
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–water covalency in the photo-aquated ferrocyanide complex as seen by multi-edge picosecond X-ray absorption†
Vinícius
Vaz da Cruz‡
 *a,
Eric J.
Mascarenhas‡
*ab,
Robby
Büchner
ab,
Raphael M.
Jay§
b,
Mattis
Fondell
a,
Sebastian
Eckert
a and
Alexander
Föhlisch
ab
*a,
Eric J.
Mascarenhas‡
*ab,
Robby
Büchner
ab,
Raphael M.
Jay§
b,
Mattis
Fondell
a,
Sebastian
Eckert
a and
Alexander
Föhlisch
ab
aHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Institute for Methods and Instrumentation for Synchrotron Radiation Research, 12489 Berlin, Germany. E-mail: vinicius.vaz_da_cruz@helmholtz-berlin.de; eric.mascarenhas@helmholtz-berlin.de
bUniversität Potsdam, Institut für Physik und Astronomie, 14476 Potsdam, Germany
First published on 25th October 2022
Abstract
In this work, we investigate the photo-aquation reaction of the ferrocyanide anion with multi-edge picosecond soft X-ray spectroscopy. Combining the information of the iron L-edge with nitrogen and oxygen K-edges, we carry out a complete characterization of the bonding channels in the [Fe(CN)5(H2O)]3− photo-product. We observe clear spectral signatures of covalent bonding between water and the metal, reflecting the mixing of the Fe dz2 orbital with the 3a1 and 4a1 orbitals of H2O. Additional fingerprints related to the symmetry reduction and the resulting loss in orbital degeneracy are also reported. The implications of the elucidated fingerprints in the context of future ultra-fast experiments are also discussed.
Introduction
Triggering the breaking of chemical bonds with light constitutes one of the most basic photo-chemical transformations in transition metal complexes.1–3 In particular, in the cases where the broken bond may be replaced by a new one, which could not otherwise be formed on the ground state potential energy surface. Such light-driven ligand substitution reactions are ubiquitous in coordination photo-chemistry and draw much scientific interest for their potential technical applications. Controlling these reactions, however, remains a major challenge as they are often intricate, involving complicated electronic and nuclear relaxation pathways.4In this context, the photo-reactivity of the model octahedral complex [Fe(CN)6]4− in aqueous solutions has been a major testing ground for several fundamental concepts in coordination chemistry.5 In the ground state, it has an octahedral geometry, where the Fe(II) central atom is surrounded by 6 strong-field and backbonding cyanide ligands. In this highly symmetric Oh ligand field, the d-orbitals are split into eg and t2g orthogonal subsets. The bonding in this system is well described by the standard σ-donation, π-donation and -backdonation channels.6 Solute–solvent interactions, specially in aqueous solutions, also play a crucial role in the properties and stability of this species, much owing to its high negative charge.7–10
Numerous pioneering investigations were devoted to the photo-chemistry of the aqueous ferrocyanide anion,11–16 in which flash-photolysis was used to establish the two wavelength-dependent photo-chemical transformations in this system: (I) photo-oxidation accompanied by solvated electron formation and (II) photo-aquation. The latter process is a fundamental ligand-substitution reaction in which a cyanide ligand is replaced by a water molecule to form [Fe(CN)5(H2O)]3−, as schematized in Fig. 1. The ultra-fast photo-reaction mechanisms leading to the photoaquated product [Fe(CN)5(H2O)]3− were addressed in recent time-resolved studies with different spectroscopic techniques.17–19 However, many details, e.g. the picosecond dynamics in the pentacoordinated intermediate, remain under debate.
 | ||
| Fig. 1 Photo-aquation of ferrous hexacyanide induced by excitation at λ > 300 nm. | ||
Surprisingly, little attention has been paid to the chemical and electronic properties of the [Fe(CN)5(H2O)]3− product itself. This peculiar species, which persists in solution for minutes once it is formed,11,12,16 is fundamentally interesting for numerous reasons, among which we can mention: its symmetry lowering from the Oh point group to a local C4v symmetry at the metal, the replacement of a strong-field ligand for a weak field one, and the formation of a metal–water chemical bond. The ability to, even if transiently, break the strong Fe–CN bond and replace it with Fe–OH2 is interesting from a photo-catalytic point of view. Hence, the so far uncovered details of the electronic changes upon replacement of the cyanide ligand by a water molecule require investigation.
In this study, we focus on the elucidation of the electronic properties of the aquated photo-product. To that end, transient soft X-ray absorption spectroscopy, with picosecond time resolution, is used as an ideal element-specific probe. A complete picture of chemical bonding can be obtained by investigating complementary X-ray absorption edges as demonstrated before for the system under study.20,21 The Fe L-edge probes transition from the metal 2p core-orbitals into the valence orbitals. These transitions monitor the projection of the unoccupied levels around the metal, being highly sensitive to the 3d orbital occupation, as well as the back-bonding character.22–26 On the other hand, N K-edge and O K-edge probes transition from the N 1s level and O 1s level into the valence orbitals, respectively, monitoring the unoccupied levels around the ligands.27,28 Thereby, we are able to track the changes in the electronic structure upon the replacement of a cyanide ligand with a water molecule with site-selectivity. The information distilled from these spectra is combined into a detailed description of the bonding channels in the [Fe(CN)5(H2O)]3− photo-product. Special attention is paid to the chemical bonding between water and iron, which our measurements demonstrate to be significantly covalent. The ligand field effects associated with the reduced symmetry of this complex are also discussed. Lastly, the implications of our results for the mechanism of the photo-reaction are discussed based on the derived properties of the product and predicted soft X-ray spectral signatures of intermediate species involved in the photo-aquation pathway.
