
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using diketopyrrolopyrroles to stabilize double excitation and control internal conversion†
Mariana T.
do Casal
 a,
Josene M.
Toldo
*a,
Felix
Plasser
b and
Mario
Barbatti
*ac
a,
Josene M.
Toldo
*a,
Felix
Plasser
b and
Mario
Barbatti
*ac
aAix Marseille University, CNRS, ICR, Marseille, France. E-mail: mario.barbatti@univ-amu.fr; josene-maria.toldo@univ-amu.fr
bLoughborough University, Loughborough, LE11 3TU, UK
cInstitut Universitaire de France, 75231, Paris, France
First published on 19th September 2022
Abstract
Diketopyrrolopyrrole (DPP) is a pivotal functional group to tune the physicochemical properties of novel organic photoelectronic materials. Among multiple uses, DPP–thiophene derivatives forming a dimer through a vinyl linker were recently shown to quench the fluorescence observed in their isolated monomers. Here, we explain this fluorescence quenching using computational chemistry. The DPP–thiophene dimer has a low-lying doubly excited state that is not energetically accessible for the monomer. This state delays the fluorescence allowing internal conversion to occur first. We characterize the doubly excited state wavefunction by systematically changing the derivatives to tune the π-scaffold size and the acceptor and donor characters. The origin of this state's stabilization is related to the increase in the π-system and not to the charge-transfer features. This analysis delivers core conceptual information on the electronic properties of organic chromophores arranged symmetrically around a vinyl linker, opening new ways to control the balance between luminescence and internal conversion.
Introduction
Photoexcitation deposits large amounts of energy in a single molecule, which can be released as light or converted without radiation to produce work, charge shifts, or heat. In most applications, the interplay between radiative and nonradiative rates is critical. For example, in organic light-emitting diodes1 and fluorescent probes,2 one wants to maximize luminescence and prevent energy dissipation through secondary processes. On the other hand, efficient internal conversion is crucial when designing materials for photocatalysis3 (used to initiate chemical reactions), photovoltaics4 (used to induce charge separation), or photothermics5–7 (used to heat microenvironments).In searching for optimal organic photomaterials, diketopyrrolopyrrole (DPP) became a jack-of-all-trades. DPPs are stable and inexpensive. They can be easily synthesized and modified by introducing different functional groups and side chains, making them attractive options for adjusting emission and fluorescent wavelengths. They strongly absorb visible light, show significant electron-withdrawing effects, and possess high electron mobility.8–12 Due to these properties, DPPs are often used as an electron-acceptor unit to design optoelectronic devices13,14 such as new push–pull polymers,15,16 organic light-emitting diodes,17 and more recently, singlet-fission chromophores.8,18,19
Motivated by the design of new intramolecular singlet-fission materials, Mukhopadhyay et al.20 proposed a combination of a thiophene-DPP derivative (with charge-transfer (CT) character) with a vinyl linker (polyene character) (Fig. 1b). Although they did not observe singlet fission in this new material, they did detect fast internal conversion from the initial bright state into a short-lived dark singlet state, with no evidence of substantial conformational change, triplet formation, or charge separation. Also, a remarkable fluorescence quenching was observed for the dimer (Fig. 1b) but not for the monomer (Fig. 1d).
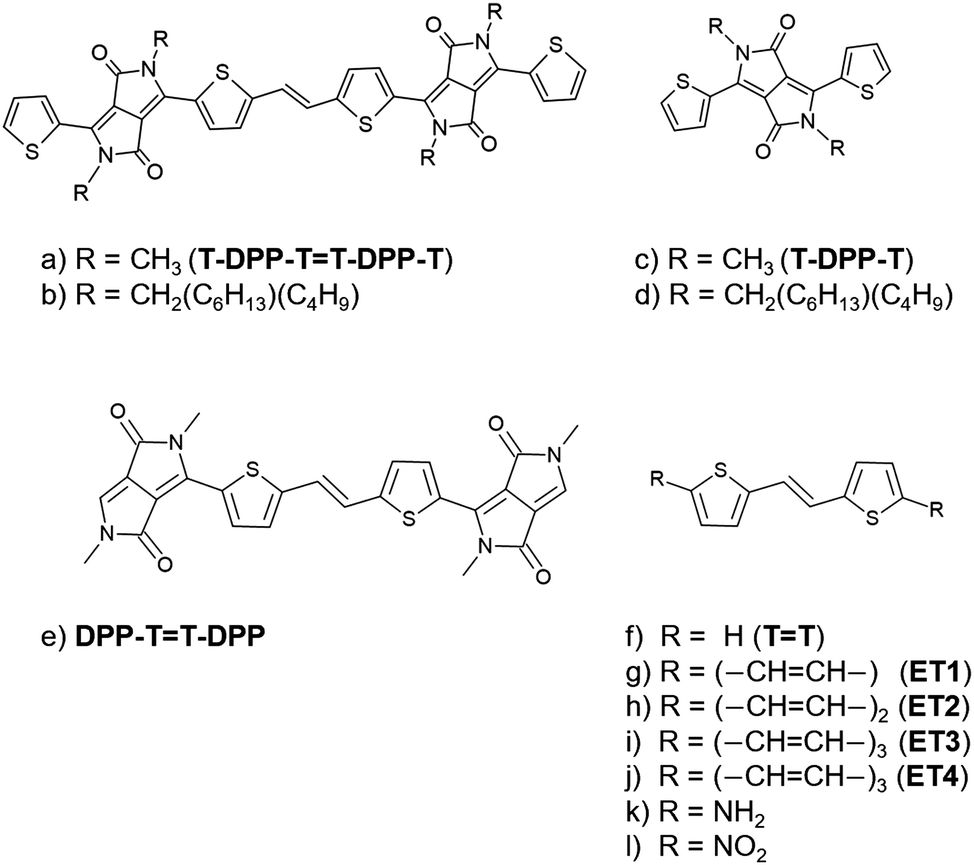 | ||
| Fig. 1 Molecular structures investigated in this work. | ||
In this work, we investigate the role of the π-scaffold in this fluorescence quenching. We show that the dimer T-DPP-T![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) T-DPP-T (Fig. 1a) has a low-lying 2Ag state with a significant doubly excited character that mediates the population transfer from the bright state to the ground state. In contrast, the doubly excited state in the monomer T-DPP-T (Fig. 1c) is higher in energy and, therefore, not accessible. To understand such a difference, we explored the effect of introducing different functional groups attached to the central thiophene-vinyl linker unit. First, we focused on the dimer's building blocks, namely DPP-TT-DPP and TT, shown in Fig. 1e and f. Second, we isolate the effect of the number of π-electrons from the donor-acceptor effects by substituting the DPP-thiophene units with long-chain polyenes (Fig. 1g–j), –NH2 (Fig. 1k), and –NO2 (Fig. 1l). Lastly, we quantify the charge-transfer character of the DPP derivatives.
T-DPP-T (Fig. 1a) has a low-lying 2Ag state with a significant doubly excited character that mediates the population transfer from the bright state to the ground state. In contrast, the doubly excited state in the monomer T-DPP-T (Fig. 1c) is higher in energy and, therefore, not accessible. To understand such a difference, we explored the effect of introducing different functional groups attached to the central thiophene-vinyl linker unit. First, we focused on the dimer's building blocks, namely DPP-TT-DPP and TT, shown in Fig. 1e and f. Second, we isolate the effect of the number of π-electrons from the donor-acceptor effects by substituting the DPP-thiophene units with long-chain polyenes (Fig. 1g–j), –NH2 (Fig. 1k), and –NO2 (Fig. 1l). Lastly, we quantify the charge-transfer character of the DPP derivatives.
