
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of high photoluminescence quantum yield of melem†
Hiroki
Kiuchi
 a,
Yoriko
Sonoda
b,
Yuto
Miyake
a,
Fumiya
Kobayashi
c,
Jun’ya
Tsutsumi
b,
Makoto
Tadokoro
c and
Kaname
Kanai
*a
a,
Yoriko
Sonoda
b,
Yuto
Miyake
a,
Fumiya
Kobayashi
c,
Jun’ya
Tsutsumi
b,
Makoto
Tadokoro
c and
Kaname
Kanai
*a
aDepartment of Physics, Faculty of Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan. E-mail: kaname@rs.tus.ac.jp
bResearch Institute for Advanced Electronics and Photonics, National Institute of Advanced Industrial Science and Technology (AIST), Higashi 1-1-1, 305-8565 Tsukuba, Ibaraki, Japan
cDepartment of Chemistry, Faculty of Science, Tokyo University of Science, Kagurazaka 1-3, Shinjuku-ku, Tokyo 162-8601, Japan
First published on 16th September 2022
Abstract
To produce high-efficiency organic light-emitting diodes, materials that exhibit thermally activated delayed fluorescence (TADF) are attracting attention as alternatives to phosphorescent materials containing heavy metallic elements. Melem, a small molecule with a heptazine backbone composed only of nitrogen, carbon, and hydrogen, is known to emit light in the near-ultraviolet region and exhibit high photoluminescence (PL) quantum yield and delayed fluorescence. However, the mechanism underlying the high PL quantum yield remains unclear. This study aimed to elucidate the mechanism of the high PL quantum yield of melem by examining its optical properties in detail. When the amount of dissolved oxygen in the melem solution was increased by bubbling oxygen through it, the PL quantum yield and emission lifetime decreased significantly, indicating that the triplet state was involved in the light-emission mechanism. Furthermore, the temperature dependence of the PL intensity of melem was investigated; the PL intensity decreased with decreasing temperature, indicating that it increases thermally. The experimental results show that melem is a TADF material that produces an extremely high PL quantum yield by upconversion from the triplet to the singlet excited state.
1. Introduction
Two-dimensional materials, such as graphene, have a wide range of potential applications, including new electronic functional materials, photocatalysts, and optical materials.1,2 In recent years, one of the 2D-like materials that have attracted particular attention as next-generation functional materials is carbon nitride (CN) polymers. CN-polymers are organic molecules consisting mainly of nitrogen, carbon, and hydrogen and have a metal-free structure. Furthermore, owing to their low material and fabrication costs, they can be synthesized using a simple thermal polymerization method, making them attractive. Melon is a representative CN-polymer that is a linear polymer composed of a one-dimensional chain of 2,5,8-triamino-tri-s-triazine (melem) units, as shown in Fig. 1(a). Melon can generate hydrogen in sunlight because of its ability as a photocatalyst to decompose water and generate hydrogen using visible light.3 Additionally, melon is a direct-gap semiconductor as expected from its energy band structure, and is known to emit blue fluorescence, as shown in Fig. 1(b). Therefore, several previous studies used melon as a blue light-emitting layer in organic light-emitting diode (OLED) devices.4,5 Conversely, small-molecule melem, a typical precursor of CN-polymers, such as melon,6–8 has a larger energy gap than melon and emits in the near-ultraviolet (UV) region. The photoluminescence (PL) quantum yield of melem crystals is very high (approximately 80%).5,9,10 Although there have been reports of OLED devices that use melem as the light-emitting layer, stable and high-brightness devices have not yet been successfully realized.11 The realization of highly efficient near-UV-emitting OLEDs is expected for new white-light-emitting devices, information storage applications, backlight sources for full-color OLED displays, applications in sterilization technology, and medical applications.12,13 However, the mechanism by which melem exhibits high quantum yield remains unclear. Melem exhibits delayed fluorescence,4,14,15 which may contribute to the high PL quantum yield; however, the specific mechanism of the PL has not been clarified. | ||
| Fig. 1 (a) Molecular structures of melamine, melem, melem tetramer (MT), and melon. (b) Photographs of melem, MT, and melon powdery samples under ultraviolet (mercury lamp) irradiation. | ||
In OLEDs, based on the spin statistical law, singlet and triplet-excited states are produced with probabilities of 25% and 75%, respectively. Therefore, only 25% of the excitons in fluorescent materials are involved in the emission of light. Besides, a portion of that 25% of excitons will be lost by thermal deactivation. Therefore, the extraction of triplet excitons is the key to improving the efficiency of OLEDs. For many years, there have been many efforts to realize highly efficient OLED devices that use phosphorescence, which is light emitted from triplet-excited states; however, to use phosphorescence, it is necessary to emit light at very low temperatures, such as 77 K,16 or to use heavy atoms, such as Ir or Pt that have large spin-orbit interactions.17,18 However, the Ir reserves are limited and device manufacturing is costly. Thermally activated delayed fluorescence (TADF) materials have been reported as third-generation light-emitting materials.19 TADF upconverts the triplet-excited state to the singlet excited state via thermal activation and uses the fluorescence process from the S1 excited state to the ground state. The use of TADF materials in light-emitting layers enables almost 100% upconversion,19 and because they do not require low temperatures and heavy metals as phosphorescent materials, they have attracted much attention as new OLED materials in recent years. In addition, hyperfluorescence, a fourth-generation luminescence technology that combines third-generation TADF materials and first-generation fluorescent materials, has been intensively studied in recent years. Hyperfluorescence can achieve a 100% internal quantum yield, and because it emits light from fluorescent molecules, a narrow emission spectrum with high color purity can be obtained.20
A possible mechanism for the high PL quantum yield exhibited by melem is TADF that has also been reported for molecules with a heptazine skeleton similar to that of melem.21–25 Since melem has a very high quantum yield at room temperature, phosphorescence contribution is unlikely, and TADF is a possible origin. Furthermore, melem has been reported to exhibit delayed fluorescence,4,14,15 but the specific mechanism of PL, such as whether this is because of TADF and how it relates to the quantum yield, has not been clarified. Understanding the origin of the high PL quantum yield of melem should provide important information for the future development of light-emitting materials for third- and fourth-generation luminescence technologies for OLEDs.
