
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photooxidation of PC60BM: new insights from spectroscopy†
Iulia Emilia
Brumboiu‡§
 ad,
Leif K.E.
Ericsson‡
b,
Vanja
Blazinic
b,
Rickard
Hansson
b,
Andreas
Opitz
c,
Barbara
Brena
d and
Ellen
Moons
*b
ad,
Leif K.E.
Ericsson‡
b,
Vanja
Blazinic
b,
Rickard
Hansson
b,
Andreas
Opitz
c,
Barbara
Brena
d and
Ellen
Moons
*b
aDepartment of Theoretical Chemistry and Biology, KTH Royal Institute of Technology, SE-10691, Stockholm, Sweden
bDepartment of Engineering and Physics, Karlstad University, SE-65188 Karlstad, Sweden. E-mail: ellen.moons@kau.se
cInstitut für Physik, Humboldt-Universität zu Berlin, 12489 Berlin, Germany
dDepartment of Physics and Astronomy, Uppsala University, SE-75120 Uppsala, Sweden
First published on 14th September 2022
Abstract
This joint experimental–theoretical spectroscopy study of the fullerene derivative PC60BM ([6,6]-phenyl-C60-butyric acid methyl ester) aims to improve the understanding of the effect of photooxidation on its electronic structure. We have studied spin-coated thin films of PC60BM by X-ray Photoelectron Spectroscopy (XPS), Near-edge X-ray Absorption Fine Structure (NEXAFS) spectroscopy, and Fourier Transform Infrared Spectroscopy (FTIR), before and after intentional exposure to simulated sunlight in air for different lengths of time. The π* resonance in the C1s NEXAFS spectrum was found to be a very sensitive probe for the early changes to the fullerene cage, while FTIR spectra, in combination with O1s NEXAFS spectra, enabled the identification of the oxidation products. The changes observed in the spectra obtained by these complementary methods were compared with the corresponding Density Functional Theory (DFT) calculated single-molecule spectra of a large set of in silico generated oxidation products of PC60BM where oxygen atoms were attached to the C60 cage. This comparison confirms that photooxidation of PC60BM disrupts the conjugation of the fullerene cage by a transition from sp2 to sp3-hybridized carbon and causes the formation of several oxidation products, earlier proposed for C60. The agreement between experimental and calculated IR spectra suggests moreover the presence of dicarbonyl and anhydride structures on the fullerene cage, in combination with cage opening at the adsorption site. By including PC60BM with physisorbed O2 molecules on the cage in our theoretical description in order to model oxygen diffused through the film, the experimental O1s XPS and O1s NEXAFS spectra could be reproduced.
Introduction
The fullerene derivative PC60BM has for two decades been the prototypical electron acceptor molecule used in organic photovoltaics (OPVs). Although, in the quest to improve the power conversion efficiencies (PCE) of OPV devices, PC60BM has been to a large extent replaced by non-fullerene acceptors,1,2 it is still being used in ternary solar cells to improve the open circuit voltage (VOC) and electron mobility.3,4 In perovskite solar cells, PC60BM, alongside other fullerene derivatives, is routinely employed as an electron transport layer.5 Additionally, thanks to the favourable energy position of the lowest unoccupied molecular orbital (LUMO) and good charge transport properties, PC60BM and other fullerene derivatives have further applications in several areas of organic electronics.6 As stability is one of the most important challenges for organic electronics and photovoltaics,7,8 insights into the photodegradation mechanisms of organic materials such as PC60BM are highly relevant and a pre-requisite to the development of new mitigation strategies, as well as more stable active layer materials and charge transport layers.When it comes to organic photovoltaics, stability testing of device operation under normal solar illumination conditions9 as well as concentrated sunlight10 has provided some insights in the types of degradation that can occur in some commonly studied types of polymer solar cells. Contributions to the performance from electrode degradation, degradation of the organic molecules making up the active layer of the solar cell, as well as morphology changes in this layer have been reported.7,8,11
The active layer of polymer solar cells typically has a bulk heterojunction structure consisting of a conjugated polymer as electron donor and a fullerene derivative as electron acceptor. Investigations of the photodegradation mechanisms in such polymer-based solar cells have been focused on the light-absorbing component, the conjugated polymer, with the purpose of identifying and synthesizing more stable donor molecules,12–14 while the photodegradation of the fullerene has been addressed in detail more recently.15–21 We have previously shown that for the blend of the polymer TQ1 (poly[2,3-bis-(3-octyloxyphenyl)quinoxaline-5,8-diyl-alt-thiophene-2,5-diyl]) with PC60BM, the LUMO of PC60BM is much more affected by photooxidation than the LUMO of TQ1, contributing to the performance degradation.22,23
It is known that solid C60 is air-sensitive and can quickly convert into a product with reduced solubility in apolar organic solvents, upon exposure to light at room temperature.24 The triplet excited state of C60-based fullerenes readily transfers energy to an oxygen molecule, forming singlet molecular oxygen, 1O2, or, alternatively, it transfers an electron to an oxygen molecule and forms a radical ion.25,26 The formation of several oxidation products has been reported for unsubstituted fullerenes, from epoxides to carbonyls, dicarbonyls, and cyclic anhydrides. IR spectroscopy and mass spectrometry have been useful methods to identify these products. Taliani et al. have shown that, upon illumination, the fullerene C60-cage can open due to a reaction with excited molecular oxygen (i.e. singlet oxygen 1O2), generated via C60 triplet energy transfer with molecular oxygen, 3O2. This singlet oxygen can cleave a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the C60 cage and, via the suggested dioxetane mechanism, open up the cage and form a fullerene dioxide with dicarbonyl structures on the edges of the opening.26 Werner et al. suggested that the mechanism for thermal oxidation of C60 (in the absence of light) consists of several stages and differs depending on the oxygen partial pressure and the temperature. They suggest that when C60 is exposed to O2 at temperatures below 370 K, the intercalated molecular oxygen forms fullerene oxides with an epoxide structure, while at elevated temperatures above 500 K and at high O2 partial pressure, cyclic anhydrides are formed, evolving CO2 and ring opening. The authors also mention that several metastable intermediate products may coexist.27–29
C bond of the C60 cage and, via the suggested dioxetane mechanism, open up the cage and form a fullerene dioxide with dicarbonyl structures on the edges of the opening.26 Werner et al. suggested that the mechanism for thermal oxidation of C60 (in the absence of light) consists of several stages and differs depending on the oxygen partial pressure and the temperature. They suggest that when C60 is exposed to O2 at temperatures below 370 K, the intercalated molecular oxygen forms fullerene oxides with an epoxide structure, while at elevated temperatures above 500 K and at high O2 partial pressure, cyclic anhydrides are formed, evolving CO2 and ring opening. The authors also mention that several metastable intermediate products may coexist.27–29
By mass spectrometry, Reese et al. have found a series of fullerene oxides in PC60BM films that were exposed 1000 h to light in air,18 and Yamane et al. have reported several oxidation products of PC60BM with subsequent addition of oxygen atoms to the cage.30 Under inert conditions, in the absence of oxygen, photo-induced dimerization of PC60BM has been reported.21,31–34 However, in the presence of oxygen, the photopolymerization reaction is inhibited and photooxidation is the dominant photodegradation mechanism. Dettinger et al. have shown by FTIR spectroscopy that the photodegradation of PC60BM in O2 can be classified in three stages: partial loss of conjugation in the C60 leaving the tail intact, followed by the oxidation of sites on or close to the side chain, and finally the saturation stage when an average number of about three carbonyl groups have been formed per degraded PC60BM molecule. This leads to a series of different degradation products.35 We have earlier shown that exposure to simulated sunlight in air affects both the valence band spectrum and the C1s Near Edge X-ray Absorption Fine Structure (NEXAFS) spectrum of solid PC60BM films and that the effect of photooxidation of PC60BM on its electronic structure is similar to that of C60.15 In the same study, we observed a strong decrease in the valence density of states of both C60 and PC60BM when exposed to light in air. Moreover, we have recently demonstrated that PC60BM photodegrades faster than PC70BM.36
In the present study, we follow the photooxidation of PC60BM in air over time, using complementary spectroscopy techniques, C1s X-ray Photoelectron Spectroscopy (XPS), O1s XPS, C1s NEXAFS, O1s NEXAFS and Fourier-Transform Infrared Spectroscopy (FTIR). From these methods, in combination with Density Functional Theory (DFT), we identify the oxidation products of PC60BM and the impact of photooxidation on the electronic properties of the material, especially on the unoccupied electronic levels, which are of crucial importance for its role as electron acceptor and electron transporting material in the photoactive layer of organic solar cells.