The effect of DPP units on the performance of photovoltaic materials and the closed or open-shell nature of DPP's ground state has already been investigated.8,12,21,22 Given the large size of these molecules, discussions about their electronic structure properties are often based on the density functional theory (DFT) HOMO–LUMO gaps or linear-response time-dependent DFT (TDDFT) excitations.19,21,23,24 Nevertheless, depending on the molecular architecture, TDDFT cannot correctly describe DPPs derivatives as they have a low-lying doubly-excited state. Formally, this state has a significant proportion of doubly-excited Slater determinants, i.e., promotion of two electrons from occupied into virtual orbitals.25 Doubly-excited states have transition dipole moment close to zero and, therefore, are optically dark. Spectroscopically, they are ascribed to 2Ag states in linear polyenes26–29 and carotenes,30–33 for example, where they play a decisive role in the decay pathways.
Furthermore, while the characterization of the singly-excited states (as the bright 1Bu state in this work) is well described by TDDFT, the description of doubly-excited states on large molecules is still a challenge for computational chemistry. We face this challenge, delivering a methodological road map for further studies, based on a combination of spin-flip TDDFT and multireference configuration interaction density functional theory (DFT/MRCI), some of the few available quantum chemical methods that can be used for large molecules and describe doubly-excited states. In the combined DFT/MRCI, DFT recovers part of the dynamic correlation and MRCI part of the static correlation.34 However, gradients are still not available for this method. Thus, we used SF-TDDFT to optimize geometries. Here, lower spin-states are obtained from a high-spin Kohn–Sham reference by spin-flipping the electrons of the reference during the transition.35 This procedure leads to closed-shell, singly, and doubly excited configurations, which can describe doubly excited states, although the incomplete excitation space can lead to spin-contamination.36,37
This deep-level electronic-structure characterization goes much beyond the particular systems focused here. We expect similar effects whenever the molecular system has a symmetry inversion center at a carbon–carbon double bond (vinyl linker), either using DPP units or not. Thus, the size of the π-scaffold can be used to tune the relative stability between the bright and dark states, opening a way to control the ratio between radiative and nonradiative rates. We stress that our primary goal was not to benchmark vinyl-linked chromophores but to explain the quenching of fluorescence in the aforementioned DPPs derivatives. Many ethylene spaced molecules have been synthesized in the past,38–40 mostly in the context of push–pull chromophores (where the olefinic unit acts in order to decrease the HOMO–LUMO gap),39 but their particular investigation is outside the scope of this paper.
Computational methods
Ground-state geometry optimizations for all systems were performed with DFT using the B3LYP41,42 functional and D3 Grimme's dispersion correction,43 as this functional is well-known for providing satisfactory molecular geometries for the ground state of organic molecules. All geometry optimizations considered a closed-shell ground state. For comparison, optimizations of ground and first excited states along with computation of excitation energies were also performed with TDDFT using the B3LYP and CAM-B3LYP functional (see Tables S1 and S2, ESI†).44 All (TD)DFT calculations were done with the 6-31G(d,p) basis set using Gaussian 16 rev A03.45 The impact of diffuse functions in the basis set and solvation effects (using PCM/ethanol) were also explored (see Tables S4 and S5, ESI†). Our TDDFT calculations showed only a small influence of the implicit solvation on the absorption and emission energies. Thus, performing the calculations in vacuum seems reasonable to explain the main features controlling the differences in the deactivation mechanism of the monomer and dimer. Therefore, our subsequent DFT/MRCI and SF-TDDFT were done in vacuum.Bright and doubly excited state minima were optimized using spin-flip time-dependent density functional theory (SF-TDDFT) in the collinear approximation35 with restricted open-shell reference, as implemented in GAMESS-US 2019 R2.46 SF-TDDFT calculations were performed with BHHLYP hybrid functional47 since benchmark calculations suggest the better performance of functionals with a larger fraction of Hartree–Fock exchange in collinear schemes.48 Dispersion corrections were not applied because they are not available for SF-TDDFT as implemented in GAMESS-US. All geometry optimizations were done using the 6-31G(d,p) basis set. For T-DPP-T, excited state geometry optimizations were considered converged when the root mean square (RMS) and maximum gradient variation were below 0.0008 and 0.0038 Hartree Å−1, respectively, while for T-DPP-TT-DPP-T, the default thresholds implemented in GAMESS were used (the largest component of the gradient should be below 10−4 Hartree Å−1 and the RMS below 10−4/3 Hartree Å−1). The assignment of the spin-flip states was done based on the symmetry, spin expectation value, and configurations of each root (Table S6, ESI†).
Ground and excited-state energies were calculated with DFT/MRCI, as developed by Grimme and Waletzke49 and redesigned by Marian et al.,34,50 at either B3LYP or SF-TDDFT geometries. In this approach, the major part of dynamic electron correlation comes from the DFT, while static electron correlation effects are computed by considering a short configuration interaction expansion. In this way, this method allows computing extended π-systems where double excitations play an important role in obtaining the correct order of excited states. We used the R2018 redesigned Hamiltonian34 with optimized threshold parameter esel = 0.8 Hartree and def2-SV(P) basis set. The initial reference space includes all possible configurations generated by up to doubly exciting 6 electrons within 6 orbitals. The DFT/MRCI code was used along with the Kohn Sham orbitals and integrals obtained with TURBOMOLE v.7.5.51,52 All calculations were performed using C2h symmetry, except for T-DPP-T, where C1 symmetry was used.
TheoDORE 2.453,54 was used to analyze the state densities, transition densities, and attachment/detachment densities of the excited-state computations. Specifically, we considered three descriptors: (i) the promotion number53,55 (p), (ii) the squared norm of the 1-particle transition density (1TDM) matrix Ω,53 and (iii) the number of unpaired electrons53,56 (nu,nl). p is obtained as the sum over the positive (or negative) eigenvalues of the 1-particle difference density matrix and can be understood as a measure of how many electrons were promoted during the electronic excitation. Ω measures the amount of single excitation character in a state. nu,nl is computed as the sum over the occupation (np) over all natural orbitals of the system, as shown in eqn (1).