The purpose of this study is to elucidate the mechanism underlying the high PL quantum yield of melem. Bubbling oxygen, a triplet quencher, into the melem solution significantly decreased the quantum yield and fluorescence lifetime, indicating that the triplet state is involved in the luminescence mechanism. The temperature dependence of the PL intensity was measured, and it was confirmed that the PL intensity increased with increasing temperature. In fluorescent materials, as the temperature increases, intramolecular motions, such as vibration and rotation, tend to cause nonradiative deactivation, resulting in a decrease in PL intensity. However, the opposite phenomenon was observed in melem, indicating that the PL process was thermally activated. These results show that TADF, owing to upconversion from the triplet-excited state, is responsible for the high PL quantum yield of melem. Furthermore, comparing the PL quantum yields of melem crystals and melem solutions showed that the PL quantum yield of melem crystals was larger than that of melem solutions, indicating that the hydrogen bonds between molecules in the crystal suppressed intramolecular motions, such as vibration and rotation, resulting in an improved PL quantum yield.
2. Experimental and calculation methods
2.1. Synthesis of melem crystals (c-melem)
1,3,5-Triazine-2,4,6-triamine (melamine, Fig. 1(a)) (5 g; purity: 99.0%; FUJIFILM Wako Pure Chem. Co., Ltd; 139-00945) was placed in a tube furnace (KTF035N1; Koyo Thermo Systems Co., Ltd) in an N2 atmosphere (purity: 99.99995%). The temperature of the material was increased from room temperature at 1 °C min−1 and maintained at 310 °C for 5 h. The material was subsequently cooled naturally to obtain melem. The as-synthesized melem (500 mg) was washed with 50 ml of N,N-dimethylformamide (DMF; purity: 99.5%; FUJIFILM Wako Pure Chem. Co., Ltd; 045-02916) in which melamine is easily soluble and melem is less soluble.26,27 The mixture was sonicated and centrifuged twice for 15 min before adding 50 ml of acetone (purity: 99.5%; FUJIFILM Wako Pure Chem. Co., Ltd.; 012-00343)28 to remove DMF. Thereafter, 2 sets of 15 min sonication at 42 kHz and centrifugation at approximately 2500 rpm were carried out. The powdered melem sample was subsequently air-dried to obtain c-melem.2.2. Synthesis of melem tetramer (MT) and melon
Melamine (5 g; purity: 99.0%; FUJIFILM Wako Pure Chem. Co., Ltd; 139-00945) was placed in a tube furnace (KTF035N1; Koyo Thermo Systems Co., Ltd) in an N2 atmosphere (purity: 99.99995%). The temperature of the material was increased from room temperature at 1 °C min−1 and maintained at 415 °C and 450 °C for 5 h. After reducing the temperature to room temperature at a rate of 2 °C min−1, the powdered product in the quartz test tube was removed and ground in a mortar to obtain uniform particle sizes.Regarding MT, the procedures in a vacuum were performed as follows, based on the previous study:5 the sample was heated in an alumina crucible using the resistive heat generated by a tungsten wire wrapped around the periphery of the crucible. The average vacuum pressure during calcination was approximately 1 × 10−4 Pa. During heating, a thermocouple was inserted into the powder in the crucible in the chamber to determine the temperature, and the temperature was increased to 180 °C at a rate of approximately 1.5 °C min−1. After a predetermined time (4.0 or 5.3 h), the heating was stopped and the sample was removed from the chamber after it cooled to room temperature. The obtained sample was ground using a mortar and pestle.
2.3. Preparation of melem solution
c-Melem was dissolved in superdehydrated dimethyl sulfoxide (DMSO, s.d.; purity: 99.0%; FUJIFILM Wako Pure Chem). The solution was subsequently placed in a quartz cell (T-23-UV-10, Tosoh Quartz Corporation), covered with a septum (Precision Seal Z565776, Sigma-Aldrich), and bubbled. Nitrogen was bubbled through for 10 min followed by oxygen for 3 min. An oxygen can (oxygen 95%; Taiyo Nippon Sanso Gas & Welding Co.) was used for oxygen bubbling. Measurements of solution samples bubbled with gas were completed within approximately 10 minutes after bubbling.2.4. Characterization
PL spectra and absolute PL quantum yields of the powdery samples were measured using a spectrometer (Quantaurus-QY, C11347-01; Hamamatsu Photonics, Ltd) with the samples in quartz Petri dishes (A10095-03; Hamamatsu Photonics, Ltd). The excitation wavelength used in the measurements of the c-melem and melem solution was λex = 320 nm, whereas that of MT and melon was λex = 340 nm. PL measurements were performed in the emission wavelength range of 330–600 nm for all samples. A SHIMADZU RF-6000 was used to measure the temperature dependence of the PL spectra, and a CoolSpeK UV/CD USP-203 cryostat was used for cooling. The measurements were performed at an excitation wavelength of 310 nm with an interval of 1.0 nm while decreasing the temperature from 100 °C to −191 °C. To measure at a stable sample temperature, 3 measurements were taken at 15, 20, and 25 min after the desired temperature was reached.The samples for the emission lifetime measurements were prepared by sandwiching a powder sample between 2 quartz plates (20 × 20 mm2). The measurements were performed using a compact fluorescence lifetime measurement system (Quantaurus-Tau, C11367-01; Hamamatsu Photonics, Ltd) at excitation and monitoring emission wavelengths of 340 nm and 370 nm, respectively.
