
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Branching out: redox strategies towards the synthesis of acyclic α-tertiary ethers
Benjamin D. A.
Shennan†
 ,
Diana
Berheci†
,
Jessica L.
Crompton†
,
Timothy A.
Davidson†
,
Joshua L.
Field†
,
Benedict A.
Williams‡
and
Darren J.
Dixon
*
,
Diana
Berheci†
,
Jessica L.
Crompton†
,
Timothy A.
Davidson†
,
Joshua L.
Field†
,
Benedict A.
Williams‡
and
Darren J.
Dixon
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: darren.dixon@chem.ox.ac.uk
First published on 30th June 2022
Abstract
Acyclic α-tertiary ethers represent a highly prevalent functionality, common to high-value bioactive molecules, such as pharmaceuticals and natural products, and feature as crucial synthetic handles in their construction. As such their synthesis has become an ever-more important goal in synthetic chemistry as the drawbacks of traditional strong base- and acid-mediated etherifications have become more limiting. In recent years, the generation of highly reactive intermediates via redox approaches has facilitated the synthesis of highly sterically-encumbered ethers and accordingly these strategies have been widely applied in α-tertiary ether synthesis. This review summarises and appraises the state-of-the-art in the application of redox strategies enabling acyclic α-tertiary ether synthesis.
 Diana Berheci, Jessica Crompton, Joshua Field, Benedict Williams, Timothy Davidson, Benjamin Shennan, Darren Dixon | The following six authors are pursuing their DPhil studies in the EPSRC Synthesis for Biology and Medicine Centre for Doctoral Training (SBM CDT) at the University of Oxford. Benjamin D. A. Shennan: Ben received his MChem degree from the University of Oxford, completing his Master's project with Prof. Darren Dixon investigating new synthetic routes towards spirocyclic pyrrolidines. He now continues research with Prof. Dixon, focusing on the total synthesis of complex diamine natural products. Diana Berheci: Diana completed her MSci in Chemistry at Imperial College London, working on alkene metathesis chemistry under the supervision of Prof. Christopher Braddock for her final year project. She is now working on oxonium ion chemistry within Prof. Jonathan Burton's group. Jessica L. Crompton: Jessica received her MChem degree from the University of Oxford, completing her Master's project under the supervision of Prof. Angela Russell investigating a one-pot synthesis of a novel fragment in Medicinal Chemistry. She is now pursuing a DPhil under the supervision of Prof. Timothy Donohoe, working on the synthesis of new motifs using hydrogen borrowing catalysis. Timothy A. Davidson: Tim completed his undergraduate studies at the University of Cambridge, investigating the photochemistry of EDA complexes under the supervision of Dr Manuel Nappi and Prof. Matthew Gaunt. He is now pursuing doctoral studies with Prof. Darren Dixon, working towards the total synthesis of complex alkaloids. Joshua L. Field: Josh completed his undergraduate studies at the University of Cambridge, undertaking his masters project with Prof. David Spring. He is now pursuing his DPhil in the group of Prof. Harry Anderson, exploring the synthesis, aromaticity and photophysics of porphyrin nanorings and nanobelts. Benedict A. Williams: Ben received his MChem degree from the University of Oxford, completing his Master's project with Prof. Michael Willis investigating the reactivity of sulfonimidoyl radicals. He now continues research with Prof. Michael Willis, focussing on the development of new catalytic sulfonylation methods. |
Darren J. Dixon: Darren studied at the University of Oxford where he received a Masters degree in 1993 and DPhil in 1997 under the supervision of Prof. Stephen Davies. After postdoctoral work with Prof. Steven Ley CBE, FRS, he joined the faculty at the Department of Chemistry in Cambridge in 2000. In 2004 he took a Senior Lecturership at The University of Manchester and in 2007 he was promoted to Reader. In 2008 he moved to his current post at the University of Oxford where he is Professor of Chemistry and the Knowles–Williams Fellow in Organic Chemistry at Wadham College. |
1. Introduction
As the demand for ever greater three-dimensional complexity in target molecule synthesis increases,1–3 hindered ethers, in particular α-tertiary ethers, have assumed a role of fundamental importance. Notoriously challenging to synthesise by classical methods, the past decades have witnessed a concerted effort to improve access to this unique functionality and rectify the dearth of widely-applicable and mild methodologies for their synthesis.Driving this interest in hindered ether synthesis is the widespread occurrence of this motif within synthetic chemistry (Fig. 1).4–7 While familiar components in fuels and commodity chemicals,8–10 α-tertiary ethers have also become key motifs in pharmaceuticals, agrochemicals and functional materials and are frequently observed in natural products.11–14 Within medicinal chemistry programs, the combination of Lewis basicity, enhanced polarity, resistance to α-oxidation and steric influence, offered by α-tertiary ethers, can confer profound effects on a lead compound's physical and biochemical properties.15–17
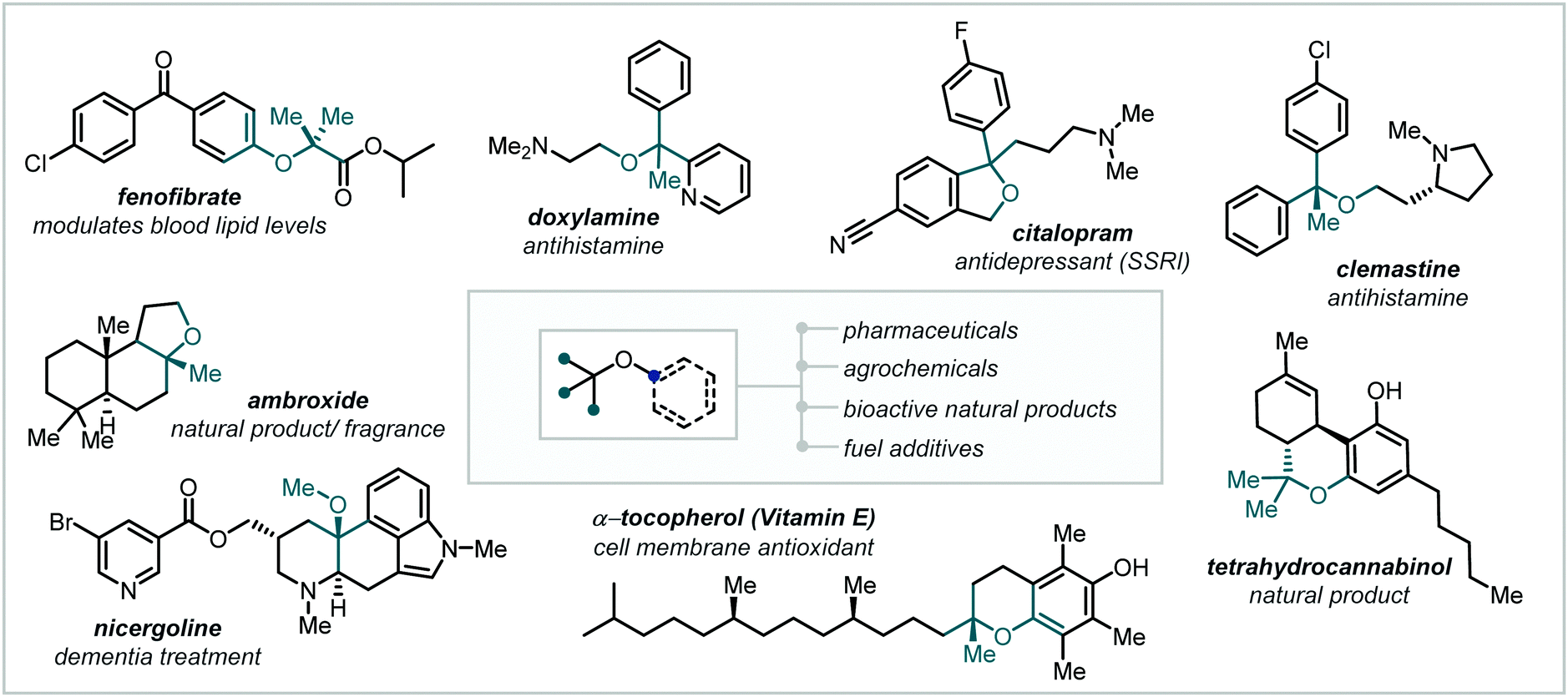 | ||
| Fig. 1 The prevalence of (α-tertiary) ethers. | ||
As a result of this, such structural modifications are apparent in a preponderance of active pharmaceutical ingredients (APIs) in a broad range of medical fields, including antidepressants, antibiotics, and antihistamines, among others.18,19
Etherification has also been the focus of intense study in natural product synthesis with numerous highly potent macrocyclic antibiotics containing hindered alkyl-aryl ether linkages.20 Notably, both tetrahydrocannabinol (THC),21 a prominent cannabinoid with promising and broad therapeutic applications, and tocopherols and tocotrienols (vitamin E), implicated as antioxidants in cell membrane protection,22 feature α-tertiary alkyl-aryl cyclic ethers.
It is noteworthy that alongside the prevalence of such ethers in high-value compounds, the importance of ethers as versatile synthetic intermediates is beginning to be recognised, transitioning from unreactive moieties or protecting groups10,23 to key synthetic handles.24,25
The harsh conditions traditionally required for the synthesis of α-tertiary ethers have limited their facile construction.6 While historical ether syntheses exhibit broad applicability and tolerance, it is noteworthy that most of these methods demonstrate significantly lower efficiency as steric bulk increases and, in some cases, are totally inapplicable for the synthesis of α-tertiary ethers. The venerable Williamson ether synthesis, involving nucleophilic substitution of an alkyl halide by a deprotonated alcohol (Scheme 1), is largely limited to primary or secondary systems with only a few notable tertiary cases – typically enabled by activation from an adjacent group or forcing conditions.26,27 An alternative traditional strategy involves the treatment of an alkene with strong acid to form the corresponding carbocation which can be trapped by a wide range of alcohol nucleophiles.28 While this strategy has seen widespread adoption in hindered ether synthesis, a highly acid-tolerant substrate is required given the forcing conditions. Expanding upon these early methods, the Mitsunobu reaction has been widely applied in ether synthesis however classically is not applicable to the synthesis of α-tertiary ethers (see Section 5).29,30
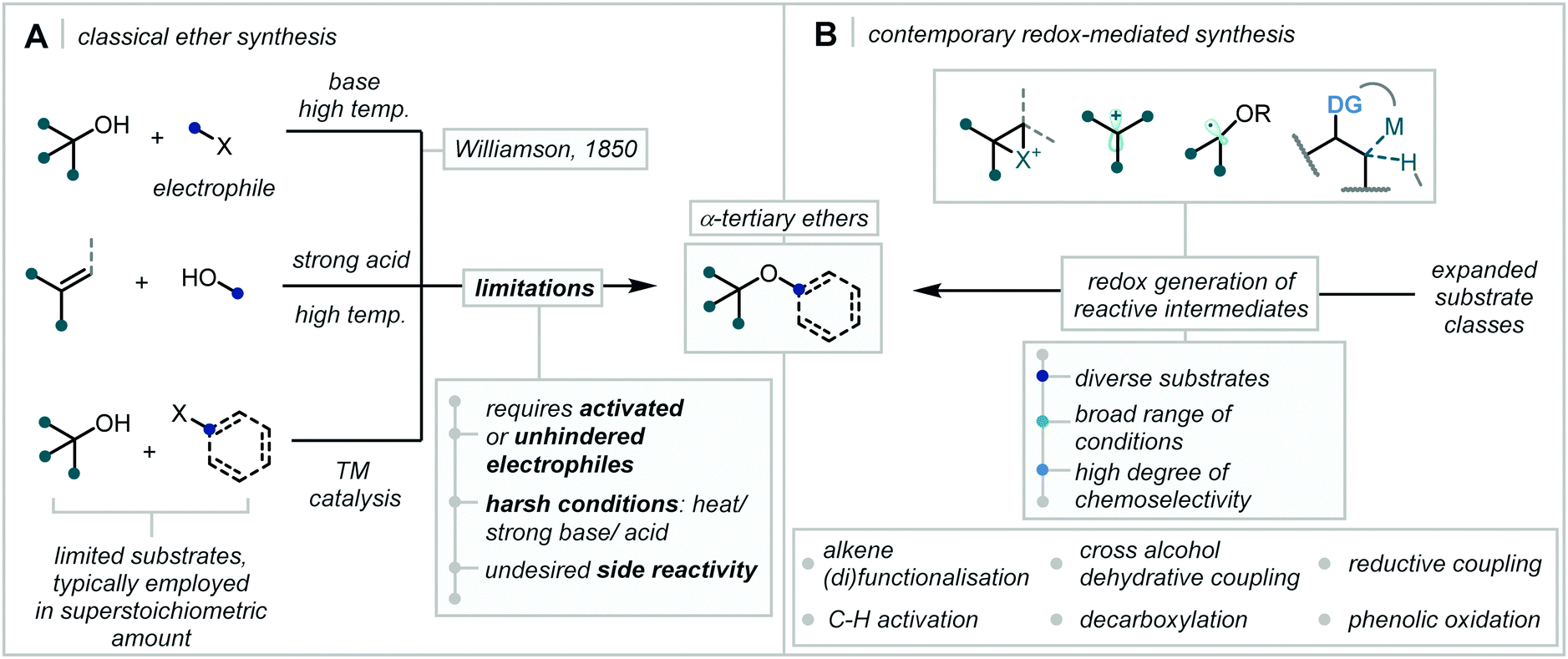 | ||
| Scheme 1 (A) Classical synthesis of α-tertiary ethers; (B) contemporary redox-mediated synthesis of α-tertiary ethers. | ||
As the drawbacks and challenges integral to these traditional techniques have become more apparent and more limiting, the synthetic community has responded with a host of powerful, enabling etherification strategies for the synthesis of hindered ethers.
It is no surprise that shortly after the disclosure of the Buchwald–Hartwig amination,31,32 now commonplace in the synthesis of C–N bonds, the transformation was adapted to C–O bond formation, rapidly enabling access to a plethora of ether-containing motifs.33,34 Notably, in order to prevent competitive and deleterious β-H-elimination, the early reports of Pd-catalysed C–O etherification opted for, and almost exclusively reported, the use of tertiary alcohols as coupling partners.35 The application of the Buchwald–Hartwig coupling systems to etherification obviated the use of the Ullmann reaction and the associated harsh conditions.36
Additionally, the traditional base-mediated Williamson ether synthesis and the acid-mediated activation approaches have continued to be refined and a suite of catalytic – Brønsted and Lewis, acidic and basic – methodologies have been reported vastly expanding the scope of accessible α-tertiary ethers.
However, most striking is the vast expansion of etherification strategies that employ redox chemistry to generate reactive intermediates, or facilitate new mechanistic frameworks, capable of affording hindered ethers and, in most cases, resulting in formal oxidation or reduction of the substrate. It is these methods that will be the focus of this review, namely, etherification reactions that are driven by an associated reduction or oxidation process, typically accompanied by a change in the formal oxidation state of one of the ether linked carbons.
Given the traditional limitations associated with ether bond formation, the review will focus on the formation of acyclic α-tertiary ethers thereby collating proven methods that are applicable in cases of high steric hindrance and able to synthesise the most challenging ethers. It is notable that in the acyclic case, the C–O bond formation is typically intermolecular, asserting even greater kinetic and thermodynamic challenges on the associated etherification. With this in mind it is worth noting that the broad and exciting applications of and synthesis of cyclic ethers, among other oxygen-containing heterocycles, has been elegantly reviewed elsewhere.37–39
The vast expansion in new methodologies employing contemporary redox chemistry has dramatically increased the diversity of suitable substrates for α-tertiary ether synthesis. This has enabled alkene di- and mono-functionalisation, C–H activation, cross-alcohol coupling, decarboxylative strategies, phenolic oxidations and a plethora of more specialised approaches to dramatically diminish the once considerable challenge associated with intermolecular α-tertiary ether formation. Due to the extensive review literature covering the application of metal-catalysed cross-coupling to C–O bond formation, this important class of reaction is considered outside the scope of this review.40–43
2. Alkene difunctionalisation
The synthesis of substituted alkenes and their subsequent oxy-functionalisation is well-established in synthetic chemistry. This mode of reactivity has proven particularly well-suited to the installation of α-tertiary ethers due to the highly reactive intermediates that are readily accessed from alkenes under oxidative or reductive activation. The following section will detail such approaches and broadly classify them by the mode of alkene activation and the nature of functionalisation coupled with alkoxylation.2.1 Photochemical alkene difunctionalisation
In recent years, the application of photochemistry in synthetic organic chemistry has seen a significant resurgence, becoming an important and widely applicable reaction manifold.44,45 Novel photochemical reaction manifolds have enabled the generation of reactive chemical species from accessible precursors under mild conditions, greatly expanding the toolbox of reactions available to the synthetic chemist and enabling diverse access to single-electron chemistry and mechanistic frameworks.In the context of alkene difunctionalisation for α-tertiary ether synthesis, many of the transformations discussed in this review share a similar reaction design or mechanistic proposal (Scheme 2). An initiation event leads to the formation of a reactive radical species, capable of adding to the π-bond of an alkene. This generates a carbon-centred radical, usually at the most-stabilised position. Oxidation of the resultant radical then yields the corresponding carbocation and often enables completion of the catalytic cycle of a photoredox catalyst, or additives such as metal salts can also mediate this process. This involvement of both radical and polar species in a mechanism is described as radical-polar crossover (RPC).46–50 The carbocation can finally be trapped by an external nucleophile, such as an alcohol, to form the ether product.
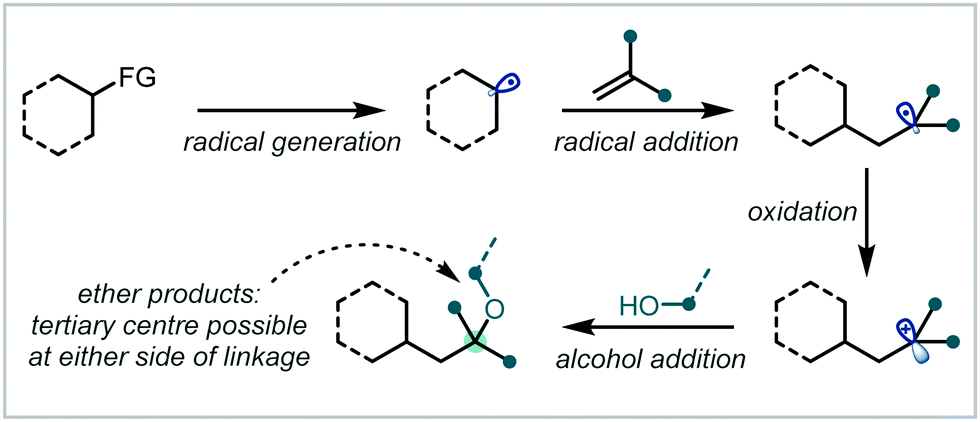 | ||
| Scheme 2 General mechanism for alkene difunctionalisations involving RPC. | ||
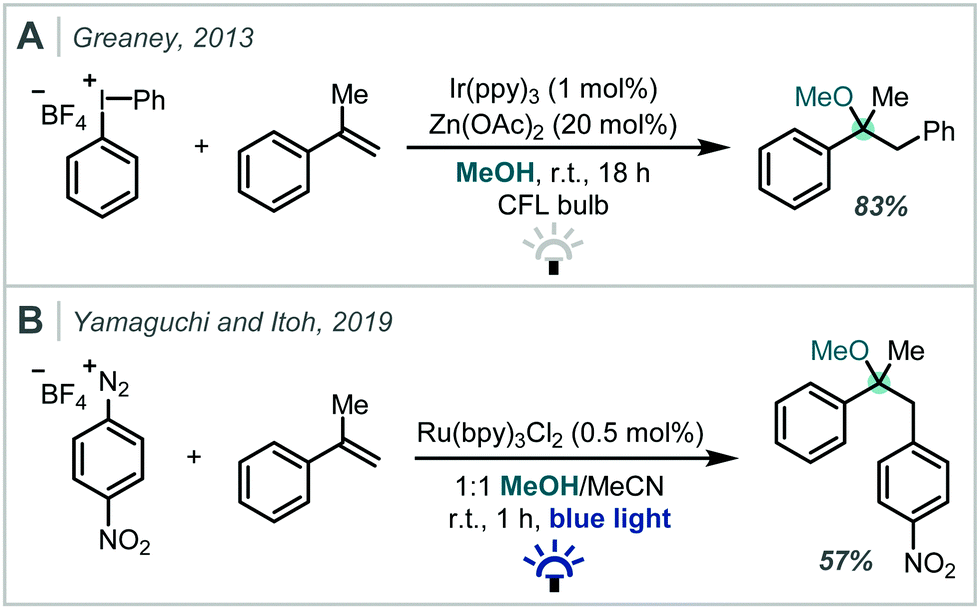 | ||
| Scheme 3 Photoredox-catalysed oxyarylation of styrenes using: (A) diaryliodonium salts, and; (B) aryl diazonium salts. | ||
Recently, Heinrich published a catalyst-free carboetherification of α-methylstyrene using aryl diazonium salts and alcohols under visible light irradiation (Scheme 4).53 A broad scope of primary alcohols was used to form α-tertiary ethers in good yields with useful pendant functionality, including olefins. The reaction was found to proceed without irradiation, albeit in lower yields, which is consistent with prior work on this transformation.54 The beneficial effect of irradiation was attributed to the formation of very weak electron donor–acceptor complexes between the substrates, which increased the efficiency of the initiation step. Once initiated, a radical chain mechanism propagated, which yielded ethers upon oxidation of the benzylic radical and subsequent alcohol addition.
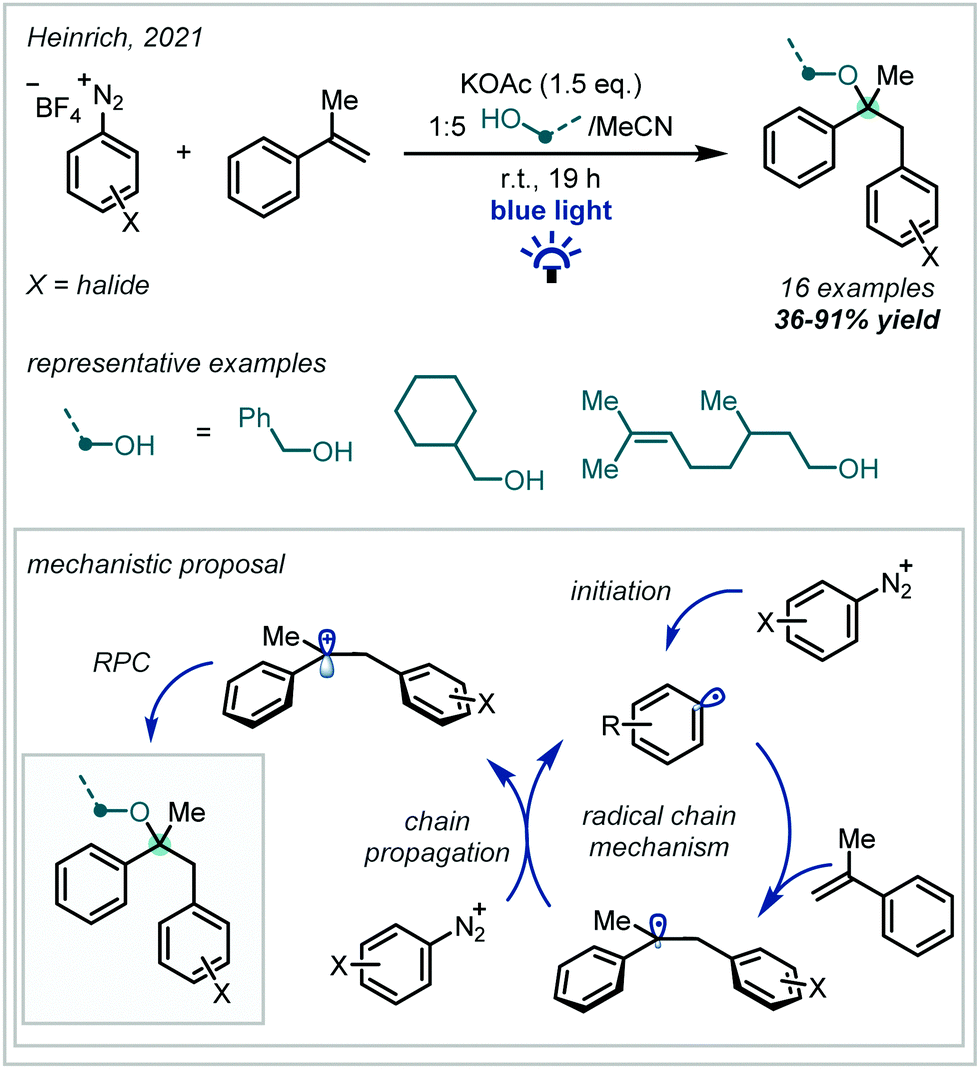 | ||
| Scheme 4 Styrene oxyarylation mediated by visible light irradiation of weak electron donor–acceptor complexes. | ||
Under photoredox catalysis, the generation of C(sp3)-centred radicals for addition into π-systems has become a widely explored concept. In 2017, Glorius reported a multicomponent reaction for the decarboxylative oxyalkylation of styrenes using N-(acyloxy)phthalimides (Scheme 5).55 The reduction of N-(acyloxy)phthalimides has previously been used as a method to generate alkyl radical precursors under photoredox catalysis,56,57 but this required strongly reducing conditions which limited the scope to net reductive transformations. Glorius showed that the reduction of the N-(acyloxy)phthalimide substrate could be made more favourable by exploiting hydrogen bonding interactions, which removed the requirement for strongly reducing conditions and thus enabled the reported redox–neutral process. This reaction between various primary alcohols, α-methylstyrene and the cyclohexyl radical proceeded in good yields to afford α-tertiary ether products. However, analogous reactions using more hindered alcohols such as isopropanol proceeded in lower yields, even though higher catalyst loadings were employed, emphasising the challenge associated with forming sterically-hindered α-tertiary ethers.
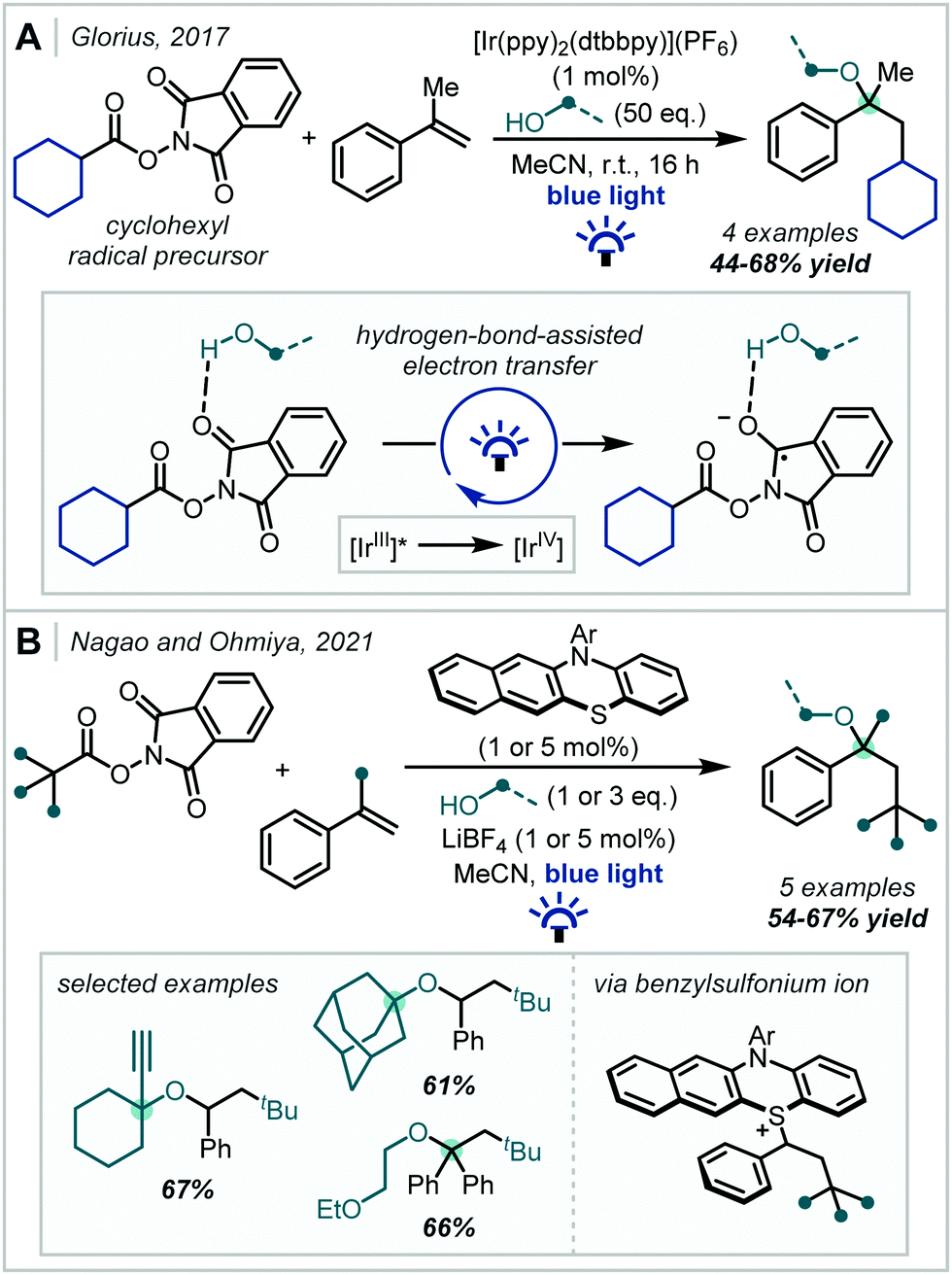 | ||
| Scheme 5 (A) Photochemical alkoxyalkylation of styrenes using N-(acyloxy)phthalimides, enabled by hydrogen bond-assisted electron transfer. (B) Organophotoredox-catalysed alkoxyalkylation of styrenes. dtbbpy = 4,4′-di-tert-butyl-2,2′-dipyridyl, Ar = 4-trifluoromethylphenyl. | ||
N-(acyloxy)phthalimides were later used by Nagao & Ohmiya to enact a similar transformation (Scheme 5) using a benzo[b]phenothiazine organic photocatalyst (see Section 3.2, Scheme 65 for leading studies on benzo[b]phenothiazine-catalysed decarboxylative etherification).58 A wide scope of alcohols and alkyl radicals could participate in the reaction, yielding a range of α-tertiary ether examples in typically good yields. Since the alcohol was used in stoichiometric quantities, more elaborate alcohols with pendant functionality could be incorporated. Moreover, tertiary alcohols were competent nucleophiles in contrast to previously reported methods. An RPC mechanism was invoked for the transformation, featuring a benzylsulfonium intermediate, displaced in a nucleophilic substitution by the alcohol to yield the ether products.
Seeking to introduce additional functional handles in tandem with etherification, in 2014, Lei published the synthesis of γ-alkoxynitriles containing tertiary methyl ethers, mediated by reduction of bromoacetonitrile by Ir(ppy)3 and subsequent radical addition into styrenes (Scheme 6).59 Zhou later reported a cyanopropylation strategy using O-acyl oximes as radical precursors. Upon photochemical activation, β-scission yielded a reactive alkyl radical which added to the alkene π-bond of substituted styrenes, affording α-tertiary ether products in good to excellent yields.60
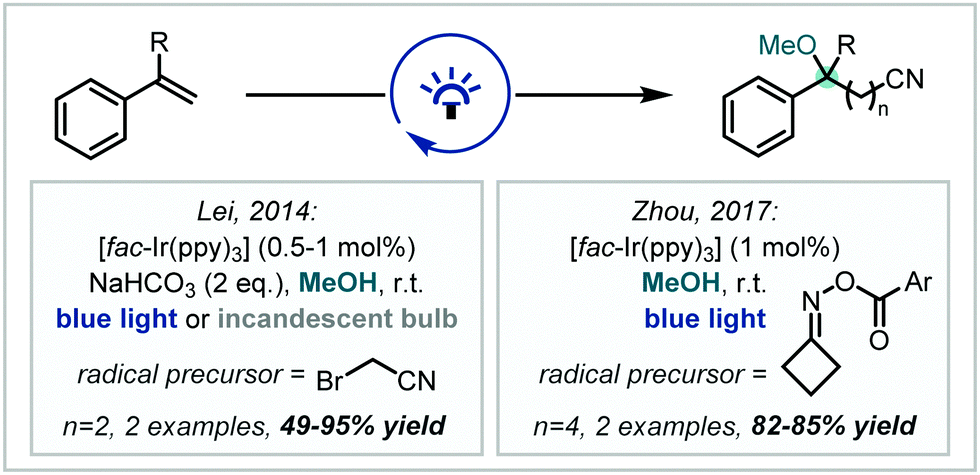 | ||
| Scheme 6 Strategies for the photochemical synthesis of α-tertiary ethers containing nitrile functionality from styrenes. Ar = 4-trifluoromethylphenyl. | ||
In 2017, Glorius reported the synthesis of γ-hydroxy-α-tertiary ethers via radical addition into aryl carbonyl compounds (Scheme 7).61 Such a transformation is rare because the reaction is readily reversible – it was shown that this limitation could be overcome using hole catalysis.62,63 Mechanistically, this involved oxidation of a styrene derivative by a strongly oxidising photo-excited acridinium species,64 with subsequent interception of the radical cation by methanol. It was suggested that the protonated ether intermediate was able to form an intermolecular hydrogen bond with the incoming carbonyl, facilitating the subsequent radical addition by enhancing the rate of alkoxy radical quenching and thus preventing the reverse reaction. An impressive 30 α-tertiary ethers were reported, formed in excellent yields of up to 90%. Although yields were generally higher when aldehydes were used as the radical acceptor, ketones were nonetheless effective, allowing the efficient formation of α-tertiary ethers containing a hydroxylated γ-quaternary centre.
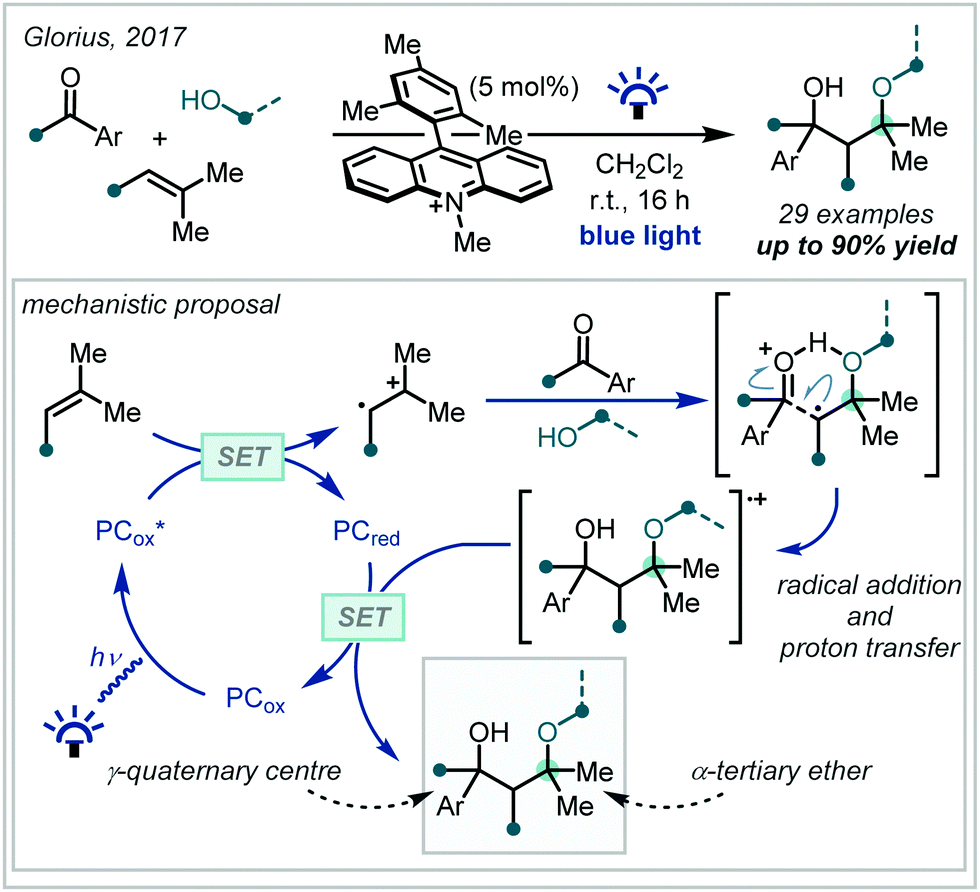 | ||
| Scheme 7 Multicomponent synthesis of γ-hydroxy-α-tertiary ethers via hydrogen bond-assisted radical addition into carbonyl compounds. SET = single electron transfer. | ||
The introduction of halogen atoms, particularly fluorine, into pharmaceuticals and other bioactive molecules has well-documented benefits on pharmacokinetics and efficacy.65–70 Oxyhaloalkylation of alkenes mediated by photoredox catalysis therefore represents a step-economical method for the simultaneous introduction of valuable haloalkyl and α-tertiary ether functionalities into a molecule (Scheme 8). Since Koike & Akita's report of alkoxytrifluoromethylation using Umemoto's reagent in 2012,71 several reaction manifolds have been published that achieve this transformation using alternative haloalkyl radical precursors.72–75 In general, these reactions proceed via the RPC mechanism depicted in Scheme 2, yielding halogenated α-tertiary ethers in moderate to excellent yields. A noteworthy example was disclosed by Masson in 2019, involving a four-component coupling to forge a nitrile-substituted α-tertiary ether in 59% yield.76
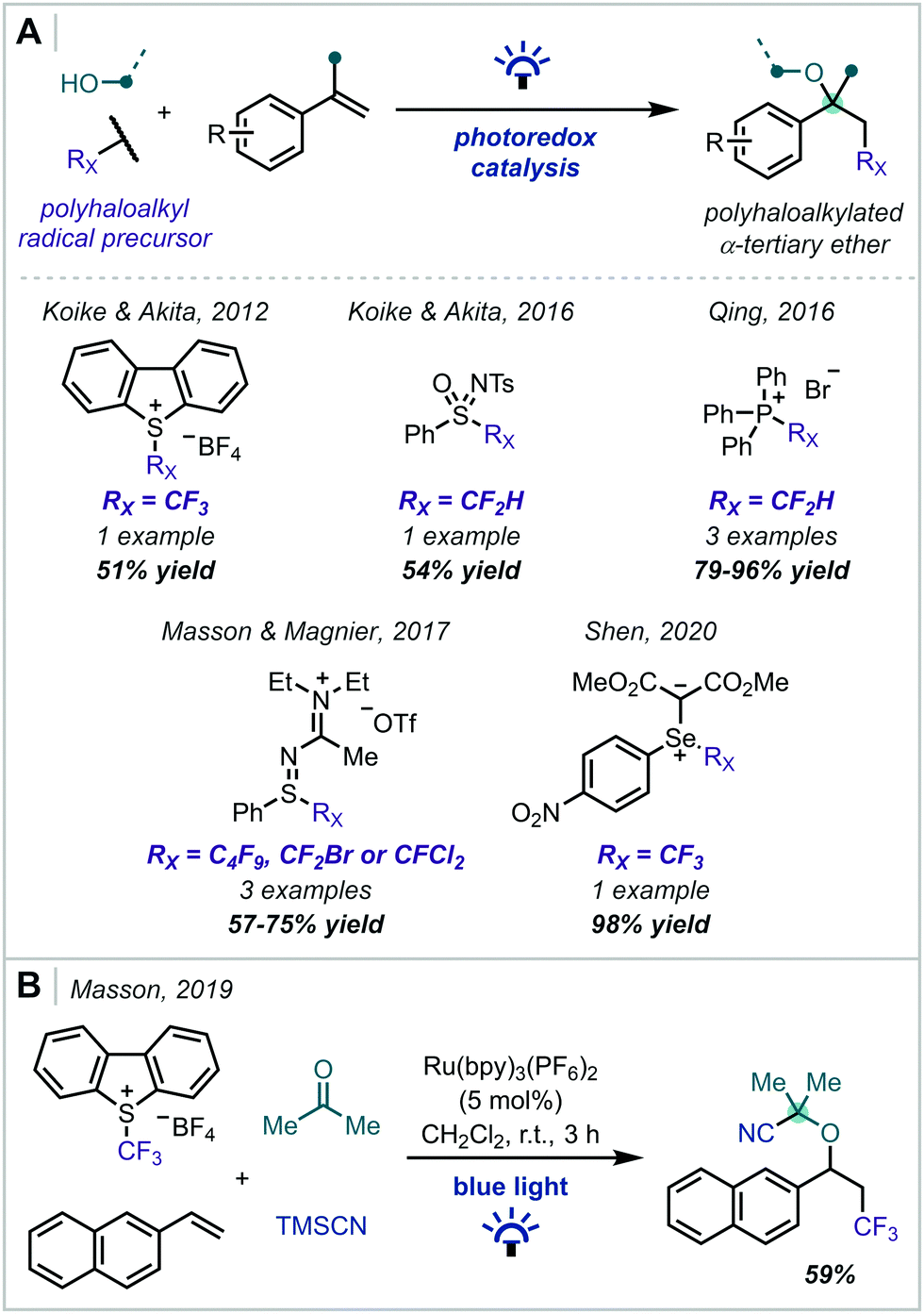 | ||
| Scheme 8 (A) Photochemical synthesis of polyhaloalkylated α-tertiary ethers from styrene precursors. (B) Multicomponent reaction enabling the synthesis of trifluoromethylated α-cyano-α-tertiary ethers. | ||
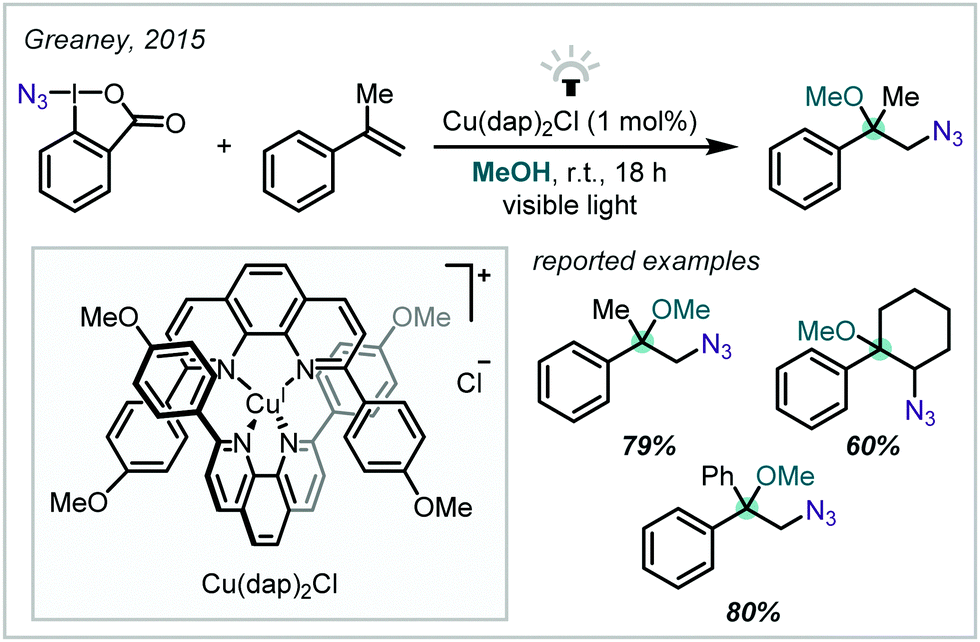 | ||
| Scheme 9 Alkoxyazidation of styrenes mediated by a Cu-based photocatalyst. | ||
In 2018, Xu & Yu disclosed a radical relay strategy towards the synthesis of protected β-amino alcohols and ethers (Scheme 10).79 This hinged upon the use of Tfoc [Tfoc = –CO2CH2CF3] protected hydroxylamine derivatives as photolabile nitrogen radical precursors. Upon radical addition across an enol ether, the α-oxy radical could react further with a styryl boronic acid, forming cinnamyl α-tertiary ether products after oxidation of the resulting radical and elimination of boron. It was also shown that the geometry of the cinnamyl olefin could be subsequently isomerised under photocatalytic conditions. Soon afterwards, Yu reported a similar transformation, instead enacting a Kornblum-type oxidation of the intermediate carbocation to yield ketones at the benzylic position.80
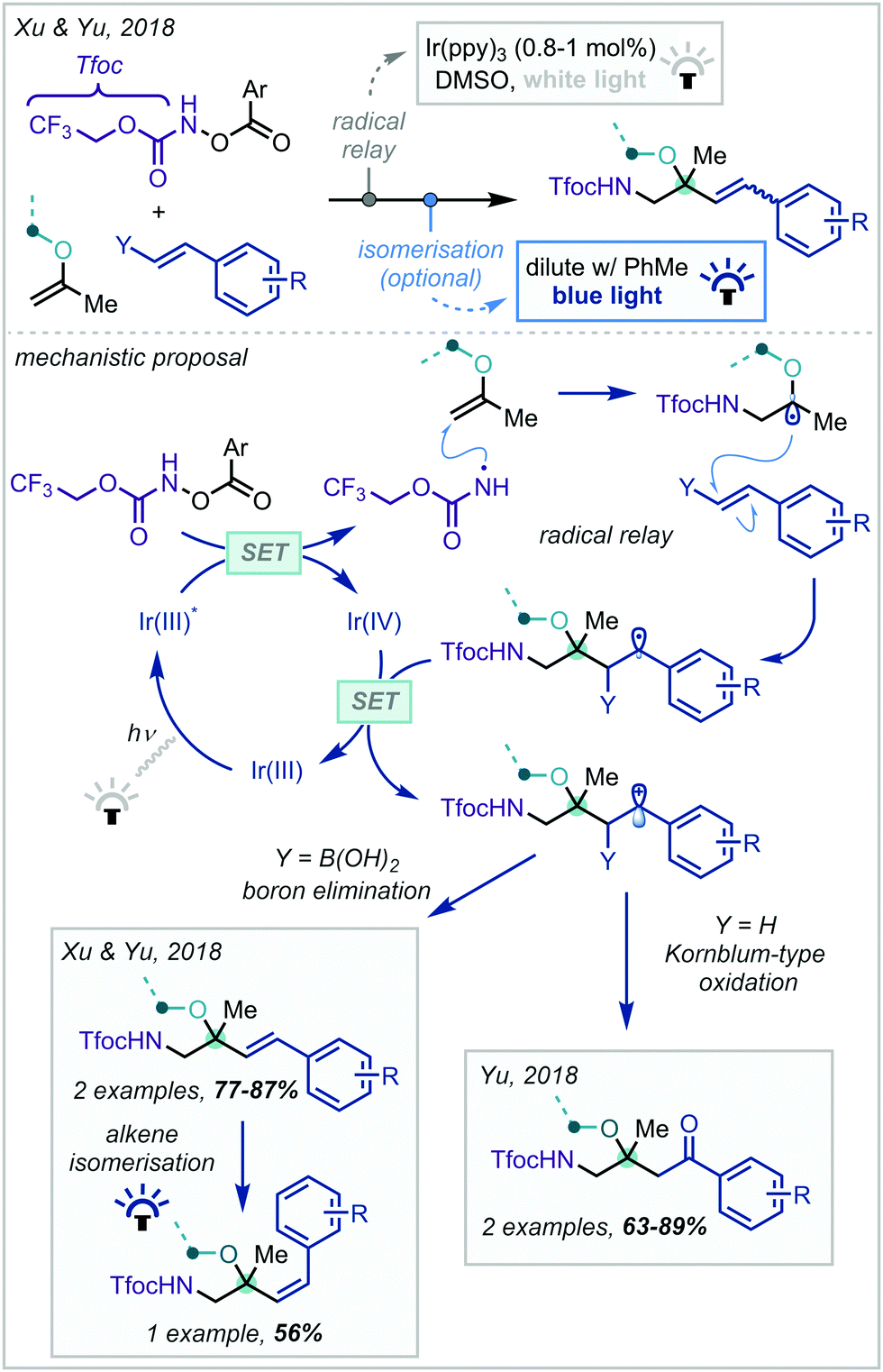 | ||
| Scheme 10 Synthesis of Tfoc-protected β-amino-α-tertiary ethers from styrenes and enol ethers. Ar = 4-trifluoromethylphenyl. | ||
In 2019, Ye & Wu reported a four-component reaction of cyclobutanone-derived O-acyl oximes with K2S2O5-derived sulfur dioxide, styrene derivatives and primary alcohols to produce α-tertiary ethers with sulfones in the β-position (Scheme 11).81 This proceeded via reductive decomposition of the O-acyl oxime precursor, forming an alkyl radical. The radical was subsequently intercepted by in situ generated SO2, affording a sulfur-centred radical that added to the alkene π-bond of aromatic olefins. A variety of 1,1-disubstituted styrenes, both electron-poor and electron-rich, were tolerated in the reaction, and it was demonstrated that O-acyl oximes of substituted cyclobutanones were competent radical precursors, allowing the introduction of aromatic groups, an ester, and a protected piperidine moiety onto the alkyl chain.