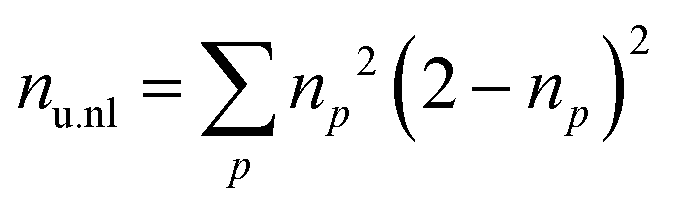 | (1) |
Due to the doubly excited character of the wavefunction, previously developed charge-transfer (CT) descriptors53,54 that rely on the partition of the one-electron transition density matrix are not appropriate. Therefore, we turned to the detachment and attachment densities. These densities are derived from the diagonalization of the difference density matrix55 between the ground state and excited state. They contain information about shifts in the electron density associated with the excitation process. Therefore, a quantitative index δ for the charge-transfer character can be obtained from a Mulliken population analysis of these quantities. More details about how δ was calculated and the fragmentation schemes considered are available in Section S7 of the ESI.†
Results and discussion
Photophysics of the monomer and dimer
Our starting point was to use TDDFT to characterize the excited states of the dimer (Fig. 1b) and monomer (Fig. 1d). First, we investigated the effect of the alkyl chains (R group in Fig. 1) attached to the nitrogen atoms of the DPP scaffold on the excited-state properties of the monomer and dimer. At the S0 minimum, the DPP and thiophene units remain on the plane in both molecules, independently of the alkyl chains (ESI,† Fig. S1 and Tables S1, S2). More generally, we find a negligible impact of the alkyl groups on the geometry and the photophysical properties of these molecules. Our TDDFT calculations show that their absorption wavelength and main excitations remain nearly the same when comparing monomer and dimer with and without alkyl groups (Tables S1 and S2, ESI†). This result agrees with that reported in the literature, which points out the small effect of the side alkyl chains on the photophysics of DPPs derivatives in solution (although this effect becomes significant in the solid-state).57 Thus, to decrease the computational cost and turn the subsequent DFT/MRCI calculations feasible, the alkyl chains were substituted by methyl groups (Fig. 1a and c).The TDDFT excitation energies and oscillator strengths computed for the dimer 1b and the monomer 1d show that the bright state of both molecules is the S1 state, with a much larger oscillator strength for the dimer (f = 2.3) than for the monomer (f = 0.5) (Tables S1 and S2, ESI†). The S2 state is dark in both molecules, with oscillator strength equal to zero. At the ground-state equilibrium geometry, the S1–S2 energy gap is at least twice as big for the monomer than for the dimer. This difference increases at their respective TDDFT S1 equilibrium geometry. A bright S1 state with a large gap to S2 suggests strong fluorescence emission in both molecules, contradictory to the experimental results.20 Yet, there is no experimental evidence of triplet state population on the DPP dimer,20 and our DFT calculations confirm that T1 state is energetically inaccessible; besides, it is forbidden by El-Sayed rules.58
We ascribe this failure of linear-response TDDFT to describe the photophysics of the DPP-dimer to its intrinsic limitations in dealing with doubly excited and multireference states.59 Due to the adiabatic approximation used in this method, only single excitations are included in the time-dependent exchange–correlation potential.60 Thus, we resort to DFT/MRCI, which shows good performance for the computation of excited-state properties of polyaromatic hydrocarbons61,62 and polyenes.63
DFT/MRCI revealed a significant doubly excited character for the dark state (2Ag) that is not captured by TDDFT. Also, the state's order shown by DFT/MRCI differs from that previously assigned by TDDFT. Table 1 shows the excitation energies, symmetries, and amount of doubly excited configurations on the description of S1 and S2 states of the monomer 1c (T-DPP-T) and dimer 1a (T-DPP-TT-DPP). At DFT/MRCI level, the monomer's bright state 1Bu lies 0.93 eV below its dark state 2Ag. In contrast, these states are nearly degenerated (ΔE[2Ag − 1Bu] = 0.09 eV), and the 2Ag lies below the 1Bu in the dimer.
T-DPP-T)
| Minimum | T-DPP-T | T-DPP-TT-DPP-T |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sym | VE | AE | f | %D | Sym | VE | AE | f | %D | ||
| S0 | S0 | 1Ag | 0.00 | 8.8 | 1Ag | 0.00 | 17.0 | ||||
| S1 | 1Bu | 2.34 | 0.60 | 7.4 | 2Ag | 1.57 | 0.00 | 57.0 | |||
| S2 | 2Ag | 3.27 | 0.00 | 39.4 | 1Bu | 1.66 | 2.10 | 10.4 | |||
| Bright state | S0 | 1Ag | 0.00 | 0.10 | 9.5 | 1Ag | 0.00 | 0.00 | 18.5 | ||
| S1 | 1Bu | 2.10 | 2.19 | 0.63 | 8.4 | 2Ag | 1.30 | 1.30 | 0.00 | 53.1 | |
| S2 | 2Ag | 2.92 | 3.02 | 0.00 | 45.3 | 1Bu | 1.46 | 1.46 | 2.13 | 14.0 | |
| Dark state | S0 | 1Ag | 0.00 | 0.26 | 10.6 | 1Ag | 0.00 | 0.24 | 23.3 | ||
| S1 | 1Bu | 1.98 | 2.24 | 0.66 | 9.4 | 2Ag | 0.98 | 1.23 | 0.00 | 50.5 | |
| S2 | 2Ag | 2.65 | 2.92 | 0.00 | 48.4 | 1Bu | 1.26 | 1.50 | 2.05 | 18.0 | |
The reversal in S1 character between the monomer (1Bu) and dimer (2Ag) may be responsible for the different luminescence properties of the two molecules. To substantiate this hypothesis, we proceed to compute the relative position of the dark and bright states at their equilibrium geometries. Since energy gradients are unavailable in DFT/MRCI, we turned to SF-TDDFT to optimize these states (see also ESI,† Section S2). Both minima are still planar but with different bond length alternation patterns, as shown in ESI,† (Table S7 and Fig. S2, ESI†). For the monomer, the 1Bu state remains at least 0.67 eV lower than 2Ag after geometry relaxation of both excited states (Table 1). On the other hand, this order is reversed for the dimer: the 2Ag state lies below the 1Bu state at all geometries considered.
These results confirm that the monomer's deactivation pathway is fundamentally different from the dimer, as illustrated in Fig. 2. After excitation to the optically active 1Bu, the deactivation of the dimer is mediated by an S2/S1 internal conversion, which populates the dark doubly excited state, quenching the fluorescence as seen experimentally.20 As the 1Bu → 2Ag internal conversion is ultrafast, it is expected that it occurs through a conical intersection mediated by displacements along the bond length alternation (BLA) coordinate, as found in polyenes64 and carotenes.33 For the monomer, the dark state is not energetically accessible in the gas phase, and, therefore, its deactivation pathway is confined to the relaxation in the bright state potential energy surface and subsequent fluorescence from this state.
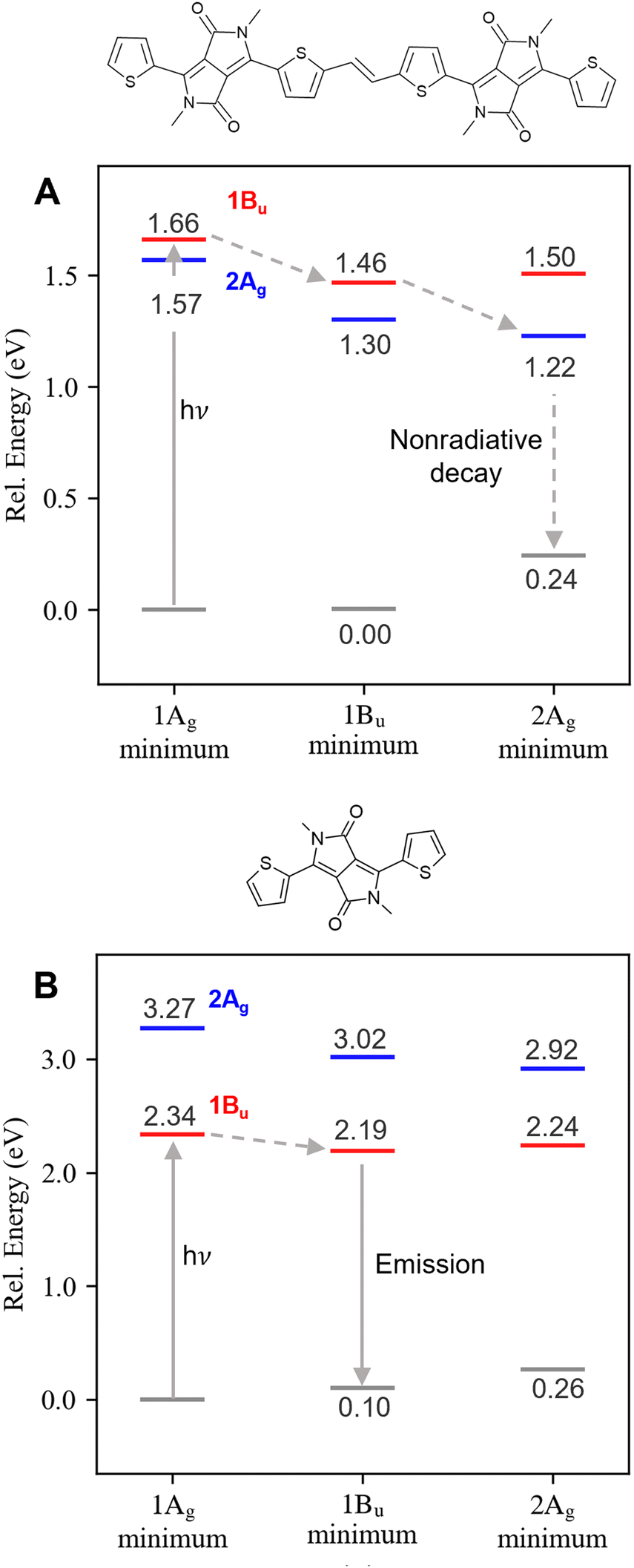 | ||
| Fig. 2 Relative energies in eV of the bright 1Bu (red) and dark 2Ag (blue) states at the ground state (1Ag), bright state (1Bu), and dark (2Ag) state minima (horizontal axis) of T-DPP-TT-DPP-T (A) and T-DPP-T (B). The arrows indicate the possible deactivation pathway in each case. | ||
The fluorescence quenching in the dimer depends on the nonradiative decay rate being larger than the fluorescence rate. The small value of the vertical gap (below 1.0 eV) certainly suggests that nonradiative decay is likely. The similarity of the DPP dimer with long-chain polyenes and carotenes (discussed in the next section) suggests that the 2Ag → 1Ag internal conversion will be likely mediated by vibronic coupling between the states65,66 (although conical intersections involving two coordinates has also been proposed for polyenes),64 rather than torsional motion around the central double bond.67 A mechanistic description of this decay process goes beyond the scope of this work, and we focus on our primary goal, discussing why the doubly excited state is stabilized.