Materials and methods
Materials
PC60BM (purity 99.5%) was purchased from Solenne B.V., the Netherlands. Thin films of PC60BM for X-ray spectroscopy were prepared by spin-coating a 12 mg ml−1 PC60BM solution in chloroform on Si substrates. Chloroform (analytical grade) was purchased from Merck, Germany. The spin speed was 3000 rpm during 60 s. The substrates were n-type Si(001) with a resistivity of 0.001–0.003 Ω cm that were cleaned according to the standard RCA method without the final hydrofluoric acid etching step.37–39 Thin films for FTIR spectroscopy were prepared by spin-coating a 15 mg ml−1 PC60BM solution in chlorobenzene on KBr plates, at 750 rpm for 100 s. Chlorobenzene (purity 99%) was purchased from Alfa Aesar, Germany. The KBr plates (∅ 13 × 2 mm) were purchased from Thermo Fisher Scientific, UK. All solution preparations and spin-coating were performed under yellow light in N2 atmosphere (O2 < 0.1 ppm, H2O < 0.1 ppm) inside a glove-box (MB200MOD from MBraun, Germany). Prior to spin-coating the solutions were stirred overnight at 60 °C.The photodegradation of the samples was performed by exposing the thin PC60BM films in air (non-zero humidity) to the simulated solar light (AM1.5) of a solar simulator (Sol2A, model 94022A, Oriel Instruments, USA), calibrated by a silicon photodiode reference cell (Kimo SL100, Kimo Instrument, Sweden). Except during the intentional exposure to simulated solar light for specified amounts of time, the films prepared for X-ray spectroscopy were protected from ambient light and air until the measurements were completed. For this purpose, a N2-filled ultra-high vacuum compatible metal container was used for sample transport to the synchrotron facility.
Experimental methods
The thickness of the films was measured by making a scratch in a spin-coated PC60BM film on a Si wafer and scanning in tapping mode across the scratch using the Si tip of an atomic force microscope (Multimode Nanoscope IIIa/8, Bruker, France). The film thickness of samples for X-ray spectroscopy was (90 ± 10) nm and for the FTIR samples it was (70 ± 10) nm.NEXAFS spectra both at the C1s-edge and at the O1s-edge were collected at beamline D1011 of the MAX IV National Laboratory for Synchrotron Radiation in Lund, Sweden. Total electron yield (TEY) spectra were obtained from the sample drain current. For partial electron yield (PEY) spectra, a multichannel plate detector was used with a retarding voltage of −150 V applied to the entrance grid, to obtain a higher surface sensitivity. Spectra were measured at 55° incident light relative to the sample surface, close to the so-called magic angle, to avoid dependence of absorption on molecular orientation. The spectra were divided by the corresponding spectrum of a freshly in situ Ar-sputtered Au/Mica sample and then normalized in the high photon energy region according to the “stable monitor method”.40,41 The photon energy scale was calibrated by measuring the spectrum of highly ordered pyrolytic graphite and using the position of the exciton resonance at 291.65 eV as an energy reference.42
The XPS measurements of the C1s and O1s core levels were performed using the same equipment as for NEXAFS and the photoelectrons were collected with a VG Scienta SES200 analyzer using a pass energy of 50 eV, in the sample normal direction. The binding energy (EB) scale in the XPS spectra was referenced to the Fermi level at EB = 0 eV as measured from a freshly in situ Ar-sputtered Au/Mica sample. The spectral intensities were normalized to the photon flux and a Shirley background43 was removed.
Infrared spectra were recorded in transmission mode using a Nexus 470-FTIR spectrometer, working with OMNIC 6.0a software from Thermo Fisher Scientific, UK. Spectra were obtained using 32 scans at 2 cm−1 resolution. The spectra were recorded for unexposed PC60BM and for cumulative exposure times of 15 min, 2, 10, 19, 30 and 47 hours in air.
Computational methods
The molecular structure of PC60BM (inset of Fig. 1a) and of several possible photodegradation products was relaxed in Gaussian 09,44 at the DFT level, using the B3LYP functional,45 in combination with the 6-31G(d,p) basis set.46 The optimized geometry of each single molecule was then used to determine the C1s XPS and NEXAFS spectra, as well as the molecular vibrations and the corresponding IR intensities.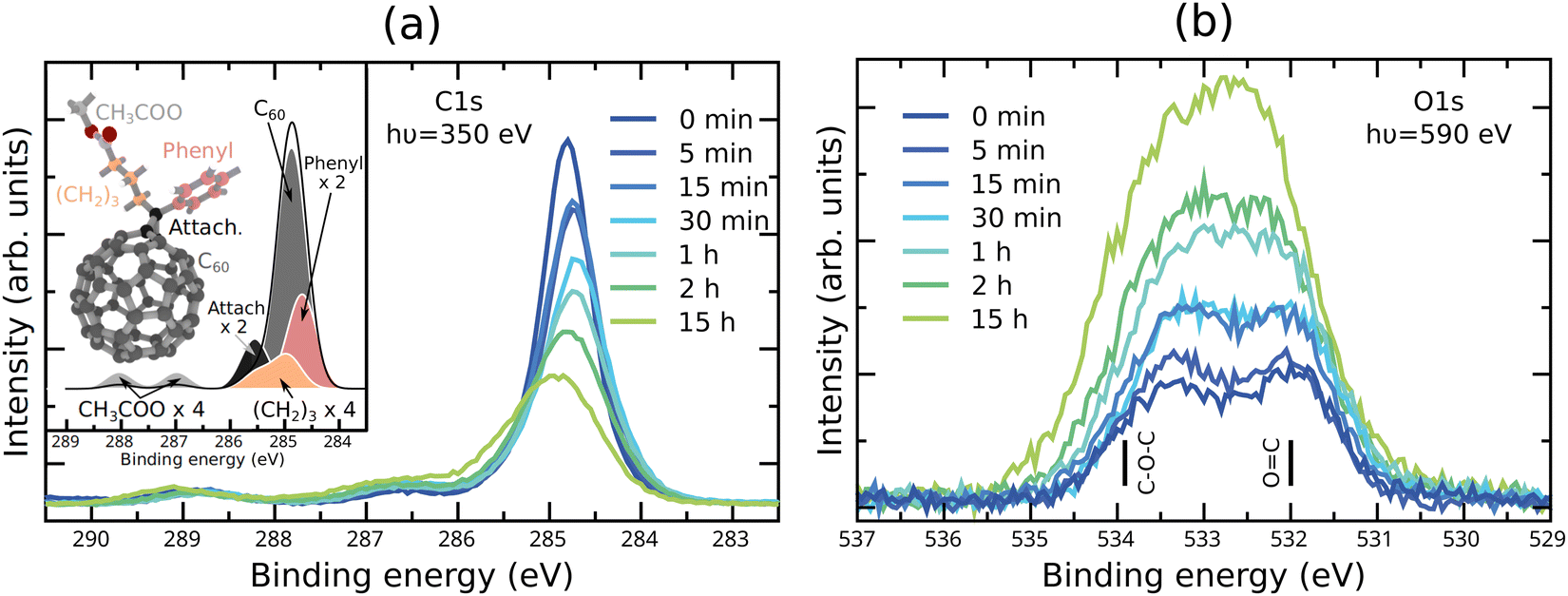 | ||
| Fig. 1 Experimental (a) C1s XPS spectra and (b) O1s XPS spectra of the unexposed PC60BM film and PC60BM films exposed to light in air for different durations. The inset of (a) shows the calculated spectrum of pristine PC60BM separated into components originating from different molecular moieties, marked with the same colour on the molecular structure. In (b) the binding energies for C–O–C and OC are indicated, as calculated for pristine PC60BM. | ||
Calculated O1s XPS and NEXAFS spectra have already been reported for several configurations of PC60BM with oxygen in our previous article.47 Several new configurations, described below, were calculated in the same way and the full list of possible photodegradation products computed in this study is included in the Fig. S1 and S2 (ESI†). Besides products with intact number of C atoms in the cage, we have included products with C58 cages, to be able to model the possible loss of C atoms from the fullerene. Since the PC60BM films were photodegraded in non-zero humidity, we considered photodegradation products with OH groups attached to the fullerene cage. However, these products could be excluded based on the clear pre-edge peak in their calculated C1s NEXAFS spectrum, which is not present in the measured spectrum (see Fig. S3, ESI†).
The O1s and C1s XPS and NEXAFS calculations were performed at the DFT level in StoBe,48 using the exchange functional by Becke,45 and the correlation functional by Perdew,49 in combination with an augmented diffuse basis set (19s, 19p, 19d).50 The core-excited atom was described by an IGLO-III triple zeta basis set,51 while the remaining atoms of the same species in the molecule were described by an effective core potential of 6 electrons (O), respectively 4 electrons (C), provided by the StoBe package. The atoms of a different species than the core-excited atom were described by a double zeta plus valence polarization basis set. The ionization energy (IE) for each individual oxygen and, respectively, carbon atom in the single molecule was determined as the difference between the ground state energy and the energy calculated with a full core hole on the 1s state of the core-excited atom (ΔKohn–Sham). Discrete O1s, respectively, C1s XPS spectra were obtained by collecting the calculated 1s ionization energies. The discrete O1s XPS spectra were broadened using Gaussian functions of 1.0 eV full width at half maximum (FWHM) and were shifted by 4.7 eV towards lower binding energies, so that the calculated energy position of the low binding energy peak of pristine PC60BM matched the position of the same peak measured for the unexposed film. The C1s XPS spectra were broadened using Gaussian functions of 0.6 eV FWHM and shifted by 5.0 eV towards lower binding energies, in order to match the energy position of the main experimental peak of pristine PC60BM. These shifts account to a large extent for the fact that the calculations and experiments have different reference energies, i.e. the vacuum level (calculations), respectively the Fermi level (experiments).
