
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroscopic investigation of photophysics and tautomerism of amino- and nitroporphycenes†
Idaresit
Mbakara
 a,
Agnieszka
Gajewska
a,
Arkadiusz
Listkowski
ab,
Michał
Kijak
a,
Krzysztof
Nawara
ab,
Tatu
Kumpulainen‡
c,
Eric
Vauthey
c and
Jacek
Waluk
*ab
a,
Agnieszka
Gajewska
a,
Arkadiusz
Listkowski
ab,
Michał
Kijak
a,
Krzysztof
Nawara
ab,
Tatu
Kumpulainen‡
c,
Eric
Vauthey
c and
Jacek
Waluk
*ab
aInstitute of Physical Chemistry, Polish Academy of Sciences, 01-224 Warsaw, Kasprzaka 44/52, Poland. E-mail: jwaluk@ichf.edu.pl
bFaculty of Mathematics and Science, Cardinal Stefan Wyszyński University, Dewajtis 5, 01-815 Warsaw, Poland
cPhysical Chemistry Department, Sciences II, University of Geneva, 30, Quai Ernest Ansermet, CH-1211 Geneva 4, Switzerland
First published on 28th November 2022
Abstract
Parent, unsubstituted porphycene and its two derivatives: 2,7,12,17-tetra-n-propylporphycene and 2,7,12,17-tetra-t-butylporphycene were substituted at the meso position with amino and nitro groups. These two families of porphycenes were characterized in detail with respect to their spectral, photophysical, and tautomeric properties. Two trans tautomers of similar energies coexist in the ground electronic state, but only one form dominates in the lowest excited singlet state. Absorption, magnetic circular dichroism (MCD), and emission anisotropy combined with quantum-chemical calculations led to the assignment of S1 and S2 transitions in both tautomers. Compared with the parent porphycene, the S1–S2 energy gap significantly increases; for one tautomeric form, the effect is twice as large as for the other. Both amino- and nitroporphycenes emit single fluorescence; previously reported dual emission of aminoporphycenes is attributed to a degradation product. Introduction of bulky t-butyl groups leads to a huge decrease in fluorescence intensity; this effect, arising from the interaction of the meso substituent with the adjacent t-butyl moiety, is particularly strong in the nitro derivative.
Introduction
Porphycene (Pc), the first reported constitutional isomer of porphyrin,1 is an important molecule for fundamental research and various possible applications. Spectral and photophysical parameters of porphycenes, such as strong absorption in the red part of the visible range and considerable triplet population yield, make this class of compounds promising agents in photodynamic therapy of cancer2 or photoinactivation of microbes.3,4 Other possible uses include application of porphycenes as artificial heme components,5 building blocks in molecular electronics,6 catalysts,7–10 or liquid crystals.11Regarding fundamental studies, porphycenes have been widely utilized as model systems for intramolecular hydrogen bond (HB) and tautomerism involving single or double hydrogen transfer.12 The rectangular shape of the inner cavity composed of four nitrogen atoms leads to strong HBs, and, in consequence, low tautomerization barriers. Under such conditions, tautomerization in solution is very fast (femto- and picoseconds);13 moreover, it is governed by tunneling, both “deep” (occurring from the vibrational ground state) and thermally activated after excitation of specific vibrational modes.14 Tautomerization in porphycenes has been studied in various experimental regimes: ensemble studies in condensed phases,13–24 investigations of ultracold molecules isolated in supersonic jets25,26 or helium nanodroplets,27 and, finally, single molecule techniques involving fluorescence,28–31 Raman,32,33 and scanning probe microscopy.34–40
It has been demonstrated that photophysics of porphycenes and their tautomeric properties are strongly related. Even though most porphycene derivatives are good or moderate emitters, some spectacular exceptions have been reported. For instance, substitution with alkyl groups at the four meso positions (9,10,19,20-) lowers the fluorescence quantum yield by three orders of magnitude,20 whereas 9,20-doubly substituted porphycenes do not reveal such an effect.24 Fluorescence can be recovered by placing the chromophore in a rigid environment,20 which suggests that rapid S1 radiationless deactivation involves a large amplitude geometry distortion. A good correlation was found between the fluorescence quantum yield of porphycenes and the distance between the nitrogen atoms linked by the intramolecular H-bond.23 The latter is a reliable measure of the HB strength. Similar behavior has been reported for other porphycenes.41
Interestingly, large changes in photophysics caused by modification of the geometry of the inner cavity can be induced even by “mild” substituents, e.g., fluorine or alkyl. This raises a question of the influence on the photophysics of strong electron donating and electron accepting substituents, such as amino or nitro groups. Several amino-substituted porphycenes have been reported in the literature.19,42–45 Nonell and coworkers studied 9-amino derivatives of porphycenes tetrasubstituted at the β positions (2,7,12,17-) with phenyl,19,43,44 propyl,45 or methoxyethyl45 moieties. They reported dual fluorescence, which was attributed to two trans tautomeric forms. Different decay times (in the range of a few nanoseconds), as well as different excitation spectra, indicated lack of excited state equilibrium between the two excited species. In particular, slow excited state tautomerization from the higher energy form was a rather unexpected result, given the rates previously reported for other porphycenes.
Nitro-substituted porphycenes have not been explored much. Absorption spectra are available for 9-nitro-2,7,12,17-tetra-n-propylporphycene46 and 9-nitro-2,7,12,17-tetraphenylporphycene,43 and 9-acetoxy-19-nitro-2,7,12,17-tetra-n-propylporphycene.46 Arad et al. reported a single emission of 9-nitro-2,7,12,17-tetraphenyl porphycene.43 Interestingly, while the decay time of 3.9 ns is only 2.5 times shorter than that reported for bare porphycene in the same solvent (toluene), the difference in fluorescence quantum yield is dramatic: the emission is 15 times stronger in the parent, unsubstituted compound.
The photophysical data reported for 9-aminoporphycenes bearing different substituents at the β positions indicate that the photophysical parameters can be strongly affected by the substituents. For example, fluorescence quantum yield in toluene is over an order of magnitude stronger in the tetrapropyl derivative42 than in the tetraphenyl analogue43 (0.06 vs. 0.004, respectively). Therefore, in order to accurately determine the influence of nitro and amino derivatives, singly substituted porphycenes are required. This was our motivation for the present work. We have synthesized 9-aminoporphycene (APc) and 9-nitroporphycene (NPc), as well as their corresponding 2,7,12,17-tetra-n-propyl derivatives (tprAPc and tprNPc, respectively), and determined their spectral and photophysical parameters. In order to assess the additional role of bulky substituents at the β positions on the spectra and photophysical parameters, tetra-t-butyl analogs of APc and NPc (ttAPc and ttNPc, respectively, see Scheme 1) have also been investigated and characterized. Combination of the experimental results with DFT modeling was used to understand the spectral and tautomeric properties of these two classes of compounds.