Reasons for doubly excited state stabilization
The state order discussed in the previous section explains the fluorescence quenching in the dimer, and we will now proceed to discuss why the doubly excited state is more stabilized in the dimer than in the monomer. What is the DPP's role in this stabilization? Would we have a similar effect if we changed the DPP units for other acceptor or donor groups? We address these questions in the following subsections.T, and DPP-TT-DPP) for stabilizing the doubly excited state and their contribution to the double excitation of the dimer (T-DPP-TT-DPP-T). All of them are planar in the ground state and have C2h symmetry (ESI,† Table S3).
In Table 2 and Fig. 3, we show the calculated excitation energies of all building blocks and the dimer. In Fig. 3, the substituents (R) to the R-TT-R core are defined on the horizontal axis. For instance, TT is represented by “H” and DPP-TT-DPP by “DPP”. TT and T-DPP-T yield a similar picture, where the 2Ag state lies well above the 1Bu. Adding DPP units to TT to form DPP-TT-DPP reverses 2Ag and 1Bu order even without the terminal thiophenes. This DPP addition stabilizes the 2Ag and 1Bu states by 2.6 and 1.7 eV, respectively. In comparison, adding the terminal thiophenes to DPP-TT-DPP stabilizes these states by only 0.28 eV and 0.26 eV. Therefore, the main stabilization effect is due to the addition of the second DPP unit.
T, DPP-TT-DPP, and T-DPP-TT-DPP-T
| T-DPP-T | TT |
DPP-TT-DPP |
T-DPP-TT-DPP-T |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sym | VE | f | %D | Sym | VE | f | %D | Sym | VE | f | %D | Sym | VE | f | %D | |
| S0 | 1Ag | 8.8 | 1Ag | 6.2 | 1Ag | 14.6 | 1Ag | 17.0 | ||||||||
| S1 | 1Bu | 2.34 | 0.60 | 7.4 | 1Bu | 3.60 | 1.04 | 3.9 | 2Ag | 1.85 | 0.00 | 53.8 | 2Ag | 1.57 | 0.00 | 57.0 |
| S2 | 2Ag | 3.27 | 0.00 | 39.4 | 2Ag | 4.43 | 0.00 | 35.9 | 1Bu | 1.92 | 1.35 | 16.4 | 1Bu | 1.66 | 2.10 | 10.4 |
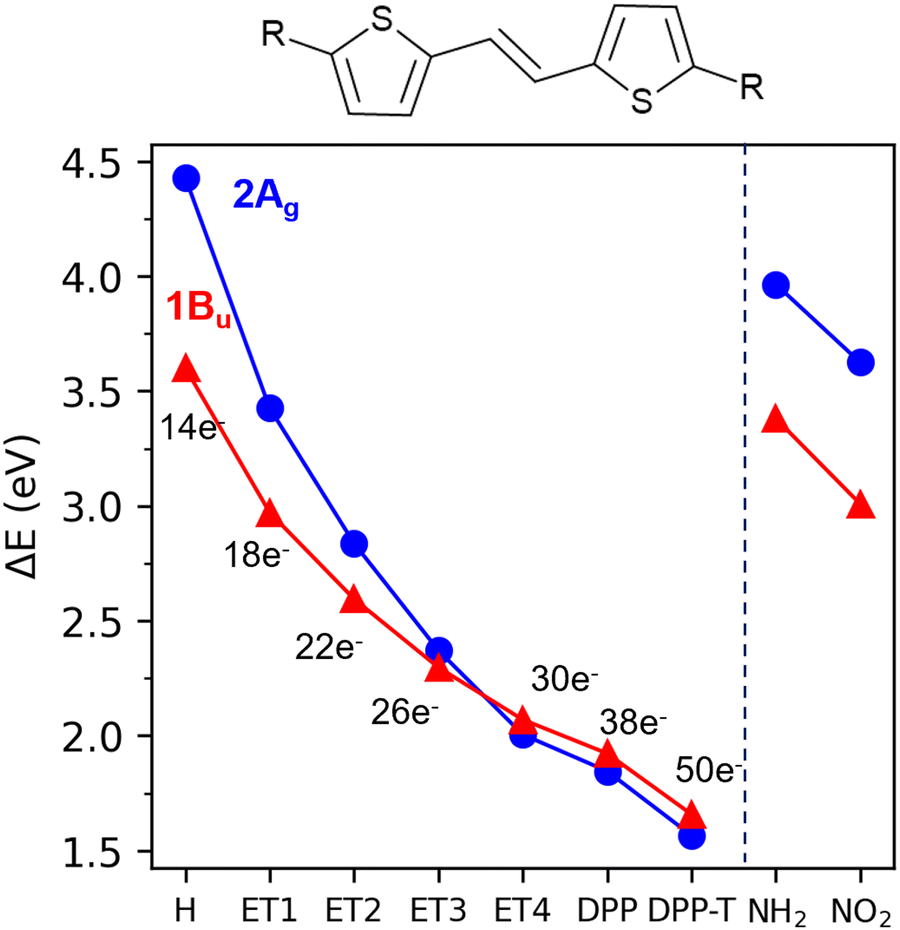 | ||
| Fig. 3 π-Electrons (left) and donor-acceptor (right) effects on the excitation energies (eV) of 1Bu and 2Ag states at the ground-state minima of R-TT-R according to the number of electrons in the π-system. The R substituents are defined in the horizontal axis. | ||
Turning now to the contribution of the building blocks to the doubly excited character, we analyze the 2Ag state in more detail using systematic wavefunction analysis techniques. We focus on three main descriptors: Ω,68 which accounts for the amount of singly excited configurations;68 the number of unpaired electrons53 (nu,nl); and the promotion number55 (p), which indicates the number of electrons promoted in the photoexcitations.55Ω is obtained from the one-particle transition density matrix (1TDM) between the ground and excited state, nu,nl is computed from the excited-state density matrix, and p is computed from the difference density matrix. Thus, three different viewpoints into the wavefunction are obtained. Typically, a doubly excited state shows Ω ∼ 0 and p ∼ 2. Furthermore, the limiting case of a pure, closed-shell HOMO2 → LUMO2 excitation would be reflected with nu,nl ∼ 0, whereas a doubly excited state with four open shells (e.g., HOMO−1 HOMO → LUMO LUMO+1) would yield nu,nl ∼ 4.