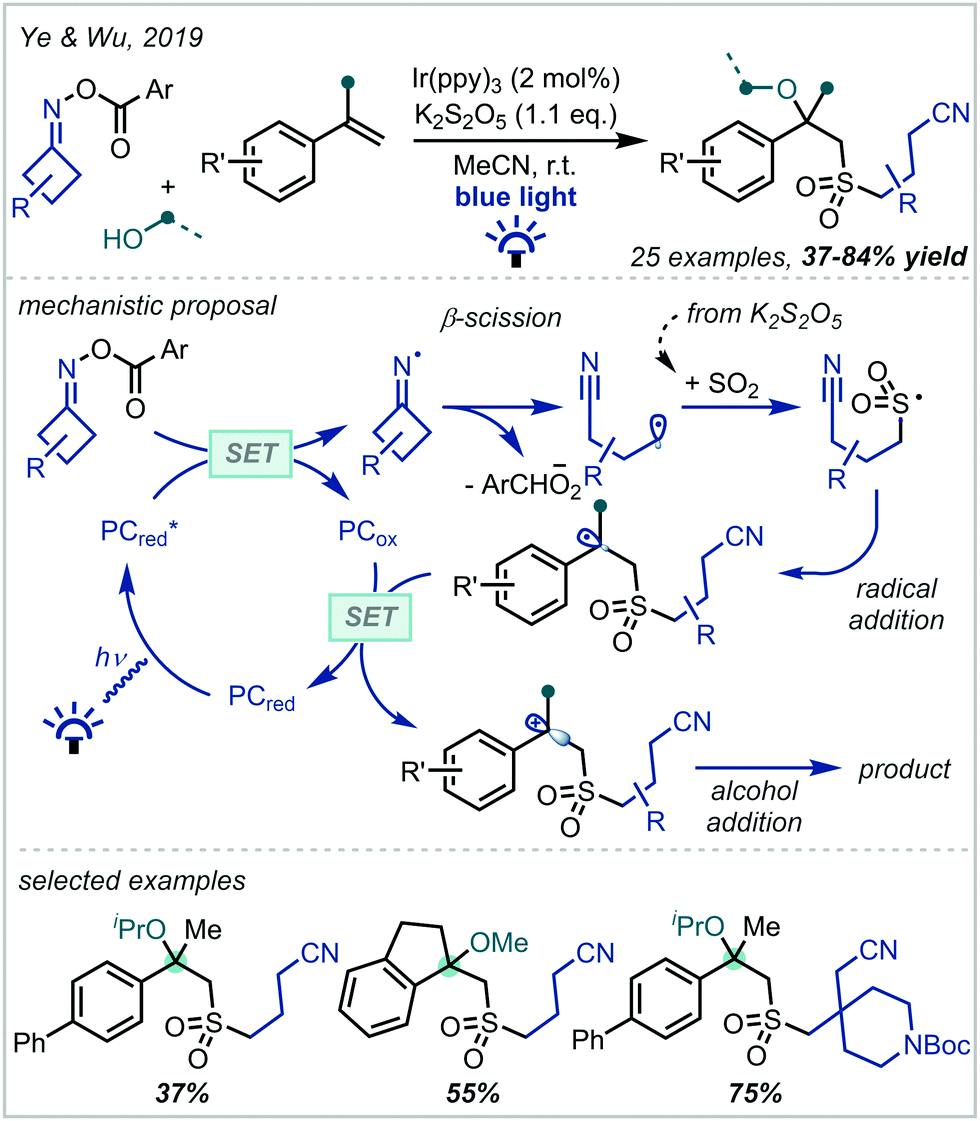 | ||
| Scheme 11 Multicomponent synthesis of sulfonylated α-tertiary ethers using O-acyl oximes as alkyl radical precursors and K2S2O5 as a sulfur dioxide source. Ar = 4-trifluoromethylphenyl. | ||
In 2018, Knowles reported an efficient two-step procedure for the enantioselective synthesis of substituted pyrroloindolines (Scheme 12).82 Inspired by the reactivity of tryptophan radical cations in enzymatic systems, hydrogen bonding between a tryptamine substrate and a chiral phosphoric acid catalyst enabled proton-coupled electron transfer (PCET) from Ir(ppy)3 under visible light irradiation, resulting in a chiral radical cation–phosphate anion pair. This could be enantioselectively intercepted by 2,2,6,6-tetramethyl piperidine 1-oxyl (TEMPO) to give an iminium ion intermediate, upon which the pendant Cbz-protected amine functionality could cyclise in a diastereoselective manner. The inclusion of the stoichiometric oxidant triisopropylsilyl-ethynyl benziodoxolone (TIPS-EBX) maintained optimal reaction efficiency, since the carboxylate base formed upon reduction negated the formation of deleterious TEMPO-H. In a subsequent photoinduced step, the TEMPO moiety could be efficiently substituted to yield the corresponding α-tertiary ethers while maintaining good enantiomeric excess. Impressively, t-BuOH was a proficient nucleophile in the reaction, giving access to synthetically challenging α,α′-tertiary ether products.
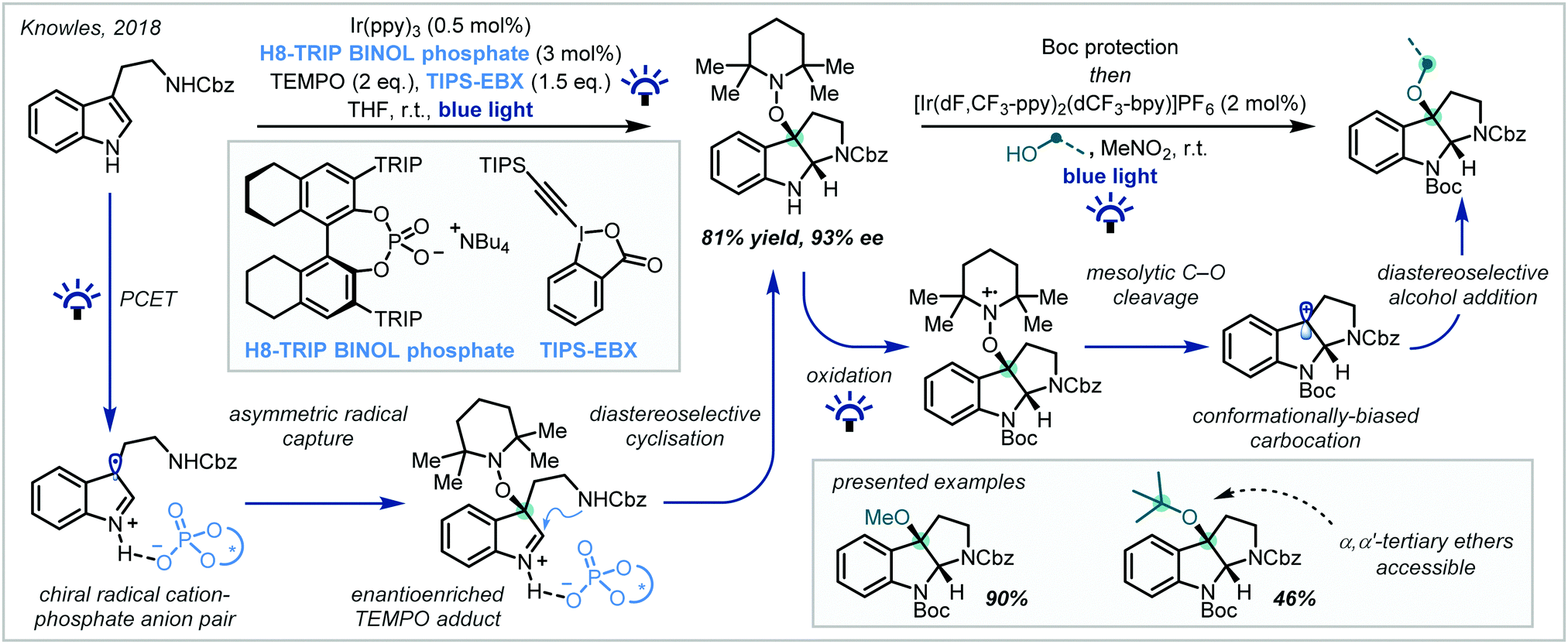 | ||
| Scheme 12 Biomimetic, enantioselective synthesis of substituted pyrroloindolines under cooperative photoredox/organocatalysis. TRIP = (2,4,6-triisopropyl)phenyl, TIPS = triisopropylsilyl, [dF,CF3-ppy] = 3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl]phenyl, [dCF3-bpy] = 5,5′-bis(trifluoromethyl)-2,2′-bipyridine. | ||
In 2021, Gutierrez and Molander reported a dicarbofunctionalisation methodology that exemplifies the synthetic utility of this disconnection (Scheme 13).83 It was shown that benzophenone-derived hydrogen atom transfer (HAT) catalysts, under irradiation at 390 nm, enacted hydrogen atom abstraction from the α-position of cyclopentylmethyl ether (CPME) or diisopropyl ether to yield a nucleophilic α-oxy radical. Giese addition across activated alkenes ensued and subsequent Ni-mediated cross-coupling with an aryl bromide afforded a variety of arylated α-tertiary ethers. Although a significant excess of the ether precursor was necessary, excellent product yields could be obtained.
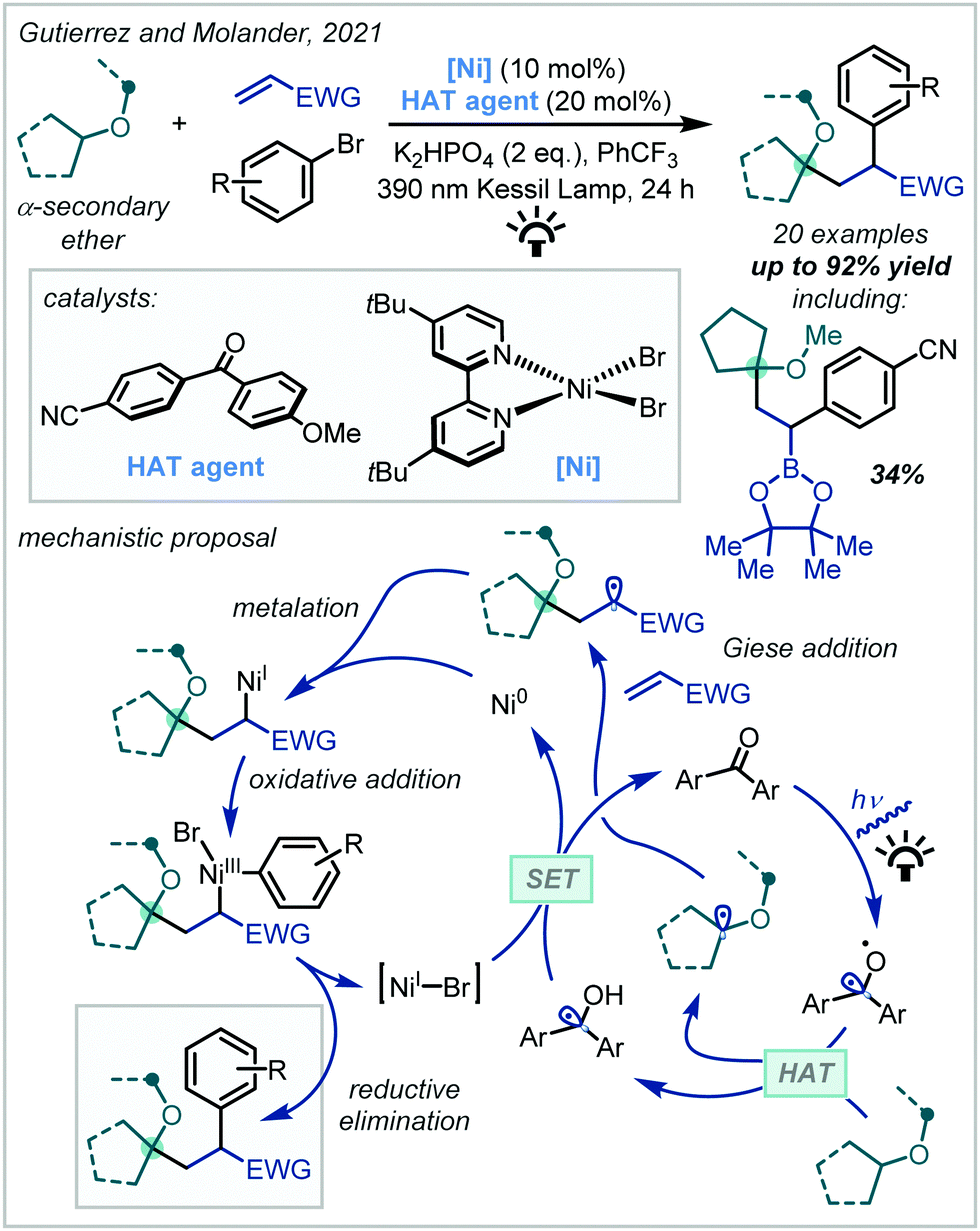 | ||
| Scheme 13 Synthesis of α-tertiary ethers via α-functionalisation of secondary ethers. EWG = electron-withdrawing group. | ||
In a conceptually distinct approach, in 2017, Yoon reported that enol ethers could participate in such a transformation with thiophenol-substituted alkenes to yield ether products (Scheme 14).84 The thiophenol moiety acted as a redox auxiliary, making the alkene more susceptible to oxidation by the Ru(bpz)3(BArF)2 photocatalyst [bpz = 2,2′-bipyrazine, BArF = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate]. The resultant radical cation then underwent a [2+2] cycloaddition, followed by photoinduced reduction allowing catalyst turnover and release of the cyclobutane product. After the reaction, the sulfur moiety could be removed using standard reductive methods. Notably, the reaction was tolerant of steric bulk at the enol ether, and vinyl tert-butyl ether reacted to form the corresponding α-tertiary ether in high yield.
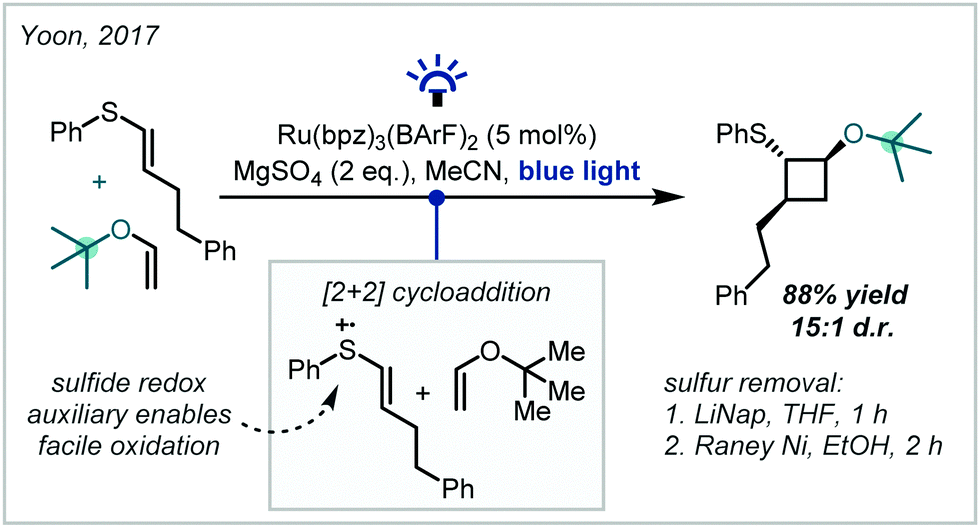 | ||
| Scheme 14 Radical cation-mediated [2+2] cycloadditions promoted by the installation of a sulfur-based redox auxiliary. | ||
2.2 Electrochemical alkene difunctionalisation
Electrochemistry offers a highly versatile framework in which to enact redox-mediated transformations and has been widely cited as offering more sustainable and atom-economical solutions to these reactions.85 Accordingly, electrochemistry has been widely applied in the synthesis of ethers and notably shows broad applicability to acyclic α-tertiary ether synthesis. The difunctionalisation of alkenes to afford the corresponding β-functionalised ethers has predominated in the applications of electrochemistry for ether synthesis.Among these approaches, the various reports can be broadly divided by the mechanistic framework in operation and the specific role the electrochemical cell is performing (Scheme 15). In this manner, we can view the role of the electrochemical cell as either generating a radical cation from anodic oxidation of the alkene; oxidative generation of an electrophilic species which engages in polar reactivity with the substrate; or oxidative generation of an electrophilic radical which is able to react further with the alkene.
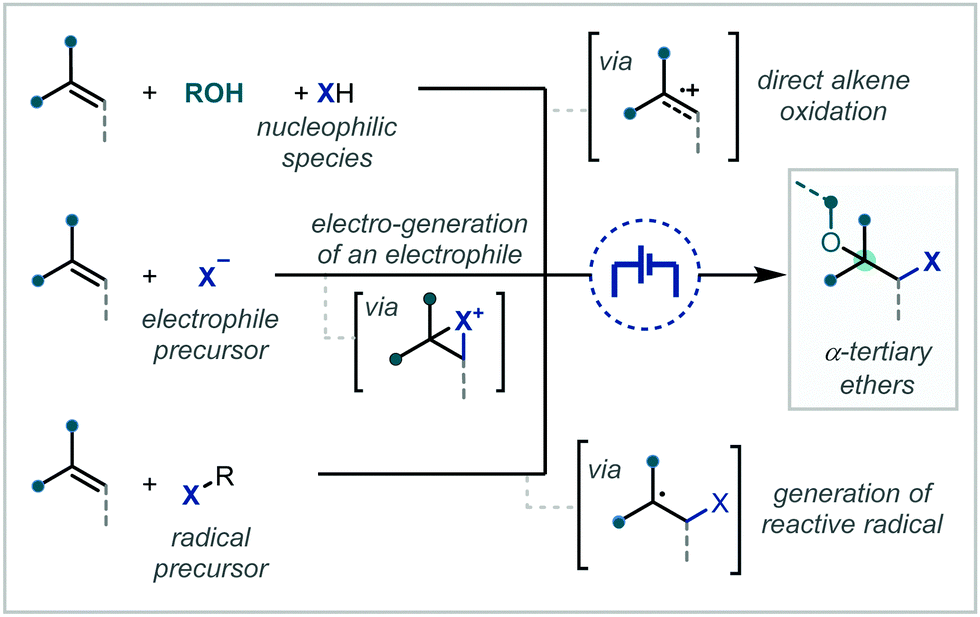 | ||
| Scheme 15 Mechanistic approaches to the electrochemically-mediated synthesis of α-tertiary ethers via alkene difunctionalisation. | ||
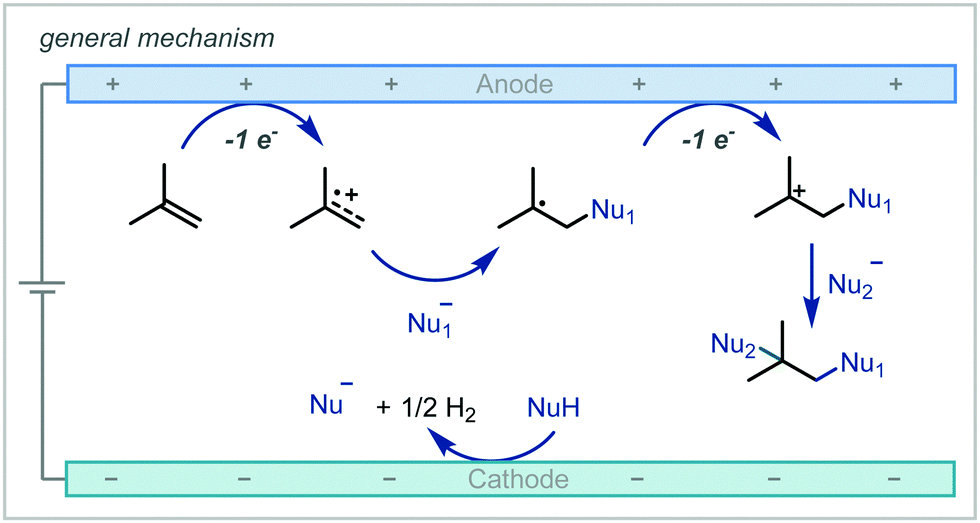 | ||
| Scheme 16 General mechanism for alkene difunctionalisation via electrochemical oxidation. | ||
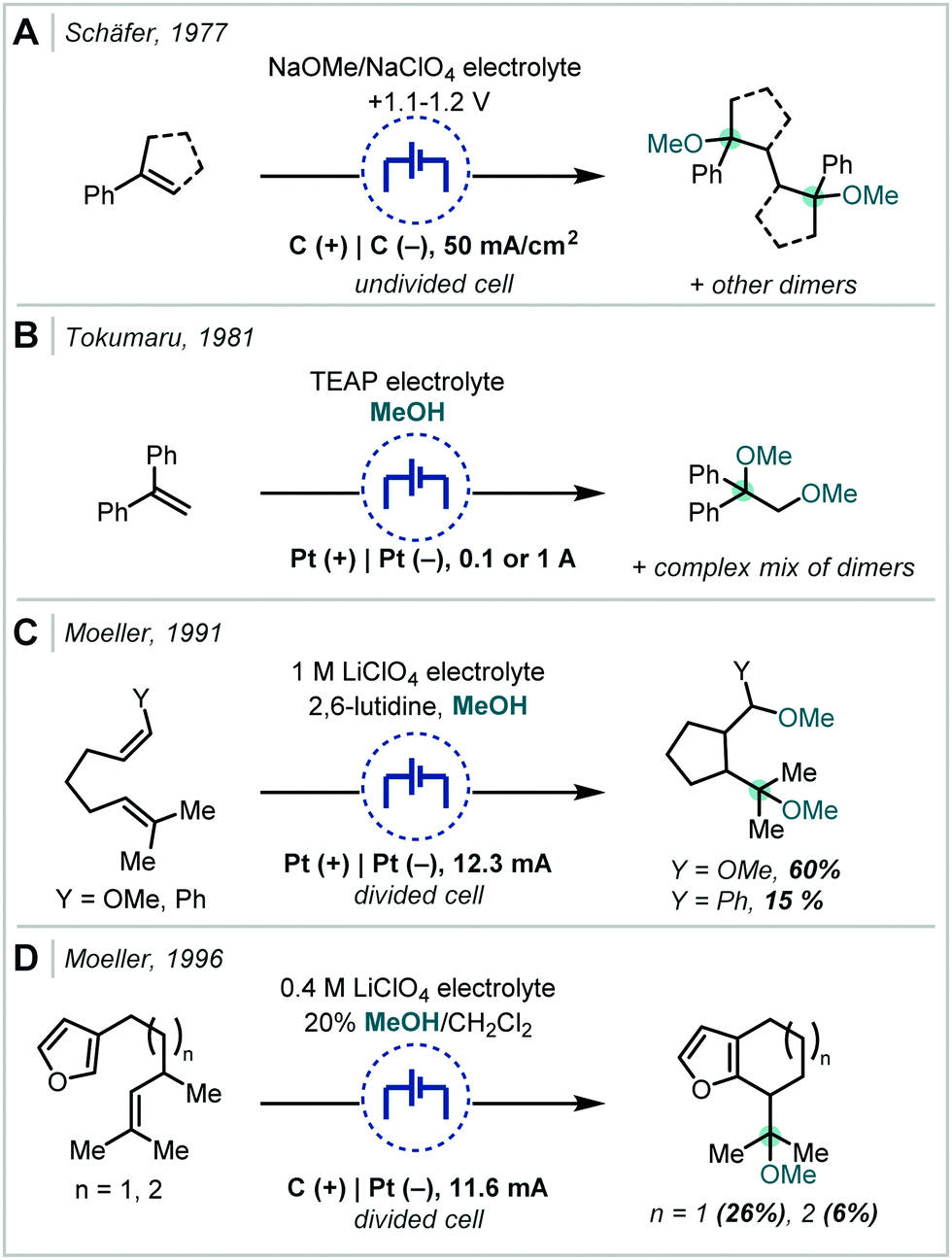 | ||
| Scheme 17 Early electrochemical ether syntheses via direct alkene oxidation; TEAP = tetraethylammonium perchlorate. | ||
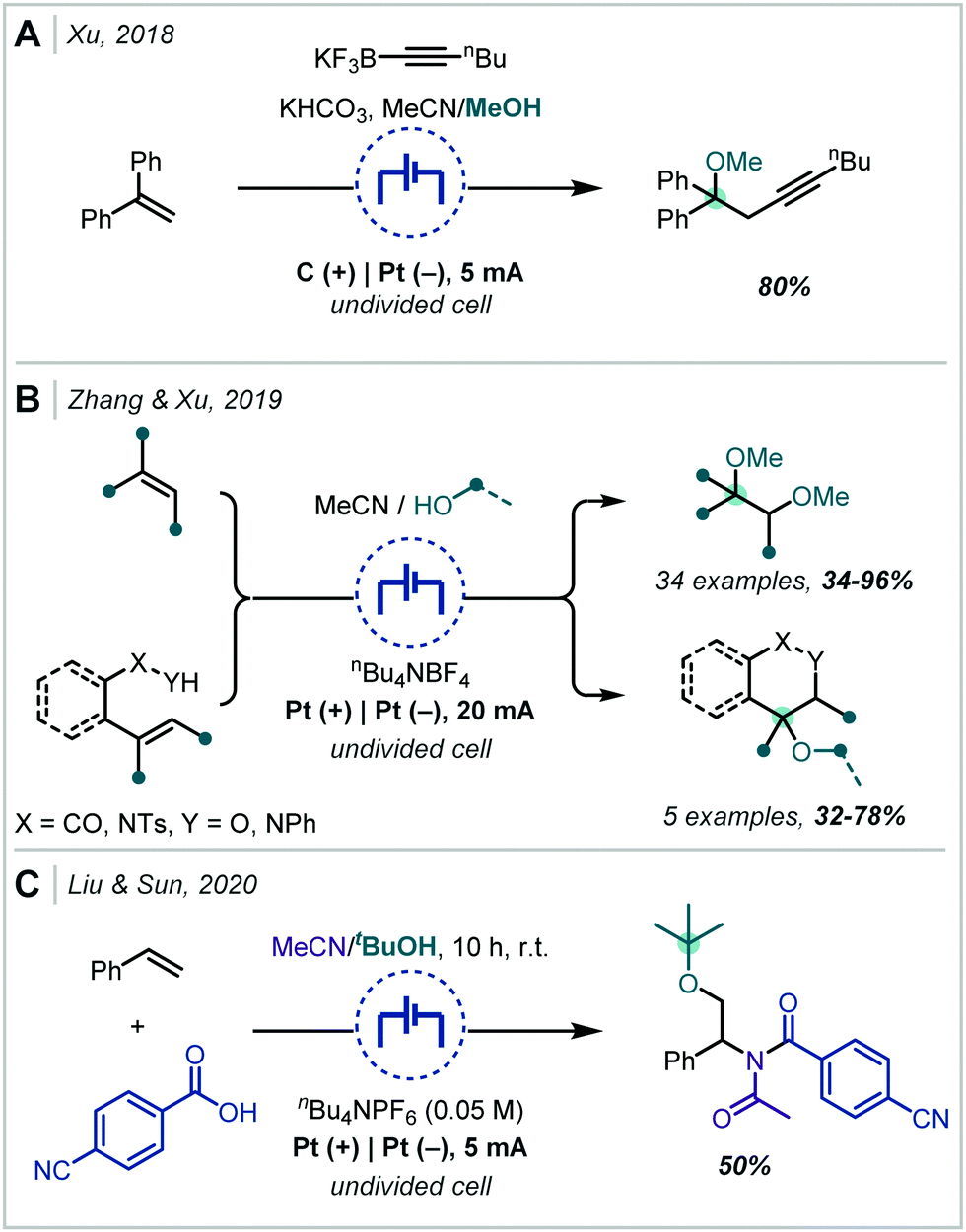
Following the recent renewal of interest in electrochemical transformations, this approach towards the synthesis of hindered ethers has been revisited and significantly developed. In 2018, Xu reported a broad scope strategy towards the electrochemical carbohydroxylation of alkenes and successfully demonstrated an extension of this method in the synthesis of α-tertiary ethers (Scheme 18A).90 Organotrifluoroborates were employed as the nucleophilic component which could intercept the electro-generated alkene radical cation. The resulting radical could be further oxidised and intercepted with MeOH. The etherification occurred in a high yield employing 1,1-diphenylethylene as the substrate and a hexynyl trifluoroborate as the nucleophile.
 | ||
| Scheme 18 Contemporary applications of electrochemical alkene oxidation. | ||
In an alternative difunctionalisation approach, Zhang and Xu described the dimethoxylation of alkenes, in which methoxide reacts with both the alkene radical cation and the resulting carbocation after each anodic oxidation event (Scheme 18B).91 The procedure demonstrated very broad applicability to α-tertiary ether synthesis and could be up-scaled to >5 g. Notably in a number of cases when secondary ether synthesis was attempted, the correspondingly less stable carbocation could undergo rearrangement, resulting in acetal synthesis. Liu & Sun extended this strategy to interception with acetonitrile, generating a nitrilium ion which could react with a carboxylic acid (Scheme 18C).92 The resulting acyl imidate was capable of undergoing a Mumm rearrangement to afford the imide. When t-BuOH was employed as co-solvent, an α-tertiary ether was afforded in 50% yield.
Terent’ev applied this strategy, using a bromide source, to the synthesis of both bromohydrins and β-bromoethers (Scheme 19A).93 In the latter case, MeOH was employed as a co-solvent and was proposed to open the bromonium ion resulting from reaction with α-methyl styrene.
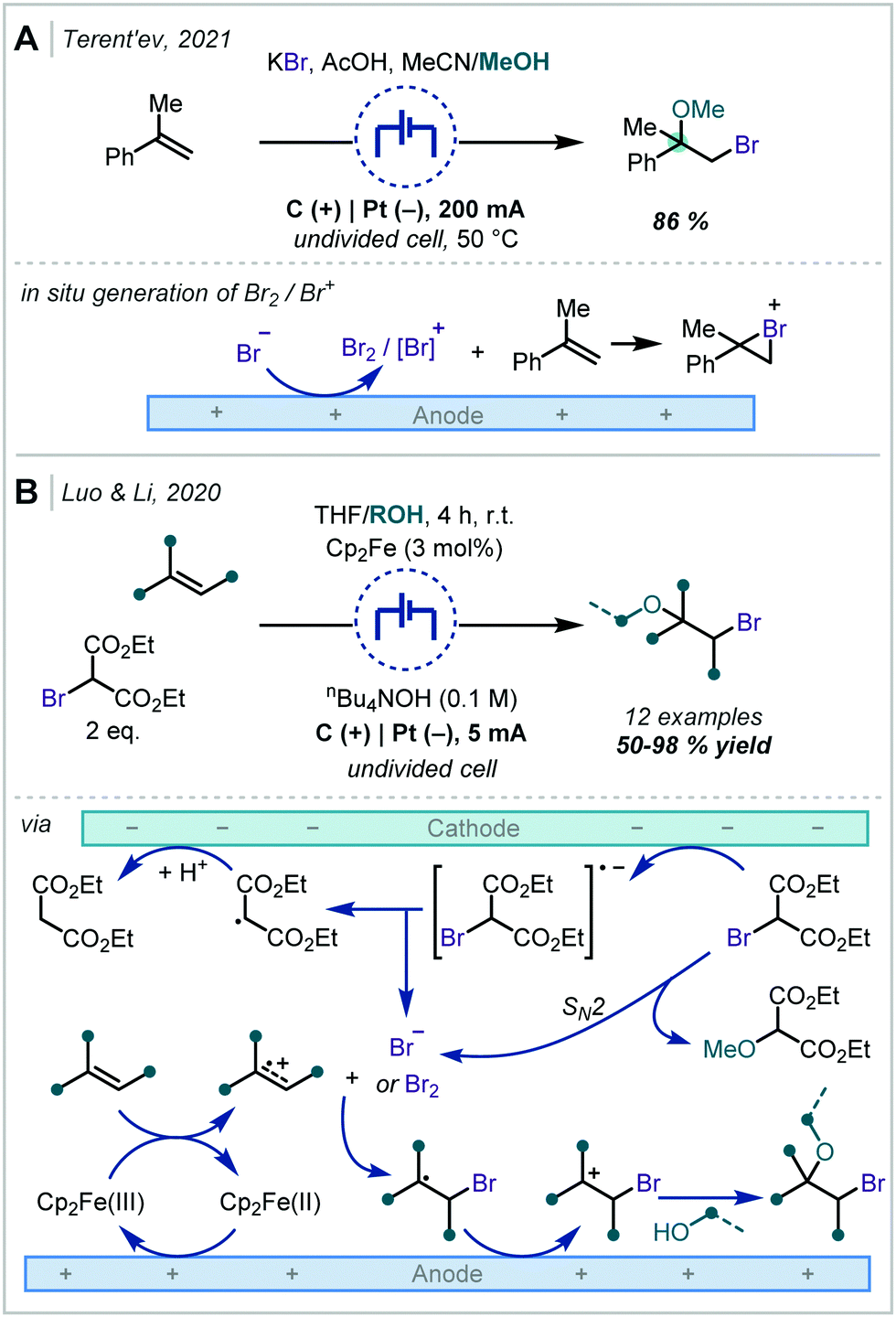 | ||
| Scheme 19 Electrochemical generation of halogen electrophiles capable of reaction with alkenes. Note: proposed anodic oxidation of bromide ions to bromine has been omitted for clarity. | ||
In a modified approach, Luo and Li reported a similar transformation (Scheme 19B); however, the authors employed Cp2Fe as an electro-catalyst capable of effecting the direct oxidation of an alkene.94 This could undergo further reaction either with in situ generated bromide or with bromine and the resulting radical followed the same course as previously described. Notably, bromomalonates were employed as both the bromide source and implicated in the reaction at the cathode. The authors demonstrated that α-tertiary ether synthesis was possible by use of either α-substituted styrenes or by changing the incoming nucleophile to a tertiary alcohol such as t-BuOH.
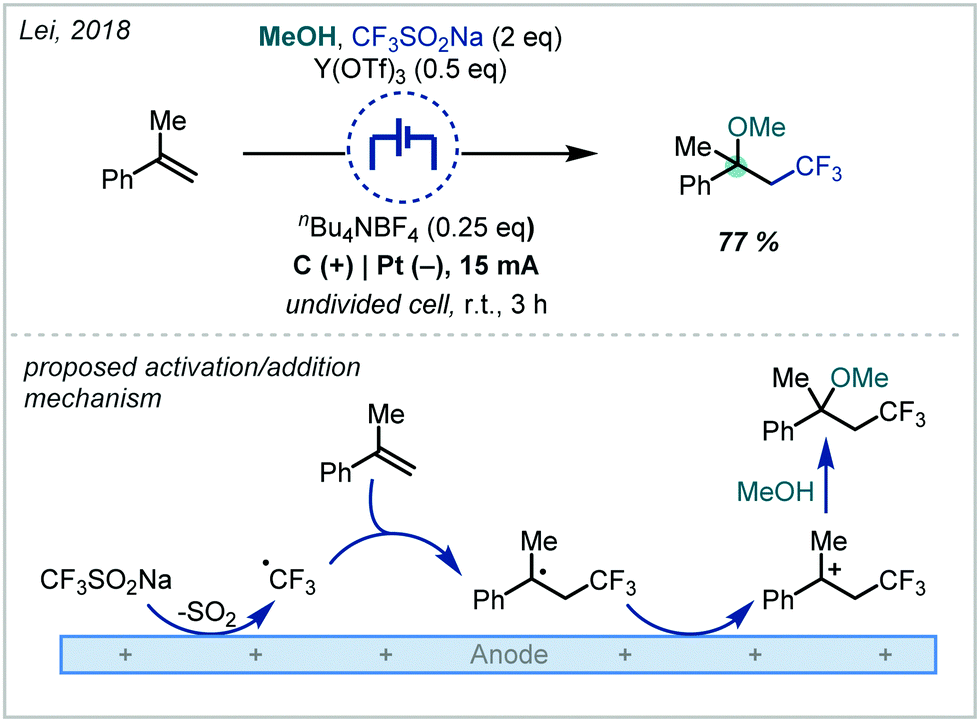 | ||
| Scheme 20 Electrochemical generation of the trifluoromethyl radical and subsequent reactivity to afford α-tertiary ethers. | ||
In an analogous fashion, sulfonyl radicals have been widely applied to such difunctionalisation reactions (Scheme 21A). Lei has demonstrated the application of sulfonyl hydrazides in this transformation, which, upon oxidation, afford the desired sulfonyl radical;98 and subsequently Sun and Han reported the use of sulfinic acids and sulfonate salts, respectively.99,100 Han's report is notable given the extensive range of α-tertiary ethers afforded.
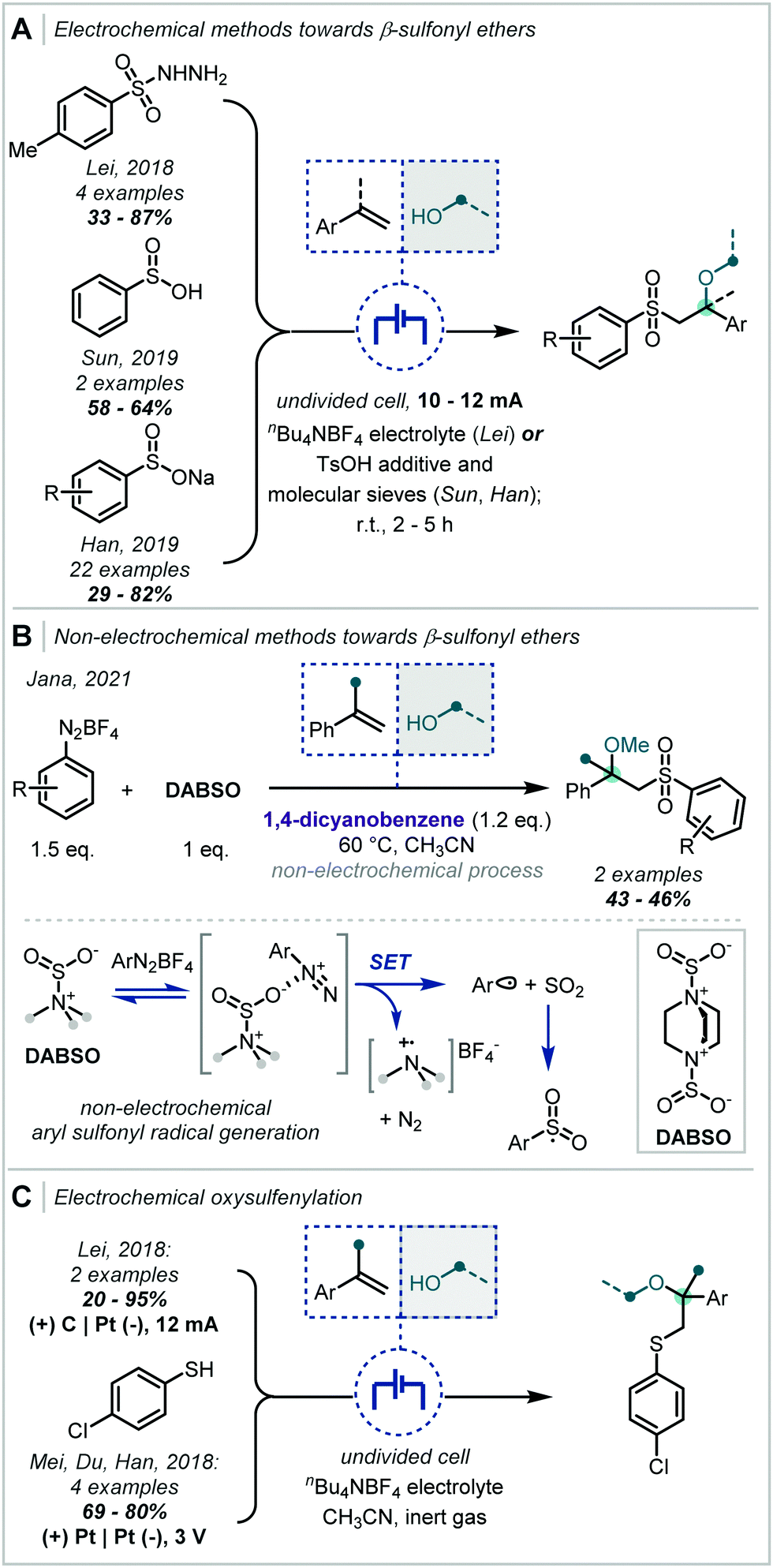 | ||
| Scheme 21 Alkoxy-sulfonylation and -sulfenylation routes to α-tertiary ethers. | ||
It should be noted that a related alkoxysulfonylation was reported by Jana without the use of electrochemical activation (Scheme 21B).101 In this work, the authors employed 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) (DABSO), in the presence of aryl diazonium salts, to generate in situ the desired aryl sulfonyl radical. Mechanistically, it was proposed that SET from DABSO brought about fragmentation of the diazonium salt and produced an aryl radical capable of combination with SO2. Subsequent reactivity was as described above, however, the oxidation of the alkyl radical, resulting from sulfonyl addition to the alkene, was effected by 1,4-dicyanobenzene (DCB).
Through minor modification to the electrochemical system described for oxidation of sulfonyl hydrazides, Lei additionally reported the oxysulfenylation of alkenes (Scheme 21C).102 Employing thiols in this case, the authors observed Markovnikov regioselectivity following the addition of a thiyl radical. The study demonstrated that the subsequently formed carbocations could be trapped with a broad range of alcohols and even extended this to amine nucleophiles. α-Tertiary ethers could be afforded in variable yields, with variation on either side of the ether linkage. Shortly after, Mei, Du & Han reported the oxysulfenylation of alkenes and demonstrated that this system could be extended to etherification, with α-tertiary ether examples afforded in good yields.103 Notably, Lei proposed a putative alternative mechanism in which an electrophilic sulfur species could be generated following oxidation of transiently formed disulfides.
2.3 Transition metal-mediated alkene difunctionalisation
The extensive redox chemistry of transition metals presents significant opportunity for catalysis under both one- and two-electron manifolds. Accordingly, this behaviour has been applied to the redox-mediated synthesis of α-tertiary ethers. The following section will highlight such methods, and discuss their pertinent mechanistic aspects. Due to their inclusion in the Section 2.1, however, reactions mediated by transition metal-based photocatalysts will not be included. In addition, cross coupling reactions mediated by Pd and other metals will not be covered, since they have been extensively reviewed elsewhere.40–43,104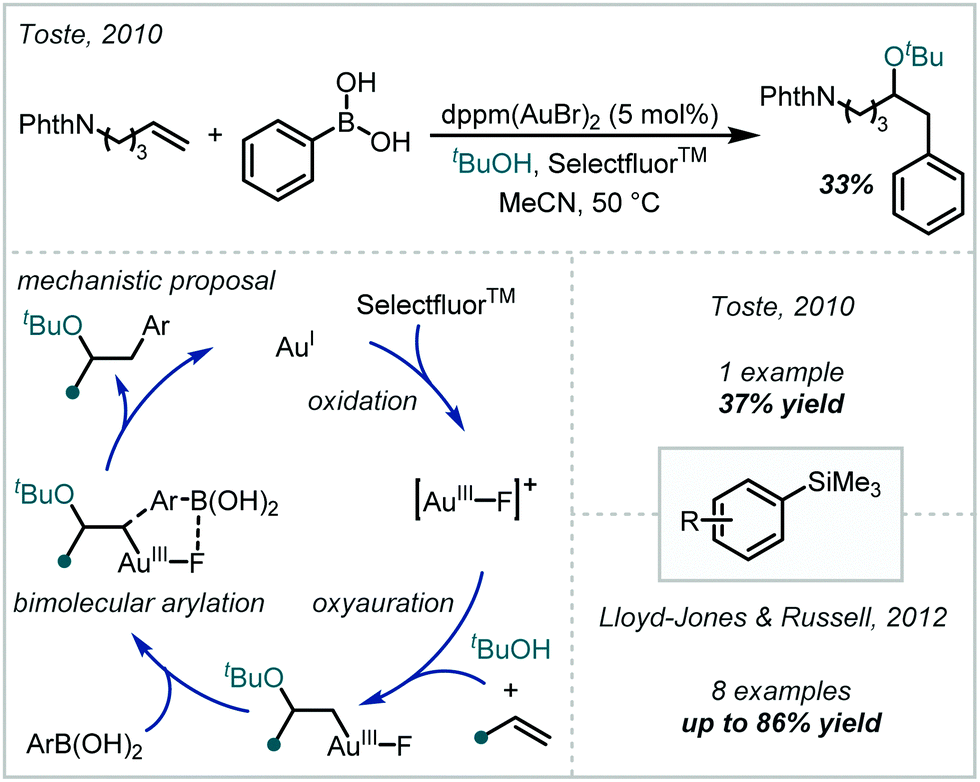 | ||
| Scheme 22 Au-catalysed oxyarylation of alkenes with aryl boronic acids, and extension to aryltrimethylsilanes. | ||
The realisation that no transmetalation occurred during this reaction inspired the use of aryltrimethylsilanes in analogous works.106,107 Consequently, stable and readily accessible silane reagents could be used to good effect without the need for exogenous fluoride or hydroxide, which is typically required to facilitate their transmetalation to metal centres. Additionally, the use of silanes reduced the formation of biaryl side-products associated with Toste's boronic acid protocol. Reports by Toste and Lloyd-Jones & Russell established the efficacy of the silane protocol for forming α-tertiary ethers when 1,1-disubstituted olefins were employed, with slight modifications to the catalytic systems in each case.
In 2017, Wolfe detailed an oxidative carboalkoxylation of alkenes for the synthesis of carbocyclic scaffolds (Scheme 23).108 The authors proposed that upon oxidative addition of a Pd catalyst into a preinstalled alkenyl or aryl triflate, pendant alkenes underwent oxypalladation with phenol nucleophiles to install the ether moiety. Subsequent reductive elimination formed the C–C bond and released the bicyclic products. This protocol was shown to be effective for the formation of a collection of carbocyclic α-tertiary ethers, which were afforded in good yield with high levels of diastereoselectivity.
 | ||
| Scheme 23 Pd-catalysed carboalkoxylation of alkenes. | ||
Additionally, the single-electron redox chemistry of transition metal salts has been effectively employed in alkene difunctionalisation reactions for the formation of α-tertiary ethers. In 2014, Deng and Lei reported that CuCl can facilitate the single electron reduction of α-bromonitriles to yield the corresponding alkyl radical.109 This then participated in a radical addition/RPC mechanism with styrenes (Scheme 24A), which yielded an α-tertiary ether in 30% yield. A mechanistically similar transformation was later reported by Nishikata, in which α-bromoesters were used as the alkyl radical precursor.110 However, the yields of α-tertiary ether products were low: a single example with t-BuOH as the nucleophile was reported in 16% yield.
