
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptide probes for proteases – innovations and applications for monitoring proteolytic activity
Maria
Rodriguez-Rios
 a,
Alicia
Megia-Fernandez
a,
Daniel J.
Norman
b and
Mark
Bradley
*a
a,
Alicia
Megia-Fernandez
a,
Daniel J.
Norman
b and
Mark
Bradley
*a
aEaStCHEM School of Chemistry, University of Edinburgh, David Brewster Road, EH9 3FJ Edinburgh, UK. E-mail: Mark.Bradley@ed.ac.uk
bTechnical University of Munich, Trogerstrasse, 30, 81675, Munich, Germany
First published on 21st February 2022
Abstract
Proteases are excellent biomarkers for a variety of diseases, offer multiple opportunities for diagnostic applications and are valuable targets for therapy. From a chemistry-based perspective this review discusses and critiques the most recent advances in the field of substrate-based probes for the detection and analysis of proteolytic activity both in vitro and in vivo.
 Maria R. Rios | Maria Rodriguez-Rios received her bachelor's degree in Biochemistry from the Universitat Autonoma de Barcelona in 2015. She obtained a MSc in Drug Design and Biomedical Sciences from Edinburgh Napier University, working on the development of optical probes for cancer imaging. Following a short period working in industry, she started her PhD studies at the University of Edinburgh under the supervision of Professor Mark Bradley working on the synthesis of fluorogenic peptide-based probes for imaging inflammation. |
 Alicia Megia-Fernandez | Alicia Megia-Fernandez, MRSC, obtained her MSc degree in Chemistry and PhD from the University of Granada, Spain, under the guidance of Professor Santoyo-Gonzalez, where she was awarded the Extraordinary PhD Award in Experimental Science. This was followed by the award of Fundación Ramón Areces and Marie Curie IEF Fellowships to work as a postdoctoral researcher at the University of Edinburgh with Professor Bradley. In 2022 she was appointed to a Maria Zambrano Senior Fellowship at the University of Granada. Her current research interests are in Chemical Biology and include chemical methodology and molecular imaging probes for diagnosis and treatment of different diseases. |
 Daniel J. Norman | Dr Daniel Norman obtained his BSc (Hons) at the University of Dundee in Medicinal Chemistry and then worked on the development of peptide-based hydrogels at Biogelx. He obtained his PhD in Chemistry at the University of Edinburgh under the guidance of Prof. Mark Bradley with a research focus on novel prodrug activation methods for cancer treatment. Following a post-doctorate with Professor Bradley developing diagnostics for tuberculosis, he began a post-doc position at the Technical University of Munich with Dr Hannelore Meyer in drug discovery of novel antibiotics and β-lactamase inhibitors for resistant infections. |
 Mark Bradley | Professor Mark Bradley has led research groups in the UK for over 30 years with funding from across the research councils and a variety of commercial and translational concerns. He has published over 440 papers and book chapters, has an H-factor of 62, filed >25 patents and spun-out five (still active) companies. The main focus of the Bradley group is in the area of life-sciences, specifically the application of the tools of chemistry to allow the manipulation, control and understanding of specific biological processes and functions and to address specific biological questions, problems in the areas of fluorescent reporters, smart materials/polymers and in vivo chemistry (see http://www.combichem.co.uk). |
Introduction
Proteases are enzymes with wide-ranging and crucial activities that mediate the hydrolysis of amide bonds. The residues within the active site of the enzyme responsible for the biological activity allow the partitioning of these enzymes into the five major classes: so-called serine, threonine, cysteine, aspartic acid or metallo-based proteases. Cleavage by proteases typically occurs on well-defined substrates to allow for discrete and precise regulation of biological functions and takes place between two amino acids (P1–P1′) (by endopeptidases) or at the N- or C-terminus of the peptide (by exopeptidases). Proteases need to recognise their substrate to allow exertion of its function, with the most crucial amino acid being that located on the P1 position of the scissile bond. The degree of substrate specificity is variable, with some proteases accepting a broad spectrum of substrates and others accepting very few.The function of proteases is essential for an abundance of cellular processes, that includes matrix remodelling by metalloproteinases during cell growth, angiogenesis and tissue remodelling;1 regulation of cell division by the calpain family2 and the control of programmed cell death by caspases.3 These physiological proteolytic functions are tightly regulated and dysfunction of proteases can be highly detrimental and has been linked to a variety of diseases with examples including cancer,4 diabetes,5 inflammation,6 vascular diseases7 and Alzheimer's.8 Indeed, Alzheimer's disease, which is characterised by a cognitive decline, is associated with an accumulation of amyloid-β (Aβ) plaques produced by a sequence of proteolytic events of the amyloid precursor protein by α-, β- and γ-secretases that result in aggregation and generation of Aβ.8 The disease-causing proteolytic network of Alzheimer's disease exemplifies the need for specific and versatile protease probes in order to distinguish and elucidate the biochemical processes responsible, such as chemical probes that enable the specific identification of the aspartyl protease γ-secretase.9 Proteases have emerged as excellent disease biomarkers. For instance, thrombin is widely used as a biomarker in the diagnosis of blood disorders and prostate cancer. Proteases are also attractive drug targets, with a variety of inhibitors developed used to treat diseases such as the HIV-1 protease inhibitors to treat AIDS,10 and captopril to treat hypertension.11
Understanding the role of proteases in disease and the evaluation of treatment efficacy are crucial for the success of the drug development process, but current gold-standard methods typically rely on in vitro detection by genetic expression measurements or immunostaining. However, these methods base their detection on indirect measurement of protease concentration instead of proteolytic activity and they cannot distinguish between active and inactive (pro-forms) of the protease. Peptide-based probes have emerged as an alternative to these methods to allow the evaluation of the activity of a variety of proteases.12,13 A key advantage is that these probes rely solely on “real” proteolytic activity and allow both in vitro and in vivo evaluation in real time. These probes typically contain a peptide-targeting moiety (that is recognised by the protease) and a reporter (e.g. a fluorophore, a contrast agent, an electrochemical tag or a mass tag). Peptides are ideal as the recognition element as they mimic the endogenous substrate and offer high natural specificity, while providing a scaffold for the attachment of labels or signal transducers.
In vivo imaging of proteolytic activity is becoming extremely useful in the area of diagnostics or for optical-image-guided surgery.14 Fluorescent peptide-based probes are widely used in optical imaging, with many efforts pushing into the near infrared imaging window of the spectrum,15 where biological tissues are silent and penetration of light is enhanced. In recent years, other approaches have made an appearance as promising alternatives to fluorescence-based technologies. These include chemiluminescent probes, that do not need an external light source and provide virtually no background signal, and photoacoustic imaging that offers great promise for deep-tissue imaging, with the main advantage of being able to image tissue at depths of more than 1 cm.
Bench-top or in vitro technologies for laboratory analysis of proteolytic activity have seen advances in the area of colorimetric detection; with applications for in vitro, low cost and easy-to read analysis, electrochemical probes, with redox-based detection of proteases offering an alternative to other methods where complex media samples might affect other detection methods such as fluorescence. The area of MS and proteomics has seen proteolytic profiling appearing with applications in disease diagnostics and comparative analysis of proteolytic activity in health and disease. In this review, we discuss recent advances in the area of probes for the detection of proteases and new strategies in the area of FRET, pro-fluorophores, bioorthogonal reactions, aggregation-induced emission and chemiluminescence based optical probes as well as methods based on photoacoustic contrast agents, electrochemical detection, mass spectrometry and colorimetric reporters.
Comprehensive reviews have been published in the field of protease activity and we would like to highlight those of Ong,16 Oliveira-Silva17 and Zhang.14
1. Optical imaging
1.1 Fluorescence
Cleavage stops the energy transfer (the quenching) of the fluorophore, “turning on” the fluorescence (Scheme 1). It is vital that peptide cleavable “spacers” display high specificity towards the target protease, while the signal to noise ratio of the cleaved and non-cleaved FRET pair is high. Many routes to discover optimal substrates/spacers for FRET protease assays exist. These include a variety of combinatorial approaches (both solid21,22 and solution based,23 MS substrate profiling methods,24,25 phage libraries26 or substrate modification of known substrates, including direct screening on complex tumour tissues.27 Many FRET peptide probes have been developed28–30 and there are reviews covering this area so here we highlight some key and novel examples14,19 with FRET based probes developed for legumain,31 thrombin,30 SARS-CoV protease,32 Granzyme B33 and Cathepsins,34 among others.
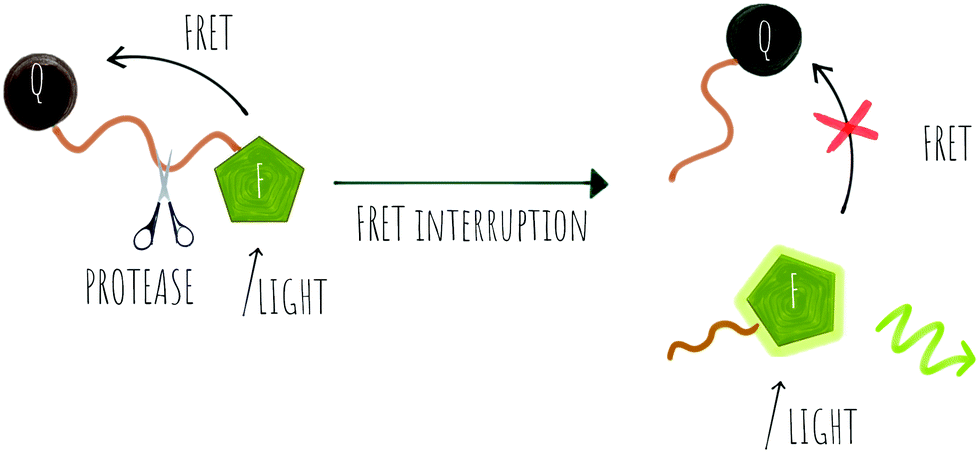 | ||
| Scheme 1 FRET probes where the light emitted by the fluorophore (F) is transferred to the quencher (Q) until the FRET is disrupted by the action of a protease causing the release of the fluorophore and restoring its emission. | ||
Key successes in this area include improvement of the signal to noise ratio of conventional linear FRET-probes, shifting the emission into the NIR range35 or by addition of chains of positively and negatively charged amino acids on opposite sides of the cleavage site that result in an “electrostatic zipper” whereby folding of the peptide brings the fluorophore and quencher into closer proximity via a hairpin-type structure to facilitate activity-dependent imaging.28,36 In these probes, not only does the hairpin enhance the quenching but the polycationic species promotes cellular entry upon cleavage. A useful review covering this field was published by Tsien,37 and clearly the concept could be extended to include oppositely charged fluorophores, that by themselves promote hairpin formation.
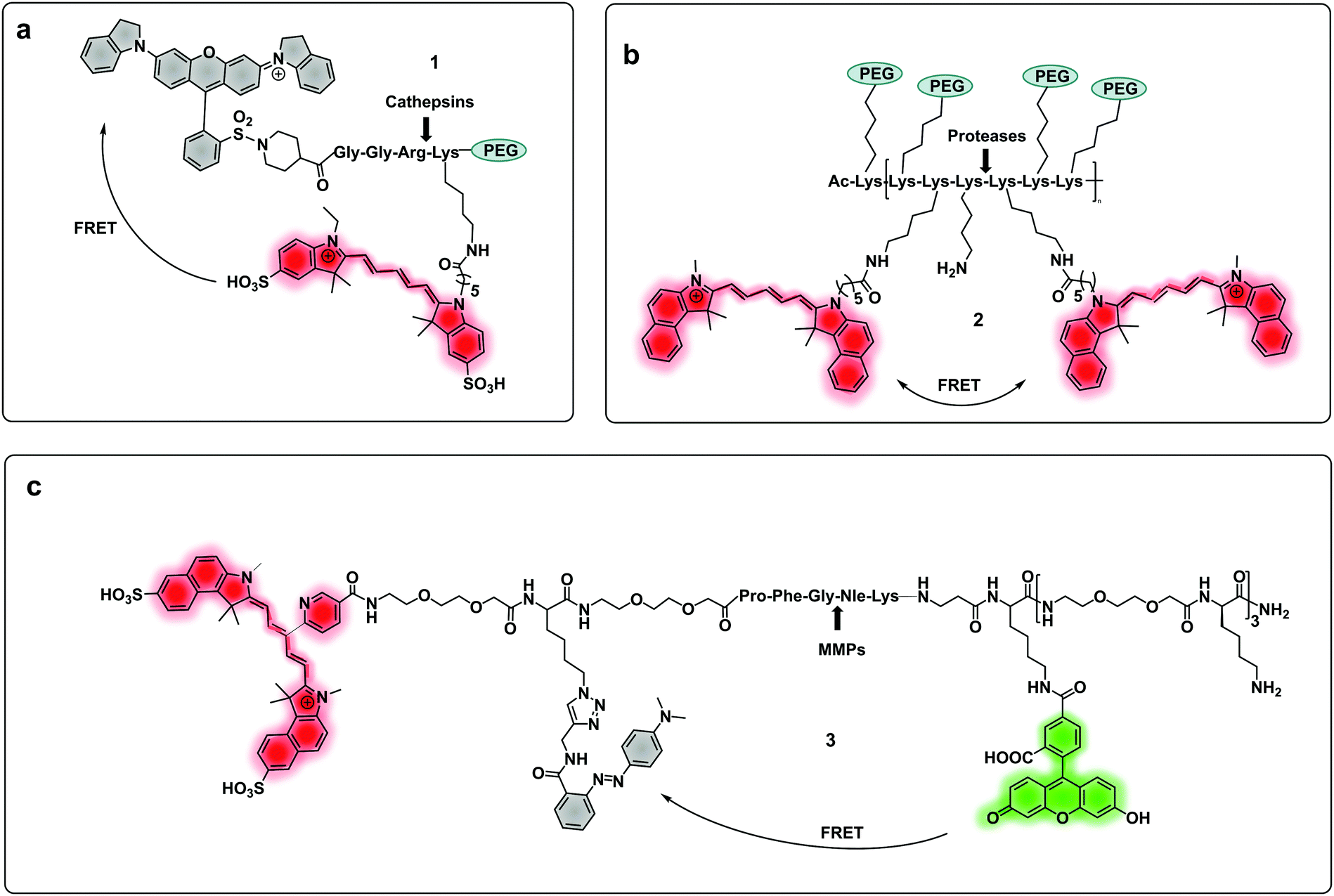 | ||
| Fig. 1 (a) A cathepsin reporter using the ICG/QSY21 FRET pair probe in clinical studies.40 (b) Structure of the generic “ProSense” probe using Cy5.5 homo-labelling.44 (c) MMP dual probe for painting tumours,46 with cleavage generating a Cy5.5 fragment that sticks to the tumor tissue. Also note that this probe is terminated with a D-Lys residue to provide proteolytic stability. | ||
Some FRET probes have shown excellent applicability in vivo. Thus, a broad spectrum probe for detection of cathepsins using sulfo-Cy5 and QSY21 as a FRET pair was developed38 and later optimized incorporating the FDA-approved dye ICG, as the NIR-emitting dye and was successfully used for fluorescence-guided-surgery in mouse and ex vivo in keratinocyte carcinoma.39 One probe40 (1), was recently validated for intraoperative assessment of cancer margins during surgery. It uses the same FRET pair and was designed for the detection of activated cathepsins K, L, S and B (Fig. 1a).41–43 A series of fluorescent FRET based probes (2) (ProSense), based on self-quenching of multiple copies of the same fluorophore (Cy5.5) within a repeating graft copolymer44 have successfully been developed for in vivo imaging in animal models for the detection of MMPs, cathepsins, renin and neutrophil elastase (Fig. 1b). Recently, a tribranched FRET based probe for sensing MMP activity45 in patients with fibroproliferative lung disease allowed in situ imaging of MMP activity and allowed the assessment of pharmacological action in human disease using optical molecular imaging. A “tumour painting probe” (3)46 (Fig. 1c) has also been developed that contained a green FRET pair (FAM/Methyl Red) on opposite sides of an MMP 2/9 cleavable sequence, that produced a signal upon protease cleavage. The construct also contained a NIR fluorophore that served as an “always on” reference (sulfoCy5). Thus, activation of the probe by MMPs resulted in two signals, the reference NIR dye and the liberated carboxyfluorescein. Interestingly, once the construct was cleaved, the reference NIR dye fragment, “painted” the tumour tissue allowing tumour margin visualization on ex vivo tissue (Fig. 3a).
Two-photo excitation (TPE) fluorescence has attracted much attention in the area of optical imaging as it allows for deeper and more accurate localisation of the probe (the focal volume for TPE is small) but the method also needs higher power laser sources compared to one-photon technology (although at far longer wavelengths). Probes based on TPE technology have been designed for the detection of caspase 3 using the generic substrate (DEVD)47 with the naphthalene–pyrazoline two-photon absorber coupled to a dabcyl quencher via a caspase 3 cleavable peptide. The construct also allowed for tuneable targetability with modification of the pyrazoline substituent accommodating a cRGD motif to target cancer cell uptake. Yan48 developed a two-photon fluorogenic reporter for caspase-3 using two-photon absorbing nanomicelles, with the β-cyclodextrin based micelles contained the two-photon absorbing dye trans-4-[p-(N,N-diethylamino)styryl]-N-methylpyridinium iodide. The micelles were modified with caspase 3 cleavable peptides labelled with the Black Hole Quencher (BHQ-2) that quenched the emission from the micelles until cleaved by the protease.
Multi-branched scaffolds37,49–52 have been used to reduce probe background signals and enhance signal amplification via the generation of multiples copies of the same fluorophore. This strategy initially used “self-quenching” between identical fluorophores of homo-labelled probes, where each chromophore had a dual role of being a donor as well as an acceptor.53,54 A self-quenching green emitting tri-branched dendrimer probe for human neutrophil elastase (HNE) was reported,50 with the multivalent scaffold consisting of three-peptide branches specifically cleaved by HNE, each capped with a 5-carboxyfluorescein group.55 In 2018, the concept was modified to include a quencher on each of the peptide sequences to allow the synthesis of a broad-spectrum serprocidin probe, yielding a so-called “super-silent probe” with negligible background fluorescence and much higher signal amplification.49 More recently, optimisation of the system allowed the synthesis of a highly specific HNE probe (4, Fig. 2a), with excellent signal amplification and negligible background fluorescencethat allowed visualisation of Neutrophil Extracellular Traps55 without the need of antibodies56 (Fig. 3b). Multivalent or dendrimeric FRET reporters have been reported where scaffolds are modified with FRET-peptide reporters and targeting moieties on a central core. Nagai used anionic carboxy-terminal polyamidoamine (PAMAM) based-dendrimers (5), known to accumulate in the lymph nodes, as a core for the incorporation of a FRET substrate peptide allowing the detection of MMP-257 (Fig. 2b). Brennecke used a tetravalent DOTAM scaffold to prepare probe (6) for the detection of Cathepsin S, each scaffold being modified to accommodate two copies of a cRGD tumour-directing peptide on two of the four carboxylic acids, a Cy3 on a third functionality and finally a Cathepsin S cleavable sequence containing a BHQ-2 quencher (Fig. 2c),58 on the fourth arm. The incorporation of FRET-reporters onto/into nanoparticles as carriers, offers opportunities for novel diagnostic applications. A recent example is the development of a non-invasive nanoprobe capable of detecting granzyme B dependent transplant rejection via analysis of urine samples. Exposure to iron oxide nanoparticles functionalised with FRET-labelled peptides, releases the fluorescent reporter into the urine and allows identification of the onset of rejection with high sensitivity and specificity.59,60 Detection of multiple target proteases offers the possibility of establishing molecular signatures of diseases and therapy monitoring.61 Polydopamine nanoparticles (PDANPs) have been used as broad-spectrum quenchers and carriers of multiple fluorophore-labelled peptides.62 Thus, a 4-colour nano-reporter (7) was developed by assembly of four differently labelled fluorescent peptide substrates (urokinase-type plasminogen activator (uPA), MMP-2, cathepsin B, and MMP-7) labelled with AF405, FITC, Cy3 and Cy5 respectively, onto the PDANPs via π–π interactions with aromatic amino acid residues in the peptides/dyes (Fig. 2d). Negatively charged fluorescent PDANPs have been used to build a system with protamine to form an aggregation-based quenching system (via a static quenching mechanism).63 The fluorescence quenching mechanism relying here on electrostatic interactions between the positively charged protamine and the negatively charged PADNPs with Trypsin cleavage de-quenching the system.
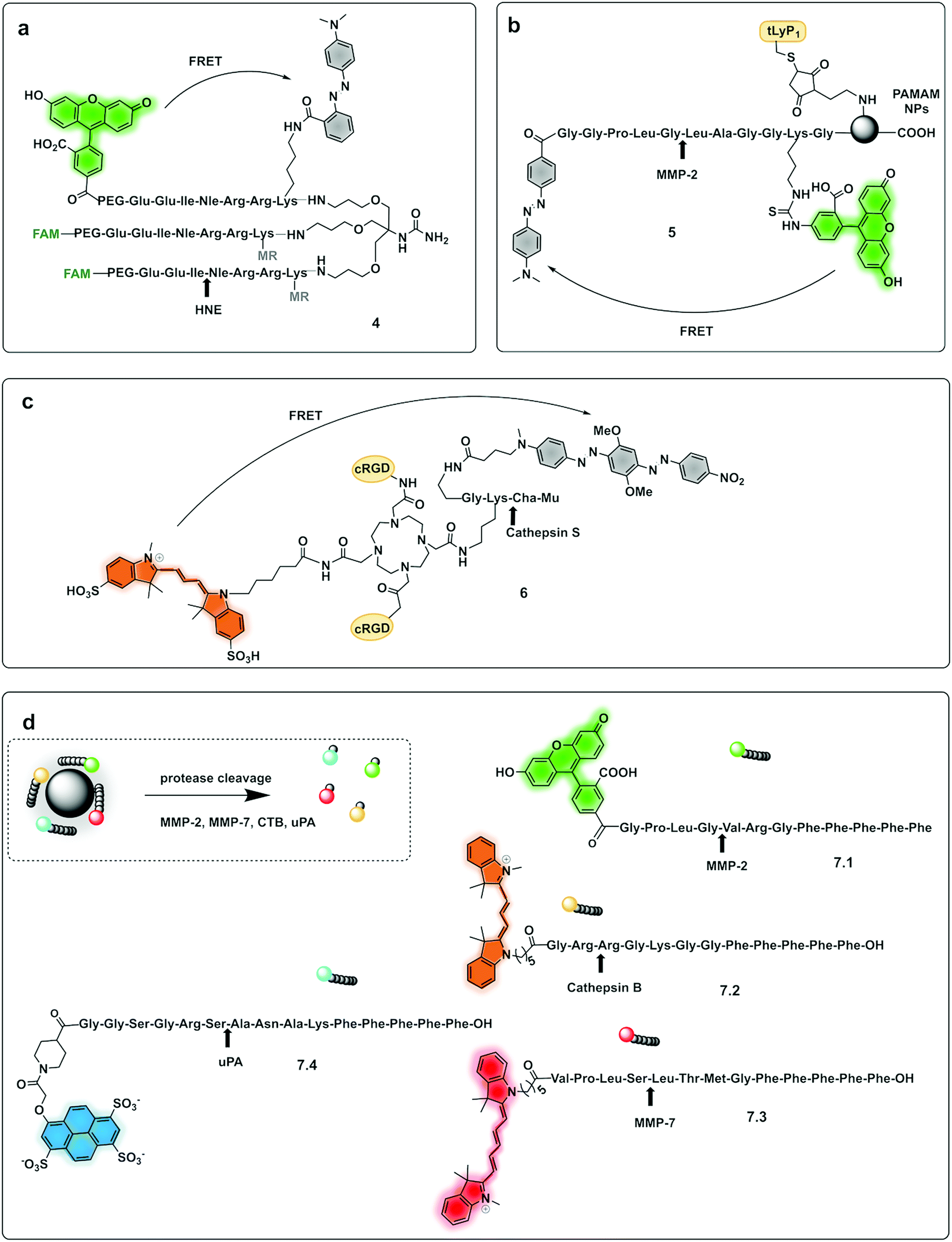 | ||
| Fig. 2 Dendrimer, multibranched and nanoparticle based FRET probes. (a) Tribranched probe for detection of human neutrophil elastase.56 (b) PAMAM-based fluorogenic dendrimers for detection of MMPs. tLyP-1: tumor homing peptide.57 (c) Tetravalent DOTAM-based probe for detection of cathepsin S.58 Mu: morpholine carboxamide; Cha: cyclohexylalanine. (d) 4-Colour nano-reporter for detection of uPA, MMP-2, cathepsin B and MMP-7.63 | ||
 | ||
| Fig. 3 (a) Fluorescence images delineating tumour margins using the MMP FRET probe 3.46 Upper row: Bright field microscopy image and fluorescence image of freshly excised lung slice, with pathologically identified adenocarcinoma (Ad), transition zone (Tr) and normal (N) tissue, following incubation with the probe. Lower row: Control tissue from the same patient sample without the addition of compound 3 was used as a measure of tissue autofluorescence within this spectral window. Reproduced from ref. 46 with permission from the Royal Society of Chemistry, copyright 2020. (b) Fluorescence microscopy image of a Neutrophil Extracellular Trap stained with HNE FRET probe 456 (green), DAPI (blue) and SYTOX orange (red). Arrows indicate chromatin studded with activated probe, indicative of chromatin release by activated cells. Reproduced from ref. 56 with permission from the Royal Society of Chemistry, copyright 2021. | ||
A probe (8) that allowed multiple molecular targets to be simultaneously analysed was demonstrated with a dual-enzyme probe64 for thrombin and MMPs, allowing detection of two distinct classes of proteolytic extracellular proteins that play important roles in early carcinogenesis. The probe construct was assembled by copper mediated “click” conjugation of two FRET-labelled substrates with orthogonal excitation–emission wavelengths (Fig. 4a). Widen recently reported an AND-gate (9) that relies on activation by two proteases for a fluorogenic response,65 in this system, a central fluorophore was flanked by two quencher-labelled peptides targeting different proteases, cleavage of both substrates and release of the two quenchers was required to restore the fluorescence of the central fluorophore (Fig. 4b).
