
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correction: Simulating X-ray photoelectron spectra with strong electron correlation using multireference algebraic diagrammatic construction theory
Carlos E. V.
de Moura
 and
Alexander Yu.
Sokolov
*
and
Alexander Yu.
Sokolov
*
Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: vieirademoura.2@osu.edu; sokolov.8@osu.edu
First published on 23rd March 2022
Abstract
Correction for ‘Simulating X-ray photoelectron spectra with strong electron correlation using multireference algebraic diagrammatic construction theory’ by Carlos E. V. de Moura and Alexander Yu. Sokolov, Phys. Chem. Chem. Phys., 2022, 24, 4769–4784, DOI: 10.1039/D1CP05476G.
Shortly after the publication of our paper,1 we discovered an error in the implementation of core–valence separation (CVS) for the single-reference algebraic diagrammatic construction (SR-ADC) methods, which incorrectly excluded the contributions from the core–core–external excitations in the effective Hamiltonian matrix M (denoted as |ϕaIJ〉 in Fig. 2 of ref. 1, where I and J correspond to the CVS orbitals and a labels an external orbital). The multireference ADC (MR-ADC) results were not affected by this error. Here, we report the ionization energies and simulated spectra computed using the correct CVS-SR-ADC implementation, along with the corrected Electronic Supplementary Information (ESI). Importantly, correcting for the error does not change the main conclusions of our paper.1 We provide a short summary of the changes in the CVS-SR-ADC results below. In short, we replace Tables 1 and 2, as well as Fig. 3, 5, 6, 8 and 12 in the original article1 with the corresponding Tables and Figures provided in this Correction. Changes in the CVS-SR-ADC results in the ESI are highlighted in dark green color. The corrected version of the paper has been also posted on the arXiv preprint server.2
| Molecule | SR-ADC(2) | SR-ADC(2)-X | SR-ADC(3) | MR-ADC(2) | MR-ADC(2)-X | EOM-CCSDT | Experiment | ||
