Boc protecting groups at side chain lysine. The binding of the purified 90 amino acid long protein to DNA was measured. Interestingly, while phosphorylation at Ser378 within the 67 amino acid long p53(326–393) peptide enhanced the specificity of binding to supercoiled DNA over linearized plasmid DNA, this phosphorylation had little, if any, effect when the 90 amino acid p53(303–393) was studied.Tan and colleagues investigated phosphorylation in the N-terminal domain of p53, a 70 amino acid long protein that was accessed through two segments.223 Both were obtained in the fully protected form by Fmoc-SPPS on a trityl TentaGel resin and cleavage with CH2Cl2/TFE/AcOH. The N-terminal segment was coupled in solution with a side chain protected glutamine thioester. Subsequent TFA treatment provided the unprotected peptide thioester. The extended cleavage time required to deprotect the phosphoserine benzyl ester resulted in methionine oxidation, which had to be reduced in a separate step. The fully protected C-terminal segment was coupled with biotin hydrazide. After NCL, the Cys at the ligation junction was converted to alanine. The method was used to prepare the unphosphorylated p53 transactivation domain and phosphoforms featuring phosphorylation at one or five Ser residues.
Lu and colleagues assessed the function of phosphorylation at serine 17 of MDM2 in the activation of p53.148 They applied native chemical ligation with three fragments and synthesized the p53 binding domain as the unphosphorylated, pS17, and S17D forms. The middle and the C-terminal fragments were prepared by Boc-SPPS and connected at a Tyr-Cys (which replaced the naturally occurring Tyr-Lys) junction via NCL. Before the second NCL, Acm protection was removed from the N-terminal Cys of the middle fragment. The N-terminal segment contained the pSer residue introduced by coupling Fmoc-Ser(PO(OBzl)OH)-OH during Fmoc-SPPS on Dawson's Dbz linker. Interactions of the purified MDM2(1–109) protein with p53-derived ligands were analyzed with surface plasmon resonance and fluorescence polarisation. However, despite the proposed importance of phosphorylation on this residue, it was found that it did not affect binding to p53 peptides.
Müller and colleagues used a semisynthetic approach to investigate the role of phosphorylation at serine 15 and 20 in the p53 N-terminal domain.224 Recombinant expression was used to synthesise the C-terminal fragment (40–393) bearing an N-terminal cysteine. The ligation junction was mutated to the cysteine for ligation because there are no cysteines in this section of the peptide; normally, this residue would be a methionine, though the change was thought to make little difference. The N-terminal 39-mer fragment was synthesized by Fmoc SPPS using benzyl-protected Fmoc serine to introduce the phosphorylated residues. For the fragment bearing a phosphate at serine 15, the best method for synthesis used couplings with DIC/Oxyma/DIPEA to introduce the building block, and in all subsequent couplings as well as deprotection with 5% piperazine. The fragment was synthesized as a hydrazide, which was converted to a thioester in situ using sodium nitrite. Following ligation, the fragments were allowed to fold and assembled into heteromeric tetramers. To investigate the crosstalk between phosphorylation and acetylation, the semisynthetic tetramers were incubated with the acetyltransferase, p300, and acetyl-CoA and overall acetylation and acetylation at Lys373 were measured by western blotting. This assay found that phosphorylation at serine 20 increased acetylation by 2.2-fold, while phosphorylation at serine 15 only increased it by 1.5-fold. Furthermore, the double phosphorylated tetramers did not lead to enhanced acetylation, only 2.3 times higher, comparable to phosphorylation only at serine 20. The authors proposed that phosphorylation at Ser15 and Ser20 would not result in an all-or-none response but instead provides a means for a graded response.
Ubiquitin is a covalent tag to target a protein for proteasomal degradation. Three enzymes are involved in the attachment process: firstly, E1 activates ubiquitin as a thioester for ligation. Next, the ubiquitin thioester is transferred to E2 via transthioesterification, where E3 catalyses the attachment of ubiquitin to the target. Ubiquitin is linked to the target protein through either an isopeptide bond to a lysine side chain of the target protein or one of the lysine residues of protein-appended ubiquitin. In addition, ubiquitination occurs at the N-terminus or lysine residues of another ubiquitin. Deubiquitinases cleave ubiquitin from the target protein. Ubiquitin itself can be post-translationally modified, including by phosphorylation, to modulate its effect.225,226
Bondalapati et al.227 reported the first chemical synthesis of phosphorylated ubiquitin and lysine 63 linked diubiquitin and examined the effect of phosphorylation on deubiquitinases (Fig. 19A). Diubiquitin was phosphorylated at serine 65, either on both, one, or neither of the monomers. Initially, the 76 amino acid sequence was attempted as one long peptide using the monobenzyl-protected phosphoserine building block. However, they observed a side product that resulted from the formation of the H-phosphonate, which could not be avoided. Therefore, monoubiquitin was accessed by NCL of two fragments. The N-terminal segment was prepared as a thioester on the Dbz linker. The C-terminal segment contained the phosphorylation site (pSer65) and an S-nitrobenzyl-protected C-terminal N-methyl cysteine. In this case, the side product was not observed. Following NCL, the masked thioester was converted to the ubiquitin(1–76) thioester upon photolysis and treatment with mercaptopropionic acid at pH 1 and used in the synthesis of diubiquitin. Diubiquitin was prepared similarly, however, with the inclusion of a thiazolidine-protected δ-mercapto lysine residue to enable ligation through Lys63. Treatment with methoxyamine liberated the thiol of δ-mercapto lysine, which was subsequently used in an NCL with a ubiquitin(1–78) thioester. Radical desulfurization restored the native Ala46 and Lys63 residues. The purified proteins were then assayed against USP2 and AMSH, amongst other deubiquitinases (Fig. 19B). AMSH has selectivity for the Lys63 linkage,228 whereas USP2 has been described as a promiscuous deubiquitinase.229 Double phosphorylation impaired the activity of both enzymes. Interestingly, while AMSH tolerated phosphorylation at the distal site but not at the proximal site, USP2 showed the opposite behaviour. The results corroborated that phosphorylation can influence the dynamics of the ubiquitin signal.
 | ||
| Fig. 19 (A) Synthesis of ubiquitin and diubiquitin phosphoforms by the convergent assembly of phosphorylated and unphosphorylated fragments. (B) The diubiquitin phosphoforms were assayed against a range of deubiqutinases (e.g. AMSH, USP2) to see how the position of the phosphate affected deubiquitinase specificity.227 | ||
Ubiquitin is also phosphorylated at other serine residues, many of which have an unknown effect. Ubiquitin monomers phosphorylated at serine residues 20, 57, or 65 were produced using genetic code expansion (Fig. 20). On the one hand, reactions with 18 different E2 ubiquitin ligases showed how phosphorylation affects linkage selectivity and rate of diubiquitination by ubiquitin ligases and, on the other hand, provided a library of ubiquitin dimers linked through isopeptide bonds at different sites.230 The authors showed that phosphorylation at Ser20 converts UBE3C from a dual-specificity to a Lys48-specific ligase. The dimers were screened against 31 deubiquitinases to examine how the phosphorylation at this site affected their specificity and activity. It was shown that phosphorylation of both Ser65 residues inhibited cleavage of the Lys63 linkage. However, phosphorylation at this site accelerated cleavage of the Lys48 linkage by the enzymes OTUB1 and USP21. Phosphorylation can exert both repression and activation of ubiquitin processing.
 | ||
| Fig. 20 Ubiquitin monomers carrying phosphates on different serine residues were prepared by genetic code expansion. Phosphoubiquitin homodimers bearing different serine phosphorylations and linked through different isopeptide bonds were prepared enzymatically by treatment of (phospho)ubiquitin monomers with an E1 (Ube1), ATP and 18 different E2s. The dimers were screened against various deubiquitinases to uncover how different phosphoforms and isomers affect deubiquitinase specificity.230 | ||
Mann et al.96 created a phosphatase resistant phosphoubiquitin probe for cellular imaging. The ubiquitin probe carried a dye at the N-terminus, a cysteine carrying a transiently linked cell-penetrating peptide, and a phosphonoserine at position 65, introduced as the dibenzylphosphonoserine building block (see Section 2.7). The cyclic-decaarginine DABCYL tag enabled the delivery of the phosphorylated probe into the cell, which bears two additional negative charges. The probe was delivered into cells expressing the E3 ligase, Parkin, and localisation, and conjugation to Parkin was evaluated by using confocal laser scanning microscopy. This approach highlights the utility of stable mimics to investigate the role of PTMs in live-cell settings.
Wang and colleagues examined the role of tyrosine phosphorylation in ubiquitin. A method to introduce bis-dimethylamino protected phosphotyrosine with amber codon suppression was developed and applied to the synthesis of phospho-ubiquitin.231 This derivative, which also finds use in Fmoc SPPS (Fig. 4), is not cleaved by cellular phosphatases. For deprotection, the purified recombinant protein was treated with 0.4 M HCl. NMR studies suggested that phosphorylation of Tyr59 changes the conformation of ubiquitin. The derivative was incorporated at Y59 of ubiquitin to examine how it influenced the transthioesterification of ubiquitin between E1 and E2. By using an E2 loading assay, the authors showed that Ube1-catalyzed conjugation of p59-Ub with UbE2D3 did not proceed whereas non-phosphorylated Ub subjected to the same treatment as p59-Ub was still conjugated. The authors concluded that also in cells, Y59 phosphorylation could negatively regulate ubiquitination.
The protein p62 recognises ubiquitinated substrates and delivers them to the autophagosome. Phosphorylation of p62 has been shown to enhance binding to ubiquitinated substrates. A 116 amino acid long construct spanning the LIR and UBA domains of p62 was accessed from three fragments (Fig. 21A).189 The middle segment harboured an Acm-protected Cys at the N-terminus, two pSer residues (introduced as Fmoc-Ser(PO(OBzl)OH)-OH) and a C-terminal hydrazide, which was converted to a thioester prior to NCL with the C-terminal segment. In the event, the ligation occurred at a Leu-Cys junction. Alkylation of the cysteine residue with bromoacetamide was used to mimic the native Gln residue. The N-terminal segment was prepared by recombinant synthesis with a C-terminal LPETG motif which introduced two mutations, (Pro–Glu, Gly–Thr), to enable a sortase-mediated hydrazinolysis affording a 69 amino acid long peptide hydrazide. Transformation to the thioester set the stage for the next NCL, and subsequent desulfurization converted the Cys at the ligation junction to the natural Ala residue. SPR studies exposed that phosphorylation of Ser403 and Ser407 increased the binding of K63diUb 240-fold (Fig. 21B). By comparison, 11-fold or 34-fold binding enhancements were obtained for monophosphorylated p62. Of note, the dramatic affinity enhancement was not observed when the Ser residues were mutated to glutamate.