Methods
Experimental
K4[Fe(CN)6]·3H2O was purchased from Sigma Aldrich and used without further purification. The salt was dissolved in deionized water to a concentration of 150 mM. The experiment was performed using the transmission NEXAFS end-station at the UE52_SGM beamline29 of the synchrotron BESSY II. The operational details of the station were previously reported.30 In short, the sample was delivered into the experimental vacuum chamber using a flat-jet system at flow rates between 1.5 and 3 mL min−1 and excited with optical pulses at a wavelength of 343 nm and a repetition rate of 208 kHz. The laser beam was focused to a spot size on the order of 80 × 80 μm2 and the pulse energy was adjusted to ∼10 μJ. The pulse arrival time was synchronized with the arrival time of X-ray pulses originating from the BESSY II hybrid bunch to enable an optical pump X-ray absorption probe experimental scheme. The X-ray signal transmitted through the flat-jet was detected using an avalanche photo diode, amplified at 20 dB and recorded using a UHFLI boxcar averager (Zurich Instruments). The transmitted signal through the sample in its ground state is recorded 800 ns prior to the probe pulse used to detect the photo-induced dynamics. This yields a differential measurement of the transient absorption cross-section. The static spectra were recorded using the average flux from all bunches in the storage ring. Supporting measurements were performed in the EDAX experiment at the beamline UE49_SGM.The energy scale of the spectra at the Fe L-edge and N K-edge was calibrated to match previously reported spectra for [Fe(CN)6]4−,20–22 while the O K-edge was shifted so that the maximum of the pre-edge feature lies at 535 eV, according to ref. 31.
Computational
All quantum chemical simulations were performed using the ORCA32 package at version 5.0.1. The molecular structures were optimized at the DFT level using the B3LYP functional, the def2-TZVP(-f)33 basis set and the def2/J34 auxiliary basis set. The D3BJ35,36 correction was considered. Two geometries were computed for [Fe(CN)5(H2O)]3−, one without symmetry restrictions and an idealized C2v geometry. Since the spectral calculations were not very sensitive to this choice, the results in the article are included for the C2v geometry. All simulations used the conductor-like polarizable continuum model37 (CPCM) to account for the effect of the water solvent.The core-level spectra were computed at the time-dependent density functional theory (TD-DFT) level by restricting the donor orbital excitation window including only the core orbitals of interest. For N K-edge simulations, all N 1s orbitals were included in the donor space. For the O K-edge XAS, only excitations from the oxygen 1s orbital were considered. For the Fe L-edge XAS, only excitations from the three 2p orbitals were considered. For simulations at the Fe L-edge, the effect of spin–orbit coupling was accounted for using the spin–orbit mean field approach implemented for a singlet Kohn–Sham reference.38 The only exception is the predicted spectra of the pentacoordinate intermediate species. In that case, spin–orbit interactions had to be neglected; however, for the system under study, the results are still expected to be qualitatively correct, as demonstrated in the ESI,† and also demonstrated for a related iron complex investigated by Hua et al.39
In order to robustly assign the transient features at the O K-edge, we simulated the pre-edge peak of liquid water. This was done based on snapshots obtained from classical molecular dynamics (MD) simulations performed with the Gromacs40 package using the SPC/Fw force field.41 The details of these simulations are available in the ESI.† The spectral calculations were performed by picking a single water molecule from each snapshot and selecting all neighbouring molecules up to the second solvation shell. This threshold was defined as the second minimum in the computed oxygen–oxygen radial distribution function. Then, a CPCM layer was applied around this reduced cluster. The XAS spectrum was then computed considering only excitations from the O 1s orbital of the central water molecule. The liquid spectrum was then computed as the average of 100 sampled spectra.
The simulated spectra were shifted for alignment with the experimental data by 13.3 eV, 11.9 eV and 15.0 eV at the iron, nitrogen and oxygen edge, respectively. The simulated transition amplitudes were broadened with a Voigt profile using Lorentzian and Gaussian broadenings of 0.2 eV and 0.7 eV for the Fe L-edge, 0.12 eV and 0.7 eV for the N K-edge and 0.16 eV and 0.5 eV for the O K-edge based on best agreement with the experimental spectra to account for a life-time, vibrational, solvent and instrumental broadening effects.
Results and discussion
Ground state measurements
We shall briefly recollect the interpretation of the ground state X-ray absorption spectra of [Fe(CN)6]4−. There is a general consensus in the literature regarding the signatures at the Fe L-edge and N K-edge. The measurements are shown in Fig. 2 along with an orbital diagram illustrating the transitions. At the Fe L-edge, we are sensitive to the occupation of the Fe 3d orbitals as well as their covalent mixing with ligand orbitals. As shown in Fig. 2a, we observe two similar bands, split by about 13 eV due to the strong spin–orbit coupling of the 2p core holes, each band consists of two main features. Concerning the L3 edge, Hocking et al.22 attributed the lower energy sharp feature to transitions to the metal-centered eg orbitals, while the higher energy intense satellite is attributed to unoccupied t2g (π*) orbitals, the intensity of this feature is proportional to the magnitude of ligand-to-metal π back-bonding. The intensity of the second feature is also related to strong configuration interaction with the eg states. This assignment has been supported by several later studies, including the studies by Engel et al.42 and Kunnus et al.,20 both of which employed multi-configurational ab initio calculations. Our TD-DFT calculations also support this assignment of the Fe L-edge XAS of [Fe(CN)6]4−. | ||
| Fig. 2 Information content of the investigated X-ray absorption edges. XAS of aqueous [Fe(CN)6]4− at the Fe L-edge (a), N K-edge (b) and O K-edge (c). (d) Qualitative orbital diagram displaying the relevant transitions observed in the spectra for the Fe L-edge and N K-edge. The O K-edge spectrum (c) displays only the pre-edge feature of liquid water, related to O1s → 4a1 transitions. | ||
The N K-edge XAS, presented in Fig. 2b, is dominated by a single intense resonance. This feature is associated with transitions from the N 1s orbital located at a cyanide ligand into the manifold of unoccupied π* orbitals. Most of the intensity is associated not only with transitions to ligand-centered unoccupied orbitals but also with contributions from the back-bonding t2g (π*) orbitals, also seen at the Fe L-edge.
