
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical-induced hetero-nuclear mixing and low-field 13C relaxation in solid pyruvic acid†
Hana
Kouřilová
*a,
Michael
Jurkutat
 *a,
David
Peat
b,
Karel
Kouřil
a,
Alixander S.
Khan
b,
Anthony J.
Horsewill
b,
James F.
MacDonald
b,
John
Owers-Bradley
b and
Benno
Meier
*ac
*a,
David
Peat
b,
Karel
Kouřil
a,
Alixander S.
Khan
b,
Anthony J.
Horsewill
b,
James F.
MacDonald
b,
John
Owers-Bradley
b and
Benno
Meier
*ac
aInstitute of Biological Interfaces 4, Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen, 76344, Germany. E-mail: hana.kourilova@kit.edu; michael.jurkutat@kit.edu; benno.meier@kit.edu
bSchool of Physics and Astronomy, University of Nottingham, Nottingham, NG7 2RD, UK
cInstitute of Physical Chemistry, Karlsruhe Institute of Technology, Karlsruhe, 76131, Germany
First published on 14th November 2022
Abstract
Radicals serve as a source of polarization in dynamic nuclear polarization, but may also act as polarization sink, in particular at low field. Additionally, if the couplings between the electron spins and different nuclear reservoirs are stronger than any of the reservoirs’ couplings to the lattice, radicals can mediate hetero-nuclear polarization transfer. Here, we report radical-enhanced 13C relaxation in pyruvic acid doped with trityl. Up to 40 K, we find a linear carbon T1 field dependence between 5 mT and 2 T. We model the dependence quantitatively, and find that the presence of trityl accelerates direct hetero-nuclear polarization transfer at low fields, while at higher fields 13C relaxation is diffusion limited. Measurements of hetero-nuclear polarization transfer up to 600 mT confirm the predicted radical-mediated proton–carbon mixing.
1 Introduction
Nuclear magnetic resonance (NMR) spectroscopy and magnetic resonance imaging (MRI) are powerful tools in various fields of science and industry, but their versatility is limited by the weak nuclear polarization. The low sensitivity may be alleviated by dynamic nuclear polarization (DNP), where the large polarization of electron spins is transferred to nuclear spins. In dissolution-DNP,1 this process is carried out at temperatures near 1 K and fields of several Tesla. Under these conditions, the electron spins are almost fully polarized, and it is possible to achieve near unity polarization also for nuclear spins. The hyperpolarized sample is then dissolved with a jet of hot solvent, and the solution is transferred to a second magnet for use in sensitized NMR spectroscopy2–4 or MRI.5–8In bullet-DNP, the order of dissolution and transfer is reversed.9,10 Here, the solid sample is transferred rapidly to the second magnet, and dissolved only near the NMR tube. This procedure limits dilution, avoids the use of hot solvents, and may be beneficial for hyperpolarization of moieties with a short T1 in the liquid state.
During the bullet transfer at low field (100 ms at currently about 70 mT) the radical spins that are needed as a polarization source for DNP, however, may act as polarization sink. A previous study by Niedbalski et al. at low temperature in pyruvic acid doped with trityl between 0.9 and 9 T found a cubic field-dependence of the carbon T1.11 If this trend were to continue to lower fields, relaxation would be prohibitively fast for bullet-DNP. Indeed, we reported that the low-temperature low-field 1H relaxation in pyruvic acid doped with trityl is linear in field, and showed that a spin temperature model (STM) by Wenckebach12,13 yields a quantitative description of the proton relaxation data over two orders of magnitude in field.14 This STM indicated a significant heat-capacity of the electron Non-Zeeman reservoir, which suggests that the latter may be used to mediate hetero-nuclear polarization transfer.15
Here, we present 13C relaxation data as well as thermal mixing experiments in non-degassed neat 1-13C pyruvic acid (neat PA, °) and 1-13C pyruvic acid doped with 15 mM OX063 (doped PA, ●) for fields up to 2 T. We find that the low-temperature low-field 13C relaxation time constant  is linear in field, similar to that we reported for protons. The data show that in presence of trityl, relaxation during the low field transfer is not critical for either nuclear species in bullet-DNP.
is linear in field, similar to that we reported for protons. The data show that in presence of trityl, relaxation during the low field transfer is not critical for either nuclear species in bullet-DNP.
The data are interpreted by extending the previously reported STM to account for the carbon Zeeman reservoir. This extended STM predicts an enhanced carbon relaxation due to effective trityl-mediated proton–carbon coupling from 20 mT to above 1 T.
For a quantitative description of the observed carbon relaxation, the extended STM takes into account enhanced low-field direct hetero-nuclear mixing due to trityl-induced nuclear line-broadening, and a field-dependent carbon spin diffusion process that limits relaxation at higher fields. Hetero-nuclear thermal mixing experiments confirm the predicted trityl-mediated polarization transfer at higher fields.