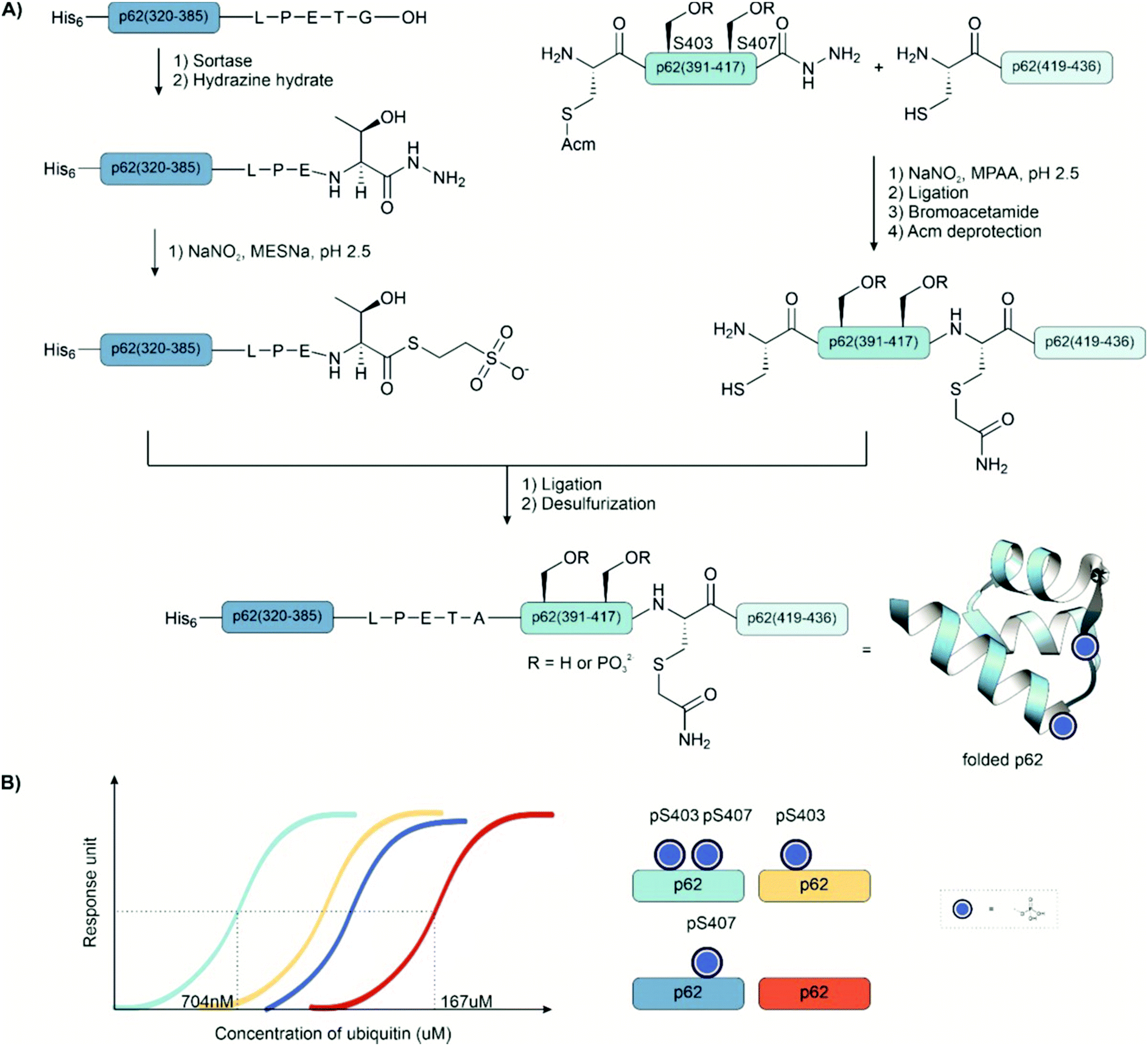 | ||
| Fig. 21 (A) Synthesis of p62 phosphoforms (pS401, pS403) from three segments. The N-terminal fragment p62(320–385) was prepared recombinantly and converted to the peptide hydrazide through a sortase-mediated hydrazinolysis. (B) The binding of different phosphoforms of p62 to diubiquitin as measured by surface plasmon resonance. In a surface plasmon resonance assay, the ligand is immobilized on a chip and the analyte flows through the flow cell. Mass changes on the surface of the chip affect the refractive index of the material, which in turn affects the SPR angle. The binding of the analyte to the ligand correlates to a change in the SPR angle. PDB: 3B0F232 | ||
Xu et al. investigated the same phosphorylations of the p62 UBA domain through total chemical synthesis using an auxiliary-mediated ligation (see Section 3.3).175 Four variants were prepared: the native unphosphorylated sequence, pSer403, pSer407 and the diphosphorylated sequence. The 48 amino acid long domain was divided into two fragments: a diphosphorylated N-terminal fragment bearing a C-terminal hydrazide and an auxiliary-bearing C-terminal fragment. The phosphorylated peptide was assembled with Fmoc SPPS on hydrazide substituted 2-chlorotrityl resin with the phosphoserine residues introduced as the benzyl-protected building block. Following cleavage, an impurity with M + 90 was observed that they attributed to incomplete deprotection of the benzyl group from the phosphate. The dimethoxybenzyl auxiliary was preloaded onto a glycine residue and coupled at the N-terminus of the C-terminal fragment. The hydrazide peptide was activated with sodium nitrite and thiolyzed, followed by the addition of the auxiliary peptide. After ligation, the auxiliary was removed by treatment with acid. Isothermal titration calorimetry (ITC) was used to measure the binding of the different phosphoforms to monoubiquitin. Phosphorylation at Ser407 or both Ser403 and Ser407 enhanced binding affinity between the UBA domain and ubiquitin almost three-fold. In contrast to the previous example, pSer403 did not affect binding. However, the previous study measured binding to K63-diubiquitin as opposed to the ubiquitin monomer.
p19INK4d is a member of the family of INK4 proteins, which inhibit the activity of D-type cyclin-dependent kinases and can arrest cells in the G1 phase. For one member of this protein family, p19INK4d, it was shown that phosphorylation affected the stability of the protein as well as influenced how it was ubiquitinated (Fig. 22).233,234 In these studies, aspartate substitution and enzymatic phosphorylation had been applied to access phosphoforms of p19INK4d. However, the singly phosphorylated, pSer76, phosphoform could not be produced enzymatically as it only occurred following phosphorylation at serine 66. Using total chemical synthesis, Brik and co-workers235 synthesized the unphosphorylated, mono-, and di-phosphorylated variants of p19INK4d to investigate their stability and cross-talk with ubiquitination (Fig. 22A). The 166 amino acid sequence was divided into three segments. The 52 amino acid middle segment had the N-terminal Cys protected as thiazolidine, contained one or two pSer residues and offered a C-terminal thioester prepared on Dawson's MeDbz linker for NCL with the 58 amino acid C-terminal segment. After NCL, thiazolidine protection was removed by treatment with a Pd(II) complex. Subsequently, the 51 amino acid N-terminal segment thioester, again prepared on the MeDbz linker, was ligated using a Gly–Cys junction. Final desulfurization afforded the (2–166) p19INK4d. Melting experiments showed that the 3D structures of the non-phosphorylated and pSer66 variants were far more stable than the pSer76 and diphosphorylated protein folds, with over a 10 °C difference in melting temperature (Fig. 22B). As no E3 ligase is known for this protein, the different proteins were incubated in cell lysates, and their ubiquitination was measured through a western blot (Fig. 22C). Phosphorylation increased ubiquitination, with diphosphorylation leading to the most significant increase. This supports previously published results. Phosphorylation can destabilize protein structure and thereby facilitate ubiquitination.
 | ||
| Fig. 22 (A) Synthesis of unphosphorylated, mono-, and di-phosphorylated variants ofp19INK4d. (B) The stability of the different p19INK4d phosphoforms was compared by melting curve measurements. (C) To assess how phosphorylation affects ubiquitination p19INK4d, phosphoforms were incubated in cell lysates, and ubiquitination was characterized by western blotting. PDB: 1BLX.236 | ||
Lahav et al.237 examined the role of phosphorylation in the WW domain of the tumour suppressor protein WWOX by total chemical synthesis. WW domains are important for protein-protein interaction in many biological systems. Phosphorylation of Tyr33 within the WW domain of WWOX is known to decrease affinity for p73, a tumour suppressor protein similar to p53. Using the defined phosphorylated material produced here, the change in affinity was quantified. Unphosphorylated fragments of WW1 and the larger WW1–WW2 fusion were expressed and purified. Next, the Tyr33-phosphorylated pY33-WW1 and pY33-WW1–WW2 were accessed by chemical synthesis. The latter was divided into two fragments: the N-terminal thioester fragment bearing the phosphotyrosine, and the C-terminal fragment bearing a cysteine residue at the N-terminus. Peptides were prepared by microwave SPPS and the thioester was prepared on the Dbz linker. Following ligation, the cysteine was desulfurized to the native alanine residue. The binding of the phosphorylated domain to a short p73 fragment was measured by fluorescence anisotropy. The recombinant WW1–WW2 had a five-fold higher affinity for the p73 fragment than the chemically synthesized pY33-WW1–WW2.