Individual O1s and C1s NEXAFS spectra were determined separately for each O, respectively, C atom in the molecule. The O1s NEXAFS spectra were computed as described in ref. 47. Specifically, the dipole transition matrix elements were determined in the presence of a half core hole created at a specific oxygen atom site. Each individual spectrum obtained this way was broadened using Gaussian functions of variable FWHM and shifted according to the calculated ionization energy (IE). The initial FWHM of 1.0 eV was linearly increased to 2.65 eV in the photon energy range between IE–6 eV and IE. The total O1s NEXAFS spectrum for a particular photodegradation product was determined as the sum of all individual O atom contributions and further shifted by 0.8 eV towards lower photon energies to align the calculated and measured positions of the π* peak of pristine and unexposed PC60BM, respectively.
The C1s NEXAFS spectra were determined as in our previous study,52 using a full core hole to describe the core-excited atom and account for the core hole relaxation effects.53 Similarly to O1s NEXAFS, the individual C1s NEXAFS spectra were shifted according to the calculated ionization energy and broadened using Gaussian functions of variable FWHM, as described in ref. 52. A total C1s NEXAFS spectrum was determined as the sum of all individual C atom spectra and it was shifted by 2.05 eV towards higher photon energies. This shift was required to align the π* peak in the calculated C1s NEXAFS spectrum of pristine PC60BM with the experimental π* peak measured for unexposed films.
Finally, the molecular vibrations and corresponding IR intensities were calculated using Gaussian 09, the B3LYP functional and the 6-31G(d,p) basis set in the harmonic approximation. A scaling by 0.9627 was performed for the vibrational frequencies larger than 1000 cm−1 as recommended for the functional and basis set used.54 A Gaussian broadening of 20 cm−1 constant FWHM was added to the discrete IR spectrum in order to facilitate the comparison to the experiment. A shift of 22 cm−1, required to align the calculated carbonyl peak position of pristine PC60BM with the experimental unexposed PC60BM carbonyl peak, was applied to all calculated spectra.
By comparing the experimental data with single molecule calculated spectra, two possible photodegradation products were identified as likely. These are (1) a PC58BM where three additional oxygen atoms are bonded to cage C atoms, forming an anhydride structure (PC58BM-anhydride), and (2) a PC58BM where two additional oxygen atoms are attached with double bonds to the cage C atoms forming a dicarbonyl structure (PC58BM-dicarbonyl), similar to that proposed for C60 by Taliani et al.26 Besides these two products, PC60BM with an O2 molecule in the vicinity of the cage (PC60BM:O2) was required to describe the O1s XPS and NEXAFS spectra. The samples used in the experiments are solid films and, depending on the exposure times, only a portion of the molecules in the film will undergo modifications as discussed in detail in the Discussion/theory–experiment comparison section. Considering this, we further modelled the photodegraded spectra (T) by taking a series of different weighted averages between the spectra of pristine PC60BM (A), PC58BM-anhydride (B), PC58BM-dicarbonyl (C), and PC60BM:O2 (D):
| T = w·A + x·B + y·C + z·D |
In the following, only these weighted averaged spectra will be shown and the complete set of individual calculated spectra of all the modified PC60BM molecules and oxidation products considered in this study are included in the Fig. S4–S13 (ESI†).
As a final note, we mention that we are here modelling the effects of photodegradation on a film by using several single molecule calculations. To simulate the actual film, including all the possible photodegradation products, would be computationally very expensive within DFT, especially if we want to achieve the same accuracy levels. Since the properties of interest here are mainly molecular, as confirmed by the agreement between the calculated spectra of the pristine PC60BM molecule and the measured spectra of the unexposed films (see below), this is a reasonable approximation for the scope of our study.
Experimental results
XPS
The C1s and O1s XPS spectra obtained for unexposed PC60BM films and for films exposed to light in air with gradually increasing exposure times, are shown in Fig. 1a and b, respectively. The C1s XPS spectrum of pristine PC60BM (Fig. 1a) shows a main peak at 284.8 eV and two side features at 286.6 eV and 288.8 eV. Through comparison with the calculated C1s XPS spectrum (inset in Fig. 1a), we assign the 284.8 eV peak mainly to the sp2 carbon of the fullerene cage. Around the main peak, there are contributions of the phenyl at the low binding energy (BE) side, and of the sp3 hybridized carbons of the side chain and the connecting carbons between the cage and the side chain at the high BE side. The two separate high BE peaks at 286.6 and 288.8 eV, often assigned to shake-up peaks in the literature,55 are also present in the calculated spectrum, but slightly shifted to 287.0 and 288.0 eV, respectively. The calculations of the individual spectra demonstrate that these two side peaks can be assigned to the carbons in the ester group at the end of the PC60BM tail. The O1s XPS spectrum of pristine PC60BM (Fig. 1b) was published in our earlier report.47 Two peaks can be distinguished for pristine PC60BM assigned to the oxygen atoms in the ester group of the PC60BM tail, one high BE component at 533.4 eV (C–O–C) and one low BE component at 532.0 eV (CO), i.e. with a binding energy difference of 1.4 eV in close agreement with the values reported by Guan et al.56 The single molecule calculation predicts a larger separation (1.9 eV) between the two peaks, but is nonetheless in reasonable agreement with the experiment.
As the exposure time increases, the spectra show that the total C1s intensity strongly decreases, while the total O1s intensity strongly increases. This could suggest that an oxygen rich surface layer is formed that strongly attenuates the carbon signal from the surface layer. Interestingly, slight shifts in peak positions are also observed. The C1s main peak maximum shifts slightly to lower binding energies (and becomes slightly wider) after 30 min exposure and starts to shift back to higher binding energies after 1 h exposure. Such shifts might be an indication for changes in the band bending at the surface of the film. This also suggests that the photooxidation process consists of different stages, in which different processes dominate. Also, the two high BE peaks in the C1s spectrum shift in the same direction after long exposure times, but do not change significantly in intensity. Changes in the line shape are observed for the C1s spectrum after long exposure times, with the formation of an asymmetric shoulder at the high BE side of the main peak. This is in agreement with an increasing portion of the sp2 hybridized carbon transforming to sp3 hybridized carbon.
The O1s spectra undergo clear shape changes, making the two peaks, present in the pristine PC60BM and assigned to the ester group in the tail, less distinct already after 30 min exposure. After short exposure times the high BE side increases and widens slightly more than the low BE side. After 15 h exposure, the O1s spectrum shows one single peak that has clearly become wider and slightly asymmetric, with increased intensity on the high BE side, although the low BE side of the resulting peak is the highest in intensity.
As a basis for the interpretation of the changes in the O1s spectrum of photodegraded PC60BM film, we make use of the large set of oxidation products calculated in our earlier theoretical study.47 These are shown in the ESI† in Fig. S8 and S9, together with additional products. Several oxidation products that were calculated exhibit new contributions to the O1s XPS spectrum that fall between the two peaks of pristine PC60BM. In particular, a good match between the experimental O1s XPS spectrum of photodegraded PC60BM and the calculated single molecule spectra was obtained for physisorbed O2, i.e. PC60BM:O2, Fig. S9m (ESI†). However, XPS alone cannot provide a conclusive chemical assignment in this study, and complementary methods are needed to identify the photooxidation products of PC60BM.
C1s NEXAFS
In Fig. 2, C1s NEXAFS spectra collected in the PEY and TEY modes are shown for a series of exposure times. For the complete assignment of the absorption resonances in the C1s NEXAFS spectrum of pristine PC60BM we refer to our previous study of the molecule that includes a complete first principles analysis.52 In fullerenes, the C1s NEXAFS shows a very distinct and strong π* absorption. We have earlier reported15 that the π* resonances are notably weakened upon exposure of PC60BM to light in air for 30 min and 19 h. This was interpreted by the transformation of sp2 carbon of the C60 cage to sp3 carbon, with the consequent loss of conjugation, in analogy with the observed changes in the C1s NEXAFS spectrum of photodegraded C60.15 The loss of conjugation is most pronounced in the three first peaks in the π* region, at 284.5, 285.8 and 286.2 eV (marked as π*, 2, 3 in Fig. 2a), and stronger at the surface (PEY) than in the sub-surface region (TEY), or the rest of the film. The decrease in π* peak intensity is very strong in C1s NEXAFS because any change in the hybridization of a single C atom in the cage will affect the conjugation of the whole cage and hence the effect on the intensity of the transitions making up the main NEXAFS resonances associated with the cage will be emphasized. We note that the peak marked “S” in Fig. 2, whose origin is discussed in detail in our earlier work,52 remains unaffected by photodegradation. | ||
| Fig. 2 C1s NEXAFS spectra in (a) partial electron yield (PEY) mode, and (b) total electron yield (TEY) mode of pristine PC60BM and PC60BM exposed to light in air for different durations, shown in comparison to the calculated NEXAFS spectrum of pristine PC60BM (black line). | ||
The C1s NEXAFS spectra assure us that it is mainly the carbon atoms of the fullerene cage (responsible for the π* peak) that are affected by the photooxidation of the surface PC60BM molecules. No significant changes in the tail of the PC60BM molecule can be identified from the evolution of the σ* region of the NEXAFS spectrum (peak 4), except for a small increase in the shoulder to this peak at 287 eV. This feature appears with a low intensity in the PEY spectrum after 2 hours exposure. In agreement with this, also the tail fingerprints in the C1s XPS spectra remain unaffected after exposure.