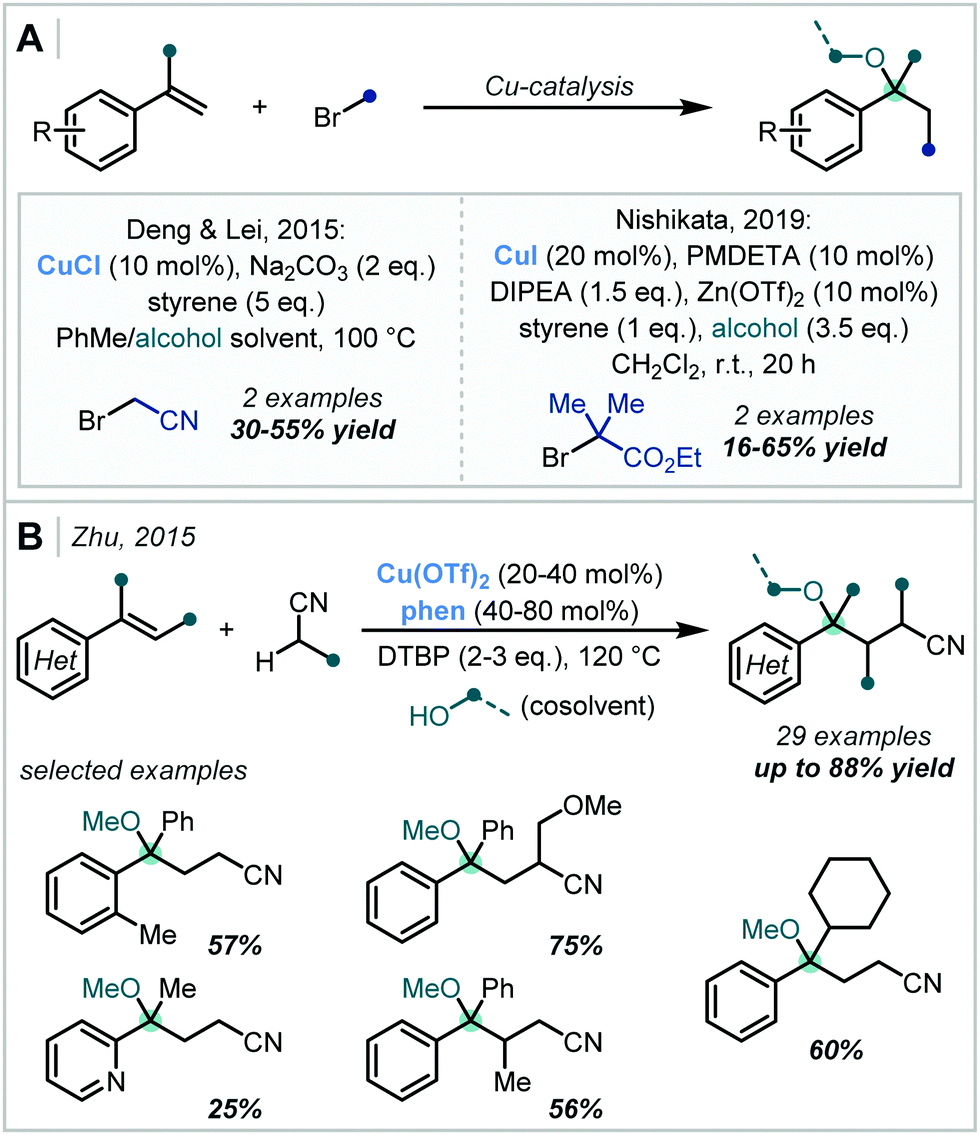 | ||
| Scheme 24 Cu-catalysed carboetherification reactions via: (A) single-electron reduction of alkyl bromides; (B) C–H activation at the α-position of alkyl nitriles. PMDETA = N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine; phen = 1,10-phenanthroline. | ||
In 2015, Zhu reported a method for the formation of a wide scope of styrene-derived α-tertiary ethers (Scheme 24C).111 It was shown that a Cu(II) catalyst could selectively metalate the α-C–H bond of alkyl nitriles via a deprotonation–metalation pathway, facilitating addition of the nitrile moiety to the alkene π-bond of styrene derivatives. Following Cu-mediated oxidation of the resultant alkyl radical, reaction with an alcohol co-solvent yielded the final products – di-tert-butyl peroxide (DTBP) was used to enable turnover of the catalytic cycle. The intermediate steps were proposed to proceed either via free radical species or Cu-bound intermediates. A variety of examples were reported with excellent yields, including those containing both electron-rich, poor and ortho-substituted aromatics, heterocycles and hindered trisubstituted alkenes.
In 2017, Bao demonstrated that Fe salts were capable of single electron reduction of diacyl peroxides, affording nucleophilic alkyl radicals (Scheme 25A).112 Addition of the alkyl radical to the alkene π-bond of styrene derivatives, RPC, and methanol addition yielded an α-tertiary ether in good yield. Recently, Guo reported the Fe-catalysed carboetherification of styrenes using silyl peroxides as the alkyl radical source (Scheme 25B).113 In this case, radical generation proceeded via peroxide reduction and subsequent β-scission of the alkoxy radical. The formation of a single α-tertiary ether was reported employing t-BuOH as the nucleophile.
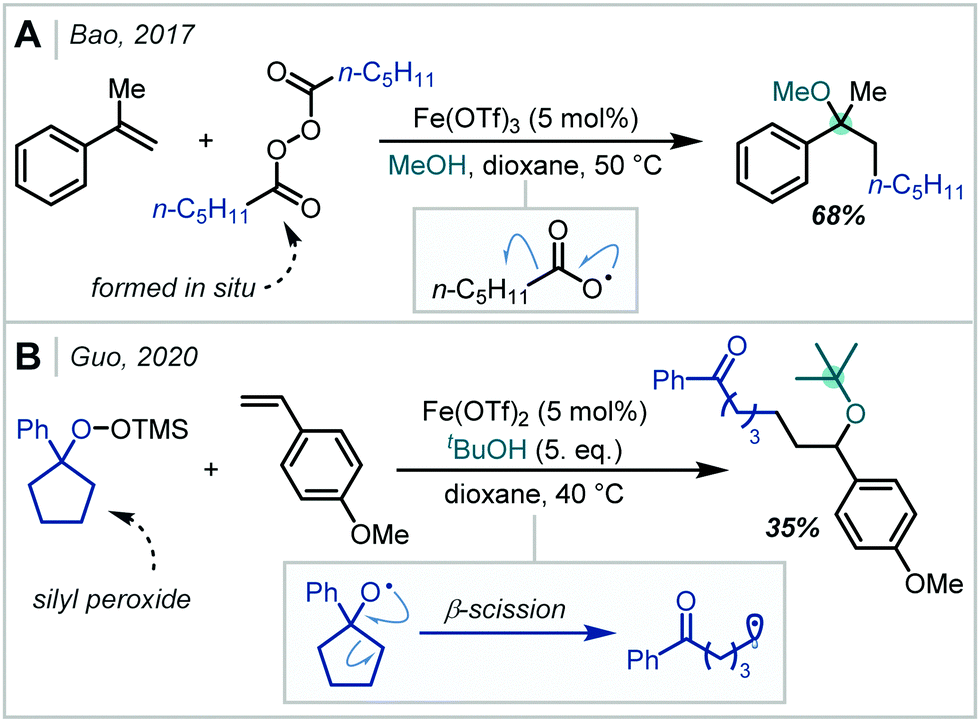 | ||
| Scheme 25 Fe-catalysed formation of α-tertiary ethers using: (A) diacyl peroxides; (B) silyl peroxides. | ||
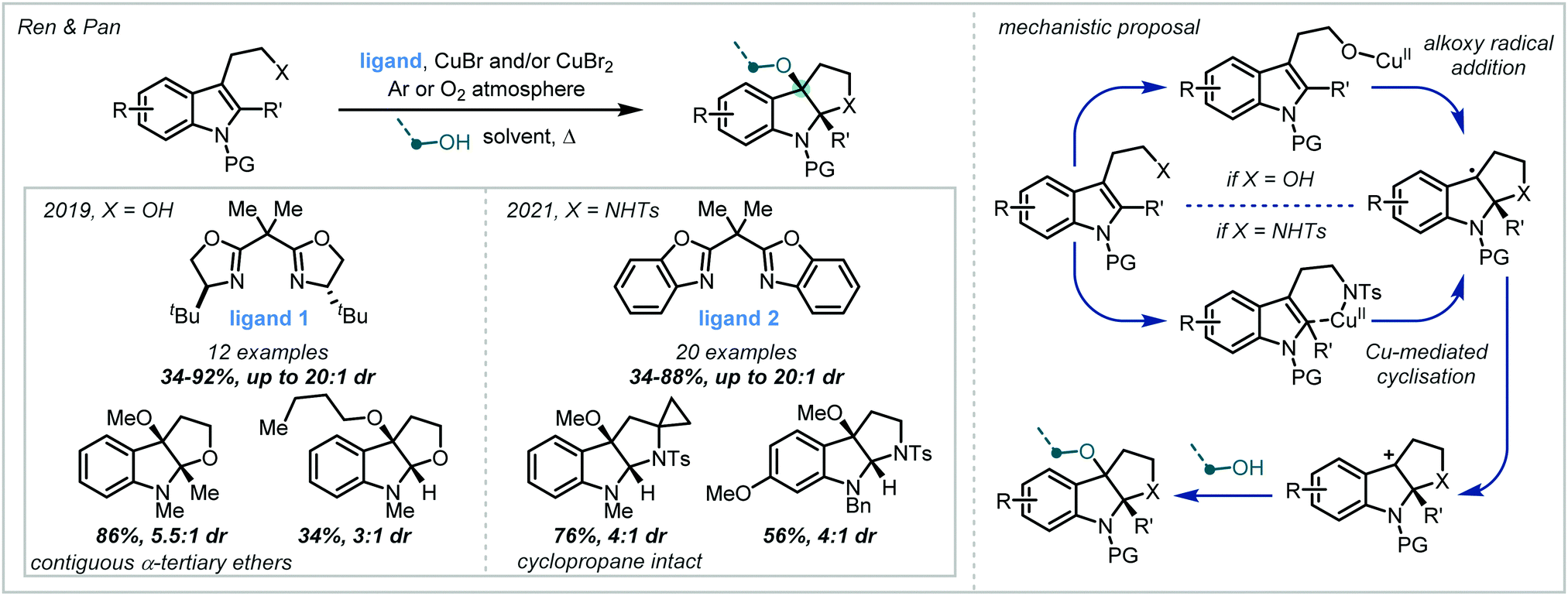 | ||
| Scheme 26 Cu-catalysed synthesis of furoindolines and pyrroloindolines from tryptophan derivatives. | ||
Oxidation of the resultant radical by a Cu(II) species generated a stabilised carbocation that could be intercepted by a primary alcohol in a diastereoselective manner. In 2021, the authors extended this methodology to the synthesis of the related pyrroloindolines.115 The proposed mechanism was quite different, implicating Cu-mediated delivery of the pendant amine rather than the generation of free aminyl radicals. Conducting the reactions under an atmosphere of O2 was shown to increase product yields and it was therefore proposed that O2 acted as the terminal oxidant, responsible for turning over the copper catalyst and closing the catalytic cycle. A wide scope of α-tertiary ether-containing products were reported, many of which were formed with excellent diastereoselectivity.
Transition metal catalysis has also found use in the oxidative formation of β-amino-α-tertiary ethers from alkene starting materials. In 2014, following investigations into tethered aminohydroxylation reactions, Robertson developed a Rh-catalysed oxyamination of allylic carbamates (Scheme 27A).116 It was proposed that in the presence of (diacetoxyiodo)benzene (PIDA) and Rh2(oct)4 [oct = octanoate], a Rh-nitrenoid was formed at the carbamate nitrogen, which was capable of cyclising onto a pendant alkene to yield a bicyclic aziridine intermediate. In a single example, the authors showed that when an excess of methanol was used in the reaction, regioselective ring opening of the aziridine was achieved, yielding the α-tertiary ether product. Following this report, the application of aziridines in oxidative alkoxyamination reactions has been expanded upon. In 2020, Sun, Yu & Dodd reported the intermolecular aminoalkoxylation of 1,3-dimethyl-5-vinyluracil with t-BuOH, utilising a similar Rh-based catalytic system, with phenyl sulfamate to generate an electrophilic nitrogen centre (Scheme 27A).117 However, competitive elimination of the alkoxide moiety in the product severely limited the yield in this case.
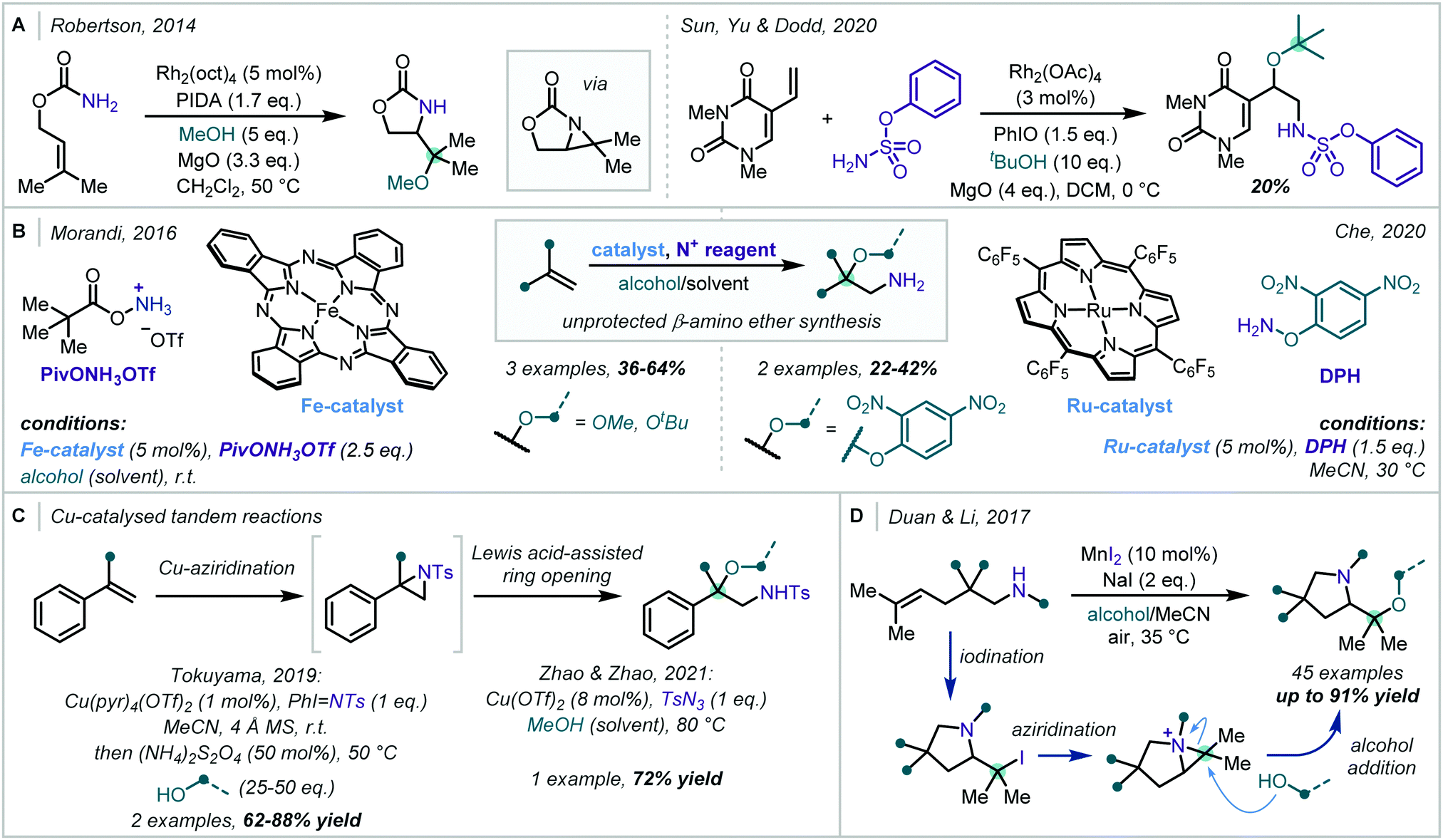 | ||
| Scheme 27 Metal-catalysed oxyamination of alkenes utilising: (A) rhodium and hypervalent iodine; (B) metal-porphyrin catalysts; (C) Cu-catalysed tandem reactions; (D) a one-pot iodination-aziridination approach. | ||
Porphyrin-derived metal catalysts have also been employed to good effect in oxyamination reactions (Scheme 27B). In 2016, Morandi reported that unprotected β-amino-α-tertiary ethers could be forged from unactivated alkenes under Fe catalysis.118 This challenging transformation was realised using a macrocyclic Fe(II)-phthalocyanine catalyst and PivONH3OTf (Piv = pivaloyl) as an electrophilic nitrogen source. Three examples of α-tertiary ethers were reported in moderate to good yield and, impressively, t-BuOH was found to be a competent nucleophile in this reaction. Later, Che reported a similar methodology that used O-(2,4-dinitrophenyl)hydroxylamine (DPH) as the aminating reagent alongside a Ru–porphyrin catalyst.119 A few examples of α-tertiary ethers were synthesised in this manner, however, no exogenous alcohol could be incorporated into the product – only ethers derived from the 2,4-dinitrophenol moiety could be formed.
Cu catalysis has also been used in related transformations, particularly since the Lewis acidity of copper salts can be used to assist the subsequent ring-opening reaction of the aziridine (Scheme 27C). Tokuyama and Zhao & Zhao exploited this dual catalytic role of copper in recent reports and detailed the synthesis of a small number of α-tertiary ether examples in good to excellent yield.120,121
In 2017, Duan and Li employed aziridinium ions as reactive intermediates in a one-pot synthesis of valuable prolinol ethers from unfunctionalised olefins (Scheme 27D).122 This was realised using a combination of NaI and catalytic MnI2, under an air atmosphere, to iodinate the double bond with concomitant formation of the pyrrolidine moiety. Subsequent aziridinium formation and ring opening by an exogenous alcohol afforded a diverse array of α-tertiary prolinol ethers in moderate to excellent yields.
Investigating the reactivity and selectivity of allenes, Lee disclosed the use of Au-catalysis in the formation of iodinated α-tertiary ethers from the parent allene (Scheme 28).123,124 Based upon the authors’ earlier work, it was shown that a combination of (IPr)AuCl [IPr = bis-2,6-diisopropylphenyl imidazolylidene], AgOTf and a ten-fold excess of alcohol enabled the regioselective oxyauration of substituted allenes to yield β-metalated-α-tertiary ethers. Terminal oxidation by N-iodosuccinimide (NIS) then formed the iodinated products; notably, NIS could not facilitate the reaction in the absence of the Au catalyst. The authors also demonstrated that the alkenyl iodide could be used as a functional handle for further elaboration via Pd-catalysed Sonogashira and Heck reactions.
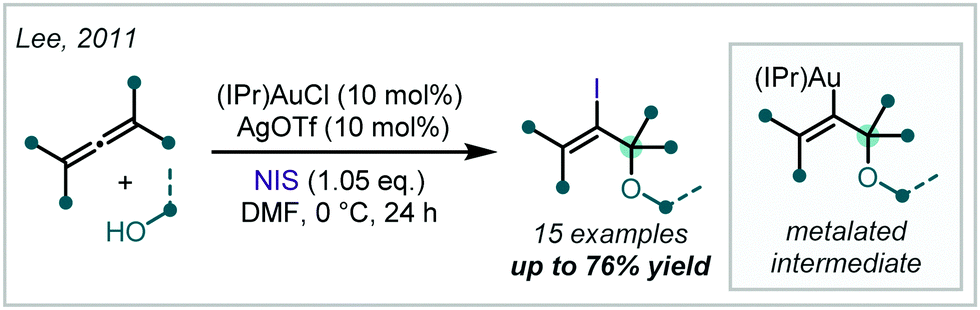 | ||
| Scheme 28 Au-catalysed synthesis of β-iodo-α-tertiary ethers from allenes. | ||
2.4 Halogen-mediated alkene difunctionalisation
The direct application of halogens in the oxidation of alkenes has become well-established as a versatile method for installing β-haloether moieties (Scheme 29). The appropriate combination of an alcohol nucleophile in the presence of oxidising halogen compounds can enable the synthesis of acyclic α-tertiary ethers, and is often dramatically improved by the application of additives and catalysts. This synthetically useful transformation gives a variety of halogenated products which are used in pharmaceuticals,65–67 agrochemicals,125,126 and as important synthons in the synthesis of complex scaffolds.127–130 These methods often employ inexpensive, commercially available reagents, with short reaction times and operate under ambient conditions.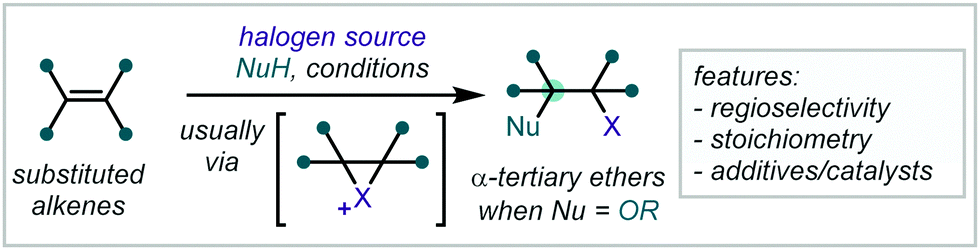 | ||
| Scheme 29 Co-halogenation of alkenes to afford β-haloethers. | ||
In order to ensure high yields, such reactions are more commonly observed for substrates that result in regioselective Markovnikov reactions, typically featuring 1,1-di- or 1,1,2-tri-substituted alkenes. Moreover, these strategies typically rely on superstoichiometric, if not solvent, quantities of alcohol, thus limiting widespread application of these methods to high-value alcohols, or to substrates that are insoluble in an alcohol or alcohol–cosolvent mixture. Nonetheless, the methods presented in this chapter offer efficient and robust routes to access important α-tertiary β-haloethers.
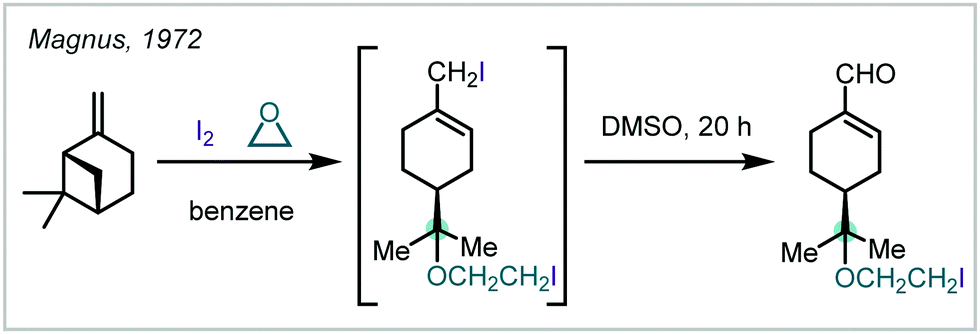 | ||
| Scheme 30 Ring opening of (−)-pin-2(10)-ene with I2 to give an α-tertiary ether. | ||
Despite early studies detailing the capability of weakly Lewis acidic cations, for example Tl(I), to promote the ring opening of transiently formed iodonium intermediates (Scheme 31A),132 there has been a concerted effort to avoid the use of potentially toxic metal salts and move towards additives which are environmentally benign or that give by-products which are more easily removed on work-up.
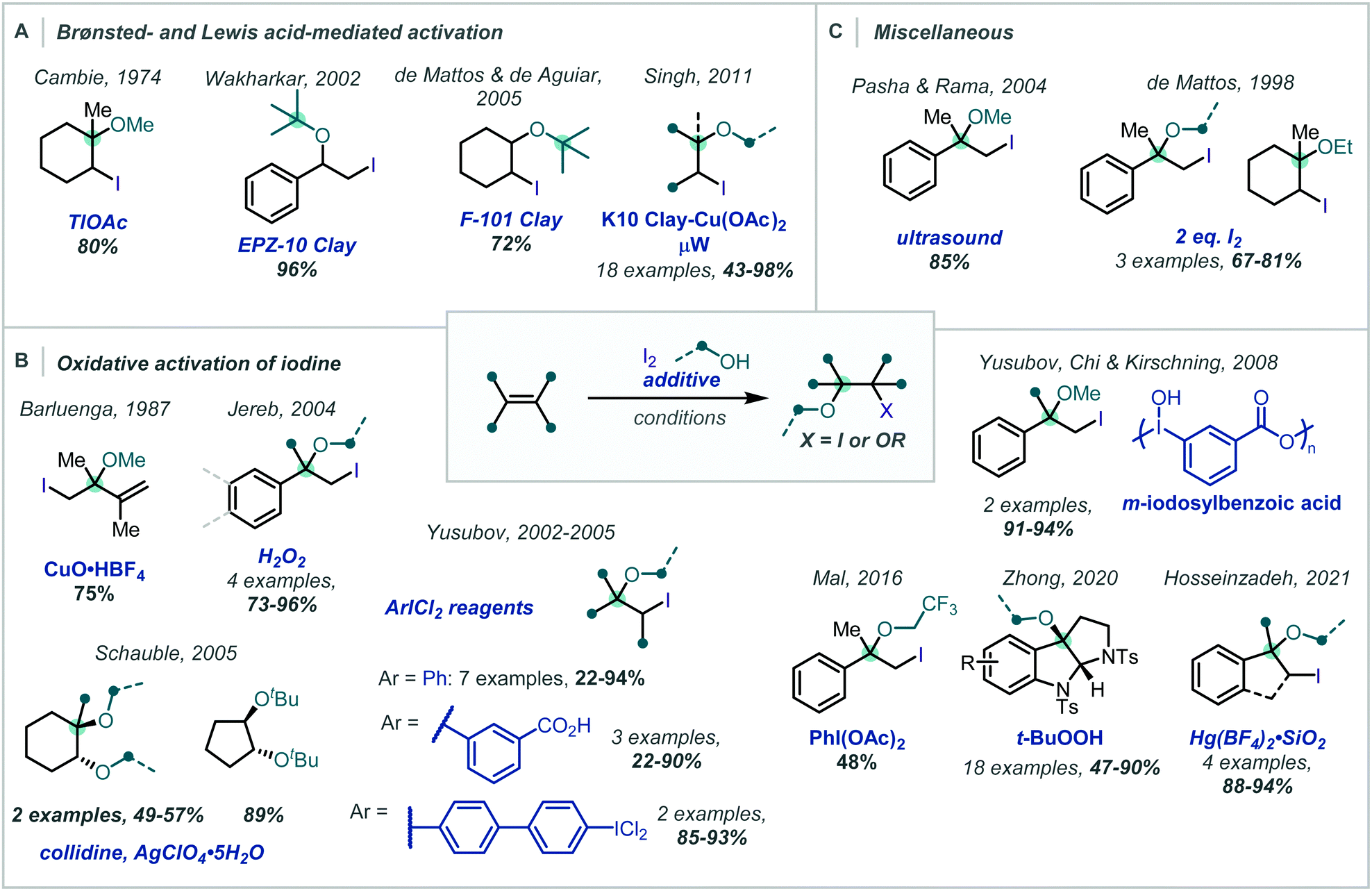 | ||
| Scheme 31 Iodine-mediated synthesis of α-tertiary ethers facilitated by: (A) acidic additives, (B) oxidative additives, and (C) miscellaneous activation strategies. | ||
In the spirit of this, de Mattos showed, in 1998, that simply using two equivalents of iodine in an alcohol solvent afforded the corresponding β-iodoethers in good to excellent yields, several bearing an α-tertiary ether (Scheme 31C).133 It is proposed that activation of one molecule of iodine by another occurs via halogen–halogen bonding in an alkene-I2 complex.
This drive for more environmentally-benign reagents has led to considerable investigation into the beneficial effects of aluminosilicates present in clays (Scheme 31A). Wakharkar, de Mattos, de Aguiar and Singh have independently highlighted this effect in the synthesis of α-tertiary ethers in good yields.134–136
Methods which employ oxidants as additives, in combination with iodine, also afford β-iodoethers in good to excellent yields. In 1987, Barluenga disclosed a CuO·HBF4 promoted iodoetherification of 2,3-dimethylbuta-1,3-diene with quantitative recovery of CuI from the crude reaction mixture (Scheme 31B).137 Recently, Hosseinzadeh reported the use of Hg(BF4)2 supported on SiO2 to facilitate the reaction of molecular iodine with α-methylstyrene and various alcohols in over 88% yield, including an α,α′-tertiary ether.138
Yusubov demonstrated the use of hypervalent iodine reagents, such as PhICl2, as oxidative additives to give more electrophilic “IX” species in situ (Scheme 31B).139,140 These reacted smoothly with alkenes, giving two examples of α-tertiary ethers in good yield; this method was also applied to halomethoxylation of monoterpenes.141 This concept was further extended to a number of modified ArICl2 reagents142 and polymeric m-iodosyl benzoic acid species,143 capable of facile recovery and recycling of the reduced species. Notably, exclusion of iodine in these approaches leads to the β-chloroethers as demonstrated in the reports from Yusubov and in subsequent work from Karade (Scheme 32), employing a modified triazine “ICl2” reagent.144
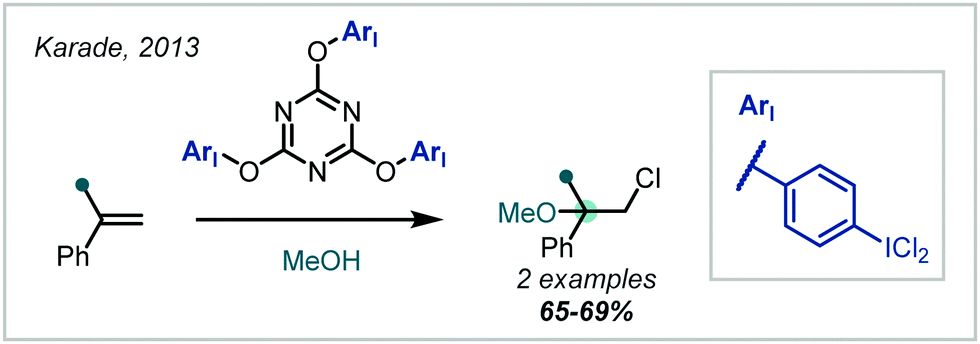 | ||
| Scheme 32 A novel I(III)–Cl reagent for the chloroetherification of alkenes. | ||
In 2016, Mal demonstrated that PIDA can react with I2, giving IOAc (Scheme 31B).145 This stronger electrophile reacted in a facile manner with α-methylstyrene in CF3CH2OH to give the corresponding β-iodoether, albeit in only 48% yield.
A related strategy had previously been employed by Jereb, using H2O2 as an oxidant to form β-iodoethers from α-methylstyrene and 1,1-diphenylethene in methanol or ethanol.146
An isolated report from Schauble involved 1,2-dialkoxylation of cyclopentene and cyclohexene-type olefins using I2, silver perchlorate, and collidine to give a β-iodoether intermediate (Scheme 31B).148 Ag+-assisted iodide abstraction facilitated substitution – notably even with hindered t-BuOH; however, undesired 1,2-hydroxyethers were frequently observed unless anhydrous silver perchlorate was used.
Zhong reported an oxidative cascade towards pyrroloindoline derivatives, employing alkyl peroxides as terminal oxidants in an etherification of the tryptamine moiety (Scheme 31B).149 This constitutes a complementary strategy in which the addition of an oxidant enables catalytic turnover of the halogenating reagent, rather than its activation. Notably PIDA was reportedly unsuccessful, with the authors identifying t-butyl hydroperoxide as a superior oxidant to re-oxidise the iodide to molecular iodine. Once the iodonium ion was formed, the pendant amino group of the tryptamine moiety cyclised onto this to give the tertiary alkyl iodide. Nucleophilic substitution of the newly generated alkyl iodide gave the α-tertiary ether and the released iodide ion was re-oxidised to molecular iodine by the peroxide, facilitating the use of catalytic I2 (10 mol%).
Interestingly, there has been one report, by Pasha & Rama, where ultrasound sonication was demonstrated to promote the reaction of iodine with α-methylstyrene in methanol (Scheme 31C).147
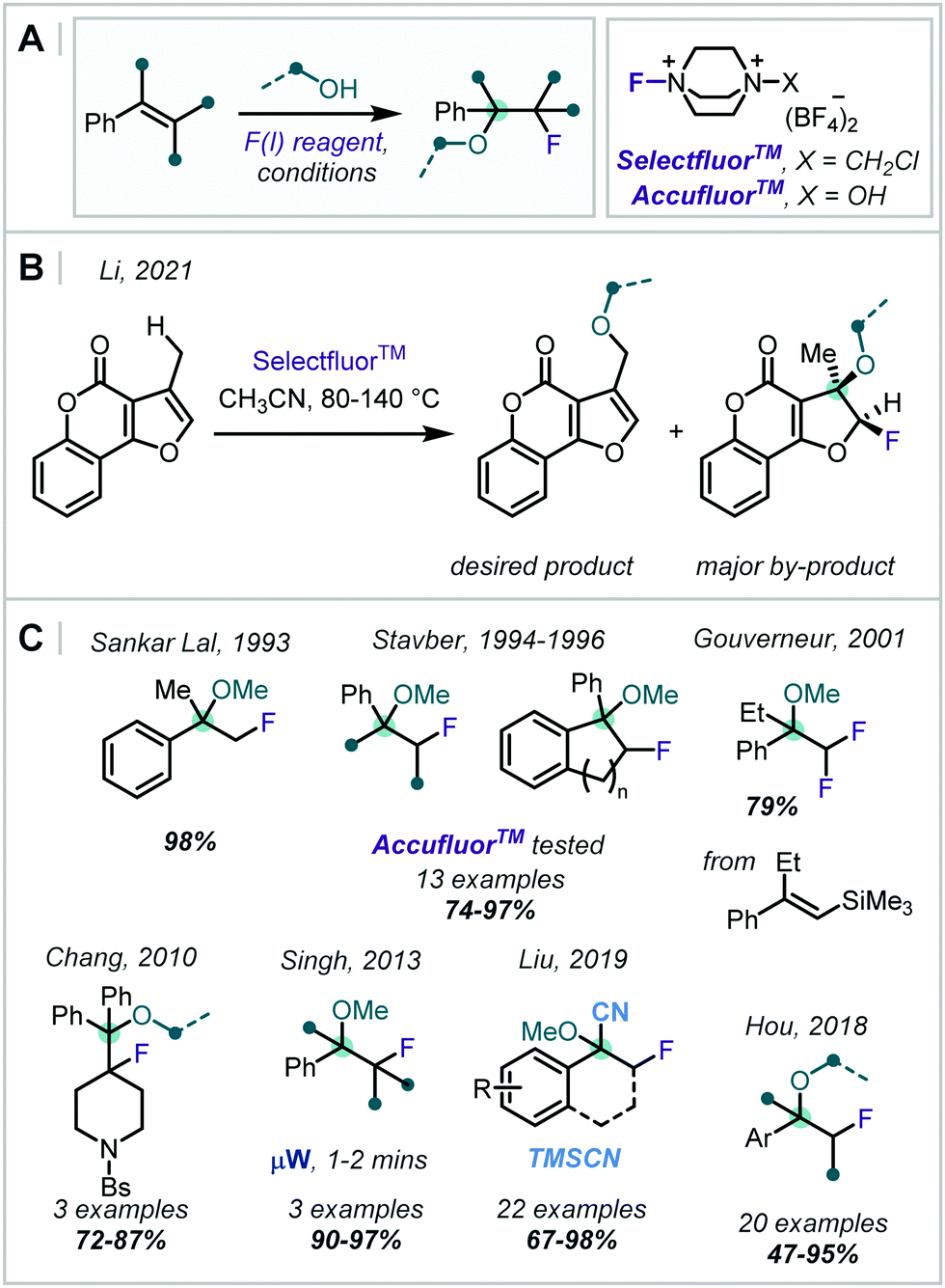 | ||
| Scheme 33 Methods for the formation of β-fluoro-α-tertiary ethers employing SelectfluorTM and AccufluorTM. Bs = SO2C6H4Br. | ||
Sankar Lal reported the use of SelectfluorTM to synthesise a β-fluoro-α-tertiary ether in very high yield, and Stavber subsequently investigated the diasteroselectivity for the same transformation with benzocyclenes (Scheme 33C).153,154 It was shown that ring size and steric hindrance of the alcohol played a significant role in the stereochemical outcome. Later, Stavber reported an analogous reaction on acyclic alkenes but with the alternative fluorinating reagent AccufluorTM and demonstrated similar yields and rates.155 The kinetics and stereochemical course of this transformation with SelectfluorTM were also thoroughly investigated and compared with analogous reactions utilising XeF2 and CsSO4F.156
In 2010, Chang applied this transformation to piperidinyl olefins, affording pharmaceutically relevant 4-functionalised piperidines (Scheme 33C).157 Later, Singh showed that by applying microwave irradiation, the reaction time for such transformations was drastically reduced.158 Investigating alternative substrates, Gouverneur disclosed that the application of two equivalents of SelectfluorTM with alkenyltrimethylsilanes gave the difluorodesilyation product.159
In 2018, Hou reported that the use of SelectfluorTM with 10 equivalents of MeOH in MeCN afforded β-fluoro-α-tertiary ethers (Scheme 33C).160 An impressive 20 examples were demonstrated, including a range of olefins and alcohols, with yields in the range 47–95%.
In an alternative approach, Liu reported cyanofluorination of a range of enol ethers using SelectfluorTM.161 However, instead of the usual direct interception by SelectfluorTM as an electrophile, a single-electron-transfer from the olefin to SelectfluorTMvia an electron donor–acceptor (EDA) complex was suggested. F-Atom transfer from SelectfluorTM to this radical cation intermediate gave an oxocarbenium ion, which was intercepted by TMSCN, forming the α-oxynitrile product.
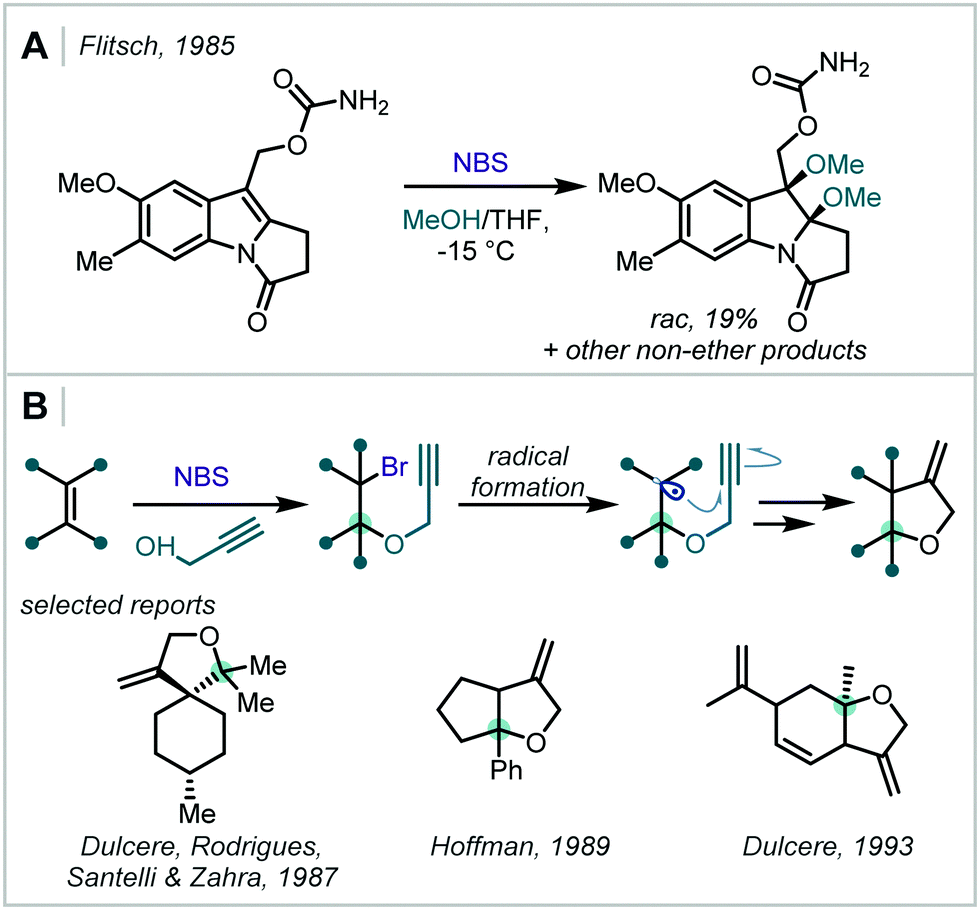 | ||
| Scheme 34 (A) Dimethoxylation of an indole-type structure with NBS in MeOH. (B) NBS-mediated bromoalkynoxylation of alkenes to afford propargylated β-bromoethers. | ||
Several groups have since employed these stable halogen sources to form β-halo-α-tertiary ethers. They have been effectively utilised in the synthesis of β-bromoprop-2-ynyl ethers which were applied in radical cyclisations towards high-value oxygen-containing heterocycles, for example β-methylene tetrahydrofurans (Scheme 34B).164–167 The olefin scope of such reactions includes enones, styrenes, 1,1-diarylethenes and cycloalkenes. In 1993, Dulcère extended this method through use of other alkene- and alkyne-containing alcohols to investigate further reactivity and intramolecular Diels–Alder reactions of the products.168,169 Similar haloetherifications have also been utilised by others in the synthesis of 2-alkoxy-3-bromoindolines and substituted deltacyclenes.170,171
It has since been shown that, by introducing Brønsted acid catalysts in NXS-mediated haloalkoxylation reactions, times could be reduced and the scope of the reactions increased, all while retaining Markovnikov selectivity. Such halogenation reactions have been demonstrated to be promoted by H2SO4,172 NH4OAc,173 thiourea174 and a more elaborate recyclable polyfluorinated hydrazine 1,2-bis(carbothioate) organocatalyst175 (Scheme 35). Typically, the Brønsted acidity of the additive is implicated in activation of the succinimide carbonyl oxygen, via a hydrogen bonding interaction, to promote electrophilic halogen formation.
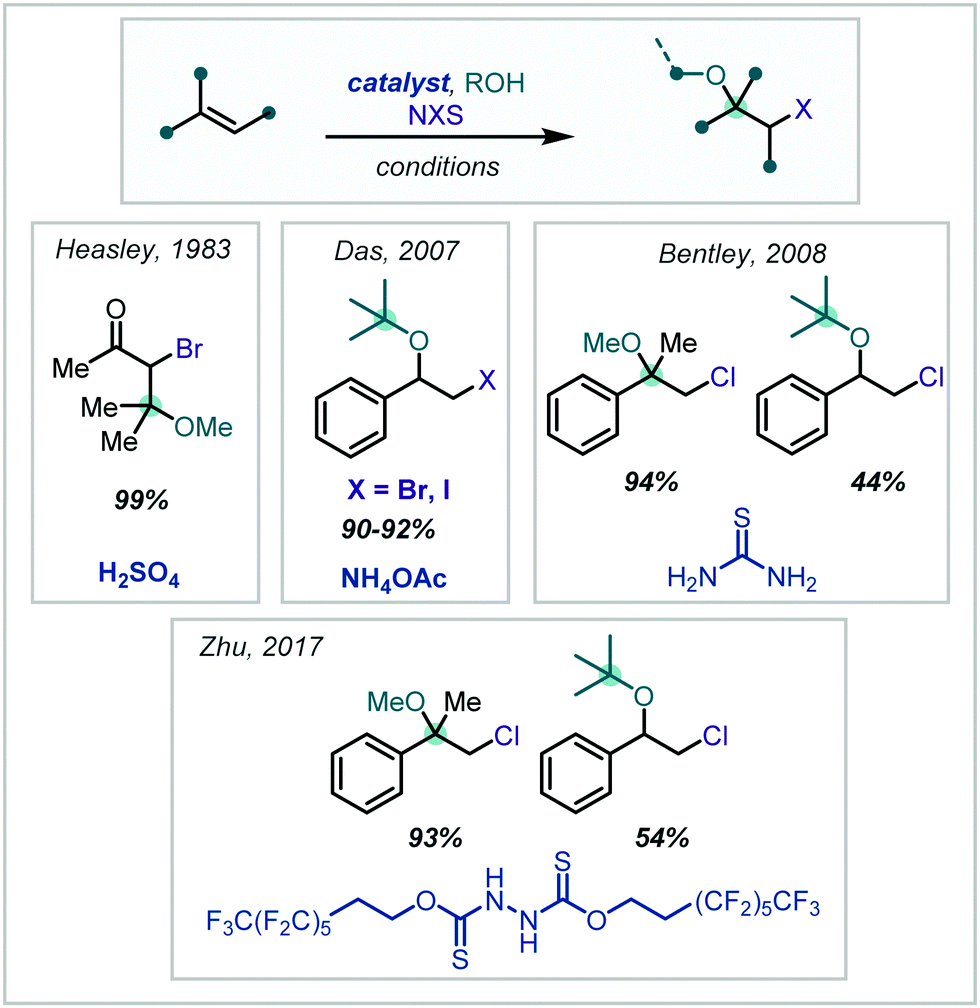 | ||
| Scheme 35 NXS-mediated methods affording β-halo-α-tertiary ethers under Brønsted acid catalysis. | ||
Multicomponent reactions (MCRs) comprise of three or more reagents incorporated into a single product molecule in a one-pot operation and represent an atom-economic approach to access complex scaffolds directly. Early work in 1983 by Dulcère, Rodriguez, Bertrand and Zahra investigated the MCR of alkenes with solvent participation, either ethylene oxide or THF, and either Br2 or Cl2 to give β-haloethers (Scheme 36).176,177
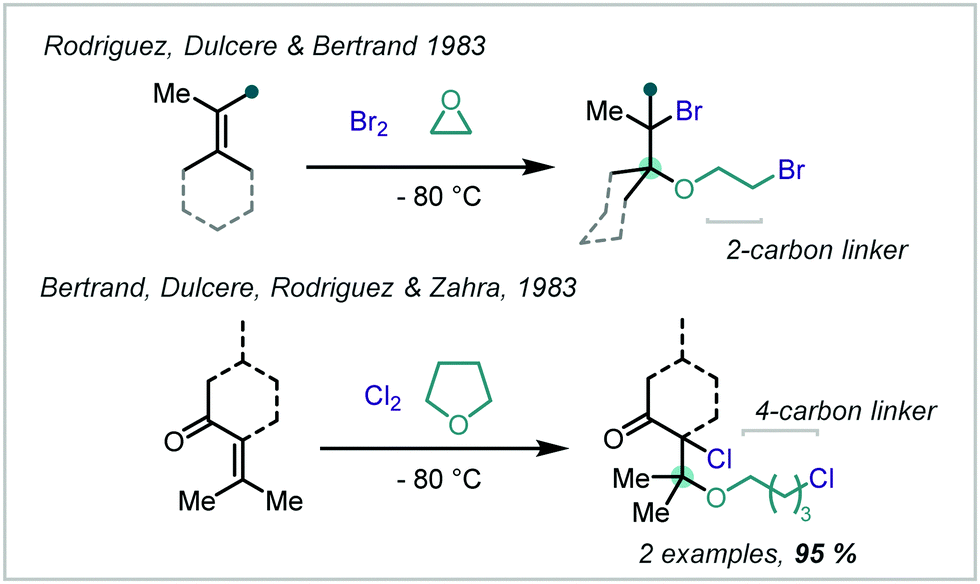 | ||
| Scheme 36 Application of MCR reactions to haloetherification reactions. | ||
In 2003, Serguchev noted a side-product formed via a similar MCR with solvent participation when NBS and a bridged bicyclic diene were reacted in THF with the fluoride source Bu4NH2F3 (Scheme 37).178 The authors also employed an analogous method without solvent participation when the solvent was changed to CH2Cl2 and a perfluorinated alcohol nucleophile was added.
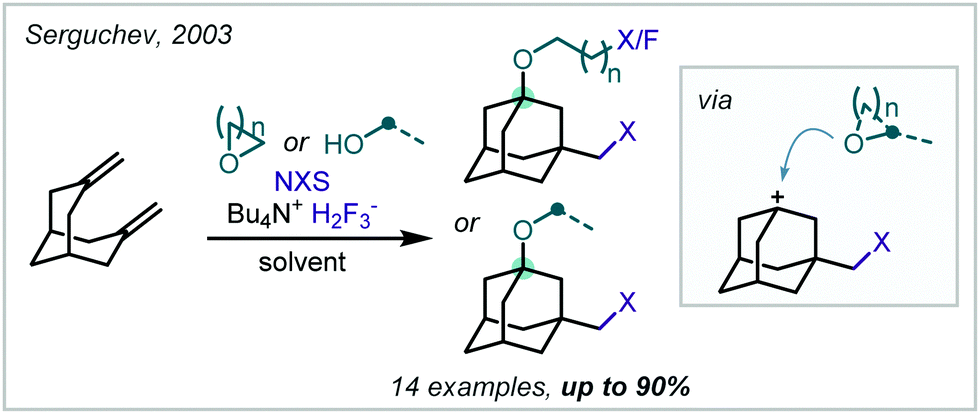 | ||
| Scheme 37 MCR reaction investigated by Serguchev, giving an α-tertiary ether. | ||
This class of MCRs involving NBS and solvent participation has continued to be expanded and in 2010, Yeung disclosed the use of electron-poor sulfonamide nucleophiles in such a reaction, forming α-tertiary ethers in moderate to excellent yields (Scheme 38).179 Yeung also demonstrated in 2011 and 2013 that carboxylic acids and phenols could also act as the nucleophile in this transformation.180,181 Following mechanistic investigations, the acidic proton was reported to play a key role, and was suggested to either activate NBS in a Brønsted acidic manner, or via full bromine atom transfer to form the more electrophilic RCOOBr species, which could rapidly brominate the alkene. With phenols, bromination at any unsubstituted ortho and para positions occurred first, leading the authors to simply use the brominated phenols, giving the desired products in excellent yields.