|---|---|---|---|---|---|---|---|---|---|
| CAS[Small] | CAS[Large] | CAS[Small] | CAS[Large] | ||||||

|
292.30 | 290.25 | 293.55 | 292.28 | 293.31 | 290.69 | 290.59 | 290.85 | 290.82 |
| C*H4 | 292.18 | 290.32 | 293.20 | 292.24 | 293.73 | 290.29 | 291.42 | 290.86 | 290.91 |
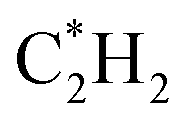
|
292.47 | 290.61 | 294.05 | 292.84 | 293.62 | 290.60 | 290.77 | 291.36 | 291.14 |
| CH3NC* | 294.73 | 292.42 | 294.69 | 294.56 | 294.83 | 291.96 | 292.37 | 292.35 | 292.37 |
| C*H3OH | 294.03 | 291.98 | 294.78 | 294.40 | 294.88 | 292.02 | 292.58 | 292.47 | 292.43 |
| CH3C*N | 294.10 | 292.23 | 295.14 | 294.23 | 293.98 | 292.06 | 292.19 | 292.81 | 292.45 |
| C*H3CN | 294.25 | 292.40 | 295.19 | 294.29 | 294.81 | 292.28 | 292.77 | 292.90 | 292.98 |
| C*H3NC | 294.83 | 292.94 | 296.12 | 295.12 | 294.94 | 293.11 | 293.00 | 293.41 | 293.35 |
| HC*N | 294.88 | 292.91 | 295.66 | 295.00 | 295.51 | 292.82 | 293.60 | 293.59 | 293.40 |
| C*H2O | 296.58 | 294.30 | 296.56 | 296.14 | 297.18 | 294.23 | 294.79 | 294.62 | 294.47 |
| C*O | 298.58 | 296.39 | 297.80 | 298.10 | 298.31 | 295.63 | 296.37 | 296.47 | 296.21 |
| C*O2 | 300.53 | 298.35 | 299.48 | 299.76 | 299.62 | 297.12 | 297.27 | 298.03 | 297.69 |
| N*H3 | 405.73 | 404.64 | 409.18 | 405.69 | 407.46 | 404.86 | 405.47 | 405.55 | 405.52 |
| CH3CN* | 405.91 | 404.73 | 410.22 | 407.25 | 408.10 | 404.87 | 405.58 | 405.71 | 405.64 |
| HCN* | 406.88 | 405.74 | 411.10 | 408.06 | 409.71 | 405.85 | 406.95 | 406.88 | 406.78 |
| CH3N*C | 406.23 | 405.45 | 411.61 | 407.48 | 409.50 | 405.36 | 407.12 | 407.02 | 406.67 |
| N*NO | 409.58 | 408.30 | 412.57 | 411.16 | 411.19 | 408.60 | 408.07 | 408.92 | 408.71 |
| N2* | 410.31 | 408.93 | 413.20 | 411.91 | 412.14 | 409.71 | 409.01 | 410.03 | 409.98 |
| NN*O | 413.98 | 412.77 | 416.11 | 413.85 | 415.77 | 412.01 | 412.80 | 413.15 | 412.59 |
| CH3O*H | 538.00 | 537.83 | 544.62 | 538.65 | 539.16 | 538.39 | 538.19 | 539.00 | 539.11 |
| CH2O* | 538.18 | 538.09 | 545.83 | 539.34 | 543.02 | 539.44 | 540.10 | 539.44 | 539.48 |