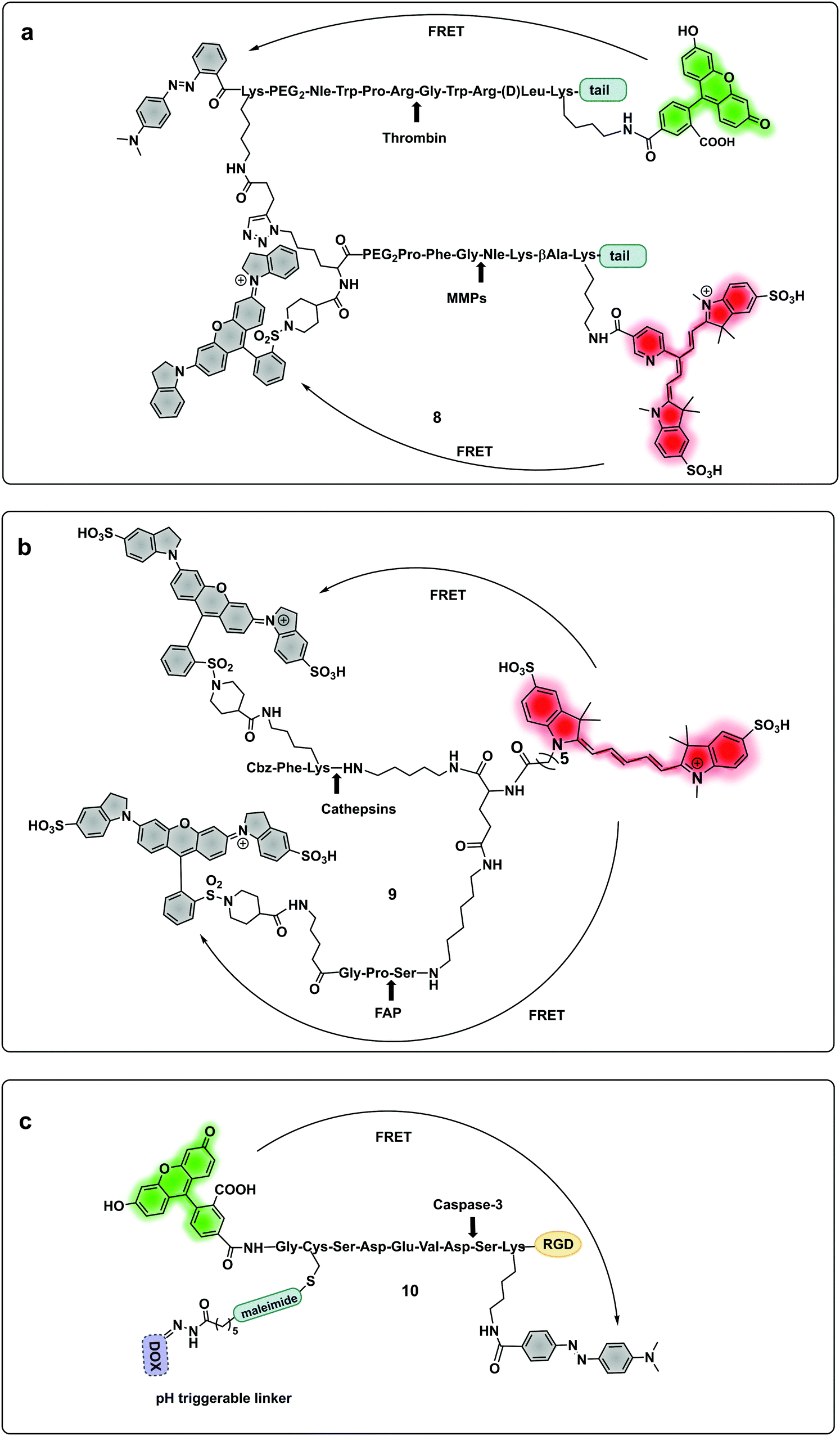 | ||
| Fig. 4 Multimodal probes. (a) Dual targeting probe for detection of thrombin and MMP.64 (b) AND-gate system based on dual quenching of one fluorophore activated following cleavage by cathepsins and FAP.65 (c) Theragnostic probe for detection of caspase-3.66 DOX: doxorubicin. | ||
Some protease-FRET probes initially used for diagnostics have been combined with therapeutic moieties to generate theragnostic entities allowing both diagnosis and therapy. Following this principle, Li66 developed a FRET-based (carboxyfluorescein–dabcyl) caspase 3 cleavable probe (10) conjugated to doxorubicin (DOX) and a targeting peptide (Fig. 4c). Enhanced delivery to the tumour tissue was enabled by the directing peptide, where the acidic pH in the cancer microenvironment (and during uptake in the endosome) triggered the release of DOX from the construct while caspase-3 cleaved the peptide resulting in a fluorescent signal. Transcription-dependent fluorogenic probes have been used for specific protease dependent activation or amplification of signal,67,68 while analysis kits for plasma and peripheral blood testing have been developed using polymer immobilization of FRET-based protease substrates.69
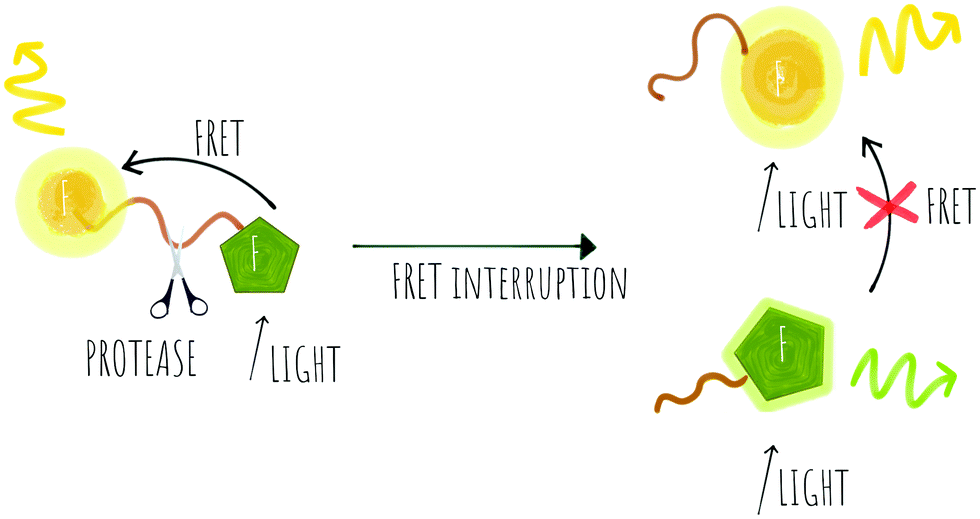 | ||
| Scheme 2 Concept of a probe that can be used for ratiometric FRET with energy transfer between two different fluorophores that is removed upon cleavage by proteases. | ||
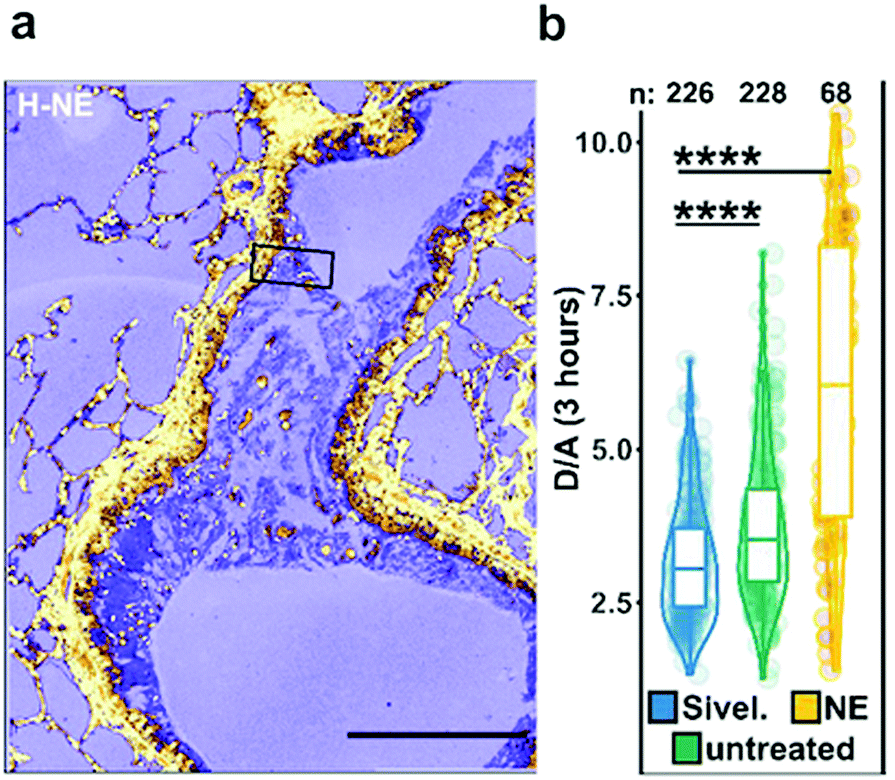 | ||
| Fig. 5 Ex vivo mouse lung slices stained with ratiometric elastase probe 15. (a) Confocal images of 5 μm thick lung slices from Scnn1b-Tg mice stained with compound 15 [2 μM] for 3 h. Scale bar: 200 μm. (b) Quantification of NE activity using the probe donor/acceptor (D/A) fluorescence on lung slices either untreated, pre-treated with Sivelestat or with elastase for 30 min before adding the probe. Reproduced from ref. 71 with permission from American Chemical Society, copyright 2020. | ||
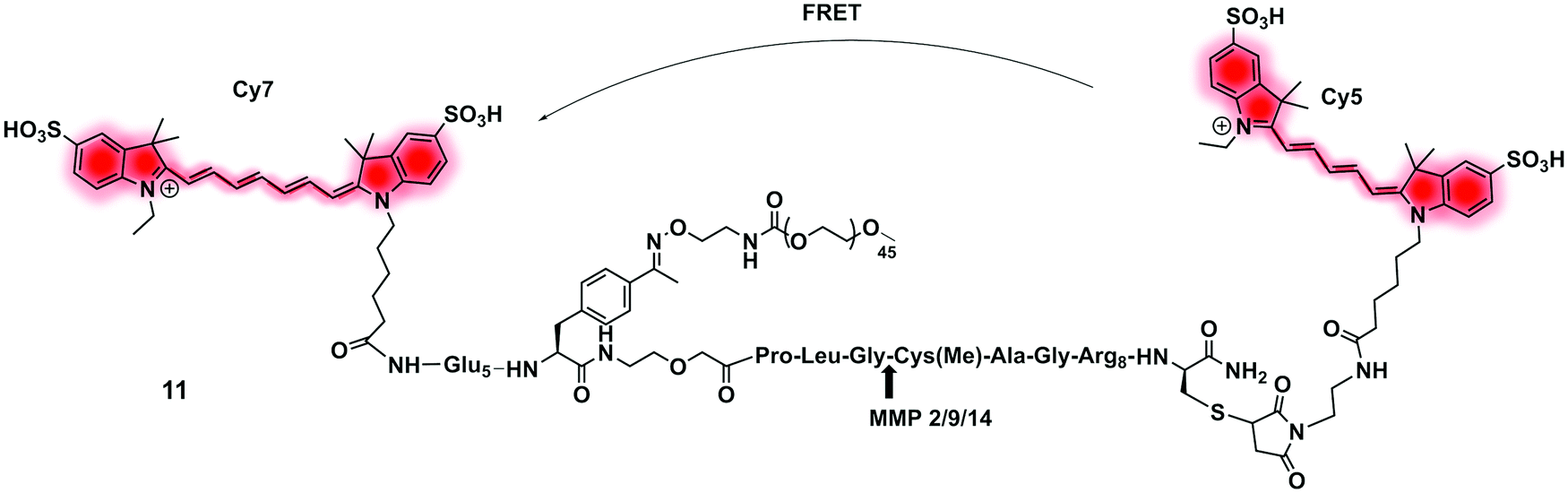 | ||
| Fig. 6 Structure of the clinically validated ratiometric probe AVB-620 for detection of MMPs based on the FRET pair Cy5/Cy7.73 | ||
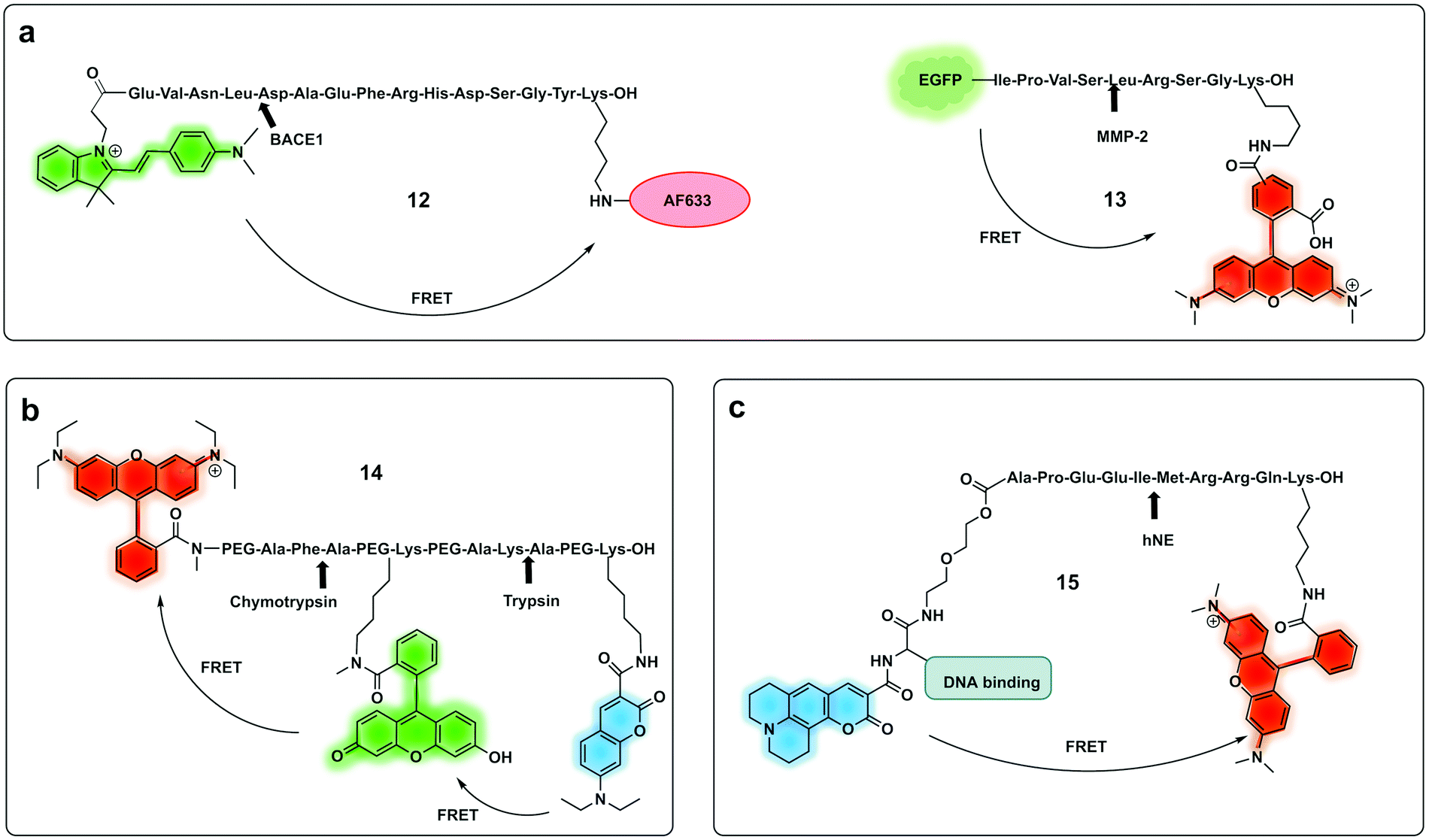 | ||
| Fig. 7 Ratiometric fluorescence detection of proteases. (a) Ratiometric FRET based probes for BACE1 (12)74 and MMP-2 (13).75 (b) dual activation-3-dye-FRET sensor providing three orthogonal fluorescence signals upon full activation of the probe.77 (c) DNA targeting ratiometric probe for human neutrophil elastase.71 | ||
A ratiometric protease probe (12) was designed74 and applied for ex vivo imaging of β-Secretase (BACE1) in an Alzheimer's disease mouse brain model. The reporter used the two-photon absorbing merocyanine (mCyd) as a donor conjugated to the acceptor fluorophore Alexa Fluor 633 (a sulfonated rhodamine derivative) as the acceptor via a β-Secretase peptide sequence (Fig. 7a).
A sophisticated assay for MMP sensing in the extracellular matrix in vitro (13) has been reported75 with collagen-immobilized FRET reporters allowing the visualisation of time-dependent secreted protease activity using an extracellular matrix (ECM) collagen anchor, conjugated to enhanced green fluorescent protein (EGFP). In this case TAMRA was attached, by intein-mediated splicing to form a collagen-adherent MMP-2 probe with a FRET pair TAMRA/GFP (Fig. 7a), linked by an MMP cleavable peptide. This strategy allowed the probe to bind to collagen in the ECM where it could interact with the secreted protease of interest, interrupting the FRET pair. The probe was validated in a 3D cell spheroid model to analyse secreted MMP-2 activity. Similarly, but by fusing the cyan fluorescent protein (CFP) and the yellow fluorescent protein (YFP) through a NE-specific cleavable linker, a neutrophil elastase fluorescent ratiometric reporter was assembled.76
The combination of three dyes in a dual FRET system yielded a dual-target probe (14) for detection of trypsin and chymotrypsin. Using a peptide-like structure (Fig. 7b) a cascade FRET system was generated using 7-diethylaminocoumarin-3-carboxylic acid, fluorescein and rhodamine B.77 This allowed for multiplexed assaying of protease activity.
Other strategies for ratiometric probes include a single fluorophore with tunable emission following activation by a protease, which uses the pro-fluorophore principle, where decaging of the fluorophore leads to changes in emission profile.78–80 A y-glutamyltranspeptidase-triggered theragnostic probe was designed for cancer detection using a caged NIR photosensitiser.81 The construct when caged emits fluorescence in the NIR region of the spectrum and cannot produce ROS. Upon decaging by y-glutamyltranspeptidase there is a shift in fluorescence emission into the yellow region which allows photodynamic therapy (PDT) due to the activity of the photosensitizer with the production of ROS at the site of activation. This field has been extensively reviewed by Huang.82 Du reported a ratiometric fluorescent probe83 for the detection of leucine aminopeptidase (LAP) based on a quinoline derived fluorophore for imaging LAP on liver tumour cells. The probe contained a galactose moiety for targeting active tumours and a caged quinolone. The probe was internalised in cancer cells, where it was decaged by LAP, producing a red shift in emission of the construct (425 nm to 510 nm). Using a cell penetrating peptide and a substrate for caspase-3 to cage cresyl violet, a probe was generated that gave a red-shift in fluorescence emission upon caspase-3 cleavage. Recently, Cao developed the first non-peptide based ratiometric pro-fluorophore probe for hNE84 using a pentafluoropropanoic acid caging group.
The use of biocompatible nanocrystals has been widely explored to build protease probes, acting as scaffolds and carriers with many offering the ability to act as spectrally broad light quenchers. Fe3O4 nanocrystals and quantum dots (QDs) have been widely used as FRET quenchers and carriers for the synthesis of several protease probes. Mingyuan developed a series of probes combining pH sensing with protease detection for tumour optical imaging.85,86 The probes (16, 17) simultaneously mapping MMP-9 activity and the extracellular pH of tumours were able to target tumour cells via conjugation to an antibody. These were based on a modified naphthalimide as a ratiometric pH sensitive fluorophore and Fe3O4 nanoparticles as a quencher by covalent attachment through an MMP-9 cleavable peptide. Upon cleavage of the peptide, naphthalimide fluorescence was switched on with the ratio of the two pH-dependent emission maxima used to quantitatively determine pH (Fig. 8a). In a second-generation reporter that was used for in vivo imaging, folic acid was incorporated as a targeting moiety, and an “always on” Cy5.5 dye with an orthogonal fluorescence signal serving as an MMP-9 reporter.85,86
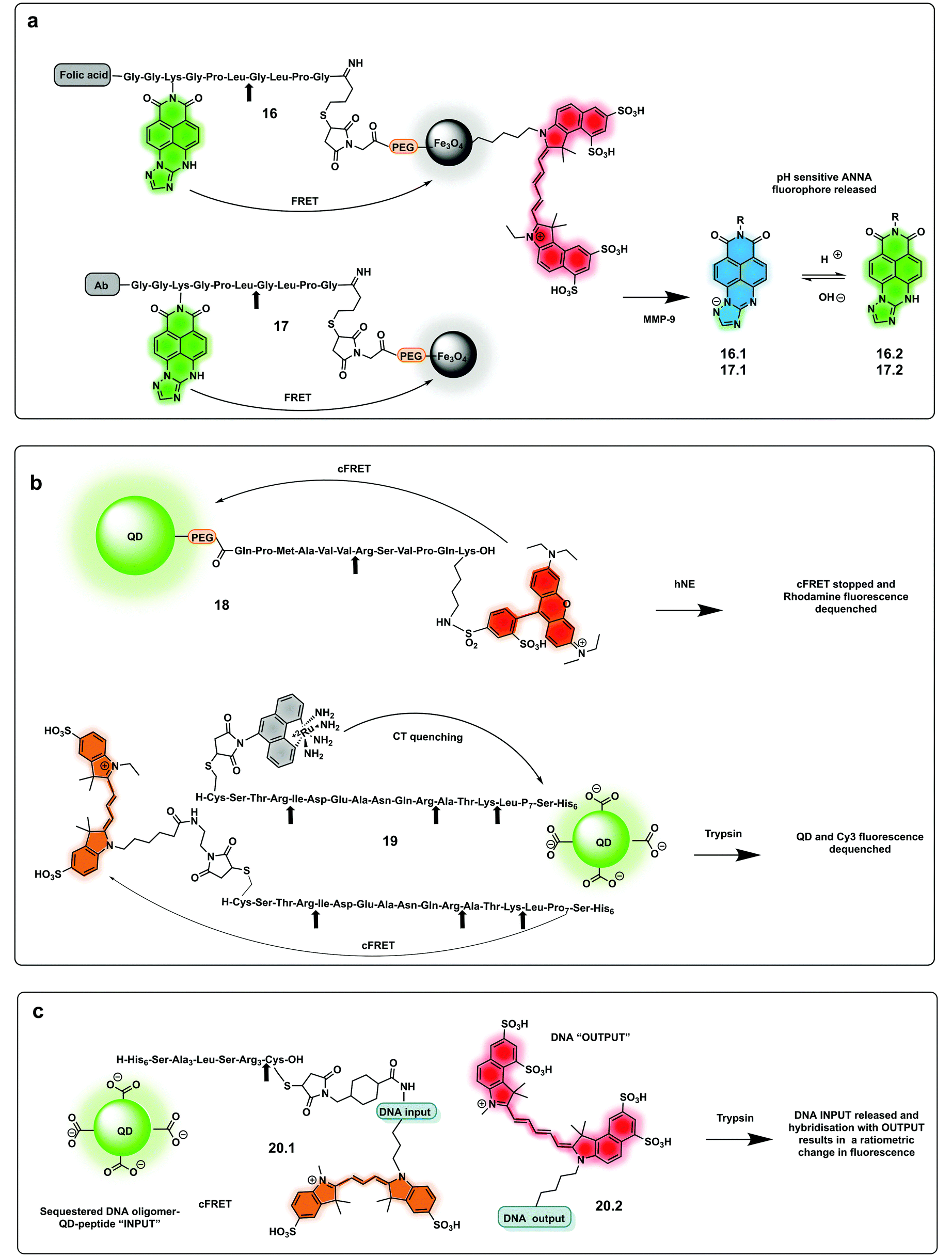 | ||
| Fig. 8 Nanoparticle and QDs based ratiometric probes. (a) Fe3O4 nanocrystals FRET based probes systems based on quantum dots (QDs) with tumour targeting capacity using folic acid or antibody targeting and generating a pH ratiometric sensor upon cleavage by MMP-9.85,86 (b) QD based FRET system with a rhodamine fluorophore donor (18)88 and a QD conjugated system allowing multiplexed sensing combining FRET quenching (using Cy3 as the acceptor and QD as a donor) and charge transfer quenching (using a ruthenium dye as a donor with the QD as an acceptor (19).89 (c) DNA-QD-conjugated approach relying on DNA hybridisation of complementary strands, with dyes inserted into the DNA via double phosphoramidite modifications.90 | ||
Quantum dots (QDs) have been used as FRET donors for ratiometric sensing of proteases with fluorophore-labelled cleavable peptides immobilised as acceptors.87 Concentric Förster Resonance Energy Transfer (cFRET) imaging is a novel application of semiconductor QDs where a central emissive QD is conjugated with multiple copies of two different biomolecular probes. Each of these probes is labelled with one or two, similar or different, fluorophores that engage in energy transfer with the QD and, in many cases, with one another. These configurations are referred to as “concentric” because the dyes on the surface form a sphere that shares the same centre as the quantum dot. A recently reported cFRET QD system88 comprised green-emitting QDs and peptides labelled with Alexa Fluor 555 (a bis-sulfonated carboxy-rhodamine) (18) and Alexa Fluor 647 (a bis-sulfonated Cy5). The two peptide sequences were selected as substrates for trypsin and chymotrypsin, whose cleavage modulate signal output by decreasing the number of dyes per QD. A bi-modal QD-type probe (19), combining charge transfer (CT) and a FRET quenching system was recently developed,89 using QDs with a hydrophilic coating assembled with multiple copies of a number of peptides. Some were labelled with ruthenium(II) phenanthroline (Ru-phen), which quenched the QD via a CT mechanism, others had a distal fluorescent label, which are well-known to quench QD emission via FRET (Fig. 8b). Acceptor dye-labelled peptide-DNA conjugates were assembled onto the QD donors as an input gate. The addition of trypsin or chymotrypsin cleaved the peptide and altered the efficiency of FRET with the QD, and liberated a DNA output (20.1) which then interacted with a tetrahedral output gate (20.2). Downstream output gate rearrangement resulted in FRET sensitization of a new acceptor dye90 (Fig. 8c).
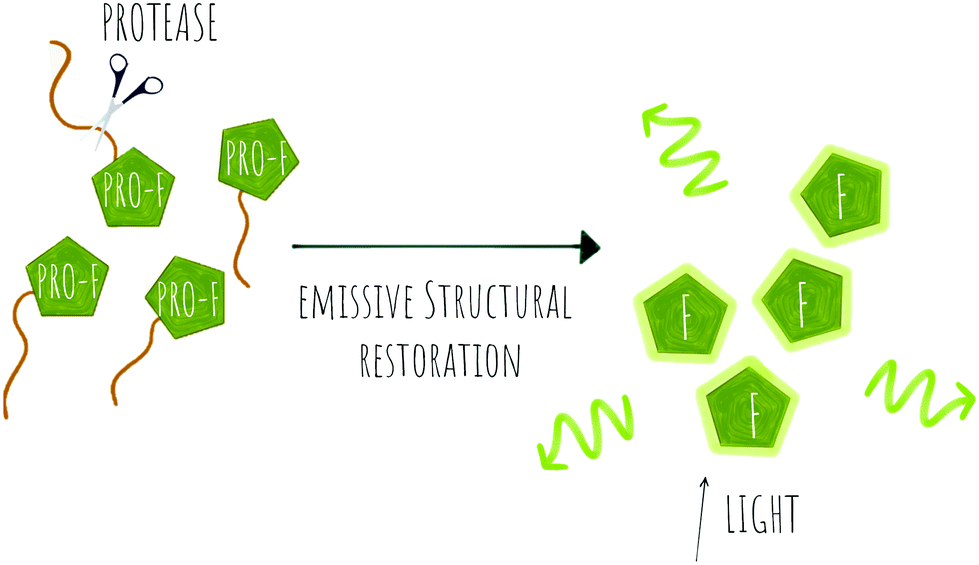 | ||
| Scheme 3 Concept of pro-fluorophore (PRO-F) activation by a protease to generate emissive molecules (F). | ||
Several fluorogenic probes have been designed for the detection of N-terminal exopeptidases such as dipeptidyl peptidase (DPP-IV),95–97 leucine aminopeptidase (LAP), pyroglutamate aminopeptidase (PGP1)98 or aminopeptidase N (APN)99,100 with attachment of the appropriate amino acid residues to a fluorophore that affects its internal charge transfer (ICT) and yields probes that switch-on upon peptidase exposure.