Liu and co-workers176 examined how phosphorylation of diubiquitin affects its interaction with the wild-type and phosphorylated E3 ubiquitin ligase, Parkin (Fig. 23). They produced four phosphoforms of K6-linked diubiquitin: unphosphorylated, phosphorylated at the distal or proximal units, or phosphorylated on both units. These phosphoforms were prepared from recombinant protein. The distal ubiquitin unit was prepared as a peptide hydrazide via Cys-promoted C-terminal hydrazinolysis and subsequently phosphorylated at Ser65 by the kinase, PINK1. The proximal unit was phosphorylated at Ser65 with the same enzyme, and subsequently, Cys6 was converted to dehydroalanine by alkylation-elimination with DBHDA (see Section 2.7). The protected glycyl-cysteamine-dimethoxybenzyl auxiliary was added to this residue through a Michael addition. Following deprotection of the auxiliary thiazolidine, the fragments were ligated by oxidation of the hydrazide with sodium nitrite and thiolysis with MPAA. Auxiliary removal afforded diubiquitin linked through a cysteine-derived lysine mimic. In a Parkin autoubiquitination assay, the dimers bearing a phosphorylated distal unit were able to activate wild-type Parkin, whereas the dimer bearing a single proximal phosphorylation could only activate phosphorylated Parkin. Isothermal titration calorimetry revealed that their binding affinity did not cause this difference. However, an E2 discharge assay and an E3 activation assay showed that the respective ubiquitin dimers activate the unphosphorylated and phosphorylated Parkins through different mechanisms.
 | ||
| Fig. 23 Four phosphoforms of K6-linked diubiquitin were synthesized from recombinant protein. Each fragment was phosphorylated with the kinase, PINK1. The proximal and distal fragments were ligated using an auxiliary installed via a Michael addition on dehydroalanine. | ||
Kulkarni et al.145 extended their DSL method into expressed protein ligation. To access the selenoester they modified Knorr-Pyrazole chemistry. This method was applied to synthesize three phosphoforms of heat-shock protein 27 (Hsp27). Hsp27 is a molecular chaperone that responds to cellular stress and prevents protein aggregation. The C-terminus can be phosphorylated at three sites. The N-terminal fragment was prepared recombinantly from the corresponding Hsp27-MxeGyrA fusion protein. The hydrazide was formed by cleavage of the fusion protein with hydrazine and then purified by reversed-phase HPLC. The C-terminal fragment bearing an N-terminal selenocysteine and phosphothreonine residue was prepared by Fmoc SPPS using benzyl-protected phosphothreonine and PMB protected selenocysteine. Following cleavage from the resin, the PMB group was deprotected with TFA and DMSO to afford the peptide diselenide. Subsequent treatment of the N-terminal fragment with acetylacetone, diphenyl diselenide, and TCEP, afforded the selenoester. The phosphorylated diselenide fragment was added to the selenoester peptide mixture to provide the ligated product. After extraction of DPDS, the selenocysteine residue was deselenized with TCEP and GSH to the native alanine residue without desulfurizing the native cysteine. The peptide bearing a phosphate at Ser199 had a different minimum in the circular dichroism spectrum, which suggested that it had a more random coil content compared to those with phosphorylation at other sites. Reduced chaperone activity was observed for all phosphoforms in an assay with citrate synthase as the client protein.
4.5 Transmembrane proteins, membrane receptors and their ligands
A strength of total chemical synthesis is the range of methods available to help dissolve hydrophobic membrane proteins. Chemical synthesis provides flexibility in the choice of solvent and the use of other additives or tags to enhance solubility. For example, native chemical ligation can tolerate the addition of polar organic solvents like DMSO or detergents to aid solubility. Recombinant expression of hydrophobic proteins is notoriously challenging, typically low yielding and does not offer the same level of flexibility as chemical synthesis with regard to solvents and the use of chaotropic agents or detergents.Liu and co-workers employed a removable arginine tag to solubilize a hydrophobic protein.238 The utility of the tag was demonstrated with the synthesis of unphosphorylated and Ser64 phosphorylated M2 proton channels from influenza A. The protein was synthesized from two fragments: the N-terminal fragment, an arginine tagged, phosphate bearing, peptide hydrazide, and the C-terminal fragment, carrying an N-terminal cysteine. The phosphoserine residue was introduced during SPPS with the Fmoc-Ser(HPO3Bzl)–OH building block. The ligation proceeded through the activation of the hydrazide with NaNO2 and thiolysis with MPAA. The synthetic protein was embedded into artificial membranes, and the measured generated potential showed little difference between non-phosphorylated and phosphorylated protein, nor did phosphorylation at this site affect inhibition by amantadine.
The KAHA ligation (see Section 3.3.2) offers properties that can facilitate the synthesis of membrane proteins.178 An ester linkage at the ligation site increases solubility compared to the native amide. The ligation (Fig. 24) is carried out under acidic conditions and tolerates high levels of organic co-solvent. The synthesis of interferon-induced transmembrane protein 3 (IFITM3), a 133 amino acid membrane protein, was achieved by ligating three fragments using KAHA chemistry. First, the C-terminal and middle fragments were ligated, followed by treatment with UV light to remove the nitrobenzyl-type protection at the N-terminal oxaproline residue. The phosphate-bearing N-terminal segment was synthesized on the cyanosulfurylide linker with the phosphotyrosine residue introduced using the monobenzyl-protected building block. Following cleavage from the resin, the peptide was treated with OXONE® to form the α-hydroxyacid at the C-terminus. This fragment was ligated as the final fragment in a mixture of NMP and water containing 0.1M oxalic acid. Finally, the full peptide was treated with base to afford the two homoserine amide junctions. IFITM3-pY20 and carboxyfluorescein-IFITM3 were successfully incorporated into artificial membranes to investigate their antiviral activity later.
 | ||
| Fig. 24 Synthesis of phosphorylated IFITM3 using the KAHA ligation. Uniprot accession code: Q01628. PDB file from AlphaFold.239 | ||
Premdjee et al.240 synthesized the 290 residue phosphorylated insulin-like growth factor binding protein 2 (IGFBP-2) using a strategy that combined native chemical ligation and diselenide–selenoester ligation and deselenization, developed in the Payne lab (see Section 3.3.4) (Fig. 25). The diselenide–selenoester ligation can overcome difficulties associated with hindered junctions and even enables ligation using proline selenoester that would otherwise be unreactive as a thioester. The phosphorylated protein was divided into three larger fragments, each constructed from multiple smaller fragments, to be ligated via native chemical ligation. Firstly, an N-terminal fragment bearing a C-terminal thioester, secondly a middle fragment bearing an N-terminal cysteine and a latent C-terminal thioester, and finally a C-terminal fragment with an N-terminal cysteine.
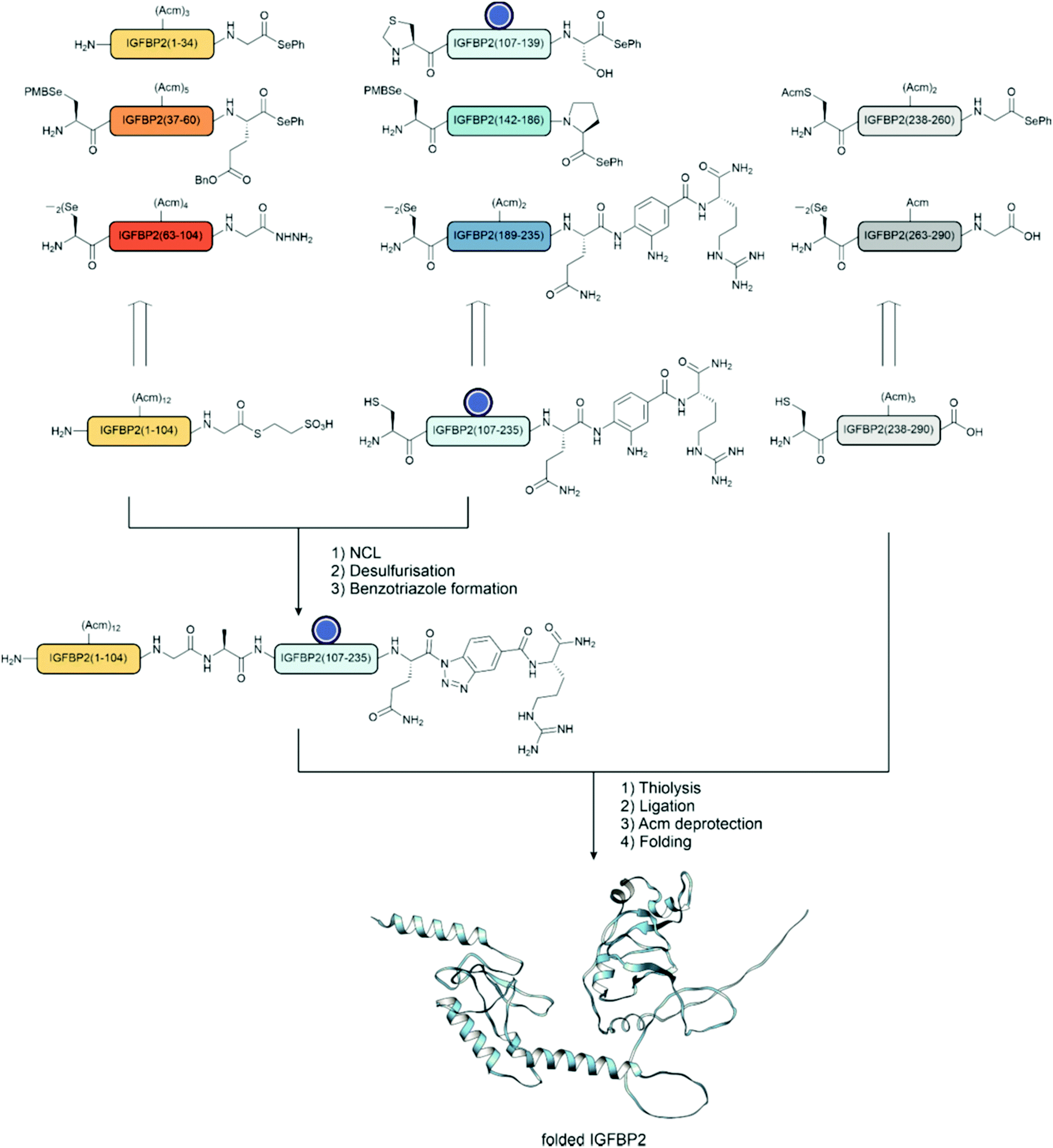 | ||
| Fig. 25 IGFBP-2 was synthesized from 3 key fragments. Fragment A (IGFBP-2 1–104) was prepared from three fragments in a C to N fashion such that a hydrazide remained available for subsequent ligation. Fragment B (IGFBP-2 107–235) was prepared from three fragments in a C to N fashion with a C-terminal Dbz moiety available for later activation. Fragment C (IGFBP2 238–290) was prepared from two fragments by DSL. The three fragments were then ligated in an N to C fashion, where the hydrazide was activated and converted to a thioester and ligated. Next, the Dbz group of the former middle fragment was converted to an acyl benzotriazole and thiolysed to the thioester and ligated with fragment C to obtain the 290-mer protein bearing one phosphorylation. Uniprot accession code: P18065. PDB file from AlphaFold.239 | ||
Each fragment was assembled using a diselenide–selenoester ligation and deselenization strategy and was designed to bear the appropriate reactive groups for the subsequent native chemical ligations. All cysteine residues were Acm protected to prevent side reactions during ligation or deselenization. The first 104 amino acid N-terminal fragment was prepared from three fragments using two diselenide–selenoester ligations in a C to N direction. This achieved a fragment with a C-terminal hydrazide available for ligation. The C-terminal hydrazide was activated with sodium nitrite and converted to the MesNa thioester before ligation. The middle fragment was also assembled from three fragments using two diselenide–selenoester ligations in a C to N direction, and finally, the thiazolidine was deprotected. In this fragment, for example, a prolyl phenylselenoester was applied at one ligation junction due to the lack of available cysteines for ligation. This afforded a fragment bearing an N-terminal cysteine with a C-terminal Dbz available for activation. The C-terminal fragment was assembled by ligation to afford a fragment bearing an N-terminal cysteine. Following ligation, selenocysteine residues were deselenized by treatment with TCEP and DTT to afford the native alanine residues.