The measurements for temperature dependence on the emission lifetime were determined using an ANCF003 nanosecond visible and near-infrared emission lifetime measurement system. The wavelengths for excitation light and emission used for the measurements were 278 nm and 375 nm, respectively. The measurements were performed using a short-pass filter at 330 nm and a band-pass filter cut below 350 nm.
2.5. Calculation method
Materials Studio (BIOVIA) was used as the calculation software, and energy level calculations were performed using the CASTEP29 plane wave basis set and general gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) as the correlation functional30 (Dassault Systemes BIOVIA). The details of the calculation are as follows: SCF tolerance threshold: 1 × 10−6 eV per atom; core treatment: all electrons were included in the calculation; basis set: DNP; basis file: 4.4.3. Results and discussions
First, the melem, MT, and melon samples used in the following experiments were obtained by a synthetic method based on previous study,5 and the obtained samples were confirmed to be the target materials by X-ray diffraction method (XRD) and other methods. The data of the XRD of the melem, MT, and melon samples we obtained are shown in Fig. S1 in the ESI.†3.1. Differences in delayed luminescence due to different degrees of polymerization of CN compounds
Transient PL decay for melem, MT, and melon was measured to investigate the cause of the high quantum yield of melem. Melon is a typical example of CN compounds that are active under visible light, and Fig. 1(a) shows that it is formed from the layered polymerization of a 1D polymer with melem as a unit. MT is a small molecular weight CN compound obtained from thermally polymerizing melem, and, as shown in Fig. 1(a), it is an oligomer consisting of 4 linearly linked melem units. In this section, we discuss how different degrees of polymerization result in different luminescent properties.5The emission lifetime of PL produced by a transition process is defined as τ, the PL intensity at t = 0 s is A, and PL intensity after t s is I(t). In reality, PL can be considered as consisting of the contributions from multiple components with different lifetimes owing to several different transition processes; thus, if there are n luminescent components contributing to the PL, the PL intensity can be obtained as follows:
 | (1) |
 | ||
| Fig. 2 Transient PL decay of melem, MT, and melon powdery samples. τ represents the average fluorescent lifetime of the fast and slow components of each sample. The dots represent the experimental data and the solid lines represent the fitted results. The vertical axis is a logarithmic scale. | ||
| Sample | Prompt | Delay | ||||
|---|---|---|---|---|---|---|
| A 1 | τ 1/ns | R p | A 2 | τ 2/ns | R d | |
| c-Melem | 2793 | 31.5 | 0.29 | 803 | 266 | 0.71 |
| MT | 221 | 3.23 | 0.54 | 38 | 16 | 0.46 |
| Melon | 241 | 2.43 | 0.60 | 44 | 9 | 0.40 |
Fig. 2 shows the results of the transient PL decay measurements of melem, MT, and melon. τ represents the average emission lifetime of the fast (Prompt) and slow (Delay) components for each sample. τ is calculated using eqn (2):31
 | (2) |
The results in Table 1 also show that the value of τ2 for delayed luminescence decreases as the polymerization degree increases from melem to MT and melon. The weighted averages, Rp and Rd, are expressed in eqn (3). Rp and Rd are the relative ratios of emission intensity Ai at t = 0 s for each component for i = 1 and 2, respectively; because we performed the fitting analysis with two components, Rp represents the fraction of prompt component with a short lifetime and Rd represents the fraction of delayed component with a long lifetime. Rp and Rd are calculated using the values in Table 1, and the delayed component accounts for approximately 70% of the total luminescence, which is the majority of the luminescence of melem. The values of Rp and Rd are listed in Table 1.
 | (3) |
The origin of the delayed luminescence remains unclear, but similarly delayed luminescence has been reported in previous studies.4,14,15 However, a previous study reported that the absolute PL quantum yields of melem, MT, and melon are very different, with the quantum yield of melem being very high compared with others.5 This trend is consistent with the trend in the emission lifetime shown in Fig. 2. Thus, the difference between the emission lifetimes of MT and melon is not as large as the difference between them and melem, which is similar to the trend in the change in the PL quantum yield. In general, delayed luminescence increases the number of photons observed for longer periods, and there is a direct relationship between delayed luminescence and higher PL quantum yields. Therefore, the delayed luminescence of melem may significantly contribute to its high PL quantum yield.