Dipeptidyl peptidase-IV cleaves X-Pro or X-Ala dipeptides from the amino-terminus of peptides and is known to be overexpressed in diabetes101 and cancer.102,103 Probes for detection of this protease95 were constructed by the incorporation of a DPP-IV substrate (an amino acid104 or dipeptide96) onto a fluorophore, with quenching of its fluorescence. Urano reported a series of probes for DPP-IV detection using hydroxymethyl rhodamine green (HMRG) as a green fluorophore scaffold in combination with a series of X-Pro dipeptides,105 with one of the compounds (21.1, Fig. 10a) used successfully for imaging of head and neck squamous cell carcinoma106 and esophagic adenocarcinoma107 (Fig. 9). Another example includes a probe that uses the NIR fluorophore SiR600,97 conjugated to the dipeptide Glu-Pro, with the quenching mechanism relying on PeT (22.1).97 Leucine aminopeptidase (LAP) has attracted significant attention, as it is known to be overexpressed in ovarian and breast cancer and several probes have been developed to detect its activity. Such probes are generally based on modification of the amino group of a fluorophore through an amide bond to a leucine residue (Fig. 10b). NIR hemicyanines99 (23.1), crysol violet108 (24.1), dicyanoisophorone derivatives (25.1)109 or rhodamine (26.1)110 have all been modified to generate fluorogenic probes to detect this peptidase, with new approaches adding targeting capacity.111
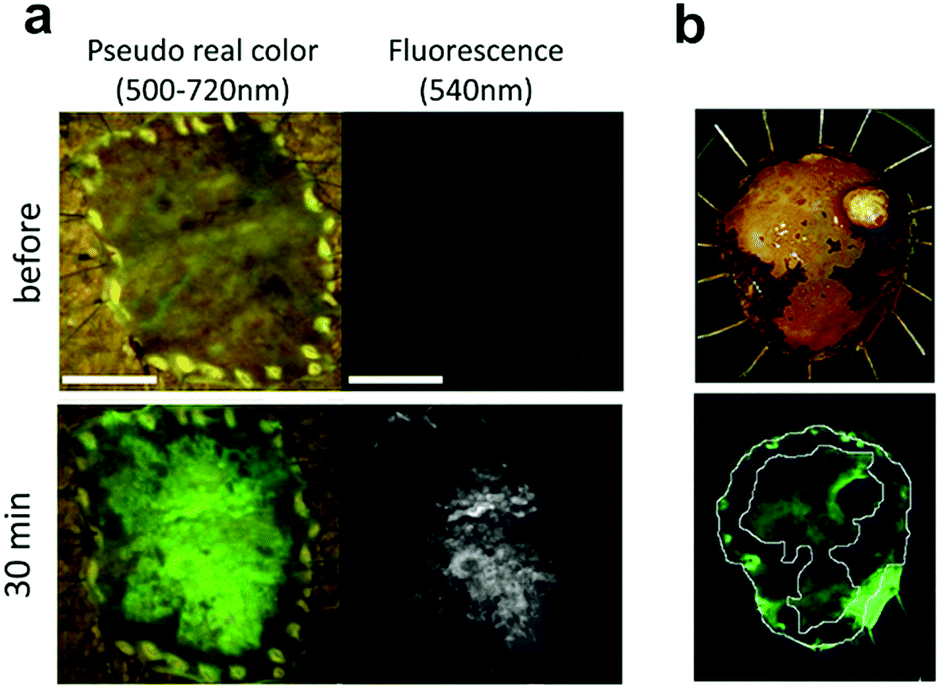 | ||
Fig. 9
Ex vivo fluorescent imaging of tumor tissue following spraying of DPPIV profluorophore 21.1 (a) esophagic carcinoma: a rapid fluorescent increase was observed in the tumor lesion after 30 min. Scale bar, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mm (b) head and neck squamous cell carcinoma tissue, upper: resected specimen observed with iodine staining shows normal mucosa (darker, periferic) vs. tumor mucosa (central area), lower: fluorescent imaging after spraying 21.1. Reproduced from ref. 105 with permission from Nature, copyright 2016 and ref. 106 with permission from Head Neck, copyright 2018. mm (b) head and neck squamous cell carcinoma tissue, upper: resected specimen observed with iodine staining shows normal mucosa (darker, periferic) vs. tumor mucosa (central area), lower: fluorescent imaging after spraying 21.1. Reproduced from ref. 105 with permission from Nature, copyright 2016 and ref. 106 with permission from Head Neck, copyright 2018. | ||
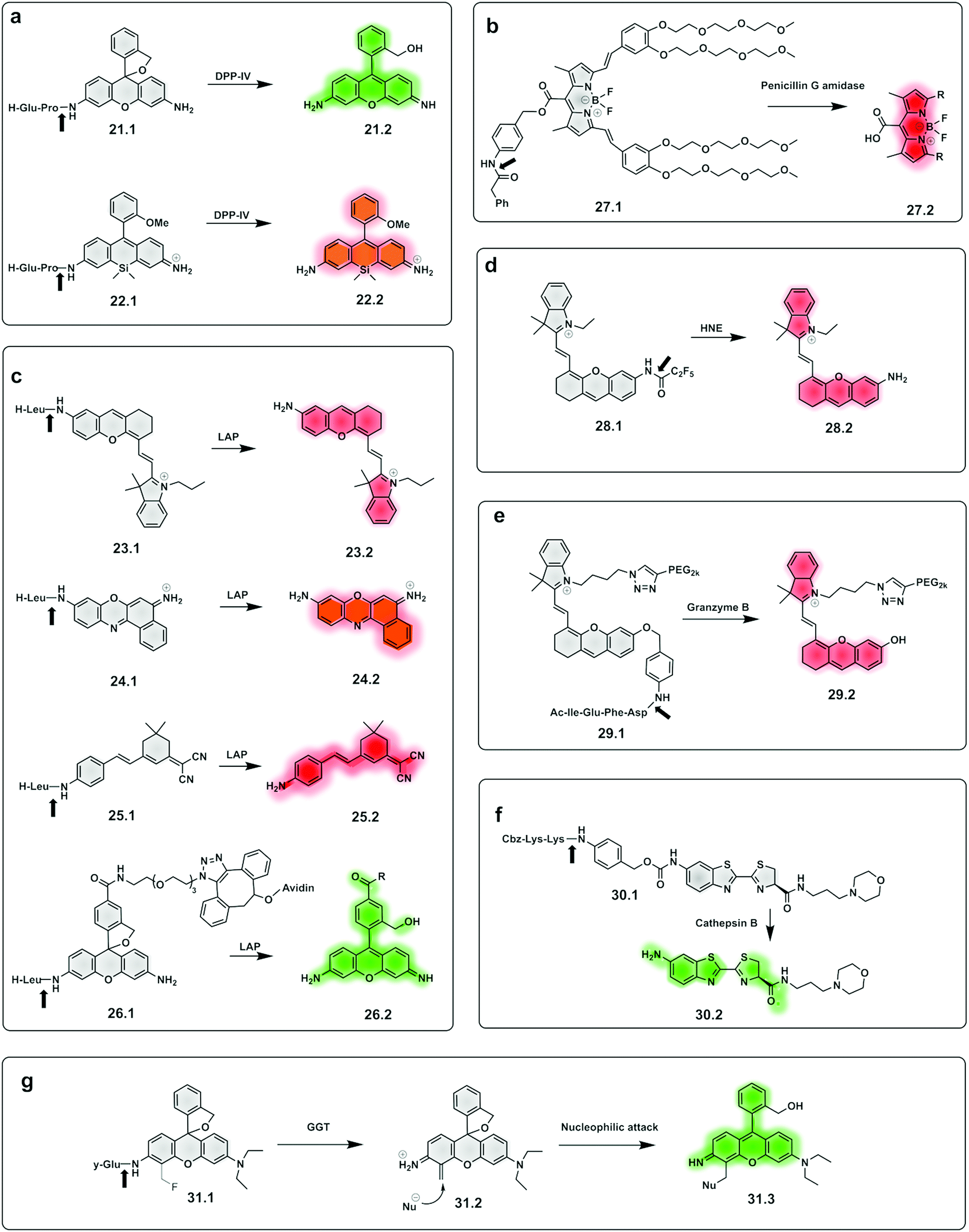 | ||
| Fig. 10 Examples of fluorogenic probes for proteases based on pro-fluorophores for detection of (a) dipeptidyl peptidase-IV (DPP-IV) using Glu-Pro97,105 (21.1 and 22.1) dipeptide substrates (b) penicilin G amidase93 (27.1) (c) leucine aminopeptidase (LAP) based on NIR hemicyanines (23.1),76 crysol violet87 (24.1), dicyanoisophorone derivatives (25.1)89 and rhodamine88 (26.1) (d) human neutrophil elastase (28.1)112,113 (e) granzyme B94 (29.1) (f) cathepsin B115 (30.1) and (g) γ-glutamyl-transferase (GGT)117 (31.1). | ||
A small-molecule probe targeting hNE (28.1),112,113 a serine protease expressed in activated neutrophils in inflammatory processes, was designed where the amino group of the NIR hemicyanine dye was conjugated to a pentafluoropropanoic acid caging group, and was optically silent until de-caged by the protease (Fig. 10d).
The same concept has been extended to include so-called safety catch or self-immolative linkers, with p-hydroxy or p-amino-benzyl alcohol the most widely used. Here enzymatic cleavage releases the conjugated fluorophores and restores fluorescence.114 Shasha94 developed a Granzyme B probe based on this approach for detecting T cell activation. The probe (29.1) consisted of a short peptide conjugated to a PEGylated NIR hemicyanine dye via a self-immolative linker. Using the same principle, a probe (30.1) for cathepsin B was developed,115 bearing a morpholine targeting moiety and a specific peptide connected to aminoluciferin. Based on the same self-immolative linker strategy, Chen synthesised a BODIPY-derived fluorogenic probe (27.1) with phenyl acetamide as a triggerable motif for penicillin G amidase detection.93
Urano also reported γ–glutamyl–transferase (GGT) fluorogenic probes based on spirocyclic caging of γ-glutamyl hydroxymethyl rhodamine green,116 with activation occurring by a rapid one-step cleavage of glutamate to release hydroxymethyl rhodamine green in its ring-open, fluorescent form. A modification of this strategy117 added a fluoromethyl group at the 4-position of the xanthene ring (31.1). Cleavage by the protease liberates the fluoride anion to produce an azaquinone methide intermediate (31.2) that can be attacked by intracellular nucleophiles (e.g. a group on a protein) to restore the hydroxymethyl diethylrhodamine (31.3) fluorescence and also trap the compound within the intracellular space, via conjugation to an intracellular protein.
 | ||
| Scheme 4 Fluorescent activation by proteases via aggregation-induced emission. | ||
A novel probe for real-time in vivo detection of MMP-13 activity in osteoarthritis was synthesized using a NIR emissive AIEgen (33) based on a cyanine–pyrene unit, which introduced a donor–acceptor system where a dimethylaminophenyl group act as an electron-donor, while nitrile and pyridinium containing units functioned as electron acceptors.129
Yuan, developed a probe for apoptosis (34) based on the sequential detection of caspases 8 and 3.130 The probe consisted of a central peptide containing the two substrates functionalised with two AIEgens with distinctive green (caspase 8) and red (caspase 3) emissions (Fig. 11b). The green and red fluorescence were sequentially turned on when the peptide substrate was cleaved by the action of caspase-8, cleaving the TPS containing fragment, followed by caspase-3, cleaving the TPETH containing fragment, with fluorophore aggregation in early apoptotic cells (Fig. 12). In the same area, a theragnostic compound (36) targeting cathepsin B/caspase-3131 was synthesised, which consisted of three components: an RGD targeting moiety, a cathepsin B-activatable gemcitabine prodrug, and a caspase-3 specific reporter based on tetraphenylene (TPE) to give AIE.
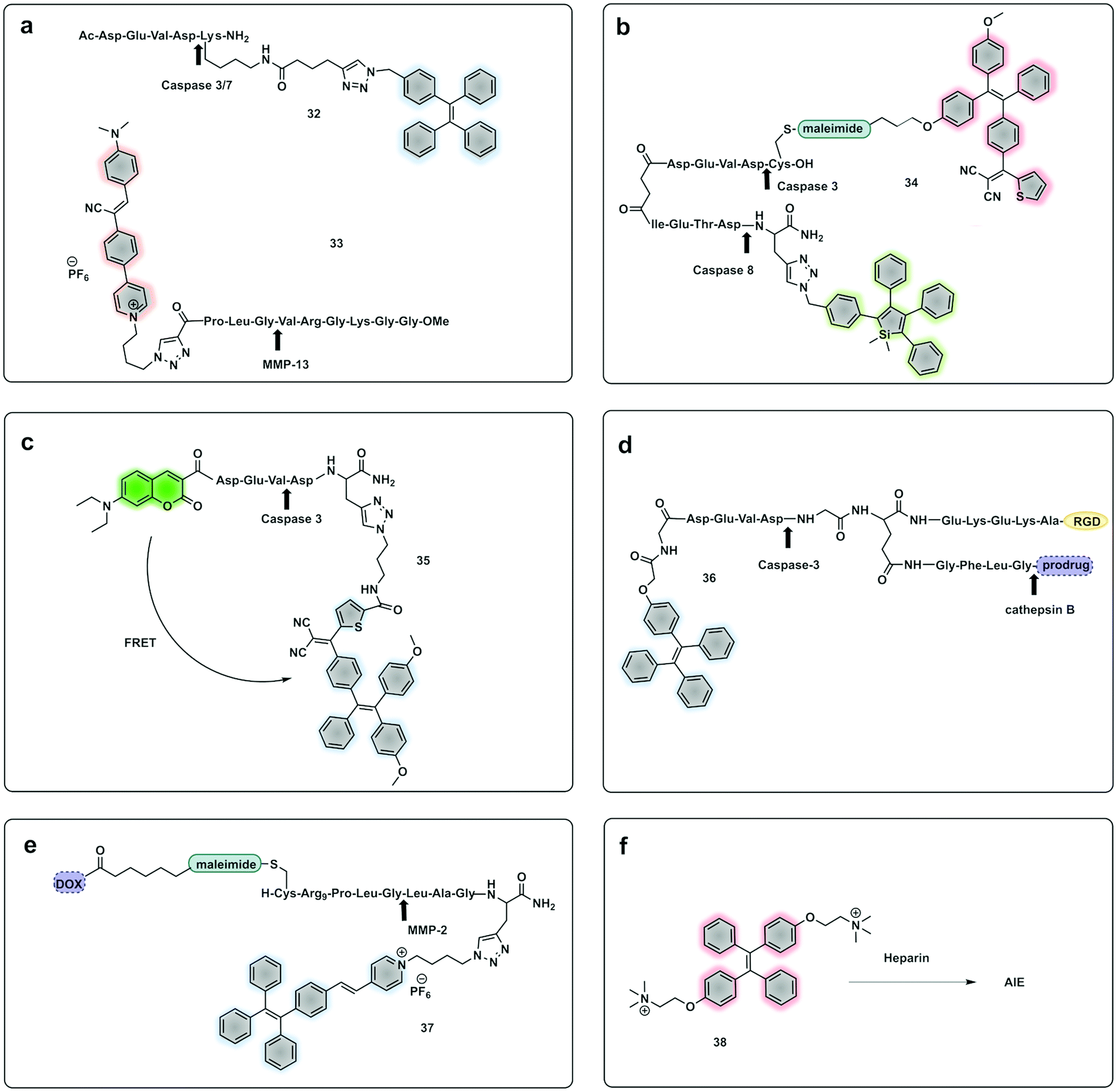 | ||
| Fig. 11 Example of fluorogenic probes where protease cleavage activates AIE fluorescence. Arrows indicate cleavage site. (a) Probes for detection of Caspase 3/7 based on TPE (32)122 or MMP-13 based on cyanine-derivates (33)129 AIEgens. (b) Cascade activatable probe with sequential fluorescence signal in the green and red.130 (c) FRET-AIEgen using a coumarin green emitting dye coupled to a TPETP AIEgen providing a dual signal upon cleavage (35).132 (d) Theragnostic AIEgen protease probe with a tumour targeting moiety, and different proteases mediating signal and therapeutic agent release.131 (e) Theragnostic probe that releases the drug doxorubicin (DOX) and a TPE derivative that generates an AIEgen upon cleavage by MMP-2.135 (f) An AIEgen detection system based on negatively charge heparin and the positively charged TPE fluorophore that allows aggregation induced emission.136 | ||
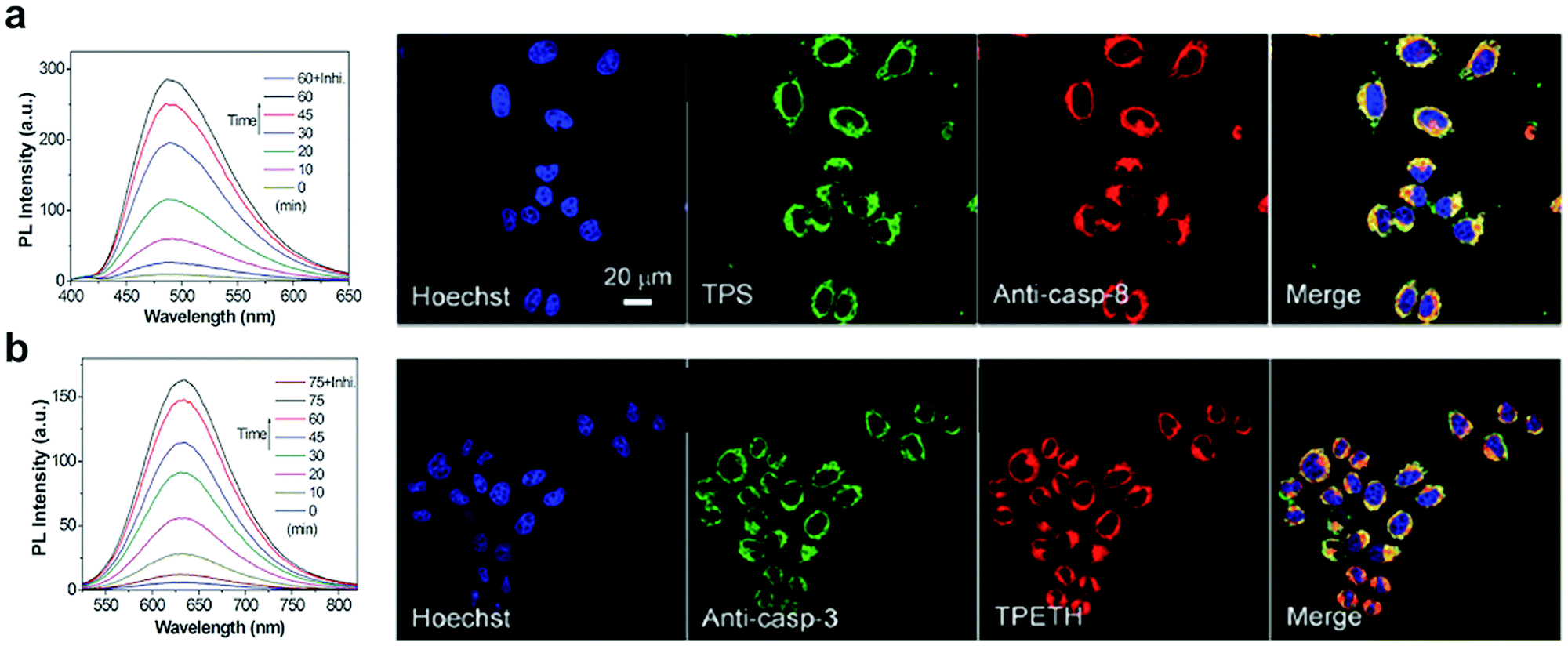 | ||
| Fig. 12 Detection of apoptotic cells using the AIEgen probe (34).130 (a) Left: Fluorescence spectra of probe 34 incubated with caspase-8 with or without inhibitor. Right: Confocal images of HeLa cells pretreated with probe 34 and further treated with H2O2 and stained with Hoechst (blue channel, for nuclear DNA), Texas red/anti-casp-8 antibodies (red channel). The probe's fluorescence is detected in the green channel (by TPS AIEgen). (b) Left: Fluorescence spectra of probe 34 incubated with caspase-3 with or without inhibitor. Right: Confocal images of HeLa cells pretreated with probe 34 and further treated with H2O2 and stained with Hoechst (blue channel, for nuclear DNA), Texas red/anti-casp-3 antibodies (artificially labeled with green color). The probe's fluorescence is detected in the green channel (by TPETH AIEgen). Reproduced from ref. 130 with permission from Royal Society of Chemistry, copyright 2017. | ||
Yuan developed a FRET-AIEgen probe (35) where a caspase-3 peptide substrate was modified with a FRET pair,132 with the donor being a green emitting coumarin capping the amino terminus. The acceptor, tetraphenylethenethiophene (TPETP), was attached onto the carboxy terminus of the caspase-3 peptide. The probe was silent as the acceptor fluorophore (TPETP) absorbs the energy from the coumarin, however, when caspase-3 cleaves the peptide both fluorophores are released and recover the emission of fluorescence at two different wavelengths simultaneously. The same group developed theragnostic probe combining a therapeutic moiety and an AIEgen linked though the caspase-3 cleavable peptide using a photosensitiser123,133 or a platinum(IV) prodrug.134 Caspase-mediated apoptosis, induced by the concurrently released therapeutic agents, could then be measured based on the fluorescence of the released AIEgen and aggregation.
Cheng published an MMP2 activatable reporter (37) for cancer theragnostics,135 based on a doxorubicin hydrophilic cell penetrating peptide conjugate linked through an MMP2-cleavable peptide conjugated to a PyTPE fluorophore (Fig. 11e). Upon cleavage by MMP2 the hydrophilic fragment containing DOX enters cells, while the hydrophobic part self-aggregates giving a strong yellow fluorescence emission.
An in vitro assay for protease activity136 has been reported (38) that uses heparin to drive aggregation of the TPE fluorophore (Fig. 11f). Heparin, a highly sulfonated (hence negatively charged) aminoglycan induces the aggregation of the positively charged AIEgen fluorophores by electrostatic interactions, to give fluorescent enhancement. Histones have a high affinity to heparin, and can displace the fluorophores, reducing the fluorescence signal. If trypsin is added, the histones are hydrolysed and the heparin-TPE retains the fluorescence signal. Kaur developed a trypsin AIEgen probe137 based on electrostatic interaction driven aggregation induced emission combining a negatively charged TPE dye and the positively charged protein protamine. The tetra-anionic dye, a sulphonyl-derivative of tetraphenyl ethylene (Su-TPE) is almost non-fluorescent when free in aqueous solution but protamine sulphate (PrS), an overall cationic protein, induces aggregation of Su-TPE by electrostatic interactions and leads to a highly emissive Su-TPE/PrS supramolecular complex with AIE characteristics. In the presence of trypsin, the cationic protein is digested and the supramolecular complex disassembles, reducing the fluorescent emission.
A bimodal reporter using Gadolinium as a contrast agent for MRI combined with an AIEgen fluorophore for optical imaging was developed.138 The probe used a DOTA-Gd(III) chelate that provides MRI signal enhancement, and TPE as the AIEgen linked to a caspase 3/7 cleavable peptide. In response to the protease, the Gd-AIEgen conjugate is released and aggregates leading to increased fluorescence and MRI signals.
 | ||
| Scheme 5 The concept of protease activation followed by a subsequent bioorthogonal fluorogenic labelling reaction. | ||
Romieu developed a reporter (39.1) whose mechanism of activation was based on pyronin assembly triggered by a protease.142,143 Following peptidase cleavage and decaging of an amino group, the mechanism involves in situ generation of an unsymmetrical pyronin via cyclisation/aromatization cascade (39.3). This strategy offers advantages over some pro-fluorophore strategies as the fluorophore is not pre-formed (so no background signal), but one potential disadvantage is that the kinetics of the reaction might impact on the time the system takes to assemble the dye. The design principle was successfully applied to leucine aminopeptidase (LAP) and penicillin amidase (PGA). Huo developed a γ-glutamyl transpeptidase (GGT) activating two-photon fluorescent probe (40.1)144 using a similar strategy. The probe could selectively detect GGT, upon cleavage, with intramolecular cyclisation resulting in a two-photon absorbing green emitting fluorophore.
Wu reported an approach using a “labelling after recognition” mechanism with phosphorescence lifetime analysis.145 The approach was based on a caspase 3 specific substrate modified with two independent bioorthogonal reactive sites that allowed FRET-labelling (41.1). The strategy takes advantage of labelling the cleaved fragments and the uncleaved full peptide in the presence or absence of the protease. The novelty of this approach is that the FRET labels are added by bioorthogonal reactions following cleavage by the protease, with the rationale that conventional FRET-labelled peptides used for protease detection are pre-labelled and the bulky fluorophores may hinder recognition and cleavage. Here, the FRET pair used was an iridium(III) complex as a donor (41.2) (attached by strain promoted alkyne–azide cycloaddition (SPAAC) and RhoB as an acceptor (41.3) (attached via an iEDDAC tetrazine–norbornene click reaction). In the absence of the protease, the intact peptide with the bioorthogonally reactive moieties undergoes bis-labelling of the uncleaved peptide and leads to a change in lifetime via FRET, with the Ir(III) complex emission quenched due to the energy transfer to rhodamine B. However, the phosphorescence lifetime of the Ir complex was not quenched when labelling occurred after cleavage.145 Potential limitations of this approach could come from incomplete labelling of the full or fragmented peptide or by differing labelling kinetics in/during the bioorthogonal labelling, an issue especially on more complex samples.
Demonstration of molecules that undergo aggregation upon activation and bioorthogonal cyclization were applied in molecular imaging (Fig. 13c) using caspase-3/7-controlled self-assembly of molecules into “nanoparticles”146 as a measure of the response of cancer tissue to chemotherapy. The intramolecular cyclization was possible by reaction between a free Cys residue and a 2-cyano-4-hydroxy-quinoline (42.1) that proceeded under physiological conditions to form a thiazoline linkage. This biocompatible reaction was possible due to the reductive environment that generates the free thiol and enzyme cleavage releasing a free amino group, which then mediate the cyclisation reaction. The cyclic molecule (42.2) then aggregates forming in situ fluorescent NPs. This aggregation enhances retention inside the cell, increasing the in vivo half-life, while the precursor units, in the absence of the protease undergo rapid clearance. The probe accumulated extensively in the cytosol allowing visualisation of apoptotic bodies in cells and tumour tissue using three-dimensional structured illumination microscopy (3D-SIM). A combination of AIE and bioorthogonal strategies were used in a probe designed for fluorescence sensing of furin activity in vitro.127 The probe (43.1) was cleaved by furin exposing a free 1,2-aminothiol unit (43.2) that reacts with the cyano group on a cyanobenzothiazole motif yielding the more hydrophobic dimer (43.3) which self-assembles into fluorescent NPs.