2 Fast-field-cycling experiments
2.1 Samples
One sample of neat 1-13C-pyruvic acid (CortecNet, FR), 14 M, referred to as neat PA, was measured during a single session. Two experimental sessions were required to record the data on 1-13C pyruvic acid doped with 15 mM OX063 (Oxford Instruments, UK), referred to as doped PA, and a fresh sample was prepared for each session. All samples were used without degassing and flash-frozen by insertion of the NMR probe into the cold variable temperature insert of the fast-field-cycling magnet.2.2 Fast-field-cycling apparatus
The measurements were performed using a fast-field-cycling (FFC) setup16,17 with a range of stable temperatures from 3 to 300 K and magnetic fields from 0 to 2.5 T, with an absolute error of 2 mT. In the experiments described here, the field is changed at a rate of 4 T s−1. A Tecmag Apollo NMR spectrometer (Tecmag, TX) enables control of the magnet from within the NMR pulse sequence. We used a home-built NMR probe equipped with a solenoid tuned to a frequency of 22.83 MHz, which corresponds to resonant magnetic fields Bres of approximately 0.536 and 2.167 T for proton and carbon spins, respectively.2.3 Fast-field-cycling NMR experiments
The field-dependence of the carbon relaxation time constant was measured using two variations of the sequence shown in Fig. 1. For field strengths of 0.5 T and higher a saturation recovery sequence is used. For fields up to 0.2 T the equilibrium polarization signal would be too small, and so a polarization decay sequence is used. This includes an additional polarization stage (stage II) of 70 s at 2 T. | ||
| Fig. 1 Fast-field-cycling experiments were performed in three variations of the displayed sequence. Polarization decay (⋯): for measurements of T1 at evolution fields Bevo below 0.5 T the magnetization is saturated with a train of n π/2-pulses in stage I, to ensure reproducibility. Then in stage II the nuclei polarize at Bpol = 2 T (red dashed buildup curve). In stage III the field is changed to the evolution field Bevo and the magnetization relaxes (red dashed decay curve). In stage IV the NMR signal is read out at the resonant field Bres. Saturation recovery(—): for evolution fields of 0.5 T and higher only stages I, III and IV are used, i.e. stage II (highlighted) is dropped. For these fields the magnetization is saturated in stage I and recovers (blue buildup curve) in stage III at Bevo to the respective equilibrium value. In stage IV the NMR signal is read out. Thermal mixing(--): the sequence for thermal mixing is similar to saturation recovery, except that stage I lasts for tsat = 70 s to ensure protons establish equilibrium polarization at 2.167 T while carbon spins are saturated. | ||
Thermal mixing is measured using another variation of the sequence shown in Fig. 1. Here, stage I is prolonged (tpol = 70 s ≫ T1,H) such that the proton magnetization reaches thermal equilibrium at the 13C resonance field of 2.167 T, while the carbon spins are saturated. Stage II is skipped, such that for stage III the magnetic field is changed to the mixing field Bevo where no pulses are applied. In this period, carbon spins relax towards their thermal equilibrium polarization. Additionally, if fast direct or indirect proton–carbon exchange at Bevo is possible, thermal mixing leads to a transfer of spin polarization from proton to carbon spins. This gives rise to an observable maximum in carbon polarization or, if protons relax faster to the lattice, to carbon reaching equilibrium polarization faster than its spin-lattice relaxation could account for. In stage IV the NMR signal is read out at the 13C resonance field of 2.167 T.
3 Low-field 13C relaxation
3.1 Low-field 13C relaxation data
The field-dependence of the carbon relaxation time constant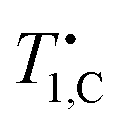 for doped PA for temperatures between 4.2 K and 40 K is shown in Fig. 2(a). At low fields, we find an approximately linear increase of
for doped PA for temperatures between 4.2 K and 40 K is shown in Fig. 2(a). At low fields, we find an approximately linear increase of  with field. This low-field linearity holds up to 40 K, although
with field. This low-field linearity holds up to 40 K, although  expectedly decreases with increasing temperature.