C1s NEXAFS spectra were calculated for pristine PC60BM, shown in Fig. 2, as well as for a large number of possible oxidation products that are presented in Fig. S10 and S11 of the ESI.† The calculated spectrum of the pristine molecule is in very good agreement with the measured spectrum of the unexposed film, suggesting that the NEXAFS features originate at the molecular level and confirming that the single molecule is a suitable model for this spectroscopy.
O1s NEXAFS
O1s NEXAFS spectra shed more light on the fine structure of the O atoms in the PC60BM molecule and its photooxidation products. Fig. 3 shows measured O1s NEXAFS spectra in PEY mode of pristine PC60BM and PC60BM exposed to light in air for different lengths of time. The peak assignment of the O1s NEXAFS spectrum for pristine PC60BM was given in ref. 47, based on the very good agreement between the calculated spectrum of the pristine molecule and the measured spectrum of the unexposed film. Briefly, peak 1 at 530.8 eV is assigned to the CO from the side-chain of PC60BM, while peaks 2 (533.5 eV) and 3 (535.3 eV) are assigned mainly to C–O–CH3. Both CO and C–O–CH3 contribute to peak 4.
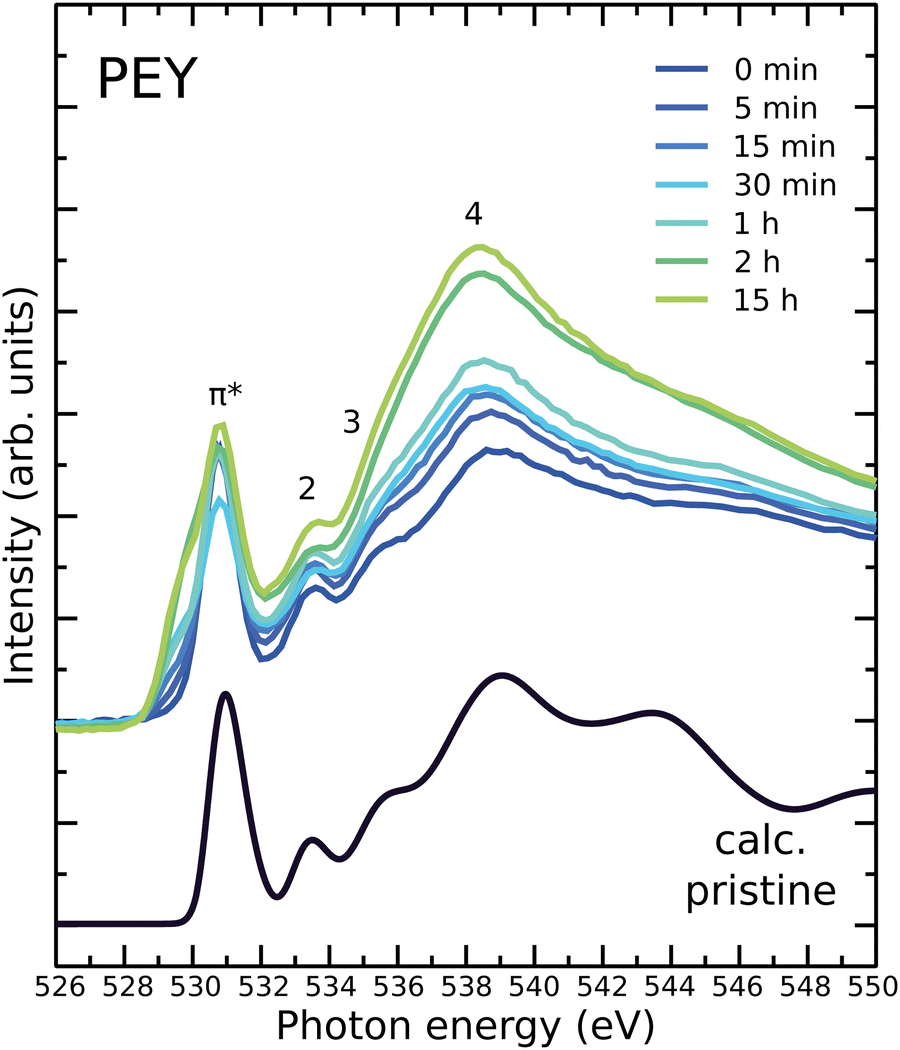 | ||
| Fig. 3 Measured O1s NEXAFS spectra of unexposed PC60BM and PC60BM samples exposed to light in air for different durations, shown in comparison to the calculated spectrum of pristine PC60BM (black line). | ||
When exposed to light in air, a new resonance is observed at 529 eV in the π* region of the O1s NEXAFS spectrum, providing a clear indication that double bonded oxygen is formed. O1s NEXAFS spectra were calculated for the possible oxidation products that are presented in Fig. S12 and S13 of the ESI.† The calculated spectra of four different structures show new features in the pre-edge where the experimental spectrum exhibits an increased absorption. These are PC60BM:O2, PC60BM-dicarbonyl, PC58BM-dicarbonyl, and PC58BM-anhydride. We therefore limit our discussion in the manuscript to these four structures.
FTIR
Fig. 4a shows FTIR spectra in the range 1000–2000 cm−1 of fresh and altered PC60BM films. The main peaks are marked and their positions and assignments listed in Table 1. Of interest are peaks 11 and 14, located at 1429 cm−1 and 1188 cm−1, respectively, and assigned to the fullerene cage.57,58 The intensity of these peaks decreases with increasing exposure of PC60BM films to light in air. Additionally, we note peak 2, located at 1737 cm−1, whose intensity remains unchanged. This peak is assigned to the carbonyl stretch of the methyl ester in the side chain of PC60BM. After 2 h light exposure in air, two features around 1780 cm−1 and 1710 cm−1 start to appear (peaks 1 and 3), and grow with increasing exposure time, as shown in Fig. 4b. These results are in agreement with those reported by Chambon et al.17 and Speller et al.19 for photooxidation of PC60BM in air, and by Dettinger et al.35 for photooxidation of PC60BM in O2.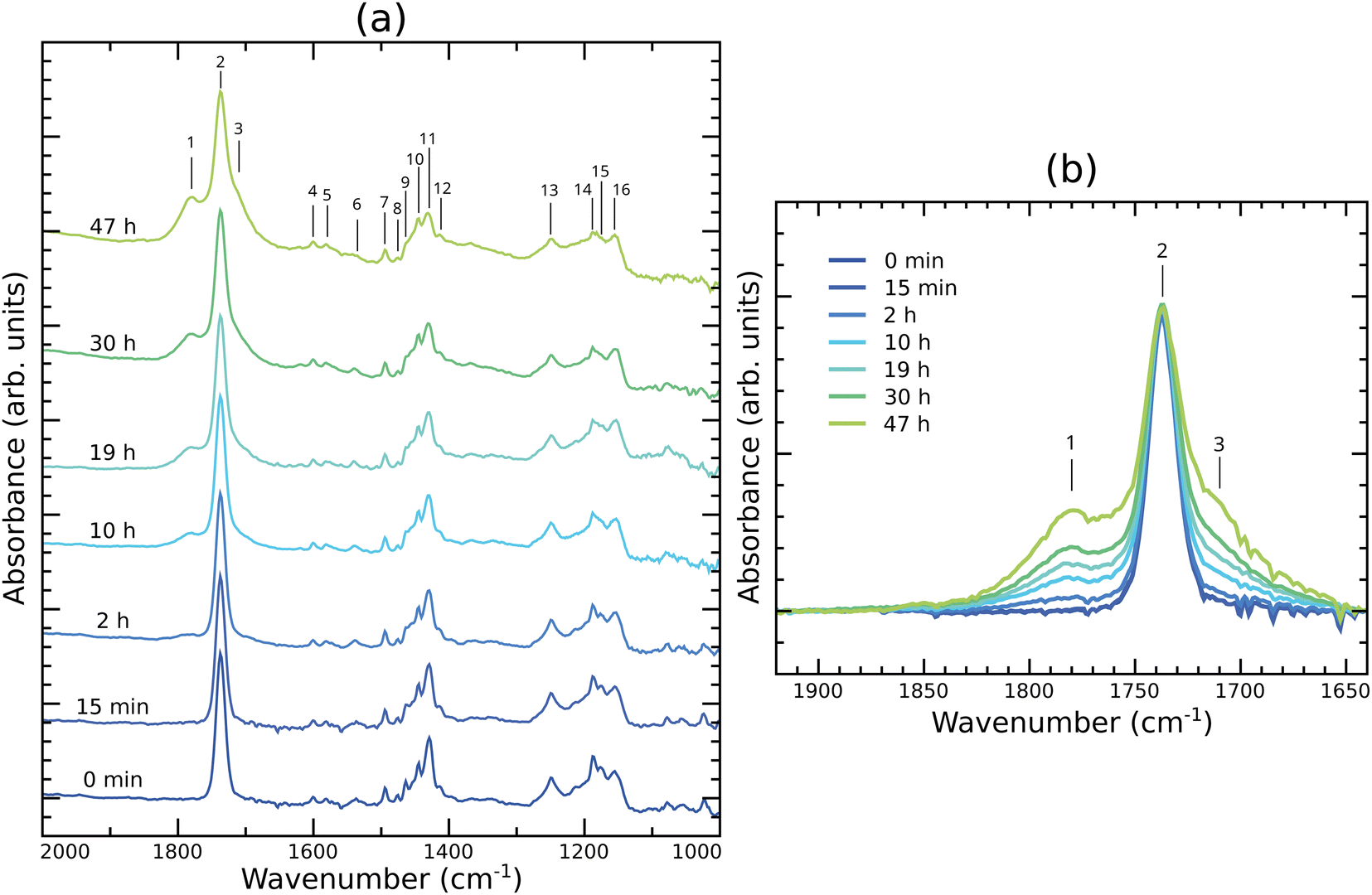 | ||
| Fig. 4 FTIR absorption spectra of PC60BM thin films exposed to light in air for different exposure times (a) in the 1000–2000 cm−1 region, and (b) zoomed in to the spectral region around the carbonyl peak. | ||
| Peak index | Frequency (cm−1) | Vibrational assignment: | Ref. |
|---|---|---|---|
| 1 | 1780 | CO stretch of anhydride |
28 |
| 2 | 1737 | CO stretch of methyl ester |
17 |
| 3 | 1710 | CO stretch of ketone or dicarbonyl |
26 |
| 4 | 1600 | CC stretch (aromatic) |
59 |
| 5 | 1581 | CC stretch (aromatic) |
28 |
| 6 | 1537 | CC stretch (aromatic) |
59 |
| 7 | 1495 | CC stretch (aromatic) |
59 |
| 8 | 1475 | C–H scissoring | 59 |
| 9 | 1464 | CC stretch (aromatic) |
59 |
| 10 | 1444 | sp3 C–H bending | 60 |
| 11 | 1429 | C60 cage | 57 |
| 12 | 1413 | sp3 C–H (bending) | 60 |
| 13 | 1250 | Acyl or phenyl C–O | 59 |
| 14 | 1188 | C60 cage | 57 |
| 15 | 1174 | Acyl or phenyl C–O | 59 |
| 16 | 1155 | Acyl or phenyl C–O | 60 and 61 |
The rising feature located at 1710 cm−1 (peak 3) could be attributed to the formation of dicarbonyl on the fullerene cage, which implies cage opening, as proposed for photooxidized C60 by Taliani et al.26 Our calculations of such dicarbonyl structures, shown in Fig. S5n and q (ESI†), yield peaks located in this region, corresponding to the symmetric and asymmetric CO vibrations, where the two CO stretch modes are in phase or out of phase, respectively.
