
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A benchmark for non-covalent interactions in organometallic crystals†
José Eduardo Zamudio
Díaz Mirón
and
Matthias
Stein
 *
*
Molecular Simulations and Design Group, Max Planck Institute for Dynamics of Complex Technical Systems, Sandtorstrasse 1, 39106 Magdeburg, Germany. E-mail: matthias.stein@mpi-magdeburg.mpg.de
First published on 23rd November 2022
Abstract
Organometallic complexes are the basis for homogeneous catalysis, have applications in materials science and are also active pharmaceutical ingredients. The interaction between transition metal complexes in the solid state is determining their thermodynamics and bio-availability. Non-covalent interactions such as hydrogen bonding and van der Waals are stabilizing crystals of transition metal complexes. The variation of ligand field, central metal atoms and their oxidation and spin states are determinants of the magnitude of their inter-molecular interactions. A comparison of a set of 43 manually curated experimental heats of sublimation (the new XTMC43 set) and results from periodic DFT calculations shows that an agreement to within 9% can be achieved using GGA or mGGA functionals with atom-centred Gaussian-type basis functions. The need for careful assessments of consistency, calibration and reproducibility of experimental and computational data is discussed. Results regarding the new XTMC43 benchmark set are suggested to serve as a starting point for further method development, systematic screening and crystal engineering.
Introduction
Transition metal complexes are the ingredients for catalysis in chemical, biochemical and bioinorganic systems. They play essential roles in industrial processes,1 organic synthesis,2 material science,3 but also as in drug discovery and development.4 The concept of metal–ligand coordination and three-dimensional complex shape determining their properties and reactivities is common ground since Alfred Werner.5,6 For transition metal complexes, in particular, molecular orbital theory can be used to rationalize coordination geometries as a function of oxidation states, spin states and ligand field strength.Single crystal X-ray crystallography in organometallic chemistry is of relevance since the complex structural features of transition metal compounds and clusters are hard to determine by other experimental techniques.7 The structures of organometallic complexes and clusters in single crystals are molecular in nature so their assemblies must be stabilized by weak, non-covalent intermolecular interactions that are, in principle, the same as in purely organic molecular crystals.8
The cohesive, non-covalent energy that is stabilizing the crystal over its individual entities, is referred to as the ‘lattice energy’ Elatt. It can, in principle, be obtained from two hypothetical thermodynamic cycles:9,10
(i) The transfer of the molecule from crystal to the sub-cooled melt and then into the gas phase. Cycle (i), the ‘melt cycle’, requires knowledge of the experimental melting temperature and enthalpies of melting.
Cycle (ii), the ‘sublimation cycle’, the focus of this paper, approximates the solubility differences by taking the molecule from the crystal to the gas phase and then into solution (see Scheme 1).
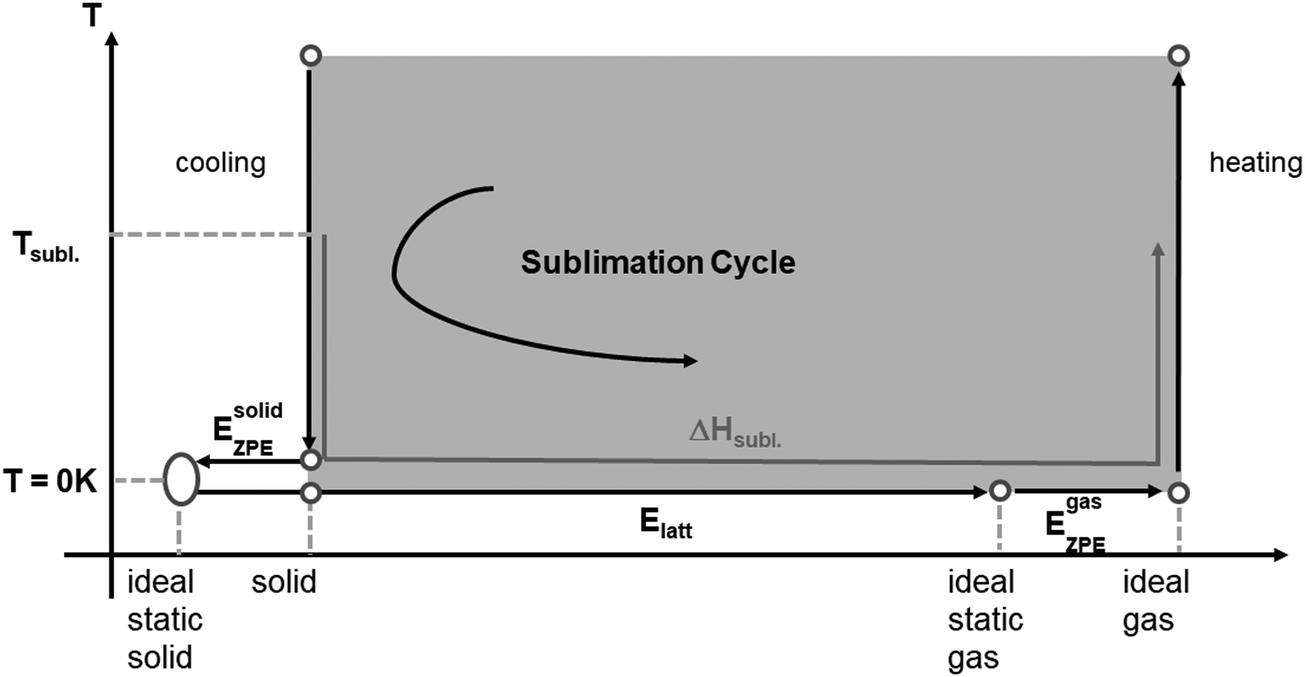 | ||
| Scheme 1 The sublimation cycle that takes the molecules in the crystal from its ideal, static state at 0 K to the ideal static gas at the same temperature. Heats of sublimation ΔHsubl have to be corrected to obtain the lattice energy Elatt which is computationally accessible10 (see text for details). | ||
Both cycles consider properties from the crystals (melting or sublimation free energy) and are thus dependent on the crystal structures. The sublimation cycle is preferred as it makes use of the computationally accessible thermodynamic states from the gas and the crystalline solid.
The applicability of the ‘sublimation cycle’ has been evaluated for the computational predictions of the solubilities11,12 and sublimation enthalpies of organic crystals.13
Sublimation enthalpies are important thermodynamic properties of the solid phase. In order to obtain reliable results, experiments have to be conducted at temperatures that deviate significantly from the standard reference temperature, 298 K. They can be adjusted to the reference state by making use of simplifications regarding the temperature dependence of the heat capacities Cxtalp and Cgasp (see below).