expectedly decreases with increasing temperature.
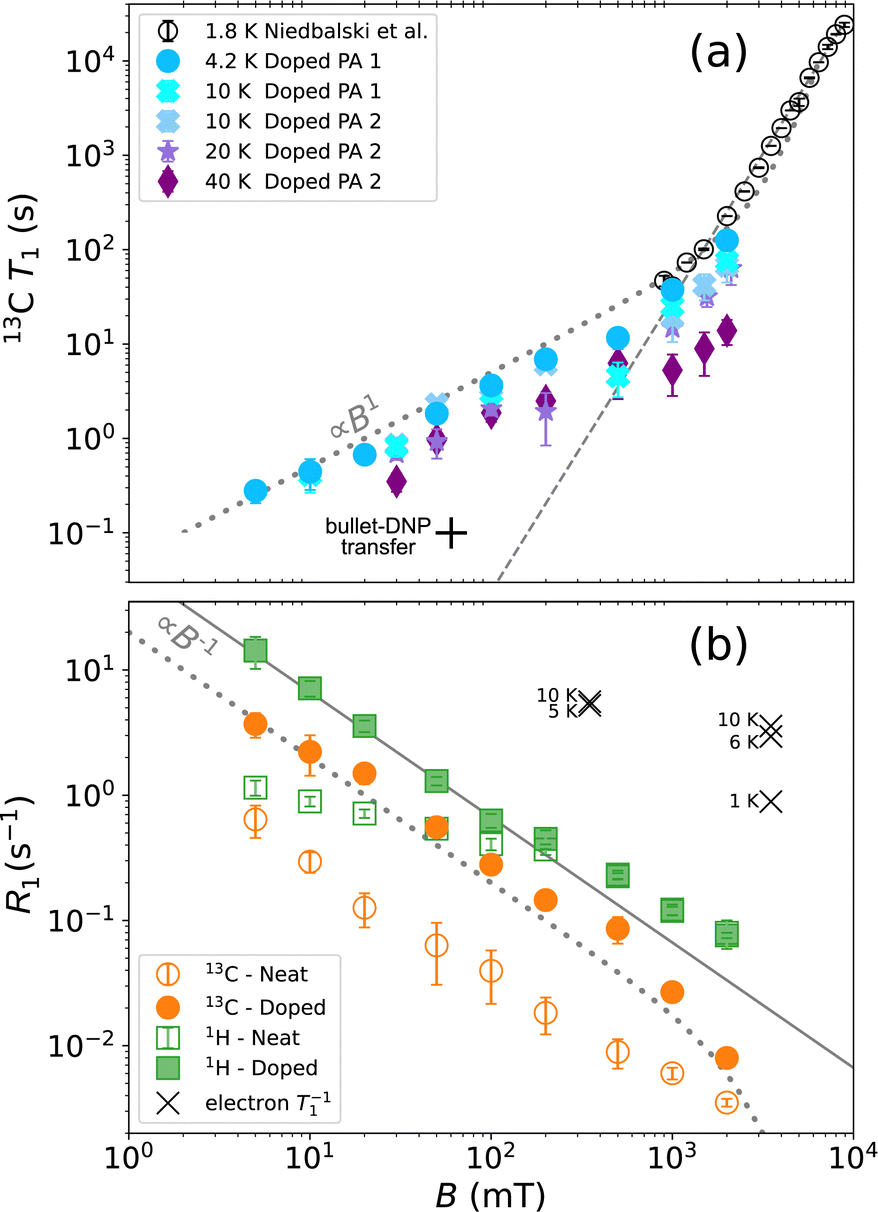 | ||
Fig. 2 Field-dependent relaxation in 1-13C PA doped with 15 mM trityl (OX063). (a) 13C relaxation time constants for different temperatures together with 1.8 K data (gray) reproduced from ref. 11. The dashed gray line corresponds to a power law with an exponent α = 3.1 as reported by Niedbalski et al. based on measurements at 1.8 K and fields from 0.9 to 9 T. The dotted gray line is computed using T1 = cB/(1 − P02), where c = 50 s T−1, and P0 = tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h(ħγSB/(2kBT)) is the electron spin polarization. (b) The 4.2 K data on doped PA (solid symbols) plotted as rates together with 1H data. For comparison the relaxation rates measured on neat PA (open symbols) are shown as well. The presence of OX063 increases the 13C relaxation rate throughout the observed field range, and causes faster 1H relaxation up to approximately 200 mT. Also shown are EPR relaxation data reported by Lumata et al.18 near 4.2 K. h(ħγSB/(2kBT)) is the electron spin polarization. (b) The 4.2 K data on doped PA (solid symbols) plotted as rates together with 1H data. For comparison the relaxation rates measured on neat PA (open symbols) are shown as well. The presence of OX063 increases the 13C relaxation rate throughout the observed field range, and causes faster 1H relaxation up to approximately 200 mT. Also shown are EPR relaxation data reported by Lumata et al.18 near 4.2 K. | ||
Also shown in Fig. 2(a) are data at 1.8 K recorded by Niedbalski, Lumata and co-workers.11 They found that their data are well described by a power law T1 = CBα, with an exponent of α = 3.1, indicated by the dashed gray line. If persistent to lower fields, this trend would correspond to critical relaxation during the transfer in bullet-DNP (the transfer takes 0.1 s at 70 mT) marked in Fig. 2(a)). While we observe a linear rather than a cubic field dependence at lower fields, our data at 4.2 K between 1 and 2 T (corresponding to the upper limit of our field range) are nonetheless in good agreement with the data by Niedbalski et al. at 1.8 K (corresponding to the lower limit of their field range).
The steep increase in 13C  , as observed by Niedbalski et al. at 1.8 K, may be attributed to the substantial electron spin polarization P0 with increasing field, which leads to a suppression of triple-spin flips. As shown in Fig. 2(a), a corresponding correction of the observed linear dependence (dotted gray line) gives a satisfactory description over more than three orders of magnitude in field and more than four orders of magnitude in T1.
, as observed by Niedbalski et al. at 1.8 K, may be attributed to the substantial electron spin polarization P0 with increasing field, which leads to a suppression of triple-spin flips. As shown in Fig. 2(a), a corresponding correction of the observed linear dependence (dotted gray line) gives a satisfactory description over more than three orders of magnitude in field and more than four orders of magnitude in T1.
To assess the effect of trityl radicals on nuclear relaxation, it is requisite to compare the relaxation of both 1H and 13C nuclear spins in the presence of trityl to that in neat PA. Relaxation rates R1 = 1/T1 of both nuclei, recorded at 4.2 K, are shown for neat and doped PA in Fig. 2(b). We note for neat PA that the rates 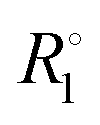 of protons and carbons converge at low field, which is expected based on reports of direct low-field thermal mixing in neat PA.17,19 This direct exchange is negligible for B > 10 mT but becomes relevant as the field approaches zero. The presence of trityl significantly accelerates the relaxation of carbon over the entire field range, while the effect on proton relaxation is only observable for fields below 200 mT.
of protons and carbons converge at low field, which is expected based on reports of direct low-field thermal mixing in neat PA.17,19 This direct exchange is negligible for B > 10 mT but becomes relevant as the field approaches zero. The presence of trityl significantly accelerates the relaxation of carbon over the entire field range, while the effect on proton relaxation is only observable for fields below 200 mT.