 | ||
| Scheme 1 Structures of porphycenes and the acronyms used. | ||
Results
Theoretical predictions of the relative energies of tautomeric forms
Six possible tautomeric forms are possible for porphycenes (Scheme 2), pairwise degenerate in Pc and ttPc, but not in the 9-substituted derivatives. While discussing relative stabilities in the ground state, we do not take into account the nonplanar cis3 and cis4 forms, whose energy is estimated to be much higher. DFT calculations predict that in the ground electronic state the two lowest energy forms in both amino and nitro derivatives are nearly degenerate: two trans tautomers are separated by less than 0.5 kcal mol−1 (Tables 1 and 2). One should note, however, that the predicted energy ordering changes: the lowest energy tautomeric species correspond to trans1 and trans2 in the amino and nitro derivatives, respectively. Upon excitation to S1, the trans2–trans1 energy difference becomes large, more than ten times that of S0 for the APc and five times for NPc. Moreover, the cis2 form of APc and the cis1 form of NPc are calculated for S1 at a lower energy than that of the less stable trans species. One should therefore consider a possible presence of the cis species, in particular in the lowest excited state. | ||
| Scheme 2 Possible tautomeric forms. | ||
| S0a | S1b | μ(S0) [D] | μ(S1)bc [D] | |
|---|---|---|---|---|
| a In parentheses, ZPVE-corrected values. b First row, B3LYP/6-31+G(d,p), second row, CAM-B3LYP/6-31+G(d,p) results. c Calculated for the optimized S1 geometries. | ||||
| Pc | ||||
| trans | 0.00 (0.00) | 0.00 (0.00) | 0.00 | 0.00 |
| 0.00 (0.00) | ||||
| cis | 2.30 (1.74) | 1.99 (1.67) | 1.31 | 1.20 |
| 2.13 (1.55) | ||||
| APc | ||||
| trans1 | 0.00 (0.00) | 0.00 (0.00) | 2.30 | 3.63 |
| 0.00 (0.00) | 3.14 | |||
| trans2 | 0.28 (0.24) | 3.20 (2.69) | 2.47 | 3.79 |
| 3.01 (2.37) | 3.39 | |||
| cis1 | 2.03 (1.51) | 5.54 (4.36) | 2.99 | 3.95 |
| 5.40 (4.22) | 3.45 | |||
| cis2 | 2.55 (1.91) | 2.01 (1.67) | 2.40 | 3.72 |
| 2.15 (1.77) | 3.63 | |||
| NPc | ||||
| trans1 | 0.42 (0.37) | 2.35 (2.08) | 6.54 | 6.77 |
| 2.12 (2.00) | 6.38 | |||
| trans2 | 0.00 (0.00) | 0.00 (0.00) | 6.85 | 8.68 |
| 0.00 (0.00) | 7.70 | |||
| cis1 | 2.60 (1.98) | 2.29 (1.85) | 6.66 | 8.23 |
| 2.43 (1.98) | 7.06 | |||
| cis2 | 2.16 (1.64) | 3.76 (3.10) | 7.05 | 7.72 |
| 3.78 (2.90) | 7.67 | |||
| S0a | S1b | μ(S0) [D] | μ(S1)bc [D] | |
|---|---|---|---|---|
| a In parentheses, ZPVE-corrected values. b First row, B3LYP/6-31+G(d,p), second row, CAM-B3LYP/6-31+G(d,p) results. c Calculated for the optimized S1 geometries. | ||||
| ttPc | ||||
| trans | 0.00 (0.00) | 0.00 (0.00) | 0.00 | 0.00 |
| 0.00 (0.00) | 0.00 | |||
| cis | 2.07 (1.53) | 1.68 (1.28) | 1.44 | 1.19 |
| 1.90 (1.28) | 1.63 | |||
| ttAPc | ||||
| trans1 | 0.00 (0.00) | 0.00 (0.00) | 2.09 | 3.04 |
| 0.00 (0.00) | 2.50 | |||
| trans2 | 0.06 (0.00) | 3.22 (2.82) | 2.40 | 3.41 |
| 3.06 (2.88) | 3.03 | |||
| cis1 | 1.63 (1.14) | 5.26 (4.21) | 2.98 | 3.39 |
| 5.29 (4.16) | 3.11 | |||
| cis2 | 2.10 (1.53) | 1.69 (1.50) | 2.39 | 3.14 |
| 1.90 (1.65) | 3.03 | |||
| ttNPc | ||||
| trans1 | -0.01 (0.04) | −0.55 (−0.69) | 6.91 | 7.96 |
| 0.12 (−0.07) | 7.39 | |||
| trans2 | 0.00 (0.00) | 0.00 (0.00) | 6.89 | 8.43 |
| 0.00 (0.00) | 7.61 | |||
| cis1 | 2.20 (1.72) | 1.84 (1.34) | 6.66 | 8.12 |
| 1.96 (1.36) | 7.24 | |||
| cis2 | 1.51 (1.15) | 1.40 (0.88) | 7.32 | 8.49 |
| 1.70 (1.06) | 8.39 | |||
According to the theoretical predictions, adding four propyl groups at the β positions does not lead to significant changes in the relative tautomer energies and their dipole moments (Table S1, ESI†). Also the calculations performed for the t-butyl derivatives yield for ttAPc the same pattern as for APc. Interestingly, for ttNPc the theory suggests that the two trans species retain similar energies in S1.
Aminoporphycenes. Since the two lowest energy forms – trans1 and trans2 tautomers – are predicted to be nearly degenerate, one can expect that the absorption should correspond to the sum of approximately equal contributions from both species. Calculations of the transition energies in the region of Q bands (S1 and S2 states, Table 3) indicate that the lowest energy band should correspond to the absorption from trans1, followed by a somewhat stronger transition from trans2, located ca. 900 cm−1 higher. The opposite pattern is obtained for the absorption to the second excited singlet electronic state. Now, the lower energy, weaker transition should occur from trans2, whereas the corresponding band of trans1 is expected to lie about 700 cm−1 higher.
| S1 ← S0a | S1 → S0b | S2 ← S0a | |
|---|---|---|---|
| a Optimized S0 geometry. b Optimized S1 geometry, first row: B3LYP/6-31+G(d,p), second row: CAM-B3LYP/6-31+G(d,p). | |||
| Pc | |||
| trans | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 904 (0.13) 904 (0.13) |
17084 (0.15) |
18804 (0.21) |
| 16649 (0.22) |
|||
| cis | 17935 (0.14) |
16883 (0.16) |
18804 (0.18) |
| 16638 (0.23) |
|||
| APc | |||
| trans1 | 15792 (0.18) |
14801 (0.18) |
18810 (0.16) |
| 14621 (0.23) |
|||
| trans2 | 16718 (0.25) |
15904 (0.25) |
18081 (0.10) |
| 15733 (0.33) |
|||
| cis1 | 16927 (0.22) |
16133 (0.22) |
18435 (0.13) |
| 16054 (0.30) |
|||
| cis2 | 15746 (0.17) |
14491 (0.17) |
18375 (0.14) |
| 14183 (0.23) |
|||
| NPc | |||
| trans1 | 17616 (0.09) |
16560 (0.12) |
18126 (0.12) |
| 16433 (0.19) |
|||
| trans2 | 17085 (0.10) |
15876 (0.10) |
18464 (0.19) |
| 15986 (0.15) |
|||
| cis1 | 17146 (0.11) |
15603 (0.12) |
18160 (0.17) |
| 15692 (0.18) |
|||
| cis2 | 17559 (0.09) |
16466 (0.10) |
18414 (0.09) |
| 16718 (0.16) |
|||
Parent porphycene and 2,7,12,17-tetraalkyl-substituted derivatives exhibit a characteristic absorption pattern, consisting of three main bands in the Q region. The lowest energy band corresponds to the origin of the S1 transition, the highest energy one to a vibronic feature of S2, while the middle band contains contributions from the origin of S2 and the vibronic transitions of S1. These contributions could be distinguished by measuring the spectra in rare gas matrices.47 One can therefore expect for APc a quite complex absorption pattern, consisting of (possibly overlapping) features due to different tautomers and different electronic states. In order to reliably assign the electronic transitions observed in absorption, we combined two techniques that rely on polarized light: magnetic circular dichroism (MCD) and fluorescence anisotropy. For the assignments of electronic transitions, MCD spectroscopy is of great help. Porphycenes are “hard” chromophores,48 of which the (+,−) MCD signal pattern of the lowest two electronic transitions is retained upon substitution or intramolecular tautomerization. Therefore, a positive/negative MCD sign indicates the S1/S2 absorption, independent of which tautomer it comes from. Moreover, MCD allows to separate the vibronic components of S1 from the origin of S2, since the vibronic features usually exhibit the same MCD sign as the electronic origin.