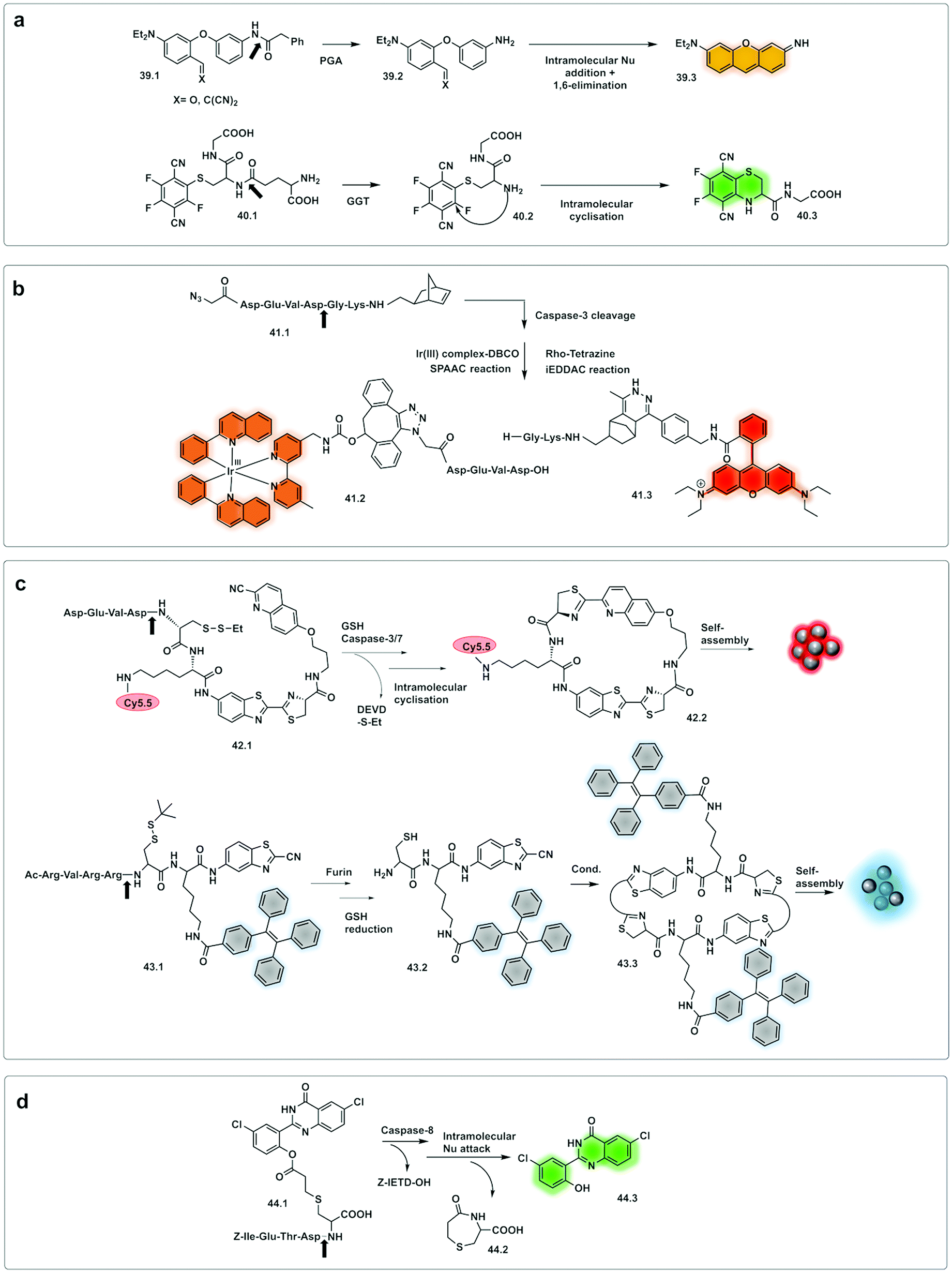 | ||
| Fig. 13 Examples of bioorthogonal strategies for protease detection. (a) After cleavage by the protease, caged precursor (39.1 and 40.1) undergo bioorthogonal addition/elimination to yield the fluorophore pyronin (39.3)142,143 or the two-photon-absorbing fluorophore (40.3).144 (b) FRET labelling after cleavage by caspase-3.145 (c) Assembling strategies based on thiazoline linkage formation in response to GSH and Caspase 3/7146 or Furin.127 (d) Excited-state intramolecular photon transfer based strategy, with cyclative cleavage following caspase-8 activation liberates the phenolic ester (44.3).148 | ||
A fluorescent activatable caspase-8 probe, consisting of an ESIPT (excited-state intramolecular photon transfer147) fluorophore attached to a Cys-residue has been reported.148 These ESIPT fluorophores produce insoluble fluorescent aggregates with fluorophores localized in close proximity to the enzyme with enhancement of fluorescent emission – a process that is similar, but different to AIE. The protease probe was synthesized by a thiol-1,4 addition reaction149 of a Cys containing peptide (caging group) with an acryloylated fluorophore (44.1). Upon internalisation of the probe in cells, cleavage of the peptide by activated caspase-8 results in intramolecular bioorthogonal cyclisation, liberating the amino group which reacts with an ester to release a 1,4-thiazepine seven-membered ring (44.2).150 Decaging the fluorophore (44.3) switches on bright green fluorescence by excited-state intramolecular photon transfer, allowing high-sensitivity and high-resolution imaging (Fig. 14).
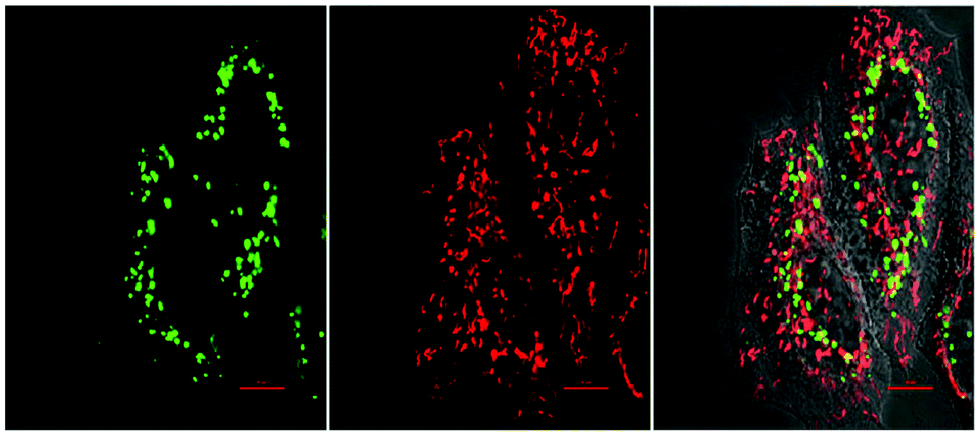 | ||
| Fig. 14 Fluorescence images of HeLa cells treated with the bioorthogonal Caspase-8 probe 44.1,148 followed by treatment with the apoptotic inducer Mitomycin C for 120 min. Left to right: Green channel (probe 44.1 (see Fig. 13d)), red channel (Mitotracker) and merged channel. Reproduced from ref. 148 with permission from American Chemical Society, copyright 2016. | ||
1.2 Chemiluminescence
Chemiluminescence (CL) is a powerful imaging technology that requires no external light source, does not suffer from photobleaching and has negligible background luminescence, but each molecule will only produce one photon. The phenomena of chemiluminescence can be defined as the emission of light resulting from a chemical reaction (Scheme 6) and differs from fluorescence in that the generation of the excited state arises thanks to the energy of a chemical reaction and does not depend on light irradiation.151 The field of chemiluminescence for imaging applications has grown rapidly with the discovery of several natural and new synthetic luminogens.152,153 | ||
| Scheme 6 The concept of protease activatable chemiluminescence. | ||
Synthetic chemiluminescent systems take inspiration from natural bioluminescent systems that generate light following two sequential reactions, firstly oxidation of a substrate by an oxidizing agent or an enzyme into a “high energy intermediate” and secondly, the rapid decomposition of this intermediate in a process that produces light, with the Luciferin/Luciferase/O2 system being the most studied.154
In the last decade, the field of chemiluminescence for imaging in vivo has expanded with synthetic modification of the natural luminogens to move beyond the limitations of natural luciferin.155,156 This includes tuning the emission properties of the natural luciferin substrate to improve its quantum yield and give NIR emission by increasing the conjugation of natural D-luciferin157–160 or by exploiting FRET with red shifted fluorophores.161 Caging of luciferin by the addition of a triggerable protecting group to the 6′-OH/NH2 group generates a non-recognisable substrate for luciferase (45.1) allowing for turn-on of signal upon protecting group removal by the given trigger (e.g. enzyme or analyte). This strategy has been adopted to develop luminogenic probes for proteases such as cathepsin B (46),162 chymotrypsin (47),163 carboxypeptidases A and B (48),164 Caspase-1 (49)165 and furin (50 and 51)166 (Fig. 15a). A novel strategy was devised with the in situ bioorthogonal assembly of firefly luciferin,167 the design consisting of two complementary caged precursors of luciferin, a peptide-based probe (52.1) that released D-Cys in the presence of Caspase-8 and a boronic acid probe (52.3) that released 6-hydroxy-2-cyanobenzothiazole upon reaction with H2O2 (Fig. 15b). Once decaged, the two precursors undergo a bioorthogonal condensation reaction to form D-luciferin (52.5), with cyclisation between the carbonitrile and the thiol group of D-cysteine, thus reporting simultaneously oxidative stress (H2O2) and inflammation (caspase-8). Another approach focused on the in situ assembly of the enzyme luciferase (instead of the luciferin substrate) was engineered by Talley.168 A modified luciferase specifically activated by Caspase-1 was generated by inserting a known target sequence into a “circular luciferase”. Cleavage by Caspase-1 allowed the generation of a functional luciferase thus, generating a bioluminescent system.
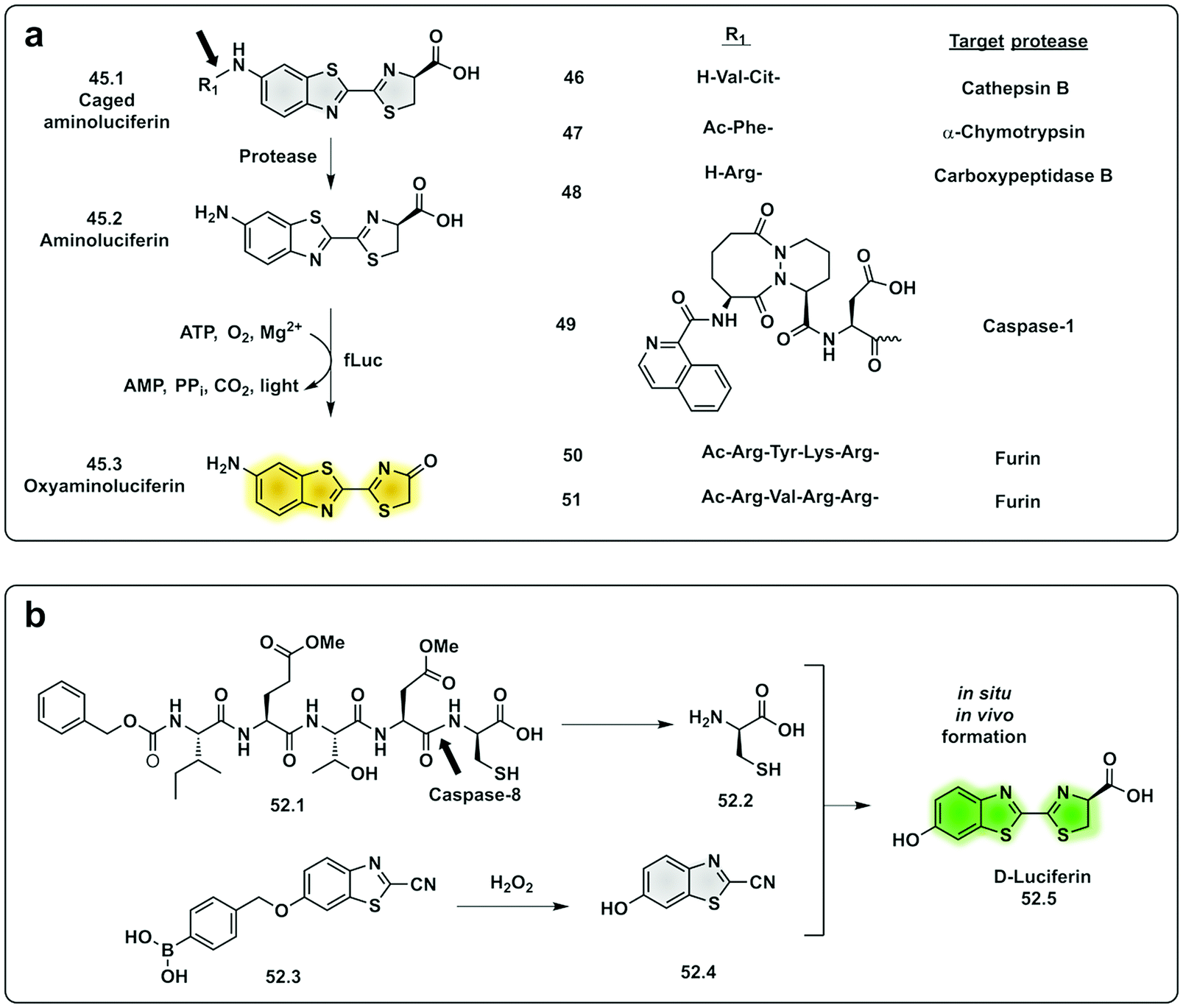 | ||
| Fig. 15 Examples of protease probes based on chemiluminescence. (a) Probes based on decaging of aminoluciferin, a substrate for fLuciferase that produces luminescent oxyaminoluciferin (b) bioorthogonal assembly of D-luciferin in situ requiring dual activation by Caspase-8 and hydrogen peroxide.167 fLuc: firefly luciferase. | ||
Natural bioluminescent organisms can modulate the colour of light emitted by the same principles as FRET by coupling luciferase to fluorescent proteins with overlapping spectral properties.169 Bioluminescence resonance energy transfer (BRET) systems for protease sensing take advantage of the luciferin–luciferase system as an energy/light donor for fluorescent acceptors (generally GFP) and commonly use an optimised luciferase-substrate system coupled to a fluorescent protein through a protease cleavable linker. Upon cleavage and energy transfer, emission from the fluorescent molecule stops and the emission corresponds solely to the bioluminescent luciferase.170–173 The original systems used a blue emitting luciferase and GFP, but in 2020 the first red-shifted BRET probe for detection of plasmin was reported.174
In an effort to simplify the system and remove the need of the enzyme luciferase for oxidation of D-luciferin, Shabat developed a series of caged 1,2-dioxetane phenols (53.1 and 53.2), highly stable compounds that rely solely on decaging of the dioxetane phenol-protecting group to produce luminescence.175,176 Upon interaction with the decaging target trigger, an unstable phenolate–dioxetane intermediate (53.3) is released producing the excited ester (53.4) that relaxes with photon emission (Fig. 16a). Caging of 1,2-dioxetane intermediates have been widely used for protease sensing and generally use a protease sensitive peptide conjugated to the 1,2-dioxetane via a safety catch or self-immolative linker (54 and 55).177,178 The emission properties of the 1,2-dioxetane can be tuned by increasing the conjugation of the phenolate, adding a conjugated π system and an electron withdrawing group (EWG) at the ortho-position.179 Using this strategy, chemiluminescent Cathepsin B probes were designed (56–58), that showed high sensitivity180 with enhanced tumour penetration by linking to a tumour targeting peptide.181 Recently, the speed of chemi-excitation was also improved, allowing for greater light emission efficiency and better signal/noise ratios, by modifying the ortho-phenol EWG functionality to an α,β-unsaturated carboxylic acid functionality. Using this system, a Granzyme B probe (59), that could detect natural killer cell activity in live mouse models tumours,182 was developed (Fig. 17). Using a similar strategy, Bogyo developed a system for detection of M. Tuberculosis,183 that uses a chemiluminogenic FLASH probe bearing the substrate of Hip1, a protease of Mtb (Ac-Igl-(4-Cl-Phe)-Lys-Leu), where Igl is a the non-natural amino acid H-(2-indanyl)Gly-OH. This probe (60) allows detection and quantification of Mtb in human sputum samples in vitro and, importantly, the system can differentiate between live and dead bacteria. As such it shows real potential as a point-of-care sensing platform, as do the PSA probes (61) developed by Shabat that allows on-site measurements using a small portable luminometer.184,185
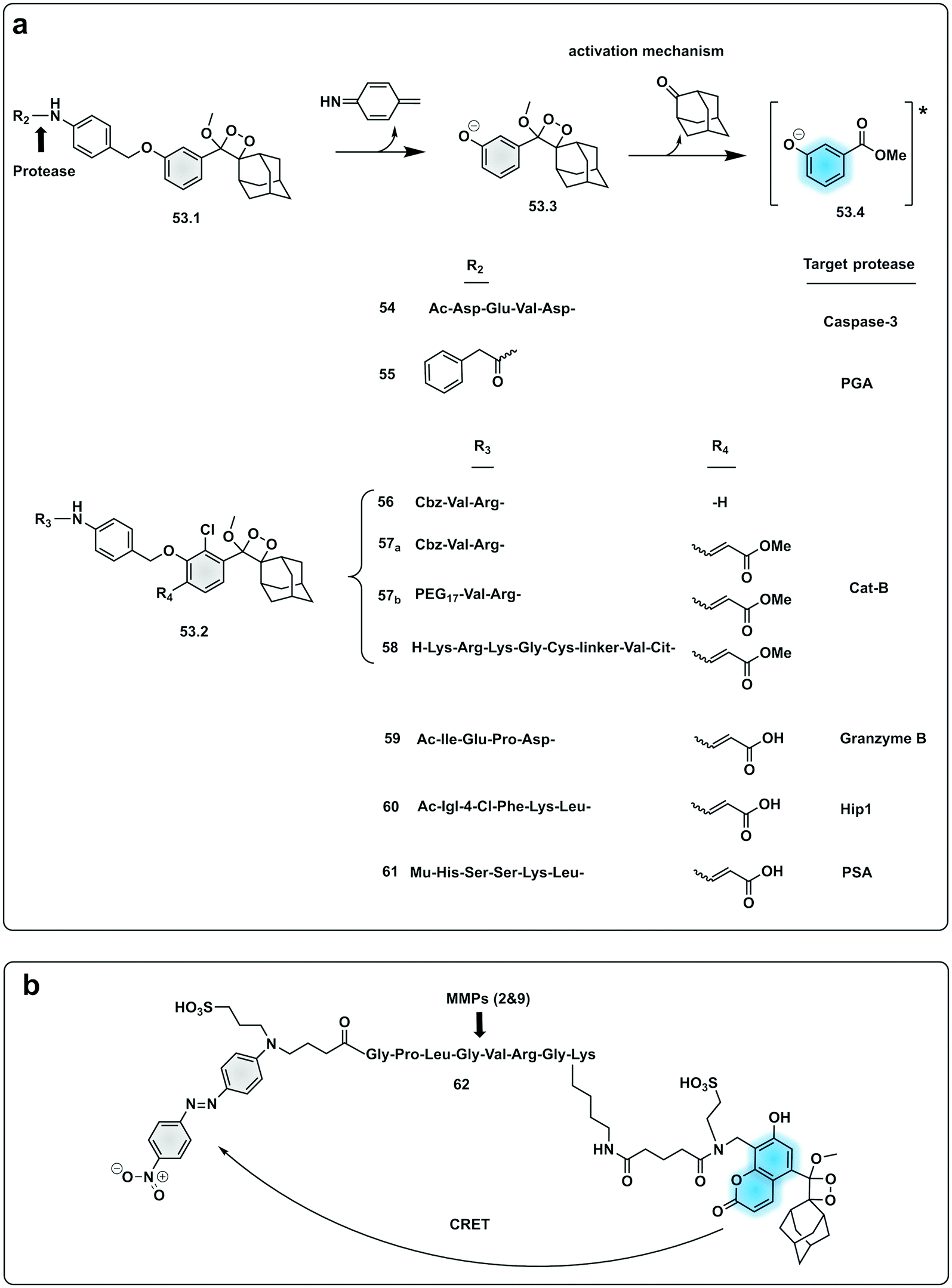 | ||
| Fig. 16 Examples of protease probes based on chemiluminescence. (a) 1,2-Dioxetane phenols relying on decaging of the dioxetane phenol-protecting group to produce luminescence from initial probes for Caspase 3 and PGA.177,178 Optimisation of light emission efficiency by increasing the conjugation of the phenolate in probes for cathepsin B,180,181 Granzyme B182 or PSA.184,185 Ac: acetyl group; Mu: morpholine carboxamide moiety. (b) Chemiluminescence Resonance Energy Transfer (CRET) strategy for detection of MMP activity utilising a phenoxy-dioxetane luminophore and a nitro-dabsyl quencher.187 | ||
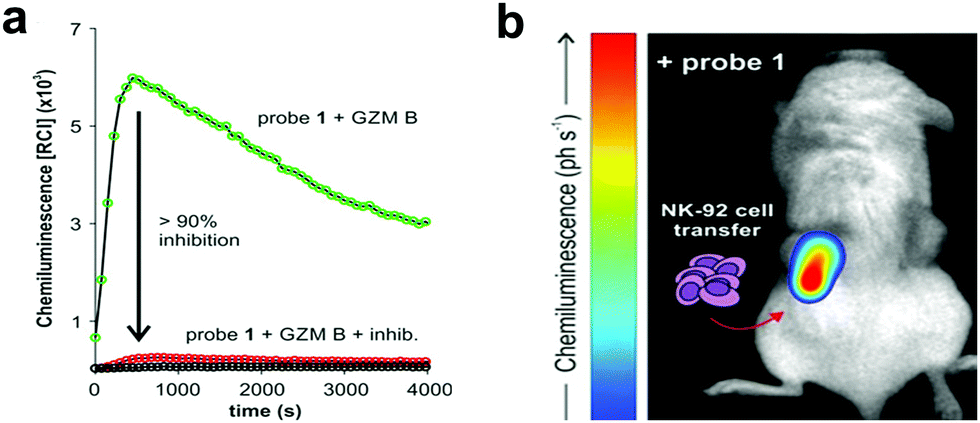 | ||
| Fig. 17 (a) Chemiluminescence spectra of granzyme B chemiluminogenic probe 59182 following incubation with granzyme B (green line) or without enzyme (black line) or with a granzyme B inhibitor (red line). (b) In vivo representative chemiluminescence image of a NSG mouse containing MDA-MB-231-xenograft tumours where NK-92 cells were adoptively transferred. Only the right tumour (red arrow) was injected with NK-92 cells with the left tumour being NK cell-free. After 8 h of the NK cell injection, probe was injected into both tumours. Reproduced from ref. 182 with permission John Wiley and Sons, copyright 2021. | ||
 | ||
| Fig. 18 Examples of some contrast agents and strategies for photoacoustic imaging. | ||
Modifications of the emission capabilities of 1,2-dioxetane probes by the addition of a fluorophore substituent at the meta-position of the benzoate unit allowed significant amplification of the signal thanks to transfer of light from the emission of the chemiluminogen to the fluorophore by Chemiluminescence Resonance Energy Transfer (CRET) (Fig. 16b). A model galactosidase triggerable probe186 has been reported with great potential for applications in protease sensing by replacing the galactosidase moiety by a cleavable peptide.187 Using CRET, a phenoxy-dioxetane luminophore attached to a 7-hydroxycoumarin scaffold was developed and linked to a quencher-peptide construct. The coumarin-dioxane luminogen exhibits intense and persistent chemiluminescence (62). Following the same principles as FRET conjugation of a non-emissive quencher acceptor molecule to the luminogen quenches any chemiluminescence emission until the peptide is cleaved by the protease.
 | ||
| Scheme 7 The concept of photoacoustic probes activated by proteases. | ||
1.3 Optoacoustics
Photoacoustic imaging (PAI) is an imaging strategy that bridges the traditional limited penetration depth and the resolution limits of diffuse optical imaging. Some reviews have been recently published188–190 and thus here we will only report on very recent papers, applications and novel concepts.In photoacoustic imaging, a laser pulse is used to “illuminate” specific structures in the body by virtue of them containing molecules that absorb photons. This causes local heating that generates pressure waves (the thermoelastic effect due to vibrational and translational relaxation). These pressure waves are in the ultrasound frequency range and as such can be measured by traditional ultrasonic transducers to generate an image. Photoacoustic imaging has rapidly been adopted in preclinical in vivo imaging in small animals for a range of disease indications, where it has provided in a non-invasive manner, images that in essence show where optical energy has been absorbed by detection of the acoustic waves generated. A key attribute is that it can do this at depths of several centimetres while offering a resolution of ∼100 μm.
PAI can exploit the absorption capacity of naturally occurring molecules, such as haemoglobin191 and melanin,192 however, probe-based contrast agents are now being used to allow directed “molecular photoacoustic imaging” at greater depths (Fig. 18). Typical molecules used in PAI have included indocyanine green (ICG) and methylene blue (MB) and some take advantage of both the photoacoustic and the fluorescence signals. The combination of optical and photoacoustic imaging has become a widely implemented strategy (Table 1).193–195
Traditional non-fluorescent dyes of various types are actually more efficient in terms of conversion of light absorption into heat and acoustic signals196,197 (simplistically it is better that light is converted to heat and not emitted as a photon). Thus, dyes offer greater thermoelastic expansion than fluorochromes, making quenchers e.g. dabsyl and the so-called black-hole quenchers (BHQ) good candidates for photoacoustic contrast agents but any dye with a high extinction coefficient and good conversion of light to heat should work well as contrast agents for PAI. One PA contrast agent reported recently used BHQ-1 conjugated to a cRGD targeting moiety.198
Recently, activatable photoacoustic imaging contrast agents have appeared. Design of these probes utilize similar approaches to those discussed for fluorescence base probes (FRET, self-assembly/bioorthogonal, pro-PAI, AIEgens), generating a specific switchable signal upon alteration of their structure, with proteases allowing disease-specific targeting/activation (Scheme 7).
NIR fluorescent dyes can act also as photoacoustic agents. MMP-Prosense 680,199 the NIR probe previously described, was evaluated for photoacoustic imaging, achieving high-resolution mapping of MMP activity deep in vulnerable plaques of intact human carotid specimens. Multispectral optoacoustic tomography allowed three-dimensional reconstruction of targeted structures in vivo200 with morphologies similar to heterogeneous MMP activity but with better than 200 μm resolution throughout intact plaque tissues and allowing volumetric images of activatable molecular probe distribution deep within optically diffuse tissues.
Levi201 developed a FRET-based protease activatable PAI probe using an AF750/BHQ-3 labelled MMP-2 cleavable peptide that under physiological conditions generates two photoacoustic signals. Cleavage/activation generates a BHQ-3 labelled cell penetrating peptide that results in accumulation of the PA contrast agent into cancer cells and a NIR dye labelled fragment that produces orthogonal photoacoustic signals.
Using a similar approach to that covered in the pro-fluorophores section, a urokinase-type plasminogen activator (uPa) activatable probe was designed202 that provides a turn-on in fluorescence and photoacoustic signals and optimal renal clearance due to the hydrophilicity dextran backbone. The probe consists of a “caged” dextran functionalised-Cyanine dye, connected to a cleavable uPA peptide sequence via a self-immolative linker. Upon cleavage by the protease, the dye is decaged and its fluorescent and photoacoustic properties restored. Using the same strategy, a probe for dual imaging of acute kidney injury, cleavable by γ-glutamyl transferase (GGT), was developed by Chen (63, Fig. 19a),194 allowing real-time imaging of kidney function at a molecular level (Fig. 20).