Lastly, Fig. 2(c) shows the O K-edge spectrum of aqueous [Fe(CN)6]4−. Evidently, this spectrum nearly perfectly coincides with that of bulk liquid water,31 since the complex itself contains no oxygen. The spectrum in the energy range beyond the 4a1 transition is saturated due to the rather high sample thicknesses essential for the transient measurements discussed in the following. The spectrum shows the well-characterized pre-edge feature of water, which is related to the excitonic O 1s → 4a1 transition, under heavy influence from the hydrogen bond network of liquid water. This spectrum is important because it allows us to directly investigate the nature of the chemical bond between a water molecule and the penta-coordinated [Fe(CN)5]3−.
Time-resolved measurements
The transient Fe L3-, N K- and O K-edge difference spectra recorded subsequent to excitation of [Fe(CN)6]4− (aq.) at a wavelength of 343 nm and a pump–probe delay of 100 ps, as well as the temporal evolution of the transient signal at selected photon energies, are presented in Fig. 3. We compare the experimental results with TD-DFT spectrum simulations of [Fe(CN)6]4− and [Fe(CN)5(H2O)]3−. The simulated difference spectra for the two species agree well with the experimentally detected transient signatures, further confirming that the X-ray absorption spectra at all three edges are sensitive to the photo-aquation reaction. | ||
| Fig. 3 Picosecond time-resolved transient absorption changes induced by photoaquation [Fe(CN)6]4− → [Fe(CN)5(H2O)]3− at 343 nm excitation. Experimental difference spectra at a pump–probe delay of 100 ps at the iron L3- (a), the nitrogen K- (c) and the oxygen K-edge (e). (b, d and f) Theoretical predictions of transient absorption changes induced by the formation of the photoaquated species at the respective edges. Ground state spectra of [Fe(CN)6]4− and the water solvent at the oxygen edge are shown as shaded curves for comparison. (g–i) Temporal evolution of the transient absorption changes at selected energies, marked in (a, c and e) reflecting the formation of the long-lived [Fe(CN)5(H2O)]3− species. | ||
Starting with the Fe L-edge, shown in Fig. 3a and b, we observe an increase in intensity in the region before the eg resonance of the ground state absorption spectrum, this transient feature is followed by a depletion around the aforementioned resonance, a return to the baseline, and lastly a depletion at the t2g (π*) resonance. Similarly, at the N K-edge (Fig. 3c and d), an absorption increase is seen just below the π* resonance, this feature is followed by a depletion around the main resonance, and a subsequent absorption increase. Lastly, at the O K-edge (Fig. 3e and f), we can robustly assign an absorption increase located at the slope of the 4a1 pre-edge of liquid water. It should be noted that beyond 535 eV, this spectrum becomes affected by saturation effects due to the thickness of the liquid-jet. The temporal evolution of the transient absorption spectra (Fig. 3g–i), within our experimental conditions, has a trivial step-function behavior, which directly reflects the formation of the stable [Fe(CN)5(H2O)]3− complex, which remains in solution on a time scale of minutes.12,16