Fig. 4 shows how those descriptors vary in the building blocks discussed above. Similar to Fig. 3, they are defined in the horizontal axis, represented by the substituent (R) to the R-TT-R core. The Ω-value of 0.61 and p of 1.51 already identify the 2Ag state of TT as partially doubly excited. Adding the DPP units (TT → DPP-TT-DPP) lowers Ω strongly (by 0.24), and p becomes approximately 2. The addition of the terminal thiophenes to this building block (DPP-TT-DPP → T-DPP-TT-DPP-T) has virtually no effect on Ω, p, and nu,nl. (Table S9, ESI†). These changes imply that the terminal thiophenes have only a minor impact on the 2Ag character and most of the double excitation is related to the vinyl linker, DPP, and core thiophene units.
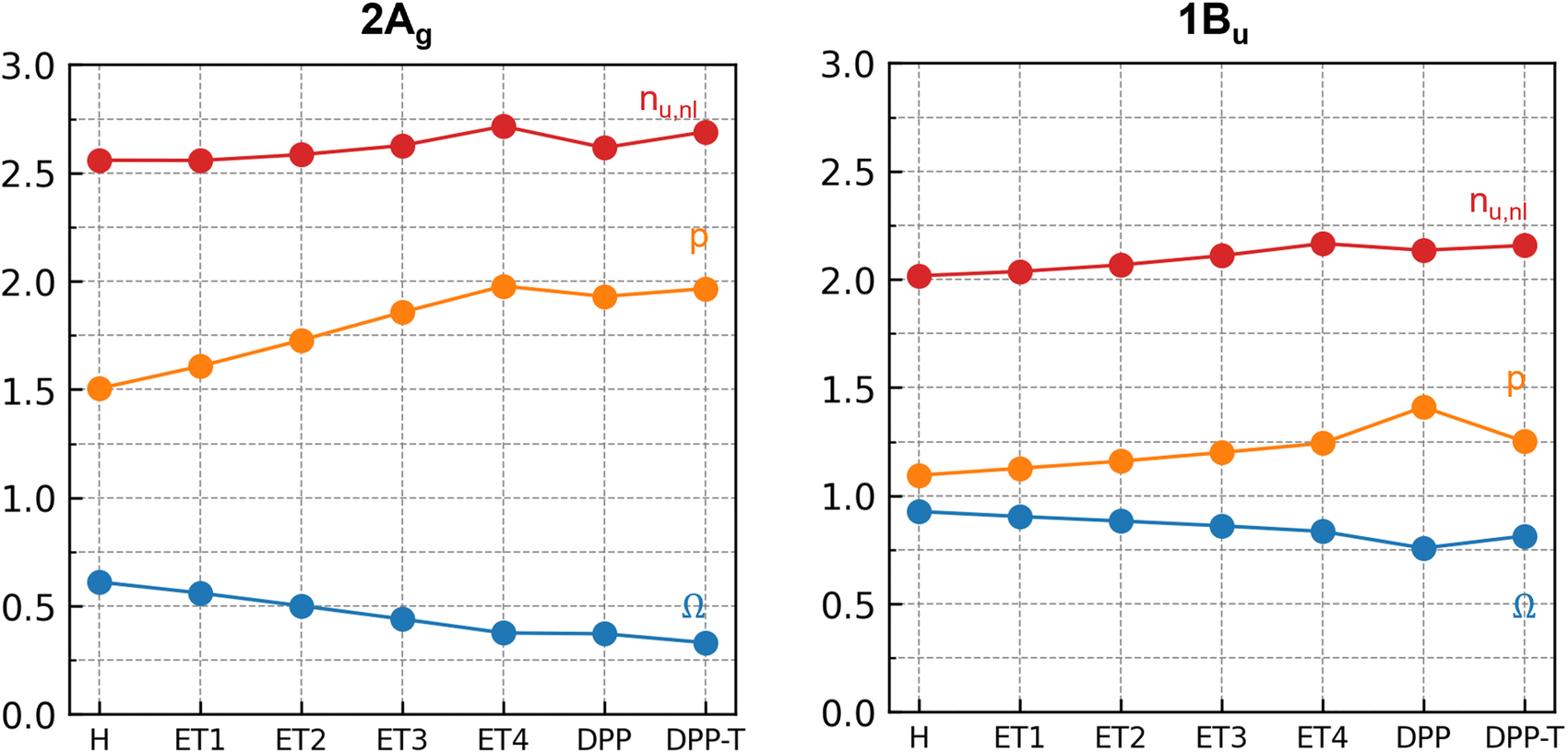 | ||
| Fig. 4
Ω (blue), promotion number (p, orange), and the number of unpaired electrons (nu,nl, red) of the 2Ag dark and 1Bu bright states at the ground-state minima of R-TT-R according to the number of electrons in the π-system. The substituents R are defined in the horizontal axis. | ||
A closer look at the configurations describing the 2Ag state in the different building blocks can give further insight into which type of double excitation is associated with each building block. For simplicity, we considered only the dominating configurations, i.e., those with squared weights larger than 0.1. All building blocks and the dimer have a significant contribution from the closed-shell (HOMO2 → LUMO2) configuration (Table S8, ESI†). DPP-TT-DPP and the dimer also have contributions with an equivalent weight from the open-shell (HOMO−1 HOMO → LUMO LUMO+1) configuration, which is absent in TT. Therefore, the DPP units cause an increase of doubly excited character through open-shell configurations.
T by adding up to four (–CHCH–)n units (Fig. 1g–j). Those derivatives are referred to as ETn, where n is the number of (–CHCH–) units. Note that ET1 and ET2 are substructures of DPP-TT-DPP while all four ETn are substructures of T-DPP-TT-DPP-T. Fig. 3 shows their impact on the calculated vertical excitation energies of 2Ag and 1Bu states. Notably, the number of electrons in the π-system in the DPP derivatives is larger than in the ETn series due to the contributions of carbonyl groups and nitrogen atoms in the DPP unit.
Fig. 3 shows how the 1Bu and 2Ag states' vertical excitation energies change with the increase of electrons in the π-system. Overall, the increase in the number of π-electrons leads to a smooth stabilization of both excited states. The 2Ag state's robust stabilization with the rise in π-electrons corresponds to a ΔE[2Ag − 1Bu] gap decrease from 0.83 eV in TT to below 0.1 eV in ET4, DPP-TT-DPP, and the dimer. It reveals that the increasing number of electrons in the π-system stabilizes the 2Ag state, similarly to polyenes.26,69 In fact, when the number of (–CHCH–) units increases to four, i.e., 30 π-electrons, the order of the excited states is already inverted, and ΔE[2Ag − 1Bu] is below 0.1 eV. This small energy gap should facilitate the internal conversion between S2 and S1 states through displacements along the BLA coordinate.33,64,67 Furthermore, another aspect of the DPP-dimer resembling the photophysics of long-chain polyenes is its 1Ag and 2Ag BLA patterns.64 Like in the linear polyenes, the BLA of these two states match at the extremes of the dimer and alternates in the center (see Fig. S2, ESI†).
This behavior indicates that the increasing π-electron number plays a crucial role in reversing these molecules' dark and bright states. As in polyenes, the reverse order of the states should be, in part, a consequence of the electronic interaction, as the ionic states (Bu) are destabilized, and the 2Ag state is stabilized due to local couplings.28,29,70–72 Despite the more seemingly complex electronic structure, which allows electron delocalization over the heterocyclic rings, the addition of DPP units is a helpful strategy to increase the π-scaffold.