In addition to the nuclear relaxation rates, Fig. 2(b) also shows electron spin-lattice relaxation rates reported by Lumata et al.18 for temperatures near 4.2 K. For our analysis below, we note that R1,S(T = 4 K) ≈ 5 s−1 is nearly independent of field, which is expected since trityl relaxes predominantly via oxygen.20
3.2 Thermodynamic model
We have recently shown that the proton relaxation can be described quantitatively14 using a thermodynamic spin temperature model.15,21,22 In this model, as indicated in the sketch in Fig. 3, the different nuclear spin species and radical electron spins are described as reservoirs with distinct inverse temperatures βi = ħ/(kBTi) that couple to one another and the lattice. In particular, we showed that the relaxation of proton spins is described by their coupling to the electron Non-Zeeman (NZ) or dipolar reservoir. This exchange is via energy-conserving triple-spin flips (TSFs), in which a nuclear spin flip and an electron–electron flip-flop occur simultaneously.23 The TSF rate was calculated from first principles,12,13 after determining the NZ heat capacity from proton relaxation data. This lead to a nearly quantitative agreement between the model and the measured proton relaxation rates over two orders of magnitude in magnetic field.14 | ||
Fig. 3 Top: 13C relaxation is described by three reservoirs coupled to the lattice and each other. The vertical axis gives the reservoirs’ heat capacities at a field of 200 mT. (a) Heat capacities of nuclear Zeeman reservoirs grow quadratically with field, while the NZ heat capacity is field-independent. (b) Exchange rates used for modelling of nuclear relaxation rates. The TSF rates τNZ–H/C−1 have previously been calculated14 and the electron T1,S−1 is known from literature data.18 Also shown are the calculated carbon core-bulk diffusion rate τC–C−1 and the proton–carbon mixing rate τH–C−1. (c) The resulting coefficient-weighted relaxation rates for protons 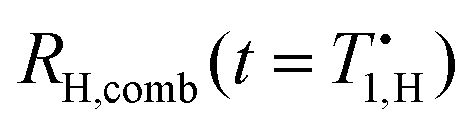 describe 1H data well.14 The predicted rate for carbon describe 1H data well.14 The predicted rate for carbon  is slower than experimental rates is slower than experimental rates  for fields up to 30 mT and faster above. Between 0.1 and 1 T the high TSF rates for both nuclei cause the predicted relaxation rate for carbon to be tied to the proton relaxation rate. Taking into account faster low-field mixing with protons and carbon–carbon spin-diffusion, the 13C data are well described (dash-dot line). for fields up to 30 mT and faster above. Between 0.1 and 1 T the high TSF rates for both nuclei cause the predicted relaxation rate for carbon to be tied to the proton relaxation rate. Taking into account faster low-field mixing with protons and carbon–carbon spin-diffusion, the 13C data are well described (dash-dot line). | ||
In our previous analysis of the proton relaxation, the carbon reservoir could be ignored, as its heat capacity CC is about  times smaller than that of the proton reservoir CH for pyruvic acid at any field, as shown in Fig. 3(a). We now extend the analysis to the carbon reservoir, which is coupled to the lattice with the rate
times smaller than that of the proton reservoir CH for pyruvic acid at any field, as shown in Fig. 3(a). We now extend the analysis to the carbon reservoir, which is coupled to the lattice with the rate  measured in neat PA and to the NZ reservoir with the TSF rate τNZ–C−1. The latter is calculated14 and compared to the TSF rate for protons in Fig. 3(b). The field-dependence of the carbon TSF rate is shifted relative to that of protons by a factor of γH/γC = 4 and its amplitude is approximately two orders of magnitude smaller.
measured in neat PA and to the NZ reservoir with the TSF rate τNZ–C−1. The latter is calculated14 and compared to the TSF rate for protons in Fig. 3(b). The field-dependence of the carbon TSF rate is shifted relative to that of protons by a factor of γH/γC = 4 and its amplitude is approximately two orders of magnitude smaller.
Details on the extension of the previous analysis14 to three reservoirs and its numerical solution can be found in the ESI.† The relaxation of three coupled reservoirs to the lattice temperature is generally given by a tri-exponential decay. Since the experimental data do not warrant an extraction of three coefficients and three decay rates, we compare the experimental data in Fig. 3(c) with effective relaxation rates Ri,comb(t) = −![[small beta, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e14d.gif) i(t)/βi(t), evaluated at the time of measurement
i(t)/βi(t), evaluated at the time of measurement  . Further details are given in the ESI.†
. Further details are given in the ESI.†
3.3 Model vs. experimental data
As expected, the presence of the carbon reservoir does not affect the resulting proton relaxation (solid green line in Fig. 3(c)), i.e., the three-reservoir description traces the proton data for doped PA (full squares) as well as the previous analysis,14 which had considered only protons and the NZ reservoir.The solution for the expected carbon rate (solid orange line in Fig. 3(c)) predicts accelerated relaxation throughout the field range, but does not describe the measured carbon rates well. At fields up to 20 mT, the predicted relaxation rates are smaller than the ones observed in experiment. For fields above 50 mT, the predicted relaxation rates are larger than the ones observed in experiment.
The discrepancy at low fields may be attributed to direct thermal mixing between the 1H and 13C reservoirs. The rate for this process, denoted by τH–C−1 in Fig. 3(b), is negligible above 20 mT but becomes significant at lower fields.17 Note that this direct mixing process causes the convergence, noted above, of the experimental carbon and proton relaxation rates at low fields in neat PA. Therefore, in neat PA below 20 mT, τH–C−1 should correspond to the experimentally observed carbon relaxation rate. We find that the low-field relaxation rates of 13C in doped PA are well described by  , i.e. the direct proton–carbon relaxation rates scale with the neat rates, but are accelerated five-fold. We attribute this increase to nuclear line-broadening due to the presence of trityl in doped PA.
, i.e. the direct proton–carbon relaxation rates scale with the neat rates, but are accelerated five-fold. We attribute this increase to nuclear line-broadening due to the presence of trityl in doped PA.
The discrepancy observed at fields above 50 mT may be attributed to carbon spin diffusion. In the model, the protons are in the fast thermal mixing limit in this field range, i.e., the NZ reservoir exchanges faster with the protons than with the lattice (τNZ–H−1 ≫ T1,S−1) and the NZ heat capacity does not affect the proton reservoir relevantly (CNZ ≪ CH). Therefore the proton reservoir sets the temperature of the Non-Zeeman reservoir. As the field is increased, the carbon reservoir increasingly couples to the NZ reservoir, and so the model predicts the same spin temperatures and hence relaxation rates for both protons and carbons.
However, only a minute portion of nuclear spins in the radical vicinity, referred to as core spins, are in direct exchange with the electron NZ reservoir. Carbon spins in the bulk only exchange with the radicals indirectly via carbon spin diffusion. From our experiments we cannot distinguish whether a slow exchange between core and bulk carbon spins impedes the relaxation, or if the diffusion through the bulk is field-dependent, and we discuss both possibilities in Section 5.