Calibration is a fundamental requirement for every sublimation enthalpy measurement. Unlike other thermochemical measurements, uncertainties in sublimation enthalpies can be large, often several kJ mol−1 or more. The compendium of Chickos and Acree14 that contains experimentally determined sublimation enthalpies for organic and organometallic compounds also discusses challenges to obtain accurate values. In a statistical analysis of 451 measurements on 80 different entities, a standard deviation between the mean and each experimental values was ±6.7 kJ mol−1.15 The absence of use of standard or reference compounds was identified as one source of systematic error. For heats of sublimation >100 kJ mol−1, when the compounds usually have a low vapor pressure and experiments have to be performed at elevated temperature, there are no standards for the calibration of instruments under these conditions available. A discussion of different experimental techniques and their intrinsic accuracies is presented in ref. 15. The uncertainty between mean and individual points of experimental sublimation enthalpies of organometallic compounds is particularly high (±23.9 kJ mol−1) and almost three times higher than for organic molecules.15
Periodic Density Functional Theory (DFT) calculations with dispersion corrections are the current state-of-the art for crystalline states but some of their pitfalls are extensively discussed in ref. 16. There are a number of computational benchmark sets for organic molecular crystals. The C21 set is used for non-covalent interactions,17 its augmented version X23,18–20 the POLY59 set for 9 possible polymorphs of 5 drug-like molecules21 and the ICE10 set for ten different ice polymorphs22 and probably more.
Transition metal complexes are a challenge due to the larger variety of central metal atom from the periodic table of elements, variability in ligand coordination sphere, oxidation and spin state. This multitude of parameters has limited the development of general, accurate models, such as force-fields, in particular for open-shell transition-metal complexes.23 There are several factors that contribute to the ‘structural variability’ of organometallic compounds, namely the number and type of ligands and their combinations, the coordination geometry and the number of metal atoms.8
The quantum chemical characterization of periodic systems can be done with many different approaches, see ref. 24 and 25 for reviews. For example plane-wave (PW)26 or atom-centered Gaussian basis functions,24,27,28 a linearized augmented plane-wave ansatz (LAPW)26 or a hybrid Gaussian and plane-wave scheme29 are possible. For a comparison of results from PWDFT vs. all-electron Gaussian atomic orbitals see ref. 30. The description of the localized electron density in molecular crystals requires a large number of plane-wave functions. As an alternative, atom-centered basis functions, i.e., Gaussian type orbitals (GTOs) may be able to give an accurate electron density. For sparsely packed molecular crystals GTOs were shown to provide a high computational efficiency and accuracy.31–33
We here present the first evaluation of the accuracy of periodic DFT calculations for a set of organometallic crystals. The use of atom-localized GTOs allows an elegant and consistent treatment of molecules and periodic systems of any dimensionality on equal footing. A systematic increase in the number of atomic GTOs allows to extrapolate the accuracy of results and shows that already medium-sized GTOs give reliable results.
The approach of PIXEL34–36 relies on a physical interpretation of intermolecular interactions in molecular crystals from terms describing pairwise coulombic, polarisation, dispersion and repulsion. Computed molecular electron densities are fitted to a combination of empirical terms such as number of electrons, the number of valence electron, the first ionization energy among others and can be applied to estimate lattice energies of organic crystals from 3D replicates of those molecular descriptors.37 The method was extended to a set of transition metal compounds38 by defining new atomic values for d-block transition metals such as ionization energy, covalent radii and van der Waals interactions. Five tested parameter sets gave very similar results.
The XTMC43 (Crystal Transition Metal Complex) benchmark set is based on manually curated experimental heats of sublimation from the collections of Chickos and Acree,14 Pilcher and Skinner39 and previous work deriving parameters for molecular electron densities.38
It consists of a series of 43 transition metal complexes covering a range of 3d-, 4d-, and 5d-transition metal elements in various oxidation and spin states that are coordinated by different ligands. There are 18 open shell and 25 closed-shell complexes in the data set. It serves as a benchmark to assess the accuracy of periodic DFT calculations of lattice energies for organometallic crystals.
Here, we are using one representative of the GGA (PBE40,41 with dispersion corrections) and meta-GGA families (M06-L42) to obtain consistent data for non-covalent weak interactions that are stabilizing organometallic crystals (see below for computational details).
The calculated lattice energies are only basis-set dependent to a very minor degree and can be calculated with a mean deviation of 11 and 10 kJ mol−1 from experiment for PBE and M06-L (∼9%). The consistency of results allows to identify experimental outliers and the generation of a high-quality data set for possible subsequent data-driven (machine learning) applications. It also becomes possible now to perform in silico crystal engineering for transition metal compounds.
Materials and methods
Crystal structures
Experimental transition metal crystal structures were retrieved from the Cambridge Crystallographic Data Centre (CCDC)43 as crystallographic information files (cif.). Unit cell parameters |a|, |b|, |c|, α, β and γ were taken from the crystal structures. |a|, |b| and |c| are lengths of the appropriate cell vectors, α is the angle between vectors b and c, β is the angle between vectors a and c, and γ is the angle between vectors a and b. Missing hydrogen atoms were added using TmoleX19, when necessary.44Computational details
All calculations were performed with TURBOMOLE45,46 version 7.547 with PBE40,41 as a representative GGA and the M06-L mGGA.42 For PBE, the D3 dispersion correction term48 with Becke–Johnson damping49,50 was included.51 The Minnesota M06-L functional is devoid of an explicit definition of London dispersion since it was already considered during the derivation and parametrization. For all calculations Ahlrichs’ def2-TZVP and def2-TZVPP basis sets52 were used in the resolution-of-identity approximation with suitable auxiliary basis sets.53,54 For atoms beyond Kr, effective core potentials (ECPs) which include scalar relativistic corrections, are used to replace the explicit treatment of core electrons.Non-hybrid density functionals were found to be reliable for the thermochemistry of transition metal complexes and outperform hybrid functionals, such as B3LYP for example.55
Lattice energies for organic molecular crystals of the X23 benchmark set and related chiral compounds were found to be almost converged using the medium-sized triple-zeta def2-TZVP basis set and the larger def2-TZVPP basis set did not significantly affect the calculated lattice energies for representatives of either the vdW, mixed and hydrogen-bonded interaction classes.33 Counterpoise corrections also showed that a basis set error (BSSE) for the def2-TZVP basis set was at most 7% of the interaction energy of molecular compounds in the crystal. This reduced the calculated lattice energy and brought it even closer to experiment.33
The fitting of the Coulomb density and continuous fast multipole methods (CFMM)56,57 enables the calculation of energies and structural optimizations using atom-centered Gaussian basis functions for molecular and periodic systems on the same grounds. For calculations on very large molecular systems a low-memory modification of the RI approximation has been implemented in the riper module.58 Thus, the lattice energy can directly be calculated using the same atom-centered Ahlrichs’ Gaussian basis functions for the ideal crystal and the isolated, fully relaxed molecule.
The periodic DFT implementation in Turbomole makes use of Γ-point centered mesh of k points. In 3D periodic systems each sampling point is defined by its components k1, k2 and k3 along the reciprocal lattice vectors b1, b2 and b3 as
| k = k1b1 + k2b2 + k3b3 | (1) |
All calculations were performed for a mesh of 3 × 3 × 3 k-points to sample the Brillouin zone in the triclinic space group (P1) and not considering molecular or crystal symmetry operations. A fine multiple integration grid m5 was used in all calculations. Structural optimizations for crystals and relaxed molecules were performed at the PBE-D3(BJ)/def2-TZVP and PBE-D3(BJ)/def2-TZVPP levels. M06-L results are from single point calculations at the PBE-D3(BJ)/def2-TZVPP optimized structures (M06-L/def2-TZVPP//PBE-D3(BJ)/def2-TZVPP). Tight convergence criteria for SCF 10−8Eh, energy 10−7Eh and gradients 10−4Eh/a0 were applied throughout.