Fig. 4 presents how the wavefunction descriptors change with the π-electrons number. A closer look at the 2Ag state shows that the increase of (–CHCH–) units reduces Ω and increases the promotion number p, indicating a more significant contribution of highly excited configurations and a higher number of electrons promoted upon photoexcitation. On the other hand, Fig. 4 shows that for the 1Bu state, most descriptors are only slightly affected by the increase in the number of π-electrons. Adding the terminal thiophene led to a slight decrease in the contribution of doubly excited configuration to this state since p decreased and Ω increased. Overall, Fig. 4 shows a smooth trend with the π-electrons number, which indicates that the character of excited states does not radically change with the addition of those substituents.
The results above show that increasing the number of π-electrons plays a decisive role in stabilizing the 2Ag state compared to 1Bu in T-DPP-TT-DPP-T. Nevertheless, DPP may act as an electron acceptor when connected to thiophene.73 Does this acceptor character also play a role in state inversion? And if it had a donor character instead, would it matter?
To address these questions, we should add acceptor and donor groups to the central TT without increasing the π-system. Thus, we replaced DPP with a strong donor (−NH2, Fig. 1k) and a strong acceptor (−NO2, Fig. 1l). As shown in Fig. 3, neither leads to a state inversion. Although both excited states are stabilized in these systems, the energy gap between them is still significant (>0.8 eV). Thus, the isolated effect (i.e., without increasing the conjugation) of donor and acceptor groups seems negligible in these molecules.
The simultaneous presence of the electron-deficient DPP moiety and the electron-rich thiophene is often reported as yielding molecules with strong intramolecular charge transfer.57,74 However, no CT effects were reported in an experimental study with thiophene-DPP dimers connected by a vinyl linker (Fig. 1b)20 in solution and computational studies with DPP dimers linked via dithienylphenylene spacers in the gas phase.19 A qualitative analysis of the natural difference orbitals (see ESI,† Section S6) does not indicate such effect in either 1Bu or 2Ag, at least in the gas phase. However, a systematic evaluation of the charge-transfer character of these systems is needed.
Well-established charge-transfer descriptors are based on partitioning the 1TDM.75 They can be successfully applied to excited states mainly described by singly excited configurations. However, they do not account for contributions of multiply excited configurations, which are of foremost importance in doubly excited states. An alternative approach is to analyze the detachment and attachment densities since they reveal how the electron density is removed from the ground-state density (detachment density) and rearranged in the respective excited state (attachment density). Those quantities are derived from the specific state densities of a reference state55—in this case, the ground state—and the respective excited state, either 2Ag or 1Bu.
Here, we applied a Mulliken population analysis on the detachment and attachment densities to obtain a quantitative index δ for the charge-transfer character of the investigated molecules.53 Section S7 of the ESI,† explains how this index was calculated and the fragmentation schemes used. The 2Ag and 1Bu states of the DPP derivatives show no charge-transfer character within the fragmentation schemes considered since the δ-values are smaller than 0.08. This result is consistent with the conclusions obtained experimentally by Mukhopadhyay et al.20 The similarity in the emission properties of the DPP dimer and long polyenes stems from the high stability of the doubly-excited state. However, the origin of this stability may be different in both cases because it may have a CT contribution for polyenes.29,66
Conclusions
In this work, we investigated how modifications of the DPP scaffold (Fig. 1) lead to a different interplay between the bright 1Bu state and the dark 2Ag states. In thiophene-DPP-thiophene dimers (Fig. 1d), the fluorescence quenching is mediated by the low-lying 2Ag state with a high doubly excited character, which is lower in energy than 1Bu state. In the monomer (Fig. 1b), the doubly excited state is above 1Bu. Thus, the monomer mostly stays in the bright state and returns to the ground state emitting light, while the dimer returns to the ground state via a nonradiative relaxation pathway mediated by the dark state.A systematic investigation of the dimer's building blocks (Fig. 1) reveals that the inversion between the dark 2Ag and bright 1Bu states occurs with the addition of the DPP units. The effect of the DPP units is due to the increase of π-electrons and does not depend on its acceptor character. Furthermore, our wavefunction analysis confirms that the doubly excited character, already present in TT, is intensified by adding the DPP units. The DPP units primarily increase the open-shell doubly excited character of the system. Lastly, the analysis revealed that those excited states do not have a charge-transfer character in the gas phase.
DPP derivatives are major players in developing new and affordable photoelectronic organic materials. However, their use has been empirically guided without much high-level quantum-chemical information about their electronic structure. This work changes this situation by setting a computational strategy to compute and analyze their excited states. We applied it to characterize a specific chromophore architecture where DPP derivatives are symmetrically arranged around a vinyl linker. Our main conclusion is that, within this architecture, the DPPs create a π-scaffold controlling the relative stability of the bright and dark (doubly excited) states, which finally impacts the competition between luminescent and internal conversion rates. Thus, the fluorescence quenching in this particular molecular architecture could be explored to design new materials where efficient internal conversion is mandatory, such as photothermal materials and molecular heaters.
Author contributions
M. T. do C. is responsible for data curation, investigation, and visualization. M. T. do C., J. M. T., and M. B. contributed with conceptualization and methodology. J. M. T., F. P., and M. B. contributed for supervision. M. B. contributed with funding acquisition and resources. All authors contributed to writing the original draft, reviewing, and editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
M. T. do C., M. B., and J. M. T. received funds from the European Union's Horizon 2020 research and innovation programme (grant agreement No 828753), project BoostCrop. The Centre de Calcul Intensif d'Aix-Marseille is acknowledged for granting access to its high-performance computing resources. This work was performed using HPC/AI resources from GENCI-TGCC (Grant 2022-A0110813035). M. T. do C., J. M. T., and M. B. thank Prof. Mike Ashfold, Dr Tom Oliver, and Dr Daniel Polak for the discussions. M. T. do C. is thankful for the support of the PhD mobility program funded by AMUtech.References
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, A Brief History of OLEDs—Emitter Development and Industry Milestones, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- S. B. Wagh, V. A. Maslivetc, J. J. La Clair and A. Kornienko, Lessons in Organic Fluorescent Probe Discovery, ChemBioChem, 2021, 22, 3109–3139 CrossRef CAS PubMed.
- M. Melchionna and P. Fornasiero, Updates on the Roadmap for Photocatalysis, ACS Catal., 2020, 10, 5493–5501 CrossRef CAS.
- Z. Jiang, F. Wang, K. Fukuda, A. Karki, W. Huang, K. Yu, T. Yokota, K. Tajima, T. Q. Nguyen and T. Someya, Highly efficient organic photovoltaics with enhanced stability through the formation of doping-induced stable interfaces, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 6391–6397 CrossRef CAS PubMed.
- H. S. Jung, P. Verwilst, A. Sharma, J. Shin, J. L. Sessler and J. S. Kim, Organic molecule-based photothermal agents: an expanding photothermal therapy universe, Chem. Soc. Rev., 2018, 47, 2280–2297 RSC.
- H. Wang, J. Chang, M. Shi, W. Pan, N. Li and B. Tang, A Dual-Targeted Organic Photothermal Agent for Enhanced Photothermal Therapy, Angew. Chem., Int. Ed., 2019, 58, 1057–1061 CrossRef CAS PubMed.
- T. T. Abiola, B. Rioux, J. M. Toldo, J. Alarcan, J. M. Woolley, M. A. P. Turner, D. J. L. Coxon, M. Telles Do Casal, C. Peyrot, M. M. Mention, W. J. Buma, M. N. R. Ashfold, A. Braeuning, M. Barbatti, V. G. Stavros and F. Allais, Towards developing novel and sustainable molecular light-To-heat converters, Chem. Sci., 2021, 12, 15239–15252 RSC.
- L. Shen, Z. Tang, X. Wang, H. Liu, Y. Chen and X. Li, Effects of aromatic substituents on the electronic structure and excited state energy levels of diketopyrrolopyrrole derivatives for singlet fission, Phys. Chem. Chem. Phys., 2018, 20, 22997–23006 RSC.