3.2. Effect of triplet quenching material on luminescence properties
Delayed luminescence is generally observed in transitions between states with different spin multiplicities. Therefore, we investigated whether the triplet-excited state was involved in the emission process of the delayed luminescence of melem. The changes in the PL quantum yield and emission lifetime caused by bubbling oxygen, a triplet quencher,32–34 into a melem solution (superdehydrated DMSO) were investigated. The ground state of oxygen is a triplet, but there is an excited singlet state with very low energy (Esinglet = 95 kJ mol−1); thus, molecules in the triplet-excited state with energies above Esinglet are efficiently quenched by the exchange mechanism. Bubbling oxygen into the solution causes a change in the emission lifetime owing to the nonradiative deactivation of excitons in the excited triplet state of melem. To evaluate the optical properties with and without oxygen, 3 samples were prepared in the following order, sample with oxygen bubbling (O2 bubbling), sample without bubbling (air), and sample with nitrogen bubbling (N2 bubbling). Bubbling with N2 gas can remove the oxygen originally dissolved in the melem solution.35 The emission lifetime and intensity measurements are shown in Fig. 3 and the PL quantum yields for each sample are summarized in Table 2. | ||
| Fig. 3 (a) Transient PL decay of air, N2 bubbling, and O2 bubbling melem solutions. The vertical axis is a logarithmic scale. The initial luminescence intensity was normalized to 3000 counts. The solvent for the melem solution was superdehydrated DMSO. (b) PL spectra of air, N2 bubbling, and O2 bubbling melem solution samples. The vertical axis represents the PL emission intensity, with the peak at 320 nm because of photoexcitation. (c) PL spectra of c-melem and melem in DMSO solution (melem solution; N2 bubbling). The peak around 320 nm is caused by photoexcitation. Φ represents the PL quantum yield of each sample and is summarized in Table 2. | ||
| Sample | Φ /% | τ /ns |
|---|---|---|
| a The absolute PL quantum yield was obtained by integrating the PL spectral intensity at wavelengths between 330 and 600 nm. b The wavelength of the excitation light is 340 nm and the measuring wavelength is 370 nm. | ||
| c-Melem | 77.7 | 176 |
| MT | 10.45 | 9.1 |
| Melon | 7.45 | 5.1 |
| Melem soln. (N2 bubbling) | 66.8 | 330 |
| Melem soln. (Air) | 24.7 | 158 |
| Melem soln. (O2 bubbling) | 5.8 | 35 |
Fig. 3(a) shows that the emission lifetime and PL quantum yield decrease in the order of the amount of dissolved oxygen, that is, O2 bubbling, air, and N2 bubbling. These results indicate that the delayed luminescence of melem involves an emission process via triplet-excited states. Furthermore, comparing the results for N2 bubbling and O2 bubbling confirm that the PL quantum yield of melem is improved by approximately 60% owing to the emission process through the triplet-excited states. These results indicate that the emission process involving triplet-excited states is a major factor in the high PL quantum yield of melem. From Fig. 3(a), the slopes of the spectra of air and N2 bubbling change after a delay of approximately 0.1 μs. This shows that the PLs of these samples have 2 components: prompt and delayed emission components. Oppositely, the spectrum for the O2 bubbling sample has a single slope, indicating that it has only a prompt emission component. From this result, the luminescence of the O2 bubbling sample is considered to be that of the S1–S0 transition and the PL quantum yield calculated from the experimental results is Φ(S1–S0) = 5.8%.
Fig. 3(b) shows the PL spectra for O2 bubbling, air, and N2 bubbling samples. The shape of the PL spectrum does not depend on the presence or absence of an emission process via the triplet, this is more clearly shown in Fig. S2 in the ESI,† confirming that the delayed luminescence is not a room-temperature phosphorescence. This is because, generally, luminescence changes from fluorescence to phosphorescence, changing the shape of the spectrum and emission wavelength. This suggests that the prompt and delayed emission components observed in the spectra of the air and N2 bubbling samples in Fig. 3(a) are emissions from the transition that produces fluorescence. Therefore, the delayed luminescence of melem is attributed to the delayed fluorescence that is responsible for its high PL quantum yield. Furthermore, comparing the prompt emission component of the luminescence of O2 bubbling with that which appears in the emission lifetime spectra of the air and N2 bubbling samples in Fig. 3(a), the slopes of the prompt component versus time do not coincide. This may indicate that the transitions that produce the prompt component of the air and N2 bubbling samples may involve intersystem crossings (ISCs) between the higher-order Tn states and S1.
3.3. Differences in the luminescence properties of melem powders and solutions
