
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interactions of limonene and carvone on titanium dioxide surfaces†
Hanyu
Fan‡
a,
Elianna S.
Frank‡
 b,
Douglas J.
Tobias
*b and
Vicki H.
Grassian
*a
b,
Douglas J.
Tobias
*b and
Vicki H.
Grassian
*a
aDepartment of Chemistry and Biochemistry, University of California San Diego, La Jolla, California 92093, USA. E-mail: vhgrassian@ucsd.edu
bDepartment of Chemistry, University of California, Irvine, California 92697, USA. E-mail: dtobias@uci.edu
First published on 19th September 2022
Abstract
Limonene, a monoterpene, found in cleaning products and air fresheners can interact with a variety of surfaces in indoor environments. An oxidation product of limonene, carvone, has been reported to cause contact allergens. In this study, we have investigated the interactions of limonene and carvone with TiO2, a component of paint and self-cleaning surfaces, at 297 ± 1 K with FTIR spectroscopy and force field-based molecular dynamics and ab initio simulations. The IR absorption spectra and computational methods show that limonene forms π-hydrogen bonds with the surface O–H groups on the TiO2 surface and that carvone adsorbs on the TiO2 surface through a variety of molecular interactions including through carbonyl oxygen atoms with Ti4+ surface atoms, O–H hydrogen bonding (carbonyl O⋯HO) and π-hydrogen bonds with surface O–H groups. Furthermore, we investigated the effects of relative humidity (RH) on the adsorption of limonene and carvone on the TiO2 surface. The spectroscopic results show that the adsorbed limonene can be completely displaced by water at a relative humidity of ca. 50% RH (∼2 MLs of water) and that 25% of carvone is displaced at ca. 67% RH, which agrees with the calculated free energies of adsorption which show carvone more strongly adsorbs on the surface relative to limonene and thus would be harder to displace from the surface. Overall, this study shows how a monoterpene and its oxidation product interact with TiO2 and the impact of relative humidity on these interactions.
Introduction
Improving indoor air quality (IAQ) has gained increasing public awareness as people spend most of their time (>90%) indoors compared to outdoors. During the COVID-19 pandemic this was notable, and many people spent more time at home.1,2 Indoor air pollutants, such as volatile organic compounds (VOCs, e.g., alkanes, alkenes, aromatics, carboxylic acids, alcohols and esters), oxides of nitrogen and sulfur (NOx and SO2), carbon monoxide (CO), and particulate matter (PM), including bacteria and viruses, come from a variety of sources. These sources include consumer products, building materials and furnishings, occupant-related activities (e.g., cleaning, cooking, smoking, breathing), exchange with outdoor air and exhaled breath.3–6 Secondary products generated from chemical reactions occurring in the gas phase, on particles and on indoor surfaces also contribute to the overall indoor environment.7,8 Many of these pollutant species and their secondary products can cause allergic reactions, asthma, respiratory diseases, heart disease and lung cancer.9,10 To improve indoor air quality, besides source control and increasing ventilation, another option is air cleaning.11,12 Current air treatment techniques include active carbon or zeolite adsorption, biofiltration, ozonation, and advanced oxidation process (AOP) such as non-thermal plasma and photocatalytic oxidation (PCO).13–15 Among these techniques, PCO is regarded as a potential cost-effective method because its superior features of operation at room temperature and atmospheric pressure to mineralize ppb level of many hazardous gaseous organics to CO2 and H2O from indoor air under solar light irradiation without chemical additives required.16 However, it is important to note that there are unexpected consequences of using these air cleaning processes.17 Thus, there needs to be testing of these under realistic conditions to further our understanding of the impact on any air cleaning processes.Titanium dioxide (TiO2) is by far the most widely available semiconductor material used in PCO to its nature abundance, low cost, high chemical stability and safety, and high photocatalytic activity.18,19 Rutile and anatase are the two main crystalline structures of TiO2; rutile is the most stable TiO2 phase whereas anatase is more photoactive.20,21 TiO2-based self-cleaning materials, such as paint films, coated glasses, and tiles, are used both in indoors and outdoors on walls, floors, rooftops and facades.22 In the state of California, self-cleaning ceramic TiO2 titles are widely used in public places such as hospitals and restrooms.23 The adsorption and desorption of air pollutants on the surface of TiO2 films are key steps in these heterogeneous photocatalytic oxidation techniques. Adsorption is an initial and critical step in the efficient catalytic treatment of all pollutants. In addition, desorption is an essential step to restore the active surface catalytic sites.24,25 Therefore, to study interaction mechanisms of various VOCs with TiO2 surfaces under relevant indoor environment conditions such as the impact of relative humidity is essential to help enhance the photocatalyst performance and understand air pollutants chemistry on indoor surfaces at the molecular level.26–28