As detailed in the ESI† and indicated in the sketch in Fig. 3, the relaxation model may be extended with a second carbon reservoir and a field-dependent carbon core-bulk time constant of 33 s T−1. This leads to a satisfactory description (dash-dot line in Fig. 3(c)) of the experimental data.
In the following section we show that thermal mixing experiments are consistent with the predicted indirect proton–carbon coupling mediated by the NZ reservoir, as well as the inferred (i) five-fold accelerated direct hetero-nuclear exchange at low field, and (ii) the slowing of indirect hetero-nuclear coupling by a field-dependent carbon diffusion.
4 Hetero-nuclear mixing
In the thermal mixing experiments the carbon spins are saturated while the proton polarization is allowed to achieve thermal equilibrium at 2.167 T. The field is then ramped to the mixing field for the duration of the mixing delay, and subsequently the field is ramped back to 2.167 T, where the 13C spin polarization is read out. In the following thermal mixing data at 4.2 K are presented, further data are given in the ESI.†4.1 Thermal mixing – neat vs. doped PA
In order to quantify the effectiveness of the thermal mixing step we define the thermal mixing efficiency η as the observed signal divided by the thermal equilibrium signal of 13C at 2.167 T. The heat load of the carbon reservoir on the protons is negligible, so that a thermal mixing efficiency of 100% implies that the carbon spins attain the spin temperature of the proton spins without any losses, which corresponds to the theoretical maximum attainable in absence of any relaxation. The dependences of TM efficiency on mixing delay at different fields measured in neat and doped PA at 4.2 K are shown in the Fig. 4(a and b), respectively.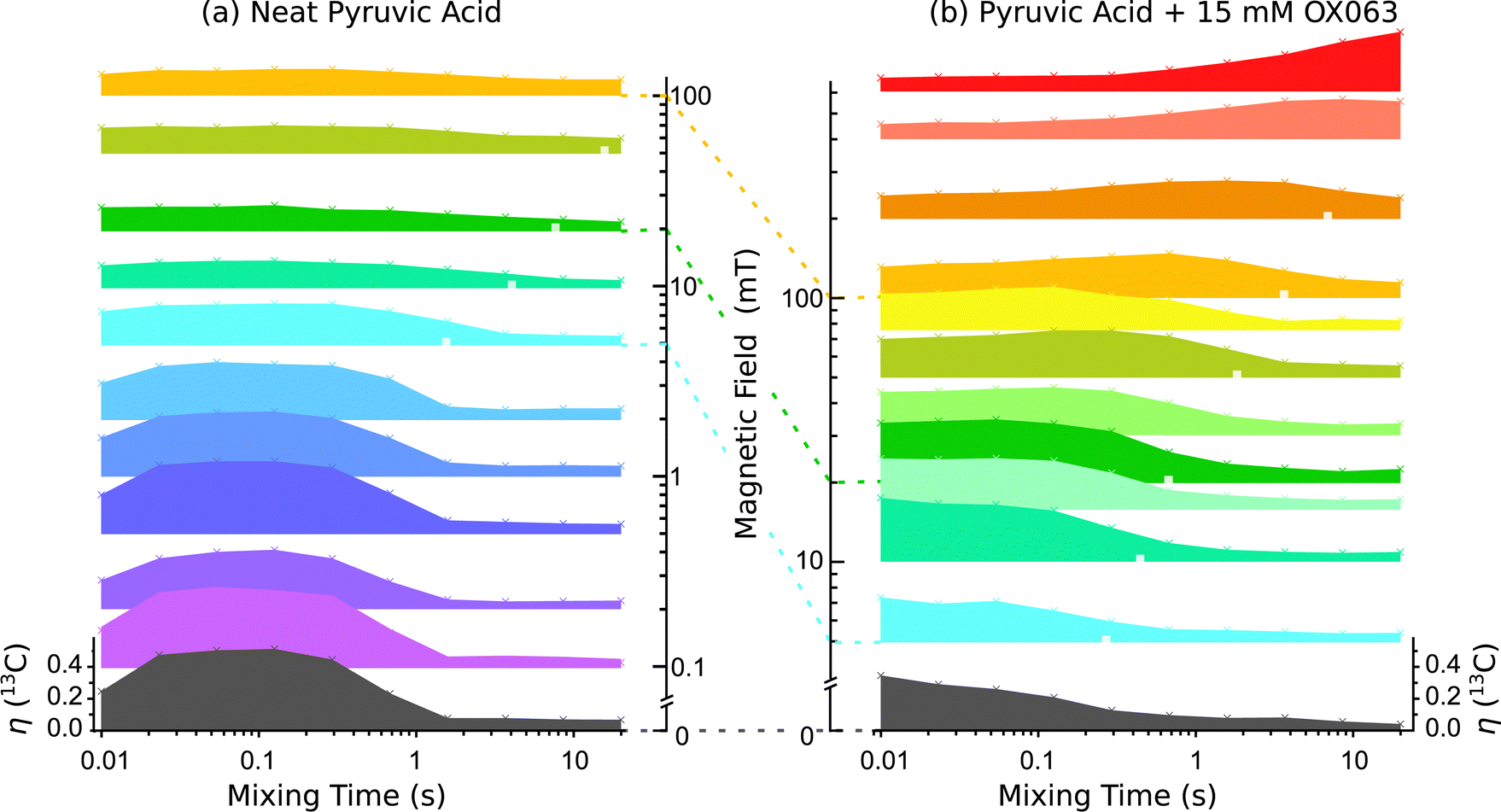 | ||
| Fig. 4 Thermal mixing in (a) neat and (b) doped PA at 4.2 K for fields from zero up to 100 mT and 600 mT, respectively. All data were normalized to the thermal equilibrium 13C signal at 4.2 K and 2 T (see ESI†), with the scale indicated for the respective zero-field data (lower left and right). TM efficiency is shown as a function of mixing delay. Also the measured 13C T1 are indicated, where available, by white squares in the respective datasets. | ||
For neat PA we find thermal mixing is most efficient well below 10 mT, in agreement with a previous study,17 and the converging  s for protons and carbons pointed out above. We note, however, a limited polarization transfer to carbon in neat PA at fields up to 100 mT, cf. top of Fig. 4(a), corresponding to η ≈ 15%. This occurs even for the shortest mixing times and has similarly been observed in previous FFC experiments.17 We infer that this transfer occurs during the field ramp up to the resonance field, and it may be associated with exchange with quantized rotational states of the methyl group.24,25
s for protons and carbons pointed out above. We note, however, a limited polarization transfer to carbon in neat PA at fields up to 100 mT, cf. top of Fig. 4(a), corresponding to η ≈ 15%. This occurs even for the shortest mixing times and has similarly been observed in previous FFC experiments.17 We infer that this transfer occurs during the field ramp up to the resonance field, and it may be associated with exchange with quantized rotational states of the methyl group.24,25
For doped PA in Fig. 4(b) we observe, as predicted, efficient thermal mixing also for fields above 20 mT. Maxima indicating hetero-nuclear mixing can be observed within the measured mixing time range for fields up to 400 mT.