| H2O* | 538.68 | 538.53 | 544.74 | 538.99 | 541.26 | 538.53 | 540.18 | 539.79 | 539.90 |
| CO2* | 540.22 | 540.38 | 547.73 | 541.17 | 543.09 | 539.98 | 540.02 | 541.40 | 541.28 |
| NNO* | 539.95 | 540.33 | 548.86 | 540.87 | 544.16 | 540.07 | 540.93 | 541.63 | 541.42 |
| CO* | 540.95 | 541.07 | 549.06 | 542.99 | 545.67 | 541.03 | 543.05 | 542.57 | 542.55 |
| HF* | 691.99 | 692.75 | 699.99 | 693.13 | 697.10 | 692.86 | 695.13 | 694.22 | 694.23 |
| F2* | 695.33 | 695.07 | 702.59 | 695.00 | 698.96 | 695.22 | 697.02 | 696.72 | 696.69 |
| ΔMAE | 1.26 | 0.84 | 3.77 | 1.22 | 2.20 | 0.82 | 0.44 | ||
| ΔSTD | 1.40 | 0.51 | 1.75 | 1.10 | 0.68 | 0.47 | 0.55 | ||
| Ionization | SR-ADC(2) | SR-ADC(2)-X | SR-ADC(3) | EOM-CCSD | MR-ADC(2) | MR-ADC(2)-X | MRCISD | Experiment |
|---|---|---|---|---|---|---|---|---|
| OT (1a−11) | 540.64 (+0.38) | 540.49 (+0.38) | 548.17 (+0.39) | 544.15 | 543.47 (+0.38) | 540.62 (+0.38) | 545.92 | 541.5 |
| OT (1b−12) | 541.64 (+0.38) | 540.49 (+0.38) | 548.17 (+0.39) | 544.16 | 543.47 (+0.38) | 540.63 (+0.38) | 545.92 | |
| OC (2a−11) | 546.46 (+0.37) | 546.09 (+0.37) | 551.37 (+0.38) | 549.23 | 548.11 (+0.38) | 545.06 (+0.37) | 550.31 | 546.2 |
| ΔCT | 5.82 (−0.01) | 5.60 (+0.01) | 3.20 (+0.00) | 5.07 | 4.64 (+0.00) | 4.43 (+0.00) | 4.39 | 4.7 |
 | ||
| Fig. 1 Mean absolute errors (ΔMAE) in the K-edge core ionization energies of weakly-correlated molecules computed using the CVS-SR-ADC and CVS-MR-ADC methods, relative to CVS-EOM-CCSDT results.3 Error bars show the corresponding standard deviation of errors (ΔSTD). All calculations used the cc-pCVTZ-X2C basis set and the X2C description of scalar relativistic effects. | ||
 | ||
| Fig. 2 Potential energy curves for the K-edge core-ionized excited state of molecular nitrogen computed using the CVS-SR-ADC, CVS-MR-ADC, and MRCISD methods with the cc-pCVTZ basis set. Multiconfigurational calculations were performed using a CASSCF(10e,8o) reference wavefunction. | ||
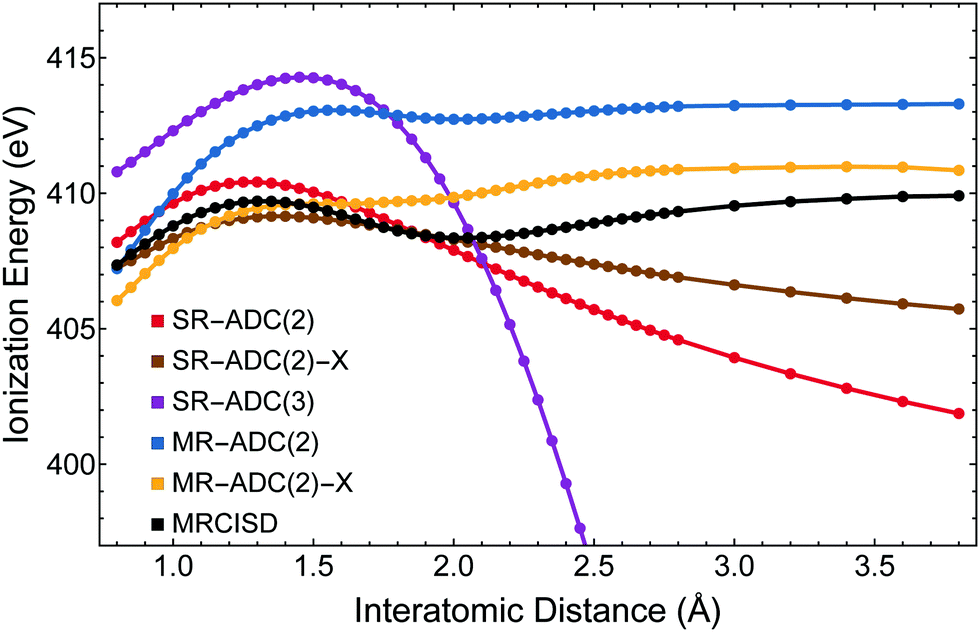 | ||