There are two general modes for VOC adsorption on a TiO2 surface: a weaker physisorption bonding mode with surface Ti–OH groups and a stronger chemisorption bonding mode on Ti4+ atoms (Lewis acid sites) or reduced Ti3+ sites near oxygen vacancies.29,30 For example, earlier studies have shown that benzene, toluene, and chlorobenzene can be adsorbed on the TiO2 (rutile) surface via Ti–OH⋯π-electron interaction and additional Ti–OH⋯Cl hydrogen bonds for chlorobenzene on the hydroxylated surface and all the above compounds can have Ti4+⋯π-electron interaction on the non-hydroxylated surface.31 The IR absorption spectrum of acetaldehyde onto TiO2 (P25) at 233 K reported that hydrogen bonding with surface OH groups (CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HO–Ti) and carbonyl bonding with a Ti4+ surface atom (CH3CHO⋯Ti4+) are the dominant surface interactions.32 Pyruvic acid adsorbs onto the TiO2 (rutile) surface by the interaction between the carboxylic acid group with the surface hydroxyl groups.33 Similar results were proposed in the study of the TiO2 (P25) surface chemistry with alcohols, organic acids and aldehydes that can be adsorbed onto TiO2 surface via hydrogen bonding to the surface OH groups.34 The degree of interaction strength between the VOCs and TiO2 surface depends on the relative bonding energies of the hydrogen bond and is expected to be stronger than π-hydrogen bond. This implies that polar VOCs are more strongly adsorbed onto TiO2 surface.35–38 For example, the adsorption of different classes of VOCs on TiO2 (P25) surface follows the order of the strength of the corresponding intermolecular forces: i-octane < toluene < acetone < methyl ethyl ketone < isopropanol < methanol.36 Additional insights from the studies of C5–C7 alkanes interactions with TiO2 (P25) surface demonstrated that the larger long-chain and more branched molecules have stronger interactions with the TiO2 surface based on the order of the strength of adsorption: pentane < hexane < i-pentane < i-hexane < heptane.39 In another study, the importance of steric hindrance of different compounds played a role in the interaction strength; it was shown that the adsorption of aromatic species on the TiO2 (P25) surface via following sequence: ethylbenzene < benzene < o-xylene < p-xylene < m-xylene < toluene, while ethyl substitution showed a larger steric hindrance compared to the methyl functional group, so that ethylbenzene is more weakly adsorbed on the TiO2 surface.40
O⋯HO–Ti) and carbonyl bonding with a Ti4+ surface atom (CH3CHO⋯Ti4+) are the dominant surface interactions.32 Pyruvic acid adsorbs onto the TiO2 (rutile) surface by the interaction between the carboxylic acid group with the surface hydroxyl groups.33 Similar results were proposed in the study of the TiO2 (P25) surface chemistry with alcohols, organic acids and aldehydes that can be adsorbed onto TiO2 surface via hydrogen bonding to the surface OH groups.34 The degree of interaction strength between the VOCs and TiO2 surface depends on the relative bonding energies of the hydrogen bond and is expected to be stronger than π-hydrogen bond. This implies that polar VOCs are more strongly adsorbed onto TiO2 surface.35–38 For example, the adsorption of different classes of VOCs on TiO2 (P25) surface follows the order of the strength of the corresponding intermolecular forces: i-octane < toluene < acetone < methyl ethyl ketone < isopropanol < methanol.36 Additional insights from the studies of C5–C7 alkanes interactions with TiO2 (P25) surface demonstrated that the larger long-chain and more branched molecules have stronger interactions with the TiO2 surface based on the order of the strength of adsorption: pentane < hexane < i-pentane < i-hexane < heptane.39 In another study, the importance of steric hindrance of different compounds played a role in the interaction strength; it was shown that the adsorption of aromatic species on the TiO2 (P25) surface via following sequence: ethylbenzene < benzene < o-xylene < p-xylene < m-xylene < toluene, while ethyl substitution showed a larger steric hindrance compared to the methyl functional group, so that ethylbenzene is more weakly adsorbed on the TiO2 surface.40