The three fragments were combined in two ligations in the C to N direction to afford the full length phosphorylated IGFBP-2. In the first ligation, the N-terminal and middle fragments were ligated, followed by desulfurization of the cysteine at the ligation junction. In the next ligation step, the C-terminal Dbz was activated with sodium nitrite to form the acyl benzotriazole, which was transformed into the MesNa thioester in situ. Following the final ligation step, all 14 Acm groups were deprotected with silver acetate, and the complete protein was folded. In a competitive assay, the binding of IGF-1 was compared between the synthetic phosphorylated material and recombinant unphosphorylated materials, which showed that phosphorylation had little effect on binding.
Shiraishi et al.186 investigated the formation of the phosphorylated β2 adrenoreceptor (β2AR)–β-arrestin 1 complex. Upon binding of a GPCR agonist, the C-terminal domain of a GPCR is phosphorylated and binds to arrestin to initiate a response. In this study, NMR was used to analyse the structure of the phosphorylated complex. Protein trans-splicing was used to prepare 2H, 13C, 15N segmentally-labelled β2AR for NMR studies. The C-terminal region has seven serines and four threonines that have been identified as sites of phosphorylation. The protein was prepared from two fragments that were ligated using PTS: an unlabelled TM region bearing the C-terminal IntN tag was ligated with the isotopically labelled C-terminal fragment bearing an IntC tag and embedded into a lipid nanodisc. The phosphorylated version was prepared by treatment with the kinase, GRK2, following splicing. The authors found that phosphorylation causes adhesion to the intracellular membrane. Furthermore, it was found that the phosphorylated conformation closely resembles that of the β-arrestin bound state. However, one downside of this study is that enzymatic phosphorylation leads to a mixture of products. The authors note that some residues are over 95% phosphorylated, however, in other cases only less than 10% are.
Li et al.190 used a sortase ligation to prepare phosphorylated and ubiquitinated variants of β2AR. The N-terminal fragment bearing the C-terminal LPETG tag was prepared recombinantly. The C-terminal fragment bearing an N-terminal glycine and eight phosphorylated residues were prepared by Fmoc SPPS and ligation. As the octaphosphorylated peptide posed synthetic challenges due to the high number of phosphorylated residues, this had to be prepared as two fragments and ligated. The phosphorylated residues were introduced in the benzyl protected form and required the use of HATU for their successful coupling and all subsequent residues. Following purification, the fragments were ligated with sortase to afford the full-length octaphosphorylated β2AR. Furthermore, the monoubiquitinated-octaphosphorylated form as well as five different phosphoforms were synthesized. This library of differential modified proteins was used to investigate the protein's interaction with ligands and other proteins. For example, a competitive binding assay showed that phosphorylation largely did not affect orthosteric ligand binding but that phosphorylation could cause up to a 10-fold increase in the affinity of the allosteric β-arrestin 1 mediated agonist binding. They were also able to show that the position of phosphorylation clusters had a strong influence on the strength of the β2AR–β-arrestin 1 interaction.
Khoo et al.187 applied tandem trans-splicing to determine the effect of phosphorylation on the voltage-gated sodium channel, NaV1.5. Tandem trans-splicing uses two orthogonal split intein pairs. In this example, a synthetic peptide bearing a phosphonylated tyrosine mimic was spliced into the middle of two recombinantly expressed fragments in order to synthesize the modified NaV1.5. The peptide was prepared by Fmoc-SPPS and ligation, bearing the phosphonotyrosine residue and flanked with the appropriate intein tags at the N- and C-termini. The three fragments were coinjected into a Xenopus laevis oocyte and the product was detected by immunoblotting. The phosphonylated variant caused a 15 mV shift in the channel's inactivation properties.
5 Outlook and summary
Over 100 years ago, the first attempt was made to phosphorylate a protein by treating egg albumin with phosphorus oxychloride.241 Nowadays, we have far more refined tools to generate specific phosphoforms of a given protein thanks to important developments in phosphorylated building blocks, powerful ligation chemistry, and analytical methods.Firstly, the currently available phosphorylated amino acid building blocks and the methods for their incorporation are essential to access a wide variety of phosphopeptides. Developments in protecting groups have largely overcome the fundamental problems of using phosphorylated building blocks, and where they cannot be applied other methods for phosphorylation exist. Secondly, ligation chemistry has enabled access to larger and more complex targets. Native chemical ligation (NCL) and extended methods are now extremely fast even at hindered junctions, allowing for a wide choice of retrosynthetic disconnections. Where NCL is not applicable, other ligation chemistries such as the KAHA ligation may excel and have some advantages over NCL. Ligation methods are also capable of joining multiple fragments allowing access to even larger proteins. Similarly, expressed protein ligation (EPL) exploits NCL and is an incredibly powerful tool to combine the atomic-level control of chemical synthesis with the ease of creating large proteins through recombinant expression. Finally, modern HPLC and MS technology enables real-time monitoring of ligation reactions and protein folding and can confirm the homogeneity of fragments, ensuring the success of a synthesis. Together, this has enabled access to phosphoforms that were previously impossible to reach by other means. Methods that enable ligations at low concentration of fragments or increase solubility of hydrophobic peptides will help access notoriously difficult transmembrane proteins, such as GPCRs, which were noticeably deficient in the literature.
However, despite all these advances, synthesizing any protein and all its phosphoforms remains an enormous task. Synthetic methods face a twofold challenge: firstly, to simply be able to incorporate multiple phosphorylated residues and, secondly, to make the significant number of possible phosphoform combinations. Furthermore, each protein sequence presents its own set of challenges for synthesis.
The synthesis of highly phosphorylated peptides and proteins is a considerable challenge for both chemical synthesis and genetic code expansion. In both cases, the incorporation of each subsequent phosphorylated residue becomes more difficult. With regard to chemical synthesis, it is clear that new protecting groups or new coupling reagents will be required to face this challenge. The problems stem from the free acid groups on protected phosphoserine and phosphothreonine building blocks. In a similar fashion to phosphotyrosine, phosphorodiamidate protected versions of these building blocks may provide a solution. With appropriate building blocks, access to the target is only limited by the capabilities of peptide synthesis. Alternatively, improved methods for on-line or post-synthetic phosphorylation could facilitate access.
A second key challenge is the synthesis of the number of possible phosphoforms. It is clear that different combinations of phosphorylation lead to distinct outcomes. Therefore, each is of interest to investigate. In combination with many other post-translational modifications, the number of isoforms then exponentially increases. There are two key bottlenecks in a typical chemical protein synthesis workflow: synthesis steps and HPLC purification steps. Microwave synthesis and flow technologies have reduced the time needed for peptide synthesis, yet peptide purification remains cumbersome, especially for insoluble fragments. With regard to purification and solubility issues, a growing number of labs have taken an interest in these particular challenges. Solubility-enhancing tags and solid-supported protein synthesis may provide solutions. We described how using a self-purifying thioester eliminated an HPLC purification step, and multiple ligations could easily be performed in a 96 well plate on an immobilized peptide. Additionally, we have also demonstrated the utility of purification tags and capture and release resins in chemical protein synthesis, which could find wider use with high throughput and to purify fragments for ligation.242,243 The purification of fragments and intermediates will be accelerated in the future with similar techniques as well as through automation of HPLC. Improved syntheses will reduce the number of side products, and additionally, one-pot methods reduce the number of purification steps needed in a synthesis.
It is particularly important to be able to investigate post-translationally modified proteins in cellulo because it is the authentic context of the protein. Live cell experiments can reveal a larger network of interactions that may not have been previously expected. Furthermore, using synthetic proteins overcomes the need to perform any genetic modification.
In future research, access to different phosphoforms will allow screening to find compounds that can selectively target a particular phosphoform. Given the role of phosphorylated proteins in pathogenesis, this may represent an important therapeutic strategy. We also expect that there will be an increased focus on non-canonical phosphorylations. O-Phosphorylation has held the spotlight primarily due to its importance in humans, however, phosphorylation at non-canonical sites is also important. For example, in the case of phosphohistidine, it has been estimated that it is between 10 to 100 times more abundant than phosphotyrosine.244 This will require similar developments in building blocks, stable mimics or methods to phosphorylate site-specifically.
In summary, methods have evolved to enable the study of more complex targets and a greater number of possible phosphoforms. The examples we discussed highlight the broad range of tools to access phosphoforms of interest. Their functional evaluation revealed a diversity of outcomes phosphorylation leads to in different biological systems. This knowledge contributes to our understanding of protein structure and function in health and disease. Yet, investigations of this type have only covered a small set of proteins and a narrow set of their phosphoforms, typically studied in the absence of other posttranslational modifications. The field is, therefore, awaiting methodological advances that accelerate the throughput of phosphoprotein synthesis.
Author contributions
T. B. and E. P. wrote the initial draft. O. S. was involved in funding acquisition, supervision, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – RTG2473 Project 392923329 and Se 819/20-1. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).245Notes and references
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS PubMed.
- P. V. Hornbeck, B. Zhang, B. Murray, J. M. Kornhauser, V. Latham and E. Skrzypek, Nucleic Acids Res., 2015, 43, D512–520 CrossRef CAS PubMed.
- G. A. Khoury, R. C. Baliban and C. A. Floudas, Sci. Rep., 2011, 1, 90 CrossRef CAS PubMed.