Definition of the lattice energy and thermal corrections
The lattice energy, Elatt, is defined as the energy needed for breaking up the static crystal lattice. More specifically it is defined as the energy difference between a static perfect infinite crystal (ideal static solid – iss) and its related ideal static gas (isg) of non-interacting molecules in their lowest energy conformation both at 0 K (see eqn (2)) where Z is the number of molecules in the unit cell) | (2) |
For a comparison to experimental sublimation enthalpies at finite temperature, thermal corrections to the ideal static solid have to be added. The corrections are comprised of the zero-point vibrational energy and thermal corrections between 0 K and the reference temperature. Contributions from the zero-point vibrational energy difference between the ideal solid and gas and the integral of the difference in heat capacities between gas and crystal at the reference temperature are summed in a correction term ΔHcorr.
A simplified expression to ΔHcorr, is the molecule and crystal structure-independent 2RT-approximation which is often used in computational chemistry (see eqn (3)).36
| ΔHsubl = −Elatt − 2RT | (3) |
For an ideal gas, the translational (CgP,trans = 5/2R) and rotational (CgP,rot = 3/2R) degrees of freedom are considered to give ΔHcorr = (5/2R − 3/2R − 6R) = –2RT.
Differences between back-corrected ‘experimental’ lattice energies using vibrational frequencies or the 2RT-approximation from eqn (3) were between 0.7 and 1.4 kJ mol−1.33 Recently, it was confirmed from plane wave DFT calculations that vibrational free-energy corrections are small with a mean value of 1.0 kJ mol−1 for a test set of organic polymorphs.59
However, calculations using on the X23 benchmark set show that the ‘−2RT’ thermal correction is a simplified approximation, since the magnitude of correction is dependent on the crystal structure studied. In addition, different electronic structure methods (PBE-XDM, PBE-TS, DFTB3-D3) were not converging to give these values. Differences between the “−2RT” approximation and quantum chemical, crystal structure-dependent thermal corrections are non-systematic and between −5 kJ mol−1 and +3 kJ mol−1.17,60,61
Source of experimental heats of sublimation
Chickos and Acree compiled a compendium of heats of sublimation of organic and organometallic compounds.14 The data reported for the same compound show variations due to possible lack of careful calibration studies with respect to reference compounds or different experimental temperatures. Substantial differences in sublimation values are revealed which establish the need for documentation of accuracy of measurements.Simplified empirical methods are used to determine ΔCgas–solidP = CgasP − CsolidP which are independent of the molecular structure. The suggested theoretical value of ΔCgas–solidP = 6RT (∼28 J mol−1 K−1)62 is close to that of an experimental data set of 284 measurements on 102 different solids with an optimal temperature coefficient of 32 J mol−1 K−1 and a large standard deviation of 23 J mol−1 K−1 ΔCgas-solidP = 32 ± 23 J mol−1 K−1.63
Experimental heat capacities for many solids are available at 298 K, whereas those of gases at that temperature have to be estimated. The 6R approximation (see above) relies on an ideal gas assumption and the Dulong–Petit value for the solid.
The manually curated entries of the XTMC43 data set are given in the ESI.† The lattice energy Elatt,exp is obtained using the ‘2RT’ approximation. In case of several experimental data points, an average of the values at or close to 298 K is taken while the largest estimated experimental error is kept in order to have a careful estimate of experimental uncertainty.
In particular, the following decision route was taken:
(i) Choice of experimental heats of sublimation at or close to 298 K.
(ii) In case of several entries at or close to 298 K, removal of outliers that differ by more than ±10%.
(iii) When multiple entries remain, take the average, calculate average deviation and consider this as ‘experimental uncertainty’.
(iv) When ‘experimental error’ is stated, retain largest value. Otherwise use 4 kJ mol−1.
Results and discussion
The XTMC43 benchmark set
The transition metal complexes of the XTMC43 benchmark set are shown in Fig. 1. It consists of a series of 43 transition metal complexes covering a range of 3d-, 4d-, and 5d-transition metal elements in various oxidation and spin states which are coordinated by different ligands (see Fig. 1). There are 18 open shell and 25 closed-shell complexes in the data set. The number of atoms per unit cell is smallest for complex 32 (25 atoms) and largest for complex 20 (344 atoms). The number of molecules per unit cell Z ranges from 1 (complexes 27, 31 and 32) to 8 (complex 20) but most of them have 2 or 4 molecules per unit cell (21 and 18 entries, respectively). | ||
| Fig. 1 Transition metal complexes 1–43 in the XTMC43 benchmark set for which single crystal structures and experimental heats of sublimation are known. | ||
This set of complexes serves as a benchmark to assess the accuracy of periodic DFT calculations for lattice energies of organometallic crystals.
We validated for a set of complexes for which several single crystal structures are available in the CCDC (compound entries 08, 09, 13, 31, 32) that the differences in lattice energies between them were below 4 kJ mol−1 and the choice of CCDC entry does not significantly affect our discussion of results (see Table S1, ESI†). It was also shown for entry 02 that the energy differences between the 1977 X-ray structure obtained at room temperature (295 K)64 and the 2020 low temperature structure (120 K)65 were 2–3 kJ mol−1 only (see Table S2, ESI†). It becomes apparent, that the choice of CCDC entry has only a very minor effect on the calculated lattice energies. We did not consider the polymorphism of organometallic crystals in this study.
Table 1 reports the selected single crystal structures together with temperature for structure determination, metal and oxidation state plus the number of molecules per unit cell.