We now consider the field-dependent maximum TM efficiency ηmax and the corresponding mixing time tmax, displayed in Fig. 5(a and b), respectively.
 | ||
Fig. 5 (a) The field-dependent maximum mixing efficiency ηmax shows that trityl mediates hetero-nuclear mixing above 5 mT, since ηmax is above equilibrium polarization throughout the investigated field range. (b) The corresponding times for TM maxima, tmax, show that trityl accelerates TM below 20 mT, uncertainty of neat tmax/5 (gray error bars) at corresponding fields (0, 5 and 10 mT) overlaps with uncertainty of tmax for doped PA. Above 20 mT tmax(B) for doped PA confirms TM,  (orange dash-dot line). Also, for B ≥ 20 mT we find tmax grows with field, consistent with TM slowed down by diffusion. Beyond 200 mT the mixing process becomes slower than the proton relaxation, tmax > T1,H (green dashed line), such that ηmax in (c) only slightly above equilibrium can be achieved by TM for 400 and 600 mT. (orange dash-dot line). Also, for B ≥ 20 mT we find tmax grows with field, consistent with TM slowed down by diffusion. Beyond 200 mT the mixing process becomes slower than the proton relaxation, tmax > T1,H (green dashed line), such that ηmax in (c) only slightly above equilibrium can be achieved by TM for 400 and 600 mT. | ||
For neat PA (open circles) we see that ηmax in Fig. 5(a) falls from about 50% at zero field and levels off at ηmax ≈ 15% for 10 mT and above. The corresponding mixing times Fig. 5(b) appear to be about 0.1 s rather independent of field.
For doped PA (full circles) in Fig. 5(a) we find mixing efficiency exceeds that of neat PA for B ≥ 5 mT, and for all fields values of ηmax greater than or equal to equilibrium polarization (gray line in Fig. 5(a)) are achieved. These maxima are established at shorter mixing times than the measured spin-lattice relaxation could account for, i.e.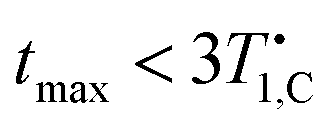 (orange dash-dot line in Fig. 5(b)), providing robust evidence for hetero-nuclear thermal mixing throughout the investigated field range.
(orange dash-dot line in Fig. 5(b)), providing robust evidence for hetero-nuclear thermal mixing throughout the investigated field range.
4.2 Mixing data and relaxation model
For comparison with the results of our relaxation data analysis, it is instructive to consider relevant field domains marked in Fig. 5.(I) Up to 20 mT, a five-fold accelerated direct hetero-nuclear exchange is attributed to radical induced line-broadening. In Fig. 5(b)tmax is indeed drastically reduced compared to neat PA, so much so that the actual mixing maxima appear to be reached for mixing times below our experimental range or even during the ramp, cf.Fig. 4(b).
(II) Above 20 mT, the relaxation model predicts efficient indirect proton–carbon mixing and we attribute the observed 13C relaxation to trityl-mediated hetero-nuclear exchange that is slowed down by field-dependent carbon core-bulk diffusion. The increase in tmax with field in Fig. 5(b) is consistent with this scenario, as is the broadening of mixing maxima with increasing field apparent in Fig. 4(b).
(III) Above 300 mT the observed indirect proton–carbon mixing becomes slower than the proton relaxation,  (green dashed line in Fig. 5(b)), such that no pronounced maxima at 400 and 600 mT are observable, cf.Fig. 4, but rather ηmax values slightly above thermal equilibrium, cf.Fig. 5(a). We can nonetheless distinguish the observed mixing from spin-lattice relaxation, since the maxima are achieved faster than spin-lattice relaxation could account for,
(green dashed line in Fig. 5(b)), such that no pronounced maxima at 400 and 600 mT are observable, cf.Fig. 4, but rather ηmax values slightly above thermal equilibrium, cf.Fig. 5(a). We can nonetheless distinguish the observed mixing from spin-lattice relaxation, since the maxima are achieved faster than spin-lattice relaxation could account for,  (orange dash-dot line in Fig. 5(b)).
(orange dash-dot line in Fig. 5(b)).
The TM data are qualitatively consistent with the results from the model used to describe the carbon relaxation. They show that the presence of trityl causes the predicted (indirect) mixing for fields above 20 mT. Additionally they indicate that (i) trityl accelerates (direct) mixing at low field, and that (ii) the (indirect) mixing above 20 mT slows down with increasing field.