Another factor in these studies is the effect of relative humidity. Relative humidity is known to be an important factor in the surface chemistry of environmental interfaces, i.e., interfaces under ambient conditions.41 There is competition between water vapor and VOCs as well as synergisms.42–45 For example, there is a respective decrease of 50% and 30% for the adsorption of toluene (80 ppb) and 2-propanol (250 ppb) on TiO2 (anatase) surface at 60% RH compared to the dry conditions due to the direct replacement of water adsorption.42 Other studies reported that the adsorptions of chlorobenzene and cyclohexane on TiO2 (P25) surface significantly decrease with increasing relative humidity, which explains the competition from the water adsorption and further hindrance from the adsorbed water layers.43,44
Atomistic simulations provide detailed insight into surface interactions, and in our previous work, we have presented studies of hydroxylated SiO2 (a model for glass surfaces) and indoor relevant organic compounds with great agreement between theory and experiment.45–47 However, TiO2 presents a unique challenge as there is still outstanding debate over how to model the interactions of water on the TiO2 surface in a simulation, to either represent the water via a molecular or dissociative association on the surface.48–53 In this study, we have constructed both a non-hydroxylated TiO2 surface with surface Ti atoms and free water molecules, and a fully hydroxylated TiO2 with all surface Ti atoms capped with OH groups. We use these two surfaces to represent the extrema of a real world TiO2 surface. As we find robust agreement between our computational results and the experimental measurements, we are motivated to use our atomistic simulations to suggest relevant surface interactions between TiO2 and organic molecules, however these insights only serve to deepen the conclusions made from the experimental results.
Among the many VOCs present indoors, limonene is widely used in cleaning products and air fresheners to provide a pleasant smell. Our previous studies showed that limonene forms one or two Si–OH⋯π-hydrogen bonds on the hydroxylated SiO2 surface, and more than 50% of limonene remains adsorbed within the water and hydroxylated SiO2 surface through H–OH or Si–OH⋯π-hydrogen bonding at a relatively high 78% RH when the water coverage is close to three monolayers (MLs); carvone forms a Si–OH⋯O hydrogen binding and an oxygen silicon association with siloxane bridge (O⋯Si–O–Si) on the hydroxylated SiO2 surface, and instead of replacing the adsorbed carvone on SiO2 surface, water adsorbs onto the carvone coverage via H–OH⋯O hydrogen binding.45–47 To the best of our knowledge, there are no systematic studies with respect to the adsorption of limonene and its oxidation product carvone on TiO2 surface or the impact of relative humidity over a wide range of relative humidity conditions. In the present work, we combined transmission FTIR spectroscopy, classical force field-based and ab initio molecular dynamics simulations to investigate the types of surface interactions and relative strengths of limonene and carvone adsorbed on the hydroxylated TiO2 surface. The effect of relative humidity ranging from <1% to 67% RH on the interactions of limonene and carvone on the hydroxylated TiO2 surface was also investigated.
Experimental and theoretical methods
Experimental methods
TiO2 (rutile, high purity, 99.9+%, Stock#: US3520) was purchased from US Research Nanomaterials, Inc. The surface area measurements of TiO2 particles were done using a Quantachrome Nova 4200e multipoint Brunauer–Emmett–Teller (BET) surface analyzer. First the TiO2 particles were degassed overnight at 120 °C prior to a BET measured. The average BET surface area and standard deviation was determined to be 17 ± 1 m2 g−1. The limonene (D-limonene, >99%, Fisher Scientific) and carvone (D-carvone, >98%, TCI) samples were prepared by degassing three times with consecutive freeze–pump–thaw cycles.The experimental infrared setup has been described previously.45–47 Approximately 8 mg of TiO2 was pressed onto to a tungsten grid with a stainless-steel press. The TiO2 particles were calcined at 200 °C in an oven for 2 hours prior to pressing onto the grid. The grid with particles was held in position by nickel jaws inside of the stainless-steel IR cell with BaF2 windows. The rest of the experimental setup consists of a stainless IR cell (310 ± 3 ml, the optical path length of the IR cell is 7 cm), a glass mixing chamber (1329 ± 2 ml), a stainless-steel connection tubing (length: 36 inch and I.D. 1/4 inch) and a two-stage vacuum system (a turbomolecular pump (Agilent TwisTorr 74 FS) with a backup mechanical pump (Adixen Pascal 2010 SD)). Two absolute pressure transducers (MKS Instruments, Inc., 10 and 1000 Torr) were used to record the equilibrium pressure data.