The high wavenumber feature, growing at 1780 cm−1 (peak 1), is more difficult to assign because carbonyl peaks at higher wavenumbers than 1750 cm−1, corresponding to esters, are rare. Aldehydes, ketones, carboxylic acids, and amides correspond with carbonyl peak positions at lower wavenumbers. The CO stretch vibrations of anhydrides, however, correspond with wavenumbers that exceed those of esters. An anhydride would generate two peaks, from the symmetrical and asymmetrical stretches of the two anhydride carbonyls, respectively. We assign the growing absorption peak near 1780 cm−1 to the formation of an anhydride on the fullerene cage.
Our observation is in agreement with Dettinger et al., who have observed two new carbonyl vibrations upon photooxidation of PC60BM in O2, one at 1784 cm−1 and one, that appeared after larger photon doses, at 1854 cm−1, together with the pristine ester carbonyl of the PC60BM side chain at 1735 cm−1.35 Analogously, two new carbonyl peaks have also been reported by Yamane et al.62 on indene-C60 monoadduct and indene-C60 bisadduct fullerene derivatives that do not have a carbonyl in the side chain. FTIR peaks at 1725 cm−1 and 1785 cm−1 were also found by Wohlers et al. for thermally oxidized C6027,63 and assigned to an anhydride structure.
To further identify the photooxidation products, IR spectra were calculated for a large number of possible oxidation products and are presented in Fig. S4 and S5 of the ESI.† Among those, we have calculated the IR spectra for two structures where the anhydride is formed onto the cage (Fig. S5o and r, ESI†), leading to the cage opening.
To summarize, the two features, at 1710 cm−1 and 1780 cm−1, observed in the carbonyl region of the IR spectrum of PC60BM films exposed to light and air, can be assigned to two different photooxidation products, dicarbonyls and anhydrides, respectively, as seen from the comparison with the calculated spectra. As both features are rather broad, it is reasonable to assume that both symmetric and asymmetric CO stretching modes of each of the photooxidation products contribute. In addition, the formation of these products implies not only the scission of CC bonds, but also the breaking of the cage, as supported by the intensity decrease of peaks 11 and 14 in the measured IR spectra, leading to the loss of the conjugation of the cage, in agreement with the C1s NEXAFS results.
Discussion/theory–experiment comparison
From the analysis of experimental spectra presented in the previous section, we conclude that the chemical modifications of the fullerene cage due to the exposure of PC60BM to light in air include (1) diffusion of O2 into the film (O1s XPS), (2) adsorption of oxygen (O1s XPS, O1s NEXAFS) to the fullerene cage (C1s NEXAFS) and (3) loss of some carbon atoms due to cage opening (C1s XPS). Thereby, oxygen adsorption on the fullerene cage must include the formation of double bonds between cage C atoms and oxygen (FTIR, O1s NEXAFS), especially after long exposure times. Considering the difference in information depth for the different measurement techniques, we can also conclude that the oxidation is much further developed at the surface of the film (XPS, NEXAFS) than in the bulk (FTIR).With these observations in mind, we have attempted to determine the most likely photodegradation products by computing the X-ray and infrared spectra of several possible oxidation products of PC60BM and by comparing the calculated spectra to the experimentally measured ones. The list of all considered molecular structures and corresponding calculated spectra can be found in the Supplementary Information. Although many of these final products were able to reproduce the features of the XPS and NEXAFS spectra, in the case of IR spectroscopy, it was necessary to combine several molecular structures to reproduce the experimental spectra. The calculated IR spectra of the products with intact cages, i.e. PC60BM-anhydride and PC60BM-dicarbonyl, present the features we observe in the carbonyl region, however shifted towards higher wavenumbers compared to the experimental peaks (see Fig. S14 of the ESI†). It is known that reducing ring strain leads to the shift of carbonyl IR peak positions towards lower wavenumbers.64 A straightforward way to reduce ring strain is to make the ring larger, e.g. by removing C atoms from the oxygen adsorption site. Removing C atoms from the cage would keep in line with the modifications observed in the C1s XPS spectra after long exposure times. We have, therefore, considered what happens if we remove two C atoms from each of the products with intact cages. In fact, the combination of these two modified molecular structures, shown together with pristine PC60BM in Fig. 5, could reproduce the experimental findings in FTIR. The structure PC60BM:O2 also contributes (see O1s XPS), but its IR spectrum is identical to pristine PC60BM and therefore it is not included in Fig. 6. The first degradation product, denoted PC58BM-anhydride in Fig. 5b, consists of an anhydride formed on the fullerene cage, as proposed by Wohlers et al.,28 and removing two C atoms from the adsorption site. The second modified structure PC58BM-dicarbonyl (Fig. 5c) consists of forming double bonded oxygens on two adjacent carbons of the cage, as described by Taliani et al.,26 and removing two other cage C atoms from the vicinity of the adsorption site.
 | ||
| Fig. 5 Molecular structures of (a) pristine PC60BM and two possible photooxidation products, (b) PC58BM-anhydride, and (c) PC58BM-dicarbonyl. | ||
 | ||
| Fig. 6 Measured IR spectrum in the carbonyl region of PC60BM exposed to light in air for 47 h (black line, upper part), shown in comparison to the calculated IR spectrum (lower part) of pristine PC60BM (black line, shaded grey), PC58BM-anhydride (green line) and PC58BM-dicarbonyl (blue line). These spectra have been broadened using Gaussian functions of 10 cm−1 FWHM. The bar graphs represent the calculated IR peak positions and intensities. | ||
The calculated IR spectra of these two possible photodegradation products are shown in Fig. 6, alongside the calculated IR spectrum of pristine PC60BM and the measured spectrum of PC60BM exposed to light in air for 47 h. The peak marked “2” in Fig. 6 is common to all three molecules (pristine PC60BM, PC58BM-anhydride, and PC58BM-dicarbonyl) and is related to the CO stretch of the ester group in the side chain. The intensity of this CO stretch peak remains unchanged in the experimental spectra, even after long exposure times. Both photodegradation products present two additional peaks, one related to a symmetric stretch (peaks “1S”, respectively “3S”) and the other to an antisymmetric stretch mode (peaks “1A”, respectively “3A”) of the carbon–oxygen double bonds. The peaks corresponding to the anhydride contribute in the higher wavenumber region of the IR spectrum, while the peaks corresponding to PC58BM-dicarbonyl contribute in the lower wavenumber region of the spectrum. The combination of these two photodegradation products gives a reasonable agreement between the experimental and calculated data and will be used to explain the experimental observations in the following.