In this section, the luminescence properties of melem powder and solution are compared. The measurements were performed in solution to avoid the formation of aggregates and to evaluate the luminescence properties of isolated melem molecules. In the PL measurements shown in Fig. 3(b), the concentration of the solution sample was adjusted such that the absorbance value of melem is maintained at approximately 0.4 at 320 nm of the wavelength of the excitation light. As shown in Fig. 3(c), comparing the wavelengths of the maximum intensities of the PL spectra, the melem solution (N2 bubbling) has a longer wavelength (6 nm) compared with c-melem, resulting from the solvent effect of DMSO, a polar solvent.36 As shown in Table 2, a comparison of the PL quantum yield values of c-melem and melem solution (N2 bubbling) evaluated using PL measurements show that the PL quantum yield of c-melem is 10.9% higher. This difference in quantum yield can be explained by considering intermolecular interactions in the crystalline state. Intermolecular interactions, such as hydrogen bonding, suppress nonradiative deactivation of the excited state of molecules after light absorption because of their inhibitory effect on molecular rotation and vibration. This phenomenon is a type of aggregation-induced emission (AIE), which does not emit light in dilute solution but emits strongly in the solid or aggregated state. Consequently, in some cases, the PL quantum yields differ between the solution and crystalline states.37,38 The crystal structures of c-melem are shown in Fig. 4(a) and (b). In the bc-plane of the c-melem crystal, strong hydrogen bonds exist between melem molecules and π-stacks of melem molecules in the a-axis direction.6 The NH⋯H hydrogen bond between the hydrogen atom of a melem molecule and the nitrogen atom of a neighboring melem molecule is a strong enough intermolecular interaction to inhibit molecular motion and deformation to some extent. Therefore, the PL quantum yield of c-melem is considered to be higher because the hydrogen bonds and other intermolecular interactions suppress molecular motions, such as rotation and vibration, thus reducing nonradiative deactivation. | ||
| Fig. 4 (a) Crystal structure of melem. (b) Arrangement of melem molecules in the bc-plane of melem crystals. The gray dotted lines in the figure represent intermolecular hydrogen bonds. In the crystal, a melem molecule forms 8 hydrogen bonds with 5 surrounding melem molecules. | ||
3.4. Mechanism of the delayed luminescence of melem
Delayed fluorescence has been observed in heptazine derivatives with a heptazine skeleton similar to that of melem and has been reported to exhibit TADF.21–24 TADF is produced by a mechanism in which excitons that are photoexcited to the S1 level and subsequently deactivated through the ISC to the T1 level transition back to the S1 level through thermal upconversion via T1 → S1 reverse ISC (RISC), and generate fluorescence via the S1 → S0 transition. Compared with the normal S1 → S0 transition, TADF transitions between states with different spin multiplicities in the emission process result in delayed fluorescence. To achieve high efficiency in TADF materials, RISC must be induced efficiently, and its rate, kRISC, must be faster than the nonradiative deactivation rate, knr, of the T1 → S0 transition. Therefore, kRISC must be increased to satisfy this condition; kRISC can be expressed using eqn (4) and (5):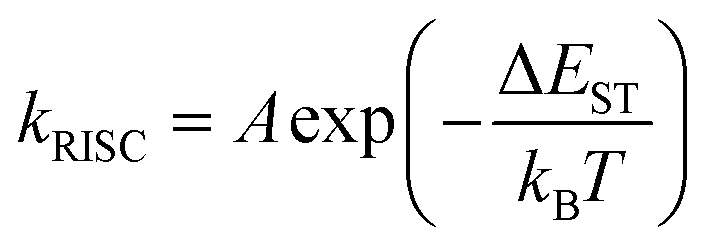 | (4) |
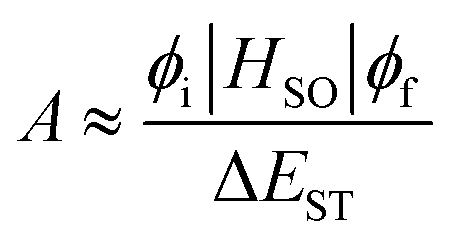 | (5) |
Regarding melem, the energy difference between the singlet and triplet states of the nπ* transition is expected to be small owing to the small spatial overlap between the n and π* orbitals. Fig. 5(a) shows the wavefunctions of the HOMO and LUMO of melem. As shown in Fig. 5(a), similar to other molecules with heptazine skeletons,21–25 melem also has a small ΔEST, since the HOMO and LUMO are localized at the nitrogen and carbon atoms, respectively. Fig. 5(b) shows the Jablonski diagram of melem obtained by density functional theory (DFT) calculations that confirms that the energy difference between S1 and T1, ΔES1–T1, is indeed small (0.1 eV) and comparable with that of other molecules exhibiting TADF. Since the energies of S1 and the second-lowest triplet-excited state, T2, of melem are also very close, an ISC from S1 to T2 upon thermal excitation is also expected. To increase the value of the coefficient, A, expressed by the overlap integral in eqn (5), the symmetry of the wavefunction, ϕi, of the initial state and the wavefunction ϕf of the final state must be significantly different according to the El-Sayed rule.40 For example, electronic transitions between orthogonal electron orbitals, such as n-orbitals (px, py) and π-orbitals (pz), are more likely to allow spin-forbidden transitions through spin-orbit interactions. Based on the El-Sayed rule, the (1nπ*) configuration of S1 and the (3ππ*) configuration of T2 indicate that the transition from S1 to T2 is a spin-allowed transition via spin-orbit coupling and ISC from S1 to T2 becomes possible. Indeed, several examples have been reported in which TADFs involve Tn slightly mixed with the S1 state through spin-orbit interactions or hyperfine coupling.41–48 This indicates that TADF is one of the likely causes of delayed luminescence in melem.
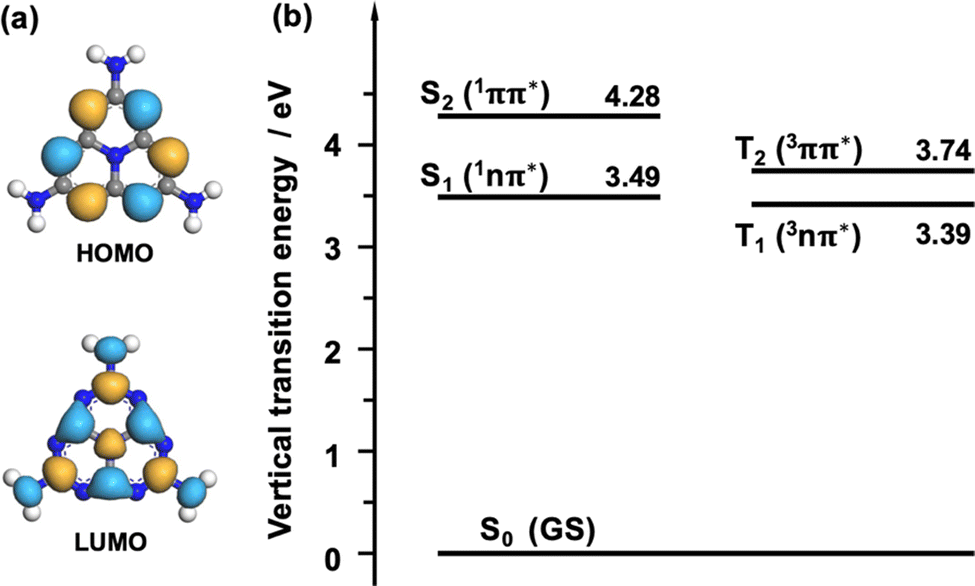 | ||
| Fig. 5 (a) Wave functions of the HOMO and LUMO of the melem molecule. The blue and yellow parts represent different wavefunction signs. (b) Jablonski diagram of the isolated melem molecule. The numbers represent the excited state energy measured from the ground state energy in eV. The descriptions in parenthesis indicate the configuration of the molecular orbitals. | ||
Fig. 6(a) shows the temperature dependence of c-melem PL intensity. Regarding fluorescent materials, higher temperatures induce more intense molecular motions, such as vibration and rotation, increasing the rate of nonradiative deactivation and decreasing the PL intensity.19 However, for TADF materials, as the temperature increases, the T1–S1 transition is promoted, increasing the PL intensity.19,49Fig. 6(a) shows that the PL intensity of c-melem decreases with decreasing temperature. In particular, the PL intensity decreases rapidly from above room temperature to room temperature; therefore, the PL intensity of c-melem increases thermally. From these results, it can be concluded that TADF is the origin of the high PL quantum yield of melem.