Using emission anisotropy, we exploit two characteristic features of the transition moments in porphycenes: (a) for each of trans tautomers, the S0–S1 and S0–S2 transitions are nearly orthogonally polarized; (b) the transition moments in trans1 and trans2 form a large angle, both for S1 and S2. Therefore, for the emission occurring from trans1, positive anisotropy values indicate that the initially excited species corresponds to either S1(trans1) or S2(trans2); negative anisotropy implies S1(trans2) or S2(trans1). We note, however, that for some unsymmetrically substituted porphycenes the situation may be more complicated; in particular, the transition moment directions in trans1 and trans2 need not form a large angle in S1 or S2. Such behavior, first discussed for 9-amino-2,7,12,17-tetraphenylporphycene,19 was confirmed by calculations of the presently studied amino derivatives (Fig. S1–S3, ESI†). It makes the distinction between two trans tautomeric forms based on emission anisotropy rather difficult. On the other hand, the calculations indicate that the pattern of nearly orthogonal S0–S1 and S0–S2 transition moments is retained after amino substitution.
The theoretical predictions are in excellent agreement with the experiment (Fig. 1). Even though the calculations overestimate the S1 ← S0 and S2 ← S0 transition energies (as is also the case for bare Pc), the calculated absorption pattern matches exactly the observed one. The lowest transition is observed (in toluene) at 13755 cm−1; the next, more intense band is located at 14760 cm−1. Weak bands at 15800 and 16860 cm−1 are barely observed in absorption, but they can be readily detected by emission anisotropy and MCD. Finally, an intense transition is observed at 17650 cm−1. These three bands exhibit negative MCD signals, indicating that they belong to S2 ← S0 transitions. The negative emission anisotropy at 17650 cm−1 leaves little doubt about the assignment to trans1. Since it is well known that the vibronic feature in the S2 ← S0 absorption in porphycenes is more intense than the 0-0 transition, the band at 17650 is assigned to the vibronic feature of S2(trans1). The energy difference between the S2 ← S0 bands observed at 17650 and 16860 cm−1, 790 cm−1, is in perfect agreement with theoretical prediction of the difference between S2 ← S0 transitions in trans1 and trans2. We therefore assign the band at 16860 cm−1 to the vibronic feature of S2(trans2). The determination of the positions of the electronic origin of S2 in trans1 and trans2 is not straightforward, since both the MCD and anisotropy show complex character, indicating mixing of S1 and S2 spectral features from both tautomers. Assuming that the dominant vibronic features lie, similarly as in other porphycenes, about 900 cm−1 to the blue from the S2 origin, we estimate the S2(0-0) energies of trans1 and trans2 as 16750 and 15960 cm−1, respectively. The latter value is close to the shoulder observed at 15900 cm−1 in the MCD spectrum. Regarding the former, it is difficult to find a spectral feature that would clearly correlate with this value. The negative MCD indicates the S2 origin. Small positive anisotropy values in this spectral region suggest contributions from both, the origin of S2(trans1) and the vibronic features of S1(trans2).
 | ||
| Fig. 1 Bottom, absorption of APc in toluene (a) and of ttAPc in paraffin (b). Middle, MCD of APc in toluene. Top, anisotropy of fluorescence excitation of ttAPc in paraffin, monitored at 735 nm. | ||
In conclusion, we assign the bands observed at 13755 and 14760 cm−1 to the origins of transitions to S1 in trans1 and trans2 forms, respectively. The corresponding values for S2 are 16750 and 15900 cm−1. The least accurate is the value of 16750 cm−1; we estimate that the maximum error in this assignment should not exceed ±200 cm−1.
As mentioned below, these assignments are in perfect agreement with the calculated S1–S2 energy splittings. An observation that strongly reinforces the assignments is a much higher MCD intensity in trans2 than in trans1, an effect clearly seen in the comparison of relative absorption and MCD intensities of the bands at 16860 and 17650 cm−1. It is caused by the smaller S1–S2 energy separation in trans2, which leads to a larger MCD signal. A similar effect has been recently reported for 9-fluoroporphycene.22
Our assignments agree with the results of a theoretical paper in which the absorption of 9-amino-2,6,12,17-tetraphenylporphycene has been modelled using the nuclear ensemble method.49 The authors concluded that the first absorption band originates from trans1, but the second band is dominated by transition from trans2.
DFT calculations performed for ttAPc (Table 4) yield a pattern very similar to that obtained for APc. The experiment (Fig. 1 and Fig. S4, ESI†) confirms the strong similarity of absorption and MCD in the two molecules. The tetra-t-butyl derivative exhibits in S1 a small blue shift with respect to APc for both trans forms. In S2, a red shift is observed for trans2, whereas for trans1 the transition energy remains the same within experimental error. Except for the latter, these shifts are correctly predicted by calculations. We assign the transitions observed in toluene at 14000 and 17650 cm−1 to the S1 and S2 transitions in trans1, whereas the corresponding bands in trans2 are located at 14915 and 16950 cm−1. Based on the analysis carried out for APc, the S2 values most likely correspond to the vibronic features.
| S1 ← S0a | S1 → S0b | S2 ← S0a | |
|---|---|---|---|
| a Optimized S0 geometry. b Optimized S1 geometry, first row: B3LYP/6-31+G(d,p), second row: CAM-B3LYP/6-31+G(d,p). | |||
| ttPc | |||
| trans | 17539 (0.15) |
16496 (0.18) |
18564 (0.26) |
| 16180 (0.23) |
|||
| cis | 17493 (0.15) |
16314 (0.18) |
18635 (0.25) |
| 16209 (0.24) |
|||
| ttAPc | |||
| trans1 | 15837 (0.21) |
14588 (0.21) |
18548 (0.20) |
| 14208 (0.26) |
|||
| trans2 | 16786 (0.31) |
15826 (0.30) |
17868 (0.11) |
| 15692 (0.41) |
|||
| cis1 | 16991 (0.26) |
15903 (0.24) |
18118 (0.16) |
| 15833 (0.36) |
|||
| cis2 | 15816 (0.21) |
14352 (0.21) |
18162 (0.18) |
| 13889 (0.26) |
|||
| ttNPc | |||
| trans1 | 16543 (0.12) |
13283 (0.11) |
17421 (0.13) |
| 13665 (0.18) |
|||
| trans2 | 16373 (0.12) |
14015 (0.13) |
17914 (0.26) |
| 14074 (0.19) |
|||
| cis1 | 16260 (0.13) |
13930 (0.14) |
17831 (0.24) |
| 14132 (0.21) |
|||
| cis2 | 16437 (0.11) |
13807 (0.11) |
17928 (0.10) |
| 14295 (0.16) |
|||
Absorption of tprAPc strongly resembles that of ttAPc; the positions of the main bands differ by ∼100 cm−1 or less (Fig. S4, ESI†); such behavior is also predicted by calculations (Table S2, ESI†). The intensity ratio of the bands assigned to S1(trans2) and S2(trans1) increases somewhat in the order: APc, tprAPc, ttAPc, suggesting a slightly higher population of trans2 in the alkylated derivatives. In APc, the population of trans2 seems to increase in a polar solvent, as indicated by the relative increase of the 16860 cm−1 peak, assigned to S2(trans2). The intensity of this transition is stronger in acetonitrile and methanol than in toluene or n-hexane.