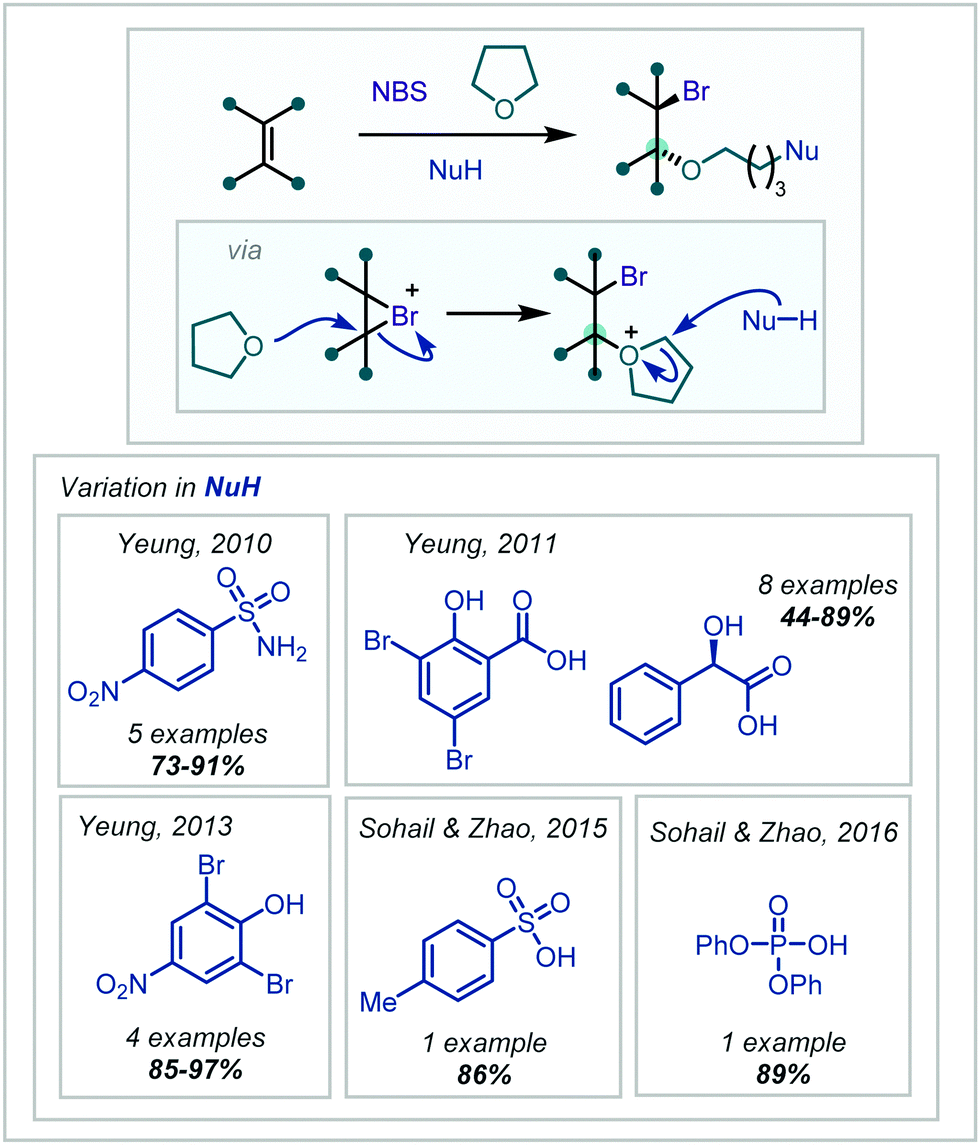 | ||
| Scheme 38 MCR employing THF to install α-tertiary ethers, with terminal incorporation of a variety of nucleophiles. The highest-yielding nucleophile used in each case is shown. | ||
Sohail and Zhao extended this principle to sulfonic and phosphoric acids (Scheme 38).182,183 Interestingly, under more concentrated conditions, there was an observed tendency for a second THF molecule to attack the initial oxonium ion intermediate, with the nucleophile opening the second oxonium ion formed.
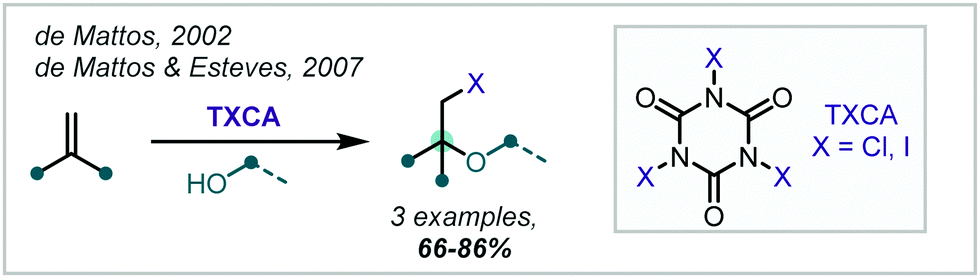 | ||
| Scheme 39 TCCA and TICA-mediated haloetherification of styrene-type olefins. | ||
Reagents containing iodine in the formal +1 oxidation state have been used in the reaction with alkenes to give β-iodoethers (Scheme 40). Glover disclosed that treatment of ICl with t-BuOK generated t-BuOI in situ, which enabled the β-iodo-tert-butoxylation of a range of alkenes in modest to excellent yields.190 Later, Alikarami reacted α-methylstyrene with alcohol nucleophiles in the presence of benzyl(triphenyl)phosphonium dichloroiodate (BTPPICl2), (Scheme 40) to give the corresponding products in very good yields.191 In 2016, Kashyap reported similar transformations of α-methylstyrene and 1,1-diphenylethene with methanol in 70–90% yield when using the in situ-generated iodinating reagent Me3SI(OAc)2.192
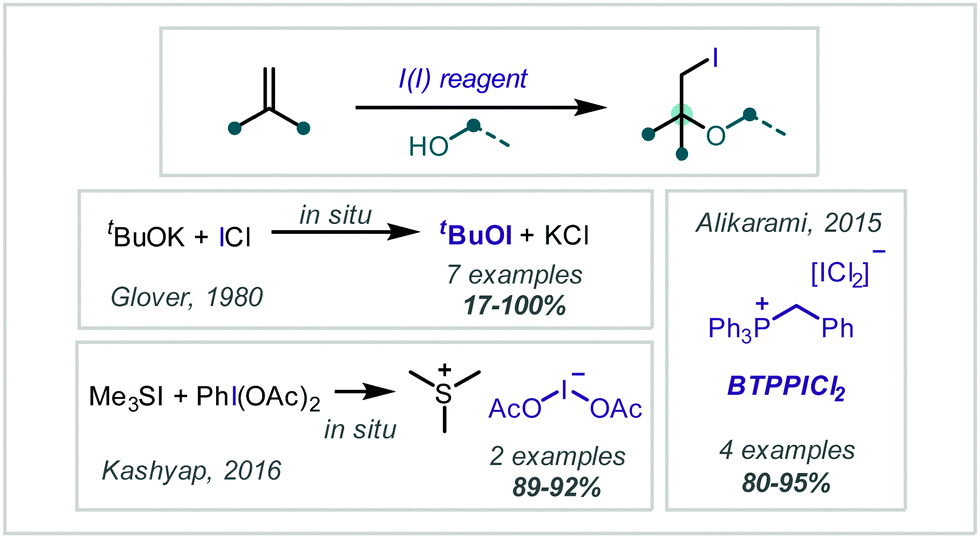 | ||
| Scheme 40 A range of I(I) reagents employed in α-tertiary ether synthesis. | ||
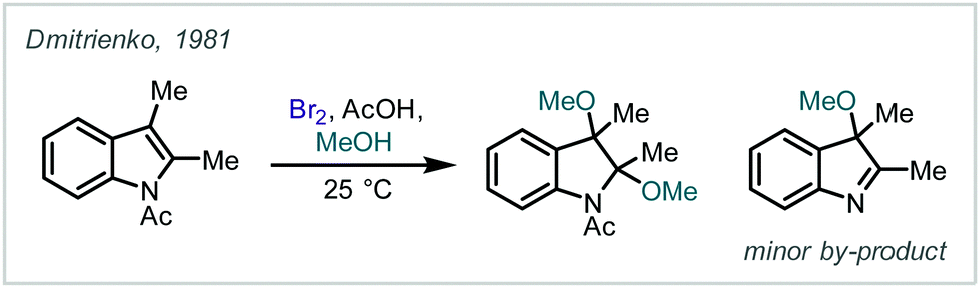 | ||
| Scheme 41 Conversion of di-alkylindoles to β-bromo-α-tertiary ethers using Br2. | ||
A few reports involve the use of a halide source, where the halogen is in the −1 oxidation state, with an oxidant to generate an electrophilic halogenating reagent in situ. In 2009, Ghosh investigated the use of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratios of X−:XO3, (X = Br or I) to generate X2in situ, in the synthesis of β-haloethers, including one α-tertiary example.194
:1 ratios of X−:XO3, (X = Br or I) to generate X2in situ, in the synthesis of β-haloethers, including one α-tertiary example.194
In contrast, Majee reported that NaIO4 in the presence of NH2OH·HCl oxidises olefins to form β-iodoethers.195 In this case, the electrophilic halogenating reagent is generated via introduction of a terminal reductant and the authors suggest I2 is generated in situ while hydroxylamine is oxidised (Scheme 42).196 Reaction with α-methylstyrene and 1,1-diphenylethene in ethanol or ethylene glycol gave the corresponding ethers in around 80% yield.
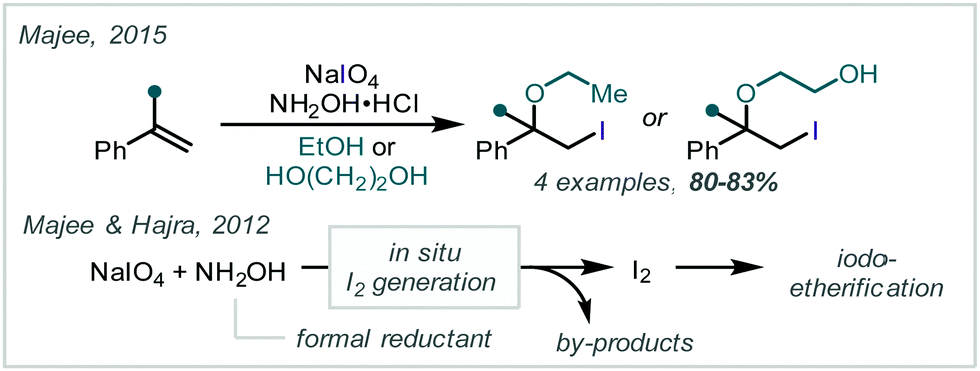 | ||
| Scheme 42 In situ generation of I2 to facilitate the iodoalkoxylation of styrenes. | ||
In 2014, Narender disclosed that the use of environmentally benign, commercially available and inexpensive Oxone® (2KHSO5:KHSO4:K2SO4) with either NH4Br or NH4Cl transformed selected olefins to β-bromo-α-tertiary ethers and β-chloro-α-tertiary ethers respectively in good yields.197,198 In a complementary fashion, Le Bras reported that omission of a halide source resulted in formation of β-alkoxy alcohols.199
In 2019, Shen & Zhang reported a related transformation employing PIDA as the oxidant and the use of a proximal oxime as an intramolecular trap, concurrently forming an isoxazoline (Scheme 43).200 A two-electron pathway is proposed, whereby PIDA oxidised the alkene directly, with subsequent cyclisation of the adjacent oxime moiety (see Section 6.4 for related transformations). Loss of PhI and an acetate anion generated a carbocation, which was intercepted by an alcohol. Many examples containing an α-tertiary ether were afforded in good to excellent yield.
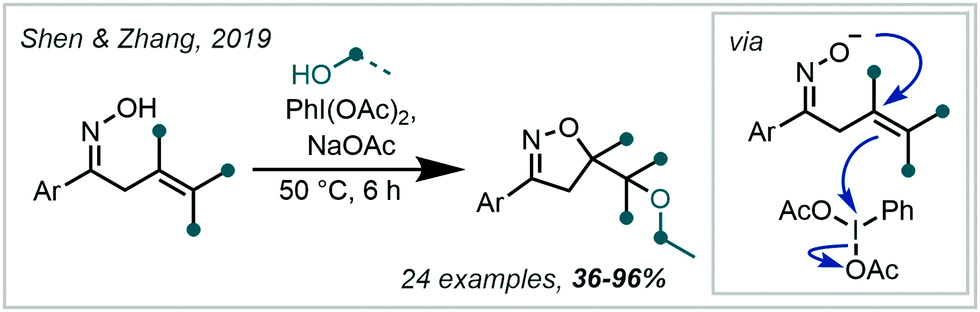 | ||
| Scheme 43 Direct oxidation of an alkene with PIDA to facilitate formation of an α-tertiary ether. | ||
Employing a related approach, Yang reported a MCR, in which an alternative hypervalent I–Cl reagent was proposed to oxidise directly an unsaturated lactam which could follow a related reaction course, albeit incorporating a butyl-oxy linker via THF trapping of the proposed iodonium intermediate (Scheme 44).201
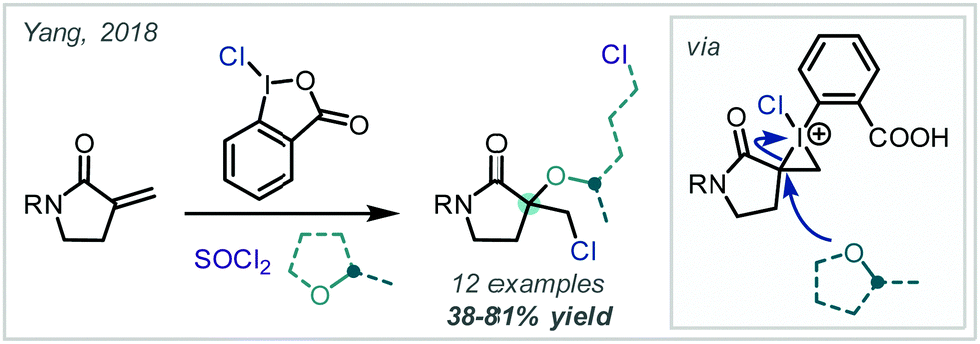 | ||
| Scheme 44 MCR reaction to give an α-tertiary ether from the unsubstituted lactam. | ||
Taking a different approach, mimicking the enzyme vanadium bromoperoxidase, Jacobs reported that a tungstate-exchanged layered double hydroxide ((Ni,Al)-LDH-WO42−) could serve as an effective oxidant of bromide (Scheme 45).202 In the presence of hydrogen peroxide, the WO42− is oxidised to the peroxotungstate species, WO4−n(O2)n2−, which oxidised bromide ions to electrophilic bromine species. These facilitated the β-bromo-α-tertiary etherification of olefins in the presence of MeOH. Hammett analysis confirmed a positively charged transition state and the observed product distributions ruled out a radical mechanism. A wider synthetic scope was subsequently published with a range of relevant examples utilising both α-methylstyrene and linear and cyclic alkenes to give β-bromo-methyl-ethers in good yields.203 Markovnikov selectivity was observed, and the anti-product was exclusively formed, however some alkenes gave 8–28% of the dibromo product as a by-product. Later in 2005, Jacobs applied this system to the oxidation of phenols to quinones, with interception by an alcohol, giving α-tertiary ethers (see Section 6.2).
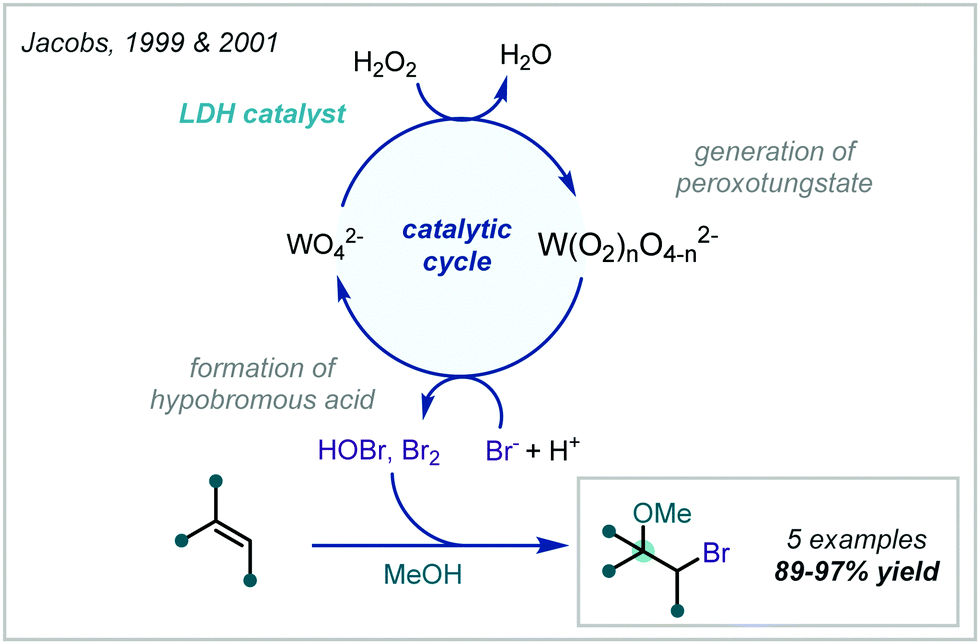 | ||
| Scheme 45 Oxidative bromination of olefins utilising a tungstate-exchanged layered double hydroxide oxidant. | ||
2.5 Oxyselenation
In a similar approach to the halogenation reactions discussed previously, the electrophilic properties of organic selenyl halides have been applied to the functionalisation of alkenes followed by subsequent nucleophilic opening of the seleniranium ion. Notably this transformation extends to nucleophilic opening with co-solvent alcohols and as such the approach has been applied to α-tertiary ether synthesis (Scheme 46).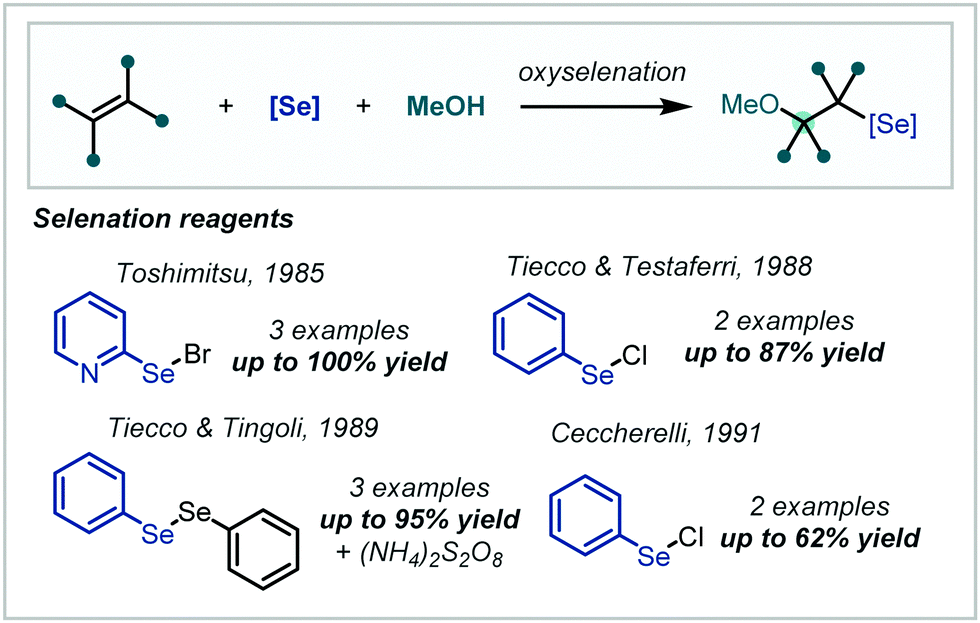 | ||
| Scheme 46 Early studies on the synthesis of β-selenyl-α-tertiary ethers. | ||
In a contemporary development to this system, Braga employed catalytic quantities of iodine as a redox catalyst for the in situ generation of arylselenyl iodides (Scheme 47).208 Their subsequent reactivity followed that of the previous reports and the iodine catalyst was regenerated via acidic activation of DMSO and subsequent iodide oxidation. α-Tertiary ethers could be afforded by the use of α-methyl styrene but notably extension to t-BuOH as the nucleophile was unsuccessful.
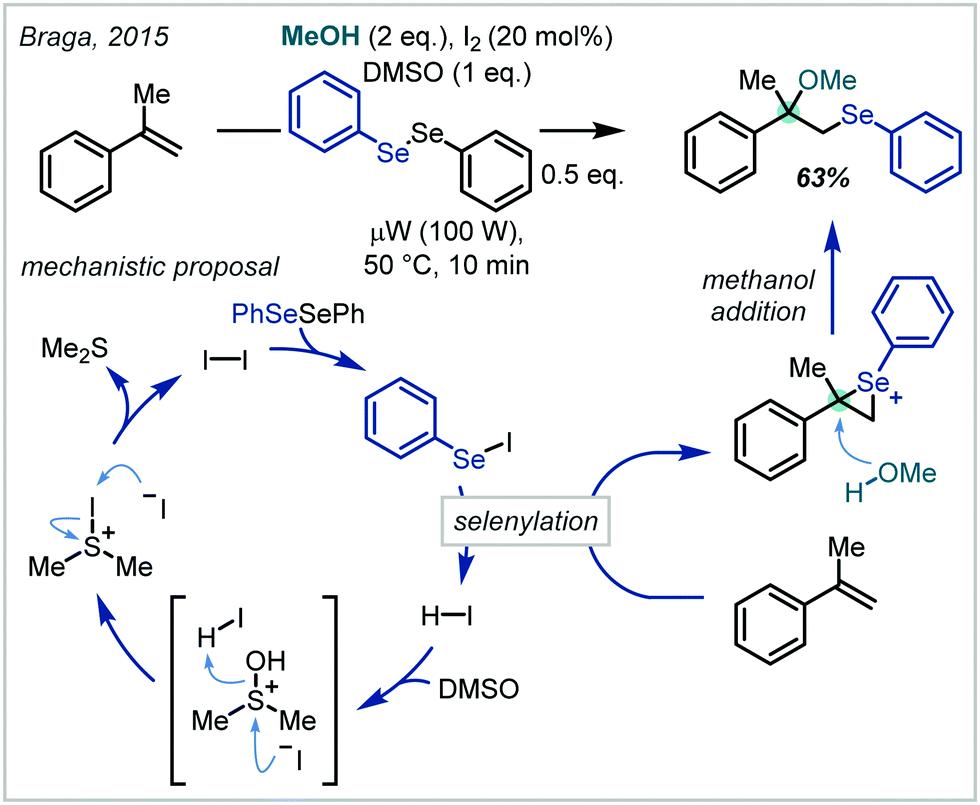 | ||
| Scheme 47 Modern application of halogen-mediated oxyselenation. | ||
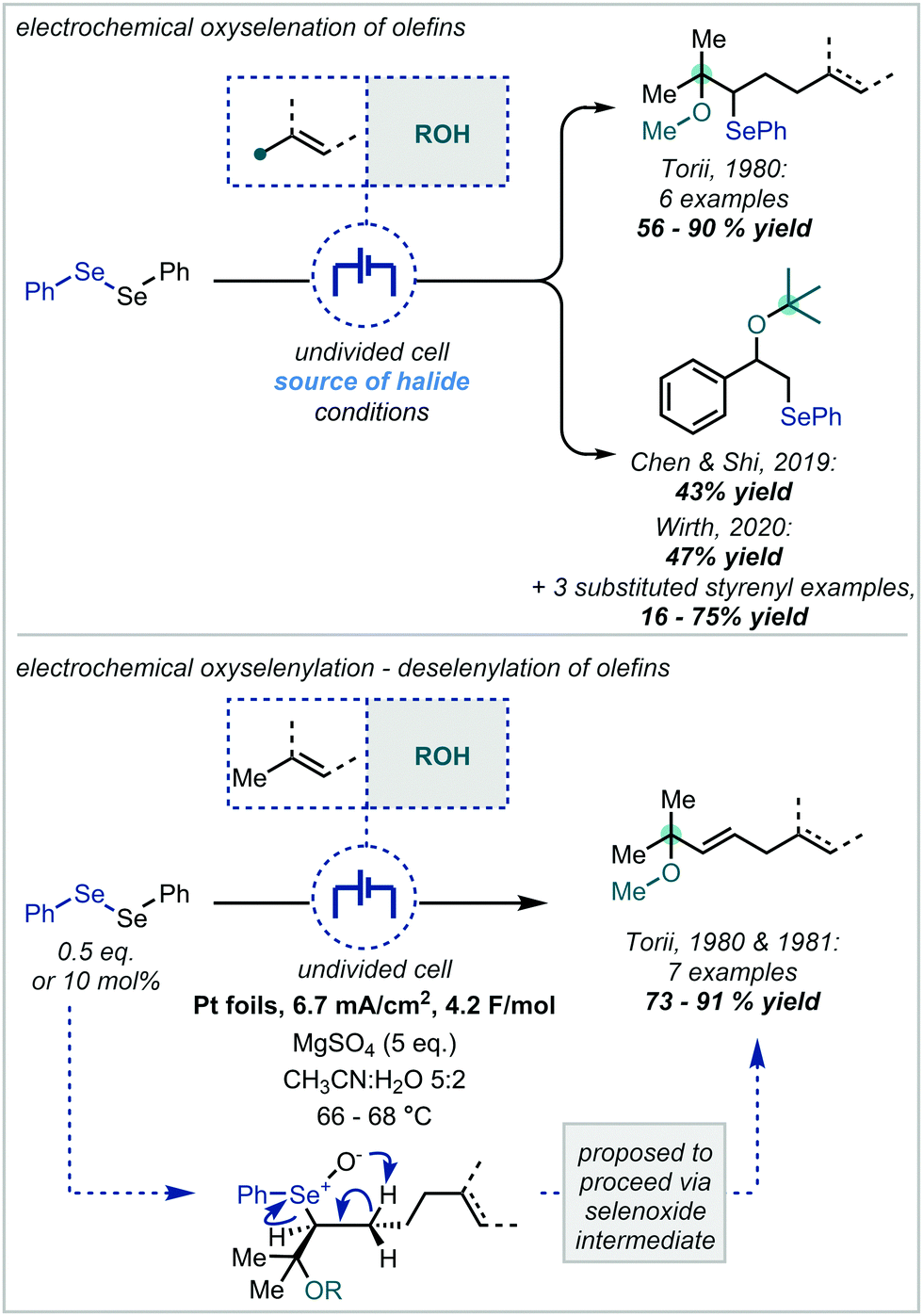 | ||
| Scheme 48 Electrochemically-mediated oxyselenation. | ||
More recently, this approach has been expanded by Chen & Shi via the use of KI as a substoichiometric electrolyte, implicated in generating iodine cations to effect the oxidation of the diselenide (Scheme 48).213 Despite mainly focussing on the hydroxyselenation of alkenes, a single example was reported employing t-BuOH as the co-solvent and therefore the nucleophilic component.
Further demonstrating the robustness of this chemistry, Wirth applied the electrochemical methoxylation to high-throughput automated flow synthesis.214 Employing tetrabutylammonium iodide as the halide source, a library of β-selenyl ethers was afforded by systematic variation of the alkene. In this variation, three tertiary ethers were exhibited from the corresponding α-methyl, -isopropyl and -phenyl styrenes.
A prominent recent development, reported by Xia & Lin, demonstrated that metal-metallaaromatic catalytic systems are capable of efficiently mediating the oxyselenation of alkenes (Scheme 49).215 The authors reported a novel Os–Cu bimetallic species in which the Os centre was incorporated into an aromatic system. The catalytic system was implicated in both nucleophile and alkene activation and in bringing the substrates into close proximity. The reaction was shown to be highly general for α-tertiary ether synthesis with 26 examples reported in good yields and exceptional regioselectivities, and the authors went on to demonstrate the late-stage derivatization of bioactive molecules. Light and oxygen were both shown to be critical for the transformation. Notably, the Os–Cu bimetallic catalytic system was compared to traditional oxyselenation procedures and demonstrated exceptional efficiency.
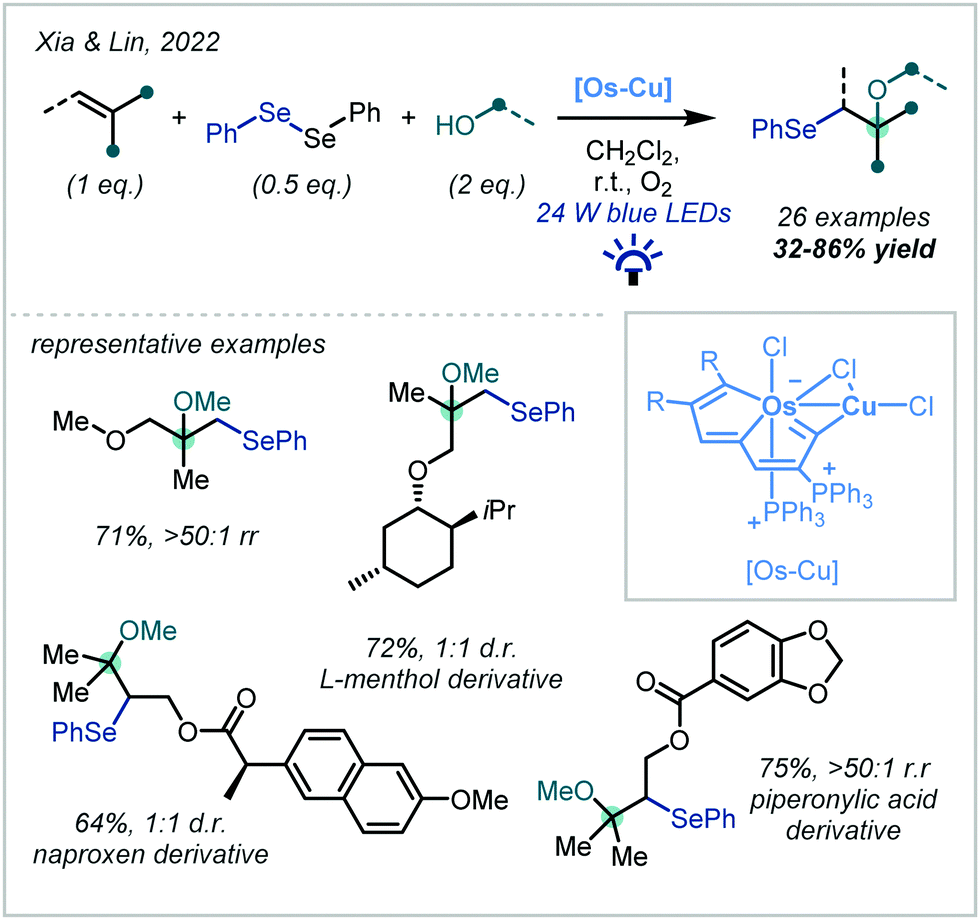 | ||
| Scheme 49 Application of metal-metallaaromatic systems to the photochemically-mediated oxyselenation of alkenes. | ||
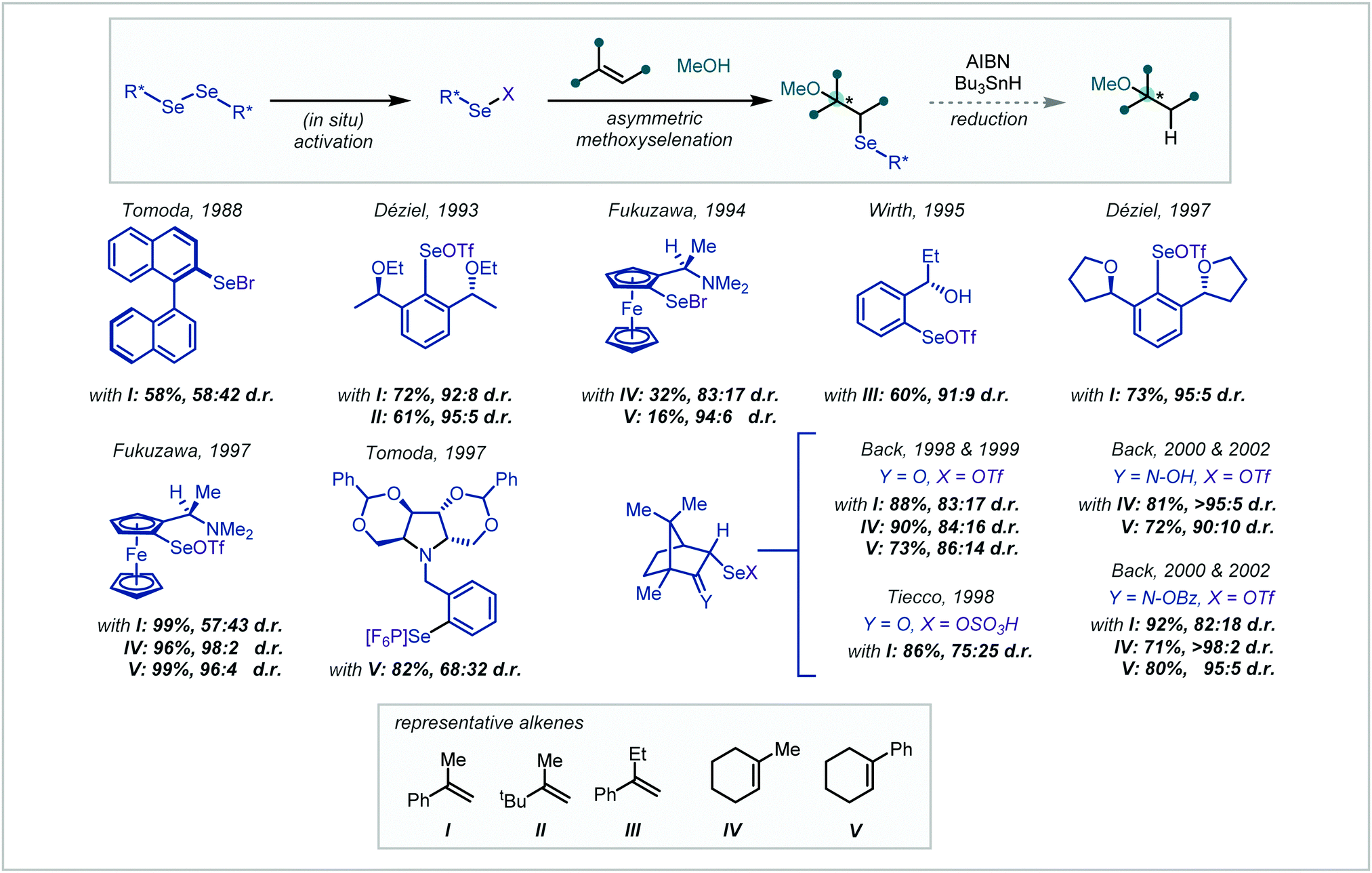 | ||
| Scheme 50 Asymmetric methoxyselenation for the synthesis of α-tertiary ethers (d.r.s rounded to integer values). | ||
Leading the field in 1988, Tomoda reported the use of a binaphthyl skeleton.216 It was proposed that this could retain the known regioselectivity in elimination of aryl selenoxides while introducing a backbone capable of asymmetric induction. Unfortunately, both the yield and the observed selectivity were low. Employing similar logic, Déziel synthesised a C2 symmetric diaryl diselenide, featuring flanking stereogenic ether units.217,218 The diselenides were activated in situ by treatment with Br2 and AgOTf and the corresponding aryl selenyl triflate afforded diastereomeric adducts with alkenes in the presence of MeOH. The yields in the methoxyselenations were typically high and showed good diastereoselectivity in the synthesis of α-tertiary ethers.
Shortly afterwards, Fukuzawa reported the use of chiral ferrocenyl diselenides from the corresponding ferrocenylamines.219,220 While the selectivities observed in α-tertiary ether examples were good, the yields were correspondingly lower for these more hindered substrates. Activation and reaction as the selenyl triflate resulted in a marked improvement in yield and selectivity.
Developing upon the systems reported by Déziel, in 1995, Wirth applied aryl selenides bearing ortho-stereogenic centres, featuring a range of ether and alcohol functionality.221 The methoxyselenation of α-ethylstyrene, to afford the corresponding α-tertiary ether, was reported in good yield and selectivity. Differing from Déziel's approach, Tomoda developed an alternative aryl diselenide bearing a chiral pyrrolidine sidechain.222 This system imparted good selectivity in the methoxyselenation of simple styrenes however the selectivity was significantly lower when employing phenylcyclohexene.
Subsequently, Back and Tiecco independently reported the use of camphor-derived diselenides. Tiecco's report employed the corresponding selenyl sulfate, produced in situ from treatment of the diselenide with ammonium persulfate.223 Despite high yields, Tiecco's report showed low selectivity for α-tertiary ether synthesis. On the other hand, Back explored a wide range of derivatized selenyl camphor reagents however the simple ketone-containing substrate performed best.224,225 Further exploration of the selenyl reagent highlighted the camphor-derived oxime as a superior methoxyselenation reagent, with improved selectivity.225,226
The continued improvement of this asymmetric methoxylation regime combined with the electrochemical oxyselenation–deselenation described represents a powerful potential tool for the synthesis of enantio-enriched allylic ethers.
2.6 Oxymercuration
Complementing the traditional halogen and chalcogen-mediated oxidative difunctionalisation of alkenes, the oxymercuration of alkenes is a fundamental and well-studied transformation.227 Typically associated with the nucleophilic opening of a transiently formed mercurinium ion by water or an alcohol, this transformation has been successfully applied to α-tertiary ether synthesis, as exemplified by Traylor's tert-butoxymercuration of cyclohexene.228 Probing this elusive 3-membered mercurinium ion, Waters & Kiefer applied a range of alcohols in the reaction between Hg(OAc)2 and 1,1-dimethylallene (Scheme 51).229 α-Tertiary ether formation showed a strong dependence on the steric hindrance associated with the incoming nucleophile, with t-BuOH affording the corresponding allylic α,α′-tertiary ether in only 10% yield. Developing this application in the functionalisation of allenes, Georgoulis demonstrated that iodine could be employed as a terminal oxidant, capable of affording vinyl iodides with adjacent tertiary ether moieties.230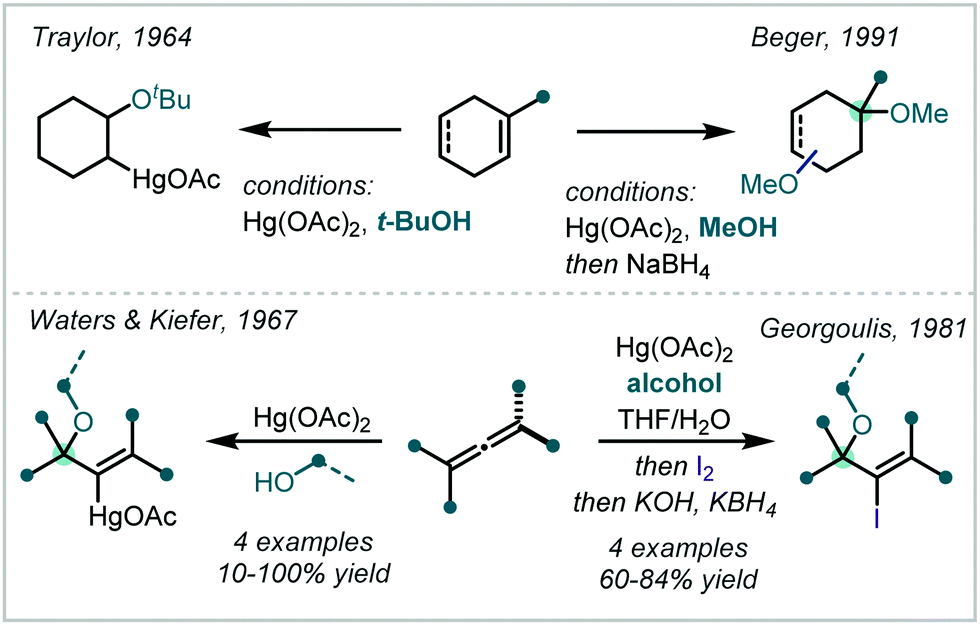 | ||
| Scheme 51 Application of oxymercuration to α-tertiary ether synthesis. | ||
Employing an appropriate cyclohexadiene and Hg(OAc)2, in 1991, Beger demonstrated the synthesis of a methyl α-tertiary ether, following a reductive cleavage of the C–Hg bond (Scheme 51).231 The products of such oxymercuration reactions corresponded to Markovnikov opening of the mercurinium ion.
2.7 Alkene difunctionalisations enabled by alternative activation modes
In 2019, Mandal reported a transition metal-free carboalkoxylation of styrene derivatives by exploiting the chemistry of phenalenyl organocatalysts such as PLY-N,O (Scheme 52).232 Without external stimuli such as light, it was shown that they could facilitate the reduction of aryl diazonium salts, yielding aryl radicals which added into styrenyl π-systems. DFT calculations suggested that alcohol addition into the resultant benzylic radical in fact preceded oxidation, representing a lower energy pathway than direct oxidation to the benzylic cation. Use of t-BuOH as the nucleophile led to the formation of the α-tertiary ether in 23% yield.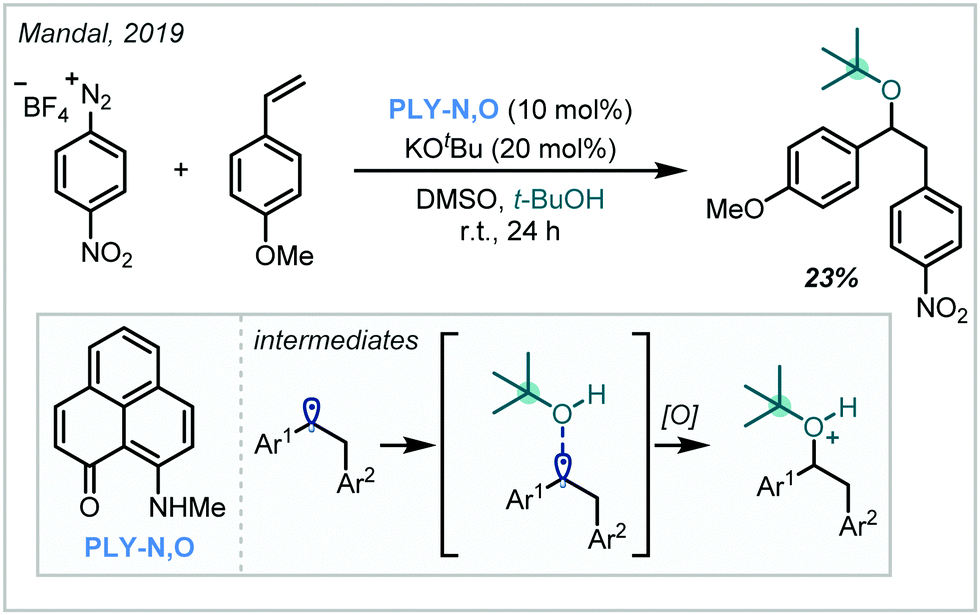 | ||
| Scheme 52 Transition metal-free synthesis of arylated α-tertiary ethers from aryl diazonium salts using a phenalenyl-derived catalyst. | ||
Reactive radical species can also be generated by the thermal decomposition of suitable precursors. In 2020, Klussmann disclosed that under acidic conditions, thermal decomposition of benzoyl peroxide (BPO), to the benzoyl radical could promote hydrogen-atom abstraction from the benzylic position of thioxanthene (Scheme 53).233 The formed radical was subsequently able to participate in a C–C bond forming event with styrene, and following RPC, an α-tertiary ether was formed upon trapping with t-BuOH. Recently, Ouyang & Li achieved the alkoxypolyhaloalkylation of styrenes using a similar catalytic manifold.234 Upon thermal decomposition, an aryl diazonium salt generated the corresponding aryl radical which enacted hydrogen atom abstraction from polyhaloalkanes such as chloroform. Following radical addition and oxidation, a variety of polyhalogenated α-tertiary ethers were formed. Depending on the substrate and alcohol used, adducts with tertiary centres at either side of the ether linkage could be formed. The method was also shown to be useful for the late-stage functionalisation of bioactive molecules, such as estrone which smoothly reacted in 72% yield.
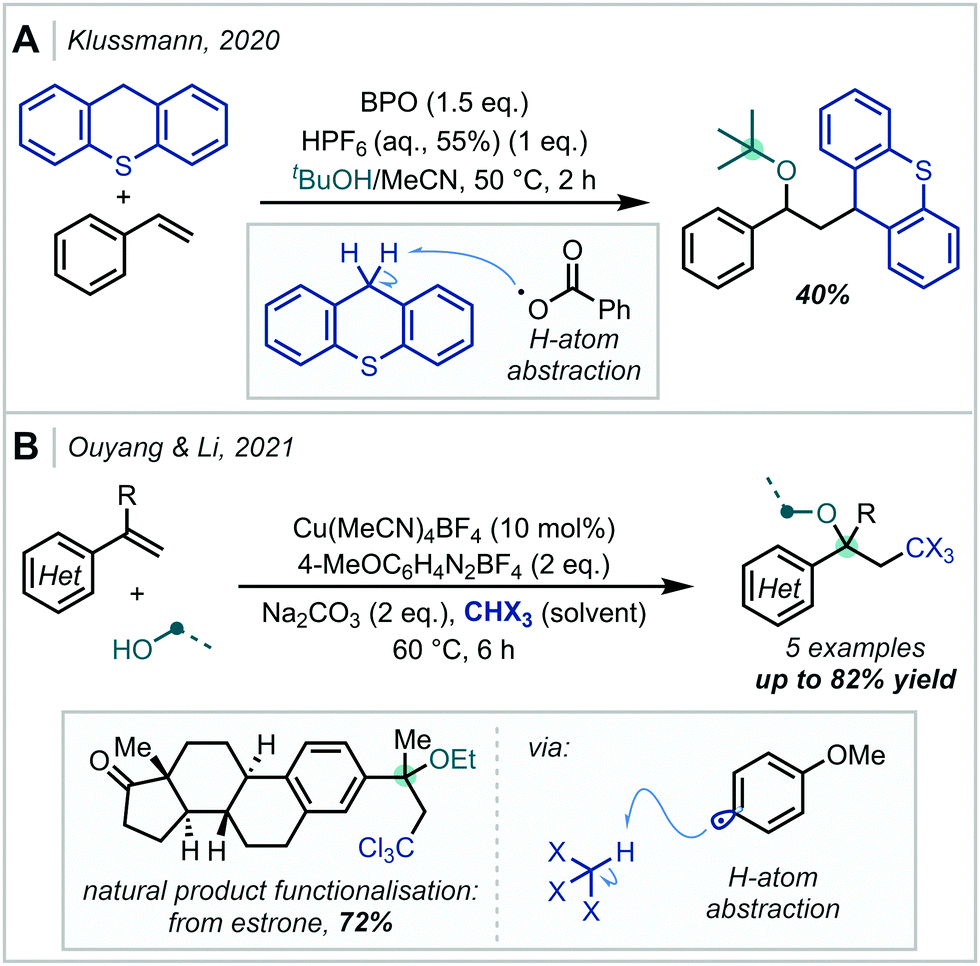 | ||
| Scheme 53 (A) Radical carboalkoxylation of styrenes. (B) Alkoxypolyhaloalkylation of styrenes initiated by the thermal decomposition of aryl diazonium salts. BPO = benzoyl peroxide. | ||
2.8 Alkene hydrofunctionalisations
Alkene hydrofunctionalisation reactions involve the addition of a hydrogen atom and another functional group across the π-bond of an olefin. A variety of reaction designs and manifolds have been developed to achieve this transformation in the context of α-tertiary ether synthesis, enabling retrosynthetic disconnections at various positions around the ether moiety; these will be the focus of the following discussion.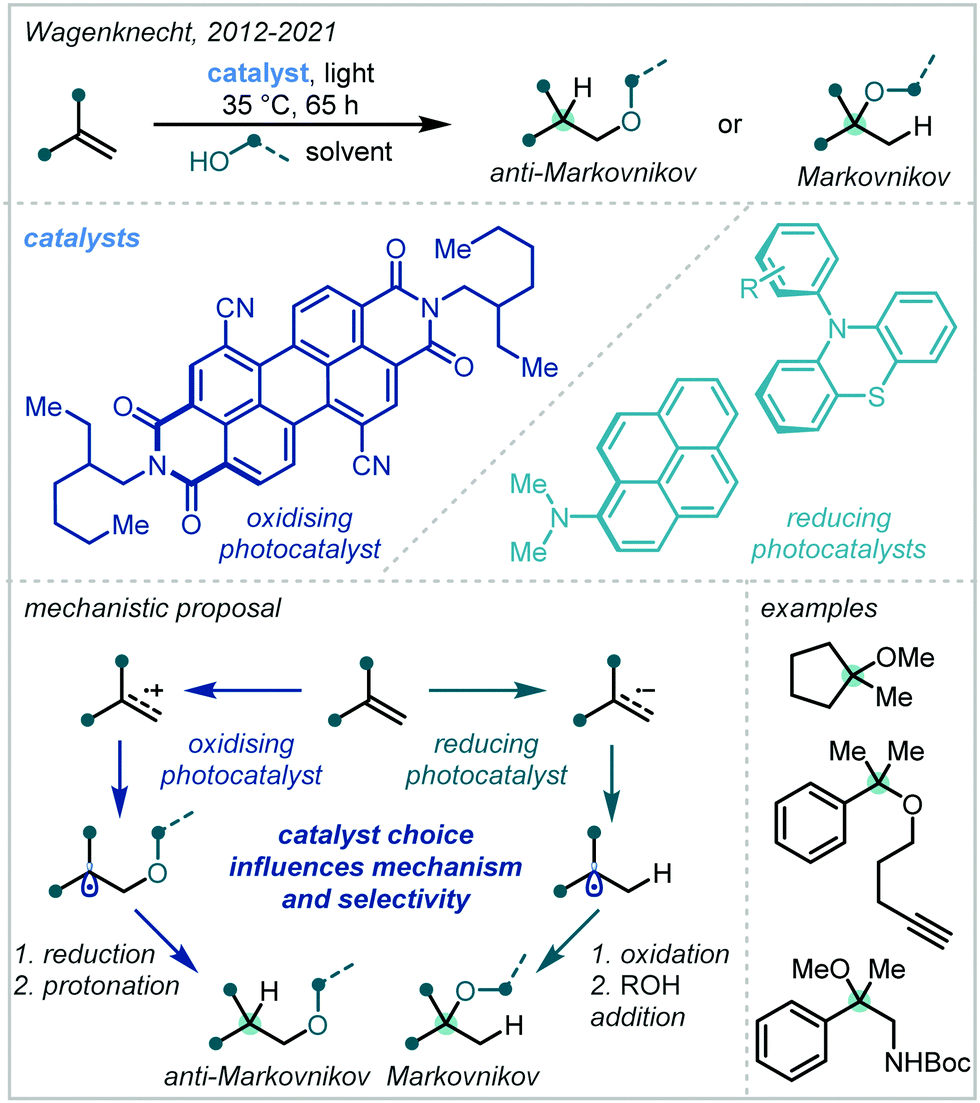 | ||
| Scheme 54 Regiodivergent, photocatalytic hydroalkoxylation of alkenes. | ||
In 2018, Yoshimi reported a protocol for the Giese addition of α-oxy radicals into electron-deficient alkenes, leading to a variety of α-tertiary ether products (Scheme 55).243 In this case, radical generation was achieved by photocatalytic oxidation of enol ethers by a combination of 1,4-dicyanobenzene and biphenyl to afford a radical cation. Depending on the concentration of alcohol used, one of two pathways dominated: either the cation was directly trapped by a nucleophile to yield bis-ether or hydroxy-ether products, or the cation reacted with starting material, and the adduct was subsequently trapped to produce acetal-containing ether products. In either case, the α-oxy radical subsequently reacted with an electron-deficient alkene, giving rise to the hydrofunctionalised products after termination. t-Butyl enol ethers were very effective in the reaction, providing a range of t-butyl ether products in good yield; this was attributed to the increased nucleophilicity of the electron-rich α-oxy radicals. In contrast, using t-BuOH as a nucleophile led to lower yields due to the increased steric hindrance associated with this alcohol.
