
Activation and catalytic transformation of methane under mild conditions
Yu
Tang
 *a,
Yuting
Li
b and
Franklin
(Feng) Tao
*b
*a,
Yuting
Li
b and
Franklin
(Feng) Tao
*b
aInstitute of Molecular Catalysis and In situ/operando Studies, College of Chemistry, Fuzhou University, Fujian, 350000, China. E-mail: yu.tang@fzu.edu.cn
bDepartment of Chemical and Petroleum Engineering, University of Kansas, KS 66045, USA. E-mail: frankin.tao.2017@gmail.com
First published on 14th December 2021
Abstract
In the last few decades, worldwide scientists have been motivated by the promising production of chemicals from the widely existing methane (CH4) under mild conditions for both chemical synthesis with low energy consumption and climate remediation. To achieve this goal, a whole library of catalytic chemistries of transforming CH4 to various products under mild conditions is required to be developed. Worldwide scientists have made significant efforts to reach this goal. These significant efforts have demonstrated the feasibility of oxidation of CH4 to value-added intermediate compounds including but not limited to CH3OH, HCHO, HCOOH, and CH3COOH under mild conditions. The fundamental understanding of these chemical and catalytic transformations of CH4 under mild conditions have been achieved to some extent, although currently neither a catalyst nor a catalytic process can be used for chemical production under mild conditions at a large scale. In the academic community, over ten different reactions have been developed for converting CH4 to different types of oxygenates under mild conditions in terms of a relatively low activation or catalysis temperature. However, there is still a lack of a molecular-level understanding of the activation and catalysis processes performed in extremely complex reaction environments under mild conditions. This article reviewed the fundamental understanding of these activation and catalysis achieved so far. Different oxidative activations of CH4 or catalytic transformations toward chemical production under mild conditions were reviewed in parallel, by which the trend of developing catalysts for a specific reaction was identified and insights into the design of these catalysts were gained. As a whole, this review focused on discussing profound insights gained through endeavors of scientists in this field. It aimed to present a relatively complete picture for the activation and catalytic transformations of CH4 to chemicals under mild conditions. Finally, suggestions of potential explorations for the production of chemicals from CH4 under mild conditions were made. The facing challenges to achieve high yield of ideal products were highlighted and possible solutions to tackle them were briefly proposed.
Professor Yu Tang received his PhD from University of Kansas. His expertise is in situ and operando studies of catalysts and fundamental studies of molecular catalysis including the transformation of methane. In 2018, he was offered a faculty position by Fuzhou University for building the Institute of Molecular Catalysis and In situ/operando Studies. Currently, he is an associate professor in chemistry and leading the Institute of Molecular Catalysis and In situ/operando Studies at the College of Chemistry at Fuzhou University. Two of the main instruments in his group at Fuzhou University are AP-XPS and HP-STM purchased from Europe. He has co-authored nearly 60 publications in international journals of chemistry. |
Dr. Yuting Li received her Bachelor's degree from Tianjin University and PhD degree from University of Kansas. Her expertise is in situ and operando studies of catalysis and fundamental studies of transformation of small molecules to chemicals through heterogeneous catalysis at high temperature and under mild conditions. Her research interests include heterogeneous catalysis through in situ and operando characterization and development of catalysts for chemical transformation through chemical synthesis. |
Professor Franklin (Feng) Tao received his PhD from Princeton University. He worked as a postdoctoral research fellow at University of California at Berkeley in Prof. Gabor Somorjai's group and Prof. Miquel Salmeron's group before starting his independent career. He has been a tenured associate professor at University of Kansas working on nonproprietary research since 2014. He was selected as a fellow of Royal Society of Chemistry (RSC) in 2013 and a fellow of American Association for the Advancement of Sciences (AAAS) in 2017. His research interests are fundamental understanding of catalysis at the molecular level including single atom catalysis, bimetallic catalysis, reducible oxide catalysis, and catalytic chemistry for environmental climate remediations as well as in situ/operando characterizations using surface analytical instruments which are whole-set and assembled AP-XPS instruments purchased from manufacturers. He has published about 190 peer-reviewed articles in important international journals. |
1. Introduction
1.1 Availability of worldwide methane and chemical transformation of methane at high temperature versus low temperature
Methane has been used for the production of numerous chemicals through thermal catalysis at high temperature or relatively high temperature for decades (Fig. 1). Unfortunately, these practices exhibit significant disadvantages. One is the high cost of transportation of natural gas in liquid due to its extremely low boiling point of −164 °C. The other is heavy consumption of energy in the current high-temperature catalytic conversions of CH4 in industries as these conversion processes are mostly performed at high temperature.1,2 For instance, both steam and dry reforming of CH4 are heavily endothermic; thus, production of one mole of CO through CH4 reforming requires a heat supply of at least 206–247 kJ.1 In addition, the short lives of the catalysts working at high temperature have largely increased the cost of CH4-based production of value-added chemicals. To overcome these limits, the transformation of CH4 to valuable chemicals under mild conditions has been explored in the last decades toward the efficient utilization of CH4 economically and in an environment-friendly manner. Here, mild condition is defined for the activation or catalytic transformation of CH4 at a temperature lower than 250 °C. In general, it covers biocatalysis at room temperature, homogeneous catalysis at near room temperature, and heterogenous catalysis mostly at a low temperature (<150 °C) or sometimes at a relatively low temperature (150–250 °C).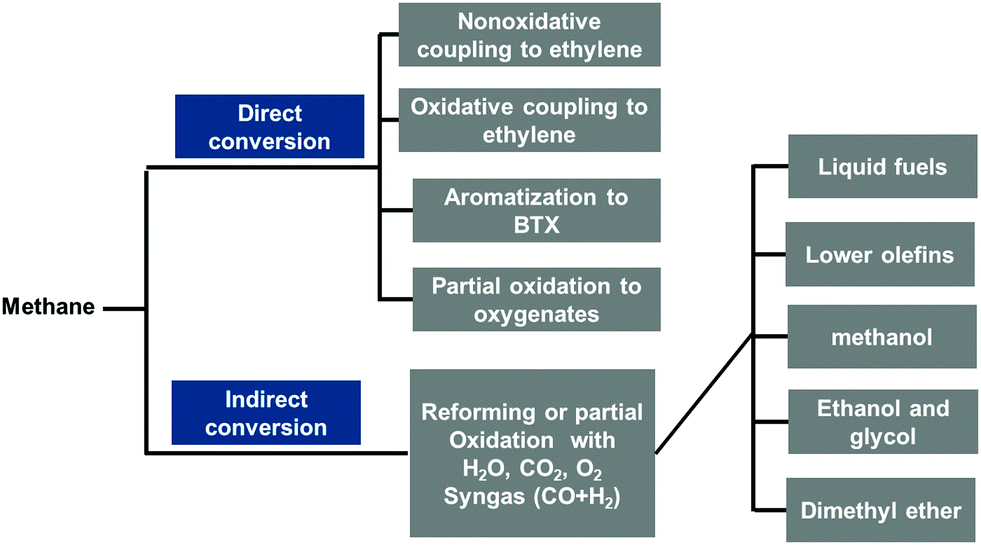 | ||
| Fig. 1 Developed main production routes using CH4 as the starting material to produce important intermediate compounds or value-added chemicals through direct or indirect transformations at high temperature. | ||
The enormous advantages of transformation of CH4 to value-added chemicals under mild conditions have attracted significant efforts in the last decades. It is significant for both producing chemicals with low energy consumption and remediating global climate through minimizing methane emission. Scientists have made significant progress in the formation of high value chemicals from CH4 under mild conditions. The feasibility of transforming CH4 to important intermediate compounds under mild conditions has been demonstrated. Through these efforts, numerous new catalysts have been developed although neither an acceptable durability nor a reasonably high activity has been obtained from these catalysts. From the fundamental science point of view, a significant amount of knowledge on noncatalytic oxidation and catalytic transformation has been obtained. Thus, it is timely to review this vital topic, i.e., the activation and catalytic transformation of CH4 under mild conditions. In this review article, we discussed the progresses on this topic achieved recently, reviewed the fundamental understanding of the reaction mechanisms in these transformations, highlighted the insights gained in developing new catalysts, present the great challenges that the scientific community are still faced with, and brief promising solutions to tackle them.
1.2 Brief of catalytic transformation of CH4 at high temperature
Fig. 1 presents these catalytic routes for producing important intermediate compounds in chemical industries by starting from CH4 for the production of valuable chemicals. These high temperature transformations of CH4 can be primarily categorized into direct and indirect conversions. Through direct conversion, CH4 is transformed to important intermediate compounds such as ethylene, benzene, toluene, xylene, and oxygenates. In terms of indirect conversion, CH4 is first transformed to syngas, a mixture of CO and H2, then, syngas is converted to several types of intermediate compounds or valuable chemicals including different hydrocarbons, alcohols, dimethyl ether, and other oxygenates, typically through Fischer–Tropsch synthesis.3–20Alternatively, these high temperature catalytical reactions of CH4 presented in Fig. 1 can be classified into oxidation and dehydrogenation. Through the oxidative process, CH4 can be transformed to alcohol, aldehyde, acid, ester, and many other organic oxygenates. In general, the catalytic oxidation of CH4 includes steam reforming,21–25 dry reforming,1,26–36 and partial oxidation37–42 to form syngas, oxidative coupling of CH4 to form ethylene,43–47 and even complete oxidation to form CO2 and H2O.48–52 Notably, the partial oxidation of CH4, oxidative coupling of CH4, and oxidative dehydrogenation of C2H6 or C3H8 at high temperature involves both the catalysis reaction performed on the surface of catalysts and the radical-based reaction occurring in the gas phase at high temperature. Typically, these high-temperature radical-based steps are more out of control; thus, there has been very limited understanding of these high-temperature radical reactions.45,47,53,54 Inspiringly, the characterization methods of radicals formed at high temperature have been developed recently53,55 and successfully used in fundamental studies of high-temperature transformations of CH4.53,55 As complete oxidation is always more thermodynamically favorable compared to partial oxidation, oxidative coupling, and oxidative dehydrogenation, CO2 and H2O are typically by-products of incomplete oxidation that occurs at high temperatures.
Unlike oxidative transformation, another category of high temperature catalysis is dehydrogenation. Compared to the exothermic nature of incomplete oxidation of CH4, C2H6, and C3H8, direct dehydrogenation of CH4 to C and H2 or C2H6 and C3H8 to unsaturated hydrocarbons and H2 is endothermic. With different dehydrogenation paths, hydrogen, benzene, toluene, xylene, ethylene, and even carbon can be produced. Unfortunately, these dehydrogenation reactions are mostly performed at quite high temperatures. Coke formation is a prevailing problem in these dehydrogenations performed at high temperature although the complete dissociation of CH4 to carbon and H2 on liquid-metal catalysts has been demonstrated as a promising route for the efficient use of CH4 at high temperatures.56–59
Most productions of important intermediate compounds involve at least one of or many of these high-temperature catalytic processes, as listed in Fig. 1. For instance, CH3OH and CH3COOH are two of the most important intermediate compounds for chemical production. As shown in Fig. 2, the production chain of CH3OH suffers from high energy cost and ready deactivation of the catalysts in the production process.60–63 The first step of this process is CH4 reforming at high temperature to produce syngas, which is a mixture of CO and H2. Then, CH3OH can be synthesized from CO and H2 at a high temperature. The catalyst of the high temperature reforming step is readily deactivated through the formation of coke on the catalyst surface.
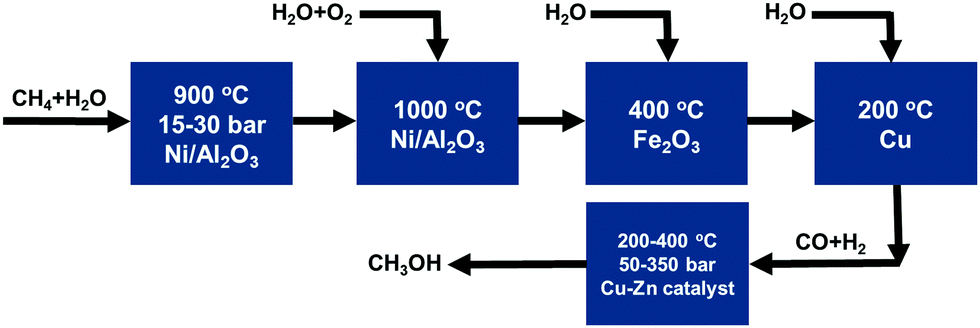 | ||
| Fig. 2 Industrial high-temperature catalytic processes for the synthesis of CH3OH using CH4 as a raw material. Reproduced from ref. 199, copyright 2013, with permission from Springer Nature. | ||
Although the development of catalysts with long durability and high thermal stability for the selective production of important intermediate compounds or valuable chemicals is a significant field and even a dominant topic in heterogeneous catalysis, this review does not cover the topic of high-temperature catalytic conversion of CH4, as listed in Fig. 1. An excellent review on this topic published by Bao et al. can be found in the literature.64
1.3 Limit to the activation of CH4 at low temperature
CH4 is one of the most inert molecules due to its strong C–H bonds with a C–H dissociation energy of 438.8 kJ mol−1, high ionization energy of 1206.1 kJ mol−1, low proton affinity of 424.5 kJ mol−1, and low acidity (pKa = 48).65,66 From the electronic structure point of view, the inertness of CH4 results from the localized C–H bonds, which makes it lack high-energy occupied orbitals and low-energy unoccupied orbitals to couple with other molecules or a catalytic site electronically. The direct dissociation of the C–H bond of CH4 is endothermic. A quite high temperature is typically required, as demonstrated in the preparation of carbon materials and molecular hydrogen on high-temperature liquid catalysts, as reported in the literature.56The design of a catalyst with high activity for oxidation of CH4 to organic oxygenates under mild conditions has been challenging, although tremendous efforts have been made in the last decades. Here, a mild condition is defined in terms of temperature. It is in the temperature range of 20–250 °C. From the surface science point of view, one reason for the existence of this challenge is that the active surface of a catalyst working for the oxidation of CH4 at low temperature can be readily poisoned through the oxidation of the active catalyst by O2 or H2O since they are the most commonly available oxidants or impurities in the reactant gas or/and solvent. For instance, even at a low temperature, O2 and H2O can readily deactivate the catalytic sites through the immediate oxidation of the supported metal clusters.67–69 From this point of view, support metal catalysts are not advantageous for the oxidative transformation of CH4 using O2 or H2O under mild conditions since they cannot retain their high surface energy state in environments containing O2 or/and H2O. Compared to bare metal clusters supported on oxide particles, the encapsulation of active sites in the microporous environment could be a better choice for the oxidative transformation of CH4 under mild conditions.61 In fact, majority of catalysts active for transformation of CH4 under mild conditions reported in the literature have catalytic sites of the metal atoms encapsulated in the micropores of zeolites. Alternatively, the preservation of the bare metal or oxide nanoclusters in inert gas or high vacuum is another approach to avoid poisoning them by O2 or/and H2O; the preservation of active sites in high vacuum has been used in the early exploration regarding the activation of CH4 at room temperature, which is reflected in the recent discovery of activation of CH4 on iridium oxide at a temperature as low as 150 K.70–74 Therefore, a balance between the high reactivity of a catalytic site and its resistance to potential poisonous molecules is an important guidance in the design of new catalysts active for CH4 transformation under mild conditions.
1.4 Organization of this article
Both activation and catalytic transformation of CH4 are covered in this review as both of them chemically transform CH4 to value-added chemicals. Here, the activation of CH4 is referred to as chemical reaction between CH4 and the oxidant; the oxidant could be oxygen atoms in the MO+ clusters in the gas phase, oxygen atoms anchored in the micropores of a zeolite, oxygen atoms of a reactant in the solute of an aqueous solution such as H2SO4, or oxygen atoms of dissolved molecules such as O2 in a solvent. In terms of the catalytic transformation of CH4, this review covers biocatalysis, homogeneous catalysis, and heterogeneous catalysis but focuses on heterogeneous catalysis. Notably, there has been increasing effort in the transformation of CH4 through electrocatalysis and photocatalysis in the field of catalysis. Due to the limited space, this review does not discuss any results of electrocatalysis and photocatalysis.There have been a great number of catalysts designed for the transformation of CH4 to value-added chemicals under mild conditions in the literature. From the reaction point of view, scientists have studied many transformative chemical reactions including these reactions adopted from other fields of chemistry for producing value-added chemicals from CH4 under mild conditions. Instead of the arranging materials to be reviewed by different categories of catalysts, this review was organized by discussing materials on the basis of different reactions transforming CH4 to value-added chemicals. We started by introducing biocatalysis for converting CH4 to CH3OH on soluble CH4 monooxygenase (sMMO) and particulate CH4 monooxygenase (pMMO), briefly discussed molecular catalysis for transforming CH4 to chemicals in the solution, discussed the oxidative activation of CH4 with MO+ existing in an inert gas or vacuum, reviewed the formation of CH3OH through oxidation of CH4 by active oxygen atoms bound to metal atoms anchored in zeolites, and discussed the catalytic production of oxygenates including CH3OH, acetic acid, and aldehydes through the catalytic oxidation of CH4 with H2O2. In each section reviewing a specific chemical reaction, its subsections were arranged according to different catalysts or samples performing oxidative activation. One feature of this review is the inclusion of comparison of different catalysts active for the same reaction and comparison of the same catalyst for different reactions. Another feature is that it offers specific suggestions on some topics to readers; these suggestions are shown in italics in the text.
Although significant efforts have been made in the last decades, actually, there has been a lack of a catalyst and a catalytic process that can be utilized for the production of oxygenates under mild conditions at a large scale. The current status results from several challenges to be tackled. At the end of this review, these challenges were presented and promising solutions for them were discussed.
2. Feature and advantage in the transformation of CH4 under mild conditions
The current industrial processes of utilizing CH4 are mainly catalytic transformations at high temperature performed on supported metal or oxide catalysts.75 The disadvantage of these transformations performed at high temperatures is the formation of coke and the rapid deactivation of the catalysts.22,23,31 In addition, these transformations performed at high temperatures consume a significant amount of energy.1 In terms of the significant amount of heat input in the current high-temperature catalytic process, it can be exemplified by at least 4.0 × 1013 kJ heat needed for the production of 6.5 million metric tons of CH3COOH in USA in 2017 based on the endothermic nature of steaming reforming of CH4 to produce CO (ΔHr = 206 kJ mol−1), which is the reactant to synthesize CH3OH (CO + 2H2 → CH3OH) and the following methanol carbonylation reaction (CH3OH + CO → CH3COOH).The chemical transformation of CH4 under mild conditions is the chemical conversion of CH4 to valuable chemicals at low temperature (≤150 °C) or relatively low temperature (150–250 °C). Thermodynamically, transformation through partial, selective, or even complete oxidation at a temperature lower than 300 °C is feasible. Thus, most transformations of CH4 under mild conditions are oxidative reactions of CH4. Compared to the current high temperature processes (>600 °C), one important feature of the transformation of CH4 under mild conditions is the low energy cost. Another feature of these transformations of CH4 under mild conditions is that the pressure of the reactants such as CH4 or O2 is high, typically several to tens of bars. High pressure is necessary since more molecules of the reactants at a higher pressure can be dissolved in the solvent to access the active sites of the catalyst particles at the solid (catalyst)–liquid (solvent) interface.
Such chemical transformations under mild conditions offer significant advantages. Low temperature catalysis can save tremendous amount of energy compared to the current high-temperature catalysis processes.1 The footprint cost of catalysis processes performed under mild conditions is compellingly lower in contrast to the facilities of current high temperature reactors and related maintenance needed for the current high temperature catalytic processes. In addition, as low temperature oxidative catalysis is expected to prevent ideal products from being further oxidized, a higher selectivity for forming an ideal product under mild conditions of CH4 transformation is highly promising in contrast to CH4 transformation through high-temperature catalysis. In addition, low-temperature catalysis can avoid deactivation as coke cannot form under mild conditions.
3. Important experimental methods
3.1 Brief overview of characterization for exploring the reaction mechanism at the molecular level
The identification of catalytic sites at an atomic scale is vital for a fundamental understanding of the mechanisms of catalytic reactions. Characterization is an essential component of the catalytic studies. To provide information on the structure and chemistry of a catalyst, characterization is typically performed under ex situ conditions. An ex situ characterization can be performed through two routes: one is the characterization of an as-prepared catalyst or a used one at room temperature in high vacuum; another route is the characterization of an as-prepared catalyst or a used one at room temperature in air. Compared to in situ or operando characterization, an ex situ characterization does not require complicated instrumentation and sample preparation. However, to reach a fundamental understanding of catalysis at the molecular level, the relationship between the measured catalytic performance and the information obtained from ex situ characterization does not necessarily reflect the intrinsic correlation between the authentic catalyst structure during catalysis and its corresponding function in terms of the catalytic activity, selectivity, and durability.76–82 A theoretical simulation based on a relationship derived from an ex situ characterization may not rationalize how a catalytic reaction is performed.The characterization of a catalyst during catalysis has always been a challenging task. It would never be as simple as an advertisement of an equipment manufacturer that a turn-on key machine of in situ/operando characterization is ready and the only thing to do for a researcher is to just push a button to collect the data. In truth, there are numerous issues existing in the characterization of oxidation and catalytic transformation of CH4 under mild conditions. The lack of appropriate methods and techniques to characterize catalysts under the working conditions of the catalyst or even semi-working conditions is still a major challenge in the community of catalysis science.
Regarding the characterization of the catalysts for CH4 transformation under mild conditions, X-ray absorption (XAS), vibrational spectroscopy, and nuclear magnetism resonance (NMR) are three main techniques valuable for the characterization of metal atoms anchored on a solid catalyst surface or encapsulated in the micropores of the solid catalyst distributed in a liquid environment under gas phase at a high pressure. The integration of isotope labelling methods with mass spectrometry, vibrational spectroscopy, and NMR can largely assist in the endeavor of achieving a profound understanding of the reaction mechanisms at a molecular level. Other than these characterization techniques, UV-vis spectroscopy and X-ray photoelectron spectroscopy are important characterizations on the topic of CH4 catalytic transformation. In addition, computational studies play an increasing role in the fundamental understanding of catalysis at the molecular level.
3.2 Quantitative analysis of products through NMR
In general, 1H NMR is the technique to identify products such as methanol, formaldehyde, formic acid, ethanol, acetic acid, and other oxygenates. Here, the quantitative analysis of methanol with NMR was used to demonstrate how NMR identifies and quantifies products of CH4 formed under mild conditions.202 After the catalytic transformation of CH4 under mild conditions, the reactor contains both the solid catalyst powder and the solution of solvent and products. The first step is to filter the mixture of the catalyst and the solution before a test solution is prepared. The test solution can be a part of the original filtrate collected from the reactor after filtration. Alternatively, it can be a part of a diluted filtrate if the concentration of the chemicals in the original filtrate is too high. For the purpose of calibrating the chemical shift of these chemicals to be analyzed, 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS) is used as a chemical shift standard. Its peak was set at δ = 0.0 ppm. A solution of 0.020 wt% DSS was made by diluting DSS in D2O solvent. 0.10 mL solution of 0.020 wt% DSS was mixed with a test solution, typically 0.70 mL in an NMR tube, for analysis. To measure the concentration of a product such as methanol in the test solution, a standard curve of the product such as methanol needs to be established. The concentration range of the standard curve should cover the concentration of all the test solutions. To establish a standard curve of methanol, a series of standard solutions with different concentrations of methanol but the same concentrations of solvent and all other solutes were prepared. The NMR spectra of these standard solutions are collected using exactly the same parameters of NMR measurements. For each standard solution, the ratio of the area of the methanol peak to the DSS peak in the spectrum was calculated. The ratios obtained from these standard solutions were plotted as a function of the known concentration of methanol in these solutions. Fitting these data points in the plot gives a linear equation. Thus, the concentration of a test solution can be readily measured by introducing the ratio of the methanol NMR peak of the test solution to its DSS peak into this linear equation. If the concentration of the test solution is beyond the range of the standard curve, another standard curve needs to be established. If the test solution is taken from a solution obtained through the dilution of the original filtrate, a dilution factor should be multiplied to calculate the concentration of the original filtrate. By multiplying the obtained concentration of the original filtrate by its volume, the amount of methanol formed in the reactor in mol or mass in gram is measured. Furthermore, the reaction rate in terms of the amount or mass of methanol produced per hour can be calculated under the assumption that this reaction is performed under a kinetics-controlled regime. The amount in mol or mass of the CH4 in gram introduced to the batch reactor before the reaction can be evaluated with the ideal gas law. Thus, the yield of methanol through this transformation of CH4 under mild conditions can be evaluated by dividing the amount of methanol produced by the amount of CH4 introduced into the batch reactor before the reaction. With the same method, the yields of other products can be readily calculated. The selectivity can be calculated through either dividing the amount in mol of produced methanol by the amount of the converted CH4 or dividing the amount (in mol) of the carbon atoms in methanol produced by the total amount (in mol) of carbon atoms in all the products. With the same method, the selectivities of other products can be readily measured.1883.3 XANES and EXAFS for revealing the electronic state and coordination environment of metal atoms
X-Ray absorption spectroscopy, including X-ray absorption near edge structure (XANES) spectroscopy and extended X-ray absorption fine structure (EXAFS) spectroscopy, has been used in studies of catalysis since the modern understanding of post-edge absorption oscillation was proposed by Sayers, Stern, and Lytle in the 1970s’.83–87 It uses X-ray with energy typically higher than 5 keV to excite the sample and then the transmitted X-ray or generated fluorescent X-ray is collected after the catalyst absorbs a certain amount of X-ray in a specific energy window. The high energies of incident X-ray and collected X-ray make this spectroscopy capable of studying catalyst particles in the gas or liquid phase. In particular, it is an appropriate technique for identifying specific chemical and coordination environments of metal atoms in the catalytic sites of single-atom catalysts, catalysts of nanoparticles with a size smaller than 3 nm, and catalysts of supported sub-nanometer clusters. Comprehensive reviews regarding the use of XAS in catalysis studies can be found in the literature.82,88–92XANES is a technique that identifies the electronic structure of elements in a sample and even the crystal structure; EXAFS is a technique that uncovers the chemical and coordination environments of metal atoms.93–96 EXAFS often provides key information for building an appropriate structural model before simulating the reaction pathway of a catalytic reaction with computational studies. It is widely acknowledged that obtaining the coordination number of atoms A around an atom M at high temperatures is challenging due to the temperature-dependent Debye–Waller factor.97–99 Thus, most in situ or operando characterization techniques published in the literature have been performed through a semi-in situ or semi-operando approaches. In other words, most reported XAS studies termed in situ or operando studies have in fact been performed by collecting data at the room temperature or a relatively low temperature <200 °C in the gas phase of the reactants after catalysis was performed at a high temperature. Thus, the catalyst was not catalyzing a reaction when the chemical and structural information of the catalyst were being extracted with XAS in such a semi-in situ or semi-operando approach. From this point of view, the development of analytical methods to extract chemical and structural information of metal atoms from the XAS data during catalysis at the catalysis temperature instead of a low temperature of data collection is needed for uncovering the catalyst structure during catalysis for establishing a direct structure-catalysis correlation. Notably, some valuable modifications of the reaction cell have been made in the literature100–105 for the collection of the spectra for XANES and EXAFS while catalysis is performed in liquid under a gas phase of high pressure. These modifications are valuable for characterizing the catalytic sites of catalysts during CH4 transformation under mild conditions.
3.4 NMR for identifying the adsorbate and catalytic sites
NMR is a technique extensively used in catalysis.106–108 Other than the quantification of products briefed in Section 3.2, it can characterize catalysts, adsorbates, or intermediates on a catalyst during catalysis in terms of the operando approach other than routine characterizations of a catalyst under an ex situ condition. Significant efforts in the instrumentation and development of methodology have been made in the last decades for characterizing a catalyst in a batch-reactor mode108–114 or a flowing reaction-cell mode.115–122 Some applications of this technique have been well developed including the identification of Brønsted acid sites with 1H MAS NMR by the adsorption of different probing molecules including deuterated pyridine and perfluorotributyl amine, the observation of Lewis acidic sites with 31P MAS NMR through the adsorption of trimethylphosphine oxide123,124 or trimethylphosphine,125–127 and the differentiation of Brønsted and Lewis acidic sites through 1H, 31P, 15N, 13C MAS NMR by choosing appropriate probing molecules.108 Bao et al. reviewed how solid-state NMR can be used in studies of catalysts under in situ conditions.108As discussed in the subsequent sections of this article, zeolite-based catalysts are the main players in the chemical transformations of CH4 to value-added chemicals under mild conditions. MAS NMR has been widely used in tracking the evolution of Brønsted acid sites of a zeolite with the change in the pre-treatment or catalysis conditions such as temperature.128In situ MAS NMR can be used to observe the active sites. One example is the use of 95Mo MAS NMR for tracking the chemical environment of Mo atoms in MoO3/ZSM-5 during the aromatization of CH4129–131 and even distinguishing different Mo species with ultrahigh field 95Mo NMR.132,133 Unquestionably, NMR is an excellent technique for studying the deactivation mechanism of catalysts.134–136 Another type of important application is the investigation of the reaction kinetics using 1H,13713C,138 or 129Xe,134 providing important information for understanding the catalytic mechanisms at the molecular level. As represented in some of the following sections of this review, NMR is a vital approach for gaining a fundamental understanding of the catalytic transformation of CH4 at the molecular level.
3.5 Vibrational spectroscopy
Vibrational spectroscopy is one of the most widely used characterization methods in situ/operando studies in the field of catalysis. It mainly includes infrared and Raman spectroscopy, which can provide significant information on the adsorbates and catalytic sites on the surface of a catalyst. As they are photon-in and photon-out techniques, in situ and operando studies have been readily performed using different types of reaction cells developed a few decades ago.139–142 Recently, a unique cell was reported for studying catalyst particles in the liquid phase and adsorbates at the solid–liquid interface.143In situ/operando studies using transmission infrared spectroscopy can track the vibrational signatures of the reactants and the products, analyze the composition, and study the kinetics during catalysis. Its instrumentation is relatively straightforward.144,145 It is the integration of an optical path mainly including the window for introduction and the exit of infrared light with an assured sealing of the liquid at a certain temperature, typically 200 °C or lower in a high-pressure reaction cell.Compared to transmission infrared spectroscopy, attenuated total reflection (ATR) spectroscopy exhibits a great advantage.139,143,146–149 It can study adsorbates on the catalyst surface during catalysis at the solid–liquid interface even while the liquid phase is below the high-pressure gas phase in a high-pressure reactor. One function of this spectroscopy is the analysis of adsorbates on the catalyst surface while the signal of liquid containing the reactant, products, by-products, and spectator is collected under the catalytic conditions. Thus, it exhibits significant potential in the exploration of the mechanism of a reaction performed at the solid–liquid interfaces while the liquid phase is under a high-pressure gas phase. We have to acknowledged that it is challenging to directly study a heterogeneous catalytic reaction occurring at the interface between the dispersed catalyst particles in the liquid and the liquid phase containing the solvent, liquid reactants, and dissolved gaseous molecules using attenuated total reflection spectroscopy, although the applications of ATR spectroscopy to in situ or operando studies of catalysis have been demonstrated in the literature.139,143,145–149 To use ATR spectroscopy to perform in situ or operando studies of catalysis at a solid–liquid interface, the catalyst particles are immobilized on an internal reflection element (IRE) crystal, typically germanium or diamond. In ATR spectroscopy, it is assumed that the catalytic reaction performed at the interface of the liquid phase and the immobilized catalyst particles is similar to the catalytic reaction on the catalyst particles freely dispersed in the liquid. Reactors have been built for a fundamental understanding of catalysis performed at the solid–liquid interface in the liquid phase under high-pressure gas phase. For instance, Baiker et al. built a high-pressure reactor with integrated view cell for in situ studies of catalysis performed at the solid–liquid interface in liquid under high-pressure gas phase.145 It can provide valuable information on this type of catalysis at a temperature up to 200 °C under a gas phase up to 200 bar. It has been used for the in situ studies of hydrogenation of ethyl pyruvate on Pt/Al2O3 catalyst particles (heterogenous catalysis) and the formylation of morpholine with CO2 and H2 on a bidentate ruthenium complex (homogeneous catalysis).145 More information on how to use ATR spectroscopy to study catalysis in the liquid phase can be found from different reviews.139,143,146–149 Even some works relevant to the chemical transformation of CH4 on active sites anchored in microporous aluminosilicate have been carried out. For instance, IR spectroscopy and mapping were used to identify the Si–O–B vibrational signature of boron in partially substituted zeolite particles MFI through an ex situ model.150 Unfortunately, very limited studies have used ATR spectroscopy for in situ or operando studies of chemical transformation of CH4 at the solid–liquid interface under high-pressure reactants during catalysis. One reason could be the difficulty of using the complicated instrument.145 It is expected that more in situ studies on the activation and catalytic transformation of CH4 in high-pressure gas phase or liquid phase under a high-pressure phase will be reported for providing insights into the activation or catalytic transformation of CH4 under these conditions.
The significance of infrared and Raman spectroscopic imaging techniques is worthy of emphasis, which have been used for the in situ studies of catalysis.151 For instance, IR microscopy can track the evolution of a specific species such as CH3 and CH2 groups in the MFI crystal along with the increase in the experimental temperature.152 Weckhuysen et al. demonstrated earlier that coherent anti-Stokes Raman scattering (CARS) can be used to track the catalytic conversion of thiophene on the ZSM-5 particles, in which H–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) stretching vibrational signature appearing at 3115 cm−1 in the Raman band was used to represent the reactant thiophene.153 The intensity of this signature was taken as a measure of the local concentration of thiophene, by which the 3-D concentration profiles of the concentration of the reactant were successfully constructed for helping to understand the catalytic conversion of thiophene.
stretching vibrational signature appearing at 3115 cm−1 in the Raman band was used to represent the reactant thiophene.153 The intensity of this signature was taken as a measure of the local concentration of thiophene, by which the 3-D concentration profiles of the concentration of the reactant were successfully constructed for helping to understand the catalytic conversion of thiophene.
3.6 UV-vis spectroscopy
UV-vis spectroscopy is a photon-in photon-out technique that examines the electronic transitions of the catalyst or the reactants and products under the reaction or catalytic conditions. It can measure a catalyst in the presence of a fluid.154,155 In general, it can provide information on the oxidation states, band gaps of the semiconducting support, particle size, and dispersion of the supported oxide moieties by measuring the electron transitions. UV-vis spectroscopy has been used in the characterization of catalysts through the ex situ or in situ model.UV-vis spectroscopy has been used in the investigations of catalysts under catalytic conditions in terms of in situ or operando UV-vis spectroscopy. For instance, heteropoly acids containing Mo and V in the mixture of CH3OH and O2 were investigated with UV-vis spectroscopy with a setup containing a homemade quartz cell combined with an integrating sphere;154,155 this in situ study revealed that the degree of reduction of V4+–O–Mo6+ was a function of the O2/CH3OH ratio. Another example is the in situ study of ethane and O2 system at 723 K on the VOx species supported on ZrO2, Al2O3, and SiO2; the intensities of d–d transitions of V4+/V3+ (17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1) in the VOx species supported on different oxides were tracked; it was found that the degree of reduction is influenced by the support of VOx, which increases in the order SiO2 < Al2O3 < ZrO2. In addition, several other catalytic reactions on supported VOx were studied with in situ UV-vis spectroscopy.156–162 Other than the supported VOx, this technique has been used for the identification of catalysts under the catalytic conditions of many different reactions including CaCO3 for ethene or propene oxidation at 473 K163 and MnO42−-exchanged layered double hydroxides for the decomposition of H2O2.164 Relevant to the topic of transformation of CH4 by aluminosilicate microporous catalysts under mild conditions, UV-vis spectroscopy was used in identifying the state of iron in Fe@ZSM-5 under the catalytic oxidation of NH3; the in situ UV-vis studies found that isolated Fe3+ ions in ZSM-5 were reduced in a mixture of 0.1% NH3 and 0.1% NO.165 More examples of UV-vis studies on catalysts under reaction and catalytic conditions have been discussed in these published reviews.166–168
000 cm−1) in the VOx species supported on different oxides were tracked; it was found that the degree of reduction is influenced by the support of VOx, which increases in the order SiO2 < Al2O3 < ZrO2. In addition, several other catalytic reactions on supported VOx were studied with in situ UV-vis spectroscopy.156–162 Other than the supported VOx, this technique has been used for the identification of catalysts under the catalytic conditions of many different reactions including CaCO3 for ethene or propene oxidation at 473 K163 and MnO42−-exchanged layered double hydroxides for the decomposition of H2O2.164 Relevant to the topic of transformation of CH4 by aluminosilicate microporous catalysts under mild conditions, UV-vis spectroscopy was used in identifying the state of iron in Fe@ZSM-5 under the catalytic oxidation of NH3; the in situ UV-vis studies found that isolated Fe3+ ions in ZSM-5 were reduced in a mixture of 0.1% NH3 and 0.1% NO.165 More examples of UV-vis studies on catalysts under reaction and catalytic conditions have been discussed in these published reviews.166–168
Another important application of in situ UV-vis spectroscopy is the acquisition of data of the catalysts and products for establishing a direct correlation between the catalyst structure and its corresponding catalytic performance for understanding catalysis. Weckhuysen et al. investigated butane dehydrogenation on chromium supported on silica alumina using a Harrick setup.169–173 In these studies, the conversion of butane was measured under the same condition on the catalysts with different loadings of chromium, by which the correlation between the catalytic activity and the amount of reduced chromium was established, and it is concluded that Cr3+ is the most active species for the oxidation of butane.169–173 Similarly, this approach was used for establishing the correlation between the catalytic performance and the catalyst structure of chromium supported on alumina for propane dehydrogenation,166,167,174 or sulphated zirconia for n-butane isomerization, and WO3 supported on ZrO2 for n-pentane isomerization.175 More examples can be found in the published review.176
Though the introduction of a fiber optical probe in a UV-vis spectrophotometer, UV-vis spectroscopic imaging can be realized.177,178 For instance, with time-resolved measurements, it has been used in the studies of kinetics of both redox reactions of supported oxides VOx on TiO2 and the reduction of WOx/ZrO2 by H2.179 Temporarily, there has been a lack of in situ studies using UV-vis spectroscopy or spectroscopic imaging techniques for in situ or operando studies of chemical transformation of CH4 under mild conditions during catalysis.