FTIR spectra were recorded with a Nicolet iS50 FTIR spectrometer (Thermo Fisher Scientific). Prior to and under the equilibrium exposure of organic compound vapor, single-beam spectra (300 scans) of the respective particles surface and gas phase were acquired using at a spectral resolution of 4 cm−1 over the spectral range of 1200–4000 cm−1. The surface absorbance spectra were obtained by subtracting the FTIR spectra of gas-phase organic compound from the particles surface plus gas-phase spectra. To investigate the effects of RH, the particles sample was first exposed to the organic vapor for 50 min under dry condition (<1% RH); followed by the introduction of water vapor into the IR cell for 1 minute; and then the IR cell was closed for another 50 minutes for the equilibrium before the particles surface were evacuated for the desorption measurement. The % RH values were recorded based on the water vapor pressure reading after 1 minute water injection. The water vapor reached equilibrium in the IR cell within 1 minute once the water vapor was injected.45
Computational methods
Force field-based MD simulations and ab initio MD (AIMD) simulations were used to investigate the adsorption of limonene and carvone on a TiO2 surfaces. For both simulation methods, a 110 rutile TiO2 unit cell was replicated in the x, y and z directions to create the initial structure.54 Due to the outstanding debate over the nature of the interactions of water on the TiO2 surface, force field-based MD simulations for both a non-hydroxylated TiO2 surface with surface Ti atoms and free TIP3P water molecules and a fully hydroxylated TiO2 with no surface Ti atoms have been prepared.48–52 For our AIMD simulations, we have prepared three surfaces, a non-hydroxylated TiO2 surface, a TiO2 surface with 4 water molecules between the adsorbate and the surface, and a fully hydroxylated TiO2 surface. However, because these AIMD simulations were all run at 300 K, some of the O–H groups on the fully hydroxylated surface underwent dissociations during the course of the AIMD simulation.For the force field-based MD simulations, a 35 × 40 × 21 Å3 TiO2 slab was created and 77 Å of vacuum was placed separating the upper and lower surfaces in the z direction. From this initial surface with 720 Ti atoms and 1440 O atoms, the non-hydroxylated and hydroxylated TiO2 surfaces were produced. For clarity, we can consider all TiO2 surfaces consisting of two parts, the frozen core Ti and O atoms and the surface atoms. In the case of the non-hydroxylated surface, the surface atoms are the bridging oxygen atoms and 5-coordinated Ti atoms. In the case of the hydroxylated surface, all atoms on the top and bottom of the surface were hydroxylated, thus the surface atoms are the surface OH groups. To investigate the role of %RH, TIP3P water was added to both sides of the TiO2 surface, which in the text we will refer to as the 0.5 monolayer and 1 monolayer systems. The 0.5 monolayer system was represented by a total of 204 water molecules, corresponding to number of bridging oxygen atoms in the non-hydroxylated surface. The 1 monolayer system was represented by 408 water molecules.