- S. Qu and H. Tian, Diketopyrrolopyrrole (DPP)-based materials for organic photovoltaics, Chem. Commun., 2012, 48, 3039–3051 RSC.
- Y. Li, P. Sonar, L. Murphy and W. Hong, High mobility diketopyrrolopyrrole (DPP)-based organic semiconductor materials for organic thin film transistors and photovoltaics, Energy Environ. Sci., 2013, 6, 1684–1710 RSC.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Diketopyrrolopyrrole Polymers for Organic Solar Cells, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS.
- D. Molina, M. J. Álvaro-Martins and Á. Sastre-Santos, Diketopyrrolopyrrole-based single molecules in photovoltaic technologies, J. Mater. Chem. C, 2021, 9, 16078–16109 RSC.
- S. Ghosh, S. Shankar, D. S. Philips and A. Ajayaghosh, Diketopyrrolopyrrole-based functional supramolecular polymers: next-generation materials for optoelectronic applications, Mater. Today Chem., 2020, 16, 100242 CrossRef CAS.
- N. Luo, G. Zhang and Z. Liu, Keep glowing and going: recent progress in diketopyrrolopyrrole synthesis towards organic optoelectronic materials, Org. Chem. Front., 2021, 8, 4560–4581 RSC.
- T. In Ryu, Y. Yoon, J.-H. Kim, D.-H. Hwang, M. Jae Ko, D.-K. Lee, J. Young Kim, H. Kim, N.-G. Park, B. Kim and H. Jung Son, Simultaneous Enhancement of Solar Cell Efficiency and Photostability via Chemical Tuning of Electron Donating Units in Diketopyrrolopyrrole-Based Push–Pull Type Polymers, Macromolecules, 2014, 47, 6270–6280 CrossRef.
- F. Khan, Y. Jang, Y. Patil, R. Misra and F. D’Souza, Photoinduced Charge Separation Prompted Intervalence Charge Transfer in a Bis(thienyl)diketopyrrolopyrrole Bridged Donor-TCBD Push–Pull System, Angew. Chem., Int. Ed., 2021, 60, 20518–20527 CrossRef CAS.
- A. Shukla, S. K. M. McGregor, R. Wawrzinek, S. Saggar, E. G. Moore, S. C. Lo and E. B. Namdas, Light Amplification and Efficient Electroluminescence from a Solution-Processable Diketopyrrolopyrrole Derivative via Triplet-to-Singlet Upconversion, Adv. Funct. Mater., 2021, 31, 1–11 Search PubMed.
- P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, Effects of Crystal Morphology on Singlet Exciton Fission in Diketopyrrolopyrrole Thin Films, J. Phys. Chem. B, 2016, 120, 1357–1366 CrossRef CAS PubMed.
- I. Papadopoulos, M. J. Álvaro-Martins, D. Molina, P. M. McCosker, P. A. Keller, T. Clark, Á. Sastre-Santos and D. M. Guldi, Solvent-Dependent Singlet Fission in Diketopyrrolopyrrole Dimers: A Mediating Charge Transfer versus a Trapping Symmetry-Breaking Charge Separation, Adv. Energy Mater., 2020, 10, 1–10 Search PubMed.
- T. Mukhopadhyay, A. J. Musser, B. Puttaraju, J. Dhar, R. H. Friend and S. Patil, Is the Chemical Strategy for Imbuing ‘Polyene’ Character in Diketopyrrolopyrrole-Based Chromophores Sufficient for Singlet Fission?, J. Phys. Chem. Lett., 2017, 8, 984–991 CrossRef CAS.
- A. Riaño, P. Mayorga Burrezo, M. J. Mancheño, A. Timalsina, J. Smith, A. Facchetti, T. J. Marks, J. T. López Navarrete, J. L. Segura, J. Casado and R. Ponce, Ortiz, The unusual electronic structure of ambipolar dicyanovinyl-substituted diketopyrrolopyrrole derivatives, J. Mater. Chem. C, 2014, 2, 6376–6386 RSC.
- S. Ray, S. Sharma, U. Salzner and S. Patil, Synthesis and Characterization of Quinoidal Diketopyrrolopyrrole Derivatives with Exceptionally High Electron Affinities, J. Phys. Chem. C, 2017, 121, 16088–16097 CrossRef CAS.
- S. J. Bradley, M. Chi, J. M. White, C. R. Hall, L. Goerigk, T. A. Smith and K. P. Ghiggino, The role of conformational heterogeneity in the excited state dynamics of linked diketopyrrolopyrrole dimers, Phys. Chem. Chem. Phys., 2021, 23, 9357–9364 RSC.
- R. S. Szabadai, J. Roth-Barton, K. P. Ghiggino, J. M. White and D. J. D. Wilson, Solvatochromism in diketopyrrolopyrrole derivatives: experimental and computational studies, Aust. J. Chem., 2014, 67, 1330–1337 CrossRef CAS.
- N. T. Maitra, Double and Charge-Transfer Excitations in Time-Dependent Density Functional Theory, Annu. Rev. Phys. Chem., 2022, 73, 117–140 CrossRef PubMed.
- J. H. Starcke, M. Wormit, J. Schirmer and A. Dreuw, How much double excitation character do the lowest excited states of linear polyenes have?, Chem. Phys., 2006, 329, 39–49 CrossRef CAS.
- Y. Shu and D. G. Truhlar, Doubly Excited Character or Static Correlation of the Reference State in the Controversial 21Ag State of trans-Butadiene?, J. Am. Chem. Soc., 2017, 139, 13770–13778 CrossRef CAS PubMed.
- S. Mazumdar, Diagrammatic exciton-basis valence-bond theory of linear polyenes, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 4822–4838 CrossRef.
- W. Barford, Theory of the dark state of polyenes and carotenoids, Phys. Rev. B, 2022, 106, 035201 CrossRef CAS.
- T. Takaya and K. Iwata, Relaxation Mechanism of β-Carotene from S2 (1Bu+) State to S1 (2Ag−) State: Femtosecond Time-Resolved Near-IR Absorption and Stimulated Resonance Raman Studies in 900–1550 nm Region, J. Phys. Chem. A, 2014, 118, 4071–4078 CrossRef CAS PubMed.
- T. A. A. Oliver and G. R. Fleming, Following Coupled Electronic-Nuclear Motion through Conical Intersections in the Ultrafast Relaxation of β-Apo-8′-carotenal, J. Phys. Chem. B, 2015, 119, 11428–11441 CrossRef CAS PubMed.
- V. Šebelík, C. D. P. Duffy, E. Keil, T. Polívka and J. Hauer, Understanding Carotenoid Dynamics via the Vibronic Energy Relaxation Approach, J. Phys. Chem. B, 2022, 126, 3985–3994 CrossRef.
- M. Bondanza, D. Jacquemin and B. Mennucci, Excited States of Xanthophylls Revisited: Toward the Simulation of Biologically Relevant Systems, J. Phys. Chem. Lett., 2021, 12, 6604–6612 CrossRef CAS PubMed.
- C. M. Marian, A. Heil and M. Kleinschmidt, The DFT/MRCI method, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, 1–31 CAS.
- D. Casanova and A. I. Krylov, Spin-flip methods in quantum chemistry, Phys. Chem. Chem. Phys., 2020, 22, 4326–4342 RSC.
- S. Lee, M. Filatov, S. Lee and C. H. Choi, Eliminating spin-contamination of spin-flip time dependent density functional theory within linear response formalism by the use of zeroth-order mixed-reference (MR) reduced density matrix, J. Chem. Phys., 2018, 149, 104101 CrossRef PubMed.