| Fig. 3 K-Edge core ionization energy along the dissociation pathway of molecular nitrogen computed using the CVS-SR-ADC, CVS-MR-ADC, and MRCISD methods with the cc-pCVTZ basis set. Multiconfigurational calculations were performed using a CASSCF(10e,8o) reference wavefunction. | ||
 | ||
| Fig. 4 Oxygen K-edge photoelectron spectra of ozone computed using five ADC approximations compared to the experimental spectrum from ref. 6. The simulated spectra used a 0.8 eV broadening parameter and were shifted to align with the first peak of the experimental spectrum. All calculations were performed using the cc-pCVTZ-X2C basis set and the X2C scalar relativistic effects. MR-ADC calculations used the CASSCF(12e,9o) wavefunction as a reference. | ||
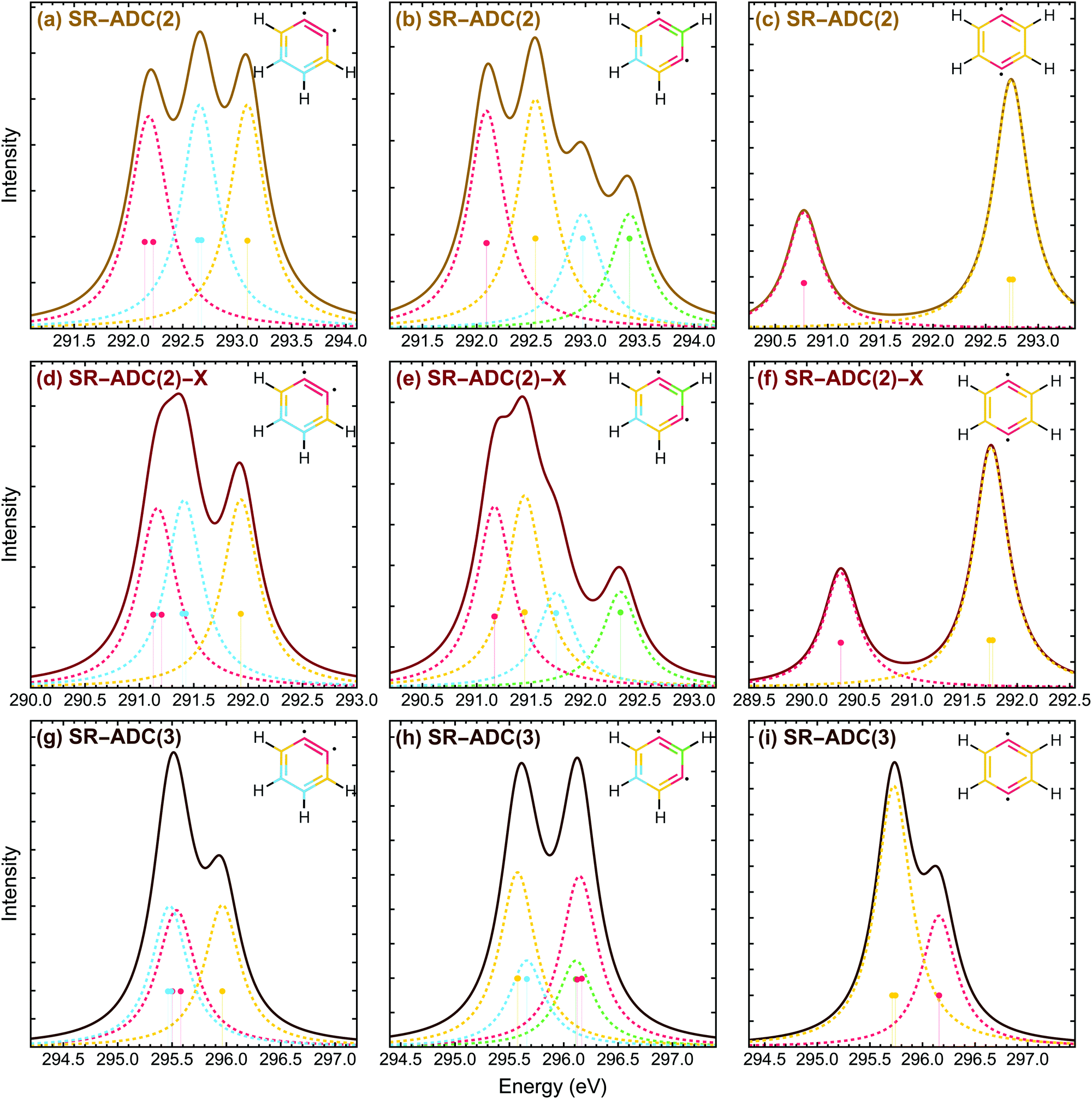 | ||
| Fig. 5 Carbon K-edge photoelectron spectra of ortho-, meta-, and para-benzyne molecules computed using CVS-SR-ADC(2) (a–c), CVS-SR-ADC(2)-X (d–f), and CVS-SR-ADC(3) (g–i), respectively. Solid lines show XPS spectra calculated using the 0.2 eV broadening. Dashed lines show spectral contributions from symmetry-equivalent carbon sites, color-coded as shown in each molecular structure. Calculations used the cc-pCVDZ-X2C basis set and the X2C description of scalar relativistic effects. | ||
In Table 1, we present the corrected CVS-SR-ADC core ionization energies for a benchmark dataset of 27 K-edge transitions in 16 small molecules. Mean absolute errors (ΔMAE) and standard deviations of errors (ΔSTD) for each method are shown in Fig. 3. Including the missing excitations in the CVS approximation reduces ΔMAE of CVS-SR-ADC(2) and CVS-SR-ADC(3) by 0.35 and 0.85 eV, respectively, but increases ΔMAE of CVS-SR-ADC(2)-X by 0.40 eV. The standard deviations of all three methods are largely unaffected. The corrected ΔMAE of CVS-SR-ADC(2) and CVS-SR-ADC(2)-X (1.26 and 0.84 eV) are very close to ΔMAE of CVS-MR-ADC(2) and CVS-MR-ADC(2)-X computed with the small active space CAS[Small] (1.22 and 0.82 eV, respectively).