3.7 X-Ray photoelectron spectroscopy
X-Ray photoelectron spectroscopy (XPS) is a routine analytical method for the surface of a material. It can qualitatively identify the elements in the surface region, quantitatively analyze the surface composition or atomic fraction of one element, characterize the oxidation state, or even deduce electronic transfer between different atoms. As the mean free paths of the photoelectrons generated with soft X-ray (hν < 1000 eV) Mg Kα or Al Kα are in the range of 0.5–2 nm, the signal of the XPS data is mainly contributed from the surface region with a depth of about 1.5 nm. In addition, as the contribution to the signal decays exponentially as a function of the depth, atoms at a region closer to the surface have much more contributions to the overall signal. Thus, XPS is an analytical technique with high surface sensitivity.Ambient pressure X-ray photoelectron spectroscopy (AP-XPS) is an XPS spectroscopy that is specific for the studying surface buried in the gas phase. Its development can be tracked back to the 1970s. Continuous development in the last decades has made it a very useful analytical method for a sample in the gas phase, particularly for a catalyst in the gas phase at the Torr pressure range or higher. A catalyst can be retained in either a static gaseous environment by filling gas in an existing chamber or a dynamic gaseous environment by flowing gas through a reaction cell, in which a catalyst is placed. Various reaction cells of AP-XPS are available from manufactures including SPECS in Germany or homemade with the same principle as a variety of reaction cells of in situ/operando spectroscopies such as infrared spectroscopy and X-ray absorption spectroscopy which were developed and reported in literature decades ago.78,180–184 In terms of AP-XPS, the X-ray source is isolated from either the reaction chamber through an aluminium foil transparent for X-ray or a reaction cell having a thin window transparent for X-ray. Unlike the electrostatic energy analyzer of a vacuum XPS, an aperture has to be installed between the reaction cell (or the UHV chamber) of AP-XPS and the electrostatic energy analyzer; in addition, additional focusing lenses working at near ambient pressure have to be added at the front of an electrostatic energy analyzer. AP-XPS has exhibited significant value in uncovering the active surface of a catalyst under the reaction conditions and during catalysis. More information on the instrumentation of AP-XPS and the application of AP-XPS to fundamental studies of catalysis and surface science can be found from the literatures.78,180–184 For instance, AP-XPS using Al Kα has been used to track the surface of catalysts during the activation of CH4 by a catalyst in the gas phase. For active catalytic sites encapsulated in microporous materials; however, a hard X-ray source is necessary for generating high-energy photoelectrons from the buried metal atoms in the micropores of the catalyst particles. Hard X-ray AP-XPS instruments are available in some synchrotron centers and on the market. It has been applied to explore various materials185 although no studies on the activation and catalytic transformation of CH4 to CH3OH with hard X-ray AP-XPS have been reported in the literature.185 It is expected that a hard X-ray AP-XPS will be used for uncovering the chemical and electronic state of the metal atoms anchored in the micropores of zeolite catalysts during the transformation of CH4 in the gas phase under mild conditions.
In the field of photoelectron spectroscopy, efforts have been for tackling the challenging task of using XPS to characterize the surface of the catalyst particles in a liquid in the last few years.186–188 This challenging task has been made feasible by retaining or even flowing a liquid containing the well dispersed catalyst particles through a graphite membrane;187,188 in these methods, a portion of photoelectrons generated from the surface of the catalyst particles can penetrate the liquid layers between the catalyst particles and graphite membrane, and then transmit the graphene membrane to enter the vacuum environment where the photoelectrons were collected by a routine energy anlzyer.186–188 As the catalytic transformation of CH4 to CH3OH is typically performed at a solid–liquid interface while the liquid phase is under a high-pressure gas phase of CH4, it is extremely challenging, if not impossible, to use XPS to observe the surface of a catalyst in a liquid under a high-pressure gas during catalysis. More information on instrumentation of AP-XPS can be found from literature published in the last a few decades. In many cases, AP-XPS instruments were prepared by assembling commercial parts including an energy analyzer, a X-ray source, a monochromator, chambers, and pumps; in addition, some groups prepared some parts such as an X-ray source or a monochromator through assembling commercial components/parts including an electron gun, power supply and circulation loop of water with the knowledge of assembly publicly available in textbooks and a large number of papers published as early as 1970s such as ref. 184 and 186. These assemblies are routine practices in surface science groups instead of design of AP-XPS or its parts.
3.8 Integration of the isotope labelling method with characterization
Isotope labelling is a powerful approach for the identification of products, stable intermediates, and catalytic sites through its integration with mass spectrometry, NMR, and vibrational spectroscopy. By replacing 11H with 21H, 126C with 136C, or 168O with 188O for a specific atom of a reactant molecule or catalytic sites of a catalyst, whether these replaced atoms participate in the catalytic reaction can be readily elucidated. For instance, the integration of isotope substitution with mass spectrometry can assist in the identification of the active sites of a catalytic reaction. Whether lattice oxygen atoms on the surface of the NiCo2O4 catalyst directly participate in the complete oxidation of CH4 can be elucidated using the isotope labelled catalyst NiCo2168O4−x188Ox, which can be prepared by annealing NiCo2O4 in 188O2.48 If surface lattice oxygen participates in the oxidation of CH4 (CH4 + 2O2 → CO2 + 2H2O), C168O188O (m/z = 46) and C188O2 (m/z = 48) must be observed in the mass spectra. By tracking the formation of products with mass spectrometry, whether or not surface lattice oxygen atoms of the NiCo2O4 catalyst participate in the reaction was determined.48
The integration of isotope labelling with NMR analysis can help to identify the reaction pathway. For instance, Rh1O5 single atom site encapsulated in ZSM-5 can catalyze the coupling of CH4, CO, and O2 to form acetic acid.188 One of the potential reaction paths is the oxidation of CH4 by O2 to form CH3OH and then the carboxylation of CH3OH to generate acetic acid. In order to elucidate whether this is a reaction path taken by this catalyst (Rh1O5@ZSM-5), isotope-substituted methanol, CH3188OH, was added into an aqueous solution containing the catalyst particles under the mixture of CH4, CO, and O2. If the reaction path taken by Rh1O5@ZSM-5 is the carboxylation of methanol, CH3C168O188OH should be produced in this isotope experiment. If no CH3C168O188OH could be observed in NMR, the path of CH3OH carboxylation should be excluded.
The integration of isotope substitution with infrared spectroscopy can assist in identifying whether the C–Y bond of X–C(R2)–Y is activated or dissociated. In this case, Y of X–C(R2)–Y can be labelled by the isotope of Y′. Then, the isotope-labelled X–C(R2)–Y′ is used to replace X–C(R2)–Y to perform the same catalytic reaction. If a shift in the vibrational peak of C–Y′ could be observed with IR or Raman spectroscopy, the activation of C–Y′ would be confirmed. If no shift in the C–Y′ vibrational peak could be observed, it shows that C–Y of X–C(R2)–Y cannot be activated.
The isotope labelling method plays a unique role in testing the binding strength of the molecules on the surface of a catalyst. For instance, binding strengths of CO on a single-atom catalyst (Pt1/SiO2) and a nanoparticle catalyst (Pt NP/SiO2) were investigated experimentally through isotope-labelled IR experiments.189 Pt1/SiO2 and Pt NP/SiO2 were exposed to 12CO gas to reach the equilibrium of chemisorption at 100 °C. Then, the two Pt catalysts with chemisorbed 12CO at 100 °C were exposed to 13CO gas phase to allow exchange between chemisorbed 12CO and free 13CO of the gas phase. The number of replaced 12CO molecules pre-chemisorbed on the catalyst can be quantized with the intensity of 13CO chemisorbed on the catalyst since ν(13C–O) of 13CO is ∼50 cm−1 lower than that of 12CO. As the ratio of the peak intensity of ν(13C–O) to ν(12C–O) for the single-atom catalyst Pt1/SiO2 is obviously lower than the ratio for the nanoparticle catalyst Pt NP/SiO2, it is concluded that the binding strength of CO on Pt1/SiO2 is much stronger than that on Pt NP/SiO2. This difference in the binding of CO confirmed by isotope-labelling IR studies rationalized the distinct difference in the catalytic activity for CO oxidation on Pt1/SiO2 and Pt NP/SiO2. Thus, the integration of isotope labelling with mass spectrometry, NMR, and infrared spectroscopy has largely strengthened the functions of these spectroscopic techniques in fundamental studies of oxidation and transformation of CH4 under mild conditions.
3.9 Computational study for simulating the reaction pathway
Computational study using density functional theory (DFT) has become a significant approach in the fundamental understanding of the catalytic reactions at a molecular level. It searches the transition states and optimizes the structure of the intermediates. Though systematic simulations, a complete catalytic cycle comprising all the elementary steps and a corresponding energy profile can be proposed. These simulations can also offer an energy profile of all the elementary steps from the reactants to the products. Through this energy profile, the rate-determining step(s) can be identified. If the active sites of the catalyst can be identified experimentally, a correlation between the activation barrier of the transition state of the rate-determining step and its corresponding chemical and coordination environments of the catalytic site can be established. With this intrinsic correlation, the descriptor of this catalytic performance can be proposed. Based on this descriptor, a new catalytic site offering a higher activity can be uncovered. Then, such a new favorable catalytic site “proposed” through the integration of experimental and computational studies is significantly valuable for researchers, who can design a synthetic route to prepare a catalyst consisting of such catalytic sites to test this prediction. Thus, theoretical simulation definitely plays a significant role in interpreting the catalytic performance of a tested catalyst and proposing a new catalyst for the transformation of CH4 under mild conditions. A significant part of this review is devoted to the molecular understanding of CH4 transformation under mild conditions based on computational studies.4. Brief overview of the catalytic conversion of CH4 by enzymes under ambient conditions
In contrast to the current high-temperature catalytic processes with low selectivity for ideal products and the facile deactivation of catalysts, CH4 monooxygenase (MMO) enzymes can oxidize CH4 to CH3OH at room temperature and ambient pressure. The understanding of efficient biocatalysis for producing methanol from CH4 is significant for designing artificial catalysts mimicking this amazing biofunction of nature. In nature, methanotrophic bacteria can oxidatively activate the C–H bond of CH4 under ambient conditions since these bacteria contain MMO.190 Methanotrophs of such bacteria can metabolize CH4 with MMO by the oxidation of CH4 to CH3OH. Two types of MMOs were reported to be active for this oxidation at room temperature. They are in soluble and particulate forms, thus called sMMO and pMMO, respectively. sMMO is in the bacterial cytoplasm in the environment with a low concertation of iron. The catalytic activity of pMMO requires copper-based catalytic sites.Methylococcus capsulatus (Bath) and Methylosinus trichosporium OB3b belong to sMMO. They have been studied well in the literature.191–197 sMMO consists of three components including reductase (MMOR), hydroxylase (MMOH), and regulatory protein (MMOB). It has been found that all the three components are necessary for the aerobic oxidation of CH4.198 Electron transfer between CH4 and O2 is performed using hydroxylase and reductase. Fig. 3a presents the main components of am sMMO enzyme system having a dimeric structure with the feature of an (αβγ)2 dimer architecture.199 These studies198 suggested that the active sites of the hydroxylase are responsible for the reduction of O2 and the activation and oxidation of CH4. The sMMO in Fig. 3 has a dimeric structure consisting of two αβγ-promoters. The feature of each αβγ-promoter is its nearly complete α-helical secondary structure. Each α-subunit has a four-helix bundle. The bundle is the di-iron active site in hydroxylase, which is responsible for hydroxylation. The di-iron site within the hydroxylase is the catalytic site where O2 is activated and reduced and CH4 is oxidized. Notably, MMOB in Fig. 3a, which is a protein cofactor, can tailor the structure and activity of the di-iron site of MMOH in terms of the activation of MMOH. MMOR in Fig. 3a is responsible for transferring electrons from the electron donor NADPH/NADH to MMOH.200 Obviously, MMOH, MMOR, and MMOB are consolidated to form a well-functioning enzyme to selectively transport four species including molecular CH4, molecular O2, electrons, and protons to the catalytic center, the di-iron site. The hydroxylase-regulatory protein controls the pathways of delivering CH4 to the active site of sMMO. Fig. 3b schematically presents the proposed mechanism of sMMO.
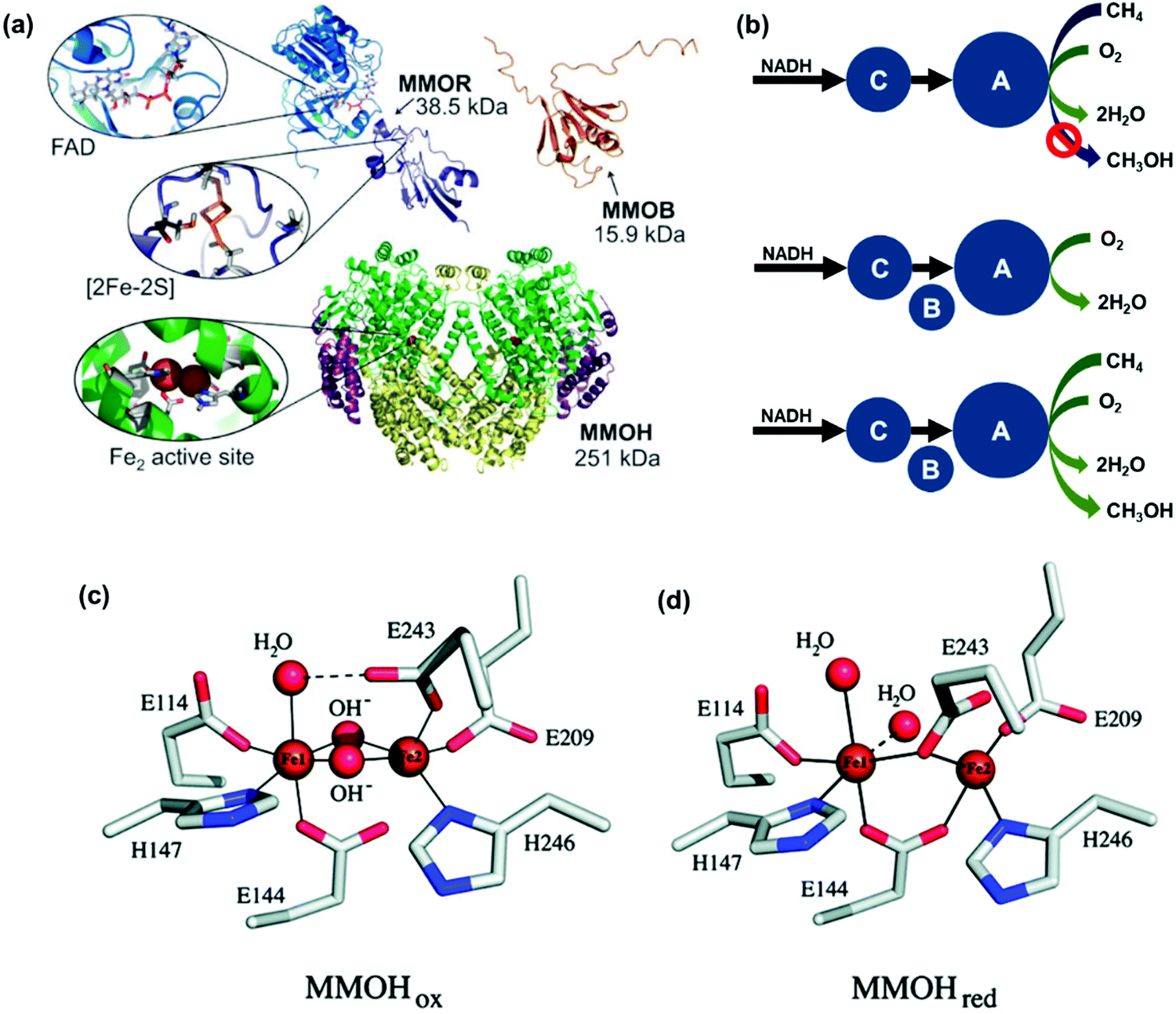 | ||
| Fig. 3 Representation of the mechanism of the catalytic oxidation of CH4 to CH3OH by sMMO. (a) Structure of sMMO enzyme obtained from Methylococus capsulatus (Bath).200 It consists of three components including (1) a hydroxylase [MMOH (PDB reference 1MTY)]. (2) An oxidoreducase [MMOR consisting of FAD domain (PDB reference 1TVC) and [2Fe2S]-Fd domain (PDB reference 1JQ4)], and (3) a regulatory protein [MMOB (PDB reference 1CKV)]. Reproduced from ref. 200 with permission from the Royal Society of Chemistry. (b) Schematic showing the mechanism of oxidation of CH4;198 A, B, and C in the schematic are three components of a soluble CH4 monooxygenase of Methylococcus capsuhtus, which was reported in the literature.203–207 Reproduced from ref. 198, copyright 1985, with permission from Elsevier. (c) Oxidation form of the di-iron site with coordination unlike the oxidation form in (c).202 (d) Reduction form of the di-iron structure.202 Reproduced from ref. 202, copyright 2011, with permission from ACS. | ||
The MMOH complex containing an active di-iron site consists of four glutamates (E114, E144, E209, and E243) and two histidines (H147 and H246). As seen in the oxidation form of MMOH (Fig. 3c), two OH ligands bridge the two Fe atoms in the oxidation state and the coordination sphere of Fe atoms contains solvent H2O molecules. The confirmed structure of the di-iron site bridging two OH groups has driven significant efforts in designing catalysts containing a di-μ-oxo structure, which are discussed in Sections 9 and 13 of this review. The di-μ-oxo structure encapsulated in zeolite was found to be reactive in the oxidative activation of C–H of CH4 to form CH3OH at a temperature of 50–100 °C in an aqueous solution. When electrons are transferred to the oxidation form of MMOH, it is changed to the reduction form (Fig. 3d); in the reduction form of MMOH, the bridging OH groups depart and one oxygen atom of E243 likely coordinates with two Fe atoms, through which the distance between the two Fe atoms increases and thus an open coordination shell in the reduction form is formed for being readily accessed by O2. The activation of molecular O2 by the reduction form of MMOH results in the breakage of the O–O bond and also the restructuring of the di-iron site, through which the restructured di-iron site is ready for the activation of C–H of CH4 to form CH3OH. More details on the oxidation of CH4 to CH3OH by sMMO can be found in the literature.200–202
In contrast to sMMO, less understanding was achieved for pMMO.208–210 pMMO is an integral membrane protein with the function of transformation of CH4 to methanol in methanotrophic bacteria. Although nature does have more pMMO than sMMO, it is more difficult to purify pMMO than sMMO. The difficulty in the purification of pMMO results from the unstable lipid bilayer and easy loss of the metal cofactors. Rosenzweig et al. reported the crystal structure of pMMO.211 Compared to sMMO, the transformation of light alkanes on pMMO is highly regiospecific and stereoselective.212–214 For the synthesis of a biomimic catalyst with a function similar to pMMO in nature, significant efforts in identifying the structure of the active site of pMMO have been made in the last two decades.209
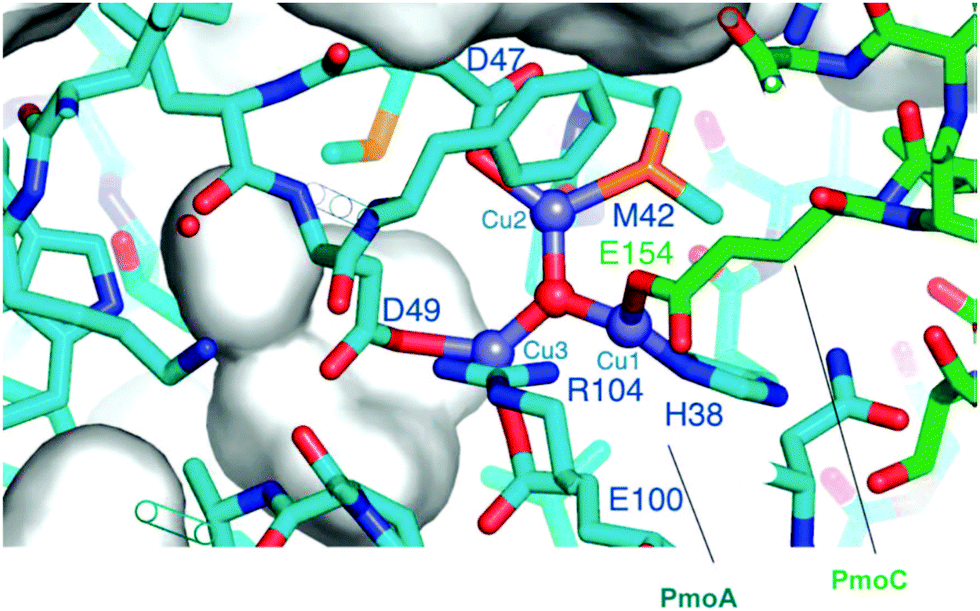
Fig. 4 presents the architecture of pMMO. pMMO consists of pmoA, pmoB, and pmoC in a trimer (α3β3γ3) of αβγ monomers. Each monomer has three copper ions and one zinc ion. The three units including pmoA, pmoB, and pmoC in Fig. 4 are shown in yellow, green, and pink, respectively. How Cu atoms and their ligands coordinate the oxy-transfer chemistry was a crucial question that was extensively explored a decade ago.209,215–218 It was suggested that a tri-copper structure located in the pmoB subunit should be the catalyst site. Recently, Martinho et al. indicated that a nonheme di-iron center is also active in this transformation although the Fe/Cu atomic ratio is only 1:80.219,220 As suggested by later studies, all 14–15 Cu atoms in a purified pMMO participate in the elementary step of CH4 oxidation; thus, the early proposal of the direct participation of the di-iron center in the hydroxylation of CH4 is implausible.209
 | ||
| Fig. 4 Representation of architecture of pMMO. PmoA, PmoB, and PmoC are shown in pink, yellow, and green, respectively.209 Reproduced from ref. 209, copyright 2008, with permission from ACS. | ||
Chan et al. divided the 14–15 Cu atoms in the pMMO into two groups since 6 of them are oxidized by O2 and the other 9 Cu atoms remain reduced.221 The group of the 6 Cu atoms and their surroundings were assessed as the catalytic clusters as they are active in the activation of both O2 and hydroxylation. The 6 Cu atoms related to the catalytic sites are two trinuclear copper clusters.218,221,222 The existence of Cu3 clusters was supported by studies using electron paramagnetic resonance (EPR).223 In the proposed mechanism,218,221,222 one Cu3-cluster is responsible for both the activation of O2 and the hydroxylation of CH4 but the other Cu3-cluster only plays a role in the activation of O2. As shown in Fig. 4, PmoA and PmoC are primarily transmembrane. Based on the reported enzyme crystal structure,211 the N- and C-terminal subdomains in PmoB are surrounded by the cytosol and buried at the water-membrane interface. Remarkably, there is a cavity comprising a hydrophilic cluster in the transmembrane domain at site D. Notably, an earlier study did not consider this hydrophilic cluster of residues as the site to bind the metal atom.211 Chan et al. suggested that site D should bind to metal ions as the electrostatic energy of the cavity of hydrophilic residues could be exceptionally high.217 Studies of Chan et al. using redox potentiometry provided unambiguous evidence for the existence of CuIICuIICuII in pMMO (Fig. 5).217 The existence of CuIICuIICuII in pMMO was further confirmed by the fact that the EPR observed from pMMO is the same as the tricopper complexes synthesized by Chan et al.217 Surprisingly, the synthesized [CuICuICuI(L)]+ complex can couple with molecular O2 to form an intermediate that is beneficial for the addition of oxygen atoms to the C–C bond at room temperature.217 In addition, Chan et al. found that the [CuICuICuI(L)]+ complex can accelerate the transfer of atomic oxygen to the C–H bond of CH3. These observations strengthened the hypothesis that the Cu3 cluster of pMMO is responsible for oxygen activation.
 | ||
| Fig. 5 Representation of the environment of CuIICuIICuII by capping “oxo” in site D. PmoA His38 and PmoC Glu154 coordinate Cu1 ion; PmoA Asp47 and Met41 surround the Cu2 ion; PmoA Asp49 and Glu100 coordinate the Cu3 ion.217 Reproduced from ref. 217, copyright 2007, with permission from John Wiley and Sons. | ||
The computational study was performed for modelling the trinuclear CuIICuIICuII cluster in order to fundamentally understand the role of this Cu3 cluster at site D in pMMO. By considering the structural parameters of the species in the widely defined coordination environment, the geometries of the residues and metal ions were optimized for minimizing the total energy of the Cu3 site, as shown in Fig. 5. The distances of Cu–O and Cu–Cu provided by this optimization show that the accommodation of the Cu3 cluster at site D is reasonable. Modelling suggests that the amino acid side chains coordinating to the Cu ions are PmoA His38 and PmoC Glu154 for Cu1′ ion, PmoA Asp47 and Met41 for Cu2′ ion, and PmoA Asp49 and Glu100 for Cu3′ ion.217 Unquestionably, site D in pMMO holding the hydrophilic cluster without the Cu ions is clearly unstable, exhibiting high activity.
Based on the assumption that the oxidation of CH4 is performed through a direct concerted O-atom insertion mechanism, Chan et al. expected a binding pocket near the Cu3 cluster for CH4.209 In addition, a hydroxylation site must include the copper site and the hydrophobic cavity since the Cu site can activate molecular O2 and the hydrophobic cavity can host CH4 molecules. They used a pentane molecule as a probe for identifying any possible binding sites of hydrocarbon molecules in the αβγ monomer. Their studies show that the most probable site binding a hydrocarbon molecule is the hydrophobic channel lined by the aromatic residues Trp48, Phe50, Trp51, and Trp54 of PmoA. Thus, this binding site of the hydrophobic substrate plays a role in the direct concerted O atom insertion into a C–H bond of light hydrocarbon.209,211,216,217 It is proposed that the active site catalyzing the insertion of the O atom into the C–H of light hydrocarbons is the intermediate [CuIICuII(μ-O)2CuIII]3+, as shown in Fig. 6a.215 This intermediate is formed through the oxidation of the reduced [CuICuICuI]3+ cluster.
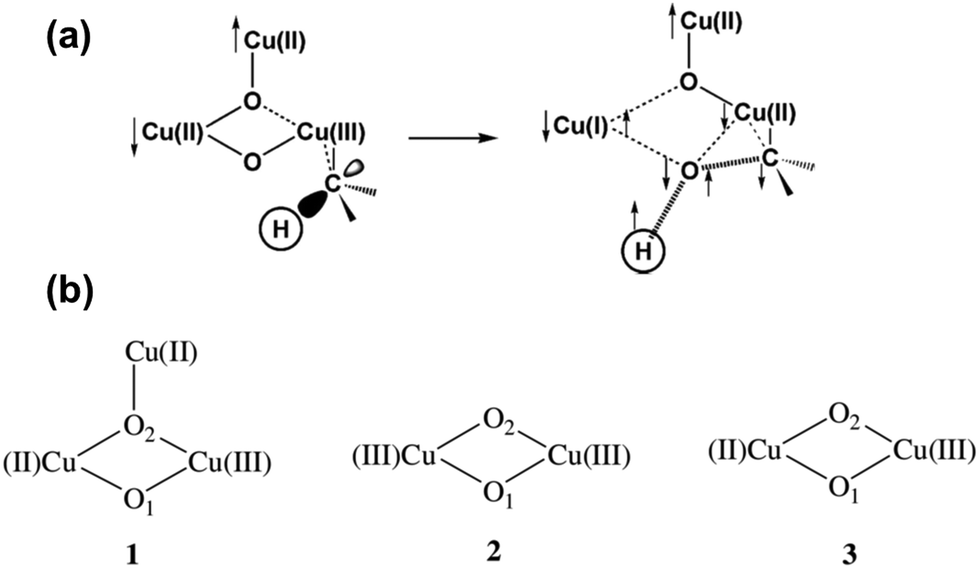 | ||
| Fig. 6 Representation of the details of the adiabatic “singlet oxene” transfer from a dioxygen activated Cu3 cluster to CH4 and the formation of the transition state. (a) The O2-activated tricopper cluster and its transition state predicted by the DFT calculation.218 The arrows show the up and down directions of the unpaired electron spins. Reproduced from ref. 218, copyright 2004, with permission from ACS. (b) Three possible structural models of the activated pMMO considered in the DFT calculation.215 Reproduced from ref. 215, copyright 2006, with permission from Elsevier. | ||
It is reported that a di-nuclear copper cluster, bis(μ-oxo)CuIII–CuIII species (2 in Fig. 6b), can catalyze the transfer of the O atom to a C–H bond.216,217,220,224 A singlet oxene could transfer from bis(μ-oxo)CuIII–CuIII to a light hydrocarbon when the μ-oxo atom is at an appropriate position for forming a transition state complex to couple with the C–H of a light hydrocarbon.217 However, DFT calculation suggested that the μ-oxo atom has to stretch much to form a transition state with a high activation barrier. Notably, the energy barrier for activating the C–H on the bis(μ-oxo)CuIII–CuIII-based intermediate (2 of Fig. 6b) is much higher than that of the [CuIICuII(μ-O)2CuIII]3+-based intermediate (1 of Fig. 6b).215 The DFT calculation suggested three most probable models including mono-valent bis(μ-oxo)CuIIICuIII complex, multi-valent bis(μ-oxo)CuIICuIII complex, and [CuIICuII(μ-O)2CuIII]3+ in Fig. 6b.215 Obviously, the calculated rate constant of CH4 consumption in the transformation of CH4 to CH3OH on [CuIICuII(μ-O)2CuIII]3+ (1 in Fig. 6b) at room temperature is 2.91 × 104 s−1, which is much larger than 0.57 s−1 for bis(μ-oxo)CuIIICuIII (2 in Fig. 6b) and is 119 times of the rate constant of bis(μ-oxo)CuIICuIII (3 in Fig. 6b). Clearly, the trinuclear cluster (1 in Fig. 6b) is the most kinetically favorable.209,218 Overall, these DFT calculations support that trinuclear Cu clusters (1 in Fig. 6b) in site D play a crucial role in the hydroxylation of CH4 in pMMO.
In terms of using the metabolic process of methanotrophic bacteria to transform CH4 to CH3OH, there is an issue of selectivity in producing methanol in the oxidation. This is because formaldehyde instead of methanol is the final product of the metabolites of bacteria.202 Thus, if natural bacteria are the “catalyst”, it must be challenging to produce a large scale of methanol due to the limit of selectivity in the production of CH3OH. Based on this fact, it is necessary to engineer or modify the structure of the CH4 monooxygenase to prevent the further oxidation of methanol to formaldehyde. However, such an engineering must be based on a deep understanding of the mechanism of enzyme-catalyzed oxidation of CH4. For instance, understanding of the pathway through which CH4 accesses the di-iron site within the hydroxylase of sMMO is a prerequisite for developing an efficient biocatalyst with high selectivity for producing CH3OH. Similarly, a deep understanding of oxidation of CH4 by pMMO is a requirement for developing a biocatalyst with high selectivity for producing CH3OH. Upon deep understanding of CH4 oxidation by sMMO and pMMO, the modifications of MMO structures could be done through chemical or/and biological approaches toward developing biocatalysts for the production of CH3OH with high selectivity. Due to the low selectivity and the limited available amount of MMO and the high cost of MMO, it is too early to claim production of CH3OH from CH4 at a large scale with biocatalytic processes. Inspired by these insights gained from experimental exploration and computational analysis, it is expected that numerous efforts in synthesizing artificial catalysts for mimicking the di-iron and the tri-nuclear Cu clusters could be made in the near future.
5. Brief overview of the organometallic approach and related homogeneous catalysis
Compared to the activation of O–O of O2 and C–H of CH4 to generate CH3OH with sMMO and pMMO, CH4 can be activated and oxidized in many reactions accelerated with molecular catalysis in liquid. Periana and his colleagues published a comprehensive review on how CH4 can be functionalized through homogeneous reactions.225 They categorized all homogeneous functionalizations reported in the literature into 12 types of reactions based on (1) the specific consideration in terms of whether O2 can be directly utilized and (2) the reaction mechanism of the elementary step reacting with CH4 with regard to whether the CH4-involved elementary step is proceeded with chain or no-chain reactions, with stoichiometric reagents, or catalytic species.225 Here, we only briefly discuss C–H activation of CH4 with organometallic approaches in homogeneous systems under mild conditions. The progress and achievement in the catalytic transformation of CH4 can be found in this excellent review.225As reviewed in the literature,226 C–H activation of light alkanes can be performed through σ-bond metathesis, electrophilic activation, oxidation addition, 1,2-addition, and metalloradical activation. Regarding oxidative addition, it is performed on [LnMy]. The M atom is coordinatively unsaturated in [LnMy] as it needs to bond to additional alkyl (–R) and H atom. [LnMy] is typically generated from a stable precursor such as LnMy+2X2. Fig. 7a represents the oxidation activation of the light alkane by [LnMy]. For this type of activation, M is typically a metal atom with rich d electrons such as Rh, Ir, and Pt with low valence.
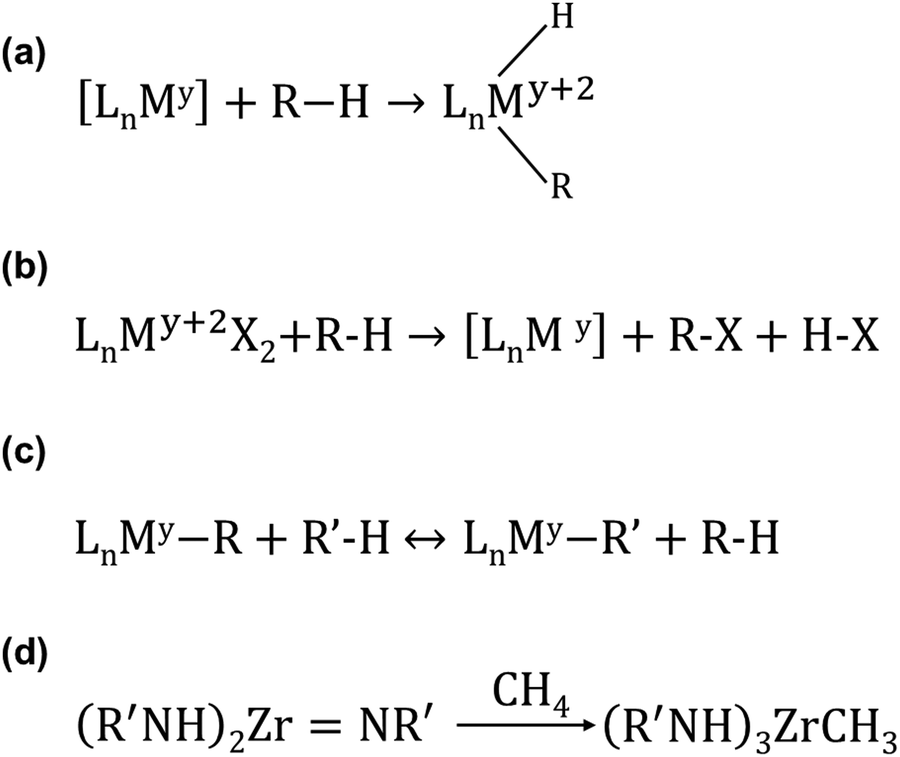 | ||
| Fig. 7 Representation of promising organometallic reactions for the activation of C–H of light alkanes.226 (a) Oxidative addition. (b) Electrophilic activation. (c) σ-bond metathesis. (d) 1,2-Addition. Reproduced from ref. 226, copyright 2002, with permission from Springer Nature. | ||
Fig. 7b represents the reaction of electrophilic activation performed on LnMy+2X2. [My+2] in Fig. 7b is typically a late transition metal such as Pd2+ or Pt2+. In this activation, an intermediate [LnMy+2(R)(X)] is first formed by coupling [LnMy+2X2] with alkane. Then, the R and X atoms on [LnMy+2(R)(X)] couple with each other to form R–X, releasing and regenerating [LnMy]. Electrophilic activation is typically performed in a highly polar solvent.
Regarding σ-bond metathesis (Fig. 7c), it is performed on LnMy. M is commonly an early 3d transition metal such as Sc. This reaction is more like an interchange of alkyl fragments (R and R′) instead of net alkane activation.
Regarding 1,2-addition, it is the addition of the R and H groups of the alkane RH to the double bond MY (M: transition metal, Y: non-metal element such as C and N) (Fig. 7d). Literature reports the addition of R–H to MC and MN.227 It is necessary to perform further studies on how this addition reaction can be used to produce value-added chemicals from CH4.