 | ||
| Fig. 6 (a) Temperature dependence of c-melem PL intensity. The vertical axis is the integrated intensity of the PL spectrum from 325–525 nm. The data were acquired 3 times at 15, 20, and 25 minutes after the sample reached the target temperature. (b) Arrhenius plot of PL intensity of c-melem. The vertical axis is the natural logarithm of the average intensity of c-melem in (a). The horizontal axis is the reciprocal of temperature. The dotted lines represent the fitted results. The data could be fitted separately in two temperature regions and were fitted in dotted lines A and B. Ea in the figure represents the activation energy calculated from the slope of the line A. (c) The PL component, represented by A, is identified as TADF, while B is due to normal fluorescence and phosphorescence. | ||
The wave functions involved in the transition of MT fluorescence may provide a clue as to why the delayed component decreases with increasing degree of polymerization as discussed in Fig. 2. As shown in Fig. S3 in ESI,† the wavefunctions involved in the transition for the fluorescence from MT have a small spatial overlap. As discussed above, in general, a small overlap of the wavefunctions tend to give a small ΔES1–T1 and is favorable for TADF, while the transition dipole moment is dramatically reduced. Therefore, if the overlap between the wave functions is excessively small, the delayed component is expected to decrease because the rate of the fluorescence emission process is reduced and the rate of nonradiative inactivation is increased.
Fig. 6(b) shows the Arrhenius plot of PL intensity of c-melem.50 The slope of the line clearly changes after room temperature (T−1 ∼ 3.5 × 10−3 K−1). Fitting the straight line at temperatures above room temperature with straight line A yields an activation energy Ea = 31.2 ± 10.4 meV, which can be considered to correspond to the activation energy of T1–S1 transition as shown in Fig. 6(c). This value is equivalent to the thermal energy at room temperature and comparable to Ea of other TADF materials.19 In contrast, at temperatures below room temperature, the temperature dependence of PL intensity is very slight; Ea = 0.87 ± 0.19 meV. This result shows the emission pathway changing from T1–S1 to the normal fluorescence from the S1–S0 transition and phosphorescence from the T1–S0 transition shown in Fig. 6(b), which will be discussed later.
Fig. 7 shows the PL decay curves of c-melem in the time range up to 1.0 μs measured at various temperatures. The slope of the prompt component of the PL decay curve up to 0.1 μs in Fig. 7 significantly depends on the temperature. At 98 K, the PL decay curve slope is smaller than at 348 K, and it increases with increasing temperature. This indicates that the value of kp, which is the rate constant of the prompt component, increases with temperature. In general, kp and kd, which is the rate constants of the delayed components, are expressed by the following equations:35,51–53
 | (6) |
 | (7) |
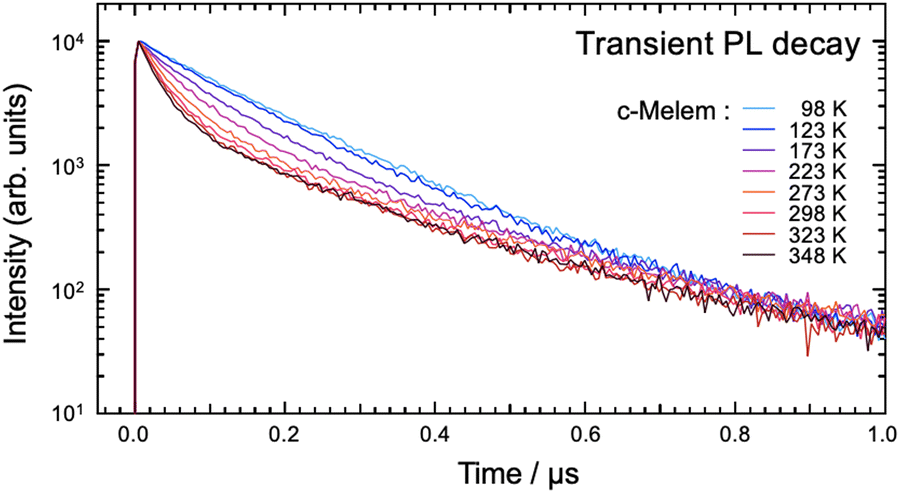 | ||
Fig. 7 Transient PL decay curve of c-melem as a function of temperature. The vertical axis is on a logarithmic scale. The initial intensity of luminescence is normalized to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 00 counts. 00 counts. | ||
Here, kS = kSr + kSnr and kT = kTr + kTnr. kISC represent the rate of ISC, kSr represents the radiative deactivation constant from the S1 state, and kSnr represents the nonradiative deactivation constant from the S1 state. kTr is the radiative deactivation constant from the T1 state and kTnr is the nonradiative deactivation constant from the T1 state. Assuming the relationship, kS + kISC ≫ kRISC+ kT that is generally known for TADF materials, eqn (6) can be expressed simply as follows:54
| kp = kS + kISC. | (8) |
In cases where the shape of the PL spectrum of the prompt component is independent of temperature, the values of kSr and kSnr can be considered temperature-independent.54,55 In fact, there are known TADF materials, such as a series of carbazolyl-dicyanobenzene-based CT molecules, in which kS is independent of temperature.54,55 Thus, if there is no temperature dependence on kS, the temperature dependence on kp can be attributed to the temperature dependence on kISC.