The observed spectroscopic signatures encode the changes in the electronic structure of the complex upon the replacement of a cyanide ligand by a water molecule. To shed light on these changes, let us examine closer the transient signatures in the framework of the spectral simulations in combination with the molecular orbital diagrams and orbital illustrations of [Fe(CN)6]4− and [Fe(CN)5(H2O)]3− in Fig. 4. Although it can be expected that the symmetry lowering will introduce new spectroscopic features, the observed signatures carry deeper insight. The pre-edge features seen in all of the transient spectra are directly related to Fe-OH2 covalency. In more detail, the mixing of the metal dz2 orbital (formerly eg) with the 4a1 and 3a1 water orbitals, which are higher in energy than the CN− σ orbitals, lowers the energy of the dz2 orbital. Meanwhile, the dx2–y2 orbital does not mix with the water orbitals, shifting down in energy only slightly. The drastic energy reduction of the dz2 orbital and the only minor changes of the dx2–y2 orbital are reflected in the red-shift and the splitting of the former eg absorption line in the iron L3-edge spectrum in Fig. 4a into two components. These effects can also be framed under the expected effect that replacing a strong field CN− by the weak field H2O will lead to a reduction in ligand-field splitting. This reduced ligand field splitting is also supported by the red shift of the visible absorption band peaking at ∼330 nm in [Fe(CN)6]4− to ∼420 nm for the photoaquated [Fe(CN)5H2O]3− species.18
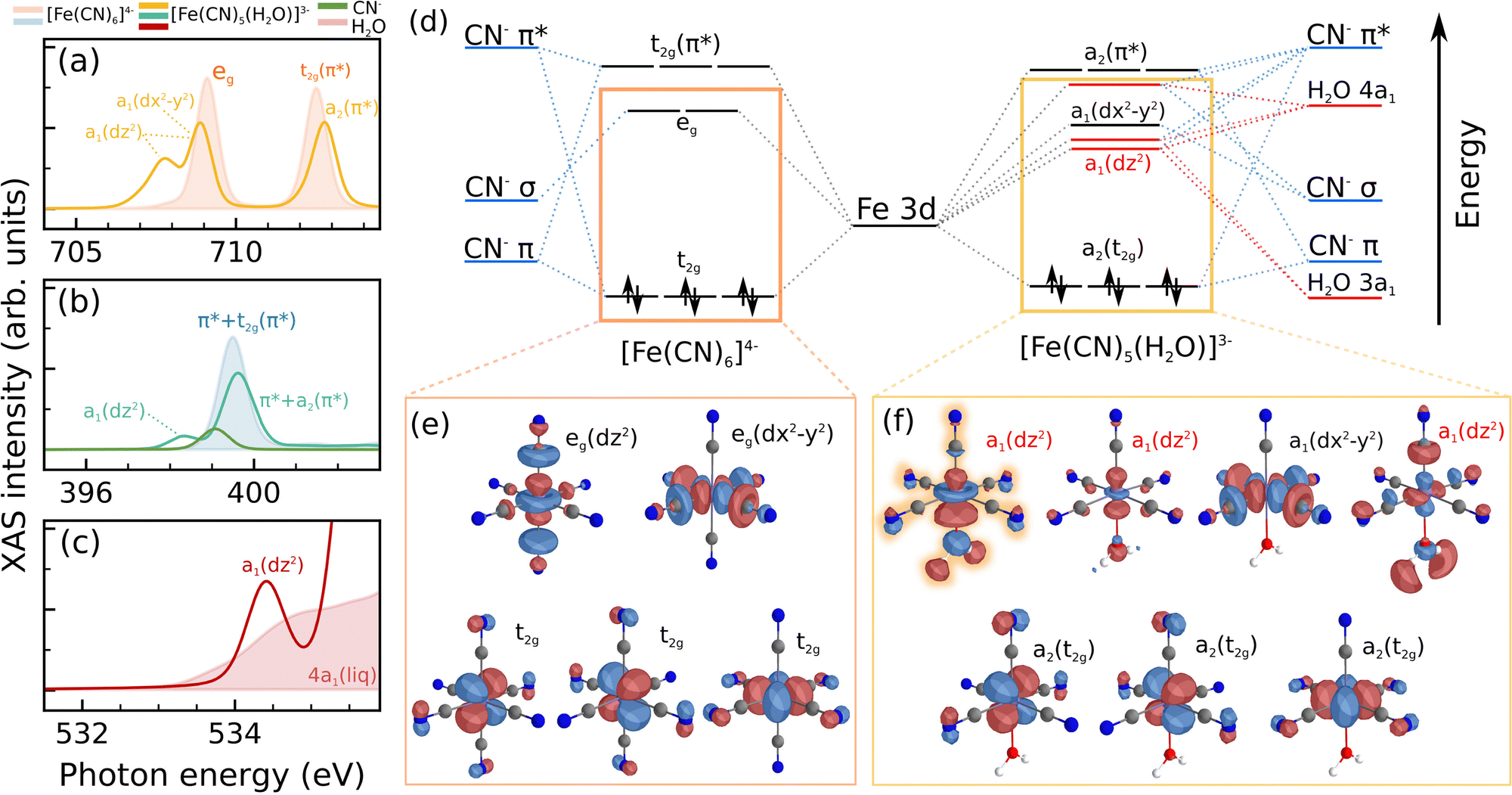 | ||
| Fig. 4 Electronic structure changes induced by photoaquation [Fe(CN)6]4− → [Fe(CN)5(H2O)]3−. Simulated soft X-ray absorption spectra of [Fe(CN)6]4− (shaded) and [Fe(CN)5(H2O)]3− (solid) at the iron L3- (a), nitrogen K- (b) and the oxygen K-edge (c, shaded: water MD-sampled spectrum). Additional resonances in the pre-edge region at all edges result from covalent interactions and symmetry reductions in [Fe(CN)5(H2O)]3−, as reflected in the molecular orbital diagram (d) and the orbital visualizations for [Fe(CN)6]4− (e) and [Fe(CN)5(H2O)]3− (f). Orbital diagrams are drawn qualitatively, roughly based on the calculated Kohn–Sham energies. The highlighted orbital in (f) directly evidences the covalency of the metal–water bond and contributes to the transient pre-edge features at all three edges. | ||
Strikingly, the signatures of the O K-edge spectrum directly support the picture described above. Namely, the mixing of the 4a1 orbital with the Fe dz2 red-shifts the oxygen K-edge absorption of [Fe(CN)5(H2O)]3− below the 4a1 absorption band of bulk liquid water, as seen in Fig. 4c. The shift between the experimentally detected maximum of the absorption difference and the maximum of the oxygen 1s → 4a1 band of the ground state absorption spectrum in Fig. 3e nicely coincides with the energetic separation of the lowest lying absorption line of [Fe(CN)5(H2O)]3− and the 1s → 4a1 band simulated for snapshots from a molecular dynamics simulation of liquid water in Fig. 3f. Hypothetically, if the bond between water and iron was purely ionic, no transient features should be expected below the liquid water pre-edge. In that scenario, the absorption resonance of the water would shift towards higher energies due to the core-level shift arising from coordination to a positively charged center. This argument further evidences the covalent nature of the observed feature. It is also worth mentioning that similar covalent features below the water pre-edge have been reported for free ions in water by Näslund et al.43
The low energy transition in the nitrogen K-edge spectrum in Fig. 4b is also related to excitations into the dz2 orbital. Although in this case, the signatures arise due to a combination of two factors: First, a sizable admixture of nitrogen 2p-character, via the π* manifold, to the dz2 orbital, due to the symmetry lowering in the [Fe(CN)5(H2O)]3− species, which is absent in the case of [Fe(CN)6]4−; second, intensity borrowing from more intense transitions into the higher lying cyanide-centered π* orbitals via configuration interaction.
The photo-aquation additionally affects the energetically higher lying t2g (π*) and ligand-centered π* orbitals. Naturally, the replacement of CN− by water means that the octahedral ligand field loses one ligand capable of π-donation and -backdonation, this contributes to a reduction in intensity at the N K-edge absorption as well as a lowering in the energy of the orbitals with π*-character. The reduction of intensity is associated with the collapse of the π* states into those of the isolated cyanide ligand, which lie lower in energy. Moreover, the lowering of symmetry lifts the degeneracy of π* orbitals in the aquated species. The resulting energy splitting causes a broadening of the main N K-edge resonance for the aquated species (Fig. 4b). These effects explain the oscillatory shape of the transient spectrum in Fig. 3c and d.
As mentioned, signatures associated with the dissociated cyanide ligand are also present, albeit less intense and overlap with the signal of [Fe(CN)5(H2O)]3−. However, upon inspection of our data within a pump–probe delay range of 20 ns, we observe a weak decay of the main depletion (lifetime of 7 ± 1 ns) of the transient signal, associated with the protonation kinetics of the cyanide anion12 (pKa = 9.2). This analysis is included in the ESI.†
Combining the different edges, a complete picture of the chemical bonding in [Fe(CN)5(H2O)]3− arises. The water molecule introduces a perturbation to the octahedral ligand field initially composed of 6 cyanide ligands. Direct mixing of water orbitals with the formerly eg orbitals, specially with the dz2 orbitals is the main defining feature of this complex, characterizing σ donation from the water to the metal. This interaction is also responsible for the lower absorption energy in the visible spectrum. At an orbital level, the eg manifold is split into a subset of dz2-derived orbitals with significant water contribution and a relatively unaffected dx2–y2 orbital (Fig. 4d and e). In the parent system [Fe(CN)6]4−, the dz2 orbital only mixes with the eg linear combination of cyanide σ orbitals, arising from the strong σ-donation abilities of these ligands. In [Fe(CN)5(H2O)]3−, however, the dz2 orbital mixes not only with the σ-orbital of the remaining axial cyanide ligand, but also with the a1 orbitals of water. Additionally to the σ-channels, the reduced symmetry enables π*–dz2 mixture, further splitting the levels. Hence, we display three of these orbitals in Fig. 4f, of which the first is most prominent, which contributes to the observed spectral signatures, resulting from the mixing of the metal dz2 with the water 3a1 and 4a1 orbitals, the cyanide σ and π* orbitals.
The t2g manifold is only weakly affected by the replacement of CN− by water. The dxz – and dyz-derived orbitals can overlap with the occupied 1b1 orbital of water, but this effect is not as pronounced, and it is comparatively smaller than π-backdonation via the cyanide ligands. It should be noted that the stability of the complex is only weakly affected by these interactions because it takes place between occupied orbitals. However, in a distorted solution environment, additional mixing between the unoccupied dz2 and the occupied 1b1 orbitals can also be expected.
Implications for the photo-reaction mechanism
In the paragraphs above, we have characterized the electronic properties of the [Fe(CN)5(H2O)]3− species, based on multi-edge soft X-ray spectroscopy. The spectroscopic signatures proved to be exceptionally sensitive to changes in electronic structure and evidenced the covalent nature of the water–metal bond. In light of these findings, a question arises whether soft X-ray spectroscopy could also help to shed light on additional open questions regarding the photo-aquation reaction of [Fe(CN)6]4−. In the following, we would like to discuss how the information from these spectroscopic techniques could be used to determine the mechanism leading to the formation of [Fe(CN)5(H2O)]3−.Let us recapitulate the proposed decay pathway for excitation of [Fe(CN)6]4− (aq.) at wavelengths λ > 300 nm. Based on the findings of Reinhard et al.,18 the initial population of the ligand-field state 1T1g relaxes to the lowest triplet state 3T1g within 100 fs. The dissociative character of the triplet state leads to the detachment of one of the axial CN− ligands. After dissociation, the system is still in a triplet state and exhibits a square pyramidal (SP) geometry, which then relaxes to a trigonal bipyramidal (TBP) structure within 3–4 ps. From the 3TBP, a water molecule coordinates within 20 ps to yield the stable photo-aquated species.
March et al.19 proposed a dynamical role of the pentacoordinated intermediate, in their study presenting picosecond Fe K-edge XAS measurements and QM/MM simulations. They found instead that the two pentacoordinated species interconvert on a 3 ps time scale. However, the incoming water molecule is only able to attack and bind to the 3SP species. Although their experimental measurements evidence the existence of the pentacoordinated species, the Fe K-edge spectra are not sensitive enough to differentiate between the SP and TBP species.17,19 One should keep in mind that interconversion between SP and TBP structures is generally expected for pentacoordinated species due to a prevalent pseudo-rotation mechanism,44,45 which connects the SP and TBP geometries.
Based on the paragraphs above, we can schematize the current understanding of the photoaquation pathway in Fig. 5a. The qualitative potential energy curves and relative energies are based on calculations shown in the ESI.† The energy order of the possible reaction intermediates was computed to be 3TBP < 1SP < 3SP, in agreement with Reinhard et al.18
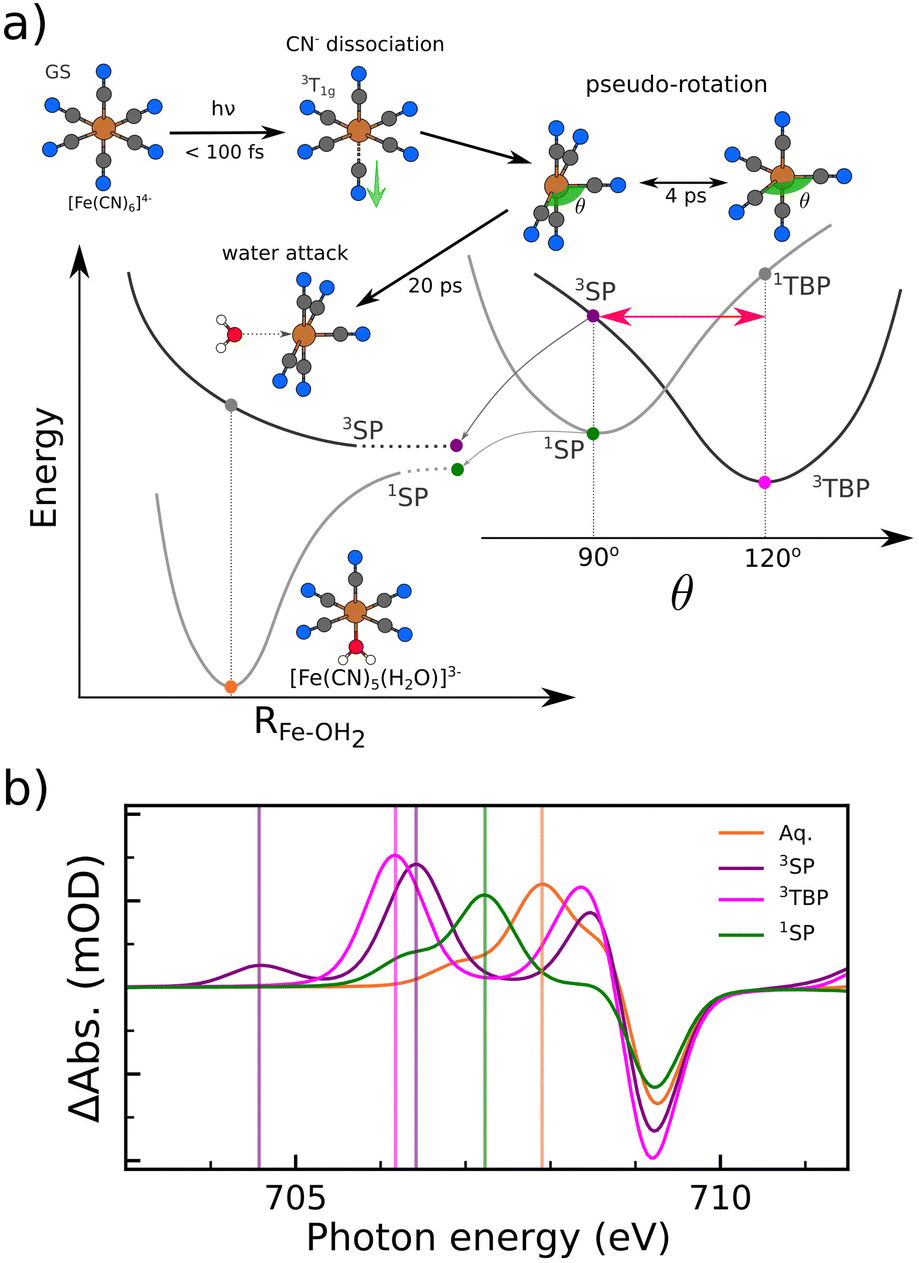 | ||
| Fig. 5 Current understanding of the photo-aquation mechanism of [Fe(CN)6]4− (a) and calculated spectral fingerprints of possible pentacoordinated intermediate and the aquated species (b). | ||
An elusive question concerns whether the singlet ground state of [Fe(CN)5(H2O)]3− is reached through inter-system crossing coupled to the water attack or if the system crosses to the singlet manifold in the course of the pseudo-rotation, before water coordination. Attachment of a water molecule to a triplet [Fe(CN)5]3− is highly unfavourable as the anti-bonding dz2–4a1 orbital is populated. This picture is supported by the dissociative potential energy scans along the Fe–OH2 in the triplet state (see ESI†). The attack of a water molecule on the 1SP structure is the lowest energy option among the possible pentacoordinated intermediates, followed by the 3SP one. Currently, neither the 3SP-3TBP oscillations/pseudo-rotation nor the possible inter-system crossing pathways have been experimentally confirmed.
In this context, let us examine the predicted Fe L3-edge spectra in the different [Fe(CN)5]3− configurations presented in Fig. 5b. Interestingly, the 3SP species displays an unambiguous signature through an absorption resonance at 704.6 eV well-separated from the 3TBP spectral features. This isolated resonance is associated with transitions to the lowest half-filled d orbital. In contrast, the resonances of the 3TBP species lie at higher energies and are associated with the two degenerate half-filled d orbitals. The resonances associated with transitions to the highest-lying empty d-orbital in both structures lie close to the ground state bleach. We expect that oscillations between these triplet structures would be detectable through intensity modulation at the energetically lowest resonance and the resonance shift in the range between 705.5 and 706.5 eV. Another advantage is the clear signature associated with the 1SP species, which could potentially play a role in the formation of the [Fe(CN)5(H2O)]3− product. Its main absorption feature lies at 707.2 eV, in the spectral region between the signature of the aquated photo-product and the ones of the pentacoordinated species.
Conclusion
In this article, we have investigated the photo-aquation reaction in [Fe(CN)6]4− by means of picosecond multi-edge soft X-ray spectroscopy. A transient population of [Fe(CN)5(H2O)]3− was generated by excitation with a laser pulse and subsequently probed at the Fe L3-, N K- and O K-edges. The complementary elemental selectivity of metal and ligand edges allowed for a complete characterization of the bonding in this unusual chemical system. The transient spectral signatures at the investigated edges yielded direct access to the covalent metal–water bond, arising from the σ-donation from the water to the metal, through the absorption into the dz2/a1 molecular orbital. Additionally, the signatures at the nitrogen K-edge reflected the loss of degeneracy among the ligand π*-system and revealed the protonation dynamics of the dissociated CN−.In [Fe(CN)5(H2O)]3−, the formerly eg orbitals are split into a subset of dz2-derived orbitals and a mostly unaffected dx2–y2 orbital. The dz2-derived subset arises from mixing with the water 3a1 and 4a1 orbitals as well as the axial CN− σ and π* orbitals. The formerly t2g set of orbitals did not exhibit any significant mixing with the orbitals of the coordinating water molecule.
Lastly, an outlook included in this article pertains to the ongoing debate in the literature on the precise mechanism of photo-aquation. The remarkable sensitivity observed in our soft X-ray measurements, combined with the predicted spectral signatures of the possible intermediates indicates that future sub-picosecond measurements could resolve the remaining questions surrounding this important photo-chemical reaction.
Author contributions
V. V. C. and E. J. M. contributed equally. V. V. C. conceptualization, supervision, investigation, formal analysis, and writing – original draft; E. J. M. data curation, investigation, and writing – original draft; R. B. software and investigation; R. M. J. conceptualization; M. F. supervision, investigation, and data curation; S. E. software, investigation, and writing – original draft; A. F. conceptualization, funding acquisition, and supervision; all authors contributed to writing – review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Helmholtz-Zentrum Berlin for the allocation of synchrotron radiation beamtime. R. B., R. M. J. and A. F. acknowledge funding from the ERC-ADG-2014 Advanced Investigator grant no. 669531 EDAX under the Horizon 2020 EU Framework, Programme for Research and Innovation.References
- L. Moggi, F. Bolletta, V. Balzani and F. Scandola, J. Inorg. Nucl. Chem., 1966, 28, 2589 CrossRef CAS , 0022-1902.
- J. Sykora and J. Sima, Coord. Chem. Rev., 1990, 107, 1 CrossRef , 0010-8545.
- J. Chen and W. R. Browne, Coord. Chem. Rev., 2018, 374, 15 CrossRef CAS , 0010-8545.
- P. Wernet, K. Kunnus, I. Josefsson, I. Rajkovic, W. Quevedo, M. Beye, S. Schreck, S. Grübel, M. Scholz and D. Nordlund, et al. , Nature, 2015, 520, 78 CrossRef CAS PubMed , 1476-4687.
- M. Chergui, Coord. Chem. Rev., 2018, 372, 52 CrossRef CAS.
- H. B. Gray and N. A. Beach, J. Am. Chem. Soc., 1963, 85, 2922, DOI:10.1021/ja00902a014 , 0002-7863.
- A. O. Tirler, I. Persson, T. S. Hofer and B. M. Rode, Inorg. Chem., 2015, 54, 10335, DOI:10.1021/acs.inorgchem.5b01701.
- G. Prampolini, P. Yu, S. Pizzanelli, I. Cacelli, F. Yang, J. Zhao and J. Wang, J. Phys. Chem. B, 2014, 118, 14899, DOI:10.1021/jp511391b , 1520-6106.
- M. Ross, A. Andersen, Z. W. Fox, Y. Zhang, K. Hong, J.-H. Lee, A. Cordones, A. M. March, G. Doumy and S. H. Southworth, et al. , J. Phys. Chem. B, 2018, 122, 5075, DOI:10.1021/acs.jpcb.7b12532 , 1520-6106.
- T. J. Penfold, M. Reinhard, M. H. Rittmann-Frank, I. Tavernelli, U. Rothlisberger, C. J. Milne, P. Glatzel and M. Chergui, J. Phys. Chem. A, 2014, 118, 9411, DOI:10.1021/jp5055588 , 1089-5639.
- S. Asperger, Trans. Faraday Soc., 1952, 48, 617 RSC , 0014-7672.
- A. G. MacDiarmid and N. F. Hall, J. Am. Chem. Soc., 1953, 75, 5204, DOI:10.1021/ja01117a017 , 0002-7863.
- S.-i Ohno, Bull. Chem. Soc. Jpn., 1967, 40, 1765, DOI:10.1246/bcsj.40.1765.
- S.-i Ohno, Bull. Chem. Soc. Jpn., 1967, 40, 1770, DOI:10.1246/bcsj.40.1770.
- M. Shirom and G. Stein, J. Chem. Phys., 1971, 55, 3372, DOI:10.1063/1.1676587. , 0021-9606.
- M. Shirom and G. Stein, J. Chem. Phys., 1971, 55, 3379, DOI:10.1063/1.1676588. , 0021-9606.
- M. Reinhard, T. J. Penfold, F. A. Lima, J. Rittmann, M. H. Rittmann-Frank, R. Abela, I. Tavernelli, U. Rothlisberger, C. J. Milne and M. Chergui, Structural Dynamics, 2014, 1, 024901, DOI:10.1063/1.4871751.
- M. Reinhard, G. Auböck, N. A. Besley, I. P. Clark, G. M. Greetham, M. W. D. Hanson-Heine, R. Horvath, T. S. Murphy, T. J. Penfold and M. Towrie, et al. , J. Am. Chem. Soc., 2017, 139, 7335, DOI:10.1021/jacs.7b02769 , 0002-7863.
- A. M. March, G. Doumy, A. Andersen, A. A. Haddad, Y. Kumagai, M.-F. Tu, J. Bang, C. Bostedt, J. Uhlig and D. R. Nascimento, et al. , J. Chem. Phys., 2019, 151, 144306 CrossRef PubMed.
- K. Kunnus, W. Zhang, M. G. Delcey, R. V. Pinjari, P. S. Miedema, S. Schreck, W. Quevedo, H. Schröder, A. Föhlisch and K. J. Gaffney, et al. , J. Phys. Chem. B, 2016, 120, 7182 CrossRef CAS PubMed.
- R. M. Jay, S. Eckert, R. Mitzner, M. Fondell and A. Föhlisch, Chem. Phys. Lett., 2020, 754, 137681 CrossRef CAS , 0009-2614.
- R. K. Hocking, E. C. Wasinger, F. M. F. de Groot, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 10442, DOI:10.1021/ja061802i , 0002-7863.
- N. Huse, T. K. Kim, L. Jamula, J. K. McCusker, F. M. F. de Groot and R. W. Schoenlein, J. Am. Chem. Soc., 2010, 132, 6809, DOI:10.1021/ja101381a.
- M. Kubin, M. Guo, T. Kroll, H. Lochel, E. Kallman, M. L. Baker, R. Mitzner, S. Gul, J. Kern and A. Föhlisch, et al. , Chem. Sci., 2018, 9, 6813 RSC.
- R. M. Jay, S. Eckert, V. Vaz da Cruz, M. Fondell, R. Mitzner and A. Föhlisch, Angew. Chem., Int. Ed., 2019, 58, 10742 CrossRef CAS PubMed.
- R. M. Jay, V. Vaz da Cruz, S. Eckert, M. Fondell, R. Mitzner and A. Föhlisch, J. Phys. Chem. B, 2020, 124, 5636, DOI:10.1021/acs.jpcb.0c00638.
- B. E. Van Kuiken, H. Cho, K. Hong, M. Khalil, R. W. Schoenlein, T. K. Kim and N. Huse, J. Phys. Chem. Lett., 2016, 7, 465, DOI:10.1021/acs.jpclett.5b02509.
- E. J. Mascarenhas, M. Fondell, R. Buchner, S. Eckert, V. Vaz da Cruz and A. Föhlisch, Phys. Chem. Chem. Phys., 2022, 24, 17979–17985 RSC.
- P. S. Miedema, W. Quevedo and M. Fondell, Journal of Large-Scale Research Facilities, 2016, 2, A27 Search PubMed , 2364-091X.
- M. Fondell, S. Eckert, R. M. Jay, C. Weniger, W. Quevedo, J. Niskanen, B. Kennedy, F. Sorgenfrei, D. Schick and E. Giangrisostomi, et al. , Structural Dynamics, 2017, 4, 054902, DOI:10.1063/1.4993755.
- J. Niskanen, M. Fondell, C. J. Sahle, S. Eckert, R. M. Jay, K. Gilmore, A. Pietzsch, M. Dantz, X. Lu and D. E. McNally, et al. , Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4058 CrossRef CAS PubMed.
- F. Neese, WIREs Computational Molecular Science, 2022, e1606 Search PubMed , 1759-0876.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- B. de Souza, G. Farias, F. Neese and R. Izsak, J. Chem. Theory Comput., 2019, 15, 1896 CrossRef CAS PubMed.
- W. Hua, G. Tian, G. Fronzoni, X. Li, M. Stener and Y. Luo, J. Phys. Chem. A, 2013, 117, 14075, DOI:10.1021/jp408776p.
- E. Lindahl, M. J. Abraham, B. Hess and D. van der Spoel, Gromacs 2019 source code DOI:10.5281/zenodo.2424363.
- Y. Wu, H. L. Tepper and G. A. Voth, J. Chem. Phys., 2006, 124, 024503 CrossRef PubMed.
- N. Engel, S. I. Bokarev, E. Suljoti, R. Garcia-Diez, K. M. Lange, K. Atak, R. Golnak, A. Kothe, M. Dantz and O. Kuhn, et al. , J. Phys. Chem. B, 2014, 118, 1555, DOI:10.1021/jp411782y.
- L.-A. Naslund, M. Cavalleri, H. Ogasawara, A. Nilsson, L. G. M. Pettersson, P. Wernet, D. C. Edwards, M. Sandstrom and S. Myneni, J. Phys. Chem. A, 2003, 107, 6869, DOI:10.1021/jp034296h.
- S. Trushin, K. Kosma, W. FuB and W. Schmid, Chem. Phys., 2008, 347, 309 CrossRef CAS , 0301-0104.
- S. A. Trushin, W. Fuss, W. E. Schmid and K. L. Kompa, J. Phys. Chem. A, 1998, 102, 4129, DOI:10.1021/jp973133o.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04084k |
| ‡ These authors contributed equally. |
| § Current address: Department of Physics and Astronomy, Uppsala University, Box 516, SE-751 20 Uppsala, Sweden. |
| This journal is © the Owner Societies 2022 |