| Entry | Compound name | CCDC ID (Tcryst)a | Metal oxidation state | Z |
|---|---|---|---|---|
| a Temperature at which crystal structure was determined. b r.t. room temperature (283–303 K). | ||||
| 1 | Tris(2,4-pentanedionato)-chromium(III) | ACACCR07 (290 K) | Cr3+ | 4 |
| 2 | Bis(2,4-pentanedionato)-chromium(II) | ACACCS (r.t.)b | Cr2+ | 2 |
| 3 | Bis(2,4-pentanedionato)–copper(II) | ACACCU02 (r.t.) | Cu2+ | 2 |
| 4 | Tris(2,4-pentanedionato)-manganese(III) | ACACMN21(r.t.) | Mn3+ | 4 |
| 5 | Bis(2,4-pentanedionato)-oxovanadium(IV) | ACACVO12 (r.t.) | V4+ | 2 |
| 6 | (η6-Benzene)-tricarbonyl-chromium | BZCRCO14 (r.t.) | Cr0 | 2 |
| 7 | (η6-Toluene)-tricarbonyl)-chromium | CCRTOL01 (r.t.) | Cr0 | 4 |
| 8 | Bis(η5-cyclopentadienyl)-titanium(IV) dichloride | CDCPTI04 (r.t.) | Ti4+ | 4 |
| 9 | (η5-Cyclopentadienyl)-titanium(IV) trichloride | CEHPIO01 (r.t.) | Ti4+ | 2 |
| 10 | Tris(2,4-pentanedionato)-cobalt(III) | COACAC10 (r.t.) | Co3+ | 4 |
| 11 | Bis(η5-cyclopentadienyl)-vanadium(II) | CPNDYV07 (r.t.) | V2+ | 2 |
| 12 | trans-Bis(1-phenylbutane-1,3-dionato)-copper(II) | CUBEAC01 (r.t.) | Cu2+ | 2 |
| 13 | Bis(8-hydroxyquinolinato-N,O)-copper(II) | CUQUIN05 (r.t.) | Cu2+ | 2 |
| 14 | Bis(η6-benzene)chromium | DBENCR11 (r.t.) | Cr0 | 4 |
| 15 | Bis(η5-cyclopentadienyl)cobalt(II) | DCYPCO04 (r.t.) | Co2+ | 2 |
| 16 | Bis(2,2,6,6-tetramethyl-3,5-heptane-dionato)-copper(II) | DERNOD05 (r.t.) | Cu2+ | 2 |
| 17 | Bis(N,N-dimethyldithiocarbamato)-copper(II) | DMTCCU (r.t.) | Cu2+ | 4 |
| 18 | Bis(2,2,6,6-tetramethylheptane-3,5-dionato)-nickel(II) | DPIMNI (r.t.) | Ni2+ | 2 |
| 19 | Bis(2,4-pentanedionato)-nickel(II) | DURHEE (r.t.) | Ni2+ | 2 |
| 20 | Tris(2,4-pentanedionato)-iron(III) | FEACAC03 (r.t.) | Fe3+ | 8 |
| 21 | Bis(η5-cyclopentadienyl)-iron(II) | FEROCE27 (r.t.) | Fe2+ | 2 |
| 22 | Hexacarbonyl-chromium | FOHCOU02 (r.t.) | Cr0 | 4 |
| 23 | Tris(1,1,1,5,5,5-hexafluoro-2,4-pentanedionato)-chromium(III) | IGAGEC (r.t.) | Cr3+ | 4 |
| 24 | Bis(2,2,6-trimethylheptane-3,5-dionato)-copper(II) | IPEZOS (r.t.) | Cu2+ | 2 |
| 25 | Bis(N,N-di-isopropyldithiocarbamato)-nickel complex | IPTCNI10 (r.t.) | Ni2+ | 4 |
| 26 | Bis(2,4-pentanedionato)-cobalt(II) | LIYLIO (190 K) | Co2+ | 2 |
| 27 | Bis(3-methylpentane-2,4-dionato)-copper(II) | MACACU10 (180 K) | Cu2+ | 1 |
| 28 | Bis(η5-cyclopentadienyl)-nickel(II) | NCKLCN01 (101 K) | Ni2+ | 2 |
| 29 | Bis(N,N-diethyldithiocarbamato)-nickel(II) | NIDCAR06 (r.t.) | Ni2+ | 2 |
| 30 | Bis(salicylaldoximato-N,O)-nickel(II) | NISALO01 (r.t.) | Ni2+ | 2 |
| 31 | trans-Bis(1,1,1-trifluoro-2,4-pentanedionato)-copper(II) | QQQBWP03 (173 K) | Cu2+ | 1 |
| 32 | Bis(N,N-dimethyldithiocarbamato)-nickel(II) | TCBMNI (r.t.) | Ni2+ | 1 |
| 33 | Bis(2,4-pentanedionato)-palladium(II) | ACACPD01 (100 K) | Pd2+ | 2 |
| 34 | bis(η5-cyclopentadienyl)-ruthenium(II) | CYCPRU06 (r.t.) | Ru2+ | 4 |
| 35 | Hexacarbonyl-molybdenum | FUBYIK01 (98 K) | Mo0 | 4 |
| 36 | Dihydrido-bis(η5-cyclopentadienyl)-molybdenum(IV) | HCYPMO02 (r.t.) | Mo4+ | 4 |
| 37 | Bis(8-hydroxyquinolinato)-palladium(II) | HQUIPD01 (100 K) | Pd2+ | 2 |
| 38 | Dichloro-bis(η5-cyclopentadienyl)-hafnium(IV) | KOKPEF (r.t.) | Hf4+ | 4 |
| 39 | Hexacarbonyl-tungsten | KOVSOD02 (r.t.) | W0 | 4 |
| 40 | Tris(2,4-pentanedionato)-iridium(III) | QQQCXJ02 (273 K) | Ir3+ | 4 |
| 41 | Tetrakis(1,1,1-trifluoro-2,4-pentanedionato)-hafnium(IV) | REGSAY (243 K) | Hf4+ | 2 |
| 42 | Dihydrido-bis(η5-cyclopentadienyl)tungsten(IV) | REPKIH (200 K) | W4+ | 4 |
| 43 | Bis(η5-cyclopentadienyl)-osmium(II) | SINWER (r.t.) | Os2+ | 4 |
Heats of sublimation
Data for experimental heats of sublimation for entries of the benchmark set were extracted using the procedure explained above. For the 43 entries, 106 individual experimental data points remain of which only 5 were obtained at elevated temperatures between 331 and 549 K. However, even those did not differ by more than 5 (entry 29) to 12 (entry 32) kJ mol−1 from experiments at room temperature. The collection of experimental heats of sublimation plus experimental details are given in the Table S3 (see ESI†).Fig. 2 shows the correlation between the individual data points and their mean values which is excellent. The variation of individual measurements from the mean value for each compound, can be considered as ‘experimental uncertainty’ (see Table S3, ESI†).