Although there are limited examples in the catalytic conversion of CH4 or C2H6 to value-added chemicals through the organometallic approaches of C–H activation, they are promising paths to catalytically transform CH4 under mild conditions through homogeneous catalysis. An example of the electrophilic activation of alkanes and oxidation was early proposed by Bercaw et al.228 In this activation, the [PtL3Cl] complex replaces a proton of the light alkane H–R, forming a [PtIIL3R] complex (1 in Fig. 8). Then, [PtIIL3R] is reduced to form the PtIV complex (2 in Fig. 8) upon reaction with [PtIVCl6]2−; in this step, [PtIIL3R] transfers two electrons to [PtIVCl6]2−, which binds with two additional Cl atoms, forming complex 2 in Fig. 8, [PtIVL3RCl2]. Then, the H2O molecule performs nucleophilic attack on complex 2, which produces a CH3OH molecule and releases a HCl molecule; meanwhile, the molecular catalyst, [PtIIL3Cl] is regenerated. Electrophilic activation follows a non-radical path, similar to C–H activation of alkenes in organometallic chemistry. As [PtIVCl6]2− is transformed to [PtIICl4]2−, it acts as an oxidant instead of a recoverable catalyst; therefore, the regeneration of [PtIICl4]2− to [PtIVCl6]2− needs another reaction. This route was illustrated in the transformation of CH4 to CH3OH by Shilov et al.229 Notably, the relatively low selectivity and quite low activity in the oxidation of CH4 to CH3OH through this electrophilic activation make this route more of an ideal instead of a promising application. Efforts for replacing [PtIVCl6]2− with an economic oxidant such as copper salts230 or even electrochemical approach have been made.231
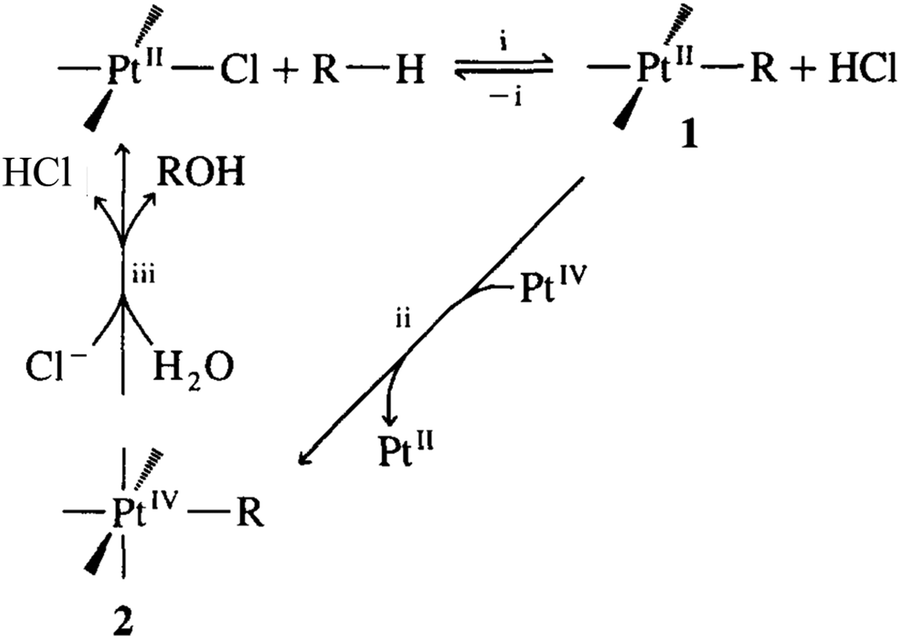 | ||
| Fig. 8 Representation of activation and oxidation of alkanes.228 Reproduced from ref. 228, copyright 1995, with permission from Elsevier. | ||
One successful example of oxidation of CH4 through electrophilic activation with high activity and selectivity is the catalytic oxidation of CH4 to CH3OSO3H by the oxidant H2SO4 and the catalyst (bpym)Pt(OP)2 (bpym: 2,2′-bipyrimidine) at 100 °C, as reported by Periana et al.232 The key role of bpym is to increase the solubility of the Pt species. One reason for using H2SO4 is the protection of the bi-sulphate group, which is a ligand inert toward oxidation. Although the product of organometallic chemistry, CH3OSO3H is not a value-added chemical without further hydrolysis; this close catalytic cycle of oxidation of CH4 with H2SO4 to CH3OSO3H with high activity and selectivity distinctly demonstrates the success of the organometallic approach in the transformation of CH4 to value-added chemicals under mild conditions. Fig. 9 represents the proposed reaction mechanism for the oxidation of CH4 through (bpym)Pt(OP)2 to form CH3OSO3H.232 Periana et al. concluded that C–H activation on (bpym)Pt(OP)2 is performed through electrophilic activation and occurs with the highly electrophilic, largely uncoordinated 14-electron complex with a T-shape (Fig. 9). The product, methyl bisulfate CH3OSO3H, has steric and electronic effects in preventing it from being further oxidized.232 The –OSO3H group of CH3OSO3H prevents the C–H bond of the CH3 group from being accessed. More importantly, the electronic withdrawing effect of the OSO3H group makes the CH3 of CH3OSO3H electronically deficient. Thus, the electrophilic attack of metal complex on the electron-deficient C–H bond of CH3 is unfavorable. In other words, using H2SO4 as a specific oxidant can effectively prevent CH3 from being further oxidized since the control oxidation of CH4 is the key for the transformation of CH4 to CH3OH with high selectivity. The formed CH3OSO3H can be transformed to CH3OH through additional hydrolysis.
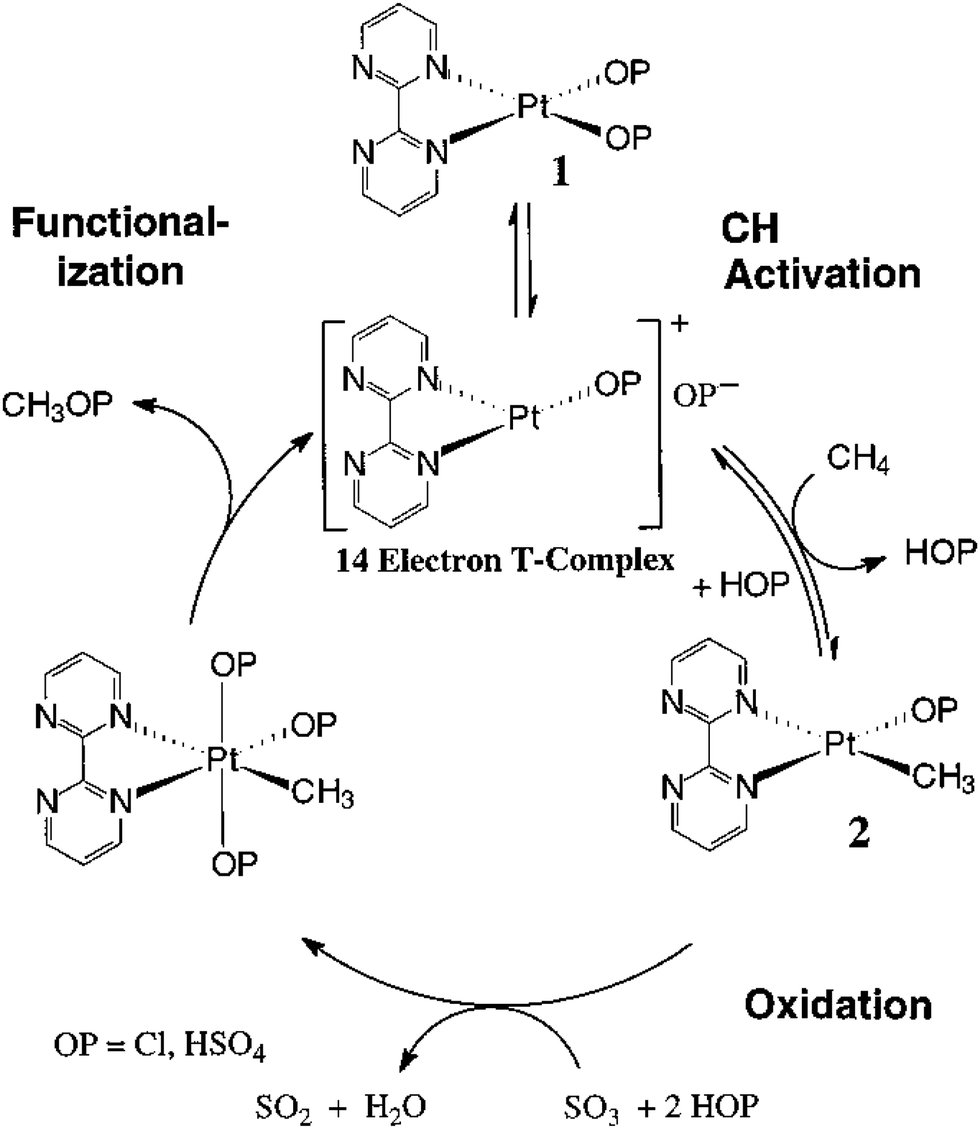 | ||
| Fig. 9 Representation of the proposed reaction mechanism of the oxidation of CH4 to methyl bisulfate CH3OSO3H by methyl bisulfate CH3OSO3H at 100 °C through electrophilic activation. This mechanism consists of C–H activation, oxidation, and functionalization.233 Reproduced from ref. 233, copyright 1998, with permission from AAAS. | ||
A significant advance in the catalytic oxidation of C–H of CH4 with molecular catalysts in the liquid phase has been made in the last decades. All these transformations catalyzed by molecular catalysis are performed under mild conditions. More information on this approach can be found from the comprehensive review published by Periana et al.225
6. Activation and oxidation of CH4 on bare MO+ to CH3OH
Some bare transition-metal cations (Mn+) in gas phase can activate C–H or/and cleave C–C of light alkanes.234–236 The oxidation of bare transition-metal cations forms bare transition-metal oxide cations (MO+). Similar to the ligand effect in tuning the reactivity of the metal center of a molecular catalyst discussed in Section 5, bare transition-metal oxide cations (MO+) exhibit high activity in the oxidation of organic molecules. Here, MO+ acts as an oxidant.Driven by the significance of low-temperature oxidation of light hydrocarbons, the oxidative reactions of CH4 on bare transition-metal oxide cations (MO+)237–241 such as FeO+,71,242–248 CoO+,72,249 and OsO+250 in gas phase were studied extensively.73 These studies of oxidation of light alkanes by MO+ in gas phase offered insights into the reactivity patterns, roles of the ligands on transition metal atoms, chemo- and regioselectivities, and potential intermediates at the molecular level.73,250 These insights are significant for developing catalysts active and selective in the oxidative transformation of CH4 to value-added chemicals. It built the foundation for the later exploration of single atom cluster M1On anchored in zeolites, which will be reviewed in later sections.
In general, MO+ clusters were generated with ion cyclotron resonance mass spectrometry.73 The theoretical base for the generation of MO+ clusters with this spectrometry has been reported in the literature.251 MO+ clusters were trapped in a magnetic field of an ion cyclotron setup and then they were selected with tunable radio-frequency pulses on the basis of their masses. The mass-selected ions can be slowed to an ideal kinetic energy before using them for reaction with CH4. Once a type of specific MO+ clusters with a specific kinetic energy was created, they were leaked into a glass chamber containing CH4 at a low pressure in the range of 10−2–0.5 Torr; the reason to maintain a low pressure of CH4 is to minimize multiple molecule-ion scatterings. This reaction between the MO+ clusters and CH4 molecules is the direct oxidation of CH4 by MO+ such as CH4 + CoO+ → CH3OH + Co+. The reaction occurred at room temperature. Thus, exothermic or nearly thermoneutral reactions can occur under this condition. The products formed in the glass chamber were collected by a quadruple mass filter for identifying their masses and analyzing their intensities.73
These oxidations reviewed in Section 6 or to be reviewed in Sections 7–13 can be categorized into three types of oxides, including oxidation by MO+ clusters in the gas phase of CH4 at sub-Torr pressure, oxidation by active oxygen atoms bound to the catalytic sites in the micropores of zeolites in 1 bar pressure of CH4, and oxidation by catalytic sites anchored in the zeolites dispersed in a liquid under the high-pressure gas phase of CH4. The operational condition of the oxidative activation of CH4 by the MO+ clusters is very similar to the catalytic oxidation of CH4 by PtO+, as reviewed in Section 7. In both the cases, the pressure of the reactant CH4 is in the sub-Torr pressure or lower range for the minimization of multiple molecule-ion scatterings. Unlike the operational condition used in Sections 6 and 7, the oxidative activation of CH4 by oxygen atoms bound to metal atoms anchored in the micropores of zeolite or oxygen atoms on metal oxide particles, as reviewed in Sections 8–10, was performed by introducing the reactant CH4 at ambient pressure to a zeolite at the room temperature or a temperature in the range of 20–250 °C. Obviously, the operational condition of oxidative activation and catalytic oxidation of CH4 on the MO+ clusters is unlike the oxidative activation of CH4 through active oxygen atoms in the zeolite or on the metal oxide.
The operational conditions of oxidation of CH4 by MO+ clusters (Sections 6 and 7) and the oxygen atoms in the micropores or on the surface of metal oxide (Sections 8–10) are unlike the operational conditions of catalytic oxidation of CH4 at the solid–liquid interface, as reviewed in Sections 11–13. The oxidation of CH4 in Sections 6 and 7 is performed in the gas phase at sub-Torr pressure of CH4 where the oxidant in terms of MO+ clusters is dispersed in the gas phase of CH4. Unlike the operational conditions of oxidation of CH4 by MO+ clusters, the oxidation of CH4 by modified zeolites in Sections 9 and 10 is performed at the solid–gas interface at 100–250 °C in the gas phase of CH4 at 1 bar; an important feature is that the zeolite needs to be regenerated through oxidation at 300–500 °C. Different from these non-catalytic oxidation reactions of CH4, as reviewed in Sections 9 and 10, the catalytic oxidation reactions of CH4 under mild conditions, as discussed in Sections 11–13, are actually performed at the solid–liquid interfaces in the temperature range of 20–250 °C; in these catalytic oxidation reactions, only dissolved CH4 molecules can access the catalytic site of the catalyst particles dispersed in the solvent. This is why the liquid phase containing the catalyst particles must be under a gas phase of CH4 with a high pressure (1–100 bar).
Compared to the oxidation reactions of CH4, as reviewed in Sections 8–13, the oxidation of CH4 by MO+ clusters dispersed in the gas phase are performed at 25 °C in the gas phase of CH4 in the low pressure range of 10−2–0.5 Torr. Obviously, the oxidation of CH4 by the MO+ clusters at low pressure of CH4 does not lead to the production of CH3OH at a large scale. In addition, the oxidation of CH4 by oxygen atoms bound to metal atoms anchored in the zeolites is a noncatalytic process, which needs the regeneration of the oxygen atom after the consumption of these active oxygen atoms; thus, it is not a feasible process to produce value-added chemicals at a large scale potentially. It is expected that the catalytic oxidation of CH4 at the solid–liquid interface by the catalyst dispersed in the liquid under a high-pressure gas phase of CH4 is a potential process for the transformation of CH4 to the oxygenates at a large scale in the future. From the point of view of a fundamental understanding of the science behind the oxidation of CH4 to CH3OH, the oxidation of CH4 by the MO+ clusters is significant as the MO+ clusters can be well characterized for elucidating the reaction mechanism at the atomic scale and its insight gained from these studies can inspire the design of catalysts for producing CH3OH.
CH4 can be transformed to oxygenates by MO+ in the gas phase. Here, the transformation of CH4 to CH3OH on MO+ is taken as an example to demonstrate the approach of converting hydrocarbons to organic oxygenates by free MO+ in the gas phase. Experimental exploration showed that late 3d-metal oxide clusters including MnO+,74 FeO+,71 CoO+,72 and NiO+73 are active for this reaction. However, the MO+ clusters of early 3d transition metals such as ScO+,252–254 ZrO+,255 and CrO+256 are not active for the oxidation of CH4 to CH3OH. Based on theoretical simulations,237 such an oxidation is processed through adsorbed CH4 on MO+, a four-membered ring-like transition state (TS1), an intermediate, a three-membered ring transition state (TS2), formed CH3OH on MO+ before desorption to the gas phase, CH4 + MO+ → (CH4)MO+ → [TS1]# → CH3-MO+-OH → [TS2]# → (CH3OH)M+ → CH3OH + M+.
The electronic structure of these MO+ clusters active for the oxidation of CH4 to CH3OH varies largely from early to late 3d metals (Fig. 10a).237 The M–O distance increases along with the increase in the d-electrons of the M metal and the dissociation energy of the M–O bond decreases with the increase in d-electrons. As shown in Fig. 10a, the electron configurations of ScO+ and FeO+ are d0 and d5. At the 1Σ+ ground state of ScO+, four pairs of electrons of ScO+ occupy a nonbonding 1σ and three bonding orbitals (2σ and 1π), thus forming a strong triple bond similar to N2, resulting in a high dissociation energy of the Sc–O bond, which rationalizes the low reactivity in the activation of C–H and the oxidation of CH4 on ScO+.252–254 However, in terms of FeO+, the bonding orbitals are doubly occupied and each 2π orbital is singly occupied (Fig. 10b), similar to the electronic configuration of a triplet O2. Thus, the dissociation energy of Fe–O is definitely lower than Sc–O, which is responsible for the higher activity of FeO+ in contrast to ScO+. The difference in the electronic states of ScO+ and FeO+ indicated the observed difference in the activity in the oxidation of CH4 to CH3OH between ScO+ and FeO+.
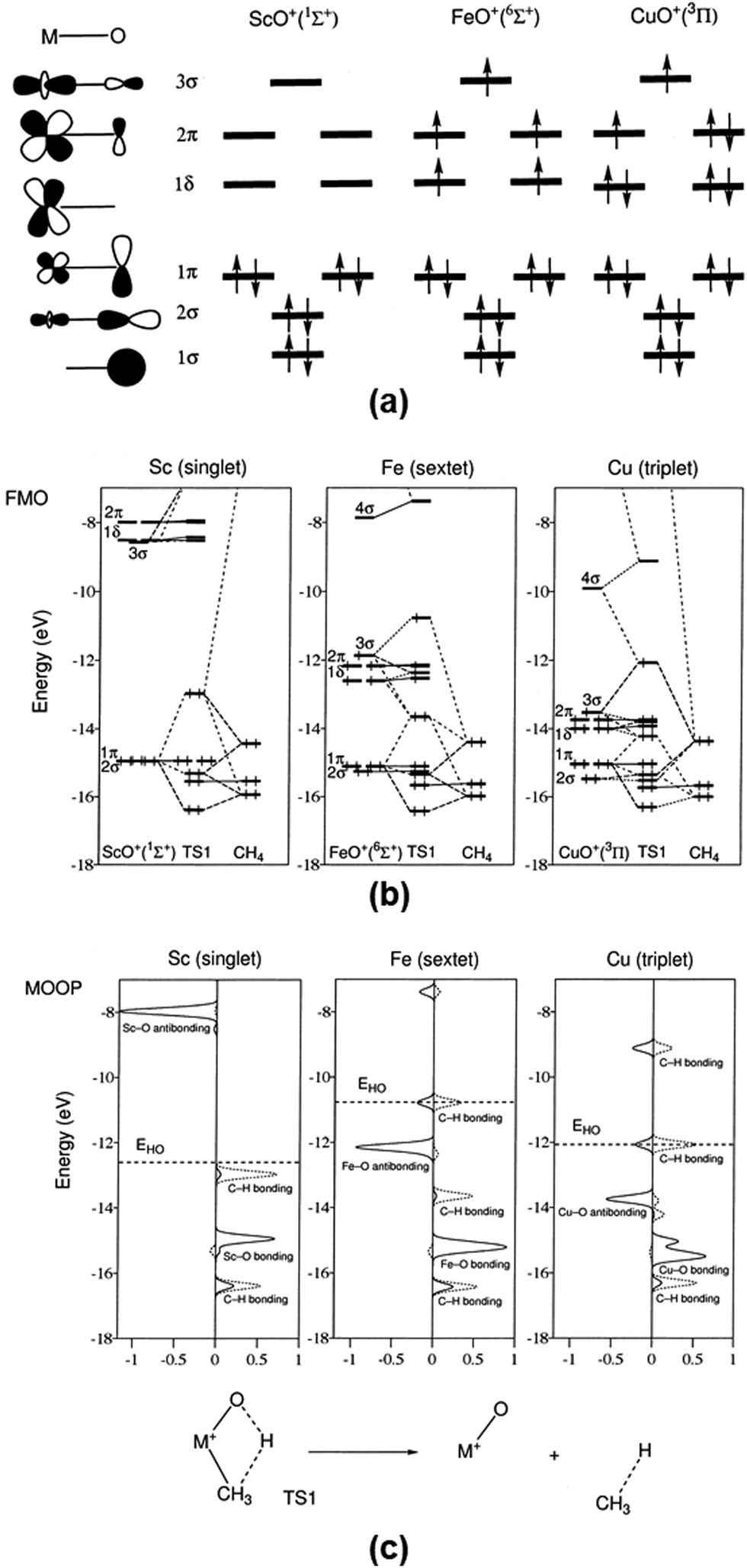 | ||
| Fig. 10 Representation of interaction of free MO+ clusters and CH4 in the transformation of CH4 to CH3OH in the gas phase. (a) Molecular orbital of the MO+ clusters. (b) Analysis of fragment molecular orbital (FMO) of CH4 and the representative MO+ including ScO+, FeO+, and CuO+. (c) Analysis of molecular orbital overlap population (MOOP) of [TS1]# formed from CH4 and representative MO+ including ScO+, FeO+, and CuO+.237 Reproduced from ref. 237, copyright 2000, with permission from ACS. | ||
The activity of the MO+ cluster of the late 3d metals in the transformation of CH4 to CH3OH originates at the orbital interaction of MO+ and CH4 in [TS1]# and the change in the fragment molecular orbital of C–H of CH4 upon the formation of [TS1]#. As shown in Fig. 10b, MO+ fragments have high-lying d-block orbitals of 3σ, 1σ, and 2π, and low-lying ligands of 2σ and 1π; the CH4 fragment has three C–H bonding orbitals, resulting from the 3-fold degenerate HOMO of Td of CH4. The molecular orbital of [TS1]# (Fig. 10b) can be constructed from these fragment molecular orbitals. As shown at the left in Fig. 10b, unfortunately, the high-lying 3σ of ScO+ cannot interact with the C–H bonding orbitals. It makes sense that ScO+ is not active for the activation of the C–H bond of CH4.
Unlike the MO+ of early 3d metals, there is a significant interaction between the molecular orbitals of MO+ of the late 3d metal and CH4. This is because the d-block orbital and 3σ of FeO+ and CuO+ are not fully occupied but are lower in energy than ScO+ (Fig. 10b). Thus, the interaction between low-lying 3σ orbitals of late 3d metal MO+ and CH4 is effective, leading to the energy stabilization of [TS1]#. Compared to the four-electron two-orbital interaction in [TS1]# formed from the coupling between ScO+ and CH4, the formed two-electron two-orbital interaction between the low-lying 3σ orbital of FeO+ and HOMO of the CH4 fragment can stabilize the [TS1]# formed from FeO+ and CH4. Similarly, the interaction between the 3σ orbital of CuO+ and HOMO of the CH4 fragment is significant, making the formed orbitals in [TS1]# low-lying.
Molecular orbital overlap population (MOOP) analysis can qualitatively explain why the MO+ of late 3d metals can weaken the C–H bond but the MO+ of the early 3d metals cannot. The weakening of C–H in [TS1]# is evidenced by pushing the highest occupied level of C–H up or down. Specifically, if the C–H bonding orbitals in [TS1]# are unoccupied or partially unoccupied, the C–H bond is effectively weakened. As shown on the left of Fig. 10c, the energy level of the highest C–H level in the [TS1]# of ScO+ is below the energy level of the HOMO of ScO+ and thus they are still fully occupied in the [TS1]#. Consequently, the interaction of the orbitals of ScO+ and CH4 does not vary the occupancy of the electrons in the HOMO of CH4; thus, such an interaction is not effective in the activation of CH4. Unlike ScO+, the orbital interaction between FeO+ and CH4 pushes the energy level of C–H up and thus makes the HOMO of CH4 half-occupied (Fig. 10c). In terms of the orbital interaction between CuO+ and CH4, in the formed orbitals of [TS1]#, the C–H orbitals at −9 eV and −12 eV are completely unoccupied and half-occupied, respectively. Since the energy level of C–H bonding in the [TS1]# of CuO+ and CH4 is definitely above EHOMO, these highest levels of CH4 are unoccupied or partially occupied. It further suggests that the C–H bond of CH4 has been effectively weakened.
The analysis of molecular orbitals of MO+, CH4, and their [TS1]# provided a profound understanding of the lack of activity in the activation of CH4 on MO+ of early 3d metals but the presence of activity on the MO+ of late 3d metals. The high activity of CuO+ and FeO+ for the oxidation of CH4 to form CH3OH is in good agreement with the later finding that Fe–O and Cu–O clusters encapsulated in the zeolite are active in the oxidation of CH4 to CH3OH under mild conditions. The oxidation of CH4 to CH3OH by Fe–O and Cu–O clusters encapsulated in zeolite is reviewed in Section 9. These insights gathered from experimental and computational studies at the level of molecular orbitals provide a foundation for understanding the activity of Fe–O and Cu–O clusters anchored in the zeolite in the oxidation of CH4 to CH3OH.
These systematic studies of free MO+ clusters of all the 3d metals have rationalized the evolution of the activity of the free MO+ clusters in the oxidation of CH4 to CH3OH at the level of molecular orbitals. However, there is a lack of experimental studies for M–O clusters of all 3d metals anchored in the zeolite such as Sc@ZSM-5, Ti@ZSM-5, V@ZSM-5, and Mn@ZSM-5, and a lack of exploration of potential evolution of activity in oxidizing CH4 to CH3OH by M@ZSM-5 (M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu) as a function of the electronic structure and molecular orbitals of the M–O clusters. Such studies are highly recommended as they will allow to uncover the correlation between the electronic state of the anchored M–O clusters and their reactivities in oxidizing CH4 to CH3OH.
7. Catalytic oxidation of CH4 on bare MO+ and M+
Earlier studies have suggested that MO+ can catalyze the oxidation of hydrocarbons to form oxygenates including alcohols and aldehydes.71,237–241,243 One example of catalytic oxidation involving MO+ is the catalytic oxidation of CH4 by O2 on Pt+ ions with a turnover rate of about 6.0 for the Pt+-mediated oxidation of methane to produce multiple products including CH3OH, formaldehyde, and formic acid.73,257 PtO+ is not an initial reactant. But PtO+ clusters are formed based on the temporal evolution of the species involved (Fig. 11a).257 Thus, PtO+ clusters do play an important role in the catalytic oxidation of CH4. In addition, the Pt[CH2]+ complex was formed in the catalytic oxidation of CH4 by Pt+. Thus, three sequential reactions involving Pt+, [PtCH2]+, and PtO+ are proposed (Fig. 11b).257 Reaction III includes two parallel reactions: one is the direct oxidation of CH4 by PtO+ to form methanol. In fact, this catalysis is more complicated than simply understanding it as three sequential reactions because all the three Pt-based species can activate the CH4 molecules, as shown in Fig. 11b. In addition, as reaction II in Fig. 11b is the slowest step in this catalytic oxidation, a large excess of O2 has to be used if a high turn-over rate of CH4 is required. In the study of Fig. 11a, an O2:CH4 ratio of 1:20 was used. A consequence of the high O2/CH4 ratio used is the high selectivities for the deeper oxidations of CH4 in terms of the formations of formaldehyde and formic acid in this catalytic oxidation process.
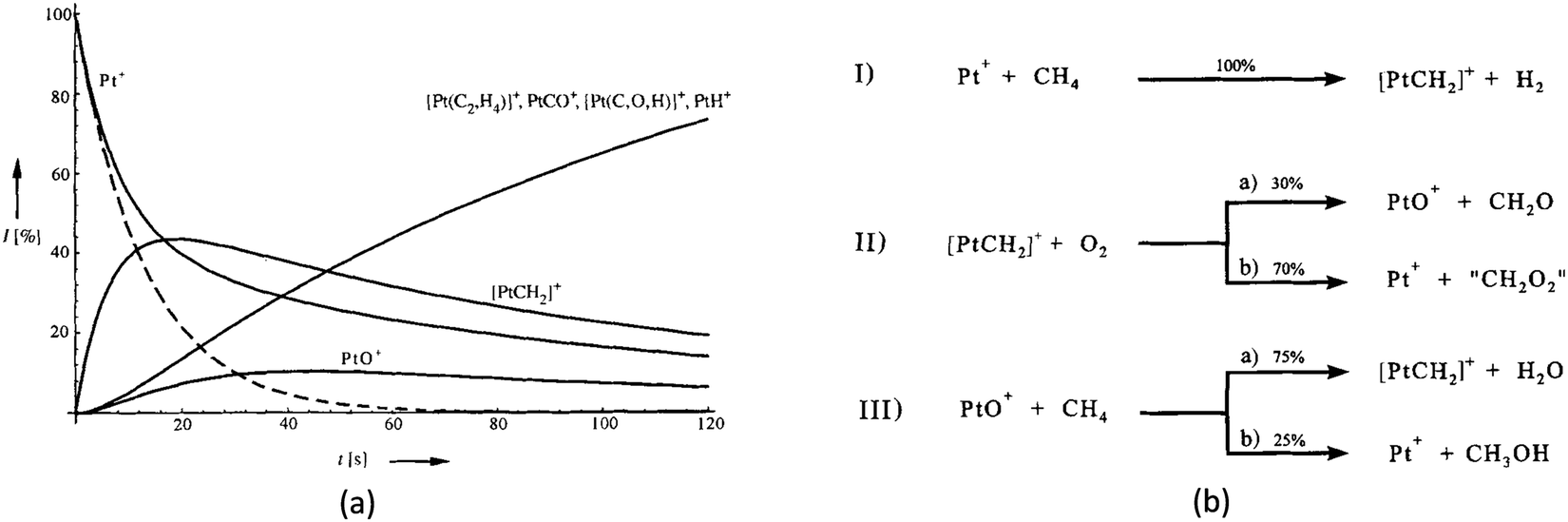 | ||
| Fig. 11 Catalytic oxidation of CH4 to methanol, formaldehyde, and formic acid through Pt+ involving the formation of PtO+ and oxidation of CH4 to form CH3OH by PtO+.257 (a) Temporal evolution of the intensities of Pt-based species including Pt+, [PtCH2]+, and PtO+ in the mixture of O2 and CH4 in the molar ratio of 20:1. (b) Three reactions involved in the catalytic oxidation of CH4. Reproduced from ref. 257, copyright 1994, with permission from John Wiley and Sons. | ||
8. Activation and oxidation of CH4 on the M–O cluster anchored on the oxide
Bare MO+ such as MO+ (M = Mn, Fe, Co, Ni, Cu, Pt) exhibits high reactivity in the activation or oxidation of CH4, as reviewed in Section 6. As discussed in Section 7, some of them can even oxidize light alkanes including CH4 to oxygenates such as CH3OH through a close cycle of catalysis. In such activation reactions or catalysis, MO+ cations are free standing and distributed in the gas phase. Consistent with the high reactivity of free or called free standing FeO+ or NiO+ in the oxidation of CH4 at room temperature, as reviewed in Section 6,71,72,242–249 the oxidation of CH4 by NiO species anchored on the surfaces of NiCo2O4 and NiFe2O4 has been reported recently.48,49Ni cations are introduced into Co3O4 through the partial replacement of cobalt ions. Thus, the surface of this catalyst consists of Ni–O and Co–O species. As shown in Fig. 12a, the anchored NiO cations can activate the C–H bond of CH4 at a temperature as low as 60 °C.48,49 The surface of NiCo2O4 during catalysis was tracked with AP-XPS. C 1s photoemission features of carbon-containing species on NiCo2O4 during the oxidation of CH4 in the temperature range of 60–400 °C are presented in Fig. 12a. Two carbon-containing species were clearly observed on the basis of the two C 1s peaks at 288.5 eV and 285.8 eV (Fig. 12a). As there is a lack of any carbon species of potential contaminants before exposure to the mixture of CH4 and O2, the surface of NiCo2O4 is carbon-free before the introduction of CH4. The two carbon-containing species must result from the activation of CH4 by the NiCo2O4 surface. Peak 2 of Fig. 12a was assigned to an OCHO species based on in situ infrared spectroscopy.48 As the evolution of C 1s intensity as a function of temperature of the catalyst NiCo2O4 exhibits a volcano-like evolution, OCHO is the intermediate, which was formed at a low temperature and observed by infrared spectroscopy.48
 | ||
| Fig. 12 Representation of photoemission features of C 1s collected from Ambient Pressure X-ray Photoelectron Spectroscopy (AP-XPS) and vibrational signatures collected with Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectroscopy from surface adsorbates formed through the activation of CH4 on the charged Ni–O species anchored on the surface of NiCo2O4.48 (a) C 1s spectra of the surface adsorbates formed on NiCo2O4 at 60 °C in CH4. (b) Vibrational spectrum of the adsorbate after exposing NiCo2O4 at 60 °C to CH4. Reproduced from ref. 48, copyright 2015, with permission from Springer Nature. | ||
This observation of OCHO species at 60 °C on the NiCo2O4 surface suggests that CH4 can be activated under mild conditions even at a temperature as low as 60 °C. Computational studies show that Ni–O on the surface is active for activating CH4 to form OCHO and CH3 bonded to the surface of NiCo2O4.48 Thus, the high activity of the charged Ni–O cluster anchored on Co3O4 is consistent with the reported high activity of bare NiO+ for the activation of CH4. Similarly, the charged Ni–O species anchored on the surface of Fe3O4 are active for the oxidation of CH4 at a temperature as low as 60 °C.49 Similar to NiCo2O4, the activation of CH4 on NiFe2O4 forms similar OCHO and CH3 species.
9. Activation and oxidation of CH4 by the M–O cluster anchored in the zeolite
To generate organic oxygenates by the activation and oxidation of CH4, the source of oxygen atoms can be the free-standing MO+ cations in the gas phase or the anchored MO+ on the surface of the oxide nanoparticles. From the thermodynamic point of view, these activation and oxidation reactions to transform CH4 to CH3OH can be performed under mild conditions or even at the room temperature. Upon the transformation of these pre-anchored oxygen atoms to CH3OH, this type of specific oxygen atoms has to be regenerated before more CH4 molecules can be oxidized. Strictly speaking, it is an oxidative reaction instead of a close cycle of catalysis. Notably, these transformations discussed in Sections 6–8 and this section use pre-existing oxygen atoms of the M–O clusters freely in the gas phase or anchored on a support instead of the direct use of any routine oxidant of chemical reactions such as O2, H2O2, or H2SO4. Other than these free-standing MO+ and supported M–O clusters supported on Co3O4, a few transition metal cations including Cu, Fe, and Ni can form unique oxide clusters with specific structures such as the μ-oxo structure in ZSM-5 upon pre-treatment in O2 or N2O at a high temperature. Notably, these reactions reviewed in this section are activation and oxidation of CH4 instead of catalytic oxidation. They are unlike the catalytic oxidations of CH4 with H2O2 on catalysts such as zeolite with anchored metal oxide clusters, as reviewed in Section 13.9.1 Confinement effect of microporous aluminosilicate on the activation of molecules in the micropores
As the confinement effect is an important concept for understanding the activation of C–H of CH4 in the M–O clusters anchored in ZSM-5, it has been briefed here. Olson et al. reported that sub-nanometer pores of ZSM-5 exhibit spatially limited effect for molecules with a kinetic diameter larger than about 7.0 Å.258 CH4 with a kinetic diameter of about 3.7 Å can readily diffuse in these micropores since the pore size of the 10-membered-ring of ZSM-5 is 5.4–5.6 Å. Based on Zicovich-Wilson and Corma's reports,259,260 unlike a molecule in the gas phase, the orbitals of the molecule encapsulated in a micropore of the zeolite do not extend over all space, which was called the confinement effect by Zicovich-Wilson and Corma.259,260One consequence of the confinement effect is the large distortion of the bond geometry. As shown in Fig. 13c, this confinement effect was supported by the difference in the bond angle between 85° and 111° of ∠OMC (M: metal atom) such as ∠OFeC, ∠OCoC, ∠ONiC, and ∠OCuC in the optimized geometry of CH4 adsorbed on metal atoms anchored in the confined space of ZSM-5 and the nearly 180° angle on an metal oxide cluster in open space.261
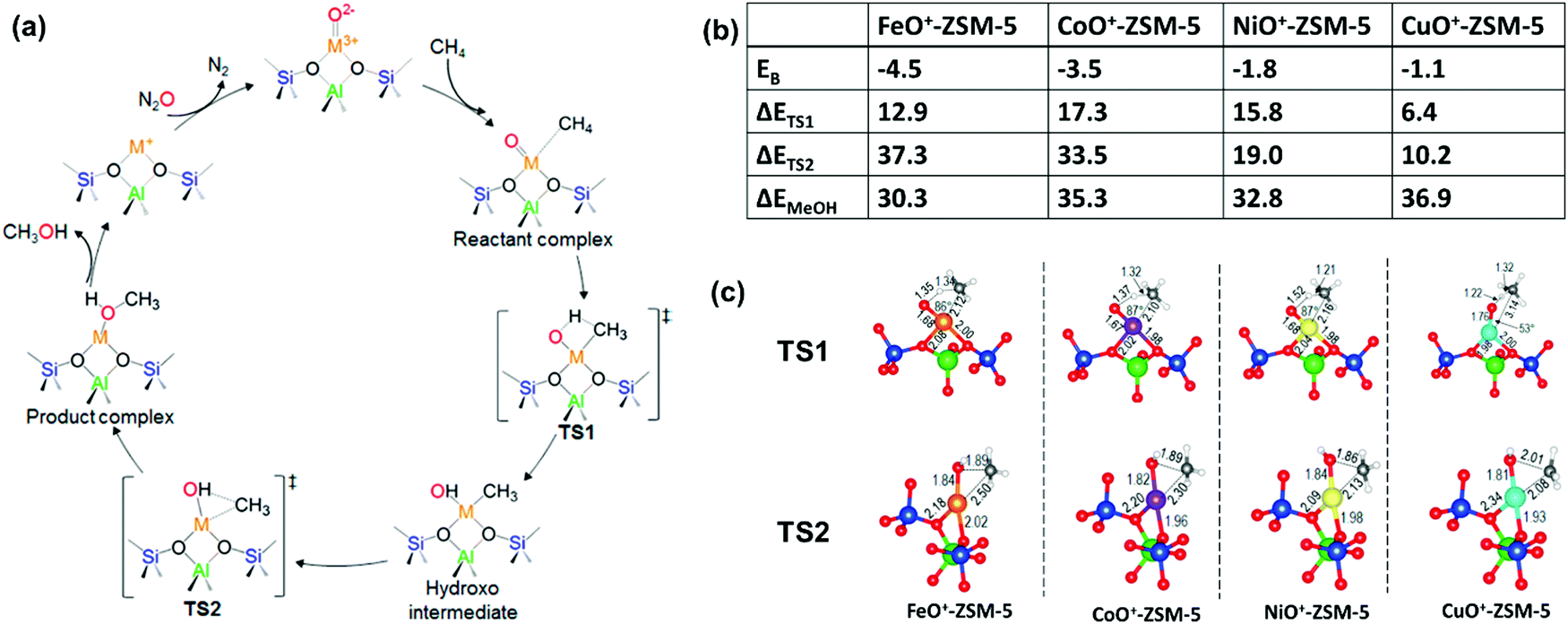 | ||
| Fig. 13 Representation of computational studies of oxidation of CH4 by MO+ (M = Fe, Co, Ni, and Cu) anchored in ZSM-5 to CH3OH.261 (a) Proposed catalytic cycle. (b) List of energies of intermediates and transition state in the oxidation of CH4 by MO+ anchored in ZSM-5 to CH3OH. (c) Optimized structures of TS1 and TS2 on the four catalysts. Reproduced from ref. 261, copyright 2016, with permission from ACS. | ||
A surprising consequence of this confinement effect261 is the low binding energy of CH4 on an MO+ confined in a microporous silicate such as CuO+-ZSM-5, which is only −1.1 kcal mol−1, much lower than −45.8 kcal mol−1 for the adsorption of CH4 on free-standing CuO+ in the gas phase.237 In addition, systematic DFT calculations on different MO+ clusters encapsulated in ZSM-5 show that the binding energies of CH4 on the MO+ clusters in ZSM-5 are correlated with the configurations of O–M–C atoms;237 a smaller ∠OMC angle on the adsorbed CH4 on MO+ in ZSM-5 in terms of a large distortion of the molecular geometry of the adsorbed CH4 corresponds to a lower binding energy of CH4 in terms of a larger confinement effect. In other words, the limited extension of the molecular orbital of CH4 in the sub-nanometer porous space forces CH4 to adopt a bent configuration, which is at an unstable state in terms of having higher energy than that in open space.