While the DFT calculations are performed for single molecules, the IR measurements were performed on films. We need to take into account that the film will consist of molecules in different stages of the degradation process, since not all molecules in the bulk and at the surface of the film undergo the same modifications at the same time. To model this situation in a straightforward way, we have calculated IR spectra for different mixtures of pristine PC60BM with photodegraded molecules in different amounts. For this purpose, we have performed weighted averages by linear combination of the calculated spectra of the pristine molecule and of the degradation products, as described in the Computational methods. The calculated IR spectra obtained this way are shown in Fig. 7, in comparison to spectra measured after different exposure times, and reproduce the experimentally observed features well. Including up to 10% PC58BM-anhydride and up to 10% PC58BM-dicarbonyl results in the rise of a clear shoulder to the pristine carbonyl peak in the high wavenumber region of the IR spectrum and to the emergence of a broad feature on the low wavenumber side of the pristine carbonyl peak. Since the IR spectrum of PC60BM:O2 is identical to the spectrum of pristine PC60BM, the proportion of molecules with molecular oxygen in their vicinity may be higher than that considered in our weighted average.
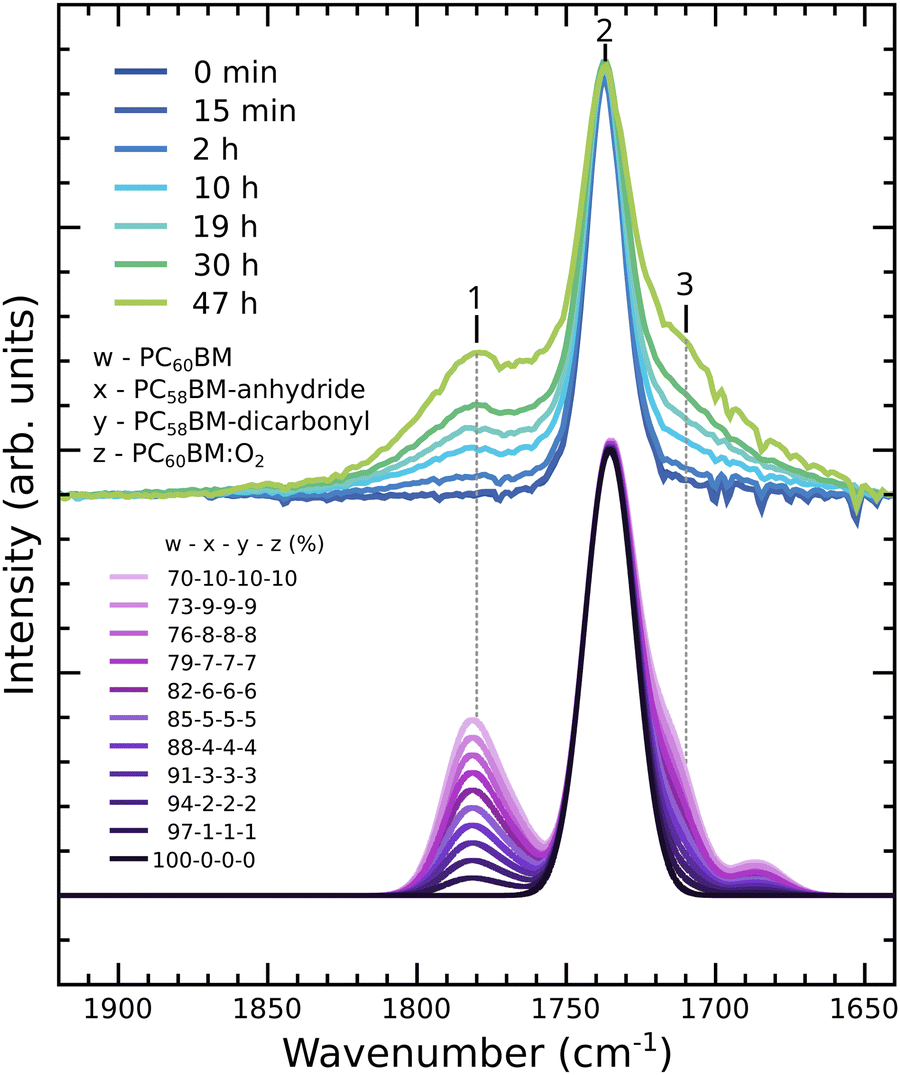 | ||
| Fig. 7 Experimental IR spectra in the carbonyl region of unexposed PC60BM and PC60BM exposed to light in air for different exposure times (upper part) compared to calculated spectra with different weighted averages between pristine PC60BM, PC58BM-anhydride, PC58BM-dicarbonyl, and PC60BM:O2 (lower part). | ||
Whereas peak 2 has a constant width, the side peaks appear broader in the experimental than in calculated spectra. The observed broadening in the experimental spectra could be explained by contributions from other oxidation products and products where the oxygen has different positions on the cage.
Having in mind that the IR measurements probe the entire film, while the X-ray spectroscopy measurements probe only the film surface, we have performed similar weighted averages between pristine PC60BM and the three selected photodegradation products for those spectra.
Fig. 8 shows the weighted averages obtained for the C1s and O1s XPS spectra, considering up to 100% photodegradation products. This would correspond to a film surface where all PC60BM molecules have undergone photodegradation, and have converted into one of the three products in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 stoichiometry. In the case of C1s XPS, the weighted average spectra reproduce the experimentally observed trends, i.e. the intensity reduction of the main peak, its broadening on the high binding energy side and the slight shift towards higher binding energies. However, the changes in the experimental spectrum are much more drastic compared to the calculated weighted averages, suggesting much stronger modifications of the PC60BM molecules at the surface of the film. These could include larger losses of C atoms from the cage and possibly the adsorption of oxygen atoms in higher numbers than considered by our simple molecular models. A similar conclusion may be drawn from the O1s XPS, where the changes in the measured spectra are much more drastic than in the case of the calculated ones. In the experimental O1s XPS spectra we observed a large increase in intensity such that the two side chain peaks become indistinct, where the low binding energy side is higher in intensity. The combination of PC58BM-dicarbonyl and PC58BM-anhydride results in a more intense peak at low binding energies, while the growth of intensity in the region between the two pristine peaks is explained by diffused O2 in the film. As discussed previously, calculated O1s XPS of PC60BM:O2 reproduces the increase in intensity in this intermediate binding energy region. This is in agreement with the finding that O2 diffusion into the film occurs before photooxidation.65,66 Since O2 is IR inactive and the presence of O2 in the vicinity of PC60BM does not affect the C1s XPS and C1s NEXAFS, diffused molecular oxygen cannot be observed using these spectroscopies and the spectra of PC60BM and PC60BM:O2 are identical.
:1:1 stoichiometry. In the case of C1s XPS, the weighted average spectra reproduce the experimentally observed trends, i.e. the intensity reduction of the main peak, its broadening on the high binding energy side and the slight shift towards higher binding energies. However, the changes in the experimental spectrum are much more drastic compared to the calculated weighted averages, suggesting much stronger modifications of the PC60BM molecules at the surface of the film. These could include larger losses of C atoms from the cage and possibly the adsorption of oxygen atoms in higher numbers than considered by our simple molecular models. A similar conclusion may be drawn from the O1s XPS, where the changes in the measured spectra are much more drastic than in the case of the calculated ones. In the experimental O1s XPS spectra we observed a large increase in intensity such that the two side chain peaks become indistinct, where the low binding energy side is higher in intensity. The combination of PC58BM-dicarbonyl and PC58BM-anhydride results in a more intense peak at low binding energies, while the growth of intensity in the region between the two pristine peaks is explained by diffused O2 in the film. As discussed previously, calculated O1s XPS of PC60BM:O2 reproduces the increase in intensity in this intermediate binding energy region. This is in agreement with the finding that O2 diffusion into the film occurs before photooxidation.65,66 Since O2 is IR inactive and the presence of O2 in the vicinity of PC60BM does not affect the C1s XPS and C1s NEXAFS, diffused molecular oxygen cannot be observed using these spectroscopies and the spectra of PC60BM and PC60BM:O2 are identical.
 | ||
| Fig. 8 Experimental (a) C1s XPS and (b) O1s XPS spectra of unexposed PC60BM and PC60BM exposed to light in air for different exposure times (upper part) compared to calculated spectra with different weighted averages between pristine PC60BM, PC58BM-anhydride, PC58BM-dicarbonyl, and PC60BM:O2 (lower part). | ||
Finally, the measured C1s and O1s NEXAFS measurements are compared to weighted averages of the calculated spectra of the oxidation products in Fig. 9 and 10, respectively. As was the case for C1s XPS, the calculated spectra containing up to 100% contributions from photodegradation products, reproduce the experimentally observed trends well. The π* peak is more reduced in intensity, the higher the amount of photodegradation products included. Peaks 2 and 3 also become less and less intense, with peak 2 quenching faster than peak 3. The region of shoulder S and the lower photon energy side of peak 4 increase slightly, while the region of peaks 5 and 6 remains rather unchanged. The modifications are, also in this case, more drastic in the experiment, and more drastic in the partial electron yield than in the total electron yield NEXAFS spectrum. This is expected, since PEY probes a thinner layer of the surface than TEY, where the very top layer is in direct contact with oxygen and would degrade faster, whereas oxygen needs to diffuse through the film to reach further down in the film.
 | ||
| Fig. 9 Experimental C1s NEXAFS in (a) partial electron yield mode, and (b) total electron yield mode of unexposed PC60BM and PC60BM exposed to light in air for different exposure times (upper part) compared to calculated spectra with different weighted averages between pristine PC60BM, PC58BM-anhydride, PC58BM-dicarbonyl, and PC60BM:O2 (lower part). | ||
 | ||
| Fig. 10 Experimental O1s NEXAFS of unexposed PC60BM and PC60BM exposed to light in air for different exposure times (upper part) compared to calculated spectra with different weighted averages between pristine PC60BM, PC58BM-anhydride, PC58BM-dicarbonyl, and PC60BM:O2 (lower part). | ||
The C1s NEXAFS spectra for the weighted averages show a reasonable agreement with the experiment. The reasons for the deviation between experimental and calculated spectra are the same as for the C1s XPS data, namely more oxygen atoms adsorbed to the same PC60BM molecule and stronger loss of carbon atoms from the fullerene cage.