- G. Hardman, S. Perkins, P. J. Brownridge, C. J. Clarke, D. P. Byrne, A. E. Campbell, A. Kalyuzhnyy, A. Myall, P. A. Eyers, A. R. Jones and C. E. Eyers, EMBO J., 2019, 38, e100847 CrossRef CAS PubMed.
- R. Seger and E. G. Krebs, FASEB J., 1995, 9, 726–735 CrossRef CAS PubMed.
- T. Pawson and G. D. Gish, Cell, 1992, 71, 359–362 CrossRef CAS PubMed.
- P. Blume-Jensen and T. Hunter, Nature, 2001, 411, 355–365 CrossRef CAS PubMed.
- S. B. Liggett, Sci. Signaling, 2011, 4(185), pe36 CrossRef CAS PubMed.
- J. Needham Elise, L. Parker Benjamin, T. Burykin, E. James David and J. Humphrey Sean, Sci. Signaling, 2019, 12(565), eaau8645 CrossRef PubMed.
- X. Zhang, R. M. Johnson, I. Drulyte, L. Yu, A. Kotecha, R. Danev, D. Wootten, P. M. Sexton and M. J. Belousoff, Structure, 2021, 29, 963–974 CrossRef CAS PubMed .e966.
- A. C. Joerger, S. Rajagopalan, E. Natan, D. B. Veprintsev, C. V. Robinson and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17705 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- J. W. Perich, P. F. Alewood and R. B. Johns, Aust. J. Chem., 1991, 44, 233–252 CrossRef CAS.
- T. J. Attard, N. O’Brien-Simpson and E. C. Reynolds, Int. J. Pept. Res. Ther., 2007, 13, 447–468 CrossRef CAS.
- N. M. O’Brien-Simpson, T. J. Attard, A. Loganathan, N. L. Huq, K. J. Cross, P. F. Riley and E. C. Reynolds, Int. J. Pept. Res. Ther., 2007, 13, 469–478 CrossRef.
- S. Li and C. Dass, Anal. Biochem., 1999, 270, 9–14 CrossRef CAS PubMed.
- H. Steen, J. A. Jebanathirajah, J. Rush, N. Morrice and M. W. Kirschner, Mol. Cell. Proteomics, 2006, 5, 172–181 CrossRef CAS PubMed.
- M. Samarasimhareddy, G. Mayer, M. Hurevich and A. Friedler, Org. Biomol. Chem., 2020, 18, 3405–3422 RSC.
- J. W. Perich and E. C. Reynolds, Synlett, 1991, 577–578 CrossRef CAS.
- J. W. Perich, N. J. Ede, S. Eagle and A. M. Bray, Lett. Pept. Sci., 1999, 6, 91–97 CAS.
- W. C. Chang and P. D. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, New York, 2000 Search PubMed.
- T. Wakamiya, K. Saruta, J.-I. Yasuoka and S. Kusumoto, Chem. Lett., 1994, 1099–1102 CrossRef CAS.
- P. W. R. Harris, G. M. Williams, P. Shepherd and M. A. Brimble, Int. J. Pept. Res. Ther., 2008, 14, 387–392 CrossRef CAS.
- D. Imhof, D. Nothmann, M. S. Zoda, K. Hampel, J. Wegert, F. D. Bohmer and S. Reissmann, J. Pept. Sci., 2005, 11, 390–400 CrossRef CAS PubMed.
- R. M. Valerio, A. M. Bray, N. J. Maeji, P. O. Morgan and J. W. Perich, Lett. Pept. Sci., 1995, 2, 33–40 CrossRef CAS.
- G. K. Tóth, Z. Kele and G. Váradi, Curr. Org. Chem., 2007, 19 Search PubMed.
- M. Ueki, M. Goto, J. Okumura and Y. Ishii, Bull. Chem. Soc. Jpn., 1998, 71, 1887–1898 CrossRef CAS.
- M. Ueki, J. Tachibana, Y. Ishii, J. Okumura and M. Goto, Tetrahedron Lett., 1996, 37, 4953–4956 CrossRef CAS.
- H.-G. Chao, B. Leiting, P. D. Reiss, A. L. Burkhardt, C. E. Klimas, J. B. Bolen and G. R. Matsueda, J. Org. Chem., 1995, 60, 7710–7711 CrossRef CAS.
- H. Eberhard and O. Seitz, Org. Biomol. Chem., 2008, 6, 1349–1355 RSC.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, J. Biol. Chem., 1994, 269, 10444–10450 CrossRef CAS PubMed.
- M. E. Hahn and T. W. Muir, Angew. Chem., Int. Ed., 2004, 43, 5800–5803 CrossRef CAS PubMed.
- A. Nguyen, D. M. Rothman, J. Stehn, B. Imperiali and M. B. Yaffe, Nat. Biotechnol., 2004, 22, 993–1000 CrossRef CAS PubMed.
- T. Kawakami, H. Cheng, S. Hashiro, Y. Nomura, S. Tsukiji, T. Furuta and T. Nagamune, ChemBioChem, 2008, 9, 1583–1586 CrossRef CAS PubMed.
- W. J. Qian, C. C. Lai, J. A. Kelley and T. R. Burke, Jr., Chem. Biodivers., 2014, 11, 784–791 CrossRef CAS PubMed.
- C. Mathé, C. Périgaud, G. Gosselin and J.-L. Imbach, J. Org. Chem., 1998, 4 Search PubMed.
- W. Q. Liu, M. Vidal, C. Mathe, C. Perigaud and C. Garbay, Bioorg. Med. Chem. Lett., 2000, 10, 669–672 CrossRef CAS PubMed.
- W. Lu, K. Shen and P. A. Cole, Biochemistry, 2003, 42, 5461–5468 CrossRef CAS PubMed.
- R. B. Terrence, Curr. Trends Med. Chem., 2006, 6, 1465–1471 CrossRef PubMed.
- R. Hamilton, G. Kay, R. E. Shute, J. Travers, B. Walker and B. J. Walker, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 76, 127–130 CrossRef.
- J. W. Perich, D. P. Kelly and E. C. Reynolds, Int. J. Pept. Protein Res., 1992, 40, 81–88 CrossRef CAS PubMed.
- Y. Ueno, F. Suda, Y. Taya, R. Noyori, Y. Hayakawa and T. Hata, Bioorg. Med. Chem. Lett., 1995, 5, 823–826 CrossRef CAS.
- N. Mora, J. M. Lacombe and A. A. Pavia, Tetrahedron Lett., 1993, 34, 2461–2464 CrossRef CAS.
- T. Wakamiya, R. Togashi, T. Nishida, K. Saruta, J. Yasuoka, S. Kusumoto, S. Aimoto, K. Y. Kumagaye, K. Nakajima and K. Nagata, Bioorg. Med. Chem., 1997, 5, 135–145 CrossRef CAS PubMed.
- A. Arendt, K. Palczewski, W. T. Moore, R. M. Caprioli, J. H. McDowell and P. A. Hargrave, Int. J. Pept. Protein Res., 1989, 33, 468–476 CrossRef CAS PubMed.
- J. W. Perich, E. Terzi, E. Carnazzi, R. Seyer and E. Trifilieff, Int. J. Pept. Protein Res., 1994, 44, 305–312 CrossRef CAS PubMed.
- J. M. Lacombe, F. Andriamanampisoa and A. A. Pavia, Int. J. Pept. Protein Res., 1990, 36, 275–280 CrossRef CAS PubMed.
- J. Lukszo, D. Patterson, F. Albericio and S. A. Kates, Lett. Pept. Sci., 1996, 3, 157–166 CrossRef CAS.
- D. E. Petrillo, D. R. Mowrey, S. P. Allwein and R. P. Bakale, Org. Lett., 2012, 14, 1206–1209 CrossRef CAS PubMed.
- D. R. Mowrey, D. E. Petrillo, S. P. Allwein, S. Graf and R. P. Bakale, Org. Process Res. Dev., 2012, 16, 1861–1865 CrossRef CAS.
- R. Pascal, P.-O. Schmit, C. Mendre, M.-N. Dufour and G. Guillon, Peptides 2000 - Proceedings of the Twenty-Sixth European Peptide Symposium, Paris, 2001, pp. 263–264 Search PubMed.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
- T. J. Attard, N. M. O’Brien-Simpson and E. C. Reynolds, Int. J. Pept. Res. Ther., 2009, 15, 69–79 CrossRef CAS.
- J. T. Du, Y. M. Li, Q. F. Ma, W. Qiang, Y. F. Zhao, H. Abe, K. Kanazawa, X. R. Qin, R. Aoyagi, Y. Ishizuka, T. Nemoto and H. Nakanishi, Regul. Pept., 2005, 130, 48–56 CrossRef CAS PubMed.
- W. C. Chang and P. D. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, New York, 1999 Search PubMed.
- D. S. King, C. G. Fields and G. B. Fields, Int. J. Pept. Protein Res., 1990, 36, 255–266 CrossRef CAS PubMed.
- R. M. Valerio, A. M. Bray, N. J. Maeji, P. O. Morgan and J. W. Perich, Lett. Pept. Sci., 1995, 2, 33–40 CrossRef CAS.
- L. A. Carpino, H. Imazumi, B. M. Foxman, M. J. Vela, P. Henklein, A. El-Faham, J. Klose and M. Bienert, Org. Lett., 2000, 2, 2253–2256 CrossRef CAS PubMed.
- S. L. Pedersen, A. P. Tofteng, L. Malik and K. J. Jensen, Chem. Soc. Rev., 2012, 41, 1826–1844 RSC.
- M. Brandt, S. Gammeltoft and K. J. Jensen, Int. J. Pept. Res. Ther., 2006, 12, 349–357 CrossRef CAS.
- W. Li, N. M. O’Brien-Simpson, M. A. Hossain and J. D. Wade, Aust. J. Chem., 2020, 73(4), 271–276 CrossRef CAS.
- E. A. Kitas, J. D. Wade, R. B. Johns, J. W. Perich and G. W. Tregear, J. Chem. Soc., Chem. Commun., 1991,(5), 338–339 RSC.
- P. White and J. Beythien, Innovation and perspectives in solid phase synthesis & combinatorial libraries: peptides, proteins and nucleic acids – small molecule organic chemical diversity. Collected papers, 4th International Symposium, Birmingham, 1996, pp. 557–560.
- C. García-Echeverría, Lett. Pept. Sci., 1995, 2, 93–98 CrossRef.
- J. S. McMurray, D. R. t Coleman, W. Wang and M. L. Campbell, Biopolymers, 2001, 60, 3–31 CrossRef CAS PubMed.
