
Reaction of lithium hexamethyldisilazide (LiHMDS) with water at ultracold conditions†
Svenja
Jäger‡
a,
Philipp
Meyer‡
a,
Kai-Stephan
Feichtner
b,
Stefan
Henkel
 a,
Gerhard W.
Schwaab
a,
Viktoria H.
Gessner
*b and
Martina
Havenith
*a
a,
Gerhard W.
Schwaab
a,
Viktoria H.
Gessner
*b and
Martina
Havenith
*a
aLehrstuhl für Physikalische Chemie II, Ruhr-Universität Bochum, 44780, Bochum, Germany. E-mail: martina.havenith@rub.de
bLehrstuhl für Anorganische Chemie II, Ruhr-Universität Bochum, 44780, Bochum, Germany
First published on 22nd September 2022
Abstract
Alkali metal amides are highly reactive reagents that are broadly applied as strong bases in organic synthesis. Here, we use a combined helium nanodroplet IR spectroscopic and theoretical (DFT calculation) study to show that the reaction of the model compound lithium hexamethyldisilazide (LiHMDS) with water is close to barrierless even at ultra-cold conditions. Upon complex formation of dimeric (LiHMDS)2 with water in helium nanodroplets as ultra-cold nano-reactors (0.37 K) we observed the reaction product (LiOH)2(HMDS)2. This can be rationalized as aggregation induced reation upon stepwise addition of water. With increasing water partial pressure, only the product (LiOH)2(HMDS)2 is observed experimentally. This implies that the large interaction energy (69 kJ mol−1) of (LiHMDS)2 with water is sufficient to overcome the follow-up reaction barriers, in spite of the rapid cooling rates in He nanodroplets.
Introduction
Organometallic compounds of the s-block metals, such as organolithium or Grignard reagents as well as alkali metal amides are among the most useful organometallic compounds which are routinely used in various fields of modern chemistry including the synthesis of complex molecules and even in industrial applications on a large scale.1–5 Alkali metal amides are frequently applied as strong non-nucleophilic bases in organic synthesis to realize difficult deprotonation reactions. Prominent examples, which are also commercially available, are lithium diispropylamide (LDA) or lithium hexamethyldisilazide (LiHMDS). However, the metallation power of these compounds comes at a cost: owing to the ionic Li–N bond,6 these metal amides are highly reactive and sensitive toward moisture, thus making them difficult to handle under ambient conditions. Therefore, they are usually applied under strictly anhydrous reaction conditions to exclude undesired protonation or decomposition reactions.7 This means that characterization of their specific properties, intermediates, and reaction mechanisms is challenging.With the growing demand for sustainable reaction protocols in recent years, however, we have seen the development of reaction protocols using alkali metal compounds in “unconventional” solvents, including water.8–10 Moreover, first crystal structure analyses of organometallic compounds have been reported, in which water acts as a ligand, coordinating to the alkali metal ion rather than instantaneously inducing protonation.11–14 This suggests that coordination may precede the protonation process, with the relative rates of these processes being influenced by the nature of the involved aggregates and the reaction conditions. This led us to assume that there is a non-negligible activation barrier even for the protonation of simple amides such as LiHMDS, which might allow us to spectroscopically detect the corresponding water complexes under ultracold reaction conditions.
Herein we present infrared (IR) spectroscopic studies to investigate the reaction of LiHMDS with water (H2O) and deuterium oxide (D2O) (Scheme 1). The experiment was carried out in our Helium Nanodroplet Isolation Spectroscopy (HeNDI) setup. HeNDI allows the step-by-step addition of pre-cooled reaction partners under controlled conditions at a temperature of 0.37 K. The experimental studies are complemented by density functional theory (DFT) quantum chemical calculations on the B3LYP-D3/def2-TZVP level of theory.
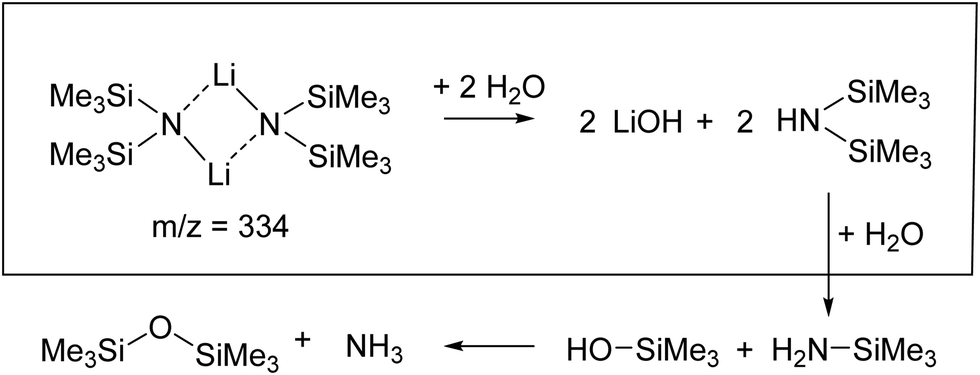 | ||
| Scheme 1 Reaction of dimeric lithium hexamethyldisilazide with water to give several hydrolyzed products: lithium hydroxide and bis(trimethylsilyl)amine (HMDS), trimethylsilylamine and trimethylsilanol and hexamethyldisiloxane and ammonia. In this work, the first step in this chain of reactions was investigated. | ||
Experimental and computational details
Helium nanodroplet isolation spectroscopy
Here, only a short summary is given, further details regarding the experimental setup can be found in ref. 15. Using a 5 μm nozzle pre-cooled to a temperature between 14 and 16 K, helium (Air Liquide, 99.9999%) was supersonically expanded with a stagnation pressure of 45 bar, yielding helium nanodroplets with a log-normal size distribution with an average size of approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms per droplet. The speed of the droplets along the optical axis is approximately 400 m s−1. They enter two individual, 4 cm long, pick-up regions, where LiHMDS and H2O or D2O (Sigma-Aldrich, 99.9%) are present in gaseous form at partial pressures between 0.5 × 10−6 and 1 × 10−5 mbar. After doping the droplets, the molecules are cooled down to 0.37 K within approximately a few tens of ns.16 This is much faster than the time of several 10 to 100 microseconds in between doping events at the given pressures and speed of the droplet beam. Once cooled down, solutes will form aggregates or reaction products if more than one molecule is present in the droplet. These will then interact with an IR laser beam, which overlaps the helium droplet beam in an antiparallel configuration. If the laser frequency is in resonance with an absorption band of the complex, the energy of the excitation is transferred to the surrounding helium, which results in an evaporation of helium from the droplet, decreasing the droplet size. In the detection chamber, the droplet is ionized by electron impact, and the resulting ions are detected by a Pfeiffer QMG 442 quadrupole mass spectrometer. Any decrease in droplet size results in a decreased ionization cross-section which can be detected using phase-sensitive detection via a lock-in amplifier and the frequency of the chopper inserted into the beam path of the laser. LiHMDS was purchased from Sigma-Aldrich with a purity of 99.9%, and sublimated at 90–100 °C and 1 × 10−2 mbar for further purification. It was transferred to a sample container under exclusion of oxygen or moisture using a glovebox. The sample container was then directly connected to the pick-up region of the helium nanodroplet setup, preventing contact with air. For the frequency range of 2590–3100 cm−1, a Lockheed-Martin Aculight Argos 2400 OPO was used, while the range of 900–1300 cm−1 is covered by a Daylight Solutions MIRcat Quantum Cascade Laser.
000 atoms per droplet. The speed of the droplets along the optical axis is approximately 400 m s−1. They enter two individual, 4 cm long, pick-up regions, where LiHMDS and H2O or D2O (Sigma-Aldrich, 99.9%) are present in gaseous form at partial pressures between 0.5 × 10−6 and 1 × 10−5 mbar. After doping the droplets, the molecules are cooled down to 0.37 K within approximately a few tens of ns.16 This is much faster than the time of several 10 to 100 microseconds in between doping events at the given pressures and speed of the droplet beam. Once cooled down, solutes will form aggregates or reaction products if more than one molecule is present in the droplet. These will then interact with an IR laser beam, which overlaps the helium droplet beam in an antiparallel configuration. If the laser frequency is in resonance with an absorption band of the complex, the energy of the excitation is transferred to the surrounding helium, which results in an evaporation of helium from the droplet, decreasing the droplet size. In the detection chamber, the droplet is ionized by electron impact, and the resulting ions are detected by a Pfeiffer QMG 442 quadrupole mass spectrometer. Any decrease in droplet size results in a decreased ionization cross-section which can be detected using phase-sensitive detection via a lock-in amplifier and the frequency of the chopper inserted into the beam path of the laser. LiHMDS was purchased from Sigma-Aldrich with a purity of 99.9%, and sublimated at 90–100 °C and 1 × 10−2 mbar for further purification. It was transferred to a sample container under exclusion of oxygen or moisture using a glovebox. The sample container was then directly connected to the pick-up region of the helium nanodroplet setup, preventing contact with air. For the frequency range of 2590–3100 cm−1, a Lockheed-Martin Aculight Argos 2400 OPO was used, while the range of 900–1300 cm−1 is covered by a Daylight Solutions MIRcat Quantum Cascade Laser.
DFT calculations
Calculations were performed by using Becke's three-parameter hybrid functional17 and the correlation functional of Lee, Yang and Parr18 (B3LYP) with the D3 correction by Grimme19 as implemented in Gaussian 09.20 The def2-TZVP was used for geometry optimizations and vibrational frequency calculations.21 Minima and transition states were identified by their number of imaginary frequencies. Structures were visualised with CYLview.22Results and discussion
In this study we want to characterize the hydrolysis reaction mechanism of LiHMDS, a highly reactive organometallic base.7,23,24 As a first step, we recorded mass spectra of LiHMDS-doped He nanodroplets. Previous investigations by Fjdeödnerg and Lappert and coworkers25,26 show that LiHMDS is evaporating as a dimer.27–30Based on the mass spectra (see S1, ESI†) we can confirm that (LiHMDS)2 is present, by observing signals at m/z = 174 (Li(LiHMDS)+) and m/z = 160 (N(SiMe3)2+), which are embedded into the droplets. As a second step, we recorded IR spectra of (LiHMDS)2.
Upon exposure to even small amounts of moisture in the sample container and transfer lines, LiHMDS easily decomposes into LiOH and HMDS. While LiOH precipitates as white solid, the volatile HMDS is also embedded in the helium droplets. Therefore, special care was taken to avoid leaks. As a control experiment, we independently measured the depletion IR spectrum of the decomposition product HMDS in the frequency region from 900 to 1300 cm−1 (Fig. 1). For HMDS we observe a strong signal at 935 cm−1, which is assigned to the asymmetric νSi–N mode (Fig. 1b and Table 1). Furthermore, we recorded multiple peaks between 1260 and 1300 cm−1, which we assign to the δC–H bending modes by comparison to the predictions.
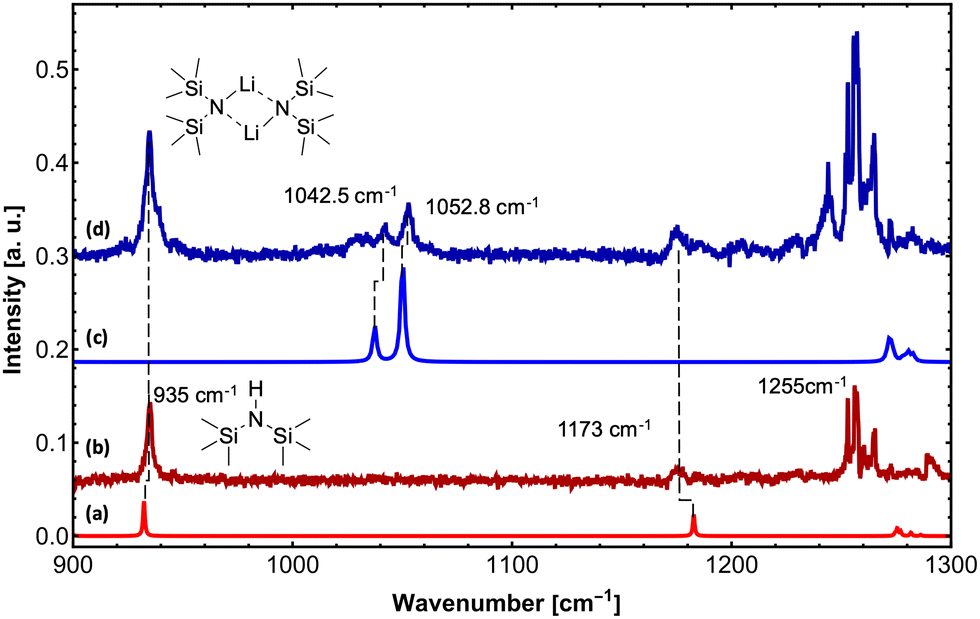 | ||
| Fig. 1 High-resolution IR spectra of (LiHMDS)2 and HMDS in the frequency region of 900–1300 cm−1 at m/z ≥ 129. (a) Calculated IR spectrum of HMDS (B3LYP-D3/def2-TZVP, scaled by 0.992). (b) IR spectrum of HMDS. (c) Calculated IR spectrum of (LiHMDS)2 (B3LYP-D3/def2-TZVP, scaled by 0.992). (d) IR spectrum of (LiHMDS)2. | ||
| Compound | Experimental | Calculation | ||||
|---|---|---|---|---|---|---|
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) [cm−1] [cm−1] |
Rel. Int. [%] | # |
[cm−1] |
Rel. Int. [%] | Assignment | |
| HMDS | 935.0 | 100 | 40 | 932.3 | 100 | Sym. νSi–N |
| 1173.0 | 12 | 41 | 1182.8 | 49 | Asym. νSi–N | |
| HMDS-d1 | 1036.0 | 100 | 41 | 1032.1 | 100 | Asym. νSi–N |
| (LiHMDS)2 | 1042.5 | 50 | 89 | 1037.5 | 42 | Sym. νSi–N |
| 1052.8 | 100 | 90 | 1050.2 | 100 | Asym. νSi–N | |
For (LiHMDS)2, we can clearly assign two bands at 1042.5 and 1052.8 cm−1 to the symmetric and asymmetric νSi–N stretching modes (Fig. 1d and Table 1), which are blue-shifted compared to the computational predictions. An additional broad feature starting at 1025 cm−1 could not be assigned. When comparing the IR spectra of HMDS and (LiHMDS)2, we can conclude that HMDS, a decomposition product of (LiHMDS)2, is present in both spectra, precluding further assignment of the IR bands of (LiHMDS)2.
In a second step, H2O and D2O were added to the (LiHMDS)2 using a second pick-up chamber, forming (LiHMDS)2(H2O)n and (LiHMDS)2(D2O)n aggregates inside the droplets (Fig. 2). Here, a partial pressure of 2.5 and 3.0 × 10−6 mbar was chosen for H2O and D2O, respectively. At these pressures, approximately two H2O or D2O molecules are being picked up by the helium droplets.
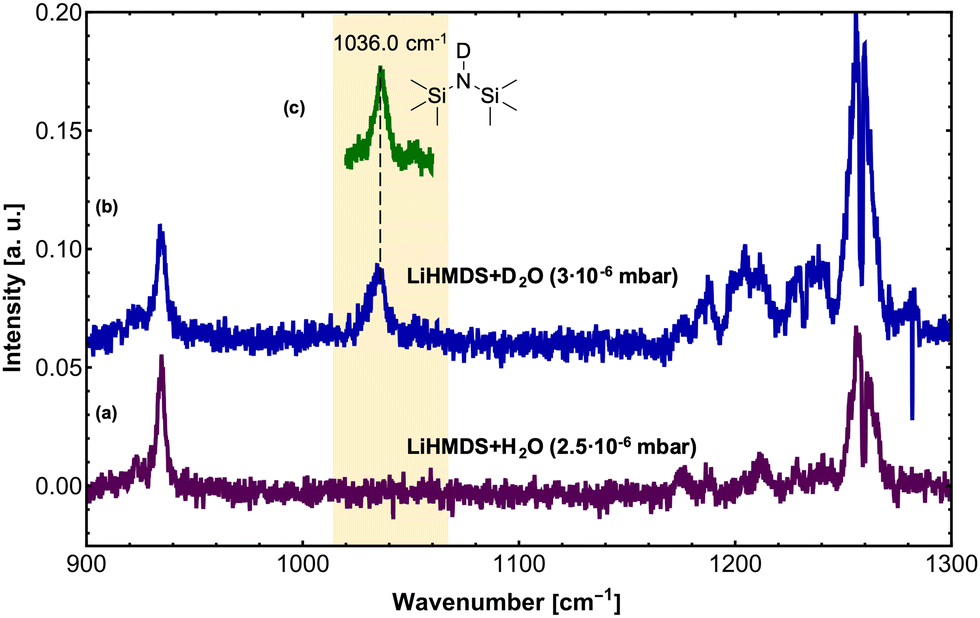 | ||
| Fig. 2 High-resolution IR spectra of (LiHMDS)2 with H2O and D2O in the frequency region of 900–1300 cm−1 at m/z ≥ 129. (a) IR spectrum of (LiHMDS)2 and H2O (partial pressure of H2O = 2.5 × 10−6 mbar) (b) IR spectrum of (LiHMDS)2 and D2O (partial pressure of D2O = 3.0 × 10−6 mbar). (c) IR spectrum of HMDS-d1 showing the νSi–N asymmetric stretching mode at 1036 cm−1. | ||
On addition of H2O, the key features of (LiHMDS)2 at 1042.5 and 1052.8 cm−1 as well as the broad feature at 1025 cm−1 are no longer observed and the spectrum in Fig. 2a closely resembles the IR spectrum of HMDS (Fig. 1b). When the N–H hydrogen in HMDS is exchanged for deuterium, a red-shift of 152 cm−1 is predicted for the asymmetric νSi–N stretching mode. The experimental data shows a shift of 137 cm−1 from 1173 cm−1 for HMDS to 1036 cm−1 for HMDS-d1. Since this new feature lies in the frequency range of the broad feature of (LiHMDS)2, we also investigated separately, HMDS-d1 (Fig. 2c and Fig. S2, ESI†), to verify these findings. Here we found clear agreement between the observed reaction product and the neat HMDS-d1 sample. Based on this result we attribute this to a reaction product, indicating a spontaneous reaction between (LiHMDS)2 and two D2O.
The isotopic substitution will also affect the νN–H stretching mode: this mode is predicted to be red-shifted by 952 cm−1, from 3538 cm−1 for HMDS to 2586 cm−1 for HMDS-d1. However, the former lies outside our experimentally accessible frequency range. Based on our result, we propose that even at temperatures of 0.37 K the reaction products (HMDS)2(LiOH)2 and (HMDS-d1)2(LiOD)2 are formed. This is further supported by the fact that with H2O no signals are observed in the 1000–1100 cm−1 region, where we would expect a band for any (LiHMDS)2(H2O)1 compound.
To check for any (LiHMDS)2(D2O)1 products, we decreased the D2O pressure inside the pickup chamber to 0.5 × 10−6 mbar, where less D2O molecules are being picked up by each droplet (Fig. 3e). At this pressure we expect to find, according to the Poisson statistics, a preferred pick-up for zero, one and up to two D2O molecules.16 Experimentally, we observe a signal of (LiHMDS)2 at 1042.5 and 1052.8 cm−1 (no pick-up of D2O) as well as the band at 1036.0 cm−1 assigned to (HMDS-d1)2(LiOD)2 (pick-up of two D2O). Based on the calculation we expect the νSi–N mode of the (LiHMDS)2(D2O)1 product in the very same frequency range (see Fig. 3b). Due to line broadening in the spectra, we are unable to deconvolute the IR signals into their partial contributions, and we assume that multiple signals are overlapping. Based on our predictions, we speculate that the νSi–N mode of the (LiHMDS)2(D2O)1 intermediate structure also contributes to the signal around 1036.0 cm−1 in Fig. 3e.
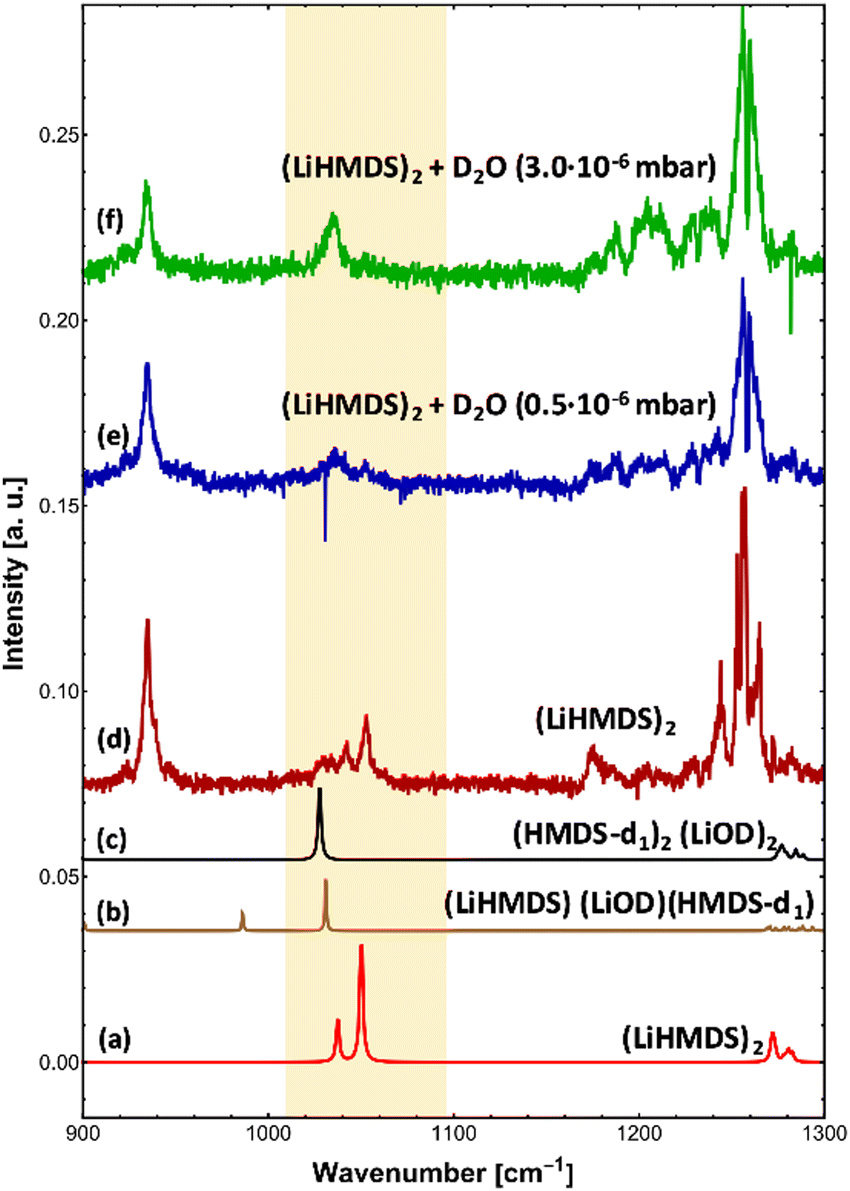 | ||
| Fig. 3 Infrared spectra of LiHMDS-D2O complexes. (a) Calculated IR spectrum of (LiHMDS)2 (B3LYP-D3/def2-TZVP, scaled by 0.992). (b) Calculated IR spectrum of (LiHMDS)(LiOH)(HMDS-d) (B3LYP-D3/def2-TZVP, scaled by 0.992). (c) Calculated IR spectrum of (HMDS-d)2(LiOD)2 (B3LYP-D3/def2-TZVP, scaled by 0.992). (d) IR spectrum of (LiHMDS)2. (e) IR spectrum of (LiHMDS)2 + D2O with a partial pressure of to 0.5 × 10−6 mbar. (f) IR spectrum of (LiHMDS)2 + D2O with a partial pressure of to 3.0 × 10−6 mbar. | ||
In order to further support our findings, we recorded the IR spectrum of (LiHMDS)2(D2O)n mixtures at 2700–3100 cm−1 to cover the νOD and symmetric and asymmetric δCH modes of the reaction products (Fig. 4). Overall, three sets of signals were found that are in good agreement with predictions for (HMDS-d)2(LiOD)2 (Table 2). These results confirm that LiHMDS undergoes a rapid reaction with water even at ultra-cold temperatures of 0.37 K.
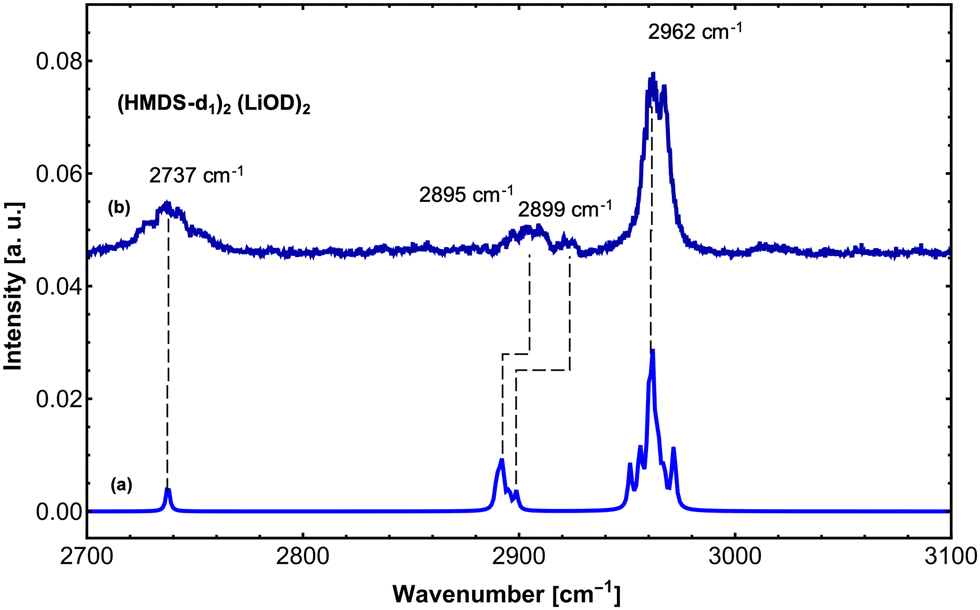 | ||
| Fig. 4 IR spectrum revealing the reaction of (LiHMDS)2 with two D2O molecules. (a) Calculated IR spectrum of (LiOD)2(HMDS-d1)2 (B3LYP-D3/def2-TZVP, scaled by 0.957). (b) IR spectrum of (LiHMDS)2 and D2O (partial pressure of D2O = 3.0 × 10−6 mbar). | ||
| Compound | Experimental | Calculation | ||||
|---|---|---|---|---|---|---|
|
[cm−1] |
Rel. int. [%] | # |
[cm−1] |
Rel. int. [%] | Assignment | |
| a Separate measurement using another laser. | ||||||
| (HMDS-d1)2(LiOD)27 | 1036.8 | 104 | 1027.7 | ν Si–N | ||
| 2737.0 | 32 | 144 | 2737.5 | 24 | ν O–D | |
| 2899.0 (broad) | 24 | 146 | 2889.5 | 16 | Sym. δCH | |
| 148 | 2890.5 | 16 | ||||
| 149 | 2891.8 | 1 | ||||
| 150 | 2891.8 | 23 | ||||
| 151 | 2892.6 | 22 | ||||
| 153 | 2895.4 | 14 | ||||
| 155 | 2898.8 | 16 | ||||
| 2962.1 (broad) | 100 | 157 | 2951.4 | 38 | Asym. δCH | |
| 159 | 2955.6 | 25 | ||||
| 162 | 2956.4 | 32 | ||||
| 164 | 2959.3 | 6 | ||||
| 166 | 2960.1 | 76 | ||||
| 168 | 2961.7 | 100 | ||||
| 169 | 2962.4 | 25 | ||||
| 171 | 2964.2 | 47 | ||||
| 173 | 2965.3 | 10 | ||||
| 176 | 2967.1 | 30 | ||||
| 178 | 2971.0 | 14 | ||||
| 179 | 2971.8 | 44 | ||||
| 180 | 2971.8 | 1 | ||||
To supplement the experimental results, DFT calculations were performed to gain a more detailed insight into the reaction mechanism. The interaction energy of (LiHMDS)2 with H2O is calculated to be −69.2 kJ mol−1 in the gas phase at the B3LYP-D3/def2-TZVP level of theory. The resulting complex 1 shows that the water acts as a Lewis base, forming a bond to the lithium atom (O⋯Li, 1.9 Å, Fig. 5).31 For the next step, the insertion of H2O in the N⋯Li bond, we found a small entrance barrier of 6.9 kJ mol−1. In our previous work, we have shown that albeit the temperature is 0.37 K, prohibiting any thermal activation, for the total free energy balance of the reaction, the interaction energy, i.e., the energy which is released via complex formation has to be taken into account. If this exceeds the barrier, we observe reactions even under ultracold conditions in helium nanodroplets, which are denoted as aggregation induced reactions, a behaviour previously observed for the dissociation of HCl upon the step-by-step addition of water.32
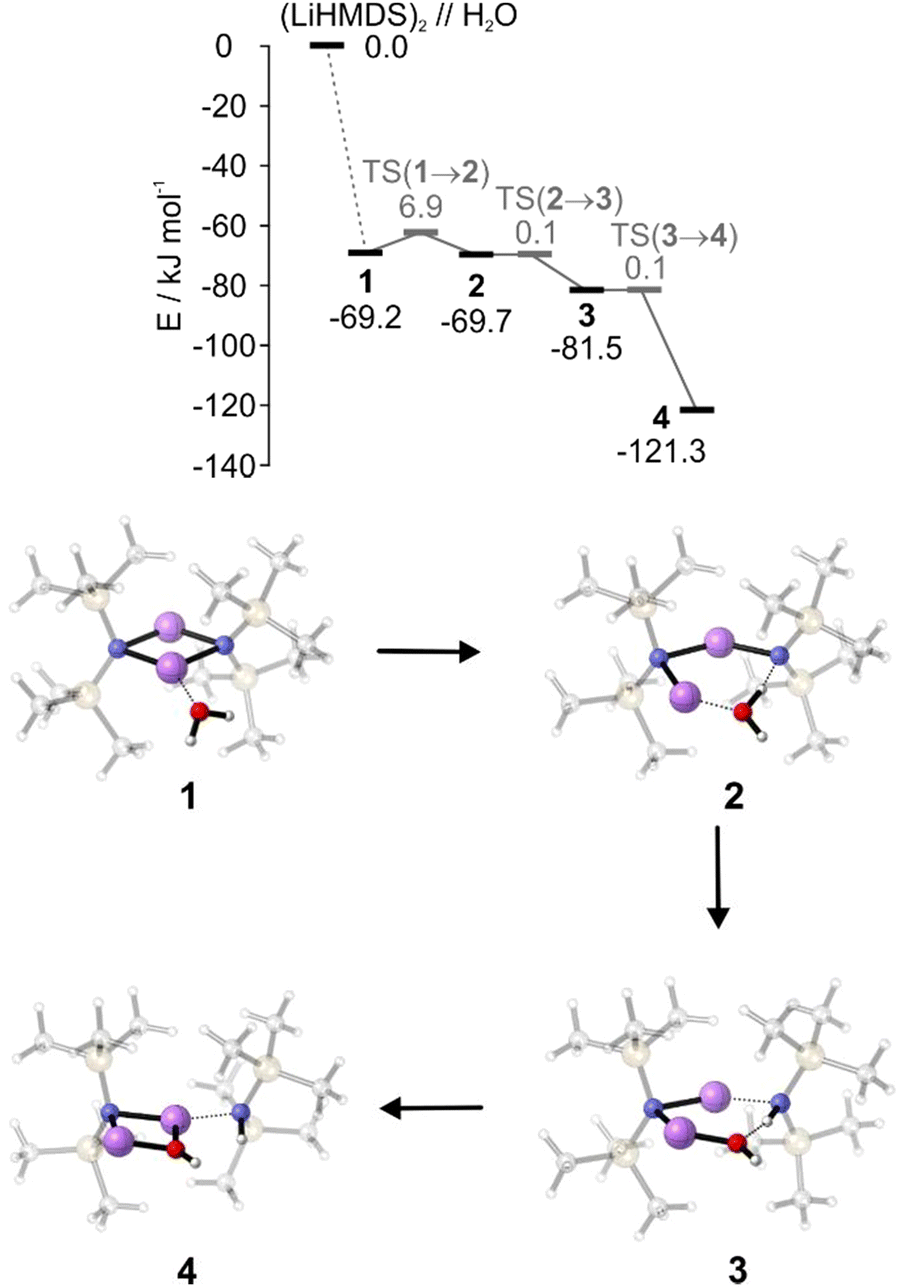 | ||
| Fig. 5 Potential energy surface (PES) of the reaction of (LiHMDS)2 with H2O and minima 1–4. The PES energies of the minima are given relative to non-interacting (LiHMDS)2 and H2O. Energies of transition states are given relative to the starting material. All calculations are carried out at the B3LYP-D3/def2-TZVP level of theory (without zero-point energy correction). | ||
In case of (LiHMDS)2 with H2O the barrier can indeed be overcome by the interaction energy (−69.2 kJ mol−1), which explains our experimental observation. Surprisingly, we predict a second minimum 2, with the OH group of water being shared between the Li and the N atom, which is even lower by 0.5 kJ mol−1 compared to 1. However, with almost no barrier (<0.1 kJ mol−1), the reaction proceeds by splitting the OH group to form 3, which in turn forms LiOH, complexed by one LiHMDS and one HMDS, 4 ((LiHMDS)(LiOH)(HMDS)).
The second addition of water, which is necessary to form product (HMDS)2(LiOH)2, is harder to capture. Here, two encounter complexes were investigated, starting from complex 4, one with the water attacking from the front (5, E = −68.6 kJ mol−1 relative to 4) and one with the water attacking from behind (6, E = −83.5 kJ mol−1 relative to 4) (Fig. 6). In both cases, the water forms a hydrogen bond to the LiHMDS nitrogen atom, but in 6 the O–H bond is weakened more, indicating stronger hydrogen bonding interaction (5, d(O⋯H) = 0.98 Å, d(H⋯N) = 2.09 Å, a(O⋯H⋯N) = 133.7°; 6, d(O⋯H) = 1.05 Å, d(H···N) = 1.59 Å, a(O···H···N) = 172.9°). Finally, the final product, (HMDS)2(LiOH)27, is calculated, releasing −241.6 kJ mol−1 compared to non-interacting (LiHMDS)2//2 H2O (Fig. 6). The spectrum of 7 is shown in Fig. 3f and 4. Further information about the calculated structures can be found in the ESI.†
 | ||
| Fig. 6 Calculated structures of (LiHMDS)2(H2O)2 structures 5–7 at the B3LYP-D3/def2-TZVP level of theory. | ||
Conclusions
We used helium droplets as nano-reactors to investigate the step-by-step aggregation of LiHMDS with water. Our mass spectroscopic results show that the compound evaporates as dimer, (LiHMDS)2. In spite of the high cooling rate of helium droplets, the large reaction energy set free in the first reaction step is sufficient to overcome the small barriers of the follow-up transition states (7 kJ mol−1). We found that even at 0.37 K (LiHMDS)2 is efficiently converted by water molecules to (HMDS)2(LiOH)2. We searched for indications of the formation of (LiHMDS)2(D2O)1. However, no clear signature of this intermediate structure was found when the amount of water was restricted. The fast reaction under ultra-cold conditions is supported by isotopic substitution measurements of (LiHMDS)2 with D2O. Spectroscopically, we observe the disappearance of the bands typical for (LiHMDS)2 and the appearance of bands typical for HMDS-d1 and LiOD as reaction products in the reaction (LiHMDS)2 + (D2O)2 → (HMDS-d1)2(LiOD)2.Author contributions
M. H. and V. G. designed the study, S. J. and P. M. measured all spectra, K.-S. F. provided all samples, S. H. carried out the theroretical predictions, S. J., P. M. and G. S. analysed the data, S. J., P. M., S. H., G. S., M. H. and V. G. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV. We thank Roberto Zambon for helping with the calculations.References
- M. P. Coles, Coord. Chem. Rev., 2015, 2, 297–298 Search PubMed.
- R. W. Hoffmann, Chem. Soc. Rev., 2003, 32, 225 RSC.
- M. F. Lappert, Metal amide chemistry, Wiley, Chichester, West Sussex (England), 2009 Search PubMed.
- V. C. R. Luisi, Lithium Compounds in Organic Synthesis – From Fundamentals to Applications, 2014 Search PubMed.
- R. E. Mulvey and S. D. Stuart, Angew. Chem., Int. Ed., 2013, 11470 CrossRef CAS PubMed.
- R. E. Mulvey, Chem. Soc. Rev., 1991, 20, 167–209 RSC.
- U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194 CrossRef CAS PubMed.
- C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 5969 CrossRef CAS PubMed.
- C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2016, 16145 CrossRef CAS PubMed.
- G. Dilauro, M. Dell’Aera, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., 2017, 129, 10334 CrossRef.
- P. Bai, S. Sun, Z. Li, H. Qiao, X. Su, F. Yang, Y. Wu and Y. Wu, J. Org. Chem., 2017, 82, 12119 CrossRef CAS PubMed.
- C. Lambert, P. v R. Schleyer, U. Pieper and D. Stalke, Angew. Chem., Int. Ed. Engl., 1992, 77 CrossRef.
- D. Barr, P. R. Raithby, P. von Raguk Schleyer, R. Snaith and D. S. Wright, J. Chem. Soc., Chem. Commun., 1990, 643 RSC.
- I. Koehne, S. Bachmann, R. Herbst-Irmer and D. Stalke, Angew. Chem., 2017, 56, 15141 CrossRef CAS PubMed.
- K. von Haeften, S. Rudolph, I. Simanovski, M. Havenith, R. E. Zillich and K. B. Whaley, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 054502 CrossRef.
- J. P. Toennies and A. F. Vilesov, Angew. Chem., 2004, 43, 2622 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 5648 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, et al., Gaussian 09, Revision D.1, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009 Search PubMed.
- M. Power, E. Alcock and G. P. McGlacken, Org. Process Res. Dev., 2020, 24, 1814 CrossRef CAS.
- L. Cicco, S. Sblendorio, R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Sci., 2016, 7, 1192 RSC.
- T. Fjdeödnerg, P. B. Hitchcock, M. F. Lappert and A. J. Thorne, J. Chem. Soc., Chem. Commun., 1984, 822 Search PubMed.
- T. Fjeldberg, M. F. Lappert and A. J. Thorne, J. Mol. Struct., 1984, 265 CrossRef CAS.
- K. W. Henderson, A. E. Dorigo, Q.-Y. Liu and P. G. Williard, J. Am. Chem. Soc., 1997, 119, 11855 CrossRef CAS.
- A. I. Ojeda-Amador, A. J. Martínez-Martínez, A. R. Kennedy and C. T. O’Hara, Inorg. Chem., 2016, 55, 5719 CrossRef CAS PubMed.
- G. J. Reyes-Rodríguez, R. F. Algera and D. B. Collum, J. Am. Chem. Soc., 2017, 139, 1233 CrossRef PubMed.
- P. Schüler, H. Görls, M. Westerhausen and S. Krieck, Dalton Trans., 2019, 48, 8966 RSC.
- S. Popenova, R. C. Mawhinney and G. Schreckenbach, Inorg. Chem., 2007, 46, 3856 CrossRef CAS PubMed.
- A. Gutberlet, G. Schwaab, O. Birer, M. Masia, A. Kaczmarek, H. Forbert, M. Havenith and D. Marx, Science, 2009, 324, 1545 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03372k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2022 |