A very favorable consequence of the confinement effect of the micropore of zeolite to the CH4 molecule is the low activation barrier for cleaving the C–H bond of CH4. The computational studies suggest that the activation barrier for C–H of CH4 on a confined CuO+ in ZSM-5 is only 6.4 kcal mol−1,261 which is much lower than 26.4 kcal mol−1 on bare CuO+.237 This consequence is also supported from the comparison of the activation of C–H of CH4 on the open surface of CuO. The activation barrier for cleaving the first C–H of CH4 in CuO+ confined in ZSM-5, which is 6.4 kcal mol−1, is obviously lower than the activation barrier of 11.3 kcal mol−1 for cleaving the first C–H of CH4 on the open surface of CuO(110), where the C atom of CH4 in the transition state binds to the cationic Cu atom of the CuO(110) surface.262
Other than the distortion of the binding configuration of a reactant molecule and the decrease in the activation barrier of the molecule confined in the zeolite, another significant impact from the confinement of the micropores of the zeolite is the largely increased repulsion between the reactant molecule and the O and Si atoms of the sub-nanometer pore. Thus, these consequences of the confinement effect of the zeolite to the molecules adsorbed on MO+ encapsulated in ZSM-5 suggest the significance of anchoring the MO+ clusters in the micropores for a facile activation of CH4 under mild conditions.
9.2 Computational studies for the transformation of CH4 to CH3OH by MO+ in ZSM-5
Transformation of CH4 to CH3OH on free-standing first-row transition metal oxide ions (MO+) in the gas phase was investigated with the computational approach (M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, and Cu).237 Although no experimental studies for the activity of oxidation of CH4 on MO+ anchored on ZSM-5 were performed, Yoshizawa et al. performed parallel computational studies of MO+@ZSM-5 (M = Fe, Co, Ni, Cu) for the investigation of the influences of different metals on the oxidation of CH4 to CH3OH.261 They proposed that this transformation is performed through three intermediates including the reactant complex, hydroxyl, and the product complex and two transition states (Fig. 13a). The energy profiles of the four “catalysts” are very different (Fig. 13b). The binding energy of the reactant complex in terms of adsorbed CH4 on CuO+@ZSM-5 is the highest among the four analogous “catalysts”, MO+@ZSM-5 (M = Fe, Co, Ni, Cu). This high binding energy originates at the significant interaction between the low-lying 3σ orbitals of CuO+ and the HOMO of the CH4 fragment.237,261Fig. 13a illustrates the catalytic cycle for the catalytic oxidation of CH4 with N2O to form CH3OH and N2. The activation barriers of the two transition states on CuO+@ZSM-5 are the lowest compared to the other three catalysts (Fig. 13b). In terms of TS1, the lowest activation barrier on CuO+@ZSM-5 among MO+@ZSM-5 (M = Fe, Co, Ni, Cu) results from the shortest O–C distance and the smallest ∠OMC in the configuration of the transition state among the four catalysts (Fig. 13c). Regarding the TS2, the lowest activation energy to cross TS2 on CuO+@ZSM-5 can be rationalized with the shortest M–C bond (2.50 Å for Fe catalyst, 2.30 Å for Co catalyst, 2.13 Å for Ni catalyst, 2.08 Å for Cu catalyst) (Fig. 13d).As pointed out by Shiota et al., the number of d-electrons and the energy of the d-block orbitals are two dominant factors affecting the barrier of C–H activation of CH4 on free-standing MO+.237 In terms of the activation of C–H of CH4 on anchored MO+ in ZSM-5, we predict that the number of d-electrons and the energy of the d-block orbitals are the descriptors playing the controlling roles in governing the barrier of C–H activation of CH4. Thus, here, a computational study is suggested to investigate whether the two descriptors of free-standing MO+ reported in the literature237 could be applied to MO+ anchored on ZSM-5. In addition, such a proposed computational study can help to elucidate whether the confinement effect of ZSM-5 on MO+ could dominate the descriptors of C–H activation of CH4 on MO+ (M = Fe, Co, Ni, Cu) anchored in ZSM-5 on comparing the descriptors of anchored MO+ in ZSM-5 to those of free-standing MO+.
9.3 Anchored bis(μ-oxo)diiron in ZSM-5 for the activation of CH4
The early studies reported the oxidation of CH4 at a high temperature by the anchored oxygen atoms on Fe-ZSM-5, which was prepared through the pre-treatment of the precursor in N2O; the oxidation of CH4 by Fe-ZSM-5 formed different products.263–268 Among these studies, CH3OH was produced from the oxidation of CH4 at the room temperature by the anchored oxygen atoms on FeIII in Fe-ZSM-5.267,268 Notably, the formed CH3OH was strongly adsorbed in the micropores of ZSM-5. The strong adsorption results from the van de Waals forces, particularly the hydrogen bonding of CH3OH in the micropores enhanced by the confinement effect of the aluminosilicate pores. In general, the formed CH3OH is extracted from the micropores with a mixture of acetonitrile and water269 or through water steam.60 A creative method developed by van Bokhoven is that CH3OH formed in Cu-MOR can be extracted in an on-line mode in steam.270The activity of the activated Fe-ZSM-5 originates at the oxygen atom bonded to FeIII, which is called the α-oxygen in the literature.264–266,271–273 The (FeII)α complex, sometimes called the α-site, can be formed through the calcination of Fe-ZSM-5 at a temperature higher than 500 °C in O2. Based on the literature,274 the FeIII of Fe-ZSM-5 evolves to FeII at 500 °C along with the release of O2. Some FeII atoms were stabilized in the zeolite matrix,275 making the oxidation of this species thermodynamically unfavorable even at a temperature higher than 700 °C.276 Thus, an appropriate activation temperature in O2 is key for gaining reactivity in the oxidation of CH4 to CH3OH. In addition, active Fe-ZSM-5 with (FeIII–O˙)α sites can be prepared through the oxidation of the precursor in terms of Fe-loaded ZSM-5 in N2O in the range of 200–250 °C since Oα desorbs in the form of molecular O2 at a temperature higher than 300 °C. Based on the measured stoichiometric ratio of Oα to Fe never exceeding 1:2,277 driven by the coincidence of Mössbauer spectroscopy of Fe in sMMO and Fe in ZSM-5,268 inspired by the modelling of oxygen activation on CH4 monooxygenase278 and supported by the computational studies of Fe-ZSM-5,277 Panov et al. suggested that Fe atoms in Fe-ZSM-5 could exist in the form of a binuclear complex, bis(μ-oxo) in which two Fe atoms are linked through bis(μ-oxo) although there was a lack of EXAFS data to support this.277 The first successful construction of the structural model of the active site of Fe-ZSM-5 in terms of the oxo-bridged di-iron complex can be tracked to an early study in 1993.273 The analysis of the electronic structure of the oxo-bridged di-iron complex shows that the lowest unoccupied molecular orbital (LUMO) has a very low eigenvalue (−0.79 eV), suggesting that this complex is a strong oxidant. This low-lying LUMO is mainly contributed from the d-orbitals of one Fe atom and has admixtures of the orbital centered on adjacent oxygen atoms.277 Thus, the Fe atom in the oxo-bridged di-iron complex efficiently acted as the actual oxidizing site at the atomic scale.
Yoshizawa et al. simulated the reaction mechanism of CH4 hydroxylation on the α-oxygen of a simplified structure model (Fig. 14a).279 This study suggests that the coordinatively unsaturated iron–oxo is the species activating C–H, which is consistent with the analysis of the electronic structure of a derivative of bis(μ-oxo)diiron in terms of the contribution of LUMO.277 Based on this computation represented in Fig. 14, a CH4 complex is formed; in this complex, CH4 weakly binds to Fe(III), which coordinates with three oxygen atoms. Upon formation of the CH4 complex, one C–H bond is cleaved through a Fe–C–H–O four-member-ring-like transition state, as shown in Fig. 14b, which is similar to the first intermediate formed on FeO+ in the activation CH4 on free FeO+.237 Upon traversing this first transition state, the hydroxy intermediate is formed; it binds to the FeIII atom (Fig. 14b). Subsequently, the CH3 and OH groups bound to the same FeIII atom couple with each other, forming a three-center transition state (TS2 in Fig. 14b). Upon dissociation of the Fe–O bond, a CH3OH molecule adsorbed on the Fe atom is formed. Fig. 14c is the energy profile of direct CH4 hydroxylation on Fe–Oα in ZSM-5 (Fig. 14c),279 which is very similar to that of the gas phase reaction between CH4 and free FeO+.280,281 Notably, the reaction mechanism of formation of CH3OH through the direct oxidation of CH4 by the Fe–Oα of ZSM-5279 is distinctly different from the radical mechanisms in the catalytic oxidation of CH4 by H2O2 on di-iron species anchored in ZSM-5, which has been reviewed in Section 13.
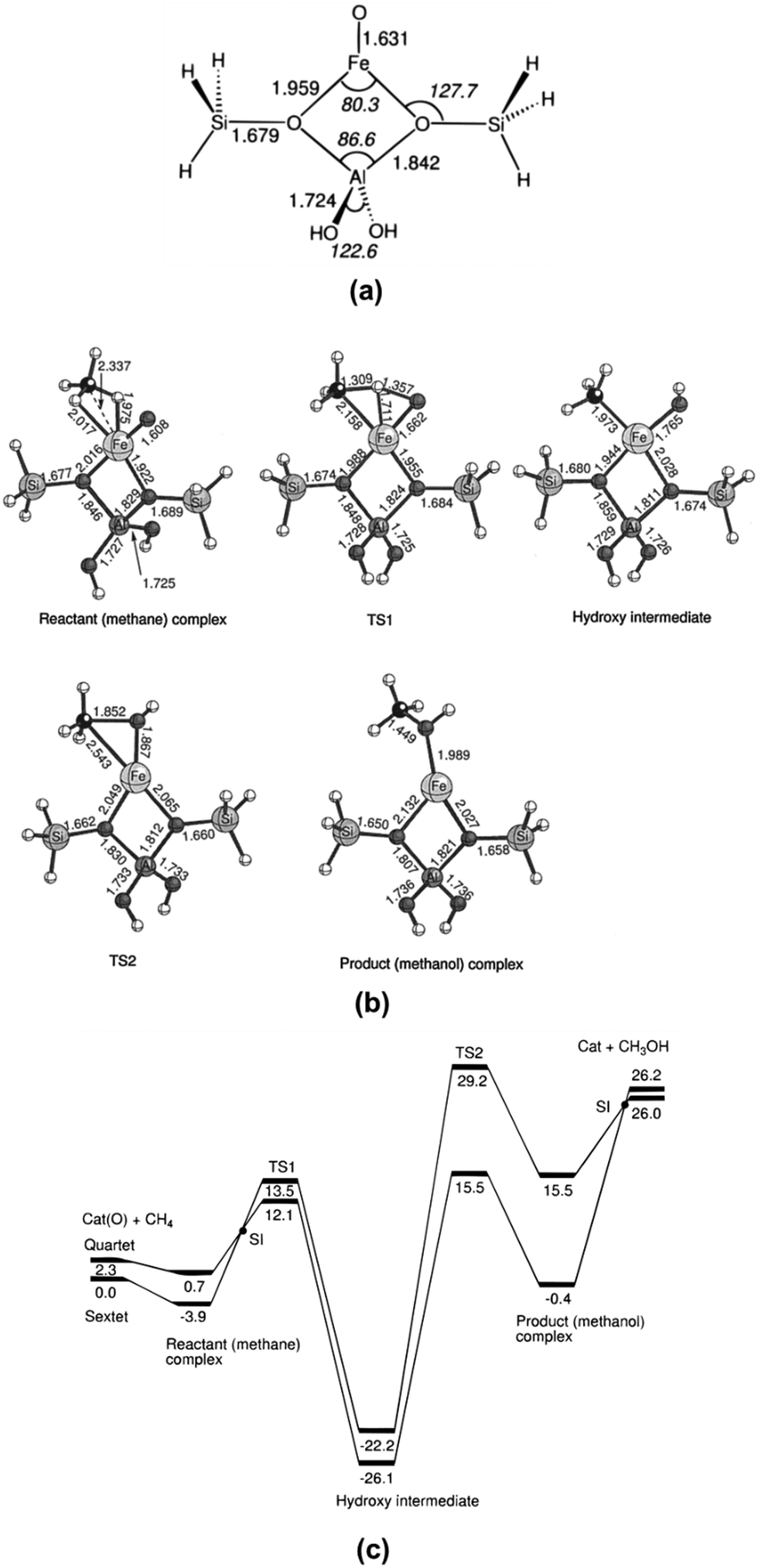 | ||
| Fig. 14 Representation of the results of the computational studies of the mechanism for the oxidation of CH4 with the α-oxygen of Fe-ZSM-5 to form CH3OH.279 (a) Scheme of (Fe–O)α. (b) Optimized intermediates and transition state for the reaction of CH4 with α-oxygen to form the CH3OH molecule. (c) Energy profile for direct CH4 hydroxylation on the structural model (a) of α-oxygen of Fe-ZSM-5 in the sextet and quartet states. Reproduced from ref. 279, copyright 2000, with permission from ACS. | ||
9.4 Anchored oxygen over the Cu sites for the activation of CH4
700 cm−1 in the UV-vis spectrum.269 This assignment was supported by the correlation between the oxidation of more CH4 and the simultaneous decay of the intensity at 22700 cm−1, as clearly shown in Fig. 15 (left and right panel). The formation of this bis(μ-oxo)dicopper species was supported by other characterizations including EXAFS, TEM, and EPR.286 More importantly, the time-dependent attenuation of the intensity of the peak at 22700 cm−1 during reaction with CH4 clearly shows that the Cu complex is the site for the oxidation of CH4. With the extraction method using a solution of 1:1 water/acetonitrile, 82 μmol of CH3OH was generated from 1 g of Cu-ZSM-5 with Si/Al = 12 and Cu/Al = 0.58 (CZ-12-0.58).269
 | ||
| Fig. 15 Representation of fiber-optic UV-vis spectra of O2-activated CZ-12-0.58 during the reaction with CH4 (5% in N2; 25 mL min−1) at 448 K (left panel) and at 398 K (right panel). Reproduced from ref. 269, copyright 2005, with permission from ACS. | ||
The selectivity for producing CH3OH on CZ-12-0.58 is as high as 98%. Unlike the low selectivity for producing CH3OH in the catalytic oxidation of CH4 with H2O2, the high selectivity for producing CH3OH by non-catalytic oxidation benefits from the lack of excessive oxidant since only these oxygen atoms anchored to the metal atoms in the zeolite act as the oxidant for the oxidation of CH4. Similar to Fe-ZSM-5,267,268 the formation of CH3OH through the oxidation of CH4 by Cu-ZSM-5 is not a catalytic process as Cu-ZSM-5 was not exposed to CH4 and an oxidant, and CH3OH was not produced continuously. Section 13 extensively discusses the catalytic oxidation of CH4 to form CH3OH using H2O2 as an oxidant under mild conditions.
The chemical state and coordination environments of Cu atoms in the micropores of Cu-ZSM-5 is quite complicated. Many endeavors have been made in the elucidation of the structure of the Cu-based active sites at an atomic scale. It is suggested that the reactivity of Cu-ZSM-5 for the oxidation of CH4 to form CH3OH mainly stems from the anchored binuclear oxygen-containing the Cu complex, [Cu(μ-O)Cu]2+.287–291 However, early studies proposed that the active site could be bis(μ-oxo)dicopper, [Cu(μ-O)2Cu]2+, which is an analogue of a catalytic site of pMMO hypothesized early without approval.292–297 These ambiguous assignments probably resulted from the challenges in experimentally distinguishing bis(μ-oxo)dicopper, [Cu(μ-O)2Cu]2+, and the binuclear species, [Cu(μ-O)Cu]2+. In fact, the analysis of the lobes of the HOMO of [Cu(μ-O)2Cu]2+ and [Cu(μ-O)Cu]2+anchored in 10-membered rings suggests that (1) both of them are in a hybridization of μ-O atom(s), two Cu atoms, and two O atoms between Cu and Al, and (2) both of them have a complicated d-type bonding.298 In addition, bis(μ-oxo)dicopper, [Cu(μ-O)2Cu]2+ and the binuclear species, [Cu(μ-O)Cu]2+ exhibit very similar stability. Despite these early studies, the recent resonance Raman spectroscopy studies of Cu-ZSM-5 suggested that the binuclear species [Cu(μ-O)Cu]2+ plays a significant role in the oxidation of CH4 to form CH3OH.299
Inspired by the importance of multi-nuclear Cu species (Cu3O4) (1 in Fig. 6b) in the oxidation of CH4 in pMMO218,300–302 and the significant heterogeneity of the extra-framework species formed in the cation-exchanged microporous aluminosilicate, Pidko et al. proposed that multi-nuclear Cu complexes are active sites for the oxidation of CH4 to form CH3OH in their computational study combining periodic DFT calculations and ab initio thermodynamic analysis.303 This computational study shows that the formation of the Cu complexes depends on the conditions of activation of Cu-ZSM-5 in terms of the temperature and gas phase composition used for activation, particularly the partial pressure of O2.303 As shown in Fig. 16a, a low partial pressure of O2 is favorable for the formation of binuclear [Cu(μ-O)Cu]2+ species and the trinuclear Cu complex [Cu3(μ-O)3]2+ can be formed at a relatively high oxygen pressure. Fig. 16b suggests that [Cu3(μ-O)3]2+ exhibits a higher stability than mono-nuclear [Cu(μ-O)]2+ and binuclear [Cu(μ-O)Cu]2+ from the thermodynamic point of view. In addition, the calculated Bader atomic charges of binuclear [Cu(μ-O)Cu]2+ shows the nature of an anion radical of μ-O (−0.77 e). Similarly, the Bader charge on the O atom of [Cu3(μ-O)3]2+ is about −0.70e to −0.73e, exhibiting the nature of an anion radical.303
 | ||
Fig. 16 Representation of the ab initio thermodynamic analysis of the Cu complexes formed at different conditions. (a) 2D projection of the lowest Gibbs free energy CuxOmHn species in ZSM-5 (DG (eV/U.C.)), 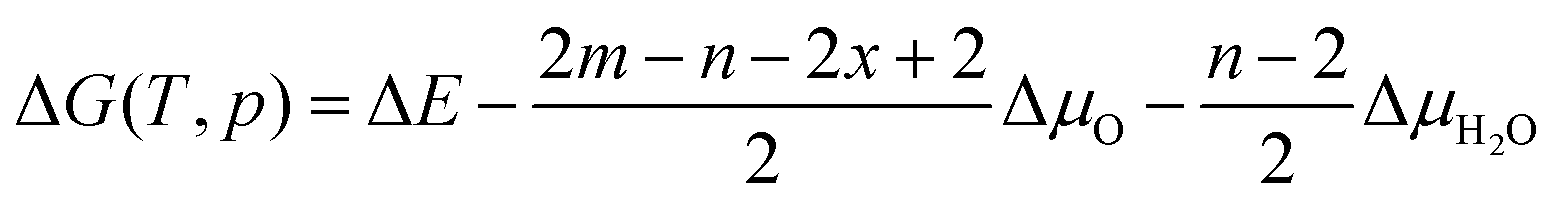 as a function of oxygen (ΔμO) and water ΔμH2O chemical potentials. (b) A cross section of the 3D phase diagram at a fixed ΔμH2O (indicated with a dashed line in (a)), corresponding to 10−2 mbar H2O at 700 K. (c) Reaction path and energy profile for the selective oxidation of CH4 with binuclear [Cu(μ-O)Cu]2+ to form CH3OH. (d) Reaction path and energy profile for the selective oxidation of CH4 with trinuclear [Cu3(μ-O)3]2+ to form CH3OH.299,303 Reproduced from ref. 303, copyright 2016, with permission from Elsevier. as a function of oxygen (ΔμO) and water ΔμH2O chemical potentials. (b) A cross section of the 3D phase diagram at a fixed ΔμH2O (indicated with a dashed line in (a)), corresponding to 10−2 mbar H2O at 700 K. (c) Reaction path and energy profile for the selective oxidation of CH4 with binuclear [Cu(μ-O)Cu]2+ to form CH3OH. (d) Reaction path and energy profile for the selective oxidation of CH4 with trinuclear [Cu3(μ-O)3]2+ to form CH3OH.299,303 Reproduced from ref. 303, copyright 2016, with permission from Elsevier. | ||
The activations of C–H by [Cu(μ-O)Cu]2+ and [Cu3(μ-O)3]2+ were simulated with DFT approaches. These computational studies suggested that the activation of C–H of CH4 on [Cu(μ-O)Cu]2+ is performed through homolytic C–H bond dissociation, followed by direct radical rebound.299 The dissociated H atom binds to μ-O, forming an OH group bridging the two Cu atoms. The activation energy for the homolytic C–H dissociation step is 68–78 kJ mol−1.299,303 Alternatively, heterolytic dissociation results in H and CH3 bond to Cu and μ-O, respectively, forming [CH3–Cu(μ-OH)Cu]2+. Other than the two paths, a more thermodynamically favorable path is homolytic cleavage on one lattice oxygen atoms of the zeolite neighboring to the Cu complex (Fig. 16c), forming a framework-anchored CH3O group.299,303 Unfortunately, the formation of CH3OH from this framework-anchored CH3O group is unfavorable since the transfer of the OH group to CH3 in this case is significantly endothermic. For the radical rebound route, as shown in Fig. 16c, about 156 kJ mol−1 energy is needed to desorb CH3OH adsorbed on the two Cu atoms through the O atoms of the CH3OH molecule. Unquestionably, this is due to the strong chemisorption of CH3OH, which is unfortunately favorable for further oxidation to form formaldehyde, formic acid, or even CO2.
In terms of [Cu3(μ-O)3]2+, direct rebound, absorption, and heterolytic routes on trinuclear [Cu3(μ-O)3]2+ were investigated for the oxidation of CH4 to form CH3OH. Compared to the strong binding of CH3OH to Cu atoms of [Cu(μ-O)Cu]2+ (156 kJ mol−1 in Fig. 16c), the binding energy of CH3OH on trinuclear [Cu3(μ-O)3]2+ in the rebound route and heterolytic routes is 86 kJ mol−1 (Fig. 16d). It suggests that the oxidation of CH4 with [Cu3(μ-O)3]2+ is thermodynamically favorable. Kinetically, on the basis of computational studies of Li et al.,303 the activation barrier for C–H cleavage by trinuclear [Cu3(μ-O)3]2+ of about 10 kJ mol−1 is much lower than that of binuclear [Cu(μ-O)Cu]2+, which is 64–68 kJ mol−1.303 These DFT studies299,303 predicted that [Cu3(μ-O)3]2+ formed in ZSM-5 is an active site for the selective oxidation of CH4 to form CH3OH, although exclusive experimental evidences supporting this prediction have not been reported.303 The reactivity of tri-nuclear [Cu3(μ-O)3]2+ anchored in MOR reported by Grundner et al.304 supported the suggestion made by Pidko et al. that [Cu3(μ-O)3]2+ in ZSM-5 is the active site for the oxidation of CH4 to CH3OH.303
The inconsistency between the structure suggested by the experimentalists and that proposed by theoreticians very likely results from (1) the heterogeneity of the sites of the sample prepared experimentally, (2) the lack of information on the authentic sites due to the limited access of current characterization techniques, (3) difficulty in differentiating [Cu(μ-O)Cu]2+ and [Cu3(μ-O)3]2+. Further efforts from the experimentalists are necessary for elucidating the reaction mechanism of oxidation of CH4 to CH3OH on Cu@ZSM-5 under mild conditions.
Inspired by the reported activity for the oxidation of CH4 to CH3OH on Cu-ZSM-5,269 the test of activity for the oxidation of CH4 on Cu-MOR was done by Schoonheydt et al. in 2005,293 although the elucidation of the chemical and coordination environments of Cu atoms of Cu-MOR was quite challenging. Cu-MOR is active for the oxidation of CH4 to form CH3OH. The yields of CH3OH are in the range of 1–7 μmol g−1 of Cu-MOR.293 In this early study, Cu-MOR was activated with O2 and then CH4 was introduced to react with the activated Cu-MOR. The formed CH3OH was extracted. The close correlation between the amount of CH3OH produced and the intensity of the UV-vis peak at 22700 cm−1 was established, suggesting that the activity of Cu-MOR originates at the species contributing to this peak.293
The peak at 22700 cm−1 observed in the UV-vis spectrum results from the transition of the Obridge → Cu charge transfer.293 A few CuxOy2+ complexes including mono (μ-oxo) dicopper species, bi(μ-oxo) dicopper species, or even tri(μ-oxo)dicopper species could contribute to the 22700 cm−1 band. In fact, a later study using resonant Raman spectroscopy and other approaches propounded that the active Cu-based species is a bent mono(μ-oxo)dicopper complex.299 As the intensity of this peak decays rapidly along with the reduction by CH4, a Cu-based species containing a Cu–O–Cu bridge structure must be the active oxidant for transforming CH4 to CH3OH. This deduction was further supported by the reinstallation of this active bridge structure through the calcination of a used Cu-MOR in O2 at 500 °C.
Although UV-vis and resonant Raman spectroscopy could not provide the atomic-scale structure of the Cu-based sites, CuxOy2+ in MOR, extensive XAS studies on the evolution of the chemical and coordination environments of Cu in the MOR have provided significant insights toward understanding the active sites for the partial oxidation of CH4 to CH3OH.296,308 Smeets et al. suggested that the activation of Cu-exchanged MOR in O2 at 450 °C results in the formation of the (μ-η2:η2-peroxo)dicopper species, which is interchangeable to mono(μ-oxo)dicopper with the involvement of two electrons provided by the Cu(I) species.296Fig. 17a schematically shows the evolution of the chemical and coordination environments of the Cu atoms of the Cu-exchanged MOR along with the increase in the activation temperature in O2.296,308 The in situ observation of the structural parameters of the Cu coordination environment derived from the features of the energy space spectra and the r-space spectra of the Cu K-edge as a function of the activation temperature in 30–450 °C in O2 or helium was plotted in Fig. 16e and 17b. One important experimental finding in the r-space spectra is the contribution of an additional fractional oxygen. The coordination numbers of the fractional O to Cu and O–Cu distance are 0.3 and 2.42 Å, respectively. The additional fraction oxygen atom was observed in O2 in the temperature range of 300–450 °C (Fig. 16b and c). Notably, the contribution of this additional fractional oxygen was not observed when the Cu-exchanged MOR was treated at 300–450 °C in 1 bar helium (Fig. 16d and e). These extensive XAS studies suggest that the high-temperature treatment (300–450 °C) of Cu-MOR in O2 is necessary for the formation of the μ-oxo dicopper species although whether the μ-oxo dicopper species could be an active phase of the oxidation of CH4 to form CH3OH was not concluded.