- J. W. Perich and R. B. Johns, Tetrahedron Lett., 1988, 29, 2369–2372 CrossRef CAS.
- D. M. Andrews, J. Kitchin and P. W. Seale, Int. J. Pept. Protein Res., 1991, 38, 469–475 CrossRef CAS PubMed.
- H. B. A. de Bont, J. H. van Boom and R. M. J. Liskamp, Tetrahedron Lett., 1990, 31, 2497–2500 CrossRef CAS.
- H. B. A. de Bont, J. H. van Boom and R. M. J. Liskamp, Recl. Trav. Chim. Pays-Bas, 1990, 109, 27–28 CrossRef CAS.
- A. A. Bielska and N. J. Zondlo, Biochemistry, 2006, 45, 5527–5537 CrossRef CAS PubMed.
- G. Shapiro, R. Swoboda and U. Stauss, Tetrahedron Lett., 1994, 35, 869–872 CrossRef CAS.
- W. Bannwarth, E. Küng and T. Vorherr, Bioorg. Med. Chem. Lett., 1996, 6, 2141–2146 CrossRef CAS.
- E. A. Kitas, R. Knorr, A. Trzeciak and W. Bannwarth, Helv. Chim. Acta, 1991, 74, 1314–1328 CrossRef CAS.
- J. W. Perich, Lett. Pept. Sci., 1998, 5, 49–55 CAS.
- Q. Xu, E. A. Ottinger, N. A. Solé and G. Barany, Lett. Pept. Sci., 1997, 3, 333–342 CrossRef CAS.
- F. Daus, E. Pfeifer, K. Seipp, N. Hampp and A. Geyer, Org. Biomol. Chem., 2020, 18, 700–706 RSC.
- J. W. Perich, Lett. Pept. Sci., 1996, 3, 127–132 CrossRef CAS.
- Z. Kupihár, G. Váradi, É. Monostori and G. K. Tóth, Tetrahedron Lett., 2000, 41, 4457–4461 CrossRef.
- Z. Kupihar, Z. Kele and G. K. Toth, Org. Lett., 2001, 3, 1033–1035 CrossRef CAS PubMed.
- T. Johnson, L. C. Packman, C. B. Hyde, D. Owen and M. Quibell, J. Chem. Soc., Perkin Trans. 1, 1996, 719 RSC.
- V. V. Kalashnikov, Y. Tang and J. H. Pascal, in Understanding Biology Using Peptides, ed. S. E. Blondelle and S. E. Blondelle, New York, NY, 2006, pp. 222–224 Search PubMed.
- M. Samarasimhareddy, D. Mayer, N. Metanis, D. Veprintsev, M. Hurevich and A. Friedler, Org. Biomol. Chem., 2019, 17, 9284–9290 RSC.
- D. Grunhaus, A. Friedler and M. Hurevich, Eur. J. Org. Chem., 2021, 3737–3742 CrossRef CAS.
- C. C. Lechner and C. F. W. Becker, Chem. Sci., 2012, 3, 3500–3504 RSC.
- S. Peyrottes and C. Perigaud, Current Protocols in Nucleic Acid Chemistry, 2007, ch. 15, Unit 15, p. 13 Search PubMed.
- G. M. Rankin, D. Vullo, C. T. Supuran and S. A. Poulsen, J. Med. Chem., 2015, 58, 7580–7590 CrossRef CAS PubMed.
- A. Sickmann and H. E. Meyer, Proteomics, 2001, 1, 200–206 CrossRef CAS PubMed.
- K. F. Medzihradszky, N. J. Phillipps, L. Senderowicz, P. Wang and C. W. Turck, Protein Sci., 1997, 6, 1405–1411 CrossRef CAS PubMed.
- P. V. Attwood, K. Ludwig, K. Bergander, P. G. Besant, A. Adina-Zada, J. Krieglstein and S. Klumpp, Biochim. Biophys. Acta, 2010, 1804, 199–205 CrossRef CAS PubMed.
- U. M. Hohenester, K. Ludwig and S. Konig, Curr. Drug Delivery, 2013, 10, 58–63 CrossRef CAS PubMed.
- A. M. Marmelstein, J. Moreno and D. Fiedler, Top. Curr. Chem., 2017, 375, 22 CrossRef PubMed.
- A. M. Marmelstein, L. M. Yates, J. H. Conway and D. Fiedler, J. Am. Chem. Soc., 2014, 136, 108–111 CrossRef CAS PubMed.
- L. M. Yates and D. Fiedler, ChemBioChem, 2015, 16, 415–423 CrossRef CAS PubMed.
- Alan M. Marmelstein, J. A. M. Morgan, M. Penkert, D. T. Rogerson, J. W. Chin, E. Krause and D. Fiedler, Chem. Sci., 2018, 9, 5929–5936 RSC.
- D. T. Rogerson, A. Sachdeva, K. Wang, T. Haq, A. Kazlauskaite, S. M. Hancock, N. Huguenin-Dezot, M. M. K. Muqit, A. M. Fry, R. Bayliss and J. W. Chin, Nat. Chem. Biol., 2015, 11, 496–503 CrossRef CAS PubMed.
- G. Mann, G. Satish, P. Sulkshane, S. Mandal, M. H. Glickman and A. Brik, Chem. Commun., 2021, 57, 9438–9441 RSC.
- J. Xie, L. Supekova and P. G. Schultz, ACS Chem. Biol., 2007, 2, 474–478 CrossRef CAS PubMed.
- J. Dadová, S. R. G. Galan and B. G. Davis, Curr. Opin. Chem. Biol., 2018, 46, 71–81 CrossRef PubMed.
- K. P. Chooi, S. R. G. Galan, R. Raj, J. McCullagh, S. Mohammed, L. H. Jones and B. G. Davis, J. Am. Chem. Soc., 2014, 136, 1698–1701 CrossRef CAS PubMed.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS PubMed.
- A. Yang, S. Ha, J. Ahn, R. Kim, S. Kim, Y. Lee, J. Kim, D. Söll, H.-Y. Lee and H.-S. Park, Science, 2016, 354, 623 CrossRef CAS PubMed.
- T. H. Wright, B. J. Bower, J. M. Chalker, G. J. Bernardes, R. Wiewiora, W. L. Ng, R. Raj, S. Faulkner, M. R. Vallee, A. Phanumartwiwath, O. D. Coleman, M. L. Thezenas, M. Khan, S. R. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, aag1465 CrossRef PubMed.
- R. Serwa, I. Wilkening, G. Del Signore, M. Mühlberg, I. Claußnitzer, C. Weise, M. Gerrits and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2009, 48, 8234–8239 CrossRef CAS PubMed.
- J. Bertran-Vicente, R. A. Serwa, M. Schümann, P. Schmieder, E. Krause and C. P. R. Hackenberger, J. Am. Chem. Soc., 2014, 136, 13622–13628 CrossRef CAS PubMed.
- J.-M. Kee, B. Villani, L. R. Carpenter and T. W. Muir, J. Am. Chem. Soc., 2010, 132, 14327–14329 CrossRef CAS PubMed.
- R. L. Saxl, G. S. Anand and A. M. Stock, Biochemistry, 2001, 40, 12896–12903 CrossRef CAS PubMed.
- P. E. Paz, S. Wang, H. Clarke, X. Lu, D. Stokoe and A. Abo, Biochem. J., 2001, 356, 461–471 CrossRef CAS PubMed.
- K. E. Paleologou, A. W. Schmid, C. C. Rospigliosi, H.-Y. Kim, G. R. Lamberto, R. A. Fredenburg, P. T. Lansbury, C. O. Fernandez, D. Eliezer, M. Zweckstetter and H. A. Lashuel, J. Biol. Chem., 2008, 283, 16895–16905 CrossRef CAS PubMed.
- W. Zheng, Z. Zhang, S. Ganguly, J. L. Weller, D. C. Klein and P. A. Cole, Nat. Struct. Mol. Biol., 2003, 10, 1054–1057 CrossRef CAS PubMed.
- N. Balasuriya, M. T. Kunkel, X. Liu, K. K. Biggar, S. S. C. Li, A. C. Newton and P. O'Donoghue, J. Biol. Chem., 2018, 293, 10744–10756 CrossRef CAS PubMed.
- A. M. Pasapera, I. C. Schneider, E. Rericha, D. D. Schlaepfer and C. M. Waterman, J. Cell Biol., 2010, 188, 877–890 CrossRef CAS PubMed.
- H. S. Park, M. J. Hohn, T. Umehara, L. T. Guo, E. M. Osborne, J. Benner, C. J. Noren, J. Rinehart and D. Soll, Science, 2011, 333, 1151–1154 CrossRef CAS PubMed.
- M. S. Zhang, S. F. Brunner, N. Huguenin-Dezot, A. D. Liang, W. H. Schmied, D. T. Rogerson and J. W. Chin, Nat. Methods, 2017, 14, 729–736 CrossRef CAS PubMed.
- T. Arslan, S. V. Mamaev, N. V. Mamaeva and S. M. Hecht, J. Am. Chem. Soc., 1997, 119, 10877–10887 CrossRef CAS.
- J. W. Chin, Nature, 2017, 550, 53–60 CrossRef CAS PubMed.
- H. Chen, S. Venkat, P. McGuire, Q. Gan and C. Fan, Molecules, 2018, 23, 1662 CrossRef PubMed.
- S. B. H. Kent, Protein Sci., 2019, 28, 313–328 CrossRef CAS PubMed.
- T. Wöhr, F. Wahl, A. Nefzi, B. Rohwedder, T. Sato, X. Sun and M. Mutter, J. Am. Chem. Soc., 1996, 118, 9218–9227 CrossRef.
- V. Cardona, I. Eberle, S. Barthélémy, J. Beythien, B. Doerner, P. Schneeberger, J. Keyte and P. D. White, Int. J. Pept. Res. Ther., 2008, 14, 285–292 CrossRef CAS.
- H. M. Yu, S. T. Chen and K. T. Wang, J. Org. Chem., 1992, 57, 4781–4784 CrossRef CAS.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Science, 2020, 368, 980–987 CrossRef CAS PubMed.
- H. Sun, S. M. Mali, S. K. Singh, R. Meledin, A. Brik, Y. T. Kwon, Y. Kravtsova-Ivantsiv, B. Bercovich and A. Ciechanover, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7805 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- J. J. Ling, C. Fan, H. Qin, M. Wang, J. Chen, P. Wittung-Stafshede and T. F. Zhu, Angew. Chem., Int. Ed., 2020, 59, 3724–3731 CrossRef CAS PubMed.