5 Discussion
The relaxation data reported here explain the viability of bullet-DNP experiments.9,10 Earlier work at fields down to 1 T indicated a scaling of 13C relaxation rates with field according to T1 ∼ B3,11 that would lead to very fast relaxation at low fields. Conversely, our data show that below 1 T the relaxation time constants scale only linearly with the applied field.An extended STM predicts indirect proton–carbon exchange mediated by the NZ reservoir. Qualitatively, this is consistent with the observed enhanced carbon relaxation and with the enhanced mixing observed above 20 mT.
At fields up to 20 mT, a quantitative description of the carbon relaxation needs to include enhanced direct proton–carbon mixing. This is consistent with the shortened times tmax to attain maximum mixing in doped PA, and we attribute the enhanced direct hetero-nuclear mixing to nuclear line broadening due to the presence of trityl.
At fields above 50 mT, the experimentally observed relaxation rates are substantially smaller than those predicted. Here the model predicts efficient indirect coupling via the NZ reservoir such that carbon relaxation rates are equal to those of protons. Since only core 13C nuclei in the radical vicinity exchange directly with the NZ reservoir, we attribute this discrepancy to a field-dependent carbon diffusion process. This is also consistent with the thermal mixing data, where tmax increases with increasing field. A diffusion process that slows with increasing field is also compatible with long DNP build-up times at high field.26 There are two different diffusion processes that may each limit the polarization exchange between carbon bulk spins and the radical NZ reservoir.
One possibility is that this reduction arises from slow carbon spin diffusion through the bulk. The carbon line in 1-13C PA has a contribution from chemical shift anisotropy (CSA),27 and one may expect spin diffusion to limit the overall 13C relaxation as the field, and thereby the carbon linewidth, is increased.28 It should be noted, however, that at the fields investigated in this study, the carbon line is dominated by dipolar interactions.
An alternative explanation of the observed slower carbon relaxation rates is that the proximity to the radicals causes a shift in the resonance frequency of the core spins, which in turn impedes diffusion to the bulk spins. The diffusion barrier separates the carbon spins into NMR-invisible core nuclei in the radical vicinity, and visible bulk nuclei outside the barrier. The diffusion among the bulk nuclei is fast, and so the carbon relaxation rate is limited by the exchange between core and bulk nuclei. As measured recently by Stern et al. on protons,29 the core spins do nonetheless exchange polarization with the bulk spins. This exchange was modelled with two coupled reservoirs representing the core and bulk spins, respectively, where the diffusion barrier limits the exchange of Zeeman energy between these reservoirs.29 The energy flow across the barrier is frequently attributed to dipolar interactions,30–33 and it is conceivable that the coupling of the two reservoirs is field-dependent, since more Zeeman energy has to be transferred across the barrier at higher fields. However, for 13C no experimental studies of this coupling have have been presented to date.
6 Conclusion
In conclusion, we have found that at low fields and low temperatures the carbon T1,C in pyruvic acid doped with 15 mM OX063 scales linearly with the applied field, and is long compared to the transfer times in bullet-DNP. The relaxation data may be described with a spin temperature model that predicts radical-mediated, triple-spin-flip driven hetero-nuclear polarization transfer at fields from 20 mT to beyond 1 T. The model comprises a carbon spin diffusion process that hampers polarization exchange between the radical and bulk carbon spins. Long build-up times in DNP of low-γ nuclei are a possible manifestation of this process, although its microscopic origin has yet to be determined.Author contributions
HK performed experiments with help from DP, KK, ASK, AJH, JFM, JOB and BM. Experimental data were analysed by HK, BM and MJ. MJ and BM devised the thermodynamic description and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Tom Wenckebach for numerous discussions and assistance with the relaxation model. We thank Lloyd Lumata for discussions and for providing the high-field 13C T1 data, and David Gadian for help with experiments. We thank Geoffrey Bodenhausen, Matthias Ernst and Malcolm Levitt for discussions. This work has been supported by the EPSRC (EP/R031959/1), by the “Impuls- und Vernetzungsfonds of the Helmholtz-Association” (grant VH-NG-1432), and by the DFG (grant number 454252029 - SFB 1527). This project has received funding from the European Research Council (ERC) under the European Unions Horizon 2020 research and innovation programme (grant agreement No 951459).References
- J. H. Ardenkjær-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef PubMed.
- J. H. Ardenkjær-Larsen, Handbook of High Field Dynamic Nuclear Polarization, 2019, 239 Search PubMed.
- W. Köckenberger, in Dissolution Dynamic Nuclear Polarization, John Wiley & Sons, Ltd; 2014, pp. 161–170 Search PubMed.
- D. Kurzbach and S. Jannin, in Dissolution Dynamic Nuclear Polarization Methodology and Instrumentation, American Cancer Society; 2018, pp. 117–132 Search PubMed.
- S. J. Nelson, J. Kurhanewicz, D. B. Vigneron, P. E. Z. Larson, A. L. Harzstark, M. Ferrone, M. van Criekinge, J. W. Chang, R. Bok, I. Park, G. Reed, L. Carvajal, E. J. Small, P. Munster, V. K. Weinberg, J. H. Ardenkjær-Larsen, A. P. Chen, R. E. Hurd, L.-I. Odegardstuen, F. J. Robb, J. Tropp and J. A. Murray, Sci. Transl. Med., 2013, 5, 198ra108 Search PubMed.
- Z. J. Wang, M. A. Ohliger, P. E. Z. Larson, J. W. Gordon, R. A. Bok, J. Slater, J. E. Villanueva-Meyer, C. P. Hess, J. Kurhanewicz and D. B. Vigneron, Radiology, 2019, 291, 273–284 CrossRef PubMed.
- F. A. Gallagher, R. Woitek, M. A. McLean, A. B. Gill, R. M. Garcia, E. Provenzano, F. Riemer, J. Kaggie, A. Chhabra, S. Ursprung, J. T. Grist, C. J. Daniels, F. Zaccagna, M.-C. Laurent, M. Locke, S. Hilborne, A. Frary, T. Torheim, C. Boursnell, A. Schiller, I. Patterson, R. Slough, B. Carmo, J. Kane, H. Biggs, E. Harrison, S. S. Deen, A. Patterson, T. Lanz, Z. Kingsbury, M. Ross, B. Basu, R. Baird, D. J. Lomas, E. Sala, J. Wason, O. M. Rueda, S.-F. Chin, I. B. Wilkinson, M. J. Graves, J. E. Abraham, F. J. Gilbert, C. Caldas and K. M. Brindle, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2092–2098 CrossRef CAS PubMed.
- R. Woitek, M. A. McLean, S. Ursprung, O. M. Rueda, R. M. Garcia, M. J. Locke, L. Beer, G. Baxter, L. Rundo, E. Provenzano, J. Kaggie, A. Patterson, A. Frary, J. Field-Rayner, V. Papalouka, J. Kane, A. J. Benjamin, A. B. Gill, A. N. Priest, D. Y. Lewis, R. Russell, A. Grimmer, B. White, B. Latimer-Bowman, I. Patterson, A. Schiller, B. Carmo, R. Slough, T. Lanz, J. Wason, R. F. Schulte, S.-F. Chin, M. J. Graves, F. J. Gilbert, J. E. Abraham, C. Caldas, K. M. Brindle, E. Sala and F. A. Gallagher, Cancer Res., 2021, 81, 6004–6017 CrossRef CAS PubMed.
- K. Kouřil, H. Kouřilová, S. Bartram, M. H. Levitt and B. Meier, Nat. Commun., 2019, 10, 1733 CrossRef PubMed.
- K. Kouřil, M. Gramberg, M. Jurkutat, H. Kouřilová and B. Meier, Magn. Reson., 2021, 2, 815–825 CrossRef.
- P. Niedbalski, Q. Wang, C. Parish, F. Khashami, A. Kiswandhi and L. Lumata, J. Phys. Chem. B, 2018, 122, 1898–1904 CrossRef CAS PubMed.
- W. Wenckebach, J. Magn. Reson., 2019, 299, 124–134 CrossRef CAS PubMed.
- W. Wenckebach, J. Magn. Reson., 2019, 299, 151–167 CrossRef CAS PubMed.
- M. Jurkutat, H. Kouřilová, D. Peat, K. Kouřil, A. S. Khan, A. J. Horsewill, J. F. MacDonald, J. Owers-Bradley and B. Meier, J. Phys. Chem. Lett., 2022, 13, 10370–10376 Search PubMed.
- S. F. J. Cox, V. Bouffard and M. Goldman, J. Phys. C: Solid State Phys., 1973, 6, L100–L103 CrossRef CAS.
- A. J. Horsewill and Q. Xue, Phys. Chem. Chem. Phys., 2002, 4, 5475–5480 RSC.
- D. T. Peat, M. L. Hirsch, D. G. Gadian, A. J. Horsewill, J. R. Owers-Bradley and J. G. Kempf, Phys. Chem. Chem. Phys., 2016, 18, 19173–19182 RSC.
- L. Lumata, Z. Kovacs, A. D. Sherry, C. Malloy, S. Hill, J. van Tol, L. Yu, L. Song and M. E. Merritt, Phys. Chem. Chem. Phys., 2013, 15, 9800 RSC.
- M. L. Hirsch, N. Kalechofsky, A. Belzer, M. Rosay and J. G. Kempf, J. Am. Chem. Soc., 2015, 137, 8428–8434 CrossRef CAS PubMed.
- C. Hess, J. Herick, A. Berlin, W. Meyer and G. Reicherz, Nucl. Instrum. Methods Phys. Res., Sect. A, 2012, 694, 69–77 CrossRef CAS.
- A. Abragam and W. G. Proctor, Phys. Rev., 1957, 106, 160–161 CrossRef CAS.
- A. Abragam and W. G. Proctor, Phys. Rev., 1958, 109, 1441–1458 CrossRef.
- W. Wenckebach, Essentials of Dynamic Nuclear Polarization, Spindrift Publications, 2016 Search PubMed.
- A. Horsewill, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 35, 359–389 CrossRef CAS.
- B. Meier, Magn. Reson. Chem., 2018, 56, 610–618 CrossRef CAS PubMed.
- J. H. Ardenkjær-Larsen, S. Bowen, J. R. Petersen, O. Rybalko, M. S. Vinding, M. Ullisch and N. C. Nielsen, Magn. Reson. Med., 2018, 81, 2184 CrossRef.
- S. Macholl, H. Jóhannesson and J. H. Ardenkjaer-Larsen, Phys. Chem. Chem. Phys., 2010, 12, 5804 RSC.
- Y. Wang and K. Takeda, New J. Phys., 2021, 23, 073015 CrossRef CAS.
- Q. Stern, S. F. Cousin, F. Mentink-Vigier, A. C. Pinon, S. J. Elliott, O. Cala and S. Jannin, Sci. Adv., 2021, 7, eabf5735 CrossRef CAS PubMed.
- A. Z. Genack and A. G. Redfield, Phys. Rev. B: Solid State, 1975, 12, 78–87 CrossRef CAS.
- S. F. J. Cox, S. F. J. Read and W. T. Wenchebach, J. Phys. C: Solid State Phys., 1977, 10, 2917–2936 CrossRef CAS.
- A. E. Dementyev, D. G. Cory and C. Ramanathan, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 024413 CrossRef.
- G. B. Furman, S. D. Goren, V. M. Meerovich and V. L. Sokolovsky, Appl. Magn. Reson., 2021, 52, 781–796 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: T1 measurements, additional TM data, multi-reservoir relaxation. See DOI: https://doi.org/10.1039/d2cp04535d |
| This journal is © the Owner Societies 2022 |