
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correction: Gauging van der Waals interactions in aqueous solutions of 2D MOFs: when water likes organic linkers more than open-metal sites
Mohammad R.
Momeni
 *,
Zeyu
Zhang
,
David
Dell'Angelo
and
Farnaz A.
Shakib
*
*,
Zeyu
Zhang
,
David
Dell'Angelo
and
Farnaz A.
Shakib
*
Department of Chemistry and Environmental Science, New Jersey Institute of Technology, Newark 07102, NJ, USA. E-mail: momeni@njit.edu; shakib@njit.edu
First published on 17th October 2022
Abstract
Correction for ‘Gauging van der Waals interactions in aqueous solutions of 2D MOFs: when water likes organic linkers more than open-metal sites’ by Mohammad R. Momeni et al., Phys. Chem. Chem. Phys., 2021, 23, 3135–3143, https://doi.org/10.1039/D0CP05923D.
In the originally published article, a few parameters were missing from the force fields affecting the reported results for the classical molecular dynamics simulation data in Section 3.1. (i.e., Fig. 2 and Table 1). Specifically, a number of dihedral angles were missing from the force field. Also, errors were found in the implementation of the q-TIP4P/F water potential which are fixed. The simulations were repeated to correct this error, along with optimisation of the simulation details, which have been outlined here. The corrected figure and table are reported below. The corresponding tables of force field parameters along with data related to the MD results have been updated in the ESI. The input and output files of all simulations have also been provided as part of the Supplementary Materials to the original article.
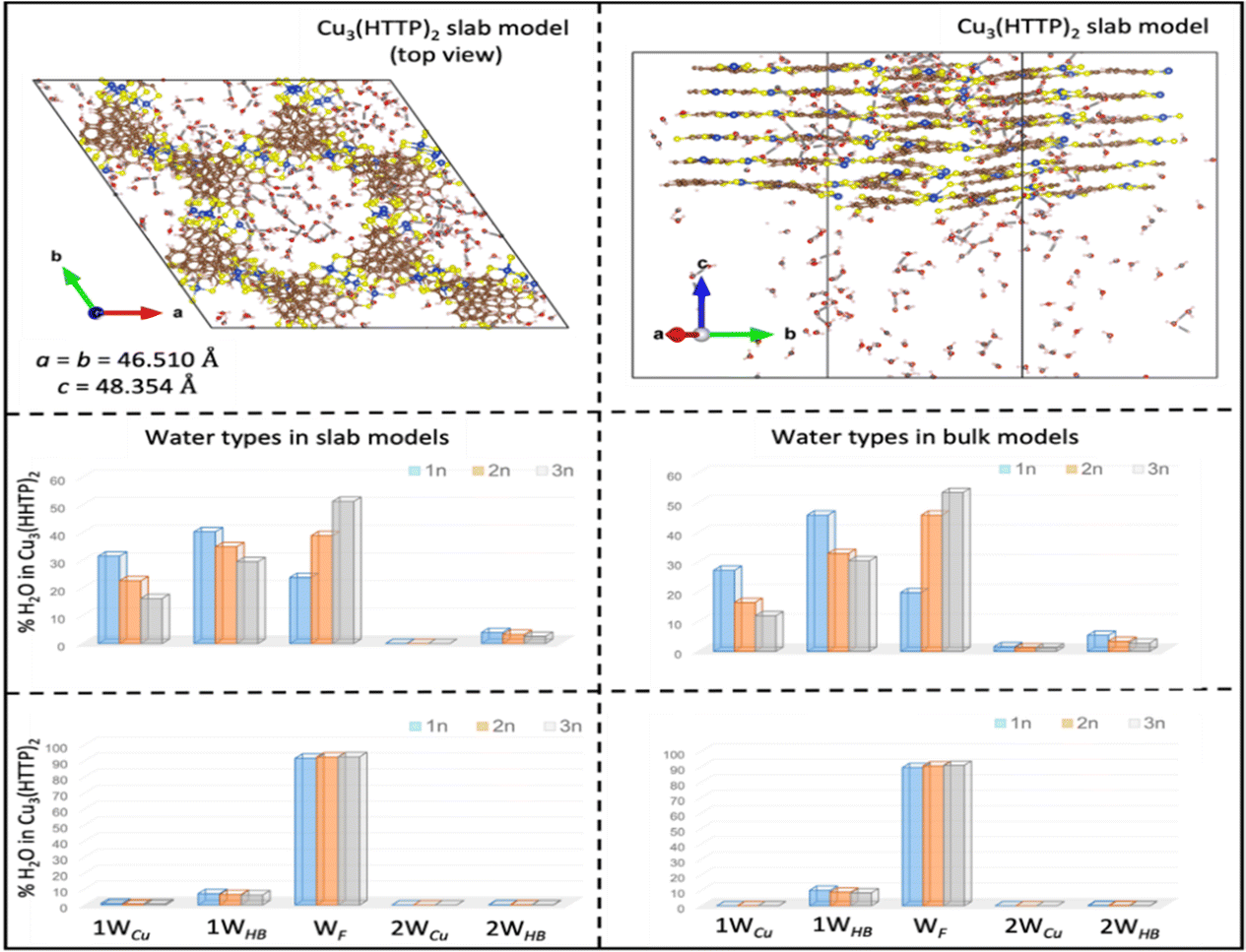 | ||
| Fig. 1 Top: Top and side views of representative snapshots for equilibrated slab models of Cu3(HTTP)2 with the highest water loading of 288 H2O and with the equilibrated dimensions of the simulations box given. Calculated percentage different types of water molecules in the slab (left) and bulk (right) models of Cu3(HHTP)2 (middle) and Cu3(HTTP)2 (bottom) MOFs with different water loadings. n refers to the number of Cu2+ centers in the unit cell which is equal to 72 in both the bulk and the slab models. Calculated percentage water molecules coordinated to one or two open-Cu2+ sites (1WCu and 2WCu), hydrogen bonded (HB) to one or two oxygen or sulfur atoms of the linkers (1WHB and 2WHB) as well as free water molecules (WF) for both bulk and slab models are shown. | ||