- F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232–1240 CrossRef CAS PubMed.
- S. Aimoto, Biopolymers, 1999, 51, 247–265 CrossRef CAS PubMed.
- T. Kawakami, K. Hasegawa, K. Teruya, K. Akaji, M. Horiuchi, F. Inagaki, Y. Kurihara, S. Uesugi and S. Aimoto, J. Pept. Sci., 2001, 7, 474–487 CrossRef CAS PubMed.
- N. Stuhr-Hansen, T. S. Wilbek and K. Strømgaard, Eur. J. Org. Chem., 2013, 5290–5294 CrossRef CAS.
- F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232–1240 CrossRef CAS PubMed.
- G. W. Kenner, J. R. McDermott and R. C. Sheppard, J. Chem. Soc. D, 1971, 636 RSC.
- B. J. Backes and J. A. Ellman, J. Org. Chem., 1999, 64, 2322–2330 CrossRef CAS.
- R. Ingenito, E. Bianchi, D. Fattori and A. Pessi, J. Am. Chem. Soc., 1999, 121, 11369–11374 CrossRef CAS.
- M. Huse, M. N. Holford, J. Kuriyan and T. W. Muir, J. Am. Chem. Soc., 2000, 122, 8337–8338 CrossRef CAS.
- R. R. Flavell, M. Huse, M. Goger, M. Trester-Zedlitz, J. Kuriyan and T. W. Muir, Org. Lett., 2002, 4, 165–168 CrossRef CAS PubMed.
- F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2007, 46, 4577–4580 CrossRef CAS PubMed.
- R. Zitterbart and O. Seitz, Angew. Chem., Int. Ed., 2016, 55, 7252–7256 CrossRef CAS PubMed.
- A. R. Mezo, R. P. Cheng and B. Imperiali, J. Am. Chem. Soc., 2001, 123, 3885–3891 CrossRef CAS PubMed.
- S. Warriner, A. Nagalingam and S. Radford, Synlett, 2007, 2517–2520 CrossRef.
- G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS PubMed.
- Y. Wang and Y.-M. Li, in Expressed Protein Ligation: Methods and Protocols, ed. M. Vila-Perelló, Springer US, New York, NY, 2020, pp. 119–140 Search PubMed.
- J. S. Zheng, S. Tang, Y. K. Qi, Z. P. Wang and L. Liu, Nat. Protoc., 2013, 8, 2483–2495 CrossRef CAS PubMed.
- S. W. Pedersen, L. Albertsen, G. E. Moran, B. Levesque, S. B. Pedersen, L. Bartels, H. Wapenaar, F. Ye, M. Zhang, M. E. Bowen and K. Stromgaard, ACS Chem. Biol., 2017, 12, 2313–2323 CrossRef CAS PubMed.
- P. A. Cistrone, M. J. Bird, D. T. Flood, A. P. Silvestri, J. C. J. Hintzen, D. A. Thompson and P. E. Dawson, Curr. Protoc. Chem. Biol., 2019, 11, e61 CrossRef PubMed.
- D. T. Flood, J. C. J. Hintzen, M. J. Bird, P. A. Cistrone, J. S. Chen and P. E. Dawson, Angew. Chem., Int. Ed., 2018, 57, 11634–11639 CrossRef CAS PubMed.
- S. S. Kulkarni, E. E. Watson, J. W. C. Maxwell, G. Niederacher, J. Johansen-Leete, S. Huhmann, S. Mukherjee, A. R. Norman, J. Kriegesmann, C. F. W. Becker and R. J. Payne, Angew. Chem., Int. Ed., 2022, e202200163 CAS.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- J. B. Blanco-Canosa, B. Nardone, F. Albericio and P. E. Dawson, J. Am. Chem. Soc., 2015, 137, 7197–7209 CrossRef CAS PubMed.
- C. Zhan, K. Varney, W. Yuan, L. Zhao and W. Lu, J. Am. Chem. Soc., 2012, 134, 6855–6864 CrossRef CAS PubMed.
- K. P. Chiang, M. S. Jensen, R. K. McGinty and T. W. Muir, ChemBioChem, 2009, 10, 2182–2187 CrossRef CAS PubMed.
- J.-X. Wang, G.-M. Fang, Y. He, D.-L. Qu, M. Yu, Z.-Y. Hong and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 2194–2198 CrossRef CAS PubMed.
- J. Tailhades, N. A. Patil, M. A. Hossain and J. D. Wade, J. Pept. Sci., 2015, 21, 139–147 CrossRef CAS PubMed.
- P. Botti, M. Villain, S. Manganiello and H. Gaertner, Org. Lett., 2004, 6, 4861–4864 CrossRef CAS PubMed.
- K. P. Chiang, M. S. Jensen, R. K. McGinty and T. W. Muir, ChemBioChem, 2009, 10, 2182–2187 CrossRef CAS PubMed.
- K. Nakamura, T. Kanao, T. Uesugi, T. Hara, T. Sato, T. Kawakami and S. Aimoto, J. Pept. Sci., 2009, 15, 731–737 CrossRef CAS PubMed.
- J. Vizzavona, F. Dick and T. Vorherr, Bioorg. Med. Chem. Lett., 2002, 12, 1963–1965 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity, M. Morgan, C. Wolberger and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 4972–4976 CrossRef CAS PubMed.
- N. Ollivier, J. Vicogne, A. Vallin, H. Drobecq, R. Desmet, O. El Mahdi, B. Leclercq, G. Goormachtigh, V. Fafeur and O. Melnyk, Angew. Chem., Int. Ed., 2012, 51, 209–213 CrossRef CAS PubMed.
- J. Dheur, N. Ollivier, A. Vallin and O. Melnyk, J. Org. Chem., 2011, 76, 3194–3202 CrossRef CAS PubMed.
- H. Hojo, Y. Kwon, Y. Kakuta, S. Tsuda, I. Tanaka, K. Hikichi and S. Aimoto, Bull. Chem. Soc. Jpn., 1993, 66, 2700–2706 CrossRef CAS.
- H. Hojo, S. Yoshimura, M. Go and S. Aimoto, Bull. Chem. Soc. Jpn., 1995, 68, 330–336 CrossRef CAS.
- T. Kawakami, S. Yoshimura and S. Aimoto, Tetrahedron Lett., 1998, 39, 7901–7904 CrossRef CAS.
- K. Teruya, A. C. Murphy, T. Burlin, E. Appella and S. J. Mazur, J. Pept. Sci., 2004, 10, 479–493 CrossRef CAS PubMed.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- E. E. Watson, L. R. Malins and R. J. Payne, in Total Chemical Synthesis of Proteins, ed. P. D. A. L. L. A. Brik, 2021 Search PubMed.
- S. Bondalapati, M. Jbara and A. Brik, Nat. Chem., 2016, 8, 407–418 CrossRef CAS PubMed.
- C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef CAS PubMed.
- L. E. Canne, S. J. Bark and S. B. H. Kent, J. Am. Chem. Soc., 1996, 118, 5891–5896 CrossRef CAS.
- J. Offer and P. E. Dawson, Org. Lett., 2000, 2, 23–26 CrossRef CAS PubMed.
- S. F. Loibl, Z. Harpaz and O. Seitz, Angew. Chem., Int. Ed., 2015, 54, 15055–15059 CrossRef CAS PubMed.
- S. F. Loibl, A. Dallmann, K. Hennig, C. Juds and O. Seitz, Chem. – Eur. J., 2018, 24, 3623–3633 CrossRef CAS PubMed.
- O. Fuchs, S. Trunschke, H. Hanebrink, M. Reimann and O. Seitz, Angew. Chem., Int. Ed., 2021, 60, 19483–19490 CrossRef CAS PubMed.
- L. Xu, Y. Zhang, Y.-M. Li and X.-F. Lu, Org. Biomol. Chem., 2020, 18(42), 8709–8715 RSC.
- M. Pan, Q. Zheng, S. Gao, Q. Qu, Y. Yu, M. Wu, H. Lan, Y. Li, S. Liu and J. Li, CCS Chem., 2019, 1, 476–489 CrossRef CAS.
- T. J. Harmand, C. E. Murar and J. W. Bode, Nat. Protoc., 2016, 11, 1130–1147 CrossRef CAS PubMed.
- T. J. Harmand, V. R. Pattabiraman and J. W. Bode, Angew. Chem., Int. Ed., 2017, 56, 12639–12643 CrossRef CAS PubMed.
- X. Li, H. Y. Lam, Y. Zhang and C. K. Chan, Org. Lett., 2010, 12, 1724–1727 CrossRef CAS PubMed.
- T. Li, H. Liu and X. Li, Org. Lett., 2016, 18, 5944–5947 CrossRef CAS PubMed.
- N. J. Mitchell, L. R. Malins, X. Liu, R. E. Thompson, B. Chan, L. Radom and R. J. Payne, J. Am. Chem. Soc., 2015, 137, 14011–14014 CrossRef CAS PubMed.
- M. Cargoët, V. Diemer, B. Snella, R. Desmet, A. Blanpain, H. Drobecq, V. Agouridas and O. Melnyk, J. Org. Chem., 2018, 83, 12584–12594 CrossRef PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS PubMed.
- R. E. Thompson and T. W. Muir, Chem. Rev., 2020, 120, 3051–3126 CrossRef CAS PubMed.
- H. Wu, Z. Hu and X.-Q. Liu, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9226–9231 CrossRef CAS PubMed.
- Y. Shiraishi, M. Natsume, Y. Kofuku, S. Imai, K. Nakata, T. Mizukoshi, T. Ueda, H. Iwaï and I. Shimada, Nat. Commun., 2018, 9, 194 CrossRef PubMed.
- K. K. Khoo, I. Galleano, F. Gasparri, R. Wieneke, H. Harms, M. H. Poulsen, H. C. Chua, M. Wulf, R. Tampé and S. A. Pless, Nat. Commun., 2020, 11, 2284 CrossRef CAS PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS PubMed.
- X. L. Tan, M. Pan, Y. Zheng, S. Gao, L. J. Liang and Y. M. Li, Chem. Sci., 2017, 8, 6881–6887 RSC.
- Y. Li, J. Heng, D. Sun, B. Zhang, X. Zhang, Y. Zheng, W.-W. Shi, T.-Y. Wang, J.-Y. Li, X. Sun, X. Liu, J.-S. Zheng, B. K. Kobilka and L. Liu, J. Am. Chem. Soc., 2021, 143, 17566–17576 CrossRef CAS PubMed.