In general, the transition from the initial state to the intermediate or final state caused by light absorption requires the system to overcome a barrier at the intersection of two potential energy surfaces. The transition from the S1 state to the T1 state is also caused by the crossing of the potential energy barrier. In the high-temperature region, thermal energy makes the system more likely to exceed the potential barrier between S1 and T1, resulting in a larger kISC value, whereas, in the low-temperature region, the system is unable to exceed the potential barrier between S1 and T1, resulting in a smaller kISC value. Therefore, in the high-temperature region, the slope of the prompt component up to 0.1 μs in the PL decay curve becomes steeper because of the larger value of kp; in contrast, in the low-temperature region, it becomes slower owing to the smaller value of kp.54
The spectral shapes of the PL spectra of c-melem measured at 82 K and 298 K shown in Fig. 8(a) are similar but not identical; the PL spectrum at 298 K has a broad peak at 363 nm, while the PL spectrum at 82 K has a peak at 366 nm. Additionally, the shape of the PL spectrum at 82 K is narrower than that measured at 298 K. This can be attributed to the suppression of molecular vibration at low temperatures. As the temperature decreases, the width of the distribution of the PL spectrum is suppressed because the molecule no longer occupies highly vibrational excited states, and the transitions between excited and ground states that cause deexcitation become limited. The ratio of the c-melem PL intensities at 366 nm and 363 nm shown in Fig. 8(b) indicates that the ratio of the intensity at 366 nm increases at lower temperatures. The peak wavelength of the PL spectrum shifts to 366 nm at lower temperatures, although the shape of the spectrum does not change significantly. This shift in the peak wavelength is thought to indicate a change in the PL component, indicating that the fluorescent component is reduced, and the phosphorescent component is enhanced as the temperature is lowered. Therefore, at lower temperatures, upconversion from T1 to S1 is no longer possible, resulting in phosphorescence being observed. Several examples of similar phenomena have been reported, such as molecules that no longer exhibit TADF and become phosphorescent at low temperatures.21,49,56 In general, the T1 state of the molecule is lower in energy than the S1 state; thus, phosphorescence originating from the T1–S0 transition has a longer PL wavelength than that originating from the S1–S0 transition. However, regarding melem, the Jablonski diagram shown in Fig. 4 shows that the energy difference between S1 and T1 is very small, approximately 10 nm, which is consistent with the small peak wavelength shift observed in Fig. 8(a) to an enhancement in the phosphorescence component. The shift in the peak wavelengths of the PL spectra measured at 82 K and 298 K in Fig. 8(a) may be caused by the enhancement in the phosphorescent component with a decrease in temperature, in addition to the temperature-dependent effect of ISC, as described above.
 | ||
| Fig. 8 (a) PL spectra of c-melem measured at 82 K and 298 K. The intensity of each spectrum is normalized to the respective maximum intensity. (b) The ratio of the PL intensity at 366 nm to that at 363 nm for PL spectra at each measuring temperature. | ||
4. Conclusions
In this study, the specific luminescence mechanism of melem was elucidated and the origin of the high PL quantum yield was investigated. The melem solution was bubbled with oxygen, which is a triplet quenching agent, and its optical properties were investigated. The PL quantum yield and emission lifetime of the melem solution were significantly reduced by bubbling oxygen to quench the triplet state, indicating that the triplet-excited state was involved in the luminescence mechanism. DFT calculations also revealed that the energy difference ΔES1–T1 between S1 and T1 of melem is small (0.1 eV) and comparable to that of other molecules exhibiting TADF. Furthermore, measurements of the temperature dependence of the PL intensity of melem showed that the PL intensity of melem decreased with decreasing temperature. Thus, the PL intensity of c-melem increased thermally. These experimental results indicate that melem is a TADF material whose PL quantum yield is considerably enhanced by upconversion from the triplet to the singlet excited state upon thermal activation.The PL quantum yield of c-melem was approximately 10% higher than that of the solution state. This is because, in c-melem, there are many intermolecular interactions between melem molecules. Intermolecular interactions, such as hydrogen bonds, tend to suppress molecular motions, such as rotation and vibration of the molecules, preventing the nonradiative deactivation of the excited states of the molecules after light absorption.
Understanding the origin of the high PL quantum yield of melem, which is a basic molecule, should afford important insights for the future development of light-emitting materials for OLEDs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research (Grant No. 22K05259) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT). This study was also supported by the AIST Nanocharacterization Facility (ANCF) platform as a program of the “Nanotechnology Platform” of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan (Grant Number JPMXP09A21AT0091).References
- H. Nakano, H. Tetsuka, M. J. S. Spencer and T. Morishita, Sci. Technol. Adv. Mater., 2018, 19, 76–100 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- V. W. hei Lau and B. V. Lotsch, Adv. Energy Mater., 2022, 12, 2101078 CrossRef.
- Y. Zhang, Q. Pan, G. Chai, M. Liang, G. Dong, Q. Zhang and J. Qiu, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- Y. Miyake, G. Seo, K. Matsuhashi, N. Takada and K. Kanai, Mater. Adv., 2021, 2, 6083–6093 RSC.
- B. Jürgens, E. Irran, J. Senker, P. Kroll, H. Müller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef.
- X. Yuan, K. Luo, N. Liu, X. Ji, C. Liu, J. He, G. Tian, Y. Zhao and D. Yu, Phys. Chem. Chem. Phys., 2018, 20, 20779–20784 RSC.
- N. Liu, T. Li, Z. Zhao, J. Liu, X. Luo, X. Yuan, K. Luo, K. Luo, J. He, D. Yu and Y. Zhao, ACS Omega, 2020, 5, 12557–12567 CrossRef CAS PubMed.
- H. B. Zheng, W. Chen, H. Gao, Y. Y. Wang, H. Y. Guo, S. Q. Guo, Z. L. Tang and J. Y. Zhang, J. Mater. Chem. C, 2017, 5, 10746–10753 RSC.
- H. Zheng, Z. Zhao, J. B. Phan, H. Ning, Q. Huang, R. Wang, J. Zhang and W. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 2145–2151 CrossRef CAS PubMed.
- K. Aoyama, K. Akaike and K. Kanai, Chem. Phys. Lett., 2020, 749, 137475 CrossRef CAS.
- J. Na, S. Bi, C. Jiang and J. Song, Org. Electron., 2020, 82, 105718 CrossRef CAS.
- A. Rezaie, G. Y. Melmed, G. Leite, R. Mathur, W. Takakura, I. Pedraza, M. Lewis, R. Murthy, G. Chaux and M. Pimentel, Adv. Therapy, 2021, 38, 4556–4568 CrossRef CAS PubMed.
- J. Wen, R. Li, R. Lu and A. Yu, Chem. – Asian J., 2018, 13, 1060–1066 CrossRef CAS PubMed.
- T. Dai, H. Kiuchi, H. Minamide, Y. Miyake, H. Inoki, Y. Sonoda, J. Tsutsumi and K. Kanai, Phys. Chem. Chem. Phys., 2022, 24, 13922–13934 RSC.
- S. Hoshino and H. Suzuki, Appl. Phys. Lett., 1996, 69, 224–226 CrossRef CAS.
- M. A. Baldo, D. F. O’brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- C. Y. Chan, M. Tanaka, Y. T. Lee, Y. W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photonics, 2021, 15, 203–207 CrossRef CAS.
- J. Li, T. Nakagawa, J. Macdonald, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Adv. Mater., 2013, 25, 3319–3323 CrossRef CAS PubMed.
- J. Li, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2014, 105, 013301 CrossRef.
- J. Li, H. Gong, J. Zhang, S. Zhou, L. Tao, L. Jiang and Q. Guo, Front. Chem., 2021, 9, 693813 CrossRef CAS PubMed.
- J. Li, L. Tao, Y. Wang, Y. Yao and Q. Guo, Front. Chem., 2021, 9, 717569 CrossRef CAS.
- J. Li, J. Zhang, H. Gong, L. Tao, Y. Wang and Q. Guo, Photonics, 2021, 8, 293 CrossRef CAS.
- L. M. Pedroso, M. M. C. A. Castro, P. Simões and A. Portugal, Polymer, 2005, 46, 1766–1774 CrossRef CAS.
- H. Wang, Elektronische Hochschulschriften, 2013 DOI:10.5282/edoc.16549.
- H. Zuo, Y. Li and Y. Liao, ACS Appl. Mater. Interfaces, 2019, 11, 39201–39208 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard Ii, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 77, 3865 CrossRef PubMed.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- J. R. Lakowicz and G. Weber, Biochemistry, 1973, 12, 4161–4170 CrossRef CAS PubMed.
- W. R. Ware, J. Phys. Chem. A, 1962, 66, 455–458 CrossRef CAS.
- M. Okamoto and F. Tanaka, J. Phys. Chem. A, 2002, 106, 3982–3990 CrossRef CAS.
- R. Ishimatsu, Y. Kirino, C. Adachi, K. Nakano and T. Imato, Chem. Lett., 2016, 45, 1183–1185 CrossRef CAS.
- H. McConnell, J. Chem. Phys., 1952, 20, 700–704 CrossRef CAS.
- Y. Gong, L. Zhao, Q. Peng, D. Fan, W. Z. Yuan, Y. Zhang and B. Z. Tang, Chem. Sci., 2015, 6, 4438–4444 RSC.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- K. Shizu, T. Miwa, Y. Wada, I. Ogata and H. Kaji, J. Photopolym. Sci. Technol., 2017, 30, 475–481 CrossRef CAS.
- F. B. Laforge, N. Green and W. A. Gersdorff, J. Am. Chem. Soc., 1948, 70 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- T. Ogiwara, Y. Wakikawa and T. Ikoma, J. Phys. Chem. A, 2015, 119, 3415–3418 CrossRef CAS.
- H. Noda, H. Nakanotani and C. Adachi, Sci. Adv., 2018, 4, eaao6910 CrossRef PubMed.
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282 CrossRef PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef.
- H. Zhang, G. Li, X. Guo, K. Zhang, B. Zhang, X. Guo, Y. Li, J. Fan, Z. Wang, D. Ma and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 22241–22247 CrossRef CAS PubMed.
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Inorg. Chem., 2017, 56, 13274–13285 CrossRef CAS PubMed.
- P. Pander, F. Dias and F. B. Dias, Display Imaging, 2017, 1–15 Search PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- M. A. Baldo and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10958 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- T. Kobayashi, D. Kawate, A. Niwa, T. Nagase, K. Goushi, C. Adachi and H. Naito, Phys. Status Solidi A, 2019, 217, 1900616 CrossRef.
- T. Kobayashi, A. Niwa, K. Takaki, S. Haseyama, T. Nagase, K. Goushi, C. Adachi and H. Naito, Phys. Rev. Appl., 2017, 7, 034002 CrossRef.
- X. Wei, Y. Chen, R. Duan, J. Liu, R. Wang, Y. Liu, Z. Li, Y. Yi, Y. Yamada-Takamura, P. Wang and Y. Wang, J. Mater. Chem. C, 2017, 5, 12077–12084 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03693b |
| This journal is © the Owner Societies 2022 |