Fig. 5 and 6 show the corrected CVS-SR-ADC potential energy curves and core ionization energies plotted along the dissociation pathway of molecular nitrogen (N2), respectively. In Fig. 5, correcting for the error shifts all CVS-SR-ADC curves to lower energies. While this somewhat reduces the errors of CVS-SR-ADC(2) and CVS-SR-ADC(3) near equilibrium, all three curves still exhibit unphysical divergence at longer distances. The best agreement with MRCISD is demonstrated by the MR-ADC(2)-X method. Similar changes are observed in Fig. 6 where the CVS-SR-ADC(2) and CVS-SR-ADC(3) core ionization energies are found to be closer to the MRCISD energies near equilibrium, but show large errors at dissociation. At 5 Å, the CVS-SR-ADC(2)-X error in ionization energy relative to MRCISD is −5.26 eV.
Table 2 and Fig. 8 show the corrected CVS-SR-ADC core ionization energies and X-ray photoelectron spectra of the ozone molecule. Removing the error in our CVS-SR-ADC implementation lowers all computed SR-ADC core ionization energies, but has very little effect (≤0.03 eV) on the spacing of peaks corresponding to ionization of the central and terminal oxygen atoms (ΔCT). Corrected core ionization energies and spectroscopic factors are presented in Table S2 of the ESI.
Corrected carbon K-edge X-ray photoelectron spectra of ortho-, meta-, and para-benzynes simulated using CVS-SR-ADC are presented in Fig. 12. As for the ozone molecule, all transitions have lower core ionization energies, but no significant changes are observed in the relative position of peaks in the simulated spectra. Corrected core ionization energies and spectroscopic factors are presented in Tables S5, S7 and S9 of the ESI.
The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.
Acknowledgements
This work was supported by the National Science Foundation, under Grant No. CHE-2044648. Computations were performed at the Ohio Supercomputer Center under Project No. PAS1583 and PAS1963.7References
- C. E. V. de Moura and A. Y. Sokolov, Phys. Chem. Chem. Phys., 2022, 24, 4769–4784 RSC.
- C. E. V. de Moura and A. Y. Sokolov, Simulating X-ray Photoelectron Spectra With Strong Electron Correlation Using Multireference Algebraic Diagrammatic Construction Theory, arXiv, 2022, preprint, arXiv:2112.00505.
- J. Liu, D. Matthews, S. Coriani and L. Cheng, J. Chem. Theory Comput., 2019, 15, 1642–1651 CrossRef CAS PubMed.
- W. Jolly, K. Bomben and C. Eyermann, At. Data Nucl. Data Tables, 1984, 31, 433–493 CrossRef CAS.
- D. B. Beach, C. J. Eyermann, S. P. Smit, S. F. Xiang and W. L. Jolly, J. Am. Chem. Soc., 1984, 106, 536–539 CrossRef CAS.
- M. Banna, D. C. Frost, C. A. McDowell, L. Noodleman and B. Wallbank, Chem. Phys. Lett., 1977, 49, 213–217 CrossRef CAS.
- Ohio Supercomputer Center, http://osc.edu/ark:/19495/f5s1ph73.
| This journal is © the Owner Societies 2022 |