For MD simulations of bulk models of 2D MOFs, larger 2 × 2 × 3 periodic cells, composed of 1512 atoms and 72 metal centers, were used along with a larger cutoff of 10 Å for treating long-range electrostatic interactions. This is opposed to the 2 × 2 × 2 periodic cells in the original paper with a 6 Å cutoff. The simulations were carried out using our in-house modified software package coined as DL_POLY Quantum v1.0 which is publicly available through our GitHub page.1 The corrected Fig. 2 is provided below. The trends observed and the related discussions in the main text on the importance of hydrogen bond formation for the adsorption of water are not changed. The corrected Table 1 is given below. To determine the orientational relaxation time (τreor), we used a bi-exponential function2 as in:
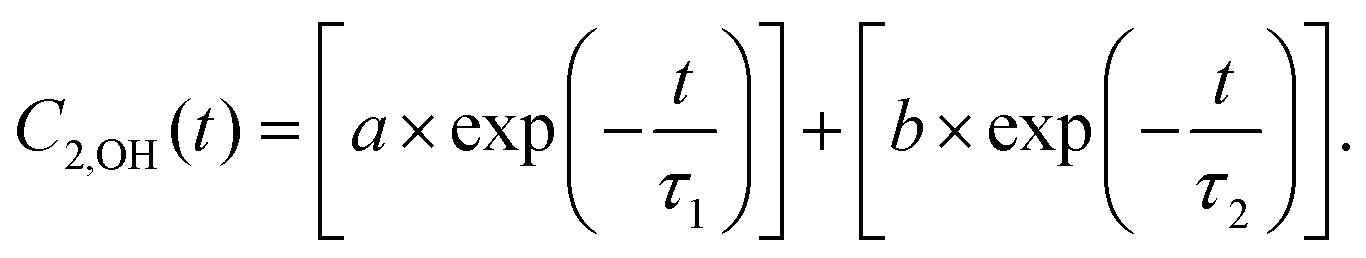
The first 100 fs of the simulations which correspond to the fast liberational motion of the water molecules2 were excluded from these fits. The mean square displacement (MSD) plots for calculating diffusion coefficients (D) are updated in Fig. S12 and S13 of the new Supplementary Materials. The reported trends and main conclusions on the freer nature of water in Cu3(HTTP)2 compared to Cu3(HHTP)2 remain unchanged. The noticeable difference of the new results compared to the original data is the increasing trend of both Dz and Dxy with respect to water concentration which is in line with the increasing trend of Dtot.

| n | Cu3(HHTP)2 | |||
|---|---|---|---|---|
| τ reor | D tot | D xy | D z | |
| 1H2O/Cu2+ | 55.3 (77.1) | 0.032 (0.027) | 0.020 (0.022) | 0.054 (0.036) |
| 2H2O/Cu2+ | 44.3 (53.7) | 0.039 (0.042) | 0.032 (0.036) | 0.054 (0.055) |
| 4H2O/Cu2+ | 31.1 (29.6) | 0.049 (0.052) | 0.042 (0.050) | 0.062 (0.055) |
| n | Cu3(HTTP)2 | |||
|---|---|---|---|---|
| τ reor | D tot | D xy | D z | |
| 1H2O/Cu2+ | 5.9 (3.5) | 0.126 (0.271) | 0.120 (0.235) | 0.139 (0.344) |
| 2H2O/Cu2+ | 9.5 (3.3) | 0.113 (0.257) | 0.095 (0.238) | 0.149 (0.295) |
| 4H2O/Cu2+ | 5.0 (4.1) | 0.158 (0.178) | 0.119 (0.171) | 0.235 (0.193) |
| Exp. bulk water | 1.7–2.6 | 0.229 | ||
The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.
References
- M. R. Momeni and F. A. Shakib, The DL_POLY Quantum molecular simulation package. Available from: https://github.com/fashakib/DL_POLY-Quantum-v1.0.
- S. Kim, X. Wang, J. Jang, K. Eom, S. L. Clegg, G.-S. Park and D. Di Tommaso, ChemPhysChem, 2020, 21, 2334–2346 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2022 |