300 cm−1, followed by the maximum at 15810 (in toluene). The same pattern is observed in acetonitrile solution. The anisotropy of fluorescence excitation rapidly decreases between 15300 and 15810 cm−1, corroborating the assignment of these two bands to the S1 transition in two different tautomeric forms.
 | ||
| Fig. 2 Bottom, absorption of NPc in toluene (a) and of ttNPc in paraffin (b). Middle, MCD of NPc in toluene. Top, anisotropy of fluorescence excitation of ttNPc in paraffin, monitored at 710 nm. | ||
The S2 assignment is more challenging. Two peaks are observed at 16475 and 17650 cm−1. Both exhibit a negative MCD signal, indicating that they correspond to S2. The same pattern is observed in bare porphycene, where the higher energy peak corresponds to the vibrational feature of the S2 ← S0 transition.47 In fact, except for low energy shoulder in NPc, absorption spectra of Pc and NPc are very similar with respect to the location and intensity of the bands.
Absorption spectra of tprNPc and ttNPc are analogous to that of NPc (Fig. S5, ESI†). The bands in the spectra of ttNPc are distinctively broader than in NPc and tprNPc, but the overall shape remains similar. The low energy shoulder, visible in NPc, is not observed, in agreement with calculations that predict that in ttNPc the S1 origins of trans1 and trans2 forms are spaced by less than 200 cm−1. The experiment also confirms the theoretically predicted red shifts of transition energies in the t-butyl derivative.
Fig. 3 summarizes the S1 ← S0 and S2 ← S0 transition energies obtained experimentally and predicted by theory. It is evident that nitro and amino substitutions lead to different spectral patterns and excited states ordering. The lowest energy transition corresponds to trans1 in the amino and to trans2 in the nitro derivatives. In aminoporphycenes, the S1–S2 energy gap is larger in trans1 than in trans2, whereas the opposite occurs in nitroporphycenes. It should be recalled that the reversal of relative tautomer energies is predicted also for the ground electronic state, but the effect becomes much stronger upon excitation (Table 1). Based on the predicted relative energies in the trans1 and trans2 forms, one could expect the dominance of only one tautomer in S1. As shown below, this is indeed the case, but, since in S0 the other species is also present, the dynamics of its conversion into the lower energy form should also be observed.
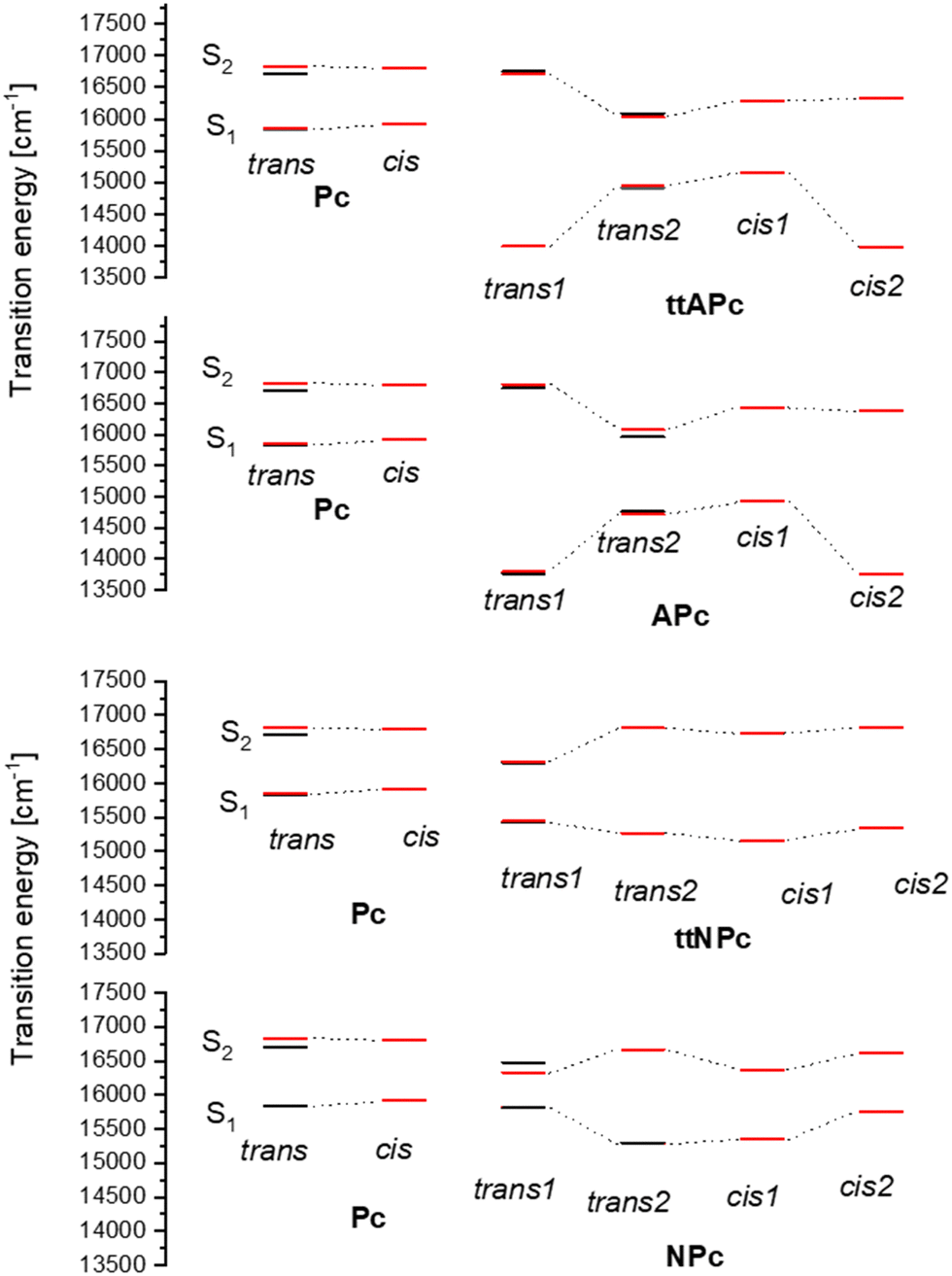 | ||
| Fig. 3 Comparison of calculated (red bars) and experimentally observed (black bars) S1 ← S0 and S2 ← S0 transition energies in different tautomeric forms of Pc and its amino and nitro derivatives. The calculated values (see Tables 3 and 4) have been shifted to lower energies, so that the experimental and calculated values of the S0–S1 transition coincide within less than 50 cm−1. | ||
 | ||
| Fig. 4 Room temperature fluorescence spectra: APc (a), tprAPc (c), and ttAPc (e) in acetonitrile, APc (b) and tprAPc (d) in toluene, and ttAPc (f) in paraffin. The spectra were normalized to the F1 maximum. Excitation wavelengths have been color-coded. | ||
Previous studies of derivatives of 9-aminoporphycene, substituted at the β positions with phenyl, propyl or methoxyethyl groups,44,45 reported F1 and F2 (but not F3) emissions of comparable intensity. These emissions were assigned to trans1 and trans2 tautomers, respectively. Our present results suggest a different interpretation. We noticed that for freshly prepared samples (measured one or two days after synthesis or just after sample purification by chromatography), the F2 and F3 emissions are barely observable. However, their intensity steadily grows with time (Fig. S7, ESI†) and, for samples that are a few weeks old, F2 and F3 bands become (for certain excitation wavelengths) comparable or even stronger than F1. These results leave no doubts that F2 and F3 do not originate from APc. The final proof was provided by applying chromatography for a sample of tprAPc. In addition to the main spot, two other ones were observed on the chromatographic plate. Fluorescence spectra recorded for the species present in these additional spots coincided with F2 and F3 emissions. In a separate HPLC experiment carried out for APc, we separated, in addition to the main component, two other species, each of them showing single emission corresponding to F2 and F3. We conclude that the previous assignment of F2 emission in aminoporphycenes to the trans2 tautomer has to be abandoned. Regarding F1, the assignment to trans1 seems safe. Still, time-resolved measurements reported below show that the high energy portion of F1 contains a fraction of short-lived fluorescence from trans2.