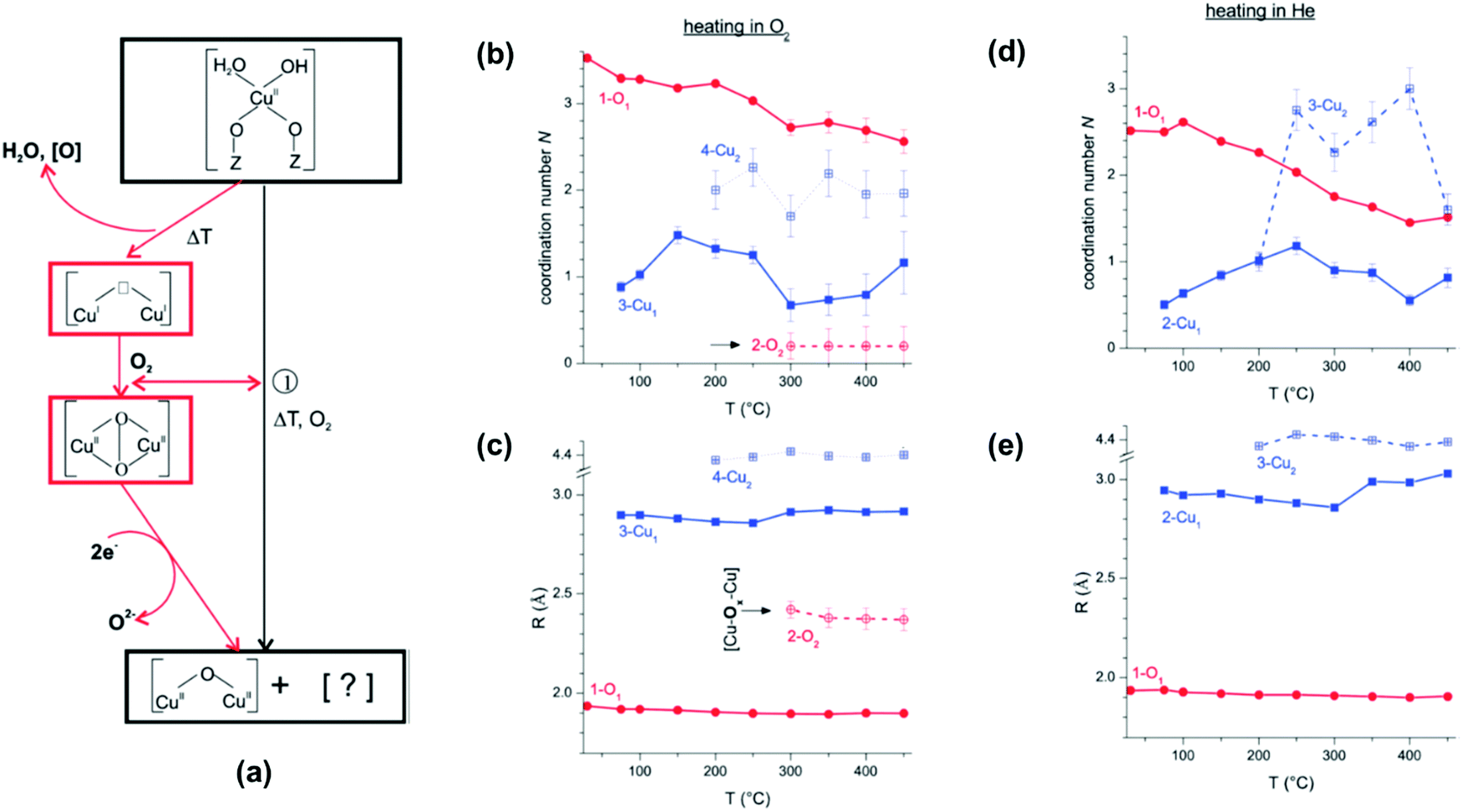 | ||
| Fig. 17 Representation of the schematic showing the changes in the chemical and coordination environments during the activation of Cu-exchanged MOR in O2 at 450 °C and the in situ observation of the coordination environment of Cu during activation in O2 and H2 in the temperature range of 30–450 °C. (a) Schematic of the chemical change. (b) Coordination number of O around Cu during the activation of Cu-exchanged MOR in O2 at different temperatures. (c) Distance between O and Cu during the activation of Cu-exchanged MOR in O2 at different temperatures. (d) Coordination number of O around Cu during the activation of Cu-exchanged MOR in helium at different temperatures. (e) Distance between O and Cu during the activation of Cu-exchanged MOR in helium at different temperatures.296,308 Reproduced from ref. 308, copyright 2015, with permission from Royal Society of Chemistry. | ||
Due to the intricate factors, the atomic details of the sites responsible for the activation and oxidation of CH4 to CH3OH on Cu-MOR were not conclusively identified. A very recent work of Lercher et al. made an exclusive assignment to the CuxOy site responsible for the activity it exhibited.304 A single-site trinuclear copper oxygen cluster Cu3O32− anchored in the pocket channel of MOR was reported as an active site for the oxidation of CH4 to form CH3OH with unprecedented activity by Lercher et al.304 Their preparation of a single type of Cu3O32− was realized by strictly following two requirements. Firstly, the species of Cu precursor must not form Cu(OH)2 in an aqueous solution. Secondly, the anchoring sites should be homogeneous. They chose MOR instead of ZSM-5 to anchor the Cu atoms since the protons of the BAS sites in the more constrained side pockets of MOR can preferentially exchange with the cations of a solution.309 The formation of Cu3O32− in MOR was confirmed with comparative fittings of the k2-weighted Fourier transform EXAFS with potential structural models of binuclear [Cu(μ-O)Cu]2+ and [Cu3(μ-O)3]2+. In fact, the r-space spectrum of the Cu K-edge can be better fit when [Cu3(μ-O)3]2+ was used as the structural model of fitting. This fitting shows that [Cu3(μ-O)3]2+ instead of [Cu(μ-O)Cu]2+ is formed through this specific preparation protocol.304
The fitting of the r-space spectra of the Cu K-edge of the Cu–O clusters anchored in MOR, [Cu3(μ-O)3]2+, suggests that there are two types of oxygen atoms in the second shell of Cu (Fig. 18a): two of them are extra-framework oxygen atoms (OEF) and the other two are framework oxygen atoms (OF). These parameters are quite consistent with the coordination environment of the structure provided by the DFT calculation.304Fig. 18a is the optimized structure of [Cu3(μ-O)3]2+ localized in the side-pocket of MOR. In fact, the two paired type I (AlI) atoms at the pocket mouth shown in yellow anchored the cluster [Cu3(μ-O)3]2+ (Fig. 18b). The formation of Cu–O–Cu through activation in O2 at 450 °C was confirmed by the appearance of the peak at ∼2.4 Å (distance before phase correction) in the red spectrum in Fig. 18c. In the black spectrum in Fig. 18c, the preservation of this peak at 200 °C in CH4 suggests that the product molecule is adsorbed on the cluster after the O atom(s) of Cu3O32+ participated in the formation of CH3OH. Upon desorption of CH3OH through steam treatment at 135 °C (blue spectrum), the disappearance of the peak at ∼2.4 Å suggests the removal of oxygen atoms of [Cu3(μ-O)3]2+. Upon consumption of oxygen atoms bonded to the Cu atoms, [Cu3(μ-O)3]2+ clusters can be regenerated through activation at 500 °C in O2. The catalytic activity of the catalyst recovered after seven tests exhibited the same activity in the production of CH3OH as the original catalyst. Lercher et al. proposed that C–H activation of CH4 on the extra framework [Cu3(μ-O)3]2+ cluster is facilitated through the coupling of the C–H bond with a formally radical-anionic extra-framework oxygen center.304,310 They further suggested that electron transfer from the molecular orbital of Cu3O32+ to the antibonding C–H orbital of CH4 weakens the C–H bond of CH4, thus leading to C–H cleavage under mild conditions.304,310
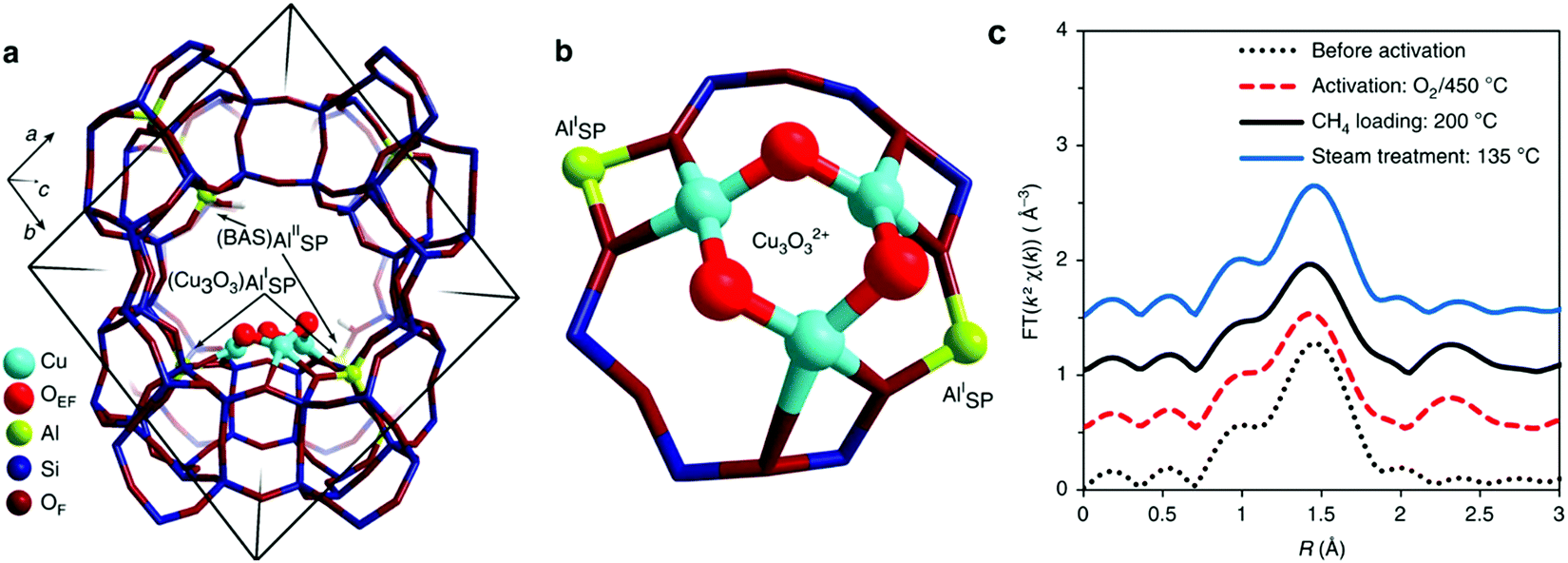 | ||
| Fig. 18 Representation of the optimized structure of [Cu3(μ-O)3]2+ in MOR and in situ EXAFS of the Cu K-edge of the catalyst precursor during pre-treatment and catalyst during catalysis.304 (a) MOR encapsulated with a [Cu3(μ-O)3]2+ cluster; two types of Al atoms are marked; type I (AlI) is paired Al atoms, which anchor [Cu3(μ-O)3]2+; type II (AlII) is the isolated Al atoms. (b) [Cu3(μ-O)3]2+ stabilized by two anionic centers formed on two AlI lattice sites at the entrance of the side pocket of MOR. (c) r-Space spectra of the Cu K-edge of the catalyst precursor during activation and catalysis. Reproduced from ref. 304, copyright 2015, with permission from Springer Nature. | ||
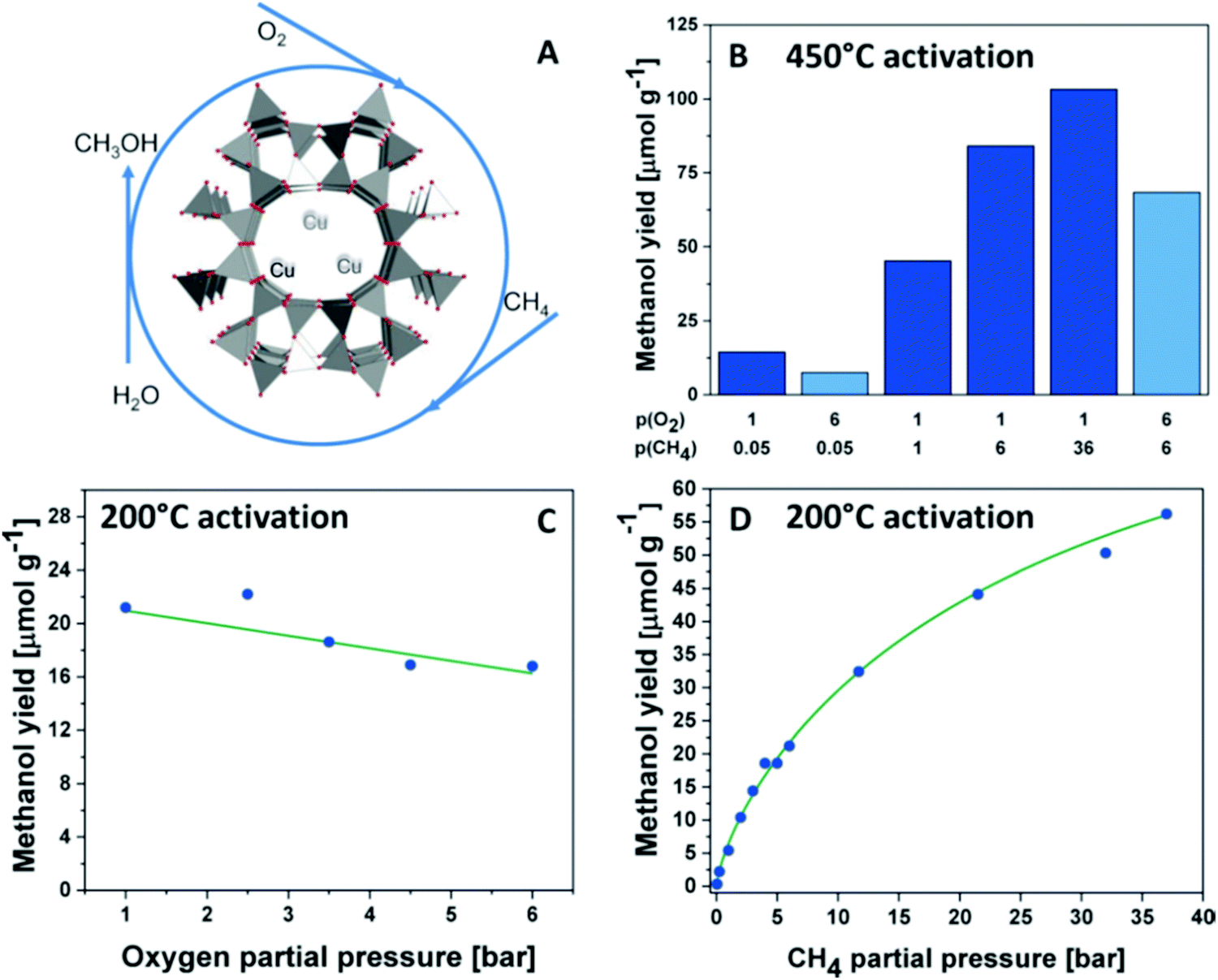 | ||
| Fig. 19 Representation of the schematic of isothermal approaches and the influence of the partial pressures of O2 and CH4 on the yields of CH3OH from Cu-MOR (Si/Al = 6, 4.7 wt% Cu).311 (A) Schematic showing the main step of the isothermal approach of the continuous production of CH3OH from Cu-MOR without the off-line extraction of CH3OH. (B) Yields of CH3OH after activation at 450 °C, reaction and off-line extraction as a function of the partial pressures of O2 and CH4. (C) Influence of the partial pressures of O2 used in the activation at 200 °C on the yield of CH3OH. (D) Influence of the partial pressure of CH4 used in the reaction on the yield of CH3OH (activation was done at 1 bar O2 and 200 °C). Reproduced from ref. 311, copyright 2016, with permission from John Wiley and Sons. | ||
An intriguing finding of this study311 is the exclusion of the mono-μ-oxo dicopper complex as the active site of the Cu-MOR in the oxidation of CH4 to form CH3OH because there is lack of the signature of the mono-μ-oxo dicopper complex (22700 cm−1) in the UV-vis spectrum after the activation of Cu-MOR before using it for oxidizing CH4 to CH3OH.311 Previous studies confirmed that the mono μ-oxo dicopper complex was formed from either the activation of Cu-exchanged ZSM-5 in N2O at a temperature higher than 350 °C or the activation of the Cu-exchanged MOR in O2 at a temperature higher than 350 °C,269,270,293,312,313 although it has been continuously debated whether μ-oxo dicopper complex(es) could be the active site for the oxidation of CH4 to CH3OH. The observation of evolution of the chemical and coordination environments of the Cu atoms using in situ EXAFS suggests that the mono μ-oxo dicopper complex species are formed from Cu-exchanged MOR in O2 at 300 °C or higher.308 To check whether the mono-μ-oxo dicopper complex is a necessary site for oxidizing CH4 to CH3OH, the Cu-exchanged MOR was activated at 200 °C in O2 before being used for the oxidation of CH4 to CH3OH.311 Clearly, this Cu-exchanged MOR did not have any mono-μ-oxo dicopper complex since it was activated at a temperature lower than 300 °C. However, notably, this specific Cu-exchanged MOR, which does not have the mono-μ-oxo dicopper complex, in fact exhibited high activity in the oxidative transformation of CH4 to CH3OH. Thus, these studies concluded that the mono-μ-oxo species is not a necessary site for the activity of Cu-MOR in the isothermal approach.311 Actually, control experiments designed by van Bokhonven et al. suggested that the active sites for the oxidation of CH4 to CH3OH in the isothermal approach are dehydrated copper oxide clusters, which are less active at a low pressure of CH4 but quite active at a high pressure of CH4.311
The conclusion of the active sites of oxidizing CH4 to CH3OH by Cu-MOR at 200 °C suggests the high complexity of the active sites of Cu-MOR in the oxidation of CH4 to CH3OH. The complication results from multiple interacting factors determining the reactivity in the activation and oxidation of CH4 to CH3OH. These factors are closely related to the preparation step and temperature of activation of Cu-MOR.
Other than the size-dependent stability of the CunOn2+ clusters, a size-dependent reactivity in the activation of C–H of CH4 was suggested in the computational studies. The formation of a Cu–O–H and CH3 species is the first step in the oxidative transformation of CH4 to CH3OH. For a mono(μ-oxo)dicopper cluster in MOR and ZSM-5, the free total energies of the formed Cu-O-H and CH3 species are higher than the reactants by 76 kJ mol−1 for MOR and 61 kJ mol−1 for ZSM-5, respectively.303,315 Interestingly, in terms of a trimer cluster [Cu3(μ-O)3]2+ in ZSM-5, the formation of the Cu–O–H and CH3 species only results in an increase in the free energy by 37 kJ mol−1. In other words, the Cu–O–H and CH3 species formed on [Cu3(μ-O)3]2+ in ZSM-5 are more stable than that on the mono(μ-oxo)dicopper cluster [Cu(μ-O)Cu]2+ in ZSM-5.298 Similarly, the cost of free energy for the formation of Cu–O–H and CH3 species on the dimer (Cu2O22+), trimer (Cu3O32+), tetramer (Cu4O42+), and pentamer (Cu5O52+) in MOR are 11.6 kJ mol−1, 41.4 kJ mol−1, 34.7 kJ mol−1, and 28.9 kJ mol−1, respectively;298 it suggests that the increase in the reactivity of the CunOn2+ clusters along with the increase in the size. In other words, the higher stabilization of a larger cluster make it better stabilize the formed HO and CH3 upon C–H activation. The predicted higher activity of a larger cluster CunOn2+ suggests the origin of the activity of Cu-MOR and even Cu-ZSM-5 in the activation of C–H of CH4 could be more complicated than that hypothesized or even deduced in the literature. The complexity was supported by the proposal of Tomkins et al. that the activity of CH4 oxidation to form CH3OH on Cu-exchanged MOR could result from some copper oxide clusters that have not been identified.311 An excellent review published by van Bokhoven et al. summarized these insights and highlighted the complexity of oxidative transformation of CH4 to CH3OH on Cu-ZSM-5 and Cu-MOR.60
000–22700 cm−1 in the UV-vis spectra.
| Sample | Source | Topology | Si/Al | #mL 0.01 M Cu solution per gram | Cu/Al | Cu wt% |
|---|---|---|---|---|---|---|
| a In this case, the molarity of the Cu solution amounted to 0.001 M. b In this case, the molarity of the Cu solution amounted to 0.6 mM. c In this case, the molarity of the Cu solution amounted to 0.4 mM. | ||||||
| CZ-12-0.54 | ALSI-PENTA | MFI | 12 | 63 | 0.54 | 4.3 |
| CZ-25-0.51 | PQ-zeolites | MFI | 25 | 32 | 0.51 | 2.0 |
| CZ-30-0.47 | ExxonMobil | MFI | 30 | 25 | 0.47 | 1.5 |
| CZ-77.5-0.55 | PQ-zeolites | MFI | 77.5 | 97.5 | 0.55 | 0.68 |
| CZ-120-0.88 | PQ-zeolites | MFI | 120 | 6.3 | 0.88 | 0.77 |
| CM-5.3-0.39 | Norton | MOR | 5.3 | 144 | 0.39 | 6.2 |
| CM-8.0-0.50 | TRICAT | MOR | 8.8 | 78 | 0.50 | 5.1 |
| CE-4-0.36 | ExxonMobil | EMT | 4 | 189 | 0.36 | 7.3 |
| CF-6.2-0.42 | Toyo Soda | FER | 6.2 | 126 | 0.42 | 5.9 |
| CB-9.8-0.50 | ZEOCAT™ | BEA | 9.8 | 100 | 0.50 | 4.7 |
| CY-2.7-0.45 | ZEOCAT™ | FAU | 2.7 | 664 | 0.45 | 11 |
| CU-13.5-0.32 | PQ-zeolites | FAU | 13.5 | 500a | 0.32 | 2.3 |
| CU-14.5-0.42 | ZEOCAT™ | FAU | 14.5 | 500a | 0.42 | 2.9 |
| CU-27.5-0.34 | PQ-zeolites | FAU | 27.5 | 500b | 0.34 | 1.2 |
| CU-36.9-0.34 | PQ-zeolites | FAU | 36.9 | 500c | 0.34 | 0.9 |
Compared to the inactive CS-1-2.0, CY-2.7-0.45, CU-2.75-0.34, and CE-4-0.36 and CA-1-1.7, CF-6.1-0.42, and CB-9.8-0.50 are obviously active in the oxidation of CH4 to CH3OH. Yields of CH3OH of 12 μmol g−1 and 4.2 μmol g−1 were obtained from CF-6.1-0.42 and CB-9.8-0.50 upon their reactions with CH4 at 200 °C, respectively. Surprisingly, no band in the region of 22000–22700 cm−1 was observed for the Cu-exchanged CF-6.1-0.42 and CB-9.8-0.50. Thus, the observation of high yields from CF-6.1-0.42 and CB-9.8-0.50 and the absence of a peak in the region of 22000–22700 cm−1 in the UV-vis spectra clearly show that both CF-6.1-0.42 and CB-9.8-0.50 must have other types of active sites, which can provide oxygen atoms to partially oxidize CH4 to form CH3OH.293 This finding has urged the further exploration of the chemical and coordination environments of Cu atoms in Cu-based catalytic sites anchored in CF- and CB-type zeolites.
9.5 Anchored oxygen atoms over Co sites for the activation of CH4
Other than zeolite loaded with Fe or Cu, the O2-activated Co-ZSM-5 was reported to be reactive in the oxidative transformation of CH4 to CH3OH at 150 °C.317 Co-ZSM-5 was prepared through impregnation or ion exchange. Co-ZSM-5 exhibits activity for oxidizing CH4 to form CH3OH and formaldehyde although it is much lower than that of Cu-ZSM-5. The yield of CH3OH on Co-ZSM-5 is in the range of 0.1–0.6 μmol per gram of the catalysts.316, 317Although the chemical and coordination environments of Co atoms in Co-ZSM-5 have not been characterized thoroughly, it is extrapolated that the Co atoms in Co-ZSM-5 prepared with impregnation mainly exist in the form of CoO or Co3O4 and cobalt oxide clusters are responsible for the activity in the production of CH3OH through the oxidation of CH4.317,318 It even further propounded that the activity in the oxidation of CH4 to form CH3OH on Co-ZSM-5 does not originate at the μ-oxo dicobalt species. More studies on this system are necessary for uncovering the origin of the activity on Co-ZSM-5. In addition, it would be valuable if the structure and activity in the oxidative transformation of CH4 to CH3OH on Co-MOR and even other zeolites loaded with Co can be studied in a comparative manner toward gaining a fundamental understanding of the correlation between the chemical and coordination environment of Co atoms in different zeolites and their corresponding activity in the activation and oxidation of CH4 to CH3OH.9.6 Anchored mono(μ-oxo)dinickel site for the activation of CH4
As reviewed in Sections 6–8, Ni atoms in the form of free-standing NiO+ cations and anchored Ni–O species on the support oxide are quite active in the oxidative transformation of CH4 to CH3OH. Consistent with them, Ni-based mono(μ-oxo)dinickel species anchored in ZSM-5 exhibits high reactivity in the oxidation of CH4 to CH3OH under mild conditions.Microporous aluminosilicate consisting of bent mono(μ-oxo)dinickel species on the internal wall was prepared through first impregnation and then filtration.319 In the filtration step, the sample precursor obtained upon impregnation was washed with distilled water for several cycles. Fig. 20b is 2.5 wt% Ni-ZSM-5 catalyst, which does not have NiO nanoparticles formed on the external surface of microporous aluminosilicate particles. The TEM images excluded the formation of NiO nanoparticles on microporous aluminosilicate. The lack of NiO nanoparticles on the surface of microporous aluminosilicate particles is further supported by the lack of satellite peaks of Ni 2p3/2, which are 3d10L−2 and 3d9LLfar−1 (Fig. 20c). The absence of satellite peaks must be related to an unusual bonding environment of the Ni atom anchored to ZSM-5 [mono(μ-oxo)dinickel in Fig. 20d]. It is widely acknowledged that the satellite peaks of Ni 2p3/2 of Ni2+ of NiO originate from the charge-transfer effect during photoionization.320 After the photoionization of the electrons of Ni 2p, the lowest final state is 3d9L−1, which is formed through electron transfer from ligand L to the Ni 3d orbital.320 Here, L−1 in 3d9L−1 denotes a hole generated on the nearest ligand due to the transfer of one electron from the ligand to the 3d orbital of the Ni atom. This 3d9L−1 electronic state gives the main photoemission peak at 853.8 eV (Fig. 20c). In addition, two electrons of the nearest oxygen atoms could be transferred to the photoionized Ni atom, creating another electronic state (d10L−2), which gives a satellite peak at 861.0 eV for NiO particles. In addition, the photoionized Ni atom can be neutralized by transferring an electron from one of the oxygen atoms of the second-nearest Ni atoms, Ni*–O(L1)–Ni(L2)–O(Lfar)–Ni(L4)–O(L5), forming a different final state labelled as 3d9LLfar−1, which corresponds to the strong satellite peak at 856.0 eV of the NiO particle (Fig. 20c). The absence of 3d9LLfar−1 at 856.0 eV and 3d10L−2 at 861.0 eV in the Ni 2p photoemission feature of 10 wt% Ni-ZSM-5 in Fig. 20c clearly shows the lack of long-range crystal structure of NiO. Thus, XPS studies clearly show the lack of a lattice of NiO nanoparticles in the 2.5 wt% Ni-ZSM-5. The XPS peak intensity in Fig. 20c was contributed from the singly dispersed Ni atoms anchored in these micropores near the surface of a ZSM-5 particle, although these Ni atoms singly dispersed deep in the pores of ZSM-5 could not be observed by XPS.
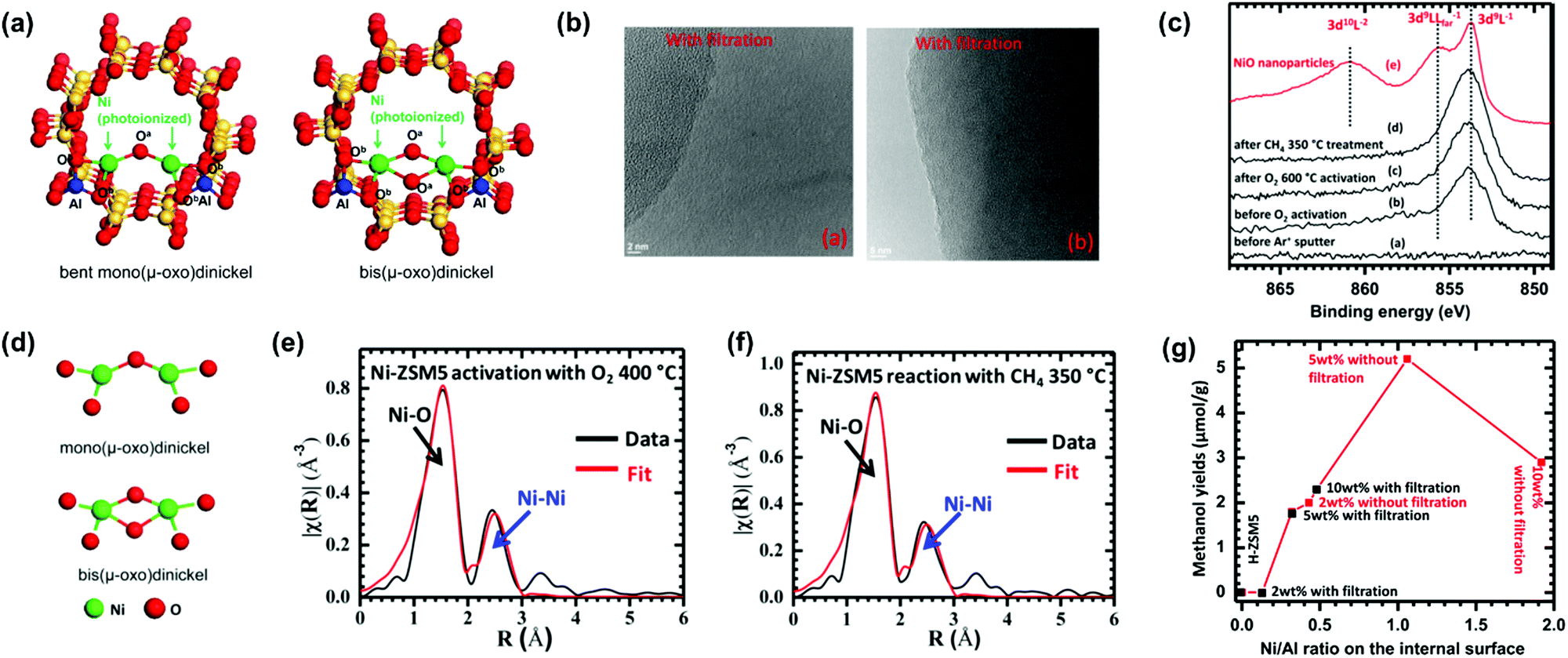 | ||
| Fig. 20 Representation of the structure and characterizations of the Ni-ZSM5 catalysts consisting of the bent mono(μ-oxo) dinickel.319 (a) Structural mode [a1: bent mono(μ-oxo)dinickel; a2: bis(μ-oxo)dinickel], (b) TEM images, (c) Ni 2p photoemission feature of 10 wt% Ni-ZSM-5, (d) bonding environment of Ni atoms, (e) Ni K-edge of 5 wt% Ni-ZSM-5 activated with O2 at 400 °C, (f) Ni K-edge of 5 wt% Ni-ZSM-5 reacted with CH4 at 350 °C, (g) Yield of methanol as a function of Ni/Al ratio on the internal surface of Ni/ZSM-5. Reproduced from ref. 319, copyright 2014, with permission from ACS. | ||
The existence of Ni–O–Ni bonds in the micropores was confirmed with the in situ studies of the Ni K-edge of 5 wt% Ni-ZSM5. As shown in Fig. 20e1, the peak contributed from Ni–O–Ni was observed in Ni-ZSM-5, which was activated with O2 at 400 °C. The Ni–O–Ni species was marked with Ni–Ni in Fig. 20e for the convenience of showing the direct distance of Ni to Ni, although there is an oxygen atom bonding with the two Ni atoms. The fitting of the r-space spectrum of the Ni K-edge of Ni-ZSM-5 at 350 °C in CH4 (Fig. 20f) shows that the coordination number of O atoms to Ni is 3.0 ± 0.3. Thus, the in situ EXAFS studies suggest that the obtained structure is a bent mono(μ-oxo)dinickel (Fig. 20e and f).
This Ni-ZSM-5 exhibits activity in the activation and oxidation of CH4 to CH3OH, HCOOH, and HOCH2CH2OH. The activity originated at the bent mono(μ-oxo)dinickel since microporous aluminosilicate without the bent mono(μ-oxo)dinickel is inert for the formation of these product molecules.
9.7 Necessity of parallel studies of different metal atoms anchored in the zeolite
The activation of the C–H of CH4, followed by oxidation to form CH3OH and even products of deep oxidation was studied by several types of catalysts, which are mainly Fe and Cu-anchored in ZSM-5 and MOR. Significant insights on the oxidation of CH4 to CH3OH on this type of catalysts were achieved, although a complexity in the environment of this catalysis and in the structure of the catalyst were suggested. Inspired by the systematic studies of activation and oxidation of CH4 on free-standing MO+ clusters of 3d transition metals, parallel studies of a type of MaOa2+ such as [M2(μ-O)2]2+ of different transition metals such as Mn, Fe, Co, Ni, Cu, and Zn anchored in the same zeolite such as ZSM-5 or MOR could provide valuable insights for uncovering the descriptor for reactivity in the oxidation of CH4 to CH3OH. Another set of parallel studies could be M atoms in [M3(μ-oxo)3]2+ anchored in ZSM-5; here, M is a transition metal such as Mn, Fe, Co, Ni, Cu, and Zn. By replacing ZSM-5 with MOR, the systematic studies of [M2(μ-O)2]2+ anchored in MOR and [M3(μ-oxo)3]2+ anchored in MOR would be valuable. Notably, the complexity in catalyst preparation and challenges in characterization could make these comparisons quite challenging. To save the cost of these explorations, a specific suggestion is to perform parallel computational studies on [M2(μ-O)2]2+ anchored in MOR and [M3(μ-oxo)3]2+ anchored in MOR (= Mn, Fe, Co, Ni, Cu, or Zn) to obtain a list of prioritized metals before any experimental explorations are planned. We expect that these sets of proposed parallel studies can provide profound knowledge for developing zeolite-based catalysts for the oxidative transformation of CH4 to CH3OH.10. Activation and oxidation of CH4 with anchored oxygen atoms bonding with Cu in zeolite to form acetic acid
Acetic acid is an important intermediate compound for the production of high-value chemicals. It is produced from the catalytic carboxylation of CH3OH at an industrial scale. CH3OH is produced from high-temperature processes of syngas production from CH4 or coal and then the synthesis of CH3OH from syngas or a mixture of CO, CO2, and H2, as shown in Fig. 2. It was reported recently that Cu–H-MOR is reactive for the formation of acetic acid through tandem reactions in terms of first the formation of a methoxy species and then coupling between the methoxy species and CO at 200 °C although the reported formation of acetic acid is not a catalytic cycle.321 Correspondingly, the role of Cu–H-MOR in the formation of acetic acid is the oxidant instead of a catalyst. Oxygen atoms anchored on the Cu clusters in Cu–H-MOR oxidize CH4 to form the methoxy species and then the formed methoxy species on Cu–H-MOR can couple with CO through carboxylation, forming acetic acid. Notably, Cu–Na-MOR with Cu/Al = 0.22 is also active for the production of acetic acid. But its activity is much lower than that of Cu–H-MOR. The observed formation of acetic acid in the tandem reactions suggests that the CuxOyn+ clusters are active for the formation of acetic acid from CH4 although the chemical and coordination environments of Cu atoms of the CuxOyn+ clusters of the Cu–H-MOR and Cu–Na-MOR were not reported.321The Cu/Al ratio of Cu–Na-MOR is an important factor for determining whether a Cu–Na-MOR has activity in the formation of acetic acid or not. Notably, Cu–Na-MOR with Cu/Al > 0.36 is not active for the formation of acetic acid although Cu–Na-MOR with Cu/Al ≤ 0.36 is active for the formation of acetic acid. However, the Cu/Al ratio is not a factor in determining the activity in the formation of acetic acid for Cu–H-MOR. Cu–H-MORs in both the ranges of Cu/Al > 0.36 and Cu/Al ≤ 0.36 are active for the formation of acetic acid. Thus, the mechanism for carboxylation of methoxy to acetic acid on Cu–Na-MOR is different from Cu–H-MOR. The difference in the reaction mechanisms between Cu–Na-MOR and Cu–H-MOR was supported by UV-vis spectroscopy. For Cu–H-MOR, upon being activated in O2, the d–d transition at 9600 cm−1 was observed before the introduction of CH4; more importantly, the intensity of this d–d transition decayed upon simultaneous reaction with CH4 at 200 °C for 2 h; this coherent observation clearly shows that the species contributing to 9600 cm−1 is responsible for the formation of acetic acid on Cu–H-MOR.
Parallel studies were performed on Cu–H-MOR and Cu–Na-MOR for further understanding their different reaction mechanisms. Cu–H-MOR (Cu/Al = 0.35) and Cu–Na-MOR (Cu/Al = 0.36) have the same Cu/Al ratio but obviously Cu–H-MOR (Cu/Al = 0.35) produced a larger amount of acetic acid than Cu–Na-ZSM-5 (Cu/Al = 0.36). The high activity in the transformation of CH4 to acetic acid on Cu–H-MOR (Cu/Al = 0.35) suggests that the Brønsted acid sites of Cu–H-MOR (Cu/Al = 0.35) can enhance the formation of acetic acid.321 Since the Brønsted acid sites in Cu–H-MOR that play a significant role in the carbonylation of methoxy and CuxOy sites are reactive in the activation of CH4 to form methoxy, methoxy formed on the CuxOy sites must be transferred from the CuxOy sites to the Brønsted acid sites so that methoxy could be carbonylated to form acetic acid. Upon the consumption of oxygen atoms of the CuxOy clusters for the formation of acetic acid, Cu–H-MOR and Cu–Na-MOR can be regenerated a few times through pre-treatment in O2 at 600 °C, although the reported tandem reactions are not a catalytic cycle.
11. Catalytic oxidation of CH4 with O2 at low temperature in aqueous solutions
Unlike the oxidation of CH4 with O2 catalyzed by molecular catalysts,225 Yamanaka et al. reported one exciting catalytic oxidation of CH4 with molecular O2 at 40 °C by their unique catalytic system consisting of Zn powder, EuCl3, and CF3CO2CH3.322,323 Although the preparation of this catalyst is not intricate, there has definitely been a lack of a specific understanding on the reaction mechanism of this amazing catalytic oxidation of CH4 with inexpensive molecular O2 by an inexpensive catalyst at near room temperature. The hypothesized mechanism is as follows.322,323 Eu2+ is a key reductant, which is generated in situ through the reduction of Eu3+ with metallic Zn powder. Then, Eu2+ activates molecular O2 to form a type of active oxygen species O*. The active oxygen species O* is expected to exhibit strong electrophilicity. It activates CH4 to form CH3OH. The choice of Zn powder to reduce Eu3+ is that its redox potential ψ(Zn2+/Zn) (−0.77 V) is lower than that of ψ(Eu3+/Eu2+) (−0.35 V). In addition, the use of a strong acid CF3COOH to stabilize the product CH3OH through the formation of the ester CF3COOCH3 drove the thermodynamics of CH4 + O2 → CH3OH to the direction favorable for the production of CH3OH since the oxidation of CH4 to CH3OH is only weakly exothermic at 298 K (ΔH° = −26 kcal mol−1). Another hypothesis could be that the catalytic transformation of CH3OH from CH4 is due to the oxidation of CH4 by H2O2. This hypothesized path is feasible since H2 can be readily formed by the reduction of H+ with Zn and then H2 can couple with O2 to form H2O2. However, it is not clear how the formation of H2O2 from H2 and O2 was catalyzed. A fundamental understanding of these fascinating catalysts is expected to be necessary for providing insights into the design of active catalysts for the catalytic oxidation of CH4 to CH3OH under mild conditions.12. Catalytic oxidation coupling of CH4, CO, and O2 to synthesize acetic acid
Compared to the oxidation of CH4 with an oxidant such as H2O2 or O2, as reviewed in Section 13, the reaction of coupling CH4 with CO and O2 is quite a different path for utilizing CH4 to generate high value chemicals. It uses inexpensive oxidant, O2, or even cost-free air as the oxidant instead of costly H2O2. This coupling is a carbon-addition reaction, unlike other reactions reported in the literature. Unlike the tandem reactions in Cu-MOR, the coupling of CH4 with CO and O2 on Rh1O5 clusters anchored in ZSM-5 is a catalytic reaction that continuously transforms CH4 to acetic acid with high activity at 150 °C.18812.1 Advantage of the direct conversion of CH4 to acetic acid under mild conditions
Acetic acid is an important intermediate compound used for the production of high value chemicals. Currently, acetic acid is produced from CH3OH carbonylation, in which CO reacts with CH3OH to form acetic acid. As shown in Fig. 2, CH3OH is synthesized from CO/CO2 and H2 at a relatively high temperature in the range of 250–300 °C; the mixture of CO/CO2 and H2 is produced from steam reforming of either coal or CH4 at quite high temperatures of 600–800 °C. Unfortunately, high-temperature reforming of coal or CH4 has made the synthesis of acetic acid a high energy-demanding and a high-cost process. It would be ideal if a catalytic reaction could directly transform CH4 to acetic acid through one-step catalysis, instead of the current three-step synthesis, under mild conditions instead of high-temperature harsh conditions, through heterogeneous catalysis instead of the current homogeneous catalysis, using a reusable catalyst instead of a current Ir or Rh-based molecular catalyst without recycling.Motivated by these expected advantages, a carbon-addition reaction was used for the purpose of direct transformation of CH4 to acetic acid. Inspired by the use of precious metal catalyst in the Badische Anilin und Soda Fabrik (BASF) process developed in the 1960s and the Monsanto process developed in the 1990s, a Rh-based single atom catalytic site anchored in microporous aluminosilicate has been reported recently.188 It was used for the synthesis of acetic acid through a carbon-addition reaction under mild conditions at 150 °C and lower.188
12.2 Direct conversion of CH4 to acetic acid on a Rh1-based single-atom site in microporous aluminosilicate
A single-atom catalyst was used for catalyzing this carbon addition reaction.188 For a reliable and repeatable preparation of a catalyst with singly dispersed metal atoms, microporous aluminosilicate with isolated protons was used as a support to anchor the specific catalytic sites Rh1O5.188 Through ion-exchange, followed by calcination, a catalyst with singly dispersed Rh1 atoms was prepared. In the micropores, Rh coordinates with 5 oxygen atoms (Fig. 21a).188 This site is highly active for the synthesis of acetic acid molecules from CH4 through carbon addition and oxidation using O2 at 150 °C. Extensive isotope experiments confirmed the participation of the three reactants in the catalytic cycle for the formation of acetic acid. The Rh–O bond protruding to the center of a micropore shown in Fig. 21a acts as the site to activate the first C–H bond of CH4 with an activation barrier of 124 kJ mol−1, which is the rate-determining step among the 18 elementary steps of synthesis of two acetic acid molecules from two CH4, two CO, and one O2 molecules, 2CH4 + 2CO + O2 → 2CH3COOH (Fig. 21b). The second rate-determining step is the coupling of CH3CO and OH adsorbed on Rh1 to form weakly adsorbed acetic acid through a barrier of 107 kJ mol−1.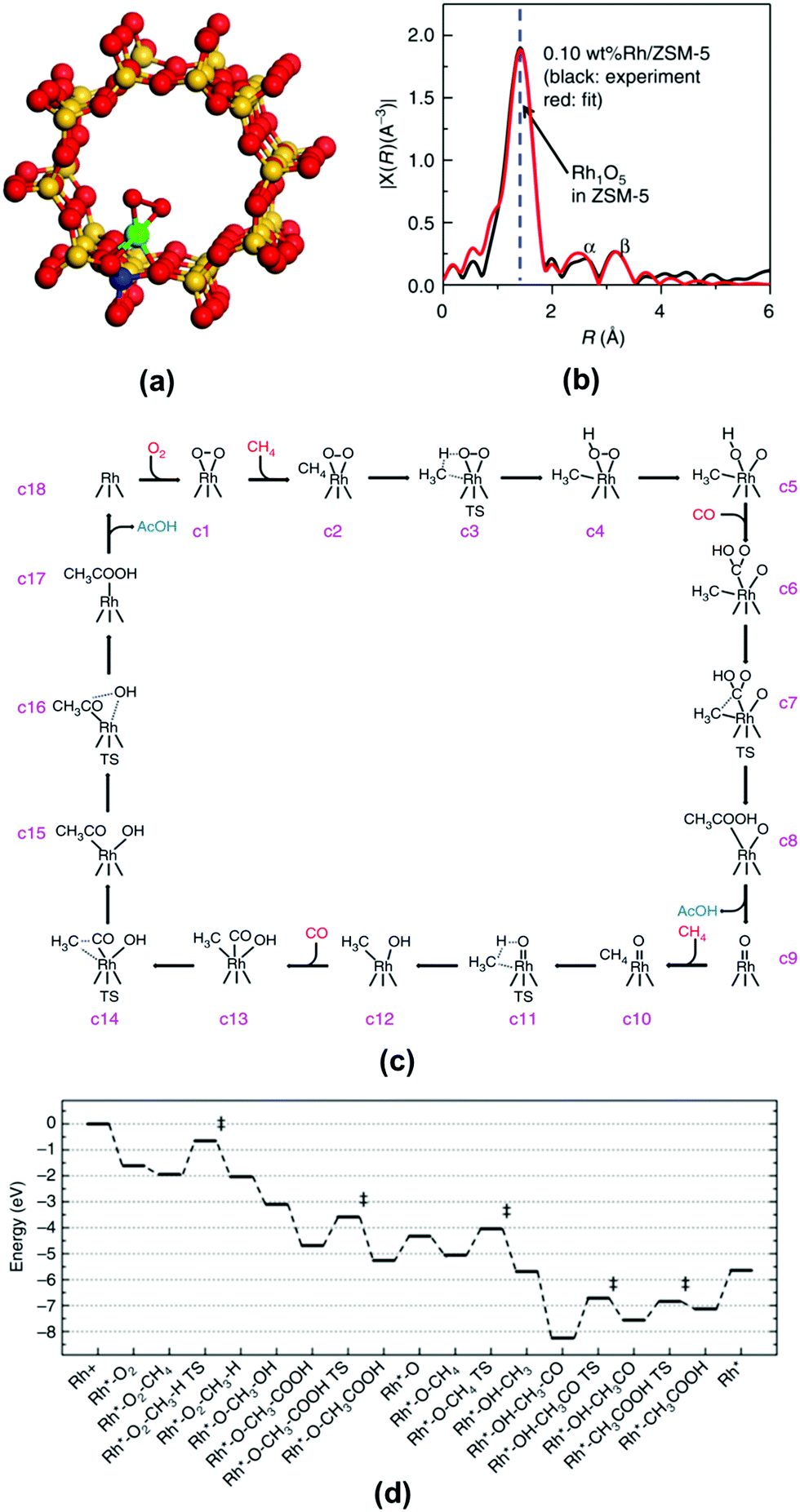 | ||
| Fig. 21 Representation of EXAFS characterization of Rh1O5 anchored in ZSM-5 and the results of the computational studies for the catalytic transformation of CH4, CO, and O2 to acetic acid molecules.188 (a) Structural model of microporous aluminosilicate with the anchored Rh1O5 sites. (b) EXAFS studies of the Rh K-edge. (c) Catalytic cycle consisting of 18 elementary steps for 2CH4 + 2CO + O2 → CH3COOH. (d) Energy profile of the catalytic cycle in (c). Reproduced from ref. 188, copyright 2018, with permission from Springer Nature. | ||
Parallel studies show that single atom sites of precious metals supported on the open surface of reducible oxide Rh1/Co3O4,324 Rh1/TiO2,188 and inert oxide Rh1/SiO2325 and Rh1/Al2O3 are neither active for this carbon addition reaction to produce acetic acid nor for the oxidation of CH4 to CH3OH. Close comparison between single-atom sites anchored in the micropores and the ones supported on the open surface of oxide nanoparticles suggests the significance of micropores in terms of the confinement of the molecules for providing distorted molecules, which has been briefed in Section 9.1. The capability of the micropores for forcing the reactant molecules to distort structurally was further evidenced by the observed activity of other microporous aluminosilicates encapsulating the singly dispersed precious metal atoms.60,269,326 The structural confinement effect of the micropores on this reaction is supported by the DFT calculation of the activation of CH4 by ZSM-5, which suggests that the sub-nanometer pores of ZSM-5 exert an effect to destabilize the adsorption and thus decrease the activation barrier for C–H cleavage in the pores.261
12.3 Mechanistic understanding of this catalytic carbon-addition transformation of CH4
It is well known that not many catalytic reactions involve three different reactants. The direct participation of the three reactants CH4, CO, and O2 for the production of acetic acid was confirmed through extensive isotope experiments.188 Based on the stoichiometry of this carbon-addition reaction, 2CH4 + O2 + 2CO → 2CH3COOH, a complete catalytic cycle requires the participation of five molecules. Computational studies suggest that this catalytic cycle involves 18 elementary steps. Interestingly, the two CH4 molecules were activated through two different sites, Rh1O5 (c1 in Fig. 21b) and Rh1O4 (c9 in Fig. 21b). Compared to Rh1O5, Rh1O4 is highly under-coordinated. Rh1O4 is generated after the formation of the first acetic acid molecule. Compared to the formation of the first acetic acid molecule (c1–c8 in Fig. 21b), the pathway for the formation of the second acetic acid molecule (c9–c17) is different. In the formation of the first acetic acid molecule, CO was inserted into the Rh–O of the Rh–OH species as the Rh atom is fully coordinated with six atoms (c4), forming a COOH group on Rh1. A subsequent coupling between HOOC- and CH3-forms an acetic acid molecule (c8). Upon the desorption of the first acetic acid molecule, the Rh1O4 site was formed (c9). On Rh1O4, CH4 was activated through the RhO bond (c10–c12). As the Rh atom in c12 only bonds with five atoms, CO can directly bond with the Rh1 atom and then couple with its adjacent CH3 to form an acetyl group, –COCH3 (c16), which subsequently couples with a hydroxyl group, forming the second acetic acid molecule (c17). The energy profile of the proposed pathway shows that the rate-determining steps are the activation of C–H of the first CH4 molecule by the Rh–O bond and the coupling of CO and CH3 to form the CH3CO-intermediate (c15).