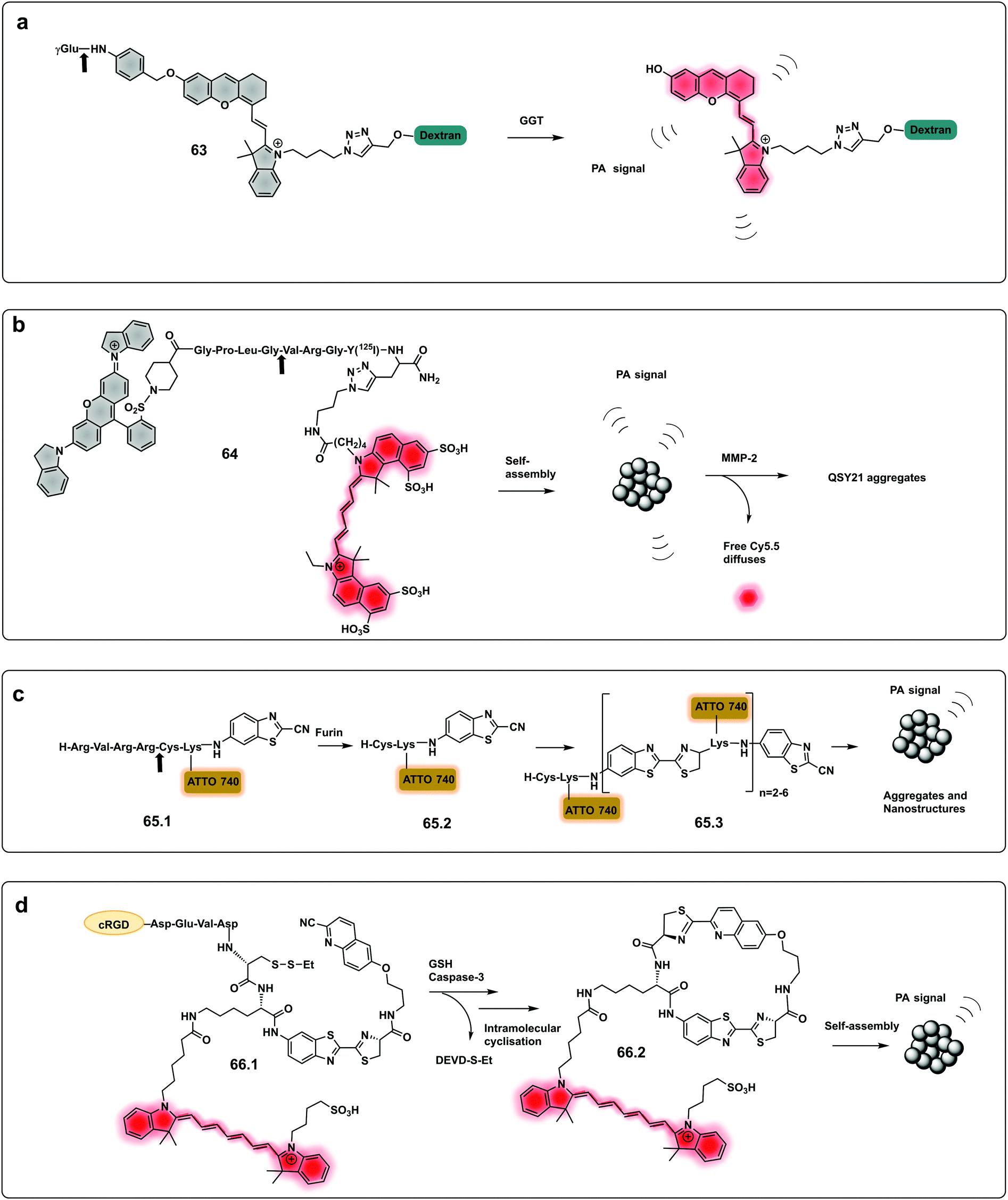 | ||
| Fig. 19 Dual and aggregation based optoacoustic activatable probes. (a) Dual imaging, fluorescence/photoacoustic probe for detection GGT, cleavage by the protease decages a cyanine dye with fluorescence and photoacoustic properties.194 (b) Amphiphilic construct using QSY21 as a photoacoustic agent and sulfonated Cy5.5 as a fluorophore. Amphiphilicity of the construct drives nanoparticle formation, while cleavage by MMP-2 disassembles the nanoparticles and results in a change in photoacoustic signal.205 (c) Bioorthogonal photoacoustic emissive oligomer forming probe for furin.207 (d) Dual activation dependent probe for sensing of GSH and caspase-3 that results in photoacoustic emissive nanoparticle assembly.208 | ||
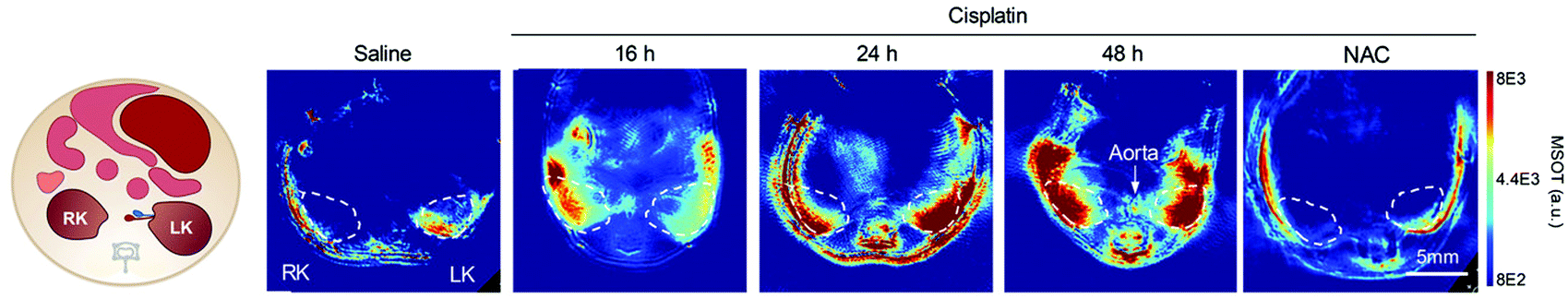 | ||
| Fig. 20 PA imaging of cisplatin-induced Acute Kidney Injury using a γ-glutamyl transferase optoacoustic probe (63).194 Mice transverse section after i.v. injection of the probe in different treatment groups (detection at 700 nm). The white circles indicate the two kidneys. RK, right kidney; LK, left kidney, NAC: N-acetyl-l-cysteine protected. Reproduced from ref. 194 with permission John Wiley and Sons, copyright 2021. | ||
CuS nanoparticles, exhibit strong (broad spectrum) absorbance, and have been used as photothermal agents for tumour ablation. As might be anticipated from their “black colour” they absorb broadly and show strong photoacoustic signals at 930 nm. A probe based on a BHQ-3 labelled MMP-cleavable-peptide conjugated to CuS nanoparticles was developed,203 that provided a combination of two PA signals in absence of the MMP. Upon cleavage by the protease, only the PA signal from the nanoparticles remained. The probe successfully sensed MMP-activity in mice in a promising alternative to optical imaging techniques with improved detection depth. Due to their strong and tunable optical absorptions gold-based nanoprobes have been widely used as PAI agents. Probes have been developed that produce a change in the emitted PA signal and produce combined fluorescence signals in response to proteases. Using a nanocage, a probe for MMP-2 was developed,204 which produces a strong acoustic signal at 800 nm. In the intact construct fluorescence emission by the Alexa Fluor 680-conjugated peptide was quenched by the gold nanocage and producing a PA signal by illumination at 680 nm indirectly. Upon MMP cleavage, the NIR dye is released and cleared from the tumour tissue which results in changes in the photoacoustic spectral signature, going from a signal contributed by both reporters to one generated by the gold nanocage alone.
An optoacoustic activatable probe205 with capacity for simultaneous photoacoustic and NIR optical imaging based on a self-assembly strategy activated by protease cleavage, driven by hydrophobic interactions has been reported. The probe was constructed with a near-infrared dye (Cy5.5) and a quencher (QSY21) linked through a peptide substrate of MMP-2. The construct is amphiphilic as QSY21 is highly hydrophobic while sulfonated Cy5.5 is hydrophilic, which drives self-assembly into nanoparticles. When cleaved by the protease enhanced NIR fluorescence is generated, simultaneously with photoacoustic signal ratiometric changes as the nanoparticle-assemblies disaggregate. Photoacoustic emissions from the intact construct were detectable with high intensity at 680 nm and at 730 nm. Upon cleavage, a reduction in the 680 nm photoacoustic signal was observed in an MMP concentration dependent manner with the corresponding increase in fluorescent emission by the de-quenching of Cy5.5 (the photoacoustic signal at 730 nm remained constant (MMP independent) and served as a reference signal (64, Fig. 19b). Following the same mechanistic principle (but in reverse), a self-assembling gelatinase probe used Purpurin 18 as the functional photoacoustic stimulator206 with self-assembly upon protease cleavage leading to the formation of photoacoustic emitting nanofibers. Another PAI self-assembling probe (65.1) for detection of furin has been developed where cleavage leads to the formation of photoacoustic emissive oligomers via a bioorthogonal reaction.207 The activation of the probe via furin cleavage of the peptide substrate released (ATTO740)-1,2-aminothiol that undergoes self-condensation to form oligomers (65.3) containing ATTO740 (Fig. 19c). These nanostructures enhance the quenching effect of the fluorophores thus minimizing radiative emission, which increases the photoacoustic signal. The same bioorthogonal reaction was used to design a caspase-3 probe.208 In this case dual activation was dependent on caspase-3 and glutathione (GSH). The construct (66.1) contained a cyano-6-hydroxyquinoline (CHQ), a D-Cys residue, a caspase-3-cleavable peptide substrate (DEVD), ICG and contained a cRGD targeting peptide (Fig. 19d). Owing to aggregation-caused quenching (ACQ) the nonradiative relaxation process was favoured, which increased the PA signal and decreased the fluorescence of ICG.
Anupama recently developed photoacoustic probes for the detection of proteases209 consisting of engineered gas vesicle based biosensors, air-filled protein nanostructures that change their ultrasound contrast in response to the activity of different biomolecules. In this case, they used tobacco etch virus protease (TEV). In presence of the protease, the gas vesicle shell becomes less stiff, thereby allowing it to undergo buckling and produce enhanced nonlinear ultrasound signals that can be easily distinguished from the intact, non-digested “gas vesicles”.
A theranostic approach based on gold nanostars was developed210 with multiple and simultaneous imaging modalities. The probe consisted of a gold nanostars core (photoacoustic and photothermal effector), conjugated to the MMP-2 peptide (Ac-GPLGIAGQ) via BSA loaded with a NIR dye IR-780, for optical imaging and photodynamic therapy.
| Modality | Contrast agents | Strategy | Protease | Sequence | Ref. |
|---|---|---|---|---|---|
| Optoacoustics | Cy5.5 dimer | FRET | MMPs | Ac-K(PEG)-[K(PEG)-K(Cy5.5))K/K(Cy5.5)-K(PEG)]n- | 199 |
| Alexa750/BHQ-3 | FRET | MMPs | Not reported | 201 | |
| Hemicyanine | De-caging | GGT | GGR | 194 | |
| uPa | γ-Glu | 202 | |||
| Cy5.5/QSY21 | Self-assembly | MMPs | KCPLGVRGY | 205 | |
| Photoacoustics | CuS nanoparticles/BHQ-3 | FRET | MMPs | GLPGVRGKGG | 203 |
| Gold nanocages/Dye 680 | FRET | MMPs | GKGPLGVRGC | 204 | |
| Purpurin 18 | Self-assembly | Gelatinase | PLGVRG | 206 | |
| ATTO740 | Bioorthogonal | Furin | RVRRC | 207 | |
| ICG | Bioorthogonal | Caspase-3 | DEDV | 208 | |
| Gas Vesicle Protein (GvpC) | Buckling of the GvpC | TEV | ENLYFQG | 209 | |
| PA/PTT/PDT | Gold nanostars/IR – 780 | FRET | MMPs | GPLGIAGQ | 210 |
2 Bench-top or in vitro applications
2.1 Colorimetric
Colorimetric sensing determines the presence or absence of target analytes by measuring optical density (Beer–Lambert law) and depends on the generation of coloured species. Due to the advantages of simple operation with no need for complicated instrumentation, colorimetric sensing offers a good strategy for point-of-care diagnosis with test results that can be interpreted with the naked eye (classic examples are Gram staining of bacteria). This is useful in terms of speed of analysis, and the reduction in the cost of testing in resource-limited settings where sophisticated technologies and time delays would limit application. In the field of protease sensing, there are two main types of colorimetric probes used: colorimetric probes based on enzyme-catalyzed organic chromogenic substrates that form coloured products211 and biosensors based on modification of the surface plasmon resonance (SPR) of noble metal nanoconstructs.212 Indeed gold nanoparticle (AuNPs) colorimetric assays are the most widely used with many publications over the past 30 years taking advantage of the AuNPs excellent tunable properties as simple chromophores213 with the optical properties of AuNPs depending on the SPR of the nanoparticles.214 Modifications of the surface of the AuNPs can induce or prevent aggregation, which correlates with changes in the SPR that leads to a colour dependence (Scheme 8). The surface of the AuNPs are usually modified using thiol-gold chemistry to simultaneously add specific molecular recognition moieties and to tune the stability of the particle suspension. Surface modification of the construct by targeted biomarkers can trigger changes in the aggregation behaviour of the AuNPs that can be visible by the naked eye as a colorimetric response. Generally the aggregated particles give rise to a distinct purple colour, while the dispersed AuNPs look red. A simple protease colorimetric probe can thus use peptides conjugated to AuNPs215e.g. via thiol chemistry or by electrostatic interactions, with cleavage of the peptide resulting in either induction or prevention of aggregation that results in a change in colour. The systems based on AuNPs aggregation can use a “mix and detect” system where standard unmodified gold nanoparticles (generally stabilised with citrate) are mixed with a peptide. Alternatively, gold nanoparticles may be pre-modified, where the protease substrate peptide is immobilised on the surface of the nanoparticles which can then generate a visual response based on the exposure to (or lack of) the protease. | ||
| Scheme 8 The concept of colorimetric sensing based on gold nanoparticle aggregation in response to the presence of proteases. | ||
A probe for MMP-7216 was synthesised (67) using pre-modified gold nanoparticles using a C terminal Cys peptide substrate for the protease. The negatively charged peptide sequence prevented aggregation of the AuNP until cleavage by the protease, that removed the negatively charged residues causing a reduction of the net charge and decrease in nanoparticle size, leading to a loss in stability of the nanoparticles that would aggregate and shift from red to purple; a colour shift detectable by the naked eye. A mix and detect system (68) used unmodified free gold nanoparticles (citrate stabilised, negatively charged) and a caspase-3 cleavable peptide.217 The peptide contained two adjacent Cys at the C terminus, three negatively charged residues and one positively charged arginine. When the peptide was pre-incubated with the protease, the three negatively charged residues were cleaved off, leaving a net positively charged thiol fragment that would be immobilised on the gold nanoparticle via the cysteine side-chains and induce aggregation (due to interaction between the positively charged arginine and remaining negatively charged areas of the AuNPs). When the free AuNPs were incubated with the mixture of peptide and protease, a blue colour would be observed due to aggregation whilst the incubation with the intact peptide and no protease prevented aggregation retaining the initial red colour.217 Chen218 reported a probe for MMP-2 (69) following a similar strategy with the negatively-charged fragment on the N terminus (as a Glu4 moiety) and the positively charged residue being Lys on the thiol containing fragment.
Conjugation by electrostatic interactions has used peptides containing high levels of positively charged residues that coordinate with the free negatively charged AuNPs (citrate stabilised) in suspension. Using this approach, a colorimetric assay for trypsin was reported (72)219 that used a hexa-Arg peptide as both a substrate for trypsin and to allow conjugation to the nanoparticles. When the free (negatively charged) nanoparticles are exposed to the positively charged peptide, the peptide intercalates between gold nanoparticles and induces cross-linking and aggregation of Au-NPs leading to the red-shift. If, however, the peptide is pre-incubated with enzyme prior to exposure to the nanoparticles, the peptide is digested, and the nanoparticles remain dispersed. Another probe using electrostatic conjugation was reported220 which used negatively charged carboxyl-functionalised AuNPs (using a carboxy-PEG12-thiol) in combination with a MMP substrate with hexa-His tags at both ends (metal ions drive metal-affinity coordination between the carboxyl groups and the His-tags). The intact peptide therefore drives aggregation whilst the cleaved peptide promotes the dispersion of the nanoparticles.
Ding221 used a “mix and detect” trypsin (70) assay with a peptide bearing a Cys residue at the C-terminus and a positively charged Lys at the P1 position. Since the oligopeptide contains both Cys and a positively charged Lys residue, it causes aggregation of the citrate-stabilised, negatively charged, AuNPs. However, since trypsin cleaves the peptide, separating the Cys and Lys residues, the cleaved oligopeptide no longer causes aggregation of the AuNPs.
Meng-Qi reported a novel strategy222 allowing the properties of the SPR to be tunable via the modification of gold nanorods (AuNRs) (Fig. 21c). This protease detector (71.1, 71.2, 71.3) was based on a protease specific peptide with two central Cys residues at the cleavage site. Cleavage by the protease thus releases two fragments exposing the monothiol groups from the Cys, that are otherwise non-accessible, and react with the AuNRs causing morphological changes, with “new spikes” formed by addition of the peptides fragments to the surface, resulting in unique SPR peak shifts that were also seen by the naked eye (red-to-blue). Using this strategy, the group developed a probe for the detection of trypsin activity in a label and instrument-free manner and with ultrahigh sensitivity, with a LoD of 60 fM.
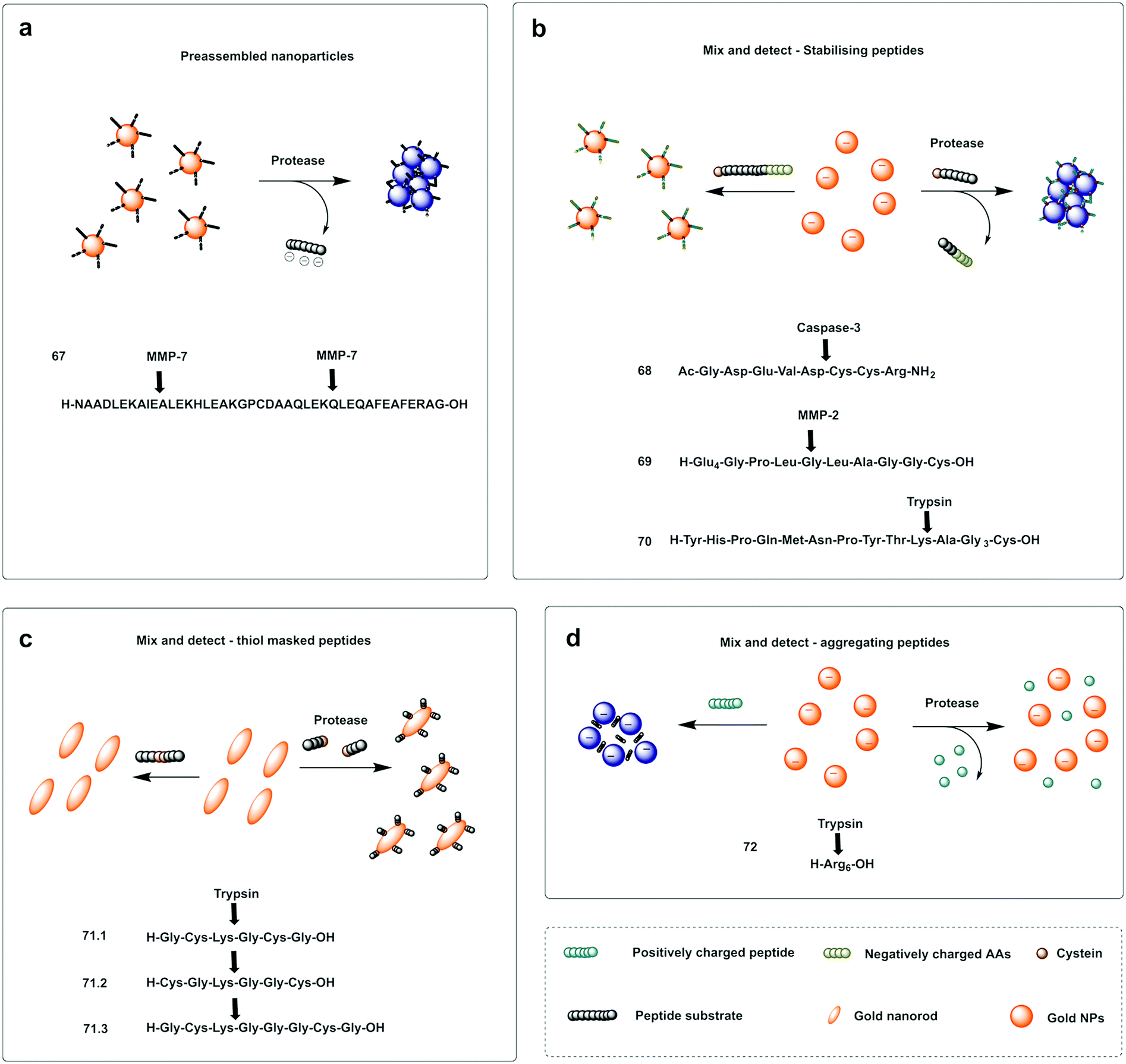 | ||
| Fig. 21 Examples of protease colorimetric assays based on gold nanoparticles. (a) Premodified gold nanoparticles conjugated to a cysteine terminal cleavable peptide for MMP-7.216 (b) Mix and detect strategies based on gold nanoparticles and stabilising peptides (with destabilising effect if protease is present) for detection of caspase-3,217 MMP-2218 or trypsin.221 (c) Mix and detect method based on tunable SPR of gold nanorods (AuNRs)222 using a protease specific peptide with two central Cys residues at the cleavage site. Cleavage by the protease causes morphological changes, with “new spikes” formed by addition of the peptides fragments to the surface via newly exposed thiol groups. (d) Colorimetric assay for trypsin219 that uses a hexa-Arg peptide as both a substrate for trypsin and to allow aggregation of the nanoparticles. When nanoparticles are exposed to the positively charged peptide, the peptide intercalates between Gold nanoparticles and induces aggregation of Au-NPs leading to the red-shift, which is prevented when the peptide is digested by trypsin. | ||
A colorimetric method for prostate-specific antigen (PSA) detection was developed by Liu223 using the formation or inhibition of formation of AuNPs as a colorimetric indicator. The method was based on ascorbic acid induced formation of AuNPs, a process that can be inhibited by the presence of Cu2+. HAuCl4 (74.1) can be reduced to form AuNPs (74.2) by ascorbic acid (73.1), however, in the presence of Cu2+, ascorbic acid is oxidised (73.2), and can no longer reduce the HAuCl4. The designed construct was based on peptide coated gold magnetic microbeads containing a Cu2+ binding triad (Asp-Ala-His) followed by a PSA cleavable sequence (Fig. 22a). When the protease is present, the peptide is cleaved and the Cu2+ binding triad released. Consequently, the copper ions are not trapped, resulting in oxidation of ascorbic acid and no AuNP formation. Alternatively, the Cu2+ binding triad can be localized internally so that it remains masked (Fig. 22b), but can be demasked by proteolytic hydrolysis (β-secretase) exposing the Cu2+ chelating ligand. In this design, formation of AuNPs only happens in the presence of the protease.224
Bhatia reported on the design and synthesis of protease probes for pre-clinical use developing a series of colorimetric assays/platforms as diagnostics tools for detection of disease related proteases. They used a variety of peptide-modified platforms that can be administered intravenously, activated at the site of the disease and detected in the urine. The urinary, colorimetric, in vivo assay225 conjugates gold nanoclusters (AuNC) to a carrier protein (neutravidin) tethered to a protease cleavable peptide with a Cys residue for binding to the AuNC (76–79). Exposure to the protease (thrombin or MMP) at the disease site, following IV administration, resulted in the release of the AuNC from the construct enabling passage into the urine to produce a direct colorimetric readout of the disease state. The strategy exploited the peroxidase capacity of the ultra-small AuNC, with a size <2 nm that also showed efficient filtration capacity through the kidneys into the urine. These AuNCs had an intrinsic catalytic peroxidase activity, catalysing the oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB) (75.1), that could be monitored by absorbance at 652 nm (Fig. 23a).