All three aminoporphycenes exhibited qualitatively the same behavior, but the appearance of F2 and F3 was significantly slower in the alkylated derivatives than in APc. We also noticed that F3/F2 ratio seemed to be smaller in nonpolar toluene or paraffin than in polar acetonitrile. The origin of the species responsible for F2 and F3 is now under investigation and will be a subject of a separate article.
When excited into the lowest energy absorption band, the F1 decay in APc is monoexponential (2.6 ± 0.1 ns in n-hexane, 2.00 ± 0.05 ns in toluene, 1.40 ± 0.05 ns in acetonitrile, and 1.00 ± 0.05 ns in methanol). Similar behavior was found for tprAPc (2.40 ± 0.05 ns in toluene, 1.90 ± 0.05 ns in acetonitrile). When the fluorescence was probed at its high energy edge (670 nm), both molecules demonstrated the same kinetic feature (Fig. S8 and S9, ESI†): in addition to the main component, identical to that obtained for monitoring the low energy part, a short decay was found, indicating a rapid excited state transformation. It is natural to assign this component to trans2 → trans1 tautomerization, since trans2, upon excitation, is destabilized with respect to trans1. Such interpretation allows to explain the coincidence of F1 excitation spectrum with that of absorption (which contains contributions from trans1, trans2, and, possibly, also cis1). The unusual behavior results from the fact that most of the excited population finally ends in the S1 state of trans1, which emits the dominant fluorescence.
The behavior of ttAPc is qualitatively similar to that of APc and tprAPc, but the fluorescence quantum yield is lower (Table 5) and the decay time is shorter. Interestingly a large difference is found between the decay times in toluene (0.15 ± 0.03 ns) and paraffin (1.01 ± 0.05 ns). The longer decay in more viscous solvent indicates a radiationless channel involving a large amplitude motion, such as distortion from planarity. Similar viscosity dependence has been reported for other porphycenes.20
| Solvent | ϕ fl | |
|---|---|---|
| a Estimated maximum error: ±20% for ϕfl > 10−2, ±30% for lower values. | ||
| APc | Toluene | 0.03 |
| Acetonitrile | 0.01 | |
| tprAPc | Toluene | 0.04 |
| Acetonitrile | 0.02 | |
| ttAPc | n-Hexane | 3.6 × 10−3 |
| Acetonitrile | 6 × 10−4 | |
| NPc | Toluene | 0.10 |
| Acetonitrile | 0.06 | |
| tprNPc | Toluene | 0.03 |
| Acetonitrile | 0.009 | |
| ttNPc | n-Hexane | 4 × 10−4 |
| Toluene | 3 × 10−4 | |
| Acetonitrile | 6 × 10−4 | |
| Ethanol | 3 × 10−4 | |
| DMSO | 5 × 10−4 | |
:water 80:20 v/v).
 | ||
| Fig. 5 Room temperature fluorescence spectra: NPc, tprNPc, and ttNPc in toluene (a, c and e) and acetonitrile (b, d and f). Excitation wavelengths have been color-coded. | ||
The emission of ttNPc is shifted to the blue with respect to NPc (by about 15 nm). The quantum yield is extremely low: the NPc:ttNPc intensity ratio is ∼200 (Table 5). The decay time, a few tens of picoseconds, was too short to be accurately measured with our setup. We also observed that the shape of the emission varies somewhat for different excitation wavelengths. This may indicate the presence of various conformers, but we cannot exclude contribution from impurities, since the fluorescence of ttNPc is extremely weak, which precludes reliable analysis of excitation spectra monitored at different emission wavelengths.
Discussion
Absorption spectra
The large difference between the absorption spectra of amino and nitroporphycenes can be understood using the calculated pattern of frontier electronic orbitals (Fig. 6). Substitution of porphycene with the electron donating amino moiety results in the destabilization of orbital energies; the strongest effect is induced in the HOMO−1 orbital, which has the largest LCAO coefficient at position 9. The other occupied orbital is only weakly affected, as it has a node at this position. In consequence, the two occupied orbitals, nearly degenerate in parent Pc, become split in APc by a significant amount, 0.4 eV. In addition, their ordering is reversed, as the more affected, destabilized orbital (HOMO in APc) corresponds to HOMO−1 in Pc.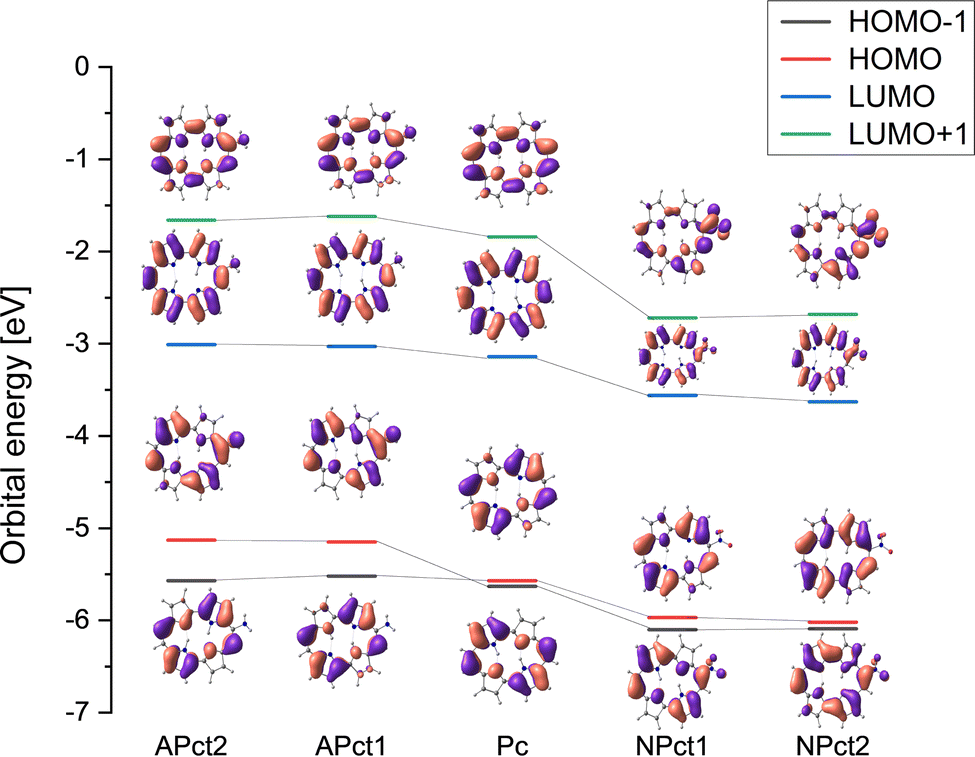 | ||
| Fig. 6 Calculated frontier orbital energy patterns. Suffixes t1 and t2 indicate trans1 and trans2 tautomers, respectively. | ||
Substitution by the electron-accepting nitro group leads to the stabilization of all the orbitals. The most affected orbitals are the unoccupied ones, but the HOMOs also shift to lower energies. The energy ordering is not inverted, and the spacing increases in comparison with Pc. Still, the effect is much weaker than in APc, resulting in the observed S1–S2 splittings of 665 cm−1 in trans1 and 1500 cm−1 in trans2.
The above analysis explains the increasing spectral range of the Q absorption upon passing from Pc to NPc and APc. One should note that simple consideration of orbital energies cannot account for different splittings of transition energies in trans1 and trans2. On the other hand, DFT calculations accurately reproduce the experimental results (Fig. S10, ESI†).
Photophysical properties
The radiative rate constant of S1 depopulation in Pc has been previously measured for different solvents. After correction for the square of the refractive index, the same value of (2.3 ± 0.1) × 107 s−1 is obtained for each solvent.24 We obtained practically the same values for n-hexane solutions of APc (2.3 ± 0.2) × 107 s−1 and NPc (1.9 ± 0.2) × 107 s−1. On the other hand, the sum of nonradiative rate constants in APc (3.4 ± 0.4) × 108 s−1 and NPc (1.8 ± 0.2) × 108 s−1 is definitely higher than in Pc (5.7 ± 0.6) × 107 s−1. The radiative rate constants do not seem to change significantly in the alkyl derivatives, but a large increase of nonradiative deactivation rate is observed for ttAPc (about an order of magnitude) and ttNPc (more than two orders of magnitude). Sevenfold difference in the fluorescence lifetimes measured for ttAPc in toluene (0.15 ns) and paraffin (1.01 ns) indicates that the efficient nonradiative process may be associated with geometry changes in S1, an effect reported for other porphycenes.24 In order to analyze this process in more detail, we compare the optimized geometries in the ground and lowest excited singlet states. The macrocycle of NPc is predicted to be planar in S0 and S1 (Fig. S11, ESI†). However, the NO2 plane forms an angle of 38° (trans1) or 30° (trans2) with the plane of the macrocycle. Upon excitation to S1, coplanarity is predicted for trans2, which is also the emitting form. Interestingly, trans1 retains its S0 structure in the S1 state. Introduction of t-butyl substituents that leads to ttNPc (Fig. S12, ESI†) induces nonplanarity already in S0, but the distortion becomes much larger in S1. In both states, the effect is stronger in trans1.For APc, planar macrocycle is obtained for both S0 and S1, with a slightly less pyramidalized amino group in the excited state (Fig. S13, ESI†). ttAPc is slightly nonplanar in S0. The distortion increases in S1 and consists of the deviation of the pyrrole ring closest to the amino group from the macrocycle plane (Fig. S14, ESI†). The effect is definitely stronger for the (emitting) trans1 tautomer.
The above results show that the huge decrease in fluorescence quantum yield in ttNPc is associated with steric interactions between t-butyl and nitro moieties. In agreement with experiment, a similar, but less pronounced effect is expected for ttApc. The finding that geometry distortions are different for different tautomers brings up an interesting issue: the possibility of influencing the tautomerization rate by changing the rigidity of the environment.
We finally note that, upon geometry optimization of the S1 state of the cis3 form, a conical intersection was found for both ttAPc and ttNPc, which may explain the low emission intensity.
Tautomeric equilibria
Photophysical studies demonstrate that both, amino and nitroporphycenes behave similarly to other unsymmetrically substituted porphycenes, such as 9-acetoxy18,50 or 9-fluoro22 derivatives. In the ground electronic state, two trans tautomers of similar energy coexist, whereas in S1 the equilibrium is shifted towards the structure which was already more stable in S0 (trans1 in amino-, trans2 in nitroporphycenes).Excited state tautomerization is a unidirectional “downhill” process leading to the most stable form. Determination of the rates requires techniques with better time resolution than used in the present work; we estimate, based on the value of the fast component appearing in the emission probed at its blue edge, that the double hydrogen transfer in S1 takes a few tens of picoseconds.
The mechanisms of tautomerization can be quite complex. In the ground electronic states, three different pathways should be considered for trans2 → trans1 conversion. The first two involve cis1 and cis2 as intermediates in the stepwise process. The third is the concerted asynchronous transfer of two hydrogens; a synchronous process is very unlikely due to lack of symmetry. In the lowest excited singlet state, tautomerization may be even more complicated, possibly involving high energy cis3 and cis4 species, because their energy is calculated as being close to that of the S1 energies of the other four species.
The calculations (Fig. 7) indicate similar barriers for stepwise trans1–trans2 conversions involving either cis1 or cis2 in the ground electronic state of both APc and NPc. The barrier for the first step is always higher than for the second one. This is also true for S1. This means that the experimental observation of the cis form (which would demonstrate a stepwise mechanism) may be difficult, as the decay of this intermediate species would be faster than its formation.
 | ||
| Fig. 7 Calculated relative energies (kcal mol−1) of four tautomeric forms of APc (left) and NPc (right) in the ground (bottom) and lowest excited singlet (top) states, and of the transition states involved in tautomerization. In parentheses, zero-point-vibrational-energy corrected values. In red, imaginary frequencies (cm−1); in blue, calculated permanent dipole moments (D). | ||
The barriers for concerted transfer are about 50% higher than those obtained for the stepwise process. A similar pattern was obtained for Pc and its other symmetrically substituted derivatives. However, the experimental evidence in the case of Pc favors the concerted mechanism, involving activation of a specific, low frequency vibrational mode.12 It remains to be checked whether the symmetry lowering in amino and nitro derivatives leads to the change in the tautomerization path. It seems likely, since the calculations predict that the two intramolecular H-bonds are no longer equivalent. The difference between the HB strengths is both substituent- and tautomer-specific. It can also vary between S0 and S1. For instance, in the ground electronic state of trans1 tautomer of APc, the N21H⋯N24 HB is predicted to be stronger than the other one, N22H⋯N23, as evidenced by the calculated values of the NH stretching frequencies, 2917 and 2944 cm−1, respectively. The opposite occurs for trans2 (2861 vs. 2844 cm−1). Electronic excitation enhances these patterns: the S1 frequencies calculated for trans1 are 2945 and 3090 cm−1, whereas for trans2 we obtain 2894 and 2761 cm−1. For the trans1 tautomer of NPc, the S0 frequencies are 2876 and 2973 cm−1, whereas the S1 calculation yields the values of 2923 and 2999 cm−1. Thus, contrary to the case of APc, the difference between the two HBs decreases in S1. Finally, in the trans2 form of NPc, the two HBs are different in S0 (2892 vs. 2848 cm−1), but in S1 both protons participate in the antisymmetric and symmetric combinations of NH stretches, separated by only 2 cm−1 (2911 and 2913 cm−1, respectively). This result suggests an attractive possibility to switch from stepwise to concerted tautomerization mechanism by a suitable combination of structure, tautomeric form, and electronic state.
However, the predictive power of calculations of unsymmetrical porphycenes seems to be lower than in the case of symmetrical derivatives. In particular, the correlation between the parameters that characterize the HB strength, which has been very useful for symmetrically substituted porphycenes,13 becomes rather weak. As an example, the plot of calculated NH stretching frequencies vs. the N–N distances (Fig. S15, ESI†) shows that for practically the same N–N separation (266.1–266.3 pm), frequencies that differ by more than 200 cm−1 are obtained. Evidently, the distance between the HB donor and acceptor is no longer a good index when the electron density distribution is not symmetrical. This is true even when considering two HBs of the same tautomeric form in the same electronic state.
Summary and conclusions
Substitution of porphycene at the meso position with the amino or nitro group leads to spectral changes that can be rationalized by the shifts of frontier orbital energies caused by electron donating or accepting moieties. The spacing between S1 and S2 states, ca. 900 cm−1 in unsubstituted Pc, increases to 3000 cm−1 in the trans1 form of APc and to 1500 cm−1 in the trans2 tautomer of NPc. For the other trans species the S2–S1 separation is about two times smaller in both, amino and nitro derivatives.Two trans tautomers of similar energies coexist in the ground state. Upon electronic excitation, one form (trans1 in APc, trans2 in NPc) is strongly stabilized with respect to the other. The least stable species rapidly converts into the lower energy one. As a result, fluorescence occurs mainly from one tautomeric form. Support for one-way rapid excited state trans–trans conversion is provided by a fast-decaying component in the blue part of the emission.
APc and NPc, as well as their 2,7,12,17-tetrapropyl derivatives emit with moderate quantum yields, but fluorescence becomes very weak for tetra-t-butyl substituted porphycenes. The effect is the strongest for the nitro derivative and is most likely caused by the steric repulsion between the nitro and t-butyl moieties.
The two classes of porphycenes studied in this work are attractive models for more detailed studies of ground and excited state tautomerization mechanisms. Unfortunately, such investigation may be difficult to perform because of instability, which is a problem especially in the case of aminoporphycenes. Our ongoing studies focus on the identification of the species produced both in the dark and after photoirradiation.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Polish National Science Center, grant 2016/22/A/ST4/00029, and by the grants from the PL-Grid infrastructure and the Interdisciplinary Centre for Mathematical and Computational Modeling for a computing grant (grant no. G17-14).References
- E. Vogel, M. Köcher, H. Schmickler and J. Lex, Porphycene - a novel porphin isomer, Angew. Chem., Int. Ed. Engl., 1986, 25, 257–259 CrossRef.
- J. C. Stockert, M. Cañete, A. Juarranz, A. Villanueva, R. W. Horobin, J. Borrell, J. Teixidó and S. Nonell, Porphycenes: facts and prospects in photodynamic therapy of cancer, Curr. Med. Chem., 2007, 14, 997–1026 CrossRef CAS PubMed.
- L. Polo, A. Segalla, G. Bertoloni, G. Jori, K. Schaffner and E. Reddi, Polylysine–porphycene conjugates as efficient photosensitizers for the inactivation of microbial pathogens, J. Photochem. Photobiol., B, 2000, 59, 152–158 CrossRef CAS PubMed.
- N. Masiera, J. Ostapko, A. Gorski, A. Bojarska, I. Gawryszewska, E. Sadowy, W. Hryniewicz and J. Waluk, Photoeradication of bacteria with porphycenes: substituent effects on the efficiency, Eur. J. Med. Chem., 2020, 200, 112472 CrossRef CAS PubMed.
- T. Matsuo, D. Murata, Y. Hisaeda, H. Hori and T. Hayashi, Porphyrinoid chemistry in hemoprotein matrix: detection and reactivities of iron(IV)-oxo species of porphycene incorporated into horseradish peroxidase, J. Am. Chem. Soc., 2007, 129, 12906–12907 CrossRef CAS PubMed.
- C. M. Che, H. F. Xiang, S. S. Y. Chui, Z. X. Xu, V. A. L. Roy, J. J. Yan, W. F. Fu, P. T. Lai and I. D. Williams, A high-performance organic field-effect transistor based on platinum(II) porphyrin: peripheral substituents on porphyrin ligand significantly affect film structure and charge mobility, Chem. – Asian J., 2008, 3, 1092–1103 CrossRef CAS PubMed.
- T. Hayashi, K. Okazaki, N. Urakawa, H. Shimakoshi, J. L. Sessler, E. Vogel and Y. Hisaeda, Cobaltporphycenes as catalysts. The oxidation of vinyl ethers via the formation and dissociation of cobalt–carbon bonds, Organometallics, 2001, 20, 3074–3078 CrossRef CAS.
- W. C. Lo, C. M. Che, K. F. Cheng and T. C. W. Mak, Catalytic and asymmetric cyclopropanation of styrenes catalysed by ruthenium porphyrin and porphycene complexes, Chem. Commun., 1997, 1205–1206 RSC.
- A. Berlicka and B. König, Porphycene-mediated photooxidation of benzylamines by visible light, Photochem. Photobiol. Sci., 2010, 9, 1359–1366 CrossRef CAS PubMed.
- K. Oohora, H. Meichin, L. Zhao, M. W. Wolf, A. Nakayama, J.-Y. Hasegawa, N. Lehnert and T. Hayashi, Catalytic Cyclopropanation by Myoglobin Reconstituted with Iron Porphycene: Acceleration of Catalysis due to Rapid Formation of the Carbene Species, J. Am. Chem. Soc., 2017, 139, 17265–17268 CrossRef CAS PubMed.
- M. Stępień, B. Donnio and J. L. Sessler, Discotic liquid-crystalline materials based on porphycenes: a mesogenic metalloporphycene-tetracyanoquinodimethane (TCNQ) adduct, Chem. – Eur. J., 2007, 13, 6853–6863 CrossRef PubMed.
- J. Waluk, Spectroscopy and tautomerization studies of porphycenes, Chem. Rev., 2017, 117, 2447–2480 CrossRef CAS PubMed.
- P. Ciąćka, P. Fita, A. Listkowski, M. Kijak, S. Nonell, D. Kuzuhara, H. Yamada, C. Radzewicz and J. Waluk, Tautomerism in Porphycenes: Analysis of Rate-Affecting Factors, J. Phys. Chem. B, 2015, 119, 2292–2301 CrossRef PubMed.
- P. Ciąćka, P. Fita, A. Listkowski, C. Radzewicz and J. Waluk, Evidence for Dominant Role of Tunneling in Condensed Phases and at High Temperatures: Double Hydrogen Transfer in Porphycenes, J. Phys. Chem. Lett., 2016, 7, 283–288 CrossRef PubMed.
- M. Gil, J. A. Organero, J. Waluk and A. Douhal, Ultrafast dynamics of alkyl-substituted porphycenes in solution, Chem. Phys. Lett., 2006, 422, 142–146 CrossRef CAS.
- M. Gil and J. Waluk, Vibrational gating of double hydrogen tunneling in porphycene, J. Am. Chem. Soc., 2007, 129, 1335–1341 CrossRef CAS PubMed.
- P. Fita, N. Urbańska, C. Radzewicz and J. Waluk, Ground and excited state tautomerization rates in porphycenes, Chem.– Eur. J., 2009, 15, 4851–4856 CrossRef CAS PubMed.
- P. Fita, P. Garbacz, M. Nejbauer, C. Radzewicz and J. Waluk, Ground and excited state double hydrogen transfer in symmetric and asymmetric potentials: comparison of 2,7,12,17-tetra-n-propylporphycene with 9-acetoxy-2,7,12,17-tetra-n-propylporphycene, Chem. – Eur. J., 2011, 17, 3672–3678 CrossRef CAS PubMed.
- P. Fita, M. Pszona, G. Orzanowska, D. Sánchez-García, S. Nonell, E. Vauthey and J. Waluk, Tautomerization in 2,7,12,17-tetraphenylporphycene and 9-amino-2,7,12,17-tetraphenylporphycene: influence of asymmetry on the transition moment directions, Chem. – Eur. J., 2012, 18, 13160–13167 CrossRef CAS PubMed.
- M. Gil, J. Dobkowski, G. Wiosna-Sałyga, N. Urbańska, P. Fita, C. Radzewicz, M. Pietraszkiewicz, P. Borowicz, D. Marks, M. Glasbeek and J. Waluk, Unusual, solvent viscosity-controlled tautomerism and photophysics: meso-alkylated porphycenes, J. Am. Chem. Soc., 2010, 132, 13472–13485 CrossRef CAS PubMed.
- P. Kasprzycki, P. Kopycki, A. Listkowski, A. Gorski, C. Radzewicz, D. J. S. Birch, J. Waluk and P. Fita, Influence of local microenvironment on the double hydrogen transfer in porphycene, Phys. Chem. Chem. Phys., 2020, 22, 17117–17128 RSC.
- A. Listkowski, A. Kharchenko, P. Ciąćka, M. Kijak, N. Masiera, R. Rybakiewicz, R. Luboradzki, P. Fita and J. Waluk, Fluorinated Porphycenes: Synthesis, Spectroscopy, Photophysics, and Tautomerism, ChemPlusChem, 2020, 85, 2197–2206 CrossRef CAS PubMed.
- A. Listkowski, N. Masiera, M. Kijak, R. Luboradzki, B. Leśniewska and J. Waluk, Controlling Emissive Properties by Intramolecular Hydrogen Bonds: Alkyl and Aryl meso-Substituted Porphycenes, Chem. – Eur. J., 2021, 27, 6324–6333 CrossRef CAS PubMed.
- M. Kijak, K. Nawara, A. Listkowski, N. Masiera, J. Buczyńska, N. Urbańska, G. Orzanowska, M. Pietraszkiewicz and J. Waluk, 2 + 2 Can Make Nearly a Thousand! Comparison of Di- and Tetra-Meso-Alkyl-Substituted Porphycenes, J. Phys. Chem. A, 2020, 124, 4594–4604 CrossRef CAS PubMed.
- E. T. Mengesha, A. Zehnacker-Rentien, J. Sepioł, M. Kijak and J. Waluk, Spectroscopic Study of Jet-Cooled Deuterated Porphycenes: Unusual Isotopic Effects on Proton Tunneling, J. Phys. Chem. B, 2015, 119, 2193–2203 CrossRef CAS PubMed.
- A. Vdovin, J. Sepioł, N. Urbańska, M. Pietraszkiewicz, A. Mordziński and J. Waluk, Evidence for two forms, double hydrogen tunneling, and proximity of excited states in bridge-substituted porphycenes: supersonic jet studies, J. Am. Chem. Soc., 2006, 128, 2577–2586 CrossRef CAS PubMed.
- A. Vdovin, J. Waluk, B. Dick and A. Slenczka, Mode-selective promotion and isotope effects of concerted double-hydrogen tunneling in porphycene embedded in superfluid helium nanodroplets, Chem. Phys. Chem., 2009, 10, 761–765 CrossRef CAS PubMed.
- H. Piwoński, A. Sokołowski, M. Kijak, S. Nonell and J. Waluk, Arresting Tautomerization in a Single Molecule by the Surrounding Polymer: 2,7,12,17-Tetraphenylporphycene, J. Phys. Chem. Lett., 2013, 4, 3967–3971 CrossRef.
- H. Piwoński, C. Stupperich, A. Hartschuh, J. Sepioł, A. Meixner and J. Waluk, Imaging of tautomerism in a single molecule, J. Am. Chem. Soc., 2005, 127, 5302–5303 CrossRef PubMed.
- L. Piatkowski, C. Schanbacher, F. Wackenhut, A. Jamrozik, A. J. Meixner and J. Waluk, Nature of Large Temporal Fluctuations of Hydrogen Transfer Rates in Single Molecules, J. Phys. Chem. Lett., 2018, 9, 1211–1215 CrossRef CAS PubMed.
- A. Bednarz, I. Kamińska, A. Jamrozik, K. Zielonka, A. Listkowski and J. Waluk, Substituent screening effect on single-molecule photostability: comparison of three differently substituted porphycenes, Methods Appl. Fluoresc., 2021, 9, 035004 CrossRef CAS PubMed.
- S. Gawinkowski, M. Pszona, A. Gorski, J. Niedziółka-Jönsson, I. Kamińska, W. Nogala and J. Waluk, Single molecule Raman spectra of porphycene isotopologues, Nanoscale, 2016, 8, 3337–3349 RSC.
- M. Pszona, S. Gawinkowski, R. Jager, I. Kaminska and J. Waluk, Influence of bulky substituents on single-molecule SERS sensitivity, J. Chem. Phys., 2022, 156, 014201 CrossRef CAS PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Thermally and vibrationally induced tautomerization of single porphycene molecules on a Cu(110) surface, Phys. Rev. Lett., 2013, 111, 246101–246105 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Controlling intramolecular hydrogen transfer in a porphycene molecule with single atoms or molecules located nearby, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- J. Ladenthin, T. Frederiksen, M. Persson, J. Sharp, S. Gawinkowski, J. Waluk and T. Kumagai, Force-induced tautomerization in a single molecule, Nat. Chem., 2016, 8, 935–940 CrossRef CAS PubMed.
- M. Koch, M. Pagan, M. Persson, S. Gawinkowski, J. Waluk and T. Kumagai, Direct Observation of Double Hydrogen Transfer via Quantum Tunneling in a Single Porphycene Molecule on a Ag(110) Surface, J. Am. Chem. Soc., 2017, 139, 12681–12687 CrossRef CAS PubMed.
- J. N. Ladenthin, L. Grill, S. Gawinkowski, S. Liu, J. Waluk and T. Kumagai, Hot Carrier-Induced Tautomerization within a Single Porphycene Molecule on Cu(111), ACS Nano, 2015, 9, 7287–7295 CrossRef CAS PubMed.
- T. Kumagai, J. Ladenthin, Y. Litman, M. Rossi, L. Grill, S. Gawinkowski, J. Waluk and M. Persson, Quantum tunneling in real space: tautomerization of single porphycene molecules on the (111) surface of Cu, Ag, and Au, J. Chem. Phys., 2018, 148, 102330 CrossRef PubMed.
- H. Böckmann, S. Liu, J. Mielke, S. Gawinkowski, J. Waluk, L. Grill, M. Wolf and T. Kumagai, Direct Observation of Photoinduced Tautomerization in Single Molecules at a Metal Surface, Nano Lett., 2016, 16, 1034–1041 CrossRef PubMed.
- D. Koga, T. Ono, H. Shinjo and Y. Hisaeda, Hydrogen Bond Engineering Visualized by Picometer-Level Distortion of Planar Porphyrin Isomers, J. Phys. Chem. Lett., 2021, 12, 10429–10436 CrossRef CAS PubMed.
- S. E. Braslavsky, M. Müller, D. O. Mártire, S. Pörting, S. G. Bertolotti, S. Chakravorti, G. Koç-Weier, B. Knipp and K. Schaffner, Photophysical properties of porphycene derivatives (18 π porphyrinoids), J. Photochem. Photobiol., B, 1997, 40, 191–198 CrossRef CAS.
- O. Arad, N. Rubio, D. Sánchez-García, J. I. Borrell and S. Nonell, Asymmetric porphycenes: synthesis and photophysical properties of 9-substituted 2,7,12,17-tetraphenylporphycenes, J. Porphyrins phthalocyanines, 2009, 13, 376–381 CrossRef CAS.
- M. Duran-Frigola, R. Tejedor-Estrada, D. Sánchez-García and S. Nonell, Dual fluorescence in 9-amino-2,7,12,17-tetraphenylporphycene, Phys. Chem. Chem. Phys., 2011, 13, 10326–10332 RSC.
- O. Planas, R. Tejedor-Estrada and S. Nonell, Tautomerism and dual fluorescence in 9-substituted n-propyl- and methoxyethyl-porphycenes, J. Porphyrins phthalocyanines, 2012, 16, 633–640 CrossRef CAS.
- M. Taneda, A. Tanaka, H. Shimakoshi, A. Ikegami, K. Hashimoto, M. Abe and Y. Hisaeda, Synthesis and characterizations of meso-disubstituted asymmetric porphycenes, Tetrahedron Lett., 2013, 54, 5727–5729 CrossRef CAS.
- A. Starukhin, E. Vogel and J. Waluk, Electronic Spectra in Porphycenes in Rare Gas and Nitrogen Matrices, J. Phys. Chem. A, 1998, 102, 9999 CrossRef CAS.
- J. Waluk, M. Müller, P. Swiderek, M. Köcher, E. Vogel, G. Hohlneicher and J. Michl, Electronic States of Porphycenes, J. Am. Chem. Soc., 1991, 113, 5511–5527 CrossRef CAS.
- Z. G. Lan, S. Nonell and M. Barbatti, Theoretical Characterization of Absorption and Emission Spectra of an Asymmetric Porphycene, J. Phys. Chem. A, 2012, 116, 3366–3376 CrossRef CAS PubMed.
- M. Gil, J. Jasny, E. Vogel and J. Waluk, Ground and excited state tautomerization in 9-acetoxy-2,7,12,17-tetra-n-propylporphycene, Chem. Phys. Lett., 2000, 323, 534 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04555a |
| ‡ Present address: Department of Chemistry/Nanoscience Center, University of Jyväskylä, Survontie 9 C, 40014, Jyväskylä, Finland. |
| This journal is © the Owner Societies 2022 |