- S. Lahiri, R. Seidel, M. Engelhard and C. F. W. Becker, Mol. BioSyst., 2010, 6, 2423–2429 RSC.
- E. Buchdunger, J. Zimmermann, H. Mett, T. Meyer, M. Müller, B. J. Druker and N. B. Lydon, Cancer Res., 1996, 56, 100 CAS.
- M. Huse and J. Kuriyan, Cell, 2002, 109, 275–282 CrossRef CAS PubMed.
- M. A. Young, S. Gonfloni, G. Superti-Furga, B. Roux and J. Kuriyan, Cell, 2001, 105, 115–126 CrossRef CAS PubMed.
- S. S. Taylor, M. M. Keshwani, J. M. Steichen and A. P. Kornev, Philos. Trans. R. Soc. London, Ser. B, 2012, 367, 2517–2528 CrossRef CAS PubMed.
- J. J. Ottesen, M. Huse, M. D. Sekedat and T. W. Muir, Biochemistry, 2004, 43, 5698–5706 CrossRef CAS PubMed.
- W. Lu, D. Gong, D. Bar-Sagi and P. A. Cole, Mol. Cell, 2001, 8, 759–769 CrossRef CAS PubMed.
- Z. Zhang, K. Shen, W. Lu and P. A. Cole, J. Biol. Chem., 2003, 278, 4668–4674 CrossRef CAS PubMed.
- A. Camara-Artigas and J. M. Martin-Garcia, 2014 DOI:10.2210/pdb4jjc/pdb.
- W. Chen and P. ten Dijke, Nat. Rev. Immunol., 2016, 16, 723–740 CrossRef CAS PubMed.
- M. Huse, T. W. Muir, L. Xu, Y.-G. Chen, J. Kuriyan and J. Massagué, Mol. Cell, 2001, 8, 671–682 CrossRef CAS PubMed.
- M. Huse, Y.-G. Chen, J. Massagué and J. Kuriyan, Cell, 1999, 96, 425–436 CrossRef CAS PubMed.
- K. Luger, A. W. Mader, R. K. Richmond, D. F. Sargent and T. J. Richmond, Nature, 1997, 389, 251–260 CrossRef CAS PubMed.
- S. Vijay-Kumar, C. E. Bugg and W. J. Cook, J. Mol. Biol., 1987, 194, 531–544 CrossRef CAS PubMed.
- M. T. Morgan, M. Haj-Yahya, A. E. Ringel, P. Bandi, A. Brik and C. Wolberger, Science, 2016, 351, 725 CrossRef CAS PubMed.
- W.-S. Lo, R. C. Trievel, J. R. Rojas, L. Duggan, J.-Y. Hsu, C. D. Allis, R. Marmorstein and S. L. Berger, Mol. Cell, 2000, 5, 917–926 CrossRef CAS PubMed.
- M. A. Shogren-Knaak, C. J. Fry and C. L. Peterson, J. Biol. Chem., 2003, 278, 15744–15748 CrossRef CAS PubMed.
- S. Lee, S. Oh, A. Yang, J. Kim, D. Soll, D. Lee and H. S. Park, Angew. Chem., Int. Ed., 2013, 52, 5771–5775 CrossRef CAS PubMed.
- S. Dovat, T. Ronni, D. Russell, R. Ferrini, B. S. Cobb and S. T. Smale, Genes Dev., 2002, 16, 2985–2990 CrossRef CAS PubMed.
- D. Jantz and J. M. Berg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7589–7593 CrossRef CAS PubMed.
- S. Chen, R. Maini, X. Bai, R. C. Nangreave, L. M. Dedkova and S. M. Hecht, J. Am. Chem. Soc., 2017, 139, 14098–14108 CrossRef CAS PubMed.
- G. Niederacher, D. Urwin, Y. Dijkwel, D. J. Tremethick, K. J. Rosengren, C. F. W. Becker and A. C. Conibear, RSC Chem. Biol., 2021, 2, 537–550 RSC.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10 Suppl, S10–S17 CrossRef PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- M. Haj-Yahya, P. Gopinath, K. Rajasekhar, H. Mirbaha, M. I. Diamond and H. A. Lashuel, Angew. Chem., Int. Ed., 2020, 59, 4059–4067 CrossRef CAS PubMed.
- F. Liu, B. Li, E. J. Tung, I. Grundke-Iqbal, K. Iqbal and C.-X. Gong, Eur. J. Neurosci., 2007, 26, 3429–3436 CrossRef PubMed.
- D. Ellmer, M. Brehs, M. Haj-Yahya, H. A. Lashuel and C. F. W. Becker, Angew. Chem., Int. Ed., 2019, 58, 1616–1620 CrossRef CAS PubMed.
- S. Brahmachari, P. Ge, S. H. Lee, D. Kim, S. S. Karuppagounder, M. Kumar, X. Mao, J. H. Shin, Y. Lee, O. Pletnikova, J. C. Troncoso, V. L. Dawson, T. M. Dawson and H. S. Ko, J. Clin. Investig., 2016, 126, 2970–2988 CrossRef PubMed.
- I. Dikiy, B. Fauvet, A. Jovičić, A.-L. Mahul-Mellier, C. Desobry, F. El-Turk, A. D. Gitler, H. A. Lashuel and D. Eliezer, ACS Chem. Biol., 2016, 11, 2428–2437 CrossRef CAS PubMed.
- B. Pan, E. Rhoades and E. J. Petersson, ACS Chem. Biol., 2020, 15, 640–645 CrossRef CAS PubMed.
- M. Hejjaoui, S. Butterfield, B. Fauvet, F. Vercruysse, J. Cui, I. Dikiy, M. Prudent, D. Olschewski, Y. Zhang, D. Eliezer and H. A. Lashuel, J. Am. Chem. Soc., 2012, 134, 5196–5210 CrossRef CAS PubMed.
- A. Ansaloni, Z. M. Wang, J. S. Jeong, F. S. Ruggeri, G. Dietler and H. A. Lashuel, Angew. Chem., Int. Ed., 2014, 53, 1928–1933 CrossRef CAS PubMed.
- Z. Tan, X. Guan, P. Chaffey, Y. Ruan, C. Hurd and D. Taatjes, Synlett, 2017, 1917–1922 CrossRef.
- S. Margiola, K. Gerecht and M. M. Müller, Chem. Sci., 2021, 12, 8563–8570 RSC.
- L. Cappadocia and C. D. Lima, Chem. Rev., 2018, 118, 889–918 CrossRef CAS PubMed.
- M. H. Glickman and A. Ciechanover, Physiol. Rev., 2002, 82, 373–428 CrossRef CAS PubMed.
- S. Bondalapati, W. Mansour, M. A. Nakasone, S. K. Maity, M. H. Glickman and A. Brik, Chemistry, 2015, 21, 7360–7364 CrossRef CAS PubMed.
- Y. Sato, A. Yoshikawa, A. Yamagata, H. Mimura, M. Yamashita, K. Ookata, O. Nureki, K. Iwai, M. Komada and S. Fukai, Nature, 2008, 455, 358–362 CrossRef CAS PubMed.
- M. Renatus, S. G. Parrado, A. D'Arcy, U. Eidhoff, B. Gerhartz, U. Hassiepen, B. Pierrat, R. Riedl, D. Vinzenz, S. Worpenberg and M. Kroemer, Structure, 2006, 14, 1293–1302 CrossRef CAS PubMed.
- N. Huguenin-Dezot, V. De Cesare, J. Peltier, A. Knebel, Y. A. Kristaryianto, D. T. Rogerson, Y. Kulathu, M. Trost and J. W. Chin, Cell Rep., 2016, 16, 1180–1193 CrossRef CAS PubMed.
- C. Hoppmann, A. Wong, B. Yang, S. Li, T. Hunter, K. M. Shokat and L. Wang, Nat. Chem. Biol., 2017, 13, 842–844 CrossRef CAS PubMed.
- S. Isogai, D. Morimoto, K. Arita, S. Unzai, T. Tenno, J. Hasegawa, Y.-S. Sou, M. Komatsu, K. Tanaka, M. Shirakawa and H. Tochio, J. Biol. Chem., 2011, 286, 31864–31874 CrossRef CAS PubMed.
- C. Löw, N. Homeyer, U. Weininger, H. Sticht and J. Balbach, ACS Chem. Biol., 2008, 4, 53–63 CrossRef PubMed.
- A. Kumar, M. Gopalswamy, A. Wolf, D. J. Brockwell, M. Hatzfeld and J. Balbach, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3344–3349 CrossRef CAS PubMed.
- M. Msallam, H. Sun, R. Meledin, P. Franz and A. Brik, Chem. Sci., 2020, 11, 5526–5531 RSC.
- D. H. Brotherton, V. Dhanaraj, S. Wick, L. Brizuela, P. J. Domaille, E. Volyanik, X. Xu, E. Parisini, B. O. Smith, S. J. Archer, M. Serrano, S. L. Brenner, T. L. Blundell and E. D. Laue, Nature, 1998, 395, 244–250 CrossRef CAS PubMed.
- N. Lahav, S. Rotem-Bamberger, J. Fahoum, E.-J. Dodson, Y. Kraus, R. Mousa, N. Metanis, A. Friedler and O. Schueler-Furman, ChemBioChem, 2020, 21, 1843–1851 CrossRef CAS PubMed.
- J. S. Zheng, M. Yu, Y. K. Qi, S. Tang, F. Shen, Z. P. Wang, L. Xiao, L. Zhang, C. L. Tian and L. Liu, J. Am. Chem. Soc., 2014, 136, 3695–3704 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 Search PubMed.
- B. Premdjee, A. S. Andersen, M. Larance, K. W. Conde-Frieboes and R. J. Payne, J. Am. Chem. Soc., 2021, 143, 5336–5342 CrossRef CAS PubMed.
- H. Bechhold, Biol. Chem., 1901, 34(2), 122–127 CrossRef CAS.
- R. Zitterbart, N. Berger, O. Reimann, G. T. Noble, S. Lüdtke, D. Sarma and O. Seitz, Chem. Sci., 2021, 12, 2389–2396 RSC.
- S. F. Loibl, Z. Harpaz, R. Zitterbart and O. Seitz, Chem. Sci., 2016, 7, 6753–6759 RSC.
- H. R. Matthews, Pharmacol. Ther., 1995, 67, 323–350 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |