
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the electronic structure and photophysics of thiophene–diketopyrrolopyrrole derivatives in solution†
Daniel W.
Polak
 a,
Mariana T.
do Casal
b,
Josene M.
Toldo
b,
Xiantao
Hu
c,
Giordano
Amoruso
a,
Olivia
Pomeranc
a,
Martin
Heeney
c,
Mario
Barbatti
bd,
Michael N. R.
Ashfold
a and
Thomas A. A.
Oliver
*a
a,
Mariana T.
do Casal
b,
Josene M.
Toldo
b,
Xiantao
Hu
c,
Giordano
Amoruso
a,
Olivia
Pomeranc
a,
Martin
Heeney
c,
Mario
Barbatti
bd,
Michael N. R.
Ashfold
a and
Thomas A. A.
Oliver
*a
aSchool of Chemistry, Cantock's Close, University of Bristol, Bristol, BS8 1TS, UK. E-mail: tom.oliver@bristol.ac.uk
bAix Marseille Université, CNRS, ICR, Marseille, France
cDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, White City Campus, London, W12 0BZ, UK
dInstitut Universitaire de France, 75231, Paris, France
First published on 15th August 2022
Abstract
Diketopyrrolopyrroles are a popular class of electron-withdrawing unit in optoelectronic materials. When combined with electron donating side-chain functional groups such as thiophenes, they form a very broad class of donor–acceptor molecules: thiophene–diketopyrrolopyrroles (TDPPs). Despite their widescale use in biosensors and photovoltaic materials, studies have yet to establish the important link between the electronic structure of the specific TDPP and the critical optical properties. To bridge this gap, ultrafast transient absorption with 22 fs time resolution has been used to explore the photophysics of three prototypical TDPP molecules: a monomer, dimer and polymer in solution. Interpretation of experimental data was assisted by a recent high-level theoretical study, and additional density functional theory calculations. These studies show that the photophysics of these molecular prototypes under visible photoexcitation are determined by just two excited electronic states, having very different electronic characters (one is optically bright, the other dark), their relative energetic ordering and the timescales for internal conversion from one to the other and/or to the ground state. The underlying difference in electronic structure alters the branching between these excited states and their associated dynamics. In turn, these factors dictate the fluorescence quantum yields, which are shown to vary by ∼1–2 orders of magnitude across the TDPP prototypes investigated here. The fast non-radiative transfer of molecules from the bright to dark states is mediated by conical intersections. Remarkably, wavepacket signals in the measured transient absorption data carry signatures of the nuclear motions that enable mixing of the electronic-nuclear wavefunction and facilitate non-adiabatic coupling between the bright and dark states.
1. Introduction
Molecules with alternating electron rich and electron deficient moieties are known as donor–acceptor (DA) chromophores. DA molecules are an important category of material due to their tuneable and low energy band gaps which originate from alternating electron densities across the molecular framework.1,2 Diketopyrrolopyrroles (DPPs) are a popular class of electron acceptor unit, that display a range of excited state dynamics.3–6 The judicious choice of connecting donor structures between DPP repeat units can have a major influence on the associated fluorescence quantum yield, non-radiative rates, probability of charge-transfer, and excited state lifetimes of the resulting molecule. This semi-empirical tunability of molecular parameters means DPP-containing systems can support processes such as exciton dissociation, long-range charge-separation, singlet fission7–16 and even symmetry-breaking charge-transfer, which has led to their application in a range of optical applications such as biosensors17–19 or photoelectronic/photovoltaic devices.3,20–29Due to their wide-ranging applications, many studies have investigated the structure–function relationship of DPP-containing thin films.30–34 A greater understanding of how the molecular structure and deposition method affects film morphology has allowed for the creation of 12% efficient solar cells with DPP-based materials as the active photovoltaic component.3,24–26,35–37 The interdependence of microscopic and mesoscopic phenomena in such solid samples hampers the development of reliable mechanistic insights into their operation. Solution-phase studies of DPP-containing systems offer a way of circumventing morphological complexities associated with thin films and allow for systematic investigations of the excited state dynamics under conditions more amenable to comparison with electronic structure calculations. In the solution phase studies conducted to date, changes in chemical structure and environment have reportedly led to drastic changes in photodynamical behaviour.9,12,13,15,38–41
DPP-containing molecules typically have nanosecond excited state lifetimes with appreciable fluorescence quantum yields (>50%).7,8,12,14,19,40 The corresponding thiophene–diketopyrrolopyrrole (TDPP) family of molecules are no exception to this observation.12 Counter to this general trend, a dimer of TDPP bridged by a vinylene linker (TDPP-v-TDPP) has no measurable fluorescence and a very short excited state lifetime.38 Many experimental and computational studies of other DPP-containing monomers,7,8,12,14,40 polymers,23,31,39 and dimers9,13,41 have failed to identify similar behaviour to that of the vinylene linker containing dimer. This inconsistency in the excited state dynamics motivated the present fundamental, ‘bottom up’ investigation of the electronic structure and photophysics of the prototypical TDPP-based systems. The chemical structures of the compounds studied experimentally, along with their respective absorption spectra in toluene solution, are given in Fig. 1.
 | ||
| Fig. 1 Chemical structures of (a) monomer TDPP-Br, (b) dimer TDPP-v-TDPP and (c) polymer DPPDTT. (d) Steady state absorption spectra of the three studied compounds in toluene solution. | ||
The present study used transient absorption (TA) spectroscopy to unravel the strikingly different dynamics of a monomer (TDPP-Br), dimer (TDPP-v-TDPP) and polymer (DPPDTT42). Experimental results are compared to recent multireference configuration interaction density functional theory (DFT/MRCI) and spin-flip time-dependent DFT (SF-TD-DFT) calculations of TDPP and TDPP-v-TDPP molecules.43 These studies demonstrate that two photoexcited states, the optically ‘bright’ and ‘dark’ excited states, are key to determining the visible photochemistry of the different TDPP systems. The observed photophysical differences are shown to be dominantly governed by the energetic re-ordering of the first two excited states, which greatly impacts the excited state dynamics, changing them from predominantly fluorescent decay (monomer) to non-fluorescent (dimer). Further, transient absorption data contain signatures of the nuclear motions responsible for driving ultrafast conversion between the critical bright and dark electronic states – a phenomenon rarely observed in ultrafast condensed phase dynamics of polyatomic molecules.
2. Results and discussion
2.1. TDPP-Br
 | ||
| Fig. 2 Steady state and time resolved spectroscopic characterisation of TDDP-Br in toluene solution. (a) Linear absorption and fluorescence spectra. (b) Transient absorption spectral slices measured at four illustrative pump–probe time delays. Probe wavelengths close to the 800 nm fundamental have been removed. (c) Normalised late time dynamics associated with five different probe wavelengths. (d) Normalised early time dynamics (out to 1.6 ps) and overlaid fits to data (dashed lines) associated with 615 nm and 750 nm probe wavelengths. | ||
The negative transient features were assigned by examining the kinetics associated with the maximal intensity of each signal, which are displayed in Fig. 2(c) and scaled for comparison. Two distinct decay regimes were found within the 1.1 ns time measurement window: a fast dynamical component occurring on the timescale of a few picoseconds, and a much slower component spanning hundreds of picoseconds through to nanoseconds. The kinetic traces presented in Fig. 2(c) for the positive GSB and SE features and for the negative ESA transients at 710 and 820 nm all show very similar, if not identical, kinetics. It was not possible to accurately determine the lifetime of the long-lived component, as the 5 ns time constant returned by fitting is far longer than the optical delay window, but this value is in accord with a previous study of (Br-free) TDPP.9
The most prominent ESA transient, peaking at 750 nm, decays far more rapidly than all other negative features. The first 1.6 ps of dynamics for the 615 nm SE and the 750 nm ESA signals, overlaid with fits to the data, are shown focusing on the earliest time delays in Fig. 2(d). Section S3 of the ESI,† gives further results from the kinetic analysis. During the earliest time delays, the 615 nm SE signal was observed to decay with a time constant τ = 103 ± 30 fs (that accounted for 59% of the total signal amplitude), and the 750 nm ESA feature was found to rise with a matching time constant (105 ± 5 fs). The strong agreement between the time constants of the transient species responsible for these features implies direct conversion from the initial photo-prepared population into another excited state, the latter with a distinct ESA spectrum centred at 750 nm. After this initial decay/rise, the 615 nm SE feature decays slowly, on an identical timescale to the GSB recovery, whereas the 750 nm ESA feature decays more rapidly. The latter decay is well described by a single exponential (with τ = 175 ± 5 ps (73%)), together with an additional long-time offset (τ ∼ 5 ns, 27%). We note that the 5 ns lifetime component is similar to the long-lived population observed in Fig. 2(c), suggesting this component is attributable to the long wavelength tail of the same parent ESA revealed in the kinetic fits to ESA signals at 710 and 820 nm.
The prompt but incomplete population transfer from the photo-prepared state to another excited state implies that multiple electronic states contribute to the very early time dynamics of TDPP-Br. To gain further insights into the electronic structure of TDPP-Br, a discussion and comparison with recent DFT/MCRI calculations by do Casal et al. of very similar molecules to those studied experimentally is helpful43 (see Table 1). To reduce computational costs, the prior computational study truncated the pendant branched alkyl chains (see Fig. 1) with methyl groups and the Br atoms were replaced with H atoms (henceforth referred to as simplified structures); moreover, the calculations were performed on isolated molecules (thus, solvation effects are absent). In this previous study, the simplified structures of TDPP-Br and TDPP-v-TDPP were optimized with DFT in the ground state and SF-TD-DFT in the excited singlet states. Subsequent single point energy calculations of the resulting structures were carried out using DFT/MRCI. The validity of truncating the alkyl chains and the effects of this simplification on the ground state geometry/symmetry are explored with additional DFT and TDDFT calculations discussed in the ESI,† Section S7. For the sake of simplicity, we refer to electronic states as labelled by their corresponding symmetry in the C2h point group throughout.
| State | VEE/eV (nm) | AEE/eV (nm) | Sn ← S0f | % D (S0 min) |
|---|---|---|---|---|
| S1 (1Bu) | 2.34 (530) | 2.19 (566) | 0.60 | 7.4 |
| S2 (2Ag) | 3.27 (379) | 2.92 (425) | 0.00 | 39.4 |
Geometry optimisation returned a planar ground (S0) state equilibrium geometry for the simplified TDPP, with Ag electronic symmetry (in the C2h point group), as depicted in Fig. S6 of the ESI.† The lowest excited singlet state (S1) also has a planar equilibrium geometry but Bu symmetry. Single point calculations on these structures returned corresponding vertical and adiabatic excitation energies of 2.34 and 2.19 eV, respectively (equivalent to ∼530 and ∼566 nm) and identified the S1 state as optically ‘bright’, formed predominantly via a π* ← π excitation. Henceforth, the ground electronic state will be described interchangeably as the S0 or 1Ag state, and the first singlet excited state of TDPP (and TDPP-Br) will be referred to as the 1Bu state.
The S2 state of the simplified TDPP has Ag symmetry and a vertical excitation energy of 3.27 eV (∼379 nm). The S2–S0 transition dipole moment is predicted to be zero; the S2 state is thus optically ‘dark’. This state has a large contribution from two-electron promotions (i.e., it has a substantial doubly excited character, %D in Table 1). Minimising the structure of 2Ag state confirms that it too has a planar equilibrium geometry and a subsequent single point energy calculation yields an adiabatic excitation energy of 2.92 eV (equivalent to a wavelength ∼425 nm).
Given that theory predicts the optically ‘dark’ 2Ag state of the simplified variant of TDPP-Br to be at higher energy than the ‘bright’ 1Bu state in the vertical Franck–Condon region,43 the most likely explanation for the early time experimental data (Fig. 2(d)) is that the short wavelength edge of the broadband pump pulse populates vibrationally excited levels of the 1Bu state with sufficient energy to access the 2Ag state. At some point subsequent to vibrational cooling, the 2Ag and 1Bu state populations are no longer able to interconvert, locking-in the respective populations, e.g. the linking conical intersection can only be accessed via vibrationally excited levels of the 1Bu state. This is reinforced by the wavelength dependence of the fluorescence quantum yield (ΦF) of TDPP-Br: as the excitation wavelength is tuned from 530 to 500 and then 475 nm, the ΦF decreases by 13% (from 61, to 57 and then 48%, respectively), consistent with the hypothesis that an alternative non-radiative decay pathway becomes more prominent at shorter wavelengths.
Thus, we arrive at a picture wherein photoexcitation of TDPP-Br populates a broad spread of 1Bu state vibrational levels. Some of the most highly excited vibrational states of the 1Bu state couple to 2Ag state levels (τ = 105 fs), but the remainder vibrationally cool to the 1Bu state minimum from whence they radiatively decay (τ ∼ 5 ns). Population that relaxes into the 2Ag minimum is tracked via the 750 nm ESA feature, which is found to decay on a faster (τ = 175 ps) timescale, with no associated recovery of the ground state bleach. The decay of this ESA is mirrored by the growth of a small negative transient on the blue edge of the probe window (400–475 nm), indicating the ‘dark’ state decays to a further excited state rather than the ground state. Hartnett et al. reported intensity in a similar wavelength region in previous triplet sensitisation studies of a (Br-free) TDPP in solution and ascribed the feature to triplet excited state absorption.12 We similarly assign the τ ∼ 175 ps decay component in the TDPP-Br data to intersystem crossing from the 2Ag state into the triplet manifold. Hartnett et al. did not observe triplets in solution without the addition of a sensitizer12 and we suggest that, in the present case, triplet production is likely facilitated by the presence of the heavy Br atoms.
 | ||
| Fig. 3 Wavepacket dynamics for TDPP-Br dissolved in toluene. False colour contour map of transient absorption data (a) before and (b) after subtraction of electronic population dynamics. | ||
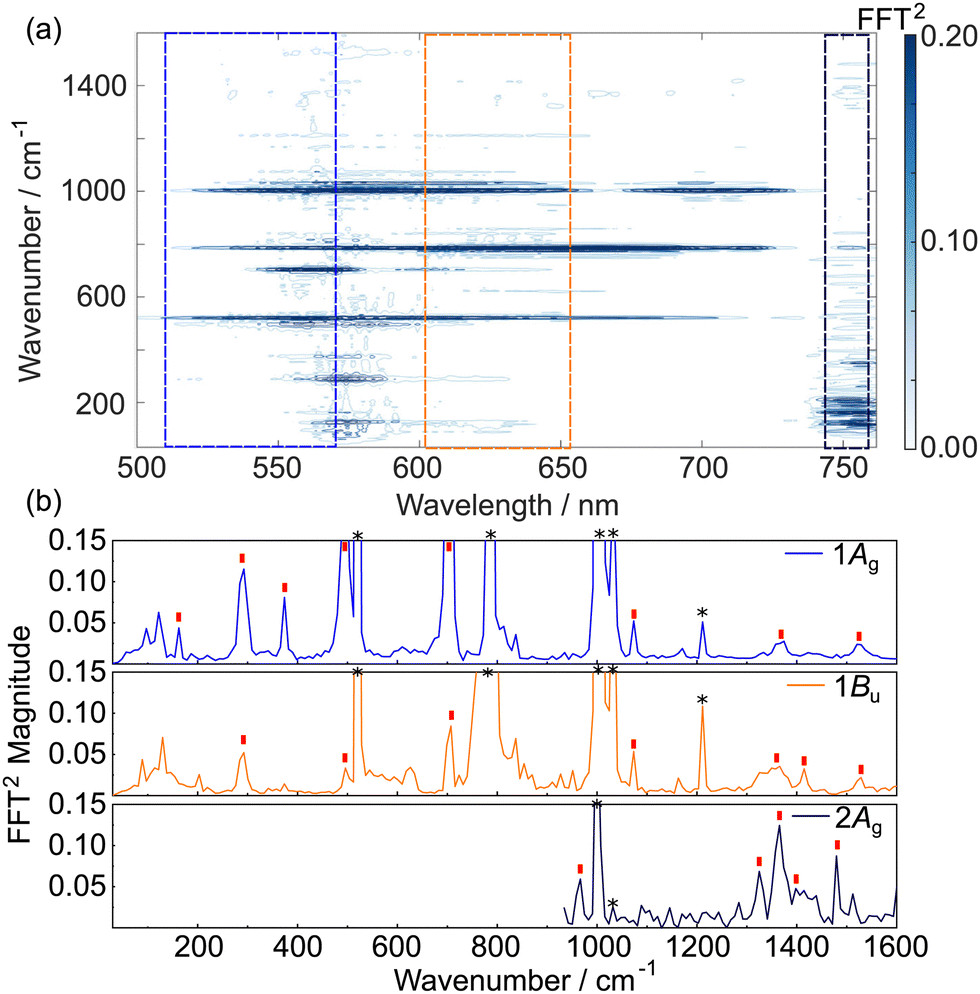 | ||
| Fig. 4 TDPP-Br wavepacket dynamics in toluene. (a) Fourier-Transform map of wavepacket-modulated transient absorption spectra of TDDP-Br in toluene. (b) Vibrational wavenumbers associated with the 1Ag (510–560 nm), 1Bu (600–650 nm), and 2Ag (745–755 nm) states. The trace associated with the 2Ag state was truncated below 800 cm−1 to remove low frequency noise associated with the probe supercontinuum generation. Red ticks in panel (b) indicate assigned TDDP-Br vibrational wavenumbers discussed in the main text and/or in Section S8 of the ESI.† Asterisks indicate major peaks associated with toluene solvent. | ||
Definitive assignment of the solute modes is very challenging given the high density of vibrational states (the full TDPP-Br molecule has 438 vibrational degrees of freedom). However, DFT/B3LYP/6-311G(d,p) calculations of the ground state normal modes of the core model system using methyl side chains and including both bromine atoms (96 vibrational degrees of freedom) provide many useful insights.
Vibrational wavepackets associated with the TDDP-Br transient GSB were observed at wavenumbers throughout the range 50–1550 cm−1 (Fig. 4(a)). For wavenumbers ≤700 cm−1, the observed features can be plausibly assigned using DFT calculations to in-plane ring breathing and/or in-plane twisting modes involving the thiophene rings and the DPP core, while features at >700 cm−1 are consistent with vibrational modes involving C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–N–C stretching motions along the π-conjugated backbone extending through flanking thiophene units and the DPP core. A full list of the observed wavenumbers and proposed assignments for the GSB wavepackets observed in the TDPP-Br TA data is given in Table S4 of the ESI.† All assignments were made by considering calculated wavenumbers within a 5% tolerance of the experimentally observed value. In most instances, this method generated between one and three plausible assignments due to the vast density of vibrational states; however, the associated nuclear motions of the candidate modes were quite similar.
C/C–N–C stretching motions along the π-conjugated backbone extending through flanking thiophene units and the DPP core. A full list of the observed wavenumbers and proposed assignments for the GSB wavepackets observed in the TDPP-Br TA data is given in Table S4 of the ESI.† All assignments were made by considering calculated wavenumbers within a 5% tolerance of the experimentally observed value. In most instances, this method generated between one and three plausible assignments due to the vast density of vibrational states; however, the associated nuclear motions of the candidate modes were quite similar.
Analysis of the 600–650 nm probe window associated with the 1Bu excited state, also returned several vibrational wavepackets in the 250–1550 cm−1 range (Fig. 4(b) and listed in Table S5, ESI†). These features were assigned using vibrational wavenumbers calculated for the smaller TDPP-Br analogue with truncated alkyl sidechains using TD-DFT/B3LYP/6-311G(d,p) calculations of the 1Bu minimum energy structure. B3LYP can be reliably employed in this case due to the minor charge-transfer character of this state. The feature appearing at 288 cm−1 aligns closely with the same peak found in the GSB trace and, again, is likely attributable to in-plane breathing and/or in-plane twisting distortions of the DPP and/or thiophene rings. The appearance of such a feature in both traces is consistent with Franck–Condon activity in the 1Bu ← 1Ag transition. Wavepackets at higher wavenumbers, including one at ∼1362 cm−1 that matches well with the vibrational peak separation in the TDPP-Br electronic absorption spectrum (Fig. 2(a)), are plausibly assigned to C–C/CC and/or C–N–C stretching motions. In the trace associated with the 1Bu state, much of the vibrational wavepacket activity identified in the 600–650 nm probe window appears to reflect nuclear motions centred on the core DPP unit, consistent with the SF-TD-DFT predicted geometry changes43 upon photoexcitation (see Fig. S6 and S7, ESI† for the reproduced minimum energy structures and associated bond lengths).
High frequency vibrational wavepackets were also observed in the 745–755 nm region associated with the 2Ag transient. These are more noteworthy, given that the 2Ag ← 1Ag transition has zero-oscillator strength (see Table 1) and thus any population in the 2Ag state must arise by ultrafast coupling from (high vibrational levels of) the photoexcited 1Bu state. Wavepacket activity in the 2Ag state could therefore in principle arise in several ways:
(i) The 1Bu and 2Ag electronic states have significantly different equilibrium geometries, and if 1Bu → 2Ag transfer is sufficiently ultrafast, the nascent 2Ag molecules would be created far from equilibrium, launching semi-ballistic coherent nuclear motion reminiscent of the wavepacket dynamics recently observed in a model electron transfer reaction.47
(ii) The wavepackets could be created impulsively in the 1Bu state by 1Bu ← 1Ag photoexcitation but involve nuclear motions orthogonal to the coordinate that drives the prompt 1Bu → 2Ag surface crossing. These motions then survive as ‘spectators’ to the fast coupling, mapping into the corresponding vibrations in the 2Ag state, as observed previously in a range of ultrafast non-radiative processes of various molecules;48–51
(iii) The wavepackets could reflect nuclear motions that mediate the non-adiabatic transfer in the regions of conical intersection between the states.52–55 If the crossing between the two states is sufficiently fast, coupling nuclear motions could be created coherently at the intersection and map into 2Ag state wavepackets.
Unfortunately, it is not possible to calculate the vibrational normal modes of the 2Ag state with SF-TD-DFT- a level of theory required to describe this doubly excited state accurately. Thus, another strategy had to be used to allow insights into the nature of the 2Ag state wavepackets. Given the similar calculated minimum energy geometries of the 1Bu and 2Ag states43 (cf. Fig. S7 and S8, ESI† reproduced from do Casal et al.), we assume that their normal modes of vibration and the associated wavenumbers are also similar. The similar excited state geometries mean that the above scenario (i) is unlikely to be the origin of 2Ag wavepackets for TDPP-Br. Scenario (ii) involving wavepackets created upon photoexcitation of the 1Bu state and survive the surface crossing to the 2Ag state is likely. Indeed, several 2Ag peaks can be ascribed to this category: wavenumbers 1365 and 1410 cm−1 have credibly assigned motions similar to those identified for the 1Bu state. As before, these are plausibly assigned to in-plane C–N–C stretching modes and C–C/CC stretching motions extending across the DPP and thiophene rings.
Surprisingly, examination of Fig. 4(b) also reveals wavepackets in the 2Ag data (at 965, 1325 and 1479 cm−1) that have no analogues in the 1Bu or S0 transients. As these wavenumbers do not appear in any other trace, the wavepacket motion must have been produced upon 1Bu → 2Ag interconversion. As such, it is most likely that these reflect nuclear motions created at a 1Bu/2Ag conical intersection, i.e., the coupling mode(s) themselves. The overall symmetry of the electronic-nuclear wavefunction must be conserved upon transfer from the 1Bu state to the 2Ag state, meaning that the coupling nuclear motions must have bu symmetry (as Bu ⊗ bu = Ag), i.e. in-plane (preserving the C2h mirror plane symmetry), but anti-symmetric (breaking the inversion symmetry) motions. Consistent with this symmetry requirement, the 965 cm−1 wavepacket can be plausibly assigned to a nuclear mode involving anti-symmetric C–S–C stretching on the thiophene units coupled to C–C/CC stretching motions over the whole π-conjugated system, the 1325 cm−1 feature could arise from anti-symmetric DPP and thiophene ring breathing motions, and the 1479 cm−1 feature could be assigned to a high amplitude anti-symmetric stretching motion of the central CC bonds of the DPP unit.
2.2. TDPP-v-TDPP
 | ||
| Fig. 5 Transient absorption spectra and kinetics of TDPP-v-TDPP in toluene solution. (a) Linear absorption spectrum, (b) spectrally resolved TA spectra at selected pump–probe time delays in the range 30 fs ≤ t ≤ 10 ps. (c) Scaled wavelength averaged dynamics over the first 50 ps associated with key spectral features and (d) scaled early time dynamics showing the population transfer between the optically bright and lower energy states. | ||
Again, comparison with recent DFT/MRCI calculations of a TDPP-v-TDPP variant with truncated alkyl chains43 proved insightful. For the simplified TDPP-v-TDPP dimer, calculations reveal two excited states at similar excitation energies (see reproduced data from ref. 43 in Table 2): as in TDPP, a bright, primarily singly excited 1Bu state and a dark predominantly doubly excited 2Ag state. In contrast to TDPP, do Casal et al. found that the energetic ordering of these two states is reversed: the 2Ag state is predicted to be ∼0.1 eV lower in energy in the Franck–Condon region and to have an adiabatic excitation energy ∼0.5 eV less than that of the 1Bu state. The systematic study showed that the 1Bu/2Ag state ordering is greatly influenced by the length of the π-system;43 increasing the number of π electrons stabilizes both states, but the 2Ag state more significantly, in agreement with the inversion of excited states observed experimentally (vide infra). By analogy with the preceding discussion of TDPP-Br and theoretical calculations, the 500 nm ESA feature is assigned to the optically bright 1Bu state prepared by the initial photoexcitation, which decays rapidly into the lower-energy 2Ag state.
| State | VEE/eV (nm) | AEE/eV (nm) | Sn ← S0f | % D (S0 min) |
|---|---|---|---|---|
| S1 (2Ag) | 1.57 (790) | 1.23 (1008) | 0.00 | 57.0 |
| S2 (1Bu) | 1.66 (747) | 1.46 (849) | 2.10 | 10.4 |
No further spectral evolution was observed at pump–probe time delays beyond 1 ps, and both the GSB and near-IR ESA features were found to decay on the same ∼5.5 ps timescale (GSB: τ = 5.3 ± 0.4 ps, ESA: τ = 5.7 ± 0.4 ps). The kinetics reported here are consistent with a previous TA study of TDPP-v-TDPP in chlorobenzene solution using ∼150 fs pump pulses, except that the prior study was unable to fully capture the early time 1Bu/2Ag interconversion dynamics.38
The early time dynamics have clear parallels with those displayed by the monomer, i.e., very fast coupling between the photoexcited state and the dark doubly excited state. Thereafter the monomer and dimer exhibit strikingly different photophysics. TDPP-Br molecules that remain in the 1Bu state survive for nanoseconds in a fluorescent state. Those that couple into the 2Ag state display a somewhat shorter lifetime (τ = 175 ps), determined by the rate of intersystem crossing. In contrast, for TDPP-v-TDPP we observe a full depopulation of the 1Bu state, and a much shorter 2Ag lifetime of 5.5 ps. The fast and efficient non-radiative decay of the 2Ag state necessitates a low-lying, accessible within thermal energies, conical intersection with the S0 state – a pathway seemingly unavailable in monomer TDDP-Br.
 | ||
| Fig. 6 Wavepacket dynamics of TDPP-v-TDPP in toluene solution. (a) False colour map of vibrational wavenumber vs. probe wavelength extracted from transient absorption measurements. (b) Vibrational wavenumbers associated with the 1Ag (550–600 nm), 2Ag (850–900 nm) and 1Bu (480–530 nm) electronic states. Due to the short lifetime of the 1Bu state, the wavepacket intensity is notably weaker. Asterisks indicate solvent vibrations; red labels indicate modes assigned in the main text and in Section S8 of the ESI.† Asterisks indicate major peaks associated with toluene solvent. | ||
For the S0 and 1Bu states, associated wavepacket spectra contain peaks throughout the 200–1550 cm−1 range, which can be plausibly assigned to in-plane ring breathing motions and C–C/CC stretching motions over the whole molecular structure. These assignments are consistent with the predicted geometry changes from the ground to excited state minimum. Full assignment of these wavepackets is given in Tables S7 and S8 of the ESI.†
Transient data in the near-IR probe region associated with the 2Ag dark intermediate state also exhibits vibrational wavepacket activity and, similar to the TDPP-Br monomer, has two origins. Vibrational activity was observed at 1422 and 1502 cm−1, at similar wavenumbers as wavepackets observed in the 1Bu or 1Ag states, but slightly shifted, suggesting that these wavenumbers correspond to spectator vibrational modes to the 1Bu/2Ag surface crossing, and correspond to ring breathing motions of the thiophene and DPP units. At lower wavenumbers three peaks with significant intensity unique to the 2Ag state are evident at 54, 1107 and 1258 cm−1. As the recent computational study revealed the minimum energy geometries of the 2Ag and 1Bu states of TDPP-v-TDPP are very similar43 (associated bond lengths reproduced in Fig. S10 and S11, ESI†), and thus these new wavepackets must arise from coherent nuclear motion associated with coupling modes created at the 1Bu/2Ag conical intersection of the required bu symmetry which map into 2Ag state wavepackets. With these criteria and rationale, it was possible to suggest the following wavepackets assignments: an in-plane anti-symmetric rocking of the DPP units relative to the thiophene rings (with a calculated wavenumber of 50 cm−1); DPP/thiophene anti-symmetric ring breathing motions (1113 cm−1) and in-plane anti-symmetric CC stretching motions localised to the central thiophene rings/vinyl linker (1230 cm−1).
2.3. DPPDTT
The commercially obtained DPPDTT polymer sample had a reported polydispersity index of 2.05 and, unlike TDPP-Br and TDPP-v-TDDP, lacks high order C2h symmetry. The linear absorption spectrum of DPPDTT in chloroform (Fig. 7(a)) is red-shifted relative to that of TDPP-Br, but is similar to that of TDPP-v-TDPP (see comparison in Fig. 1(d)). This similarity hints that the delocalisation length scale in the DPPDTT polymer sample is close to that of the dimer, i.e., 2 repeat units. Fig. 7(b) shows TA spectra for DPPDTT dissolved in chloroform at four representative pump–probe delays, the analysis of which yields the population dynamics reported in this section. Similar TA measurements in toluene (in which the polymer is less soluble) yield poorer signal-to-noise data but return similar kinetics, as described in the ESI† (Fig. S3 and S4).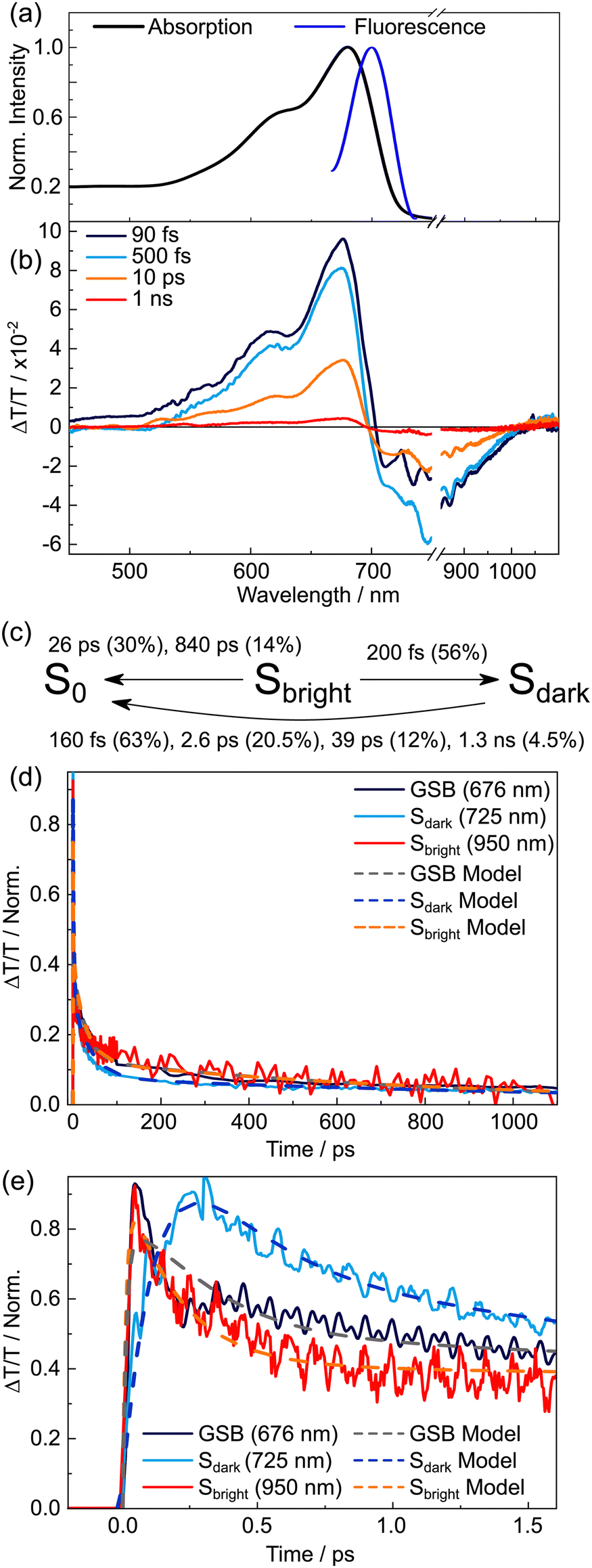 | ||
| Fig. 7 Transient absorption spectra and kinetics of DPPDTT in chloroform solution. (a) Linear absorption and fluorescence spectra of DPPDTT in chloroform. (b) Spectrally resolved transient absorption spectra for four different pump–probe time delays. (c) Summary of the rate model used to describe DPPDTT kinetics after excitation to the bright state. Errors associated with lifetimes are reported in the text; the associated amplitudes (given as percentages) are stated in brackets. Normalised kinetics for (d) nanosecond and (e) picosecond dynamics, and include overlaid kinetics returned from the rate model. | ||
The TA spectra for DPPDTT are dominated by two major spectral features, with assignments analogous to the other systems. A large positive signal that spans most of the visible probe window can be assigned to a combination of ground state bleach and stimulated emission, and the negative band peaking in the near-IR to an ESA transient. All these time traces require multiple exponential decay components to fully describe the kinetics, and the ESA feature clearly embodies (at least) two behaviours. Fig. 7(d) and (e) compare kinetic data for the two ESAs (725 and 950 nm) and the GSB (675 nm) displayed on two different time axes. The 950 nm ESA rises instantaneously within the instrument response function (IRF), consistent with it being the signature of a directly photoexcited state and thus optically bright in nature (henceforth simply labelled Sbright). In contrast, the 725 nm ESA rises on a timescale slower than the IRF, consistent with a state formed after the initial photoexcitation. In tandem with the growth of this feature, we note a blue-shift of the isosbestic point at ∼700 nm – at probe wavelengths where SE from the bright state would be expected to contribute. The time-dependent shift in the isosbestic point thereby tracks the decay of the fluorescent Sbright state. In analogy with TDPP-Br and TDPP-v-TDPP, the slow rise and loss of SE intensity suggest dynamics wherein photoexcitation populates the Sbright state (probed by ESA at 950 nm), which rapidly converts to an optically ‘dark’ state (henceforth labelled Sdark) that is responsible for the 725 nm ESA. Despite the 725 nm ESA maximising within ∼300 fs, some fraction of the 950 nm ESA signal persists out to beyond 1 ns. Such behaviour has parallels with that observed for the TDPP-Br monomer and suggests incomplete interconversion between the Sbright and Sdark states, leaving some emissive population in the Sbright state.
The observed kinetics are very rich, making fitting individual lifetimes difficult. The TA data at different probe wavelengths could be fit using 3 (or 4) different exponential components, yielding time constants similar to those reported previously for donor–acceptor polymers such as PTB7.56–58 Such an analysis is difficult to underpin with a physical model but is consistent with polymer samples involving a distribution of chain conformations, but maintaining a delocalisation length of ∼2 repeat units, each with different local environments.56,57 To confirm if a three state model is sufficient to describe the complex kinetics of DPPDTT, a rate model solving a set of coupled differential equations representing the time-dependent populations of the Sbright, Sdark and S0 states was used. These modelled populations are compared to kinetics representing each state to confirm its validity (Fig. 7(d) and (e)). Full details of the modelling procedure are given in Section S2(c) of the ESI.†
The initial population of the Sbright state is generated by a term which describes the Gaussian IRF. The model allows for several independent populations of the Sbright state, each representative of a distribution of polymer conformations in the sample which exhibit different photophysical behaviours. Further, to simplify the model, no exciton diffusion between these domains is allowed on the order of picoseconds, enforcing static inhomogeneity, and thereby allowing the sub-populations to be treated independently. In the model, the Sbright state decayed with three-time constants: 200 ± 8 fs (56%), 26 ± 1 ps (30%) and 840 ± 40 ps (14%) where the numbers in parenthesis report the respective normalised amplitudes. The shortest decay component (200 fs) results in formation of the dark state, while the other components of the Sbright state decay are associated with direct return to the S0 state via radiative or non-radiative mechanisms.
After its formation, Sdark decays to S0 as described by four time constants: 160 ± 7 fs (63%), 2.6 ± 0.1 ps (20.5%), 39 ± 15 ps (12%) and 1.3 ± 0.1 ns (4.5%). The kinetics associated with the GSB are described by an IRF limited rise, and a decay to zero based on a weighted combination of four Sdark and two Sbright state decay constants. Combining these elements enables prediction of the population relaxation kinetics for the three key states, as summarised in Fig. 7(c). The results returned by the three-state model are overlaid on the experimental data in Fig. 7(d) and (e), and a striking agreement between the predicted and experimentally recorded kinetics is found on both picosecond (Fig. 7(d)) and nanosecond (Fig. 7(e)) timescales. The complex kinetics are not unexpected given the heterogeneous nature of the polymer sample, as noted in previous studies of similar systems.57
To unravel a physical mechanism underpinning this proposed model and the nature of the Sdark state, inspiration was taken from the analysis of other donor–acceptor polymers,59–62 and guided by our analysis of monomer TDDP-Br. The long wavelength wing of the pump pulses populates low-vibrational levels of the DPPDTT polymer near the Sbright minimum. These levels vibrationally relax to the minimum of the Sbright potential and then radiatively decay back to S0. The short-lived lifetimes associated with these components (26 and 840 ps), which are similar to those observed for P3HT,57 hint at non-radiative processes back to S0 that limit the excited state lifetime. This observation explains the weak, yet measurable, fluorescence for DPPDTT as opposed to the non-fluorescent dimer. In analogy with TDDP-Br, DPPDTT polymers excited with the short wavelength edge of the laser pulse, which populates highly vibrationally excited levels of the Sbright state, are assumed to follow a similar mechanism to TDDP-Br. The excess vibrational energy within the Sbright state, prior to vibrational cooling, allows for interconversion into the Sdark state within 200 fs. The Sdark state then decays (with τ = 160 fs, 2.6 ps, 39 ps and 1.3 ns) directly back to S0. Unlike TDPP-Br, however, the DPPDTT polymer is asymmetric, and therefore the dark state can take charge-transfer or even charge-separated character,57 which is essential to mediating electron/hole migration. Given there are a myriad of non-radiative decay rates observed in DPPDTT kinetics, with similar timescales and magnitudes to other similar organic polymers such as P3HT and PTB7,56–58 we tentatively ascribe these to charge-recombination processes, which inevitably depend on the local environment specific to the polymer conformation, that in turn dictates the initial electron-hole separation distance and energetic barrier to recombination.
As Fig. 7(d) shows, the DPPDTT transient absorption kinetics also show oscillations attributable to vibrational wavepackets from the polymer along with ISRS from the chloroform solvent. In chloroform, the solvent ISRS signal dwarfs the intensity of solute wavepackets. Therefore, to probe DPPDTT wavepackets, the early time dynamics were measured in toluene solution. The wavepacket analysis for DPPDTT in toluene is reported in Fig. S5 of the ESI.† Unlike the monomer and dimer, the size and polydispersity of the polymer sample prevent accurate calculations and thus makes assignment of modes difficult. Wavepacket wavenumbers associated with the Sdark state are noteworthy (618, 734, 859, 946 cm−1), and consistent with the deduced ultrafast coupling of Sbright and Sdark states. Three wavepackets (734, 859 and 946 cm−1) are evident in both Sbright, S0 and Sdark traces, a signature that the surface crossing is prompt and that coherent nuclear motion can carry through the surface crossing into the dark state. A single wavepacket, 618 cm−1, however, is only evident in the Sdark trace and therefore is a likely signature of nuclear motions created at the conical intersection linking the two states.
2.4. Summary
The energetic ordering of dark (2Ag) and bright (1Bu) electronic states has a profound influence on the photophysics of the TDPP monomer and dimer. The multireference computational results by do Casal et al. using SF-TD-DFT and DFT/MRCI43 were critical to elucidating and characterising the dark 2Ag state. This recent computational study highlights the need to use methods which can properly describe double excitations (or to benchmark where appropriate) electronic structure methods, especially when there is no a priori knowledge of the nature of the electronically excited states, which may be poorly characterised by standard TD-DFT methods.To the best of our knowledge, wavepackets originating from coherent nuclear motions associated with coupling modes at conical intersections have only been reported twice before for molecular systems,54,55 and only one of these studies offered mode assignments based on high-level calculations.54 The assignment of the wavepacket vibrational modes in the present study was only possible with the use of the corresponding theory and consideration of symmetry requirements. Such an approach could be applied to many existing data in the literature and offer deeper insights into coherent motions in the region of conical intersections. It is important to note that observation of these dynamics in TDPPs is also in part due to three key other fortuitous factors: there is minimal spectral overlap between 2Ag and 1Bu ESA transients, which greatly simplifies spectral assignments and means wavepacket signatures can be readily associated with a single electronic state; the minimal spectral shifting present in the TA data, presumably due to the small/zero structural re-organisation between the 2Ag and 1Bu states; the prompt surface crossings ensured vibrational dephasing/intra-molecular vibrational distribution is incomplete by the time the 1Bu/2Ag conical intersection is encountered and that the decoherence of nuclear motion is sufficiently minimal that new coherences created at the conical intersection map into observable 2Ag wavepackets.
3. Conclusions
The photophysics of TDPP-Br, TDPP-v-TDPP and DPPDTT upon broadband visible excitation are dictated by two electronically excited states. Photoexcitation populates a dipole-allowed excited state, which in the C2h point group is of 1Bu symmetry for monomer TDPP-Br and the TDPP-v-TDPP dimer. In the monomer, the 1Bu state is the lowest energy singlet excited state, with a ∼5 ns lifetime. A small fraction of the 1Bu population that is created in vibrationally excited levels, are able to convert into a different electronic state (2Ag), which has significant doubly excited character.43 In TDPP-Br, the 2Ag state acted as a precursor for rapid intersystem crossing into a triplet state (Fig. 8(a)). In the dimer, the dark 2Ag state lies below the 1Bu state in the vertical Franck–Condon region. Analysis of the transient kinetics shows that the vast majority of excited molecules are converted into the lower-lying 2Ag state within <100 fs and subsequently couple back to the S0 state on a tens of picosecond timescale, with high efficiency (Fig. 8(b)). | ||
| Fig. 8 Schematic potential energy curves and illustrative pathways representing the major excited state dynamics for (a) TDPP-Br, and (b) TDPP-v-TDPP. | ||
The DPPDTT polymer exhibits some dynamical and electronic structural similarities with the monomer and dimer. The delocalisation length scale of the polymer sample is deduced to be similar to that of the dimer, spanning ∼2 repeat units. However, the excited state dynamics and kinetics of the polymer more closely resemble those of TDPP-Br, but with two notable differences. The interconversion of the optically bright state into the dark state is more efficient in DPPDTT than the monomer, and the electronic character of the dark state in the polymer is charge-transfer in nature, supporting subsequent electron/hole transport within polymer chains. Furthermore, due to the inhomogeneity of the polymer sample, each dynamical process exhibits a range of lifetimes originating from differences in the surrounding environment.
The ultrashort laser pulses used in these studies inevitably launched excited state wavepacket dynamics. Wavepackets were observed in the bright 1Bu state and, surprisingly, also the optically dark 2Ag state. These wavepackets are deduced to be formed via two different mechanisms: (i) wavepackets generated upon photoexcitation of the 1Bu state but have associated nuclear motions that lie orthogonal to the 1Bu/2Ag crossing, meaning the vibrational coherence is unaffected by the crossing and transferred into the second electronic state. This phenomenon has been observed previously in other ultrafast condensed phase studies involving fast non-radiative transfer between two electronic states on a timescale faster than the associated vibrational dephasing.48–51 (ii) More remarkable, is the observation of new wavepackets unique to the optically dark 2Ag state, born from coherent nuclear motions generated at the 1Bu/2Ag conical intersection. These motions, the so-called coupling modes, are created out of necessity to couple the electronic state wavefunctions and map into 2Ag vibrational wavepackets. This phenomenon has only been reported twice before in molecular condensed phase studies.54,55
Our study emphasises how the excited state dynamics of a family of prototypical TDPP molecules is finely tuned by the relative energetic ordering of two electronic states of very different character. The present experimental study, along with recent computational results,43 also highlights the need for caution when invoking models to explain the excited state phenomena of TDPPs, without strong experimental and/or theoretical evidence. Finally, the wavepacket dynamics observed here further illustrate the exquisite detail that can be extracted about the non-radiative relaxation dynamics in condensed phase studies of modest sized molecular systems.
4. Methods
4.1. Sample preparation
TDPP-Br (3,6-bis(5-bromo-2-thienyl)-2,5-bis(2-octyldodecyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione, >98%) and DPPDTT (poly[2,5-(2-octyldodecyl)-3,6-diketopyrrolopyrrole-alt-5,5-(2,5-di(thien-2-yl)thieno [3,2-b]thiophene)], PDI: 2.03) were purchased from Osilla and used without any further purification (DPPDTT is also known as pDPPT-TT). TDPP-v-TDPP (E)-6,6′-(ethene-1,2-diylbis(thiophene-5,2-diyl))bis(2,5-bis(2-ethylhexyl)-3-(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione) was synthesized by the Pd-catalysed Stille cross-coupling of bis(tributylstannyl)ethene with mono-brominated TDPP, following the methodology of Patil et al.63 The reaction afforded a mix of the desired product and a major by-product, which were separated by a mixture of column chromatography over silica followed by preparative recycling gel permeation chromatography. TDPP-v-TDPP was characterized by 1H NMR, 13C NMR and mass spectra (see Fig. S14–S19, ESI†). The by-product was identified as the homo-coupled TDPP product (TDPP-TDPP) based on NMR and mass spectroscopy measurements, full details of the synthesis and purification procedures are given in Section S6 of the ESI.†All molecules were dissolved in either toluene or chloroform depending on their solubility. Solvents were purchased from Sigma Aldrich at HPLC grade (99.8% purity). For TA measurements, the solutions were diluted to have an absorbance between ∼0.3–0.4 at the peak wavelength of the pump laser in static 1 mm pathlength cuvettes. Fresh solutions were prepared before each measurement, and no sign of degradation was observed in visible absorption spectra acquired before and after ultrafast spectroscopic measurements. The fluorescence quantum yield of TDDP-Br was determined using a commercial fluorimeter and integrating sphere (Edinburgh Instruments, FS5).
4.2. Ultrafast transient absorption spectroscopy
A 1 kHz, 1 W, 800 nm commercial laser system (Coherent, Libra) was used to pump a homebuilt non-collinear optical parametric amplifier (NOPA) to produce the pump pulses. The homebuilt NOPA was tuned to produce broadband pump pulses centred at 580 nm with a ∼75 nm FWHM (Fig. S1, ESI†). Pump pulses were compressed using chirped mirrors (Layertec) and fine-tuned with a pair of fused-silica wedges (FemtoOptics, Newport). The pulse duration was determined using polarisation gated frequency resolved optical gating to be 22 fs (FWHM) and limited by third order dispersion (Fig. S1, ESI†). The compressed pump pulses were attenuated to below 100 nJ using an achromatic polariser–waveplate pair and focused into a ∼150 μm spot size at the sample with a 25 cm focal length curved mirror. A very small portion of the Libra output was used to generate white light supercontinuum probe pulses in sapphire or yttrium aluminium garnet crystals, producing probes spanning the visible and near-IR wavelength regions. The pump–probe time delay was controlled by a mechanical delay stage (Physik Instrumente, M-521.DD1). After the sample, the probe and collinear signal was collimated and focused into a Czerny–Turner spectrograph (Shamrock 163, Andor) coupled to a linear CCD array detector (Entwicklungsbüro Stresing) and digitised. All transient measurements were conducted under the magic angle condition. Data were chirp corrected by fitting the solvent only transient response to a high order polynomial.4.3. Density functional theory calculations
DFT and TD-DFT calculations were performed using the Gaussian 16 computational suite.64 Ground state DFT calculations were used to investigate the effect of alkyl group length (R = –H, –CH3, –CH2CH(CH3)2) and symmetry point group (C2h or Ci) on multiple conformers of the TDPP-Br monomer and TDPP-v-TDPP dimer. The full results from these calculations are detailed in Section S7 of the ESI.† The vibrational normal modes and associated wavenumbers were calculated at the determined global minimum energy geometries. TD-DFT was used to minimise the bright electronic state geometry of both the monomer and dimer, and associated normal modes to assign experimentally observed wavepackets. A full list of vibrational assignments of wavepacket data are given in Section S8 of the ESI.† All calculations used the B3LYP exchange-correlation function and a 6-311G(d,p) basis set, and did not include any effect of solvation.Author contributions
D. W. P., G. A. and O. P. acquired ultrafast and steady state experimental data. M. T. do C. and J. M. T. analysed the data in the light of DFT/MRCI and SF-TD-DFT calculations. X. H. synthesised, purified and characterised TDPP-v-TDPP. D. W. P. analysed all time-resolved data and wrote the first and subsequent major drafts of the manuscript. M. H., M. B., M. N. R. A. and T. A. A. O. supervised the project. All authors contributed to the writing of the manuscript and approved the final version.Notes
The raw data supporting this article are archived in the University of Bristol's Research Data Storage Facility (https://doi.org/10.5523/bris.280mpmf2l7ihj2jl1x2k9qwdw0).Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The authors at Bristol and Aix-Marseille acknowledge funding from the European Union's Horizon 2020 Research and Innovation programme, under grant agreement 828753 (Boostcrop). T. A. A. O. acknowledges financial support from the Royal Society for a University Research Fellowship (UF1402310 and URF\R\201007) and Research Fellows Enhancement Awards (RGF\EA\180076 and RF\ERE\210045). The authors at Aix-Marseille Université acknowledge the Centre de Calcul Intensif d'Aix-Marseille for granting access to its high-performance computing resources and the HPC/AI resources from GENCI-TGCC (Grant 2022 – A0110813035). M. H. thanks the Royal Society and the Wolfson Foundation for financial support.References
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Spectral engineering in π-conjugated polymers with intramolecular donor–acceptor interactions, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed
.
- J. Roncali, Molecular engineering of the band gap of π-conjugated systems: Facing technological applications, Macromol. Rapid Commun., 2007, 28, 1761–1775 CrossRef CAS
- Q. Liu, S. E. Bottle and P. Sonar, Developments of diketopyrrolopyrrole-dye-based organic semiconductors for a wide range of applications in electronics, Adv. Mater., 2020, 32, 1903882 CrossRef CAS PubMed
- H. Bürckstümmer, A. Weissenstein, D. Bialas and F. Würthner, Synthesis and characterization of optical and redox properties of bithiophene-functionalized diketopyrrolopyrrole chromophores, J. Org. Chem., 2011, 76, 2426–2432 CrossRef PubMed
- M. Grzybowski and D. T. Gryko, Diketopyrrolopyrroles: Synthesis, reactivity, and optical properties, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS
- C. B. Nielsen, M. Turbiez and I. McCulloch, Recent advances in the development of semiconducting DPP-containing polymers for transistor applications, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed
- L. Shen, L. Jiato, L. Heyuan, M. Qingguo and L. Xiyou, Evaluation of fused aromatic-substituted diketopyrrolopyrrole derivatives for singlet fission sensitizers, J. Phys. Chem. A, 2020, 124, 5331–5340 CrossRef CAS PubMed
- C. E. Miller, M. R. Wasielewski and G. C. Schatz, Modeling singlet fission in rylene and diketopyrrolopyrrole derivatives: the role of the charge transfer state in superexchange and excimer formation, J. Phys. Chem. C, 2017, 121, 10345–10350 CrossRef CAS
- I. Papadopoulos, M. J. Álvaro-Martins, D. Molina, P. M. McCosker, P. A. Keller, T. Clark, Á. Sastre-Santos and D. M. Guldi, Solvent dependent singlet fission in diketopyrrolopyrrole dimers – a mediating charge transfer versus a trapping symmetry breaking charge separation, Adv. Energy Mater., 2020, 10, 2001496 CrossRef CAS
- D. Rais, P. Toman, J. Pfleger, U. Acharya, Y. R. Panthi, M. Menšík, A. Zhigunov, M. A. Thottappali, M. Vala, A. Marková, S. Stříteský, M. Weiter, M. Cigánek, J. Krajčovič, K. Pauk, A. Imramovský, A. Zaykov and J. Michl, Singlet fission in thin solid films of Bis(thienyl)diketopyrrolopyrroles, ChemPlusChem, 2020, 85, 2689–2703 CrossRef CAS PubMed
- A. M. Levine, G. He, G. Bu, P. Ramos, F. Wu, A. Soliman, J. Serrano, D. Pietraru, C. Chan, J. D. Batteas, M. Kowalczyk, S. J. Jang, B. L. Nannenga, M. Y. Sfeir, E. H. R. Tsai and A. B. Braunschweig, Efficient free triplet generation follows singlet fission in diketopyrrolopyrrole polymorphs with goldilocks coupling, J. Phys. Chem. C, 2021, 125, 12207–12213 CrossRef CAS PubMed
- P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y. L. Wu, T. J. Marks and M. R. Wasielewski, Effects of crystal morphology on singlet exciton fission in diketopyrrolopyrrole thin films, J. Phys. Chem. B, 2016, 120, 1357–1366 CrossRef CAS PubMed
- C. M. Mauck, Y. J. Bae, M. Chen, N. Powers-Riggs, Y.-L. Wu and M. R. Wasielewski, Charge-transfer character in a covalent diketopyrrolopyrrole dimer: implications for singlet fission, ChemPhotoChem, 2018, 2, 223–233 CrossRef CAS
- C. M. Mauck, P. E. Hartnett, Y. L. Wu, C. E. Miller, T. J. Marks and M. R. Wasielewski, Singlet fission within diketopyrrolopyrrole nanoparticles in water, Chem. Mater., 2017, 29, 6810–6817 CrossRef CAS
- S. J. Bradley, M. Chi, J. M. White, C. R. Hall, L. Goerigk, T. A. Smith and K. P. Ghiggino, The role of conformational heterogeneity in the excited state dynamics of linked diketopyrrolopyrrole dimers, Phys. Chem. Chem. Phys., 2021, 23, 9357–9364 RSC
- M. Chakali, H. Mandal, M. Venkatesan, B. Dyaga, V. J. Rao and P. R. Bangal, Charge separation and singlet fission in covalently linked diketopyrrolopyrrole derivatives and triphenylamine triad in solution, J. Photochem. Photobiol., A, 2021, 406, 113017 CrossRef CAS
- Y. Qu, J. Hua and H. Tian, Colorimetric and ratiometric red fluorescent chemosensor for fluoride ion based on diketopyrrolopyrrole, Org. Lett., 2010, 12, 3320–3323 CrossRef CAS PubMed
- Y. N. Zheng, W. Bin Liang, C. Y. Xiong, Y. L. Yuan, Y. Q. Chai and R. Yuan, Self-enhanced ultrasensitive photoelectrochemical biosensor based on nanocapsule packaging both donor–acceptor-type photoactive material and its sensitizer, Anal. Chem., 2016, 88, 8698–8705 CrossRef CAS PubMed
- M. A. Auwalu and S. Cheng, Diketopyrrolopyrrole Fluorescent Probes, Photophysical and Biological Applications, Chemosensors, 2021, 9, 44 CrossRef CAS
- J. D. Yuen and F. Wudl, Strong acceptors in donor–acceptor polymers for high performance thin film transistors, Energy Environ. Sci., 2013, 6, 392–406 RSC
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Diketopyrrolopyrrole polymers for organic solar cells, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS PubMed
- F. S. Kim, X. Guo, M. D. Watson and S. A. Jenekhe, High-mobility ambipolar transistors and high-cain inverters from a donor–acceptor copolymer semiconductor, Adv. Mater., 2010, 22, 478–482 CrossRef CAS PubMed
- S. Otep, Y. Lin, H. Matsumoto, T. Mori, K. Wei and T. Michinobu, Diketopyrrolopyrrole – thiophene – methoxythiophene based random copolymers for organic field effect transistor applications, Org. Electron., 2020, 87, 105986 CrossRef CAS
- C. Zhao, Y. Guo, Y. Zhang, N. Yan, S. You and W. Li, Diketopyrrolopyrrole-based conjugated materials for non-fullerene organic solar cells, J. Mater. Chem. A, 2019, 7, 10174–10199 RSC
- K. Gao, S. B. Jo, X. Shi, L. Nian, M. Zhang, Y. Kan, F. Lin, B. Kan, B. Xu, Q. Rong, L. Shui, F. Liu, X. Peng, G. Zhou, Y. Cao and A. K. Y. Jen, Over 12% efficiency nonfullerene all-small-molecule organic solar cells with sequentially evolved multilength scale morphologies, Adv. Mater., 2019, 31, 1807842 CrossRef PubMed
- M. Privado, P. Malhotra, P. de la Cruz, R. Singhal, J. Cerdá, J. Aragó, E. Ortí, G. D. Sharma and F. Langa, Ternary organic solar cell with a near-infrared absorbing selenophene–diketopyrrolopyrrole-based nonfullerene acceptor and an efficiency above 10%, Sol. RRL, 2020, 4, 1900471 CrossRef CAS
- X. Jiang, L. Wang, H. Tang, D. Cao and W. Chen, Diketopyrrolopyrrole: an emerging phototherapy agent in fighting, Dyes Pigm., 2020, 181, 108599 CrossRef CAS
- Z. Fei, L. Chen, Y. Han, E. Gann, A. S. R. Chesman, C. R. McNeill, T. D. Anthopoulos, M. Heeney and A. Pietrangelo, Alternating 5,5-dimethylcyclopentadiene and diketopyrrolopyrrole copolymer prepared at room temperature for high performance organic thin-film transistors, J. Am. Chem. Soc., 2017, 139, 8094–8097 CrossRef CAS PubMed
- Y. Li, P. Sonar, L. Murphy and W. Hong, High mobility diketopyrrolopyrrole (DPP)-based organic semiconductor materials for organic thin film transistors and photovoltaics, Energy Environ. Sci., 2013, 6, 1684–1710 RSC
- J. Podlesný, L. Dokládalová, O. Pytela, A. Urbanec, M. Klikar, N. Almonasy, T. Mikysek, J. Jedryka, I. V. Kityk and F. Bureš, Structure-property relationships and third-order nonlinearities in diketopyrrolopyrrole based D–π–A–π–D molecules, Beilstein J. Org. Chem., 2017, 13, 2374–2384 CrossRef PubMed
- J. Y. Back, H. Yu, I. Song, I. Kang, H. Ahn, T. J. Shin, S. K. Kwon, J. H. Oh and Y. H. Kim, Investigation of structure-property relationships in diketopyrrolopyrrole-based polymer semiconductors via side-chain engineering, Chem. Mater., 2015, 27, 1732–1739 CrossRef CAS
- S. Loser, S. J. Lou, B. M. Savoie, C. J. Bruns, A. Timalsina, M. J. Leonardi, J. Smith, T. Harschneck, R. Turrisi, N. Zhou, C. L. Stern, A. A. Sarjeant, A. Facchetti, R. P. H. Chang, S. I. Stupp, M. A. Ratner, L. X. Chen and T. J. Marks, Systematic evaluation of structure-property relationships in heteroacene-diketopyrrolopyrrole molecular donors for organic solar cells, J. Mater. Chem. A, 2017, 5, 9217–9232 RSC
- E. Ripaud, D. Demeter, T. Rousseau, E. Boucard-Cétol, M. Allain, R. Po, P. Leriche and J. Roncali, Structure-properties relationships in conjugated molecules based on diketopyrrolopyrrole for organic photovoltaics, Dyes Pigm., 2012, 95, 126–133 CrossRef CAS
- C. Kim, J. Liu, J. Lin, A. B. Tamayo, B. Walker, G. Wu and T. Q. Nguyen, Influence of structural variation on the solid-state properties of diketopyrrolopyrrole-based oligophenylenethiophenes: Single-crystal structures, thermal properties, optical bandgaps, energy levels, film morphology, and hole mobility, Chem. Mater., 2012, 24, 1699–1709 CrossRef CAS
- J. Liu, B. Walker, A. Tamayo, Y. Zhang and T. Q. Nguyen, Effects of heteroatom substitutions on the crystal structure, film formation, and optoelectronic properties of diketopyrrolopyrrole-based materials, Adv. Funct. Mater., 2013, 23, 47–56 CrossRef CAS
- M. Shahid, R. S. Ashraf, Z. Huang, A. J. Kronemeijer, T. McCarthy-Ward, I. McCulloch, J. R. Durrant, H. Sirringhaus and M. Heeney, Photovoltaic and field effect transistor performance of selenophene and thiophene diketopyrrolopyrrole co-polymers with dithienothiophene, J. Mater. Chem., 2012, 22, 12817–12823 RSC
- V. S. Gevaerts, E. M. Herzig, M. Kirkus, K. H. Hendriks, M. M. Wienk, J. Perlich, P. Müller-Buschbaum and R. A. J. Janssen, Influence of the position of the side chain on crystallization and solar cell performance of DPP-based small molecules, Chem. Mater., 2014, 26, 916–926 CrossRef CAS
- T. Mukhopadhyay, A. J. Musser, B. Puttaraju, J. Dhar, R. H. Friend and S. Patil, Is the chemical strategy for imbuing ‘polyene’ character in diketopyrrolopyrrole-based chromophores sufficient for singlet fission?, J. Phys. Chem. Lett., 2017, 8, 984–991 CrossRef CAS PubMed
- S. Wood, J. Wade, M. Shahid, E. Collado-Fregoso, D. D. C. Bradley, J. R. Durrant, M. Heeney and J. S. Kim, Natures of optical absorption transitions and excitation energy dependent photostability of diketopyrrolopyrrole (DPP)-based photovoltaic copolymers, Energy Environ. Sci., 2015, 8, 3222–3232 RSC
- M. Kirkus, L. Wang, S. Mothy, D. Beljonne, J. Cornil, R. A. J. Janssen and S. C. J. Meskers, Optical properties of oligothiophene substituted diketopyrrolopyrrole derivatives in the solid phase: Joint J- and H-type aggregation, J. Phys. Chem. A, 2012, 116, 7927–7936 CrossRef CAS PubMed
- T. Kim, W. Kim, O. Vakuliuk, D. T. Gryko and D. Kim, Two-step charge separation passing through thepartial charge-transfer state
in a molecular dyad, J. Am. Chem. Soc., 2020, 142, 1564–1573 CrossRef CAS PubMed
- Z. Chen, M. J. Lee, R. Shahid Ashraf, Y. Gu, S. Albert-Seifried, M. Meedom Nielsen, B. Schroeder, T. D. Anthopoulos, M. Heeney, I. McCulloch and H. Sirringhaus, High-performance ambipolar diketopyrrolopyrrole-thieno[3,2-b]thiophene copolymer field-effect transistors with balanced hole and electron mobilities, Adv. Mater., 2012, 24, 647–652 CrossRef CAS PubMed
- M. T. Casal, J. M. Toldo, F. Plasser and M. Barbatti, Using diketopyrrolopyrroles to stabilize double excitation and control internal conversion, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-2z60p
- L. R. Khundkar and A. H. Zewail, Ultrafast molecular reaction dynamics in real-time: progress over a decade, Annu. Rev. Phys. Chem., 1990, 41, 15–60 CrossRef CAS
- M. J. Rosker, F. W. Wise and C. L. Tang, Femtosecond relaxation dynamics of large molecules, Phys. Rev. Lett., 1986, 57, 321–324 CrossRef CAS PubMed
- N. F. Scherer, L. D. Ziegler and G. R. Fleming, Heterodyne-detected time-domain measurement of I2 predissociation and vibrational dynamics in solution, J. Chem. Phys., 1992, 96, 5544–5547 CrossRef CAS
- S. Rafiq, B. Fu, B. Kudisch and G. D. Scholes, Interplay of vibrational wavepackets during an ultrafast electron transfer reaction, Nat. Chem., 2021, 13, 70–76 CrossRef CAS PubMed
- T. M. Nila Mohan, C. H. Leslie, S. Sil, J. B. Rose, R. W. Tilluck and W. F. Beck, Broadband 2DES detection of vibrational coherence in the Sx state of canthaxanthin, J. Phys. Chem., 2021, 155, 035103 CrossRef PubMed
- S. Ruetzel, M. Diekmann, P. Nuernberger, C. Walter, B. Engels and T. Brixner, Multidimensional spectroscopy of photoreactivity, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4764–4769 CrossRef CAS PubMed
- R. Monni, G. Capano, G. Auböck, H. B. Gray, A. Vlcek, I. Tavernelli and M. Chergui, Vibrational coherence transfer in the ultrafast intersystem crossing of a diplatinum complex in solution, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6396–E6403 CrossRef CAS PubMed
- C. Lee, K. Seo, M. Kim and T. Joo, Coherent internal conversion from high lying electronic states to S1 in boron-dipyrromethene derivatives, Phys. Chem. Chem. Phys., 2021, 23, 25200–25209 RSC
- M. N. R. Ashfold, A. L. Devine, R. N. Dixon, G. A. King, M. G. D. Nix and T. A. A. Oliver, Exploring nuclear motion through conical intersections in the uv photodissociation of azoles, phenols and related systems, Proc. Natl. Acad. Sci. U. S. A., 2011, 105, 12701–12706 CrossRef PubMed
-
W. Domcke, D. Yarkony and H. Köppel, Conical Intersections Electronic Structure, Dynamics & Spectroscopy, World Scientific, 2004 Search PubMed
- C. Schnedermann, A. M. Alvertis, T. Wende, S. Lukman, J. Feng, F. A. Y. N. Schröder, D. H. P. Turban, J. Wu, N. D. M. Hine, N. C. Greenham, A. W. Chin, A. Rao, P. Kukura and A. J. Musser, A molecular movie of ultrafast singlet fission, Nat. Commun., 2019, 10, 4207 CrossRef PubMed
- J. Brazard, L. A. Bizimana, T. Gellen, W. P. Carbery and D. B. Turner, Experimental detection of branching at a conical intersection in a highly fluorescent molecule, J. Phys. Chem. Lett., 2016, 7, 14–19 CrossRef CAS PubMed
- B. S. Rolczynski, J. M. Szarko, H. J. Son, Y. Liang, L. Yu and L. X. Chen, Ultrafast intramolecular exciton splitting dynamics in isolated low-band-gap polymers and their implications in photovoltaic materials design, J. Am. Chem. Soc., 2012, 134, 4142–4152 CrossRef CAS PubMed
- S. Cho, B. S. Rolczynski, T. Xu, L. Yu and L. X. Chen, Solution phase exciton diffusion dynamics of a charge-transfer copolymer PTB7 and a homopolymer P3HT, J. Phys. Chem. B, 2015, 119, 7447–7456 CrossRef CAS PubMed
- B. Carsten, J. M. Szarko, H. J. Son, W. Wang, L. Lu, F. He, B. S. Rolczynski, S. J. Lou, L. X. Chen and L. Yu, Examining the effect of the dipole moment on charge separation in donor–acceptor polymers for organic photovoltaic applications, J. Am. Chem. Soc., 2011, 133, 20468–20475 CrossRef CAS PubMed
- P. B. Miranda, D. Moses and A. J. Heeger, Ultrafast photogeneration of charged polarons in conjugated polymers, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 081201 CrossRef
- V. I. Arkhipov, E. V. Emelianova and H. Bassler, Hot exciton dissociation in conjugated polymers, Phys. Rev. Lett., 1999, 82, 1321–1324 CrossRef CAS
- G. Grancini, M. Maiuri, D. Fazzi, A. Petrozza, H. J. Egelhaaf, D. Brida, G. Cerullo and G. Lanzani, Hot exciton dissociation in polymer solar cells, Nat. Mater., 2013, 12, 29–33 CrossRef CAS PubMed
- S. D. Dimitrov, A. A. Bakulin, C. B. Nielsen, B. C. Schroeder, J. Du, H. Bronstein, I. McCulloch, R. H. Friend and J. R. Durrant, On the energetic dependence of charge separation in low-band-gap polymer/fullerene blends, J. Am. Chem. Soc., 2012, 134, 18189–18192 CrossRef CAS PubMed
- Y. Patil, R. Misra, A. Sharma and G. D. Sharma, D–A–D–π–D–A–D type diketopyrrolopyrrole based small molecule electron donors for bulk heterojunction organic solar cells, Phys. Chem. Chem. Phys., 2016, 18, 16950–16957 RSC
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03238d |
| This journal is © the Owner Societies 2022 |