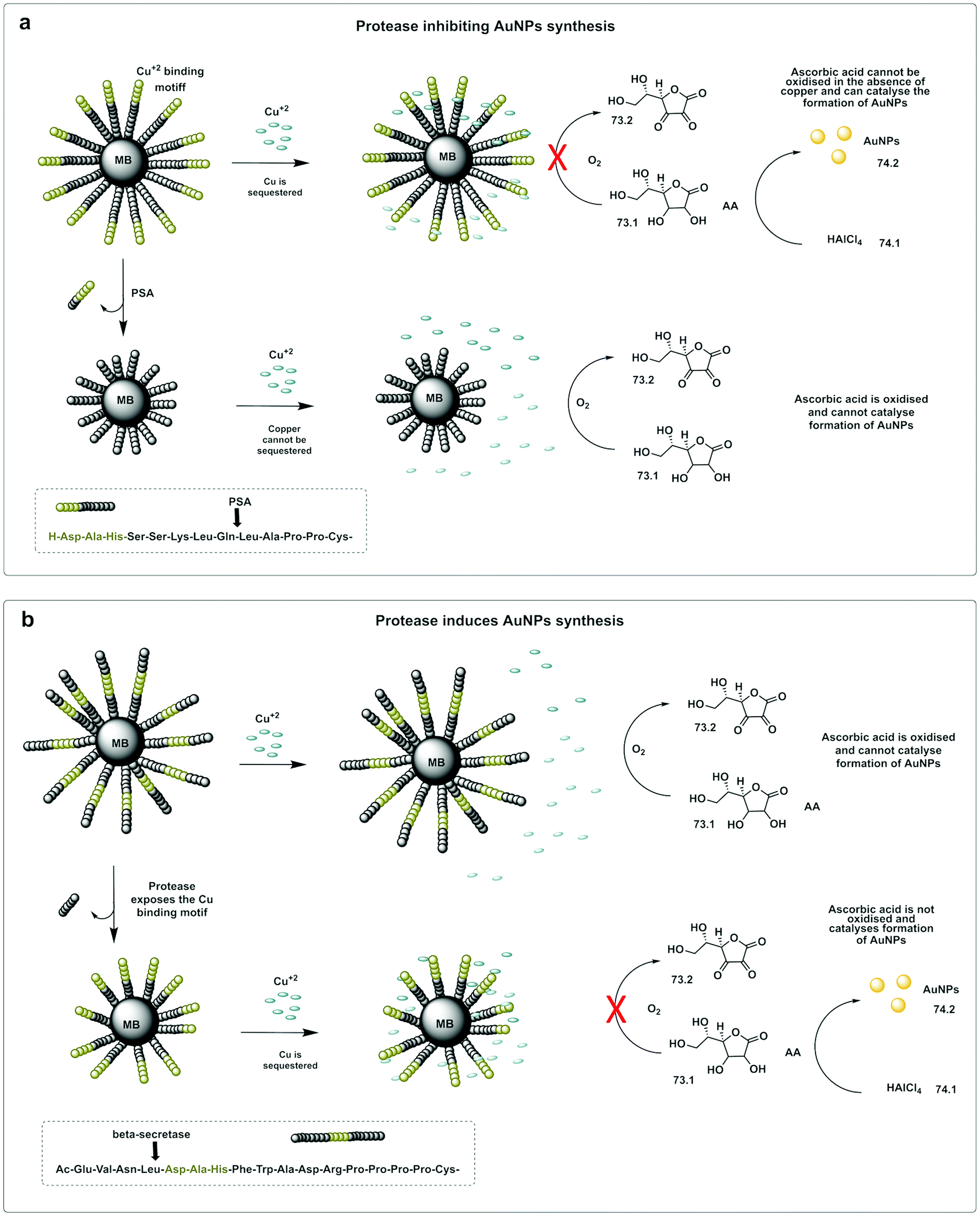 | ||
| Fig. 22 In situ formation of catalytic AuNPs. (a) Colorimetric assay for detection of PSA based on in situ synthesis of gold nanoparticles from the precursor HAuCl4 catalysed by ascorbic acid in absence of Cu(II).223 The design used peptide-modified magnetic nanoparticles containing a Cu2+ binding triad. The peptide was cleaved by PSA and the Cu2+ binding triad released. Consequently, the copper ions are not trapped, resulting in oxidation of ascorbic acid and no AuNP formation. (b) System with the Cu2+ binding triad is localized internally and demasked by proteolytic hydrolysis (β-secretase) exposing the Cu2+ chelating ligand. In this design, formation of AuNPs only happens in the presence of the protease.224 MB: magnetic beads. AA: Ascorbic Acid. | ||
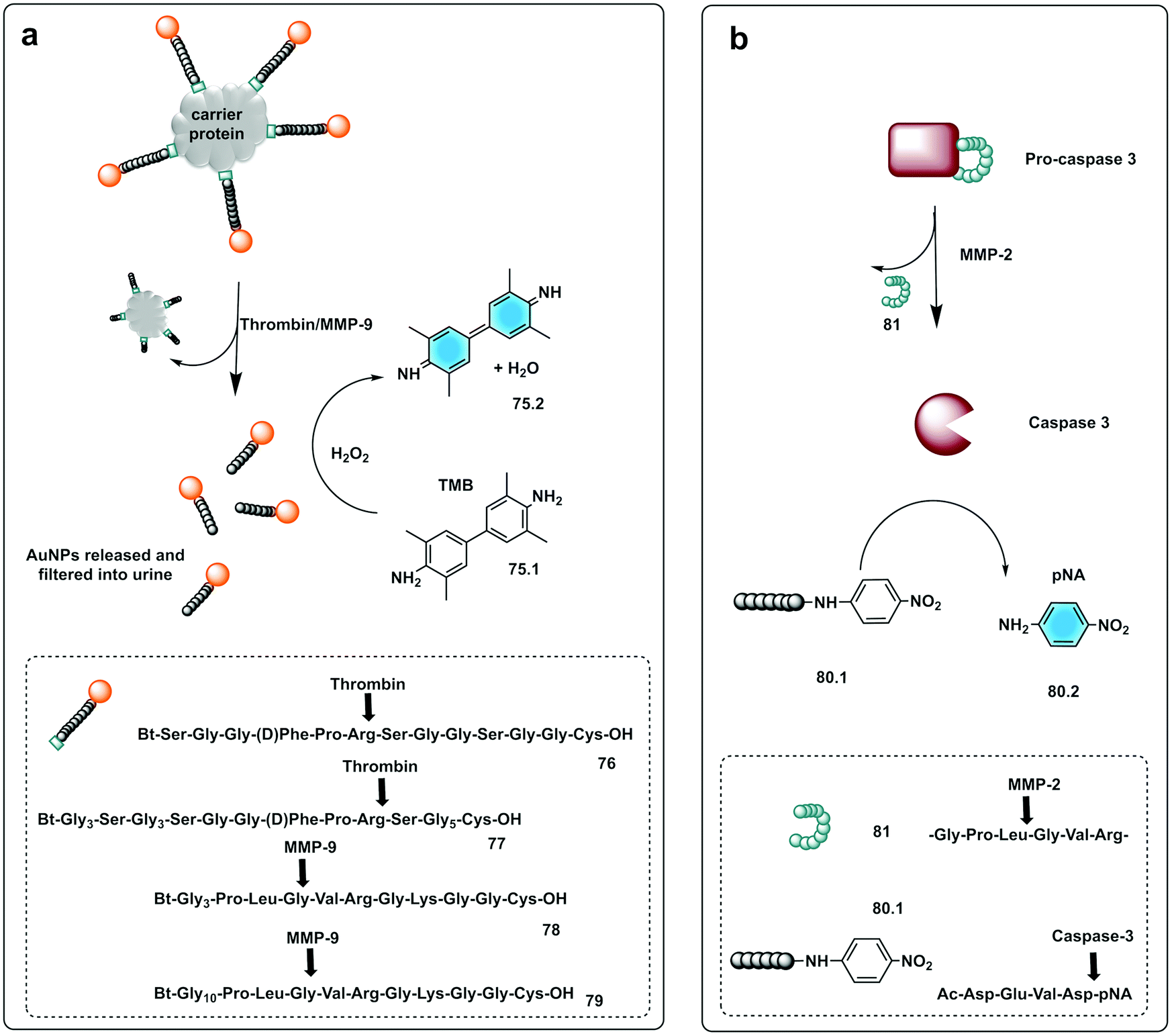 | ||
| Fig. 23 Small molecule based systems for colorimetric sensing of proteases. (a) Urinary colorimetric in vivo assay225 using gold nanoclusters (AuNC) attached to a carrier protein via a protease cleavable linker. Exposure to thrombin or MMP-9 releases AuNCs to produce a direct colorimetric readout exploiting the intrinsic catalytic peroxidase activity of AuNCs to oxidise TMB. (b) Engineered procaspase-3 is auto-inhibited in the absence of the target protease (MMP-2) by a weak reversible inhibitor attached to the enzyme by a peptide linker. Upon activation by MMP-2 active caspase-3 is generated that cleaves the substrate Ac-DEVD-pNA generating a chromogenic response.231 | ||
Zourob designed a series of low-cost, easy-to-handle, highly sensitive and portable colorimetric biosensors (Fig. 24) capable of detecting different disease-related proteases (82, Fig. 24). The method used a probe that consisted of a specific peptide substrate covalently attached to magnetic beads through its N-terminus and linked to a gold probe surface at the C-terminus via a Cys residue. This construct resulted in a layer of magnetic beads adsorbed on the probe surface masking its golden colour in the absence of the protease. Upon protease cleavage the peptide linkage between the magnetic beads and the gold probe surface is lost and the released magnetic beads were magnetically collected and analysed. This idea was applied to develop portable probes for PSA,226 HNE and Cathepsin G,227 Listeria monocytogenes protease228 and P. aeruginosa proteases.229
 | ||
| Fig. 24 Colorimetric sensing platform for HNE and Cat G.227 These probes consist of specific protease substrates covalently bound to a magnetic bead at one end and to a gold surface by the other. Cleavage by the protease results in dissociation of the magnetic bead complex. A magnet placed on the back of the strip (see arrow) attracts the cleaved beads that are displaced exposing the gold color. HNE sequence: -GSGSGGGAAPVAAKGGGSGSC- (82.1) and cathepsin-G sequence: -GPQGIWGQR- (82.2). Reproduces with permission of the American Chemical Society, copyright 2015. | ||
 | ||
| Fig. 25 Mass tagged nanocluster carriers, conjugated to cleavable peptide mass reporters for profiling of MMP protease activity overexpressed in a variety of diseases.244 The nanoclusters (each containing a different peptide sequence) are delivered to the disease site where overexpressed proteases release the isobaric MS reporter tags that are filtered into the urine. The reporters in the urine are treated with light to release the MS isobaric tags that are analysed by MS/MS. NC: nanocluster | ||
Small molecules have found successful applicability in this area with p-nitroaniline (pNA), a well-known reporter. Thus, chromogenic peptides, conjugated to pNA (80.2) have been used for colorimetric-based testing, which when conjugated are colourless, but upon cleavage result in a colorimetric response.230 An interesting strategy was developed by Dokyung, where an engineered procaspase-3 was modified to be auto-inhibited in absence of the target protease (MMP-2) by adding a weak reversible inhibitor linked to the enzyme by a peptide linker (81) that was cleaved by the protease of interest (Fig. 23b). This cascade system also used a chromogenic caspase-3 substrate (Ac-DEVD-pNA, 80.1) to generate a chromogenic response with its hydrolysis rate reporting on MMP-2 activity.231 Using pNA-peptides, immobilised on cellulose, a paper-based chromogenic probe to detect inflammation was developed.232
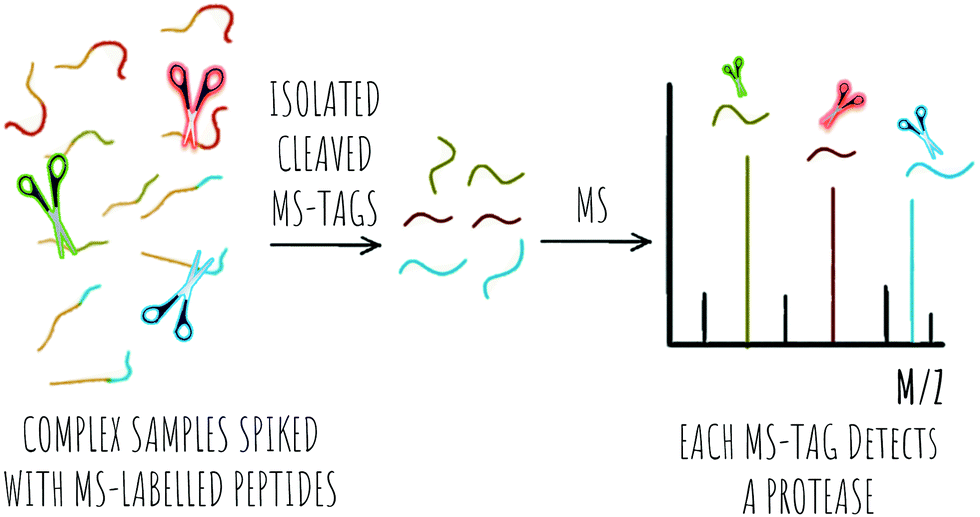 | ||
| Scheme 9 MS proteolytic profiling, where protease peptide substrates are modified with MS tags (shown here in blue, red or green) and used as reporters. Complex samples can be spiked with these mixtures of reporters and the targeted proteases cleave the peptides releasing the MS tags. Each tag has a unique MS spectrum and each reports on the activity of a different protease. | ||
2.2 MS based detection
MS has been used as a method for a variety of proteomic analysis studies including as a diagnostic tool to differentiate disease from healthy profiles as well as staging and disease monitoring. Proteome profiling for diagnostic purposes is a thriving area in proteomics, however, the complexity of the samples and the inter-individual (not disease-related) heterogeneity poses challenges.24 Analysis of proteases via activity profiling has attracted significant attention, but the complexity of samples still poses a problem. Certain proteolytic fragments, from high abundant serum proteins, are potential markers of tumour-specific proteolytic activity that can be analysed in blood/serum specimen, but they present a significant analytical challenge. This problem can be circumvented using exogenous synthetic substrates. Thus, “spiking” exogenous reporter peptides into biological samples for the characterization of protease activity offers substantial advantages over profiling of ‘native’ proteomic serum. These reporter peptides allow accumulation of signal to levels that are readily detectable with MS and eliminate the background signal provided by high-abundance ‘native’ proteins or peptides in the sample (Scheme 9).24 When reporter peptides are added to the specimen, the proteolysis of these exogenous reporters produces a unique MS spectrum that will be different in the absence or presence of the protease (excess reporter peptides are necessary not only to ensure substrate saturation, but also to displace competing natural substrates from any tumour-associated proteases).233The use of isotopic MS tags has greatly simplified analysis of complex samples with tagging of freshly generated peptide fragments prior to separation and analysis allowing quantitative MS/MS analysis of digested peptides. The first isotope-based MS tags for peptides incorporated stable heavy isotopes such as 13C, 15N,18O and 2H with initial tags consisting of a duplex system with “heavy/light” isotope tags,234 allowing comparison of two different conditions (Table 2).
ICAT (Isotope-Coded Affinity Tags)235 is an isotopic, duplex system of MS tags that contain a biotin moiety that allows isolation by affinity chromatography, allowing co-elution of isotopically labelled peptides for further analysis.236 However, screening of larger number of variable conditions was not possible.237 In 2002, isobaric tags were introduced, ITRAQ (Isobaric Tags for Relative and Absolute Quantitation)238 and tandem mass tags (TMT)239,240 as powerful tools for multiplexed MS proteomic analysis. These tags contain multiple isotopic variants that are chemically identical, and co-migrate in liquid chromatography separations but offer different MS signatures.
Optimisation of MS reporter peptides led to the design of a generation of reporters that used isobaric and non-isobaric isotope mass tagging for targeting proteases overexpressed in disease. The reporters generally contained a specific peptide-based substrate covalently linked to a MS tag. The tag-containing peptides co-eluted in chromatographic separations, while the combination of reporter peptides could be analysed by MS/MS. Findeisen developed a series of synthetic MS tagged reporter peptides25,241,242 to help address a key challenge of protease profiling in complex biological samples, namely the risk of cleavage by non-specific peptidases, that are generally very abundant in complex samples. This was achieved by incorporating a flanking aminohexanoic acid groups in the peptide substrate that successfully reduced degradation by native exopeptidases.242
Ouyang developed non-isotopic labels to screen for caspase-3 cleavable peptides,243 using MALDI-MS as the analytical method with peptides labelled by virtue of a terminal maleimide handle. This demonstrated sensitive detection of caspase-3 activity and offers a platform that would be applicable to other proteases.
Bhatia244 developed a variety of synthetic biomarkers for disease-related proteases based on photocaged tandem isobaric peptides allowing up to 10-plexed protease analysis in a single run for possible disease stratification (Fig. 25). The probes/reporters consisted of nanocluster carriers, conjugated to cleavable peptide mass reporters for proteases overexpressed in a variety of diseases. The reporters contained substrate peptides with isobaric MS tags conjugated through a photolabile linker245 with an additional peptide that enhances renal clearance. The nanocluster constructs were cleaved by disease related proteases and since the released fragments were connected to isobaric MS tags (via a photocleavable linker), this allowed comparative relative abundance quantification of all 10 conditions/tags. One biomarker approach was developed for detection of fibrosis using so-called iron oxide nanoworms as carriers for the reporters,245 with each nanoworm containing multiple copies of the same isobaric peptide, with 10 different isobaric nanoworm complexes co-administered. Cleavage of the constructs led to reporters in urine, and an indication of active protease present. Following the same approach, probes were developed for the detection the protease activity in prostate246 and lung cancer.244
| Strategy | Reporter peptide | Type of tag | Tag | Systems/conditions | Protease | Ref. |
|---|---|---|---|---|---|---|
| Unlabelled peptide | Exogenous peptide from bacteria | Non-isotopic | Tryptic digest of the N-terminal adenomatous polyposis coli protein | 2 | Non-specific | Findeisen 2008233 |
| MS encoded peptide | Synthetic protease substrate with protease resistant label | Non-isotopic | AhxAhx-HHHHHH | 2 | Cysteine-endopeptidase cancer procoagulant | Peccerella 2010241 |
| Yepes 2011242 | ||||||
| Ahx-ateevlkl | 2 | Cysteine-endopeptidase cancer procoagulant | Yepes 201225 | |||
| Dual maleimide (DuMal) | 2 | Caspase-3 | Ouyuong 2019243 | |||
| Isobaric tagged peptides (iCore) | Isobaric | One letter code | 10 | Thrombin Tissue factor FXa | Bathia 2020244 | |
| D-Amino acids lowercase | Cathepsin B | |||||
| Note: Charges in the structures below refer to isotope mass additions not formal charges | MMP2 | |||||
| e+3G+6VndneeGFfsAr | MMP7 | |||||
| e+2G+6Vndnee+1GFfsAr e+1G+6Vndnee+2GFfsAr eG+6Vndnee+2GFfs+1Ar eG+5VndneeGFfs+4Ar | MMP8 | |||||
| e+3G+1Vndnee+1GFfs+4Ar e+3GVndneeG+6FfsAr e+2GVndneeG+6Ffs+1Ar e+1GVndnee+2G+6FfsAr eGVndnee+3G+6FfsAr | MMP9 | |||||
| MMP14 | ||||||
2.3 Electrochemical
Electrochemical probes are a class of chemical probes in which an electrode is used as a transducer element in the presence of an analyte that allows generation of an electrochemical signal change. Electrochemical biosensors have been used for the last 60 years247 and were first reported was by Clark and Lyons248 to allow measurement of glucose levels. Based on their mode of operation and the type of electrode, electrochemical biosensors can be classified as potentiometric, amperometric or impedimetric but all have in common the conversion of chemical information into a measurable electrochemical signal.Electrochemical protease biosensors offer some advantages over those based on fluorescence as they can offer higher specificity and sensitivity even in turbid or intrinsically fluorescent solutions. Many of these are also compatible with miniaturisation and mass-manufacture for point-of-care devices (Fig. 27).249 Comprehensive reviews covering this area include those by Vanova,250 Ming249 and Ong.16 Typically, protease analysis probes are composed of a redox-tagged recognition peptide for the enzyme of interest, which is then attached to an electrode surface (Scheme 10). The flexibility of the peptide allows for the redox tag to come into contact with the electrode surface to elicit an electrochemical signal. Upon cleavage of the peptide by the enzyme of interest, the redox tag is released into the bulk solution and a quantifiable signal decrease can be used to determine enzyme activity. This was demonstrated by a probe (83) developed by Liu with ferrocene-labelled MMP-7-cleavable peptides immobilised onto a gold electrode.251 In the absence of MMP-7, maximal voltammetric signal was achieved as the redox labels were close to the surface allowing redox cycling with the electrode surface. Observable decreases in the signal corresponded to various levels of MMP-7 in solution with a limit of detection of 0.1 ng mL−1. p-Aminodiphenylamine (pADA) and methylene blue have been used as redox reporters in probes for thrombin (84)252 and MMP-9 (85),253 and similar approaches have been employed for a range of other proteases including the cathepsin family,254 neutrophil elastase (87, Fig. 26)255 and HIV-1 protease (88).256 Gold electrodes have been always the classical choice when designing electrochemical protease probes, however, in recent years, several novel electrode platforms have been used showing excellent performance on protease sensing. Indium Tin Oxide electrodes257 and carbon based electrodes are examples of alternatives to gold electrodes used in the field. Embedded vertical carbon nanofiber electrodes, separated from each other forming a bush-like platform, were used for the detection of Cathepsin B (86).258 These approaches reduce steric hindrance and improve temporal resolution. Novel Pt based microelectrodes for detection of trypsin were pioneered by Ucar,259 whose results demonstrate similar specificity when compared to Au electrodes and enhanced reproducibility and stability. However, all these strategies rely on a ‘signal-off’ output, which may be undesirable.
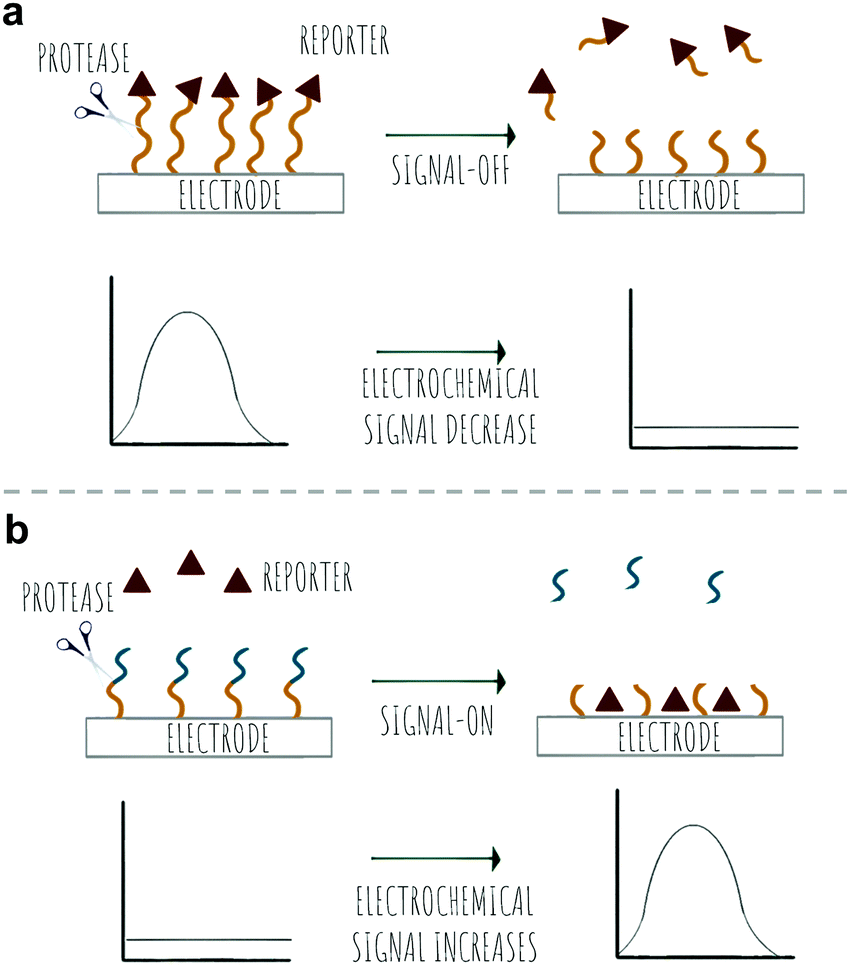 | ||
| Scheme 10 Electrochemical sensing strategies for proteases. (a) Signal-off detection. Cleavage by the protease, releases the reporter from the electrode resulting in a decrease of signal. (b) Signal-on detection. Cleavage by the peptide demasks a high-affinity sequence that promotes electrochemical reporter binding, resulting in an increase in the electrochemical signal. | ||
 | ||
| Fig. 26 Electrochemical detection of HNE (87).255 (a) % signal change-time course for substrate-modified electrodes immersed in varying HNE concentrations (0, 10, 50, 75, 100 and 150nM) in HEPES buffer. (b) Adjusted signal, A (%), after 90min plotted against the concentration of HNE. (c) Electrical signal registered at different incubation times (0, 5, 10, 20, 60 and 90min) for 100nM HNE in HEPES buffer. Reproduced from ref. 255 with permission Elsevier, copyright 2018. | ||
 | ||
| Fig. 27 Electrochemical probes for proteases where the signal deceases upon protease activation. Abbreviations: pADA: p-aminodiphenylamine; Sta: statin (4-amino-3-hydroxy-6-methylheptanoic acid); Bt: biotin; arrows indicate cleavage site. While peptide sequences are always written N → C terminal by convention (NPeptideC), we have utilised some C → N (cPeptideN) to allow ease of presentation. *Binding based assay. Abbreviations: pADA: p-aminodiphenylamine; Sta: statin (4-amino-3-hydroxy-6-methylheptanoic acid); Bt: biotin; arrows indicate cleavage sites. | ||
A label-free strategy can be employed to overcome these drawbacks, wherein a peptide sequence is used to preclude or promote the approach of a redox mediator to the electrode surface; either by steric or electrostatic repulsion. An advantage of this strategy is that peptide substrates can be used without the need of a label, allowing for more sensitive assays. Cao has demonstrated a general method using electrostatic repulsion in a system where a peptide with a cationic region is immobilised on the electrode. The peptide formed a layer that prevents the penetration of the cationic [Ru(NH3)6]Cl2]+ redox reporter due to electrostatic repulsion, and no electrical signal was transferred. Upon the cleavage of the peptide, the positively charged region was released and the reporter can approach the electrode surface to produce a “signal-on” (89).260 However, this signal requires the addition of an electrochemical reporter to the assay. Deng designed a ‘signal off’ probe, using [Fe(CN)6]3−/4− as a redox reporter and a peptide immobilised to a gold surface. A positively charged Lys in a peptide induced binding of the negatively charged redox reporter resulting in an electrical signal output. Upon the addition of the serine protease PSA, the Lys residue was cleaved and the reporter no longer binds the peptide, resulting in a decrease in the electrical signal.261 However, high background-to-signal ratios limit the sensitivity of this method compared to redox labelled-peptide strategies.
Li,262 using a gold electrode with immobilised peptides (via 11-mercaptoundecanoic acid) developed a system that contained a “seed peptide” that accelerates/catalyses amyloid β misfolding on the electrode surface. The incorporation of a cleavable sequence between the anchoring fragment and the seed sequence allowed protease mediated release of the seed motif (and prevention of amyloid formation) allowing the electrode surface to be available for interactions with the free redox reporter.
Another example that falls in between a redox-labelled and label-free design is the ‘signal on’ system designed by Ko,263 based on the triggerable interaction of a redox reporter with the electrode. The system was used to report on the presence of thrombin using ferrocene as the redox reporter and a fibrinogen coated electrode. The electrode, coated with Fc-fibrinogen, was susceptible to thrombin mediated hydrolysis of the coating fibrinogen, that “demasked” the surface of the support.
Other strategies that depend on a complex formation or secondary triggered reactions have also been developed. Thus, proteolytic activity has been measured electrochemically by use of the cleaved peptide acting as a ligand for an electrocatalyst: for example, an electrocatalyic reaction can be triggered by caspase-3.264 Cleavage of the substrate peptide (90) released a Cu(II)/Ni(II) peptide binding motif (Ser-Lys-His) that resulted in the in situ synthesis of the reporter, a copper electrocatalyst for water oxidation at the electrode surface. The use of water as a substrate negates the need for addition of other reagents and provides a straightforward and simple operating procedure with low background current for caspase-3, and a LoD of 0.2 pg mL−1. This sensing platform was shown to be robust in complex media, such as cell lysate. Following a similar principle, Wu265 developed a two-enzyme system (91) where the catalyst in this case was alkaline phosphatase (ALP) and the activating protease B lichenifromis. ALP catalyses the formation of electrochemically active phenol. The system incorporated streptavidin-alkaline phosphatase (Sav-ALP) on the electrodes through a biotin-labelled peptide substrate. In absence of the protease, streptavidin-alkaline phosphatase catalyses the formation of a redox reporter, while in its presence the biotin moiety was removed from the electrodes reducing the electrochemical signal.
Chen266 reported a system with a LoD for caspase-3 of 0.06 pg mL−1 (∼3 fM). The system used the triggerable binding of methylene blue to the electrode by immobilisation of a peptide substrate. Thus, an acetylated caspase-3 peptide substrate was anchored into an electrode via a C-terminal Cys residue (94). Cleavage exposed a free terminal amine group on the remaining anchored fragment, which was able to covalently bind graphene oxide. Electroactive methylene blue then bound through π–π stacking and resulted in an increase of electrochemical signal. Meng267 used graphene oxide for the in situ generation of silver nanoparticles on a peptide functionalised gold electrode. In the absence of PSA, the graphene oxide was immobilised on the peptide and silver nanoparticles were generated in situ, resulting in an electrochemical signal.
Xia used an immobilised peptide–heme catalyst complex268 to increase electrochemical signals in response to the β-amyloid precursor protein enzyme 1 (BACE1). The construct (93) consisted of a heme-binding segment (Aβ1 → 16), the heme being capable of the electrocatalytic reduction of O2. This sequence was bound to the BACE1 substrate whose cleavage by BACE1 released the heme binding region of the peptide, such that reduction of O2 was no longer possible.
Attempts have been made to incorporate amplification within these systems to further increase their sensitivity. Magnetic beads or nanoparticles269 functionalised with peptide-reporter systems (94), have been used to assay activity in complex media (e.g. blood or urine) and can then be extracted and deposited onto magnetic electrodes, to allow analysis in less complex media (e.g. aqueous buffers). Neutravidin coated magnetic beads (95) were used for detection of trypsin270 with enhanced sensitivity and robustness in cell lysate and clinical samples (probe consisting of neutravidin magnetic nanoparticles functionalised with substrate peptides via a biotin moiety, labelled with a FITC antigen tag detectable by a labelled antibody). In the absence of trypsin, the peptide probe was intact and a high number of redox tags providing a large amperometric response (using the hydroquinone (HQ)/HRP/H2O2 system). While the presence of trypsin decreased the amperometric signals. Fluorescence from cadmium telluride quantum dots (CdTe-QDs) was converted into measurable photocurrent by Liu,271 using electrodes functionalised with a peptide that contained a positively charged region to attract the QDs (96). Upon trypsin cleavage, the QDs were released from the surface and the signal decreased.
A related strategy was designed by Yuan to translate the peptide cleavage event to a nucleic acid-based detection platform.272 Peptide-functionalised magnetic polystyrene microspheres were coupled to DNA–AuNPs (97). The proteolytic action of the enzyme of interest (MMP-2) decouples the DNA–AuNPs from the peptide-microspheres with the DNA released by proteolytic activity hybridising to a redox-labelled complementary strand of DNA. Next, an exonuclease III assisted cycling signal amplification step was applied, whereby the duplex DNA was selectively digested to release the redox label (methylene blue), as well as the DNA-AuNP. The electrode surface, functionalised with a macrocyclic cavity (cucurbituril) binds the redox label and provides an electrical signal. The release of the DNA–AuNPs allows for another hybridisation event to occur with cyclical amplification system allowing a single DNA-AuNP to allow multiple redox labels to approach the electrode surface. The host–guest interactions at the electrode surface provided a LoD of 0.15 pg mL−1. This ‘signal-on’ sensing platform allows for robust analysis, with multiple steps preventing false positive results and could be applied to a range of proteases in human serum.
Another method for amplification has been developed by Hu273 that combined the use of substrate peptides as a recognition element and RAFT polymerization for signal amplification. Thrombin substrate peptides were immobilised onto a gold electrode via an N-terminal cysteine residue (98). Following cleavage by thrombin, the electrode was immersed into a solution containing a Zr(IV) source and a carboxylate-containing RAFT initiator. Only at the site of the cleaved peptide (free carboxylic acid), did the Zr complex form a bridge between the carboxylate of the RAFT initiator and the peptide. The addition of acrylate-ferrocene allowed polymerisation and recruitment of multiple redox reporters at the site of cleavage, allowing for very sensitive detection.
3 Summary and perspectives
In this review, we have discussed the recent advances, and high-lighted the most relevant and novel substrate-based reporters and strategies, for the detection of proteolytic activity.The design of substrate-based reporters for the detection of proteolytic activity needs to overcome a number of challenges, namely the level of sensitivity can be a challenge when trying to target relevant proteases where expression levels can be low; while specificity is another key issue. This means that the generation of highly specific probes to target a single protease within a relatively broad family (e.g. caspases or MMPs), becomes a difficult task due to overlap in substrate recognition, while by-stander cleavage by generic/non-specific proteases is a common problem. Most of the reporters described in this review, target model proteases that are well understood and have been extensively used in a variety of applications, but the targeting of novel, low abundance, protease biomarkers remains a challenge.
A key challenge in the area of optical probes remains their in vivo application, due to poor tissue penetration of light enabled by current imaging technologies and high background signals from complex biological environments. However, several reporters for proteolytic activity have entered clinical studies e.g. for margin detection during surgery, in the last decade. New strategies are driving improvements in tissue penetration and background signal reduction with the shifting of the emission of fluorescent probes into the near-infrared region and beyond.
Photoacoustic imaging allows for enhanced tissue penetration and can be used in combination with fluorescence, reaping the benefits of both worlds. Chemiluminogens show great promise as alternatives to fluorescence, with virtually no background signal, with efforts focused on improvement of probe stability and shifting of emission wavelength by tuning natural substrates. Quantitative analysis is not easily achieved with optical substrate-based probes and only a few examples exist that allows quantitative analysis in association with signal amplification. Activity-based probes, that covalently bind to the protease following activation, can overcome this problem, however, they render the protease inactive and provide no signal amplification. There has been much work in the area of theragnostics, with multimodal probes combining optical imaging-based diagnosis and therapeutic intent, using photodynamic, photothermal strategies and activatable prodrugs.
On the in vitro-based application side, the novel applications of colorimetric sensing and electrochemical sensors offer great promise as low-cost and highly sensitive chemistries for point-of-care testing, but the immobilisation of the substrate probe can still present challenges. Electrochemical probes offer great promise as a point-of-care technology with the possibility of miniaturisation with relatively simple designs, however most rely on a signal-off response. MS based protease detection is a powerful tool for protease activity profiling, but a key challenge is the complexity of biological samples, that can affect the sensitivity/specificity of the assay and the resulting read-out.
The field of protease-based chemical probes is rapidly evolving to allow further detailed analysis of proteolytic events. In this review, we have described a few from the many examples of each type of probe applied in a variety of situations. The breadth of technologies developed enable access to protease detection in almost any situation, be it in vivo real-time fluorescent imaging by applying the concept of aggregation-induced emission or low-cost easily applied, colorimetric platforms for use in resource-limited settings. The future of protease chemical probes will continue to be bright, diverse and innovative.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Engineering and Physical Sciences. Research Council (EPSRC, UK) for Interdisciplinary Research Collaboration grant EP/R005257/1.Notes and references
- R. Visse and H. Nagase, Circ. Res., 2003, 92, 827–839 CrossRef CAS PubMed.
- S. Gottesman and M. R. Maurizi, Microbiol. Rev., 1992, 56, 592–621 CrossRef CAS PubMed.
- N. A. Thornberry and Y. Lazebnik, Science, 1998, 281, 1312–1316 CrossRef CAS PubMed.
- J. E. Koblinskia, M. Ahrama and B. F. Sloane, Clin. Chim. Acta, 2000, 291, 113–135 CrossRef.
- P. V. Ravindra and T. K. Girish, in Proteases in Physiology and Pathology, ed. S. Chakraborti and N. S. Dhalla, Springer Singapore, Singapore, 2017 DOI:10.1007/978-981-10-2513-6-13, pp. 289–296.
- C. E. Reed and H. Kita, J. Allergy Clin. Immunol., 2004, 114, 997–1008 CrossRef CAS PubMed.
- M. A. Slack and S. M. Gordon, Arterioscler., Thromb., Vasc. Biol., 2019, 39, e210–e218 CrossRef CAS PubMed.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- M. S. Wolfe, M. Citron, T. S. Diehl, W. Xia, I. O. Donkor and D. J. Selkoe, J. Med. Chem., 1998, 41, 6–9 CrossRef CAS PubMed.
- Z. Lv, Y. Chu and Y. Wang, HIV AIDS, 2015, 7, 95–104 Search PubMed.
- C. G. Smith and J. R. Vane, FASEB J., 2003, 788–789 CrossRef CAS PubMed.
- M. Staderini, A. Megia-Fernandez, K. Dhaliwal and M. Bradley, Bioorg. Med. Chem., 2018, 26, 2816–2826 CrossRef CAS PubMed.
- L. E. Edgington, M. Verdoes and M. Bogyo, Chem. Biol., 2011, 15, 798–805 CAS.
- J. Zhang, X. Chai, X. P. He, H. J. Kim, J. Yoon and H. Tian, Chem. Soc. Rev., 2019, 48, 683–722 RSC.
- J. Chin and H. J. Kim, Coord. Chem. Rev., 2018, 354, 169–181 CrossRef CAS.
- I. L. H. Ong and K. L. Yang, Analyst, 2017, 142, 1867–1881 RSC.
- R. Oliveira-Silva, M. Sousa-Jeronimo, D. Botequim, N. J. O. Silva, P. M. R. Paulo and D. M. F. Prazeres, Trends Biochem. Sci., 2020, 45, 604–618 CrossRef CAS PubMed.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- B. Wallace and P. J. Atzberger, PLoS One, 2017, 12, e0177122 CrossRef PubMed.
- T. W. Liu, J. Chen and G. Zheng, Amino Acids, 2011, 41, 1123–1134 CrossRef CAS PubMed.
- J. J. Diaz-Mochon, L. Bialy, L. Keinicke and M. Bradley, Chem. Commun., 2005, 1384–1386, 10.1039/b415847d.
- P. M. S. Hilaire, M. Willert, M. A. Juliano, L. Juliano and M. Meldal, J. Comb. Chem., 1999, 1, 509–523 CrossRef PubMed.
- C. Pinilla, J. Appel, S. Blondelle, C. Dooley, B. Dorner, J. Eichler, J. Ostresh and R. A. Houghte, Biopolymers, 1995, 37, 221–240 CrossRef CAS PubMed.
- P. Findeisen and M. Neumaier, Proteomics Clin. Appl., 2012, 6, 60–78 CrossRef CAS PubMed.
- D. Yepes, A. Jacob, M. Dauber, V. Costina, R. Hofheinz, M. Neumaier and P. Findeisen, Int. J. Oncol., 2011, 39, 145–154 CAS.
- D. J. M. a. J. A. Wells, Sci. Transl. Med., 1993, 260, 1113–1117 Search PubMed.
- M. Tholen, J. J. Yim, K. Groborz, E. Yoo, B. A. Martin, N. S. van den Berg, M. Drag and M. Bogyo, Angew. Chem., Int. Ed., 2020, 59, 19143–19152 CrossRef CAS PubMed.
- M. Wartenberg, A. Saidi, M. Galibert, A. Joulin-Giet, J. Burlaud-Gaillard, F. Lecaille, C. J. Scott, V. Aucagne, A. F. Delmas and G. Lalmanach, Biochimie, 2019, 166, 84–93 CrossRef CAS PubMed.
- P. Kasperkiewicz, M. Poreba, S. J. Snipas, S. J. Lin, D. Kirchhofer, G. S. Salvesen and M. Drag, PLoS One, 2015, 10, e0132818 CrossRef PubMed.
- A. Megia-Fernandez, B. Mills, C. Michels, S. V. Chankeshwara, K. Dhaliwal and M. Bradley, Org. Biomol. Chem., 2017, 15, 4344–4350 RSC.
- S. Mathur, A. Turnbull, I. Akaev, C. Stevens, N. Agrawal, M. Chopra and D. Mincher, Int. J. Pept. Res. Ther., 2019, 26, 1965–1980 CrossRef.
- H. A. Alhadrami, A. M. Hassan, R. Chinnappan, H. Al-Hadrami, W. H. Abdulaal, E. I. Azhar and M. Zourob, Mikrochim. Acta, 2021, 188, 137 CrossRef CAS PubMed.
- T. Janiszewski, S. Kołt, D. Kaiserman, S. J. Snipas, S. Li, J. Kulbacka, J. Saczko, N. Bovenschen, G. Salvesen, M. Drąg, P. I. Bird and P. Kasperkiewicz, J. Biol. Chem., 2020, 295, 9567–9582 CrossRef CAS PubMed.
- J. J. Yim, S. Harmsen, K. Flisikowski, T. Flisikowska, H. Namkoong, M. Garland, N. S. van den Berg, J. G. Vilches-Moure, A. Schnieke, D. Saur, S. Glasl, D. Gorpas, A. Habtezion, V. Ntziachristos, C. H. Contag, S. S. Gambhir, M. Bogyo and S. Rogalla, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2008072118 CrossRef CAS PubMed.
- J. I. Scott, Q. Deng and M. Vendrell, ACS Chem. Biol., 2021, 16, 1304–1317 CrossRef CAS PubMed.
- T. Jiang, E. S. Olson, Q. T. Nguyen, M. Roy, P. A. Jennings and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17867–17872 CrossRef CAS PubMed.
- Y. Cheng, L. Zhao, Y. Li and T. Xu, Chem. Soc. Rev., 2011, 40, 2673–2703 RSC.
- L. O. Ofori, N. P. Withana, T. R. Prestwood, M. Verdoes, J. J. Brady, M. M. Winslow, J. Sorger and M. Bogyo, ACS Chem. Biol., 2015, 10, 1977–1988 CrossRef CAS PubMed.
- E. Walker, Y. Liu, I. Kim, M. Biro, S. R. Iyer, H. Ezaldein, J. Scott, M. Merati, R. Mistur, B. Zhou, B. Straight, J. J. Yim, M. Bogyo, M. Mann, D. L. Wilson, J. P. Basilion and D. L. Popkin, Cancer Res., 2020, 80, 2045–2055 CrossRef CAS PubMed.
- M. J. Whitley, D. M. Cardona, A. L. Lazarides, I. Spasojevic, J. M. Ferrer, J. Cahill, C.-L. Lee, M. Snuderl, D. G. Blazer, E. S. Hwang, R. A. Greenup, P. J. Mosca, J. K. Mito, K. C. Cuneo, N. A. Larrier, E. K. O’Reilly, R. F. Riedel, W. C. Eward, D. B. Strasfeld, D. Fukumura, R. K. Jain, W. D. Lee, L. G. Griffith, M. G. Bawendi, D. G. Kirsch and B. E. Brigman, Sci. Trans. Med., 2016, 8, 320ra324 Search PubMed.
- B. L. Smith, C. R. Lanahan, M. C. Specht, B. N. Kelly, C. Brown, D. B. Strasfeld, J. M. Ferrer, U. Rai, R. Tang, T. Rice-Stitt, A. Biernacka, E. F. Brachtel and M. A. Gadd, Ann. Surg. Oncol., 2020, 27, 1854–1861 CrossRef PubMed.
- B. L. Smith, M. A. Gadd, C. R. Lanahan, U. Rai, R. Tang, T. Rice-Stitt, A. L. Merrill, D. B. Strasfeld, J. M. Ferrer, E. F. Brachtel and M. C. Specht, Breast Cancer Res. Treat., 2018, 171, 413–420 CrossRef PubMed.
- C. R. Lanahan, B. N. Kelly, M. A. Gadd, M. C. Specht, C. L. Brown, K. S. Hughes, R. Tang, U. Rai, E. F. Brachtel, T. Rice-Stitt and B. L. Smith, Breast Cancer Res. Treat., 2021, 187, 145–153 CrossRef PubMed.
- R. Weissleder, C.-H. Tung, U. Mahmood and A. B. Jr, Nat. Biotechnol., 1999, 17, 375–378 CrossRef CAS PubMed.
- A. Megia-Fernandez, A. Marshall, A. R. Akram, B. Mills, S. V. Chankeshwara, E. Scholefield, A. Miele, B. C. McGorum, C. Michaels, N. Knighton, T. Vercauteren, F. Lacombe, V. Dentan, A. M. Bruce, J. Mair, R. Hitchcock, N. Hirani, C. Haslett, M. Bradley and K. Dhaliwal, BME Front., 2021, 2021, 1–11 CrossRef.
- B. Mills, D. Norberg, K. Dhaliwal, A. R. Akram, M. Bradley and A. Megia-Fernandez, Chem. Commun., 2020, 56, 9962–9965 RSC.
- M. Zhou, J. Hu, M. Zheng, Q. Song, J. Li and Y. Zhang, Chem. Commun., 2016, 52, 2342–2345 RSC.
- H. Yan, L. He, W. Zhao, J. Li, Y. Xiao, R. Yang and W. Tan, Anal. Chem., 2014, 86, 11440–11450 CrossRef CAS PubMed.
- T. H. Craven, N. Avlonitis, N. McDonald, T. Walton, E. Scholefield, A. R. Akram, T. S. Walsh, C. Haslett, M. Bradley and K. Dhaliwal, Sci. Rep., 2018, 8, 13490 CrossRef PubMed.
- N. Avlonitis, M. Debunne, T. Aslam, N. McDonald, C. Haslett, K. Dhaliwal and M. Bradley, Org. Biomol. Chem., 2013, 11, 4414–4418 RSC.
- S. Lebreton, S. E. How, M. Buchholz, B. E. Yingyongnarongkul and M. Bradley, Tetrahedron, 2003, 59, 3945–3953 CrossRef CAS.
- Y. Zhang, M. Ucuncu, A. Gambardella, A. Baibek, J. Geng, S. Zhang, J. Clavadetscher, I. Litzen, M. Bradley and A. Lilienkampf, J. Am. Chem. Soc., 2020, 142(52), 21615–21621 CrossRef CAS PubMed.
- A. K. Galande, S. A. Hilderbrand, R. Weissleder and C.-H. Tung, J. Med. Chem., 2006, 49, 4715–4720 CrossRef CAS PubMed.
- J. M. Ellard, T. Zollitsch, W. J. Cummins, A. L. Hamilton and M. Bradley, Angew. Chem., Int. Ed., 2002, 41, 3233–3236 CrossRef PubMed.
- V. Brinkmann, C. Goosmann, B. Fauler, Y. Uhelmann, D. S. Weiss, Y. Weinrauch and A. Zichlinsky, Science, 2004, 303, 1532–1535 CrossRef CAS PubMed.
- M. R. Rios, G. Garoffolo, G. Rinaldi, A. Megia-Fernandez, S. Ferrari, C. T. Robb, A. G. Rossi, M. Pesce and M. Bradley, Chem. Commun., 2020, 57, 97–100 RSC.
- K. Nagai, T. Sato and C. Kojima, Bioorg. Med. Chem. Lett., 2021, 33, 127726 CrossRef CAS PubMed.
- B. Brennecke, Q. Wang, W. Haap, U. Grether, H. Y. Hu and M. Nazare, Bioconjugate Chem., 2021, 32, 702–712 CrossRef CAS PubMed.
- Q. D. Mac, D. V. Mathews, J. A. Kahla, C. M. Stoffers, O. M. Delmas, B. A. Holt, A. B. Adams and G. A. Kwong, Nat. Biomed. Eng., 2019, 3, 281–291 CrossRef CAS PubMed.
- E. J. Kwon, J. S. Dudani and S. N. Bhatia, Nat. Biomed. Eng., 2017, 1, 1–10 CrossRef.
- S. Y. Li, L. H. Liu, H. Cheng, B. Li, W. X. Qiu and X. Z. Zhang, Chem. Commun., 2015, 51, 14520–14523 RSC.
- J. Xu, L. Fang, M. Shi, Y. Huang, L. Yao, S. Zhao, L. Zhang and H. Liang, Chem. Commun., 2019, 55, 1651–1654 RSC.
- F. Li, Y. Chen, R. Lin, C. Miao, J. Ye, Q. Cai, Z. Huang, Y. Zheng, X. Lin, Z. Zheng and S. Weng, Anal. Chim. Acta, 2021, 1148, 338201 CrossRef CAS PubMed.
- A. Megia-Fernandez, B. Mills, C. Michels, S. V. Chankeshwara, N. Krstajic, C. Haslett, K. Dhaliwal and M. Bradley, Org. Biomol. Chem., 2018, 16, 8056–8063 RSC.
- J. C. Widen, M. Tholen, J. J. Yim, A. Antaris, K. M. Casey, S. Rogalla, A. Klaassen, J. Sorger and M. Bogyo, Nat. Biomed. Eng., 2021, 5, 264–277 CrossRef CAS PubMed.
- S.-Y. Li, L.-H. Liu, L. Rong, W.-X. Qiu, H.-Z. Jia, B. Li, F. Li and X.-Z. Zhang, Adv. Funct. Mater., 2015, 25, 7317–7326 CrossRef CAS.
- F. Liu, M. Yang, W. Song, X. Luo, R. Tang, Z. Duan, W. Kang, S. Xie, Q. Liu, C. Lei, Y. Huang, Z. Nie and S. Yao, Chem. Sci., 2020, 11, 2993–2998 RSC.
- R. R. Jetson and C. J. Krusemark, Angew. Chem., Int. Ed., 2016, 55, 9562–9566 CrossRef CAS PubMed.
- R. Lee, S.-J. Choi, K. C. Moon, J. W. Park, K. Kim, S.-Y. Yoon and I. Youn, ACS Biomater. Sci. Eng., 2019, 5, 3039–3048 CrossRef CAS PubMed.
- S. Gehrig, M. A. Mall and C. Schultz, Angew. Chem., Int. Ed., 2012, 51, 6258–6261 CrossRef CAS PubMed.
- M. Guerra, V. S. Halls, J. Schatterny, M. Hagner, M. A. Mall and C. Schultz, J. Am. Chem. Soc., 2020, 142, 20299–20305 CrossRef CAS PubMed.
- M. Whitney, E. N. Savariar, B. Friedman, R. A. Levin, J. L. Crisp, H. L. Glasgow, R. Lefkowitz, S. R. Adams, P. Steinbach, N. Nashi, Q. T. Nguyen and R. Y. Tsien, Angew. Chem., Int. Ed., 2013, 52, 325–330 CrossRef CAS PubMed.
- M. Miampamba, J. Liu, A. Harootunian, A. J. Gale, S. Baird, S. L. Chen, Q. T. Nguyen, R. Y. Tsien and J. E. Gonzalez, Theranostics, 2017, 7, 3369–3386 CrossRef CAS PubMed.
- L. Ge, Z. Liu and Y. Tian, Chem. Sci., 2020, 11, 2215–2224 RSC.
- H. Lee, S. J. Kim, H. Shin and Y. P. Kim, ACS Sens., 2020, 5, 655–664 CrossRef CAS PubMed.
- C. Schulenburg, G. Faccio, D. Jankowska, K. Maniura-Weber and M. Richter, Analyst, 2016, 141, 1645–1648 RSC.
- Y. Okorochenkova, M. Porubsky, S. Benicka and J. Hlavac, Chem. Commun., 2018, 54, 7589–7592 RSC.
- L. Chen, W. Sun, W. Li, J. Li, L. Du, W. Xu, H. Fang and M. Li, Anal. Methods, 2012, 4, 2661–2663 RSC.
- L. W. Zou, P. Wang, X. K. Qian, L. Feng, Y. Yu, D. D. Wang, Q. Jin, J. Hou, Z. H. Liu, G. B. Ge and L. Yang, Biosens. Bioelectron., 2017, 90, 283–289 CrossRef CAS PubMed.
- X. He, Y. Xu, W. Shi and H. Ma, Anal. Chem., 2017, 89, 3217–3221 CrossRef CAS PubMed.
- Z. Yang, W. Xu, J. Wang, L. Liu, Y. Chu, Y. Wang, Y. Hu, T. Yi and J. Hua, J. Mater. Chem. C, 2020, 8, 8183–8190 RSC.
- X. Huang, J. Song, B. C. Yung, X. Huang, Y. Xiong and X. Chen, Chem. Soc. Rev., 2018, 47, 2873–2920 RSC.
- K. Du, L. Sheng, X. Luo, G. Fan, D. Shen, C. Wu and R. Shen, Spectrochim. Acta, Part A, 2021, 249, 119328 CrossRef CAS PubMed.
- T. Cao, Z. Teng, L. Zheng, J. Qian, H. Ma, J. Wang, W. Qin and H. Guo, Anal. Chim. Acta, 2020, 1127, 295–302 CrossRef CAS PubMed.
- T. Ma, Y. Hou, J. Zeng, C. Liu, P. Zhang, L. Jing, D. Shangguan and M. Gao, J. Am. Chem. Soc., 2018, 140, 211–218 CrossRef CAS PubMed.
- Y. Hou, J. Zhou, Z. Gao, X. Sun, C. Liu, D. Shangguan, W. Yang and M. Gao, ACS Nano, 2015, 9, 3199–3205 CrossRef CAS PubMed.
- S. Y. Liu, A. M. Yan, W. Y. Guo, Y. Y. Fang, Q. J. Dong, R. R. Li, S. N. Ni, Y. Sun, W. C. Yang and G. F. Yang, ACS Nano, 2020, 14, 4244–4254 CrossRef CAS PubMed.
- M. Wu and W. R. Algar, Anal. Chem., 2015, 87, 8078–8083 CrossRef CAS PubMed.
- W. R. Algar, A. Khachatrian, J. S. Melinger, A. L. Huston, M. H. Stewart, K. Susumu, J. B. Blanco-Canosa, E. Oh, P. E. Dawson and I. L. Medintz, J. Am. Chem. Soc., 2017, 139, 363–372 CrossRef CAS PubMed.
- H. Bui, C. W. Brown, 3rd, S. Buckhout-White, S. A. Diaz, M. H. Stewart, K. Susumu, E. Oh, M. G. Ancona, E. R. Goldman and I. L. Medintz, Small, 2019, 15, 1805384 CrossRef PubMed.
- J. Breidenbach, U. Bartz and M. Gutschow, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140445 CrossRef CAS PubMed.
- M. Zimmerman, B. Ashe, E. C. Yurewicz and G. Patel, Anal. Biochem., 1977, 78, 47–51 CrossRef CAS PubMed.
- H. Chen, X. He, M. Su, W. Zhai, H. Zhang and C. Li, J. Am. Chem. Soc., 2017, 139, 10157–10163 CrossRef CAS PubMed.
- S. He, J. Li, Y. Lyu, J. Huang and K. Pu, J. Am. Chem. Soc., 2020, 142, 7075–7082 CrossRef CAS PubMed.
- Q. Gong, W. Shi, L. Li, X. Wu and H. Ma, Anal. Chem., 2016, 88, 8309–8314 CrossRef CAS PubMed.
- X. Guo, S. Mu, J. Li, Y. Zhang, X. Liu, H. Zhang and H. Gao, J. Mater. Chem. B, 2020, 8, 767–775 RSC.
- A. Ogasawara, M. Kamiya, K. Sakamoto, Y. Kuriki, K. Fujita, T. Komatsu, T. Ueno, K. Hanaoka, H. Onoyama, H. Abe, Y. Tsuji, M. Fujishiro, K. Koike, M. Fukayama, Y. Seto and Y. Urano, Bioconjugate Chem., 2019, 30, 1055–1060 CrossRef CAS PubMed.
- Q. Gong, L. Li, X. Wu and H. Ma, Chem. Sci., 2016, 7, 4694–4697 RSC.
- X. He, L. Li, Y. Fang, W. Shi, X. Li and H. Ma, Chem. Sci., 2017, 8, 3479–3483 RSC.
- X. He, Y. Hu, W. Shi, X. Li and H. Ma, Chem. Commun., 2017, 53, 9438–9441 RSC.
- E. E. Mulvihill and D. J. Drucker, Endocr. Rev., 2014, 35, 992–1019 CrossRef CAS PubMed.
- A. P. Femia, L. Raimondi, G. Maglieri, M. Lodovici, E. Mannucci and G. Caderni, Int. J. Cancer, 2013, 133, 2498–2503 CrossRef CAS PubMed.
- A. Beckenkamp, J. B. Willig, D. B. Santana, J. Nascimento, J. D. Paccez, L. F. Zerbini, A. N. Bruno, D. A. Pilger, M. R. Wink and A. Buffon, PLoS One, 2015, 10, 1–17 CrossRef PubMed.
- T. Liu, J. Ning, B. Wang, B. Dong, S. Li, X. Tian, Z. Yu, Y. Peng, C. Wang, X. Zhao, X. Huo, C. Sun, J. Cui, L. Feng and X. Ma, Anal. Chem., 2018, 90, 3965–3973 CrossRef CAS PubMed.
- H. Onoyama, M. Kamiya, Y. Kuriki, T. Komatsu, H. Abe, Y. Tsuji, K. Yagi, Y. Yamagata, S. Aikou, M. Nishida, K. Mori, H. Yamashita, M. Fujishiro, S. Nomura, N. Shimizu, M. Fukayama, K. Koike, Y. Urano and Y. Seto, Sci. Rep., 2016, 6, 26399 CrossRef CAS PubMed.
- T. Mizushima, S. Ohnishi, Y. Shimizu, Y. Hatanaka, K. C. Hatanaka, Y. Kuriki, M. Kamiya, A. Homma, K. Yamamoto, S. Ono, Y. Urano and N. Sakamoto, Head Neck, 2018, 40, 1466–1475 CrossRef PubMed.
- K. Yamamoto, S. Ohnishi, T. Mizushima, J. Kodaira, M. Ono, Y. Hatanaka, K. C. Hatanaka, Y. Kuriki, M. Kamiya, N. Ehira, K. Shinada, H. Takahashi, Y. Shimizu, Y. Urano and N. Sakamoto, BMC Cancer, 2020, 20, 64 CrossRef CAS PubMed.
- Q. Gong, W. Shi, L. Li and H. Ma, Chem. Sci., 2016, 7, 788–792 RSC.
- W. Zhang, F. Liu, C. Zhang, J. G. Luo, J. Luo, W. Yu and L. Kong, Anal. Chem., 2017, 89, 12319–12326 CrossRef CAS PubMed.
- M. Sakabe, D. Asanuma, M. Kamiya, R. J. Iwatate, K. Hanaoka, T. Terai, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2013, 135, 409–414 CrossRef CAS PubMed.
- K. Yamamoto, M. Kamiya and Y. Urano, Bioorg. Med. Chem. Lett., 2019, 29, 126663 CrossRef CAS PubMed.
- S. Y. Liu, H. Xiong, R. R. Li, W. C. Yang and G. F. Yang, Anal. Chem., 2019, 91, 3877–3884 CrossRef CAS PubMed.
- Q. Sun, J. Li, W. N. Liu, Q. J. Dong, W. C. Yang and G. F. Yang, Anal. Chem., 2013, 85, 11304–11311 CrossRef CAS PubMed.
- S. Wang, B. G. Vigliarolo, M. A. Chowdhury, J. N. K. Nyarko, D. D. Mousseau and C. P. Phenix, Bioorg. Chem., 2019, 92, 103194 CrossRef CAS PubMed.
- Y. Wang, J. Li, L. Feng, J. Yu, Y. Zhang, D. Ye and H. Y. Chen, Anal. Chem., 2016, 88, 12403–12410 CrossRef CAS PubMed.
- Y. Urano, M. Sakabe, N. Kosaka, M. Ogawa, M. Mitsunaga, D. Asanuma, M. Kamiya, M. R. Young, T. Nagano, P. L. Choyke and H. Kobayashi, Sci. Transl. Med., 2011, 3, 110ra119 Search PubMed.