- S. Lee, S. Shostak, M. Filatov and C. H. Choi, Conical Intersections in Organic Molecules: Benchmarking Mixed-Reference Spin-Flip Time-Dependent DFT (MRSF-TD-DFT) vs Spin-Flip TD-DFT, J. Phys. Chem. A, 2019, 123, 6455–6462 CrossRef CAS PubMed.
- Z. Yang, C. Shao and D. Cao, Screening donor groups of organic dyes for dye-sensitized solar cells, RSC Adv., 2015, 5, 22892–22898 RSC.
- F. Bureš, Fundamental aspects of property tuning in push–pull molecules, RSC Adv., 2014, 4, 58826–58851 RSC.
- A. Cesaretti, P. Foggi, C. G. Fortuna, F. Elisei, A. Spalletti and B. Carlotti, Uncovering Structure-Property Relationships in Push–Pull Chromophores: A Promising Route to Large Hyperpolarizability and Two-Photon Absorption, J. Phys. Chem. C, 2020, 124, 15739–15748 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A. 03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- M. S. Barca, M. J. Giuseppe, B. Colleen, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina, E. Guidez, T. Harville and S. Irle, Recent developments in the general atomic and molecular electronic structure system, J. Chem. Phys., 2020, 152, 154102 CrossRef.
- A. D. Becke, A new mixing of Hartree-Fock and local density-functional theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- Y. Shao, M. Head-Gordon and A. I. Krylov, The spin-flip approach within time-dependent density functional theory: theory and applications to diradicals, J. Chem. Phys., 2003, 118, 4807–4818 CrossRef CAS.
- S. Grimme and M. Waletzke, A combination of Kohn-Sham density functional theory and multi-reference configuration interaction methods, J. Chem. Phys., 1999, 111, 5645–5655 CrossRef CAS.
- I. Lyskov, M. Kleinschmidt and C. M. Marian, Redesign of the DFT/MRCI Hamiltonian, J. Chem. Phys., 2016, 144, 034104 CrossRef PubMed.
- TURBOMOLE V7.5 2020, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007 Search PubMed.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. Van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, TURBOMOLE: modular program suite for ab initio quantum-chemical and condensed-matter simulations, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- F. Plasser, M. Wormit and A. Dreuw, New tools for the systematic analysis and visualization of electronic excitations. I. Formalism, J. Chem. Phys., 2014, 141, 024106 CrossRef PubMed.
- F. Plasser, TheoDORE: a toolbox for a detailed and automated analysis of electronic excited state computations, J. Chem. Phys., 2020, 152, 084108 Search PubMed.
- M. Head-Gordon, A. M. Grana, D. Maurice and C. A. White, Analysis of electronic transitions as the difference of electron attachment and detachment densities, J. Phys. Chem., 1995, 99, 14261–14270 CrossRef CAS.
- M. Head-Gordon, Characterizing unpaired electrons from the one-particle density matrix, Chem. Phys. Lett., 2003, 372, 508–511 CrossRef CAS.
- B. W. H. Saes, M. Lutz, M. M. Wienk, S. C. J. Meskers, R. A. J. Janssen, B. W. H. Saes, M. Lutz, M. M. Wienk, S. C. J. Meskers and R. A. J. Janssen, Tuning the Optical Characteristics of Diketopyrrolopyrrole Molecules in the Solid State by Alkyl Side Chains, J. Phys. Chem. C, 2020, 124, 25229–25238 CrossRef CAS.
- M. A. El-Sayed, Spin-Orbit Coupling and the Radiationless Processes in Nitrogen, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- M. Huix-Rotllant, N. Ferré and M. Barbatti, in Quantum Chemistry and Dynamics of Excited States: Methods and Applications, ed. L. González and R. Lindh, John Wiley & Sons, Ltd, 2020, pp. 13–46 Search PubMed.
- P. Elliott, S. Goldson, C. Canahui and N. T. Maitra, Perspectives on double-excitations in TDDFT, Chem. Phys., 2011, 391, 110–119 CrossRef CAS.
- B. Shi, D. Nachtigallová, A. J. A. Aquino, F. B. C. Machado and H. Lischka, Excited states and excitonic interactions in prototypic polycyclic aromatic hydrocarbon dimers as models for graphitic interactions in carbon dots, Phys. Chem. Chem. Phys., 2019, 21, 9077–9088 RSC.
- B. Shi, D. Nachtigallová, A. J. A. Aquino, F. B. C. Machado and H. Lischka, Emission Energies and Stokes Shifts for Single Polycyclic Aromatic Hydrocarbon Sheets in Comparison to the Effect of Excimer Formation, J. Phys. Chem. Lett., 2019, 10, 5592–5597 CrossRef CAS.
- M. R. Silva-Junior, M. Schreiber, S. P. A. Sauer and W. Thiel, Benchmarks for electronically excited states: time-dependent density functional theory and density functional theory based multireference configuration interaction, J. Chem. Phys., 2008, 129, 104103 CrossRef PubMed.
- W. Fuß, Y. Haas and S. Zilberg, Twin states and conical intersections in linear polyenes, Chem. Phys., 2000, 259, 273–295 CrossRef.
- T. Sashima, Y. Koyama, T. Yamada and H. Hashimoto, The 1Bu+, 1Bu−, and 2Ag Energies of Crystalline Lycopene, β-Carotene, and Mini-9-β-carotene as Determined by Resonance-Raman Excitation Profiles: Dependence of the 1Bu- State Energy on the Conjugation Length, J. Phys. Chem. B, 2000, 104, 5011–5019 CrossRef CAS.
- S. Santra, J. Ray and D. Ghosh, Mechanism of singlet fission in carotenoids from a polyene model system, J. Phys. Chem. Lett., 2022, 13, 6800–6805 CrossRef CAS PubMed.
- P. O. Andersson, S. M. Bachilo, R. L. Chen and T. Gillbro, Solvent and temperature effects on dual fluorescence in a series of carotenes. Energy gap dependence of the internal conversion rate, J. Phys. Chem., 1995, 99, 16199–16209 CrossRef CAS.
- F. Plasser, A. I. Krylov and A. Dreuw, libwfa: wavefunction analysis tools for excited and open-shell electronic states, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 1–15 Search PubMed.
- R. J. Cave and E. R. Davidson, Non-vertical excitation energies for low-lying singlet states of butadiene and hexatriene, Chem. Phys. Lett., 1988, 148, 190–196 CrossRef CAS.
- Z. G. Soos, S. Ramasesha and D. S. Galvão, Band to Correlated Crossover in Alternating Hubbard and Pariser–Parr–Pople Chains: Nature of the Lowest Singlet Excitation of Conjugated Polymers, Phys. Rev. Lett., 1993, 71, 1609–1612 CrossRef CAS PubMed.
- D. Mukhopadhyay, G. W. Hayden and Z. G. Soos, Molecular-exciton approach to spin-charge crossovers in dimerized Hubbard and excitonic chains, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 9476–9492 CrossRef CAS PubMed.
- M. Y. Lavrentiev and W. Barford, 1 1Bu–2 1Ag crossover in conjugated polymers: the phase diagram of the molecular-orbital mode, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 48–55 Search PubMed.
- U. Salzner, Optoelectronic properties of diketopyrrolopyrrole homopolymers compared to donor–acceptor copolymers, J. Chem. Phys., 2021, 154, 054309 CrossRef CAS PubMed.
- H. Liu, X. F. Zhang, J. Z. Cheng, A. G. Zhong, H. R. Wen and S. Y. Liu, Novel Diketopyrrolopyrrole-Based π-Conjugated Molecules Synthesized Via One-Pot Direct Arylation Reaction, Molecules, 2019, 24, 1760 CrossRef PubMed.
- F. Plasser and H. Lischka, Analysis of excitonic and charge transfer interactions from quantum chemical calculations, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03533b |
| This journal is © the Owner Societies 2022 |