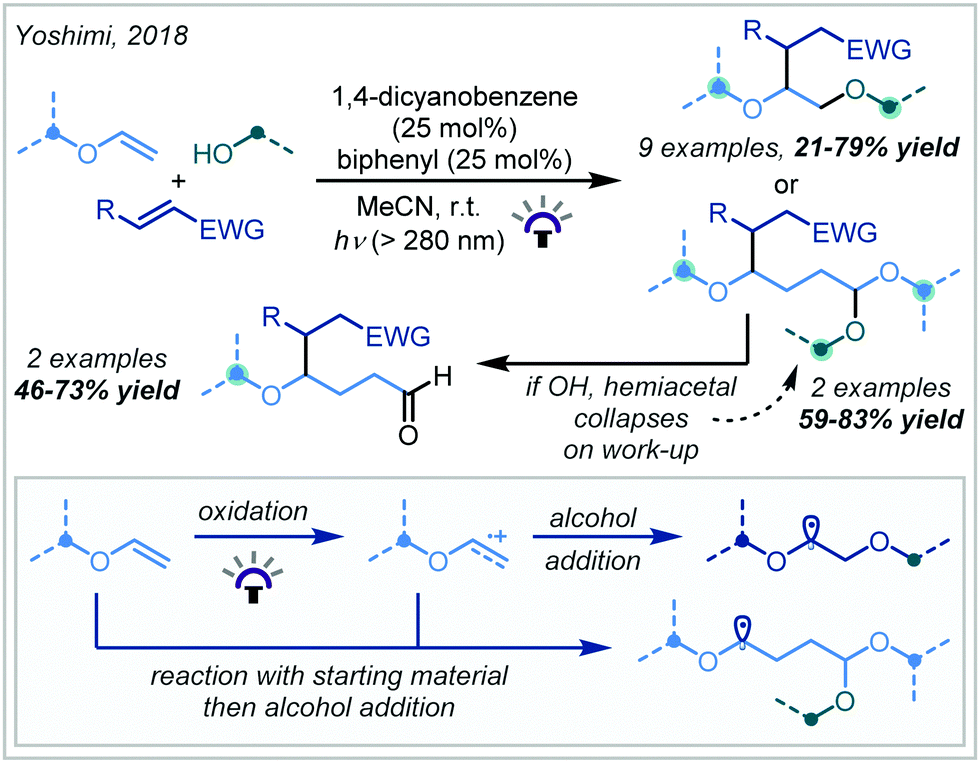 | ||
| Scheme 55 α-Tertiary ether synthesis via photochemical oxidation of enol ethers. | ||
In 2019, Dixon disclosed the photocatalytic reductive formation of diverse α-tertiary ethers from ketals (Scheme 56).244 Treatment of the ketal with Lewis acidic TMSOTf afforded the corresponding oxocarbenium ions. Umpolung reactivity was achieved through single electron reduction with an excited state iridium photocatalyst, affording nucleophilic α-oxy radicals that could engage in Giese addition chemistry with activated π-systems. Termination via HAT or a reduction/protonation sequence with a Hantzsch ester yielded the final products in good yields. Typically, yields were higher when ketals derived from electron-rich aryl ketones were employed, although electron-deficient and limited aliphatic substrates also reacted in acceptable yields. The alcohol component of the ketal could also be varied, allowing for variation at the other α-site of the ether. A key feature of this methodology was the stoichiometric quantities of ketal and radical acceptor used, allowing for potential application to more elaborate synthetic intermediates in multi-step syntheses.
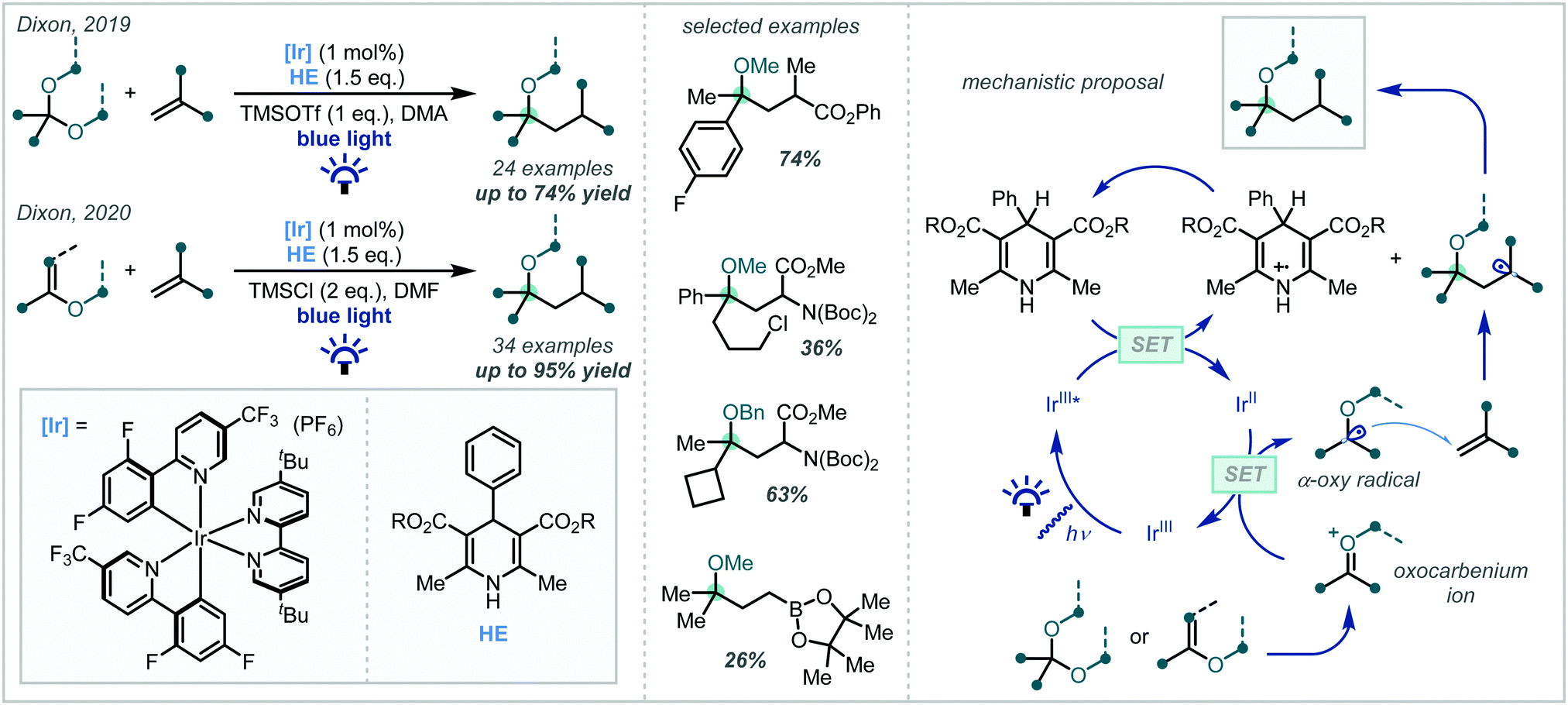 | ||
| Scheme 56 Visible light-mediated synthesis of α-tertiary ethers from ketals and enol ethers. | ||
The authors later developed this further to utilise enol ethers as precursors to the α-oxy radical.245 This significantly expanded the substrate scope of the reaction as all-alkyl substituted α-tertiary ethers, in addition to those with an aryl substituent, could now be more readily constructed: this was the most pertinent limitation of the authors’ initial report. In addition, this second report presented a much broader scope of alkene radical acceptors, including alkenes with more sensitive functional groups such as vinyl sulfones and boronate esters.
The oxidation of silyl enol ethers with quinones such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and p-chloranil has been reported to enable the synthesis of β-keto-α-tertiary ethers, with concomitant incorporation of the quinone-derived moiety.246–248 In 2021, Dagousset reported a photocatalytic protocol to access similar structures from silyl enol ethers, predicated upon the oxidation of α-oxy radical intermediates (Scheme 57).249 Alkoxy radicals were implicated as the key reactive species, formed from the reductive decomposition of alkoxypyridinium salts under photoredox catalysis. Radical addition into tetra-substituted enol ethers, followed by oxidation yielded the carbonyl-containing α-tertiary ether products in good yield.
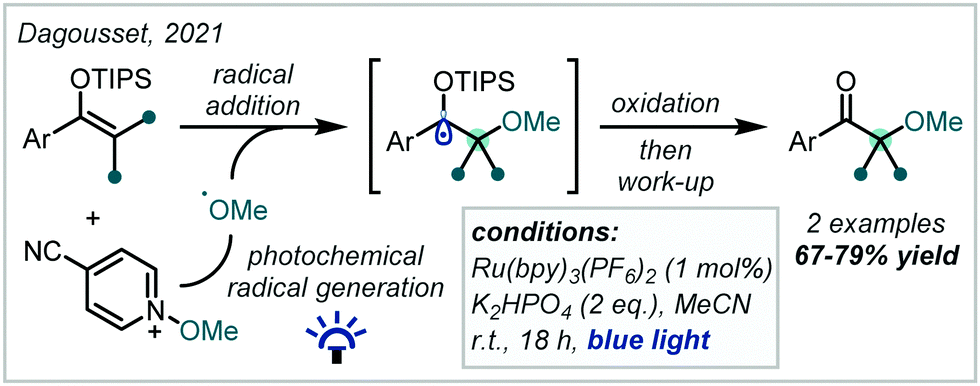 | ||
| Scheme 57 Synthesis of β-keto-α-tertiary ethers from silyl enol ethers. TIPS = triisopropylsilyl. | ||
In 2021, Shi disclosed a mechanistically-distinct method for the hydrofunctionalisation of allenes (Scheme 58).250 It was shown that energy transfer from an excited Ru-photocatalyst to the allene moiety allowed access to a reactive triplet state, which participated in selective [1,5]-HAT at the α-position of a preformed ether moiety. Subsequent radical recombination yielded the elaborated α-tertiary ether product.
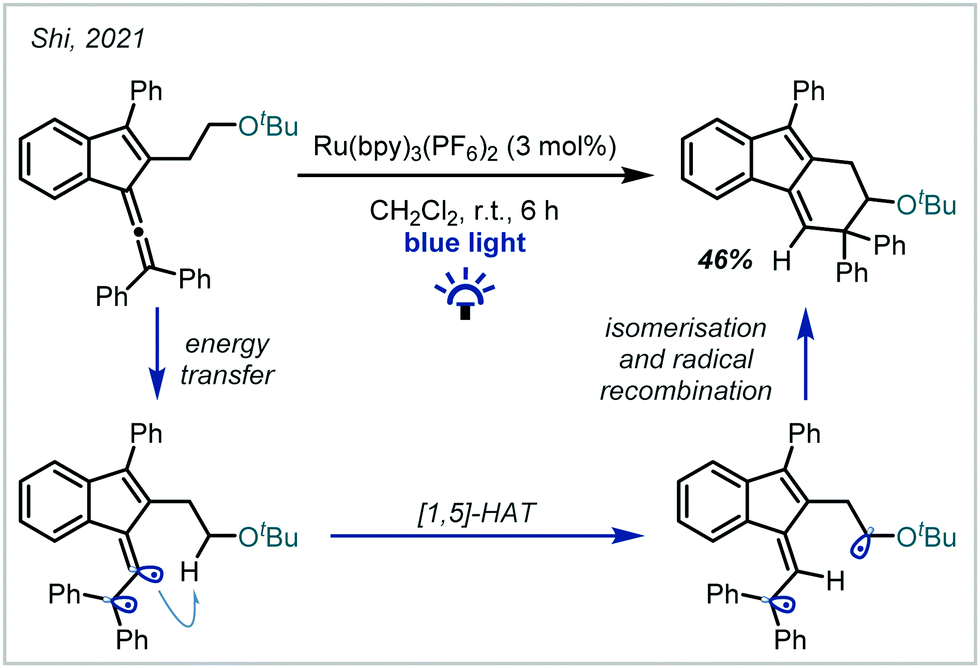 | ||
| Scheme 58 Photochemical hydrofunctionalisation of allenes enabled by energy transfer processes. | ||
 | ||
| Scheme 59 Pd-catalysed hydroalkoxylation of styrenes. | ||
In 2013, Shigehisa & Hiroya reported a Co-catalysed protocol for the hydroalkoxylation of unactivated olefins (Scheme 60).255 Mechanistically, oxidation of a Co(II) complex using a pyridinium fluoride salt followed by reaction with a hydridic silane species generated a Co-hydride in situ, which underwent hydrometalation across double bonds with Markovnikov selectivity. Homolysis of the Co–C bond, followed by oxidation of the resultant radical by Co(II) yielded a carbocation, which could be trapped by alcohol nucleophiles. Remarkably, t-BuOH was an effective nucleophile in this reaction, yielding a variety of α-tertiary ethers in excellent yields. Particularly impressive was the reaction of t-BuOH at tertiary carbocations to yield very hindered α,α′-tertiary ethers. While excess alcohol was used in the majority of scope examples, more elaborate, high-value alcohols could be used in stoichiometric quantities.
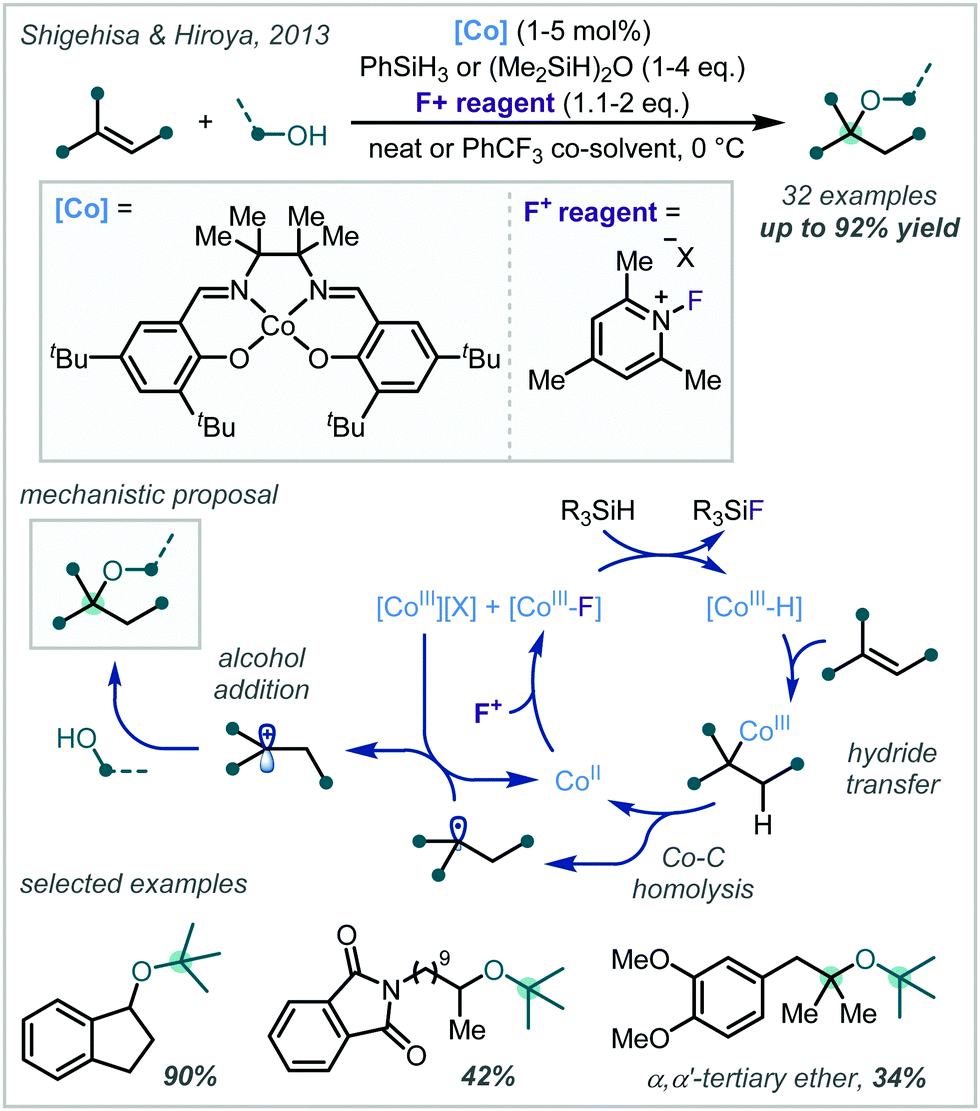 | ||
| Scheme 60 Co-catalysed hydroalkoxylation of unactivated olefins. | ||
3. Decarboxylation
Given the ubiquity of carboxylic acids within feedstock chemicals and biological molecules, the direct application of such fragments in α-tertiary ether synthesis would greatly expand the accessible chemical space. In this way, the decarboxylation of such acids offers a new mode of reactivity, building upon well-established chemistry.256–258 Fragmentation of a carboxyl radical, formed by oxidation of a carboxylate salt affords a C-centred radical capable of the same termination mechanisms previously described. An alternative mechanistic framework involves the reductive fragmentation of redox-active esters, typically derived from N-hydroxyphthalimides. Given the recent interest in such methods, it is unsurprising that both electrochemical and photochemical strategies have been employed to enable the decarboxylation of alkyl carboxylic acids to afford α-tertiary ethers. Since examples of decarboxylative alkene difunctionalisation have already been highlighted within the previous section, such methods will not be discussed further.3.1 Electrochemically-mediated decarboxylation
Taking inspiration from the oldest known electrochemical transformation, the Kolbe electrolysis, and the related Hofer–Moest reaction,86,259–261 Baran developed the synthesis of hindered dialkyl ethers, predominantly consisting of α-tertiary examples, using electrogenerated carbocations, arising from oxidative decarboxylation, (Scheme 61) and alcohol coupling partners.262 This method allowed the direct synthesis of such ethers from ubiquitous and inexpensive carboxylic acids and tolerated a wide range of functional groups in both the acid and alcohol coupling partners. The potential of the methodology to expedite target molecule synthesis was demonstrated by its application in a number of small molecule syntheses and comparison against previously reported routes. It was shown that tertiary sites could be incorporated from both the carboxylic acid and alcohol coupling partners; furthermore, the synthesis of several α,α′-bis-tertiary ethers was demonstrated in good yields and an α-tertiary ether was also prepared on ∼2 g scale in 72% yield. Extensive studies were undertaken to further understand the process and it was proposed that the mechanism likely involved rate-limiting anodic oxidation of the carboxylate, followed by decarboxylative fragmentation and an additional anodic oxidation, to generate a carbocation, which was subsequently intercepted by a nucleophilic alcohol coupling partner.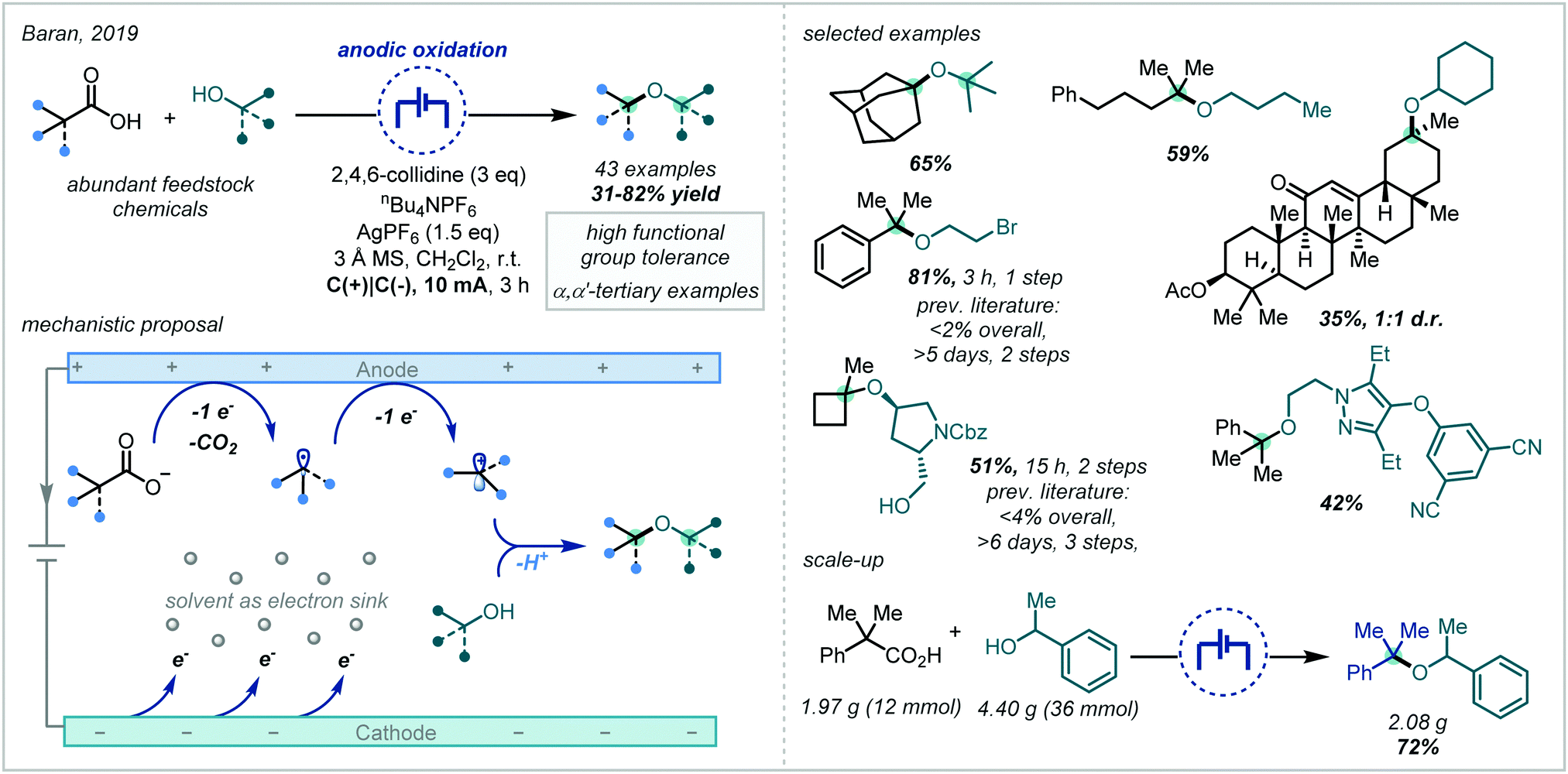 | ||
| Scheme 61 Electrochemical decarboxylative etherification. | ||
The interception of such electrogenerated carbocations by alcohol nucleophiles has been employed and developed by others, further expanding access to α-tertiary ethers (Scheme 62). Linclau and Brown incorporated a similar strategy into flow chemistry to enable the synthesis of alkoxy cubanes;263 the synthesis of an anisole bioisostere was possible by this method and the authors utilised this chemistry to make an analogue of the anticoagulant anisindione. Similarly, Zhang and Lei used electrochemical oxidative decarboxylation in their synthesis of 1,2-diaryl ethers.264 Although this process involves 1,2-aryl migration, it was shown that α-tertiary centres could be incorporated either by employing β-tertiary acids or by using a tertiary alcohol coupling partner.
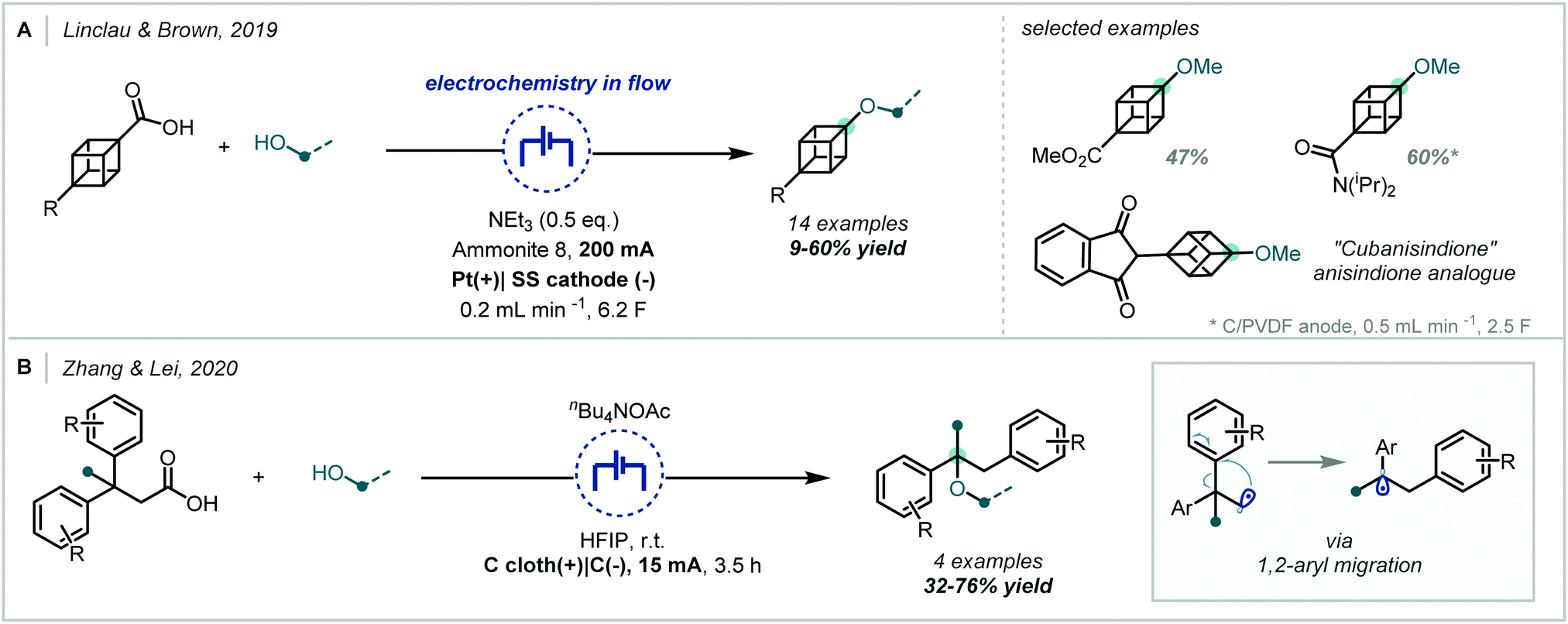 | ||
| Scheme 62 Further electrochemical decarboxylative etherification reactions. | ||
3.2 Photochemically-mediated decarboxylation
Given the recent resurgence of photoredox catalysis in organic synthesis, such methods have also been employed within the decarboxylative etherification manifold. In 2017, Gonzalez–Gomez reported the redox-neutral synthesis of an α-tertiary ether while investigating a decarboxylative Giese-type reaction promoted by visible light (Scheme 63), mediated by an acridinium photocatalyst.265 In this case, decarboxylation afforded the stabilised α-oxy radical which could successfully take part in a Giese reaction.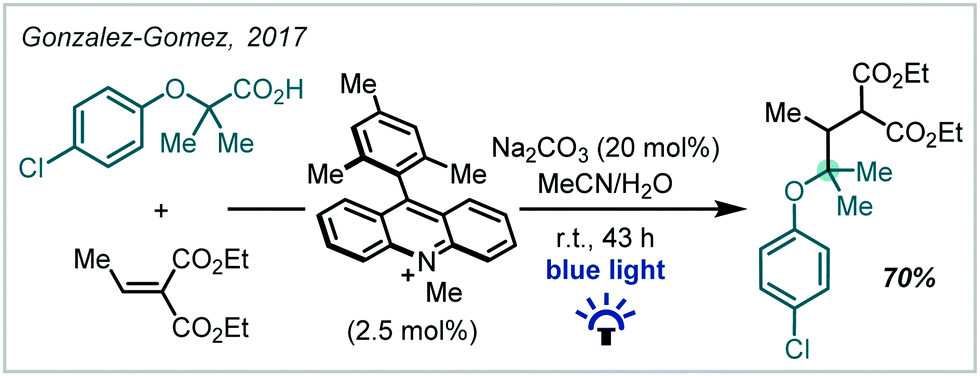 | ||
| Scheme 63 Organophotoredox decarboxylative Giese-type addition. | ||
In a mechanistically distinct approach to the oxidative decarboxylative couplings previously discussed, Hu developed a reductive catalytic system employing tandem photoredox and copper catalysis which allowed the synthesis of a broad range of ethers (Scheme 64).266 Reduction of the redox-active phthalimide ester by Ir(II) enabled decarboxylation to the corresponding radical, which was then trapped by the Cu(II)-aryloxide complex. Reductive elimination generated the alkyl-aryl ether and a Cu(I) complex, which was re-oxidised via SET from the photoexcited Ir(III), allowing catalytic turnover. While this method was limited to alkyl–aryl ethers, a variety of α-tertiary ethers were made using an α-cyclopropyl N-hydroxyphthalimide ester as a coupling partner.
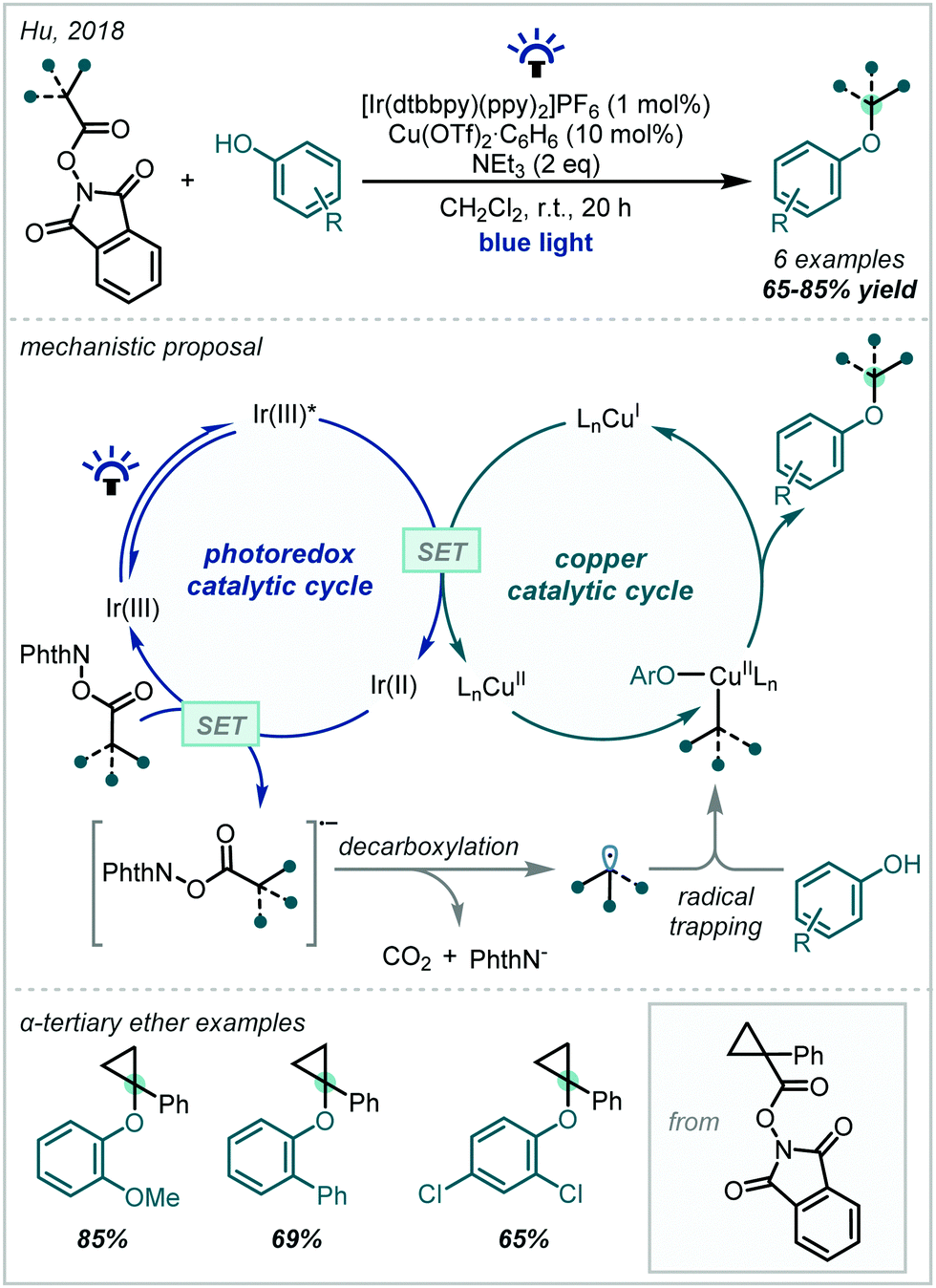 | ||
| Scheme 64 Decarboxylative etherification via dual photoredox and Cu catalysis. | ||
In a seminal report, Nagao and Ohmiya described the application of a phenothiazine photocatalyst in a decarboxylative etherification of redox-active esters, operating via a radical-polar crossover manifold (Scheme 65, see Scheme 5B for subsequent alkene difunctionalisation study).267 They demonstrated that tertiary sites could be incorporated from both the carboxylic acid-derived and alcohol coupling partners, allowing the synthesis of a variety of synthetically challenging α-tertiary ethers in fair to good yields over an extensive scope. They proposed that photo-excitation of the catalyst and subsequent SET to the phthalimide ester enabled a decarboxylation to give an intermediate (tertiary) radical II. The generated radical intermediate could then couple with the catalyst to give labile intermediate III, which underwent nucleophilic substitution to give the hindered ether product.
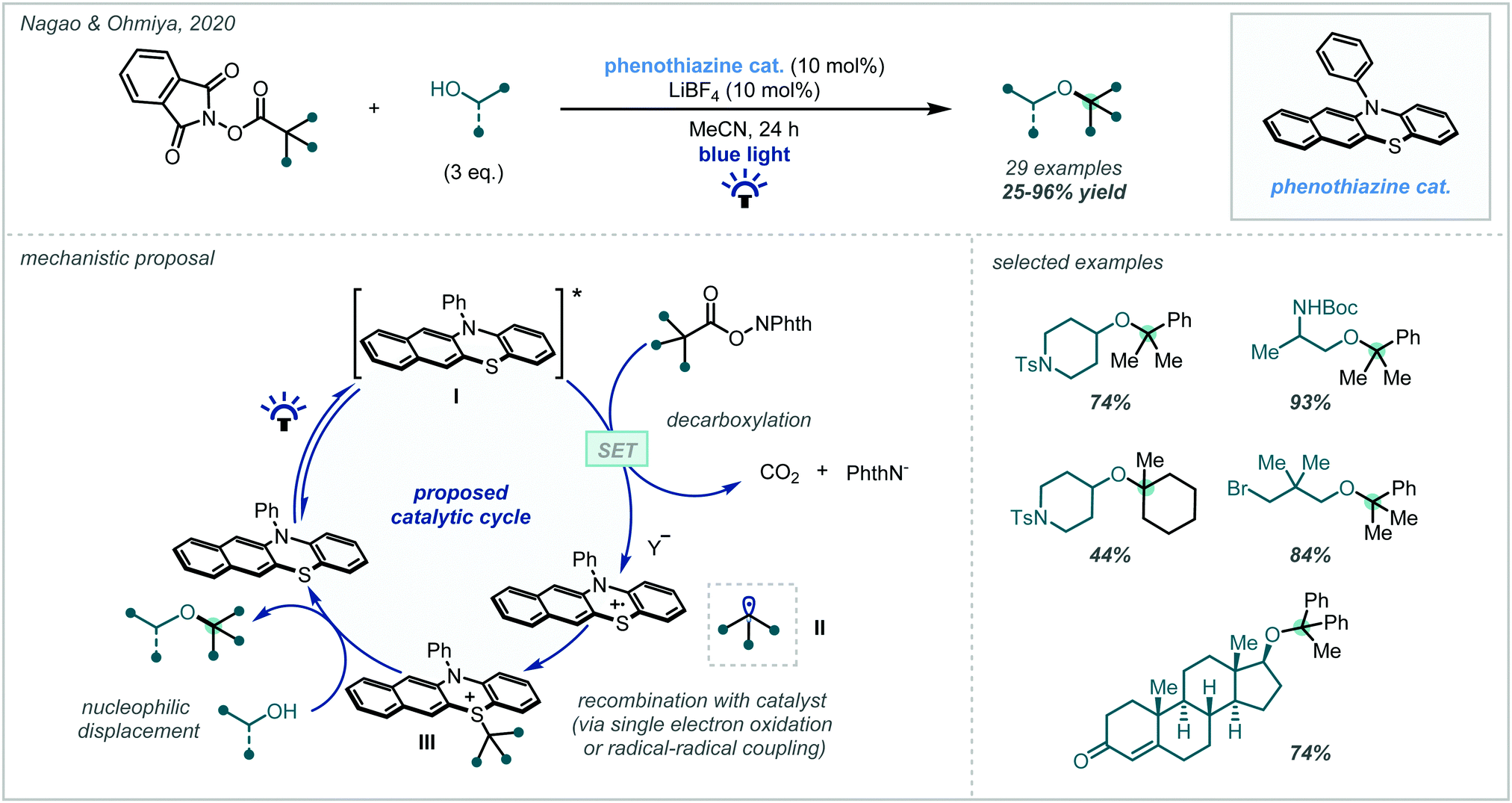 | ||
| Scheme 65 Decarboxylative etherification of N-hydroxyphthalimide esters using organophotoredox catalysis. | ||
In 2020, Zbieg & Terrett reported a method for decarboxylative C–O couplings employing photoredox catalysis (Scheme 66).268 They proposed that the iodine(III) oxidant (Ox) promoted in situ activation of the carboxylic acid to give I.268 Subsequent decarboxylation followed by SET could give a carbocation intermediate III which could then be trapped by an alcohol coupling partner. This new method displayed a large scope with many challenging α-tertiary ether motifs formed in moderate to good yields.
 | ||
| Scheme 66 Decarboxylative C–O couplings using photoredox catalysis. | ||
Recently, Yoon reported a Cu-mediated net-oxidative decarboxylative coupling of carboxylic acids directly with a range of nucleophiles under visible light irradiation.269 It was proposed that a Cu(II) carboxylate species, formed in situ, (Scheme 67) was photoexcited, facilitating ligand-metal charge transfer (LMCT), giving the corresponding carboxyl radical which could undergo rapid decarboxylation. Subsequent oxidation to the corresponding cation and interception by a wide range of nucleophiles gave the coupled products, including a wide range of ethers, including an α-tertiary example. This method demonstrated good use of diverse carboxylic acid feedstocks without the need for pre-functionalisation and exhibited a wide functional group tolerance, making it a valuable tool for late-stage functionalisation of molecules.
 | ||
| Scheme 67 Cu-catalysed net-oxidative decarboxylative coupling under visible light irradiation. | ||
4. C–H activation
C–H activation has arisen as a powerful and attractive strategy in synthetic chemistry due to the typically high atom economy, the lack of a requirement for pre-functionalisation and the applicability to late-stage functionalisation in drug discovery programs.270,271Although incredibly attractive from a synthetic point of view, C–H activation methods require delicate control of selectivity. Multiple ways to achieve this transformation have been explored, with many early methods harnessing the intrinsic difference in reactivity of C–H bonds within simple hydrocarbons.272 More recently, however, use of directing groups has been favoured due to their compatibility with more complex systems which contain multiple competing sites.273 This understanding and control of C–H reactivity has enabled the development of a broad suite of reactions, including C–C, C–O, C–N and C–S bond formations.
C–H bond alkoxylation to forge ether linkages is therefore an appealing strategy that would streamline synthetic approaches to high-value medicinal chemistry targets, natural products and other bioactive compounds (Scheme 68).274,275 Such strategies have been reviewed elsewhere for the generation of cyclic tertiary ethers.276
 | ||
| Scheme 68 Challenges in C–H activation for α-tertiary ether synthesis and conceptual approaches to overcome them. | ||
4.1 Transition metal-directed C–H activation
Transition metal catalysis has enabled a myriad of chemical transformations in the past decades,277 and has been used in the construction of C–O bonds from unactivated C–H bonds to afford α-tertiary ethers.278A leading example of Pd-mediated alkoxylation of unactivated C(sp3)–H bonds was reported in 2012 by Chen (Scheme 69A).279 The authors demonstrated that the Pd(OAc)2/PIDA couple is efficient in installing a variety of solvent alcohols at the γ-methyl site of amines bearing a picolinamide group.279 Two examples of t-butyl ethers were reported in moderate yield. In a related approach, Rao explored the use of Pd(OAc)2 and Dess-Martin periodinane (DMP) or 1-methoxy-1,2-benziodoxole-3(1H)-one (oxidant A) as oxidising agents of C–H bonds (Scheme 69B).280 They found that carboxamides bearing an 8-aminoquinoline directing group Q would undergo C(sp3)–H alkoxylation with a wide variety of alcohols, employed as solvents.280
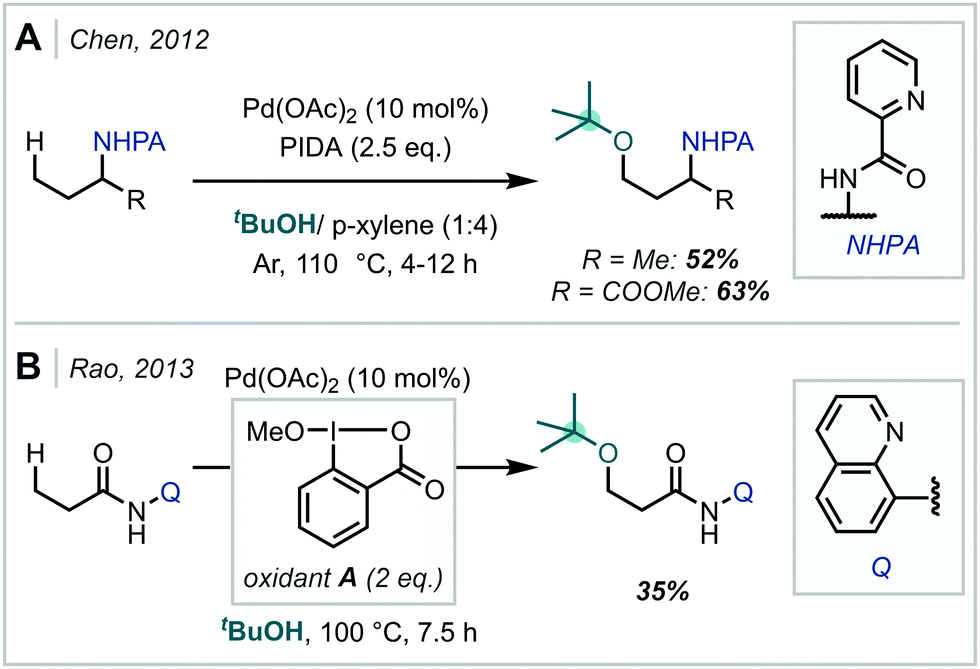 | ||
| Scheme 69 Pd-mediated remote C(sp3)–H activation utilising picolinamide (A) and quinoline (B) directing groups. | ||
In a complementary approach, peroxides have been shown to act as effective terminal oxidants under both Pd- and Cu-catalysis.281 In 2012, Warren & Cundari studied the catalytic activity of Cu(II) alkoxides, generated with peroxides.282 As part of their mechanistic studies, they demonstrated that t-butoxycyclohexane can be generated from cyclohexane in good yield, in the presence of Cu(II) and DTBP. This observation was expanded upon in 2016, when directing group-free C–H etherification method was developed by Warren (Scheme 70).283 Where both secondary and tertiary sites were available for functionalisation, etherification occurred predominantly at the tertiary site but with minor quantities of α-secondary ether products also observed. Recently, Ghosh reported a Cu(II)-mediated oxidation of C–H bonds, utilising DTBP, focussing specifically on the synthesis of hindered ethers (Scheme 70).284 This methodology made use of the ortho-directing picolinamide group and enabled the synthesis of a wide array of aryl–alkyl α-tertiary ethers.284
 | ||
| Scheme 70 Cu-catalysed C–H activations employing organic peroxides as the terminal oxidant (A) or as both the oxidant and the reagent (B). | ||
In 2016, Yu employed a thioamide α-directing group to achieve Pd-catalysed C(sp3)–H etherification with p-benzoquinone serving as both the alkoxylating reagent and a stoichiometric oxidant (Scheme 71).285 The process was exclusive to the synthesis of α-tertiary ethers, with less substituted substrates shown to be unreactive to the conditions.285
 | ||
| Scheme 71 Pd-catalysed synthesis of α-tertiary aryl–alkyl ethers from thioamides. | ||
Recently, Stahl has focused on the activation of benzylic C–H bonds towards etherification, aided by a Cu(I) catalyst and a N-fluorobenzenesulfonimide (NFSI) oxidant.286 The Cu(I)/NFSI couple generated a N-centred radical capable of HAT of a benzylic hydrogen to generate a C-centred radical (Scheme 72A).286 The procedure is highly selective for the benzylic C–H position, but notably utilises the alcohol coupling partners in excess. The synthesis of an adamantyl ether was reported in good yield as part of a wide scope, which also showcased precursors to derivatives of type 2 diabetes drug canaglifozin. The same group utilised the Cu(I)/NFSI methodology to fluorinate benzylic C–H bonds.287 The resulting benzylic fluorides were utilised in situ as electrophilic coupling partners to generate new C–O, C–N, or C–C bonds;287 the authors demonstrated that t-BuOH is a competent coupling partner in this reaction, generating an α-tertiary ether. Shortly after this work, Beng reported a one-pot hydroxylation/etherification process from dihydroisoquinolones and δ-lactams employing the same Cu(I)/NFSI C–H activation strategy (Scheme 72B).288
 | ||
| Scheme 72 (A) Cu(I)/NFSI radical relay benzylic etherification; (B) related functionalisation of dihydroisoquinolones and δ-lactams. | ||
4.2 Photoredox-mediated C–H activation
Photoredox-mediated C–H activation has received increased attention recently, and has been applied in the synthesis of α-tertiary ethers. Numerous methods involve formation of high energy radical intermediates capable of [1,n]-HAT, which then trigger formation of carbon-centred radicals capable of trapping electron-deficient moieties. These approaches feature diverse radical precursors and photocatalysts and have proved amenable to a range of coupling partners.In 2016, Meggers described an exciting merger of photoredox and enantioselective catalysis towards C–H alkoxylation, employing a chiral, Lewis acidic rhodium complex. The authors used N-alkoxyphthalimides as radical precursors capable of generating C-centred α-oxy radicals via 1,5-HAT, which then reacted with α,β-unsaturated N-acylpyrazoles to make hindered ethers (Scheme 73A).289 The protocol achieved remarkable enantioselectivities (up to 97% ee) and was applied towards a wide variety of α-tertiary ethers. Similarly, Chen used a similar strategy, but trapped the resulting C-centred radical with allyl sulfones.290 More recently, Wang and Xu used N-hydroxyphthalimide esters to achieve asymmetric C–H activation, with one report of α-tertiary ether formation.291 The phthalimide redox handle was not retained in the products of these strategies.