- R. Obara, M. Kamiya, Y. Tanaka, A. Abe, R. Kojima, T. Kawaguchi, M. Sugawara, A. Takahashi, T. Noda and Y. Urano, Angew. Chem., Int. Ed., 2021, 60, 2125–2129 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741, 10.1039/b105159h.
- Y. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- Y. Hong, Methods Appl. Fluoresc., 2016, 4, 022003 CrossRef PubMed.
- M. Chen, L. Li, H. Nie, J. Tong, L. Yan, B. Xu, J. Z. Sun, W. Tian, Z. Zhao, A. Qin and B. Z. Tang, Chem. Sci., 2015, 6, 1932–1937 RSC.
- H. Shi, R. T. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef CAS PubMed.
- Y. Yuan, C. J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780–1786 CrossRef CAS PubMed.
- D. Ding, J. Liang, H. Shi, R. T. K. Kwok, M. Gao, G. Feng, Y. Yuan, B. Z. Tang and B. Liu, J. Mater. Chem. B, 2014, 2, 231–238 RSC.
- Y. Wang, X. Wu, Y. Cheng and X. Zhao, Chem. Commun., 2016, 52, 3478–3481 RSC.
- S. Huang, Y. Wu, F. Zeng, J. Chen and S. Wu, Anal. Chim. Acta, 2018, 1031, 169–177 CrossRef CAS PubMed.
- X. Liu and G. Liang, Chem. Commun., 2017, 53, 1037–1040 RSC.
- T. I. Kim, H. Jin, J. Bae and Y. Kim, Anal. Chem., 2017, 89, 10565–10569 CrossRef CAS PubMed.
- J. Li, W. Y. Lee, T. Wu, C. W. Leung, J. Xu, D. S. Wong, R. Li, G. Li, B. Z. Tang and L. Bian, Adv. Biosyst., 2018, 2, 1800010 CrossRef.
- Y. Yuan, C. J. Zhang, R. T. K. Kwok, D. Mao, B. Z. Tang and B. Liu, Chem. Sci., 2017, 8, 2723–2728 RSC.
- H. Han, W. Teng, T. Chen, J. Zhao, Q. Jin, Z. Qin and J. Ji, Chem. Commun., 2017, 53, 9214–9217 RSC.
- Y. Yuan, R. Zhang, X. Cheng, S. Xu and B. Liu, Chem. Sci., 2016, 7, 4245–4250 RSC.
- Y. Yuan, C.-J. Zhang, R. T. K. Kwok, S. Xu, R. Zhang, J. Wu, B. Z. Tang and B. Liu, Adv. Funct. Mater., 2015, 25, 6586–6595 CrossRef CAS.
- Y. Yuan, R. T. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546–2554 CrossRef CAS PubMed.
- Y. Cheng, F. Huang, X. Min, P. Gao, T. Zhang, X. Li, B. Liu, Y. Hong, X. Lou and F. Xia, Anal. Chem., 2016, 88, 8913–8919 CrossRef CAS PubMed.
- Y. Zhang, Y. Li, N. Yang, X. Yu, C. He, N. Niu, C. Zhang, H. Zhou, C. Yu and S. Jiang, Sens. Actuators, B, 2018, 257, 1143–1149 CrossRef CAS.
- J. Kaur, J. N. Malegaonkar, S. V. Bhosale and P. K. Singh, J. Mol. Liq., 2021, 333, 115980 CrossRef CAS.
- H. Li, G. Parigi, C. Luchinat and T. J. Meade, J. Am. Chem. Soc., 2019, 141, 6224–6233 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873–877 CrossRef CAS PubMed.
- P. Shieh and C. R. Bertozzi, Org. Biomol. Chem., 2014, 12, 9307–9320 RSC.
- S. Debieu and A. Romieu, Org. Biomol. Chem., 2017, 15, 2575–2584 RSC.
- S. Debieu and A. Romieu, Tetrahedron Lett., 2018, 59, 1940–1944 CrossRef CAS.
- R. Huo, X. Zheng, W. Liu, L. Zhang, J. Wu, F. Li, W. Zhang, C. S. Lee and P. Wang, Chem. Commun., 2020, 56, 10902–10905 RSC.
- Q. Wu, K. Y. Zhang, P. Dai, H. Zhu, Y. Wang, L. Song, L. Wang, S. Liu, Q. Zhao and W. Huang, J. Am. Chem. Soc., 2020, 142, 1057–1064 CrossRef CAS PubMed.
- D. Ye, A. J. Shuhendler, L. Cui, L. Tong, S. S. Tee, G. Tikhomirov, D. W. Felsher and J. Rao, Nat. Chem., 2014, 6, 519–526 CrossRef CAS PubMed.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- W. Liu, S. J. Liu, Y. Q. Kuang, F. Y. Luo and J. H. Jiang, Anal. Chem., 2016, 88, 7867–7872 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- X. Yang, Y. Guo and R. M. Strongin, Angew. Chem., Int. Ed., 2011, 50, 10690–10693 CrossRef CAS PubMed.
- G. Zomer, Chemiluminescence and Bioluminescence, 2010 10.1039/9781849732024-00051, pp. 51–90.
- A. Roda, M. Guardigli, P. Pasini, M. Mirasoli, E. Michelini and M. Musiani, Anal. Chim. Acta, 2005, 541, 25–35 CrossRef CAS.
- Y. Yan, P. Shi, W. Song and S. Bi, Theranostics, 2019, 9, 4047–4065 CrossRef CAS PubMed.
- N. Thorne, J. Inglese and D. S. Auld, Chem. Biol., 2010, 17, 646–657 CrossRef CAS PubMed.
- A. Fleiss and K. S. Sarkisyan, Curr. Genet., 2019, 65, 877–882 CrossRef CAS PubMed.
- N. Hananya and D. Shabat, Angew. Chem., Int. Ed., 2017, 56, 16454–16463 CrossRef CAS PubMed.
- A. P. Jathoul, H. Grounds, J. C. Anderson and M. A. Pule, Angew. Chem., Int. Ed., 2014, 53, 13059–13063 CrossRef CAS PubMed.
- S. Iwano, R. Obata, C. Miura, M. Kiyama, K. Hama, M. Nakamura, Y. Amano, S. Kojima, T. Hirano, S. Maki and H. Niwa, Tetrahedron, 2013, 69, 3847–3856 CrossRef CAS.
- M. S. Evans, J. P. Chaurette, S. T. Adams, Jr., G. R. Reddy, M. A. Paley, N. Aronin, J. A. Prescher and S. C. Miller, Nat. Methods, 2014, 11, 393–395 CrossRef CAS PubMed.
- T. Kuchimaru, S. Iwano, M. Kiyama, S. Mitsumata, T. Kadonosono, H. Niwa, S. Maki and S. Kizaka-Kondoh, Nat. Commun., 2016, 7, 11856 CrossRef CAS PubMed.
- R. Kojima, H. Takakura, T. Ozawa, Y. Tada, T. Nagano and Y. Urano, Angew. Chem., Int. Ed., 2013, 52, 1175–1179 CrossRef CAS PubMed.
- Y. Ni, Z. Hai, T. Zhang, Y. Wang, Y. Yang, S. Zhang and G. Liang, Anal. Chem., 2019, 91, 14834–14837 CrossRef CAS PubMed.
- T. Monsees, W. Miska and R. Geiger, Anal. Biochem., 1994, 221, 329–334 CrossRef CAS PubMed.
- W. Miska and R. Geiger, J. Clin. Chem. Clin. Biochem., 1987, 25, 23–30 CAS.
- M. Kindermann, H. Roschitzki-Voser, D. Caglic, U. Repnik, C. Miniejew, P. R. Mittl, G. Kosec, M. G. Grutter, B. Turk and K. U. Wendt, Chem. Biol., 2010, 17, 999–1007 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, G. Liang and J. Rao, Bioconjugate Chem., 2009, 20, 1660–1666 CrossRef CAS PubMed.
- G. C. Van de Bittner, C. R. Bertozzi and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 1783–1795 CrossRef CAS PubMed.
- S. Talley, O. Kalinina, M. Winek, W. Paik, A. R. Cannon, F. Alonzo, 3rd, M. A. Choudhry, K. L. Knight and E. M. Campbell, J. Immunol., 2019, 203, 2497–2507 CrossRef CAS PubMed.
- J. G. Morin and J. W. Hastings, J. Cell. Physiol., 1970, 77, 313–318 CrossRef PubMed.
- H. Dacres, M. Michie and S. C. Trowell, Anal. Biochem., 2012, 424, 206–210 CrossRef CAS PubMed.
- F. Weihs, M. Gel, J. Wang, A. Anderson, S. Trowell and H. Dacres, Biosens. Bioelectron., 2020, 158, 112162 CrossRef CAS PubMed.
- A. den Hamer, P. Dierickx, R. Arts, J. de Vries, L. Brunsveld and M. Merkx, ACS Sens., 2017, 2, 729–734 CrossRef CAS PubMed.
- H. Dacres, J. Wangb, A. Andersonb and S. C. Trowellb, Sens. Actuators, B, 2019, 301, 127141 CrossRef CAS.
- F. Weihs, A. Peh and H. Dacres, Anal. Chim. Acta, 2020, 1102, 99–108 CrossRef CAS PubMed.
- A. P. Schaap, R. S. Handley and B. P. Gin, Tetrahedron Lett., 1987, 28, 935–938 CrossRef CAS.
- N. Hananya and D. Shabat, ACS Cent. Sci., 2019, 5, 949–959 CrossRef CAS PubMed.
- J. A. Richard, L. Jean, C. Schenkels, M. Massonneau, A. Romieu and P. Y. Renard, Org. Biomol. Chem., 2009, 7, 2941–2957 RSC.
- J.-A. Richard, L. Jean, A. Romieu, M. Massonneau, P. Noack-Fraissignes and P.-Y. Renard, Org. Lett., 2007, 9, 4853–4855 CrossRef CAS PubMed.
- O. Green, T. Eilon, N. Hananya, S. Gutkin, C. R. Bauer and D. Shabat, ACS Cent. Sci., 2017, 3, 349–358 CrossRef CAS PubMed.
- M. E. Roth-Konforti, C. R. Bauer and D. Shabat, Angew. Chem., Int. Ed., 2017, 56, 15633–15638 CrossRef CAS PubMed.
- L. S. Ryan and A. R. Lippert, Angew. Chem., Int. Ed., 2018, 57, 622–624 CrossRef CAS PubMed.
- J. I. Scott, S. Gutkin, O. Green, E. J. Thompson, T. Kitamura, D. Shabat and M. Vendrell, Angew. Chem., Int. Ed., 2021, 60, 5699–5703 CrossRef CAS PubMed.
- B. M. Babin, G. Fernandez-Cuervo, J. Sheng, O. Green, A. A. Ordonez, M. L. Turner, L. J. Keller, S. K. Jain, D. Shabat and M. Bogyo, ACS Cent. Sci., 2021, 7, 803–814 CrossRef CAS PubMed.
- N. Hananya, J. P. Reid, O. Green, M. S. Sigman and D. Shabat, Chem. Sci., 2019, 10, 1380–1385 RSC.
- S. Gutkin, O. Green, G. Raviv, D. Shabat and O. Portnoy, Bioconjugate Chem., 2020, 31, 2488–2493 CrossRef CAS PubMed.
- N. Hananya, A. Eldar Boock, C. R. Bauer, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2016, 138, 13438–13446 CrossRef CAS PubMed.
- N. Hananya, O. Press, A. Das, A. Scomparin, R. Satchi-Fainaro, I. Sagi and D. Shabat, Chemistry, 2019, 25, 14679–14687 CrossRef CAS PubMed.
- Q. Miao and K. Pu, Bioconjugate Chem., 2016, 27, 2808–2823 CrossRef CAS PubMed.
- Y. Zhang, H. Hong and W. Cai, Cold Spring Harb. Protoc., 2011 DOI:10.1101/pdb.top065508.
- J. Weber, P. C. Beard and S. E. Bohndiek, Nat. Methods, 2016, 13, 639–650 CrossRef CAS PubMed.
- Y. P. a. G. K. Xueding Wang, Opt. Lett., 2003, 28, 1739–1741 CrossRef PubMed.
- J. T. Oh, M. L. Li, H. F. Zhang, K. Maslov, G. Stoica and L. V. Wang, J. Biomed. Opt., 2006, 11, 34032 CrossRef PubMed.
- H. Wang, K. Xue, Z. Duan, Y. Yang, Z. He, C. Wu, W. Zhang, W. Zhang, P. Li and B. Tang, Sens. Actuators, B, 2019, 286, 243–249 CrossRef CAS.
- P. Cheng, W. Chen, S. Li, S. He, Q. Miao and K. Pu, Adv. Mater., 2020, 32, e1908530 CrossRef PubMed.
- L. Yin, H. Sun, M. Zhao, A. Wang, S. Qiu, Y. Gao, J. Ding, S. J. Ji, H. Shi and M. Gao, J. Org. Chem., 2019, 84, 6126–6133 CrossRef CAS PubMed.
- C. Lee, M. Jeon and C. Kim, Applications of Nanoscience in Photomedicine, 2015 DOI:10.1533/9781908818782.31, pp. 31–47.
- J. Weber, P. C. Beard and S. E. Bohndiek, Nat. Methods, 2016, 13, 639–650 CrossRef CAS PubMed.
- K. Haedicke, C. Brand, M. Omar, V. Ntziachristos, T. Reiner and J. Grimm, Photoacoustics, 2017, 6, 1–8 CrossRef PubMed.
- B. M. Wallis de Vries, J. L. Hillebrands, G. M. van Dam, R. A. Tio, J. S. de Jong, R. H. Slart and C. J. Zeebregts, Circulation, 2009, 119, e534–e536 CrossRef CAS PubMed.
- D. Razansky, N. J. Harlaar, J. L. Hillebrands, A. Taruttis, E. Herzog, C. J. Zeebregts, G. M. van Dam and V. Ntziachristos, Mol. Imaging Biol., 2012, 14, 277–285 CrossRef PubMed.
- J. Levi, S. R. Kothapalli, T.-J. Ma, K. Hartman, B. T. Khuri-Yakub and S. S. Gambhir, J. Am. Chem. Soc., 2010, 132, 11264–11269 CrossRef CAS PubMed.
- Q. Li, S. Li, S. He, W. Chen, P. Cheng, Y. Zhang, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2020, 59, 7018–7023 CrossRef CAS PubMed.
- K. Yang, L. Zhu, L. Nie, X. Sun, L. Cheng, C. Wu, G. Niu, X. Chen and Z. Liu, Theranostics, 2014, 4, 134–141 CrossRef CAS PubMed.
- C. Liu, S. Li, Y. Gu, H. Xiong, W. T. Wong and L. Sun, Mol. Imaging Biol., 2018, 20, 919–929 CrossRef CAS PubMed.
- L. Yin, H. Sun, H. Zhang, L. He, L. Qiu, J. Lin, H. Xia, Y. Zhang, S. Ji, H. Shi and M. Gao, J. Am. Chem. Soc., 2019, 141, 3265–3273 CrossRef CAS PubMed.
- D. Zhang, G. B. Qi, Y. X. Zhao, S. L. Qiao, C. Yang and H. Wang, Adv. Mater., 2015, 27, 6125–6130 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, S. R. Kothapalli, G. A. Tikhomirov, J. Rao and S. S. Gambhir, J. Am. Chem. Soc., 2013, 135, 11015–11022 CrossRef CAS PubMed.
- Y. Wang, X. Hu, J. Weng, J. Li, Q. Fan, Y. Zhang and D. Ye, Angew. Chem., Int. Ed., 2019, 58, 4886–4890 CrossRef CAS PubMed.
- A. Lakshmanan, Z. Jin, S. P. Nety, D. P. Sawyer, A. Lee-Gosselin, D. Malounda, M. B. Swift, D. Maresca and M. G. Shapiro, Nat. Chem. Biol., 2020, 16, 988–996 CrossRef CAS PubMed.
- F. Xia, J. Niu, Y. Hong, C. Li, W. Cao, L. Wang, W. Hou, Y. Liu and D. Cui, Acta Biomater., 2019, 89, 289–299 CrossRef CAS PubMed.
- S. Nakamura, T. Morita, S. Iwanaga, M. Miwa and K. Takahashi, J. Biochem., 1977, 81, 1567–1569 CrossRef CAS PubMed.
- X. Zhu and T. Gao, Nano-Inspired Biosensors for Protein Assay with Clinical Applications, 2019 DOI:10.1016/b978-0-12-815053-5.00010-6, pp. 237–264.
- H. Aldewachi, T. Chalati, M. N. Woodroofe, N. Bricklebank, B. Sharrack and P. Gardiner, Nanoscale, 2017, 10, 18–33 RSC.
- E. Petryayeva and U. J. Krull, Anal. Chim. Acta, 2011, 706, 8–24 CrossRef CAS PubMed.
- Q. Zhang, Y. Lu, S. Li, J. Wu and Q. Liu, Peptide Applications in Biomedicine, Biotechnology and Bioengineering, 2018 DOI:10.1016/b978-0-08-100736-5.00024-7, pp. 565–601.
- P. Chen, R. Selegard, D. Aili and B. Liedberg, Nanoscale, 2013, 5, 8973–8976 RSC.
- Y. Pan, M. Guo, Z. Nie, Y. Huang, Y. Peng, A. Liu, M. Qing and S. Yao, Chem. Commun., 2012, 48, 997–999 RSC.
- G. Chen, Y. Xie, H. Zhang, P. Wang, H.-Y. Cheung, M. Yang and H. Sun, RSC Adv., 2014, 4, 6560–6563 RSC.
- W. Xue, G. Zhang and D. Zhang, Analyst, 2011, 136, 3136–3141 RSC.
- G. B. Kim, K. H. Kim, Y. H. Park, S. Ko and Y. P. Kim, Biosens. Bioelectron., 2013, 41, 833–839 CrossRef CAS PubMed.
- X. Ding, D. Ge and K.-L. Yang, Sens. Actuators, B, 2014, 201, 234–239 CrossRef CAS.
- M. Q. He, S. Chen, K. Yao, J. Meng, K. Wang, Y. L. Yu and J. H. Wang, Anal. Chem., 2020, 92, 1395–1401 CrossRef CAS PubMed.
- N. Xia, D. Deng, Y. Wang, C. Fang and S. J. Li, Int. J. Nanomed., 2018, 13, 2521–2530 CrossRef CAS PubMed.
- L. Liu, D. Deng, Y. Wang, K. Song, Z. Shang, Q. Wang, N. Xia and B. Zhang, Sens. Actuators, B, 2018, 266, 246–254 CrossRef CAS.
- C. N. Loynachan, A. P. Soleimany, J. S. Dudani, Y. Lin, A. Najer, A. Bekdemir, Q. Chen, S. N. Bhatia and M. M. Stevens, Nat. Nanotechnol., 2019, 14, 883–890 CrossRef CAS PubMed.
- G. A. Suaifan, C. Esseghaier, A. Ng and M. Zourob, Analyst, 2013, 138, 3735–3739 RSC.
- S. Wignarajah, G. A. Suaifan, S. Bizzarro, F. J. Bikker, W. E. Kaman and M. Zourob, Anal. Chem., 2015, 87, 12161–12168 CrossRef CAS PubMed.
- S. Alhogail, G. Suaifan and M. Zourob, Biosens. Bioelectron., 2016, 86, 1061–1066 CrossRef CAS PubMed.
- S. Alhogail, G. Suaifan, F. J. Bikker, W. E. Kaman, K. Weber, D. Cialla-May, J. Popp and M. M. Zourob, ACS Omega, 2019, 4, 21684–21688 CrossRef CAS PubMed.
- R. I. Roth and J. Levin, J. Biochem. Biophys. Methods, 1989, 19, 129–142 CrossRef CAS PubMed.
- D. Yang, H. J. Park and T. H. Yoo, Anal. Methods, 2016, 8, 6270–6276 RSC.
- Z. Ling, F. Xu, J. V. Edwards, N. T. Prevost, S. Nam, B. D. Condon and A. D. French, Carbohydr. Polym., 2019, 216, 360–368 CrossRef CAS PubMed.
- P. Findeisen, T. Peccerella, S. Post, F. Wenz and M. Neumaier, Rapid Commun. Mass Spectrom., 2008, 22, 1223–1229 CrossRef CAS PubMed.
- J.-L. Hsu, S.-Y. Huang, N.-H. Chow and S.-H. Chen, Anal. Chem., 2003, 75, 6843–6852 CrossRef CAS PubMed.
- S. P. Gygi, B. Rist, S. A. Gerber, F. Turecek, M. H. Gelb and R. Aebersold, Nat. Biotechnol., 1999, 17, 994–999 CrossRef CAS PubMed.
- T. J. Griffin, D. K. M. Han, S. P. Gygi, B. Rist, H. Lee and R. Aebersold, J. Am. Soc. Mass Spectrom., 2001, 12, 1238–1246 CrossRef CAS PubMed.
- R. Bachor, M. Waliczek, P. Stefanowicz and Z. Szewczuk, Molecules, 2019, 24, 701 CrossRef CAS PubMed.
- K. Jia, X. Zhao and X. Dang, Oncol. Lett., 2020, 19, 4106–4114 CAS.
- N. Rauniyar and J. R. Yates, 3rd, J. Proteome Res., 2014, 13, 5293–5309 CrossRef CAS PubMed.
- A. Thompson, J. Schafer, K. Kuhn, S. Kienle, J. Schwarz, G. Schmidt, T. Neumann and C. Hamon, Anal. Chem., 2002, 75, 1895–1904 CrossRef PubMed.
- T. Peccerella, N. Lukan, R. Hofheinz, D. Schadendorf, M. Kostrezewa, M. Neumaier and P. Findeisen, Clin. Chem., 2010, 56, 272–280 CrossRef CAS PubMed.
- P. Findeisen, V. Costina, D. Yepes, R. Hofheinz and M. Neumaier, J. Exp. Clin. Cancer Res., 2012, 31, 56 CrossRef CAS PubMed.
- F. Ouyang, T. Yu, C. Gu, G. Wang, R. Shi, R. Lv, E. Wu, C. Ma, R. Guo, J. Li, A. Zaczek and J. Liu, Analyst, 2019, 144, 6751–6759 RSC.
- J. D. Kirkpatrick, A. D. Warren, A. P. Soleimany, P. M. K. Westcott, J. C. Voog, C. Martin-Alonso, H. E. Fleming, T. Tammela, T. Jacks and S. N. Bhatia, Sci. Trans. Med., 2020, 12, eaaw0262 CrossRef CAS PubMed.
- B. B. Brown, D. S. Wagner and H. M. Geysen, Mol. Diversity, 1995, 1, 4–12 CrossRef CAS PubMed.
- J. S. Dudani, M. Ibrahim, J. Kirkpatrick, A. D. Warren and S. N. Bhatia, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8954–8959 CrossRef CAS PubMed.
- M. Pohanka and P. Skládal, J. Appl. Biomed., 2008, 6, 57–64 CrossRef CAS.
- L. C. Clark, Jr. and C. Lyons, Ann. N. Y. Acad. Sci., 1962, 102, 29–45 CrossRef PubMed.
- X.-Y. Z. Ming La, Q.-L. Peng, C.-D. Chen and G.-Q. Zhao, Int. J. Electrochem. Sci., 2015, 10, 3329–3339 Search PubMed.
- V. Vanova, K. Mitrevska, V. Milosavljevic, D. Hynek, L. Richtera and V. Adam, Biosens. Bioelectron., 2021, 180, 113087 CrossRef CAS PubMed.
- G. Liu, J. Wang, D. S. Wunschel and Y. Lin, J. Am. Chem. Soc., 2006, 128, 12382–12383 CrossRef CAS PubMed.
- J. Ji, J. Gan, J. Kong, P. Yang, B. Liu and C. Ji, Electrochem. Commun., 2012, 16, 53–56 CrossRef CAS.
- D. S. Shin, Y. Liu, Y. Gao, T. Kwa, Z. Matharu and A. Revzin, Anal. Chem., 2013, 85, 220–227 CrossRef CAS PubMed.
- L. Z. Swisher, A. M. Prior, S. Shishido, T. A. Nguyen, D. H. Hua and J. Li, Biosens. Bioelectron., 2014, 56, 129–136 CrossRef CAS PubMed.
- E. González-Fernández, M. Staderini, A. Yussof, E. Scholefield, A. F. Murray, A. R. Mount and M. Bradley, Biosens. Bioelectron., 2018, 119, 209–214 CrossRef PubMed.
- K. Kerman, K. A. Mahmoud and H.-B. Kraatz, Chem. Commun., 2007, 3829–3831 RSC.
- J. Liu, H. Cheng, D. He, X. He, K. Wang, Q. Liu, S. Zhao and X. Yang, Anal. Chem., 2017, 89, 9062–9068 CrossRef CAS PubMed.
- Y. Song, H. Fan, M. J. Anderson, J. G. Wright, D. H. Hua, J. Koehne, M. Meyyappan and J. Li, Anal. Chem., 2019, 91, 3971–3979 CrossRef CAS PubMed.
- A. Ucar, E. Gonzalez-Fernandez, M. Staderini, N. Avlonitis, A. F. Murray, M. Bradley and A. R. Mount, Analyst, 2020, 145, 975–982 RSC.
- Y. Cao, J. Yu, B. Bo, Y. Shu and G. Li, Biosens. Bioelectron., 2013, 45, 1–5 CrossRef CAS PubMed.
- Y. S. Dehua Deng, H. Feng, Q. Chen, D. Li and L. Liu, Int. J. Electrochem. Sci., 2013, 8, 6933–6940 Search PubMed.
- H. Li, Y. Huang, B. Zhang, D. Yang, X. Zhu and G. Li, Theranostics, 2014, 4, 701–707 CrossRef PubMed.
- H. Ko, S. Park and K. Kim, J. Electroanal. Chem., 2015, 742, 70–73 CrossRef CAS.
- D. Deng, Y. Hao, S. Yang, Q. Han, L. Liu, Y. Xiang, F. Tu and N. Xia, Sens. Actuators, B, 2019, 286, 415–420 CrossRef CAS.
- H. Wu, S. Liu, J. Jiang, G. Shen and R. Yu, Analyst, 2012, 137, 4829–4833 RSC.
- H. Chen, J. Zhang, Y. Gao, S. Liu, K. Koh, X. Zhu and Y. Yin, Biosens. Bioelectron., 2015, 68, 777–782 CrossRef CAS PubMed.
- F. Meng, H. Sun, Y. Huang, Y. Tang, Q. Chen and P. Miao, Anal. Chim. Acta, 2019, 1047, 45–51 CrossRef CAS PubMed.
- N. Xia, Y. Zhang, P. Guan, Y. Hao and L. Liu, Sens. Actuators, B, 2015, 213, 111–115 CrossRef CAS.
- X. Miao, H. Yu, Z. Gu, L. Yang, J. Teng, Y. Cao and J. Zhao, Anal. Bioanal. Chem., 2017, 409, 6723–6730 CrossRef CAS PubMed.
- C. Muñoz-San Martin, M. Pedrero, M. Gamella, A. Montero-Calle, R. Barderas, S. Campuzano and J. M. Pingarron, Anal. Bioanal. Chem., 2020, 412, 6177–6188 CrossRef PubMed.
- Y.-F. Liu, J.-X. Chen, M.-Q. Xu and G.-C. Zhao, Int. J. Electrochem. Sci., 2014, 9, 4014–4023 Search PubMed.
- D. Wang, Y. Yuan, Y. Zheng, Y. Chai and R. Yuan, Chem. Commun., 2016, 52, 5943–5945 RSC.
- Q. Hu, Y. Bao, S. Gan, Y. Zhang, D. Han and L. Niu, Anal. Chem., 2020, 92, 3470–3476 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |