
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iodine speciation in deep eutectic solvents†
Jennifer M.
Hartley
 *a,
Sean
Scott
a,
Zarfishan
Dilruba
a,
Anthony J.
Lucio
a,
Philip J.
Bird
b,
Robert C.
Harris
b,
Gawen R. T.
Jenkin
b and
Andrew P.
Abbott
a
*a,
Sean
Scott
a,
Zarfishan
Dilruba
a,
Anthony J.
Lucio
a,
Philip J.
Bird
b,
Robert C.
Harris
b,
Gawen R. T.
Jenkin
b and
Andrew P.
Abbott
a
aSchool of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: jmh84@le.ac.uk
bSchool of Geography, Geology and the Environment, University of Leicester, Leicester, LE1 7RH, UK
First published on 27th September 2022
Abstract
Iodine has been shown to act as a good electrocatalyst for metal digestion in deep eutectic solvents (DESs) but little is known about its speciation or reactivity in these high chloride containing media. Extended X-ray absorption fine structure (EXAFS) spectroscopy measurements were made at the iodine K-edge in a range of DESs with different glycolic or acidic hydrogen bond donors (HBDs), along with examining the effect of iodine concentration between 0.01 and 0.5 mol dm−3. Three groups of speciation were detected: mixed I2Cl−/I3− (glycol and lactic acid systems), mixed I3−/I2 (oxalic acid and urea systems), and singular I3− (levulinic acid system). UV-vis spectroscopy was used to confirm the speciation. Electrochemistry showed that iodine redox behaviour was unaffected by the changing speciation. Leaching data showed that metal oxidation was related not only to changing iodine speciation, but also the reactivity and coordination ability of the HBD.
1. Introduction
Deep eutectic solvents (DESs) are mixtures of Lewis or Brønsted acids and bases that produce liquids, which can have properties that are similar to ionic liquids. They have been applied to a wide variety of applications, including electrodeposition, catalysis, lubrication, and natural product extraction.1 One critical area of interest is their use for metal processing, in particular digestion and recovery of metals from ores and electronic waste streams. In 2019 54 Mt of waste electronic equipment was produced globally, with this amount projected to increase annually up to 75 Mt by 2030.2 The predominant methods for processing this waste material involve pyrometallurgy, due to the flexibility of feedstocks and the high throughput capacity brought about by the elevated temperatures, enabling an enhancement in reaction rates. Recent developments into pyrometallurgy have focused on reducing environmental impact, such as decreasing toxic gas emissions through off-gas treatments. In their lithium ion battery processing method, Umicore have eliminated flue dust and reduced the overall gas evolution.3 However, several limitations still remain, namely the large energy requirements for pyrolysis, the low purity of resulting materials (i.e. alloy formation) and limited recovery of key metals, with notable amounts being lost in slag phases.4,5 In many cases, due to the latter two limitations, a combined pyrometallurgy/hydrometallurgy methodology is required to optimise metal extraction at a sufficient purity. While the incorporation of hydrometallurgy steps enables selective recovery of these remaining metals at an improved purity, hydrometallurgical processing comes with numerous issues of its own. These include long leaching times and the usage of harmful solvents, which can lead to a low processing capacity and safety issues respectively.5 However, it is the limitation of environmental impacts that is the focal point in metallurgy research, as both of these processing routes can lead to potentially harmful emissions to the atmosphere and waterways.DESs have significant potential as a low impact alternative to traditional processing methods and can be used alone or in combination with an (electro)chemical oxidising agent. For example, in the field of mineral processing, materials such as chalcopyrite,6 pyrites,7 scheelite,8 bastnäsite,9 goethite sludge,10 and flue dusts11 have been processed to extract the valuable metals. With regards to E-waste processing studies, the recycling of NdFeB permanent magnets has been investigated in DESs made from lactic acid with either guanidine hydrochloride12 or choline chloride13 as the quaternary ammonium salt, where the target species were recovered either through oxalate precipitation, or through solvent–solvent extraction. More recently, DESs have been used to process lithium ion battery cathode materials such as lithium cobalt, nickel and manganese oxides.14–17 In each of these cases, the DES was used without additional oxidising agents. DESs are of interest because the high chloride concentration generally dominates metal speciation.18 Metals are dissolved into DESs as chlorometallate species that have very different redox properties compared to the same metals in aqueous solutions, which enables normally electropositive metals to be relatively easily oxidised.19 Another advantage of using DESs is that the components are often bulk commodity chemicals such as glycols, simple carboxylic acids, or urea, hence no extra Reach regulations are required. This means that it is easier to employ them in a bulk industrial setting than it would be for traditional ionic liquids.
Iodine is a very efficient oxidising agent which has been used for metal and mineral digestion, as it is strong enough to dissolve gold in both aqueous20–22 and DES systems19,23–26 under mild conditions, along with other metals found in printed circuit boards27 and minerals.28–30 Phosphonium halide and trihalide ionic liquids have also been successfully used to digest metals.31–33 Iodine has a high solubility in ionic media, despite normally only being soluble in non-polar solvents if a complex anion is formed with another halide species, e.g. [I3]−, [ICl2]−, [I2Cl]−, [I2Br]−, [IBr2]−, etc.34–36 In aqueous solutions, the most stable iodine complex is the triiodide species,37 while in organic solvents iodine is present either as “free” molecular iodine, e.g. in chloroform, or as a ligand-stabilised complex, e.g. in ethanol or diethyl ether.38 In imidazolium-based ionic liquids, iodine can interact with all anions and the 1H NMR spectra change as the iodine content increases.39 Electrocatalysis has been proposed as a generic and efficient method for metal dissolution and recovery, and has already been demonstrated for the digestion and separation of gallium and arsenic from semiconductors using iodine in a choline chloride: ethylene glycol DES.24 Electrocatalysis also has promise for gold processing as it is extremely fast, atom efficient, and negates the use of reagents such as cyanide and mercury.25
Voltammetry shows the presence of two redox processes, which have previously been assigned to the I2/2I− (or 3I2/2I3−) and [I3]−/3I− redox couples,24 similarly to other iodine/iodide-containing ionic liquid systems.40–42 The redox potentials of these redox couples are also observed to vary in DESs formed with different hydrogen bond donors (HBDs).43 This could be due to a change in the speciation of the dissolved iodine complex e.g. complexation of iodine with chloride, or through solvation interactions with the HBD. It is therefore important to understand how iodine changes speciation with DES composition, as this will influence process design in both terms of the selected DES and any considerations that must be made for the DES physical properties, such as viscosity or conductivity. Additionally, it must be considered that iodine may be able to oxidise the DES components.
In this work, we aim to determine the speciation of iodine in seven DESs based on choline chloride, and identify which iodine species is the most reactive. Additionally, we identify whether iodine speciation in DESs is affected by iodine concentration, as this may alter chemical reactivity, redox properties and electrode potentials during a digestion reaction. Iodine speciation could change by aggregation at high concentrations to form [I5]− or higher order polyiodides, or through ligand exchange for solvent ligands at low concentrations. Here we undertake EXAFS measurements on solutions of 0.5, 0.1, and 0.01 mol dm−3 iodine which cover the reported working concentration range used for oxidation of metals and minerals in DESs,19,23–25 along with a concentration that is sufficiently dilute to permit UV-vis spectra to be measured. Our results suggest that multiple different iodine species are present in solution, depending on the choice of HBD used.
2. Experimental
2.1 Sample preparation
All DESs were made using the methods described in previous publications.44,45 Mixtures were made with 1 molar equivalent of choline chloride (ChCl) (Scientific Laboratory Supplies, 99%) and 2 molar equivalents of ethylene glycol (EG) (98%, Sigma Aldrich), 1,2-propanediol (12Prop) (Sigma Aldrich, 99%), urea (U) (Merck, synthesis grade), glycerol (Gly) (Fischer Scientific, ≥95%), lactic acid (LacA) (Fischer Scientific, ≥88%), levulinic acid (LevA) (Sigma Aldrich, 98%), or 1 molar equivalent of oxalic acid dihydrate (OxA) (Alfa Aesar, 98%), hereafter referred to as EG:ChCl, 12Prop:ChCl, U:ChCl, Gly:ChCl, LacA:ChCl, LevA:ChCl, and OxA:ChCl, respectively. Once the stock DES solutions were formed, 10 mL solutions of different iodine (Fischer Scientific, ≥95%) concentrations were formulated at concentrations of 0.5, 0.1, and 0.01 mol dm−3.Karl Fischer titration was used to identify the percentage of water content within the stock DESs. The experiments were carried out using a V10S Volumetric KF titrator (Mettler Toledo). Hydranal Solvent E (Honeywell) was used as the solvent and Hydranal Composite 5K (Honeywell) as the titrant. The system was calibrated before use, using an Aquastar water standard of 5 mg mL−1 (≈0.5%) (Supelco). The amount of titrant added in mg was used to calculate the water content, which was then normalised to a percentage. The water content of each DES was measured in triplicate, with an average water content and standard deviation quoted in Table S1 (ESI†).
2.2 Speciation
The speciation of iodine in the different DESs was determined via extended X-ray absorption fine structure (EXAFS), measured at B18 of the Diamond Light Source synchrotron. All measurements were carried out at the iodine K-edge, nominally 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 169 eV. Transmission data were measured using ionisation chambers, whilst fluorescence data were measured with a nine-element Ge solid-state detector operating in total fluorescence yield mode. The monochromator was a Si(311) double crystal. Potassium iodide ground into a pellet with cellulose was used as a reference sample for amplitude calibration. The 0.01, and 0.1 mol dm−3 samples were contained in plastic cuvettes with a path length of 1 cm, while the 0.5 mol dm−3 samples were contained in plastic cuvettes with a path length of 0.5 cm due to their high absorption. These cuvettes were mounted in the beam, aligned at approximately 55° with respect to the X-ray beam, with the fluorescence detector placed perpendicular to the X-ray beam in order to minimise any elastic scattering signals. Three to nine spectra were recorded for the 0.1 and 0.5 mol dm−3 samples and 27 spectra were recorded for the 0.01 mol dm−3 samples in quick EXAFS mode. These were then averaged, calibrated and background subtracted with the program Athena.46 The EXAFS spectra were fitted using the program Artemis to determine type and number of coordinating atoms, and calculate the interatomic distances and their root-mean-square variations (σ2). Quoted uncertainties on fitted parameters are equal to two standard deviations. The fitting range was ca. 3.9 to 12.0 Å−1 for EG:ChCl, 12Prop:ChCl, Gly:ChCl, LacA:ChCl, LevA:ChCl, and ca. 3.9 to 15.4 Å−1 for OxA:ChCl and U:ChCl. Note that a shorter k-range of ca. 3.9 to 9.0 Å−1 was required for the 0.01 mol dm−3 OxA:ChCl sample due to signal decay. Series fits were made for each DES using all concentrations where possible. Coordination number, reference energy (E0) and interatomic distances were always allowed to vary. Initial fits were made with σ2 for each element remaining the same, but allowed to vary during secondary fits to test if the additional parameters were necessary to improve the fit index significantly. An example fitting model is presented in the ESI,† Fig. S1, along with an explanation of the selection of the number and type of fitting parameters. Fourier transforms are presented as phase-corrected using the I–I scattering path.
169 eV. Transmission data were measured using ionisation chambers, whilst fluorescence data were measured with a nine-element Ge solid-state detector operating in total fluorescence yield mode. The monochromator was a Si(311) double crystal. Potassium iodide ground into a pellet with cellulose was used as a reference sample for amplitude calibration. The 0.01, and 0.1 mol dm−3 samples were contained in plastic cuvettes with a path length of 1 cm, while the 0.5 mol dm−3 samples were contained in plastic cuvettes with a path length of 0.5 cm due to their high absorption. These cuvettes were mounted in the beam, aligned at approximately 55° with respect to the X-ray beam, with the fluorescence detector placed perpendicular to the X-ray beam in order to minimise any elastic scattering signals. Three to nine spectra were recorded for the 0.1 and 0.5 mol dm−3 samples and 27 spectra were recorded for the 0.01 mol dm−3 samples in quick EXAFS mode. These were then averaged, calibrated and background subtracted with the program Athena.46 The EXAFS spectra were fitted using the program Artemis to determine type and number of coordinating atoms, and calculate the interatomic distances and their root-mean-square variations (σ2). Quoted uncertainties on fitted parameters are equal to two standard deviations. The fitting range was ca. 3.9 to 12.0 Å−1 for EG:ChCl, 12Prop:ChCl, Gly:ChCl, LacA:ChCl, LevA:ChCl, and ca. 3.9 to 15.4 Å−1 for OxA:ChCl and U:ChCl. Note that a shorter k-range of ca. 3.9 to 9.0 Å−1 was required for the 0.01 mol dm−3 OxA:ChCl sample due to signal decay. Series fits were made for each DES using all concentrations where possible. Coordination number, reference energy (E0) and interatomic distances were always allowed to vary. Initial fits were made with σ2 for each element remaining the same, but allowed to vary during secondary fits to test if the additional parameters were necessary to improve the fit index significantly. An example fitting model is presented in the ESI,† Fig. S1, along with an explanation of the selection of the number and type of fitting parameters. Fourier transforms are presented as phase-corrected using the I–I scattering path.
All UV-Vis spectra were recorded using an UV5Bio (Mettler Toledo) UV-Vis spectrometer between 190 and 1100 nm. These spectra were obtained using a 0.1 mm glass slide cuvette and the 0.01 mol dm−3 samples, with some diluted to 0.005 mol dm−3 for optimised peak resolution. Data presented in this study was normalised to the 362 nm peak and reference solutions of iodine were made to provide species spectra: aqueous potassium iodide (KI) for [I3]−, aqueous choline chloride (ChCl) and aqueous lithium chloride (LiCl) for [I2Cl]−, and a solution of iodine in ethanol was made for the I2 spectrum.
2.3 Electrochemical measurements
Cyclic voltammetry (CV) measurements were carried out using an IVIUMnSTAT multichannel potentiostat/galvanostat, together with the corresponding Ivium software. A 3-electrode set-up was used, consisting of a 1 mm diameter Pt-disc working electrode, a Pt-flag counter electrode, and a 0.1 mol dm−3 AgCl/Ag in EG:ChCl reference electrode. The reference electrode was calibrated to the [Fe(CN)6]3−/4− redox couple in each DES to permit comparability.47 All experiments were carried out under ambient conditions. Before each experiment, the working electrode was polished with 0.3 and 0.05 μm alumina slurry, rinsed with deionised water, and air dried. All CVs were recorded at a scan rate of 5 mV s−1. Because the two iodine redox couples were so close in energy, it was necessary to start the measurements at an oxidising potential and take the third scan to ensure the systems can be compared at equilibrium.2.4 Metal dissolution experiments
Leaching experiments were carried out for copper and nickel in solutions of 0.1 mol dm−3 iodine in EG:ChCl, Gly:ChCl, OxA:ChCl, and U:ChCl. The DESs were placed in sample tubes held in an aluminium block on a hotplate to maintain a constant temperature of 50 ± 2 °C. The copper and nickel plates were selected to have more mass than could theoretically be dissolved by the moles of iodine present. They were pre-treated by degreasing in acetone and rinsing in deionised water, before being dried in air. After dissolution, the metal plates were removed from the DES and rinsed in a dilute aqueous solution of sodium thiosulfate (99%, Avocado) or potassium iodide (99.5%, Fisher Scientific) to remove any remaining iodine. They were further washed with deionised water and dried in air. The metal pieces were weighed before and after dissolution to determine mass loss.3. Results and discussion
3.1 Effect of hydrogen bond donor on iodine speciation
EXAFS provides information on the average local coordination environment of the absorbing atoms. As all iodine atoms in the systems investigated here can act as absorbing atoms, an average predicted coordination can be calculated by dividing the number of scattering paths by the number of iodine atoms in each species. For many of the potential iodine species present, the average coordination number obtained from fitting the data may not be a whole number. Actual speciation must therefore be inferred from these results. Table 1 shows the most likely species present in ionic media with an average expected coordination, and the literature iodine–ligand bond lengths used to develop the fitting models used in the present work. It must be noted that iodine halide compounds can form both symmetric and asymmetric species, depending on their local environments (i.e. physical state, presence of hydrogen bonding, presence of cations). Datta et al. determined the bond lengths of the symmetric ion to be ca. 2.83 Å, whereas the asymmetric ion has average bond lengths of ca. 2.97 Å.48 If any hydrogen iodide is formed in these DESs, there will be no EXAFS signal distinguishable from the smooth background function for a monatomic species.| Name | Species | Average coordination | Bond length/Å | |
|---|---|---|---|---|
| Iodine34,49 | I2 | 2/2 = 1 | I*–I | 2.68 (s) |
| 2.71 (l) | ||||
| Triiodide48 | [I3]− | 4/3 = 1.3 | I*–I | ca. 2.92 (s) |
| Pentaiodide34 | [I5]− | 8/5 = 1.6 | I*–I | ca. 2.94 (s) |
| Iodine monochloride36 | ICl | 1/1 = 1 | I*–Cl | 2.32 (g) |
| Iodine dichloride36 | [ICl2]− | 2/1 = 2 | I*–Cl | 2.55 (s) |
| Diiodine monochloride35 | [I2Cl]− | 1/2 = 0.5 | I*–Cl | 2.60 |
| 2/2 = 1 | I*–I | 2.95 (ab initio) | ||
Fig. 1 shows that in general, three types of speciation environment were observed in the solutions of 0.1 mol dm−3 iodine in the DESs investigated here. In the DESs formed from the glycols or LacA, two peaks were present in the Fourier transform (FT), which could be fitted to an average of 0.1 to 0.3 chloride atoms (peak a), and 1.5 iodine atoms (peaks b′ and b′′, which are part of the same scattering contribution). This chloride coordination is indicative of the presence of [I2Cl]−. However, average iodine–iodine coordination is too high for that species alone to be present. Instead, a mixture of [I2Cl]− and [I3]− is predicted, with varying ratios in each of the DESs investigated here. These ratios would be roughly 3[I2Cl]−:2[I3]− for EG:ChCl, 1:1 for 12Prop:ChCl, 1:2 for Gly: ChCl, and 1:3 for LacA:ChCl. The proposed species, based on EXAFS data fitting, are shown in Table 2. This hypothesis of two different iodine coordination environments is supported by the high mean square relative displacement (σ2) values for the I*–I scattering paths. The iodine–iodine path lengths determined here are at least 0.1 Å shorter than would be predicted from crystalline literature data, due to the local environment being an amorphous liquid. This variation in the amount of [I2Cl]− species could be related to the relative chloride activity in the different DESs, resulting from interaction of chloride with the HBD. For example, glycerol has a stronger interaction with the chloride than ethylene glycol or 1,2-propanediol.50 This will result in a lower chloride activity and hence shift the equilibrium reaction with iodine. A lower chloride activity is also likely to result in a lower overall solubility of iodine.
 | ||
| Fig. 1 Fourier transforms of the EXAFS spectra for solutions of 0.1 mol dm−3 iodine in a range of DESs formed from ChCl with different HBDs. Measured data are shown as circles, while data fits are the lines. Spectra are offset for clarity. The I–I scattering path was used for phase correction. | ||
| Iodine conc./mM | Coordinating atom/group | Number of atoms, N | Distance from I, r (Å) | Debye–Waller factor, a (Å2) | Fit index | Proposed species |
|---|---|---|---|---|---|---|
| a Fluorescence data. | ||||||
| EG:ChCl | ||||||
| 500 | Cl | 0.30(7) | 2.552(8) | 0.005(2) | 0.0054 | 3[I2Cl]−:2[I3]− |
| I | 1.3(1) | 2.793(4) | 0.0104(8) | |||
| 100 | Cl | 0.31(7) | 2.550(6) | 0.005(2) | 3[I2Cl]−:2[I3]− | |
| I | 1.3(1) | 2.800(3) | 0.0104(8) | |||
| 10 | Cl | 0.3(1) | 2.56(3) | 0.005(2) | 3[I2Cl]−:2[I3]− | |
| I | 1.1(2) | 2.80(2) | 0.0104(8) | |||
| 12Prop:ChCl | ||||||
| 500 | Cl | 0.25(6) | 2.554(8) | 0.003(2) | 0.0060 | 1[I2Cl]−:1[I3]− |
| I | 1.4(1) | 2.798(4) | 0.0112(9) | |||
| 100 | Cl | 0.23(5) | 2.557(7) | 0.003(2) | 1[I2Cl]−:1[I3]− | |
| I | 1.4(1) | 2.812(4) | 0.0116(9) | |||
| 10 | Cl | 0.15(9) | 2.59(4) | 0.003(2) | 1[I2Cl]−:2[I3]− | |
| I | 1.6(5) | 2.84(2) | 0.014(3) | |||
| Gly:ChCl | ||||||
| 500 | Cl | 0.17(3) | 2.534(1) | 0.001(1) | 0.0056 | 1[I2Cl]−:2[I3]− |
| I | 1.3(1) | 2.769(5) | 0.0098(8) | |||
| 100 | Cl | 0.15(3) | 2.539(1) | 0.001(1) | 1[I2Cl]−:2[I3]− | |
| I | 1.4(1) | 2.779(5) | 0.0107(7) | |||
| 10 | Cl | 0.07(7) | 2.55(6) | 0.001(1) | 1[I2Cl]−:4[I3]− | |
| I | 1.5(5) | 2.82(3) | 0.014(4) | |||
| LacA:ChCl | ||||||
| 500 | Cl | 0.16(2) | 2.54(1) | 0.0009(7) | 0.0094 | 1[I2Cl]−:3[I3]− |
| I | 1.3(1) | 2.768(5) | 0.0095(7) | |||
| 100 | Cl | 0.13(2) | 2.540(8) | 0.0009(7) | 1[I2Cl]−:3[I3]− | |
| I | 1.46(8) | 2.783(4) | 0.0116(6) | |||
| 10a | Cl | 0.07(15) | 2.6(3) | 0.0009(7) | [I2Cl]− or I2? | |
| I | 1.0(7) | 2.890(8) | 0.012(4) | |||
| OxA:ChCl | ||||||
| 500 | I | 0.92(8) | 2.752(4) | 0.0053(5) | 0.0163 | I2 |
| 100 | I | 0.11(4) | 2.747(9) | 0.0010(8) | I2 and [I3]− | |
| I | 1.6(2) | 2.86(2) | 0.016(2) | |||
| 10 | I | 0.9(2) | 2.86(2) | 0.011(3) | 0.0468 | [I2Cl]− or I2? |
| U:ChCl | ||||||
| 500 | Cl | 0.11(3) | 2.55(2) | 0.001(1) | 0.0077 | 1[I2Cl]−:3[I3]− |
| I | 1.2(1) | 2.762(6) | 0.0093(8) | |||
| 100 | I | 1.1(1) | 2.800(9) | 0.0088(8) | 0.0238 | I2 and [I3]− |
| I | 0.5(1) | 2.96(2) | 0.007(1) | |||
| 10 | Bleached! | I− | ||||
| LevA:ChCl | ||||||
| 500 | I | 1.68(7) | 2.909(3) | 0.009(3) | 0.0052 | [I5]− |
| 100 | I | 1.23(7) | 2.911(4) | 0.0091(5) | 0.0089 | [I3]− |
| 10 | Bleached! | I− | ||||
In the OxA:ChCl and U:ChCl DESs, scattering path contributions b′, b′′, and c were observed in the FT. This is suggestive that a mixture of I–I species with different scattering path lengths are present, with any I–Cl coordination being only in trace amounts. This is not surprising as there is a strong hydrogen bonding interaction between the carboxylic acid and the chloride ion so the activity of free chloride will be very low. In these two systems, it was observed that not all the iodine dissolved, leaving an amount of a dense, fluid dark brown substance at the bottom of the solution. Therefore, the most likely iodine species present in these systems would be [I3]−, with a smaller amount of a suspended microemulsion of crystalline iodine, as indicated by the presence of a minor third peak at ca. 3.5 Å with a similar scattering path length to the interatomic distances found in solid iodine (3.56 Å).34 Finally, in LevA:ChCl, only a single peak was present, indicating the presence of a single iodine species with an average coordination of 1.2 iodine atoms. This species is most likely to be [I3]−, with no other species present.51 The presence of varying quantities of [I3]− in all of the solvents investigated here indicates that a part of the dissolved iodine must be being reduced to iodide, potentially via oxidation of a small amount of the solvent components. It is interesting that the acidic systems display a greater tendency towards higher triiodide content, and this can potentially be explained by the fact that some of the carboxylic acids used in DES preparation, such as citric, ascorbic, and oxalic acids, are known to act as reducing agents. In the case of the urea-based system, there is the possibility for ammonia (a strong reducing agent) to build up over time due to decomposition of the urea. However, no I–O or I–C coordination was observed (as would be identified by peaks fittable to ca. 1.8 or 2.2 Å, respectively),49 indicating that the iodine is not forming chemical bonds with any of the DES components in detectable quantities. This is important for long-term stability and safety of the solution with respect to the iodine species present, as iodinated organic compounds are not being generated. However, in the systems where triiodide is being generated, it remains unknown which part of the DES is reacting with iodine to produce iodine and further experiments would need to be carried out in order to determine the oxidation mechanisms of the solvent components, and also to determine if the process is limited by iodine concentration or if it is catalytic. This will be critical for processes where it is desirable to recover and reuse the DES, as it will inform the necessary solvent composition as much as solute/oxidising agent speciation. For example, if the HBD is the source of electrons for iodide production, the physical and chemical properties of the solvent could be compromised. If the quaternary ammonium salt is reacting, then a more stable alternative would need to be sought.
In summary, EXAFS spectroscopy has shown that there are at least three different coordination environment types present in the DESs investigated here. These include [I2Cl]−, [I3]− and potentially molecular iodine in the form of a microemulsion. This is mostly due to the relative activity of the chloride ions in the mixture and DESs with stronger hydrogen bonding moieties having lower relative chloride activities, resulting in a higher proportion of iodine existing in the I2 state. In a digestion process, the appropriate DES must be selected to obtain the correct iodine speciation for the desired task. For example, using EG:ChCl or 12Prop:ChCl will ensure the highest [I2Cl]− content, but the use of a DES with more strongly complexing ligands, such as OxA:ChCl, will enable modification of the speciation properties of any dissolved metal ions.
3.2 Effect of iodine concentration on speciation
Data fitting showed that iodine speciation in the glycol systems (EG:ChCl, 12Prop:ChCl, and Gly:ChCl) and in LacA:ChCl did not change significantly between 0.1 and 0.5 mol dm−3 (Table 2 and Fig. S2, ESI†). A slight contraction in the I*–I scattering path lengths for the 0.5 mol dm−3 iodine system is likely due to the closer proximity of the iodine species to each other. For the OxA:ChCl and U:ChCl systems, similar species were present at 0.1 mol dm−3 iodine. However, when the concentration was increased to 0.5 mol dm−3 data fitting of the OxA:ChCl spectra indicated the presence of a single species with no Cl-coordination. The fitted coordination number and path lengths indicated the presence of molecular iodine, similarly to that observed for iodine in ethanol.49 Given the poor solubility of iodine in OxA:ChCl compared to the other DESs, it is likely that the signal from molecular iodine is dominating over any of the expected trihalide complexes in solution. This is not surprising as oxalic acid is known to strongly coordinate the chloride anion, which has been observed to result in a very high surface tension.52 It could therefore be predicted that more iodine will exist in solution as I2 and this would result in iodine having a higher vapour pressure in these DESs. This is observed qualitatively by increased iodine staining of the sample tube lid in OxA:ChCl compared to the other DESs. The corresponding U:ChCl sample could be fitted to a mixture of I–Cl and I–I coordination, similarly to the glycol-based DESs. The [I2Cl]− to [I3]− ratio in the U:ChCl sample was roughly 1:3, i.e. comparable to either the Gly:ChCl or LacA:ChCl systems. The spectrum of LevA:ChCl with 0.5 mol dm−3 iodine could be fitted to an average coordination of ca. 1.7 iodine atoms, in contrast to ca. 1.2 iodine atoms in the 0.1 mol dm−3 solution. Interestingly, there was no change to the fitted values of σ2, and only the slight contraction of scattering path lengths that was observed for the other DESs with high iodine concentrations. This could indicate the presence of a higher order polyiodide complex, e.g. [I5]−.
A concentration dependence on speciation was observed when between the spectra of 0.1 and 0.01 mol dm−3 iodine, ranging from an increased average number of I–I scattering paths coupled with a substantially larger σ2 values, to a decreased average number of I–Cl scattering paths, or even solution bleaching and the suspected formation of “free” iodide. These observations could be related to an increased number of different iodine species, the presence of a higher order polyiodide, or most likely that the spectra are affected by a poorer signal-to-noise ratio within the fitting window. For the glycol-based systems, EG:ChCl appears unaffected by the decrease in concentration, whereas data fitting indicates that greater proportions of [I3]− are present in the 12Prop:ChCl and Gly:ChCl systems, despite the greater availability of chloride to coordinate per dissolved iodine molecule. Data fitting of the LacA:ChCl system shows a decrease in average I–I scattering paths from ca. 1.5 to 1.0, which could be due to the presence of increased [I2Cl]− content relative to [I3]−. This would however require an average of ca. 0.5 I–Cl scattering paths, which is not the case. The I–I scattering path length of 2.890(8) Å corresponds closer to the [I2Cl]− species35 than molecular iodine,49 hence an alternative spectroscopic technique must be used to confirm the data fit. Data fitting the 0.01 mol dm−3 iodine in OxA:ChCl spectra required a much shorter k-range than the other concentrations due to poor signal-to-noise ratio resulting in usable signal intensity dropping off quickly (see Fig. S2(f), ESI†). This meant it was not possible to easily determine if only a single or many scattering paths were present. Based on the average coordination number of 0.9(2) iodine atoms, it can be inferred that either [I2Cl]− or molecular iodine is present in solution. Similarly to the LacA:ChCl system, an I–I scattering path length of 2.86(2) Å would support the presence of [I2Cl]−, but inclusion of I–Cl scattering paths in the fitting model did not improve the fit index.
Of great importance was the observation that the 0.01 mol dm−3 solutions of iodine in U:ChCl and LevA:ChCl were seen to have bleached, either before, during, or after the first set of EXAFS measurements. As the edge step for these solutions was the same as for the other 0.01 mol dm−3 systems, this bleaching effect is not due to loss of iodine from the solution. Instead, it must be due to a reduction process that results in a colourless iodide product. In U:ChCl, this could be caused by reaction of triiodide with solvent decomposition products such as ammonia, which is a strong reducing agent. In LevA:ChCl, where no strong reducing agent is present, a reaction of iodine with the protons is possible to form e.g. hydrogen iodide. However, there is no immediately obvious corresponding bleaching effect in either LacA:ChCl (higher relative pH) or OxA:ChCl (lower relative pH). Another possibility is the acid-catalysed iodination reaction of levulinic acid with iodine, as has been shown for other keto-acids such as pyruvic acid,53 or ethyl levulinate.54 If an iodinated organic complex were present, a peak in the FT relating to I–C that could be fitted to ca. 2.1–2.2 Å should be present.49 This was not detected in the EXAFS of the 0.01 mol dm−3 solution. For the more concentrated solutions, these side reactions are also highly likely to be taking place. However, the bleaching effect will be masked by the intense colour of the other iodine species.
Overall, increasing concentration from 0.1 to 0.5 mol dm−3 generally had little effect on iodine speciation. However, at lower concentrations there appeared to a change in the [I2Cl]−:[I3]− ratio towards a higher proportion of [I3]−, and a bleaching effect was observed in the U:ChCl and LevA:ChCl systems at low iodine concentrations. This speciation behaviour is confirmed by the XANES spectra remaining the same for the higher iodine concentrations, and varying slightly upon dilution to 0.01 mol dm−3, with the main differences in local structure and oxidation states shown for the LacA:ChCl, LevA:ChCl, OxA:ChCl and U:ChCl systems (Fig. S3, ESI†). This means that when using iodine as an oxidising agent, a certain concentration of iodine must be maintained throughout any leaching process to ensure that the correct oxidising iodine species is present and that losses due to side reactions with the DES are minimised.
3.3 UV-vis spectroscopy
In addition to EXAFS, UV-vis spectroscopy can be used to characterise speciation. If molecular iodine is present, characteristic absorption maxima would be expected to appear at 450 to 510 nm, with an additional ligand–iodine charge transfer band at 230 to 250 nm, depending on the solvent polarity.38 If [I3]− is present, two absorption maxima should be present in the region of 292 and 362 nm relating to the σ → σ* and π → σ* transitions, respectively. Aqueous reference systems containing either ChCl or LiCl to complex with the iodine and form [I2Cl]− showed multiple maxima, indicating that a mixture of species are present, including both [I3]− and molecular iodine. This mixture of species arises from the poor stability of [I2Cl]− in water in comparison to [I3]− (stability constants of 1.66 and 725 at 25 °C, respectively).55 Critically, a maximum at ca. 250 nm was observed in both systems which can be linked to the presence of [I2Cl]−.In all the DESs investigated here (Fig. 2), a maximum at ca. 362 nm is present that can be linked to the presence of [I3]−, as was also indicated by EXAFS. Interestingly, there are two maxima at shorter wavelengths, at 262 and 290 nm, for all systems except LevA:ChCl. The longer wavelength transition can be assigned to the σ → σ* transition of the [I3]− complex, and based on the reference spectrum of iodine with either ChCl or LiCl in water, the shorter wavelength transition can be assigned to the [I2Cl]− complex, most likely also involving the σ → σ* transition. There is a red-shift of ca. 10 nm between the reference and DES systems due to a change in the solvation environment increasing the stability of the [I2Cl]− species. An absorption at ca. 420 nm is present in the systems containing the greatest proportions of [I2Cl]−, which can tentatively be assigned to the π → σ* charge transfer transitions. No molecular iodine is detected at these concentrations, therefore any indication from the EXAFS spectra of molecular or microemulsion iodine in the more concentrated samples must be due to iodine solubility effects. Critically, the ratios of the 262 and 290 nm maxima (Table S2, ESI†) in the UV-vis spectra corroborate the predicted speciation and composition ratio obtained from EXAFS spectroscopy (assuming that the molar extinction coefficients for the two species are similar). There is minimal solvatochromic shift between the different DESs (<5 nm), indicating that the reactivity of the solvation sheath is similar in each DES. This means that the amount and ratio of the solution species is changing but the absolute reactivity of each species is not. Therefore, the oxidation ability of iodine in DESs could be driven by the relative amounts of [I2Cl]− and [I3]− present. Importantly, for the systems of LacA:ChCl and OxA:ChCl where poor signal-to-noise ratio in the EXAFS prevented reasonable fitting of the iodine–chloride coordination shell, a significant proportion of [I2Cl]− was confirmed to be present.
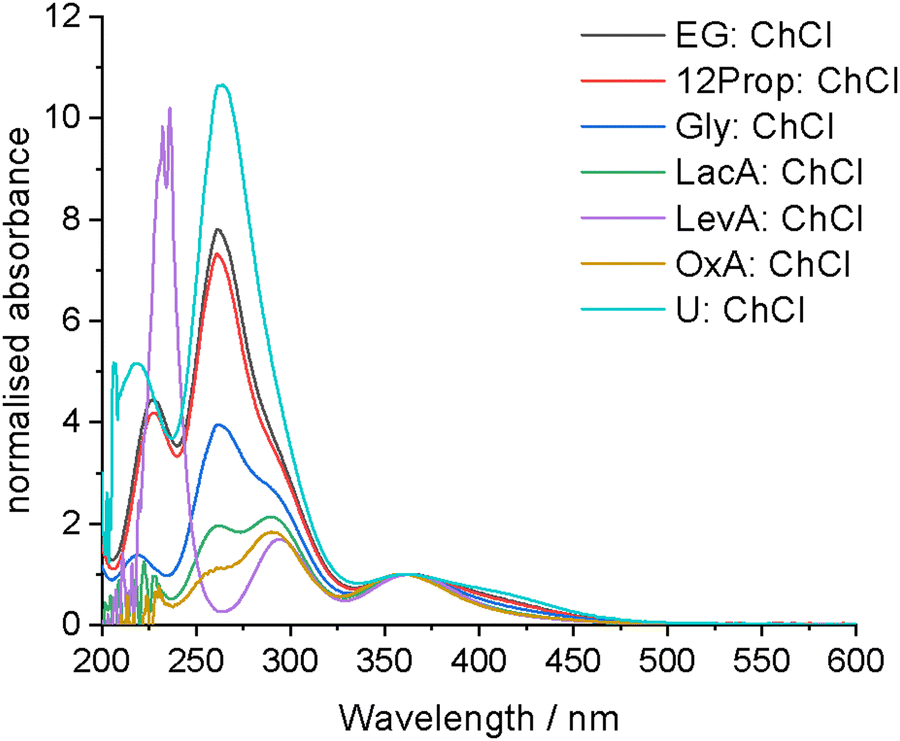 | ||
| Fig. 2 UV-vis spectra of 0.005 to 0.02 mol dm−3 iodine in the different DESs. All spectra are normalised to the 362 nm maximum to aid in comparison. | ||
To determine the cause of bleaching in the 0.01 mol dm−3 iodine in the U:ChCl system, fresh samples were made, and UV-vis spectra were recorded over a period of three days. As it was suspected that the reducing agent was ammonia produced via thermal decomposition of urea, a sample was also stored in the oven at 50 °C to accelerate formation of ammonia. From Fig. 3, it can be seen that only a small change in the spectrum is seen between the fresh and 3 day aged room temperature samples, but a significant change is observed after 3 day ageing at 50 °C. This confirms the predicted bleaching mechanism of urea thermally decomposing to produce ammonia, followed by reduction of the trihalide species to colourless iodide. UV-vis spectroscopy confirms the speciation determined by EXAFS but does show that at low iodine concentrations some side reactions occur when the HBD of the DES is easily oxidised, as is the case for urea. This finding has implications for the long-term usage of iodine as an oxidising agent in urea:ChCl, as the solvent decomposes over time and with elevated temperatures. This results in a side reaction that consumes the oxidising species. Coupled with the poorer solubility and stability of iodine in this solvent compared to the other DESs, urea:ChCl is unlikely to be used in large-scale applications for metal oxidation where reuse/recycling of the solvent will be critical.
 | ||
| Fig. 3 UV-vis spectra of 0.01 mol dm−3 iodine in U:ChCl before and after aging for three days at room temperature and 50 °C. The cuvette was 0.1 mm thick. | ||
3.4 Electrochemistry
The electrochemistry of 0.1 mol dm−3 iodine in EG:ChCl, Gly:ChCl, OxA:ChCl and U:ChCl was investigated via cyclic voltammetry (CV). These DESs were selected because of the different physical and chemical properties of the HBD. EG:ChCl has a high [I2Cl]− content, whereas Gly:ChCl has a low [I2Cl]− content. OxA:ChCl is an acidic DES, and U:ChCl is a basic DES.56 From Fig. 4 it can be seen that the relative redox potentials change with HBD type, with the most anodic redox couples being present in OxA:ChCl. This would suggest that iodine species in OxA:ChCl should be a better oxidising agent than the iodine species in EG:ChCl. The two redox couples can be assigned to:40| 3I2 + 2e− → 2[I3]− (most anodic) |
| [I3]− + 2e− → 3I− or [I2Cl]− + 2e− → 2I− + Cl− (most cathodic) |
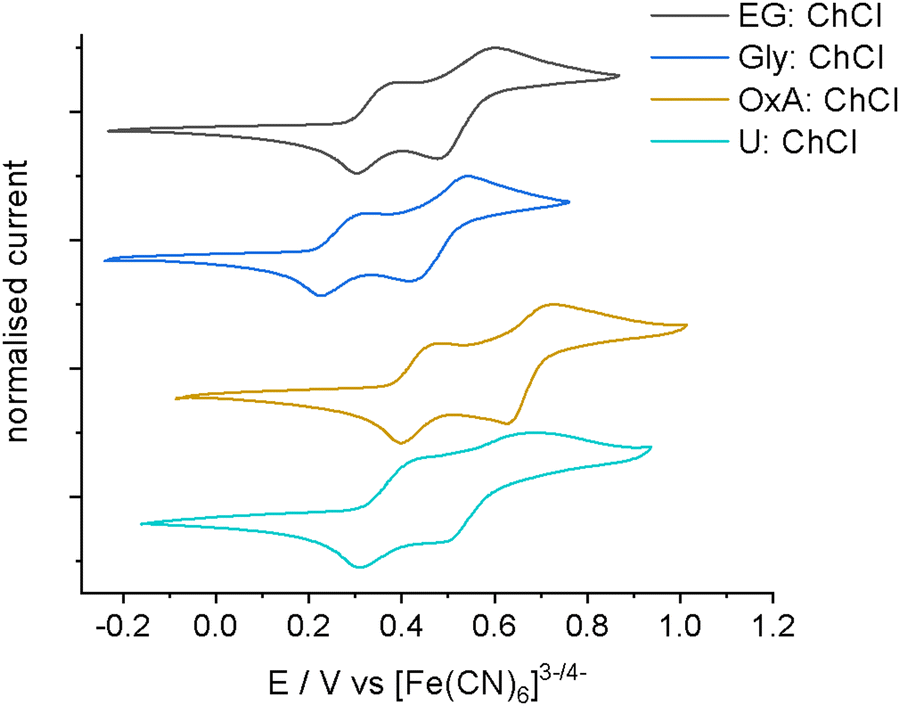 | ||
| Fig. 4 CVs showing the effect of the choice of HBD on redox potentials of 0.1 mol dm−3 iodine in four different DESs at 5 mV s−1. Scans were carried out at room temperature, with a 1 mm Pt-disc working electrode, Pt-flag counter electrode, referenced to the [Fe(CN)6]3−/4− redox couple. | ||
3.5 Comparison of iodine-in-DES leaching systems
Leaching experiments of copper and nickel were carried out in solutions of 0.1 mol dm−3 of iodine in EG:ChCl, Gly:ChCl, OxA:ChCl, and U:ChCl to identify whether the critical factor for metal leaching is iodine speciation, solvent physical properties, or the relative redox potentials. The EG:ChCl system is the most commonly used solvent for metal processing as it is significantly more fluid than the other systems, with a viscosity of 20 mPa s at 40 °C. The other systems have viscosity values of 118, 149, and 218 mPa s at 40 °C, for Gly:ChCl, OxA:ChCl, and U:ChCl respectively.57Assuming each trihalide requires two electrons to be fully reduced, then the proposed leaching reactions for the copper and nickel systems would be:
| 2Cu0 + X3− → 2Cu+ + 3X− |
| Ni0 + X3− → Ni2+ + 3X− |
| 2Cu+ + X3− → 2Cu2+ + 3X− |
Leaching experiments in 0.1 mol dm−3 iodine solution (Fig. 5) showed that the maximum theoretical concentration of 0.2 mol dm−3 copper had been reached after 6 hours in EG:ChCl, and 24 hours in Gly:ChCl at 50 °C. The difference in dissolution rate is likely due to higher solvent viscosity for Gly:ChCl, rather than the slight difference in iodine speciation. For nickel in the same DESs, the maximum predicted concentration of 0.1 mol dm−3 was reached after 24 to 48 hours. Therefore, it can be concluded that the secondary oxidation of Cu+ to Cu2+ is not taking place in significant amounts over the timescale of these experiments. Additionally, regeneration of the trihalide species (likely by oxygen) must be taking place, as the total amount of copper oxidised in EG:ChCl is higher than predicted. Regeneration of the oxidising species is important as it permits reuse of the solution and means that the process is auto-catalytic.
 | ||
| Fig. 5 Plots showing the relative dissolution rates of: copper (left), and nickel (right) in EG:ChCl, Gly:ChCl, OxA:ChCl, and U:ChCl, with an iodine concentration of 0.1 mol dm−3. Measurements were carried out under stirring conditions of 200 rpm, at 50 °C. The dashed line indicates the predicted maximum metal concentration. | ||
In OxA:ChCl, however, an excess of copper was dissolved, which when coupled with the presence of significant amounts of the solution iodine species remaining (the solution remained brown), indicated that the acidic DES is itself digesting the metal. For nickel, the opposite behaviour was observed, where the amount of metal digested was significantly lower than predicted. This was due to the formation of a thick, but non-adhesive, layer of a blue-green precipitate on the nickel surface that prevented the iodine species from reaching the metal surface. This precipitate is most likely nickel oxalate formed from reaction of nickel ions with the DES, as it is known that nickel species are poorly soluble in OxA:ChCl.58 The maximum amount of nickel oxidised by iodine in OxA:ChCl after 48 hours was 0.004 mol dm−3.
Based on the iodine bleaching effect seen in U:ChCl with time and temperature, a lower concentration of dissolved metal was anticipated. Instead, it was observed that a higher amount of copper was digested than expected, resulting in the formation of a blue solution – likely a copper(II) ammonia complex. Upon rinsing the copper foil with water, a white precipitate formed, which is thought to be copper(I) chloride. The higher than expected copper digestion is either a result of iodine regeneration, or due to the presence of ammonia. The dissolution of nickel was slower than that of copper, with not all of the iodine species being fully consumed after 48 hours.
In conclusion, the physical properties of the DESs (viscosity, conductivity, etc.) do not appear to have a significant impact on metal digestion, and neither does the redox potential of the iodine species. Within the present experiment, it is difficult to separate the effect of iodine speciation from the effect of the chemical properties of the HBD, as the systems with the most different iodine speciation (OxA:ChCl and U:ChCl) are also the systems where the HBD will coordinate metal ions the most. Therefore, the choice of HBD is the most critical part of designing a leaching experiment. For example, OxA is a good choice when dissolving copper because both iodine and OxA can oxidise the copper. However, OxA shows poor solubility for nickel because it will form a passivating layer of nickel oxalate once nickel ions have been liberated by the iodine species. This method could be used for selective leaching for applications where both copper and nickel were present. At alternate approach is to mix DESs as different HBDs can change metal speciation without necessarily increasing viscosity significantly, as has previously been observed in hybrid systems of EG:ChCl with U:ChCl or OxA:ChCl, where copper was recovered from chalcopyrite without iron contamination of the deposit.59
4. Conclusions
The speciation of iodine in seven different DESs was investigated using EXAFS and UV-vis spectroscopies. It was found that three general types of environment were possible at an iodine concentration of 0.1 mol dm−3: mixed I2Cl−/I3− (glycol and LacA systems), mixed I2/I3− (OxA and U systems), and I3− on its own (LevA system). Upon decreasing the concentration to 0.01 mol dm−3, there was a general trend towards solutions containing a greater proportion of [I3]− species with respect to the other iodine species present. Bleaching of the iodine was seen in the 0.01 mol dm−3 solutions of U:ChCl and LevA:ChCl, with the reaction in U:ChCl being catalysed by thermal decomposition of urea to ammonia, which is a strong reducing agent. The reduction mechanism of iodine in LevA:ChCl remains unresolved.Cyclic voltammetry of solutions of 0.1 mol dm−3 iodine in EG:ChCl, Gly:ChCl, OxA:ChCl, and U:ChCl were able to quantify relative redox potentials for the species present. While the iodine species in OxA:ChCl showed the most anodic redox potentials, i.e. oxidation of metals with iodine should be more thermodynamically favourable, metal oxidation was found to be more greatly affected by the species formed between the metal ions and oxalic acid and by the acidity of the DES.
Bulk dissolution of copper and nickel appeared to be mainly affected by a combination of the iodine speciation and chemical properties of the HBD. The glycol-based systems studied showed stoichiometric dissolution of the metal, whereas the systems with strongly coordinating HBDs showed sensitivity to the type of metal used and solubility of those metal-HBD products, e.g. poor solubility of nickel oxalate. Some regeneration of the iodine species during the course of the experiments via contact with atmospheric oxygen was detected in the copper-containing glycol systems, but not those containing nickel.
The type of HBD is therefore the most important factor for metal dissolution with iodine, as it controls the iodine speciation and solubility via chloride activity, and also controls solubility and stability of the dissolved metal species. Therefore, the DES composition must be carefully selected in order to obtain the necessary physical properties for a process (viscosity, conductivity, acidity) and also ensure a high solubility of both the iodine and the target metals.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Faraday Institution (Faraday Institution grant codes FIRG005 and FIRG006, project website https://relib.org.uk), the UKRI Interdisciplinary Circular Economy Centre for Technology Metals, Met4Tech project (EP/V011855/1) for funding this work. Initial work on redox potentials was funded by First Solar. This work was carried out with the support of Diamond Light Source, instrument B18 (proposal SP26622). The authors would also like to thank Nitya Ramanan for assistance at the beamline during remote working conditions.References
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- V. Forti, C. P. Balde, R. Kuehr and G. Bel, The Global E-waste Monitor 2020: Quantities, flows and the circular economy potential, United Nations University/United Nations Institute for Training and Research, International Telecommunication Union, and International Solid Waste Association, Bonn, Geneva and Rotterdam, 2020 Search PubMed.
- S. Rothermel, M. Winter and S. Nowak, in Recycling of Lithium-Ion Batteries, ed. A. Kwade and J. Diekmann, Springer, 2018, ch. 1, pp. 1–31 DOI:10.1007/978-3-319-70572-9_1.
- A. Ashiq, J. Kulkarni and M. Vithanage, Electronic Waste Management and Treatment Technology, 2019, pp. 225–246 DOI:10.1016/B978-0-12-816190-6.00010-8.
- A. Khaliq, M. Rhamdhani, G. Brooks and S. Masood, Resources, 2014, 3, 152–179 CrossRef.
- C. Carlesi, E. Cortes, G. Dibernardi, J. Morales and E. Muñoz, Hydrometallurgy, 2016, 161, 29–33 CrossRef CAS.
- A. P. Abbott, A. Z. M. Al-Bassam, A. Goddard, R. C. Harris, G. R. T. Jenkin, F. J. Nisbet and M. Wieland, Green Chem., 2017, 19, 2225–2233 RSC.
- L. Yurramendi, J. Nieto and A. Siriwardana, Mater. Proc., 2022, 5, 96 Search PubMed.
- A. Entezari-Zarandi and F. Larachi, J. Rare Earths, 2019, 37, 528–533 CrossRef CAS.
- N. Rodriguez Rodriguez, L. Machiels, B. Onghena, J. Spooren and K. Binnemans, RSC Adv., 2020, 10, 7328–7335 RSC.
- P. Zürner and G. Frisch, ACS Sustainable Chem. Eng., 2019, 7, 5300–5308 CrossRef.
- C. Liu, Q. Yan, X. Zhang, L. Lei and C. Xiao, Environ. Sci. Technol., 2020, 54, 10370–10379 CrossRef CAS PubMed.
- S. Riaño, M. Petranikova, B. Onghena, T. Vander Hoogerstraete, D. Banerjee, M. R. S. Foreman, C. Ekberg and K. Binnemans, RSC Adv., 2017, 7, 32100–32113 RSC.
- S. Wang, Z. Zhang, Z. Lu and Z. Xu, Green Chem., 2020, 22, 4473–4482 RSC.
- P. G. Schiavi, P. Altimari, M. Branchi, R. Zanoni, G. Simonetti, M. A. Navarra and F. Pagnanelli, Chem. Eng. J., 2021, 417, 129249 CrossRef CAS.
- O. M. Gradov, I. V. Zinov’eva, Y. A. Zakhodyaeva and A. A. Voshkin, Metals, 2021, 11, 1964 CrossRef CAS.
- S. Tang, M. Zhang and M. Guo, ACS Sustainable Chem. Eng., 2022, 10, 975–985 CrossRef CAS.
- J. M. Hartley, C. M. Ip, G. C. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53, 6280–6288 CrossRef CAS PubMed.
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 47, 10031–10033 RSC.
- M. Sahin, A. Akcil, C. Erust, S. Altynbek, C. S. Gahan and A. Tuncuk, Sep. Sci. Technol., 2015, 50, 2587–2595 CAS.
- Q. Meng, G. Li, H. Kang, X. Yan, H. Wang and D. Xu, Metals, 2019, 10, 50 CrossRef.
- S. S. Konyratbekova, A. Baikonurova and A. Akcil, Miner. Process. Extr. Metall. Rev., 2014, 36, 198–212 CrossRef.
- D. Jones, J. Hartley, G. Frisch, M. Purnell and L. Darras, Palaeontol. Electron., 2012, 15, 1–7 Search PubMed.
- A. P. Abbott, R. C. Harris, F. Holyoak, G. Frisch, J. Hartley and G. R. T. Jenkin, Green Chem., 2015, 17, 2172–2179 RSC.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- A. M. Popescu, V. Soare, O. Demidenko, J. M. Calderon Moreno, E. I. Neacsu, C. Donath, M. Burada, I. Constantin and V. Constantin, Rev. Chim., 2020, 71, 122–132 CrossRef CAS.
- A. M. Popescu, C. Donath, E. I. Neacsu, V. Soare, I. Constantin, M. Burada, D. V. Dumitrescu, K. Yanuskevich and V. Constantin, Rev. Chim., 2017, 68, 1963–1968 CrossRef CAS.
- R. Winarko, D. B. Dreisinger, A. Miura, C. Tokoro and W. Liu, Hydrometallurgy, 2020, 197, 105481 CrossRef CAS.
- H. A. Sabzkoohi and G. Kolliopoulos, Mater. Proc., 2021, 5, 39 Search PubMed.
- Y. Fukano and A. Miura, Hydrometallurgy, 2021, 206, 105752 CrossRef CAS.
- A. Van den Bossche, E. De Witte, W. Dehaen and K. Binnemans, Green Chem., 2018, 20, 3327–3338 RSC.
- X. Li, A. Van den Bossche, T. Vander Hoogerstraete and K. Binnemans, Chem. Commun., 2018, 54, 475–478 RSC.
- A. Van den Bossche, N. Rodriguez Rodriguez, S. Riaño, W. Dehaen and K. Binnemans, RSC Adv., 2021, 11, 10110–10120 RSC.
- P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649–1684 CrossRef CAS PubMed.
- R. Wojciechowski, J. Ulanski, S. Lefrant, E. Faulques, E. Laukhina and V. Tkacheva, J. Chem. Phys., 2000, 112, 7634–7640 CrossRef CAS.
- A. G. Maki and R. Forneris, Spectrochim. Acta, Part A, 1967, 23, 867–880 CrossRef CAS.
- P. H. Qi and J. B. Hiskey, Hydrometallurgy, 1991, 27, 47–62 CrossRef CAS.
- M. Bastianini, D. Costenaro, C. Bisio, L. Marchese, U. Costantino, R. Vivani and M. Nocchetti, Inorg. Chem., 2012, 51, 2560–2568 CrossRef CAS PubMed.
- Z. Fei, F. D. Bobbink, E. Paunescu, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2015, 54, 10504–10512 CrossRef CAS.
- E. I. Rogers, I. Streeter, L. Aldous, C. Hardacre and R. G. Compton, J. Phys. Chem. C, 2008, 112, 10976–10981 CrossRef CAS.
- A. Ejigu, K. R. J. Lovelock, P. Licence and D. A. Walsh, Electrochim. Acta, 2011, 56, 10313–10320 CrossRef CAS.
- C. L. Bentley, A. M. Bond, A. F. Hollenkamp, P. J. Mahon and J. Zhang, J. Phys. Chem. C, 2015, 119, 22392–22403 CrossRef CAS.
- A. Y. M. Al-Murshedi, Deep eutectic solvent-water mixtures, PhD thesis, University of Leicester, 2018.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- N. Frenzel, J. Hartley and G. Frisch, Phys. Chem. Chem. Phys., 2017, 19, 28841–28852 RSC.
- S. N. Datta, C. S. Ewig and J. R. Van Wazer, J. Mol. Struct., 1978, 48, 407–416 CrossRef CAS.
- M. C. Feiters, F. C. Kupper and W. Meyer-Klaucke, J. Synchrotron Radiat., 2005, 12, 85–93 CrossRef CAS.
- A. P. Abbott, R. C. Harris and K. S. Ryder, J. Phys. Chem. B, 2007, 111, 4910–4913 CrossRef CAS.
- H. Sakane, T. Mitsui, H. Tanida and I. Watanabe, J. Synchrotron Radiat., 2001, 8, 674–676 CrossRef CAS.
- A. P. Abbott, A. Y. M. Al-Murshedi, O. A. O. Alshammari, R. C. Harris, J. H. Kareem, I. B. Qader and K. Ryder, Fluid Phase Equilib., 2017, 448, 99–104 CrossRef CAS.
- W. J. Albery, R. P. Bell and A. L. Powell, Trans. Faraday Soc., 1965, 61, 1194–1198 RSC.
- R. P. Bell and P. De Maria, Trans. Faraday Soc., 1970, 66, 930–935 RSC.
- J. Li, H. Zhang, T. Xue, Q. Xiao, T. Qi, J. Chen and Z. Huang, Sep. Purif. Technol., 2021, 277, 119364 CrossRef CAS.
- A. P. Abbott, S. S. M. Alabdullah, A. Y. M. Al-Murshedi and K. S. Ryder, Faraday Discuss., 2018, 206, 365–377 RSC.
- A. P. Abbott, E. I. Ahmed, R. C. Harris and K. S. Ryder, Green Chem., 2014, 16, 4156–4161 RSC.
- I. M. Pateli, D. Thompson, S. S. M. Alabdullah, A. P. Abbott, G. R. T. Jenkin and J. M. Hartley, Green Chem., 2020, 22, 5476–5486 RSC.
- S. Anggara, F. Bevan, R. C. Harris, J. M. Hartley, G. Frisch, G. R. T. Jenkin and A. P. Abbott, Green Chem., 2019, 21, 6502–6512 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Karl Fisher titration values for DES water content, Fourier transforms and k2-weighted EXAFS spectra, normalised XANES spectra, UV-vis data tables and reference sample spectra. See DOI: https://doi.org/10.1039/d2cp03185j |
| This journal is © the Owner Societies 2022 |