The experimental O1s NEXAFS spectra shown in Fig. 10 are also well reproduced by the calculated weighted averages. All features increase in intensity, most notably the π* peak and its low photon energy shoulder. The region around peak 4 grows faster than the rest, becoming as large as the π* when all molecules are considered to have undergone one type of photodegradation or the other. In the experiment, this region increases more drastically compared to the π* peak than obtained by our simple model, as observed for the other X-ray spectroscopies as well. We should keep in mind that in the experiment several modifications may take place on the same molecule and diffused O2 will be found in many different configurations, for example at various distances from the cage, in the vicinity of the side-chain, or in the vicinity of already degraded molecules, as discussed previously.
To summarize, based on the photodegradation products calculated here, as well as IR reference data from the literature, the features observed in the experimental IR spectrum in the carbonyl region are likely explained by the formation of anhydride and dicarbonyl structures on the fullerene cage. These photodegradation products are also consistent with the C1s and O1s XPS and NEXAFS spectra, but these spectroscopies, which probe the surface layers of the films, reveal a much more drastic photodegradation. Going beyond our simple molecular models, more oxygen atoms could adsorb on one PC60BM, a larger amount of C atoms could be removed from the cage and the same PC60BM molecule could undergo several types of modifications at the same time. All measurements here, together with the photodegradation changes observed in the optical absorption spectrum of PC60BM,36 indicate damage to the fullerene cage. This damage leads to changes in the electronic structure, as shown for the unoccupied energy levels by our NEXAFS measurements. These changes, together with the loss of π delocalization in individual molecules, will reduce the charge transport properties of the film by trapping and disorder, resulting in a decrease of device performance.
Conclusions
We have shown that the surface of spin-coated PC60BM thin films exposed to simulated sunlight in air undergoes photo-induced oxidation that deteriorates the conjugated character of the fullerene molecules. XPS spectra show a strong increase of the amount of oxygen present in the top-most area of the film close to the surface and the associated attenuation of the carbon signal, as well as an indication for band bending development at the film surface. While chemical shifts and peak broadening in the XPS spectra indicate the formation of several oxidation products, the fine structure in the π* region of the C1s NEXAFS spectrum reveals the chemical changes to the fullerene cage. The strong decrease of the intensity of the resonances in the π* region of the C1s NEXAFS spectra, with increasing exposure time, demonstrates that sp2 hybridized carbons of the fullerene cage become sp3 hybridized, in particular in the first few monolayers of the film surface. The O1s NEXAFS spectra of photodegraded PC60BM exhibit a growing new pre-edge resonance in the π* region, which provides evidence for the formation of double bonded oxygen. Finally, FTIR spectra shed light on the type of bonds formed between oxygen and the fullerene cage. The spectral changes in the carbonyl region include the appearance of a new peak on the high wavenumber side, and the development of a shoulder on the low wave number side of the ester peak from the PC60BM tail. Through comparison of experimental IR, XPS and NEXAFS spectra with corresponding calculated spectra of a large number of possible oxidation products of PC60BM, we could conclude that several carbonyl-based derivatives are formed during the photooxidation process of PC60BM. The best match between experimental and calculated spectra is obtained for a weighted average of the unmodified PC60BM, the dicarbonyl structure PC58BM-dicarbonyl and the anhydride structure PC58BM-anhydride, in addition to molecular oxygen intercalated into the PC60BM film. To reproduce the experimental spectra, our molecular model required the removal of C atoms to reduce the ring strain, which shifts the IR modes of symmetric and asymmetric CO stretches to lower wavenumbers. This suggests that losing C atoms from the cage is indeed a part of the photodegradation process, as has been suggested for C60 in earlier work to occur in the form of volatile CO and CO2. We conclude that the results from complementary experimental X-ray and IR spectroscopy techniques and corresponding calculations form a strong indication that fullerene dicarbonyls and anhydrides, accompanied by physisorbed molecular oxygen, are among the photooxidation products that are formed when PC60BM films are exposed to light in ambient conditions. These insights yield an in-depth understanding of the degradation mechanisms through photooxidation that rapidly affect PC60BM and polymer:PC60BM blend films, when prepared in ambient conditions. They also suggest the detrimental consequences for the solar cell operation, when oxygen diffuses into the devices and photodegrades the electron-transporting component in the photoactive layer. Since PC60BM is commonly used in several organic electronic devices, the photooxidation discussed here is highly relevant for the functioning of these devices. As the photooxidation process affects the C60 cage, other fullerene derivatives employed in photovoltaics and organic electronics are likely to photodegrade in a similar manner.
Author contributions
E. M. and B. B. initiated the study. I. E. B. performed the DFT calculations and prepared the visualisation of the results. L. E., R. H., A. O. and E. M. planned the experiments and acquired NEXAFS and PES data. V. B. performed the IR measurements. All authors contributed to the analysis and validation of the produced data. A. O., B. B. and E. M. supervised the work. I. E. B., L. E. and E. M. wrote the original draft and all authors contributed to finalise the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge MAX IV Laboratory for time on Beamline D1011 under Proposals 20120122, 20130020, and 20140169. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. We thank Ana Sofia Anselmo, Krister Svensson, Ulrich Hörmann, Alexander Generalov, and Karsten Handrup for participation in the X-ray spectroscopy measurements and the D1011 beamline manager Alexei Preobrajenski fortechnical support. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputing Centre (NSC) in Linköping, Sweden. E. M. acknowledges financial support from the Swedish Energy Council (Grant 48598-1), the Swedish Research Council (Grant 2015-03778), the Knut and Alice Wallenberg Foundation (Grant KAW-2016.0059), and the Göran Gustafsson Foundation for Research in Natural Sciences and Medicine. B. B. and I. E. B. acknowledge financial support from Swedish Research Council (Grant 2014-03776), the Knut and Alice Wallenberg Foundation (Grant KAW-2013.0020) and the Carl Trygger Foundation. A. O. is grateful for support by Deutsche Forschungsgemeinschaft (project no. 239543752).References
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Z. Zhou, S. Xu, J. Song, Y. Jin, Q. Yue, Y. Qian, F. Liu, F. Zhang and X. Zhu, Nat. Energy, 2018, 3, 952–959 CrossRef CAS.
- M.-A. Pan, T.-K. Lau, Y. Tang, Y.-C. Wu, T. Liu, K. Li, M.-C. Chen, X. Lu, W. Ma and C. Zhan, J. Mater. Chem. A, 2019, 7, 20713–20722 RSC.
- S. Tsarev, T. S. Dubinina, S. Yu Luchkin, I. S. Zhidkov, E. Z. Kurmaev, K. J. Stevenson and P. A. Troshin, J. Phys. Chem. C, 2020, 124, 1872–1877 CrossRef CAS.
- J. Liu, L. Qiu and S. Shao, J. Mater. Chem. C, 2021, 9, 16143–16163 RSC.
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef PubMed.
- Stability and Degradation of Organic and Polymer Solar Cells, ed. F. C. Krebs, John Wiley & Sons, Chichester, UK, 2012 Search PubMed.
- E. Voroshazi, I. Cardinaletti, T. Conard and B. P. Rand, Adv. Energy Mater., 2014, 4, 1400848 CrossRef.
- T. Tromholt, E. A. Katz, B. Hirsch, A. Vossier and F. C. Krebs, Appl. Phys. Lett., 2010, 96, 073501 CrossRef.
- J. U. Lee, J. W. Jung, J. W. Jo and W. H. Jo, J. Mater. Chem., 2012, 22, 24265–24283 RSC.
- A. Tournebize, J.-L. Gardette, C. Taviot-Guého, D. Bégué, M. A. Arnaud, C. Dagron-Lartigau, H. Medlej, R. C. Hiorns, S. Beaupré, M. Leclerc and A. Rivaton, Polym. Degrad. Stabil., 2015, 112, 175–184 CrossRef CAS.
- A. Rivaton, A. Tournebize, J. Gaume, P. Bussière, J. Gardette and S. Therias, Polym. Int., 2014, 63, 1335–1345 CrossRef CAS.
- M. Manceau, A. Rivaton, J.-L. Gardette, S. Guillerez and N. Lemaître, Polym. Degrad. Stabil., 2009, 94, 898–907 CrossRef CAS.
- A. S. Anselmo, A. Dzwilewski, K. Svensson and E. Moons, Chem. Phys. Lett., 2016, 652, 220–224 CrossRef CAS.
- J. C. Hummelen, J. Knol and L. Sanchez, Proc. Soc. Photo-Opt. Instrum. Eng., 2001, 76–84 CrossRef CAS.
- S. Chambon, A. Rivaton, J.-L. Gardette and M. Firon, Sol. Energy Mater. Sol. Cells, 2007, 91, 394–398 CrossRef CAS.
- M. O. Reese, A. M. Nardes, B. L. Rupert, R. E. Larsen, D. C. Olson, M. T. Lloyd, S. E. Shaheen, D. S. Ginley, G. Rumbles and N. Kopidakis, Adv. Funct. Mater., 2010, 20, 3476–3483 CrossRef CAS.
- E. M. Speller, J. D. McGettrick, B. Rice, A. M. Telford, H. K. H. Lee, C.-H. Tan, C. S. D. Castro, M. L. Davies, T. M. Watson, J. Nelson, J. R. Durrant, Z. Li and W. C. Tsoi, ACS Appl. Mater. Interfaces, 2017, 9, 22739–22747 CrossRef CAS PubMed.
- H. K. H. Lee, A. M. Telford, J. A. Röhr, M. F. Wyatt, B. Rice, J. Wu, A. de, C. Maciel, S. M. Tuladhar, E. Speller, J. McGettrick, J. R. Searle, S. Pont, T. Watson, T. Kirchartz, J. R. Durrant, W. C. Tsoi, J. Nelson and Z. Li, Energy Environ. Sci., 2018, 11, 417–428 RSC.
- T. Heumueller, W. R. Mateker, A. Distler, U. F. Fritze, R. Cheacharoen, W. H. Nguyen, M. Biele, M. Salvador, M. von Delius, H.-J. Egelhaaf, M. D. McGehee and C. J. Brabec, Energy Environ. Sci., 2016, 9, 247–256 RSC.
- R. Hansson, C. Lindqvist, L. K. E. Ericsson, A. Opitz, E. Wang and E. Moons, Phys. Chem. Chem. Phys., 2016, 18, 11132–11138 RSC.
- V. Blazinic, L. K. E. Ericsson, I. Levine, R. Hansson, A. Opitz and E. Moons, Phys. Chem. Chem. Phys., 2019, 21, 22259–22271 RSC.
- R. Taylor, J. P. Parsons, A. G. Avent, S. P. Ranard, T. J. Dennis, J. P. Hare, H. W. Kroto and D. R. M. Walton, Nature, 1991, 351, 277 CrossRef CAS.
- L. Juha, V. Hamplová, J. Kodymová and O. Špalek, J. Chem. Soc., Chem. Commun., 1994, 2437–2438 RSC.
- C. Taliani, G. Ruani, R. Zamboni, R. Danieli, S. Rossini, V. N. Denisov, V. M. Burlakov, F. Negri, G. Orlandi and F. Zerbetto, J. Chem. Soc., Chem. Commun., 1993, 220–222 RSC.
- H. Werner, Th Schedel-Niedrig, M. Wohlers, D. Herein, B. Herzog, R. Schlögl, M. Keil, A. M. Bradshaw and J. Kirschner, J. Chem. Soc., Faraday Trans., 1994, 90, 403–409 RSC.
- M. Wohlers, H. Werner, D. Herein, T. Schedel-Niedrig, A. Bauer and R. Schlögl, Synth. Met., 1996, 77, 299–302 CrossRef CAS.
- M. Wohlers, A. Bauer, T. Belz, T. Rühle, T. Schedel-Niedrig, R. Schlögl and H. Werner, ACS Div. Fuel Chem. Prepr., 1996, 41, 108–112 CAS.
- S. Yamane, J. Mizukado, Y. Suzuki, M. Sakurai, L. Chen and H. Suda, Chem. Lett., 2015, 44, 339–341 CrossRef CAS.
- F. Piersimoni, G. Degutis, S. Bertho, K. Vandewal, D. Spoltore, T. Vangerven, J. Drijkoningen, M. K. V. Bael, A. Hardy, J. D’Haen, W. Maes, D. Vanderzande, M. Nesladek and J. Manca, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1209–1214 CrossRef CAS.
- A. Dzwilewski, T. Wågberg and L. Edman, J. Am. Chem. Soc., 2009, 131, 4006–4011 CrossRef CAS.
- J. Wang, J. Enevold and L. Edman, Adv. Funct. Mater., 2013, 23, 3220–3225 CrossRef CAS.
- A. Distler, T. Sauermann, H. Egelhaaf, S. Rodman, D. Waller, K. Cheon, M. Lee, N. Drolet and D. M. Guldi, Adv. Energy Mater., 2013, 4, 1300693 CrossRef.
- U. Dettinger, H.-J. Egelhaaf, C. J. Brabec, F. Latteyer, H. Peisert and T. Chasse, Chem. Mater., 2015, 27, 2299–2308 CrossRef CAS.
- V. Blazinic, L. K. E. Ericsson, S. A. Muntean and E. Moons, Synth. Met., 2018, 241, 26–30 CrossRef CAS.
- G. Kissinger and W. Kissinger, Phys. Status Solidi, 1991, 123, 185–192 CrossRef.
- W. Kern, J. Electrochem. Soc., 1990, 137, 1887–1892 CrossRef CAS.
- W. Kern and D. Puotinen, RCA Rev., 1970, 31, 187–205 CAS.
- B. Watts, L. Thomsen and P. C. Dastoor, J. Electron Spectrosc. Relat. Phenom., 2006, 151, 105–120 CrossRef CAS.
- J. Stöhr, NEXAFS Spectroscopy, Springer, 1992 Search PubMed.
- B. Watts and H. Ade, J. Electron Spectrosc. Relat. Phenom., 2008, 162, 49–55 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. M. Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- I. E. Brumboiu, L. Ericsson, R. Hansson, E. Moons, O. Eriksson and B. Brena, J. Chem. Phys., 2015, 142, 054306 CrossRef PubMed.
- K. Hermann, L. G. M. Pettersson, M. E. Casida, C. Dual, A. Goursot, A. Koester, E. Proynov, A. St-Amant and D. R. Salahub, StoBe-deMon, version 3.0, 2008 Search PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- L. Triguero, L. G. M. Pettersson and H. Ågren, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 8097–8110 CrossRef CAS.
- U. Fleischer, W. Kutzelnigg, H.-H. Limbach, G. J. Martin, M. L. Martin, M. Schindler, W. Kutzelnigg, U. Fleischer and M. Schindler, NMR Basic Princ. Prog., 1990, 23, 165–262 Search PubMed.
- I. E. Brumboiu, A. S. Anselmo, B. Brena, A. Dzwilewski, K. Svensson and E. Moons, Chem. Phys. Lett., 2013, 568, 130–134 CrossRef.
- M. Nyberg, Y. Luo, L. Triguero, L. G. M. Pettersson and H. Ågren, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 7956–7960 CrossRef CAS.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem., 2007, 111, 11683–11700 CrossRef CAS PubMed.
- M. P. Felicissimo, D. Jarzab, M. Gorgoi, M. Forster, U. Scherf, M. C. Scharber, S. Svensson, P. Rudolf and M. A. Loi, J. Mater. Chem., 2009, 19, 4899–4901 RSC.
- Z.-L. Guan, J. B. Kim, H. Wang, C. Jaye, D. A. Fischer, Y.-L. Loo and A. Kahn, Org. Electron., 2010, 11, 1779–1785 CrossRef CAS.
- D. M. Cox, S. Behal, M. Disko, S. M. Gorun, M. Greaney, C. S. Hsu, E. B. Kollin, J. Millar and J. Robbins, J. Am. Chem. Soc., 1991, 113, 2940–2944 CrossRef CAS.
- B. Chase, N. Herron and E. Holler, J. Phys. Chem., 1992, 96, 4262–4266 CrossRef CAS.
- J. B. Lambert, H. F. Shurvell, D. A. Lightner and R. G. Cooks, Introduction to Organic Spectroscopy, Macmillan Publishing Company, New York, 1987 Search PubMed.
- A. T. Flores and S. M. Rosales, J. Chem. Chem. Eng., 2011, 5, 1034–1040 Search PubMed.
- C.-R. Zhang, L.-H. Han, J.-W. Zhe, N.-Z. Jin, Y.-L. Shen, L.-H. Yuan, Y.-Z. Wu and Z.-J. Liu, J. Nanomater., 2013, 1–8 Search PubMed.
- S. Yamane, J. Mizukado, T. Uchimaru, Y. Suzuki, L. Chen and H. Suda, Sol. Energy Mater. Sol. Cells, 2015, 143, 135–140 CrossRef CAS.
- M. Wohlers, H. Werner, T. Belz, T. Rühle and R. Schlögl, Microchim. Acta, 1997, 125, 401–406 CrossRef CAS.
- B. Galabov, D. Simov and L. Krustev, J. Mol. Struct., 1976, 34, 235–244 CrossRef CAS.
- C. C. Eloi, D. J. Robertson, A. M. Rao, P. Zhou, K.-A. Wang and P. C. Eklund, J. Mater. Res., 1993, 8, 3085–3089 CrossRef CAS.
- P. C. Eklund, A. M. Rao, P. Zhou, Y. Wang, K.-A. Wang, G. T. Hager and J. M. Holden, Mater. Sci. Eng., B, 1993, 19, 154–161 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03514f |
| ‡ These authors have contributed equally. |
| § Present address: Faculty of Physics, Astronomy and Informatics, Nicolaus Copernicus University, 87-100 Toruń, Poland. |
| This journal is © the Owner Societies 2022 |