Cleary, the catalytic oxidation of CH4 and coupling with CO to form acetic acid has been successfully demonstrated.188 Computational studies have suggested that the Rh–O bond of the anchored single atom site plays a key role. Although the TOF of Rh1O5 is quite high, Rh is a precious metal. It would be ideal if the 3d transition metal could replace Rh to catalyze this reaction. Thus, it is highly valuable to explore the early transition metals that could replace Rh. However, due to the complexity of catalyst preparations of this type of catalysts, an inappropriate preparation of a hypothesized catalyst could easily invalidate a good idea. Thus, here, we suggest to perform computational studies first to pre-screen in-expensive transition metal atoms that are in a cluster with the same structure as that of Rh1O5. A list proposed by computational studies would be tested by the experimentalists. It is expected that such an approach could develop a cost-effective catalyst with high activity and selectivity for the synthesis of acetic acid through the coupling of CH4, CO, and O2 in aqueous solution at 150 °C or lower temperature.
13. Catalytic oxidation of CH4 with H2O2
Although H2O2 is somewhat expensive, it is one of the most active oxidants. The main products of oxidation of CH4 with H2O2 are CH3OH and formic acid. A number of catalysts active in the catalytic oxidation of CH4 with H2O2 have been reported. They are mainly singly dispersed Pd, Rh, and other metal atoms coordinated with a certain number of oxygen atoms in the micropores of the zeolite.13.1 Catalytic oxidation of CH4 to CH3OH over Fe sites anchored in ZSM-5 with H2O2
Hutchings et al. first reported the high activity of Fe-ZSM-5 in the catalytic oxidation of CH4 to CH3OH through H2O2 under mild conditions.288 In addition, although TS-1 is not active for this oxidation, TS-1 or silicaliate-1 with anchored Fe exhibits high activity for the catalytic oxidation of CH4 to CH3OH through H2O2 at 50 °C.288 These studies show that the Fe species formed in MFI is the key player in the catalytic oxidation of CH4 with H2O2. For instance, 27 mg of 0.014 wt% ZSM-5 converts 10% of dissolved CH4 in an aqueous solution of H2O2 with 96% selectivity for producing oxygenates including CH3OH, CH3OOH, and HCOOH. The 13C and 1H NMR spectra confirmed that CH4 is the sole carbon source of these products. The presence of the Si–O–Fe bond at 700–710 cm−1 in the FT-IR of the catalyst shows that Fe atoms were incorporated into the tetrahedral framework sites before calcination.327Ex situ EXAFS studies suggest that Fe atoms exist in the form of di-iron species, similar to the structures reported in the literature.328–330 Structure 1 in Fig. 22a, [Fe2(m2-OH)2(OH)2(H2O)2]2+ anchored in ZSM-5, is the structure closest to the structural parameters of the EXAFS studies of the catalyst. It catalyzes the synthesis of methanol from CH4 under mild conditions (50 °C) with a TOF > 0.6 s−1, which is three orders of magnitude greater activity in the catalytic oxidation of CH4 to CH3OH on these catalysts reported before this work.288 The significance of this work is that the production of CH3OH from CH4 can be performed in a continuous flow fixed bed stainless steel reactor with a close catalytic cycle.331,332 | ||
| Fig. 22 Representation of the catalytic oxidation of CH4 with H2O2 with the di-iron complex anchored in ZSM-5.288 (a) Proposed catalytic cycle of oxidizing CH4 to CH3OOH with H2O2. (b) DFT calculation for the elementary steps of transforming CH4 to CH3COOH on [Fe2(m2-OH)2(OH)2(HCOO)(O)]. Reproduced from ref. 288, copyright 2012, with permission from John Wiley and Sons. | ||
Time-on-line analysis suggested that CH3OOH is the primary product and CH3OH is formed consecutively. Thus, it is proposed that H2O2 replaces one H2O molecule bound to the di-iron site (1 in Fig. 22a), forming OCHO–Fe (2 in Fig. 22a). A subsequent transfer of H+ forms HOO bound to one Fe ion (3 in Fig. 22a). The replacement of another H2O molecule bound to Fe2+ by H2O2 forms a Fe4+O site (4 in Fig. 22a) based on a reported redox reaction to produce a ferryl ion in Fenton's reagent.333,334 The Fe4+O of species 4 activates the C–H of CH4, forming CH3 and Fe–OH (5 in Fig. 22a). The CH3 couples with the oxygen atoms of HOO-, forming a CH3OOH-like species on one Fe ion (5 in Fig. 22a). Fig. 22c presents the transformation of CH3OOH to CH3OH. CH3OH releases a HO·, forming CH3O bound to the Fe ion (6 in Fig. 22a). Other than the first report in the literature,288 this catalyst and the catalytic oxidation of CH4 to CH3OH were extensively studied with respect to the identification of the active component,335 kinetics studies,332 and the influence of the Si/Al ratio of H-ZSM-5 on the catalytic performance.336
The catalytic selectivity for the production of CH3OH on Fe-ZSM-5 can be obviously increased by the introduction of Cu2+ as a part of the catalyst to reduce the over-oxidation process of CH3OH to other oxygenates.288 For instance, by mixing Fe-1-silicate with Cu-1-silicate, a high selectivity of 93% for producing CH3OH from CH4 can be achieved.288 In addition, it was found that the presence of strong acidity is favorable for the formation of formic acid instead of CH3OH.337
13.2 Catalytic oxidation of CH4 to CH3OH over Fe sites incorporated in the graphene lattice with H2O2 at the room temperature
Bao et al. reported a room temperature catalysis for oxidizing CH4 to CH3OH with H2O2.338 This catalysis is performed on a unique catalyst FeN4/GN consisting of catalytic sites –FeN4–. Here, GN denotes graphene. This catalyst was prepared by the high-energy ball milling of a mixture of an iron precursor, iron phthalocyanine.338 50 mg of FeN4/GN can catalyze the oxidation of CH4 with a yield of CH3OH of nearly 120 μmol at room temperature in 10 h. The selectivity for C1 oxygenated products is as high as 94%.Evolutions of different C1 oxygenate products were tracked with the operando TOF-MS method, which extracts products during the reaction and analyzes them in real time.338 This operando study suggested that CH4 is oxidized to CH3OH and CH3OOH at the beginning and then further oxidized into HCOOH and HOCH2OOH. The deep oxidation of CH4 to HCOOH and HOCH2OOH upon CH3OH is formed as an intermediate compound was supported by the isotope-labelled 13C-NMR experiments.338
A fundamental understanding of this room temperature catalysis of CH4 to CH3OH on the unique catalyst of FeN4/GN was achieved by DFT calculations.338 As shown in Fig. 23, H2O2 can readily adsorb on an FeN4 site to decompose into atomic O and H2O (Fig. 23a). Atomic O is absorbed on the Fe atom of the FeN4 site. As the FeN4 is incorporated into the graphene lattice, each FeN4 can chemisorb two O atoms, forming a O–FeN4–O structure. Compared to bare –FeN4–, the density of electronic states near the Fermi level of O–FeN4–O is largely increased upon binding two O atoms at two sides of –FeN4– (Fig. 23b). This difference shows that O–FeN4–O is more active for electrophilic attack. The reaction pathway of oxidizing CH4 to CH3OH was computationally simulated (Fig. 23c). DFT calculations suggest that the C–H activation of CH4 is performed through a radical pathway instead of a concerted mechanism.338Fig. 23C represents the reaction pathway and the corresponding energy profile. The rate-determining step is C–H bond cleavage with a barrier of 0.79 eV. The generated CH3˙ can couple with OH or OOH groups, forming CH3OH and CH3OOH, respectively. In addition, DFT calculation shows that O–FeN4–O incorporated in the graphene lattice exhibits high activity than the free-standing O–FePc–O (FePc: iron phthalocyanine), suggesting that the graphene lattice enhances the activity in the oxidation of CH4 to CH3OH.338
 | ||
| Fig. 23 Representation of the theoretical simulation of the reaction pathway for the oxidation of CH4 to form CH3OH on –FeN4– sites and other similar sites.338 (A) Thermodynamic calculation of the decomposition of H2O2 and the binding of oxygen atoms to –FeN4– to form O–FeN4–O. (B) Density of states of FeN4, FeN4–O, and O–FeN4–O. (C) Suggested reaction pathway for the oxidation of CH4 to CH3OH, CH3OOH, HOCH2OOH, and HCOOH on O–FeN4–O and the corresponding energy profile. (D) Correlation of the rate of activation of C–H of CH4 in log(rate) with the formation energy of O–MN4–O. Reproduced from ref. 338, copyright 2018, with permission from Elsevier. | ||
DFT calculations were performed on similar sites (O–MN4–O) but different metal centers (M = Cr, Mn, Co, Ni, Cu). Based on the formation energy of the O–MN4–O structure, only O–CrN4–O, O–MnN4–O, O–FeN4–O, and O–CoN4–O can be formed. The activation rates of C–H of CH4 by the four catalysts were calculated (Fig. 23d). Bao et al. found a volcano-like correlation between the activation rate of C–H of CH4, log (rate), and the formation energy of the four O–MN4–O catalysts (Fig. 23d).338 Thus, these computational studies suggested that the formation energy of O–MN4–O can be taken as a descriptor for the catalytic oxidation of CH4 to CH3OH on this type of catalysts (O–MN4–O).338
13.3 Catalytic oxidation of CH4 to CH3OH over Pd sites with H2O2
Through ion-exchange between Pd cations and protons in microporous aluminosilicate, Pd1O4 clusters anchored in the micropore (0.01 wt% Pd) were prepared (Fig. 23a and b).339 Although the loading of Pd is extremely low, its role is crucial in the oxidation of CH4 to CH3OH and other oxygenates in the temperature range of 50–95 °C. As shown in Fig. 24c, even at 50 °C, CH3OH can be produced from the catalytic oxidation of CH4 with H2O2. The role of the singly dispersed Pd1 sites, Pd1O4 anchored in the micropores, was confirmed by the fact that the same microporous aluminosilicate but without Pd atoms under the same catalytic conditions exhibits much lower activity. Notably, the increase in the loading of Pd to the microporous aluminosilicate from 0.01 wt% to 0.1 wt% and 2 wt% does not increase the yields of oxygenates, suggesting the crucial role of the singly dispersed single-atom Pd1 sites in the catalytic oxidation of CH4 to CH3OH with H2O2. Due to the available significant excess of H2O2, products of deep oxidation including formic acid and even carbon dioxide were clearly observed.339 Thus, the control of selectivity for the product CH3OH is a challenging issue for catalytic oxidation using H2O2 as an oxidant. Compared to the catalytic oxidation of CH4 using H2O2, the oxidation of CH4 with metal atoms anchored in the zeolite without using H2O2 forms CH3OH with much higher activity. As shown in Fig. 24d, by co-loading 2.0 wt% CuO on the surface of the microporous aluminosilicate consisting of Pd1O5 sites, the selectivity for producing CH3OH was obviously increased. The promotion of selectivity using CuO suggests that CuO can catalyze the decomposition of H2O2 to H2O and O2, thus supressing the further oxidation of CH3OH to products of deep oxidation. | ||
| Fig. 24 Representation of structural characterization and the catalytic performance for the catalytic oxidation of CH4 with H2O2 to CH3OH under mild conditions.339 (a) Structural model of microporous aluminosilicate with anchored Pd1O4 sites. (b) EXAFS. (c) Catalytic performances of oxidizing CH4 to CH3OH with H2O2. (d) Increase in the selectivity for producing CH3OH by adding CuO to supress the oxidation of CH3OH. Reproduced from ref. 339, copyright 2016, with permission from John Wiley and Sons. | ||
13.4 Catalytic oxidation of CH4 to CH3OH over Cu sites with H2O2
Catalysts containing the same amount of Cu (1.2 wt%) and Fe (0.09 wt%) were prepared through different methods including ion exchange between Cu2+ and H+ of H-ZSM-5 by Yashnik et al.340,341 Similar to Fe-ZSM-5,288 the main products of CH4 oxidation with H2O2 on Cu-ZSM-5 are CH3OOH and CH3OH. After an induction period of oxidation, more formic acid and CO2 were formed. The role of the anchored Cu2+ was confirmed by careful parallel experiments performed on a series of catalysts including catalyst 1, which is H-ZSM-5, originally containing 0.09 wt% Fe in the lattice, and catalyst 2, which is H-ZSM-5, originally containing both Fe (0.09 wt%) and the lately anchored Cu (1.2 wt%) by ion exchange between the Cu2+ of the precursor and the H+ of H-ZSM-5, followed by calcination at 500 °C in air.342 Compared to catalyst 1 (H-ZSM-5 with 0.09 wt% Fe but no Cu), the introduction of Cu2+ into ZSM-5 led to an increase in the conversion of both CH4 and H2O2. It is suggested that the Cu–peroxo complexes in ZSM-5 participate in the formation of CH3OH from CH3OOH.342 When the concentration of Fe(III) in H-ZSM-5 is high, the role of Cu(II)–peroxo complexes is less important. Notably, some of the anchored Cu2+ in ZSM-5 leached to the aqueous solution during catalysis. It is reported that even half of the anchored Cu2+ cations were leached in the first half hour of catalysis. Compared to Cu2+ introduced at the cation exchange positions of H-ZSM-5, the Fe(III) cations introduced into the lattice of ZSM-5 in the synthesis of H-ZSM-5 are much more stable during catalysis.342As reviewed in Sections 13.1–13.4, ZSM-5 anchored with Fe, Pd, and Cu are active for catalyzing the oxidation of CH4 to CH3OH with H2O2. Similar to the proposals in other sections, there is a lack of systematic and parallel studies for ZSM-5 anchored with other transition metals including 3d and 4d metals. The coordination environment of metal atoms is crucial for the catalytic performance in the oxidation of CH4 with H2O2. However, it is challenging to prepare a series of catalysts of different metals that have the same coordination environment such as M1O4 (M = Zr, Nb, Mo, Ru, Rh, Pd, Ag). Thus, here, computational “experiments” were suggested to perform parallel studies on these catalysts ZSM-5 anchored with M1O4 sites (M = Zr, Nb, Mo, Ru, Rh, Ag). By applying the same reaction pathway on these catalysts, the rate determining steps and their corresponding activation barriers can be uncovered through computational studies. With this piece of information, a correlation of the activation barriers of these catalysts and the electronic factors of the active site M1O4 of these catalysts such as Bader charge and orbital contribution can be established. With this correlation, active catalysts can be proposed on the basis of these computational studies for experimental tests. Through these computational studies-driven experimental studies, catalysts with high activity and selectivity are expected to be developed.
13.5 Catalytic oxidation of CH4 to CH3OH with H2O2 over Rh single atom sites anchored on the open surface of the oxide support
As reviewed in Sections 13.1–13.3, the encapsulated metal atoms in zeolites are active in catalyzing the oxidation of CH4 with H2O2. Recently, it has been reported that single atoms Rh (SAs Rh) anchored on CeO2 nanowires (SAs Rh–CeO2 NWs) can catalyze the oxidation of CH4 with H2O2 at 50 °C.343 SAs Rh–CeO2 NWs were prepared by impregnating Na3RhCl6 on the as-prepared CeO2 NWs through a conventional wet impregnation method.343 The formation of single atoms Rh on CeO2 NWs was suggested by the lack of a peak of Rh–O–Rh in the r-space of the Rh K-edge. For comparison, Rh NPs supported on commercial CeO2 NWs (Rh/CeO2-com) were prepared through the impregnation of Rh in commercial CeO2. Although the chemical and coordination environments of Rh atoms of the Rh/CeO2-com catalysts have not been reported, Rh 3d5/2 of the Rh/CeO2-com is 308.9 eV, which is obviously higher than 307.4 eV for the metal Rh nanoparticles. It suggests that the Rh atoms of Rh/CeO2-com are in the form of RhOx nanoclusters supported on commercial CeO2.Compared to Rh/CeO2-com, SAs Rh-CeO2 NWs exhibit much higher catalytic activity and selectivity in the production of CH3OH and CH3OOH through the oxidation of CH4 with H2O2 at 50 °C. This significant difference was attributed to the distinctly different electronic states of Rh atoms in the two catalysts in terms of the projected density of states (PDOSs) of the surfaces of the two catalysts. For SAs Rh–CeO2 NWs, its Rh 4d occupation near the Fermi level is sharp. However, the Rh 4d occupation of the Rh/CeO2-com is quite broad. Unlike SAs Rh–CeO2 NWs, the significant overlap among Rh 4d, O 2p, and Ce 4f suggests a strong coupling between the RhOx cluster and the CeO2 nanowires support. This strong coupling in Rh/CeO2-com results in over-binding effects, consequently increasing the activation barrier of dehydrogenation of CH4 on Rh/CeO2-com.
Other than single atoms Rh anchored on CeO2,343 Rh anchored on ZrO2, TiO2, and SiO2 were chosen to explore the potential oxidation of CH4 with H2O2.344 Single atoms Rh anchored on ZrO2 exhibit high activity in the catalytic oxidation of CH4 to CH3OH. However, no CH3OH was produced from 0.3 wt% Rh/TiO2 under the catalytic conditions of 30 mg catalyst and 0.5 M H2O2 at 70 °C with a reaction time of 0.5 h under 30 bar CH4.
13.6 Catalytic oxidation of CH4 to CH3OH over Au–Pd alloy nanoparticles
Hutchings et al. found that 5 wt% Au–Pd/TiO2 exhibits high activity and selectivity for the oxidation of CH4 to CH3OH with H2O2.345 Notably, their studies also showed that no product could be observed from bare TiO2 under the same conditions as 5% Au–Pd/TiO2, concluding that the activity for the oxidation of CH4 with H2O2 on 5 wt% Au–Pd/TiO2 results from Au–Pd nanoparticles instead of TiO2. The time-on-line studies of the products showed that CH3OOH is the intermediate compound formed first and then it is transformed to CH3OH;345 subsequently, CH3OH is oxidized to formic acid or even CO2, which is dependent on the catalysis temperature and concentration of H2O2. The feature of sequential reactions including the transformation of CH4 to CH3OOH first, then CH3OH, and finally formic acid and CO2,345 is similar to the oxidation of CH4 with H2O2 on Fe-ZSM-5.288 Catalysis for the formation of CH3OH from CH3OOH was confirmed using isotope-labelled 13CH3OH as the reactant. In addition, the oxidation of CH3OH to HCOOH was evidenced by the observation of H13COH and H13COOH when 13CH3OH was used as the reactant.A deep understanding of the catalytic mechanisms for the oxidation of CH4 on Au–Pd/TiO2 was achieved through electron paramagnetic resonance (EPR) studies under catalytic conditions.345 Both ˙CH3 and ˙OH were tracked with 5,5′-dimethyl-1-pyrroline-N-oxide (DMPO) in the EPR studies, revealing that ˙CH3 and ˙OH are formed during the catalytic oxidation of CH4 with H2O2. Thus, it is concluded that the catalytic oxidation of CH4 with H2O2 on Au–Pd/TiO2 must involve ˙CH3 radicals. Then, the formed ·CH3 can couple with the intermediate O2 or ˙OOH in situ formed from the decomposition of H2O2, forming CH3OOH, which is the intermediate compound for the formation of CH3OH. Notably, this catalysis mechanism is different from the oxidation of CH4 with H2O2 on Fe-ZSM-5, where no ·CH3 was observed.288
Other than the catalytic synthesis of CH3OH from the oxidation of CH4 with H2O2 on Au–Pd/TiO2, Au–Pd/TiO2 is active for the synthesis of CH3OH from CH4, H2, and O2.346 This is understandable since Au–Pd/TiO2 can catalyze the synthesis of H2O2 and catalyze the oxidation of CH4 to CH3OH with H2O2 under mild conditions. In other words, a possible path for the synthesis of CH3OH from CH4, H2, and O2 on Au–Pd/TiO2 involves Au–Pd/TiO2 first catalyzing the synthesis of H2O2 from H2 and O2, and then catalyzing the synthesis of CH3OH from the oxidation of CH4 with H2O2.346,347
13.7 Catalytic oxidation of CH4 to CH3OH catalyzed by an engineered nanocomposite catalyst AuPd@ZSM-5-R
The feature of the high activity of Au–Pd nanoparticles in catalyzing the oxidation of H2 by O2 to form H2O2 at room temperature or even low temperature and the capability of H-ZSM-5 in catalyzing the oxidation of CH4 with H2O2 to form CH3OH under mild conditions were intelligently mingled in a recent work, creating a catalyst with high activity and selectivity for the synthesis of CH3OH from CH4, H2, and O2.348 In this work, Jin et al. reported that AuPd@ZSM-5-C16 exhibits high activity and selectivity for the oxidation of CH4 to CH3OH at 70 °C.348 As shown in Fig. 25a, this is a nanocomposite catalyst with an MFI framework encapsulated with Au–Pd nanoparticles internally supported with a molecular fence of C3–C16 on the external surface of the ZSM-5 particle. The conversions and yields on the catalysts with a fence such as AuPd@ZSM-5-C3 (or C6, C16) are obviously higher than those of AuPd@ZSM-5 without a molecular fence (Fig. 25b). The external organic layer anchored on the ZSM-5 particle is hydrophobic and thus prevents the migration of H2O2 formed on the surface of AuPd, which was encapsulated in the ZSM-5 particle. This is evidenced by the quantitative analyses of the amount of H2O2 in the zeolite and that in the liquid. As shown in Fig. 25c, there is a distinct difference in the distribution of H2O2 in ZSM-5 between a catalyst with a molecular fence such as AuPd@ZSM-5-C3 and a catalyst without any molecular fence such as AuPd@ZSM-5. With the protection of this molecular fence, H2O2 formed through catalysis on AuPd nanoparticles encapsulated in ZSM-5 particles mainly remains in the micropores of ZSM-5; however, without such a fence, a majority of H2O2 in the micropores migrated to the liquid, which is the external environment of the AuPd@ZSM-5 particles. Clearly, the hydrophobic nature of the organic layer (C3, C6, or C16) on the external surface of the ZSM-5 particle stopped the migration of H2O2 efficiently. Thus, due to the hydrophobic nature of the organic fence, the high local concentration of H2O2 in ZSM-5 near the catalyst sites of CH4 oxidation largely increased the reaction rate of CH4 oxidation by H2O2 on the surface of the Au–Pd nanoparticles. Notably, this hydrophobic layer on the external surface of ZSM-5 does not hinder the diffusion of hydrophobic, nonpolar CH4 from the liquid to the micropores of AuPd@ZSM-5.348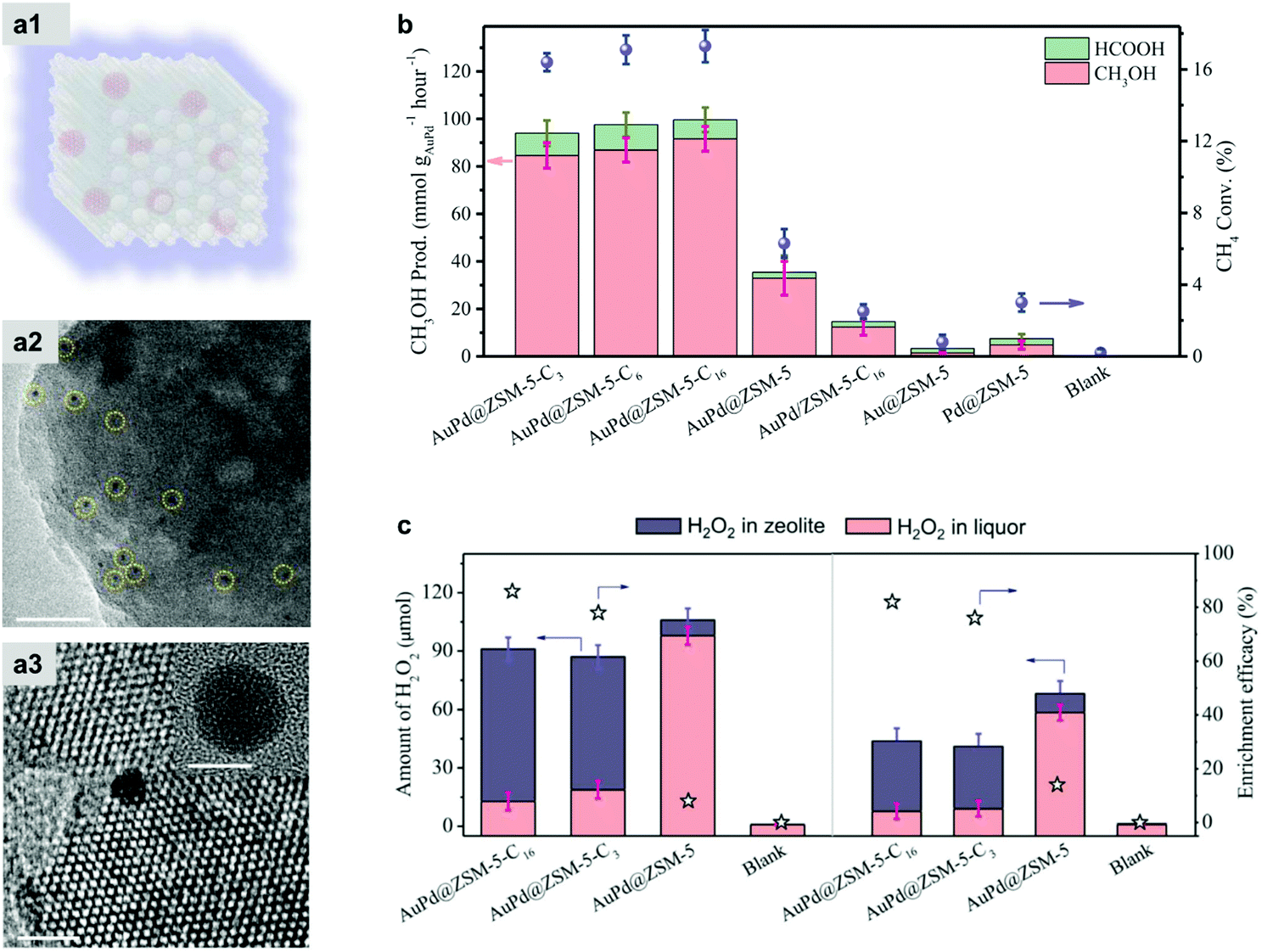 | ||
| Fig. 25 Representation of AuPd@ZSM-5-R and their catalytic performance and molecular fence effect of the external layer of Cn.348 (a) Models (a1) and TEM images (a2 and a3) of AuPd@ZSM-5-R. (b) Activity of CH3OH and the conversion of CH4 on AuPd@ZSM-5-R, AuPd@ZSM-5, and other related catalysts; here, R denotes the organic layer of the molecular fence. (c) Molecular-fence effect investigation; it shows a distinct difference in the distribution of H2O2 in ZSM-5 between a catalyst with a molecular fence such as AuPd@ZSM-5-C3 and a catalyst without a fence such as AuPd@ZSM-5. Reproduced from ref. 348, copyright 2020, with permission from AAAS. | ||
CH3OH was considered as an intermediate product of further oxidation to form HCHO and formic acid.62,349 As discussed in Section 13.3, one disadvantage of the catalytic oxidation of CH4 with H2O2 by the catalytic sites, MxOy clusters, anchored in the micropores of the zeolites, is the further oxidation of CH3OH by excess of the oxidant, H2O2. The work of Jin et al. shows that CH3OH on Au–Pd can readily migrate from the micropores of ZSM-5 to the solvent although H2O2 cannot. The efficient separation of CH3OH from the region of high concentration H2O2 in ZSM-5 prevents CH3OH from being further oxidized to HCHO and HCOOH, leading to high selectivity for the production of CH3OH. Thus, such a smart catalyst with a molecular fence, AuPd@ZSM-5-C16, can catalyze the conversion of CH4 to CH3OH with a selectivity of 60–90%, which is higher than the selectivity for the production of CH3OH through the oxidation of CH4 with H2O2 on other catalysts.348
13.8 Catalytic oxidation of CH4 to C1 oxygenates on vanadium-based complexes
Vanadium-based complexes are active in the catalytic oxidation of organic compounds with H2O2.350,351 It is reported that the acetonitrile solution of [NBu4]VO3 (or NaNO3) and pyrazine-2-carboxylic acid (PCA) is active in the catalytic oxidation of CH4 with H2O2 and O2 by groups in Russia and Switzerland.352,353 It produces methyl hydroperoxide, formaldehyde, and formic acid. Based on the control experiments using cyclohexane to substitute for CH4, it is claimed that H2O2 is only a promoter and molecular O2 is the actual oxidant. They proposed that the HO˙ radical is formed from H2O2 and then it attacks the alkane including CH4 to form alkyl radicals such as CH3˙. The formed CH3˙ can rapidly react with molecular O2 to form peroxyl radicals ROO˙ such as CH3OO˙. ROO˙ is transformed to ROOH such as CH3OOH.352,354–356 In terms of the active sites, there was no indication in this literature although it is certain that the active sites must be vanadium-based species.Another group independently reported that the H3+xPVxMo12−xO40 catalyst (x = 0, 1, 2, and 3) in (CF3CO)2O solvent at 353 K can catalyze the oxidation of CH4 with H2O2 under an inert gaseous environment.357 H4PVMollO40 gave the highest conversion of CH4. A majority of the products is HCOOCH3, while HCOOH and CH3OH are minor products.357 H3+xPVxMol2−xO40 (x = 0–3) Keggin-type heteropoly acid was used as the precursor. Among the used solvents (CF3CO)2O, CH3CN, H2O, and (CH3)2SO, (CF3CO)2O have the highest solubility of CH4,357 resulting in highest conversion among the same catalyst in different solvents. Although there was no information on the active sites for this oxidation, it seems that vanadium is a necessary element of the unknown active sites.
Other than the above two types of vanadium-based catalysts, vanadyl oxysulfate was found to be active in the oxidation of CH4 with H2O2 at 60 °C.358 The main product is formic acid. Their UV-vis spectroscopy measurements suggest that VOSO4 is oxidized to the oxoperoxo VO(O2)+ species by H2O2. Thus, the V5+ species formed under catalytic conditions was proposed to be the active site for the oxidation of CH4.
13.9 Catalytic oxidation of CH4 by H2O2 to form oxygenates in a solution of transition metal chloride
A series of transition metal chlorides (FeCl3, CoCl2, CuCl2, RuCl3, RhCl3, PdCl2, OsCl3, IrCl3, H2PtCl6, and HAuCl4) have been studied in parallel for the catalytic oxidation of CH4 with H2O2 in aqueous solution.359 An obvious difference in the catalytic activity in the oxidation of CH4 was found. FeCl3, CoCl2, CuCl2, RhCl3, PdCl2, OsCl3, IrCl3, H2PtCl6, and HAuCl4 are active for the oxidation of CH4 with H2O2. The actual active sites of the catalytic reactions under mild condition were not identified in the literature.359Among these chlorides, OsCl3 is the most active catalyst for the oxidation of CH4. Compared to other chlorides, the high activity for the catalytic oxidation of CH4 with H2O2 on OsCl3 is probably correlated with the high activity of OsO4 in the selective oxidation of alkenes,360,361 although OsO4 is likely not the real player in this catalysis. UV-vis spectral studies of OsCl3, OsCl3 + H2O2 before catalysis, OsCl3 + H2O2 after catalysis, Na2OsCl6, and OsO4 suggested that the nominal catalyst OsCl3 was evolved into a species with high oxidation state (+IV) of Os during catalysis. Compared to the spectral feature of OsO4, the UV-vis spectrum of the catalyst (OsCl3 + H2O2) after catalysis is distinctly different. Thus, OsO4 could be the active site formed during catalysis and is evolved into other species after catalysis. Undoubtedly, more studies are necessary for the elucidation of the active site of this oxidation.
At the initial period of catalysis, methyl hydroperoxide was observed.360,361 The analysis of the products as a function of time suggests that methyl hydroperoxide is an intermediate product. In addition, the oxidation of CH4 ceased once the radical scavenger hydroquinone was added to the system, showing that this catalysis is performed through a radical-based pathway. It is suggested that the active radical, HO˙ or HOO˙, activates CH4 by abstracting an H atom, forming the CH3˙ radical. Then, coupling the CH3˙ radical with HO˙ or HOO˙ forms the products.
13.10 Catalytic oxidation of CH4 with H2O2 to CH3OH over the M1On cluster in the complex encapsulated in the second coordination shell
A type of well-studied catalysts are μ-nitrido diiron phthalocyanine362–365 and porphyrin,366 which are active for the catalytic oxidation of CH4 with H2O2. These Fe- or Cu-containing catalysts exhibit high selectivity for formic acid but low selectivity for CH3OH.Complexes including FeII(TPA) and V(TKA) shown in Fig. 25a and b, respectively, exhibit low activity for the oxidation of CH4 with H2O2 and low selectivity for producing CH3OH. These complexes are soluble in aqueous solutions. Upon encapsulating such a complex into a hydrophobic hemicryptophane cage, a new catalyst was formed, as shown in Fig. 25c–e.367 These cage-like catalysts can accommodate CH4 molecules and make them physically proximal to the oxidation site of these complexes.367 Notably, this cage structure obviously improved the catalytic activity and selectivity. For instance, FeII(Hm-TPA) and V(Hm-TKA) having cages (Fig. 25c and d) exhibit much higher catalytic activity in terms of the TOF than FeII(TPA) and V(TKA) without the cages (Fig. 25a and b). Covering the TPA–iron(II) active center with a hydrophobic cage, FeII(Hm-TPA) promotes the yield of mono-oxidized products (CH3OH and CH3OOH) on FeII(TPA) by a factor or about 4. Such a promotion of selectivity results from the physical proximity between the adsorbed CH4 and the Fe-based active site of the complex FeII(TPA) in the cage, and the preferential release of hydrophilic products (CH3OH and CH3OOH) by the hydrophobic cage.
The promotion of the selectivity by caging a molecule was also supported by the increased yields of CH3OH and CH3OOH on V(Hm-TKA) (Fig. 26d) compared to V(TKA) (Fig. 26b). By replacing the phenyl spacers of the cage (Hm) of V(Hm-TKA) with biphenyl groups, a different catalyst was formed (Fig. 26e). The catalytic activity of V(Hm-BINOL-TKA) increased by 2 times due to the increase in the capability of recognition of hydrophobic substrates. Thus, obviously, the encapsulation of the molecular catalyst in an organic shell is a strategy to improve the catalytic performance of the oxidation of CH4 with H2O2 under mild conditions. It is expected that the encapsulation approach could be used in other catalyst systems for the promotion of the catalytic activity.
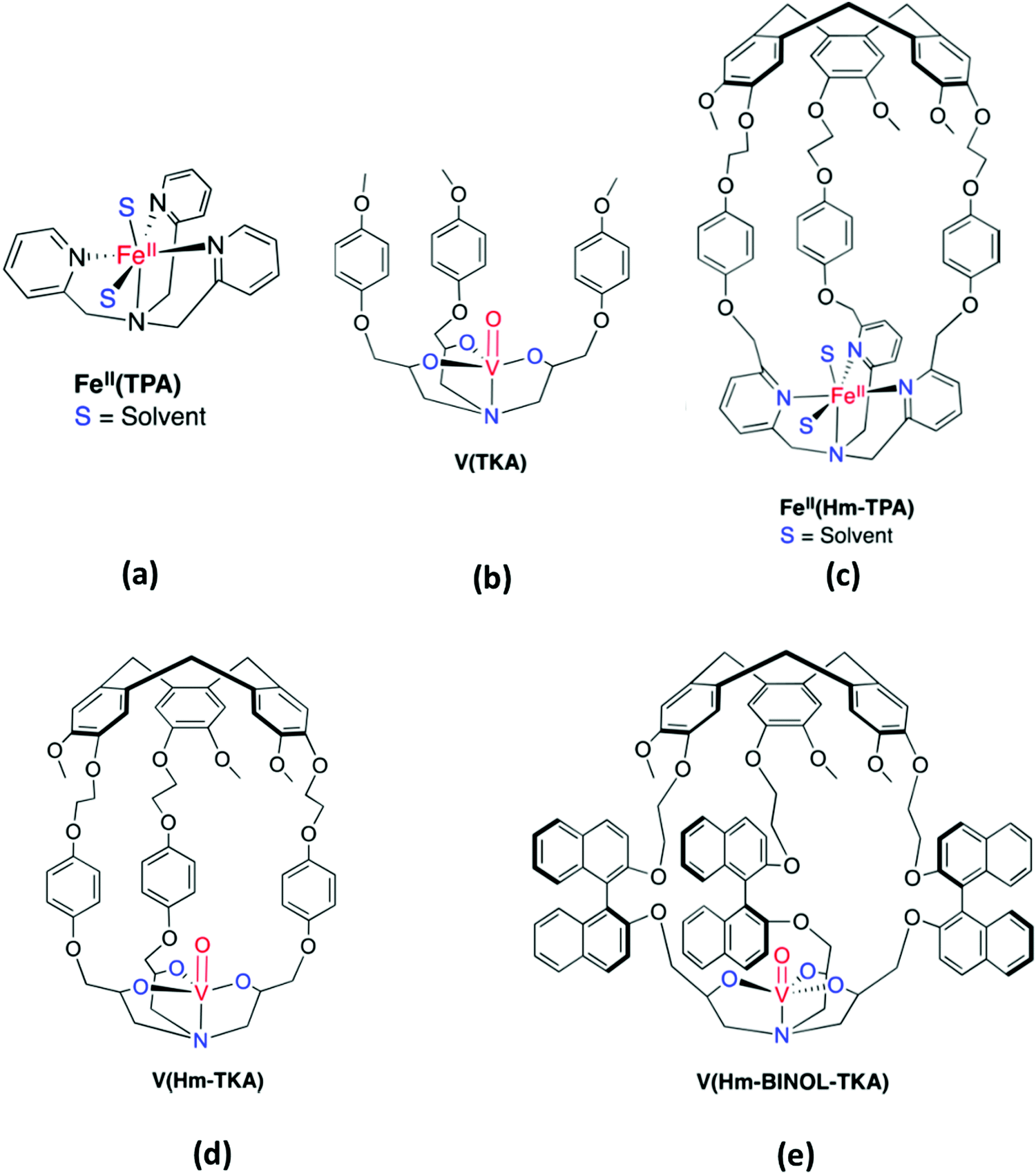 | ||
| Fig. 26 Representation of the structure of Fe and V complexes without the second coordination shell (a and b) and with the second coordination shell (c–e).367 (a) FeII(TPA), (b) V(TKA), (c) FeII(Hm-TPA), (d) V(Hm-TKA), and (e) V(Hm-BINOL-TKA). Reproduced from Ref. 367, copyright 2019, with permission from ACS. | ||
14. Facing challenges
The direct transformation of CH4 to value-added chemicals under mild conditions is one of the most challenging tasks in the field of catalysis. Compared to the well-studied high temperature processes for the catalytic transformation of CH4, no coke is formed and there is much less extent of decay of the catalysts in the transformation of CH4 under mild conditions. However, transformations under mild conditions rely on specific catalytic sites, which need to be designed and prepared with atomic precision. These sites are typically metal oxide nanocluster-like structures with a specific composition and specific coordination environment of the metal atoms. In many cases, they need to be anchored to some “protective” environment such as microporous silica since the confinement effect of the micropores is significant for performing many transformations under mild conditions. Although significant progress has been achieved in the field of activation and catalytic transformation of CH4 under mild conditions, the following challenges still remain.Challenges in the preparation with atomic precision and ready duplication
A majority of catalysts active for transforming CH4 to value-added chemicals are catalysts consisting of single-atom sites or sub-nanometer clusters encapsulated in the micropores of zeolites. The chemical and coordination environments of these metal atoms are essential for catalysis under mild conditions. Compared to the preparation of conventional catalysts such as supported oxide or oxide nanoparticles, the repeated preparation of these catalysts has brought about great challenges. From this point of view, the development of a precise preparation method including a new preparation protocol and the integration of other preparation techniques could be a solution to tackle this challenge. For instance, vapor deposition technique can be used to homogenously introduce metal precursor into the micropores, making a catalyst consisting of homogeneous sites toward the repeatable syntheses of the catalysts active for the transformation of CH4 under mild conditions.Challenges in the characterization of the catalytically sites during catalysis
It has been well acknowledged that the establishment of a correlation between the structure of the active sites and their corresponding catalytic performance is a key approach to achieve a fundamental understanding of catalytic the mechanism at the molecular level. As the catalyst consisting of active sites is dispersed in liquid, which is under a high-pressure gas phase, it is quite challenging to characterize these catalytic sites during catalysis since most electron-excitation or electron-generation-related analytical methods of solid materials such as TEM and XPS have been developed for characterizing catalyst nanoparticles in high vacuum or in the gas-phase instead of a liquid phase. The development of analytical methods with the capability of characterizing the catalysts in liquid during catalysis is a significant but challenging task.Access to the chemical and coordination environments of atoms of authentic active sites anchored in micropores during catalysis is challenging even for catalysis performed at the solid–gas interface. Unfortunately, most transformations of CH4 to value-added chemicals under mild conditions are performed at solid–liquid or solid–liquid–gas interfaces. It is extremely challenging for accessing the chemical and coordination environments of the metal atoms of the catalyst sites of the catalysts working in the liquid. There are the following challenges: (1) due to the much higher density of molecules in the liquid environment around the catalyst particles than the gas phase, most electron-based techniques are limited in probing the surface of catalyst particles in liquid. (2) Most reactions require a high pressure reactant gas above the liquid since gas molecules can only access the surface of the catalyst in the liquid by molecular diffusion; thus, the pressure of gas above the liquid is 10 bar or higher. (3) Many of the reactions and catalytic reactions are performed on the active sites embedded in the micropores of the zeolite. The thickness of the wall of the silicate limits the access of the electron or light to the active sites encapsulated in the pores and also prevents the generated electrons or photons from escaping detection. (4) The preservation of products in the micropores after reaction could make the characterization of the active sites in zeolite at the atomic scale challenging; for instance, the adsorption of CH3OH molecules on or near the active sites in the zeolite prevents EXAFS from distinguishing the coordination environment of the Cu atoms of the active sites from the coordination environment of Cu-bound reactants to the product molecules.
Al Kα X-ray photoelectron spectroscopy (XPS) is an appropriate analytical method for characterizing the chemical environment of most elements. Due to its high surface sensitivity in terms of the short inelastic scatting mean free path (λ), it is extremely challenging, if not impossible, to study metal sites anchored in the micropores of the zeolite in the liquid phase. In addition, although AP-XPS has been widely used in the observation of the chemical state of the metal atoms in the gas phase,78 the working pressure of the reactant gas is still quite low in terms of the sub-Torr or Torr pressure range. Many efforts have made with the goal of having Al Kα or soft X-ray AP-XPS for higher working pressure. Unfortunately, scientists have failed in these efforts. These challenges have made the elucidation of the chemical states of the metal elements of the catalysts functioning in the liquid phase quite difficult. Alternatively, XANES and EXAFS are appropriate spectroscopic methods for probing metal atoms in the gas phase up to a few tens of bar or in the liquid phase although they lack surface sensitivity.
To tackle this challenge, there are some possible approaches. Attenuated total reflection (ATR) spectroscopy could be used to track the adsorbate of the active site and measure the concentration of stable intermediates or products near the catalyst particles as long as the catalyst power can be immobilized on the IGE crystal robustly.
Challenge in establishing a correlation between the catalytic performance and their corresponding chemical and coordination environments
DFT calculations have been a significant approach to provide an understanding of the catalytic mechanism at the molecular level. In most cases, a reaction pathway can be proposed and the rate-determining step can be identified. A deep analysis of for the participation of the orbitals of the atoms of a catalytic site in the transition states allows to propose a descriptor of the catalyst structure at an atomic scale. Finding out the authentic chemical and coordination environments of the atoms of a catalytic site responsible for the reported catalytic performance is a primary task before establishing the intrinsic structure–property correlation. Unfortunately, this is a challenging task in the fundamental studies of the catalytic reaction performed at the solid–liquid or solid–liquid–gas interface. A possible solution to tackle this challenge is to design a catalysis-XAS reaction system that can perform both the catalytic oxidation of CH4 on catalyst particles dispersed in the liquid under high-pressure gas phase and XAS experiments during catalysis. We have designed a reactor for tracking catalyst nanoparticle dispersed in liquid at high temperature up to 350 °C under a gas phase with a pressure up to 50 bars using XAS spectroscopy. Through the modification of this type of reactor, a catalytic-XAS system could be developed, in which the liquid in the reaction cell can be periodically injected into HPLC for on-line analysis.Challenges in prompting the catalytic selectivity for the production of value-added chemicals
In terms of the transformation of CH4 to value-added chemicals, by-products are observed due to different extents of catalytic oxidation. For instance, in most cases, formic acid and CO2 are produced upon the formation of the ideal product CH3OH in the catalytic oxidation of CH4. One reason for the formation of these by-products is that deep oxidation is thermodynamically favorable compared to selective oxidation to form an ideal product. The suppression of deep oxidation is a challenging task for the selective oxidation of CH4 to CH3OH. A possible solution is to add a certain amount of a scarifying agent to consume the extra oxidant, preventing CH3OH from being further oxidized.Challenges in achieving high yield of the ideal product
Most of the yields of value-added chemicals such as CH3OH, ethanol, and acetic acid reported in the literature are lower than 10%; typically, they are in the range of 0.1–5%; many of them are in fact lower than 1%. From the measurement point of view, low yield achieved under mild conditions could result in a large error bar in the analysis of products. The limited yield results from several aspects. One is the limited density of the active sites on the support. For example, the maximum number of Rh1O5 and Pd1O4 sites anchored to one gram of microporous aluminosilicate is limited by the slow diffusion of metal precursor in the micropores of the zeolite. In addition, the thermodynamics at low temperature limits the potentially achievable conversion of CH4. To increase the conversion, the separation of products from the liquid environment of the reactants and catalyst could be a necessary step other than catalysis for gaining high kinetics. More importantly, the low yield has been the bottleneck toward any potential production of value-added chemicals at a large scale. So far, there have not been catalysts reported for production at a large scale.Challenges in the fair comparison of the catalytic activities among the reported catalysts
Most references in the literature have reported catalytic performances in the unit of number of μmol CH3OH produced through catalysis by 1 g of a catalyst per h, although most of them never used 1 g of a catalyst in their experiments. The reported number is typically 1–50 μmol CH3OH formed on 1 gram of the catalyst per h. This is a value derived from the result of the measurement such as x μmol CH3OH formed on y gram of the catalyst. Again, in most cases, the weight of a sample used in the experiments is only 0.020–0.050 g instead of 1.0 g. Thus, the amount of measured CH3OH from a sample is in fact in the range of 0.02–2.5 μmol of CH3OH. If the volume of the solution used in the test of NMR of other technique is 2 mL, the original concentration of CH3OH in the solution is only 0.46–57.5 ppm. This is an extremely low concentration. However, most studies did not report the original concentration of the ideal products in their solutions used for the measurements with NMR, GC, mass spectrometry, or other instruments. This makes the evaluation of the uncertainty and reliability of the measurements difficult. The error bars for the quantitative analysis of a solute with a concentration of 0.46–57.5 ppm is in fact quite large for many analytical techniques. To have a fair comparison and a reasonable evaluation, it is necessary to provide the concentration of methanol of the solution used for analysis. To elaborate on the accuracy of the catalytic performances, it is suggested to include the following three pieces of information. (1) The concentration of the products in the solution used for the measurements of NMR or other spectroscopic methods in each test sample and the mass of the catalyst in each path along with the yield in μmol in one gram of the catalyst can be reported as a supplementary message; (2) the amount of product in μmol in a blank experiment without using any catalyst; (3) the absolute error bar of measurements of NMR, mass spectrometry, or other techniques.15. Summary and prospect
The transformations of CH4 to value-added chemicals under mild conditions have driven the development of significant new chemistries in the activation of CH4, oxidative transformation of CH4, and catalytic oxidation of CH4 to value-added chemicals through biocatalysis, molecular catalysis, and heterogeneous catalysis. The significant progress in the fundamental understanding of these chemistries at a molecular level has been achieved. Compared to extensively studied metal catalysis in the last several decades, the topic of transformation of CH4 to oxygenates with high activity and selectivity under mild conditions is still at its incubation period although important achievements have been made. Due to the complexity in the transformation chemistry of CH4 under mild conditions, no catalysts and catalytic processes exhibit feasibility of production at a large scale. The future development of catalysts for the catalytic transformations of CH4 under mild conditions will definitely benefit from these insights achieved in the last two decades. The ultimate goal of the transformation of CH4 through catalysis under mild conditions is to develop catalysts and CH4-based catalytic processes, which can replace the current high-temperature catalytic processes. To realize this goal, a library of durable, cost-effective catalysts with high activity and selectivity for the formation of ideal products under mild conditions need to be developed.The development of an efficient catalyst relies on the fundamental understanding of the profound chemistry in the transformation of CH4 to an ideal oxygenate. Such an understanding has to be established on appropriate characterizations of the active sites at an atomic scale during catalysis. Unlike the exposure of the catalytic sites on an external surface of supported metal or oxide catalysts widely used under high temperature conditions, the catalytic sites of many catalysts active for the transformation of CH4 under mild conditions are embedded in the micropores at the solid–liquid or solid–liquid–gas interfaces under high pressure of the reactant gas. In contrast to catalysis performed at the solid–gas interface at high temperature, the transformations of CH4 to ideal oxygenates under mild conditions have to be performed at solid–liquid or solid–liquid–gas interfaces since the catalysis temperatures under mild conditions are typically lower than the boiling point of the products such as methanol, formic acid, and acetic acid under high pressure of the reactants. This feature of performing catalysis at the solid–liquid or solid–liquid–gas interface under high pressure of the reactant gases makes the characterizations of the catalytic sites at the atomic scale and during catalysis extremely challenging. In addition, the influence of the existence of solvent molecules around the catalyst particles on the catalytic activity and the selectivity in the transformation of CH4 are important issues to address. Also, the lack of kinetic studies on the catalysis performed at the solid–liquid or solid–liquid–gas interface could have limited our understanding of the catalytic process since the molecular diffusion of CH4 in the liquid phase could limit the production of the oxygenate and the diffusion of products formed at the solid–liquid interface could limit the reaction kinetics. Furthermore, compared to the lack of loss of metal atoms of a catalyst while a catalyst is at a high temperature in the gas phase, another complicated factor is the instability of the catalysts resulting from the leaching of the metal atoms of the catalyst in acidic aqueous solutions. Overall, these factors have largely impeded the accomplishment of the profound understanding of these catalytic reactions under mild conditions.
Computational studies have been a quite valuable approach in achieving an understanding of these low-temperature catalytic processes of CH4 transformation. Intermolecular interactions, particularly the interactions among reactants in confined space at sub-nanometer scale in the micropores, the interaction between the reactant molecules and the wall of the micropores, and the interaction between the reactants and the solvent in a confined space, and even the interaction between the products and solvents, must be considered in the computational studies toward proposing a pathway reflecting the actual reaction path at the molecular level.
Many characterization techniques appropriate for studying catalysis during catalysis, performed at the solid–gas interfaces, may not be applicable to low-temperature catalysis at the solid–liquid or solid–liquid–gas interface under high pressure gas phase. The development of characterization methods congruous with the catalytic conditions of CH4 transformation under mild conditions is expected to be significant. Due to these challenges, it is expected that continuous endeavors for a couple of decades could be necessary for the design of catalysts and the development of catalytic processes for the transformation of CH4 to important intermediate compounds or value-added chemicals under mild conditions at a large scale.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Chemical Catalysis Program of Division of Chemistry of National Science Foundation through the project no. NSF-CHE-1462121 of USA and the Catalysis Science Program of the Chemical Sciences, Geosciences and Biosciences Division, Office of science, U.S. Department of Chemistry under Grant No. DE-SC0014561. F. T. was supported by NSF-CHE-1462121 and DE-SC0014561. Y. L. was was partially supported by DE-SC0014561, NSF-CHE-1462121 and NSF-OIA-1539105. Y. T. was supported by his institution. FFT and YT initialized this preparation. FFT spent a few months of 2019 before August 2019 in drafting the manuscript of this article. Y. T. wrote one third of this manuscript and prepared figures. Y. L. wrote a few subsections, edited some section and placed references. This review article does not carry on any intellectual properties from institutions of authors. All information reviewed in this article is knowledge and insights of fundamental understanding of catalytic reaction mechanism published in literature instead of information of any potential applications to any industries.References
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- T. V. Choudhary and V. R. Choudhary, Angew. Chem., Int. Ed., 2008, 47, 1828–1847 CrossRef CAS PubMed.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- W. Chen, Z. L. Fan, X. L. Pan and X. H. Bao, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS PubMed.
- Y. Zhu, Y. Ye, S. Zhang, M. E. Leong and F. Tao, Langmuir, 2012, 28, 8275–8280 CrossRef CAS PubMed.
- K. Xu, B. Sun, J. Lin, W. Wen, Y. Pei, S. Yan, M. Qiao, X. Zhang and B. Zong, Nat. Commun., 2014, 5, 5783 CrossRef CAS PubMed.
- X. Pan, Z. Fan, W. Chen, Y. Ding, H. Luo and X. Bao, Nat. Mater., 2007, 6, 507–511 CrossRef CAS PubMed.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- J. P. den Breejen, P. B. Radstake, G. L. Bezemer, J. H. Bitter, V. Froseth, A. Holmen and K. P. de Jong, J. Am. Chem. Soc., 2009, 131, 7197–7203 CrossRef CAS PubMed.
- R. Liu, R. Liu, X. Ma, B. H. Davis and Z. Li, Fuel, 2018, 211, 827–836 CrossRef CAS.
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y.-W. Li, Q. Zhang, X.-D. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS PubMed.
- B. Zhao, P. Zhai, P. Wang, J. Li, T. Li, M. Peng, M. Zhao, G. Hu, Y. Yang, Y.-W. Li, Q. Zhang, W. Fan and D. Ma, Chem, 2017, 3, 323–333 CAS.
- C. Yang, H. Zhao, Y. Hou and D. Ma, J. Am. Chem. Soc., 2012, 134, 15814–15821 CrossRef CAS PubMed.
- H. M. T. Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef.
- H. M. T. Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef PubMed.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed.
- H. T. Luk, C. Mondelli, D. C. Ferre, J. A. Stewart and J. Perez-Ramirez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–87 CrossRef CAS PubMed.
- Y. Chen, B. deGlee, Y. Tang, Z. Wang, B. Zhao, Y. Wei, L. Zhang, S. Yoo, K. Pei, J. H. Kim, Y. Ding, P. Hu, F. F. Tao and M. Liu, Nat. Energy, 2018, 3, 1042–1050 CrossRef CAS.
- C.-j. Liu, J. Ye, J. Jiang and Y. Pan, ChemCatChem, 2011, 3, 529–541 CrossRef CAS.
- A. Iulianelli, S. Liguori, J. Wilcox and A. Basile, Catal. Rev.: Sci. Eng., 2016, 58, 1–35 CrossRef CAS.
- D. A. J. M. Ligthart, R. A. van Santen and E. J. M. Hensen, J. Catal., 2011, 280, 206–220 CrossRef CAS.
- L. Barelli, G. Bidini, F. Gallorini and S. Servili, Energy, 2008, 33, 554–570 CrossRef CAS.
- Y. Tang, Y. Wei, Z. Wang, S. Zhang, Y. Li, L. Nguyen, Y. Li, Y. Zhou, W. Shen, F. F. Tao and P. Hu, J. Am. Chem. Soc., 2019, 141, 7283–7293 CrossRef CAS PubMed.
- N. A. K. Aramouni, J. G. Touma, B. Abu Tarboush, J. Zeaiter and M. N. Ahmad, Renewable Sustainable Energy Rev., 2018, 82, 2570–2585 CrossRef CAS.
- L. C. Buelens, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Science, 2016, 354, 449–452 CrossRef CAS PubMed.
- Z. Liu, D. C. Grinter, P. G. Lustemberg, N.-P. Thuy-Duong, Y. Zhou, S. Luo, I. Waluyo, E. J. Crumlin, D. J. Stacchiola, J. Zhou, J. Carrasco, H. F. Busnengo, M. Veronica Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 7455–7459 CrossRef CAS PubMed.
- M. Akri, S. Zhao, X. Li, K. Zang, A. F. Lee, M. A. Isaacs, W. Xi, Y. Gangarajula, J. Luo, Y. Ren, Y.-T. Cui, L. Li, Y. Su, X. Pan, W. Wen, Y. Pan, K. Wilson, L. Li, B. Qiao, H. Ishii, Y.-F. Liao, A. Wang, X. Wang and T. Zhang, Nat. Commun., 2019, 10, 5181 CrossRef PubMed.
- Z. Wang, X. M. Cao, J. Zhu and P. Hu, J. Catal., 2014, 311, 469–480 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Science, 2020, 368 DOI:10.1126/science.abb5459.
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi and C. T. Yavuz, Science, 2020, 367, 777–781 CrossRef CAS PubMed.
- T. Margossian, K. Larmier, S. M. Kim, F. Krumeich, A. Fedorov, P. Chen, C. R. Muller and C. Coperet, J. Am. Chem. Soc., 2017, 139, 6919–6927 CrossRef CAS PubMed.
- J. Dong, Q. Fu, H. Li, J. Xiao, B. Yang, B. Zhang, Y. Bai, T. Song, R. Zhang, L. Gao, J. Cai, H. Zhang, Z. Liu and X. Bao, J. Am. Chem. Soc., 2020, 142, 17167–17174 CrossRef CAS PubMed.
- Z. Xie, B. Yan, S. Kattel, J. H. Lee, S. Yao, Q. Wu, N. Rui, E. Gomez, Z. Liu, W. Xu, L. Zhang and J. G. Chen, Appl. Catal., B, 2018, 236, 280–293 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Ind. Eng. Chem. Res., 1998, 37, 2333–2335 CrossRef CAS.
- E. Ruckenstein and Y. H. Hul, Appl. Catal., A, 1999, 183, 85–92 CrossRef CAS.
- S. Liu, G. Xiong, H. Dong, W. Yang, S. Sheng, W. Chu and Z. Yu, Stud. Surf. Sci. Catal., 2000, 130, 3567–3572 CrossRef.
- S. Sengodan, R. Lan, J. Humphreys, D. Du, W. Xu, H. Wang and S. Tao, Renewable Sustainable Energy Rev., 2018, 82, 761–780 CrossRef CAS.
- A. H. Elbadawi, L. Ge, Z. Li, S. Liu, S. Wang and Z. Zhu, Catal. Rev., 2021, 63, 1–67 CrossRef CAS.
- G. Pauletto, N. Libretto, D. C. Boffito, J. T. Miller, A. Jentys, G. S. Patience and J. A. Lercher, Appl. Catal., B, 2021, 286, 119849 CrossRef CAS.
- A. M. Arinaga, M. C. Ziegelski and T. J. Marks, Angew. Chem., Int. Ed., 2021, 60, 10502–10515 CrossRef CAS PubMed.
- S. Liu, S. Udyavara, C. Zhang, M. Peter, T. L. Lohr, V. P. Dravid, M. Neurock and T. J. Marks, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2012666118 CrossRef CAS PubMed.
- S. Zou, Z. Li, Q. Zhou, Y. Pan, W. Yuan, L. He, S. Wang, W. Wen, J. Liu, Y. Wang, Y. Du, J. Yang, L. Xiao, H. Kobayashi and J. Fan, Chin. J. Catal., 2021, 42, 1117–1125 CrossRef CAS.
- P. Wang, G. Zhao, Y. Wang and Y. Lu, Sci. Adv., 2017, 3, e1603180 CrossRef PubMed.
- K. Takanabe, A. M. Khan, Y. Tang, L. Nguyen, A. Ziani, B. W. Jacobs, A. M. Elbaz, S. M. Sarathy and F. Tao, Angew. Chem., Int. Ed., 2017, 56, 10403–10407 CrossRef CAS PubMed.
- F. F. Tao, J. J. Shan, L. Nguyen, Z. Y. Wang, S. R. Zhang, L. Zhang, Z. L. Wu, W. X. Huang, S. B. Zeng and P. Hu, Nat. Commun., 2015, 6, 7798 CrossRef CAS PubMed.
- S. Zhang, J. Shan, L. Nie, L. Nguyen, Z. Wu and F. F. Tao, Surf. Sci., 2016, 648, 156–162 CrossRef CAS.
- Y.-H. Chin, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 15958–15978 CrossRef CAS PubMed.
- Y.-H. Chin, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2013, 135, 15425–15442 CrossRef CAS PubMed.
- M. Cargnello, J. J. Delgado Jaen, J. C. Hernandez Garrido, K. Bakhmutsky, T. Montini, J. J. Calvino Gamez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- L. Luo, X. Tang, W. Wang, Y. Wang, S. Sun, F. Qi and W. Huang, Sci. Rep., 2013, 3, 1625 CrossRef PubMed.
- Y. Gao, J. Bu, Z. Peng and B. Yang, J. Spectrosc., 2014, 2014, 1–10 Search PubMed.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng and M. Wei, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- D. C. Upham, V. Agarwal, A. Khechfe, Z. R. Snodgrass, M. J. Gordon, H. Metiu and E. W. McFarland, Science, 2017, 358, 917–920 CrossRef CAS PubMed.
- Y. Q. Su, J. X. Liu, I. A. W. Filot, L. Zhang and E. J. M. Hensen, ACS Catal., 2018, 8, 6552–6559 CrossRef CAS PubMed.
- J. R. Zeng, M. Tarazkar, T. Pennebaker, M. J. Gordon, H. Metiu and E. W. McFarland, ACS Catal., 2020, 10, 8223–8230 CrossRef CAS.
- C. Palmer, D. C. Upham, S. Smart, M. J. Gordon, H. Metiu and E. W. McFarland, Nat. Catal., 2020, 3, 83–89 CrossRef CAS.
- P. Tomkins, M. Ranocchiari and J. A. van Bokhoven, Acc. Chem. Res., 2017, 50, 418–425 CrossRef CAS PubMed.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- M. A. Newton, A. J. Knorpp, V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, Chem. Soc. Rev., 2020, 49, 1449–1486 RSC.
- P. Del Campo, C. Martinez and A. Corma, Chem. Soc. Rev., 2021, 50, 8511–8595 RSC.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- J. W. M. H. Geerts, J. H. B. J. Hoebink and K. V. D. Wiele, Catal. Today, 1990, 6, 613–620 CrossRef CAS.
- G. A. Somorjai and Y. Li, Introduction to surface chemistry and catalysis, John Wiley & Sons, 2010 Search PubMed.
- S. Zhang, X.-S. Li, B. Chen, X. Zhu, C. Shi and A.-M. Zhu, ACS Catal., 2014, 4, 3481–3489 CrossRef CAS.
- K. Paredis, L. K. Ono, F. Behafarid, Z. Zhang, J. C. Yang, A. I. Frenkel and B. Roldan Cuenya, J. Am. Chem. Soc., 2011, 133, 13455–13464 CrossRef CAS PubMed.
- Z. Liang, T. Li, M. Kim, A. Asthagiri and J. F. Weaver, Science, 2017, 356, 298–301 CrossRef PubMed.
- D. Schröder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1990, 29, 1433–1434 CrossRef.
- Y.-M. Chen, D. Clemmer and P. Armentrout, J. Am. Chem. Soc., 1994, 116, 7815–7826 CrossRef CAS.
- D. Schröder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1995, 34, 1973–1995 CrossRef.
- M. F. Ryan, A. Fiedler, D. Schroeder and H. Schwarz, J. Am. Chem. Soc., 1995, 117, 2033–2040 CrossRef CAS.
- J. Hagen, Industrial catalysis: a practical approach, John Wiley & Sons, 2015 Search PubMed.
- F. Tao and M. Salmeron, Science, 2011, 331, 171–174 CrossRef CAS PubMed.
- F. Tao and P. A. Crozier, Chem. Rev., 2016, 116, 3487–3539 CrossRef CAS PubMed.
- L. Nguyen, F. F. Tao, Y. Tang, J. Dou and X.-J. Bao, Chem. Rev., 2019, 119, 6822–6905 CrossRef PubMed.
- J. Dou, Z. Sun, A. A. Opalade, N. Wang, W. Fu and F. Tao, Chem. Soc. Rev., 2017, 46, 2001–2027 RSC.
- S. Zhang, N. Luan, Y. Zhu, S. Zhan, C.-K. Tsung and F. Tao, Acc. Chem. Res., 2013, 46, 1731–1739 CrossRef CAS PubMed.
- F. Tao, S. Zhang, N. Luan and X. Zhang, Chem. Soc. Rev., 2012, 41, 7980–7993 RSC.
- J. Timoshenko and B. R. Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- D. E. Sayers, E. A. Stern and F. W. Lytle, Phys. Rev. Lett., 1971, 27, 1204 CrossRef CAS.
- F. Lytle, P. Wei, R. Greegor, G. Via and J. Sinfelt, J. Chem. Phys., 1979, 70, 4849–4855 CrossRef CAS.
- F. Lytle, G. Via and J. Sinfelt, J. Chem. Phys., 1977, 67, 3831–3832 CrossRef CAS.
- F. Lytle, G. Via and J. Sinfelt, Synchrotron Radiation Research, Springer, 1980, pp. 401–424 Search PubMed.
- J. H. Sinfelt, Rev. Mod. Phys., 1979, 51, 569 CrossRef CAS.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736–1850 CrossRef CAS PubMed.
- I. Ogino, Chin. J. Catal., 2017, 38, 1481–1488 CrossRef.
- Y. Iwasawa, K. Asakura and M. Tada, XAFS techniques for catalysts, nanomaterials, and surfaces, Springer, 2017 Search PubMed.
- X. Li, X. Yang, J. Zhang, Y. Huang and B. Liu, ACS Catal., 2019, 9, 2521–2531 CrossRef CAS.
- J. Singh, C. Lamberti and J. A. van Bokhoven, Chem. Soc. Rev., 2010, 39, 4754–4766 RSC.
- I. Yasuhiro, X-ray absorption fine structure for catalysts and surfaces, World Scientific, 1996 Search PubMed.
- J. Sá, High-resolution XAS/XES: analyzing electronic structures of catalysts, CRC Press, 2014 Search PubMed.
- J. A. Van Bokhoven and C. Lamberti, X-ray absorption and X-ray emission spectroscopy: theory and applications, John Wiley & Sons, 2016 Search PubMed.
- J. Stöhr, NEXAFS spectroscopy, Springer Science & Business Media, 2013 Search PubMed.
- H. Kuroda, T. Yokoyama, K. Asakura and Y. Iwasawa, Faraday Discuss., 1991, 92, 189–198 RSC.
- M. W. Small, S. I. Sanchez, N. S. Marinkovic, A. I. Frenkel and R. G. Nuzzo, ACS Nano, 2012, 6, 5583–5595 CrossRef CAS PubMed.
- A. Frenkel and J. Rehr, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 585 CrossRef CAS PubMed.
- N. Weiher, E. Bus, B. Gorzolnik, M. Möller, R. Prins and J. A. Van Bokhoven, J. Synchrotron Radiat., 2005, 12, 675–679 CrossRef CAS PubMed.
- L. Nguyen, Y. Tang, Y. Li, X. Zhang, D. Wang and F. Tao, Rev. Sci. Instrum., 2018, 89, 054103 CrossRef PubMed.
- Y. Li, D. Zakharov, S. Zhao, R. Tappero, U. Jung, A. Elsen, P. Baumann, R. G. Nuzzo, E. A. Stach and A. I. Frenkel, Nat. Commun., 2015, 6, 7583 CrossRef CAS PubMed.
- P. T. Kristiansen, T. C. R. Rocha, A. Knop-Gericke, J. H. Guo and L. C. Duda, Rev. Sci. Instrum., 2013, 84, 113107 CrossRef CAS PubMed.
- J. D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, Phys. Chem. Chem. Phys., 2004, 6, 3037–3047 RSC.
- I. Pettiti, D. Gazzoli, M. Inversi, M. Valigi, S. De Rossi, G. Ferraris, P. Porta and S. Colonna, J. Synchrotron Radiat., 1999, 6, 1120–1124 CrossRef CAS.
- A. T. Bell, NMR techniques in catalysis, CRC Press, 2020 Search PubMed.
- A. A. Lysova and I. V. Koptyug, Chem. Soc. Rev., 2010, 39, 4585–4601 RSC.
- W. Zhang, S. Xu, X. Han and X. Bao, Chem. Soc. Rev., 2012, 41, 192–210 RSC.
- W. Zhang, D. Ma, X. Liu, X. Liu and X. Bao, Chem. Commun., 1999, 1091–1092 RSC.
- J. F. Haw, P. W. Goguen, X. Teng, T. W. Skloss and Z. Wang, Angew. Chem., Int. Ed., 1998, 37, 948–949 CrossRef CAS PubMed.
- X. Teng, D. H. Barich, P. W. Goguen, W. Song and J. F. Haw, J. Am. Chem. Soc., 1998, 120, 4025–4026 CrossRef.
- J. F. Haw, J. B. Nicholas, W. Song, F. Deng, Z. Wang, T. Xu and C. S. Heneghan, J. Am. Chem. Soc., 2000, 122, 4763–4775 CrossRef CAS.
- D. M. McCann, D. Lesthaeghe, P. W. Kletnieks, D. R. Guenther, M. J. Hayman, V. Van Speybroeck, M. Waroquier and J. F. Haw, Angew. Chem., Int. Ed., 2008, 47, 5179–5182 CrossRef CAS PubMed.
- M. Xu, K. D. Harris, J. M. Thomas and D. E. Vaughan, Chem. Phys. Chem., 2007, 8, 1311–1313 CrossRef CAS PubMed.
- P. Goguen and J. F. Haw, J. Catal., 1996, 161, 870–872 CrossRef CAS.
- M. Hunger, M. Seiler and T. Horvath, Catal. Lett., 1999, 57, 199–204 CrossRef CAS.
- J. F. Haw, Top. Catal., 1999, 8, 81–86 CrossRef CAS.
- G. W. Haddix, A. T. Bell and J. A. Reimer, J. Phys. Chem., 1989, 93, 5859–5865 CrossRef CAS.
- M. S. Went and J. A. Reimer, J. Am. Chem. Soc., 1992, 114, 5768–5775 CrossRef CAS.
- A. Bendada, G. Chinchen, N. Clayden, B. Heaton, J. Iggo and C. Smith, Catal. Today, 1991, 9, 129–136 CrossRef CAS.
- M. Hunger and T. Horvath, J. Chem. Soc., Chem. Commun., 1995, 1423–1424 RSC.
- C. Keeler, J. Xiong, H. Lock, S. Dec, T. Tao and G. E. Maciel, Catal. Today, 1999, 49, 377–383 CrossRef CAS.
- L. Baltusis, J. S. Frye and G. E. Maciel, J. Am. Chem. Soc., 1986, 108, 7119–7120 CrossRef CAS.
- E. F. Rakiewicz, A. W. Peters, R. F. Wormsbecher, K. J. Sutovich and K. T. Mueller, J. Phys. Chem. B, 1998, 102, 2890–2896 CrossRef CAS.
- P. J. Giammatteo, W. W. Hellmuth, F. G. Ticehurst and P. W. Cope, J. Magn. Reson., 1987, 71, 147–150 Search PubMed.
- W. Zhang, X. Han, X. Liu and X. Bao, J. Mol. Catal. A: Chem., 2003, 194, 107–113 CrossRef CAS.
- J. Zhuang, D. Ma, Z. Yan, F. Deng, X. Liu, X. Han, X. Bao, X. W. Liu, X. Guo and X. Wang, J. Catal., 2004, 221, 670–673 CrossRef CAS.
- D. Ma, Y. Shu, W. Zhang, X. Han, Y. Xu and X. Bao, Angew. Chem., Int. Ed., 2000, 39, 2928–2931 CrossRef CAS PubMed.
- W. Zhang, D. Ma, X. Han, X. Liu, X. Bao, X. Guo and X. Wang, J. Catal., 1999, 188, 393–402 CrossRef CAS.
- K. J. Sutovich, A. W. Peters, E. F. Rakiewicz, R. F. Wormsbecher, S. M. Mattingly and K. T. Mueller, J. Catal., 1999, 183, 155–158 CrossRef CAS.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- H. Zheng, D. Ma, X. Bao, J. Z. Hu, J. H. Kwak, Y. Wang and C. H. Peden, J. Am. Chem. Soc., 2008, 130, 3722–3723 CrossRef CAS PubMed.
- J. Z. Hu, J. H. Kwak, Y. Wang, C. H. Peden, H. Zheng, D. Ma and X. Bao, J. Phys. Chem. C, 2009, 113, 2936–2942 CrossRef CAS.
- S. Xu, W. Zhang, X. Liu, X. Han and X. Bao, J. Am. Chem. Soc., 2009, 131, 13722–13727 CrossRef CAS PubMed.
- J. Weitkamp and S. Maixner, Zeolites, 1987, 7, 6–8 CrossRef CAS.
- J. L. White, N. D. Lazo, B. R. Richardson and J. F. Haw, J. Catal., 1990, 125, 260–263 CrossRef CAS.
- J. Huang, Y. Jiang, V. R. Marthala, Y. S. Ooi and M. Hunger, Chem. Phys. Chem., 2008, 9, 1107–1109 CrossRef CAS PubMed.
- M. Xu, W. Wang and M. Hunger, Chem. Commun., 2003, 722–723 RSC.
- T. Bürgi and A. Baiker, Adv. Catal., 2006, 50, 227–283 Search PubMed.
- J. Ryczkowski, Catal. Today, 2001, 68, 263–381 CrossRef CAS.
- T. Bürgi, R. Wirz and A. Baiker, J. Phys. Chem. B, 2003, 107, 6774–6781 CrossRef.
- J. A. Lercher, V. Veefkind and K. Fajerwerg, Vib. Spectrosc., 1999, 19, 107–121 CrossRef CAS.
- A. Vimont, F. Thibault-Starzyk and M. Daturi, Chem. Soc. Rev., 2010, 39, 4928–4950 RSC.
- G. L. Chiarello, M. Nachtegaal, V. Marchionni, L. Quaroni and D. Ferri, Rev. Sci. Instrum., 2014, 85, 074102 CrossRef PubMed.
- M. S. Schneider, J. D. Grunwaldt, T. Burgi and A. Baiker, Rev. Sci. Instrum., 2003, 74, 4121–4128 CrossRef CAS.
- J.-M. Andanson and A. Baiker, Chem. Soc. Rev., 2010, 39, 4571–4584 RSC.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
- B. L. Mojet, S. D. Ebbesen and L. Lefferts, Chem. Soc. Rev., 2010, 39, 4643–4655 RSC.
- F. Zaera, Chem. Soc. Rev., 2014, 43, 7624–7663 RSC.
- P. A. Jacobs and R. A. van Santen, Zeolites: facts, figures, future, Elsevier, 1989 Search PubMed.
- E. Stavitski and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4615–4625 RSC.
- M. Nowotny, J. A. Lercher and H. Kessler, Zeolites, 1991, 11, 454–459 CrossRef CAS.
- M. H. F. Kox, K. F. Domke, J. P. R. Day, G. Rago, E. Stavitski, M. Bonn and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2009, 48, 8990–8994 CrossRef CAS PubMed.
- J. Melsheimer and R. Schlogl, Ber. Bunsen-Ges., 1997, 101, 733–740 CrossRef.
- J. Melsheimer and R. Schlogl, Fresenius' J. Anal. Chem., 1997, 357, 397–400 CrossRef CAS.
- X. T. Gao, J. M. Jehng and I. E. Wachs, J. Catal., 2002, 209, 43–50 CrossRef CAS.
- L. J. Burcham, G. Deo, X. T. Gao and I. E. Wachs, Top. Catal., 2000, 11, 85–100 CrossRef.
- E. V. Kondratenko and M. Baerns, Appl. Catal., A, 2001, 222, 133–143 CrossRef CAS.
- A. Bruckner, Appl. Catal., A, 2000, 200, 287–297 CrossRef CAS.
- M. D. Argyle, K. D. Chen, C. Resini, C. Krebs, A. T. Bell and E. Iglesia, Chem. Commun., 2003, 2082–2083, 10.1039/b305264h.
- M. D. Argyle, K. D. Chen, C. Resini, C. Krebs, A. T. Bell and E. Iglesia, J. Phys. Chem. B, 2004, 108, 2345–2353 CrossRef CAS.
- A. Bruckner and E. Kondratenko, Catal. Today, 2006, 113, 16–24 CrossRef.
- J. Q. Lu, J. J. Bravo-Suarez, A. Takahashi, M. Haruta and S. T. Oyama, J. Catal., 2005, 232, 85–95 CrossRef CAS.
- B. F. Sels, D. E. De Vos, P. J. Grobet and P. A. Jacobs, Chem. – Eur. J., 2001, 7, 2547–2556 CrossRef CAS.
- M. Schwidder, W. Grunert, U. Bentrup and A. Bruckner, J. Catal., 2006, 239, 173–186 CrossRef.
- T. A. Nijhuis, S. J. Tinnemans, T. Visser and B. M. Weckhuysen, Chem. Eng. Sci., 2004, 59, 5487–5492 CrossRef CAS.
- T. X. Nijhuis, S. J. Tinnemans, T. Visser and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2003, 5, 4361–4365 RSC.
- F. C. Jentoft, in Advances in Catalysis, ed. B. C. Gates and H. Knozinger, 2009, vol. 52, pp. 129–211 Search PubMed.
- B. M. Weckhuysen and R. A. Schoonheydt, Catal. Today, 1999, 49, 441–451 CrossRef CAS.
- B. M. Weckhuysen, P. Voort and G. Catana, Spectroscopy of transition metal ions on surfaces, Leuven University Press, 2000 Search PubMed.
- B. M. Weckhuysen, A. A. Verberckmoes, J. Debaere, K. Ooms, I. Langhans and R. A. Schoonheydt, J. Mol. Catal. A: Chem., 2000, 151, 115–131 CrossRef CAS.
- B. M. Weckhuysen, D. Baetens and R. A. Schoonheydt, Angew. Chem., Int. Ed., 2000, 39, 3419–3422 CrossRef CAS PubMed.
- B. M. Weckhuysen, A. Bensalem and R. A. Schoonheydt, J. Chem. Soc., Faraday Trans., 1998, 94, 2011–2014 RSC.
- S. Tinnemans, M. Kox, T. Nijhuis, T. Visser and B. Weckhuysen, Phys. Chem. Chem. Phys., 2005, 7, 211–216 RSC.
- S. Kuba, P. Lukinskas, R. Ahmad, F. C. Jentoft, R. K. Grasselli, B. C. Gates and H. Knözinger, J. Catal., 2003, 219, 376–388 CrossRef CAS.
- F. C. Jentoft, Adv. Catal., 2009, 52, 129–211 CAS.
- L. G. van de Water, J. A. Bergwerff, G. Leliveld, B. M. Weckhuysen and K. P. de Jong, J. Phys. Chem. B, 2005, 109, 14513–14522 CrossRef CAS PubMed.
- L. G. van de Water, J. A. Bergwerff, T. A. Nijhuis, K. P. de Jong and B. M. Weckhuysen, J. Am. Chem. Soc., 2005, 127, 5024–5025 CrossRef CAS PubMed.
- U. Bentrup, A. Brückner, C. Rüdinger and H.-J. Eberle, Appl. Catal., A, 2004, 269, 237–248 CrossRef CAS.
- L. Nguyen and F. Tao, Rev. Sci. Instrum., 2016, 87, 064101 CrossRef PubMed.
- M. Salmeron and R. Schlogl, Surf. Sci. Rep., 2008, 63, 169–199 CrossRef CAS.
- D. E. Starr, Z. Liu, M. Haevecker, A. Knop-Gericke and H. Bluhm, Chem. Soc. Rev., 2013, 42, 5833–5857 RSC.
- Y. Han, H. Zhang, Y. Yu and Z. Liu, ACS Catal., 2021, 11, 1464–1484 CrossRef CAS.
- C. S. Fadley, Prog. Surf. Sci., 1984, 16, 275–388 CrossRef CAS.
- Y. Takagi, T. Uruga, M. Tada, Y. Iwasawa and T. Yokoyama, Acc. Chem. Res., 2018, 51, 719–727 CrossRef CAS PubMed.
- U. Gelius, E. Basiler, S. Svensson, T. Bergmark and K. Siegbahn, J. Electron Spectrosc. Relat. Phenom., 1973, 2, 405–434 CrossRef CAS.
- N. Luan, P. Tao, H. Liu, M. Al-Hada, M. Amati, H. Sezen, Y. Tang, L. Gregoratti and F. Tao, Chem. Commun., 2018, 54, 9981–9984 RSC.
- Y. Tang, Y. T. Li, V. Fung, D. E. Jiang, W. X. Huang, S. R. Zhang, Y. Iwasawa, T. Sakata, L. Nguyen, X. Y. Zhang, A. I. Frenkel and F. Tao, Nat. Commun., 2018, 9, 1231 CrossRef PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- M. A. Culpepper and A. C. Rosenzweig, Crit. Rev. Biochem. Mol. Biol., 2012, 47, 483–492 CrossRef CAS PubMed.
- H. J. Kim, J. Huh, Y. W. Kwon, D. Park, Y. Yu, Y. E. Jang, B. R. Lee, E. Jo, E. J. Lee, Y. Heo, W. Lee and J. Lee, Nat. Catal., 2019, 2, 342–353 CrossRef CAS.
- N. R. Hunter, H. D. Gesser, L. A. Morton, P. S. Yarlagadda and D. P. C. Fung, Appl. Catal., 1990, 57, 45–54 CrossRef CAS.
- G. Ljunger, P. Adlercreutz and B. Mattiasson, Biocatalysis, 1993, 7, 279–288 CrossRef CAS.
- M. Urrutigoity and J. Souppe, Biocatalysis, 1989, 2, 145–149 CrossRef CAS.
- Z. Gou, X.-H. Xing, M. Luo, H. Jiang, B. Han, H. Wu, L. Wang and F. Zhang, FEMS Microbiol. Lett., 2006, 263, 136–141 CrossRef CAS PubMed.
- R. L. Lieberman and A. C. Rosenzweig, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 147–164 CrossRef CAS PubMed.
- I. McDonald, H. Uchiyama, S. Kambe, O. Yagi and J. Murrell, Appl. Environ. Microbiol., 1997, 63, 1898–1904 CrossRef CAS PubMed.
- J. Green and H. Dalton, J. Biol. Chem., 1985, 260, 5795–5801 Search PubMed.
- D. Park and J. Lee, Korean J. Chem. Eng., 2013, 30, 977–987 CrossRef CAS.
- S. Friedle, E. Reisner and S. J. Lippard, Chem. Soc. Rev., 2010, 39, 2768–2779 RSC.
- B. Fox, W. Froland, J. Dege and J. Lipscomb, J. Biol. Chem., 1989, 264, 10023–10033 CrossRef CAS.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- H. Dalton, Advances in Applied Microbiology, Elsevier, 1980, vol. 26, pp. 71–87 Search PubMed.
- J. Lund, M. P. Woodland and H. Dalton, Eur. J. Biochem., 1985, 147, 297–305 CrossRef CAS PubMed.
- J. Colby and H. Dalton, Biochem. J., 1978, 171, 461–468 CrossRef CAS PubMed.
- J. Colby and H. Dalton, Biochem. J., 1979, 177, 903–908 CrossRef CAS PubMed.
- M. P. Woodland and H. Dalton, J. Biol. Chem., 1984, 259, 53–59 CrossRef CAS.
- R. Balasubramanian and A. C. Rosenzweig, Acc. Chem. Res., 2007, 40, 573–580 CrossRef CAS PubMed.
- S. I. Chan and S. S.-F. Yu, Acc. Chem. Res., 2008, 41, 969–979 CrossRef CAS PubMed.
- A. S. Hakemian and A. C. Rosenzweig, Annu. Rev. Biochem., 2007, 76, 223–241 CrossRef CAS PubMed.
- R. L. Lieberman and A. C. Rosenzweig, Nature, 2005, 434, 177–182 CrossRef CAS PubMed.
- K.-Y. Ng, L.-C. Tu, Y.-S. Wang, S. I. Chan and S. S. F. Yu, ChemBioChem, 2008, 9, 1116–1123 CrossRef CAS PubMed.
- S. S. F. Yu, L. Y. Wu, K. H. C. Chen, W. I. Luo, D. S. Huang and S. I. Chan, J. Biol. Chem., 2003, 278, 40658–40669 CrossRef CAS PubMed.
- B. Wilkinson, M. Zhu, N. D. Priestley, H. H. T. Nguyen, H. Morimoto, P. G. Williams, S. I. Chan and H. G. Floss, J. Am. Chem. Soc., 1996, 118, 921–922 CrossRef CAS.
- P. P. Y. Chen and S. I. Chan, J. Inorg. Biochem., 2006, 100, 801–809 CrossRef CAS PubMed.
- K. Yoshizawa and Y. Shiota, J. Am. Chem. Soc., 2006, 128, 9873–9881 CrossRef CAS PubMed.
- S. I. Chan, V. C. C. Wang, J. C. H. Lai, S. S. F. Yu, P. P. Y. Chen, K. H. C. Chen, C.-L. Chen and M. K. Chan, Angew. Chem., Int. Ed., 2007, 46, 1992–1994 CrossRef CAS PubMed.
- S. I. Chan, K. H. C. Chen, S. S. F. Yu, C. L. Chen and S. S. J. Kuo, Biochemistry, 2004, 43, 4421–4430 CrossRef CAS PubMed.
- S. S. F. Yu, K. H. C. Chen, M. Y. H. Tseng, Y. S. Wang, C. F. Tseng, Y. J. Chen, D. S. Huang and S. I. Chan, J. Bacteriol., 2003, 185, 5915–5924 CrossRef CAS PubMed.
- M. Martinho, D. W. Choi, A. A. DiSpirito, W. E. Antholine, J. D. Semrau and E. Muenck, J. Am. Chem. Soc., 2007, 129, 15783–15785 CrossRef CAS PubMed.
- H. H. T. Nguyen, K. H. Nakagawa, B. Hedman, S. J. Elliott, M. E. Lidstrom, K. O. Hodgson and S. I. Chan, J. Am. Chem. Soc., 1996, 118, 12766–12776 CrossRef CAS.
- K. H. C. Chen, C. L. Chen, C. F. Tseng, S. S. F. Yu, S. C. Ke, J. F. Lee, H. T. Nguyen, S. J. Elliott, J. O. Alben and S. I. Chan, J. Chin. Chem. Soc., 2004, 51, 1081–1098 CrossRef CAS.
- S. C. Hung, C. L. Chen, K. H. C. Chen, S. S. F. Yu and S. I. Chan, J. Chin. Chem. Soc., 2004, 51, 1229–1244 CrossRef CAS.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- J. L. Bennett and P. T. Wolczanski, J. Am. Chem. Soc., 1997, 119, 10696–10719 CrossRef CAS.
- G. Luinstra, L. Wang, S. Stahl, J. Labinger and J. Bercaw, J. Organomet. Chem., 1995, 504, 75–91 CrossRef CAS.
- A. E. Shilov and G. B. Shul'pin, Russ. Chem. Rev., 1987, 56, 442 CrossRef.
- M. Lin, C. Shen, E. A. Garcia-Zayas and A. Sen, J. Am. Chem. Soc., 2001, 123, 1000–1001 CrossRef CAS PubMed.
- M. S. Freund, J. A. Labinger, N. S. Lewis and J. E. Bercaw, J. Mol. Catal., 1994, 87, L11–L15 CrossRef CAS.
- R. A. Periana, D. J. Taube, E. R. Evitt, D. G. Loffler, P. R. Wentrcek, G. Voss and T. Masuda, Science, 1993, 259, 340–343 CrossRef CAS PubMed.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560–564 CrossRef CAS PubMed.
- J. Allison, Prog. Inorg. Chem., 1986, 627–676 CAS.
- P. Watson, J. Liebman and A. Greenberg, Selective Hydrocarbon Activation, VCH Publishers, Inc., New York, 1990 Search PubMed.
- K. Eller and H. Schwarz, Chem. Rev., 1991, 91, 1121–1177 CrossRef CAS.
- Y. Shiota and K. Yoshizawa, J. Am. Chem. Soc., 2000, 122, 12317–12326 CrossRef CAS.
- A. Gannouni, F. O. Delbecq, M. Saïd Zina and P. Sautet, J. Phys. Chem. A, 2017, 121, 5500–5508 CrossRef CAS PubMed.
- V. C.-C. Wang, S. Maji, P. P.-Y. Chen, H. K. Lee, S. S.-F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS.
- M. H. Mahyuddin, A. Staykov, Y. Shiota, M. Miyanishi and K. Yoshizawa, ACS Catal., 2017, 7, 3741–3751 CrossRef CAS.
- T. Jackson, D. Jacobson and B. Freiser, J. Am. Chem. Soc., 1984, 106, 1252–1257 CrossRef CAS.
- D. Schröder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1990, 29, 1431–1433 CrossRef.
- D. Schroeder, A. Fiedler, M. F. Ryan and H. Schwarz, J. Phys. Chem., 1994, 98, 68–70 CrossRef CAS.
- D. Clemmer, Y.-M. Chen, F. A. Khan and P. Armentrout, J. Phys. Chem., 1994, 98, 6522–6529 CrossRef CAS.
- D. Schröder and H. Schwarz, Helv. Chim. Acta, 1992, 75, 1281–1287 CrossRef.
- D. Schrder, H. Florencio, W. Zummack and H. Schwarz, Helv. Chim. Acta, 1992, 75, 1792–1797 CrossRef.
- H. Becker, D. Schroeder, W. Zummack and H. Schwarz, J. Am. Chem. Soc., 1994, 116, 1096–1100 CrossRef CAS.
- M. F. Ryan, A. Fiedler, D. Schroeder and H. Schwarz, Organometallics, 1994, 13, 4072–4081 CrossRef CAS.
- K. K. Irikura and J. Beauchamp, J. Am. Chem. Soc., 1989, 111, 75–85 CrossRef CAS.
- P. B. Grosshans and A. G. Marshall, Anal. Chem., 1991, 63, 2057–2061 CrossRef CAS.
- D. Clemmer, N. Aristov and P. Armentrout, J. Phys. Chem., 1993, 97, 544–552 CrossRef CAS.
- M. Azzaro, S. Breton, M. Decouzon and S. Geribaldi, Int. J. Mass Spectrom. Ion Processes, 1993, 128, 1–20 CrossRef CAS.
- K. Liu and J. Parson, J. Phys. Chem., 1979, 83, 970–973 CrossRef CAS.
- J. M. Parnis, R. D. Lafleur and D. M. Rayner, Chem. Phys. Lett., 1994, 218, 544–550 CrossRef CAS.
- H. Kang and J. Beauchamp, J. Am. Chem. Soc., 1986, 108, 5663–5668 CrossRef CAS PubMed.
- R. Wesendrup, D. Schroder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1994, 33, 1174–1176 CrossRef.
- D. Olson, W. Haag and R. Lago, J. Catal., 1980, 61, 390–396 CrossRef CAS.
- C. Zicovich-Wilson, A. Corma and P. Viruela, J. Phys. Chem., 1994, 98, 10863–10870 CrossRef CAS.
- A. Corma, H. García, G. Sastre and P. M. Viruela, J. Phys. Chem. B, 1997, 101, 4575–4582 CrossRef CAS.
- M. H. Mahyuddin, A. Staykov, Y. Shiota and K. Yoshizawa, ACS Catal., 2016, 6, 8321–8331 CrossRef CAS.
- Y. Jin, C. Sun and S. Su, Phys. Chem. Chem. Phys., 2015, 17, 16277–16284 RSC.
- J. Anderson and P. Tsai, J. Chem. Soc., Chem. Commun., 1987, 1435–1436 RSC.
- V. Sobolev, A. Kharitonov, Y. A. Paukshtis and G. Panov, J. Mol. Catal., 1993, 84, 117–124 CrossRef CAS.
- G. Panov, G. Sheveleva, A. E. A. Kharitonov, V. Romannikov and L. Vostrikova, Appl. Catal., A, 1992, 82, 31–36 CrossRef CAS.
- G. Pannov, V. Sobolev and A. Kharitonov, J. Mol. Catal., 1990, 61, 85–97 CrossRef.
- V. Sobolev, K. Dubkov, O. Panna and G. Panov, Catal. Today, 1995, 24, 251–252 CrossRef CAS.
- K. Dubkov, V. Sobolev, E. Talsi, M. Rodkin, N. Watkins, A. Shteinman and G. Panov, J. Mol. Catal. A: Chem., 1997, 123, 155–161 CrossRef CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- E. M. Alayon, M. Nachtegaal, M. Ranocchiari and J. A. van Bokhoven, Chem. Commun., 2012, 48, 404–406 RSC.
- G. Panov, A. Kharitonov and V. Sobolev, Appl. Catal., A, 1993, 98, 1–20 CrossRef CAS.
- A. M. Volodin, V. A. Bolshov and G. I. Panov, J. Phys. Chem., 1994, 98, 7548–7550 CrossRef CAS.
- M. Filatov, A. Pelmenschikov and G. Zhidomirov, J. Mol. Catal., 1993, 80, 243–251 CrossRef CAS.
- E. Starokon, K. Dubkov, L. Pirutko and G. Panov, Top. Catal., 2003, 23, 137–143 CrossRef CAS.
- V. I. Sobolev, O. N. Kovalenko, A. S. Kharitonov, Y. D. Pankrat'ev and G. I. Panov, Mendeleev Commun., 1991, 1, 29–30 CrossRef.
- K. Dubkov, N. Ovanesyan, A. Shteinman, E. Starokon and G. Panov, J. Catal., 2002, 207, 341–352 CrossRef CAS.
- A. Arbuznikov and G. Zhidomirov, Catal. Lett., 1996, 40, 17–23 CrossRef CAS.
- L. Que and Y. Dong, Acc. Chem. Res., 1996, 29, 190–196 CrossRef CAS.
- K. Yoshizawa, Y. Shiota, T. Yumura and T. Yamabe, J. Phys. Chem. B, 2000, 104, 734–740 CrossRef CAS.
- K. Yoshizawa, Y. Shiota and T. Yamabe, Chem. – Eur. J., 1997, 3, 1160–1169 CrossRef CAS.
- K. Yoshizawa, Y. Shiota and T. Yamabe, J. Am. Chem. Soc., 1998, 120, 564–572 CrossRef CAS.
- D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed.
- R. Hamada, Y. Shibata, S. Nishiyama and S. Tsuruya, Phys. Chem. Chem. Phys., 2003, 5, 956–965 RSC.
- M. Shelef, Chem. Rev., 1995, 95, 209–225 CrossRef CAS.
- H. Yahiro and M. Iwamoto, Appl. Catal., A, 2001, 222, 163–181 CrossRef CAS.
- M. H. Groothaert, J. A. van Bokhoven, A. A. Battiston, B. M. Weckhuysen and R. A. Schoonheydt, J. Am. Chem. Soc., 2003, 125, 7629–7640 CrossRef CAS PubMed.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, ACS Catal., 2014, 4, 16–22 CrossRef CAS.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- A. Itadani, T. Yumura, T. Ohkubo, H. Kobayashi and Y. Kuroda, Phys. Chem. Chem. Phys., 2010, 12, 6455–6465 RSC.
- P. Vanelderen, R. G. Hadt, P. J. Smeets, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, J. Catal., 2011, 284, 157–164 CrossRef CAS PubMed.
- P. Vanelderen, B. E. Snyder, M.-L. Tsai, R. G. Hadt, J. Vancauwenbergh, O. Coussens, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 6383–6392 CrossRef CAS PubMed.
- B. Wathen, M. Kuiper, V. Walker and Z. Jia, J. Am. Chem. Soc., 2003, 125, 729–737 CrossRef CAS PubMed.
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Catal. Today, 2005, 110, 303–309 CrossRef CAS.
- R. A. Himes and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18877–18878 CrossRef CAS PubMed.
- J. S. Woertink, L. Tian, D. Maiti, H. R. Lucas, R. A. Himes, K. D. Karlin, F. Neese, C. Würtele, M. C. Holthausen and E. Bill, Inorg. Chem., 2010, 49, 9450–9459 CrossRef CAS PubMed.
- P. J. Smeets, R. G. Hadt, J. S. Woertink, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2010, 132, 14736–14738 CrossRef CAS PubMed.
- R. A. Himes, K. Barnese and K. D. Karlin, Angew. Chem., Int. Ed., 2010, 49, 6714–6716 CrossRef CAS PubMed.
- D. Palagin, A. J. Knorpp, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Nanoscale, 2017, 9, 1144–1153 RSC.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- A. Burkhardt, E. T. Spielberg, H. Görls and W. Plass, Inorg. Chem., 2008, 47, 2485–2493 CrossRef CAS PubMed.
- E. I. Solomon, J. W. Ginsbach, D. E. Heppner, M. T. Kieber-Emmons, C. H. Kjaergaard, P. J. Smeets, L. Tian and J. S. Woertink, Faraday Discuss., 2011, 148, 11–39 RSC.
- S. I. Chan, V. C. C. Wang, J. C. H. Lai, S. S. F. Yu, P. P. Y. Chen, K. H. C. Chen, C. L. Chen and M. K. Chan, Angew. Chem., Int. Ed., 2007, 46, 1992–1994 CrossRef CAS PubMed.
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. Hensen and E. A. Pidko, J. Catal., 2016, 338, 305–312 CrossRef CAS.
- S. Grundner, M. A. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- G. Centi and S. Perathoner, Appl. Catal., A, 1995, 132, 179–259 CrossRef CAS.
- Y. Kuroda and M. Iwamoto, Top. Catal., 2004, 28, 111–118 CrossRef CAS.
- M. H. Groothaert, K. Lievens, H. Leeman, B. M. Weckhuysen and R. A. Schoonheydt, J. Catal., 2003, 220, 500–512 CrossRef CAS.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi, M. Ranocchiari and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2015, 17, 7681–7693 RSC.
- V. A. Veefkind, M. L. Smidt and J. A. Lercher, Appl. Catal., A, 2000, 194, 319–332 CrossRef.
- N. Dietl, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2012, 51, 5544–5555 CrossRef CAS PubMed.
- P. Tomkins, A. Mansouri, S. E. Bozbag, F. Krumeich, M. B. Park, E. M. C. Alayon, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2016, 55, 5467–5471 CrossRef CAS PubMed.
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Catal. Lett., 2010, 138, 14–22 CrossRef CAS.
- P. Vanelderen, J. Vancauwenbergh, M. L. Tsai, R. G. Hadt, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, Chem. Phys. Chem., 2014, 15, 91–99 CrossRef CAS PubMed.
- A. A. Verma, S. A. Bates, T. Anggara, C. Paolucci, A. A. Parekh, K. Kamasamudram, A. Yezerets, J. T. Miller, W. N. Delgass and W. F. Schneider, J. Catal., 2014, 312, 179–190 CrossRef CAS.
- Z.-J. Zhao, A. Kulkarni, L. Vilella, J. K. Nørskov and F. Studt, ACS Catal., 2016, 6, 3760–3766 CrossRef CAS.
- M. J. Wulfers, S. Teketel, B. Ipek and R. F. Lobo, Chem. Commun., 2015, 51, 4447–4450 RSC.
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Catal. Lett., 2010, 136, 52–56 CrossRef CAS.
- N. V. Beznis, A. N. Van Laak, B. M. Weckhuysen and J. H. Bitter, Microporous Mesoporous Mater., 2011, 138, 176–183 CrossRef CAS.
- J. Shan, W. Huang, N. Luan, Y. Yu, S. Zhang, Y. Li, A. I. Frenkel and F. Tao, Langmuir, 2014, 30, 8558–8569 CrossRef CAS PubMed.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- K. Narsimhan, V. K. Michaelis, G. Mathies, W. R. Gunther, R. G. Griffin and Y. Roman-Leshkov, J. Am. Chem. Soc., 2015, 137, 1825–1832 CrossRef CAS PubMed.
- I. Yamanaka, M. Soma and K. Oisuka, J. Chem. Soc., Chem. Commun., 1995, 2235–2236 RSC.
- I. Yamanaka, M. Soma and K. Otsuka, Res. Chem. Intermed., 2000, 26, 129–135 CrossRef CAS.
- L. Wang, S. Zhang, Y. Zhu, A. Patlolla, J. Shan, H. Yoshida, S. Takeda, A. I. Frenkel and F. Tao, ACS Catal., 2013, 3, 1011–1019 CrossRef CAS.
- S. Zhang, Y. Tang, L. Nguyen, Y.-F. Zhao, Z. Wu, T.-W. Goh, J. J. Liu, Y. Li, T. Zhu and W. Huang, ACS Catal., 2018, 8, 110–121 CrossRef CAS.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed.
- P. Ratnasamy and R. Kumar, Catal. Today, 1991, 9, 329–416 CrossRef.
- A. Battiston, J. Bitter, W. Heijboer, F. De Groot and D. Koningsberger, J. Catal., 2003, 215, 279–293 CrossRef CAS.
- J. Jia, Q. Sun, B. Wen, L. X. Chen and W. M. Sachtler, Catal. Lett., 2002, 82, 7–11 CrossRef CAS.
- A. Battiston, J. Bitter and D. Koningsberger, J. Catal., 2003, 218, 163–177 CrossRef CAS.
- J. Xu, R. D. Armstrong, G. Shaw, N. F. Dummer, S. J. Freakley, S. H. Taylor and G. J. Hutchings, Catal. Today, 2016, 270, 93–100 CrossRef CAS.
- C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, M. H. Ab Rahim, M. M. Forde, A. Thetford, D. M. Murphy, H. Hagen and E. E. Stangland, Chem. – Eur. J., 2012, 18, 15735–15745 CrossRef CAS PubMed.
- B. Ensing, F. Buda, P. E. Blöchl and E. J. Baerends, Phys. Chem. Chem. Phys., 2002, 4, 3619–3627 RSC.
- B. Ensing, F. Buda, M. C. Gribnau and E. J. Baerends, J. Am. Chem. Soc., 2004, 126, 4355–4365 CrossRef CAS PubMed.
- C. Hammond, N. Dimitratos, R. L. Jenkins, J. A. Lopez-Sanchez, S. A. Kondrat, M. Hasbi ab Rahim, M. M. Forde, A. Thetford, S. H. Taylor and H. Hagen, ACS Catal., 2013, 3, 689–699 CrossRef CAS.
- C. Kalamaras, D. Palomas, R. Bos, A. Horton, M. Crimmin and K. Hellgardt, Catal. Lett., 2016, 146, 483–492 CrossRef CAS.
- M. Shahami and D. F. Shantz, Catal. Sci. Technol., 2019, 9, 2945–2951 RSC.
- X. Cui, H. Li, Y. Wang, Y. Hu, L. Hua, H. Li, X. Han, Q. Liu, F. Yang, L. He, X. Chen, Q. Li, J. Xiao, D. Deng and X. Bao, Chem, 2018, 4, 1902–1910 CAS.
- W. X. Huang, S. R. Zhang, Y. Tang, Y. T. Li, L. Nguyen, Y. Y. Li, J. J. Shan, D. Q. Xiao, R. Gagne, A. I. Frenkel and F. Tao, Angew. Chem., Int. Ed., 2016, 55, 13441–13445 CrossRef CAS PubMed.
- S. Yashnik and Z. Ismagilov, Appl. Catal., B, 2015, 170, 241–254 CrossRef.
- S. Yashnik and Z. Ismagilov, Kinet. Catal., 2016, 57, 776–796 CrossRef CAS.
- S. A. Yashnik, V. V. Boltenkov, D. E. Babushkin, O. P. Taran and V. N. Parmon, Top. Catal., 2020, 1–19 Search PubMed.
- S. Bai, F. Liu, B. Huang, F. Li, H. Lin, T. Wu, M. Sun, J. Wu, Q. Shao, Y. Xu and X. Huang, Nat. Commun., 2020, 11, 954 CrossRef CAS PubMed.
- Y. Kwon, T. Y. Kim, G. Kwon, J. Yi and H. Lee, J. Am. Chem. Soc., 2017, 139, 17694–17699 CrossRef CAS PubMed.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223–226 CrossRef CAS PubMed.
- J. K. Edwards, B. Solsona, E. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang and C. Mesters, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- H. D. Gesser, N. R. Hunter and C. B. Prakash, Chem. Rev., 1985, 85, 235–244 CrossRef CAS.
- A. Butler, M. J. Clague and G. E. Meister, Chem. Rev., 1994, 94, 625–638 CrossRef CAS.
- G. Shul'pin, D. Attanasio and L. Suber, Russ. Chem. Bull., 1993, 42, 55–59 CrossRef.
- G. V. Nizova, G. Süss-Fink and G. B. Shul’pin, Chem. Commun., 1997, 397–398 RSC.
- G. Süss-Fink, G. V. Nizova, S. Stanislas and G. B. Shul'pin, J. Mol. Catal. A: Chem., 1998, 130, 163–170 CrossRef.
- I. Arends, K. Ingold and D. Wayner, J. Am. Chem. Soc., 1995, 117, 4710–4711 CrossRef CAS.
- J. Kim, R. G. Harrison, C. Kim and L. Que, J. Am. Chem. Soc., 1996, 118, 4373–4379 CrossRef CAS.
- R. H. Fish, M. S. Konings, K. J. Oberhausen, R. H. Fong, W. M. Yu, G. Christou, J. B. Vincent, D. K. Coggin and R. M. Buchanan, Inorg. Chem., 1991, 30, 3002–3006 CrossRef CAS.
- Y. Seki, N. Mizuno and M. Misono, Appl. Catal., A, 1997, 158, L47–L51 CrossRef CAS.
- X. Wei, L. Ye and Y. Yuan, J. Nat. Gas Chem., 2009, 18, 295–299 CrossRef CAS.
- Q. Yuan, W. Deng, Q. Zhang and Y. Wang, Adv. Synth. Catal., 2007, 349, 1199–1209 CrossRef CAS.
- M. Schroeder, Chem. Rev., 1980, 80, 187–213 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC.
- Ü. İşci, A. S. Faponle, P. Afanasiev, F. Albrieux, V. Briois, V. Ahsen, F. Dumoulin, A. B. Sorokin and S. P. de Visser, Chem. Sci., 2015, 6, 5063–5075 RSC.
- M. G. Quesne, D. Senthilnathan, D. Singh, D. Kumar, P. Maldivi, A. B. Sorokin and S. P. de Visser, ACS Catal., 2016, 6, 2230–2243 CrossRef CAS.
- P. Afanasiev and A. B. Sorokin, Acc. Chem. Res., 2016, 49, 583–593 CrossRef CAS PubMed.
- E. V. Kudrik, P. Afanasiev, L. X. Alvarez, P. Dubourdeaux, M. Clémancey, J.-M. Latour, G. Blondin, D. Bouchu, F. Albrieux and S. E. Nefedov, Nat. Chem., 2012, 4, 1024–1029 CrossRef CAS PubMed.
- S. A. Ikbal, C. Colomban, D. Zhang, M. Delecluse, T. Brotin, V. Dufaud, J.-P. Dutasta, A. B. Sorokin and A. Martinez, Inorg. Chem., 2019, 58, 7220–7228 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |