
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting the grammar of Tau aggregation and pathology formation: how new insights from brain pathology are shaping how we study and target Tauopathies
Galina
Limorenko†
 and
Hilal A.
Lashuel†
*
and
Hilal A.
Lashuel†
*
Laboratory of Molecular and Chemical Biology of Neurodegeneration, Brain Mind Institute, École Polytechnique Federal de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: hilal.lashuel@epfl.ch
First published on 10th December 2021
Abstract
Converging evidence continues to point towards Tau aggregation and pathology formation as central events in the pathogenesis of Alzheimer's disease and other Tauopathies. Despite significant advances in understanding the morphological and structural properties of Tau fibrils, many fundamental questions remain about what causes Tau to aggregate in the first place. The exact roles of cofactors, Tau post-translational modifications, and Tau interactome in regulating Tau aggregation, pathology formation, and toxicity remain unknown. Recent studies have put the spotlight on the wide gap between the complexity of Tau structures, aggregation, and pathology formation in the brain and the simplicity of experimental approaches used for modeling these processes in research laboratories. Embracing and deconstructing this complexity is an essential first step to understanding the role of Tau in health and disease. To help deconstruct this complexity and understand its implication for the development of effective Tau targeting diagnostics and therapies, we firstly review how our understanding of Tau aggregation and pathology formation has evolved over the past few decades. Secondly, we present an analysis of new findings and insights from recent studies illustrating the biochemical, structural, and functional heterogeneity of Tau aggregates. Thirdly, we discuss the importance of adopting new experimental approaches that embrace the complexity of Tau aggregation and pathology as an important first step towards developing mechanism- and structure-based therapies that account for the pathological and clinical heterogeneity of Alzheimer's disease and Tauopathies. We believe that this is essential to develop effective diagnostics and therapies to treat these devastating diseases.
 Galina Limorenko | Galina Limorenko received her BSc Degree in Genetics from Cardiff University (2018) and is currently in doctoral training for her PhD in Neuroscience in the Lashuel Laboratory at the École Polytechnique Fédérale de Lausanne. Her current research interests include elucidating the biochemical and molecular processes underlying Tau aggregation and toxicity in Alzheimer's disease and Tauopathies and developing novel methods and bioassays that recapitulate Tau pathology formation in the brain. She hosts the New Books Network podcast in Science and Technology. |
 Hilal A. Lashuel | Dr Hilal Lashuel received his BSc degree in chemistry from Brooklyn College (1994) and completed his doctoral studies at Texas A&M University and the Scripps Research Institute (2000). He joined Harvard Medical School and the Brigham and Women's Hospital (2001) as a research fellow in the Center for Neurologic Diseases. He was later promoted to an instructor in neurology at Harvard Medical School. In 2005, he joined the Brain Mind Institute at the École Polytechnique Fédérale de Lausanne where he now directs the laboratory of molecular and chemical biology of neurodegeneration. He is founder and CSO of ND BioSciences SA. |
Introduction
The microtubule-binding protein Tau is an intrinsically disordered protein, which is expressed as six isoforms in the adult human central nervous system (Fig. 1A). In cells, Tau is most prominently associated with dynamic regulation and stabilization of cytoskeletal and mitotic microtubules (MTs) through direct binding to tubulin dimers,1 however other non-canonical functions of Tau have been proposed2 (Fig. 1B). In neurons, Tau is important for regulating axon outgrowth and maintenance of axonal cytoskeletal integrity and transport.3 However, factors such as mutations,4 post-translational modifications (PTMs),5 interaction with other molecules6,7 (Fig. 1C), and changes to the biochemical composition of surrounding milieu (e.g. pH, drug compounds)8 may result in weaker interaction between Tau and its natural partners or its dissociation from MTs (reviewed in ref. 9). This is thought to lead to Tau accumulation, which creates conditions that favor its aggregation and formation of the β-sheet rich fibrillar aggregates found in the brains of individuals with Alzheimer's disease (AD) and other neurodegenerative diseases (NDDs). Hyperphosphorylated and fibrillar Tau is found in the form of paired helical filaments (PHFs) and straight filaments (SFs) in cytoplasmic neurofibrillary tangles (NFTs) and dystrophic neurites, which represents the main hallmarks of Alzheimer's disease (AD), in addition to amyloid plaques. Tau aggregates, both fibrils, and oligomers are also found in the brain of individuals that suffered from other neurodegenerative diseases (NDs), which include Pick's disease (PiD) and progressive supranuclear palsy (PSP), collectively known as Tauopathies.10–17 Several structures of brain-derived Tau fibrils have been recently solved using cryo-electron microscopy (cryo-EM), and the list currently includes Tau fibril folds structures from AD, Pick's disease (PiD), chronic traumatic encephalopathy (CTE), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), argyrophilic grain disease (AGD), primary age-related tauopathy (PART), familial British dementia (FBD), familial Danish dementia (FDD), globular glial tauopathy (GGT), and author-coined limbic-predominant neuronal inclusion body 4R tauopathy’ (LNT)18 (for recent reviews see ref. 19–21).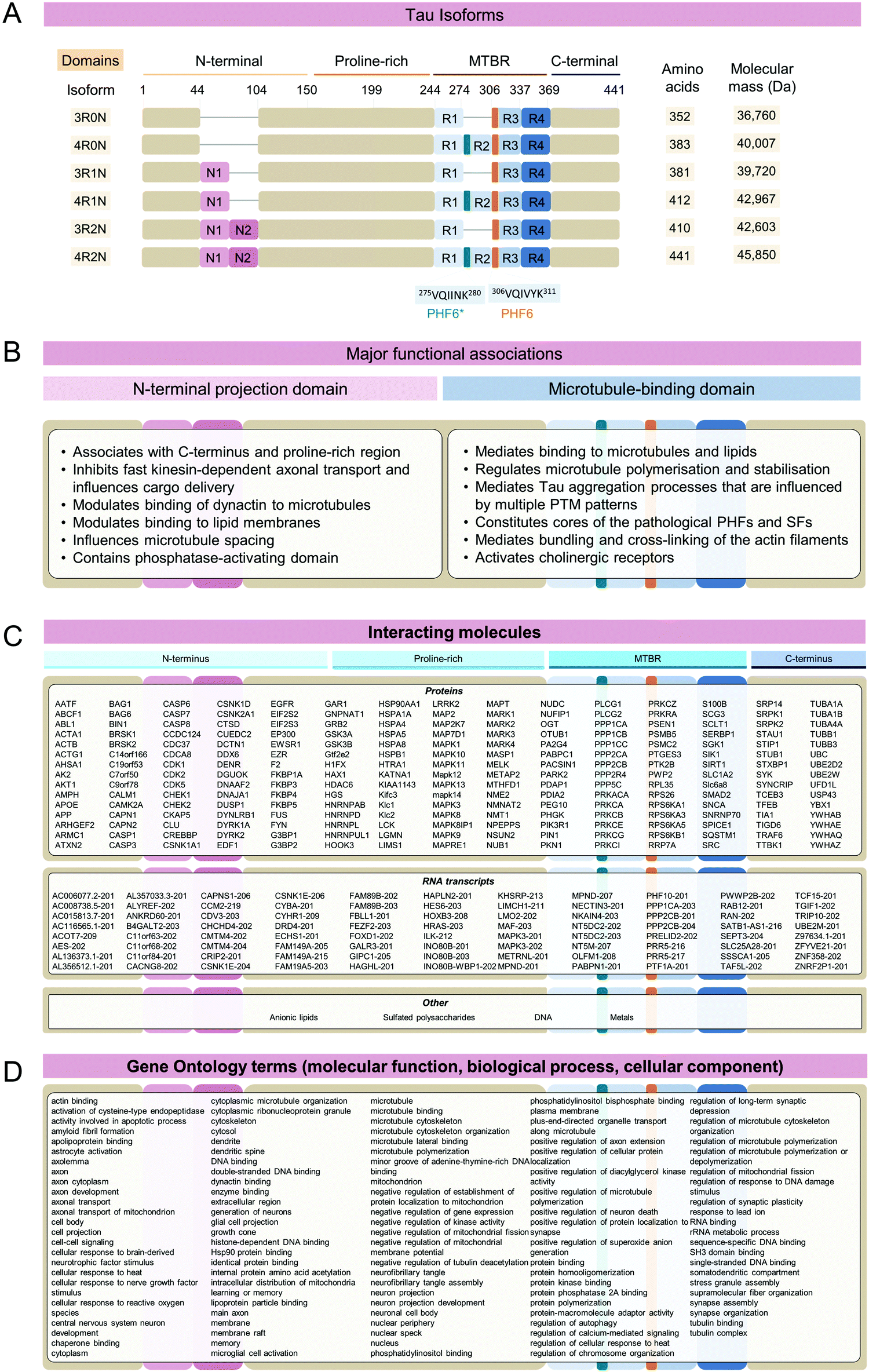 | ||
| Fig. 1 Tau sequences, functions, and interactome. (A) In the adult human central nervous system, six Tau isoforms are highly expressed. Functional domains contain N-terminal inserts (N1, N2), a proline-rich region, four microtubule-binding repeat domains (R1 aa 244–274; R2 aa 275–305; R3 aa 306–336; R4 aa 337–368) and C-terminus. Amino acid numbering is based on the sequence of the full-length human 4R2N Tau amino acids 1-441. (B) Major functions attributed to the Tau N-terminal projection domain, which include the N-terminal and first part of mid-domain, and microtubule regulation domain that includes the second part of mid-domain, MTBR, and remaining C-terminal residues. (C) A partial list of known Tau interacting proteins. Protein binding partners for Tau 4R2N were established using searches through IntAct22 and BioGRID databases23 filtering for unique direct interactors verified experimentally. Notably, a specific binding region on Tau is established only for a tiny fraction of all interactors. Tau interacting RNA transcripts were predicted using RNAct database.24 Other types of Tau-interacting molecule classes include DNA, lipids, metals, and polysaccharides (IntAct22). (D) Gene Ontology25 terms associated with Tau query. Tau is associated with multiple molecular functions, biological processes, and cellular components. | ||
Increasing evidence points to Tau aggregation (oligomerization and fibril formation) and PTMs as central events in the pathogenesis of AD and Tauopathies, which made Tau an appealing target for drug discovery and development.26–28 However, the molecular and cellular factors that trigger Tau misfolding and aggregation and drive the spreading of Tau pathology in the brain remain unknown. Furthermore, the functional significance of Tau aggregation is still unclear. Whether it is a pathogenic gain-of-function (GOF) linked to the formation of cytotoxic Tau species or represents a neuroprotective process that sequesters soluble toxic forms of Tau, or a balance of both, remains to be elucidated. These processes are also accompanied by the loss of physiological function(s) (LOF) due to the depletion of soluble and functional Tau proteins. It is likely that both GOF and LOF mechanisms contribute to the development and progression of Tauopathies. However, the relative contributions of each to the various stages of disease development remain unknown.
Crucially, which forms of Tau are the primary initiators of these processes and how PTMs influence the course of Tau aggregation and pathology formation and spreading remain subjects of intense investigation and debate. In this review article, we will (1) review how our understanding of Tau aggregation and pathology formation has evolved over the past few decades; (2) present analysis of new findings and insights from recent studies illustrating the biochemical, structural and functional heterogeneity of Tau aggregates; and (3) discuss the importance of adopting new experimental approaches that embrace the complexity of Tau aggregation and pathology as an important first step towards developing mechanism- and structure-based therapies that account for the pathological and clinical heterogeneity of Alzheimer's disease and Tauopathies. We believe that this is essential to develop effective diagnostics and therapies to treat these devastating diseases.
Tau protein expression
Six major Tau isoforms are found in the central nervous system that differs based on the inclusion and exclusion of the exons 2, 3, and 10 of the MAPT gene, resulting in the Tau protein isoforms containing 0, 1, or 2 N-terminal segments, and differential exclusion of the second MT-binding repeat29 (Fig. 1A). On the level of protein expression, Tau is most abundant in the frontal and occipital cortices, followed by white matter, whereas its levels are significantly reduced in putamen and cerebellum in concordance with gene expression data.30 The protein levels or distribution of 3R-Tau and 4R-Tau isoforms are approximately equal across brain regions, with 1N-Tau isoforms accounting for about 50%, 0N-Tau isoforms for ∼40%, and 2N-Tau isoforms for ∼10% of total Tau.30 However, in neurodegenerative diseases, the ratio of Tau protein isoforms containing 3 or 4 MT-binding repeats is perturbed.31–36Cellular processes
The Tau proteins have been implicated in multiple cellular processes (Fig. 1D). It was long thought that the primary function of Tau was to stabilize MTs. Still, more recently, the focus shifted to its abilities to regulate the MT dynamics, more so than to stabilize them37 (reviewed in ref. 38). Tau is involved in the insulin signaling pathway by inhibiting phosphatase and tensin homolog (PTEN)39, nuclear functions such as stability of heterochromatin,2 and synaptic processes such as long-term depression40,41 and potentiation42 (reviewed in ref. 43). Depletion of functional Tau leads to impairments of the processes related to the subset of highly cycling and dividing brain cells, such as hippocampally-positioned neuronal precursors, involved in neurogenesis44–47 and glial precursors to neuronal cells,48,49 as well as incomplete neuronal cell-cycle re-entry.50 This contributed to the neurodegeneration that underlies impairment in memory, increased anxiety, and reduced learning ability.51,52 Recent studies have also shown that Tau proteins are secreted and propagated from one cell to another. Although this process has been proposed to play central roles in the spreading of Tau aggregates, whether extracellular Tau also plays a physiological role in the brain remains unknown.What happens upon complete depletion of Tau in neurons? Interestingly, complete mouse Tau knockout mice that do not express any Tau isoforms showed variable pathological phenotypes at an advanced age, manifesting in cognitive and motor deficits, which varied according to the genetic background of mice.53 Another commonly used mouse line is hTau.54 It is hemizygous for overexpression of all six human Tau isoforms on mouse Tau knockout background, and the litters result in the expected half of the mice to express human Tau only, and the other half to be complete mouse and human Tau knockout, providing the isogenic experimental controls. Tau proteins are ubiquitously expressed throughout tissues, and function in processes related to cell division and dynamic cytoskeleton reorganization. Consistent with this, hTau mice showed peripheral deficits in functions related to the highly cell-cycling, dividing, and metabolically-active secreting pancreatic cells in the Tau knockouts, and overexpression of human Tau in islet cells did not rescue the deficits in glucose homeostasis.55,56 Also, impairment in memory, increased anxiety, and reduced learning ability were observed in these mice, which could not be rescued by the expression of human Tau isoforms.56 Incomplete cell-cycle re-entry (CCR) of post-mitotic neurons was also observed in hTau mice in ref. 50, suggesting that loss of physiological Tau could contribute to neurodegeneration via mechanisms independent of Tau aggregation (LOF) and formation of NFTs. Consistent with this hypothesis, aberrant CCR was implicated in the processes of neuronal loss in AD patients,51,52 and in mouse models,57,58 and was found to be independent of the Aβ plaque or NFT formation. Further, the ectopic cell CCR was found to require soluble Aβ oligomers-mediated activation of Tau kinases Fyn, PKA, and CCKII, which phosphorylated Tau at positions pY18, pS409, and pS416, respectively.59 Tau knockout neurons did not enter the CCR despite the activation of kinases. However, the expression of Tau restored the CCR, in contrast to the phosphorylation-resistant Tau mutants which failed to restore it.59
In summary, depletion of Tau may lead to impairments of the processes related to the subset of highly cycling and dividing brain cells, such as hippocampally-positioned neuronal precursors, involved in neurogenesis44,45 and glial precursors to neuronal cells.48,49
One way to gain insight into the functions of Tau in health and disease is through understanding its interactome network. Tau is involved in multiple interactions with a wide range of molecules, with different domains implicated in regulating these interactions (Fig. 1B). These include proteins, nucleic acids, lipids, and polysaccharides, many of which have been shown to regulate many aspects of Tau functions under physiological and/or pathological conditions60–62 (IntAct database,22 BIOGrid database,23 RNAct database24) (Fig. 1C). Tau interacts with more than 200 proteins (Fig. 1C, proteins). This versatility and broadness of Tau interactions with proteins are highly complex and it differs between healthy and pathological human conditions.63 Experimental approaches such as neuroproteomics,25 laser capture microdissection coupled with mass spectrometry,64,65 and interactome studies have been instrumental for the interrogation of Tau interactions with other molecules,63,66,67 and in organoid68 and mouse models.69 Tau was found to interact with deoxyribonucleic acid (DNA)70 and ribonucleic acid (RNA),71 strongly suggesting that it has important regulatory roles in the nucleus,2,72 such as wide-scale chromatin reorganization, detected in AD brain,73 as well as through post-transcriptional regulation of multiple gene transcripts (Fig. 1C, RNA transcripts).
Tau interactions with other molecules are often heavily influenced and modulated by the extensive Tau PTMs.74 One of the most prominent PTM-regulated Tau interactions is with tubulin, the subunits composing MTs. Hyperphosphorylation in the MT-binding domain (MTBR) decreases Tau affinity for MTs and leads to an increased rate of its dissociation. This may lead to a higher rate of Tau self-association conducive to aggregation.75 Further functional validations of the Tau interacting partners are necessary to understand the significance and extent of the molecular functions of Tau beyond its canonical association with the tubulin.
Tau contribution to neurodegenerative disorders
The presence and spreading of Tau-positive detection of pathology in the brain of normally-aging individuals is well-known, and is not in itself considered to be a functional Tau pathology, and may not be associated with cognitive impairment, however, could indicate the incipient pathology.76–79 In older adults, Tau accumulates predominantly in medial temporal lobes and is concurrent with low cognitive deficits that accompany the normal aging processes.80,81 Regional brain vulnerability to Tau pathology is well-known in humans and is thought to vary depending on the different Tauopathies, for example in AD the pathology tends to follow the “subcortical to cortical” spreading pattern affecting the cortex at the later stages, whereas in PSP and CBD pathology is confined to the subcortical regions, with later stages affecting basal ganglia and cerebellum.82–84 Entorhinal, temporal isocortices, and hippocampal formations are highly susceptible to Tau aggregate formation and neurodegeneration.82,85–96 Brain circuits between these areas are anatomically connected and are implicated in memory formation, consolidation, and cognition. The aggregation of Tau observed in these neocortical regions temporally follows the deposition of amyloid-β, and correlates with the loss of cognitive abilities in Alzheimer's patients.97,98 Non-neuronal and glial cell populations can also contribute to Tau-mediated pathology formation in mouse models,99,100 and microglia-expressed triggering receptor expressed on myeloid cells 2 (TREM2) gene variants contribute to the increased AD risk in humans.101 Tau pathology is thought to proceed through the primary mechanism of direct Tau aggregation in the cell cycle-arrested non-neuronal oligodendrocyte102 and astrocytic cells,103 through secondary pathology events such as gliosis and neuroinflammation via cytokine excretion,104,105 or glia-mediated Tau spreading.106Therefore, it is important to delineate the conditions which flip the switch of physiological-to-pathological Tau aggregation in the brain. These may include (1) regional vulnerability of the brain circuits to Tau aggregation, (2) neuronal and non-neuronal cell type vulnerability, such as increased susceptibility of the excitatory presynaptic neurons,107 (3) emergence of particularly toxic Tau protein subpopulations due to PTMs or associations with cofactor molecules or other amyloidogenic proteins, such as amyloid-β,108 (4) local changes in the brain and cerebrospinal fluid (CSF) acidification levels,109,110 as well as (5) subcellular mislocalization of Tau protein from axonal to somatodendritic compartments111 that may contribute to Tau displacement from microtubules in cells112 and subsequent aggregation. It is yet not fully understood which mechanisms contribute substantially to the inception and progression of human Tauopathies.
In terms of the clinical manifestations of Tau pathologies, loss of cognition coupled with the regional brain Tau NFT distribution detected by live positron emission tomography (PET) imaging may indicate the specific Tau-associated disease. These tests, combined with the post-mortem histochemical stainings and the biochemical properties of the isolated Tau, form the basis of the classification of the Tauopathy disorders, and their stages of progression.113 The Tauopathies with the known biochemical Tau profile are classified into predominantly 4R-Tauopathies containing the 4R Tau isoforms in the insoluble protein brain fractions, such as PSP, CBD, and AGD, as well as gliopathies ARTAG and GGT. Predominantly-3R Tauopathies include the neuronopathy and gliopathy PiD and neuronopathy the behavioural variant of frontotemporal dementia (bvFTD).113 The Tauopathies defined by the presence of all six Tau isoforms include AD, CTE, PART, tangle-only dementia (TOD), dementia with Lewy bodies (DLB), and primary progressive aphasia (PPA) (Table 1). Also, co-occurrence of Tau pathology may be observed in the late stages of other proteinopathies, such as Huntington's disease,114 synucleinopathies,115 and amyotrophic lateral sclerosis (ALS).116
| 3R + 4R | 4R |
|---|---|
| Alzheimer's disease (AD) | Age-related Tau astrogliopathy (ARTAG) |
| Amyotrophic lateral sclerosis/parkinsonism-dementia complex (ALS) | Argyrophilic grain disease (AGD) |
| Anti-IgLON5-related Tauopathy | Corticobasal degeneration (CBD) |
| Chronic traumatic encephalopathy (CTE) | Guadeloupean parkinsonism |
| Diffuse neurofibrillary tangles with calcification | Globular glial Tauopathy (GGT) |
| Down's syndrome | Hippocampal Tauopathy |
| Familial British dementia (FBD) | Huntington's disease |
| Familial Danish dementia (FDD) | Progressive supranuclear palsy (PSP) |
| Gerstmann–Sträussler–Scheinker disease (GSS) | Familial frontotemporal dementia and parkinsonism (FTDP, mutations P301S, intronic mutations, coding region mutations in exon 10) |
| Niemann–Pick disease, type C | |
| Nodding syndrome | |
| Non-Guamanian motor neuron disease with neurofibrillary tangles | 3R |
| Postencephalitic parkinsonism | Pick's disease (PiD) |
| Primary age-related tauopathy (PART) | Familial frontotemporal dementia and parkinsonism (FTDP, mutations G272V and Q336R) |
| Progressive ataxia and palatal tremor | |
| SLC9A6-related parkinsonism | |
| Tangle-only dementia (TOD) | |
| Familial frontotemporal dementia and parkinsonism (FTDP, mutations V337M and R406W) |
Following the findings that cognitive decline correlated with Tau pathology progression in patients117 to a better extent than amyloid β pathology, the levels of Tau or phosphorylated Tau in CSF,118 as well as Tau PET imaging119 in the brain have been studied as early biomarkers. This is especially prescient due to fact that the sporadic and secondary Tauopathies, such as AD, are not associated with specific mutations in the Tau-coding MAPT gene and therefore elude early screening. This is in contrast to familial Tauopathies (Table 1), such as autosomal dominant frontotemporal dementia and parkinsonism (FTDP) linked to chromosome 17q21,120 or Tauopathies with a clear underlying genetic component, such as Pick's.121 Therefore, early detection of incipient pathological Tau processes as diagnostics biomarkers, as well as therapeutic targets, has recently garnered much attention.
Spreading of Tau pathology
At the brain level, in AD the Tau pathology spreading follows the sequence of pathology progression (Fig. 2A), starting from entorhinal and transentorhinal cortices at stage I, progressing to hippocampal in stage II and transentorhinal cortical regions in stage III, followed by the cerebral and visual cortices in stage IV and later stages V and VI.82,122 Evidence suggests that Tau pathology in AD is transmitted along the anatomically connected brain regions (reviewed in ref. 123). Computationally-aided integration of imaging and biochemical data would allow for reconstructing the AD connectome, which, combined with computer modeling approaches, have the potential to provide higher resolution and granularity to our understanding of stages of tauopathy progression in the human brain.124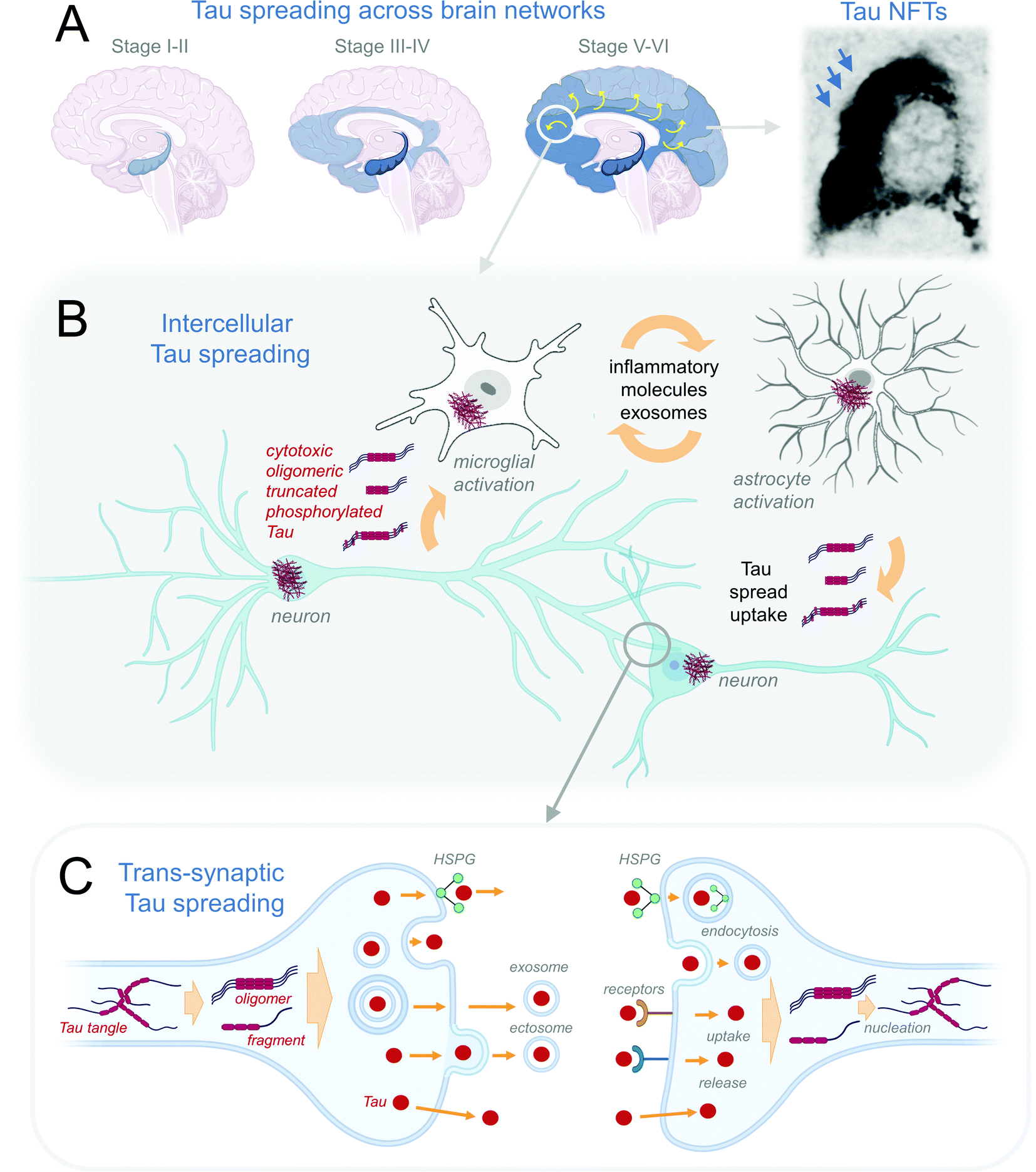 | ||
| Fig. 2 Mechanisms of Tau spreading. (A) Schematic representation of Braak stages of Tau pathology across brain networks as detected by immunohistochemistry of NFTs (reproduced from ref. 125 with permission from the Elsevier, copyright 1991). (B) Intercellular Tau pathology spreading between neuronal and neuronal, and neuronal and glial cells. (C) Trans-synaptic spreading of different pathogenic Tau proteoforms is thought to occur through their transport across membrane release, exocytosis, endosome-, exosome-, HSPG-mediated release from the affected donor cell. The uptake of putative pathological Tau species is thought to be mediated by HSPG-, receptor-coupled mechanisms, endocytosis, and direct transport across the plasma membrane. The recipient cell may develop further Tau pathology through nucleation and seeding of the endogenous Tau. | ||
On a cellular level, Tau pathology is thought to spread from one cell to the next through Tau proteoform release from the donor cell via multiple potential pathways, such as the direct transport across membrane, exocytosis, endosome-, exosome-, and heparan sulfate proteoglycan (HSPG)-mediated release (Fig. 2B). A further transmission of Tau across synaptic cleft is thought to be followed by Tau uptake by recipient cell via direct transport, endocytosis, or receptor-, or HSPG-mediated uptake. In the recipient neurons Tau is thought to nucleate and initiate recruitment and aggregation of endogeneous Tau (Fig. 2C), first affecting the distal, followed by proximal dendrites, then cell soma, eventually reaching the axonal compartment.126 It has been suggested that the spread of toxic Tau species underlying the pathologies may be mediated by one or a combination of (a) a direct prion-like spreading mechanism of misfolded Tau across the cellular membrane with the recruitment of normal Tau in the receiving cell; (b) exosome-mediated and (c) trans-synaptic spreading of toxic Tau species (reviewed in ref. 127 and 128) (Fig. 2A). Thus far, however, the evidence for the prion-like Tau propagation is only circumstantial in humans, with most work to elaborate this hypothesis done in cellular and animal models (see recent reviews26,129,130). Which forms of Tau are secreted and propagated in the human brain remains unclear, although studies have identified several truncated131,132 and post-translationally modified forms of the protein133,134 in CSF and blood of Tauopathy patients.
Composition of neurofibrillary tangles
Besides the Tau proteins in fibrillized conformation, NFTs are composed of multiple other molecules, including proteins, carbohydrates, nucleic acids, and lipids. Formation and diversity of molecular composition of NFTs are established by (1) the molecules that drive the primary formation of NFTs, such as mutant proteins, or proteins in pathological conformations, such as fibrillar Tau, (2) recruitment of other molecules and cellular components through direct binding or diffusion, and (3) accumulation of cellular waste products following cellular impairment and degeneration.Although the exact molecular composition of pathological Tau aggregates remains unknown, several studies have highlighted their complexity and diverse composition. Cytoskeletal proteins such as MAP1B,135 MAP2,136 neurofilament, and vimentin137 were detected by immunohistochemistry in AD-derived NFTs early on. Further, in AD, extracellular NFTs contained amyloid-β, amyloid-P, extracellular signal-related kinase-2, glial fibrillary acidic protein (GFAP), and ubiquitin, whereas intracellular NFTs contained apolipoprotein E (Apo E), basic fibroblast growth factor (bFGF), GFAP, heparan sulfate proteoglycan (HSPG), complement membrane attack complex C5b-9, neurofilament, synaptophysin, and ubiquitin. Other NFT-associated molecules included casein kinase II (CKII), glycogen synthase kinase-3 (GSK3), phospholipase C-δ, malondialdehyde, heme oxygenase-1, vitronectin, gangliosides C, anti-thrombin III, and lactotransferrin. Pick's bodies were shown to contain Apo E, advanced glycation end-products (AGEs), bFGF, chromogranin B, complements C1, C1q, C4, C2, C3, C5, C6 and C8, clathrin, membrane complement inhibitor CD59, clusterin, GFAP, synaptophysin, and ubiquitin (reviewed in ref. 138). NFTs in PSP contained enriched GFAP and ubiquitin, whereas CBD inclusions contained GFAP and marker for killer lymphocytes Leu-7 (reviewed in ref. 139). The proteomic profiling of NFTs using laser capture microdissection coupled to mass spectrometry revealed peptides derived from 542 proteins.140 However, the list of these 542 proteins is not openly available. 41 polypeptides were identified in AD NFTs by mass spectrometry and included GAPDH and ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) that colocalized with NFTs by immunostaining.141 Another study identified 72 proteins within the NFTs, with biochemically-verified glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a major constituent.142 S-glutathionylated GAPDH form was detected in the blood of Alzheimer's patients and is a pro-apoptotic molecule that was not present in blood under healthy conditions.143
Given such complexity of NFTs and the implications of their biochemical diversity for targeting and imaging pathological Tau, it is crucial to revisit Tau pathology with more systematic approaches to dissect the molecular compositions of intracellular, extracellular, and ghost tangle NFTs. A better understanding of the proteomic compositions of individual NFTs could provide insight into biochemical changes associated with the formation and maturation of Tau pathology. The knowledge gained from these studies, combined with the structural insight gained from cryo-EM could guide future efforts to develop diagnostic and therapeutic strategies that target specific Tau pathologies or capture their diversity in the brain. Furthermore, a better understanding of the molecular components of pathological Tau aggregates could also provide novel insight into natural cofactors responsible for triggering Tau pathology formation in different Tauopathies.
Tau aggregation
Under pathological conditions, Tau is observed in the aggregated state in the surviving cells of patients with Tauopathies (Fig. 3A). Therefore, there is considerable interest to understand the conditions and molecular processes contributing to Tau protein aggregation. In solution, all Tau isoforms are soluble and exist as an ensemble of disordered conformations, but are capable of acquiring secondary structure upon association with MTs, dimeric tubulin,144 or lipid membranes.145 The solubility of Tau proteins could be attributed to the high positive charge density surrounding the aggregation-prone microtubule-binding region, as well as long-range N-terminal interactions with the mid-domain and C-terminal domain. To aggregate and form β-sheet-rich fibrillar structures – Tau proteins must undergo conformational changes or interaction with other molecules that alter the charge distribution along the Tau molecule, leading to the formation of aggregation-competent intermediates (Fig. 3B). This can be mediated and influenced by negatively-charged cofactors, such as heparin;146 changes in the environmental solvent conditions like the presence of osmolyte urea or trimethylamine N-oxide;147 fluctuations of the proton density gradient pH;148 the introduction of specific patterns of post-translational modifications,149,150 (reviewed in ref. 151), or other yet to be identified factors.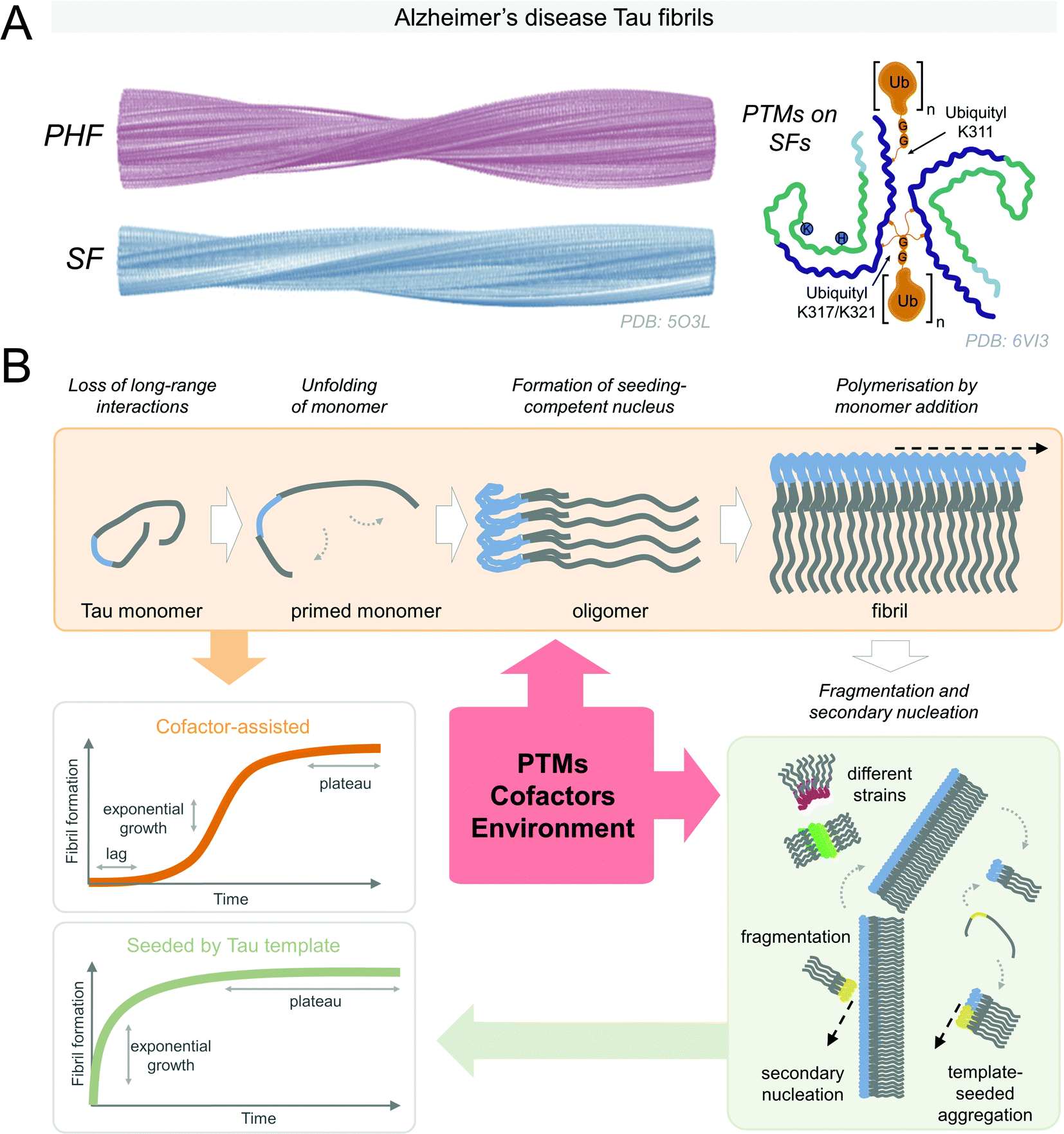 | ||
| Fig. 3 (A) Alzheimer's disease derived-Tau fibrils in the paired helical filament (PHF) and straight filament (SF) structures (reproduced with modifications from ref. 152 with permission from the Macmillan Publishers Limited, part of Springer Nature, copyright 2017). SFs may be post-translationally modified (reproduced with modifications from ref. 153 with permission from the Elsevier, copyright 2020). (B) Tau aggregation is thought to start by the loss of the long-range contacts on the soluble Tau monomers in semi-stable paperclip conformation. The formation of a seeding competent nucleus is necessary for the fibrillization to proceed. Secondary nucleation events and fragmentation of fibrils contribute to further Tau fibrillization that may result in Tau aggregates in various structural conformations. PTMs, cofactors, and environmental conditions can influence Tau fibrillization. Kinetics of cofactor-assisted Tau fibrillization generally follows the S-shaped curve kinetics, seeded Tau fibrillization follows exponential curve kinetics with the loss of the lag phase. | ||
Biophysical concepts applied to Tau aggregation
Tau is thought to aggregate through a nucleation-dependent mechanism, surpassing the kinetic barrier by forming a seeding where monomeric Tau self-assembles to form and aggregation-competent nucleus (lag phase) that undergoes rapid elongation through templated addition of monomers to to form fibrils154 (see Fig. 3B). The priming of soluble semi-stable monomers existing in a paperclip conformation155 into partially misfolded conformation is necessary for the formation of the seeding-competent Tau nucleus. The lag phase can be overcome upon the addition of the preformed fibrillar Tau seed or cofactor molecules. ΔK280, P301L, V337M, and R406W Tau mutants, found in FTDP-17 patients, were found to have higher aggregation propensity into paired helical filaments than wild type Tau156. Both Tau mutations detected in familial (P364S), and sporadic (G366R) FTDP-17 reduced MT polymerization activity of Tau, however, interestingly, only the protein with mutation detected in the sporadic case had higher aggregation propensity in vitro.157 Specific mutations associated with FTDP-17 on the sites P301 and S320 were highly dependent on the residue substitutions in their propensity to be seeded by exogeneous preformed fibrils, or facilitate aggregation in cells.158 Only the threonine substitution of proline 301 of 4R0N Tau resulted in an increase in the detergent-insoluble Tau upon overexpression in HEK293T cells, followed by K18 fibril seeding, whereas overexpressed mutants G303V, G304S, or S305N Tau remained in soluble fractions. Double mutants P301S/S320F and P301L/S320F, however, exhibited high propensity to aggregate even without the addition of the preformed seed. These mutations likely work through destabilizing the semi-stable paperclip conformation of the Tau monomer,155 leading to the formation of highly aggregation-prone intermediates, however, the exact mechanisms are unknown.Understanding of biologically relevant Tau protein aggregation is increasingly informed by the concepts commonly used in polymer science fields. Solubility and assembly properties of monomers under supersaturated conditions define the assembly characteristics, such as labile conditions favoring stochastic nuclei formation, or metastable phase, where nuclei turn into elongated structures is favored.159,160 Conversion of Tau monomers must proceed through misfolded aggregation-competent conformations to form the aggregation-competent nuclei upon transient association. Formation of the first aggregation-competent oligomers is considered the rate-limiting step in the aggregation process. This process could be induced or promoted by mutations, cellular stress factors, e.g., oxidation stress, PTMs, or interaction with aggregation promoting cofactors or changes in the cellular environment leading to disruption of the long-range intramolecular interactions that stabilize the Tau monomers. On the other hand, in “noisy” biological systems, stable Tau nuclei may also assemble into amorphous OFF-pathway aggregates, predominantly due to transient contacts of Tau molecules of heterogeneous conformations. Structural template-guided Tau fibril polymerization can be thermodynamically unfavorable in these cases, and these types of aggregates would not result in the formation of the fibrils even with cofactor molecules acting as catalysts to overcome the energetic nucleation barrier.161 This was observed for some meta-stable cofactor heparin–Tau complexes that did not polymerize, however they could be induced to form ON-pathway fibrillar structures in the presence of nucleation-competent Tau seed.162 Once the surfaces of the Tau conformational structural template are established, the assembly proceeds predominantly by end-addition of Tau monomers. This further illustrates the versatility and dynamical nature of structures that Tau is capable of adopting.
Determinants of Tau aggregation
In a recent study, Wesseling et al.133 provided the first unbiased qualitative and quantitative profiling of the diversity of the Tau proteoforms and how it changes during AD progression. Although this study revealed that the cumulative number of the detected PTMs was much less than predicted, it still further highlighted the complexity of the Tau PTM patterns in pathological samples with the identification of 55 phosphorylation, 17 ubiquitination, 19 acetylation, and 4 methylation sites. However, several other types of Tau PTMs, which have been detected in the brains of Tauopathy patients, were not investigated in this study, including nitration, oxidation, O-GlcNacylation, and glycation.
The high abundance of PTMs in pathological Tau aggregates, relative to soluble Tau, led to hypotheses implicating a central role of PTMs in triggering Tau aggregation and pathology formation. The patterns of Tau PTMs evolve and change during disease progression,133 however, the significance and relations to underlying pathology are not yet understood. It has become clear that the PTM occurrence patterns, cross-interactions, and spatial distribution contribute to the combinatorial Tau PTM code, functions of which are yet to be fully deciphered.168
Phosphorylation is one of the most common Tau PTMs and may occur on several of the 85 tyrosine, serine, and threonine residues throughout the longest Tau isoform sequence (Fig. 4, Phosphorylation). Physiologically, Tau phosphorylation patterns modulate Tau-MT interactions, generally decreasing the Tau binding affinity for tubulin subunits.169–172 However, specific sites, such as pT50 were found to promote tubulin assembly in vitro and in cells.173 Phosphorylation patterns differ between Tau derived from the brain and biological fluids of healthy individuals and neurodegenerative disease patients133,167,174,175 Early studies detected higher phosphorylation levels of Tau in AD patients’ brains by immunohistochemistry,176,177 as well as by nanoelectrospray mass spectrometry.178 Immunolabelling by AT8 antibody detects Tau phosphorylated at mid-domain residues pS199/pS202/pT205/pS208,179 which are present in all Tau isoforms (see Fig. 1) and is extensively used to assess and quantify the Tau pathology in humans and animal models. Phosphorylation at these sites is also used for histological Braak staging of Tau pathology,82 and the diagnosis of AD in living patients. Which phosphorylation sites drive Tau aggregation and the role of different phosphorylation sites or patterns in regulating different aspects of Tau aggregation, pathology formation, interactions with other contributing factors, and spreading in the brain remain subjects of active research and debate.180 Several strategies have been used to investigate the role of phosphorylation in regulating Tau functions in health and disease. However, many of the commonly used approaches have limitations that preclude deciphering the Tau phosphorylation and PTM code.
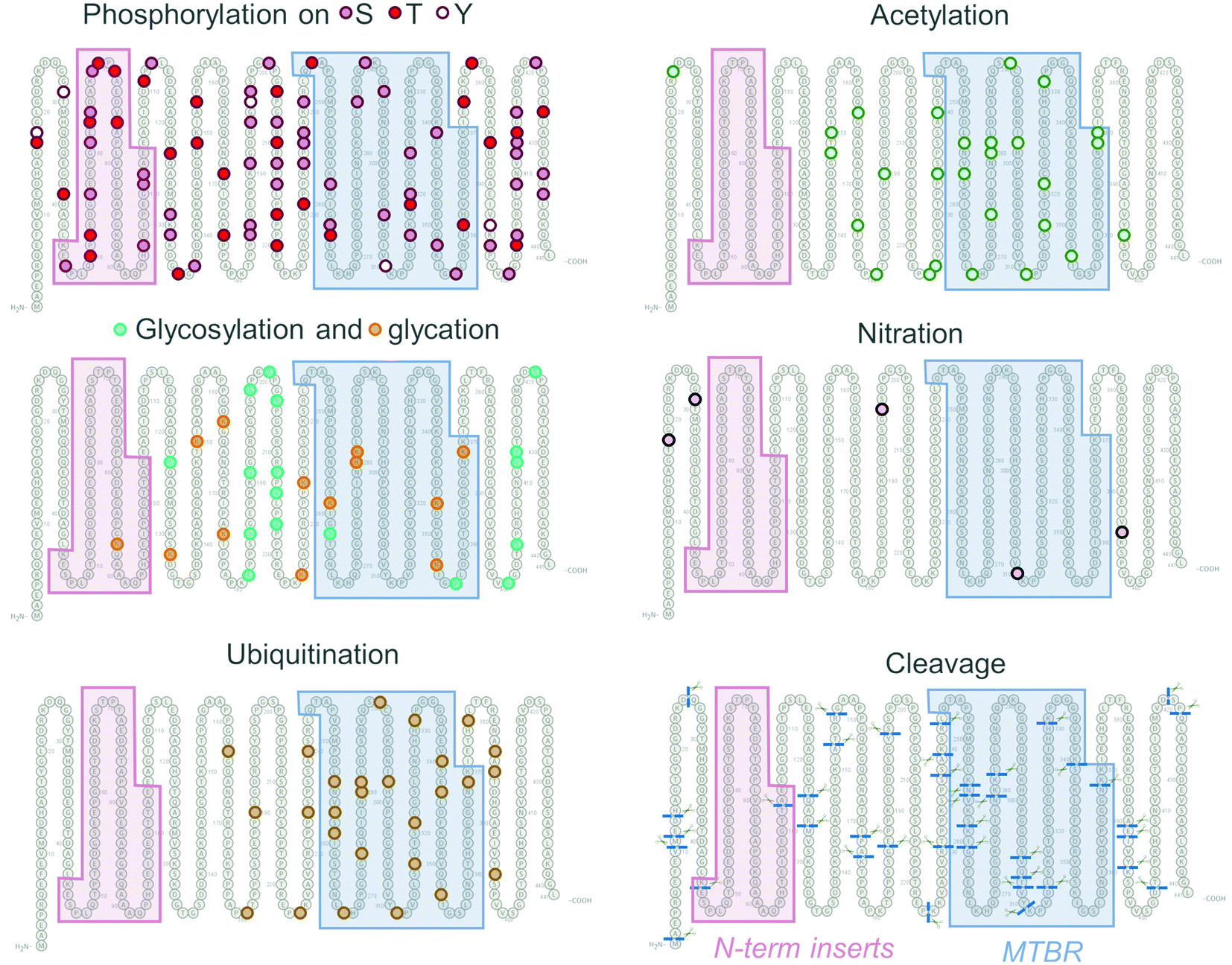 | ||
| Fig. 4 PTM patterns on Tau. Phosphorylation sites are distributed along the Tau molecule, whereas acetylation and ubiquitination sites are clustered around mid-domain and MTBR. Two cleavage sites, a single glycation site, and no acetylation, nitration, or ubiquitination sites are present in the N-terminal repeats. | ||
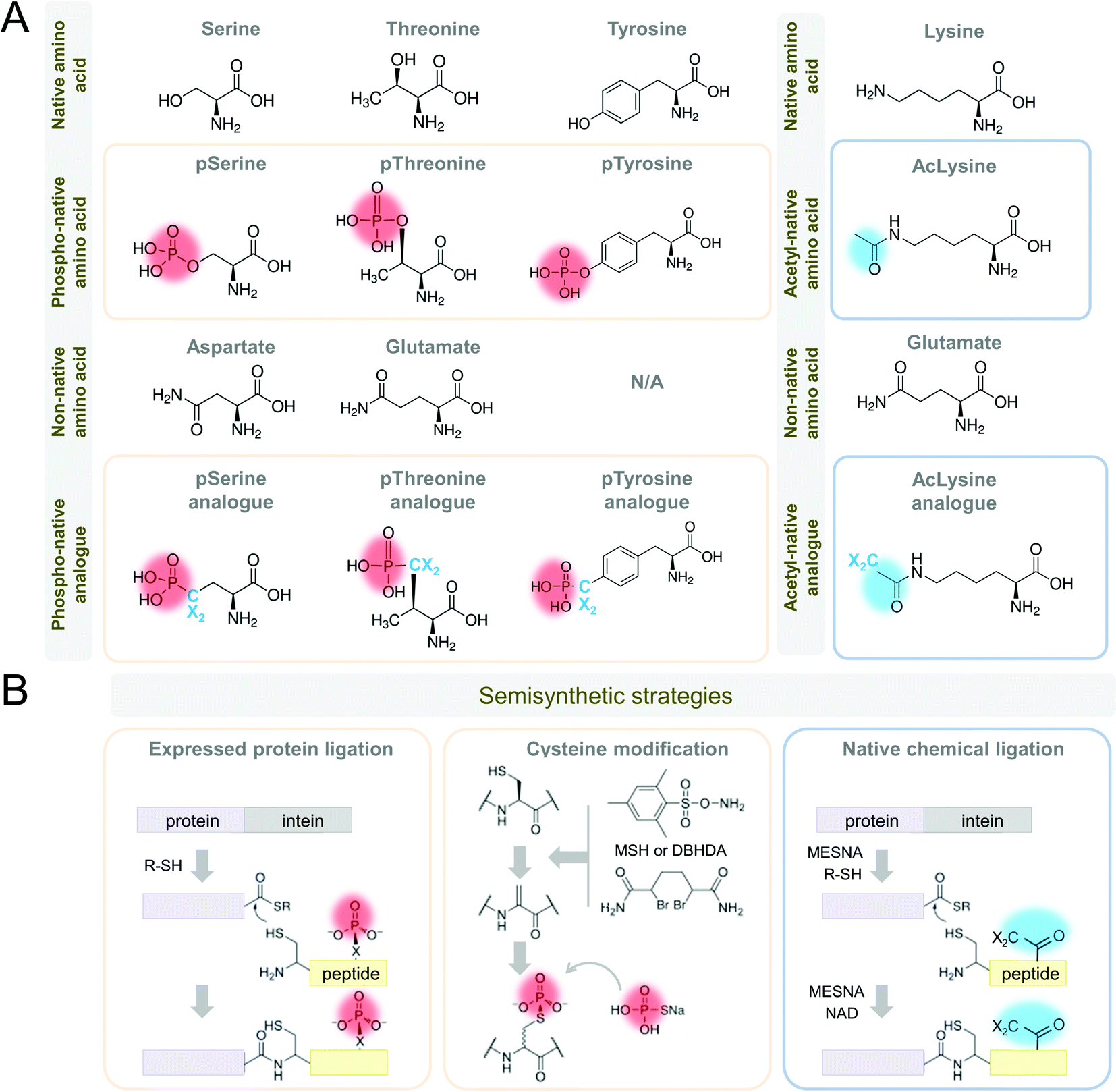 | ||
| Fig. 5 Strategies for in vitro mimics of phosphorylation and acetylation of Tau protein. (A) Amino acid substitutions for phosphorylated serine and threonine include aspartic and glutamic acid, as well as phospho-analog residues, including phospho-tyrosine. Amino acid substitutions for acetylated lysine include glutamic acid, as well as acetylation, mimics. (B) Semisynthetic strategies include protein ligation and cysteine modification (reproduced with modifications from ref. 181 with permission from the Elsevier Ltd, copyright 2015). Abbreviations: DBHDA: 2,5-dibromohexane diacetamide, MESNA: 2-mercaptoethanesulfonate, MSH: O-mesitylenesulfonylhydroxylamine, NAD: nicotinamide adenine dinucleotide, R-SH: thioester. Phospho-group is designated in red, acetyl group is designated in blue. | ||
One alternative and commonly used approach to phosphorylate Tau involves the use of kinases that have been shown to phosphorylate Tau in vitro or in cells. However, the enzymatic phosphorylation suffers from a lack of specificity or differential affinity of kinases for specific residues. Almost all the kinases that phosphorylate Tau do so at multiple residues. This can lead to variable rates of phospho-group addition and result in samples containing a mixture of differentially phosphorylated proteins. Furthermore, our knowledge of Tau kinases is still limited, and not all desired residues or their combinations may be efficiently phosphorylated in vitro. Previously, the lack of knowledge about the enzymes involved in regulating phosphorylation at specific sites precluded studies from assessing whether phosphomimetics accurately reproduce the effect of phosphorylation. However, recent advances in protein synthesis of Tau and other proteins now make this possible, therefore providing means for assessing the suitability of using phosphomimetics to investigate phosphorylation in vivo.
Alternative approaches for precise and clean in vitro Tau phosphorylation include combinations of enzymatic, synthetic, and semisynthetic strategies (Fig. 5B)186,187 that were successfully adapted to Tau.150,188–193 These strategies provide homogeneous site-specifically modified proteins, including phosphorylations, nitrations, and acetylations, in large quantities for experimental application in vitro, in vivo, and for drug screening and development. In addition to paving the way for deciphering the Tau PTM code with great precision, these approaches will allow reproducing the complexity of Tau species as they exist in vivo. This is essential for developing accurate assays to quantify and monitor changes in total Tau or specific modified Tau species.
The C-terminal domain appears to play a crucial role in modulating Tau aggregation propensity. Mimicking of phosphorylation at residues S396 and S404 by mutating serines to glutamines was found to increase Tau aggregation.198 A subsequent study by Haase et al.199 using a pseudophosphorylation mutation strategy of selected serine or threonine residues to aspartic or glutamic acids found that N-terminally positioned mutations inhibited, and C-terminally positioned mutations promoted Tau aggregation, particularly the pseudophosphorylation of residue 422. The enzymatic phosphorylation of C-terminal residues pS396, pS404, pS409 or pS422 alone, or combined with the mid-domain residues greatly enhanced Tau aggregation in vitro.172 In addition, FTDP-17 Tau mutations G272V, P301L, V337M, and R406W were found to increase the rate of Tau fibrillization in vitro when the C-terminal residues S396, S400, T403, and S404 were phosphorylated, compared to non-mutant protein, that also fibrillized when phosphorylated at these sites.171 Taken together, additions of negative-charge bulky phospho-group before aggregation onset to N-terminal domain or MTBR tend to decrease Tau aggregation; phosphorylations of C-terminal residues tend to increase Tau aggregation; whereas mid-domain phosphorylations tend to be highly context-dependent concerning the modulation of Tau aggregation.
Conversely, phosphorylation at S262 was previously associated with the inhibition of full-length200 or truncated Tau aggregation.150 Also, in a Tau overexpression cell system, the phosphorylation pattern corresponding to AT8 antibody epitope did not result in the formation of high molecular weight Tau species, or observable PHF-like fibrils, even at high amounts of phosphorylated Tau reaching up to 8% of total cellular proteins.201 Similarly, using eukaryotic Sf9 cellular Tau overexpression system coupled to native mass spectrometry of phosphorylated Tau in these cells, Drepper et al. found that even the highly-phosphorylated Tau fractions did not show self-assembly into the fibrils.202 In addition, recent work from our group showed that phosphorylation of all five tyrosine residues, three N-terminal residues Y18, Y29, and Y197, or a single tyrosine residue Y310 in the MTBR greatly reduced Tau aggregation.149
These observations suggest that the pattern of phosphorylation is a key determinant of Tau structure and aggregation. However, a more systematic investigation of the role of Tau phosphorylation using strategies that enable homogeneous and site-specific phosphorylation or hyperphosphorylation is essential for elucidating the role of phosphorylation in the pathogenesis of Tauopathies.
Interestingly, deglycosylation of PHFs in vitro promoted their conversion into straight filaments (SFs),204 illustrating the profound influence of PTMs on the structure, dynamics, and morphology of the Tau fibrils. Tau can be O- and N-glycosylated on asparagine, serine, and threonine residues,208 thus competing with phosphorylation of the same serine and threonine sites (Fig. 4, Glycosylation).209 Several sites modified by OGT were identified in vitro and included T123, S400, S409, S412, S413.210,211
An inverse correlation between Tau phosphorylation and O-GlcNAcylation levels212,213 was found in AD patients’ brains, implicating glucose metabolism impairment as a contributing factor to AD progression and pathology.214O-GlcNAcylation of Tau peptides at S400 was found to inhibit phosphorylation at the neighboring S396 and S404 sites.215 In cells, different subsets of the overexpressed Tau molecules were found to carry O-GlcNAcylation and phosphorylation, suggesting a function-related balance between these PTMs.216 In a HEK293 Tau-BiFC cellular system, inhibition of OGT resulted in increased Tau phosphorylation at S199 and S396.217
On the other hand, NMR-based studies showed that phosphorylation was not blocked by O-GlcNAcylation.211 Furthermore, more recent work using antemortem AD patient CSF samples found increased levels of total N-glycans, which correlated with an increase of total and phosphorylated Tau levels, especially in patients with subjective cognitive impairment, an early stage in AD pathology development.218 This suggests that any offset of Tau phosphorylation by Tau glycosylation may have a negligible effect on the total levels of phosphorylated Tau detected in CSF.
It has been proposed that O-linked Tau glycosylation reduces Tau aggregation through the prevention of its phosphorylation on the same residues,212,219–221 pointing to the importance of the temporal sequence of Tau modifications. The deglycosylation of already hyperphosphorylated Tau derived from the cytosolic fraction of AD patients’ brains, failed to induce its fibrillization.196 Tau phosphorylation on the PHF-1 antibody epitope, which includes residues pS396/pS404, and spans over O-GlcNAc S400 residue was found to enhance Tau aggregation, whereas glycosylation of residues S400, S412, and S413 slowed Tau aggregation rate.222 N-linked glycosylation may also contribute to the structural integrity of the AD-derived PHFs, as evidenced by the fact that deglycosylation by F/N-glycosidase F resulted in untwisting of the fibrils into thin filaments.223 In addition, N-glycosylated Tau had a lower aggregation propensity and showed shorter and thinner fibrils than deglycosylated Tau.224 Despite this, the impact of increased Tau O-GlcNAcylation may only be prominent in the physiological systems, and it may involve or require complex interactions with other Tau PTMs and/or cofactors. HEK293 Tau-BiFC cell-based studies showed that Tau aggregation was decreased by inhibition of OGA, and increased by inhibition of OGT (Lim, Haque et al. 2015), suggesting that this particular Tau PTM must be studied within the complex cellular context.
Acetylation at K280 has been reported to be associated with the early stages of AD. It is thought to occur in CTE patients’ brains before Tau hyperphosphorylation, suggesting that Tau acetylation might be an early marker of incipient Tau pathology.237 Acetylation at K174 was also detected at early stages in AD patients by mass spectroscopy,238 whereas AcK274 and AcK281 were found at the later stages of the AD progression.239In vivo rodent experiments showed that AcK174, AcK274, and AcK281 disrupted synaptic homeostasis, suggesting that Tau acetylation on specific residues leads to hippocampus-associated memory loss at the early stages of dementia.240 Contrary, Choi et al.241 used AD patient-derived organoids and the rodent model of AD to show that the inhibitor of histone deacetylase 6, which was found to deacetylate Tau and modulate its clearance,242 reduced the synaptic defects and pathology associated with Tau. In addition, acetylation of potential and canonical degron motifs, including the Tau sequence KFERQ, may be required for enhanced protein recognition and degradation by autophagy systems.243 This suggests that at least some residues and patterns of Tau acetylation may be protective. Acetylation may have cross-talk with other PTMs, such as phosphorylation. When acetylated on KXGS motifs that modulate Tau binding to MTs, the phosphorylation of these sites on Tau was reduced, and Tau MT-binding capacity was preserved, thus preventing Tau detachment and downstream aggregation.244 Acetylation modifies lysine residues, which can also be ubiquitinated, methylated, glycated and SUMOylated. This introduces the competition between modifications, which in case of decreased ubiquitination due to acetylation resulted in a decrease of Tau turnover.245
In vitro investigations of Tau acetylation at lysine residues predominantly focused on its connection to Tau aggregation. Furthermore, previous work predominantly utilized pseudoacetylation by lysine-to-glutamic acid substitution, which may not fully represent the effects of acetylation, or in vitro enzymatic acetylation, which may result in a non-specific and differentially-modified mixture of proteins.188,246 Acetylation-mimic of residues K280 and K281 was found to promote Tau fibrillization247,248 (see Fig. 5A). On the other hand, singular acetylation or mimic of K321, or in combination with other lysines, including K280 and K281, was found to decrease Tau fibrillization.244,249,250 Using precise site-specific acetylation of K280 through protein semisynthesis (Fig. 5B) to obtain homogeneously-acetylated protein,188 our group showed that AcK280 greatly enhanced the rate of Tau fibrillization and promoted the formation of short fibrils.
Overall, Tau acetylation may be a promising PTM marker of incipient Tau pathology. However, the underlying mechanisms linking it to Tau aggregation and pathology formation and spreading must be investigated further.244,245
The roles of ubiquitination in Tau fibril formation are still unclear. Ubiquitination is a complex PTM that remains understudied because of the lack of knowledge of the enzymes that regulate Tau ubiquitination in this domain. Tau is known to be ubiquitinated by E3 ligases such as axotrophin/MARCH7,258 C-terminus of the Hsc70-interacting protein (CHIP),259 and TNF receptor-associated factor 6 (TRAF6), which was also found to colocalize with sequestosome/p62 in the Alzheimer's brain-derived Tau aggregates.260 The polyubiquitination of Alzheimer's brain-derived Tau by CHIP-Hsc70 complex was dependent on its hyperphosphorylated state only in the presence of E2 ligase HbcH5B,261 indicating the process specificity and interplay between PTMs. Thus far, only one deubiquitinating Tau enzyme OTUB1 has been identified in mice.262 Further investigations must focus on at which stages of Tau aggregation and at what Tau sites ubiquitination occurs, and how it impacts fibril structure and clearance. This can be achieved using precision protein semisynthesis strategies successfully applied to study impacts of singular ubiquitination, as well as the impact of multiple additions of ubiquityl groups on α-synuclein aggregation properties.263,264
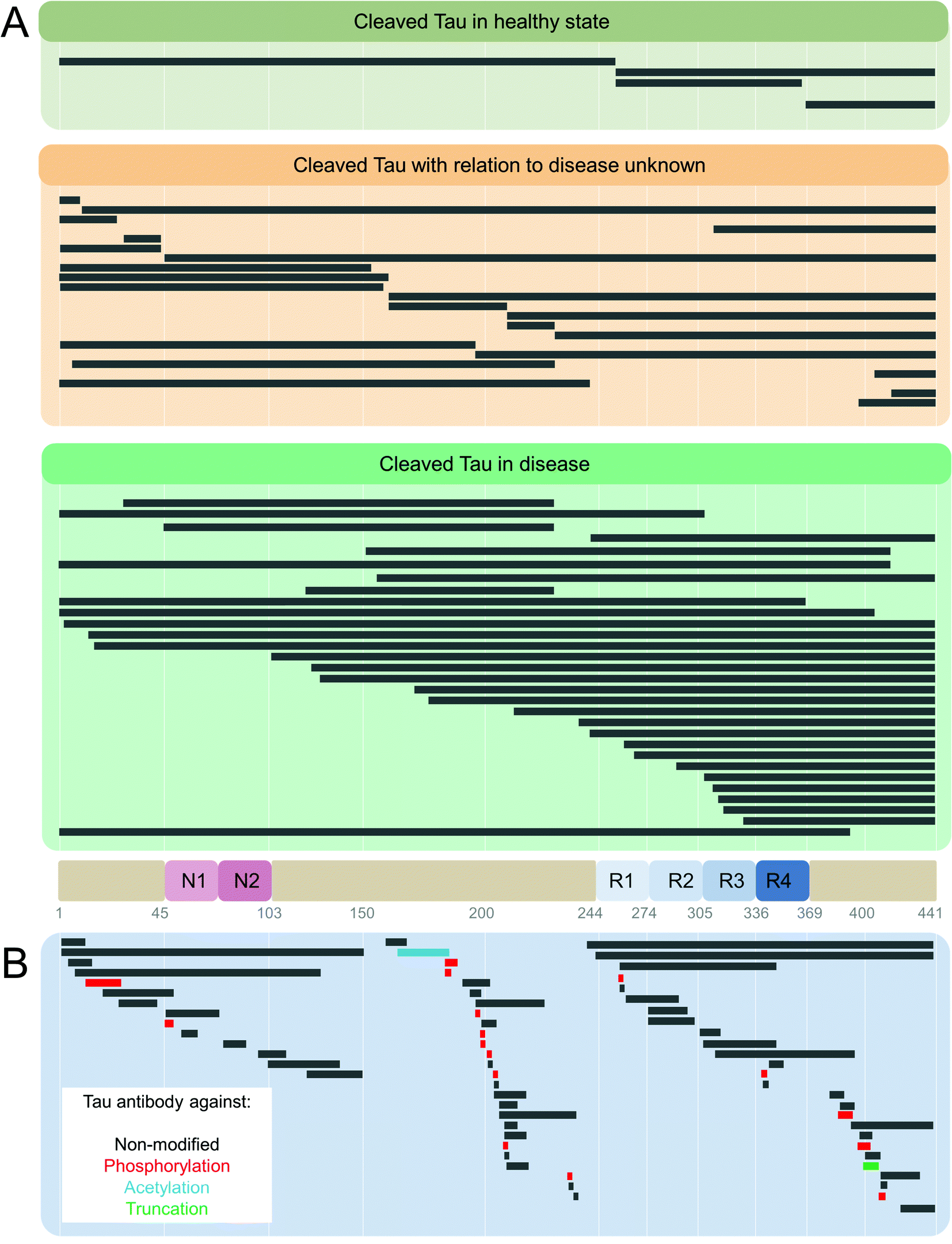 | ||
| Fig. 6 (A) Enzymatic cleavage-generated Tau truncation products identified in healthy and Tauopathy patients.131 (B) Anti-Tau antibody list from http://www.alzforum.com database. Alzforum lists a total of 572 Tau antibodies, 45 laboratory-produced, and 527 commercial. 534 are human-reactive, 360 with listed epitopes identified, with unique sites mapped in the figure. Where epitope is unknown, antibodies are raised against full-length,6 C-terminus,18 and N-terminus.14 The break in the Tau sequence coverage is at amino acids 150–159 in the proline-rich domain. | ||
Tau fragments resulting from truncations at positions D13,274 E391, and D421275,276 were reported to promote Tau aggregation, accumulate in AD brains, and their levels correlated with the progression of disease.274 46 kDa fragment was detected using mass spectrometry and primary amino acid sequencing in AD and was determined to be a result of caspase-cleaved Tau at D421.277 Multiple Tau truncations were also found in PHFs,278 illustrating the heterogeneity of Tau forms present within the Tau aggregates found in the brain. Truncated Tau fragments were found in NFTs in AD (4R + 3R Tau),10,279–282 Pick's disease (PiD) (3R Tau),283 PSP (4R Tau),284 and other neurodegenerative disorders.285
Several studies suggest that proteolytic Tau fragments may contribute directly to Tau neurotoxicity and the initiation of Tau pathology. The Tau 45–230 (∼17 kDa) fragment produced by proteolytic cleavage by calpain-1 and thrombin between residues 45 and 230, lead to functional impairments in synapses and axonal transport in cells, as well as behavioral deficits in animals.286–288 The Tau 26–230 (20–22 kDa) fragment could seed full-length Tau aggregation in vitro.289 In cells, it was enriched in mitochondria and impaired its activity,290 and induced cell death in differentiated human SH-SY5Y cell line,291 and primary neurons.292 A 24 kDa C-terminal Tau fragment produced by calpain 1 cleavage between residues R242 and L243 showed higher aggregation capacity and lower affinity for MTs in vitro.293 In the SH-SY5Y cell line, it showed enhanced propagation and seeding of Tau in the neighboring cells.294 Another 35 kDa Tau fragment, likely generated by cleavage between residues in the region of 182 and 194, and is present in 4R Tauopathies such as PSP and CBD.284 Further investigations in the Chinese hamster ovary cell line showed that overexpression of Tau 35 kDa led to its hyperphosphorylation, lower MT-binding affinity, induction of unfolded protein stress response, and aberrant insulin signaling.295 Tau truncations produced by removing from 12 to 121 C-terminal residues were also found to increase Tau aggregation.198 Finally, it has been widely known that truncated Tau fragment K18 has a higher propensity to aggregate compared to full-length Tau. These findings suggest that some of the Tau fragments are capable of acting as seeds that could initiate the aggregation of full-length Tau proteins.
Some Tau truncations resulted in subcellular mislocalization or misdirection of Tau thus impairing its normal functions. The 4R isoform of the Tau fragment 151–391 was displaced from axonal to nuclear compartment in human neuroblastoma cells and primary rat neuronal cultures.289 Tau fragment 1–314 was found to detach from MTs and promote loss of hippocampal neurons concurrent with induction of mislocalization of full-length Tau to the somatodendritic compartment and synaptic impairment.296
Other292 Tau fragments 1–255 and 369–441 showed impaired MT-binding and polymerization, whereas fragments 1–368, 256–441, and 256–368, in addition, had higher aggregation propensities as compared to full-length Tau.131,297 N-terminal fragment 1–13 resulted in degeneration of the axonal structures, whereas its counterpart Tau fragment 14–441 contributed to the formation of Tau aggregates.298,299
Other fragments290,296,298,299 seem to have no associated toxicity in cellular models, such as Tau 1–25291 and 125–230,131 however, the functional or pathogenic roles of the most known enzyme-cleaved and pathology-associated Tau fragments remain understudies. These include Tau fragments 1–242,293 45–441,286 258–372,300 315–441,296 403–441,266 1–152, 1–197, 3–124, 3–230, 156–209, 156–441, 198–441, 210–230, 210–441, 231–441 and 422–441.131 Equally unclear are the functional or protective roles, if any, of some Tau fragments. Interestingly, some truncated species may enhance proteolytic and autophagic degradation of Tau and its turnover through specific interactions with co-chaperones and ubiquitin ligases.301
Whether Tau cleavages found in pathological aggregates occur before, during, or after the onset of Tau aggregate formation279,302 needs further investigation. Some studies have reported that Tau hyperphosphorylation preceded Tau cleavage in cell cultures and human AD brain,283,303,304 whereas others reported that NFT formation followed the Tau cleavage events.305 Tau cleavage was also reported to precede Tau ubiquitination256 and glycation254 events in AD. Early and late NFT formation-specific markers were associated with Tau cleavage by caspase-3, -6, -7, and -9 in the AD brain and correlated with the scores of cognitive decline.274,306–310 Tau cleavage by calpains311,312 was detected in AD and FTPD.313 Despite these findings, multiple truncated Tau species were also detected in normal human hippocampal, cerebellar, entorhinal, prefrontal, and motor cortical brain regions across the age range of 18 to 104 years old,314 functions and significance of which alone, or in combination with other Tau PTMs, are yet to be determined (see Fig. 6A).
Taken together, these observations suggest that cleavage of Tau plays important roles in Tau aggregation, AD pathogenesis, and possibly in the disease progression and severity.309,315 This also underscores the critical importance of mapping the Tau cleavage patterns in health and disease. They also suggest that Tau fragments play important roles in the pathogenesis of AD and other Tauopathies. Finally, the abundance of Tau fragments underscores the importance of reassessing their physiologic and pathogenic roles and the extent to which assays used to quantify total Tau can capture these various fragments. These studies should consider all six Tau parent isoforms, which could be cleaved at equivalent sequence positions but produce fragments of varying lengths and sizes.
Cross-talk between Tau PTMs and impacts on aggregation
Increasing evidence suggests that the Tau PTM code is a combinatorial code that involves the co-occurrence of and cross-interactions between multiple PTMs depending on the type of neuropathology and the stage of its progression. Tau PTMs were found to cluster in specific regions of the protein in the brain samples of AD patients.133 The cross talks between Tau PTMs were and are still being investigated in the context of Tau phosphorylation, primarily due to the unavailability and poor characterization of antibodies for other PTMs. For example, Tau cleavage and Tau dephosphorylation co-occurrences have been observed in hippocampal, cortical, and cerebellar granule neurons.288,316,317 This could link the decreased cleavage of Tau hyperphosphorylated at the specific sites to impaired degradation of Tau. This may be followed by the accumulation and subsequent aggregation of Tau in neuronal and glial cells, leading to degeneration. Short Tau fragments containing MBD sequences such as the PHF6 motif were found to aggregate in vitro without the addition of cofactors,318,319 whereas PTMs in this region, such as pY310, were found to reduce the fibrillization propensity of truncated K18 or full-length Tau.149 Furthermore, Guillozet-Bongaarts et al.304 reported that dephosphorylation of residue S422 was necessary for caspase-3 mediated cleavage at D421 in vitro, emphasizing the importance of cross-talk between Tau PTMs. A recent report269 using Tau proteins truncated at various positions expressed in HEK-293T cells showed that Tau truncation after the first 50 amino acids reduced its phosphorylation at T205 and T231. Truncation after amino acid 150 enhanced its phosphorylation at T205, S212, S214, T217, T231, and S235 irrespective of the presence of the C-terminus. Truncation after amino acid 231 increased its phosphorylation at S262, S396, and S404.269Cross-talks between other PTMs were also observed, for example between the lysine-targeting PTMs such as acetylation, methylation, glycation, SUMOylation, and ubiquitination (reviewed in ref. 235). Hyperphosphorylation was associated with hypoacetylation – less acetyl PTMs – of the four KXGS motifs present in MTBR, which increased Tau aggregation propensity.242 AD patients’ brain-derived co-phosphorylated and ubiquitinated Tau peptides containing KXGS sequences were enriched compared to control-derived samples.255 In cellular system,320 Tau phosphorylation was increased by SUMOylation of K340. This was correlated with the decrease in Tau ubiquitination and degradation, resulting in enhanced levels of insoluble Tau. These findings suggest that Tau aggregation may be modulated by the synergistic effects of the multiple PTMs.
O-GlcNAcylation has also been shown to influence the site-specific phosphorylation pattern of Tau in vitro and in a cellular system.212 The crosstalk between Tau phosphorylation and O-GlcNAcylation was confirmed in cells, where the lower levels of Tau phosphorylation correlated with increased glycosylation and higher nuclear localization of Tau.216 Interestingly, an NMR study by Bourrée et al.211 found that O-GlcNAcylation did not impact Tau phosphorylation by the rat brain extract or mitogen-activated protein kinase 1 enzyme. However, phosphorylation slightly increased Tau O-GlcNAcylation by the O-linked N-acetylglucosamine transferase enzyme through indirect cross-talk mechanisms which remain to be fully understood.
Spatially, phosphorylation events seem to accrue in the N-terminal and proline-rich Tau region during the Tau NFT aggregate formation and maturation, whereas acetylated and non-ubiquitinated regions seem to dominate in the MTBR overtime.133 These studies further underscore the complexity of Tau PTMs’ cross-talks in the modulation of Tau isoforms’ normal functions, its aggregation propensities, and remodeling of the fibrils.
A complex interplay of multiple Tau PTMs in a tightly regulated spatiotemporal manner is likely to occur during the various stages of Tau aggregation and pathology formation.167 The phosphorylation patterns of Tau oligomers were heterogeneous among AD patients and correlated with the severity of the disease progression in biophysical, biochemical, cell- and animal-based functional assays.134 Further investigations of the Tau oligomer PTM landscapes are needed to understand their functional significance in vivo (reviewed in ref. 321 and 322).
Advantages and limitations of the current approaches to investigate the Tau PTM code
Similarly, most studies attempting to map and identify the naturally occurring Tau cleavage products in biofluids323 rely on mass spectrometry-based experimental approaches324 often in combination with immuno-enrichment of Tau species using antibodies, followed by the enzymatic digestion, labeling, and mass spectrometry analyses of fragments. Several antibodies, which cover virtually the whole sequence of the Tau molecule (www.alzforum.org) have been produced (Fig. 6B). However, the most often utilized Tau enrichment antibodies tend to target the proline-rich middle domain,103–188,194–246,325–327 present in all six Tau isoforms, and peptides from which tend to be overrepresented in mass spectrometry hits.328,329 Previously, N-terminally truncated Tau fragments found in AD samples by Derisbourg et al.330 were identified by immuno-enrichment using Tau5 antibody against Tau amino acids 218–225. However, the possible range of the Tau species devoid of this region, such as nucleation-competent and aggregation-prone MTBR, or conformations where this region is inaccessible was not captured. Similarly, Portelius et al.331 identified 19 tryptic Tau fragments in AD patients’ CSF by nanoflow LC-ESI mass spectrometry using immunoprecipitation with antibodies BT2 (epitope 194–198), HT7,158–162 AT120 (against phospho-threonine-181), or AT270 (phospho-dependent epitope 176–182), thus limiting the detection to Tau species containing the middle domain, whereas Tau is known to undergo extensive N-terminal271,284,291,332 and C-terminal333,334 truncations. Therefore, future studies aimed at the comprehensive mapping of the Tau PTM profile should employ approaches and a combination of antibodies that capture the diversity of Tau proteins and fragments in brain tissues and biological fluids.
Co-factor-induced Tau aggregation mechanisms determined in vitro
 | ||
| Fig. 7 Hypothetical mechanism of heparin-induced Tau fibril formation. Tau exists in solution as a disordered monomer that could undertake the paperclip conformation. Negatively-charged heparin can bind to positively-charged regions on Tau and prime it into aggregation-prone conformation. Tau forms the nucleation-competent seed, ON-pathway oligomers, and fibrils upon the addition of primed monomers. Heparin molecules may tightly associate with Tau fibrils. Tau MTBR is designated with coral, N- and C-termini in grey, heparin molecule in magenta. | ||
GAGs are a large class of long anionic unbranched polysaccharides consisting of repeating disaccharide units.336 Based on the compositional differences, GAGs comprise five classes of molecules, such as hyaluronic acid, predominantly associated with skin tissues, synovial fluid and articular cartilage;337 chondroitin sulfate, which is a major component of perineuronal nets, changes in sulfation patterns of which were is linked to cognitive decline in AD,338 as well as dermatan sulfate, keratan sulfate, and HS, all of which play important structural and signaling roles in neural and other tissues.336,339–341 Heparan sulfate proteoglycans (HSPGs) have been implicated in Tau pathology formation and spreading at the stages of its release from the affected cell, as well as in facilitating the internalization of pathogenic Tau forms by the neighboring cell (reviewed in ref. 342). Despite the popularity and ease of use of polysaccharides as cofactors for in vitro Tau fibrillization, the formed fibrils differ from pathological in several ways. The heparin-induced fibrils are highly heterogeneous, predominantly single protofibril, and composed out of the single Tau isoform, which contrasts with the highly conserved disease-specific Tau fibril fold. In addition, the heparin-induced fibrils are generated from recombinant unmodified proteins and thus lack the PTMs found on the patient brain-derived Tau fibrils.21,335 Other concerns for the downstream applications in vitro, in cells and in vivo arise when using added heparin as a cofactor to fibrillize Tau. These may include interference with the Tau fibril-targeting compounds,343 disruption of the cellular uptake of Tau fibrils,344 and causing vasodilation or immune response in animals,345,346 all of which may contribute to the modification of Tau seeding propensity non-contingent on the inherent properties of the Tau seed itself.
Mechanisms of heparin-induced Tau aggregation
At physiological pH and in the presence of a reducing agent, such as dithiothreitol (DTT), the 4R Tau MTBR formed a stable complex with ∼7 kDa heparin at a ratio of one-to-one350,351 or two Tau MTBR molecules per one 12 kDa heparin molecule.351 These interactions were slower in the absence of reducing agents, which suggests that the formation of intramolecular cysteine bridge could stabilize Tau MTBR monomer and/or disrupt its interactions with heparin, thus preventing efficient 4R Tau MTBR fibrillization even in the presence of cofactors.352–354 Interestingly, the lag phase was unaltered when the Tau MTBR to heparin molar ratio was either one-to-one or one-to-two. However, it decreased sharply at higher molar ratios of heparin. Conversely, the exponential growth rate increased with a decreasing ratio of heparin to Tau MTBR.351 Quantitation of Tau MTBR and heparin-binding kinetics, based on calculations of the stoichiometry of the molecules and association and dissociation rate constants, suggested that the interaction between Tau MTBR and heparin occurs at the nucleation step. The exponential growth phase, which proceeds by the sequential addition of Tau MTBR monomers to aggregation competent nuclei or fibril seeds, was reduced at lower heparin to Tau MTBR molar ratios and inhibited at higher ratios. This demarcates the narrow optimal range of heparin-to-Tau molecule ratios necessary for the efficient Tau fibrillization reactions. Furthermore, these studies show that the heparin association rate with Tau is higher than the dissociation rate. On average, more heparin molecules are likely to be associated with Tau than are released back into the solution.
The kinetics of heparin-induced Tau fibrillization was also dependent upon the length of heparin polysaccharide chains, with longer chains providing higher Tau-heparin binding and nucleation efficiency,355 likely due to a higher number of the binding sites along the Tau molecule. However, due to the high complexity of the glycochemistry of the heparin, the quality of the commercial heparin products depends on the purification or synthesis methods, and the origin or the raw material. This might lead to high batch-to-batch variability and large heterogeneity in terms of the heparin molecular sizes.344
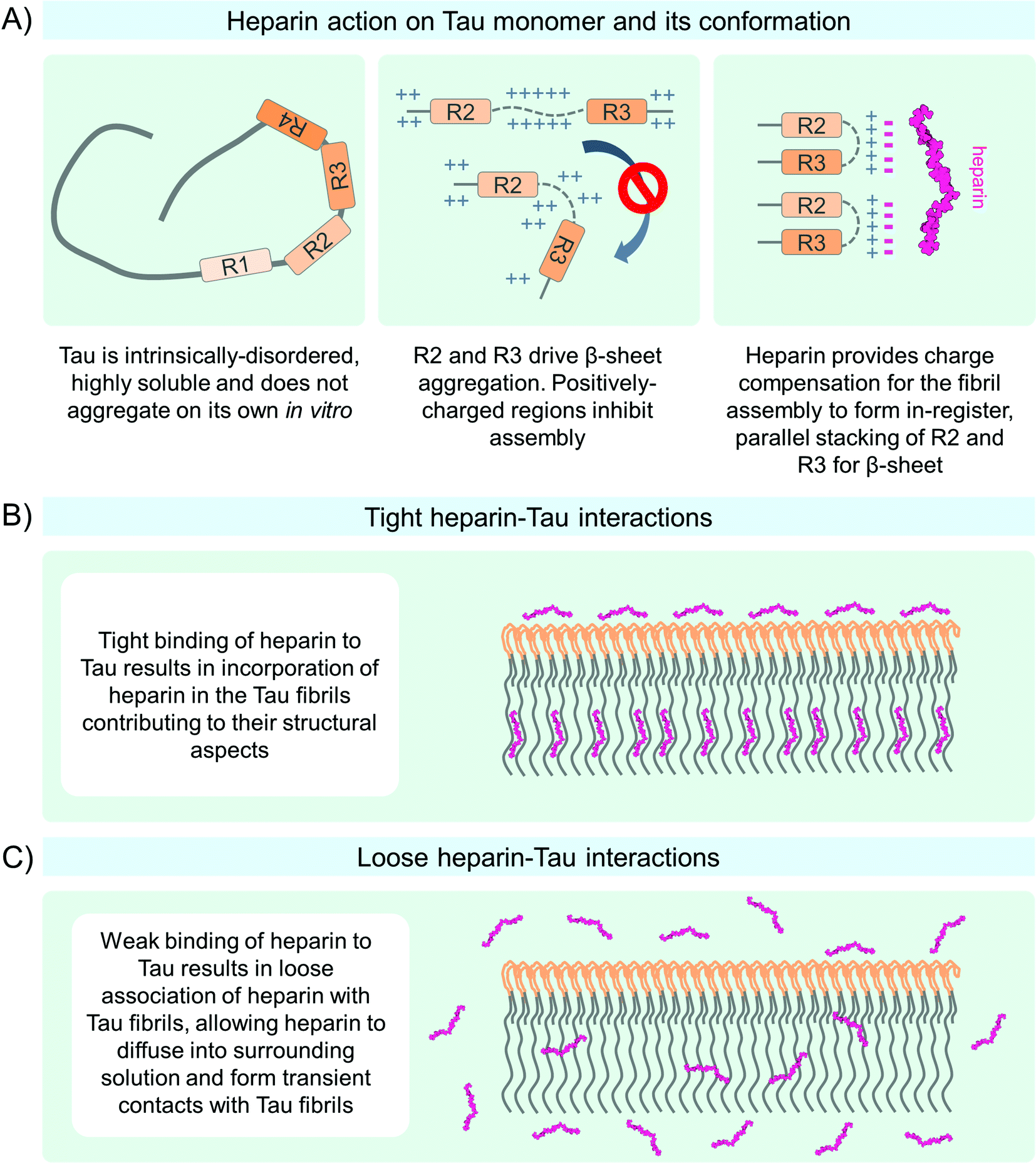 | ||
| Fig. 8 Heparin-induced Tau aggregation mechanisms. (A) Proposed mechanism of charge neutralization by heparin around R2 and R3 regions of Tau molecule conducive to Tau aggregation. (B) Tight interactions between heparin and Tau may lead to the incorporation of heparin into fibrils and contributions to their structure. (C) Loose associations of heparin with Tau may lead to the release of heparin molecules into the surrounding medium. | ||
In vitro chemical cross-linking performed at various time points during Tau aggregation showed that Tau formed stable dimers, tetramers, and higher-order assemblies in the presence of heparin.364 This suggests that in addition to direct effects on Tau molecular conformation, heparin promotes Tau aggregation and fibril formation through stabilizing or increasing the formation of nucleation-competent Tau oligomers.365 Heparin was shown to associate with the rigid core region of Tau PHFs composed of the MTBR. However, the N-terminal residues upstream of the N-terminal inserts N1 at amino acid position 45–74 and N2 75–103 were also important for Tau-heparin interactions.349,355,360,366 Studies by electrochemical impedance spectroscopy and cyclic voltammetry showed that Tau monomers immobilized on the gold electrode surface by the N-termini had a lower capacity to bind heparin compared to the Tau molecules immobilized by cysteine residues 291 and 322 in the middle of the protein, which allowed the N- and C-termini mobility.367 These studies indicate that heparin binds tightly to several regions of Tau molecules and influences its conformational and aggregation properties.
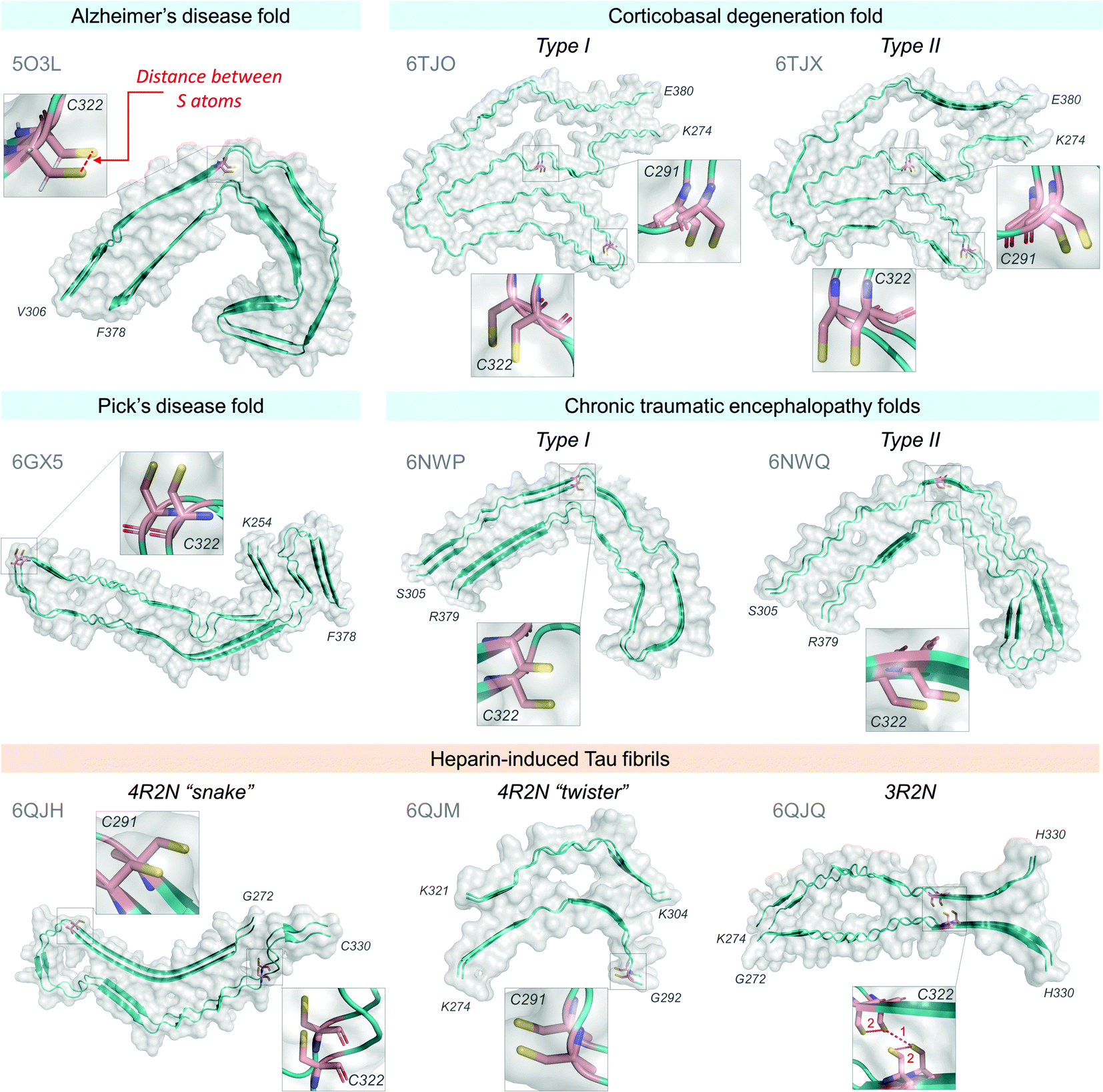 | ||
| Fig. 9 Cryo-EM structures of Tau fibrils and cysteine residue positions. Tau fibril structures available at Protein Data Bank,384 accession codes are in grey, structures were rendered using PyMOL software (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC385). Two molecule strands are shown for each structure. Atomic distances between sulfur atoms on adjacent cysteine residues (as depicted in Alzheimer's disease fold, red dashed line) were measured using the PyMOL385 Measure tool and are summarized in Table 1. All distances between cysteine residues are significantly larger than the distance required for the formation of the disulfide bond at 2.3 Å.383 | ||
| Disulfide bond distance383 | ∼2.3 Å | ||
|---|---|---|---|
| PDB | S–S distances (Å) between residues | ||
| C291–C291 | C322–C322 | ||
| Pathological Tau fibril fold | |||
| Alzheimer's disease | 5O3L | — | 4.79 |
| Corticobasal degeneration | |||
| Type I | 6TJO | 4.80 | 4.86 |
| Type II | 6TJX | 4.81 | 4.82 |
| Chronic traumatic encephalopathy | |||
| Type I | 6NWP | — | 4.72 |
| Type II | 6NWQ | — | 4.90 |
| Pick's disease | 6GX5 | — | 4.85 |
| Heparin-induced Tau fibrils | |||
| 4R2N “snake” | 6QJH | 4.80 | 4.76 |
| 4R2N “twister” | 6QJM | 4.72 | — |
| 1. C322–C322 | 2. C322–C322 | ||
| 3R2N | 6QJQ | 3.70 | 5.13 |
The heparinase-treated PHFs also showed altered biochemical properties, such as higher sodium-dodecyl-sulfate (SDS) solubility and reduced PHF-1 immunoreactive high molecular weight species by Western blotting. Although several in vitro studies have attempted to address the question of whether heparin is integrated into Tau fibrils,349,351,389–392 at this time no consensus has been reached. It is likely that heparin associates strongly with specific sites on Tau fibrils. Several aspects of the biochemical properties of heparin (e.g. molecular weight and sulfation patterns) and Tau molecules (e.g. isoform, modifications, mutations, and aggregation) have been shown to influence the interactions between these two molecules. Finally, the binding and the heparin–Tau association–dissociation equilibrium might maintain a stochastic component, i.e. not readily attributable to any predictive variable. All these observations motivated researchers to look increasingly closer at heparin–Tau interactions to disentangle the complex relationships between heparin and Tau.
Electron paramagnetic resonance (EPR) studies showed that RNA induced the formation of Tau fibrils containing in-register stacking of in-parallel β-sheet-containing monomers, conformationally resembling heparin-induced Tau fibrils. However, RNA affinity for 3R MTBR fibrils was higher than heparin, which suggests other structural differences between Tau fibrils induced by heparin or RNA. Subsequent studies by Fichou et al.390 (see below) showed that RNA was stably associated with the Tau fibrils, but could be displaced into the soluble fraction by the addition of heparin. No change in fibril morphology was detected after heparin addition and RNA displacement. The majority of fluorescein-conjugated heparin (around 80%) was directly shown to be associated with 3R or 4R MTBR fibrils, with a low amount remaining in the soluble fraction. These results show that the binding of these negatively-charged cofactors to the surfaces of Tau fibrils is mediated by electrostatic forces that are more pronounced in the case of heparin, most likely due to high negative charge density per heparin molecule.393 The main limitation of this study was the use of truncated Tau proteins with non-native amino acid residues, which could limit the translation of these results to the full-length Tau isoforms. However, increasing evidence suggests that Tau fragments are present in appreciable amounts under physiological and pathogenic conditions.265–268,270–272,329
So far, the most compelling evidence for heparin as a constituent of Tau fibrils came from the direct detection of spin-labeled heparin within Tau fibrils. Using continuous-wave EPR and biochemical approaches, Fichou et al.390 showed that heparin (∼15 kDa) and RNA (∼900 base pairs) were integral parts of the in vitro formed Tau fibrils. Upon removal or heparin, using enzymatic digestion by heparinase which removed around 20% of bound heparin, a fraction of the Tau fibrils depolymerized into the Tau monomers and dimers. Similarly, treatment with RNAse led to the removal of around 60–70% of bound RNA. The detection of spin-labeled heparin in the thioflavin-positive heparin/RNA-containing Tau fibril-containing fraction post-heparinase/RNAs treatment showed that some of the heparin/RNA bound were structurally integrated into Tau fibrils and were not easily accessible by enzymes. Nevertheless, in both cases, heparin and RNA cofactors were instrumental for maintaining the thioflavin-positive and continuous-wave electron paramagnetic resonance of paramagnetic spin spectra signatory of β-sheet arrangement within Tau fibrils. These observations suggest that these two cofactors contribute to the structural integrity of Tau fibrils. Interestingly, thus far no non-proteinaceous electron densities in cryo-EM imaging of Tau fibrils were attributed to heparin, possibly due to sample preparation procedures and reversible and dynamic nature of heparin–Tau contact. It remains unclear to what extent the level of heparin/RNA integration into the fibrils alters their morphology or stability. Also, whether the interactions happen before or after Tau fibril formation in vivo must be clarified.
Using kinetic and steady-state analyses, Carlson et al.392 suggested that heparin-to-Tau molar ratios were more important factors in Tau polymerization than the concentration of Tau molecules. These authors argued for an allosteric impact of heparin on Tau molecules resulting in a low thermodynamic barrier to nucleation.392 Based on the classical model of allosteric regulators,394 the authors concluded that heparin was not integrated into the Tau fibrils. However, the data in the study did not directly demonstrate that on the structural level. Similarly, Ramachandran and Udgaonkar351 estimated the heparin-to-Tau molecule stoichiometry at the end of fibril formation to be one-to-twenty. Based on the theoretical calculations using heparin–Tau dissociation constant and Tau and heparin molecules’ concentrations values, they argued that heparin was only a minor constituent of the formed fibrils. Analyses of the heparin-induced full-length Tau or MTBR fibrils at three different heparin concentrations by AFM and FTIR spectroscopy showed no significant size or structural differences of fibrils between these three conditions.351 One obvious limitation of this experimental design is the lack of independent controls, such as alternative inducer-generated fibrils or reassessment of the fibril biophysical properties after enzymatic heparin removal treatment.
In summary, kinetic, structural, and biochemical studies strongly suggest the possibility of tight heparin association with or integration into Tau fibrils under certain conditions in vitro. However, further investigations using complementary experimental techniques and quantitative approaches are warranted to determine the precise mechanisms of heparin–Tau fibril interactions (Fig. 7B and C) and whether these interactions occur at the monomer or fibril levels in the brain.
The tight binding of heparin or other cofactors to Tau fibrils has significant implications for using heparin-induced fibrils to investigate Tau fibril toxicity mechanisms of seeding activity in cells or in vivo as the Tau-non-contingent responses, due to the presence or release of these molecules may occur. In the case of heparin, this includes vasodilation of the blood vessels346 and immune345 or aberrant cell stress responses.395–400 Also, the presence of heparin of other cofactors might directly impact the post-translational modifications of Tau fibrils,401 alter their surface properties, secondary nucleation events, and thus their seeding activity and spreading through extracellular matrix interactions.344,348 Furthermore, the interference of the polysaccharide with the Tau-targeting molecules343 cautions the use of the current systems in the development of Tau pathology targeting drugs and tracers.
Tau molecules in physiological conformation present in the ECF might have ample opportunities to encounter heparin, and other cofactors, especially in the events of heparin-mediated neuroinflammatory responses. Thus, one may speculate that during these events, heparin exerts conformational changes on Tau molecules, priming them into fibrillization-competent forms and/or tightly binding to Tau. The subsequent cellular intake of these primed Tau molecules or Tau-heparin complexes might cascade into the pathological Tau oligomerization, fibrillization, and formation of NFTs, concomitantly sequestering the functional pool of Tau molecules and inciting cellular stress responses. Further investigations are necessary to determine whether the ECF heparin mediates the Tau conformational changes from physiological to pathological forms in vivo. Addressing this question might be achieved using techniques such as microdialysis of ECF in the brain of model animals,414–417 or recovery of the material from the brain electrode implants of neurodegenerative disease patients undergoing the procedures such as deep brain stimulation. The Tau proteoforms and heparin molecules then can be determined by highly sensitive biophysical techniques, such as mass spectrometry418 at different stages of Tau pathology formation.
Heparin activates Tau kinases. Interestingly, apart from the direct interaction of heparin with Tau to induce the formation of fibrils, heparin might also indirectly influence Tau's biochemical and biophysical properties through stimulation of Tau hyperphosphorylation by several Tau kinases. This is thought to occur due to heparin-induced conformational changes that ultimately lead to increased exposure of phosphorylation sites on Tau.364
Heparin was shown to greatly enhance Tau phosphorylation by p34/cdc28 serine-threonine kinase419 and by protein kinase A at least on serine 156.420 However, the physiological relevance of modifications by these kinases is unknown. On the other hand, heparin enhanced Tau phosphorylation by protein kinase FA/glycogen synthase kinase-3α (GSK-3α) in vitro, which phosphorylated residues found in the AD pathology, such as serines 235, 262, 404, 324 and 356, and threonines 212 and 231.401,421 Similarly, heparin was shown to enhance Tau phosphorylation by stress-activated serine-threonine kinases (SAPK1γ, SAPK2a, SAPK2b, SAPK3, SAPK4) on the serines (S202, S396, S404, S422) and threonines (T181 and T205, T231422) in vitro. Interestingly, Hasegawa et al.423 showed that the heparin concentration required to induce Tau phosphorylation by MAP kinase, NCLK, and GSK3β on multiple serine residues, and notably on serine 262, was lower than the heparin concentration required to induce Tau fibrillization. This suggests possible precedence of heparin-induced Tau phosphorylation to fibrillization, however subsequent studies showed that cofactor-induced Tau fibrillization kinetics were complex and dependent on other factors in addition to heparin–Tau concentration stoichiometry.351,392 Serine 262 is considered abnormally phosphorylated in AD.424,425 However, our group150 and others197,200 showed that phosphorylation of serine 262 in combination with other residues inhibited, rather than promoted, heparin-induced Tau aggregation in vitro. It is still unclear whether and how the predominantly extracellular heparin or plasma membrane-associated molecules of HSPGs can directly impact Tau kinase-mediated downstream fibrillization of Tau in the intracellular milieu in vivo.
Taken together, cofactors such as heparin and others, known and unknown, seem to play a much larger role in the Tau fibril formation, their biochemical characteristics, and their impact on neuropathology formation and propagation than is generally thought and should be investigated further.
RNA. Tau was first discovered to directly bind to RNA molecules in 1984 by Schröder et al.,426 later corroborated by others.389,427 RNA was also found in association with hyperphosphorylated Tau in NFTs, Pick bodies, and neuritic plaques,428,429 providing credence to the physiological relevance of investigating RNA-induced Tau fibrillization. In 1996, Kampers et al.430 showed that RNA induced Tau assembly into fibrils through the formation of the intermolecular cysteine 322 disulfide bridges, which led to the formation of Tau dimer intermediate before Tau fibrillization. Subsequently, RNA has been successfully used to induce and study Tau fibrillization mechanisms in vitro.431 Nevertheless, the formation of disulfide-mediated dimer formation is not necessary for Tau fibrillization in the brain352 as exemplified by their absence in the cryo-EM-solved fibrils from sporadic AD, Pick's, CBD, and CTE patients (see Fig. 9). The presence of RNA catalyzed the conversion of soluble Tau into insoluble forms, likely through the charge balance to allow the tighter packing of the molecules.432 Recently, the role of RNA-Tau interactions has received increased attention due to converging data implicating liquid–liquid phase separation as a key mechanism in the initiation of Tau aggregation and fibril formation.427
Another recently proposed model for the aggregation of Tau involves liquid–liquid phase separation (LLPS) (reviewed in ref. 433 and 434). In biology, LLPS defines biophysical processes of condensation of biomolecules driven by transient interactions.435 These condensates are involved in the formations of membrane-less organelles in cells, such as nucleoli, stress granules, and liquid droplets. Recent findings suggest that these condensates may be implicated in the protein aggregation processes in neurodegenerative diseases spurred interest among researchers to study LLPS processes further436 which have been reviewed in ref. 435. RNA and heparin were found to induce LLPS of Tau into reversible condensed droplets that increased local Tau concentration, where Tau retained mobile conformations.427 In cells, the transfer RNAs were the predominant RNA species that were associated with Tau droplets.427 Wegmann et al.437 showed that Tau condensation into droplets coincided with the formation of Tau fibrils in the presence of heparin or RNA cofactors in vitro, suggesting the possibility of the coacervate heparin–Tau complex formation as an intermediate to Tau fibrillization.438 However, whether these two processes were independent was unclear.
Recent studies using turbidity assays with varying salt concentrations showed that in the presence of heparin, the formation of thioflavin-positive Tau aggregates occurred under a much wider range of sodium chloride concentrations, in contrast to complex coacervation, which occurred only in a small subset of the conditions.439 These results indicated that canonical heparin-induced Tau fibrillization was preferential to electrostatically driven heparin–Tau coacervate complex formation. The two processes – Tau LLPS formation and fibrillization – were likely to occur under overlapping conditions, but were independent processes. On the contrary, another study showed that LLPS-promoting conditions increased the fibrillization kinetics of Tau.440 This was exacerbated by the presence of the disease-associated mutations ΔK280, P301L, and G272V, which, in their own right, did not influence the phase transition propensity of Tau compared to non-mutant protein. Under physiological buffer conditions and Tau concentration of 2 μM,290,441,442 resembling that of the protein levels in the neurons, Tau required molecular crowding agents such as polyethylene glycol to phase separate. However, the P301L mutation or AT8-positive phosphorylation at pS202/pT205/pS208 increased the formation of droplets.443
These studies indicate that total Tau concentration and molecular crowding conditions, isoform composition and ratios, and the presence of pathological mutations can all impact Tau's propensity to phase separate. Nevertheless, whether the LLPS and ordered amyloid assembly of Tau fibrils are co-occurring or co-dependent processes has not yet been definitively demonstrated. Furthermore, the LLPS should be modeled under physiological conditions to provide useful insights into Tau biology. Although the Tau LLPS is an exciting new line of research into mechanisms leading to Tau fibrillization, thus far the lack of robust tools to study these delicate and dynamic biophysical processes in cells and in vivo restricted studies to explore the therapeutic potential of modulating these processes and investigating their role in the pathogenesis of AD and other neurodegenerative diseases (reviewed in ref. 444).
Tau interactions with membranes could be modulated by Tau PTMs. Phosphorylation had differential effects on Tau interactions with lipid membranes in various systems. Phosphorylation of Y310 in the MTB region149 reduced its affinity for lipids in vitro. In neuronal cells casein kinase 1 and GSK3β inhibition led to increased Tau association with the membrane, whereas inhibiting protein phosphatase 2A, or mimicking phosphorylation in the N-terminal and proline regions, led to a decrease of Tau affinity for membranes.456 Similarly, mimicking phosphorylation of 3R0N fetal Tau isoform on multiple residues significantly decreased its membrane association.457 Membrane fraction-containing extracts of cell lines458,459 and neuronal cultures456 contained predominantly dephosphorylated non-aggregated Tau. On the other hand, oligomeric Tau phosphorylated by GSK3β in vitro had a higher affinity for di-palmitoylphosphatidylcholine (DPPC) or 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) unilamellar vesicles, whereas phosphorylation of monomeric 4R1N did not affect its lipid-binding affinity.460 During apoptotic cell death phosphorylated aggregated Tau was enriched in the plasma membrane-containing PC12 cell line extracts.461 Tau phosphorylation by Fyn kinase resulted in its recruitment and enrichment at the membrane in neuronal cells,462 and oligomeric phosphorylated Tau was enriched at plasma membrane of human 4R0N-overexpressing murine neuronal N2a cell line.463 As evident from the contradicting reports, the role of Tau phosphorylation and PTMs on its lipid interactions is dynamic and multifaceted, and must be investigated further in light of its impact on transmembrane Tau spreading (see Fig. 3A).128,464
From a Tau aggregation tool development perspective, unsaturated long-chain fatty acids, such as arachidonic acid (5,8,11,14-eicosatetraenoic acid (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4)) and 9-palmitoleic acid (16:1), were found to efficiently promote full-length Tau assembly into long fibrils in vitro.465 Arachidonic acid-induced Tau fibrils showed twists at the 88 ± 15 nm intervals, were thioflavin-positive and immunoreactive to PHF-specific Alz50 antibody.466 Nevertheless, the lipids were not extensively adopted for in vitro Tau fibrillization assays, likely due to the difficulty of the post-fibrillization purification and lipid removal, as well as the heterogeneity of the fibrillar assemblies.
:4)) and 9-palmitoleic acid (16:1), were found to efficiently promote full-length Tau assembly into long fibrils in vitro.465 Arachidonic acid-induced Tau fibrils showed twists at the 88 ± 15 nm intervals, were thioflavin-positive and immunoreactive to PHF-specific Alz50 antibody.466 Nevertheless, the lipids were not extensively adopted for in vitro Tau fibrillization assays, likely due to the difficulty of the post-fibrillization purification and lipid removal, as well as the heterogeneity of the fibrillar assemblies.
Furthermore, the co-occurrence of Tau and α-synuclein in neuropathological inclusions has been observed in other neurodegenerative diseases since the early 1990s, including AD with Lewy bodies475 and dementia with Lewy bodies (DLB).476–478 Only recently, however, the direct relationship between these proteins and Tau fibrillization has been investigated. Heterotypic induction of Tau fibrillization has been demonstrated upon co-incubation with pre-aggregated amyloid-β fragments469 or monomeric α-synuclein, which bound Tau through its C-terminus.479 The latter case also resulted in the formation of homotypic α-synuclein fibrils under a low concentration of α-synuclein monomers. α-synuclein oligomers were found to nucleate oligomeric Tau formation, but not β-sheet forming fibrils.480 Tau K18 was found to form co-oligomeric complexes with α-synuclein,481 indicating that this region is important in Tau interactions with α-synuclein. α-Synuclein was also found to modulate the spread of Tau pathology in the mouse brain.482 Fibrils composed from both Tau and α-synuclein monomers showed differential pathology propagation when injected into the mouse brain, where Tauopathy was induced at higher levels than synucleinopathy.483
Both Tau and α-synuclein have been implicated in pathology spreading through anatomically connected brain regions in a protein strain-specific manner, resulting in distinct disease phenotypes in humans (reviewed in ref. 484 and 485). Presence of high levels of Tau in the AD-subtype of Parkinson's disease correlated with age, early cognitive impairment scores, grey matter reduction in the frontal cortex, and APOE ε4 allele status.486 Increasingly, evidence suggests that heterotypic induction of Tau aggregation by other amyloidogenic proteins contributes to the co-occurrence of different proteinopathies found across neurodegenerative disorders.27 These findings underscore the critical importance of conducting systematic studies to determine the key sequence, structural and cellular determinants of Tau interactions with other amyloid proteins and how they influence each other's aggregation, toxicity, and pathology spreading. The fact that the pathological aggregates of Tau, α-synuclein, and other amyloid proteins such as TDP-43 are heavily modified emphasizes the importance of conducting these studies in models that reproduce the biochemical complexity of disease-relevant pathological aggregates of these proteins.
How can the new biochemical and structural insights into physiological and pathogenic Tau inform drug discovery?
Increasing evidence points to the importance of cross-talk between the different PTMs in the modulation of Tau aggregation and seeding propensities, remodeling of the fibrils, and potentially disease progression and clinical heterogeneity of Tauopathies.487 These observations underscore the importance of employing more precise tools and experimental approaches to (1) refine our knowledge of the distribution maps of the Tau PTMs in healthy individuals at different stages of aging and disease progression; (2) identify the pathology-driving or preventing PTM patterns; and (3) map co-occurring PTMs, as well as their Tau isoform origin. This will pave the way for elucidating the role of cross-talk between PTMs in regulating Tau functions, aggregation, pathology formation, and spreading during disease progression. The knowledge gained from these studies will also help guide more disease-relevant reductionist approaches by defining the precise chemical identity of the Tau proteoforms that should be used to model Tau aggregation and toxicity in vitro. Finally, the complexity of the Tau PTMs should be taken into consideration when developing therapeutic antibodies targeting specific domains of Tau, or antibody-based tools used to quantify Tau species and pathological aggregates. In the section below, we provide an overview of previous studies to assess the therapeutic potential of targeting Tau PTMs to treat AD and other tauopathies. Although there are many studies on targeting Tau kinases in preclinical models, we will focus the discussion below on kinases and drugs that have been evaluated in clinical trials.
One promising kinase candidate is glycogen synthase kinase 3β (GSK3β) (reviewed in ref. 491 and 492), which phosphorylates Tau on at least 26 residues, and its levels are increased as the disease progresses.493,494 Overactivation of GSK3β resulted in Tau hyperphosphorylation and led to neuronal death.495,496 Lithium is a potent inhibitor of the GSK3β, inhibition of which was shown to lead to lower levels of hyperphosphorylated Tau in mice.497,498 Lithium has also been shown to reduce GSK3β-mediated phosphorylation of Tau and to increase Tau binding to the microtubules and their assembly in neurons,499–501 in mice,502 and prevented NFT pathology formation.503
Lithium salts are drugs commonly used for symptom management in bipolar and other psychiatric disorders.504 Lithium has been studied as a modifying treatment for AD, mild cognitive impairment, and dementia.505,506 However, its efficacy is still unclear. One important consideration is the toxicity, poor tolerance, and side effects of lithium, which led to discontinuation of the recent clinical trial (ClinicalTrials.gov Identifier: NCT00703677) evaluating lithium for the treatment of tauopathies CBD and PSP. Epidemiological observations of lower incidence of dementia in populations with access to tap water containing higher long-term levels of lithium,507 although later contested,508 still spurred interest in the use of microdose lithium as a preventative measure to stave off dementia, as well as disease progression modifying treatment in Alzheimer's disease (ClinicalTrials.gov Identifier: NCT03185208, LATTICE trial, currently recruiting). Microdosing of lithium (NP03) at levels 400-fold lower than standard formulations is being considered for clinical trials in humans based on the preclinical studies.509 The selective GSK3β inhibitor tideglusib (NP031112)510 was also evaluated for the treatment of AD (ClinicalTrials.gov Identifiers: NCT00948259 and NCT01350362) and PSP (ClinicalTrials.gov Identifier: NCT01049399).511 However, it failed to show a clinically relevant modification of disease progression despite reducing brain atrophy in PSP,512 no levels of phosphorylated Tau were reported.
Another Tau-phosphorylating enzyme of interest is Src-family tyrosine kinase Fyn. It specifically phosphorylates the residue Y18, which was found in the Alzheimer's brain NFTs.513 Despite promising preclinical data514,515 and good drug penetration and tolerability profile (ClinicalTrials.gov Identifier: NCT01864655),516 Fyn inhibitor saracatinib (AZD0530) showed no clinical efficacy in Alzheimer's patients with no significant changes in the CSF total or phosphorylated Tau (ClinicalTrials.gov identifier: NCT02167256).517
Other Tau kinases have also been considered potential targets for pharmacological agents with expected lowering of Tau phosphorylation and aggregation. These include cyclin-dependent kinase 5,518 c-Abl tyrosine kinase (c-Abl),519 lemur tyrosine kinase 2 (LTK),520 dual specificity tyrosine-phosphorylation-regulated kinase 1A (Dyrk1A),521 and thousand-and-one amino acid kinases (TAOKs).522 However, thus far few have proceeded beyond preclinical investigations, or their clinical efficiency is not yet known, for example for the c-Abl inhibitor Nilotinib (ClinicalTrials.gov Identifier: NCT02947893). Tau kinases that (a) phosphorylate Tau in vitro or (b) promote its phosphorylation by other kinases, and (c) are implicated in the neuropathological profile of patients with Tauopathies are the potential targets for further investigations.523 These include casein kinase 1 (CK1),524 c-Jun amino-terminal kinase (JNK),525 extracellular signal-regulated kinases 1 and 2 (Erk1 and Erk2),526 adenosine-monophosphate activated protein kinase (AMPK),527 cyclic AMP (cAMP)-dependent protein kinase (PKA),528 protein kinase N1,529 tau-tubulin kinases 1 and 2 (TTBK1 and TTBK2),493,530 Ca2 +/calmodulin-dependent protein kinase II (CaMKII) and microtubule-affinity regulating kinases (MARKs).531 Also, targeting of the specific regulatory subunits of Tau phosphatases has been proposed as a viable strategy to selectively reduce pathology-associated phosphorylation of Tau. Tau phosphatases that are implicated in Tauopathies include protein phosphatases 2A and 2B (PP2A and PP2B),532,533 protein phosphatase 5 (PP5),534 and calcyclin binding protein and Siah-1 interacting protein (CacyBP/SIP).535 Recently, we proposed that focusing on inhibiting Tau phosphorylation events that lead to its disassociation from MTs may offer an alternative and more effective strategy to prevent Tau aggregation by stabilizing its native conformations.150
Preclinical studies provide insights into the possible targets for inhibition of Tau aggregation and β-sheet-containing fibrillization intermediates or oligomers (reviewed in ref. 547). Small molecules, such as methylene blue,548 orange G,549 oleocanthal,550 “molecular tweezer” CLR01,551 phthalocyanine tetrasulphonate,552 curcumin553 and diazodinitrophenol,549 thiophene,554 and polythiophene555 compounds have also been reported to inhibit Tau aggregation in vitro. Thus far, methylene blue derivative and curcumin are the only small molecule Tau aggregation inhibitors that have advanced to clinical trials, and none have been approved for clinical use.556 The most prominent attempt to target Tau aggregation directly has centered on methylene blue. Phenothiazine compound methylene blue (methylthioninium chloride) was found to prevent Tau fibrillization, but not the formation of oligomeric Tau,557 likely through oxidation of cysteines.558,559 Phenothiazine could also mitigate Tau-related neurodegeneration and pathology through autophagy induction,560 reduction of Tau phosphorylation by mitogen-associated kinase 4, reduction of synaptotoxicity,561 and upregulation of genes by NF-E2-related factor 2/antioxidant response element in mice.562 Furthermore, due to its primary function as a redox cycler, the total impact of methylene blue is thought to be the result of stimulating the mitochondrial function, metabolism, and reduction of inflammatory responses. Its oldest application in toxicology includes treatment of methemoglobinemia,563–565 and against malaria from 1891,566,567 psychiatric conditions such as depression,568,569 claustrophobia or fear,570 and ifosfamide-induced encephalopathy.571 Despite such widespread use of methylene blue in various conditions, unfortunately, no epidemiological studies capturing incidence rates of dementia or AD have been done in these patient populations. Therefore, despite the possible direct effects of methylene blue on Tau fibrillization, it is likely to have pleiotropic effects on brain pathology. Recently, despite promising preclinical data,572,573 the derivative of methylene blue in a reduced form, LMTX (hydromethanesulphonate; TRx0237) failed to show clinical efficacy in Phase III clinical trials (NCT01689246 and NCT01689233). However LMTX is being investigated as a monotherapy in early stage AD patients (NCT03446001).
Despite decades of research, it is still unclear which Tau proteoforms are driving Tau pathology. The reductionist biophysical concepts have dominated the Tau aggregation field and have been useful tools for informing on the kinetics of in vitro to advance our understanding of the basic mechanisms and molecular determinants of Tau aggregation. However, it must be stressed that they can only be applied to in vitro systems. The processes of Tau aggregation taking place under pathological conditions in cells likely involve multiple yet unidentified cofactors, complex PTMs, and pathological diversity, which could be driven by parallel or cell-type dependent mechanisms of aggregation.
The attempts to systematically identify toxic Tau species, such as oligomers or fibrils, from patient samples are confounded by the difficulties of their purification without disrupting their native structure, in addition to the possibility that there are multiple toxic forms of Tau. Although in vitro approaches allowed investigators to produce large quantities of pure Tau fibrils, their biophysical, biochemical, and structural properties substantially differed from the brain-derived material. These factors complicate the rational design of antibodies targeting specific Tau proteoforms due to our limited knowledge of the relevance of these proteoforms to pathological processes, as well as the difficulties to reconstruct the complexity of native Tau proteoforms in vitro for the antibody design and screening. Therefore, the exact species of Tau targeted by antibodies are not well defined,321 and their formation pathways and clinical relevance remain unknown.
The advent of super-resolution imaging techniques, such as cryo-electron microscopy (cryo-EM), allowed for a direct structure visualization of human brain-derived Tau fibrillar cores from postmortem brain tissues of patients with different Tauopathies (Fig. 10).19,21 These studies have enabled structure based classification of Tauopathies, thus paving the way for a rational design of disease-specific therapeutics and and diagnostics.575 Recently, Cryo-EM was used to directly map the ligand-binding sites within the AD Tau fold.576 It showed that fibril core residues in multiple diseases primarily comprise the MTBR repeats R3 and R4, which is important in designing the antibodies with epitopes directed outside of these regions. However, this technique is limited to detecting the Tau sequence that constitutes the solid core of fibrils, thus overlooking the less structured Tau domains that might bind ligands, promote Tau aggregation or regulate pathological Tau spreading, for example heavily post-translationally modified proline-rich and C-terminal regions. Furthermore, robust methodological approaches are yet to be developed to isolate the putatively pathological Tau intermediate species and oligomers from the human brain in large amounts and good enough quality for cryo-EM and related techniques.
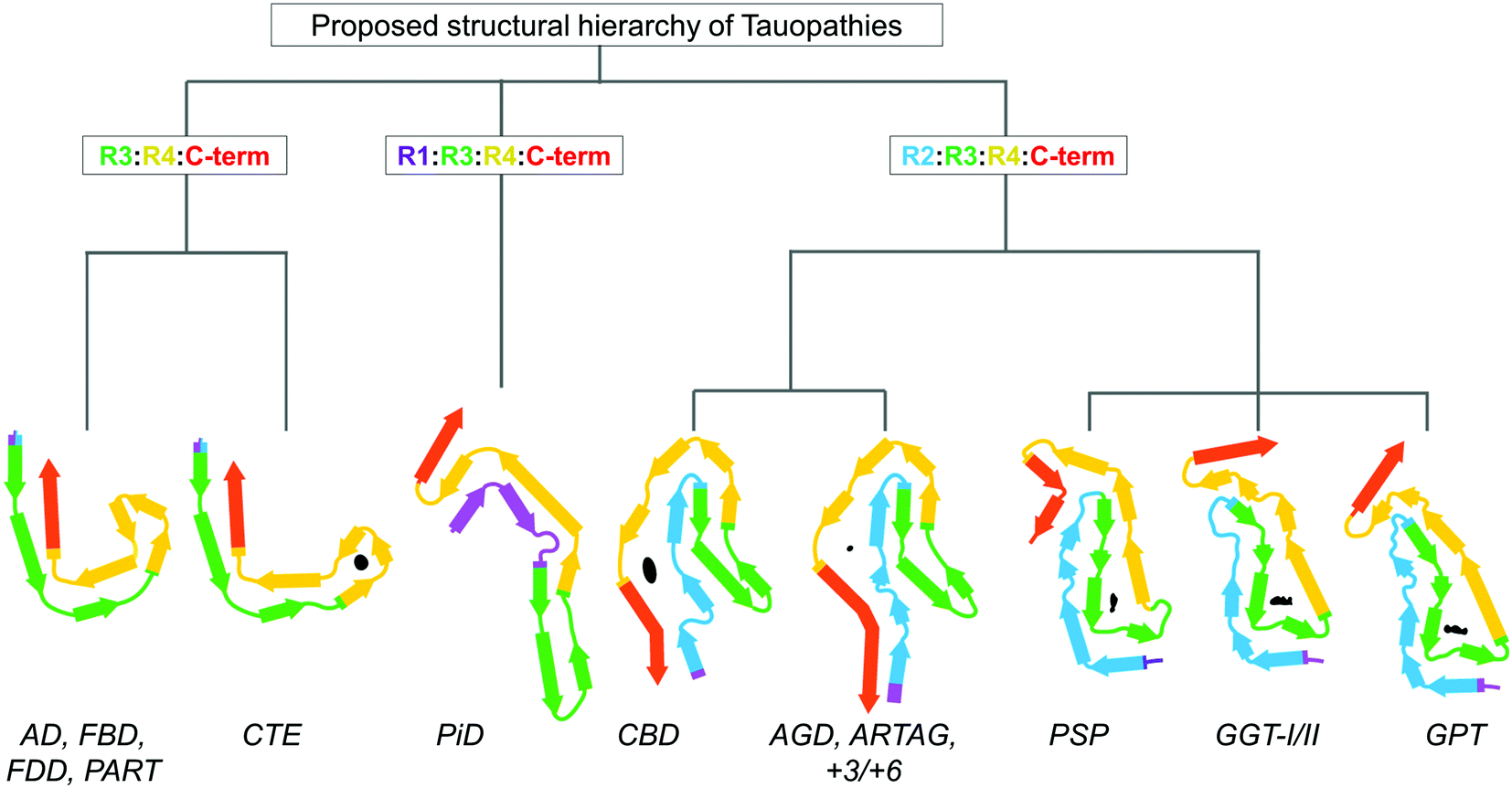 | ||
| Fig. 10 Proposed classification of Tauopathies based on the structural hierarchy of the Tau fold formations based on the similarity. Colors denote different Tau domains (reproduced with modifications from ref. 21 with permission from the author(s), under exclusive licence to Springer Nature Limited, copyright 2021). | ||
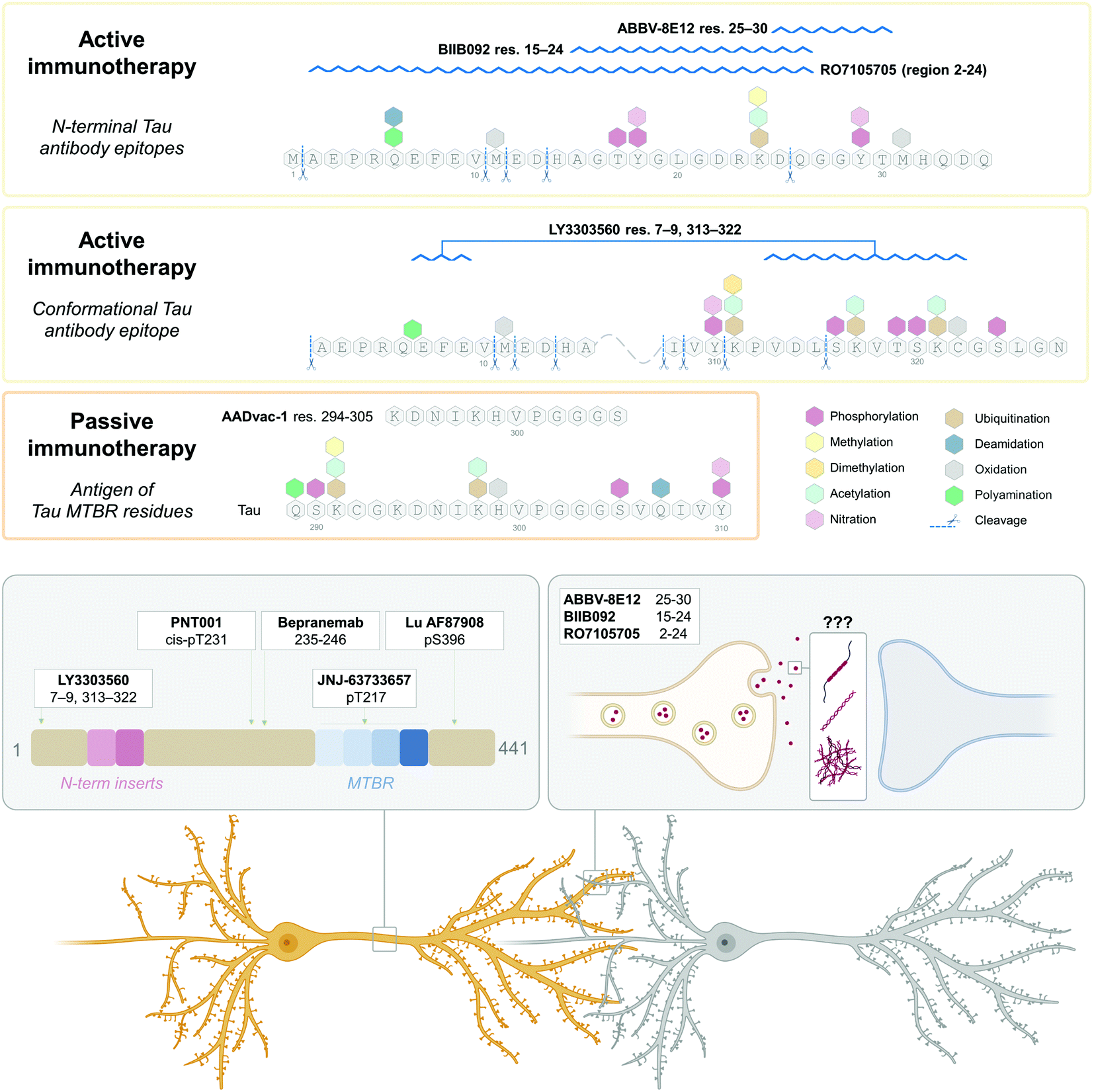 | ||
| Fig. 11 Active and passive Tau immunotherapies. Known epitopes of antibodies are aligned to the Tau sequence with highlighted PTMs. Putative intracellular and extracellular Tau targeting antibodies with epitope amino acid numbers where known. | ||
Active immunotherapies (vaccinations) for Tau include delivery of Tau sequence fragments to elicit the immune response of the recipient.577 This should arguably prevent the pathology formation, or help clear existing Tau aggregates and inclusion through their recognition by the innate immune system. The furthest therapy along the development pipeline is AADvac-1, which is a Tau peptide consisting of residues 294–305 adjunct to keyhole limpet hemocyanin.584 It was evaluated in the Phase II trial in AD (ClinicalTrials.gov Identifier: NCT02579252) with promising results that will further its evaluation in the Phase III trial. Another active Tau vaccination is ACI-35 which includes 16 copies of pS396 and pS404 Tau in a liposome carrier. This showed a robust immune response in P301L-Tau and wild-type mice, and produced antibodies that bound the NFTs, and reduced insoluble and soluble Tau levels in the brain homogenates.585 In human subjects, redesign into ACI-35.030 resulted in a higher immune response compared to the original version. Currently, no investigations in the clinical settings are underway for this therapy.
The PROTAC approach for Tau clearance has been investigated in preclinical models. TH006 peptide was designed to recruit both Tau and ubiquitin ligase von Hippel-Lindau tumor suppressor protein (Vhl), and reduce Tau levels in 3xTg-AD mice and primary neuronal cultures.601 PROTAC binding Tau and ubiquitin ligase kelch-like ECH-associated protein-1 (Keap1) efficiently reduced Tau levels in cellular SH-SY5Y human bone marrow cancer model and rodent Neuro-2a and PC-12 cell lines.602 Small molecule PROTACs that recruited ubiquitin ligase Cereblon were reported to degrade non-modified and phosphorylated Tau in human neuronal models harboring Tau mutations A152T or P301L.603 Another PROTAC recruiting molecule Cereblon was designed from the Tau PET tracer 18F-T807.604 The compound QC-01-175 cleared Tau in FTLD patient-derived neurons, resulting in a higher resilience of cells to stress. Presumably, QC-01-175 selectively bound to and degraded the monomeric aberrant FTD-associated Tau variants, as the clearance of monomeric Tau in the neurons derived from healthy controls was insignificant. Another small molecule, C004019, designed to bind both monomeric Tau and Vhl ubiquitin ligase efficiently targeted it for degradation by the proteasome in cellular and mouse models.605 C004019 was efficient at improving synaptic and cognitive deficits in hTau and 3XTg-AD mice. The reduction in the high molecular weight Tau observed was attributed to the shift in the equilibrium between soluble monomeric and oligomeric or aggregated Tau species. The Tau aggregates are not preferentially degraded by the proteasomal pathway.606,607 A related approach to targeted Tau clearance involves the direct proteolysis of bound Tau, conceptually demonstrated by the artificial synthetic apocyclen-hybrid hydrolase I2-Cu(II),608 which was designed to recognize the Tau hexapeptide PHF6, VQIVYK, present in MTBR R3 repeat. I2-Cu(II) cleaved the Tau fragments and prevented their aggregation in vitro, and reduced the overexpressed Tau-EGFP signal in the N2a cell line.
Conclusions and implications for targeting Tau
Converging evidence continues to point towards Tau aggregation as one of the central events in the pathogenesis of AD disease and other Tauopathies. The relative contribution of loss of functions vs. gain of toxic functions to the pathogenesis of Tauopathies remains unknown. However, it is clear that preventing Tau aggregation in the first place and neutralizing its seeding activity would also contribute to maintaining the normal level of Tau and thus minimize loss of Tau function. Therefore, targeting the process of Tau aggregation, instead of just specific Tau species only, remains a viable therapeutic strategy for treating Tauopathies.Despite significant advances in understanding the morphological and structural properties of Tau fibrils, many fundamental questions about what causes Tau to aggregate in the first place and the role of cofactors, Tau PTMs, and Tau interactome in regulating Tau aggregation, pathology formation, and toxicity remain unknown. In this review article, we provide an overview of our current understanding of the sequence and molecular determinants of Tau aggregation and the various experimental approaches that have shaped our understanding of its mechanisms of aggregation and role in Tauopathies. Our analyses of the literature revealed a widening gap between the complexity of Tau sequence, structure, and pathology in the brain, and the tools and model systems commonly used to investigate the mechanisms of Tau aggregation and toxicity in research laboratories. We also showed that many of the methods and assays to screen for modifiers of Tau aggregation and toxicity are carried out using unmodified Tau aggregates that do not share the same biochemical signatures or structural properties of pathological Tau. Currently, most experimental approaches do not take into account the diversity of Tau PTMs, the complexity of the Tau PTM patterns in their physiological and pathological forms, or the fact that the Tau PTM patterns change during disease progression. Therefore, it is not surprising that the conventional approaches have not borne tangible results in the clinic. We highlight the necessity to revisit the Tau aggregation processes, associated cofactors, and develop more suitable models that recapitulate the structural and biochemical diversity of pathological Tau. The extensive mechanistic studies and experimental approaches that have been used to investigate and gain insight into the role of heparin in regulating Tau aggregation are useful and can be extended to other newly discovered cofactors.
One important consideration for Tau-targeting antibody-based therapies is defining the precise epitopes or conformational states the antibodies are engineered to recognize. Tau is extensively modified, with cleavage, phosphorylation, ubiquitination, and O-GlcNAcylation potentially interfering and preventing the binding of antibodies raised and tested against non-modified forms of Tau protein produced in vitro by conventional approaches. This includes PTMs on the neighboring residues. Also, epitope masking and inaccessibility due to conformational changes in Tau might contribute to less recognition and binding by the antibodies. The design of Tau-specific antibodies should be informed by the data from patient-derived pathological Tau and using reagents that reproduce the key features of the relevant Tau proteoforms. This could be achieved using semisynthetic and synthetics strategies to produce precisely-post-translationally modified Tau, which is then fibrillized using a clean and efficient system with the addition of pathology-relevant cofactor molecules. This will allow to raise and optimize antibodies that have a higher potential to yield efficient Tau pathology modifying molecules that will be successful in clinical settings. One alternative approach is to develop cellular and animal models that recapitulate pathological Tau aggregation at the structural and biochemical levels. Today, it remains unknown whether Tau aggregates in cellular and animal models of Tauopathies possess the same core structure and morphological features of brain-derived Tau aggregates. More detailed biochemical and structural characterization of Tau aggregates in these models is essential to maximize the translation of preclinical success to more effective therapies in the clinic.
Several approaches have been employed to generate antibodies targeting different epitopes and aggregated forms of Tau species, and many are in clinical trials today. Given that many of these antibodies' design was not guided by precise information about the pathogenic Tau species in the brain, it should not surprise anyone that many of these antibodies will not succeed in clinical trials. However, the outcomes of these clinical trials will provide important insight into the potential of sequence and species-specific antibodies for the treatment of AD. They will also help guide future efforts to develop more effective antibodies and therapies. That being said, many steps could be introduced now to minimize failures or ensure that ineffective antibodies are identified early in the process, including (1) employing structure-based and computational approaches that leverage the availability of several cryo-EM structures of brain-derived Tau fibrils,609 (2) more comprehensive assessment of the biochemical properties (PTMs) of Tau species in the brain and the CSF; (3) evaluating the efficacy of antibodies in multiple animal models of Tau pathology formation and spreading, and (4) rigorous characterization of the specificity of the antibodies towards different pathologic Tau species. This could be achieved using expanded libraries of (a) recombinant Tau species (monomers, oligomers, and fibrils) bearing pathologically relevant PTMs, and (b) well-characterized soluble and insoluble Tau aggregate preparations isolated from brains of different patients with Tauopathies. This approach would determine the extent to which therapeutic antibodies can capture the diversity of pathological Tau species detected in vivo. Although cryo-EM studies of Tau fibrils suggest that the Tau fibrils in the brain are more homogeneous and possess structural features specific for each Tauopathy, it remains unclear if these studies are capturing the diversity of Tau fibrils, or mainly the dominant structures. In contrast, increasing evidence suggests that soluble oligomeric and seeding competent forms of Tau are highly heterogeneous in their size distribution and PTM patterns. Therefore, elucidating the structural properties of recombinant and brain-derived Tau oligomers is essential to any future efforts to improve the precision of therapeutic antibodies and their efficacy.
Although multiple factors may contribute to the failure of clinical trials, the wide range of Tau structures seen in the patient populations may contribute to poor targeting efficiency by the structure-based Tau aggregation inhibitors. This is especially important given recent findings revealing a high level of heterogeneity in the PTM profiles and seeding activity of Tau aggregates from AD patients' brains.118 Patient stratification based on the disease stage, informed by the robust peripheral biomarkers, may be crucial to determine the appropriate target population and the windows of therapeutic intervention for disease progress-modifying effects. This may involve the early and pre-/asymptomatic stages, where the Tau pathology is developing but has not yet fully progressed and amassed in multiple brain regions. The clearance of the already-formed Tau fibrillar burden and prevention of its spreading is being targeted by immuno-based anti-Tau therapies discussed above.
Like targeted immunotherapies, it is crucial to know precisely which Tau species should be targeted by PROTACs. These target species may include (1) monomeric Tau to reduce the total Tau levels and maintain an equilibrium of Tau species in favor of soluble monomers, (2) monomeric Tau harboring disease-associated mutations, (3) small oligomers that can be efficiently cleared by the proteasome before exerting cytotoxicity or proceeding to form larger aggregates, or (4) seeding competent fibrillar Tau aggregates. Artificially designed hydrolases targeting specific Tau oligomeric and fibrillar species, and directly degrading them, represents a promising strategy to clear the incipient or established Tau pathology.
Although this review focuses on understanding the grammar of Tau aggregation and Tau aggregate-targeting therapies, we would like to re-emphasize that more investments are needed to understand how the loss of normal Tau due to misfolding, aggregation, and seeding contributes to neurodegeneration and clinical symptoms at different stages of disease progression. It is our opinion that the most effective therapies will be combination therapies that, firstly, prevent Tau misfolding and aggregation, and, secondly, neutralize existing bioactive Tau aggregates. The clinical heterogeneity of the AD and other Tauopathies combined with increasing evidence of the possible roles of other Tau-aggregation independent mechanisms in the pathogenesis of Tauopathies suggest that future therapies will involve personalized combination therapies based on molecular mechanisms associated with each disease subtype. Such therapies are more likely to be effective at slowing the progression of the Tauopathies at different stages of disease diagnosis. The sooner we embrace the concept of combination personalized therapies, the better.
Conflicts of interest
Hilal Lashuel has received funding from industry to support research on neurodegenerative diseases, including from Merck Serono, UCB, Idorsia and Abbvie. These companies had no specific role in the in the conceptualization and preparation of and decision to publish this work. H. A. L. is also the Founder and Chief Scientific Officer of ND BioSciences SA, a company that develops diagnostics and treatments for neurodegenerative diseases based on platforms that reproduce the complexity and diversity of proteins implicated in neurodegenerative diseases and their pathologies. Galina Limorenko has no conflicts to declare.Acknowledgements
We would like to thank Laura Gasparini, Somanath Jagannath, Rajasekhar Kolla, Senthil Kumar Thangaraj, Pedro Santana Magalhães and Theodora Panagaki for providing valuable critical assessment of the manuscript.References
- D. G. Drubin and M. W. Kirschner, Tau protein function in living cells, J. Cell Biol., 1986, 103(6), 2739 CrossRef CAS PubMed.
- I. Sotiropoulos, M.-C. Galas, J. M. Silva, E. Skoulakis, S. Wegmann and M. B. Maina, et al., Atypical, non-standard functions of the microtubule associated Tau protein, Acta Neuropathol. Commun., 2017, 5(1), 91 CrossRef.
- R. Dixit, J. L. Ross, Y. E. Goldman and E. L. Holzbaur, Differential regulation of dynein and kinesin motor proteins by tau, Science, 2008, 319(5866), 1086–1089 CrossRef CAS PubMed.
- S. Barghorn, Q. Zheng-Fischhöfer, M. Ackmann, J. Biernat, M. Von Bergen and E.-M. Mandelkow, et al., Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias, Biochemistry, 2000, 39(38), 11714–11721 CrossRef CAS.
- H. Ding, T. A. Matthews and G. V. Johnson, Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation, J. Biol. Chem., 2006, 281(28), 19107–19114 CrossRef CAS.
- J. Baudier and R. D. Cole, Interactions between the microtubule-associated tau proteins and S100b regulate tau phosphorylation by the Ca2+/calmodulin-dependent protein kinase II, J. Biol. Chem., 1988, 263(12), 5876–5883 CrossRef CAS.
- M.-F. Carlier, C. Simon, R. Cassoly and L.-A. Pradel, Interaction between microtubule-associated protein tau and spectrin, Biochimie, 1984, 66(4), 305–311 CrossRef CAS PubMed.
- A. Samsonov, J.-Z. Yu, M. Rasenick and S. V. Popov, Tau interaction with microtubules in vivo, J. Cell Sci., 2004, 117(25), 6129–6141 CrossRef CAS PubMed.
- A. Venkatramani and D. Panda, Regulation of neuronal microtubule dynamics by tau: Implications for tauopathies, Int. J. Biol. Macromol., 2019, 133, 473–483 CrossRef CAS PubMed.
- C. L. Masters, R. Bateman, K. Blennow, C. C. Rowe, R. A. Sperling and J. L. Cummings, Alzheimer’s disease, Nat. Rev. Dis. Primers, 2015, 1(15056), 434 Search PubMed.
- G. G. Glenner and C. W. Wong, Alzheimer's disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein, Biochem. Biophys. Res. Commun., 1984, 120(3), 885–890 CrossRef CAS.
- V. M. Lee, M. Goedert and J. Q. Trojanowski, Neurodegenerative tauopathies, Annu. Rev. Neurosci., 2001, 24(1), 1121–1159 CrossRef CAS.
- M. Goedert, R. Jakes, M. Spillantini, M. Hasegawa, M. Smith and R. Crowther, Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans, Nature, 1996, 383(6600), 550–553 CrossRef CAS.
- M. Kidd, Paired Helical Filaments in Electron Microscopy of Alzheimer's Disease, Nature, 1963, 197(4863), 192–193 CrossRef CAS PubMed.
- B. E. Tomlinson, G. Blessed and M. Roth, Observations on the brains of demented old people, J. Neurol. Sci., 1970, 11(3), 205–242 CrossRef CAS.
- P. H. BrionJP, J. Nunez and J. Flament-Durand, Mise en evidence immunologique de la proteine tau au niveau des lesions de degenerescence neurofibril-laire de la maladie d'Alzheimer, Arch. Biol., 1985, 95, 229–235 Search PubMed.
- M. E. Murray, N. Kouri, W.-L. Lin, C. R. Jack, D. W. Dickson and P. Vemuri, Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias, Alzheimer's Res. Ther., 2014, 6(1), 1 CrossRef.
- Y. Shi, W. Zhang, Y. Yang, A. G. Murzin, B. Falcon and A. Kotecha, et al., Structure-based classification of tauopathies, Nature, 2021, 598(7880), 359–363 CrossRef CAS.
- S. H. Scheres, W. Zhang, B. Falcon and M. Goedert, Cryo-EM structures of tau filaments, Curr. Opin. Struct. Biol., 2020, 64, 17–25 CrossRef CAS.
- G. Lippens and B. Gigant, Elucidating Tau function and dysfunction in the era of cryo-EM, J. Biol. Chem., 2019, 294(24), 9316–9325 CrossRef CAS PubMed.
- Y. Shi, W. Zhang, Y. Yang, A. Murzin, B. Falcon and A. Kotecha, et al., Structure-based Classification of Tauopathies, bioRxiv, 2021, 2021.05.28.446130 Search PubMed.
- S. Kerrien, B. Aranda, L. Breuza, A. Bridge, F. Broackes-Carter and C. Chen, et al., The IntAct molecular interaction database in 2012, Nucleic Acids Res., 2012, 40(D1), D841–D846 CrossRef CAS.
- R. Oughtred, C. Stark, B.-J. Breitkreutz, J. Rust, L. Boucher and C. Chang, et al., The BioGRID interaction database: 2019 update, Nucleic Acids Res., 2019, 47(D1), D529–D541 CrossRef CAS PubMed.
- B. Lang, A. Armaos and G. G. Tartaglia, RNAct: Protein–RNA interaction predictions for model organisms with supporting experimental data, Nucleic Acids Res., 2019, 47(D1), D601–D606 CrossRef CAS PubMed.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler and J. M. Cherry, et al., Gene ontology: tool for the unification of biology, Nat. Genet., 2000, 25(1), 25–29 CrossRef CAS.
- A. Vignon, L. Salvador-Prince, S. Lehmann, V. Perrier and J. Torrent, Deconstructing Alzheimer’s Disease: How to Bridge the Gap between Experimental Models and the Human Pathology?, Int. J. Mol. Sci., 2021, 22(16), 8769 CrossRef CAS.
- N. Candelise, S. Scaricamazza, I. Salvatori, A. Ferri, C. Valle and V. Manganelli, et al., Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications, Int. J. Mol. Sci., 2021, 22(11), 6016 CrossRef CAS PubMed.
- H. A. Lashuel, Rethinking protein aggregation and drug discovery in neurodegenerative diseases: Why we need to embrace complexity?, Curr. Opin. Chem. Biol., 2021, 64, 67–75 CrossRef CAS.
- M. Goedert, M. G. Spillantini, R. Jakes, D. Rutherford and R. A. Crowther, Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease, Neuron, 1989, 3(4), 519–526 CrossRef CAS.
- D. Trabzuni, S. Wray, J. Vandrovcova, A. Ramasamy, R. Walker and C. Smith, et al., MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies, Hum. Mol. Genet., 2012, 21(18), 4094–4103 CrossRef CAS.
- A. Boutajangout, A. Boom, K. Leroy and J. P. Brion, Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer's disease, FEBS Lett., 2004, 576(1–2), 183–189 CrossRef CAS.
- C. B. Chambers, J. M. Lee, J. C. Troncoso, S. Reich and N. A. Muma, Overexpression of four-repeat tau mRNA isoforms in progressive supranuclear palsy but not in Alzheimer's disease, Ann. Neurol., 1999, 46(3), 325–332 CrossRef CAS.
- R. De Silva, T. Lashley, G. Gibb, D. Hanger, A. Hope and A. Reid, et al., Pathological inclusion bodies in tauopathies contain distinct complements of tau with three or four microtubule-binding repeat domains as demonstrated by new specific monoclonal antibodies, Neuropathol. Appl. Neurobiol., 2003, 29(3), 288–302 CrossRef CAS PubMed.
- A. Delacourte, N. Sergeant, A. Wattez, D. Gauvreau and Y. Robitaille, Vulnerable neuronal subsets in Alzheimer's and Pick's disease are distinguished by their τ isoform distribution and phosphorylation, Ann. Neurol., 1998, 43(2), 193–204 CrossRef CAS.
- N. Sergeant, A. Wattez and A. Delacourte, Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms, J. Neurochem., 1999, 72(3), 1243–1249 CrossRef CAS PubMed.
- I. D'Souza and G. D. Schellenberg, Regulation of tau isoform expression and dementia, Biochim. Biophys. Acta, Mol. Basis Dis., 2005, 1739(2), 104–115 CrossRef PubMed.
- L. Qiang, X. Sun, T. O. Austin, H. Muralidharan, D. C. Jean and M. Liu, et al., Tau does not stabilize axonal microtubules but rather enables them to have long labile domains, Curr. Biol., 2018, 28(13), 2181–2189 CrossRef CAS PubMed.
- C. W. Chang, E. Shao and L. Mucke, Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies, Science., 2021, 371(6532), eabb8255 CrossRef CAS.
- E. Marciniak, A. Leboucher, E. Caron, T. Ahmed, A. Tailleux and J. Dumont, et al., Tau deletion promotes brain insulin resistance, J. Exp. Med., 2017, 214(8), 2257–2269 CrossRef CAS.
- T. Kimura, D. J. Whitcomb, J. Jo, P. Regan, T. Piers and S. Heo, et al., Microtubule-associated protein tau is essential for long-term depression in the hippocampus, Philos. Trans. R. Soc. London, Ser. B, 2013, 369(1633), 20130144 CrossRef.
- T. Ondrejcak, N. W. Hu, Y. Qi, I. Klyubin, G. T. Corbett and G. Fraser, et al., Soluble tau aggregates inhibit synaptic long-term depression and amyloid β-facilitated LTD in vivo, Neurobiol. Dis., 2019, 127, 582–590 CrossRef CAS.
- C. A. Lasagna-Reeves, D. L. Castillo-Carranza, U. Sengupta, A. L. Clos, G. R. Jackson and R. Kayed, Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice, Mol. Neurodegener., 2011, 6, 39 CrossRef.
- M. Robbins, E. Clayton and G. S. Kaminski Schierle, Synaptic tau: A pathological or physiological phenomenon?, Acta Neuropathol. Commun., 2021, 9(1), 149 CrossRef.
- D. Dupret, J.-M. Revest, M. Koehl, F. Ichas, F. De Giorgi and P. Costet, et al., Spatial relational memory requires hippocampal adult neurogenesis, PLoS One, 2008, 3(4), e1959 CrossRef PubMed.
- A. Pino, G. Fumagalli, F. Bifari and I. Decimo, New neurons in adult brain: distribution, molecular mechanisms and therapies, Biochem. Pharmacol., 2017, 141, 4–22 CrossRef CAS PubMed.
- S. Houben, M. Homa, Z. Yilmaz, K. Leroy, J. P. Brion and K. Ando, Tau Pathology and Adult Hippocampal Neurogenesis: What Tau Mouse Models Tell us?, Front. Neurol., 2021, 12, 610330 CrossRef.
- D. D. Li Puma, R. Piacentini and C. Grassi, Does Impairment of Adult Neurogenesis Contribute to Pathophysiology of Alzheimer's Disease? A Still Open Question, Front. Mol. Neurosci., 2020, 13, 578211 CrossRef CAS PubMed.
- S. Goldman, Glia as neural progenitor cells, Trends Neurosci., 2003, 26(11), 590–596 CrossRef CAS.
- J. Morrens, W. Van Den Broeck and G. Kempermann, Glial cells in adult neurogenesis, Glia, 2012, 60(2), 159–174 CrossRef PubMed.
- C. Andorfer, C. M. Acker, Y. Kress, P. R. Hof, K. Duff and P. Davies, Cell-Cycle Reentry and Cell Death in Transgenic Mice Expressing Nonmutant Human Tau Isoforms, J. Neurosci., 2005, 25(22), 5446–5454 CrossRef CAS.
- T. Arendt, M. K. Brückner, B. Mosch and A. Lösche, Selective cell death of hyperploid neurons in Alzheimer's disease, Am. J. Pathol., 2010, 177(1), 15–20 CrossRef PubMed.
- H.-G. Lee, G. Casadesus, X. Zhu, R. J. Castellani, A. McShea and G. Perry, et al., Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer's disease, Neurochem. Int., 2009, 54(2), 84–88 CrossRef CAS.
- P. Lei, S. Ayton, S. Moon, Q. Zhang, I. Volitakis and D. I. Finkelstein, et al., Motor and cognitive deficits in aged tau knockout mice in two background strains, Mol. Neurodegener., 2014, 9, 29 CrossRef PubMed.
- C. Andorfer, Y. Kress, M. Espinoza, R. De Silva, K. L. Tucker and Y. A. Barde, et al., Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms, J. Neurochem., 2003, 86(3), 582–590 CrossRef CAS PubMed.
- N. Wijesekara, R. A. Gonçalves, R. Ahrens, F. G. De Felice and P. E. Fraser, Tau ablation in mice leads to pancreatic β cell dysfunction and glucose intolerance, FASEB J., 2018, 32(6), 3166–3173 CrossRef CAS.
- R. A. Gonçalves, N. Wijesekara, P. E. Fraser and F. G. De Felice, Behavioral abnormalities in knockout and humanized tau mice, Front. Endocrinol., 2020, 11, 124 CrossRef.
- A. C. Hradek, H.-P. Lee, S. L. Siedlak, S. L. Torres, W. Jung and A. H. Han, et al., Distinct chronology of neuronal cell cycle re-entry and tau pathology in the 3xTg-AD mouse model and Alzheimer's disease patients, J. Alzheimer's Dis., 2015, 43(1), 57–65 CAS.
- L. Li, T. Cheung, J. Chen and K. Herrup, A comparative study of five mouse models of Alzheimer’s disease: cell cycle events reveal new insights into neurons at risk for death, Int. J. Alzheimer’s Dis., 2011, 2011, 171464 Search PubMed.
- M. E. Seward, E. Swanson, A. Norambuena, A. Reimann, J. N. Cochran and R. Li, et al., Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer's disease, J. Cell Sci., 2013, 126(Pt 5), 1278–1286 CrossRef CAS PubMed.
- B. Trinczek, J. Biernat, K. Baumann, E.-M. Mandelkow and E. Mandelkow, Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules, Mol. Biol. Cell, 1995, 6(12), 1887–1902 CrossRef CAS.
- P. Barbier, O. Zejneli, M. Martinho, A. Lasorsa, V. Belle and C. Smet-Nocca, et al., Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects, Front. Aging Neurosci., 2019, 11, 204 CrossRef CAS.
- N. I. Trushina, L. Bakota, A. Y. Mulkidjanian and R. Brandt, The Evolution of Tau Phosphorylation and Interactions, Front. Aging Neurosci., 2019, 11, 256 CrossRef CAS PubMed.
- E. Drummond, G. Pires, C. MacMurray, M. Askenazi, S. Nayak and M. Bourdon, et al., Phosphorylated tau interactome in the human Alzheimer's disease brain, Brain, 2020, 143(9), 2803–2817 CrossRef.
- B. Kovacech, N. Zilka and M. Novak, New age of neuroproteomics in Alzheimer's disease research, Cell. Mol. Neurobiol., 2009, 29(6-7), 799–805 CrossRef CAS PubMed.
- E. Drummond and T. Wisniewski, Using Proteomics to Understand Alzheimer's Disease Pathogenesis, in Alzheimer's Disease, ed. Wisniewski T., Codon Publications Copyright: The Authors, Brisbane (AU), 2019 Search PubMed.
- J. Sinsky, P. Majerova, A. Kovac, M. Kotlyar, I. Jurisica and J. Hanes, Physiological Tau Interactome in Brain and Its Link to Tauopathies, J Proteome Res., 2020, 19(6), 2429–2442 CrossRef CAS.
- C. G. Gunawardana, M. Mehrabian, X. Wang, I. Mueller, I. B. Lubambo and J. E. Jonkman, et al., The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome, Mol. Cell. Proteomics, 2015, 14(11), 3000–3014 CrossRef CAS.
- H. Choi, H. J. Kim, J. Yang, S. Chae, W. Lee and S. Chung, et al., Acetylation changes tau interactome to degrade tau in Alzheimer's disease animal and organoid models, Aging Cell, 2020, 19(1), e13081 CrossRef CAS PubMed.
- P. Wang, G. Joberty, A. Buist, A. Vanoosthuyse, I. C. Stancu and B. Vasconcelos, et al., Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo, Acta Neuropathol., 2017, 133(5), 731–749 CrossRef CAS.
- A. Sultan, F. Nesslany, M. Violet, S. Bégard, A. Loyens and S. Talahari, et al., Nuclear tau, a key player in neuronal DNA protection, J. Biol. Chem., 2011, 286(6), 4566–4575 CrossRef CAS.
- M. Violet, L. Delattre, M. Tardivel, A. Sultan, A. Chauderlier and R. Caillierez, et al., A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions, Front. Cell. Neurosci., 2014, 8, 84 Search PubMed.
- L. Gil, S. A. Niño, G. Capdeville and ME. Aging Jiménez-Capdeville, and Alzheimer's disease connection: Nuclear Tau and lamin A, Neurosci. Lett., 2021, 749, 135741 CrossRef CAS.
- H.-U. Klein, C. McCabe, E. Gjoneska, S. E. Sullivan, B. J. Kaskow and A. Tang, et al., Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer's human brains, Nat. Neurosci., 2019, 22(1), 37–46 CrossRef CAS.
- A. Mietelska-Porowska, U. Wasik, M. Goras, A. Filipek and G. Niewiadomska, Tau protein modifications and interactions: their role in function and dysfunction, Int. J. Mol. Sci., 2014, 15(3), 4671–4713 CrossRef PubMed.
- D. P. Hanger, B. H. Anderton and W. Noble, Tau phosphorylation: the therapeutic challenge for neurodegenerative disease, Trends Mol. Med., 2009, 15(3), 112–119 CrossRef CAS PubMed.
- J. F. Crary, J. Q. Trojanowski, J. A. Schneider, J. F. Abisambra, E. L. Abner and I. Alafuzoff, et al., Primary age-related tauopathy (PART): a common pathology associated with human aging, Acta Neuropathol., 2014, 128(6), 755–766 CrossRef CAS.
- H. Braak and K. Del Tredici, The preclinical phase of the pathological process underlying sporadic Alzheimer's disease, Brain, 2015, 138(10), 2814–2833 CrossRef.
- T. M. Harrison, R. La Joie, A. Maass, S. L. Baker, K. Swinnerton and L. Fenton, et al., Longitudinal tau accumulation and atrophy in aging and alzheimer disease, Ann. Neurol., 2019, 85(2), 229–240 CrossRef CAS.
- V. J. Lowe, T. J. Bruinsma, H.-K. Min, E. S. Lundt, P. Fang and M. L. Senjem, et al., Elevated medial temporal lobe and pervasive brain tau-PET signal in normal participants, Alzheimer's Dementia: Diagnosis, Assess. Dis. Monit., 2018, 10, 210–216 CrossRef PubMed.
- A. Maass, S. N. Lockhart, T. M. Harrison, R. K. Bell, T. Mellinger and K. Swinnerton, et al., Entorhinal tau pathology, episodic memory decline, and neurodegeneration in aging, J. Neurosci., 2018, 38(3), 530–543 CrossRef CAS.
- V. J. Lowe, T. J. Bruinsma, H. J. Wiste, H.-K. Min, S. D. Weigand and P. Fang, et al., Cross-sectional associations of tau-PET signal with cognition in cognitively unimpaired adults, Neurology, 2019, 93(1), e29–e39 CrossRef CAS PubMed.
- H. Braak and E. Braak, Neuropathological stageing of Alzheimer-related changes, Acta Neuropathol., 1991, 82(4), 239–259 CrossRef CAS PubMed.
- D. R. Williams, J. L. Holton, C. Strand, A. Pittman, R. de Silva and A. J. Lees, et al., Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson's syndrome, Brain, 2007, 130(6), 1566–1576 CrossRef.
- M. S. Forman, V. Zhukareva, C. Bergeron, S. S. M. Chin, M. Grossman and C. Clark, et al., Signature Tau Neuropathology in Gray and White Matter of Corticobasal Degeneration, Am. J. Pathol., 2002, 160(6), 2045–2053 CrossRef CAS.
- M. Iovino, S. Agathou, A. González-Rueda, M. Del Castillo Velasco-Herrera, B. Borroni and A. Alberici, et al., Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations, Brain, 2015, 138(11), 3345–3359 CrossRef PubMed.
- D. W. Dickson, N. Kouri, M. E. Murray and K. A. Josephs, Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau), J. Mol. Neurosci., 2011, 45(3), 384–389 CrossRef CAS.
- M. Schöll, S. N. Lockhart, D. R. Schonhaut, J. P. O’Neil, M. Janabi and R. Ossenkoppele, et al., PET imaging of tau deposition in the aging human brain, Neuron, 2016, 89(5), 971–982 CrossRef PubMed.
- A. J. Schwarz, P. Yu, B. B. Miller, S. Shcherbinin, J. Dickson and M. Navitsky, et al., Regional profiles of the candidate tau PET ligand 18 F-AV-1451 recapitulate key features of Braak histopathological stages, Brain, 2016, 139(5), 1539–1550 CrossRef PubMed.
- Y. Tsuboi, Z. K. Wszolek, N. Graff-Radford, N. Cookson and D. W. Dickson, Tau pathology in the olfactory bulb correlates with Braak stage, Lewy body pathology and apolipoprotein ε4, Neuropathol. Appl. Neurobiol., 2003, 29(5), 503–510 CrossRef CAS.
- V. J. Lowe, H. J. Wiste, M. L. Senjem, S. D. Weigand, T. M. Therneau and B. F. Boeve, et al., Widespread brain tau and its association with ageing, Braak stage and Alzheimer's dementia, Brain, 2017, 141(1), 271–287 CrossRef.
- M. G. Spillantini and M. Goedert, Tau protein pathology in neurodegenerative diseases, Trends Neurosci., 1998, 21(10), 428–433 CrossRef CAS.
- W. Yang, L. C. Ang and M. J. Strong, Tau protein aggregation in the frontal and entorhinal cortices as a function of aging, Dev. Brain Res., 2005, 156(2), 127–138 CrossRef CAS PubMed.
- K. Sander, T. Lashley, P. Gami, T. Gendron, M. F. Lythgoe and J. D. Rohrer, et al., Characterization of tau positron emission tomography tracer [18F] AV-1451 binding to postmortem tissue in Alzheimer's disease, primary tauopathies, and other dementias, Alzheimer's Dementia, 2016, 12(11), 1116–1124 CrossRef.
- F. Braak, H. Braak and E.-M. Mandelkow, A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads, Acta Neuropathol., 1994, 87(6), 554–567 CrossRef.
- C. Duyckaerts, T. Uchihara, D. Seilhean, Y. He and J. J. Hauw, Dissociation of Alzheimer type pathology in a disconnected piece of cortex, Acta Neuropathol., 1997, 93(5), 501–507 CrossRef CAS PubMed.
- O. J. Olajide, M. E. Suvanto and C. A. Chapman, Molecular mechanisms of neurodegeneration in the entorhinal cortex that underlie its selective vulnerability during the pathogenesis of Alzheimer's disease, Biol. Open, 2021, 10, 1 Search PubMed.
- G. N. Bischof, H. Endepols, T. van Eimeren and A. Drzezga, Tau-imaging in neurodegeneration, Methods, 2017, 130, 114–123 CrossRef CAS.
- K. A. Johnson, A. Schultz, R. A. Betensky, J. A. Becker, J. Sepulcre and D. Rentz, et al., Tau positron emission tomographic imaging in aging and early A lzheimer disease, Ann. Neurol., 2016, 79(1), 110–119 CrossRef.
- C. E. G. Leyns, J. D. Ulrich, M. B. Finn, F. R. Stewart, L. J. Koscal and J. Remolina Serrano, et al., TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(43), 11524–11529 CrossRef CAS.
- M. Gratuze, C. E. G. Leyns, A. D. Sauerbeck, M.-K. St-Pierre, M. Xiong and N. Kim, et al., Impact of TREM2R47H variant on tau pathology–induced gliosis and neurodegeneration, J. Clin. Invest., 2020, 130(9), 4954–4968 CrossRef CAS.
- Z.-G. Zhang, Y. Li, C. T. Ng and Y.-Q. Song, Inflammation in Alzheimer's disease and molecular genetics: recent update, Arch. Immunol. Ther. Exp., 2015, 63(5), 333–344 CrossRef CAS PubMed.
- S. Narasimhan, L. Changolkar, D. M. Riddle, A. Kats, A. Stieber and S. A. Weitzman, et al., Human tau pathology transmits glial tau aggregates in the absence of neuronal tau, J. Exp. Med., 2020, 217(2), e20190783 CrossRef.
- G. G. Kovacs, Astroglia and Tau: New Perspectives, Front. Aging Neurosci., 2020, 12, 96 CrossRef CAS.
- N. Maphis, G. Xu, O. N. Kokiko-Cochran, S. Jiang, A. Cardona and R. M. Ransohoff, et al., Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain, Brain, 2015, 138(Pt 6), 1738–1755 CrossRef.
- F. Leng and P. Edison, Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nature Reviews, Neurology, 2021, 17(3), 157–172 Search PubMed.
- H. Asai, S. Ikezu, S. Tsunoda, M. Medalla, J. Luebke and T. Haydar, et al., Depletion of microglia and inhibition of exosome synthesis halt tau propagation, Nat. Neurosci., 2015, 18(11), 1584–1593 CrossRef CAS.
- H. Fu, A. Possenti, R. Freer, Y. Nakano, N. C. Hernandez Villegas and M. Tang, et al., A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology, Nat. Neurosci., 2019, 22(1), 47–56 CrossRef CAS.
- J. W. Vogel, Y. Iturria-Medina, O. T. Strandberg, R. Smith, E. Levitis and A. C. Evans, et al., Spread of pathological tau proteins through communicating neurons in human Alzheimer's disease, Nat. Commun., 2020, 11(1), 2612 CrossRef CAS PubMed.
- Y. Decker, E. Németh, R. Schomburg, A. Chemla, L. Fülöp and M. D. Menger, et al., Decreased pH in the aging brain and Alzheimer's disease, Neurobiol. Aging, 2021, 101, 40–49 CrossRef CAS PubMed.
- L. Ponto, S. Schultz, M. Jacob, Y. Menda, J. Wemmie and V. Magnotta, Brain pH and Alzheimer's pathology, J. Nucl. Med., 2014, 55(supplement 1), 192 Search PubMed.
- X. Yin, C. Zhao, Y. Qiu, Z. Zhou, J. Bao and W. Qian, Dendritic/Post-synaptic Tau and Early Pathology of Alzheimer's Disease, Front. Mol. Neurosci., 2021, 14, 671779 CrossRef PubMed.
- R. A. Charafeddine, W. A. Cortopassi, P. Lak, R. Tan, R. J. McKenney and M. P. Jacobson, et al., Tau repeat regions contain conserved histidine residues that modulate microtubule-binding in response to changes in pH, J. Biol. Chem., 2019, 294(22), 8779–8790 CrossRef.
- M. Silva and S. J. Haggarty, Tauopathies: Deciphering Disease Mechanisms to Develop Effective Therapies, Int. J. Mol. Sci., 2020, 21(23), 8948 CrossRef CAS.
- M. Fernández-Nogales, J. R. Cabrera, M. Santos-Galindo, J. J. Hoozemans, I. Ferrer and A. J. Rozemuller, et al., Huntington's disease is a four-repeat tauopathy with tau nuclear rods, Nat. Med., 2014, 20(8), 881–885 CrossRef.
- W. R. Galpern and A. E. Lang, Interface between tauopathies and synucleinopathies: a tale of two proteins, Ann. Neurol., 2006, 59(3), 449–458 CrossRef CAS PubMed.
- M. J. Strong, N. S. Donison and K. Volkening, Alterations in Tau Metabolism in ALS and ALS-FTSD, Front. Neurol., 2020, 11, 598907 CrossRef.
- T. J. Betthauser, R. L. Koscik, E. M. Jonaitis, S. L. Allison, K. A. Cody and C. M. Erickson, et al., Amyloid and tau imaging biomarkers explain cognitive decline from late middle-age, Brain, 2019, 143(1), 320–335 CrossRef.
- S. Dujardin, C. Commins, A. Lathuiliere, P. Beerepoot, A. R. Fernandes and T. V. Kamath, et al., Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease, Nat. Med., 2020, 26(8), 1256–1263 CrossRef CAS PubMed.
- S. Hameed, J. L. Fuh, V. Senanarong, E. G. M. Ebenezer, I. Looi and J. C. Dominguez, et al., Role of Fluid Biomarkers and PET Imaging in Early Diagnosis and its Clinical Implication in the Management of Alzheimer's Disease, J. Alzheimer's Dis. Rep., 2020, 4(1), 21–37 Search PubMed.
- Z. K. Wszolek, Y. Tsuboi, B. Ghetti, S. Pickering-Brown, Y. Baba and W. P. Cheshire, Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), Orphanet J. Rare Dis., 2006, 1, 30 CrossRef.
- J. Groen and L. Endtz, Hereditary Pick's disease: Second re-examination of a large family and discussion of other hereditary cases, with particular reference to electroencephalography and computerized tomography, Brain, 1982, 105(3), 443–459 CrossRef PubMed.
- H. Braak, I. Alafuzoff, T. Arzberger, H. Kretzschmar and K. Del Tredici, Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry, Acta Neuropathol., 2006, 112, 389 CrossRef PubMed.
- H. Zhang, Y. Cao, L. Ma, Y. Wei and H. Li, Possible Mechanisms of Tau Spread and Toxicity in Alzheimer's Disease, Front. Cell Dev. Biol., 2021, 9, 707268 CrossRef.
- M. Yu, O. Sporns and A. J. Saykin, The human connectome in Alzheimer disease—relationship to biomarkers and genetics, Nat. Rev. Neurol., 2021, 17(9), 545–563 CrossRef CAS PubMed.
- C. Bancher, I. Grundke-Iqbal, K. Iqbal, V. Fried, H. Smith and H. Wisniewski, Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease, Brain Res., 1991, 539(1), 11–18 CrossRef CAS PubMed.
- H. Braak and K. Del Tredici, Spreading of Tau Pathology in Sporadic Alzheimer's Disease Along Cortico-cortical Top-Down Connections, Cereb. Cortex, 2018, 28(9), 3372–3384 CrossRef PubMed.
- S. Dujardin and B. T. Hyman, Tau Prion-Like Propagation: State of the Art and Current Challenges, Adv. Exp. Med. Biol., 2019, 1184, 305–325 CrossRef CAS.
- N. Annadurai, J. B. De Sanctis, M. Hajdúch and V. Das, Tau secretion and propagation: Perspectives for potential preventive interventions in Alzheimer's disease and other tauopathies, Exp. Neurol., 2021, 343, 113756 CrossRef CAS.
- L. Song, E. A. Wells and A. S. Robinson, Critical Molecular and Cellular Contributors to Tau Pathology, Biomedicines, 2021, 9(2), 190 CrossRef CAS PubMed.
- P. d'Errico and M. Meyer-Luehmann, Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer's Disease, Front. Aging Neurosci., 2020, 12, 265 CrossRef PubMed.
- J. P. Quinn, N. J. Corbett, K. A. Kellett and N. M. Hooper, Tau proteolysis in the pathogenesis of tauopathies: neurotoxic fragments and novel biomarkers, J. Alzheimer's Dis., 2018, 63(1), 13–33 CAS.
- C. Cicognola, G. Brinkmalm, J. Wahlgren, E. Portelius, J. Gobom and N. C. Cullen, et al., Novel tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in Alzheimer's disease, Acta Neuropathol., 2019, 137(2), 279–296 CrossRef CAS.
- H. Wesseling, W. Mair, M. Kumar, C. N. Schlaffner, S. Tang and P. Beerepoot, et al., Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease, Cell, 2020, 183(6), 1699–1713 CrossRef CAS.
- S. Dujardin, C. Commins, A. Lathuiliere, P. Beerepoot, A. R. Fernandes and T. V. Kamath, et al., Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease, Nat. Med., 2020, 26(8), 1256–1263 CrossRef CAS.
- M. Hasegawa, T. Arai and Y. Ihara, Immunochemical evidence that fragments of phosphorylated MAP5 (MAP1B) are bound to neurofibrillary tangles in Alzheimer's disease, Neuron, 1990, 4(6), 909–918 CrossRef CAS.
- K. S. Kosik, L. K. Duffy, M. M. Dowling, C. Abraham, A. McCluskey and D. J. Selkoe, Microtubule-associated protein 2: monoclonal antibodies demonstrate the selective incorporation of certain epitopes into Alzheimer neurofibrillary tangles, Proc. Natl. Acad. Sci. U. S. A., 1984, 81(24), 7941–7945 CrossRef CAS.
- B. H. Anderton, D. Breinburg, M. J. Downes, P. J. Green, B. E. Tomlinson and J. Ulrich, et al., Monoclonal antibodies show that neurofibrillary tangles and neurofilaments share antigenic determinants, Nature, 1982, 298(5869), 84–86 CrossRef CAS PubMed.
- J. Sinsky, K. Pichlerova and J. Hanes, Tau Protein Interaction Partners and Their Roles in Alzheimer's Disease and Other Tauopathies, Int. J. Mol. Sci., 2021, 22(17), 9207 CrossRef CAS PubMed.
- R. A. Armstrong, P. L. Lantos and N. J. Cairns, What determines the molecular composition of abnormal protein aggregates in neurodegenerative disease?, Neuropathology, 2008, 28(4), 351–365 CrossRef.
- E. Drummond, G. Pires, C. MacMurray, M. Askenazi, S. Nayak and M. Bourdon, et al., P4-530: PHOSPHORYLATED TAU INTERACTOME IN THE HUMAN ALZHEIMER'S DISEASE BRAIN, Alzheimer's Dementia, 2019, 15(7S_Part_29), P1517 CrossRef.
- B. Minjarez, M. L. V. Rustarazo, M. M. Sanchez del Pino, A. González-Robles, J. A. Sosa-Melgarejo and J. Luna-Muñoz, et al., Identification of Polypeptides in Neurofibrillary Tangles and Total Homogenates of Brains with Alzheimer's Disease by Tandem Mass Spectrometry, J. Alzheimer's Dis., 2013, 34, 239–262 CAS.
- Q. Wang, R. L. Woltjer, P. J. Cimino, C. Pan, K. S. Montine and J. Zhang, et al., Proteomic analysis of neurofibrillary tangles in Alzheimer disease identifies GAPDH as a detergent-insoluble paired helical filament tau binding protein, FASEB J., 2005, 19(7), 1–12 Search PubMed.
- C. W. Tsai, C. F. Tsai, K. H. Lin, W. J. Chen, M. S. Lin and C. C. Hsieh, et al., An investigation of the correlation between the S-glutathionylated GAPDH levels in blood and Alzheimer's disease progression, PLoS One, 2020, 15(5), e0233289 CrossRef CAS PubMed.
- X.-H. Li, J. A. Culver and E. Rhoades, Tau Binds to Multiple Tubulin Dimers with Helical Structure, J. Am. Chem. Soc., 2015, 137(29), 9218–9221 CrossRef CAS.
- J. Majewski, E. M. Jones, C. M. Vander Zanden, J. Biernat, E. Mandelkow and E. Y. Chi, Lipid membrane templated misfolding and self-assembly of intrinsically disordered tau protein, Sci. Rep., 2020, 10(1), 13324 CrossRef CAS PubMed.
- M. Goedert, R. Jakes, M. G. Spillantini, M. Hasegawa, M. J. Smith and R. A. Crowther, Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans, Nature, 1996, 383(6600), 550–553 CrossRef CAS.
- N. A. Eschmann, T. D. Do, N. E. LaPointe, J.-E. Shea, S. C. Feinstein and M. T. Bowers, et al., Tau Aggregation Propensity Engrained in Its Solution State, J. Phys. Chem. B, 2015, 119(45), 14421–14432 CrossRef CAS.
- B. Jebarupa, M. Muralidharan, A. Arun, A. K. Mandal and G. Mitra, Conformational heterogeneity of tau: Implication on intrinsic disorder, acid stability and fibrillation in Alzheimer's disease, Biophys. Chem., 2018, 241, 27–37 CrossRef CAS.
- N. Ait-Bouziad, A. Chiki, G. Limorenko, S. Xiao, D. Eliezer and H. A. Lashuel, Phosphorylation of the overlooked tyrosine 310 regulates the structure, aggregation, and microtubule- and lipid-binding properties of Tau, J. Biol. Chem., 2020, 295(23), 7905–7922 CrossRef CAS.
- M. Haj-Yahya, P. Gopinath, K. Rajasekhar, H. Mirbaha, M. I. Diamond and H. A. Lashuel, Site-Specific Hyperphosphorylation Inhibits, Rather than Promotes, Tau Fibrillization, Seeding Capacity, and Its Microtubule Binding, Angew. Chem., Int. Ed., 2020, 59(10), 4059–4067 CrossRef CAS PubMed.
- C. Alquezar, S. Arya and A. W. Kao, Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation, Front. Neurol., 2020, 11, 595532 CrossRef.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov and H. J. Garringer, et al., Cryo-EM structures of tau filaments from Alzheimer's disease, Nature, 2017, 547(7662), 185–190 CrossRef CAS.
- T. Arakhamia, C. E. Lee, Y. Carlomagno, D. M. Duong, S. R. Kundinger and K. Wang, et al., Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains, Cell, 2020, 180(4), 633–644 CrossRef CAS .e12.
- P. Friedhoff, M. von Bergen, E.-M. Mandelkow, P. Davies and E. Mandelkow, A nucleated assembly mechanism of Alzheimer paired helical filaments, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(26), 15712–15717 CrossRef CAS.
- S. Jeganathan, M. von Bergen, H. Brutlach, H. J. Steinhoff and E. Mandelkow, Global hairpin folding of tau in solution, Biochemistry, 2006, 45(7), 2283–2293 CrossRef CAS PubMed.
- S. Barghorn, Q. Zheng-Fischhöfer, M. Ackmann, J. Biernat, M. von Bergen and E. M. Mandelkow, et al., Structure, Microtubule Interactions, and Paired Helical Filament Aggregation by Tau Mutants of Frontotemporal Dementias, Biochemistry, 2000, 39(38), 11714–11721 CrossRef CAS PubMed.
- G. Rossi, A. Bastone, E. Piccoli, G. Mazzoleni, M. Morbin, A. Uggetti, G. Giaccone, S. Sperber, M. Beeg, M. Salmona and F. Tagliavini, New mutations in MAPT gene causing frontotemporal lobar degeneration: biochemical and structural characterization, Neurobiol. Aging, 2012, 33(4), 834-e1-6 CrossRef.
- K. H. Strang, C. L. Croft, Z. A. Sorrentino, P. Chakrabarty, T. E. Golde and B. I. Giasson, Distinct differences in prion-like seeding and aggregation between Tau protein variants provide mechanistic insights into tauopathies, J. Biol. Chem., 2018, 293(7), 2408–2421 CrossRef CAS PubMed.
- N. Asherie, Protein crystallization and phase diagrams, Methods, 2004, 34(3), 266–272 CrossRef CAS.
- D. Kashchiev, Nucleation, Elsevier, 2000 Search PubMed.
- F. Oosawa, The effect of field fluctuation on a macromolecular system, J. Theor. Biol., 1975, 52(1), 175–186 CrossRef CAS.
- Y. Fichou, Z. R. Oberholtzer, H. Ngo, C. Y. Cheng, T. J. Keller and N. A. Eschmann, et al., Tau-Cofactor Complexes as Building Blocks of Tau Fibrils, Front. Neurosci., 2019, 13, 1339 CrossRef.
- L.-N. Schaffert and W. G. Carter, Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review, Brain Sci., 2020, 10(4), 232 CrossRef CAS.
- E. Ercan, S. Eid, C. Weber, A. Kowalski, M. Bichmann and A. Behrendt, et al., A validated antibody panel for the characterization of tau post-translational modifications, Mol. Neurodegener., 2017, 12(1), 87 CrossRef PubMed.
- S. Park, J. H. Lee, J. H. Jeon and M. J. Lee, Degradation or aggregation: the ramifications of post-translational modifications on tau, BMB Rep., 2018, 51(6), 265–273 CrossRef CAS.
- M. Serena, C. Massimo, I. Filomena, N. Lucia, B. Fabio and N. Robert, et al., The Involvement of Post-Translational Modifications in Alzheimer's Disease, Curr. Alzheimer Res., 2018, 15(4), 313–335 CrossRef.
- L. Martin, X. Latypova and F. Terro, Post-translational modifications of tau protein: Implications for Alzheimer's disease, Neurochem. Int., 2011, 58(4), 458–471 CrossRef CAS PubMed.
- H. A. Lashuel and G. Limorenko, Commentary on “Mounting Modifications Move Tau Toward Aggregation in Alzheimer's Brain” 2020, https://www.alzforum.org/news/research-news/mounting-modifications-move-tau-toward-aggregation-alzheimers-brain:https://www.alzforum.org.
- G. T. Bramblett, M. Goedert, R. Jakes, S. E. Merrick, J. Q. Trojanowski and V. M. Lee, Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding, Neuron, 1993, 10, 1089 CrossRef CAS PubMed.
- A. Sengupta, J. Kabat, M. Novak, Q. Wu, I. Grundke-Iqba and K. Iqbal, Maximal inhibition of tau binding to microtubules requires the phosphorylation of tau at both Thr 231 and Ser 262, Neurobiol. Aging, 1998, 19(4S), S124–S524 Search PubMed.
- A. Alonso, A. Mederlyova, M. Novak, I. Grundke-Iqbal and K. Iqbal, Promotion of hyperphosphorylation by Frontotemporal dementia Tau mutations, J. Biol. Chem., 2004, 279, 34873 CrossRef PubMed.
- F. Liu, B. Li, E. J. Tung, I. Grundke-Iqbal, K. Iqbal and C. X. Gong, Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation, Eur. J. Neurosci., 2007, 26(12), 3429–3436 CrossRef PubMed.
- C. Feijoo, D. G. Campbell, R. Jakes, M. Goedert and A. Cuenda, Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly, J. Cell Sci., 2005, 118(Pt 2), 397–408 CrossRef CAS.
- V. Duka, J.-H. Lee, J. Credle, J. Wills, A. Oaks and C. Smolinsky, et al., Identification of the Sites of Tau Hyperphosphorylation and Activation of Tau Kinases in Synucleinopathies and Alzheimer's Diseases, PLoS One, 2013, 8(9), e75025 CrossRef CAS.
- F. Kametani, M. Yoshida, T. Matsubara, S. Murayama, Y. Saito and I. Kawakami, et al., Comparison of Common and Disease-Specific Post-translational Modifications of Pathological Tau Associated With a Wide Range of Tauopathies, Front. Neurosci., 2020, 14, 581936 CrossRef PubMed.
- E. Köpke, Y.-C. Tung and S. Shaikh, Alonso AdC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease, J. Biol. Chem., 1993, 268(32), 24374–24384 CrossRef.
- I. Grundke-Iqbal, K. Iqbal, Y. C. Tung, M. Quinlan, H. M. Wisniewski and L. I. Binder, Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology, Proc. Natl. Acad. Sci. U. S. A., 1986, 83(13), 4913–4917 CrossRef CAS.
- D. P. Hanger, J. C. Betts, T. L. Loviny, W. P. Blackstock and B. H. Anderton, New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry, J. Neurochem., 1998, 71(6), 2465–2476 CrossRef CAS.
- T. J. Malia, A. Teplyakov, R. Ernst, S. J. Wu, E. R. Lacy and X. Liu, et al., Epitope mapping and structural basis for the recognition of phosphorylated tau by the anti-tau antibody AT8, Proteins: Struct., Funct., Bioinf., 2016, 84(4), 427–434 CrossRef CAS.
- D. Tosun, D. Veitch, P. Aisen, C. R. Jack Jr., W. J. Jagust and R. C. Petersen, et al., Detection of β-amyloid positivity in Alzheimer’s Disease Neuroimaging Initiative participants with demographics, cognition, MRI and plasma biomarkers, Brain Commun., 2021, 3(2), fcab008 CrossRef.
- Z. Chen and P. A. Cole, Synthetic approaches to protein phosphorylation, Curr. Opin. Chem. Biol., 2015, 28, 115–122 CrossRef CAS.
- K. E. Paleologou, A. W. Schmid, C. C. Rospigliosi, H. Y. Kim, G. R. Lamberto and R. A. Fredenburg, et al., Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein, J. Biol. Chem., 2008, 283(24), 16895–16905 CrossRef CAS.
- A. D. Alonso, J. Di Clerico, B. Li, C. P. Corbo, M. E. Alaniz and I. Grundke-Iqbal, et al., Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration, J. Biol. Chem., 2010, 285(40), 30851–30860 CrossRef CAS.
- A. Chiki, J. Ricci, R. Hegde, L. A. Abriata, A. Reif and D. Boudeffa, et al., Site-Specific Phosphorylation of Huntingtin Exon 1 Recombinant Proteins Enabled by the Discovery of Novel Kinases, ChemBioChem, 2021, 22(1), 217–231 CrossRef CAS.
- S. M. DeGuire, F. S. Ruggeri, M.-B. Fares, A. Chiki, U. Cendrowska and G. Dietler, et al., N-terminal Huntingtin (Htt) phosphorylation is a molecular switch regulating Htt aggregation, helical conformation, internalization, and nuclear targeting, J. Biol. Chem., 2018, 293(48), 18540–18558 CrossRef CAS.
- B. Fauvet and H. A. Lashuel, Semisynthesis and Enzymatic Preparation of Post-translationally Modified α-Synuclein, in Protein Amyloid Aggregation: Methods and Protocols, ed. Eliezer D., Springer New York, New York, NY, 2016, pp. 3–20 Search PubMed.
- Z. Chen and P. A. Cole, Synthetic approaches to protein phosphorylation, Curr. Opin. Chem. Biol., 2015, 28, 115–122 CrossRef CAS.
- M. Haj-Yahya and H. A. Lashuel, Protein semisynthesis provides access to tau disease-associated post-translational modifications (PTMs) and paves the way to deciphering the tau PTM code in health and diseased states, J. Am. Chem. Soc., 2018, 140(21), 6611–6621 CrossRef CAS.
- D. Ellmer, M. Brehs, M. Haj-Yahya, H. A. Lashuel and C. F. W. Becker, Single Posttranslational Modifications in the Central Repeat Domains of Tau4 Impact its Aggregation and Tubulin Binding, Angew. Chem., Int. Ed., 2019, 58(6), 1616–1620 CrossRef CAS.
- C. Smet-Nocca, M. Broncel, J. M. Wieruszeski, C. Tokarski, X. Hanoulle, A. Leroy, I. Landrieu, C. Rolando, G. Lippens and C. P. Hackenberger, Identification of O-GlcNAc sites within peptides of the Tau protein and their impact on phosphorylation, Mol. BioSyst., 2011, 7(5), 1420–1429 RSC.
- M. Broncel, E. Krause, D. Schwarzer and C. P. Hackenberger, The Alzheimer’s disease related tau protein as a new target for chemical protein engineering, Chem. - Eur. J., 2012, 18(9), 2488–2492 CrossRef CAS.
- S. Schwagerus, O. Reimann, C. Despres, C. Smet-Nocca and C. P. R. Hackenberger, Semi-synthesis of a tag-free O-GlcNAcylated tau protein by sequential chemoselective ligation, J. Pept. Sci., 2016, 22, 327–333 CrossRef CAS.
- O. Reimann, M. Glanz and C. P. Hackenberger, Native chemical ligation between asparagine and valine: application and limitations for the synthesis of tri-phosphorylated C-terminal tau, Bioorg. Med. Chem., 2015, 23(12), 2890–2894 CrossRef CAS PubMed.
- R. P. Guttmann, A. C. Erickson and G. V. Johnson, Tau self-association: stabilization with a chemical cross-linker and modulation by phosphorylation and oxidation state, J. Neurochem., 1995, 64(3), 1209–1215 CrossRef CAS.
- E. M. Mandelkow, J. Biernat, G. Drewes, B. Steiner, B. Lichtenberg-Kraag and H. Wille, et al., Microtubule-associated protein tau, paired helical filaments, and phosphorylation, Ann. N. Y. Acad. Sci., 1993, 695, 209–216 CrossRef CAS.
- A. Alonso, T. Zaidi, M. Novak, I. Grundke-Iqbal and K. Iqbal, Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(12), 6923–6928 CrossRef CAS.
- C. Despres, C. Byrne, H. Qi, F.-X. Cantrelle, I. Huvent and B. Chambraud, et al., Identification of the Tau phosphorylation pattern that drives its aggregation, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(34), 9080–9085 CrossRef CAS PubMed.
- A. Abraha, N. Ghoshal, T. C. Gamblin, V. Cryns, R. W. Berry and J. Kuret, et al., C-terminal inhibition of tau assembly in vitro and in Alzheimer's disease, J. Cell Sci., 2000, 113(21), 3737–3745 CrossRef CAS.
- C. Haase, J. T. Stieler, T. Arendt and M. Holzer, Pseudophosphorylation of tau protein alters its ability for self-aggregation, J. Neurochem., 2004, 88(6), 1509–1520 CrossRef CAS PubMed.
- A. Schneider, J. Biernat, M. von Bergen, E. Mandelkow and E. M. Mandelkow, Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments, Biochemistry, 1999, 38(12), 3549–3558 CrossRef CAS PubMed.
- L. Baum, R. Seger, J. R. Woodgett, S. Kawabata, K. Maruyama, M. Koyama, J. Silver and T. Saitoh, Overexpressed tau protein in cultured cells is phosphorylated without formation of PHF: implication of phosphoprotein phosphatase involvement, Molecular brain research, 1995, 134(1), 1–7 CrossRef.
- F. Drepper, J. Biernat, S. Kaniyappan, H. E. Meyer, E. M. Mandelkow and B. Warscheid, et al., A combinatorial native MS and LC-MS/MS approach reveals high intrinsic phosphorylation of human tau but minimal levels of other key modifications, J. Biol. Chem., 2020, 295(52), 18213–18225 CrossRef CAS.
- M. Takahashi, Y. Tsujioka, T. Yamada, Y. Tsuboi, H. Okada and T. Yamamoto, et al., Glycosylation of microtubule-associated protein tau in Alzheimer's disease brain, Acta Neuropathol., 1999, 97(6), 635–641 CrossRef CAS.
- J.-Z. Wang, I. Grundke-Iqbal and K. Iqbal, Glycosylation of microtubule–associated protein tau: An abnormal posttranslational modification in Alzheimer's disease, Nat. Med., 1996, 2(8), 871–875 CrossRef CAS PubMed.
- A. L. Aguilar, X. Hou, L. Wen, P. G. Wang and P. Wu, A chemoenzymatic histology method for O-GlcNAc detection, ChemBioChem, 2017, 18(24), 2416 CrossRef CAS PubMed.
- T. S. Pinho, S. C. Correia, G. Perry, A. F. Ambrósio and P. I. Moreira, Diminished O-GlcNAcylation in Alzheimer's disease is strongly correlated with mitochondrial anomalies, Biochim. Biophys. Acta, Mol. Basis Dis., 2019, 1865(8), 2048–2059 CrossRef CAS PubMed.
- A. C. Wang, E. H. Jensen, J. E. Rexach, H. V. Vinters and L. C. Hsieh-Wilson, Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(52), 15120–15125 CrossRef CAS.
- C. S. Arnold, G. W. Johnson, R. N. Cole, D. L.-Y. Dong, M. Lee and G. W. Hart, The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine, J. Biol. Chem., 1996, 271(46), 28741–28744 CrossRef CAS PubMed.
- L. A. Robertson, K. L. Moya and K. C. Breen, The potential role of tau protein O-glycosylation in Alzheimer's disease, J. Alzheimer's Dis., 2004, 6(5), 489–495 CAS.
- S. A. Yuzwa, A. K. Yadav, Y. Skorobogatko, T. Clark, K. Vosseller and D. J. Vocadlo, Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody, Amino Acids, 2011, 40(3), 857–868 CrossRef CAS PubMed.
- G. Bourré, F.-X. Cantrelle, A. Kamah, B. Chambraud, I. Landrieu and C. Smet-Nocca, Direct Crosstalk Between O-GlcNAcylation and Phosphorylation of Tau Protein Investigated by NMR Spectroscopy, Front. Endocrinol., 2018, 9, 595 CrossRef PubMed Available from: http://europepmc.org/abstract/MED/30386294.
- F. Liu, K. Iqbal, I. Grundke-Iqbal, G. W. Hart and C. X. Gong, O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(29), 10804–10809 CrossRef CAS.
- F. Liu, J. Shi, H. Tanimukai, J. Gu, J. Gu and I. Grundke-Iqbal, et al., Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease, Brain, 2009, 132(7), 1820–1832 CrossRef PubMed.
- C.-X. Gong, F. Liu, I. Grundke-Iqbal and K. Iqbal, Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation, J. Alzheimer's Dis., 2006, 9(1), 1–12 CAS.
- C. Smet-Nocca, M. Broncel, J.-M. Wieruszeski, C. Tokarski, X. Hanoulle and A. Leroy, et al., Identification of O-GlcNAc sites within peptides of the Tau protein and their impact on phosphorylation, Mol. BioSyst., 2011, 7(5), 1420–1429 RSC.
- T. Lefebvre, S. Ferreira, L. Dupont-Wallois, T. Bussiere, M.-J. Dupire and A. Delacourte, et al., Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins—a role in nuclear localization, Biochim. Biophys. Acta, Gen. Subj., 2003, 1619(2), 167–176 CrossRef CAS.
- S. Lim, M. Haque, G. Nam, N. Ryoo, H. Rhim and Y. K. Kim, Monitoring of intracellular tau aggregation regulated by OGA/OGT inhibitors, Int. J. Mol. Sci., 2015, 16(9), 20212–20224 CrossRef CAS.
- S. Schedin-Weiss, S. Gaunitz, P. Sui, Q. Chen, S. M. Haslam and K. Blennow, et al., Glycan biomarkers for Alzheimer disease correlate with T-tau and P-tau in cerebrospinal fluid in subjective cognitive impairment, FEBS J., 2020, 287(15), 3221–3234 CrossRef CAS.
- C. H. Yu, T. Si, W. H. Wu, J. Hu, J. T. Du and Y. F. Zhao, et al., O-GlcNAcylation modulates the self-aggregation ability of the fourth microtubule-binding repeat of tau, Biochem. Biophys. Res. Commun., 2008, 375(1), 59–62 CrossRef CAS.
- G. W. Hart, M. P. Housley and C. Slawson, Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins, Nature, 2007, 446(7139), 1017–1022 CrossRef CAS PubMed.
- F. Di Domenico, C. Lanzillotta and A. Tramutola, Therapeutic potential of rescuing protein O-GlcNAcylation in tau-related pathologies, Expert Rev. Neurother., 2019, 19(1), 1–3 CrossRef CAS.
- F.-X. Cantrelle, A. Loyens, X. Trivelli, O. Reimann, C. Despres and N. S. Gandhi, et al., Phosphorylation and O-GlcNAcylation of the PHF-1 Epitope of Tau Protein Induce Local Conformational Changes of the C-Terminus and Modulate Tau Self-Assembly Into Fibrillar Aggregates, Front. Mol. Neurosci., 2021, 14, 119 Search PubMed.
- K. Iqbal, A. D. C. Alonso, C.-X. Gong, S. Khatoon, J.-J. Pei, J. Wang, et al.,Mechanisms of neurofibrillary degeneration and the formation of neurofibrillary tangles, Springer, Ageing and Dementia, 1998, pp. 169–180.
- Y. Losev, A. Paul, M. Frenkel-Pinter, M. Abu-Hussein, I. Khalaila and E. Gazit, et al., Novel model of secreted human tau protein reveals the impact of the abnormal N-glycosylation of tau on its aggregation propensity, Sci. Rep., 2019, 9(1), 2254 CrossRef.
- L. C. Maillard, Action des acides amines sur les sucres; formation des melanoidines par voie methodique, C. R. Acad. Sci., 1912, 154, 66–68 CAS.
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Glycosylation in health and disease, Nat. Rev. Nephrol., 2019, 15(6), 346–366 CrossRef PubMed.
- S. Yan, X. Chen, A. Schmidt, J. Brett, G. Godman and Y. Zou, et al., Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(16), 7787–7791 CrossRef CAS.
- M. A. Smith, S. Taneda, P. L. Richey, S. Miyata, S.-D. Yan and D. Stern, et al., Advanced Maillard reaction end products are associated with Alzheimer disease pathology, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(12), 5710–5714 CrossRef CAS.
- N. Sasaki, R. Fukatsu, K. Tsuzuki, Y. Hayashi, T. Yoshida and N. Fujii, et al., Advanced glycation end products in Alzheimer's disease and other neurodegenerative diseases, Am. J. Pathol., 1998, 153(4), 1149–1155 CrossRef CAS.
- H.-J. Lüth, V. Ogunlade, B. Kuhla, R. Kientsch-Engel, P. Stahl and J. Webster, et al., Age-and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer's disease brains, Cereb. Cortex, 2005, 15(2), 211–220 CrossRef.
- K. Batkulwar, R. Godbole, R. Banarjee, O. Kassaar, R. J. Williams and M. J. Kulkarni, Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer's Disease, ACS Chem. Neurosci., 2018, 9(5), 988–1000 CrossRef CAS PubMed.
- M. Necula and J. Kuret, Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro, J. Biol. Chem., 2004, 279, 49694 CrossRef CAS PubMed.
- M. D. Ledesma, M. Medina and J. Avila, The in vitro formation of recombinant tau polymers: effect of phosphorylation and glycation, Mol. Chem. Neuropathol., 1996, 27(3), 249–258 CrossRef CAS.
- K. Liu, Y. Liu, L. Li, P. Qin, J. Iqbal and Y. Deng, et al., Glycation alter the process of Tau phosphorylation to change Tau isoforms aggregation property, Biochim. Biophys. Acta, Mol. Basis Dis., 2016, 1862(2), 192–201 CrossRef CAS.
- C. Kontaxi, P. Piccardo and A. C. Gill, Lysine-Directed Post-translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies, Front. Mol. Biosci., 2017, 4, 56 CrossRef.
- D. J. Irwin, T. J. Cohen, M. Grossman, S. E. Arnold, S. X. Xie and V. M.-Y. Lee, et al., Acetylated tau, a novel pathological signature in Alzheimer's disease and other tauopathies, Brain, 2012, 135(3), 807–818 CrossRef.
- B. Lucke-Wold, K. Seidel, R. Udo, B. Omalu, M. Ornstein and R. Nolan, et al., Role of Tau Acetylation in Alzheimer's Disease and Chronic Traumatic Encephalopathy: The Way Forward for Successful Treatment, J. Neurol. Neurosurg., 2017, 4(2), 140 Search PubMed.
- S.-W. Min, X. Chen, T. E. Tracy, Y. Li, Y. Zhou and C. Wang, et al., Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits, Nat. Med., 2015, 21(10), 1154–1162 CrossRef CAS.
- E. Tracy Tara, D. Sohn Peter, S. S. Minami, C. Wang, S.-W. Min and Y. Li, et al., Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss, Neuron, 2016, 90(2), 245–260 CrossRef CAS.
- T. E. Tracy and L. Gan, Acetylated tau in Alzheimer's disease: An instigator of synaptic dysfunction underlying memory loss, BioEssays, 2017, 39(4), 1600224 CrossRef.
- H. Choi, H. J. Kim, J. Yang, S. Chae, W. Lee and S. Chung, et al., Acetylation changes tau interactome to degrade tau in Alzheimer's disease animal and organoid models, Aging Cell, 2020, 19(1), e13081 CrossRef CAS.
- C. Cook, Y. Carlomagno, T. F. Gendron, J. Dunmore, K. Scheffel and C. Stetler, et al., Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance, Hum. Mol. Genet., 2014, 23(1), 104–116 CrossRef CAS.
- P. Kirchner, M. Bourdenx, J. Madrigal-Matute, S. Tiano, A. Diaz and B. A. Bartholdy, et al., Proteome-wide analysis of chaperone-mediated autophagy targeting motifs, PLoS Biol., 2019, 17(5), e3000301 CrossRef CAS.
- Y. Carlomagno, D.-C. Chung, M. Yue, M. Castanedes-Casey, B. J. Madden and J. Dunmore, et al., An acetylation–phosphorylation switch that regulates tau aggregation propensity and function, J. Biol. Chem., 2017, 292(37), 15277–15286 CrossRef CAS.
- S.-W. Min, S.-H. Cho, Y. Zhou, S. Schroeder, V. Haroutunian and W. W. Seeley, et al., Acetylation of tau inhibits its degradation and contributes to tauopathy, Neuron, 2010, 67(6), 953–966 CrossRef CAS.
- B. Fauvet, M.-B. Fares, F. Samuel, I. Dikiy, A. Tandon and D. Eliezer, et al., Characterization of Semisynthetic and Naturally Nα-Acetylated α-Synuclein in Vitro and in Intact Cells: IMPLICATIONS FOR AGGREGATION AND CELLULAR PROPERTIES OF α-SYNUCLEIN *, J. Biol. Chem., 2012, 287(34), 28243–28262 CrossRef CAS PubMed.
- H. Trzeciakiewicz, J.-H. Tseng, C. M. Wander, V. Madden, A. Tripathy and C.-X. Yuan, et al., A dual pathogenic mechanism links tau acetylation to sporadic tauopathy, Sci. Rep., 2017, 7, 44102 CrossRef CAS.
- T. J. Cohen, J. L. Guo, D. E. Hurtado, L. K. Kwong, I. P. Mills and J. Q. Trojanowski, et al., The acetylation of tau inhibits its function and promotes pathological tau aggregation, Nat. Commun., 2011, 2, 252 CrossRef.
- A. Kamah, I. Huvent, F. X. Cantrelle, H. Qi, G. Lippens and I. Landrieu, et al., Nuclear magnetic resonance analysis of the acetylation pattern of the neuronal Tau protein, Biochemistry, 2014, 53(18), 3020–3032 CrossRef CAS.
- J. C. Ferreon, A. Jain, K. J. Choi, P. S. Tsoi, K. R. MacKenzie and S. Y. Jung, et al., Acetylation Disfavors Tau Phase Separation, Int. J. Mol. Sci., 2018, 19(5), 1360 CrossRef.
- D. C. David, R. Layfield, L. Serpell, Y. Narain, M. Goedert and M. G. Spillantini, Proteasomal degradation of tau protein, J. Neurochem., 2002, 83(1), 176–185 CrossRef CAS PubMed.
- M. Morishima and Y. Ihara, Posttranslational modifications of tau in paired helical filaments, Dement. Geriatr. Cogn. Disord., 1994, 5(5), 282–288 CrossRef CAS.
- H. Mori, J. Kondo and Y. Ihara, Ubiquitin is a component of paired helical filaments in Alzheimer's disease, Science, 1987, 235(4796), 1641–1644 CrossRef CAS.
- G. Perry, R. Friedman, G. Shaw and V. Chau, Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains, Proc. Natl. Acad. Sci. U. S. A., 1987, 84(9), 3033–3036 CrossRef CAS PubMed.
- M. H. Abreha, E. B. Dammer, L. Ping, T. Zhang, D. M. Duong and M. Gearing, et al., Quantitative Analysis of the Brain Ubiquitylome in Alzheimer's Disease, Proteomics, 2018, 18(20), e1800108 CrossRef.
- M. Morishima-Kawashima, M. Hasegawa, K. Takio, M. Suzuki, K. Titani and Y. Ihara, Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments, Neuron, 1993, 10(6), 1151–1160 CrossRef CAS.
- D. Cripps, S. N. Thomas, Y. Jeng, F. Yang, P. Davies and A. J. Yang, Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation, J. Biol. Chem., 2006, 281(16), 10825–10838 CrossRef CAS.
- K. Flach, E. Ramminger, I. Hilbrich, A. Arsalan-Werner, F. Albrecht and L. Herrmann, et al., Axotrophin/MARCH7 acts as an E3 ubiquitin ligase and ubiquitinates tau protein in vitro impairing microtubule binding, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, 1842(9), 1527–1538 CrossRef CAS PubMed.
- L. Petrucelli, D. Dickson, K. Kehoe, J. Taylor, H. Snyder and A. Grover, et al., CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation, Hum. Mol. Genet., 2004, 13(7), 703–714 CrossRef CAS.
- J. R. Babu, T. Geetha and M. W. Wooten, Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation, J. Neurochem., 2005, 94(1), 192–203 CrossRef CAS PubMed.
- H. Shimura, D. Schwartz, S. P. Gygi and K. S. Kosik, CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival, J. Biol. Chem., 2004, 279(6), 4869–4876 CrossRef CAS PubMed.
- P. Wang, G. Joberty, A. Buist, A. Vanoosthuyse, I.-C. Stancu and B. Vasconcelos, et al., Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo, Acta Neuropathol., 2017, 133(5), 731–749 CrossRef CAS.
- M. Hejjaoui, M. Haj-Yahya, K. S. A. Kumar, A. Brik and H. A. Lashuel, Towards Elucidation of the Role of Ubiquitination in the Pathogenesis of Parkinson's Disease with Semisynthetic Ubiquitinated α-Synuclein, Angew. Chem., Int. Ed., 2011, 50(2), 405–409 CrossRef CAS.
- M. Haj-Yahya, B. Fauvet, Y. Herman-Bachinsky, M. Hejjaoui, S. N. Bavikar and S. V. Karthikeyan, et al., Synthetic polyubiquitinated α-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(44), 17726–17731 CrossRef CAS.
- K. Blennow and H. Hampel, CSF markers for incipient Alzheimer's disease, Lancet Neurol., 2003, 2(10), 605–613 CrossRef CAS PubMed.
- J. Ramcharitar, S. Albrecht, V. M. Afonso, V. Kaushal, D. A. Bennett and A. C. LeBlanc, Cerebrospinal Fluid Tau Cleaved by Caspase-6 Reflects Brain Levels and Cognition in Aging and Alzheimer Disease, J. Neuropathol. Exp. Neurol., 2013, 72(9), 824–832 CrossRef CAS PubMed.
- C. Vigo-Pelfrey, P. P. Seubert, R. Barbour, C. Blomquist, M. Lee and D. Lee, et al., Elevation of microtubule-associated protein tau in the cerebrospinal fluid of patients with Alzheimer's disease, Neurology, 1995, 45(4), 788–793 CrossRef CAS PubMed.
- G. V. W. Johnson, P. Seubert, T. M. Cox, R. Motter, J. P. Brown and D. Galasko, The τ Protein in Human Cerebrospinal Fluid in Alzheimer's Disease Consists of Proteolytically Derived Fragments, J. Neurochem., 2002, 68(1), 430–433 CrossRef PubMed.
- J. Gu, W. Xu, N. Jin, L. Li, Y. Zhou and D. Chu, et al., Truncation of tau selectively facilitates its pathological activities, J. Biol. Chem., 2020, 295(40), 13812–13828 CrossRef CAS.
- B. Borroni, F. Gardoni, L. Parnetti, L. Magno, M. Malinverno and E. Saggese, et al., Pattern of Tau forms in CSF is altered in progressive supranuclear palsy, Neurobiol. Aging, 2009, 30(1), 34–40 CrossRef CAS PubMed.
- T. Arai, K. Ikeda, H. Akiyama, T. Nonaka, M. Hasegawa and K. Ishiguro, et al., Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration, Ann. Neurol., 2003, 55(1), 72–79 CrossRef.
- F. P. Zemlan, E. C. Jauch, J. J. Mulchahey, S. P. Gabbita, W. S. Rosenberg and S. G. Speciale, et al., C-tau biomarker of neuronal damage in severe brain injured patients: association with elevated intracranial pressure and clinical outcome, Brain Res., 2002, 947(1), 131–139 CrossRef CAS.
- E. Barini, G. Plotzky, Y. Mordashova, J. Hoppe, E. Rodriguez-Correa and S. Julier, et al., Tau in the brain interstitial fluid is fragmented and seeding-competent, Neurobiol. Aging, 2022, 109, 64–77 CrossRef.
- P. M. Horowitz, K. R. Patterson, A. L. Guillozet-Bongaarts, M. R. Reynolds, C. A. Carroll and S. T. Weintraub, et al., Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease, J. Neurosci., 2004, 24(36), 7895–7902 CrossRef CAS.
- G. Basurto-Islas, J. Luna-Munoz, A. L. Guillozet-Bongaarts, L. I. Binder, R. Mena and F. García-Sierra, Accumulation of aspartic acid421-and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease, J. Neuropathol. Exp. Neurol., 2008, 67(5), 470–483 CrossRef CAS PubMed.
- G. Ugolini, A. Cattaneo and M. Novak, Co-localization of truncated tau and DNA fragmentation in Alzheimer's disease neurones, NeuroReport, 1997, 8(17), 3709–3712 CrossRef CAS PubMed.
- R. A. Rissman, W. W. Poon, M. Blurton-Jones, S. Oddo, R. Torp and M. P. Vitek, et al., Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology, J. Clin. Invest., 2004, 114(1), 121–130 CrossRef CAS.
- R. Mena, P. C. Edwards, C. R. Harrington, E. B. Mukaetova-Ladinska and C. M. Wischik, Staging the pathological assembly of truncated tau protein into paired helical filaments in Alzheimer's disease, Acta Neuropathol., 1996, 91(6), 633–641 CrossRef CAS PubMed.
- A. L. Guillozet-Bongaarts, F. Garcia-Sierra, M. R. Reynolds, P. M. Horowitz, Y. Fu and T. Wang, et al., Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease, Neurobiol. Aging, 2005, 26(7), 1015–1022 CrossRef CAS.
- C. M. Wischik, M. Novak, H. C. Thøgersen, P. C. Edwards, M. J. Runswick and R. Jakes, et al., Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease, Proc. Natl. Acad. Sci. U. S. A., 1988, 85(12), 4506 CrossRef CAS.
- C. M. Wischik, M. Novak, P. C. Edwards, A. Klug, W. Tichelaar and R. A. Crowther, Structural characterization of the core of the paired helical filament of Alzheimer disease, Proc. Natl. Acad. Sci. U. S. A., 1988, 85(13), 4884 CrossRef CAS.
- L. I. Binder, A. L. Guillozet-Bongaarts, F. Garcia-Sierra and RW. Tau Berry, tangles, and Alzheimer's disease, Biochim. Biophys. Acta, Mol. Basis Dis., 2005, 1739(2), 216–223 CrossRef CAS.
- S. Mondragón-Rodríguez, G. Basurto-Islas, I. Santa-Maria, R. Mena, L. I. Binder and J. Avila, et al., Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer's disease, Int. J. Exp. Pathol., 2008, 89(2), 81–90 CrossRef.
- S. Wray, M. Saxton, B. H. Anderton and D. P. Hanger, Direct analysis of tau from PSP brain identifies new phosphorylation sites and a major fragment of N-terminally cleaved tau containing four microtubule-binding repeats, J. Neurochem., 2008, 105(6), 2343–2352 CrossRef CAS.
- S. H. Jang, Traumatic Axonal Injury in Patients with Mild Traumatic Brain Injury. Traumatic Brain Injury – Pathobiology, Advanced Diagnostics and Acute Management, InTech, 2018 Search PubMed.
- L. S. Yang and H. Ksiezak-Reding, Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments, Eur. J. Biochem., 1995, 233(1), 9–17 CrossRef CAS.
- S. Garg, T. Timm, E.-M. Mandelkow, E. Mandelkow and Y. Wang, Cleavage of Tau by calpain in Alzheimer's disease: the quest for the toxic 17 kD fragment, Neurobiol. Aging, 2011, 32(1), 1–14 CrossRef CAS.
- S.-Y. Park and A. Ferreira, The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates β-amyloid-induced neurodegeneration, J. Neurosci., 2005, 25(22), 5365–5375 CrossRef CAS PubMed.
- K. Paholikova, B. Salingova, A. Opattova, R. Skrabana, P. Majerova and N. Zilka, et al., N-terminal Truncation of Microtubule Associated Protein Tau Dysregulates its Cellular Localization, J. Alzheimer's Dis., 2015, 43, 915–926 CAS.
- T. C. Gamblin, F. Chen, A. Zambrano, A. Abraha, S. Lagalwar and A. L. Guillozet, et al., Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer's disease, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(17), 10032–10037 CrossRef CAS.
- V. Corsetti, G. Amadoro, A. Gentile, S. Capsoni, M. T. Ciotti and M. T. Cencioni, et al., Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer's disease models, Mol. Cell. Neurosci., 2008, 38(3), 381–392 CrossRef CAS PubMed.
- G. Amadoro, M. T. Ciotti, M. Costanzi, V. Cestari, P. Calissano and N. Canu, NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(8), 2892–2897 CrossRef CAS.
- S. E. Matsumoto, Y. Motoi, K. Ishiguro, T. Tabira, F. Kametani and M. Hasegawa, et al., The twenty-four KDa C-terminal tau fragment increases with aging in tauopathy mice: implications of prion-like properties, Hum. Mol. Genet., 2015, 24(22), 6403–6416 CrossRef CAS.
- S. E. Matsumoto, Y. Motoi, K. Ishiguro, T. Tabira, F. Kametani, M. Hasegawa and N. Hattori, The twenty-four KDa C-terminal tau fragment increases with aging in tauopathy mice: implications of prion-like properties, Hum. Mol. Genet., 2015, 24(22), 6403–6416 CrossRef CAS.
- T. Guo, D. Dakkak, T. Rodriguez-Martin, W. Noble and D. P. Hanger, A pathogenic tau fragment compromises microtubules, disrupts insulin signaling and induces the unfolded protein response, Acta Neuropathol. Commun., 2019, 7(1), 2 CrossRef CAS.
- X. Zhao, L. A. Kotilinek, B. Smith, C. Hlynialuk, K. Zahs and M. Ramsden, et al., Caspase-2 cleavage of tau reversibly impairs memory, Nat. Med., 2016, 22(11), 1268 CrossRef CAS.
- Z. Zhang, M. Song, X. Liu, S. S. Kang, I.-S. Kwon and D. M. Duong, et al., Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease, Nat. Med., 2014, 20(11), 1254–1262 CrossRef CAS.
- S. Sengupta, P. M. Horowitz, S. L. Karsten, G. R. Jackson, D. H. Geschwind and Y. Fu, et al., Degradation of Tau Protein by Puromycin-Sensitive Aminopeptidase in Vitro†, Biochemistry, 2006, 45(50), 15111–15119 CrossRef CAS PubMed.
- J. D. Sokolowski, K. K. Gamage, D. S. Heffron, A. C. LeBlanc, C. D. Deppmann and J. W. Mandell, Caspase-mediated cleavage of actin and tubulin is a common feature and sensitive marker of axonal degeneration in neural development and injury, Acta Neuropathol. Commun., 2014, 2(1), 1–14 CrossRef.
- F. Garcia-Sierra, S. Mondragon-Rodriguez and G. Basurto-Islas, Truncation of tau protein and its pathological significance in Alzheimer's disease, J. Alzheimer's Dis., 2008, 14(4), 401–409 Search PubMed.
- A. S. Chesser, S. M. Pritchard and G. V. W. Johnson, Tau Clearance Mechanisms and Their Possible Role in the Pathogenesis of Alzheimer Disease, Front. Neurol., 2013, 4, 122 Search PubMed.
- T. F. Gendron and L. Petrucelli, The role of tau in neurodegeneration, Mol. Neurodegener., 2009, 4(1), 13 CrossRef.
- M. Saito, G. Chakraborty, R.-F. Mao, S.-M. Paik, C. Vadasz and M. Saito, Tau phosphorylation and cleavage in ethanol-induced neurodegeneration in the developing mouse brain, Neurochem. Res., 2010, 35(4), 651–659 CrossRef CAS.
- A. L. Guillozet-Bongaarts, M. E. Cahill, V. L. Cryns, M. R. Reynolds, R. W. Berry and L. I. Binder, Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo, J. Neurochem., 2006, 97(4), 1005–1014 CrossRef CAS.
- T. T. Rohn, R. A. Rissman, M. C. Davis, Y. E. Kim, C. W. Cotman and E. Head, Caspase-9 activation and caspase cleavage of tau in the Alzheimer's disease brain, Neurobiol. Dis., 2002, 11(2), 341–354 CrossRef CAS.
- S. Albrecht, N. Bogdanovic, B. Ghetti, B. Winblad and A. C. LeBlanc, Caspase-6 Activation in Familial Alzheimer Disease Brains Carrying Amyloid Precursor Protein or Presenilin I or Presenilin II Mutations, J. Neuropathol. Exp. Neurol., 2009, 68(12), 1282–1293 CrossRef CAS.
- L. Fasulo, G. Ugolini, M. Visintin, A. Bradbury, C. Brancolini and V. Verzillo, et al., The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis, J. Neurochem., 2000, 75(2), 624–633 CrossRef CAS.
- H. Guo, S. Albrecht, M. Bourdeau, T. Petzke, C. Bergeron and A. C. LeBlanc, Active Caspase-6 and Caspase-6-Cleaved Tau in Neuropil Threads, Neuritic Plaques, and Neurofibrillary Tangles of Alzheimer's Disease, Am. J. Pathol., 2004, 165(2), 523–531 CrossRef CAS.
- R. A. Rissman, W. W. Poon, M. Blurton-Jones, S. Oddo, R. Torp and M. P. Vitek, et al., Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology, J. Clin. Invest., 2004, 114(1), 121–130 CrossRef CAS.
- M. S. M. Wai, Y. Liang, C. Shi, E. Y. Cho, H.-F. Kung and D. T. Yew, Co-localization of hyperphosphorylated tau and caspases in the brainstem of Alzheimer's disease patients, Biogerontology, 2009, 10(4), 457 CrossRef CAS.
- S.-Y. Park, C. Tournell, R. C. Sinjoanu and A. Ferreira, Caspase-3-and calpain-mediated tau cleavage are differentially prevented by estrogen and testosterone in beta-amyloid-treated hippocampal neurons, Neuroscience, 2007, 144(1), 119–127 CrossRef CAS PubMed.
- R. C. Sinjoanu, S. Kleinschmidt, R. S. Bitner, J. D. Brioni, A. Moeller and A. Ferreira, The novel calpain inhibitor A-705253 potently inhibits oligomeric beta-amyloid-induced dynamin 1 and tau cleavage in hippocampal neurons, Neurochem. Int., 2008, 53(3–4), 79–88 CrossRef CAS PubMed.
- E. Adamec, P. Mohan, J. P. Vonsattel and R. A. Nixon, Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2, Acta Neuropathol., 2002, 104(1), 92–104 CrossRef CAS PubMed.
- M. G. Friedrich, A. Skora, S. E. Hancock, T. W. Mitchell, P. L. Else and R. J. Truscott, Tau Is Truncated in Five Regions of the Normal Adult Human Brain, Int. J. Mol. Sci., 2021, 22(7), 3521 CrossRef CAS PubMed.
- T. C. Gamblin, F. Chen, A. Zambrano, A. Abraha, S. Lagalwar and A. L. Guillozet, et al., Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(17), 10032–10037 CrossRef CAS.
- N. Canu, L. Dus, C. Barbato, M. T. Ciotti, C. Brancolini and A. M. Rinaldi, et al., Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis, J. Neurosci., 1998, 18(18), 7061–7074 CrossRef CAS.
- A. Rametti, F. Esclaire, C. Yardin and F. Terro, Linking alterations in tau phosphorylation and cleavage during neuronal apoptosis, J. Biol. Chem., 2004, 279(52), 54518–54528 CrossRef CAS.
- M. von Bergen, P. Friedhoff, J. Biernat, J. Heberle, E. M. Mandelkow and E. Mandelkow, Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(10), 5129–5134 CrossRef CAS PubMed.
- M. von Bergen, S. Barghorn, L. Li, A. Marx, J. Biernat and E.-M. Mandelkow, et al., Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local β-structure, J. Biol. Chem., 2001, 276(51), 48165–48174 CrossRef CAS.
- H. B. Luo, Y. Y. Xia, X. J. Shu, Z. C. Liu, Y. Feng and X. H. Liu, et al., SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(46), 16586–16591 CrossRef CAS.
- G. Niewiadomska, W. Niewiadomski, M. Steczkowska and A. Gasiorowska, Tau Oligomers Neurotoxicity, Life, 2021, 11(1), 28 CrossRef CAS.
- G. Manassero, M. Guglielmotto, D. Monteleone, V. Vasciaveo, O. Butenko and E. Tamagno, et al., Dual Mechanism of Toxicity for Extracellular Injection of Tau Oligomers versus Monomers in Human Tau Mice, J. Alzheimer's Dis., 2017, 59(2), 743–751 CAS.
- H. Zetterberg, Review: Tau in biofluids - relation to pathology, imaging and clinical features, Neuropathol. Appl. Neurobiol., 2017, 43(3), 194–199 CrossRef CAS.
- C. L. Russell, V. Mitra, K. Hansson, K. Blennow, J. Gobom and H. Zetterberg, et al., Comprehensive Quantitative Profiling of Tau and Phosphorylated Tau Peptides in Cerebrospinal Fluid by Mass Spectrometry Provides New Biomarker Candidates, J. Alzheimer's Dis., 2016, 55(1), 303–313 Search PubMed.
- H. Kadavath, R. V. Hofele, J. Biernat, S. Kumar, K. Tepper and H. Urlaub, et al., Tau stabilizes microtubules by binding at the interface between tubulin heterodimers, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(24), 7501–7506 CrossRef CAS.
- S. A. Yuzwa, X. Shan, M. S. Macauley, T. Clark, Y. Skorobogatko and K. Vosseller, et al., Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation, Nat. Chem. Biol., 2012, 8(4), 393–399 CrossRef CAS.
- S. A. Yuzwa, A. H. Cheung, M. Okon, L. P. McIntosh and D. J. Vocadlo, O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers, J. Mol. Biol., 2014, 426(8), 1736–1752 CrossRef CAS.
- N. R. Barthelemy, A. Gabelle, C. Hirtz, F. Fenaille, N. Sergeant and S. Schraen-Maschke, et al., Differential mass spectrometry profiles of tau protein in the cerebrospinal fluid of patients with Alzheimer's disease, progressive supranuclear palsy, and dementia with lewy bodies, J. Alzheimer's Dis., 2016, 51(4), 1033–1043 CAS.
- C. Sato, N. R. Barthélemy, K. G. Mawuenyega, B. W. Patterson, B. A. Gordon and J. Jockel-Balsarotti, et al., Tau Kinetics in Neurons and the Human Central Nervous System, Neuron, 2018, 97(6), 1284–1298 CrossRef CAS PubMed.
- M. Derisbourg, C. Leghay, G. Chiappetta, F.-J. Fernandez-Gomez, C. Laurent and D. Demeyer, et al., Role of the Tau N-terminal region in microtubule stabilization revealed by newendogenous truncated forms, Sci. Rep., 2015, 5(1), 1–10 Search PubMed.
- E. Portelius, S. F. Hansson, A. J. Tran, H. Zetterberg, P. Grognet and E. Vanmechelen, et al., Characterization of tau in cerebrospinal fluid using mass spectrometry, J. Proteome Res., 2008, 7(5), 2114–2120 CrossRef CAS.
- P. M. Horowitz, Early N-Terminal Changes and Caspase-6 Cleavage of Tau in Alzheimer's Disease, J. Neurosci., 2004, 24(36), 7895–7902 CrossRef CAS.
- D. Kanmert, A. Cantlon, C. R. Muratore, M. Jin, T. T. O'Malley and G. Lee, et al., C-Terminally Truncated Forms of Tau, But Not Full-Length Tau or Its C-Terminal Fragments, Are Released from Neurons Independently of Cell Death, J. Neurosci., 2015, 35(30), 10851–10865 CrossRef CAS PubMed.
- S. Sokolow, K. M. Henkins, T. Bilousova, B. Gonzalez, H. V. Vinters and C. A. Miller, et al., Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer's disease, J. Neurochem., 2015, 133(3), 368–379 CrossRef CAS PubMed.
- W. Zhang, B. Falcon, A. G. Murzin, J. Fan, R. A. Crowther and M. Goedert, et al., Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases, eLife, 2019, 8, e43584 CrossRef PubMed.
- V. H. Pomin and B. Mulloy, Glycosaminoglycans and Proteoglycans, Pharmaceuticals, 2018, 11(1), 27 CrossRef.
- R. C. Gupta, R. Lall, A. Srivastava and A. Sinha, Hyaluronic Acid: Molecular Mechanisms and Therapeutic Trajectory, Front. Vet. Sci., 2019, 6, 192 CrossRef.
- A. F. Logsdon, K. L. Francis, N. E. Richardson, S. J. Hu, C. L. Faber and B. A. Phan, et al., Decoding perineuronal net glycan sulfation patterns in the Alzheimer’s disease brain, Alzheimer’s Dementia, 2021 DOI:10.1002/alz.12451.
- A. J. Hayes and J. Melrose, Glycans and glycosaminoglycans in neurobiology: key regulators of neuronal cell function and fate, Biochem. J., 2018, 475(15), 2511–2545 CrossRef CAS.
- J. D. Esko, K. Kimata and U. Lindahl, Proteoglycans and Sulfated Glycosaminoglycans, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, et al., Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2nd edn, 2009, ch. 16 Search PubMed.
- J. R. Couchman and C. A. Pataki, An introduction to proteoglycans and their localization, J. Histochem. Cytochem., 2012, 60(12), 885–897 CrossRef.
- D. Mah, J. Zhao, X. Liu, F. Zhang, J. Liu and L. Wang, et al., The Sulfation Code of Tauopathies: Heparan Sulfate Proteoglycans in the Prion Like Spread of Tau Pathology, Front. Mol. Biosci., 2021, 8, 671458 CrossRef.
- Y. K. Al-Hilaly, S. J. Pollack, J. E. Rickard, M. Simpson, A. C. Raulin and T. Baddeley, et al., Cysteine-Independent Inhibition of Alzheimer's Disease-like Paired Helical Filament Assembly by Leuco-Methylthioninium (LMT), J. Mol. Biol., 2018, 430(21), 4119–4131 CrossRef CAS.
- B. E. Stopschinski, B. B. Holmes, G. M. Miller, V. A. Manon, J. Vaquer-Alicea and W. L. Prueitt, et al., Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus alpha-synuclein and beta-amyloid aggregates, J. Biol. Chem., 2018, 293(27), 10826–10840 CrossRef CAS.
- S. Khandelwal and G. M. Arepally, Immune pathogenesis of heparin-induced thrombocytopenia, Thromb. Haemostasis, 2016, 116(5), 792–798 CrossRef.
- T. A. Putranto, I. Yusuf, B. Murtala and A. Wijaya, Intra Arterial Heparin Flushing Increases Cereberal Blood Flow in Chronic Ischemic Stroke Patients, Indones. Biomed. J., 2016, 8(2), 119–126 CrossRef.
- J. L. Guo, S. Narasimhan, L. Changolkar, Z. He, A. Stieber and B. Zhang, et al., Unique pathological tau conformers from Alzheimer's brains transmit tau pathology in nontransgenic mice, J. Exp. Med., 2016, 213(12), 2635–2654 CrossRef CAS.
- B. E. Stopschinski, T. L. Thomas, S. Nadji, E. Darvish, L. Fan and B. B. Holmes, et al., A synthetic heparinoid blocks Tau aggregate cell uptake and amplification, J. Biol. Chem., 2020, 295(10), 2974–2983 CrossRef CAS.
- N. Sibille, A. Sillen, A. Leroy, J. M. Wieruszeski, B. Mulloy and I. Landrieu, et al., Structural impact of heparin binding to full-length Tau as studied by NMR spectroscopy, Biochemistry, 2006, 45(41), 12560–12572 CrossRef CAS PubMed.
- H. L. Zhu, C. Fernandez, J. B. Fan, F. Shewmaker, J. Chen and A. P. Minton, et al., Quantitative characterization of heparin binding to Tau protein: implication for inducer-mediated Tau filament formation, J. Biol. Chem., 2010, 285(6), 3592–3599 CrossRef CAS.
- G. Ramachandran and J. B. Udgaonkar, Understanding the kinetic roles of the inducer heparin and of rod-like protofibrils during amyloid fibril formation by Tau protein, J. Biol. Chem., 2011, 286(45), 38948–38959 CrossRef CAS.
- S. Barghorn and E. Mandelkow, Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments, Biochemistry, 2002, 41(50), 14885–14896 CrossRef CAS PubMed.
- M. M. Haque, D. Kim, Y. H. Yu, S. Lim, D. J. Kim and Y.-T. Chang, et al., Inhibition of tau aggregation by a rosamine derivative that blocks tau intermolecular disulfide cross-linking, Amyloid, 2014, 21(3), 185–190 CrossRef CAS PubMed.
- I. Huvent, A. Kamah, F. X. Cantrelle, N. Barois, C. Slomianny and C. Smet-Nocca, et al., A functional fragment of Tau forms fibers without the need for an intermolecular cysteine bridge, Biochem. Biophys. Res. Commun., 2014, 445(2), 299–303 CrossRef CAS.
- J. Zhao, I. Huvent, G. Lippens, D. Eliezer, A. Zhang and Q. Li, et al., Glycan Determinants of Heparin-Tau Interaction, Biophys. J., 2017, 112(5), 921–932 CrossRef CAS.
- S. Sarrazin, W. C. Lamanna and J. D. Esko, Heparan sulfate proteoglycans, Cold Spring Harbor Perspect. Biol., 2011, 3(7), a004952 Search PubMed.
- I. Capila and R. J. Linhardt, Heparin–protein interactions, Angew. Chem., Int. Ed., 2002, 41(3), 390–412 CrossRef CAS.
- D. Aviezer, E. Levy, M. Safran, C. Svahn, E. Buddecke and A. Schmidt, et al., Differential structural requirements of heparin and heparan sulfate proteoglycans that promote binding of basic fibroblast growth factor to its receptor, J. Biol. Chem., 1994, 269(1), 114–121 CrossRef CAS.
- M. E. Herndon, C. S. Stipp and A. D. Lander, Interactions of neural glycosaminoglycans and proteoglycans with protein ligands: assessment of selectivity, heterogeneity and the participation of core proteins in binding, Glycobiology, 1999, 9(2), 143–155 CrossRef CAS.
- M. D. Mukrasch, J. Biernat, M. von Bergen, C. Griesinger, E. Mandelkow and M. Zweckstetter, Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions, J. Biol. Chem., 2005, 280(26), 24978–24986 CrossRef CAS PubMed.
- Y. Fichou, N. A. Eschmann, T. J. Keller and S. Han, Conformation-based assay of tau protein aggregation. in Methods in cell biology, ed. Feinstein S. C. and LaPointe N. E., Academic Press, 2017, ch. 5, vol. 141, pp. 89–112 Search PubMed.
- M. von Bergen, S. Barghorn, J. Biernat, E. M. Mandelkow and E. Mandelkow, Biochim. Biophys. Acta, 2005, 1739(null), 158 CrossRef CAS.
- S. Elbaum-Garfinkle and E. Rhoades, Identification of an aggregation-prone structure of tau, J. Am. Chem. Soc., 2012, 134(40), 16607–16613 CrossRef CAS PubMed.
- H. K. Paudel and W. Li, Heparin-induced conformational change in microtubule-associated protein Tau as detected by chemical cross-linking and phosphopeptide mapping, J. Biol. Chem., 1999, 274(12), 8029–8038 CrossRef CAS.
- P. Friedhoff, M. von Bergen, E. M. Mandelkow, P. Davies and E. Mandelkow, A nucleated assembly mechanism of Alzheimer paired helical filaments, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(26), 15712–15717 CrossRef CAS.
- G. G. Moreira, J. S. Cristóvão, V. M. Torres, A. P. Carapeto, M. S. Rodrigues and I. Landrieu, et al., Zinc Binding to Tau Influences Aggregation Kinetics and Oligomer Distribution, Int. J. Mol. Sci., 2019, 20(23) DOI:10.3390/ijms20235979.
- H. Trzeciakiewicz, J. O. Esteves-Villanueva, N. Carlin and S. Martić, Electrochemistry of heparin binding to tau protein on Au surfaces, Electrochim. Acta, 2015, 162, 24–30 CrossRef CAS.
- M. Perez, J. M. Valpuesta, M. Medina, E. Montejo de Garcini and J. Avila, Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction, J. Neurochem., 1996, 67(3), 1183–1190 CrossRef CAS PubMed.
- J. G. de Ancos, I. Correas and J. Avila, Differences in microtubule binding and self-association abilities of bovine brain tau isoforms, J. Biol. Chem., 1993, 268(11), 7976–7982 CrossRef.
- E. E. Congdon, S. Kim, J. Bonchak, T. Songrug, A. Matzavinos and J. Kuret, Nucleation-dependent tau filament formation: the importance of dimerization and an estimation of elementary rate constants, J. Biol. Chem., 2008, 283(20), 13806–13816 CrossRef CAS.
- J. Díaz-Nido and F. Wandosell, Avila J. Glycosaminoglycans and β-amyloid, prion and tau peptides in neurodegenerative diseases, Peptides, 2002, 23(7), 1323–1332 CrossRef.
- P. M. Seidler, D. R. Boyer, K. A. Murray, T. P. Yang, M. Bentzel and M. R. Sawaya, et al., Structure-based inhibitors halt prion-like seeding by Alzheimer's disease-and tauopathy-derived brain tissue samples, J. Biol. Chem., 2019, 294(44), 16451–16464 CrossRef CAS PubMed.
- C. Shipps, H. R. Kelly, P. J. Dahl, S. M. Yi, D. Vu and D. Boyer, et al., Intrinsic electronic conductivity of individual atomically resolved amyloid crystals reveals micrometer-long hole hopping via tyrosines, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(2), e2014139118 CrossRef CAS.
- D. Eisenberg and M. Jucker, The amyloid state of proteins in human diseases, Cell, 2012, 148(6), 1188–1203 CrossRef CAS PubMed.
- B. Falcon, W. Zhang, A. G. Murzin, G. Murshudov, H. J. Garringer and R. Vidal, et al., Structures of filaments from Pick's disease reveal a novel tau protein fold, Nature, 2018, 561(7721), 137–140 CrossRef CAS PubMed.
- B. Falcon, J. Zivanov, W. Zhang, A. G. Murzin, H. J. Garringer and R. Vidal, et al., Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules, Nature, 2019, 568(7752), 420–423 CrossRef CAS PubMed.
- W. Zhang, B. Falcon, A. Murzin, J. Fan, A. Crowther and M. Goedert, et al., Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases, elife, 2018, 8, e43584 CrossRef PubMed.
- A. J. Dregni, V. S. Mandala, H. Wu, M. R. Elkins, H. K. Wang and I. Hung, et al., In vitro 0N4R tau fibrils contain a monomorphic β-sheet core enclosed by dynamically heterogeneous fuzzy coat segments, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(33), 16357–16366 CrossRef CAS.
- J. Yang, M. V. Agnihotri, C. J. Huseby, J. Kuret and S. J. Singer, A theoretical study of polymorphism in VQIVYK fibrils, Biophys. J., 2021, 120(8), 1396–1416 CrossRef CAS.
- I. Huvent, A. Kamah, F.-X. Cantrelle, N. Barois, C. Slomianny and C. Smet-Nocca, et al., A functional fragment of Tau forms fibers without the need for an intermolecular cysteine bridge, Biochem. Biophys. Res. Commun., 2014, 445(2), 299–303 CrossRef CAS.
- H. Wille, G. Drewes, J. Biernat, E.-M. Mandelkow and E. Mandelkow, Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro, J. Cell Biol., 1992, 118(3), 573–584 CrossRef CAS.
- W. Zhang, A. Tarutani, K. L. Newell, A. G. Murzin, T. Matsubara and B. Falcon, et al., Novel tau filament fold in corticobasal degeneration, Nature, 2020, 580(7802), 283–287 CrossRef CAS.
- R. Bhattacharyya, D. Pal and P. Chakrabarti, Disulfide bonds, their stereospecific environment and conservation in protein structures, Protein Eng., Des. Sel., 2004, 17(11), 795–808 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat and H. Weissig, et al., The protein data bank, Nucleic Acids Res., 2000, 28(1), 235–242 CrossRef CAS.
- W. L. DeLano, Pymol: An open-source molecular graphics tool, CCP4 Newsletter on Protein Crystallography, 2002, 40(1), 82–92 Search PubMed.
- F. Hernández, M. Pérez, J. J. Lucas and J. Avila, Sulfo-glycosaminoglycan content affects PHF-tau solubility and allows the identification of different types of PHFs, Brain Res., 2002, 935(1–2), 65–72 CrossRef.
- I. Santa-Maria, M. Perez, F. Hernandez, J. Avila and F. J. Moreno, Characteristics of the binding of thioflavin S to tau paired helical filaments, J. Alzheimer's Dis., 2006, 9(3), 279–285 CAS.
- M. Arrasate, M. Perez, J. M. Valpuesta and J. Avila, Role of glycosaminoglycans in determining the helicity of paired helical filaments, Am. J. Pathol., 1997, 151(4), 1115–1122 CAS.
- P. D. Dinkel, M. R. Holden, N. Matin and M. Margittai, RNA Binds to Tau Fibrils and Sustains Template-Assisted Growth, Biochemistry, 2015, 54(30), 4731–4740 CrossRef CAS PubMed.
- Y. Fichou, Y. Lin, J. N. Rauch, M. Vigers, Z. Zeng and M. Srivastava, et al., Cofactors are essential constituents of stable and seeding-active tau fibrils, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(52), 13234–13239 CrossRef CAS PubMed.
- M. von Bergen, S. Barghorn, S. A. Müller, M. Pickhardt, J. Biernat and E.-M. Mandelkow, et al., The core of tau-paired helical filaments studied by scanning transmission electron microscopy and limited proteolysis, Biochemistry, 2006, 45(20), 6446–6457 CrossRef CAS.
- S. W. Carlson, M. Branden, K. Voss, Q. Sun, C. A. Rankin and T. C. Gamblin, A complex mechanism for inducer mediated tau polymerization, Biochemistry, 2007, 46(30), 8838–8849 CrossRef CAS.
- Z. Shriver, I. Capila, G. Venkataraman and R. Sasisekharan, Heparin and heparan sulfate: analyzing structure and microheterogeneity, Handb. Exp. Pharmacol., 2012, 207, 159–176 CAS.
- M. Kasai and F. Oosawa, Removal of nucleotides from F-actin, Biochim. Biophys. Acta, 1963, 75, 223 CrossRef CAS.
- M. Tanaka, H. Xiao and K. Kiuchi, Heparin facilitates glial cell line-derived neurotrophic factor signal transduction, NeuroReport, 2002, 13(15), 1913–1916 CrossRef CAS PubMed.
- G. Daum, U. Hedin, Y. Wang, T. Wang and A. W. Clowes, Diverse effects of heparin on mitogen-activated protein kinase–dependent signal transduction in vascular smooth muscle cells, Circ. Res., 1997, 81(1), 17–23 CrossRef CAS PubMed.
- F. A. Hills, V. M. Abrahams, B. González-Timón, J. Francis, B. Cloke and L. Hinkson, et al., Heparin prevents programmed cell death in human trophoblast, MHR: Basic Sci. Reprod. Med., 2006, 12(4), 237–243 CAS.
- G. Chatzinikolaou, D. Nikitovic, A. Berdiaki, A. Zafiropoulos, P. Katonis and N. Karamanos, et al., Heparin regulates colon cancer cell growth through p38 mitogen-activated protein kinase signalling, Cell Proliferation, 2010, 43(1), 9–18 CrossRef CAS PubMed.
- Y. Chen, M. Scully, G. Dawson, C. Goodwin, M. Xia and X. Lu, et al., Perturbation of the heparin/heparin-sulfate interactome of human breast cancer cells modulates pro-tumourigenic effects associated with PI3K/Akt and MAPK/ERK signalling, Thromb. Haemost., 2013, 109(06), 1148–1157 CrossRef CAS.
- B. Mulloy and C. C. Rider, Cytokine/Proteoglycan Interactions: a Biochemical Society Focused Meeting (Royal Holloway University of London), Biochemistry, 2006, 28(4), 37–38 CrossRef.
- F. J. Moreno, M. Medina, M. Perez, E. Montejo de Garcini and J. Avila, Glycogen synthase kinase 3 phosphorylates recombinant human tau protein at serine-262 in the presence of heparin (or tubulin), FEBS Lett., 1995, 372(1), 65–68 CrossRef CAS.
- T. Kato, H. Sasaki, T. Katagiri, H. Sasaki, K. Koiwai and H. Youki, et al., The binding of basic fibroblast growth factor to Alzheimer's neurofibrillary tangles and senile plaques, Neurosci. Lett., 1991, 122(1), 33–36 CrossRef CAS PubMed.
- J. Su, B. Cummings and C. Cotman, Localization of heparan sulfate glycosaminoglycan and proteoglycan core protein in aged brain and Alzheimer's disease, Neuroscience, 1992, 51(4), 801–813 CrossRef CAS.
- D. A. DeWitt, J. Silver, D. R. Canning and G. Perry, Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer’s disease, Exp. Neurol., 1993, 121(2), 149–152 CrossRef CAS PubMed.
- D. A. Dewitt, P. Richey, D. Praprotnik, J. Silver and G. Perry, Chondroitin sulfate proteoglycans are a common component of neuronal inclusions and astrocytic reaction in neurodegenerative diseases, Brain Res., 1994, 656(1), 205–209 CrossRef CAS.
- J. Ma, C. Ma, J. Li, Y. Sun, F. Ye and K. Liu, et al., Extracellular matrix proteins involved in Alzheimer’s disease, Chem. – Eur. J., 2020, 26(53), 12101–12110 CrossRef CAS.
- A. Maiza, S. Chantepie, C. Vera, A. Fifre, M. B. Huynh and O. Stettler, et al., The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration, FEBS Lett., 2018, 592(23), 3806–3818 CrossRef CAS.
- K. Fukuchi, M. Hart and L. Li, Alzheimer's disease and heparan sulfate proteoglycan, Front. Biosci., 1998, 3, d327–d337 CrossRef CAS.
- J. N. Rauch, J. J. Chen, A. W. Sorum, G. M. Miller, T. Sharf and S. K. See, et al., Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs), Sci. Rep., 2018, 8(1), 6382 CrossRef.
- C. Pernègre, A. Duquette and N. Leclerc, Tau Secretion: Good and Bad for Neurons, Front. Neurosc., 2019, 13, 649 CrossRef.
- C. A. Brunello, M. Merezhko, R.-L. Uronen and H. J. Huttunen, Mechanisms of secretion and spreading of pathological tau protein, Cell. Mol. Life Sci., 2020, 77(9), 1721–1744 CrossRef CAS.
- J. Lewis and D. W. Dickson, Propagation of tau pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies, Acta Neuropathol., 2016, 131(1), 27–48 CrossRef CAS PubMed.
- T. Vogels, A. Leuzy, C. Cicognola, N. J. Ashton, T. Smolek and M. Novak, et al., Propagation of Tau Pathology: Integrating Insights From Postmortem and In Vivo Studies, Biol. Psychiatry, 2020, 87(9), 808–818 CrossRef CAS PubMed.
- J. K. Holth, S. K. Fritschi, C. Wang, N. P. Pedersen, J. R. Cirrito and T. E. Mahan, et al., The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans, Science., 2019, eaav2546 Search PubMed.
- K. Yamada, T. K. Patel, K. Hochgräfe, T. E. Mahan, H. Jiang and F. R. Stewart, et al., Analysis of in vivo turnover of tau in a mouse model of tauopathy, Mol. Neurodegener., 2015, 10, 55 CrossRef.
- K. Yamada, In Vivo Microdialysis of Brain Interstitial Fluid for the Determination of Extracellular Tau Levels, Methods Mol. Biol., 2017, 1523, 285–296 CrossRef CAS PubMed.
- K. Yamada, J. R. Cirrito, F. R. Stewart, H. Jiang, M. B. Finn and B. B. Holmes, et al., In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice, J. Neurosci., 2011, 31(37), 13110–13117 CrossRef CAS PubMed.
- T. Undin, S. B. Lind and A. P. Dahlin, MS for investigation of time-dependent protein adsorption on surfaces in complex biological samples, Future Sci. OA, 2015, 1(4), Fso32 Search PubMed.
- M. Mawal-Dewan, P. C. Sen, M. Abdel-Ghany, D. Shalloway and E. Racker, Phosphorylation of tau protein by purified p34cdc28 and a related protein kinase from neurofilaments, J. Biol. Chem., 1992, 267(27), 19705–19709 CrossRef CAS.
- R. Brandt, G. Lee, D. B. Teplow, D. Shalloway and M. Abdel-Ghany, Differential effect of phosphorylation and substrate modulation on tau's ability to promote microtubule growth and nucleation, J. Biol. Chem., 1994, 269(16), 11776–11782 CrossRef CAS.
- S. D. Yang, J. S. Yu, S. G. Shiah and J. J. Huang, Protein kinase FA/glycogen synthase kinase-3 alpha after heparin potentiation phosphorylates tau on sites abnormally phosphorylated in Alzheimer's disease brain, J. Neurochem., 1994, 63(4), 1416–1425 CrossRef CAS.
- M. Goedert, M. Hasegawa, R. Jakes, S. Lawler, A. Cuenda and P. Cohen, Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases, FEBS Lett., 1997, 409(1), 57–62 CrossRef CAS.
- M. Hasegawa, R. A. Crowther, R. Jakes and M. Goedert, Alzheimer-like changes in microtubule-associated protein Tau induced by sulfated glycosaminoglycans. Inhibition of microtubule binding, stimulation of phosphorylation, and filament assembly depend on the degree of sulfation, J. Biol. Chem., 1997, 272(52), 33118–33124 CrossRef CAS PubMed.
- M. Hasegawa, M. Morishima-Kawashima, K. Takio, M. Suzuki, K. Titani and Y. Ihara, Protein sequence and mass spectrometric analyses of tau in the Alzheimer's disease brain, J. Biol. Chem., 1992, 267, 17047 CrossRef CAS.
- K. Iqbal, C. del, A. Alonso, S. Chen, M. O. Chohan, E. El-Akkad and C.-X. Gong, et al., Tau pathology in Alzheimer disease and other tauopathies, Biochim. Biophys. Acta, Mol. Basis Dis., 2005, 1739(2), 198–210 CrossRef CAS PubMed.
- H. C. Schröder, A. Bernd, R. K. Zahn and W. E. Müller, Binding of polyribonucleotides and polydeoxyribonucleotides to bovine brain microtubule protein: age-dependent modulation via phosphorylation of high-molecular-weight microtubule-associated proteins and tau proteins, Mech. Ageing Dev., 1984, 24(1), 101–117 CrossRef.
- X. Zhang, Y. Lin, N. A. Eschmann, H. Zhou, J. N. Rauch and I. Hernandez, et al., RNA stores tau reversibly in complex coacervates, PLoS Biol., 2017, 15(7), e2002183 CrossRef.
- S. D. Ginsberg, J. E. Galvin, T. S. Chiu, V. M. Lee, E. Masliah and J. Q. Trojanowski, RNA sequestration to pathological lesions of neurodegenerative diseases, Acta Neuropathol., 1998, 96, 487 CrossRef CAS.
- S. D. Ginsberg, P. B. Crino, V. M. Lee, J. H. Eberwine and J. Q. Trojanowski, Sequestration of RNA in Alzheimer's disease neurofibrillary tangles and senile plaques, Ann. Neurol., 1997, 41, 200 CrossRef CAS PubMed.
- T. Kampers, P. Friedhoff, J. Biernat, E.-M. Mandelkow and E. Mandelkow, RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments, FEBS Lett., 1996, 399(3), 344–349 CrossRef CAS PubMed.
- X. S. Wang, D. L. Wang, J. Zhao, M. H. Qu, X. H. Zhou and H. J. He, et al., The proline-rich domain and the microtubule binding domain of protein tau acting as RNA binding domains, Protein Peptide Lett., 2006, 13(7), 679–685 CrossRef CAS.
- S. Ambadipudi, J. Biernat, D. Riedel, E. Mandelkow and M. Zweckstetter, Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau, Nat. Commun., 2017, 8(1), 275 CrossRef.
- A. L. Darling and J. Shorter, Combating deleterious phase transitions in neurodegenerative disease, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 1868(5), 118984 CrossRef CAS.
- A. Zbinden, M. Pérez-Berlanga, P. De Rossi and M. Polymenidou, Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force, Dev. Cell, 2020, 55(1), 45–68 CrossRef CAS.
- S. Wegmann, Liquid-Liquid Phase Separation of Tau Protein in Neurobiology and Pathology, Tau Biology, Springer, 2019, pp. 341–357 Search PubMed.
- W. M. Babinchak and W. K. Surewicz, Liquid–Liquid Phase Separation and Its Mechanistic Role in Pathological Protein Aggregation, J. Mol. Biol., 2020, 432(7), 1910–1925 CrossRef CAS.
- S. Wegmann, B. Eftekharzadeh, K. Tepper, K. M. Zoltowska, R. E. Bennett and S. Dujardin, et al., Tau protein liquid-liquid phase separation can initiate tau aggregation, EMBO J., 2018, 37(7), e98049 CrossRef.
- Y. Lin, J. McCarty, J. N. Rauch, K. T. Delaney, K. S. Kosik, G. H. Fredrickson, J. E. Shea and S. Han, Narrow equilibrium window for complex coacervation of tau and RNA under cellular conditions, eLife, 2019, 8, e42571 CrossRef PubMed.
- Y. Lin, Y. Fichou, Z. Zeng, N. Y. Hu and S. Han, Electrostatically Driven Complex Coacervation and Amyloid Aggregation of Tau Are Independent Processes with Overlapping Conditions, ACS Chem. Neurosci., 2020, 11(4), 615–627 CrossRef CAS PubMed.
- S. Boyko, K. Surewicz and W. K. Surewicz, Regulatory mechanisms of tau protein fibrillation under the conditions of liquid–liquid phase separation, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(50), 31882–31890 CrossRef CAS PubMed.
- M. R. Reynolds, R. W. Berry and L. I. Binder, Site-specific nitration and oxidative dityrosine bridging of the tau protein by peroxynitrite: implications for Alzheimer's disease, Biochemistry, 2005, 44, 1690 CrossRef CAS PubMed.
- T. C. Gamblin, R. W. Berry and L. I. Binder, Modeling tau polymerization in vitro: a review and synthesis, Biochemistry, 2003, 42, 15009–15017 CrossRef CAS PubMed.
- N. M. Kanaan, C. Hamel, T. Grabinski and B. Combs, Liquid-liquid phase separation induces pathogenic tau conformations in vitro, Nat. Commun., 2020, 11(1), 2809 CrossRef CAS PubMed.
- Y. Soeda and A. Takashima, New Insights Into Drug Discovery Targeting Tau Protein, Front. Mol. Neurosci., 2020, 13, 231 Search PubMed.
- E. G. Gray, M. Paula-Barbosa and A. Roher, Neuropathol. Appl. Neurobiol., 1987, 13, 110 CrossRef.
- D. R. Sparkman, W. J. Goux, C. M. Jones, C. L. White III and S. J. Hill, Biochem. Biophys. Res. Commun., 1991, 181, 771–779 CrossRef CAS.
- W. J. Goux, S. Rodriguez and D. R. Sparkman, Analysis of the core components of Alzheimer paired helical filaments. A gas chromatography/mass spectrometry characterization of fatty acids, carbohydrates and long-chain bases, FEBS Lett., 1995, 366(1), 81–85 CrossRef CAS . Erratum in: FEBS Lett., 1995, 375(1–2), 168.
- C. D. Surridge and R. G. Burns, The difference in the binding of phosphatidylinositol distinguishes MAP2 from MAP2C and Tau, Biochemistry, 1994, 33(26), 8051–8057 CrossRef CAS.
- R. Brandt, J. Léger and G. Lee, Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain, J. Cell Biol., 1995, 131(5), 1327–1340 CrossRef CAS.
- E. Bok, E. Leem, B. R. Lee, J. M. Lee, C. J. Yoo and E. M. Lee, et al., Role of the Lipid Membrane and Membrane Proteins in Tau Pathology, Front. Cell Dev. Biol., 2021, 9, 653815 CrossRef.
- D. Talaga, W. Smeralda, L. Lescos, J. Hunel, N. Lepejova-Caudy and C. Cullin, et al., PIP2 Phospholipid-Induced Aggregation of Tau Filaments Probed by Tip-Enhanced Raman Spectroscopy, Angew. Chem., Int. Ed., 2018, 57(48), 15738–15742 CrossRef CAS.
- E. R. Georgieva, S. Xiao, P. P. Borbat, J. H. Freed and D. Eliezer, Tau binds to lipid membrane surfaces via short amphipathic helices located in its microtubule-binding repeats, Biophys. J., 2014, 107(6), 1441–1452 CrossRef CAS PubMed.
- C. G. Barracchia, R. Tira, F. Parolini, F. Munari, L. Bubacco and G. A. Spyroulias, et al., Unsaturated Fatty Acid-Induced Conformational Transitions and Aggregation of the Repeat Domain of Tau, Molecules, 2020, 25(11), 2716 CrossRef CAS PubMed.
- A. M. Fanni, C. M. Vander Zanden, P. V. Majewska, J. Majewski and E. Y. Chi, Membrane-mediated fibrillation and toxicity of the tau hexapeptide PHF6, J. Biol. Chem., 2019, 294(42), 15304–15317 CrossRef.
- N. Ait-Bouziad, G. Lv, A. L. Mahul-Mellier, S. Xiao, G. Zorludemir and D. Eliezer, et al., Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes, Nat. Commun., 2017, 8(1), 1678 CrossRef.
- A. M. Pooler, A. Usardi, C. J. Evans, K. L. Philpott, W. Noble and D. P. Hanger, Dynamic association of tau with neuronal membranes is regulated by phosphorylation, Neurobiol. Aging, 2012, 33(2), 431 CrossRef .e27–e38.
- J. Eidenmüller, T. Fath, T. Maas, M. Pool, E. Sontag and R. Brandt, Phosphorylation-mimicking glutamate clusters in the proline-rich region are sufficient to simulate the functional deficiencies of hyperphosphorylated tau protein, Biochem. J., 2001, 357(3), 759–767 CrossRef.
- F. J. Ekinci and T. B. Shea, Phosphorylation of tau alters its association with the plasma membrane, Cell. Mol. Neurobiol., 2000, 20(4), 497–508 CrossRef CAS.
- T. Maas, J. Eidenmüller and R. Brandt, Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments, J. Biol. Chem., 2000, 275(21), 15733–15740 CrossRef CAS.
- G. S. Nuebling, E. Plesch, V. C. Ruf, T. Högen, S. Lorenzl and F. Kamp, et al., Binding of Metal-Ion-Induced Tau Oligomers to Lipid Surfaces Is Enhanced by GSK-3β-Mediated Phosphorylation, ACS Chem. Neurosci., 2020, 11(6), 880–887 CrossRef CAS PubMed.
- S. B. Shelton and GV. Tau Johnson, and HMW tau phosphorylation and compartmentalization in apoptotic neuronal PC12 cells, J. Neurosci. Res., 2001, 66(2), 203–213 CrossRef CAS.
- A. Usardi, A. M. Pooler, A. Seereeram, C. H. Reynolds, P. Derkinderen and B. Anderton, et al., Tyrosine phosphorylation of tau regulates its interactions with Fyn SH2 domains, but not SH3 domains, altering the cellular localization of tau, FEBS J., 2011, 278(16), 2927–2937 CrossRef CAS.
- M. Merezhko, C. A. Brunello, X. Yan, H. Vihinen, E. Jokitalo and R. L. Uronen, et al., Secretion of Tau via an Unconventional Non-vesicular Mechanism, Cell Rep., 2018, 25(8), 2027–2035 CrossRef CAS PubMed e4.
- T. Katsinelos, M. Zeitler, E. Dimou, A. Karakatsani, H. M. Müller and E. Nachman, et al., Unconventional Secretion Mediates the Trans-cellular Spreading of Tau, Cell Rep., 2018, 23(7), 2039–2055 CrossRef CAS PubMed.
- D. M. Wilson and L. I. Binder, Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer's disease, Am. J. Pathol., 1997, 150(6), 2181 CAS.
- M. E. King, V. Ahuja, L. I. Binder and J. Kuret, Ligand-dependent tau filament formation: implications for Alzheimer's disease progression, Biochemistry, 1999, 38(45), 14851–14859 CrossRef CAS.
- R. A. Armstrong, On the'classification'of neurodegenerative disorders: discrete entities, overlap or continuum?, Folia Neuropathol., 2012, 50(3), 201–218 CrossRef CAS PubMed.