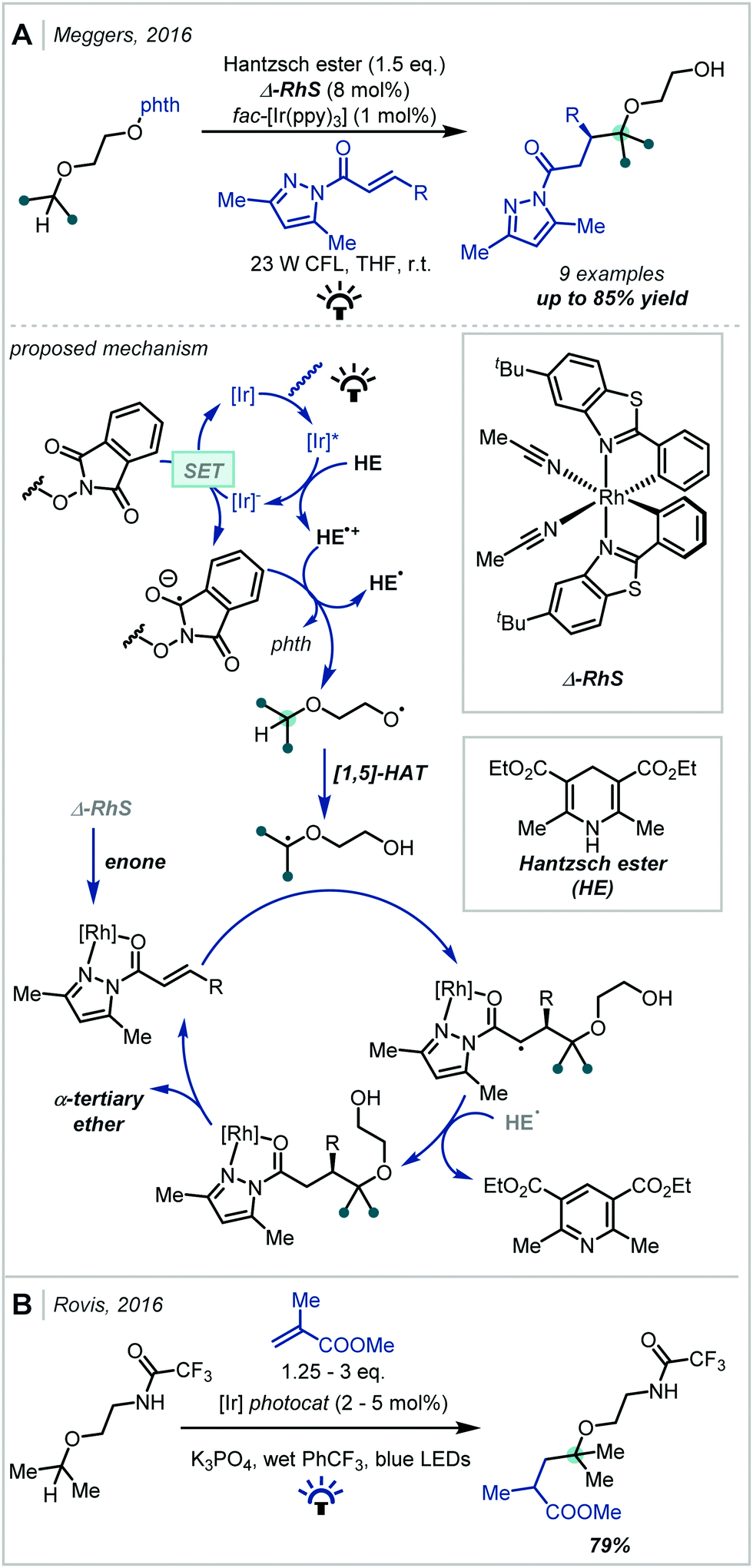 | ||
| Scheme 73 Photoredox-mediated C–H activation methodologies towards α-tertiary ethers based on C-radical generation and trapping of electron-deficient olefins. | ||
In 2016, Rovis employed olefins as the electron-deficient coupling partners (Scheme 73B).292 Through judicious choice of the amide handle, the N–H bond could be appropriately acidified to facilitate Ir(III)-mediated generation of an N-centred radical capable of reaction as above.292 Using this method, they synthesised one example of an α-tertiary ether, arising due to HAT occurring from a secondary ether site.
Alternative methods have explored the photoredox-mediated generation of C-centred radicals in a similar manner; once formed, the radicals would undergo SET to generate carbocations that would be trapped with alcohols, in a RPC approach. In 2015, Ragains focused on developing a photoredox-mediated C–H activation method using diazonium salts as redox-active moieties and reported the synthesis of two α-tertiary methyl ethers.293 This was expanded upon in 2017, when a diazonium tetrafluoroborate sulfone directing group was shown to be suitable for triggering [1,6]-HAT of tertiary C(sp3)–H bonds, followed by SET and alcohol trapping of the resultant carbocation (Scheme 74A).294 In 2018, Duan designed a temporary directing group strategy aimed towards C–H hydroxy- and alkoxylation. An iminyl radical was generated from the corresponding perfluorobenzoyl oxime upon irradiation with blue light.295 This radical triggered [1,5]-HAT to activate a remote C–H bond, and subsequent SET followed by carbocation trapping with an alkene and methanol afforded α-tertiary ethers via a MCR (Scheme 74B).
 | ||
| Scheme 74 Radical relay and SET sequences towards C–H etherification. | ||
A more recent example by Yoon focused on the benzylic C–H activation via tandem photoredox/Cu(II) catalysis.275 This is an attractive approach for activating benzylic C–H bonds as it does not require pre-functionalisation of the substrate to install a radical precursor (Scheme 75).
 | ||
| Scheme 75 Benzylic C–H activation to afford α-tertiary ethers. | ||
4.3 Metal-free methods
Given the often-higher costs, handling difficulties and sustainability issues associated with metal-catalysed methods, the development of metal-free alternatives is attractive and such approaches have found application within the synthesis of α-tertiary ethers via C–H activation.In 2010, Zhang employed p-iodotoluene difluoride and methanol as solvent to achieve C–H alkoxylation; two α-tertiary ethers were obtained (Scheme 76A).296 A report by Bao highlighted the use of DDQ as a stoichiometric oxidant to achieve propargylic C–H functionalisation with alcohols (Scheme 76B).297 Later, Kotagiri employed bis (trifluoroacetoxy)iodobenzene (PIFA) to activate the oxindole benzylic C–H bond towards t-BuOH addition (Scheme 76C).298
 | ||
| Scheme 76 Metal-free examples of C–H alkoxylation utilising hypervalent iodine (A and C) or quinone (B) terminal oxidants. | ||
Alternatively, heterocyclic C–H bonds have been oxidised into phosphonium salt moieties, which were subsequently used in SNAr etherification reactions by McNally299 and Vilotijevic300 respectively, who each reported one α-tertiary ether as part of the scope.
In 2018, List reported the application of phosphoric acids catalysts towards asymmetric α-aryloxylation of substituted ketones using p-benzoquinones (Scheme 77A).301 The procedure afforded a wide range of α-tertiary ethers under mild conditions and, notably, the use of single enantiomer chiral phosphoric acids could afford the α-tertiary ether products in excellent enantioselectivity.301 The mechanism proposed by the authors involved formation of the enol from the ketone, which was brought in close proximity to the quinone partner by the phosphoric acid catalyst. A PCET event led to a diradical complex, which underwent C–O bond formation to afford the product and regenerate the catalyst.
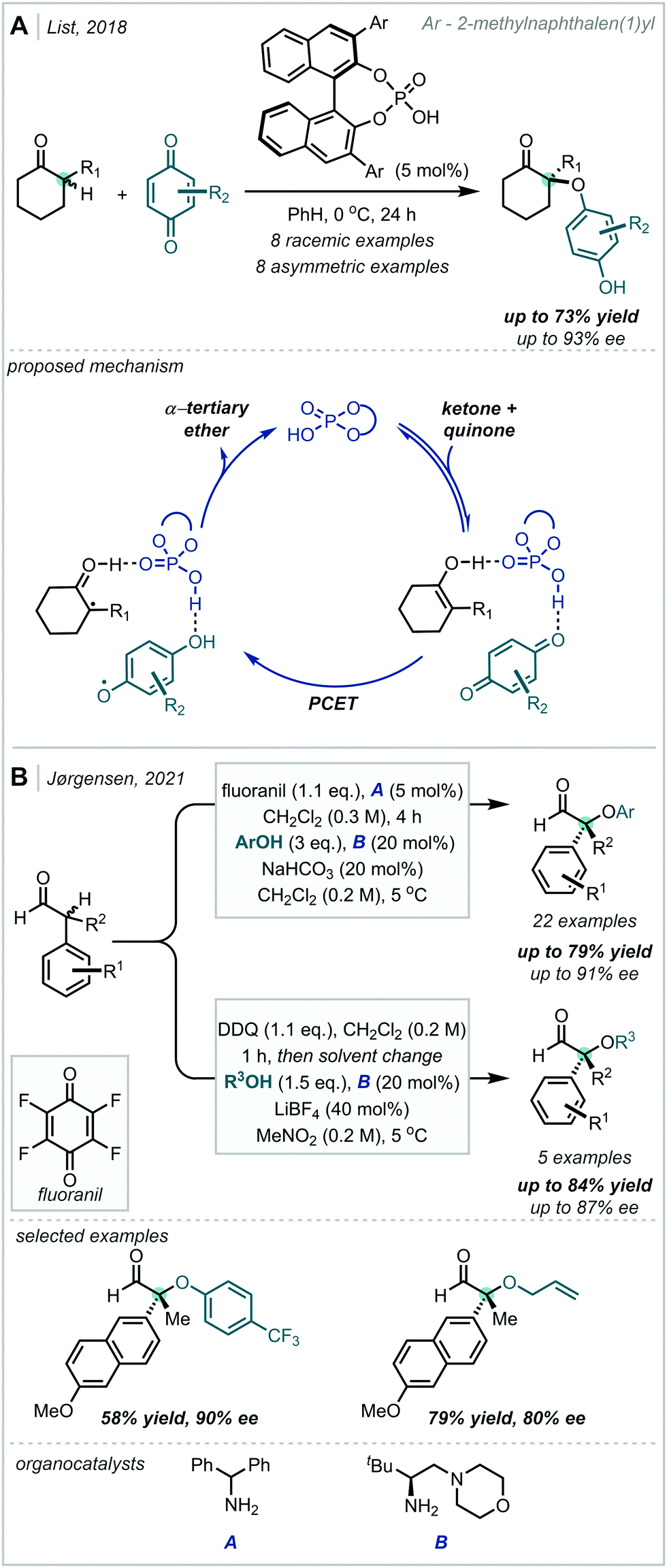 | ||
| Scheme 77 Organocatalytic approaches towards asymmetric α-tertiary ethers from racemic ketones (A) and aldehydes (B). | ||
Subsequently, Jørgensen demonstrated that aldehydes can also react with benzoquinones under primary amine catalysis via enamine intermediates to afford α-tertiary thioethers.302 Although the original method was not amenable to the synthesis of α-tertiary ethers, in 2021, the authors employed modified conditions to achieve α-aryloxylation and α-alkoxylation of α,α-disubstituted aldehydes (Scheme 77B).303 Similar to List's report, the α-alkoxylation could be rendered enantioselective by employing readily available primary amine organocatalysts (such as diamine B).
4.4 Electrochemical C–H functionalisation
Electrochemistry has also emerged as an alternative approach within the field of C–H oxidation to make hindered ethers. An early example was described by Kasch in 1979, where introduction of a methoxy group in the benzylic position of estradienes was achieved via anodic oxidation.304In 2020, Lei synthesised α-tertiary ethers derived from indane via electrochemical benzylic C(sp3)–H bond oxidation (Scheme 78). The procedure was carried out in an undivided cell and required an excess of the tertiary alcohol reaction partner. The proposed mechanism involved formation of a radical carbocation from indane via SET, which was then subjected to a hydrogen atom abstraction. A further anodic SET formed an indane carbocation, which was trapped by the alcohol nucleophile.305 In a similar approach, Li employed W2C nanocrystals as efficient electrodes for C–H activation and coupling of methanol with cumene, which proceeded in good yield to afford the corresponding α-tertiary ether.306
 | ||
| Scheme 78 Electrochemical C–H alkoxylation of the benzylic site of indanes. | ||
5. Cross-alcohol coupling
Alcohols are a naturally attractive starting material for the synthesis of ethers, and they are still employed extensively in the Williamson etherification. However, the aforementioned limitations of this approach have warranted the investigation of alternative methods. The eponymous Mitsunobu reaction, first disclosed in 1967,307 complements the classical Williamson method and expands the scope of starting materials. This reaction traditionally employs the redox couple of triphenylphosphine and diethylazodicarboxylate, which activates the alcohol towards substitution.While traditionally not amenable to the synthesis of α-tertiary ethers, there are a few successful applications of the Mitsunobu reaction to such hindered C–O bond formation. Leading reports of such strategies were investigated in efforts towards important structural motifs, such as flav-3-enes,308 spirochromenes,309 or dihydropyrano-coumarin systems310 (Scheme 79).
 | ||
| Scheme 79 Examples of the synthesis of α-tertiary ethers achieved via Mitsunobu reactions. | ||
Further work by Shi enabled the synthesis of aryl–alkyl ethers from tertiary alcohols under Mitsunobu conditions.311 The single enantiomer chiral ether products were employed as intermediates towards drug candidates. In 2010, Woggon generated α-tertiary ethers in a similar manner during efforts towards α-tocopherol.312 Bracher also reported the unexpected formation of aryl–alkyl α-tertiary ethers when employing 4-hydroxy-N-Boc piperidines as substrates under Mitsunobu conditions.313
In a strategically distinct approach, Mukaiyama focused heavily on phosphorus-mediated oxidation–reduction condensations which can be exploited to condense alcohols to the corresponding ethers (Scheme 80).314,315 While early reports demonstrated that use of triphenylphosphine as the reducing agent was less compatible with tertiary ether formation, the use of alkoxydiphenylphosphines was found to be much more successful.314,315 Utilising Ph2POtBu as both reductant and alkylating agent, and 2,6-dimethoxybenzoquinone (DMBQ) as oxidant achieved t-butylation of p-nitrophenol in good yield (conditions A, Scheme 80);314in situ synthesis of Ph2POR reagents bearing a tertiary alcohol of choice was shown to achieve aryl–alkyl α-tertiary ethers in good yields (conditions B, Scheme 80).315,316 Likewise, this methodology was further extended to the synthesis of alkyl-alkyl α-tertiary ethers when using fluoranil as the quinone oxidant.316 A key feature of Mukaiyama's work is that the reaction proceeds with exclusive inversion of stereochemical configuration as demonstrated using enantiopure substrates.
 | ||
| Scheme 80 Phosphorus-mediated oxidation–reduction condensations. | ||
Later, Mukaiyama noticed that reacting these alkyl diphenylphosphinite reagents with azides provided diphenylphosphinimidates, which were efficient alkylating agents in the presence of Lewis acids, such as TMSOTf (conditions C, Scheme 80).317 An extended study from 2006 showed that both pre-synthesised and in situ-generated diphenylphosphinimidates were able to provide α-tertiary ethers.318
A variation on this work was developed in 2022 by Schreiner, in which adamantyl phosphinites were activated towards nucleophiles using diisopropyl azodicarboxylate (DIAD)—rather than the quinones employed by Mukaiyama. This methodology was shown to be an effective route towards the synthesis of congested α-tertiary ethers and preliminary mechanistic investigations suggested the formation of carbocation intermediates. A betaine intermediate was proposed to form, from reaction between the phosphinite and DIAD, which was capable of deprotonation of a pro-nucleophile.319
6. Phenolic oxidation
The reaction of phenolic cations with alcohols has enabled the synthesis of quinolic α-tertiary acyclic ethers, following Dimroth's seminal report on stable, isolable phenolic cations in 1967.320 While originally seen as an academic curiosity, these compounds have now become valuable building blocks within modern synthesis and, additionally, can exhibit biological activity.6.1 Importance and applications
The cyclohexa-2,5-dienone products generated from phenolic oxidation of para-substituted phenols have been widely derivatised. In 1997, Russell exemplified this in the synthesis of anthraquinones from p-quinol ethers by reacting them with a cyanophthalide anion (Scheme 81A).321,322 Camps later reported a double Michael-decarboxylation sequence of dimethyl 1,3-acetonedicarboxylate to generate a three-dimensional bicyclo[3.3.1]nonane-3,7-dione scaffold from widely available phenols, affording otherwise synthetically challenging α-tertiary ether motifs (Scheme 81B).323 More recently, p-quinol ethers have been employed as substrates in asymmetric synthesis, elegantly demonstrated by Tian & Lin's Rh-catalysed desymmetrizing cyclisation, affording cis-hydrobenzofurans in excellent yield and enantioselectivity (Scheme 81C).324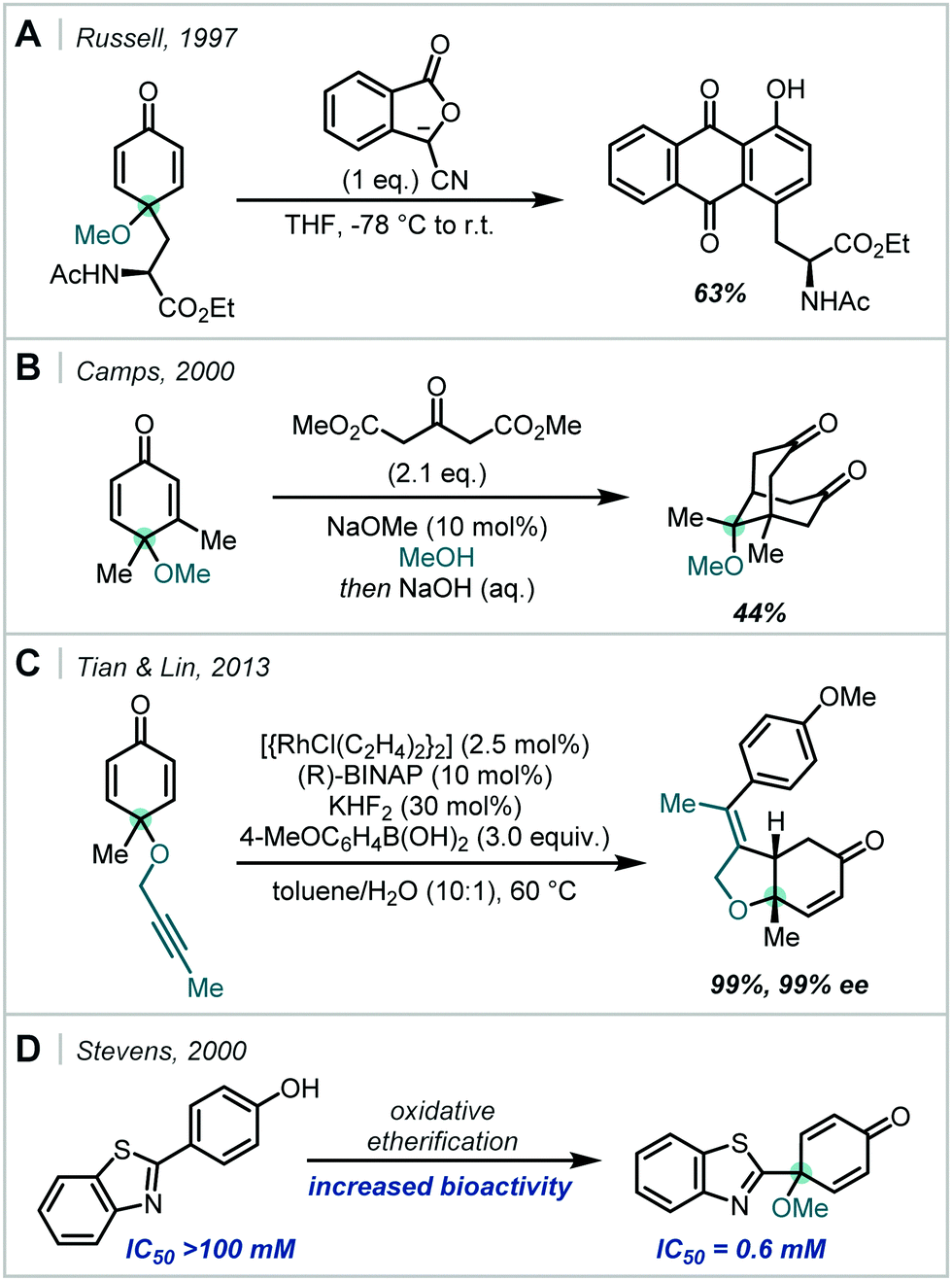 | ||
| Scheme 81 Applications of α-tertiary quinol ethers: (A) application to synthesis of anthraquinones; (B) synthesis of bicyclo[3.3.1]nonane-3,7-diones; (C) use in catalytic enantioselective synthesis; (D) biological properties. | ||
Furthermore, α-tertiary acyclic quinol ethers have been shown to exhibit anti-tumour activity.324 Stevens reported that benzothiazole quinol ethers show micromolar activity in human cancer cell lines HCT-116 and that quinol ethers show increased potency over the parent 2-(4-hydroxyphenyl)-benzothiazoles that they are derived from (Scheme 81D).325
6.2 Halogenation
One strategy for the synthesis of α-tertiary quinol ethers is the halogenation of para-alkylated phenols to generate an intermediate p-halodienone species Int1 which was able to be intercepted by an appropriate alcohol to deliver the desired tertiary ether (Scheme 82A). In 1971, Ronlán & Parker generated a bromo-quinol which was then subjected to a Ag-induced substitution with methanol to deliver the tertiary ether (Scheme 82B).326 Ronlán later reported that p-alkylphenols could be monochlorinated using excess antimony pentachloride to give the para-chlorinated quinol which was then treated with methanol to give the desired tertiary ether.327 In 1996, Omura utilised a different approach to introduce the α-tertiary motif, employing p-unsubstituted phenols, which could be intercepted by tertiary alcohols.328 By bromination of 2,6-di-tert-butyl phenol, a secondary bromide intermediate was formed, which reacted smoothly with a hindered nucleophilic tertiary alcohol to give an α-tertiary ether (Scheme 82C).328 However this tertiary ether was shown to be susceptible to irreversible re-aromatisation to the phenol on treatment with acid, base or silica gel.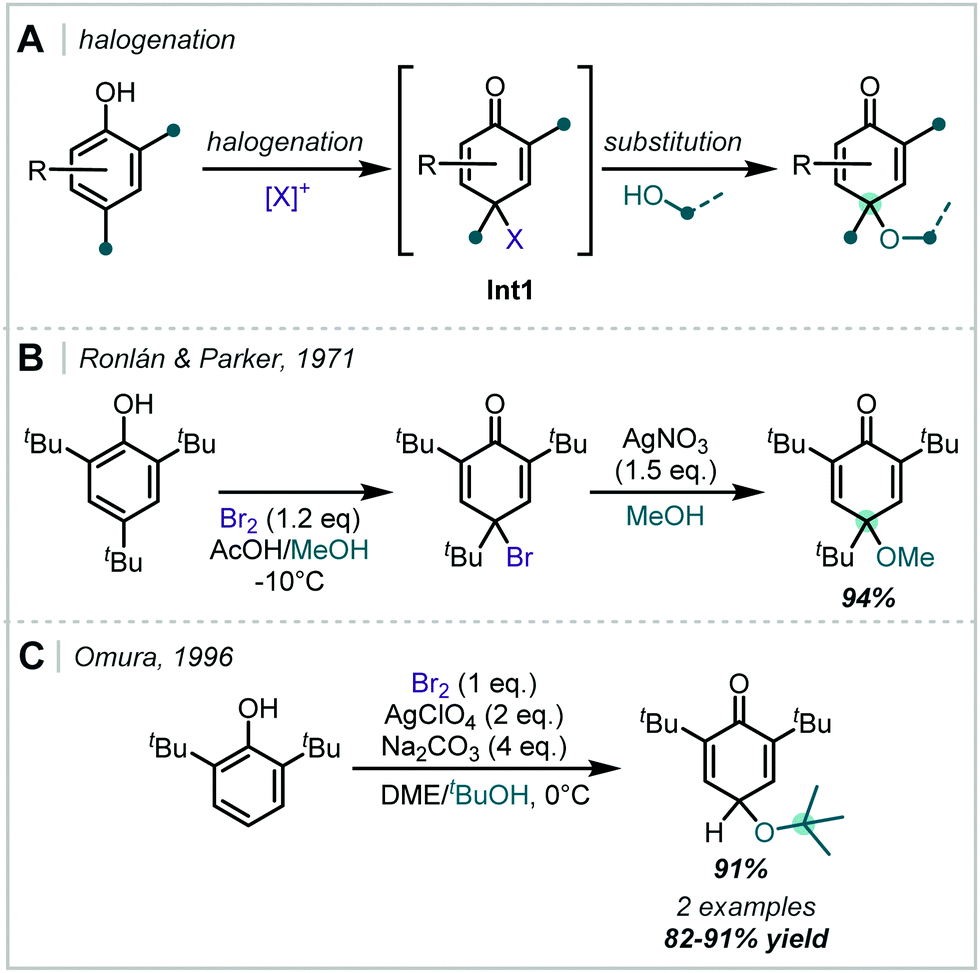 | ||
| Scheme 82 Synthesis of quinol ethers via halogenation: (A) general scheme; (B) Ag(I) mediated substitution of a tertiary alkyl bromide; (C) Ag(I) mediated substitution of an in situ-generated secondary alkyl bromide. | ||
In 2005, Jacobs developed a catalytic variant of this reaction using a WO42− – exchanged layered double hydroxide (WO42− – LDH) catalyst – an ionic solid comprised of layers of metal hydroxide with intercalated WO42− anions. WO42− – LDH catalyses the formation of hypobromite from ammonium bromide and hydrogen peroxide in situ (Scheme 83, also see Section 2.4.6). The resulting hypobromous acid was then able to brominate the phenol which was substituted with an alcohol to yield the desired α-tertiary ether and regenerate HBr to turn over the catalytic cycle.329
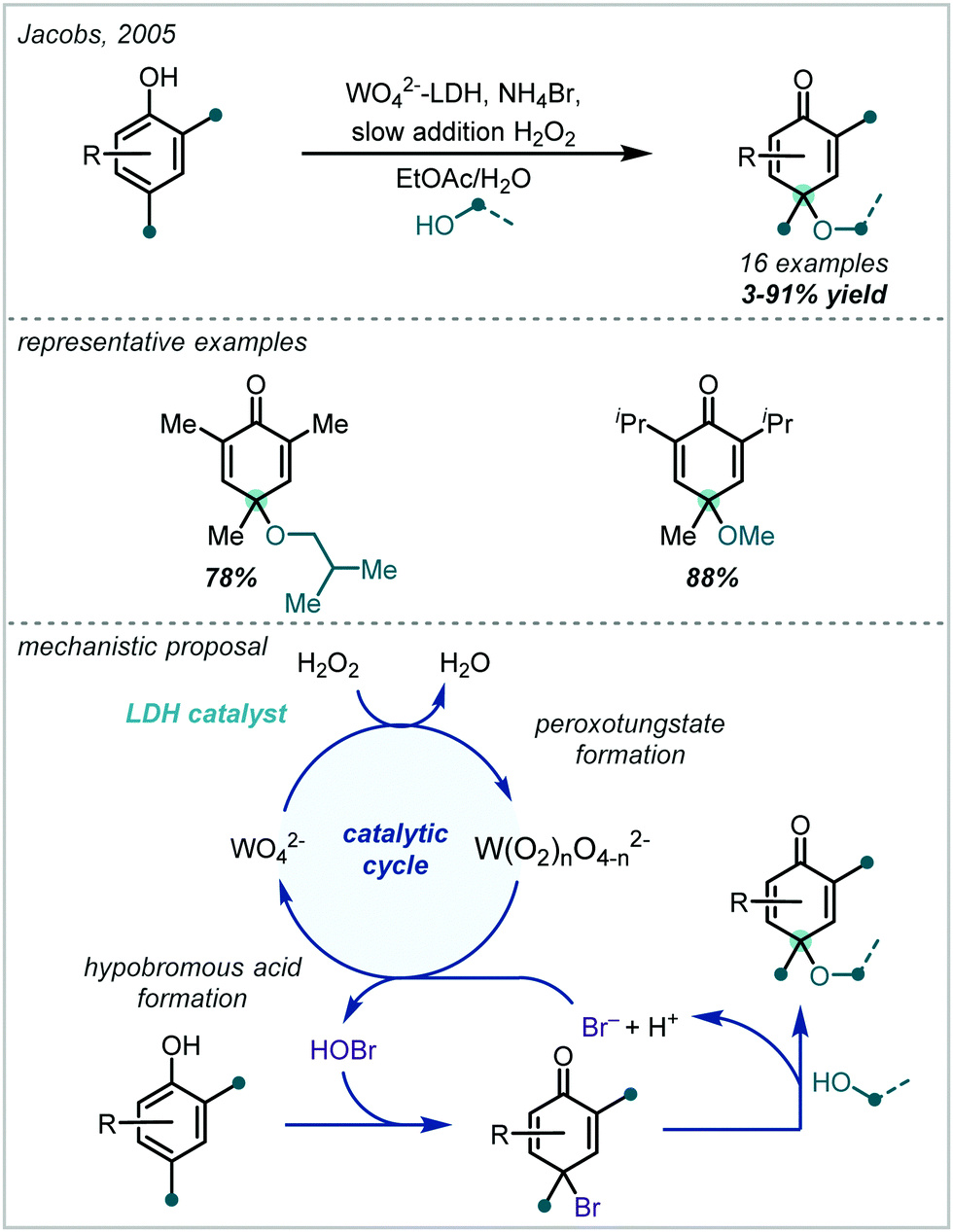 | ||
| Scheme 83 Synthesis of quinol ethers via catalytic generation of hypobromous acid. | ||
6.3 Anodic oxidation
Electrochemical methods have been shown to be particularly efficacious in the synthesis of quinol ethers (Scheme 84A), however they usually require a large excess of the nucleophile in order to generate the ether in appreciable yields. The mechanism for these reactions has been purported to go through stepwise electron-proton transfer sequences (Scheme 84B).330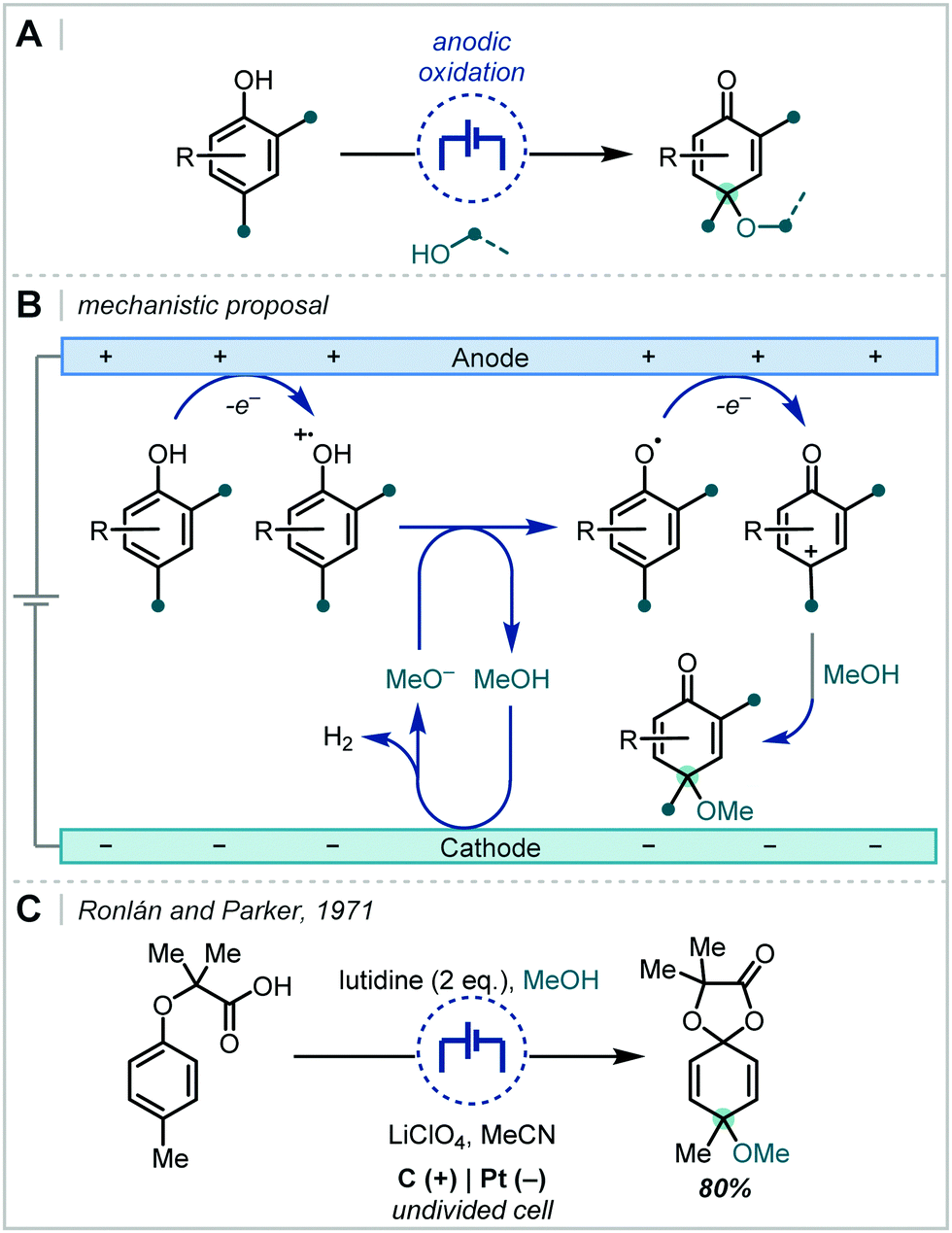 | ||
| Scheme 84 (A) General equation. (B) General mechanism for anodic oxidation of phenols to form quinol ethers. (C) Anodic oxidation of α-p-tolylisobutyric acid. | ||
An early report by Ronlán & Parker demonstrated that it was possible to trap out an electrochemically-generated phenoxonium cation with methanol in an undivided cell under constant current.326 α-p-Tolylisobutyric acids could also be anodically oxidised to give lactones bearing a tertiary ether in the para position (Scheme 84C). The authors later studied the mechanism and found that the reaction took place via direct oxidation of the phenol, not via the formation of methoxy radicals.331
In 1978, Rieker undertook a detailed investigation into the scope of the nucleophile (Scheme 85);332 a wide range of nucleophiles including primary and secondary alcohols were able to trap the phenoxonium cation. More oxidatively resistant phenols such hexachlorophenol were also employed allowing selective cross-phenol coupling. Using this methodology, Rieker was able to synthesise a synthetically challenging α,α′-tertiary ether, albeit in low yield.
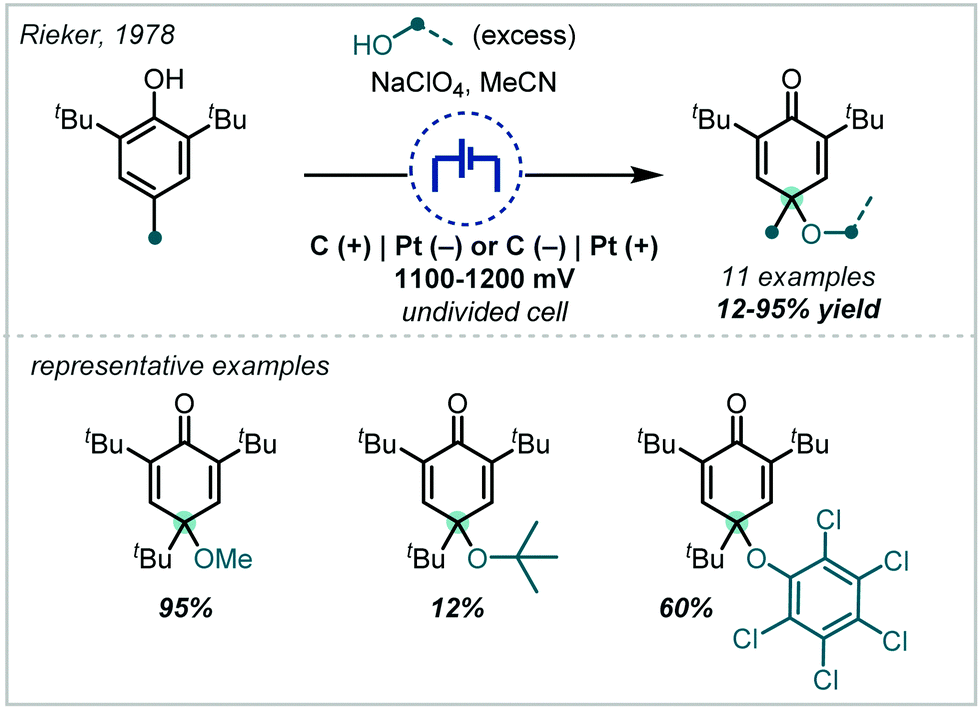 | ||
| Scheme 85 Anodic oxidation of phenols and trapping with O-nucleophiles to give quinol ethers. | ||
In 1991, Swenton applied the anodic oxidation of phenols to the synthesis of spirodienones (Scheme 86).333,334 A mechanistic investigation of this transformation demonstrated that the reaction proceeds via the phenoxonium cation,335 which is then intercepted by a pendant alkene in an intramolecular manner forging a C–C bond; the resultant carbocation was subsequently trapped by the solvent to form a tertiary ether. The scope of this transformation was later extended to include substrates with a methoxy group in the para and meta positions of the styrene.336
 | ||
| Scheme 86 Anodic oxidation of 2′-alkenyl substituted p-aryl phenols to give spirodienones bearing α-tertiary ethers. | ||
Likewise, Rieker reported a procedure for the reaction of the exocyclic primary alcohols of sugars using the anodic oxidation of phenols to form quinol-protected alcohols.330 The Pan and PChd groups could be introduced, in 78% and 50% yield respectively, in a divided cell using the pyranose in 10-fold excess (Scheme 87).330 The Pan and PChd groups were shown to be orthogonal protecting groups to traditional sugar protecting groups as they are able to be selectivity cleaved in the presence of isopropylidene and benzoyl groups.
 | ||
| Scheme 87 Pan protection of sugars via anodically generated cations. | ||
The anodic oxidation of less oxidisable biphenyls, phenolic biphenyls, anisoles and xylenes has also been reported by a number of different groups and, among other products, these reactions afforded tertiary ethers.337–343 However, these reactions typically require very strongly oxidising conditions and proceed with lower selectivity.
6.4 Hypervalent iodine
The dominant approach for generating phenoxonium cation equivalents for the synthesis of acyclic α-tertiary ethers involves oxidation of phenols by hypervalent iodine reagents (Scheme 88). Since a leading report by Becker,344 PIDA has become the most commonly used hypervalent iodine reagent, further complemented by the use of PIFA in specialised circumstances when a non-oxygen centred nucleophile is involved.345,346 In 1987, Lewis reported the synthesis of acyclic α-tertiary ethers using PIDA347 and showed a valuable proof of concept and applicability to a range of nucleophiles and phenol coupling partners. This was further expanded by Pelter's report in 1988.348,349 Swenton successfully translated their spirodieneone synthesis from an electrochemical oxidation to oxidation mediated by PIDA.350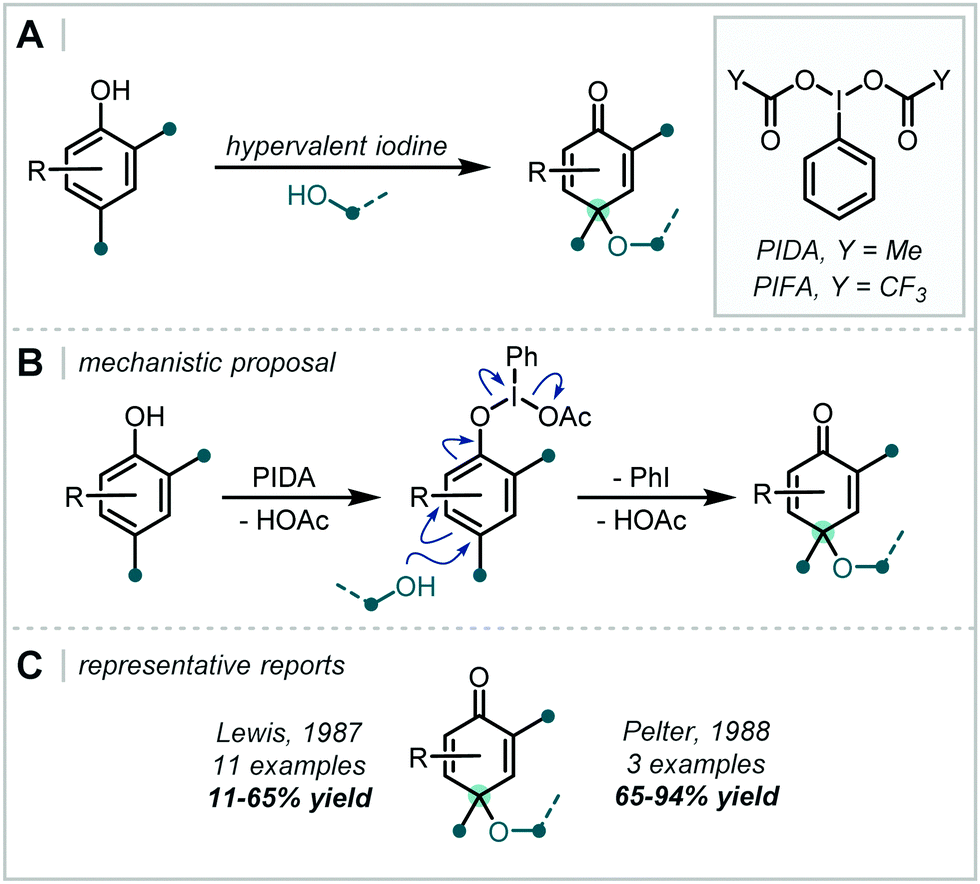 | ||
| Scheme 88 (A) General mechanism for hypervalent iodine oxidation. (B) Proposed mechanism. (C) Reports of Lewis and Pelter. | ||
Peddinti later reported a catalytic oxidative alkoxylation of phenol whereby the hypervalent iodine oxidant PhIO2 species was generated in situ from meta-peroxychlorobenzoic acid (mCPBA) and a substoichiometric amount of iodobenzene. In this case PhIO2 functions as the electrophile which was able to give the tertiary ether via intermolecular trapping with a nucleophilic solvent (Scheme 89).351
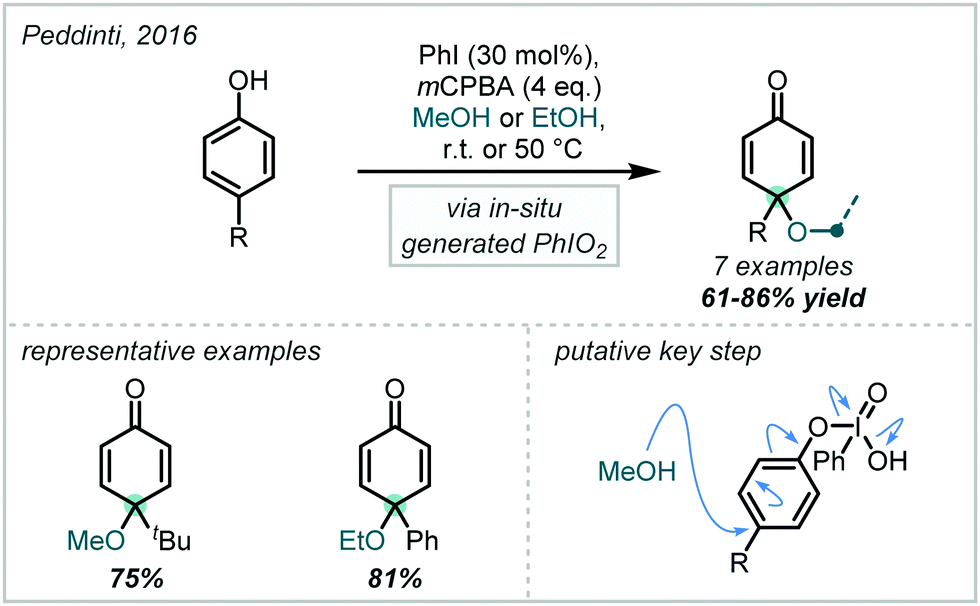 | ||
| Scheme 89 Phenolic oxidation mediated by in situ generated PhIO2. | ||
Van der Eycken & Varvounis translated this chemistry to the synthesis of α-cyano naphthones from tricyclic naphthaldehyde oximes using PIDA to trigger the dearomative oxidative ring closure and alkoxylation (Scheme 90). The nascent ring could then be cleaved under thermal or microwave conditions to give the nitrile and a corresponding phenol, constituting a C–H oxidation, nitrile formation and concurrent etherification over 2 steps.352
 | ||
| Scheme 90 Synthesis of tertiary ethers via oxidative cyclisation of naphthyl-oximes. | ||
In 1999, Quideau reported an alternative approach to synthesizing acyclic α-tertiary quinol ethers using a carbon-centred nucleophile, rather than an oxygen-centred nucleophile, to intercept the quinol intermediate with the ether oxygen already present. They reported the synthesis of naphthoid cyclohexa-2,4-dienones from naphthols via PIFA-mediated oxidative nucleophilic substitution.353 In this example PIFA was chosen instead of PIDA due to the reduced nucleophilicity of trifluoroacetate anion (Scheme 91A). In a subsequent report they extended this transformation to benzyl ethers354 and later applied it to the synthesis of the ABC tricyclic unit of aquayamycin analogues (Scheme 91B).355
 | ||
| Scheme 91 (A) PIFA mediated oxidation of 2-alkyl-and 2-alkoxynaphthols to give tertiary ether product by trapping with a C-centred nucleophile. (B) Applied to the synthesis of an aquayamycin analogue. | ||
6.5 Metal-mediated oxidation
Metal-based oxidants have also been shown to be capable of oxidising phenols to their corresponding cations, enabling the synthesis of α-tertiary ethers. The metal can be implicated either as a stoichiometric oxidant or a catalyst and both copper and iron salts have been used as catalysts for the oxidative methoxylation of phenols.356,357In 1970, Norman reported the two-electron oxidation of phenols using lead tetraacetate. This enabled the generation of the phenoxonium cation, which was subsequently trapped with methanol to afford the α-tertiary ether (Scheme 92B).358 In a related approach, McKillop later reported a procedure whereby 2,4,6-substituted phenols reacted with thallium nitrate in methanol to give p-substituted ethers in good to excellent yields (Scheme 92C).359 This approach has since been replaced by the more operationally straightforward conditions, described above, in order to avoid notoriously toxic reagents.
 | ||
| Scheme 92 (A) General scheme of metal-mediated phenolic oxidation. (B) Quinol synthesis using Pb(IV) tetraacetate. (C) Quinol synthesis using thallium(III) nitrate. | ||
7. Miscellaneous
7.1 Peroxide-mediated methods
In 1959, Lawesson reported a convenient, scalable and reproducible synthesis of t-butyl ethers via the reaction of t-butyl perbenzoate with Grignard reagents (Scheme 93).360,361 Recently, Dussault extended this synthetic strategy to peroxyacetals, employing both organolithiums and Grignard reagents in the reaction.362 This method gave a range of ethers in good yields via the transfer of primary, secondary or tertiary alkoxides through the highly regioselective substitution on the non-acetal oxygen of the peroxide. Alkoxide transfer in previous dialkylperoxide-mediated etherification methods gave lower yields with sterically hindered alkoxides. Dussault, however, proposed that the adjacent acetal moiety relatively stabilised the transition state responsible for transfer of the alkoxide via the anomeric effect,363 (see TS, Scheme 93) enabling efficient etherification.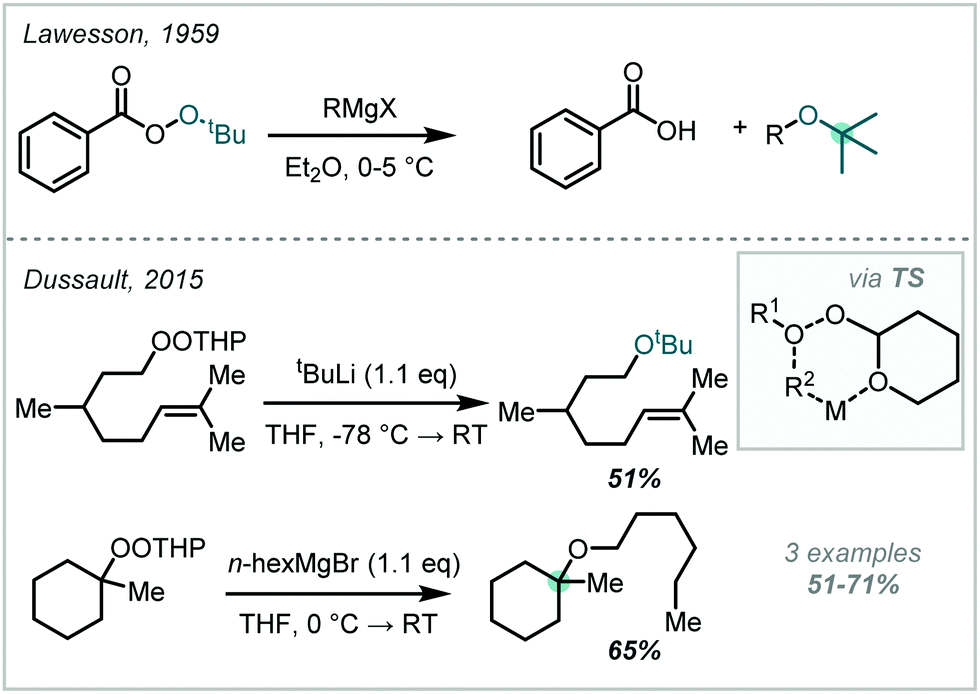 | ||
| Scheme 93 Peroxide-mediated routes to α-tertiary ethers. | ||
7.2 Miscellaneous radical-based methods
In 1973, Miller reported the reaction of Grignard and organolithium reagents with o-quinol acetates.364,365 The use of tertiary organometallic reagents enabled the synthesis of t-butyl ethers (Scheme 94). Miller proposed that this transformation was achieved by SET from the organometallic reagent to the quinone and that subsequent loss of acetate and combination of the resulting phenoxy radical with the alkyl radical resulted in ether formation. | ||
| Scheme 94 Synthesis of α-tertiary ethers from o-quinol acetates. | ||
The addition of alkyl radicals to quinones has been exploited for over 80 years as quinones are often used as free radical polymerisation inhibitors366 and, more recently, quinones have been used as radical scavengers in heterogeneous photocatalysis.367 A variety of benzoquinones have been used for this purpose and due to the prevalence of azobisisobutyronitrile (AIBN) as a radical initiator in such processes, a variety of α-tertiary aryl ethers have been synthesised as by-products.368–370 Renaud exploited this reactivity and developed the reaction of 1,4-benzoquinones with alkyl radicals generated from B-alkylcatecholboranes. This method was shown to be selective for O-addition when using sterically hindered alkyl radicals and enabled the synthesis of α-tertiary ether examples (Scheme 95).371
 | ||
| Scheme 95 Addition of tertiary radicals to quinones to give α-tertiary aryl ethers. | ||
As exemplified by these processes, the intermolecular addition of radicals to unsaturated systems has been utilised to synthesise α-tertiary ethers. In a conceptually distinct approach, Pattenden reported the intramolecular cyclisation of pendant primary alkyl radicals, generated from the primary alkyl bromides, with enol ethers, linked via an acetal tether, thus rendering the point of cyclisation an α-tertiary site (Scheme 96).372
 | ||
| Scheme 96 Cyclisation with pendant enol ethers to afford α-tertiary ethers. | ||
Alongside the alkene difunctionalisation, decarboxylative and C–H activation approaches previously discussed (see Sections 2.1, 3.2 and 4.2), numerous alternative applications of photochemistry have been developed for the synthesis of α-tertiary ethers. In 1995, Albini reported the formation of aliphatic radicals from ethers via photoinduced electron transfer (Scheme 97).373 The resulting radicals were found to add to the 1,2,4,5-tetracyanobenzene radical anion, thus forming a new alkyl-aryl ether. Notably, this constitutes an example of α-tertiary ether elaboration at the α′ site.
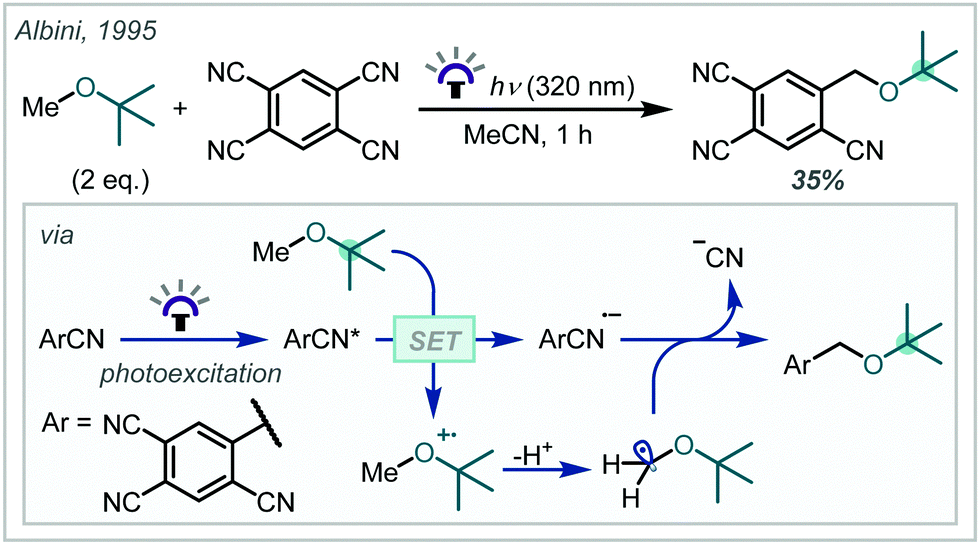 | ||
| Scheme 97 Photoinduced ether exchange. | ||
More recently, using substituted aziridines as substrates, Xia developed the regioselective ring opening of aziridines via nucleophilic addition (Scheme 98).374 Notably, the nucleophilic ring opening was promoted by photo-oxidation of the aziridine nitrogen. It was proposed that this transformation occurred via SET from the aziridine to the photo-excited Ru(III), forming a reactive amino radical cation which could undergo concerted ring opening and nucleophilic substitution to give the product ether. This constitutes an example of photoredox catalysis promoting a traditionally polar reaction pathway.
 | ||
| Scheme 98 Regioselective aziridine opening via photoredox catalysis. | ||
In 2016, Knowles demonstrated that α-tertiary ethers could be formed using photoredox catalysis-enabled mesolytic cleavage.375 In this process, electron transfer to an Ir photocatalyst from a TEMPO-derived alkoxyamine resulted in mesolytic cleavage to afford the nitroxyl radical and corresponding carbocation, which could be intercepted with a tertiary alcohol to give an α-tertiary ether in good yield and under mild conditions (Scheme 99).
 | ||
| Scheme 99 Ether formation by photoinduced mesolytic bond cleavage. | ||
Recently, Zhao reported the photoinduced etherification of cycloketoxime esters.376 It was proposed that the photoexcited Ir(III) photocatalyst was quenched via SET by the cycloketoxime substrate to give an imine radical, which subsequently rearranged via C–C bond cleavage to give a distal radical (Scheme 100). This radical could be further oxidised by the photocatalyst to give the corresponding cation, which was intercepted by an alcohol to yield the ether product. By employing these specialised substrates of varying ring sizes, the synthesis of a wide range of ethers using these mild conditions was demonstrated, including some α-tertiary examples.
 | ||
| Scheme 100 Synthesis of cyano-α-tertiary ethers via photocatalytic C–C bond activation. | ||
7.3 Carbene/carbenoid strategies
In 1989, Davies reported the formation of an undesired α-tertiary ether via 2,3-sigmatropic rearrangement while investigating the cyclopropanation of vinylcarbenoids (Scheme 101).377 Such rearrangements have been employed with a wide range of nucleophiles to access useful functionalities via ylide formation.378 In particular, the reactivity of oxygen nucleophiles with metal (usually Rh) carbenoids has since been exploited in a large variety of multicomponent reactions to yield a vast array of chemical complexity, including α-tertiary ethers, in a single reaction and such methods have been reviewed elsewhere.379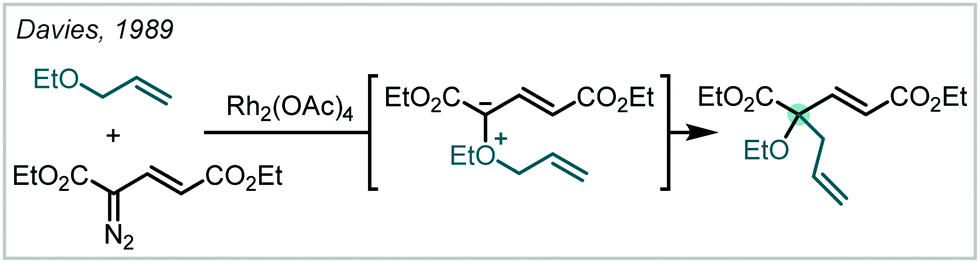 | ||
| Scheme 101 Ether synthesis via metal-carbenoid formation and subsequent 2,3-sigmatropic rearrangement. | ||
Recently, substituted benziodoxoles have been employed to enable the oxyfunctionalisation of diazo compounds (Scheme 102). In 2016, Szabó employed Togni's reagent with Rh2(OAc)4 to allow oxytrifluoromethylation of α-diazocarbonyl compounds; the α-tertiary ether centre is generated by the addition of a nucleophilic alcohol component.380 In 2016, Waser reported the oxyalkynylation of diazo compounds using a copper catalyst and EBX reagents.381 While initially only used for the synthesis of esters derived from the EBX reagent, this methodology was subsequently extended to enable the synthesis of a broad range of ethers (including α-tertiary ethers) by variation of the nucleophilic alcohol component in the reaction.382 Waser has also employed analogous vinylbenziodoxolone (VBX) reagents to enable oxyvinylation of diazo compounds and demonstrated the applicability to the synthesis of α-tertiary ethers.383 The proposed mechanisms for the oxyfunctionalisation of diazo compounds are comparable:384 the diazo compound is thought to form metal carbenoid I which is intercepted by the alcohol component to form an onium ylide II. The intermediate ylide II reacts with the electrophilic component of the appropriate benziodoxolone. Proton transfer from the resulting protonated ether III releases the product ether IV.
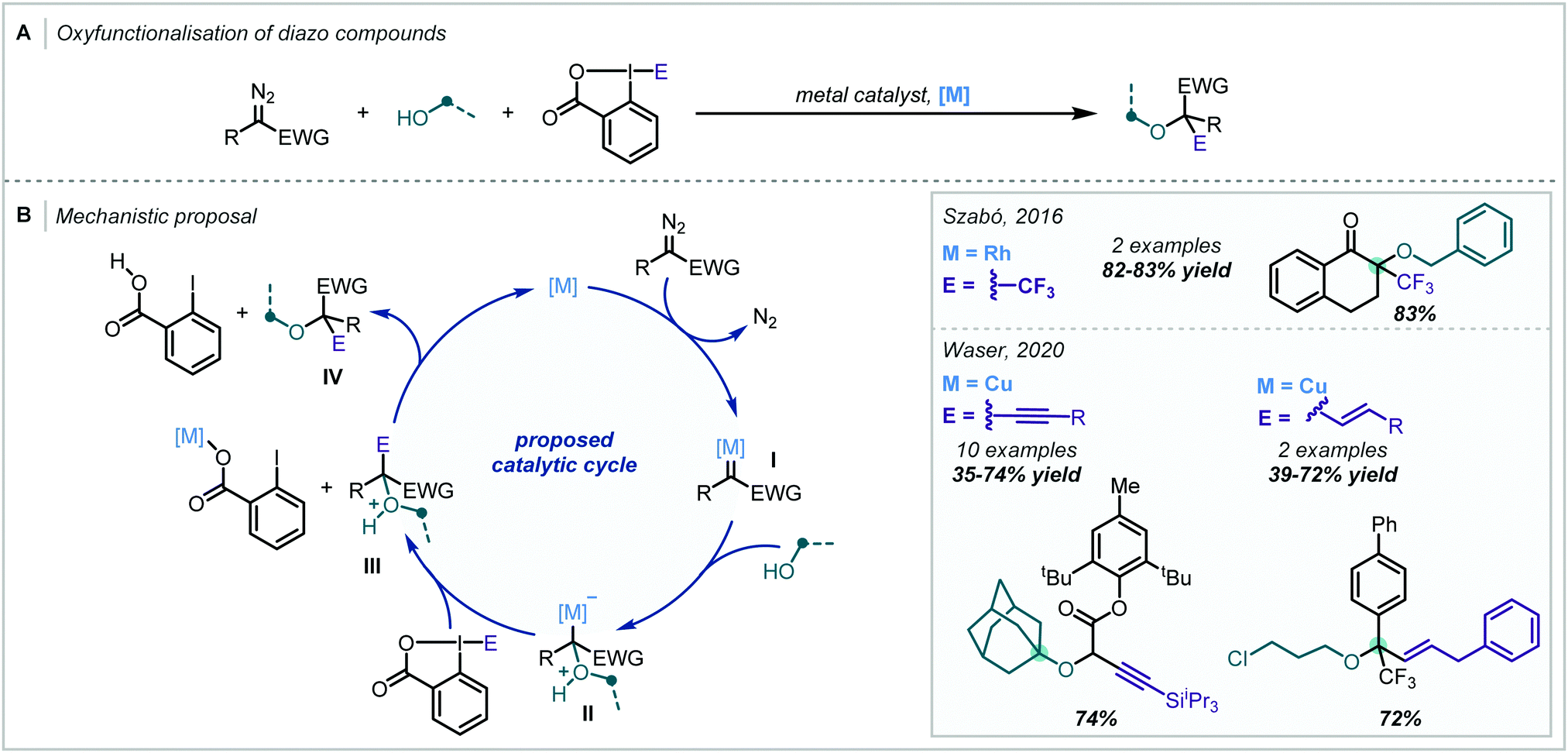 | ||
| Scheme 102 Scope and proposed mechanism of oxyfunctionalisation reactions of diazo compounds. | ||
Recently, Sun reported the reaction of N-Boc pyridones with dimethyl 2-diazomalonate-derived carbenes (Scheme 103).385 It was proposed that addition of the pyridone to the generated rhodium carbene could form an ylide intermediate, which could undergo a cyclisation onto the pendent carbamate. C–N cleavage in the resulting tetrahedral intermediate resulted in the formation of an α-tertiary pyridyl ether. Variation in the pyridone coupling partner demonstrated a wide functional group tolerance, in moderate to good yields.
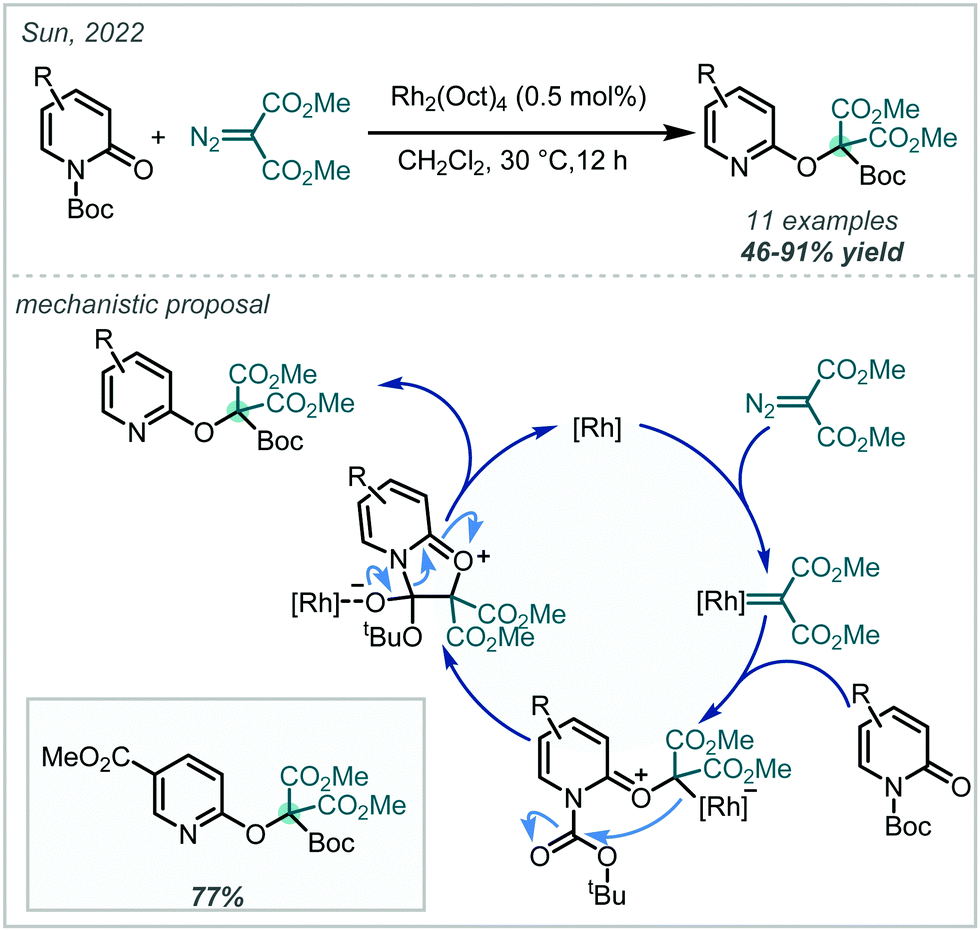 | ||
| Scheme 103 Synthesis of α-tertiary pyridyl ethers featuring a 1,4-migration of a Boc group. | ||
While many propargylic substitutions are proposed to proceed via carbocation intermediates, derived from activation by Lewis acids or through formal metal-catalysed cross-coupling,386 it has been shown that some reactions, under metal catalysis, proceed via unique metal-allenylidene or propargylic–metal intermediates. In 1994, Godfrey reported the Cu(I)-promoted reaction of propargylic carbonates with phenols to give α-tertiary ethers (Scheme 104);387 in analogous work using amine nucleophiles,388 a Cu–allenylidene intermediate had been proposed but was not detected.
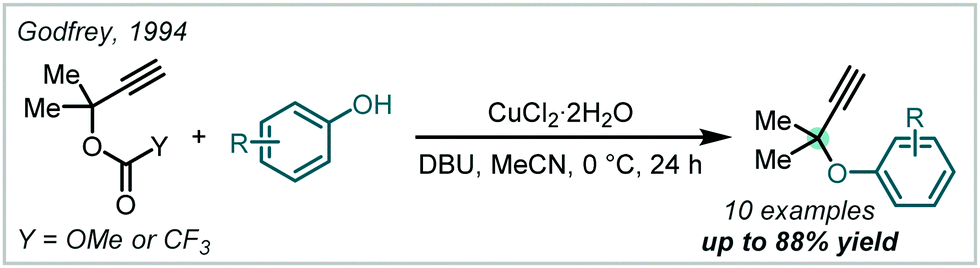 | ||
| Scheme 104 Early propargylic substitution with proposed allenylidene intermediate. | ||
Nishibayashi confirmed the intermediacy of a metal–allenylidene or metal–propargylic intermediate when using a thiolate-bridged diruthenium complex (as shown in Scheme 105) and developed a wide range of propargylic substitutions, including the synthesis of a range of α-tertiary ethers utilising a broad range of ruthenium catalysts incorporating both mono- and di-nuclear systems and featuring a range of ligand classes389–391 Bauer also developed mononuclear ruthenium catalysts for analogous processes392–394 and others have reported the applicability of a wide variety of ruthenium complexes for the activation of propargylic alcohols.395,396
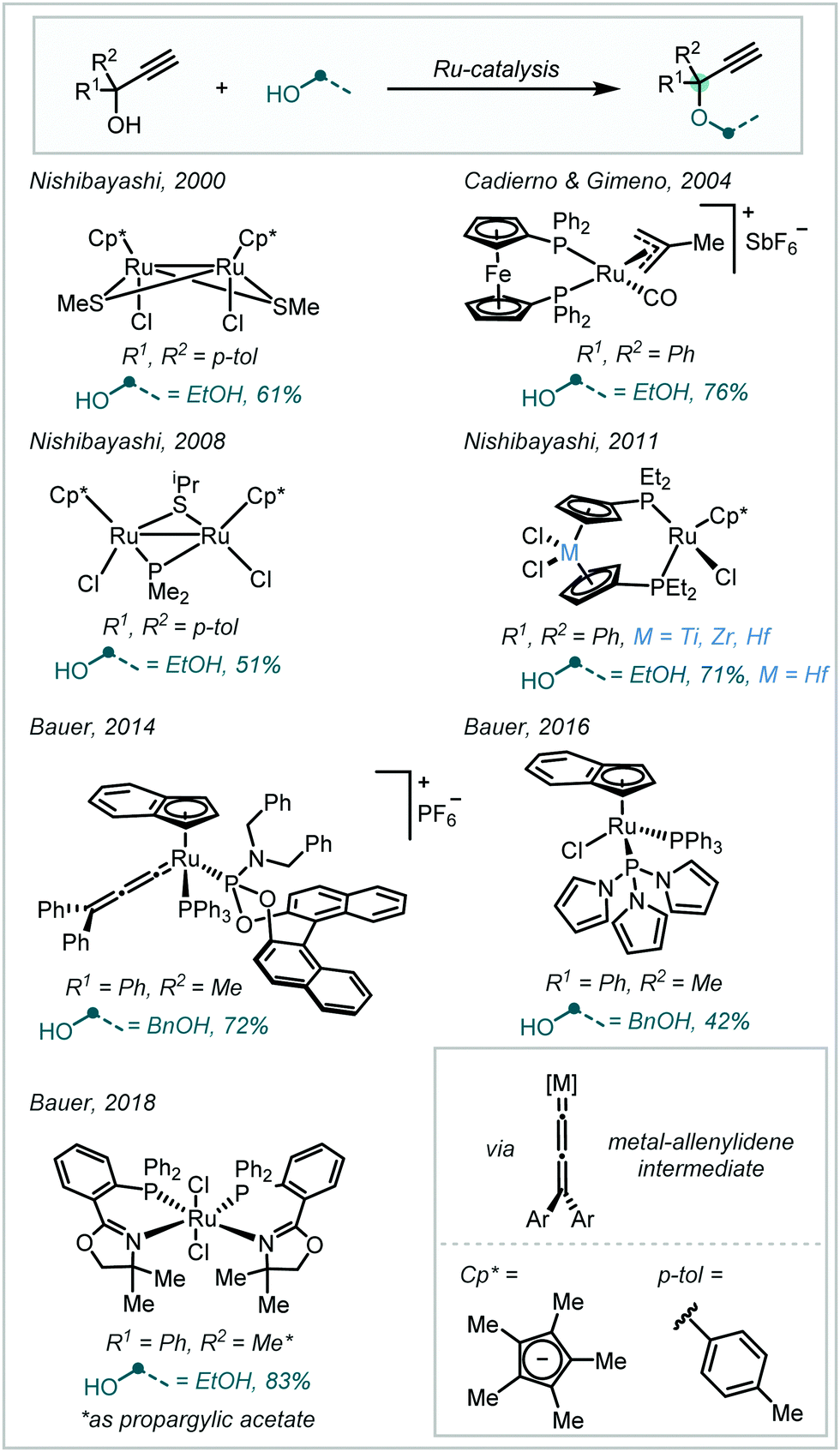 | ||
| Scheme 105 Catalysts for propargylic substitution via allenylidene intermediate. | ||
From appropriate precursors, analogous metal-free carbenoid strategies have also been successfully developed. In 2010, Barluenga & Valdés reported the metal-free reductive coupling of tosylhydrazones with alcohols or phenols, employing mechanistic principles related to the Bamford–Stevens reaction.397 This strategy allowed the synthesis of an α-tertiary ether from readily prepared and stable starting materials (Scheme 106).
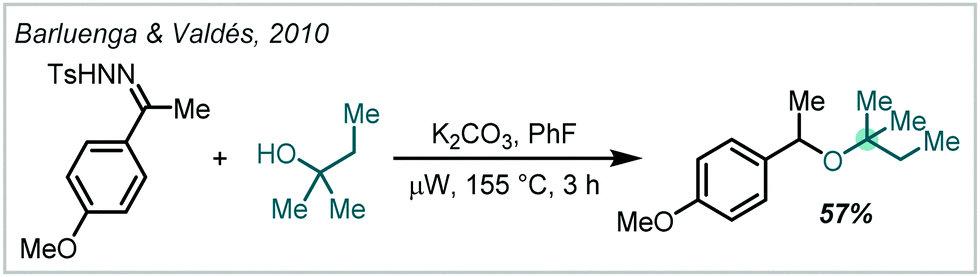 | ||
| Scheme 106 Reductive coupling of alcohols with tosylhydrazones. | ||
7.4 Reductive etherification
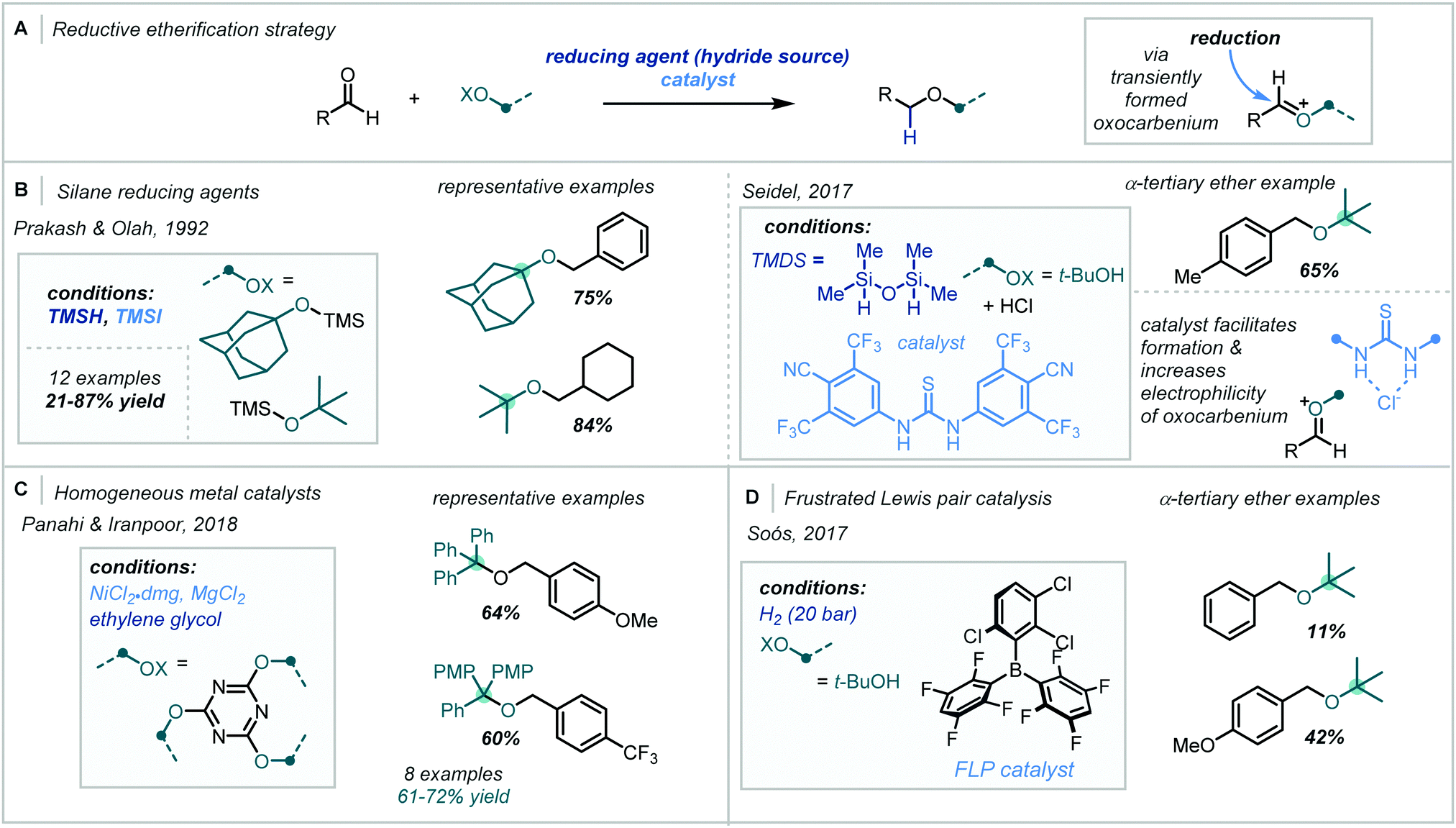 | ||
| Scheme 107 Reductive etherification strategies. (A) General reductive etherification strategy. Reductive etherifications using: (B) silane reducing agents; (C) transition metal catalysis; and (D) FLP catalysis. dmg = dimethyl glyoxime; PMP = para-methoxyphenyl. | ||
In 1972, Doyle reported the silane-mediated reduction of carbonyl compounds to give ethers, however, this reaction required excess sulfuric or trifluoroacetic acid and therefore only gave low to moderate yields of acid-sensitive α-tertiary ethers.398 Twenty years later, Prakash and Olah reported that hindered ethers could be prepared using such a reductive strategy with catalytic trimethylsilyl iodide to promote oxocarbenium ion formation (Scheme 107B).399 The mild conditions of this method allowed the synthesis of a variety of α-tertiary ethers in high yield; a similar approach using TMSOTf was also detailed by Hatakeyama and Nishizawa.400
An Fe-catalysed alternative was reported by Oriyama in 2005 which demonstrated greater operational simplicity and shorter reaction times, although the yield of α-tertiary ethers obtained by this method was moderate.401
Recently, Seidel developed a reductive etherification reaction using anion-binding catalysis.402,403 A derivative of Schreiner's thiourea catalyst was designed to facilitate the formation and increase the electrophilicity of the intermediate oxocarbenium ion and enabled the synthesis of an α-tertiary example in 65% yield.404 Boron-based reducing agents have also been employed to generate α-tertiary ethers using this reductive etherification strategy; Scheunemann utilised decaborane as a mild and fairly stable reducing agent to yield a tert-butyl ether while investigating a one-step reductive etherification of 4-[18F] fluoro-benzaldehyde for use in positron emission tomography.405
Additionally, both homogeneous and heterogeneous metal catalytic systems have been exploited in such reductive etherification strategies to yield α-tertiary ethers. In 2018, Panahi and Iranpoor reported the Ni-catalysed reductive etherification of aldehydes with activated triazine tritylating agents using ethylene glycol as both the solvent and reducing agent to give α-tertiary ethers at room temperature and in good yields (Scheme 107C).406 Heterogeneous systems such as Pd catalysts which have been modified with ethyl iodide have been used as bifunctional catalysts in the selective reductive etherification of bio-derived aldehydes with alcohols.407
Although such metal-based catalytic systems have proven useful, Soós demonstrated a metal-free approach using a frustrated Lewis pair catalyst to promote reductive etherification by enabling hydrogen activation, thus forming both a reducing agent and a Brønsted acid (Scheme 107D).408 Despite modest yields in the synthesis of α-tertiary ether examples, this represents a successful complimentary strategy to metal-based systems in achieving the synthesis of such a challenging motif.
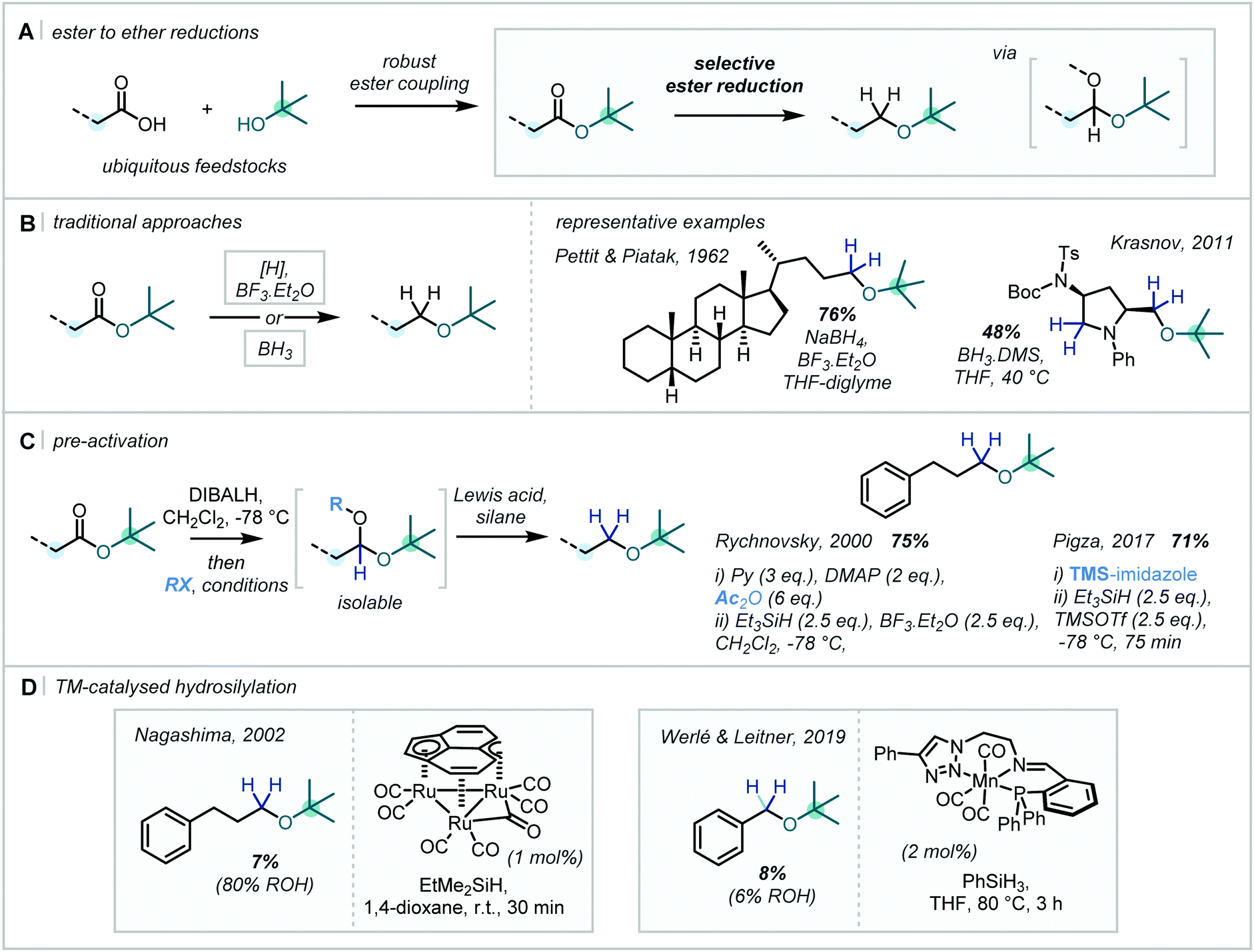 | ||
| Scheme 108 Reduction of esters to ethers: (A) general concept; (B) traditional approaches; (C) stepwise reduction-activation-reduction; (D) TM-catalysed hydrosilylation. | ||
Notably, this strategy has been more widely adopted in the synthesis of cyclic α-tertiary ethers, rather than acyclic ethers. While multi-step approaches, featuring complete reduction to the diol then subsequent nucleophilic substitution of an activated primary alcohol, have been widely adopted, single-step procedures have traditionally relied upon the combined action of a strong hydride source and a Lewis acidic activator (Scheme 108B). Most common is the combination of lithium aluminium hydride and boron trifluoride etherate, and this has been successfully applied in both cyclic and acyclic α-tertiary ether synthesis.410,411 In a similar manner, borane reductions of amides are often associated with ether side products if esters are also present.412
Alternative strategies have sought to isolate the transiently formed mixed acetals as silyl-protected or acetyl-activated hemiacetals, which can be further reduced to the desired ethers (Scheme 108C).413,414
More recently, the application of transition–metal catalysed hydrosilylation has shown significant potential in selective ester reductions.415 Early promising results were observed by Nagashima when using triruthenium carbonyl clusters in the presence of EtMe2SiH.416 While the reaction was broadly unselective for the ether product, both alcohol and ether products were observed, with the alcohol typically predominating. In a single example incorporating an α-tertiary site, the corresponding ether could be isolated in 7% yield.
Following this study, Mn-catalysed hydrosilylation has also showed great promise in ester–ether reductions. A notable report from Werlé and Leitner described the application of Mn(I) catalysts bearing triazole ligands to carbonyl hydrosilylation (Scheme 108D).417 Unfortunately, the reaction showed considerable sensitivity to sterics and, while tert-butyl benzoate could be reduced with moderate selectivity to the ether product (6:4 ether:alcohol), the conversion was very low (14%). The work was more widely applied to acid and ketone reduction.
8. Conclusion and outlook
The synthesis of acyclic α-tertiary ethers has traditionally represented an exacting challenge using classical etherification strategies. Given the unique physical and biochemical properties associated with such moieties and their potential as key motifs in high-value target molecule synthesis, it is no surprise that the synthetic community has dramatically altered this once-barren landscape. A key component enabling this resounding growth has been the multi-faceted application of redox strategies towards such challenging etherifications.The difunctionalisation of alkenes has proven to be a particularly versatile strategy in accessing such hindered ethers. In particular, the renewed use of photo- and electrochemical manifolds has driven a vast expansion in the applicable functionalisation strategies that can be coupled to alkoxylation. In tandem with these strategies, more traditional redox mediators such as transition metal catalysts, halogen compounds in many different oxidation states, and main group oxidants, such as Hg and Se compounds, have also been widely applied in this field.
As an alternative ubiquitous substrate, the decarboxylation of carboxylic acids has enabled some of the most promising strategies, in particular, the development of photo- and electrochemical regimes has vastly expanded the scope, overcoming the steric limitations of hindered etherification.
Given the striking recent advances in C–H activation, it is encouraging to see that this has also had a considerable impact in this area and a wide range of structurally distinct C–H bonds are now readily converted to α-tertiary ether linkages. Additionally, the phosphorus-mediated activation of alcohols to enable etherification has been widely adopted in the synthetic community and the unique chemistry of phenols has led to diverse strategies enabling their oxidative alkoxylation, accessing complex α-cyclic α-tertiary ethers. Numerous additional strategies have had considerable success including the application of peroxides and carbenoids to access reactive intermediates capable of facile etherification.
Notably, despite a select few promising early reports, the potential of reductive regimes to access ethers is broadly unrealised, given the preponderance of esters and analogous high oxidation state functionalities in molecular libraries.
A recurring feature of many of the methods described above that has prevented their widespread adoption by the community is the requirement for large excesses of one coupling partner, preventing the union of high-value or late-stage fragments via these approaches. This presents an opportunity to render these methods more applicable to contemporary, sustainability-driven research. Furthermore, in the coming years it will be exciting to witness how the rapidly expanding fields of photo- and electrochemistry take on the challenge of enantioselectivity in α-tertiary etherification. This challenge no longer rests on simply installing the α-tertiary ether but has extended to multicomponent reactions, cascades resulting in dramatic increases in structural complexity and pinpoint stereoselective introduction of asymmetry.
Without doubt, the α-tertiary ether is no longer the formidable and obscure exception it once was. As the strategic application of redox manipulations becomes ever more refined and broader in scope, the complexity and value of hindered ether synthesis will become truly established in synthetic chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are supported by the Centre for Doctoral Training in Synthesis for Biology and Medicine, generously supported by GlaxoSmithKline, MSD, Syngenta and Vertex. B. S. also thanks the Oxford-Leon E & Iris L Beghian Scholarship, D. B. thanks Magdalen College for a Graduate Scholarship, J. C. thanks the EPSRC for an Excellence Award, and B. W. thanks Oriel College for an Alec Bond Scholarship. Thomas Marsh is thanked for assisting in the planning of this work.References
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- J. Meyers, M. Carter, N. Y. Mok and N. Brown, Future Med. Chem., 2016, 8, 1753–1767 CrossRef PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- D. Ameen and T. J. Snape, MedChemComm, 2013, 4, 893–907 RSC.
- H. Feuer and J. Hooz, in The Ether Linkage, ed. S. Patai, John Wiley & Sons, Ltd., Chichester, UK, 1967, pp. 445–498 Search PubMed.
- S. Mandal, S. Mandal, S. K. Ghosh, P. Sar, A. Ghosh, R. Saha and B. Saha, RSC Adv., 2016, 6, 69605–69614 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- G. A. Westphal, J. Krahl, T. Brüning, E. Hallier and J. Bünger, Toxicology, 2010, 268, 198–203 CrossRef CAS PubMed.
- L. Sambri, G. Bartoli, G. Bencivenni and R. Dalpozzo, Curr. Org. Synth., 2012, 9, 137–148 CrossRef CAS.
- P. Domínguez De María, R. W. Van Gemert, A. J.-J. Straathof and U. Hanefeld, Nat. Prod. Rep., 2010, 27, 370–392 RSC.
- Y. Koiso, M. Natori, S. Iwasaki, S. Sato, R. Sonoda, Y. Fujita, H. Yaegashi and Z. Sato, Tetrahedron Lett., 1992, 33, 4157–4160 CrossRef CAS.
- Y. Koiso, Y. Li, S. Iwasaki, K. Hanaka, T. Kobayashi, R. Sonoda, Y. Fujita, H. Yaegashi and Z. Sato, J. Antibiot., 1994, 47, 765–773 CrossRef CAS PubMed.
- S. Kundu and D. Sarkar, J. Heterocycl. Chem., 2021, 58, 1741–1748 CrossRef CAS.
- C. L. Holder, W. A. Korfmacher, W. Slikker Jr, H. C. Thompson Jr and A. B. Gosnell, Biomed. Mass Spectrom., 1985, 12, 151–158 CrossRef CAS PubMed.
- Z. Zhang and W. Tang, Acta Pharm. Sin. B, 2018, 8, 721–732 CrossRef PubMed.
- G. De Martino, G. L. Regina, A. D. Pasquali, R. Ragno, A. Bergamini, C. Ciaprini, A. Sinistro, G. Maga, E. Crespan, M. Artico and R. Silvestri, J. Med. Chem., 2005, 48, 4378–4388 CrossRef CAS PubMed.
- D. Spinks and G. Spinks, Curr. Med. Chem., 2002, 9, 799–810 CrossRef CAS PubMed.
- J.-n Fischer and C. R. Ganellin, Analogue-based drug discovery, Wiley-VCH, 2006 Search PubMed.
- A. Luther, C. Bisang and D. Obrecht, Bioorg. Med. Chem., 2018, 26, 2850–2858 CrossRef CAS PubMed.
- F. Grotenhermen, Clin. Pharmacokinet., 2003, 42, 327–360 CrossRef CAS PubMed.
- X. Wang and P. J. Quinn, Mol. Membr. Biol., 2000, 17, 143–156 CrossRef CAS PubMed.
- F. M. Callahan, G. W. Anderson, R. Paul and J. E. Zimmerman, J. Am. Chem. Soc., 1963, 85, 201–207 CrossRef CAS.
- H. Lyu, I. Kevlishvili, X. Yu, P. Liu and G. Dong, Science, 2021, 372, 175–182 CrossRef CAS PubMed.
- K. C. Nicolaou, S. Pan, Y. Shelke, D. Das, Q. Ye, Y. Lu, S. Sau, R. Bao and S. Rigol, J. Am. Chem. Soc., 2021, 143, 9267–9276 CrossRef CAS PubMed.
- A. Williamson, Q. J. Indian Chem. Soc., 1852, 4, 229–239 RSC.
- A. Williamson, Liebigs Ann., 1851, 77, 37–49 CrossRef CAS.
- H. C. Beyerman and G. J. Heiszwolf, Recl. Trav. Chim. Pays-Bas, 1965, 84, 203–212 CrossRef CAS.
- K. C.-K. Swamy, N. N.-B. Kumar, E. Balaraman and K. V.-P. P. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef CAS PubMed.
- S. Fletcher, Org. Chem. Front., 2015, 2, 739–752 RSC.
- B. Schlummer and U. Scholz, Adv. Synth. Catal., 2004, 346, 1599–1626 CrossRef CAS.
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, J. Organomet. Chem., 2018, 861, 17–104 CrossRef CAS.
- M. Watanabe, M. Nishiyama and Y. Koie, Tetrahedron Lett., 1999, 40, 8837–8840 CrossRef CAS.
- G. Mann, C. Incarvito, A. L. Rheingold and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 3224–3225 CrossRef CAS.
- A. V. Vorogushin, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 8146–8149 CrossRef CAS PubMed.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525 RSC.
- T. Martín, J. I. Padrón and V. S. Martín, Synlett, 2014, 12–32 Search PubMed.
- T. Nakata, Chem. Rev., 2005, 105, 4314–4347 CrossRef CAS PubMed.
- M. D. Delost, D. T. Smith, B. J. Anderson and J. T. Njardarson, J. Med. Chem., 2018, 61, 10996–11020 CrossRef CAS PubMed.
- E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS PubMed.
- S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed.
- S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912–4924 RSC.
- R. I. Khusnutdinov and A. R. Bayguzina, Russ. J. Org. Chem., 2019, 55, 903–932 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W.-C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- C. G.-S. Lima, T. de, M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- R. J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281–293 CrossRef CAS PubMed.
- T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945–6952 CrossRef CAS PubMed.
- L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285–8291 RSC.
- S. Sharma, J. Singh and A. Sharma, Adv. Synth. Catal., 2021, 363, 3146–3169 CrossRef CAS.
- C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed.
- G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2013, 15, 4398–4401 CrossRef CAS PubMed.
- E. Yamaguchi, W. Tanaka and A. Itoh, Asian J. Org. Chem., 2019, 14, 121–124 CAS.
- L.-M. Altmann, V. Zantop, P. Wenisch, N. Diesendorf and M. R. Heinrich, Eur. J. Org. Chem., 2021, 2452–2462 CrossRef CAS PubMed.
- S. Kindt, K. Wicht and M. R. Heinrich, Angew. Chem., Int. Ed., 2016, 55, 8744–8747 CrossRef CAS PubMed.
- A. Tlahuext-Aca, R. A. Garza-Sanchez and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 3708–3711 CrossRef CAS PubMed.
- S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS.
- S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. De Sarkar and S. Murarka, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS.
- S. Shibutani, K. Nagao and H. Ohmiya, Org. Lett., 2021, 23, 1798–1803 CrossRef CAS PubMed.
- H. Yi, X. Zhang, C. Qin, Z. Liao, J. Liu and A. Lei, Adv. Synth. Catal., 2014, 356, 2873–2877 CrossRef CAS.
- L. Li, H. Chen, M. Mei and L. Zhou, Chem. Commun., 2017, 53, 11544–11547 RSC.
- L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652–13655 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- M. Julliard and M. Chanon, Chem. Rev., 1983, 83, 425–506 CrossRef CAS.
- R. A. Neunteufel and D. R. Arnold, J. Am. Chem. Soc., 1973, 95, 4080–4081 CrossRef CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- Z. Xu, Z. Yang, Y. Liu, Y. Lu, K. Chen and W. Zhu, J. Chem. Inf. Model., 2014, 54, 69–78 CrossRef CAS PubMed.
- C. Wagner, M. El Omari and G. M. König, J. Nat. Prod., 2009, 72, 540–553 CrossRef CAS PubMed.
- S. Jiang, L. Zhang, D. Cui, Z. Yao, B. Gao, J. Lin and D. Wei, Sci. Rep., 2016, 6, 34750 CrossRef CAS PubMed.
- Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567–9571 CrossRef CAS PubMed.
- Y. Arai, R. Tomita, G. Ando, T. Koike and M. Akita, Eur. J. Org. Chem., 2016, 1262–1265 CrossRef CAS PubMed.
- Y. Ran, Q.-Y. Lin, X.-H. Xu and F.-L. Qing, J. Org. Chem., 2016, 81, 7001–7007 CrossRef CAS PubMed.
- M. Daniel, G. Dagousset, P. Diter, P.-A. Klein, B. Tuccio, A.-M. Goncalves, G. Masson and E. Magnier, Angew. Chem., Int. Ed., 2017, 56, 3997–4001 CrossRef CAS PubMed.
- H. Ge, B. Wu, Y. Liu, H. Wang and Q. Shen, ACS Catal., 2020, 10, 12414–12424 CrossRef CAS.
- G. Levitre, G. Dagousset, E. Anselmi, B. Tuccio, E. Magnier and G. Masson, Org. Lett., 2019, 21, 6005–6010 CrossRef CAS PubMed.
- K. Masanobu, S. Hirochika and T. Katsumi, Bull. Chem. Soc. Jpn., 1985, 58, 521–524 CrossRef.
- G. Fumagalli, P. T.-G. Rabet, S. Boyd and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11481–11484 CrossRef CAS PubMed.
- X.-D. An, H. Zhang, Q. Xu, L. Yu and S. Yu, Chin. J. Chem., 2018, 36, 1147–1150 CrossRef CAS.
- X.-D. An, Y.-Y. Jiao, H. Zhang, Y. Gao and S. Yu, Org. Lett., 2018, 20, 401–404 CrossRef CAS PubMed.
- J. Zhang, X. Li, W. Xie, S. Ye and J. Wu, Org. Lett., 2019, 21, 4950–4954 CrossRef CAS PubMed.
- E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 3394–3402 CrossRef CAS PubMed.
- M. W. Campbell, M. Yuan, V. C. Polites, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 3901–3910 CrossRef CAS PubMed.
- S. Lin, S. D. Lies, C. S. Gravatt and T. P. Yoon, Org. Lett., 2017, 19, 368–371 CrossRef CAS PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed.
- R. Engels, H. J. Schäfer and E. Steckhan, Liebigs Ann., 1977, 1977, 204–224 CrossRef.
- M. Kojima, H. Sakuragi and K. Tokumaru, Chem. Lett., 1981, 1707–1710 CrossRef CAS.
- C. M. Hudson, M. R. Marzabadi, K. D. Moeller and D. G. New, J. Am. Chem. Soc., 1991, 113, 7372–7385 CrossRef CAS.
- C. M. Hudson and K. D. Moeller, J. Am. Chem. Soc., 1994, 116, 3347–3356 CrossRef CAS.
- P. Xiong, H. Long, J. Song, Y. Wang, J. F. Li and H. C. Xu, J. Am. Chem. Soc., 2018, 140, 16387–16391 CrossRef CAS PubMed.
- S. Zhang, L. Li, P. Wu, P. Gong, R. Liu and K. Xu, Adv. Synth. Catal., 2019, 361, 485–489 CrossRef CAS.
- X. Zhang, T. Cui, X. Zhao, P. Liu and P. Sun, Angew. Chem., Int. Ed., 2020, 59, 3465–3469 CrossRef CAS PubMed.
- O. V. Bityukov, V. A. Vil, G. I. Nikishin and A. O. Terent'ev, Adv. Synth. Catal., 2021, 363, 3070–3078 CrossRef CAS.
- T. T. Zhang, M. J. Luo, Y. Li, R. J. Song and J. H. Li, Org. Lett., 2020, 22, 7250–7254 CrossRef CAS PubMed.
- T. Inoue, K. Koyama, T. Matsuoka and S. Tsutsumi, Bull. Chem. Soc. Jpn., 1967, 40, 162–168 CrossRef CAS.
- T. Inoue and S. Tsutsumi, Bull. Chem. Soc. Jpn., 1965, 38, 661–665 CrossRef CAS.
- L. Zhang, G. Zhang, P. Wang, Y. Li and A. Lei, Org. Lett., 2018, 20, 7396–7399 CrossRef CAS PubMed.
- Y. Yuan, Y. Cao, Y. Lin, Y. Li, Z. Huang and A. Lei, ACS Catal., 2018, 8, 10871–10875 CrossRef CAS.
- Z. Zhang, J. Yan, D. Ma and J. Sun, Chin. Chem. Lett., 2019, 30, 1509–1511 CrossRef CAS.
- H. Mei, J. Liu, Y. Guo and J. Han, ACS Omega, 2019, 4, 14353–14359 CrossRef CAS PubMed.
- P. Das, S. Das, K. Varalaxmi and R. Jana, Adv. Synth. Catal., 2021, 363, 575–584 CrossRef CAS.
- Y. Yuan, Y. Chen, S. Tang, Z. Huang and A. Lei, Sci. Adv., 2018, 4, eaat5312 CrossRef CAS PubMed.
- Y. Wang, L. Deng, H. Mei, B. Du, J. Han and Y. Pan, Green Chem., 2018, 20, 3444–3449 RSC.
- D. Prim, J.-M. Campagne, D. Joseph and B. Andrioletti, Tetrahedron, 2002, 58, 2041–2075 CrossRef CAS.
- A. D. Melhado, W. E. Brenzovich Jr., A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885–8887 CrossRef CAS PubMed.
- W. E. Brenzovich Jr., J.-F. Brazeau and F. D. Toste, Org. Lett., 2010, 12, 4728–4731 CrossRef PubMed.
- L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Chem. – Eur. J., 2012, 18, 2931–2937 CrossRef CAS PubMed.
- D. R. White, M. I. Herman and J. P. Wolfe, Org. Lett., 2017, 19, 4311–4314 CrossRef CAS PubMed.
- Z. Liao, H. Yi, Z. Li, C. Fan, X. Zhang, J. Liu, Z. Deng and A. Lei, Asian J. Org. Chem., 2015, 10, 96–99 CAS.
- Y. Murata, T. Shimada and T. Nishikata, Bull. Chem. Soc. Jpn., 2019, 92, 1419–1429 CrossRef CAS.
- C. Chatalova-Sazepin, Q. Wang, G. M. Sammis and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 5443–5446 CrossRef CAS PubMed.
- W. Jian, L. Ge, Y. Jiao, B. Qian and H. Bao, Angew. Chem., Int. Ed., 2017, 56, 3650–3654 CrossRef CAS PubMed.
- M.-T. Suo, S. Yang, J.-C. Yang, Z.-Y. Liu, J.-J. Zhang and L.-N. Guo, Org. Chem. Front., 2020, 7, 2414–2418 RSC.
- H. Ren, J.-R. Song, Z.-Y. Li and W.-D. Pan, Org. Lett., 2019, 21, 6774–6778 CrossRef CAS PubMed.
- W. Wang, J.-R. Song, Z.-Y. Li, T. Zhong, Q. Chi, H. Ren and W.-D. Pan, RSC Adv., 2021, 11, 18080–18083 RSC.
- W. P. Unsworth, S. G. Lamont and J. Robertson, Tetrahedron, 2014, 70, 7388–7394 CrossRef CAS.
- Z. Wang, Z. Jin, Q. Zhong, Y. Zhang, Y. Wu, Y. Ma, H. Sun, P. Yu and R. H. Dodd, Org. Biomol. Chem., 2020, 18, 7414–7424 RSC.
- L. Legnani and B. Morandi, Angew. Chem., Int. Ed., 2016, 55, 2248–2251 CrossRef CAS PubMed.
- D. Yu, K.-P. Shing, Y. Liu, H. Liu and C.-M. Che, Chem. Commun., 2020, 56, 137–140 RSC.
- D. Kawauchi, H. Ueda and H. Tokuyama, Eur. J. Org. Chem., 2019, 2056–2060 CrossRef CAS.
- X.-F. Fu, Q. Zhao and W.-X. Zhao, Asian J. Org. Chem., 2021, 10, 1161–1166 CrossRef CAS.
- H. Sun, B. Cui, L. Duan and Y.-M. Li, Org. Lett., 2017, 19, 1520–1523 CrossRef CAS PubMed.
- M. S. Hadfield and A.-L. Lee, Org. Lett., 2010, 12, 484–487 CrossRef CAS PubMed.
- A. Heuer-Jungemann, R. G. McLaren, M. S. Hadfield and A.-L. Lee, Tetrahedron, 2011, 67, 1609–1616 CrossRef CAS.
- P. Jeschke, Pest Manage. Sci., 2017, 73, 1053–1066 CrossRef CAS PubMed.
- P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS PubMed.
- K. Maeda, H. Shinokubo and K. Oshima, J. Org. Chem., 1996, 61, 6770–6771 CrossRef CAS PubMed.
- Y. Guindon, B. Guérin, C. Chabot, N. Mackintosh and W. W. Ogilvie, Synlett, 1995, 449–451 CrossRef CAS.
- H. Shinokubo and K. Oshima, Synlett, 2001, 0322–0328 CrossRef CAS.
- D. Dolenc and M. Harej, J. Org. Chem., 2002, 67, 312–313 CrossRef CAS PubMed.
- D. H.-R. Barton, I. A. Blair and P. D. Magnus, J. Chem. Soc., Perkin Trans. 1, 1972, 614–615 RSC.
- R. C. Cambie, R. C. Hayward, J. L. Roberts and P. S. Rutledge, J. Chem. Soc., Perkin Trans. 1, 1974, 1858–1864 RSC.
- A. M. Sanseverino and M. C.-S. de Mattos, Synthesis, 1998, 1584–1586 CrossRef CAS.
- V. A. Mahajan, P. D. Shinde, A. S. Gajare, M. Karthikeyan and R. D. Wakharkar, Green Chem., 2002, 4, 325–327 RSC.
- Shallu, M. L. Sharma and J. Singh, Synth. Commun., 2012, 42, 1306–1324 CrossRef CAS.
- R. A.-S. Villegas, J. L. do Espírito Santo, A. M. Sanseverino, M. C.-S. de Mattos, M. R.-M. P. de Aguiar and A. W.-S. Guarino, Synth. Commun., 2005, 35, 1627–1631 CrossRef CAS.
- J. Barluenga, M. A. Rodríguez, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1987, 1491–1492 RSC.
- N. Hosseinzadeh, S. R. Mousavi, Y. Ahmadi, N. Batooei, Z. Sotoudehnia Korrani and M. Mahdavi, Polycyclic Aromat. Compd., 2021, 1–9 Search PubMed.
- M. S. Yusubov, R. J. Yusubova, V. D. Filimonov and K. W. Chi, Synth. Commun., 2004, 34, 443–450 CrossRef CAS.
- M. S. Yusubov, R. Y. Yusubova, V. D. Filimonov and C. Ki-Whan, Russ. J. Org. Chem., 2002, 38, 902–904 CrossRef CAS.
- M. S. Yusubov, L. A. Drygunova, A. V. Tkachev and V. V. Zhdankin, ARKIVOC, 2005, 2005, 179–188 Search PubMed.
- M. S. Yusubov, L. A. Drygunova and V. V. Zhdankin, Synthesis, 2004, 2289–2292 CrossRef CAS.
- M. S. Yusubov, R. Y. Yusubova, A. Kirschning, J. Y. Park and K.-W. Chi, Tetrahedron Lett., 2008, 49, 1506–1509 CrossRef CAS.
- P. B. Thorat, B. Y. Bhong and N. N. Karade, Synlett, 2013, 2061–2066 CAS.
- T. K. Achar, S. Maiti and P. Mal, Org. Biomol. Chem., 2016, 14, 4654–4663 RSC.
- M. Jereb, M. Zupan and S. Stavber, Green Chem., 2005, 7, 100–104 RSC.
- K. Rama and M. A. Pasha, Ultrason. Sonochem., 2005, 12, 437–440 CrossRef CAS PubMed.
- J. H. Schauble, E. A. Trauffer, P. P. Deshpande and R. D. Evans, Synthesis, 2005, 1333–1339 CrossRef CAS.
- Y. Li, J. Guo, X. Lu and F. Zhong, Org. Biomol. Chem., 2020, 18, 32–35 RSC.
- R. Filler, Isr. J. Chem., 1978, 17, 71–79 CrossRef CAS.
- G. S. Lal, G. P. Pez and R. G. Syvret, Chem. Rev., 1996, 96, 1737–1756 CrossRef CAS PubMed.
- Z. Qin, M. Zhao, K. Zhang, M. Goto, K.-H. Lee and J. Li, J. Org. Chem., 2021, 86, 7864–7871 CrossRef CAS PubMed.
- G. S. Lal, J. Org. Chem., 1993, 58, 2791–2796 CrossRef CAS.
- S. Stavber, T. Sotler and M. Zupan, Tetrahedron Lett., 1994, 35, 1105–1108 CrossRef CAS.
- S. Stavber, M. Zupan, A. J. Poss and G. A. Shia, Tetrahedron Lett., 1995, 36, 6769–6772 CrossRef CAS.
- S. Stojan, S.-P. Tjaša and Z. Marko, Bull. Chem. Soc. Jpn., 1996, 69, 169–175 CrossRef.
- M.-Y. Chang, N.-C. Lee, M.-F. Lee, Y.-P. Huang and C.-H. Lin, Tetrahedron Lett., 2010, 51, 5900–5903 CrossRef CAS.
- A. Kumar, T. V. Singh and P. Venugopalan, J. Fluorine Chem., 2013, 150, 72–77 CrossRef CAS.
- B. Greedy and V. Gouverneur, Chem. Commun., 2001, 233–234 RSC.
- H. Hou, H. Li, Y. Xu, D. Tang, Y. Han, C. Yan, X. Chen and S. Zhu, Tetrahedron, 2018, 74, 6577–6583 CrossRef CAS.
- J.-L. Liu, Z.-F. Zhu and F. Liu, Org. Chem. Front., 2019, 6, 241–244 RSC.
- I. V. Koval, Russ. J. Org. Chem., 2002, 38, 301–337 CrossRef CAS.
- W. Flitsch and P. Rußkamp, Liebigs Ann. Chem., 1985, 1985, 1422–1436 CrossRef.
- J. P. Dulcère, J. Rodriguez, M. Santelli and J. P. Zahra, Tetrahedron Lett., 1987, 28, 2009–2011 CrossRef.
- M. Okabe, M. Abe and M. Tada, J. Org. Chem., 1982, 47, 1775–1777 CrossRef CAS.
- K. Last and H. M.-R. Hoffmann, Synthesis, 1989, 901–905 CrossRef CAS.
- J. P. Dulcère, J. Crandall, R. Faure, M. Santelli, V. Agati and M. N. Mihoubi, J. Org. Chem., 1993, 58, 5702–5708 CrossRef.
- J. P. Dulcère, V. Agati and R. Faure, J. Chem. Soc., Chem. Commun., 1993, 270–271 RSC.
- J.-P. Dulcère, V. Agati and J. Rodriguez, J. Chem. Soc., Chem. Commun., 1993, 1038–1039 RSC.
- A. Tenaglia, O. Pardigon and G. Buono, J. Org. Chem., 1996, 61, 1129–1132 CrossRef CAS.
- T. Abe, Y. Kosaka, T. Kawasaki, Y. Ohata, T. Yamashiro and K. Yamada, Chem. Pharm. Bull., 2020, 68, 555–558 CrossRef CAS PubMed.
- V. L. Heasley, K. E. Wade, T. G. Aucoin, D. E. Gipe and D. F. Shellhamer, J. Org. Chem., 1983, 48, 1377–1379 CrossRef CAS.
- B. Das, K. Venkateswarlu, K. Damodar and K. Suneel, J. Mol. Catal. A: Chem., 2007, 269, 17–21 CrossRef CAS.
- P. A. Bentley, Y. Mei and J. Du, Tetrahedron Lett., 2008, 49, 2653–2655 CrossRef CAS.
- Y. W. Zhu, Y. X. Shi and Y. Q. Yin, Catalysts, 2017, 7, 66 CrossRef.
- J. Rodriguez, J. P. Dulcere and M. Bertrand, Tetrahedron Lett., 1983, 24, 4423–4424 CrossRef CAS.
- M. Bertrand, J. P. Dulcere, J. Rodriquez and J. P. Zahra, Tetrahedron Lett., 1983, 24, 1967–1970 CrossRef CAS.
- Y. A. Serguchev, M. V. Ponomarenko, L. F. Lourie and A. N. Chernega, J. Fluorine Chem., 2003, 123, 207–215 CrossRef CAS.
- L. Zhou, C. K. Tan, J. Zhou and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 10245–10247 CrossRef CAS PubMed.
- J. Chen, S. Chng, L. Zhou and Y.-Y. Yeung, Org. Lett., 2011, 13, 6456–6459 CrossRef CAS PubMed.
- Z. Ke and Y.-Y. Yeung, Org. Lett., 2013, 15, 1906–1909 CrossRef CAS PubMed.
- M. Sohail, M. Khan, Y. Zhang, C. Peng, Q. Chen and Z. K. Zhao, Tetrahedron Lett., 2015, 56, 5743–5746 CrossRef CAS.
- M. Sohail, C. Peng, S. Ning, Y. Zhang, M. Khan and Z. K. Zhao, Tetrahedron, 2016, 72, 658–663 CrossRef CAS.
- S. Stavber and M. Zupan, Tetrahedron, 1986, 42, 5035–5043 CrossRef CAS.
- S. Stavber and M. Zupan, J. Org. Chem., 1987, 52, 919–921 CrossRef CAS.
- S. Stavber, T. Sotler-Pečan and M. Zupan, Tetrahedron, 1994, 50, 12235–12244 CrossRef CAS.
- G. F. Mendonça, A. M. Sanseverino and M. C.-S. de Mattos, Synthesis, 2003, 0045–0048 Search PubMed.
- R. D.-S. Ribeiro, P. M. Esteves and M. C.-S. de Mattos, Tetrahedron Lett., 2007, 48, 8747–8751 CrossRef CAS.
- P. Phukan, P. Chakraborty and D. Kataki, J. Org. Chem., 2006, 71, 7533–7537 CrossRef CAS PubMed.
- S. A. Glover and A. Goosen, Tetrahedron Lett., 1980, 21, 2005–2008 CrossRef CAS.
- M. Alikarami and M. Farhadi, Helv. Chim. Acta, 2015, 98, 1302–1306 CrossRef CAS.
- D. S. Rao, T. R. Reddy, K. Babachary and S. Kashyap, Org. Biomol. Chem., 2016, 14, 7529–7543 RSC.
- S. F. Vice and G. I. Dmitrienko, Can. J. Chem., 1982, 60, 1233–1237 CrossRef CAS.
- M. K. Agrawal, S. Adimurthy, B. Ganguly and P. K. Ghosh, Tetrahedron, 2009, 65, 2791–2797 CrossRef CAS.
- N. Chakraborty, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov and A. Majee, RSC Adv., 2015, 5, 56780–56788 RSC.
- A. Majee, S. K. Kundu, S. Santra and A. Hajra, Tetrahedron Lett., 2012, 53, 4433–4435 CrossRef CAS.
- M. A. Kumar, M. Naresh, C. N. Rohitha and N. Narender, Synth. Commun., 2013, 43, 3121–3129 CrossRef CAS.
- P. Swamy, M. A. Kumar, M. M. Reddy, M. Naresh, K. Srujana and N. Narender, RSC Adv., 2014, 4, 26288–26294 RSC.
- J. Le Bras, D. Chatterjee and J. Muzart, Tetrahedron Lett., 2005, 46, 4741–4743 CrossRef CAS.
- C. Ye, X. Kou, G. Yang, J. Shen and W. Zhang, Tetrahedron Lett., 2019, 60, 1148–1152 CrossRef CAS.
- Y. Jia, L. Chen, H. Zhang, Y. Zheng, Z.-X. Jiang and Z. Yang, Org. Biomol. Chem., 2018, 16, 7203–7213 RSC.
- B. Sels, D. D. Vos, M. Buntinx, F. Pierard, A. Kirsch-De Mesmaeker and P. Jacobs, Nature, 1999, 400, 855–857 CrossRef CAS.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Am. Chem. Soc., 2001, 123, 8350–8359 CrossRef CAS PubMed.
- A. Toshimitsu, H. Owada, K. Terao, S. Uemura and M. Okano, J. Chem. Soc., Perkin Trans. 1, 1985, 373–378 RSC.
- M. Tiecco, L. Testaferri, M. Tingoli, D. Chianelli and D. Bartoli, Tetrahedron, 1988, 44, 2261–2272 CrossRef CAS.
- M. Tiecco, L. Testaferri, M. Tingoli, D. Chianelli and D. Bartoli, Tetrahedron Lett., 1989, 30, 1417–1420 CrossRef CAS.
- P. Ceccherelli, M. Curini, M. C. Marcotullio and O. Rosati, Tetrahedron, 1991, 47, 4211–4220 CrossRef CAS.
- A. A. Vieira, J. B. Azeredo, M. Godoi, C. Santi, E. N. Da Silva Júnior and A. L. Braga, J. Org. Chem., 2015, 80, 2120–2127 CrossRef CAS PubMed.
- S. Torii, K. Uneyama and M. Ono, Tetrahedron Lett., 1980, 21, 2653–2654 CrossRef CAS.
- S. Torii, K. Uneyama and M. Ono, Tetrahedron Lett., 1980, 21, 2741–2744 CrossRef CAS.
- S. Torii, K. Uneyama, M. Ono and T. Bannou, J. Am. Chem. Soc., 1981, 103, 4606–4608 CrossRef CAS.
- K. Uneyama, M. Ono and S. Torii, Phosphorus Sulfur Relat. Elem., 1983, 16, 35–43 CrossRef CAS.
- J. Chen, L. Mei, H. Wang, L. Hu, X. Sun, J. Shi and Q. Li, ChemistryOpen, 2019, 8, 1230–1234 CrossRef CAS PubMed.
- N. Amri and T. Wirth, Synthesis, 2020, 1751–1761 CAS.
- F.-H. Cui, Y. Hua, Y.-M. Lin, J. Fei, L.-H. Gao, X. Zhao and H. Xia, J. Am. Chem. Soc., 2022, 144, 2301–2310 CrossRef CAS PubMed.
- S. Tomoda and M. Iwaoka, Chem. Lett., 1988, 1895–1898 CrossRef CAS.
- R. Déziel, S. Goulet, L. Grenier, J. Bordeleau and J. Bernier, J. Org. Chem., 1993, 58, 3619–3621 CrossRef.
- R. Déziel, E. Malenfant, C. Thibault, S. Fréchette and M. Gravel, Tetrahedron Lett., 1997, 38, 4753–4756 CrossRef.
- S. I. Fukuzawa, K. Takahashi, H. Kato and H. Yamazaki, J. Org. Chem., 1997, 62, 7711–7716 CrossRef CAS.
- S.-i Fukuzawa, Y. Kasugahara and S. Uemura, Tetrahedron Lett., 1994, 35, 9403–9406 CrossRef CAS.
- T. Wirth, Angew. Chem., Int. Ed., 1995, 34, 1726–1728 CrossRef CAS.
- K.-i Fujita, K. Murata, M. Iwaoka and S. Tomoda, Tetrahedron, 1997, 53, 2029–2048 CrossRef CAS.
- M. Tiecco, L. Testaferri, C. Santi, F. Marini, L. Bagnoli and A. Temperini, Tetrahedron Lett., 1998, 39, 2809–2812 CrossRef CAS.
- L. Zhao, Z. Li and T. Wirth, Eur. J. Org. Chem., 2011, 7080–7082 CrossRef CAS.
- T. G. Back, Z. Moussa and M. Parvez, J. Org. Chem., 2002, 67, 499–509 CrossRef CAS PubMed.
- T. G. Back and Z. Moussa, Org. Lett., 2000, 2, 3007–3009 CrossRef CAS PubMed.
- H. C. Brown and P. Geoghegan, J. Am. Chem. Soc., 1967, 89, 1522–1524 CrossRef CAS.
- T. G. Traylor, J. Am. Chem. Soc., 1964, 86, 244–248 CrossRef CAS.
- W. L. Waters and E. F. Kiefer, J. Am. Chem. Soc., 1967, 89, 6261–6268 CrossRef CAS.
- C. Georgoulis, W. Smadja and J. M. Valery, Synthesis, 1981, 572–574 CrossRef CAS.
- J. Beger, B. Thomas, T. Vogel and R. Lang, J. Prakt. Chem., 1991, 333, 447–453 CrossRef CAS.
- R. Govindarajan, J. Ahmed, A. K. Swain and S. K. Mandal, J. Org. Chem., 2019, 84, 13490–13502 CrossRef CAS PubMed.
- S. Liu and M. Klussmann, Chem. Commun., 2020, 56, 1557–1560 RSC.
- Y.-Y. Liang, J. Huang, X.-H. Ouyang, J.-H. Qin, R.-J. Song and J.-H. Li, Chem. Commun., 2021, 57, 3684–3687 RSC.
- D. R. Arnold and A. J. Maroulis, J. Am. Chem. Soc., 1977, 99, 7355–7356 CrossRef CAS.
- D. Mangion and D. R. Arnold, Acc. Chem. Res., 2002, 35, 297–304 CrossRef CAS PubMed.
- P. J. Kropp, E. J. Reardon, Z. L.-F. Gaibel, K. F. Williard and J. H. Hattaway, J. Am. Chem. Soc., 1973, 95, 7058–7067 CrossRef CAS.
- M. W. Klett and R. P. Johnson, Tetrahedron Lett., 1983, 24, 1107–1110 CrossRef CAS.
- M. Weiser, S. Hermann, A. Penner and H.-A. Wagenknecht, Beilstein J. Org. Chem., 2015, 11, 568–575 CrossRef CAS PubMed.
- A. Penner, E. Bätzner and H.-A. Wagenknecht, Synlett, 2012, 2803–2807 CAS.
- F. Speck, D. Rombach and H.-A. Wagenknecht, Beilstein J. Org. Chem., 2019, 15, 52–59 CrossRef CAS PubMed.
- F. Seyfert, M. Mitha and H.-A. Wagenknecht, Eur. J. Org. Chem., 2021, 773–776 CrossRef CAS.
- Y. Tanaka, S. Kubosaki, K. Osaka, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2018, 83, 13625–13635 CrossRef CAS PubMed.
- T. Rossolini, B. Ferko and D. J. Dixon, Org. Lett., 2019, 21, 6668–6673 CrossRef CAS PubMed.
- J. A. Leitch, T. Rossolini, T. Rogova and D. J. Dixon, ACS Catal., 2020, 10, 11430–11437 CrossRef CAS.
- A. Bhattacharya, L. M. DiMichele, U. H. Dolling, E. J.-J. Grabowski and V. J. Grenda, J. Org. Chem., 1989, 54, 6118–6120 CrossRef CAS.
- S. Fukuzumi, M. Fujita, G.-E. Matsubayashi and J. Otera, Chem. Lett., 1993, 1451–1454 CrossRef CAS.
- X. Guo and H. Mayr, J. Am. Chem. Soc., 2013, 135, 12377–12387 CrossRef CAS PubMed.
- C. Banoun, F. Bourdreux, E. Magnier and G. Dagousset, Org. Lett., 2021, 23, 8926–8930 CrossRef CAS PubMed.
- J. Liu, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2021, 60, 12053–12059 CrossRef CAS PubMed.
- K. M. Gligorich, M. J. Schultz and M. S. Sigman, J. Am. Chem. Soc., 2006, 128, 2794–2795 CrossRef CAS PubMed.
- M. J. Schultz and M. S. Sigman, J. Am. Chem. Soc., 2006, 128, 1460–1461 CrossRef CAS PubMed.
- Y. Zhang and M. S. Sigman, Org. Lett., 2006, 8, 5557–5560 CrossRef CAS PubMed.
- M. C. Haibach, C. Guan, D. Y. Wang, B. Li, N. Lease, A. M. Steffens, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2013, 135, 15062–15070 CrossRef CAS PubMed.
- H. Shigehisa, T. Aoki, S. Yamaguchi, N. Shimizu and K. Hiroya, J. Am. Chem. Soc., 2013, 135, 10306–10309 CrossRef CAS PubMed.
- R. G. Johnson and R. K. Ingham, Chem. Rev., 1956, 56, 219–269 CrossRef CAS.
- D. H.-R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941 RSC.
- R. S.-J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- H. Kolbe, Liebigs Ann., 1849, 69, 257–294 CrossRef.
- H. Hofer and M. Moest, Liebigs Ann., 1902, 323, 284–323 CrossRef CAS.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, ed. Z. Wang, Wiley, New York, 2010, pp. 1443–1446 Search PubMed.
- J. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. Del Bel, G. M. Gallego, J. E. Spangler, J. Starr, S. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398–402 CrossRef CAS PubMed.
- D. E. Collin, A. A. Folgueiras-Amador, D. Pletcher, M. E. Light, B. Linclau and R. C.-D. Brown, Chem. – Eur. J., 2020, 26, 374–378 CrossRef CAS PubMed.
- F. Bu, L. Lu, X. Hu, S. Wang, H. Zhang and A. Lei, Chem. Sci., 2020, 11, 10000–10004 RSC.
- N. P. Ramirez and J. C. Gonzalez-Gomez, Eur. J. Org. Chem., 2017, 2154–2163 CrossRef CAS.
- R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 13624–13628 CrossRef CAS PubMed.
- S. Shibutani, T. Kodo, M. Takeda, K. Nagao, N. Tokunaga, Y. Sasaki and H. Ohmiya, J. Am. Chem. Soc., 2020, 142, 1211–1216 CrossRef CAS PubMed.
- P. Li, J. R. Zbieg and J. A. Terrett, ACS Catal., 2021, 11, 10997–11004 CrossRef CAS.
- Q. Y. Li, S. N. Gockel, G. A. Lutovsky, K. S. DeGlopper, N. J. Baldwin, M. W. Bundesmann, J. W. Tucker, S. W. Bagley and T. P. Yoon, Nat. Chem., 2022, 14, 94–99 CrossRef CAS PubMed.
- T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 1–31 CrossRef.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS.
- H. M.-L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS PubMed.
- Q. Zheng, J. Chen and G. W. Rao, Russ. J. Org. Chem., 2019, 55, 569–586 CrossRef CAS.
- B. J. Lee, K. S. DeGlopper and T. P. Yoon, Angew. Chem., Int. Ed., 2020, 59, 197–202 CrossRef CAS PubMed.
- S. Y. Zhang, F. M. Zhang and Y. Q. Tu, Chem. Soc. Rev., 2011, 40, 1937–1949 RSC.
- B. M. Trost, in Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley, New York, 2004, pp. 2–14 Search PubMed.
- B. Liu and B. F. Shi, Tetrahedron Lett., 2015, 56, 15–22 CrossRef CAS.
- S. Y. Zhang, G. He, Y. Zhao, K. Wright, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2012, 134, 7313–7316 CrossRef CAS PubMed.
- G. Shan, X. Yang, Y. Zong and Y. Rao, Angew. Chem., Int. Ed., 2013, 52, 13606–13610 CrossRef CAS PubMed.
- R. Giri, J. Liang, J. G. Lei, J. J. Li, D. H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J. Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420–7424 CrossRef CAS PubMed.
- R. T. Gephart, C. L. McMullin, N. G. Sapiezynski, E. S. Jang, M. J.-B. Aguila, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2012, 134, 17350–17353 CrossRef CAS PubMed.
- T. K. Salvador, C. H. Arnett, S. Kundu, N. G. Sapiezynski, J. A. Bertke, M. Raghibi Boroujeni and T. H. Warren, J. Am. Chem. Soc., 2016, 138, 16580–16583 CrossRef CAS PubMed.
- S. Sarkar, T. Sahoo, C. Sen and S. C. Ghosh, Chem. Commun., 2021, 57, 8949–8952 RSC.
- G. Song, Z. Zheng, Y. Wang and X. Yu, Org. Lett., 2016, 18, 6002–6005 CrossRef CAS PubMed.
- H. Hu, S. J. Chen, M. Mandal, S. M. Pratik, J. A. Buss, S. W. Krska, C. J. Cramer and S. S. Stahl, Nat. Catal., 2020, 3, 358–367 CrossRef CAS PubMed.
- A. Vasilopoulos, D. L. Golden, J. A. Buss and S. S. Stahl, Org. Lett., 2020, 22, 5753–5757 CrossRef CAS PubMed.
- T. K. Beng, V. Shearer, R. Davey and I. Redman, RSC Adv., 2020, 10, 20264–20271 RSC.
- C. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495–13498 CrossRef CAS PubMed.
- J. Zhang, Y. Li, F. Zhang, C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872–1875 CrossRef CAS PubMed.
- R. Qi, C. Wang, Y. Huo, H. Chai, H. Wang, Z. Ma, L. Liu, R. Wang and Z. Xu, J. Am. Chem. Soc., 2021, 143, 12777–12783 CrossRef CAS PubMed.
- J. C.-K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed.
- K. A. Hollister, E. S. Conner, M. L. Spell, K. Deveaux, L. Maneval, M. W. Beal and J. R. Ragains, Angew. Chem., Int. Ed., 2015, 54, 7837–7841 CrossRef CAS PubMed.
- S. Du, E. A. Kimball and J. R. Ragains, Org. Lett., 2017, 19, 5553–5556 CrossRef CAS PubMed.
- Z. Y. Ma, L. N. Guo, Y. R. Gu, L. Chen and X. H. Duan, Adv. Synth. Catal., 2018, 360, 4341–4347 CrossRef CAS.
- Y. Jun, T. Jun and Z. Chi, Adv. Synth. Catal., 2010, 352, 531–546 CrossRef.
- H. Mo and W. Bao, Tetrahedron, 2011, 67, 4793–4799 CrossRef CAS.
- R. Kotagiri and R. Adepu, Eur. J. Org. Chem., 2018, 4556–4564 CrossRef CAS.
- M. C. Hilton, R. D. Dolewski and A. McNally, J. Am. Chem. Soc., 2016, 138, 13806–13809 CrossRef CAS PubMed.
- Y. Zi, F. Schömberg, K. Wagner and I. Vilotijevic, Org. Lett., 2020, 22, 3407–3411 CrossRef CAS PubMed.
- G. A. Shevchenko, B. Oppelaar and B. List, Angew. Chem., Int. Ed., 2018, 57, 10756–10759 CrossRef CAS PubMed.
- J. Blom, G. J. Reyes-Rodríguez, H. N. Tobiesen, J. N. Lamhauge, M. V. Iversen, C. L. Barløse, N. Hammer, M. Rusbjerg and K. A. Jørgensen, Angew. Chem., Int. Ed., 2019, 58, 17856–17862 CrossRef CAS PubMed.
- J. N. Lamhauge, V. Corti, Y. Liu and K. A. Jørgensen, Angew. Chem., Int. Ed., 2021, 60, 18728–18733 CrossRef CAS PubMed.
- K. Ponsold and H. Kasch, Tetrahedron Lett., 1979, 46, 4463–4464 CrossRef.
- H. Wang, K. Liang, W. Xiong, S. Samanta, W. Li and A. Lei, Sci. Adv., 2020, 6, eaaz0590 CrossRef CAS PubMed.
- X. Lin, S. N. Zhang, D. Xu, J. J. Zhang, Y. X. Lin, G. Y. Zhai, H. Su, Z. H. Xue, X. Liu, M. Antonietti, J. S. Chen and X. H. Li, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- O. Mitsunobu and M. Yamada, Bull. Chem. Soc. Jpn., 1967, 40, 2380–2382 CrossRef CAS.
- R. S. Subramanian and K. K.-B. Balasubramanian, Synth. Commun., 1989, 19, 1255–1259 CrossRef CAS.
- K. K.-B. Balasubramanian, Tetrahedron Lett., 1989, 30, 2297–2300 CrossRef.
- A. A.-W. Voerste and J. Reisch, J. Chem. Soc., Perkin Trans. 1, 1994, 3251–3256 Search PubMed.
- Y. J. Shi, D. L. Hughes and J. M. McNamara, Tetrahedron Lett., 2003, 44, 3609–3611 CrossRef CAS.
- U. Hengartner, A. Chougnet, K. Liu and W. D. Woggon, Chem. – Eur. J., 2010, 16, 1306–1311 CrossRef CAS PubMed.
- A. Wolfgardt and F. Bracher, Lett. Org. Chem., 2013, 10, 2–7 CAS.
- T. Shintou, W. Kikuchi and T. Mukaiyama, Chem. Lett., 2003, 32, 22–23 CrossRef CAS.
- T. Shintou, W. Kikuchi and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2003, 76, 1645–1667 CrossRef CAS.
- T. Shintou and T. Mukaiyama, J. Am. Chem. Soc., 2004, 126, 7359–7367 CrossRef CAS PubMed.
- H. Aoki and T. Mukaiyama, Chem. Lett., 2005, 34, 1016–1017 CrossRef CAS.
- H. Aoki and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2006, 79, 1255–1264 CrossRef CAS.
- L. Ochmann, M. L. Kessler and P. R. Schreiner, Org. Lett., 2022, 24, 1460–1464 CrossRef CAS PubMed.
- K. Dimroth, W. Umbach and H. Thomas, Chem. Ber., 1967, 100, 132–141 CrossRef CAS.
- A. S. Mitchell and R. A. Russell, Tetrahedron Lett., 1993, 34, 545–548 CrossRef CAS.
- A. S. Mitchell and R. A. Russell, Tetrahedron, 1997, 53, 4387–4410 CrossRef CAS.
- P. Camps, A. González, D. Muñoz-Torrero, M. Simon, A. Zúñiga, M. A. Martins, M. Font-Bardia and X. Solans, Tetrahedron, 2000, 56, 8141–8151 CrossRef CAS.
- Z.-T. He, B. Tian, Y. Fukui, X. Tong, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2013, 52, 5314–5318 CrossRef CAS PubMed.
- G. Wells, T. D. Bradshaw, P. Diana, A. Seaton, D.-F. Shi, A. D. Westwell and M. F.-G. Stevens, Bioorg. Med. Chem. Lett., 2000, 513–515 CrossRef CAS.
- A. Ronlán and V. D. Parker, J. Chem. Soc. C, 1971, 3214–3218 RSC.
- A. Nilsson, A. Ronlán and V. D. Parker, Tetrahedron Lett., 1975, 16, 1107–1110 CrossRef.
- K. Omura, J. Org. Chem., 1996, 61, 7156–7161 CrossRef CAS PubMed.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, Angew. Chem., Int. Ed., 2005, 44, 310–313 CrossRef CAS PubMed.
- A. Rieker, R. Beisswenger and K. Regier, Tetrahedron, 1991, 47, 645–654 CrossRef CAS.
- A. Nilsson, U. Palmquist, T. Pettersson and A. Ronlán, J. Chem. Soc., Perkin Trans. 1, 1978, 696–707 RSC.
- A. Rieker, E. L. Dreher, H. Geisel and M. H. Khalifa, Synthesis, 1978, 851–855 CrossRef CAS.
- G. W. Morrow and J. S. Swenton, Tetrahedron Lett., 1987, 28, 5445–5448 CrossRef CAS.
- G. W. Morrow, V. Chen and J. S. Swenton, Tetrahedron, 1991, 47, 655–664 CrossRef CAS.
- J. S. Swenton, K. Carpenter, Y. Chen, M. L. Kerns and G. W. Morrow, J. Org. Chem., 1993, 58, 3308–3316 CrossRef CAS.
- J. S. Swenton, A. Callinan, Y. Chen, J. J. Rohde, M. L. Kerns and G. W. Morrow, J. Org. Chem., 1996, 61, 1267–1274 CrossRef CAS.
- A. Nilsson, U. Palmquist, T. Pettersson and A. Ronlán, J. Chem. Soc., Perkin Trans. 1, 1978, 708–715 RSC.
- R. E. DeSchepper and J. S. Swenton, Tetrahedron Lett., 1985, 26, 4831–4834 CrossRef CAS.
- I. Barba, R. Chinchilla and C. Gómez, Tetrahedron Lett., 1989, 30, 3187–3188 CrossRef CAS.
- I. Barba, R. Chinchilla and C. Gómez, Tetrahedron, 1990, 46, 7813–7822 CrossRef CAS.
- I. Barba, R. Chinchilla and C. Gomez, J. Org. Chem., 1991, 56, 3673–3676 CrossRef CAS.
- Y. Shizuri, K. Nakamura and S. Yamamura, J. Chem. Soc., Chem. Commun., 1985, 530–531 RSC.
- F. Barba, A. Guirado and I. Barba, J. Org. Chem., 1984, 49, 3022–3024 CrossRef CAS.
- H. D. Becker and K. Gustafsson, J. Org. Chem., 1979, 44, 428–432 CrossRef CAS.
- Y. Kita, H. Tohma, K. Kikuchi, M. Inagaki and T. Yakura, J. Org. Chem., 1991, 56, 435–438 CrossRef CAS.
- A. Pelter, R. S. Ward and A. Abd-El-Ghani, J. Chem. Soc., Perkin Trans. 1, 1992, 2249–2251 RSC.
- N. Lewis and P. Wallbank, Synthesis, 1987, 1103–1106 CrossRef CAS.
- A. Pelter and S. Elgendy, Tetrahedron Lett., 1988, 29, 677–680 CrossRef CAS.
- A. Pelter and S. M.-A. Elgendy, J. Chem. Soc., Perkin Trans. 1, 1993, 1891–1896 RSC.
- A. Callinan, Y. Chen, G. W. Morrow and J. S. Swenton, Tetrahedron Lett., 1990, 31, 4551–4552 CrossRef CAS.
- N. Taneja and R. K. Peddinti, Tetrahedron Lett., 2016, 57, 3958–3963 CrossRef CAS.
- C. Dolka, K. V. Hecke, L. V. Meervelt, P. G. Tsoungas, E. V.-V. D. Eycken and G. Varvounis, Org. Lett., 2009, 11, 2964–2967 CrossRef CAS PubMed.
- S. Quideau, M. A. Looney and L. Pouységu, Org. Lett., 1999, 1, 1651–1654 CrossRef CAS.
- S. Quideau, L. Pouységu, M. Oxoby and M. A. Looney, Tetrahedron, 2001, 57, 319–329 CrossRef CAS.
- N. Lebrasseur, G.-J. Fan, M. Oxoby, M. A. Looney and S. Quideau, Tetrahedron, 2005, 61, 1551–1562 CrossRef CAS.
- N. Homs, P. R. de la Piscina and F. Borrull, J. Chem. Soc., Chem. Commun., 1988, 1075–1076 RSC.
- D. G. Hewitt, J. Chem. Soc. C, 1971, 2967–2973 RSC.
- M. J. Harrison and R. O.-C. Norman, J. Chem. Soc. C, 1970, 728–730 RSC.
- A. McKillop, D. H. Perry, M. Edwards, S. Antus, L. Farkas, M. Nogradi and E. C. Taylor, J. Org. Chem., 1976, 41, 282–287 CrossRef CAS.
- S.-O. Lawesson and N. C. Yang, J. Am. Chem. Soc., 1959, 81, 4230–4233 CrossRef CAS.
- C. Frisell and S.-O. Lawesson, Org. Synth., 1965, 45, 89 CrossRef CAS.
- S. Kyasa, R. N. Meier, R. A. Pardini, T. K. Truttmann, K. T. Kuwata and P. H. Dussault, J. Org. Chem., 2015, 80, 12100–12114 CrossRef CAS PubMed.
- G. D.-P. Gomes, V. Vil, A. Terent'ev and I. V. Alabugin, Chem. Sci., 2015, 6, 6783–6791 RSC.
- B. Miller, J. Am. Chem. Soc., 1973, 95, 8458–8460 CrossRef CAS.
- B. Miller, J. Org. Chem., 1977, 42, 1402–1408 CrossRef CAS.
- P. Gijsman, in Handb. Environ. Degrad. Mater. 2nd edn, ed. M. Kutz, William Andrew Publishing, Oxford, 2012, pp. 673–714 Search PubMed.
- J. T. Schneider, D. S. Firak, R. R. Ribeiro and P. Peralta-Zamora, Phys. Chem. Chem. Phys., 2020, 22, 15723–15733 RSC.
- G. Gleixner, J. W. Breitenbach and O. F. Olaj, Makromol. Chem., 1978, 179, 73–77 CrossRef CAS.
- H. J. Hageman, Eur. Polym. J., 2000, 36, 345–350 CrossRef CAS.
- P. S. Engel, H. J. Park, H. Mo and S. Duan, Tetrahedron, 2010, 66, 8805–8814 CrossRef CAS.
- E. Kumli, F. Montermini and P. Renaud, Org. Lett., 2006, 8, 5861–5864 CrossRef CAS PubMed.
- M. Ladlow and G. Pattenden, Tetrahedron Lett., 1984, 25, 4317–4320 CrossRef CAS.
- E. Fasani, M. Mella and A. Albini, J. Chem. Soc., Perkin Trans. 2, 1995, 449–452 RSC.
- H. Sun, C. Yang, R. Lin and W. Xia, Adv. Synth. Catal., 2014, 356, 2775–2780 CrossRef CAS.
- Q. Zhu, E. C. Gentry and R. R. Knowles, Angew. Chem., Int. Ed., 2016, 55, 9969–9973 CrossRef CAS PubMed.
- M. Wang, C. Chen, M. Ma, B. Zhao and Z. Shi, J. Org. Chem., 2022, 87, 3577–3585 CrossRef PubMed.
- H. M.-L. Davies, T. J. Clark and L. A. Church, Tetrahedron Lett., 1989, 30, 5057–5060 CrossRef CAS.
- A. Padwa and S. F. Hornbuckle, Chem. Rev., 1991, 91, 263–309 CrossRef CAS.
- J. J. Medvedev and V. A. Nikolaev, Russ. Chem. Rev., 2015, 84, 737–757 CrossRef CAS.
- W. Yuan, L. Eriksson and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 8410–8415 CrossRef CAS PubMed.
- D. P. Hari and J. Waser, J. Am. Chem. Soc., 2016, 138, 2190–2193 CrossRef CAS PubMed.
- G. Pisella, A. Gagnebin and J. Waser, Chem. – Eur. J., 2020, 26, 10199–10204 CrossRef CAS PubMed.
- G. Pisella, A. Gagnebin and J. Waser, Org. Lett., 2020, 22, 3884–3889 CrossRef CAS PubMed.
- B. K. Mai, K. J. Szabó and F. Himo, ACS Catal., 2018, 8, 4483–4492 CrossRef CAS.
- J. Su, Q. Li, Y. Shao and J. Sun, Org. Lett., 2022, 24, 1637–1641 CrossRef CAS PubMed.
- Y. Miyake, S. Uemura and Y. Nishibayashi, ChemCatChem, 2009, 1, 342–356 CrossRef CAS.
- J. D. Godfrey, R. H. Mueller, T. C. Sedergran, N. Soundararajan and V. J. Colandrea, Tetrahedron Lett., 1994, 35, 6405–6408 CrossRef.
- R. J. Detz, M. M.-E. Delville, H. Hiemstra and J. H. van Maarseveen, Angew. Chem., Int. Ed., 2008, 47, 3777–3780 CrossRef CAS PubMed.
- Y. Nishibayashi, I. Wakiji and M. Hidai, J. Am. Chem. Soc., 2000, 122, 11019–11020 CrossRef CAS.
- Y. Miyake, S. Endo, M. Yuki, Y. Tanabe and Y. Nishibayashi, Organometallics, 2008, 27, 6039–6042 CrossRef CAS.
- T. Miyazaki, Y. Tanabe, M. Yuki, Y. Miyake and Y. Nishibayashi, Organometallics, 2011, 30, 3194–3199 CrossRef CAS.
- D. F. Alkhaleeli, K. J. Baum, J. M. Rabus and E. B. Bauer, Catal. Commun., 2014, 47, 45–48 CrossRef CAS.
- M. J. Stark, M. J. Shaw, N. P. Rath and E. B. Bauer, Eur. J. Inorg. Chem., 2016, 1093–1102 CrossRef CAS.
- N. Jourabchian, K. Jurkowski and E. B. Bauer, Catal. Commun., 2018, 106, 92–95 CrossRef CAS.
- M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, Organometallics, 2016, 35, 388–399 CrossRef.
- V. Cadierno, J. Díez, S. E. García-Garrido and J. Gimeno, Chem. Commun., 2004, 2716–2717 RSC.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 49, 4993–4996 CrossRef CAS PubMed.
- M. P. Doyle, D. J. DeBruyn and D. A. Kooistra, J. Am. Chem. Soc., 1972, 94, 3659–3661 CrossRef CAS.
- N. Hartz, G. K. Surya Prakash and G. A. Olah, Synlett, 1992, 569–572 CrossRef CAS.
- S. Hatakeyama, H. Mori, K. Kitano, H. Yamada and M. Nishizawa, Tetrahedron Lett., 1994, 35, 4367–4370 CrossRef CAS.
- K. Iwanami, H. Seo, Y. Tobita and T. Oriyama, Synthesis, 2005, 183–186 CAS.
- L. Schifferer, M. Stinglhamer, K. Kaur and O. G. Macheño, Beilstein J. Org. Chem., 2021, 17, 2270–2286 CrossRef CAS PubMed.
- M. D. Visco, J. Attard, Y. Guan and A. E. Mattson, Tetrahedron Lett., 2017, 58, 2623–2628 CrossRef CAS.
- C. Zhao, C. A. Sojdak, W. Myint and D. Seidel, J. Am. Chem. Soc., 2017, 139, 10224–10227 CrossRef CAS PubMed.
- U. Funke, H. Jia, S. Fischer, M. Scheunemann and J. Steinbach, J. Label. Compd. Radiopharm., 2006, 49, 745–755 CrossRef CAS.
- S. Rahimi, F. Panahi, M. Bahmani and N. Iranpoor, J. Org. Chem., 2018, 83, 973–979 CrossRef CAS PubMed.
- D. Wu, W. Y. Hernández, S. Zhang, E. I. Vovk, X. Zhou, Y. Yang, A. Y. Khodakov and V. V. Ordomsky, ACS Catal., 2019, 9, 2940–2948 CrossRef CAS.
- M. Bakos, Á. Gyömöre, A. Domján and T. Soós, Angew. Chem., Int. Ed., 2017, 56, 5217–5221 CrossRef CAS PubMed.
- I. P. Jakopović, S. Kapić, S. Alihodžić and V. Šunjić, ARKIVOC, 2015, 300–326 Search PubMed.
- G. R. Pettit and D. M. Piatak, J. Org. Chem., 1962, 27, 2127–2130 CrossRef CAS.
- V. A. Moeller, Encycl. Reagents Org. Syn., John Wiley & Sons, Ltd., New York, 2001.
- A. Y. Vigorov, I. A. Nizova, L. S. Sadretdinova, M. A. Ezhikova, M. I. Kodess, I. N. Ganebnykh and V. P. Krasnov, Eur. J. Org. Chem., 2011, 2562–2569 CrossRef CAS.
- D. J. Kopecky and S. D. Rychnovsky, J. Org. Chem., 2000, 65, 191–198 CrossRef CAS PubMed.
- A. Hart, S. A. Kelley, T. Harless, J. A. Hood, M. Tagert and J. A. Pigza, Tetrahedron Lett., 2017, 58, 3024–3027 CrossRef CAS.
- D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004–6011 CrossRef CAS PubMed.
- K. Matsubara, T. Iura, T. Maki and H. Nagashima, J. Org. Chem., 2002, 67, 4985–4988 CrossRef CAS PubMed.
- O. Martínez-Ferraté, B. Chatterjee, C. Werlé and W. Leitner, Catal. Sci. Technol., 2019, 9, 6370–6378 RSC.
Footnotes |
| † First joint authors. |
| ‡ Second authors. |
| This journal is © The Royal Society of Chemistry 2022 |