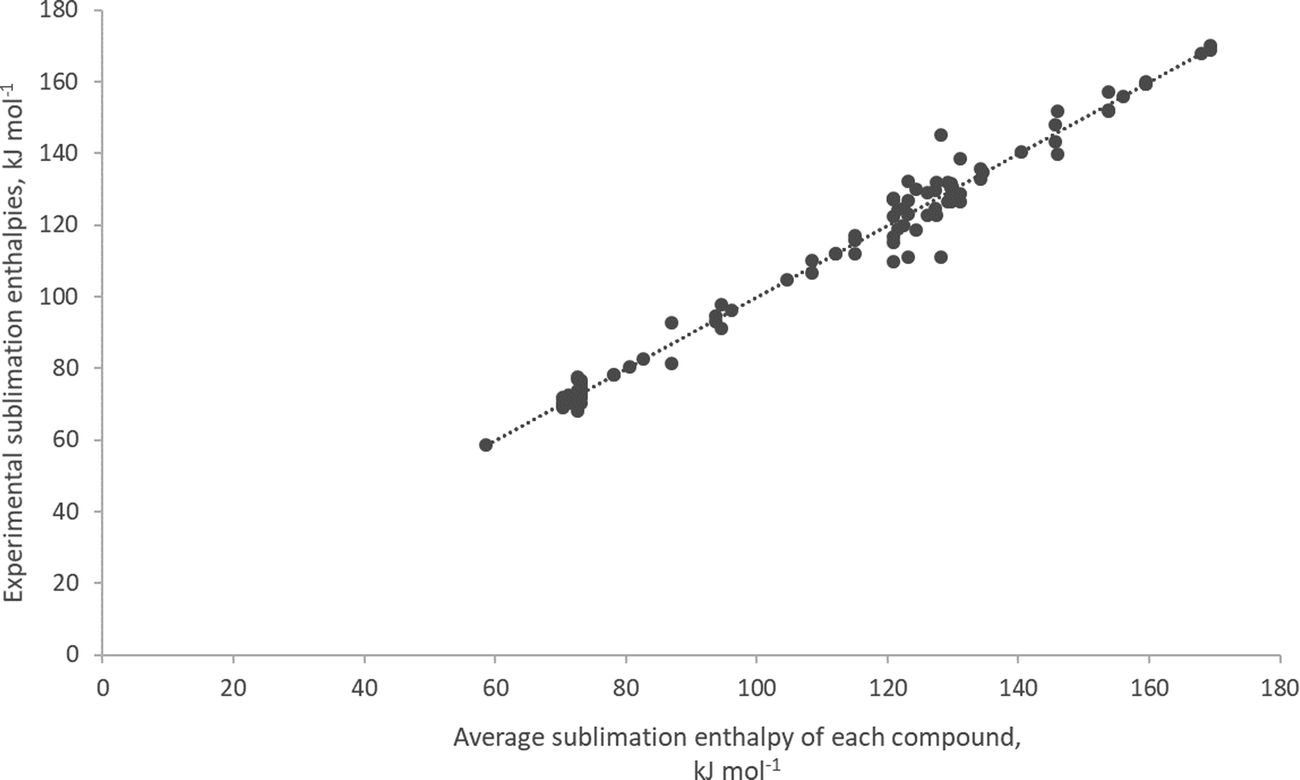 | ||
| Fig. 2 Correlation between individual measurements and mean values for ΔHsubl. A total of 106 experimental sublimation enthalpy data points for 43 organometallic compounds as a function of their mean. The equation of the line is given by: ΔHsub,expt = (0.999 ± 0.013) ΔHsub,mean + (0.001 ± 1.50). Correlation coefficient: r2 = 0.982. | ||
Each experimental technique also has its own source of ‘experimental error’ which is sometimes reported with the data and sometimes only estimated. When there are several data points for a compound, we retain the largest given ‘experimental error’ (see below). Chickos discusses the advantages and reliabilities of experimentally determined heats of sublimation.15 Knudsen or mass effusion and torsion effusion methods and calorimetric method are mentioned to be the most accurate experimental techniques. However, sublimation enthalpies reported in the literature measured calorimetrically that have been standardized only by the Joule effect appear to be lower than similar measurements made on other instruments and techniques, generally by a few per cent.
Comparison of calculated and experimental structures
Structural data of the organometallic crystals are very well reproduced from periodic PBE calculations using the smaller def2-TZVP basis set already (see Table S4, ESI†). The RMSD between experimental and optimized crystal structures is of the order of 0.1–0.16 Å. It increases to 0.3–0.4 Å when there is a rotation of methyl groups or an aromatic ring moiety. The increase of basis set to def2-TZVPP only marginally reduces the RMSD.Comparison of experimental and calculated lattice energies
Fig. 3 shows the results of calculated lattice energies for the XTMC43 set using different basis sets and exchange–correlation functionals. We report the deviation of calculated from experimental data in kJ mol−1. | ||
| Fig. 3 Deviation of calculated lattice energies from experimental values for complexes 1–43. | ||
A negative value is indicative of an overestimation of the cohesive energy and positive values are underestimating the lattice energy.
PBE tends to overestimate the stabilization or organometallic crystals by a Mean Signed Error (MSE) from −9.8 to −8.9 kJ mol−1 (see Table 2). Such an overstabilization of van der Waals complexes can be assigned to originate from the addition of a dispersion correction to an exchange functional that was designed to already mimic some dispersion-like interactions which then leads to a double counting of short-range dispersion.66 The results are hardly affected by an increase in basis set size from TZVP to TZVPP (see Table 2). The Mean Relative Error, MRE, reduces from 9.3 to 8.9%. This was also found for organic molecular crystals.33 It can thus be stated that the medium-sized Ahlrichs’ def2-TZVP basis sets are able to obtain consistent lattice energies for both organic and organometallic complexes.
| PBE-D3(BJ)/def2-TZVP | PBE-D3/def2-TZVPP | M06-L/def2-TZVPP | |
|---|---|---|---|
| a Root mean square error. b Mean signed error. c Mean absolute error. d Mean relative error. | |||
| RMSEa/kJ mol−1 | 14.6 ± 11.2 | 13.9 ± 10.8 | 11.0 ± 10.2 |
| MSEb/kJ mol−1 | −9.8 ± 8.0 | −8.9 ± 10.8 | 4.4 ± 10.2 |
| MAEc/kJ mol−1 | 11.2 ± 9.5 | 10.6 ± 9.1 | 9.1 ± 6.3 |
| MREd/% | 9.3 ± 8.1 | 8.9 ± 7.8 | 8.4 ± 6.4 |
The GGA PBE and the meta-GGA M06-L in combination with the large def2-TZVPP basis set are able to give consistent results with mean signed errors of −8.9 kJ mol−1 and +4.4 kJ mol−1, respectively (see Table 2) given an average experimental lattice energy of 115 kJ mol−1. M06-L underestimates lattice energies for 2/3 of all complexes. The MRE, however, is almost unchanged (8.9 vs. 8.4%). Table 2 gives further statistical analysis and shows that M06-L performs slightly superior to PBE and has a slightly smaller distribution of errors although the mean absolute (MAE) and relative errors (MRE) are close.
The M06-L meta-GGA is parametrized against 22 energetic databases which also include non-covalent interactions and transition metal dimer and transition metal–ligand dissociation energies.42 In a series of 31 semi-conductors, the M06-L functional performs superior for bandgaps67 but slightly less so for lattice constants.68 For the XTMC43 set, there are certain problematic cases with large deviations from experiment that will be discussed below.