- A. Serrano-Pozo, M. P. Frosch, E. Masliah and B. T. Hyman, Neuropathological alterations in Alzheimer disease, Cold Spring Harbor Perspect. Med., 2011, 1(1), a006189 Search PubMed.
- B. Vasconcelos, I.-C. Stancu, A. Buist, M. Bird, P. Wang and A. Vanoosthuyse, et al., Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo, Acta Neuropathol., 2016, 131(4), 549–569 CrossRef CAS PubMed.
- R. E. Bennett, S. L. DeVos, S. Dujardin, B. Corjuc, R. Gor and J. Gonzalez, et al., Enhanced Tau Aggregation in the Presence of Amyloid β, Am. J. Pathol., 2017, 187(7), 1601–1612 CrossRef CAS.
- S. Oddo, A. Caccamo, J. D. Shepherd, M. P. Murphy, T. E. Golde and R. Kayed, et al., Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction, Neuron, 2003, 39(3), 409–421 CrossRef CAS.
- J. Götz, Chen Fv, Van Dorpe and J. Nitsch, R. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils, Science, 2001, 293(5534), 1491–1495 CrossRef.
- Z. He, J. L. Guo, J. D. McBride, S. Narasimhan, H. Kim and L. Changolkar, et al., Amyloid-β plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation, Nat. Med., 2018, 24(1), 29–38 CrossRef CAS PubMed.
- L. Ciccone, C. Shi, D. di Lorenzo, A.-C. Van Baelen and N. Tonali, The Positive Side of the Alzheimer's Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1, Molecules, 2020, 25(10), 2439 CrossRef CAS PubMed.
- L. Hansen, D. Salmon, D. Galasko, E. Masliah, R. Katzman and R. DeTeresa, et al., The Lewy body variant of Alzheimer's disease: a clinical and pathologic entity, Neurology, 1990, 40(1), 1 CrossRef CAS PubMed.
- I. G. McKeith, D. Galasko, K. Kosaka, E. Perry, D. W. Dickson and L. Hansen, et al., Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop, Neurology, 1996, 47(5), 1113–1124 CrossRef CAS PubMed.
- N. Spotorno, D. G. Coughlin, C. A. Olm, D. Wolk, S. N. Vaishnavi and L. M. Shaw, et al., Tau pathology associates with in vivo cortical thinning in Lewy body disorders, Ann. Clin. Transl. Neurol., 2020, 7(12), 2342–2355 CrossRef CAS.
- M. Colom-Cadena, O. Grau-Rivera, L. Planellas, C. Cerquera, E. Morenas and S. Helgueta, et al., Regional Overlap of Pathologies in Lewy Body Disorders, J. Neuropathol. Exp. Neurol., 2017, 76(3), 216–224 CAS.
- B. I. Giasson, M. S. Forman, M. Higuchi, L. I. Golbe, C. L. Graves and P. T. Kotzbauer, et al., Initiation and synergistic fibrillization of tau and alpha-synuclein, Science, 2003, 300(5619), 636–640 CrossRef CAS PubMed.
- D. L. Castillo-Carranza, M. J. Guerrero-Muñoz, U. Sengupta, J. E. Gerson and R. Kayed, α-Synuclein Oligomers Induce a Unique Toxic Tau Strain, Biol. Psychiatry, 2018, 84(7), 499–508 CrossRef CAS.
- M. Iljina, A. J. Dear, G. A. Garcia, S. De, L. Tosatto and P. Flagmeier, et al., Quantifying Co-Oligomer Formation by α-Synuclein, ACS Nano, 2018, 12(11), 10855–10866 CrossRef CAS.
- F. Bassil, E. S. Meymand, H. J. Brown, H. Xu, T. O. Cox and S. Pattabhiraman, et al., α-Synuclein modulates tau spreading in mouse brains, J. Exp. Med., 2020, 218(1), e20192193 CrossRef.
- T. Williams, Z. Sorrentino, M. Weinrich, B. I. Giasson and P. Chakrabarty, Differential cross-seeding properties of tau and α-synuclein in mouse models of tauopathy and synucleinopathy, Brain Commun., 2020, 2(2), fcaa090 CrossRef PubMed.
- N. Uemura, M. T. Uemura, K. C. Luk, V. M. Y. Lee and J. Q. Trojanowski, Cell-to-Cell Transmission of Tau and α-Synuclein, Trends Mol. Med., 2020, 26(10), 936–952 CrossRef CAS.
- E. Vacchi, A. Kaelin-Lang and G. Melli, Tau and Alpha Synuclein Synergistic Effect in Neurodegenerative Diseases: When the Periphery Is the Core, Int. J. Mol. Sci., 2020, 21(14), 5030 CrossRef CAS.
- C. T. McMillan and D. A. Wolk, Presence of cerebral amyloid modulates phenotype and pattern of neurodegeneration in early Parkinson's disease, J. Neurol., Neurosurg. Psychiatry, 2016, 87(10), 1112–1122 CrossRef.
- D. C. Chung, S. Roemer, L. Petrucelli and D. W. Dickson, Cellular and pathological heterogeneity of primary tauopathies, Mol. Neurodegener., 2021, 16(1), 57 CrossRef PubMed.
- L. Martin, X. Latypova, C. M. Wilson, A. Magnaudeix, M.-L. Perrin and C. Yardin, et al., Tau protein kinases: Involvement in Alzheimer's disease, Ageing Res. Rev., 2013, 12(1), 289–309 CrossRef CAS PubMed.
- S. P. Braithwaite, J. B. Stock, P. J. Lombroso and A. C. Nairn, Protein phosphatases and Alzheimer's disease, Prog. Mol. Biol. Transl. Sci., 2012, 106, 343–379 CAS.
- H. Yadikar, I. Torres, G. Aiello, M. Kurup, Z. Yang and F. Lin, et al., Screening of tau protein kinase inhibitors in a tauopathy-relevant cell-based model of tau hyperphosphorylation and oligomerization, PLoS One, 2020, 15(7), e0224952 CrossRef CAS PubMed.
- R. V. Bhat, U. Andersson, S. Andersson, L. Knerr, U. Bauer and A. K. Sundgren-Andersson, The conundrum of GSK3 inhibitors: is it the dawn of a new beginning?, J. Alzheimer's Dis., 2018, 64(s1), S547–S554 CAS.
- C. L. Sayas and J. Ávila, GSK-3 and Tau: A Key Duet in Alzheimer’s Disease, Cells., 2021, 10(4), 721 CrossRef CAS.
- H. Yamaguchi, K. Ishiguro, T. Uchida, A. Takashima, C. A. Lemere and K. Imahori, Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3β and cyclin-dependent kinase 5, a component of TPK II, Acta Neuropathol., 1996, 92(3), 232–241 CrossRef CAS.
- K. Imahori and T. Uchida, Physiology and pathology of tau protein kinases in relation to Alzheimer's disease, J. Biochem., 1997, 121(2), 179–188 CAS.
- A. Takashima, GSK-3 is essential in the pathogenesis of Alzheimer's disease, J. Alzheimer's Dis., 2006, 9(s3), 309–317 CAS.
- T. Guo, W. Noble and D. P. Hanger, Roles of tau protein in health and disease, Acta Neuropathol., 2017, 133(5), 665–704 CrossRef CAS PubMed.
- M. Liu, T. Qian, W. Zhou, X. Tao, S. Sang and L. Zhao, Beneficial effects of low-dose lithium on cognitive ability and pathological alteration of Alzheimer’s disease transgenic mice model, NeuroReport, 2020, 31(13), 943–951 CrossRef CAS PubMed.
- R. Rubenstein, D. R. Sharma, B. Chang, N. Oumata, M. Cam and L. Vaucelle, et al., Novel Mouse Tauopathy Model for Repetitive Mild Traumatic Brain Injury: Evaluation of Long-Term Effects on Cognition and Biomarker Levels After Therapeutic Inhibition of Tau Phosphorylation, Front. Neurol., 2019, 10, 124 CrossRef PubMed.
- M. Hong, D. C. Chen, P. S. Klein and V. M.-Y. Lee, Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3, J. Biol. Chem., 1997, 272(40), 25326–25332 CrossRef CAS.
- S. Lovestone, D. R. Davis, M.-T. Webster, S. Kaech, J.-P. Brion and A. Matus, et al., Lithium reduces tau phosphorylation: effects in living cells and in neurons at therapeutic concentrations, Biol. Psychiatry, 1999, 45(8), 995–1003 CrossRef CAS.
- J. R. Muñoz-Montaño, F. J. Moreno, J. Avila and J. Díaz-Nido, Lithium inhibits Alzheimer's disease-like tau protein phosphorylation in neurons, FEBS Lett., 1997, 411(2–3), 183–188 CrossRef.
- A. Caccamo, S. Oddo, L. X. Tran and F. M. LaFerla, Lithium reduces tau phosphorylation but not Aβ or working memory deficits in a transgenic model with both plaques and tangles, Am. J. Pathol., 2007, 170(5), 1669–1675 CrossRef CAS.
- K. Leroy, K. Ando, C. Héraud, Z. Yilmaz, M. Authelet and J.-M. Boeynaems, et al., Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology, J. Alzheimer's Dis., 2010, 19(2), 705–719 CAS.
- L. Tondo, M. Alda, M. Bauer, V. Bergink, P. Grof and T. Hajek, et al., Clinical use of lithium salts: guide for users and prescribers, Int. J. Bipolar Disord., 2019, 7(1), 1–10 CrossRef PubMed.
- M. Donix and M. Bauer, Population studies of association between lithium and risk of neurodegenerative disorders, Curr. Alzheimer Res., 2016, 13(8), 873–878 CrossRef CAS.
- S. Matsunaga, T. Kishi, P. Annas, H. Basun, H. Hampel and N. Iwata, Lithium as a Treatment for Alzheimer's Disease: A Systematic Review and Meta-Analysis, J. Alzheimer's Dis., 2015, 48(2), 403–410 CAS.
- L. V. Kessing, T. A. Gerds, N. N. Knudsen, L. F. Jørgensen, S. M. Kristiansen and D. Voutchkova, et al., Association of Lithium in Drinking Water With the Incidence of Dementia, JAMA Psychiatry, 2017, 74(10), 1005–1010 CrossRef.
- W. F. Parker, R. J. Gorges, Y. N. Gao, Y. Zhang, K. Hur and R. D. Gibbons, Association between groundwater lithium and the diagnosis of bipolar disorder and dementia in the United States, JAMA Psychiatry, 2018, 75(7), 751–754 CrossRef.
- E. N. Wilson, S. Do Carmo, L. A. Welikovitch, H. Hall, L. F. Aguilar and M. K. Foret, et al., NP03, a Microdose Lithium Formulation, Blunts Early Amyloid Post-Plaque Neuropathology in McGill-R-Thy1-APP Alzheimer-Like Transgenic Rats, J. Alzheimer's Dis., 2020, 73, 723–739 Search PubMed.
- M. S. Noori, P. M. Bhatt, M. C. Courreges, D. Ghazanfari, C. Cuckler and C. M. Orac, et al., Identification of a novel selective and potent inhibitor of glycogen synthase kinase-3, Am. J. Physiol.: Cell Physiol., 2019, 317(6), C1289–C1303 CrossRef CAS.
- E. Tolosa, I. Litvan, G. U. Höglinger, D. Burn, A. Lees and M. V. Andrés, et al., A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy, Mov. Disord, 2014, 29(4), 470–478 CrossRef CAS.
- G. U. Höglinger, H. J. Huppertz, S. Wagenpfeil, M. V. Andrés, V. Belloch and T. León, et al., Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial, Mov. Disord, 2014, 29(4), 479–487 CrossRef PubMed.
- G. Lee, R. Thangavel, V. M. Sharma, J. M. Litersky, K. Bhaskar and S. M. Fang, et al., Phosphorylation of tau by fyn: Implications for Alzheimer's disease, J. Neurosci., 2004, 24(9), 2304–2312 CrossRef CAS.
- A. C. Kaufman, S. V. Salazar, L. T. Haas, J. Yang, M. A. Kostylev and A. T. Jeng, et al., F yn inhibition rescues established memory and synapse loss in A lzheimer mice, Ann. Neurol., 2015, 77(6), 953–971 CrossRef CAS.
- L. M. Smith, R. Zhu and S. M. Strittmatter, Disease-modifying benefit of Fyn blockade persists after washout in mouse Alzheimer's model, Neuropharmacology, 2018, 130, 54–61 CrossRef CAS.
- H. B. Nygaard, A. F. Wagner, G. S. Bowen, S. P. Good, M. G. MacAvoy and K. A. Strittmatter, et al., A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer's disease, Alzheimer's Res. Ther., 2015, 7(1), 1–11 CrossRef.
- C. H. Van Dyck, H. B. Nygaard, K. Chen, M. C. Donohue, R. Raman and R. A. Rissman, et al., Effect of AZD0530 on cerebral metabolic decline in Alzheimer disease: a randomized clinical trial, JAMA Neurol., 2019, 76(10), 1219–1229 CrossRef PubMed.
- T. Kimura, K. Ishiguro and S.-I. Hisanaga, Physiological and pathological phosphorylation of tau by Cdk5, Front. Mol. Neurosci., 2014, 7, 65 Search PubMed.
- S. D. Schlatterer, C. M. Acker and P. Davies, c-Abl in Neurodegenerative Disease, J. Mol. Neurosci., 2011, 45(3), 445–452 CrossRef CAS.
- G. M. Mórotz, E. B. Glennon, P. Gomez-Suaga, D. H. Lau, E. D. Robinson and É. Sedlák, et al., LMTK2 binds to kinesin light chains to mediate anterograde axonal transport of cdk5/p35 and LMTK2 levels are reduced in Alzheimer's disease brains, Acta Neuropathol. Commun., 2019, 7(1), 73 CrossRef.
- B. Melchior, G. K. Mittapalli, C. Lai, K. Duong-Polk, J. Stewart and B. Güner, et al., Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: A potential therapeutic for Alzheimer's disease, Aging Cell, 2019, 18(5), e13000 CrossRef.
- C. Giacomini, C. Y. Koo, N. Yankova, I. A. Tavares, S. Wray and W. Noble, et al., A new TAO kinase inhibitor reduces tau phosphorylation at sites associated with neurodegeneration in human tauopathies, Acta Neuropathol. Commun., 2018, 6(1), 37 CrossRef PubMed.
- A. L. Fallacara, I. M. L. Trist, S. Schenone and M. Botta, Inhibitors of Tau-Phosphorylating Kinases, in Alzheimer's Disease II, ed. Wolfe M. S., Cham, Springer International Publishing, 2017, pp. 119–158 Search PubMed.
- C. Schwab, A. J. DeMaggio, N. Ghoshal, L. I. Binder, J. Kuret and P. L. McGeer, Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases, Neurobiol. Aging, 2000, 21(4), 503–510 CrossRef CAS.
- I. Ferrer, R. Blanco, M. Carmona and B. Puig, Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38 kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies, J. Neural Transm., 2001, 108(12), 1397–1415 CrossRef CAS.
- J.-J. Pei, H. Braak, W.-L. An, B. Winblad, R. F. Cowburn and K. Iqbal, et al., Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer's disease, Mol. Brain Res., 2002, 109(1–2), 45–55 CrossRef CAS.
- V. Vingtdeux, P. Davies, D. W. Dickson and P. Marambaud, AMPK is abnormally activated in tangle-and pre-tangle-bearing neurons in Alzheimer's disease and other tauopathies, Acta Neuropathol., 2011, 121(3), 337–349 CrossRef CAS.
- F. Liu, Z. Liang, J. Shi, D. Yin, E. El-Akkad and I. Grundke-Iqbal, et al., PKA modulates GSK-3β-and cdk5-catalyzed phosphorylation of tau in site-and kinase-specific manners, FEBS Lett., 2006, 580(26), 6269–6274 CrossRef CAS.
- T. Taniguchi, T. Kawamata, H. Mukai, H. Hasegawa, T. Isagawa and M. Yasuda, et al., Phosphorylation of tau is regulated by PKN, J. Biol. Chem., 2001, 276(13), 10025–10031 CrossRef CAS.
- K. Imahori, M. Hoshi, K. Ishiguro, K. Sato, M. Takahashi and R. Shiurba, et al., Possible role of tau protein kinases in pathogenesis of Alzheimer's disease, Neurobiol. Aging, 1998, 19(1), S93–S98 CrossRef CAS PubMed.
- J. Y. Chin, R. B. Knowles, A. Schneider, G. Drewes, E.-M. Mandelkow and B. T. Hyman, Microtubule-affinity regulating kinase (MARK) is tightly associated with neurofibrillary tangles in Alzheimer brain: a fluorescence resonance energy transfer study, J. Neuropathol. Exp. Neurol., 2000, 59(11), 966–971 CrossRef CAS.
- W. Qian, X. Yin, W. Hu, J. Shi, J. Gu and I. Grundke-Iqbal, et al., Activation of protein phosphatase 2B and hyperphosphorylation of Tau in Alzheimer's disease, J. Alzheimer's Dis., 2011, 23(4), 617–627 CAS.
- H. Tanimukai, I. Grundke-Iqbal and K. Iqbal, Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease, Am. J. Pathol., 2005, 166(6), 1761–1771 CrossRef CAS.
- F. Liu, K. Iqbal, I. Grundke-Iqbal, S. Rossie and C.-X. Gong, Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer's disease, J. Biol. Chem., 2005, 280(3), 1790–1796 CrossRef CAS.
- U. Wasik, G. Schneider, A. Mietelska-Porowska, M. Mazurkiewicz, H. Fabczak and S. Weis, et al., Calcyclin binding protein and Siah-1 interacting protein in Alzheimer's disease pathology: neuronal localization and possible function, Neurobiol. Aging, 2013, 34(5), 1380–1388 CrossRef CAS.
- D. L. Graham, A. J. Gray, J. A. Joyce, D. Yu, J. O'Moore and G. A. Carlson, et al., Increased O-GlcNAcylation reduces pathological tau without affecting its normal phosphorylation in a mouse model of tauopathy, Neuropharmacology, 2014, 79, 307–313 CrossRef CAS PubMed.
- N. B. Hastings, X. Wang, L. Song, B. D. Butts, D. Grotz and R. Hargreaves, et al., Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice, Mol. Neurodegener., 2017, 12(1), 1–16 CrossRef.
- P. Borghgraef, C. Menuet, C. Theunis, J. V. Louis, H. Devijver and H. Maurin, et al., Increasing brain protein O-GlcNAc-ylation mitigates breathing defects and mortality of Tau. P301L mice, PLoS One, 2013, 8(12), e84442 CrossRef PubMed.
- Y. Yu, L. Zhang, X. Li, X. Run, Z. Liang and Y. Li, et al., Differential effects of an O-GlcNAcase inhibitor on tau phosphorylation, PLoS One, 2012, 7(4), e35277 CrossRef CAS.
- H. G. Selnick, J. F. Hess, C. Tang, K. Liu, J. B. Schachter and J. E. Ballard, et al., Discovery of MK-8719, a potent O-GlcNAcase inhibitor as a potential treatment for tauopathies, J. Med. Chem., 2019, 62(22), 10062–10097 CrossRef CAS PubMed.
- X. Wang, W. Li, J. Marcus, M. Pearson, L. Song and K. Smith, et al., MK-8719, a Novel and Selective O-GlcNAcase Inhibitor That Reduces the Formation of Pathological Tau and Ameliorates Neurodegeneration in a Mouse Model of Tauopathy, J. Pharmacol. Exp. Ther., 2020, 374(2), 252–263 CrossRef CAS.
- L. VandeVrede, A. L. Boxer and M. Polydoro, Targeting tau: Clinical trials and novel therapeutic approaches, Neurosci. Lett., 2020, 134919 CrossRef CAS.
- H. E. Lee, D. Lim, J. Y. Lee, S. M. Lim and A. N. Pae, Recent tau-targeted clinical strategies for the treatment of Alzheimer’s disease, Future Med. Chem., 2019, 11(16), 1845–1848 CrossRef CAS PubMed.
- K. Shirakawa, L. Wang, N. Man, J. Maksimoska, A. W. Sorum and H. W. Lim, et al., Salicylate, diflunisal and their metabolites inhibit CBP/p300 and exhibit anticancer activity, eLife, 2016, 5, e11156 CrossRef PubMed.
- Y. Wang and E. Mandelkow, Degradation of tau protein by autophagy and proteasomal pathways, Biochem. Soc. Trans., 2012, 40(4), 644–652 CrossRef CAS PubMed.
- C. Cook, J. N. Stankowski, Y. Carlomagno, C. Stetler and L. Petrucelli, Acetylation: a new key to unlock tau's role in neurodegeneration, Alzheimer's Res. Ther., 2014, 6(3), 29 CrossRef.
- K. Cisek, G. L. Cooper, C. J. Huseby and J. Kuret, Structure and mechanism of action of tau aggregation inhibitors, Curr. Alzheimer's Res., 2014, 11(10), 918–927 CrossRef CAS PubMed.
- C. Wischik, P. Edwards, R. Lai, M. Roth and C. Harrington, Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines, Proc. Natl. Acad. Sci. U. S. A., 1996, 93(20), 11213–11218 CrossRef CAS.
- M. Landau, M. R. Sawaya, K. F. Faull, A. Laganowsky, L. Jiang and S. A. Sievers, et al., Towards a pharmacophore for amyloid, PLoS Biol., 2011, 9(6), e1001080 CrossRef CAS.
- W. Li, J. B. Sperry, A. Crowe, J. Q. Trojanowski, A. B. Smith and V. M. Y. Lee, Inhibition of tau fibrillization by oleocanthal via reaction with the amino groups of tau, J. Neurochem., 2009, 110(4), 1339–1351 CrossRef CAS PubMed.
- S. Sinha, D. H. J. Lopes, Z. Du, E. S. Pang, A. Shanmugam and A. Lomakin, et al., Lysine-Specific Molecular Tweezers Are Broad-Spectrum Inhibitors of Assembly and Toxicity of Amyloid Proteins, J. Am. Chem. Soc., 2011, 133(42), 16958–16969 CrossRef CAS.
- E. Akoury, M. Gajda, M. Pickhardt, J. Biernat, P. Soraya and C. Griesinger, et al., Inhibition of tau filament formation by conformational modulation, J. Am. Chem. Soc., 2013, 135(7), 2853–2862 CrossRef CAS.
- S. Dolai, W. Shi, C. Corbo, C. Sun, S. Averick and D. Obeysekera, et al., “Clicked” sugar–curcumin conjugate: modulator of amyloid-β and tau peptide aggregation at ultralow concentrations, ACS Chem. Neurosci., 2011, 2(12), 694–699 CrossRef CAS PubMed.
- S. Fuse, K. Matsumura, Y. Fujita, H. Sugimoto and T. Takahashi, Development of dual targeting inhibitors against aggregations of amyloid-β and tau protein, Eur. J. Med. Chem., 2014, 85, 228–234 CrossRef CAS.
- T. Klingstedt and K. P. R. Nilsson, Luminescent conjugated poly-and oligo-thiophenes: optical ligands for spectral assignment of a plethora of protein aggregates, Biochem. Soc. Trans., 2012, 40(4), 704–710 CrossRef CAS.
- E. E. Congdon and E. M. Sigurdsson, Tau-targeting therapies for Alzheimer disease, Nat. Rev. Neurol., 2018, 14(7), 399–415 CrossRef CAS.
- Y. Soeda, M. Saito, S. Maeda, K. Ishida, A. Nakamura and S. Kojima, et al., Methylene Blue Inhibits Formation of Tau Fibrils but not of Granular Tau Oligomers: A Plausible Key to Understanding Failure of a Clinical Trial for Alzheimer's Disease, J. Alzheimer's Dis., 2019, 68(4), 1677–1686 CAS.
- A. Crowe, M. J. James, V. M. Y. Lee, A. B. Smith, J. Q. Trojanowski and C. Ballatore, et al., Aminothienopyridazines and methylene blue affect Tau fibrillization via cysteine oxidation, J. Biol. Chem., 2013, 288(16), 11024–11037 CrossRef CAS PubMed.
- C. Ballatore, A. Crowe, F. Piscitelli, M. James, K. Lou and G. Rossidivito, et al., Aminothienopyridazine inhibitors of tau aggregation: evaluation of structure-activity relationship leads to selection of candidates with desirable in vivo properties, Bioorg. Med. Chem., 2012, 20(14), 4451–4461 CrossRef CAS.
- E. E. Congdon, J. W. Wu, N. Myeku, Y. H. Figueroa, M. Herman and P. S. Marinec, et al., Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo, Autophagy, 2012, 8(4), 609–622 CrossRef CAS PubMed.
- W. Sun, S. Lee, X. Huang, S. Liu, M. Inayathullah and K.-M. Kim, et al., Attenuation of synaptic toxicity and MARK4/PAR1-mediated Tau phosphorylation by methylene blue for Alzheimer's disease treatment, Sci. Rep., 2016, 6(1), 1–10 CrossRef PubMed.
- C. Stack, S. Jainuddin, C. Elipenahli, M. Gerges, N. Starkova and A. A. Starkov, et al., Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity, Hum. Mol. Genet., 2014, 23(14), 3716–3732 CrossRef CAS.
- J. R. Williams and F. E. Challis, Methylene blue as an antidote for anilin dye poisoning: case report with confirmatory experimental study, J. Lab. Clin. Med., 1933, 19(2), 166–171 CAS.
- C. W. Steele and W. W. Spink, Methylene blue in the treatment of poisonings associated with methemoglobinemia, N. Engl. J. Med., 1933, 208(22), 1152–1153 CrossRef.
- I. Jack Clifton and J. B. Leikin, Methylene blue, Am. J. Phytomed. Clin. Ther., 2003, 10(4), 289–291 Search PubMed.
- P. Guttmann and P. Ehrlich, Ueber die wirkung des methylenblau bei malaria, The Collected Papers of Paul Ehrlich, Elsevier, 1960, pp. 9–14 Search PubMed.
- G. Lu, M. Nagbanshi, N. Goldau, M. M. Jorge, P. Meissner and A. Jahn, et al., Efficacy and safety of methylene blue in the treatment of malaria: a systematic review, BMC Med., 2018, 16(1), 1–16 CrossRef.
- A. Delport, B. H. Harvey, A. Petzer and J. P. Petzer, Methylene blue and its analogues as antidepressant compounds, Metab. Brain Dis., 2017, 32(5), 1357–1382 CrossRef CAS PubMed.
- M. Oz, D. Isaev, D. E. Lorke, M. Hasan, G. Petroianu and T. S. Shippenberg, Methylene blue inhibits function of the 5-HT transporter, Br. J. Pharmacol., 2012, 166(1), 168–176 CrossRef CAS.
- M. J. Telch, A. K. Bruchey, D. Rosenfield, A. R. Cobb, J. Smits and S. Pahl, et al., Effects of post-session administration of methylene blue on fear extinction and contextual memory in adults with claustrophobia, Am. J. Psychiatry, 2014, 171(10), 1091–1098 CrossRef PubMed.
- M. Oz, D. E. Lorke, M. Hasan and G. A. Petroianu, Cellular and molecular actions of Methylene Blue in the nervous system, Med. Res. Rev., 2011, 31(1), 93–117 CrossRef CAS.
- V. Melis, M. Magbagbeolu, J. E. Rickard, D. Horsley, K. Davidson and K. A. Harrington, et al., Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models, Behav. Pharmacol., 2015, 26(4), 353 CrossRef CAS PubMed.
- C. R. Harrington, J. M. Storey, S. Clunas, K. A. Harrington, D. Horsley and A. Ishaq, et al., Cellular models of aggregation-dependent template-directed proteolysis to characterize tau aggregation inhibitors for treatment of Alzheimer disease, J. Biol. Chem., 2015, 290(17), 10862–10875 CrossRef CAS PubMed.
- M. Colin, S. Dujardin, S. Schraen-Maschke, G. Meno-Tetang, C. Duyckaerts and J.-P. Courade, et al., From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy, Acta Neuropathol., 2020, 139(1), 3–25 CrossRef CAS PubMed.
- R. Pizzarelli, N. Pediconi and S. Di Angelantonio, Molecular Imaging of Tau Protein: New Insights and Future Directions, Front. Mol. Neurosci., 2020, 13, 586169 CrossRef CAS.
- Y. Shi, A. G. Murzin, B. Falcon, A. Epstein, J. Machin and P. Tempest, et al., Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607, Acta neuropathologica, 2021, 141(5), 697–708 CrossRef CAS.
- L. A. Sandusky-Beltran and E. M. Sigurdsson, Tau immunotherapies: Lessons learned, current status and future considerations, Neuropharmacology, 2020, 175, 108104 CrossRef CAS PubMed.
- T. West, Y. Hu, P. B. Verghese, R. J. Bateman, J. B. Braunstein and I. Fogelman, et al., Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer's Disease and Other Tauopathies, J. Prev. Alzheimer's Dis., 2017, 4(4), 236–241 CAS.
- R. J. Bateman, T. L. Benzinger, S. Berry, D. B. Clifford, C. Duggan and A. M. Fagan, et al., The DIAN-TU Next Generation Alzheimer's prevention trial: Adaptive design and disease progression model, Alzheimer's Dementia, 2017, 13(1), 8–19 CrossRef PubMed.
- B. Dunn, P. Stein and P. Cavazzoni, Approval of Aducanumab for Alzheimer Disease—the FDA’s Perspective, JAMA Int. Med., 2021, 181(10), 1276–1278 CrossRef PubMed.
- G. C. Alexander, D. S. Knopman, S. S. Emerson, B. Ovbiagele, R. J. Kryscio and J. S. Perlmutter, et al., Revisiting FDA Approval of Aducanumab, N. Engl. J. Med., 2021, 385(9), 769–771 CrossRef CAS.
- https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug .
- T. Bussiere, P. Weinreb, R. Dunstan, F. Qian, M. Arast and M. Li, Differential in vitro and in vivo binding profiles of BIIB037 and other anti-abeta clinical antibody candidates, Neurodegener. Dis., 2013, 11, 1 Search PubMed.
- E. Kontsekova, N. Zilka, B. Kovacech, P. Novak and M. Novak, First-in-man tau vaccine targeting structural determinants essential for pathological tau–tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer's disease model, Alzheimer's Res. Ther., 2014, 6(4), 1–12 Search PubMed.
- C. Theunis, N. Crespo-Biel, V. Gafner, M. Pihlgren, M. P. López-Deber and P. Reis, et al., Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau. P301L mice that model tauopathy, PLoS One, 2013, 8(8), e72301 CrossRef PubMed.
- A. Messer and D. C. Butler, Optimizing intracellular antibodies (intrabodies/nanobodies) to treat neurodegenerative disorders, Neurobiol. Dis., 2020, 134, 104619 CrossRef CAS PubMed.
- M. Visintin, G. Settanni, A. Maritan, S. Graziosi, J. D. Marks and A. Cattaneo, The intracellular antibody capture technology (IACT): towards a consensus sequence for intracellular antibodies11Edited by J. Karn, J. Mol. Biol., 2002, 317(1), 73–83 CrossRef CAS PubMed.
- R. M. Nisbet, A. Van der Jeugd, G. Leinenga, H. T. Evans, P. W. Janowicz and J. Götz, Combined effects of scanning ultrasound and a tau-specific single chain antibody in a tau transgenic mouse model, Brain, 2017, 140(5), 1220–1230 CrossRef PubMed.
- M. S. Goodwin, O. Sinyavskaya, F. Burg, V. O'Neal, C. Ceballos-Diaz and P. E. Cruz, et al., Anti-tau scFvs Targeted to the Cytoplasm or Secretory Pathway Variably Modify Pathology and Neurodegenerative Phenotypes, Mol. Ther., 2021, 29(2), 859–872 CrossRef CAS.
- C. Danis, E. Dupré, O. Zejneli, R. Caillierez, A. Arrial and S. Bégard, et al., Inhibition of Tau seeding by targeting Tau nucleation core within neurons with a single domain antibody fragment, bioRxiv, 2021, 2021.03.23.436266 Search PubMed.
- Y. Wang, X. Jiang, F. Feng, W. Liu and H. Sun, Degradation of proteins by PROTACs and other strategies, Acta Pharm. Sin. B, 2020, 10(2), 207–238 CrossRef CAS.
- H. Gao, X. Sun and Y. Rao, PROTAC Technology: Opportunities and Challenges, ACS Med. Chem. Lett., 2020, 11(3), 237–240 CrossRef CAS PubMed.
- W. McCoull, T. Cheung, E. Anderson, P. Barton, J. Burgess and K. Byth, et al., Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6, ACS Chem. Biol., 2018, 13, 3131 CrossRef CAS PubMed.
- J. Popow, H. Arnhof, G. Bader, H. Berger, A. Ciulli and D. Covini, et al., Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions, J. Med. Chem., 2019, 62, 2508 CrossRef CAS PubMed.
- T. Neklesa, L. B. Snyder, R. R. Willard, N. Vitale, J. Pizzano, D. A. Gordon, M. Bookbinder, J. Macaluso, H. Dong, C. Ferraro and G. Wang, ARV-110: an oral androgen receptor PROTAC degrader for prostate cancer, J. Clin. Oncol., 2019, 37(259.10), 1200 Search PubMed.
- J. J. Flanagan, Y. Qian, S. M. Gough, M. Andreoli, M. Bookbinder, G. Cadelina, J. Bradley, E. Rousseau, R. Willard, J. Pizzano, C. M. Crews, A. P. Crew, I. Taylor and J. Houston, ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer, Proceedings of the 2018 San Antonio Breast Cancer Symposium, San Antonio, TX, Philadelphia (PA): AACR, 2018, Cancer. Res., 2019, 79(4 Suppl.) Search PubMed , abstract no. P5-04-18.
- A. Zorba, C. Nguyen, Y. Xu, J. Starr, K. Borzilleri and J. Smith, et al., Delineating the role of cooperativity in the design of potent PROTACs for BTK, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7285 CrossRef PubMed.
- Z. I. Bassi, M. C. Fillmore, A. H. Miah, T. D. Chapman, C. Maller and E. J. Roberts, et al., Modulating PCAF/GCN5 Immune Cell Function through a PROTAC Approach, ACS Chem. Biol., 2018, 13, 2862 CrossRef CAS PubMed.
- J. Nunes, G. A. McGonagle, J. Eden, G. Kiritharan, M. Touzet and X. Lewell, et al., Targeting IRAK4 for Degradation with PROTACs, ACS Med. Chem. Lett., 2019, 10, 1081 CrossRef CAS PubMed.
- L. Bai, H. Zhou, R. Xu, Y. Zhao, K. Chinnaswamy and D. McEachern, et al., A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo, Cancer Cell, 2019, 36, 498 CrossRef CAS PubMed.
- T.-T. Chu, N. Gao, Q.-Q. Li, P.-G. Chen, X.-F. Yang and Y.-X. Chen, et al., Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation, Cell Chem. Biol., 2016, 23(4), 453–461 CrossRef CAS PubMed.
- M. Lu, T. Liu, Q. Jiao, J. Ji, M. Tao and Y. Liu, et al., Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway, Eur. J. Med. Chem., 2018, 146, 251–259 CrossRef CAS PubMed.
- R. B. Kargbo, Treatment of Alzheimer's by PROTAC-Tau Protein Degradation, ACS Med. Chem. Lett., 2019, 10(5), 699–700 CrossRef CAS PubMed.
- M. C. Silva, F. M. Ferguson, Q. Cai, K. A. Donovan, G. Nandi and D. Patnaik, et al., Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models, eLife, 2019, 8 Search PubMed.
- W. Wang, Q. Zhou, T. Jiang, S. Li, J. Ye and J. Zheng, et al., A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models, Theranostics, 2021, 11(11), 5279–5295 CrossRef CAS PubMed.
- J. L. Guo, A. Buist, A. Soares, K. Callaerts, S. Calafate and F. Stevenaert, et al., The Dynamics and Turnover of Tau Aggregates in Cultured Cells: INSIGHTS INTO THERAPIES FOR TAUOPATHIES, J. Biol. Chem., 2016, 291(25), 13175–13193 CrossRef CAS PubMed.
- M. J. Lee, J. H. Lee and D. C. Rubinsztein, Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system, Prog. Neurobiol., 2013, 105, 49–59 CrossRef CAS.
- T.-T. Chu, Q.-Q. Li, T. Qiu, Z.-Y. Sun, Z.-W. Hu and Y.-X. Chen, et al., Clearance of the intracellular high level of the tau protein directed by an artificial synthetic hydrolase, Mol. BioSyst., 2014, 10(12), 3081–3085 RSC.
- Y. Zhou, J. Li, A. Nordberg and H. Ågren, Dissecting the Binding Profile of PET Tracers to Corticobasal Degeneration Tau Fibrils, ACS Chem. Neurosci., 2021, 12(18), 3487–3496 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |