
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of the acenaphthylene cation as a common C2H2-loss fragment in dissociative ionization of the PAH isomers anthracene and phenanthrene†
Shreyak
Banhatti
*a,
Daniël B.
Rap
 b,
Aude
Simon
c,
Heloïse
Leboucher
c,
Gabi
Wenzel
d,
Christine
Joblin
e,
Britta
Redlich
b,
Stephan
Schlemmer
a and
Sandra
Brünken
*b
b,
Aude
Simon
c,
Heloïse
Leboucher
c,
Gabi
Wenzel
d,
Christine
Joblin
e,
Britta
Redlich
b,
Stephan
Schlemmer
a and
Sandra
Brünken
*b
aI. Physikalisches Institut, Universität zu Köln, Zülpicher Str. 77, 50937 Köln, Germany. E-mail: banhatti@ph1.uni-koeln.de
bInstitute for Molecules and Materials, FELIX Laboratory, Radboud University, Toernooiveld 7, 6525 ED, Nijmegen, The Netherlands. E-mail: sandra.bruenken@ru.nl
cLaboratoire de Chimie et Physique Quantiques (LCPQ), Fédération FeRMI, CNRS & Université Toulouse III – Paul Sabatier, 118 Route de Narbonne, 31062 Toulouse, France
dCenter for Interstellar Catalysis (InterCat), Department of Physics and Astronomy, Aarhus University, Ny Munkegade 120, 8000 Aarhus C, Denmark
eInstitut de Recherche en Astrophysique et Planétologie (IRAP), Université Toulouse III – Paul Sabatier, CNRS, CNES, 9 Avenue du Colonel Roche, 31028 Toulouse, France
First published on 27th October 2022
Abstract
Polycyclic aromatic hydrocarbons (PAHs) are thought to be a major constituent of astrophysical environments, being the carriers of the ubiquitous aromatic infrared bands (AIBs) observed in the spectra of galactic and extra-galactic sources that are irradiated by ultraviolet (UV) photons. Small (2-cycles) PAHs were unambiguously detected in the TMC-1 dark cloud, showing that PAH growth pathways exist even at low temperatures. The processing of PAHs by UV photons also leads to their fragmentation, which has been recognized in recent years as an alternative route to the generally accepted bottom-up chemical pathways for the formation of complex hydrocarbons in UV-rich interstellar regions. Here we consider the C12H8+ ion that is formed in our experiments from the dissociative ionization of the anthracene and phenanthrene (C14H10) molecules. By employing the sensitive action spectroscopic scheme of infrared pre-dissociation (IRPD) in a cryogenic ion trap instrument coupled to the free-electron lasers at the FELIX Laboratory, we have recorded the broadband and narrow line-width gas-phase IR spectra of the fragment ions (C12H8+) and also the reference spectra of three low energy isomers of C12H8+. By comparing the experimental spectra to those obtained from quantum chemical calculations we have identified the dominant structure of the fragment ion formed in the dissociation process to be the acenaphthylene cation for both isomeric precursors. Ab initio molecular dynamics simulations are presented to elucidate the fragmentation process. This result reinforces the dominant role of species containing a pentagonal ring in the photochemistry of small PAHs.
1 Introduction
Polycyclic aromatic hydrocarbons (PAHs) have been considered to be the carriers of the aromatic infrared bands (AIBs) for the past few decades.1–4 These emission bands in the 3–20 μm region observed in the spectra of many galactic and extra-galactic astronomical sources can be attributed to a large family of PAHs that absorb ultraviolet (UV) photons originating from astronomical sources and then subsequently relax via emission of IR photons. Models have shown that PAHs subjected to the local physical conditions of astrophysical environments can occur in different charge and hydrogenation states of varying sizes and shapes.5–9 Due to fragmentation processes, only large PAHs (∼50 or more carbon atoms) are expected to survive on long timescales in UV photon-rich environments.5,7,8Recent radio astronomical observations have opened a new window on the study of PAHs with the identification of the small PAHs indene C9H810,11 and cyanonaphthalene C10H7–CN12 in the dark cloud TMC-1. Small and medium-sized PAHs are also detected in primitive meteorites.13,14 This demonstrates the importance of small PAHs in star- and planet-forming regions and the interest to study the processes involved in their (photo)chemical evolution.
In this study, we focus on the C2H2-loss fragmentation channels (leading to C12H8+) upon dissociative ionization of C14H10 in the case of anthracene (Anth) and its isomer phenanthrene (Phen). Previous experiments include the study of dissociative or of unimolecular dissociation, of the corresponding molecular ions Anth˙+ and Phen˙+, using vacuum UV photoionization,15–17 imaging photoelectron photoion coincidence spectrometry (iPEPICO) and atmospheric pressure chemical ionization – collision induced dissociation mass spectrometry (APCI-CID).18 Using statistical theories to analyze the data, these articles report activation energies for the main dissociation channels, i.e., H, H2, and C2H2 loss channels. They also discuss possible structures for C12H8+ formed by C2H2 loss. Gotkis et al.15 proposed the acenaphthylene radical cation in the case of Phen˙+ dissociation but further studies by Ling et al.19 and Ling and Lifshitz16 favoured biphenylene for both Phen˙+ and Anth˙+. West et al.18 further discussed whether it is the biphenylene or cyclobuta[b]naphthalene cation (see Fig. 1 for an overview of fragment structures). Finally, Johansson et al.20 identified a possible low activation energy C2H2-dissociation channel for anthracene (C14H10) and acridine (C13H9N) cations. Their quantum chemical calculations predict three different cationic products for anthracene formed in their collision-induced dissociation study: 2-ethynylnaphthalene, biphenylene, and acenaphthylene (see Fig. 1).
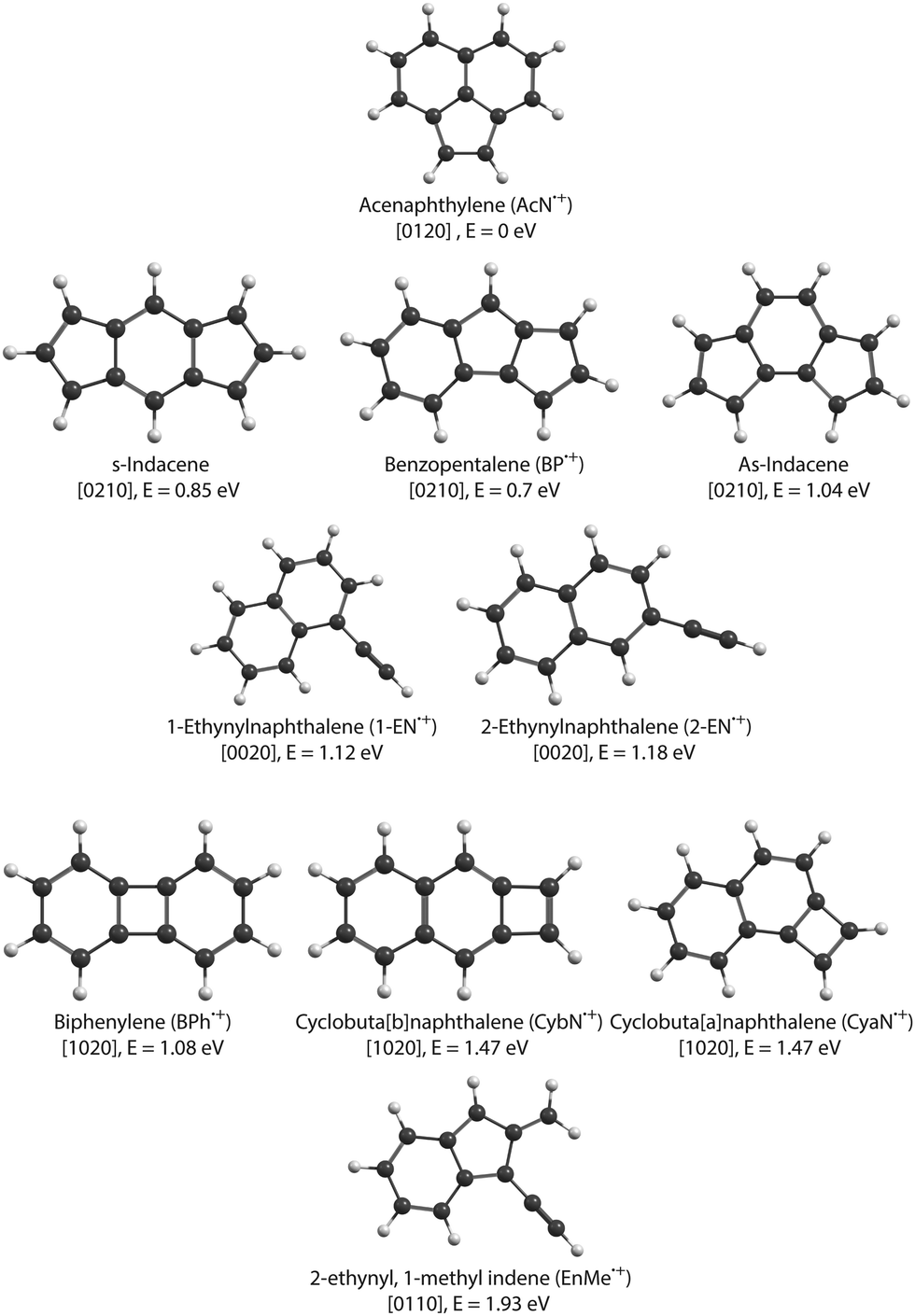 | ||
| Fig. 1 Isomers of C12H8+ radical cation. The electronic energies are shown relative to the lowest-energy isomer, the acenaphthylene cation, AcN˙+. The values noted in square brackets describe the number of n-carbon rings contained in each isomer with n = 4–7 (see Section 3.4). | ||
Vibrational spectroscopy of molecular ions is a powerful tool for their structural and isomeric characterization. Advancements in spectroscopic techniques, especially involving the use of ion traps and powerful free-electron lasers, have enabled the development of versatile infrared (IR) action spectroscopic methods such as IR multi-photon dissociation (IRMPD)21,22 and IR pre-dissociation (IRPD) of complexes with rare gas atoms. In the past, these techniques have been successfully employed to elucidate the isomeric composition of cationic fragmentation products upon dissociative ionization of PAHs and related species. Using IRPD in a cryogenic ion trap, Jusko et al.23 recorded the mid-IR spectra of Ne-tagged C7H7+ and revealed its two isomeric forms, i.e., benzylium+ and tropylium+. Ne-tagged doubly dehydrogenated pyrene (ddPy+) was spectroscopically investigated using the same technique by Panchagnula et al.24 The results showed that its spectral features correspond to a mixture of different isomers (4,5-ddPy+ and 1,2-ddPy+) favoured by the loss of hydrogen from the same aromatic ring. Bouwman et al.25 provided the first spectroscopic evidence, using IRMPD, for formation of the pentalene+ isomer in the dissociative ionization of naphthalene via loss of C2H2.
Following these studies, we perform here a direct spectroscopic investigation of the structure of C12H8+ formed by electron impact dissociative ionization of anthracene and phenanthrene. We record the mid-IR spectra of the Ne-tagged ions using IRPD action spectroscopy. The experiment is supported by detailed analysis using quantum chemical calculations which suggest the formation of mainly identical isomers for both PAHs. We also study the fragmentation process using molecular dynamics (MD) simulations with the electronic structure computed at the self-consistent charge density functional based tight binding (SCC-DFTB)26 method and discuss the branching ratios of the different isomers.
2 Methods
2.1 Studied species
The cationic PAH species studied in this work, i.e., Anth˙+, Phen˙+ (C14H10+), and several C12H8+ isomers (see Fig. 1 for the dominant expected structures and a summary of their short names used throughout the text), were generated by electron impact ionization from gaseous PAH precursors. Table 1 lists the various PAH samples used in this study along with their purity. Anthracene and phenanthrene are canonical isomers of C14H10 with molar mass M = 178.23 g mol−1, both appearing as white crystals. 1-Ethynylnaphthalene (1-EN), 2-ethynylnaphthalene (2-EN), and acenaphthylene (AcN) are some of the isomers of C12H8 with M = 152.19 g mol−1. 1-EN is a brown liquid, 2-EN and AcN are yellowish powder. All samples were obtained from Sigma Aldrich.| Sample | CAS-number | Purity |
|---|---|---|
| Anth | 120-12-7 | ≥98% |
| Phen | 85-01-8 | 98% |
| 1-EN | 5727-65-8 | 97% |
| 2-EN | 2949-26-0 | — |
| AcN | 208-96-8 | 99% |
2.2 Experimental
In this section we briefly discuss the experimental setup, FELion, used in this study, with a more detailed account given by Jusko et al.27 FELion is a cryogenic 22-pole ion trap instrument coupled to the free-electron lasers at the FELIX (Free-Electron Laser for Infrared eXperiments) Laboratory.28 In this study FEL-2, covering the 600–1700 cm−1 range, was coupled to the FELion setup, enabling us to perform IRPD spectroscopic measurements over the whole vibrational fingerprint region. Vapours of PAHs are introduced into an electron impact (EI) ionization source at pressures of several 10−5 mbar. Depending on the electron energy, which is tunable between 0 to 100 eV, several singly and doubly charged parent and fragment cations can be observed as discussed in detail in our previous study.29 A 100 ms long pulse of the target fragment cation, e.g., of mass-to-charge ratio m/z 152 corresponding to the acetylene loss fragment, C12H8+, is mass-selected with a quadrupole mass filter to then be cooled and stored (typically for 0.6 or 1.6 seconds) in the cryogenic 22-pole ion trap.30 A gas mixture of helium (He)–neon (Ne) (ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) is pulsed for 100–150 ms into the trap at the beginning of the storage cycle and collisionally cools the ions close to the nominal 6.5 K of the ion trap walls. Ternary collisions lead to the formation of van der Waals complexes of the ion with one Ne atom in the trap.
:1) is pulsed for 100–150 ms into the trap at the beginning of the storage cycle and collisionally cools the ions close to the nominal 6.5 K of the ion trap walls. Ternary collisions lead to the formation of van der Waals complexes of the ion with one Ne atom in the trap.
The IRPD spectra are recorded by counting the number of Ne-complexed ions as a function of excitation frequency with a Daly detector.31 The fragment-Ne complexes dissociate due to resonant vibrational excitation while scanning the FEL-2 IR laser. The FEL-2 laser is pulsed at 10 Hz which means up to 16 macro-pulses are admitted into the trap for storage times of up to 1.6 s. Each data point at a given wavenumber is recorded under identical conditions for a minimum of three iterations and averaged. The spectrum is normalized using
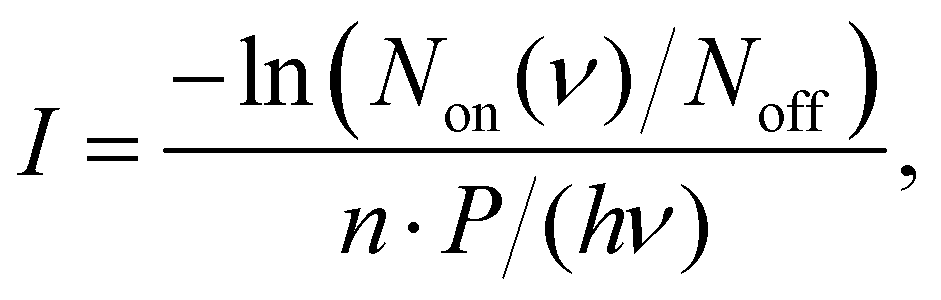 | (1) |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ≈ (0.7–1.2)%·).
≈ (0.7–1.2)%·).
In this experiment, saturation depletion measurements27 provide useful information about the isomeric ratio present in the fragment-Ne complexes, a method successfully proven in Jusko et al.23 to quantify isomeric abundances of benzylium and tropylium cations, C7H7+. Here, the relative depletion of the fragment-Ne complexes is recorded as a function of the laser energy deposited at a wavelength corresponding to a specific vibrational band. This depletion is then compared to the ion signal observed at an off-resonance frequency to account for any losses due to non-radiative processes. The saturation depletion value then directly yields the relative abundance of the isomer that is responsible for the chosen vibrational band.
2.3 Theoretical
similar to the experimentally observed FWHM is considered for convolving the quantum chemically calculated spectra throughout this study. The electronic energies and the zero-point vibrational energy corrections for the ions on the potential energy surface (PES) of the 1-EN˙+ to AcN˙+ isomerization have been calculated at the B3LYP-GD3/6-311G(d,p) level of theory. The transition states have been validated by performing intrinsic reaction coordinate (IRC) calculations. Although the presence of the Ne tag will have an effect on the vibrational band positions we expect it to be small as previously observed.29,35–38
In the present work, several hundreds (360 to 720) of MD/DFTB simulations of 1 ns to 2 ns duration were run for Anth˙+ and Phen˙+ (δt = 0.1 fs) random initial velocities in the microcanonical (NVE) ensemble at 17.5, 18.5 and 20 eV. These energies were chosen as they are the lowest values for which fragmentation is observed within the nanosecond timescale. This allowed us to gain insights into reaction kinetics, branching ratios, and mechanisms. The results are described in Section 3.4. All MD/DFTB simulations were achieved using the deMonNano code.44
3 Results and discussion
3.1 Dissociative ionization of anthracene and phenanthrene
Fig. 2 shows the breakdown curves of anthracene (top panel) and phenanthrene (bottom panel) recorded for m/z 89 (C14H102+), 152 (C12H8+), 176 (C14H8+), and 178 (C14H10+). The data was obtained by counting the number of fragment ions produced in the EI process as a function of electron energy. The appearance energy (AE) of the parent cation (C14H10+) for both precursors estimated from these scans is ∼7 eV‡ close to their previously determined EI ionization energies (IE) of 7.4(8) eV.45,46 The IEs measured via photo-ionization, i.e., 7.40(25) eV (anthracene) and 7.87(10) eV (phenanthrene)16 are also good benchmark values for the breakdown curves shown here.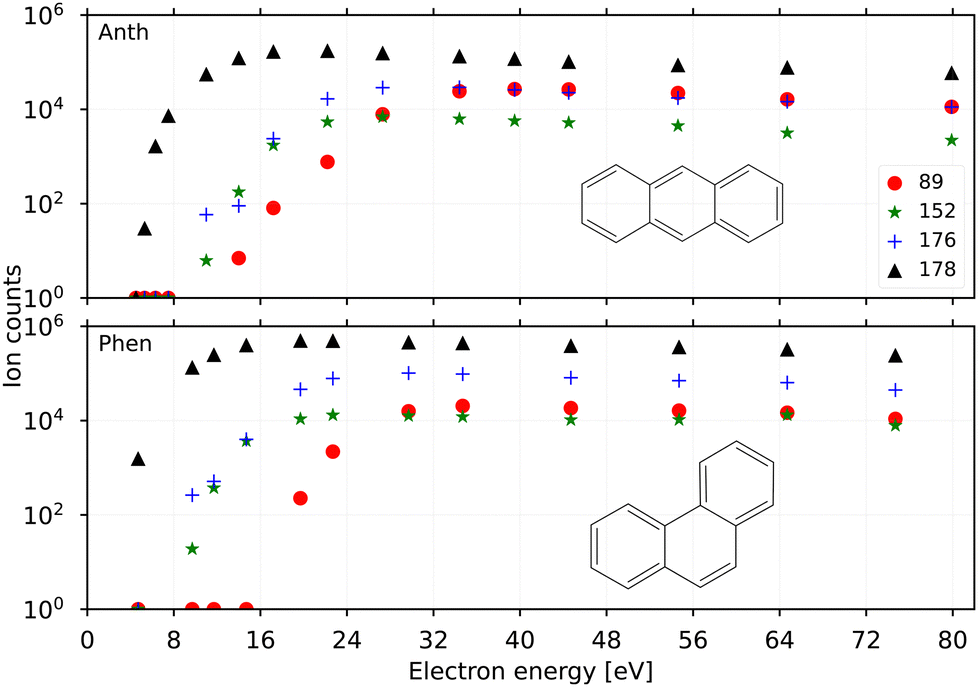 | ||
| Fig. 2 Breakdown curves for anthracene (top panel) and phenanthrene (bottom panel) showing the parent ions m/z 178, C14H10+ (black triangles), and their fragments m/z 89, C14H102+ (red dots); m/z 152, C12H8+ (green stars); m/z 176, C14H8+ (blue crosses). | ||
The AEs for fragmentation via acetylene loss and hydrogen loss in low-energy electron impact experiments were found to be similar, ∼17 eV, for anthracene by Burgt and Dunne.45 This is also reflected in the breakdown curves recorded here for the two channels at m/z 152 and 176 which follow similar breakdown behaviour. The breakdown curves indicate a slightly lower yield of the acetylene loss (m/z 152) compared to the doubly dehydrogenated m/z 176 at higher energies (>24 eV) for both precursor PAHs. It is also evident that phenanthrene fragments form more efficiently than anthracene from a comparison of their respective yields at electron energies of 8–16 eV. This is in agreement with the finding by Burgt et al.46 who measured the AE for C12H8+ from phenanthrene fragmentation to be lower at ∼15.3 eV compared to ∼17 eV for anthracene. The AE for the doubly charged m/z 89 from both precursors was predicted around 20 eV in earlier studies by Banhatti et al.29 and van der Burgt and Dunne,45 and this value agrees with the measured second ionization potential of 11.7(1) eV for the anthracene cation,17 and the present results.
3.2 Vibrational spectra and structural analysis
In order to verify the sample purity and to investigate a possible isomerization process upon ionization,20 we first recorded the IRPD spectra of Anth˙+ and Phen˙+ cations (C14H10+, m/z 178) produced by EI (Ee− = 40 eV) of their respective vapours. The spectra are shown in the two upper panels of Fig. 3 together with their calculated anharmonic spectra at the B3LYP/6-311G(d,p) level of theory. The comparison of experiment to theory shows a good match for both isomers, with typically less than 10 cm−1 deviation for the fundamental modes, which are all present in the recorded IR spectra.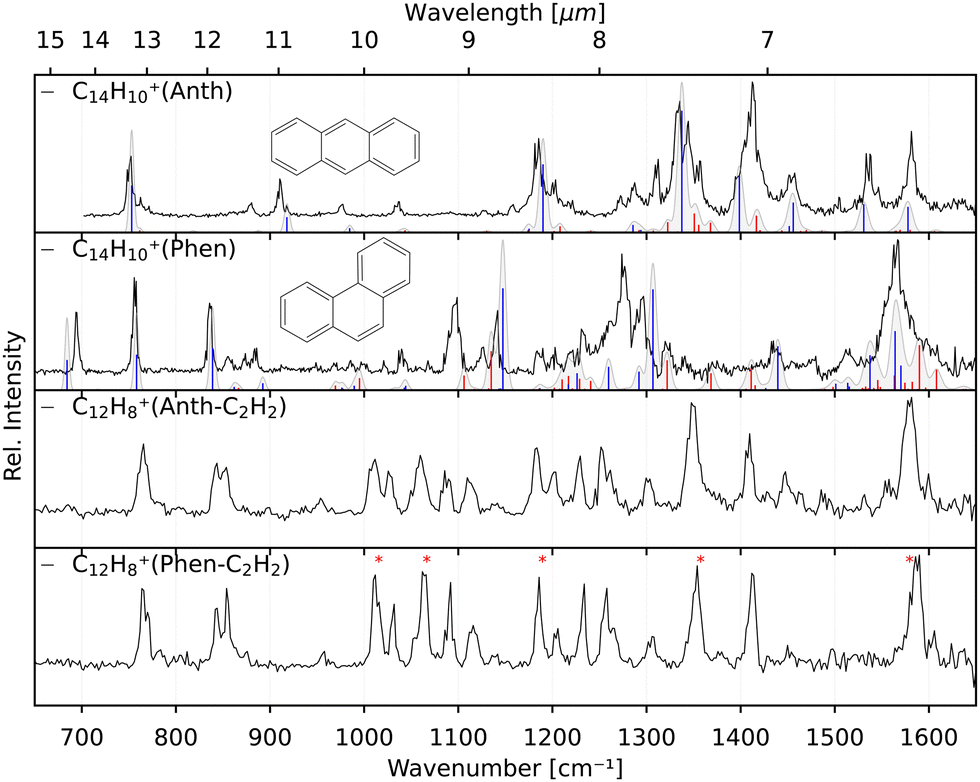 | ||
| Fig. 3 Experimental IRPD spectra of Anth˙+ and Phen˙+ and their respective C2H2 loss fragments (black). Calculated anharmonic vibrational spectra, convolved with the FEL laser linewidth, are shown for comparison for Anth˙+ and Phen˙+ including fundamental modes (blue) and combination modes (red). The spectrum of C12H8+(Phen–C2H2) presented in the bottom panel depicts several bands (*) that appear to have similar intensity which is caused by saturation. | ||
Previously measured IRPD spectra by Piest et al.38 of gas-phase Phen˙+ complexed with argon and neon also show excellent agreement with the data presented here (Fig. S1 of the ESI†), indicating a negligible effect on the vibrational frequencies between the two rare gas tags. Recent IRMPD data of Phen˙+ by Wiersma et al.47 in the far-infrared does not cover the same frequency region as our work. For Anth˙+, the data shown in Fig. 3 is the first narrow-bandwidth IRPD spectrum recorded, and it shows qualitative agreement with an earlier IRMPD spectrum by Oomens et al.48 Saturation depletion measurements at two unique band positions for Anth˙+ (746 cm−1, 1410 cm−1) and Phen˙+ (755 cm−1, 836 cm−1), respectively, prove that only one structural isomer with m/z 178, the canonical isomers, respectively, are formed in each case; i.e., no isomerization, as predicted previously as a competing channel to dissociation,16 is observed for the direct ionization products.
The two lower panels of Fig. 3 show the experimental IRPD spectra of the acetylene (C2H2)-loss fragment cations (C12H8+, m/z 152) formed via dissociative EI (Ee− = 40 eV) of anthracene and phenanthrene, respectively. Even upon first visual inspection, the two spectra look almost identical, which indicates that the same fragment ion is formed from both PAH parent species. In order to elucidate the structure of the (common) fragment ion we performed quantum chemical calculations of the vibrational spectra of the lowest-energy isomers shown in Fig. 1. The calculated spectra for some of these isomers are shown in Fig. 4 and are compared to the experimental m/z 152 fragment IRPD spectrum obtained from the anthracene precursor. The calculated spectrum of acenaphthylene (AcN˙+) clearly shows the best match with the measured IRPD spectrum of the anthracene (and phenanthrene) fragment ion. All dominant fundamental and several combination modes of AcN˙+ above 1000 cm−1 are present in the experimental fragment spectrum. At lower frequencies the assignment is more ambiguous based on the calculated band positions.
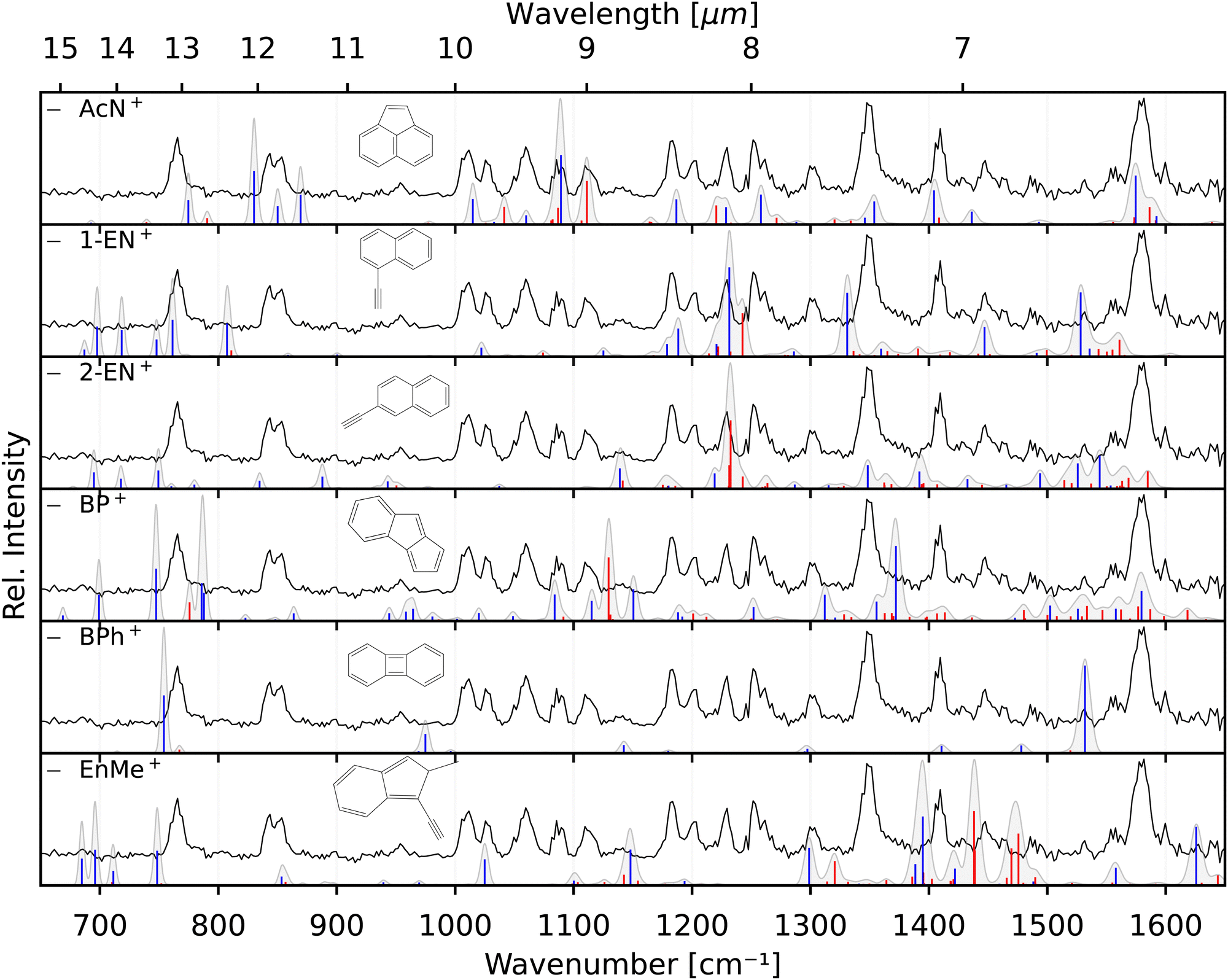 | ||
| Fig. 4 Experimental IRPD anthracene fragment spectrum (C12H8+, black, all panels) compared with calculated anharmonic vibrational spectra (blue – fundamentals, red – combination modes; convolved with the FEL laser linewidth) of AcN˙+, 1-EN˙+, 2-EN˙+, BP˙+, BPh˙+, and EnMe˙+. | ||
However, saturation depletion measurements at several bands of the fragments produced from the two precursors suggest that there may be more than one isomer formed. For example, we observe greater than 94(5)% depletion at three AcN˙+ band positions, 1090, 1111 and 1229 cm−1, for the phenanthrene fragment ion, and less than 85% saturation depletion in case of the anthracene fragment for the two latter bands, and only 70% for the 1090 cm−1 band. This indicates that phenanthrene mainly forms AcN˙+, whereas anthracene, in addition to forming AcN˙+ as the dominant isomer with about 70% relative abundance, forms another unique fragment isomer likely among those in Fig. 1. One important aspect seen in the mass spectra (see Fig. S5 of the ESI†) of Phen and Anth is the peak at m/z 179, isotopic substitution of one 12C with 13C, with a peak intensity of 16% of m/z 178, which is approximately equal to the natural abundance of the 13C isotope in Phen˙+ and Anth˙+. The peak at m/z 151 indicates the presence of a C2H3 loss channel. Thus, the fragment cation formed by the loss of C2H3 and C2H2 from m/z 179 and 178, respectively, will both appear at m/z 152. As a result we expect about 10% of the m/z 152 fragment to contain other impurities, not enough to account for the only 70% depletion observed for the anthracene fragment.
As seen in Fig. 4, Anth˙+ shows 100% depletion at 1345 cm−1 which would indicate an overlapping band with the second isomer. The calculated spectra of AcN˙+, 1-EN˙+, 2-EN˙+, and BP˙+ all predict a closely lying band around 1345 cm−1. On the other hand, EnMe˙+, BPh˙+, and other isomers within the BP family like As- and S-indacene can be ruled out easily from the lack of overlapping bands, and the absence of the calculated strong bands, e.g., at 1531 cm−1, in the experimental spectra (a comparison with calculated spectra of these isomers is shown in Fig. S4 of the ESI†). We also performed saturation depletion measurements at a lower electron energy (Ee− = 14.7 eV) to possibly shift the isomer ratios, as was demonstrated in our earlier study on tropylium and benzylium cations formed by EI of toluene.23 However, the resulting spectra did not show any noticeable change in the isomeric ratio.
To obtain additional information on the isomeric mixture we utilized Pearson's correlation coefficient49 and compared the anthracene fragment spectrum with the calculated spectra of the different isomers. The results shown in Fig. S2 of the ESI† confirm that AcN˙+ is the dominant isomer, having the highest positive correlation (0.5), followed by and 2-EN˙+ (0.15), BP˙+ (0.13), and 1-EN˙+ (0.04), and a negative correlation was found for the remaining isomers. Due to the precision of the calculated spectra, the identification of the second isomer remains elusive. As a reference, we also show the correlation values for Anth˙+ and Phen˙+ compared to calculated values in Fig. S2 (ESI†).
A likely candidate for a possible second isomer of the phenanthrene fragment is BP˙+ due to overlapping bands at 1090, 1111 and 1229 cm−1 which would account for the observed depletion value of >94%. However the concentration would have to be very low, of the order 0–10%, taking into consideration the isotopic contamination, signal-to-noise ratio and the depletion values >90(5)% observed for other unblended phenanthrene fragment bands.
3.3 Reference spectra
The identification of the additional isomer formed in both cases has proven to be difficult based on the calculated spectra alone. As can be seen in Fig. 4, there are shifts up to around 10 cm−1, and thus larger than the experimental linewidth, between the calculated and experimental band positions for the identified AcN˙+ isomer, in particular for the low-lying CH out-of-plane bending and CH2 wagging modes, for which our basic anharmonic treatment might fail.47,50,51 Similar effects are expected for the other candidate structures, making an assignment difficult in view of many overlapping vibrational bands. Therefore, we also recorded the reference IRPD spectra of the three most promising fragments 1-EN˙+, 2-EN˙+, and AcN˙+ formed by EI (Ee− = 14.7 eV) of their respective neutrals. Fig. 5 shows the fragment spectrum from the anthracene precursor together with the reference spectra of the three candidate fragment cations compared to their calculated vibrational spectra, respectively. The fragment cation spectrum presented in the top panel of Fig. 5 clearly is in large parts identical to the reference AcN˙+ spectrum, confirming our initial assignment. The relative intensities can vary widely between the spectra due to varying laser intensities leading to saturation of some strong bands, as seen at 1062 cm−1. We note in addition that strong bands below 700 cm−1 and between 1500–1600 cm−1 observed in the experimental spectra of 1-EN˙+ and 2-EN˙+ are not, or only weakly, present in the anthracene (and phenanthrene) fragment spectra. This could be due to a low abundance of specific isomers, indicating a mixture of different species, including BP˙+ for which we could not record a reference spectrum. The spectroscopic identification of the minor isomers in the dissociative ionization remains thus tentative.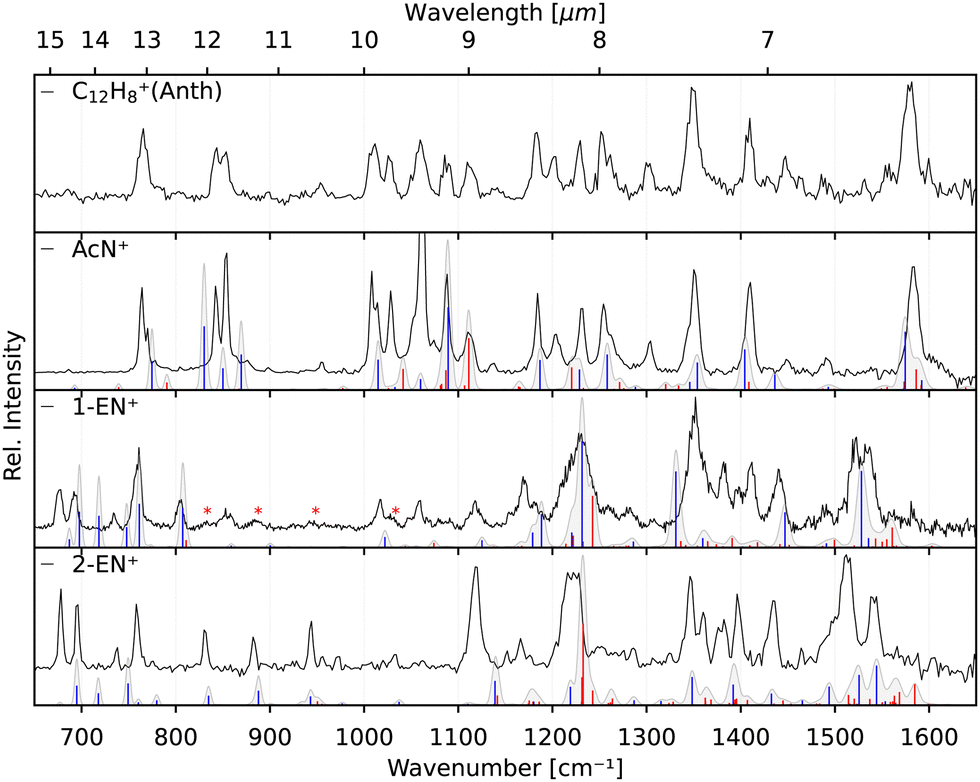 | ||
| Fig. 5 Experimental IRPD spectra of the anthracene fragment (C12H8+, black, top panel) and experimental IRPD reference spectra of its isomers, AcN˙+, 1-EN˙+, and 2-EN˙+ (black, lower three panels). Calculated anharmonic vibrational spectra, convolved with the FEL laser linewidth, are shown for comparison for the isomeric structures including fundamental modes (blue) and combination modes (red). AcN˙+: the band at 1062 cm−1 is a saturated band and out of scale on account of the normalization mentioned in Section 3.2. 1-EN˙+: minor contamination from 2-EN˙+ marked with asterisk in the 1-EN˙+ spectrum. | ||
Finally, recording the spectra of several C12H8+ isomers gives us the opportunity for further comparison with the anharmonic spectra at the B3LYP/6311G(d,p) level of theory. Overall, there is reasonable agreement between the measured and calculated spectra without the need of a scaling factor. We note that some bands show larger shifts such as the 1119 cm−1 band in 2-EN˙+, the 1345 cm−1 band in 1-EN˙+, and in general the bands below 900 cm−1. This might be due to different effects including limitations of the anharmonic calculations that provide only a very basic treatment of resonances, e.g., Mulas et al.52 and a possible role of the Ne tag on the IRPD spectrum.53
3.4 Molecular dynamics simulations: dissociation pathways
In this subsection, we report the results of the MD/DFTB simulations for Anth˙+ and Phen˙+ with internal energies of 17.5, 18.5 and 20 eV. The fraction of the parent ions as a function of time is reported in Fig. 6 (top panel). As can be seen in this figure, an energy of 17.5 eV corresponds to the lowest energy for which dissociation is observed after 1 ns. Not enough fragments are observed to deduce reliable trends for branching ratios. At 18.5 eV, the fragmentation ratio reaches 20% in the case of Anth˙+, and 35% in the case of Phen˙+. At 20 eV, the values become 80% and 90%, respectively.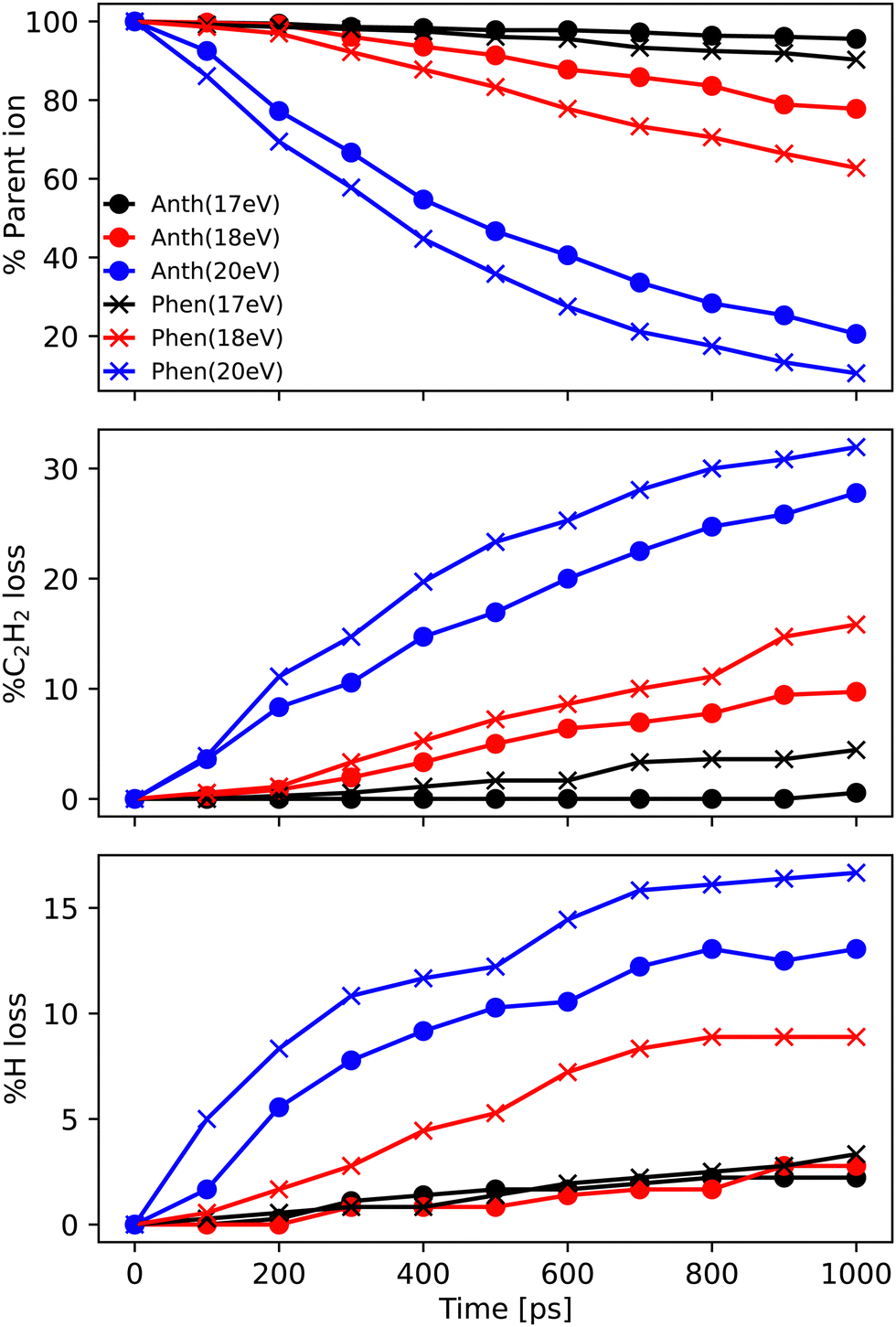 | ||
| Fig. 6 Ratio of most abundant (top panel) parent ions C14H10+, and fragment ions (middle panel) C12H8+ and (bottom panel) C14H9+ as a function of time during the MD/DFTB dissociation dynamics of Anth˙+ (dot) and Phen˙+ (cross) for internal energies of 17.5 eV (black), 18.5 eV (red), and 20 eV (blue). The reported data points are obtained from an average of (NVE) 360 simulations. | ||
At all energies, Phen˙+ was found to dissociate faster than Anth˙+ in agreement with the recorded breakdown curves. The main dissociation channel corresponds to the loss of C2H2 (see Fig. 6, middle panel) and, to a lesser extent, to the loss of H (see Fig. 6, bottom panel). As can be clearly seen in Fig. 6, fragmentation will continue after 1 ns for all energies. We chose to confirm the results and investigate the convergence of the simulations by performing additional runs at 18.5 eV of energy, with a final total of 720 simulations of 1 ns and 360 independent simulations of 2 ns (similar plots as those reported in Fig. 6 are reported in Fig. S3 of the ESI†).
Focusing on C2H2 loss, we analysed the diversity of structures for C12H8+ based on their number of n-carbon rings, n varying from 4 to 7. They are assigned hereafter as [xyzt] where x is the number of 4 carbon rings, y of 5 carbon rings, z of 6 carbon rings, and t of 7 carbon rings (see Fig. 1). The fractions corresponding to each families are reported in Table 2. At all energies the most abundant isomers correspond to [0020] (1,2-EN+) structures, followed by [0120] (AcN˙+) at 18.5 eV and 20 eV for both Phen˙+ and Anth˙+. At 17.5 eV, EnMe˙+ [0110] and BP˙+ [0210] isomers are the second most abundant isomers in the case of Anth˙+ and Phen˙+, respectively. Interestingly, regarding the [0210] structures, alternative structures to BP˙+ were obtained in MD simulations (see Fig. S6 of the ESI†), BP˙+ remaining the major one. Several [0110] isomers were also observed, differing by the lengths and positions of the linear carbon chains (see Fig. S6 of the ESI†). Several isomers were also observed in the case of the [0010] family (structures possessing a single 6 carbon ring). Regarding the [1020] family, the BPh˙+ isomer was observed only in one out of 360 MD simulations at 18.5 eV and only in the case of Phen˙+. The CyN˙+ was not formed in our simulations.
| Internal energy [eV] | 17.5 | 18.5 | 20 | ||
|---|---|---|---|---|---|
| Simulations | 360 | 360 | 720 | 360 | |
| Fragment | Parent | 1 ns | 1 ns/2 ns | 1 ns | 1 ns |
| AcN | Phen˙+ | 0.12 | 0.41/0.35 | 0.33 | 0.31 |
| 0120 | Anth˙+ | 0 | 0.18/0.18 | 0.16 | 0.11 |
| BP | Phen˙+ | 0.32 | 0.05/0.12 | 0.06 | 0.09 |
| 0210 | Anth˙+ | 0.14 | 0.18/0.18 | 0.16 | 0.06 |
| 1.2-EN | Phen˙+ | 0.44 | 0.43/0.46 | 0.44 | 0.36 |
| 0020 | Anth˙+ | 0.43 | 0.47/0.49 | 0.48 | 0.42 |
| EnMe | Phen˙+ | 0.12 | 0.05/0.15 | 0.11 | 0.17 |
| 0110 | Anth˙+ | 0.43 | 0.18/0.15 | 0.16 | 0.27 |
| Bz | Phen˙+ | 0 | 0.02/0.05 | 0.04 | 0.05 |
| 0010 | Anth˙+ | 0 | 0/0 | 0.03 | 0.08 |
Regarding the formation mechanisms, although the branching ratio (BR) for loss of C2H2 is high at 20 eV, we focus on the lower energy results (17.5 and 18.5 eV) because we expect the lower energy mechanisms to occur in experiments. In our simulations, for both Anth˙+ and Phen˙+, isomerization precedes dissociation, involving H migration, carbon skeleton rearrangement, in particular carbon ring openings and the formation of 5-carbon rings. The intermediate species often have linear carbon chains. Snapshots extracted from four MD/DFTB simulations for Anth˙+ at 18.5 eV are reported in Fig. 7. This illustrates three examples of mechanisms leading to AcN˙+, and one leading to 2-EN˙+. Interestingly, in Fig. 7(a) and (b), H migration will follow the loss of C2H2 to lead to AcN˙+. In Fig. 7(c), isomerization into Phen˙+ precedes dissociation. In Fig. 7(d), H migration precedes isomerization and dissociation to form 2-EN˙+. Examples of mechanisms leading to the formation of 2-EN˙+ and AcN˙+ from Phen˙+ at 17.5 eV are reported in Fig. S6 of the ESI.†
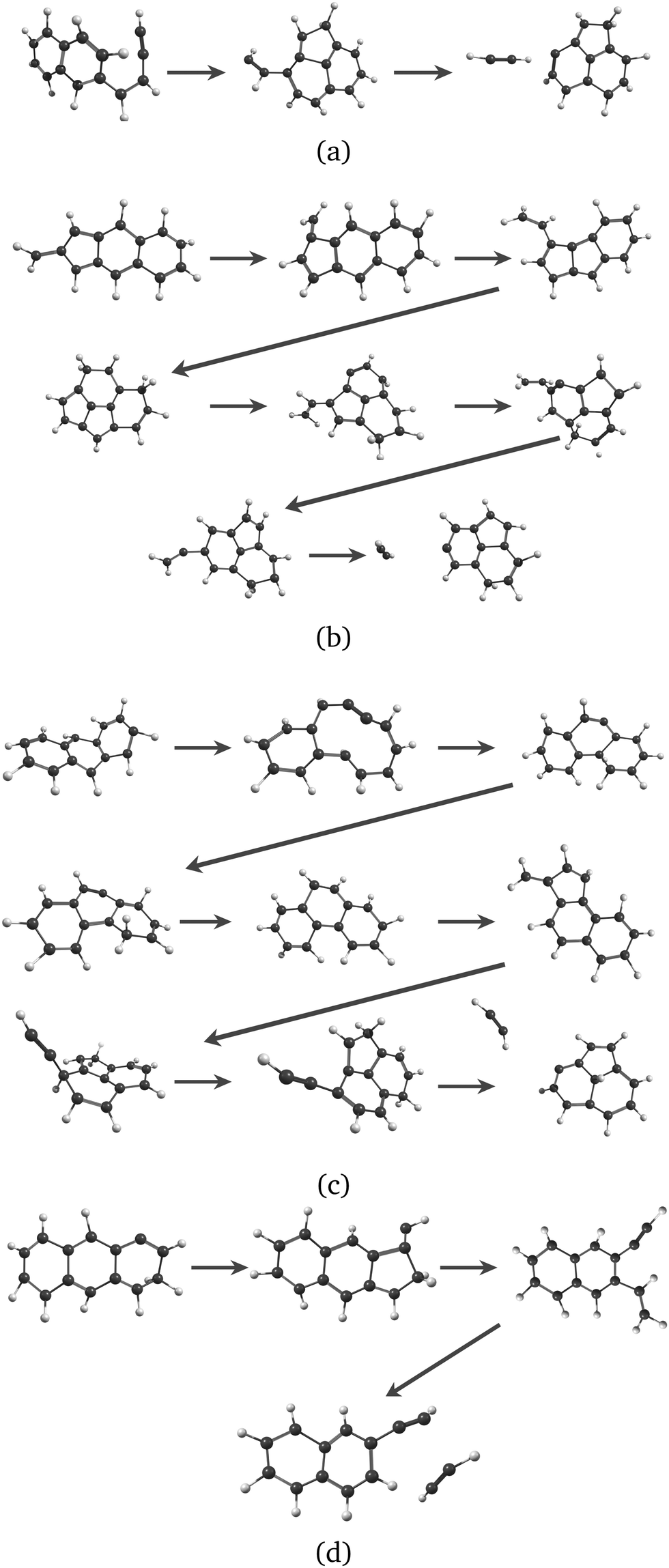 | ||
| Fig. 7 Examples of intermediates formed during the loss of C2H2 from Anth˙+ to lead to AcN˙+ (a–c) and 2-EN˙+ (d): snapshots withdrawn from 4 independent simulations at 18.5 eV. | ||
In the MD/DFTB simulations, the formation of 1,2-EN+ isomers is in competition with AcN˙+ as the major fragment, contrary to our experiments. In order to account for this discrepancy, we suggest that, with an energy of at least 17.5 eV as used in the MD simulations, all energy barriers within the paths leading to 1-EN˙+ and 2-EN˙+ can be overcome. In addition to the pathway from Anth˙+ to 2-EN˙+ shown in Fig. 7(d), an example of a mechanism leading to 1-EN˙+ from Phen˙+ at 17.5 eV is depicted in Fig. 8. It involves H migration, external ring opening, and formation of a “vinylidene” (fulvene-like) structure prior to the loss of C2H2. From such a structure, it should be possible to observe isomerization to AcN˙+ for sufficiently long simulations or sufficient internal energy in the 1,2-EN+ isomer. MD simulations from 1-EN˙+ with 10 eV of internal energy supported the latter scenario (Fig. 9). Indeed, we observed isomerization into AcN˙+ following the mechanisms reported in Fig. 7, involving C–C coupling and H migration.
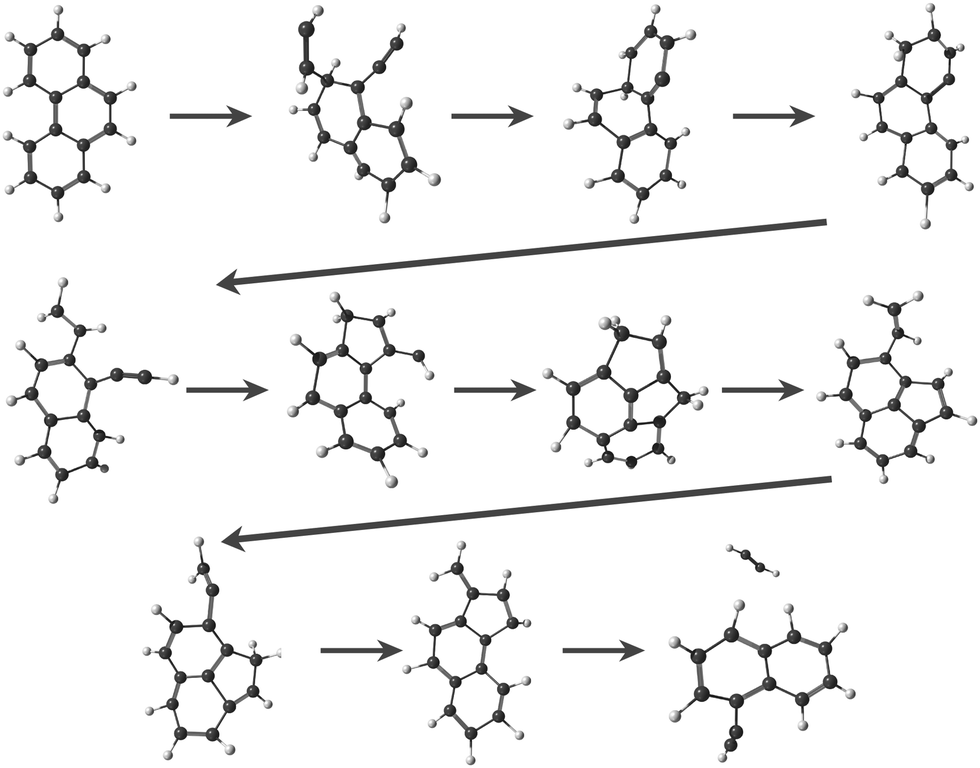 | ||
| Fig. 8 Examples of intermediates formed during the loss of C2H2 from Phen˙+ to lead to 1-EN˙+. | ||
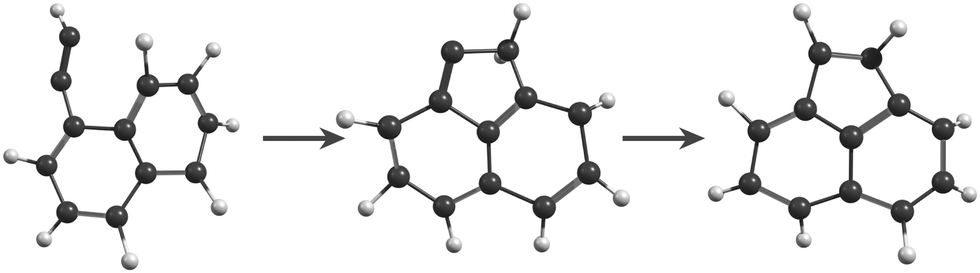 | ||
| Fig. 9 Snapshots from MD/DFTB simulations for isomerization of 1-EN˙+ (10 eV of internal energy) into AcN˙+. | ||
We should note here that in the experiment internal energies up to 30 eV might be deposited in the parent cations upon electron impact ionization. As explained in Section 2.3.2, the MD energies were chosen so as to be the lowest ones to observe the C2H2 loss, and they are not expected to mimic the experimental conditions. Furthermore, analysis of the dissociation paths in reasonable computational time is only possible at the nanosecond timescale although fragment structures are probed after several microseconds in the experiments.
3.5 Isomerization processes between 1-EN˙+ and AcN˙+
In order to elucidate a potential isomerization process between the functionalized bicyclic PAH 1-EN˙+ and the tricyclic AcN˙+, we performed additional experiments where the 1-EN and 2-EN neutral precursor were subjected to varying EI energies, thereby inducing possible additional isomerization and fragmentation processes. By measuring the IR fingerprint spectrum at different electron energies, isomerization processes within the mass channel can be identified. The experimental IRPD spectra of 1-EN˙+ and 2-EN˙+ at electron energies of 15 eV and 60 eV are shown in Fig. 10. No spectral variation with EI energy was observed for 2-EN˙+, but significant differences in several spectral features were observed for 1-EN˙+. In regions marked with gray boxes, we observe a decrease in the peak intensities at higher electron energy while regions marked with cyan boxes showed no change in peak intensities. The former regions contain rather unblended bands of 1-EN˙+, whereas the latter regions contain many overlapping bands of AcN˙+, indicating that 1-EN˙+ isomerizes into AcN˙+. This opens up the possibility of a sequential process involving 1-EN˙+ as an intermediate before forming AcN˙+, in the dissociative ionization of Anth˙+ and Phen˙+, as discussed in Section 3.4.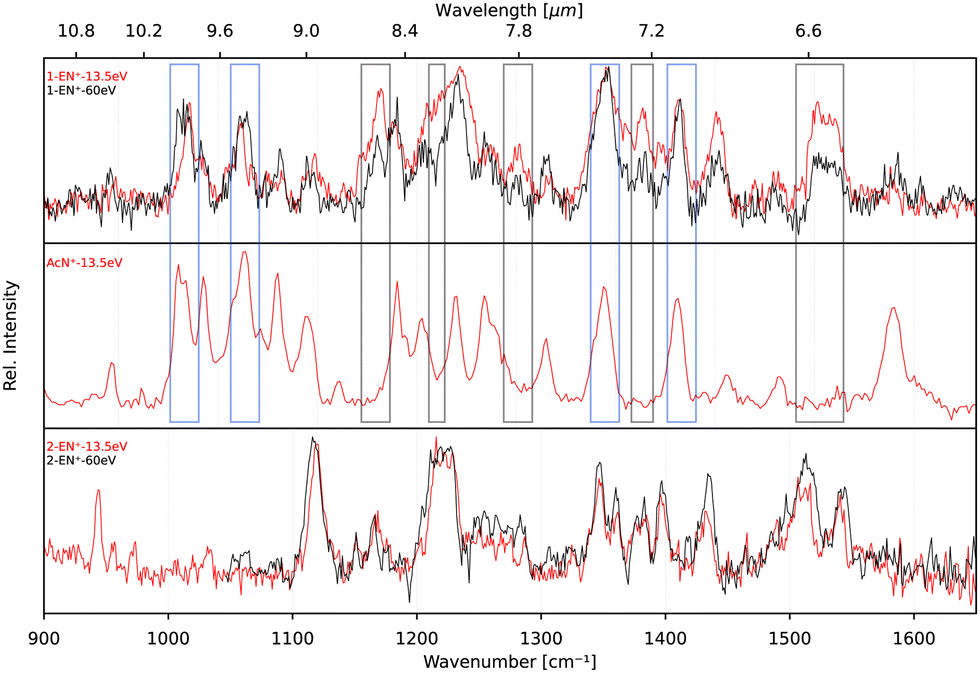 | ||
| Fig. 10 Experimental IRPD spectra of 1-EN˙+, ACN˙+, and 2-EN˙+ at electron impact ionization energies of 15 eV (red traces) and 60 eV (black traces). Regions where AcN˙+ bands overlap with 1-EN˙+ bands are indicated by cyan boxes, whereas areas where no AcN˙+ bands are present are marked with gray boxes. | ||
To look deeper into the feasibility of an isomerization pathway for 1-EN˙+, we have constructed the PES of two pathways that lead to the formation of ACN˙+ (see Fig. 11). One isomerization pathway where the first H migration takes place on the ethynyl group has been described previously.54 The overall isomerization process to form ACN˙+ is determined to be exothermic. However, a few barriers above the entrance energy exist. The highest transition state involving the ring closing reaction is calculated to be 2.5 eV higher in energy. A second pathway is found where a hydrogen atom moves from the ring to form a sp3 hybridized atom in the middle of the PAH base. This process requires one more transition state and contains an even higher lying transition state at 3.5 eV.
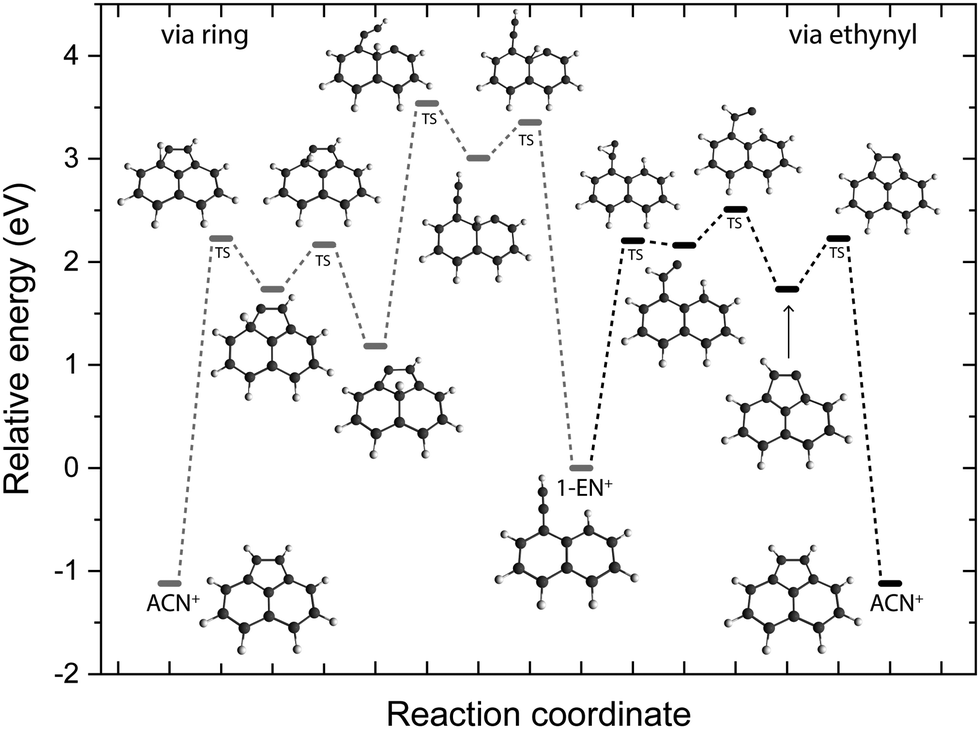 | ||
| Fig. 11 Potential energy surface of the isomerization pathways between 1-EN˙+ and ACN˙+ calculated at the B3LYP-GD3/6-311G(d,p) level of theory. The electronic energies including the zero-point vibrational energy correction are shown with respect to the 1-EN˙+. | ||
4 Astronomical implications
The formation of isomer fragments or products containing pentagonal rings such as AcN˙+ observed in this study has been previously discussed extensively in the literature. Examples include the C2H2 loss in the dissociative ionization of naphthalene, where the pentalene cation is formed25 and in that of two three-cyclic N-containing polyaromatics.55 Petrignani et al.56 also reported the formation of the fluorenyl cation C13H9+ by dissociative ionization of fluorene (C13H10), 9,10-dihydrophenanthrene (C14H12), and 9,10-dihydroanthracene (C14H12). The reaction of neutral allyl radicals (C3H5˙) with acetylene (C2H2) and o-benzyne has been shown to lead to the formation of cyclopentadiene (C5H6) and indene, respectively, as the primary isomer products,57,58 and a variety of five-membered ring structures were spectroscopically detected in benzene and naphthalene discharges.59–61We also note that in the AcN˙+ and BP˙+ spectra the bands around 1000 to 1150 cm−1 (8.7–10 μm) correspond to the in-plane C–H bending vibration involving the pentagonal ring. Features in this range were also observed in the IR spectra of rubicene˙+,62 corannulene˙+,63 and diindenoperylene˙+64 and were found to involve the pentagonal structures. At the era of the James Webb Space Telescope, these spectral features can serve as probes for detecting pentagonal carbon-ring containing PAHs in the interstellar medium (ISM).
5 Conclusions
We studied the dissociative ionization of anthracene and phenanthrene by spectroscopic analysis in the IR fingerprint region of its monocations and fragments formed via C2H2-loss. MD/DFTB simulations for Anth˙+ and Phen˙+ at different internal energies show that the main dissociation channel corresponds to the loss of C2H2 followed by the H-loss channel. The structural analysis of C2H2 loss fragments reveals several possible isomers with varying carbon rings (n = 4–7) and aliphatic chains (see Fig. 1). Strong evidence for the formation of a common product isomer AcN˙+ in the dissociation of Anth˙+ and Phen˙+ is shown by comparison of the recorded fragment vibrational spectra with anharmonic DFT calculations and recorded reference spectra. These pentagonal carbon-ring containing PAHs might be abundant in the ISM, and the present work provides laboratory reference data for their astronomical detection.Anth˙+ fragmentation also leads to the formation of a second isomer. Based on a comparison with anharmonic DFT calculations and reference spectra, in combination with the results of MD/DFTB calculations, we suggest the ethynyl-substituted naphthalene 2-EN˙+ as the second isomer for this C12H8+ fragment since it only involves hydrogen migration and bond breaking and no geometry rearrangements as seen in Fig. 7(d). The recent discovery of two isomers of ethynyl cyclopentadiene in TMC-110 suggests that PAHs containing ethynyl-chains, such as 1-EN and 2-EN, are potentially important fragments in the ISM. The presented IR spectra and calculations also revealed that 1-EN˙+ isomerizes into AcN˙+ with increasing electron energy, which might explain why phenanthrene dominantly fragments to AcN˙+ during dissociative ionization, with only minor contribution of another isomer. Through a combination of DFT calculations and experimental data we have also confirmed that anthracene does not isomerize to form a stable phenanthrene product upon ionization.
The results presented here demonstrate that high resolution action spectroscopy using rare-gas-tagging as implemented at the cryogenic ion trap user station FELion at FELIX, combined with sophisticated theoretical methods such as MD/DFTB, provides a powerful tool to investigate isomer selective fragmentation pathways of molecular ions such as the polycyclic aromatic hydrocarbons studied here.
Data availability
Source data for figures in this article is available at https://doi.org/10.34973/18wv-t810. Additional data supporting this article is available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Marie Skłodowska Curie Actions (MSCA) Innovative Training Networks (ITN) H2020-MSCA-ITN 2016 (EUROPAH project, G. A. 722346). We are grateful for the experimental support provided by the FELIX team and acknowledge Radboud University and the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. We also thank NWO Exact and Natural Sciences for the use of supercomputer facilities (Grant no. 2021.055). We thank the Cologne Laboratory Astrophysics group for providing the FELion ion trap instrument for the current experiments and the Cologne Center for Terahertz Spectroscopy (core facility, DFG grant SCHL 341/15-1) for supporting its operation. A. S. thanks the computing mesocenter CALMIP (“CALcul en MIdi Pyrénées”, UMS CNRS 3667) for generous allocation of computer resources. We also acknowledge funding from the European Research Council under the EU’s Seventh Framework Programme (FP/2007–2013) ERC-2013-SyG, Grant Agreement no. 610256 NANOCOSMOS.Notes and references
- A. Tielens, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef CAS.
- L. J. Allamandola, A. G. G. M. Tielens and J. R. Barker, Astrophys. J., Lett., 1985, 290, L25–L28 CrossRef CAS.
- L. J. Allamandola, A. G. G. M. Tielens and J. R. Barker, Astrophys. J., Suppl. Ser., 1989, 71, 733 CrossRef CAS PubMed.
- J. L. Puget and A. Léger, Annu. Rev. Astron. Astrophys., 1989, 27, 161–198 CrossRef CAS.
- V. L. Page, T. P. Snow and V. M. Bierbaum, Astrophys. J., Suppl. Ser., 2001, 132, 233–251 CrossRef.
- C. Boersma, J. D. Bregman and L. J. Allamandola, Astrophys. J., 2013, 769, 117 CrossRef.
- J. Montillaud, C. Joblin and D. Toublanc, Astron. Astrophys., 2013, 552, A15 CrossRef.
- H. Andrews, A. Candian and A. G. G. M. Tielens, Astron. Astrophys., 2016, 595, A23 CrossRef.
- M. J. Shannon and C. Boersma, Astrophys. J., 2019, 871, 124 CrossRef CAS.
- J. Cernicharo, M. Agúndez, C. Cabezas, B. Tercero, N. Marcelino, J. R. Pardo and P. de Vicente, Astron. Astrophys., 2021, 649, L15 CrossRef CAS PubMed.
- A. M. Burkhardt, K. L. K. Lee, P. B. Changala, C. N. Shingledecker, I. R. Cooke, R. A. Loomis, H. Wei, S. B. Charnley, E. Herbst, M. C. McCarthy and B. A. McGuire, Astrophys. J., Lett., 2021, 913, L18 CrossRef CAS.
- B. A. McGuire, R. A. Loomis, A. M. Burkhardt, K. L. K. Lee, C. N. Shingledecker, S. B. Charnley, I. R. Cooke, M. A. Cordiner, E. Herbst, S. Kalenskii, M. A. Siebert, E. R. Willis, C. Xue, A. J. Remijan and M. C. McCarthy, Science, 2021, 371, 1265–1269 CrossRef CAS PubMed.
- J. E. Elsila, N. P. de Leon, P. R. Buseck and R. N. Zare, Geochim. Cosmochim. Acta, 2005, 69, 1349–1357 CrossRef CAS.
- H. Sabbah, M. Carlos, P. Jenniskens, M. H. Shaddad, J. Duprat, C. A. Goodrich and C. Joblin, Astrophys. J., 2022, 931, 91 CrossRef.
- Y. Gotkis, M. Oleinikova, M. Naor and C. Lifshitz, J. Phys. Chem., 1993, 97, 12282–12290 CrossRef CAS.
- Y. Ling and C. Lifshitz, J. Phys. Chem. A, 1998, 102, 708–716 CrossRef CAS.
- J. Zhen, S. R. Castillo, C. Joblin, G. Mulas, H. Sabbah, A. Giuliani, L. Nahon, S. Martin, J.-P. Champeaux and P. M. Mayer, Astrophys. J., 2016, 822, 113 CrossRef PubMed.
- B. West, A. Sit, S. Mohamed, C. Joblin, V. Blanchet, A. Bodi and P. M. Mayer, J. Phys. Chem. A, 2014, 118, 9870–9878 CrossRef CAS PubMed.
- Y. Ling, J. M. L. Martin and C. Lifshitz, J. Phys. Chem. A, 1997, 101, 219–226 CrossRef CAS.
- H. A. B. Johansson, H. Zettergren, A. I. S. Holm, N. Haag, S. B. Nielsen, J. A. Wyer, M. B. S. Kirketerp, K. Støchkel, P. Hvelplund, H. T. Schmidt and H. Cederquist, J. Chem. Phys., 2011, 135, 084304 CrossRef CAS PubMed.
- J. Oomens, A. G. G. M. Tielens, B. G. Sartakov, G. von Helden and G. Meijer, Astrophys. J., 2003, 591, 968–985 CrossRef CAS.
- J. M. Bakker, B. Redlich, A. F. G. van der Meer and J. Oomens, Astrophys. J., 2011, 741, 74 CrossRef.
- P. Jusko, A. Simon, S. Banhatti, S. Brünken and C. Joblin, ChemPhysChem, 2018, 19, 3173 CrossRef.
- S. Panchagnula, J. Bouwman, D. B. Rap, P. Castellanos, A. Candian, C. Mackie, S. Banhatti, S. Brünken, H. Linnartz and A. G. G. M. Tielens, Phys. Chem. Chem. Phys., 2020, 22, 21651–21663 RSC.
- J. Bouwman, A. J. de Haas and J. Oomens, Chem. Commun., 2016, 52, 2636–2638 RSC.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- P. Jusko, S. Brünken, O. Asvany, S. Thorwirth, A. Stoffels, L. van der Meer, G. Berden, B. Redlich, J. Oomens and S. Schlemmer, Faraday Discuss., 2019, 217, 172–202 RSC.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CrossRef CAS.
- S. Banhatti, J. Palotás, P. Jusko, B. Redlich, J. Oomens, S. Schlemmer and S. Brünken, Astron. Astrophys., 2021, 648, A61 CrossRef CAS.
- O. Asvany, F. Bielau, D. Moratschke, J. Krause and S. Schlemmer, Rev. Sci. Instrum., 2010, 81, 076102 CrossRef PubMed.
- N. R. Daly, Rev. Sci. Instrum., 1960, 31, 264–267 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- V. Barone, J. Phys. Chem. A, 2005, 122, 014108 CrossRef PubMed.
- V. Barone, M. Biczysko and J. Bloino, Phys. Chem. Chem. Phys., 2014, 16, 1759–1787 RSC.
- J. H. Marks, E. Miliordos and M. A. Duncan, J. Phys. Chem. A, 2021, 154, 064306 CrossRef CAS PubMed.
- S. Brünken, F. Lipparini, A. Stoffels, P. Jusko, B. Redlich, J. Gauss and S. Schlemmer, J. Phys. Chem. A, 2019, 123, 8053–8062 CrossRef PubMed.
- H. Piest, G. von Helden and G. Meijer, Astrophys. J., 1999, 520, L75–L78 CrossRef CAS.
- H. Piest, J. Oomens, J. Bakker, G. von Helden and G. Meijer, Spectrochim. Acta, Part A, 2001, 57, 717–735 CrossRef.
- A. Simon, M. Rapacioli, G. Rouaut and F. Gadéa, Philos. Trans. R. Soc., A, 2017, 375, 20160195 CrossRef PubMed.
- P. Jusko, A. Simon, G. Wenzel, S. Brünken, S. Schlemmer and C. Joblin, Chem. Phys. Lett., 2018, 698, 206–210 CrossRef CAS PubMed.
- M. Rapacioli, S. Cazaux, N. Foley, A. Simon, R. Hoekstra and T. Schlatholter, Phys. Chem. Chem. Phys., 2018, 20, 22427–22438 RSC.
- A. Simon, J. P. Champeaux, M. Rapacioli, P. Moretto Capelle, F. X. Gadea and M. Sence, Theor. Chem. Acc., 2018, 137, 106 Search PubMed.
- C. Joblin, G. Wenzel, S. Rodriguez Castillo, A. Simon, H. Sabbah, A. Bonnamy, D. Toublanc, G. Mulas, M. Ji, A. Giuliani and L. Nahon, J. Phys.: Conf. Ser., 2020, 1412, 062002 CrossRef CAS.
- T. Heine, M. Rapacioli, S. Patchkovskii, J. Frenzel, A. Koster, P. Calaminici, H. A. Duarte, S. Escalante, R. Flores-Moreno, A. Goursot, J. Reveles, D. Salahub and A. Vela, deMonNano, 2009, https://demon-nano.ups-tlse.fr/.
- P. J. M. van der Burgt and M. Dunne, Eur. Phys. J. D, 2018, 72, 31 CrossRef.
- P. J. M. van der Burgt, M. Dunne and M. L. Gradziel, J. Phys.: Conf. Ser., 2019, 1289, 012008 CrossRef CAS.
- S. D. Wiersma, A. Candian, J. M. Bakker and A. Petrignani, Mon. Not. R. Astron. Soc., 2022, 516, 5216–5226 CrossRef.
- J. Oomens, B. G. Sartakov, G. Meijer and G. von Helden, Int. J. Mass Spectrom., 2006, 254, 1–19 CrossRef CAS.
- D. Freedman, R. Pisani and R. Purves, Statistics: Fourth International Student Edition, W.W. Norton & Company, 2007 Search PubMed.
- A. K. Lemmens, D. B. Rap, J. M. M. Thunnissen, C. J. Mackie, A. Candian, A. G. G. M. Tielens, A. M. Rijs and W. J. Buma, Astron. Astrophys., 2019, 628, A130 CrossRef CAS.
- B. Coutant and P. Brechignac, J. Chem. Phys., 1994, 100, 7087–7092 CrossRef CAS.
- G. Mulas, C. Falvo, P. Cassam-Chenai and C. Joblin, J. Chem. Phys., 2018, 149, 144102 CrossRef PubMed.
- G. Wenzel, A. Simon, S. Banhatti, P. Jusko, S. Schlemmer, S. Brünken and C. Joblin, J. Mol. Spectrosc., 2022, 385, 111620 CrossRef CAS.
- A. Lifshitz, C. Tamburu and F. Dubnikova, Proc. Combust. Inst., 2007, 31(1), 241–248 CrossRef.
- A. J. de Haas, J. Oomens and J. Bouwman, Phys. Chem. Chem. Phys., 2017, 19, 2974–2980 RSC.
- A. Petrignani, M. Vala, J. R. Eyler, A. G. G. M. Tielens, G. Berden, A. F. G. van der Meer, B. Redlich and J. Oomens, Astrophys. J., 2016, 826, 33 CrossRef.
- J. Bouwman, A. Bodi, J. Oomens and P. Hemberger, Phys. Chem. Chem. Phys., 2015, 17, 20508–20514 RSC.
- M. N. McCabe, P. Hemberger, E. Reusch, A. Bodi and J. Bouwman, J. Phys. Chem. Lett., 2020, 11, 2859–2863 CrossRef CAS PubMed.
- M. C. McCarthy, K. L. K. Lee, P. B. Carroll, J. P. Porterfield, P. B. Changala, J. H. Thorpe and J. F. Stanton, J. Phys. Chem. A, 2020, 124, 5170–5181 CrossRef CAS PubMed.
- A. K. Lemmens, D. B. Rap, J. M. M. Thunnissen, B. Willemsen and A. M. Rijs, Nat. Commun., 2020, 11, 269 CrossRef CAS PubMed.
- A. K. Lemmens, D. B. Rap, S. Brünken, W. J. Buma and A. M. Rijs, Phys. Chem. Chem. Phys., 2022, 24, 14816–14824 RSC.
- J. Bouwman, C. Boersma, M. Bulak, J. Kamer, P. Castellanos, A. G. G. M. Tielens and H. Linnartz, Astron. Astrophys., 2020, 636, A57 CrossRef CAS.
- H. Alvaro Galué, C. A. Rice, J. D. Steill and J. Oomens, J. Chem. Phys., 2011, 134, 054310 CrossRef PubMed.
- J. Zhen, A. Candian, P. Castellanos, J. Bouwman, H. Linnartz and A. G. G. M. Tielens, Astrophys. J., 2018, 854, 27 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03835h |
| ‡ We should note that the electron impact ion source used in this experiment is not ideally suited to measure appearance energies since our electron energy has a resolution of 2.7 eV, corresponding to the voltage drop across the filament. As a result the appearance energies are shifted to lower energies. |
| This journal is © the Owner Societies 2022 |