Discussion of systematic and non-systematic errors
Fig. 4 shows the results of periodic DFT calculations and systematic errors of experimental heats of sublimation. The grey area in Fig. 4 is the experimental error as given in the original literature, usually between 4 and 8 kJ mol−1 (1–2 kcal mol−1). This seems to be a very optimistic estimate. Only for complex 30 (NISALO01), a significantly larger error estimate of 29 kJ mol−1 is given. This is unexpected since also experimental data for 13, 19, and 37 come from the same source with significantly lower errors (3, 10, and 4 kJ mol−1, respectively; see below). According to calculations, a value between 153 kJ mol−1 (for PBE) and 132 kJ mol−1 (for M06-L) appears plausible in contrast to the given lower experimental value of 117 kJ mol−1. Given the observed overbinding for PBE and underestimation of lattice energies for M06-L these value seem to represent lower and upper boundaries.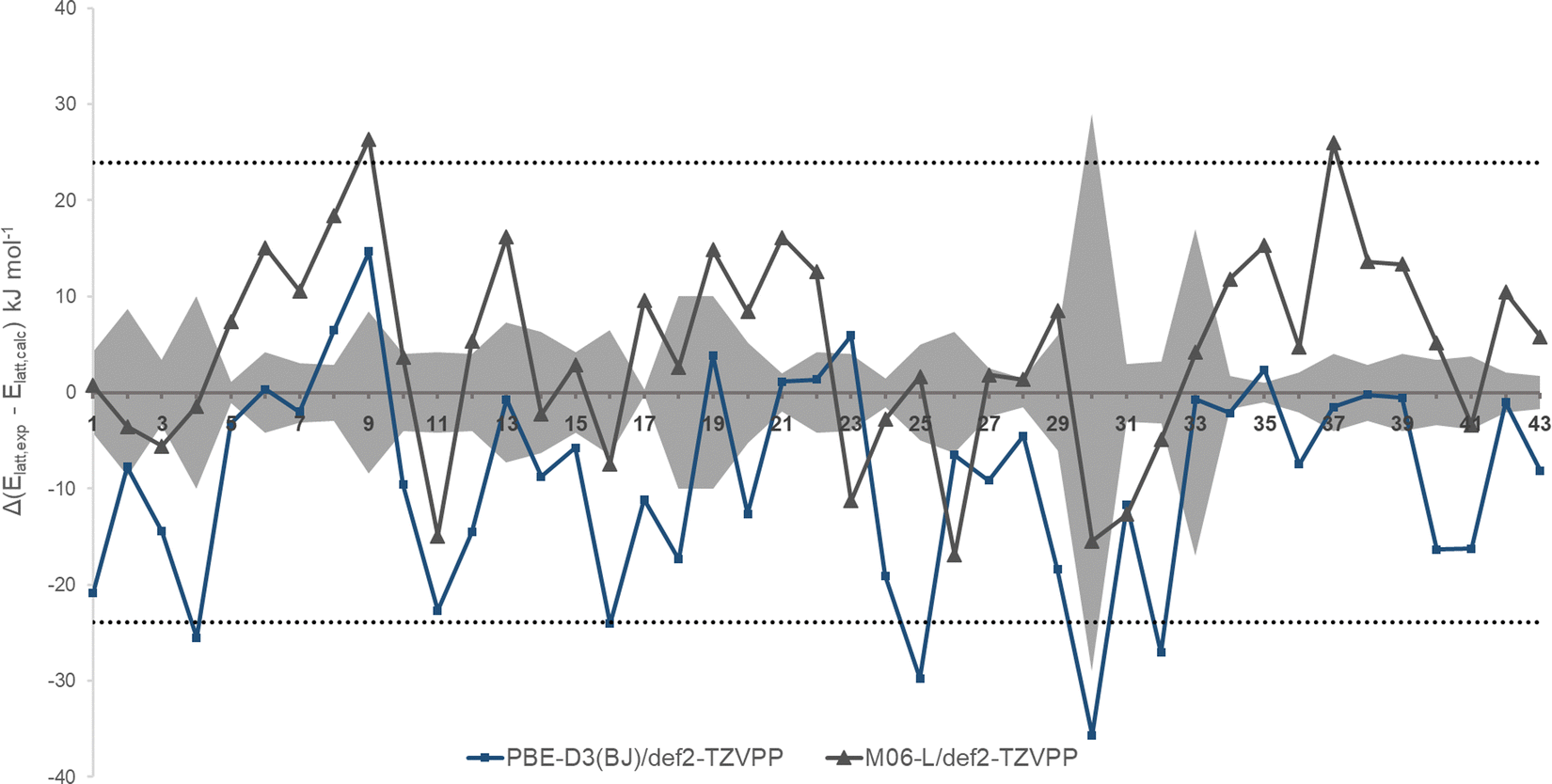 | ||
| Fig. 4 Deviation of lattice energies from periodic DFT calculations from experiment for entries of the XTMC43 set. Grey areas: experimental errors as specified. Dotted lines are the estimates of maximum experimental uncertainties (±23.9 kJ mol−1) from ref. 15. | ||
Analysis of the scattering of individual experimental heats of sublimation for a compound from its mean value gave a standard deviation of ±23.9 kJ mol−1 for a total of 395 measurements on 80 compounds which is three times what would be expected for organic compounds.15 Such a magnitude of experimental uncertainty is beyond any experimental error, temperature effects, use of different experimental techniques and the correlation of the XTMC43 experimental data (see above). However, this estimate of experimental uncertainty serves as an upper limit of error estimate (dotted lines in Fig. 4).
Even if the estimate of uncertainty of ±23.9 kJ mol−1 is too pessimistic for this set of complexes, there are several complexes with even larger deviations from experimental lattice energies.
Complexes 9 (Ti(IV)CpCl3), 13 (Cu(II)(oqu)2), 19 (Ni(II)(acac)2), and 37 (Pd(II)(hoqu)2) show unusual large deviations from experiment. M06-L is underestimating the lattice energies of these complexes by 15–20 kJ mol−1 and more.
Complexes 13 (Cu(II)(oqu)2) and 37 ((Pd(II)(oqu)2) contain the same bis(8-oxyquinolinato) ligand and experimental data come from the same original article.69 Burkinshaw and Mortimer used a Knudsen cell in which volatile complexes that sublime below 70 °C are difficult to measure. Their given experimental uncertainties of 3 and 4 kJ mol−1, respectively, seem to be too optimistic. However, in particular the deviation of the M06-L result for 37 by underestimating the lattice energy by 26 kJ mol−1 is striking. We can assign this deviation to an intrinsic error of this particular mGGA functional. By use of the TPSS-D3(BJ) meta-GGA, a smaller deviation of 14.5 kJ mol−1 (a reduction in deviation by 11 kJ mol−1) is obtained which is consistent with the PBE results. Also for complex 13, the deviation from experiment reduces from 16.1 kJ mol−1 (for M06-L) to −13.3 kJ mol−1 (for TPSS-D3(BJ)).
For compound 19, Ni(II)(acac)2, the TPSS meta-GGA significantly reduces the deviation from experiment from 14.9 kJ mol−1 (M06-L) to 3.1 kJ mol−1 (TPSS).
Apparently, the explicit consideration of atomic dispersion coefficient in DFT gives results superior to the implicit dispersion consideration during parameter fitting with M06-L for these complexes (Fig. 5).
 | ||
| Fig. 5 Investigation of the effect of choice of functional on calculated lattice energies for complexes 9, 13, 19, and 37 which showed large deviations from experiment in Fig. 4. | ||
For complex 9 (CpTiCl3), both PBE and M06-L give underestimated lattice energies with deviations of 15 and 26 kJ mol−1 which is unusually large for this set of compounds. TPSS-D3(BJ) gives a deviation of 8 kJ mol−1 from experiment. The given experimental heat of sublimation of 105 ± 8.4 kJ mol−1 in the original article70 is an estimate only based on values of compounds of similar composition and structure (here CpZrCl3). It is identical to that of the zirconium analogue compound.
Whereas CpTiCl3 adopts a structure similar to that of tetrahedral TiCl4 in which one chlorine atom has been replaced by the (η5-C5H5) ligand, ZrCl4 is a μ,μ′-dichloro-bridged polymer (Cl2(ZrCl2)Cl2) and this is also reflected in the CpZrCl3 crystal structure.71 The extra bridging μ,μ′-dichloro-zirconium interactions suggest that the heat of sublimation will be lower for CpTiCl3 compared to CpZrCl3. In our calculations for CpZrCl3, the calculated lattice energies are in good agreement with experiment (with a deviation of +6 kJ mol−1 for PBE/def2-TZVPP and +13 kJ mol−1 with M06-L/def2-TZVPP). This shows that the lattice energy of 110 kJ mol−1 for CpTiCl3 has to be corrected and will not identical to that of CpZrCl3. DFT calculations suggest a lower value between 95 (PBE) and 83 kJ mol−1 (M06-L).
Overall correlation and distribution of values
Fig. 6 displays the overall distribution of data points for experimental, PBE and M06-L lattice energies. The beanplot allows the comparison of the four different lattice energy values, their distribution as a density shape and their mean values. | ||
| Fig. 6 Beanplot of lattice energy data. Black lines show the means; white lines represent individual data points; polygons represent the estimated density of the data. Experimental lattice energy data are compared with DFT calculations. | ||
From Fig. 6 it becomes apparent, that the PBE-D3(BJ) results are slightly overestimating mean lattice energies (PBE-D3(BJ)/def2-TZVP 138.2 kJ mol−1 and PBE-D3(BJ)/def2-TZVPP 135.8 kJ mol−1) compared to experimental lattice energies (127.3 kJ mol−1). For M06-L, a mean of 128.2 kJ mol−1 is obtained. This is also reflected in the density distribution of data points and the respective quartile values (see Table S6 for more details, ESI†).
Conclusions
Atom-centred Gaussian-type basis functions in combination with GGA or meta-GGA exchange–correlation functions are able to provide useful calculated lattice energies not only for organic molecular crystals72 but also for organometallic crystals. For the X23 benchmark set of molecular organic crystals, the PBE functional with dispersion corrections had a mean relative error (MRE) of 9%73 which can also be obtained for transition metal complexes. The non-covalent interactions between compounds of the X23 and XTMC43 are molecular (van der Waals, electrostatic and hydrogen bonding) in nature and relatively weak.Overestimated delocalization in current dispersion-corrected DFT methods may lead to erroneous results in particular for molecular crystals of acids and bases.66,74–76 The development of many-body dispersion corrections for proper treatment of long-range dispersive interactions will improve the description of interactions in molecular crystals.
The demonstrated controllable systematic error with upper and lower boundaries of results for PBE and M06-L thus allows to extend investigations to more diverse sets of crystals. It can also be used to systematically assess the reliability of reported experimental heats of sublimation for a particular ligand family along the periodic table. These results will improve the consistency of reported data and can form the basis for more approximate descriptions of cohesive energies in organometallic crystals. Distinctions between non-covalent and more complex interactions is often quite intuitive in organic chemistry, however, this may not be the case in transition metal chemistry, and so caution is also necessary in this context.38
Calibration and reproducibility of experiments are fundamental requirements for every sublimation enthalpy measurement and likewise computational approach. Unlike other thermochemical measurements, uncertainties in sublimation enthalpies can be large, often several kJ mol−1 or more. The compendium of Chickos and Acree,14 that contains experimentally determined sublimation enthalpies for organic and organometallic compounds, also discusses challenges to obtain accurate values. For heats of sublimation >100 kJ mol−1, when the compounds usually have a low vapor pressure, experiments have to be performed at elevated temperature for which there are no standards for the calibration of instruments under these conditions available. The uncertainty between mean and individual points of experimental sublimation enthalpies of organometallic compounds is particularly high and almost three times higher than for organic molecules.15 For the same reason, a set of calibration and assessment of the accuracy of current computational approaches in calculating lattice energies and heats of sublimation is required. The training set ought to consider variations in central metal atoms, oxidation and spin states and ligand coordination and should encompass several independent computational methods. Likewise, the more advanced incorporation of thermodynamic and thermal corrections to experimental heats of sublimation will assist the design of an accurate database of curated data.
Computational approaches are nowadays efficient and accurate enough to process a large number of structures or complexes in a reasonable amount of time. The computational discovery of novel transition metal complexes and catalysts from high throughput screening and machine learning opens the field of a data driven approach to the design of catalysts.77 The XTMC43 set of compounds serves as a first investigation that allows making such a statement based on quantitative results.
Author contributions
MS conceptualized the study. JEDZM performed the calculations with support from MS. JEDZM and MS analyzed the results. The manuscript was written through contributions of both authors. Both authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the Max Planck Society for the Advancement of Science for financial support. This work was in part supported by the EU COST (Cooperation in Science and Technology) Action CM1305 ‘ECOSTBio’. This work is part of the Research Initiative “SmartProSys: Intelligent Process Systems for the Sustainable Production of Chemicals” funded by the Ministry for Science, Energy, Climate Protection and the Environment of the State of Saxony-Anhalt, Germany. JEZDM would like to thank the Mexican Consejo Nacional de Ciencia y Tecnología (CONACyT) in collaboration with the Deutscher Akademischer Austauschdienst (DAAD) for the founding of his studies through the awarded postgraduate scholarship. Open Access funding provided by the Max Planck Society.References
- A. Behr and P. Neubert, Applied Homogeneous Catalysis, Wiley-VCH, Weinheim, 2012 Search PubMed.
- M. Beller and C. Bolm, Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- A. A. Tedstone, D. J. Lewis and P. O’Brien, Chem. Mater., 2016, 28, 1965–1974 CrossRef CAS.
- M. L. Crawley and B. M. Trost, Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, John Wiley & Sons, Hoboken, New Jersey, 2012 Search PubMed.
- E. C. Constable and C. E. Housecroft, Chem. Soc. Rev., 2013, 42, 1429–1439 RSC.
- G. B. Kaufmann, Alfred Werner – Founder of Coordination Chemistry, Spinger, Berlin, 1966 Search PubMed.
- A. Domenicano and I. Hargittai, Accurate Molecular Structures: Their Determination and Importance, Oxford University Press, Chester, 1992 Search PubMed.
- D. Braga and F. Grepioni, Chem. Commun., 1996, 571–578 RSC.
- D. J. W. Grant and T. Higuchi, Solubility Behavior of Organic Compounds, John Wiley and Sons, New York, 1990 Search PubMed.
- D. S. Palmer, A. Llinàs, I. Morao, G. M. Day, J. M. Goodman, R. C. Glen and J. B. O. Mitchell, Mol. Pharmaceutics, 2008, 5, 266–279 CrossRef CAS PubMed.
- J. L. McDonagh, N. Nath, L. De Ferrari, T. van Mourik and J. B. O. Mitchell, J. Chem. Inf. Model., 2014, 54, 844–856 CrossRef CAS PubMed.
- D. S. Palmer, J. L. McDonagh, J. B. O. Mitchell, T. van Mourik and M. V. Fedorov, J. Chem. Theory Comput., 2012, 8, 3322–3337 CrossRef CAS PubMed.
- J. L. McDonagh, D. S. Palmer, T. V. Mourik and J. B. O. Mitchell, J. Chem. Inf. Model., 2016, 56, 2162–2179 CrossRef CAS PubMed.
- J. S. Chickos and W. Acree Jr., J. Phys. Chem. Ref. Data, 2002, 31, 537–698 CrossRef CAS.
- J. S. Chickos, Netsu Sokutei, 2003, 30, 116–124 CAS.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Chem. Rev., 2016, 116, 5105–5154 CrossRef CAS PubMed.
- A. Otero-de-la-Roza and E. R. Johnson, J. Chem. Phys., 2012, 137, 054103 CrossRef CAS PubMed.
- G. A. Dolgonos, J. Hoja and A. D. Boese, Phys. Chem. Chem. Phys., 2019, 21, 24333–24344 RSC.
- A. M. Reilly and A. Tkatchenko, J. Phys. Chem. Lett., 2013, 4, 1028–1033 CrossRef CAS PubMed.
- J. Moellmann and S. Grimme, J. Phys. Chem. C, 2014, 118, 7615–7621 CrossRef CAS.
- J. G. Brandenburg and S. Grimme, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 502–513 CrossRef CAS PubMed.
- J. G. Brandenburg, T. Maas and S. Grimme, J. Chem. Phys., 2015, 142, 124104 CrossRef PubMed.
- P.-O. Norrby and P. Brandt, Coord. Chem. Rev., 2001, 212, 79–109 CrossRef CAS.
- R. Dovesi, B. Civalleri, R. Orlando, C. Roetti and V. R. Saunders, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz, R. Larter and T. R. Cundari, John Wiley & Sons, Hoboken, New Jersey, 2005, vol. 21 Search PubMed.
- P. J. Hasnip, K. Refson, M. I. J. Probert, J. R. Yates, S. J. Clark and C. J. Pickard, Philos. Trans. R. Soc., A, 2014, 372, 20130270 CrossRef PubMed.
- P. Blaha, K. Schwarz, P. Sorantin and S. B. Trickey, Comput. Phys. Commun., 1990, 59, 399–415 CrossRef CAS.
- M. D. Towler, A. Zupan and M. Causà, Comput. Phys. Commun., 1996, 98, 181–205 CrossRef CAS.
- A. M. Burow, M. Sierka and F. Mohamed, J. Chem. Phys., 2009, 131, 214101 CrossRef PubMed.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–488 CrossRef CAS.
- G. Ulian, S. Tosoni and G. Valdrè, J. Chem. Phys., 2013, 139, 204101 CrossRef PubMed.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D'Arco, Y. Noël, M. Causà, M. Rérat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287–1317 CrossRef CAS.
- K. N. Kudin and G. E. Scuseria, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 16440–16453 CrossRef CAS.
- H. K. Buchholz and M. Stein, J. Comput. Chem., 2018, 39, 1335–1343 CrossRef CAS PubMed.
- A. Gavezzotti, Z. Kristallogr. - Cryst. Mater., 2005, 220, 499–510 CrossRef CAS.
- A. Gavezzotti, J. Phys. Chem. B, 2002, 106, 4145–4154 CrossRef CAS.
- A. Gavezzotti, CrystEngComm, 2008, 10, 367 Search PubMed.
- L. Maschio, B. Civalleri, P. Ugliengo and A. Gavezzotti, J. Phys. Chem. A, 2011, 115, 11179–11186 CrossRef CAS PubMed.
- A. G. P. Maloney, P. A. Wood and S. Parsons, CrystEngComm, 2015, 17, 9300–9310 RSC.
- G. Pilcher and H. A. Skinner, in The Chemistry of the Metal Carbon Bond, ed. F. R. Hartley and S. Patai, John Wiley & Sons, Chichester, 1982, ch. 2, pp. 43–90 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- C. Steffen, K. Thomas, U. Huniar, A. Hellweg, O. Rubner and A. Schroer, J. Comput. Chem., 2010, 31, 2967–2970 CAS.
- F. Furche, R. Ahlrichs, C. Hattig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- Turbomole and GmbH, 2017.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2005, 123, 024101 CrossRef PubMed.
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2006, 124, 174104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- J. J. Determan, K. Poole, G. Scalmani, M. J. Frisch, B. G. Janesko and A. K. Wilson, J. Chem. Theory Comput., 2017, 13, 4907–4913 CrossRef CAS PubMed.
- R. Łazarski, A. M. Burow, L. Grajciar and M. Sierka, J. Comput. Chem., 2016, 37, 2518–2526 CrossRef PubMed.
- R. Łazarski, A. M. Burow and M. Sierka, J. Chem. Theory Comput., 2015, 11, 3029–3041 CrossRef PubMed.
- L. Grajciar, J. Comput. Chem., 2015, 36, 1521–1535 CrossRef CAS PubMed.
- J. A. Weatherby, A. F. Rumson, A. J. A. Price, A. O. D. L. Roza and E. R. Johnson, J. Chem. Phys., 2022, 156, 114108 CrossRef CAS PubMed.
- J. G. Brandenburg and S. Grimme, Top. Curr. Chem., 2014, 345, 1–23 CAS.
- A. M. Reilly and A. Tkatchenko, J. Chem. Phys., 2013, 139, 024705 CrossRef PubMed.
- S. H. Neau, S. V. Bhandaka and E. W. Hellmuth, Pharm. Res., 1997, 14, 601–605 CrossRef CAS PubMed.
- J. S. Chickos, S. Hosseini, D. G. Hesse and J. F. Liebman, Struct. Chem., 1993, 4, 271–278 CrossRef CAS.
- F. A. Cotton, C. E. Rice and G. W. Rice, Inorg. Chim. Acta, 1977, 24, 231–234 CrossRef CAS.
- M. G. Vinum, L. Voigt, S. H. Hansen, C. Bell, K. M. Clark, R. W. Larsen and K. S. Pedersen, Chem. Sci., 2020, 11, 8267–8272 RSC.
- A. J. A. Price, K. R. Bryenton and E. R. Johnson, J. Chem. Phys., 2021, 154, 230902 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2009, 130, 074103 CrossRef PubMed.
- R. Peverati and D. G. Truhlar, J. Chem. Phys., 2012, 136, 134704 CrossRef PubMed.
- P. M. Burkinshaw and C. T. Mortimer, J. Chem. Soc., Dalton Trans., 1984, 75–77 RSC.
- V. I. Tel'noi and I. B. Rabinovich, Uspekhi Khimii, 1977, 46, 1337–1367 Search PubMed.
- L. M. Engelhardt, R. I. Papasergio, C. L. Raston and A. H. White, Organometallics, 1984, 3, 18–20 CrossRef CAS.
- M. Stein and M. Heimsaat, Crystals, 2019, 9, 665 CrossRef CAS.
- J. G. Brandenburg and S. Grimme, in Prediction and Calculation of Crystal Structures: Methods and Applications, ed. S. Atahan-Evrenk and A. Aspuru-Guzik, Springer International Publishing, Cham, 2014, pp. 1–23 DOI:10.1007/128_2013_488.
- D. Geatches, I. Rosbottom, R. L. M. Robinson, P. Byrne, P. Hasnip, M. I. J. Probert, D. Jochym, A. Maloney and K. J. Roberts, J. Chem. Phys., 2019, 151, 044106 CrossRef PubMed.
- L. M. LeBlanc, S. G. Dale, C. R. Taylor, A. D. Becke, G. M. Day and E. R. Johnson, Angew. Chem., Int. Ed., 2018, 57, 14906–14910 CrossRef CAS PubMed.
- Y. S. Al-Hamdani and A. Tkatchenko, J. Chem. Phys., 2019, 150, 010901 CrossRef PubMed.
- A. Nandy, C. Duan, M. G. Taylor, F. Liu, A. H. Steeves and H. J. Kulik, Chem. Rev., 2021, 121, 9927–10000 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Effect of choice of crystal structures on calculated lattice energies; collection of experimental enthalpies of sublimation; RMSD between experimental and calculated crystal structures; table of experimental and calculated lattice energies; statistical analysis of boxplots. See DOI: https://doi.org/10.1039/d2cp04160j |
| This journal is © the Owner Societies 2022 |