The force field produced by Brandt et al. was used for the non-hydroxylated surface.55 For the hydroxylated surface, the nonbonded parameters were set to 0 for hydrogen and in order to ensure a neutral surface, the partial charge of all hydrogen atoms was set to +0.44175e. And the O–H group oxygen partial charge was set to −0.80e. During the course of the simulation, all bulk atoms were frozen, and all surface atoms were allowed to move freely. There were small distortions of the TiO2 surface as seen in the molecular snapshots in Fig. 3, however the general structure of the TiO2 crystal was retained. The TIP3P water was used to describe the water molecules and details of the CHARMMM compatible force fields for limonene and carvone can be found in our previous work.46,47,56
All classical molecular dynamics simulations were done with the LAMMPS57 software package. The equations of motion were integrated using the velocity Verlet algorithm with a 1 fs time step.58 Electrostatic interactions were evaluated directly up to a cutoff of 12 Å and with a particle–particle–particle mesh solver with a relative accuracy of 10−6.59 Lennard-Jones interactions were cut off after 12 Å. The simulation temperature was maintained at 300 K using a Nosé–Hoover thermostat with a relaxation time of 100 fs.60
Umbrella sampling was used to calculate the free energy profile for the desorption of carvone and limonene from the TiO2 surfaces.61 Initial coordinates of each adsorbate on the TiO2 surface were taken after 25 ns of simulation time. The distance of the center-of-mass of adsorbate to the TiO2 surface was used as the reaction coordinate, and the process was divided into variable number of windows for each adsorbate. For limonene, 22 windows in 0.50 Å increments was sufficient, but for carvone, 0.25 Å increment windows were used. A harmonic restraining potential of 10 kcal mol−1 Å−2 was applied in each window. Each free energy profile was generated from 30 ns or longer long biased trajectories for each PMF window using the WHAM scheme.62
In the hydroxylated surface, two forms of hydrogen bonding were considered, π-hydrogen bonds formed between carbon sp2 (Csp2) atoms and the TiO2–OH surface for both limonene and carvone, and hydrogen bonds formed between the carvone oxygen atoms and the TiO2–OH surface. To calculate these hydrogen bonding probabilities, distances were measured between Csp2 centers and surrounding oxygen donors and between the carvone oxygen atom and surrounding oxygen donors. Angular measurements were made between adsorbate acceptor (Csp2 center or carvone oxygen atom), TiO2 donor H, and TiO2 donor O atoms. All measurements were made every 1 ps. Hydrogen bonds were defined to be present if the distance between donor and acceptor was less than 3.5 Å and if the angle between acceptor, donor H, and donor O was within 120–180 degrees for both types of hydrogen bonds. Additionally, in systems with free water molecules, π-hydrogen bonds between Csp2 centers and water molecules, as well as hydrogen bonds between the carvone oxygen atom and water were measured in the same way as described above.
For the ab initio simulations, three TiO2 surfaces were studied, a non-hydroxylated surface, a hydroxylated surface, and a non-hydroxylated surface with 4 water molecules placed between the surface and the adsorbate. The TiO2 surface was obtained from selecting a smaller portion of the classical MD surface. For all simulations expect for carvone on the non-hydroxylated surface with 4 water molecules, a system of 90 Ti atoms and 180 O atoms, accounting to 5 layers of Ti and O atoms, was sufficient to allow for ample simulation time (30 ps). In the case of carvone on the non-hydroxylated surface with 4 water molecules, a slightly larger size in the y direction of the TiO2 surface (125 Ti atoms and 250 O atoms) was used. This resulted in eight AIMD simulations, detailed in the ESI.† In the case of the hydroxylated surface and the non-hydroxylated surface with water molecules, only the top of the surface was either hydroxylated or had water molecules placed on top, to save computational time.
For all systems, a 30 ps AIMD simulation at 300 K with a time step of 0.5 fs was performed, except for the carvone on the non-hydroxylated surface with 4 water molecules, where at 13 ps AIMD simulation at 300 K with a time step of 0.5 fs was performed. The temperature as maintained at 300 K using a Nose–Hoover14 thermostat with a relaxation time of 100 fs. The PBE63 exchange–correlation functional with the DZVP-MOLOPT-SR-GTH64,65 basis set and the GTH pseudopotentials in the QUICKSTEP module of the CP2K66 package were used. A SCF convergence criterion of 10−6 (a.u.) with the orbital transformation67 scheme and DIIS minimization68 were used in each simulation. All systems were placed in orthorhombic boxes under periodic boundary conditions in the x and y directions with the z direction normal to the TiO2 surface. The AIMD simulation cells had sizes of 15.960 Å × 12.638 Å × 36.821 Å and for the slightly larger system for carvone on the non-hydroxylated surface with 4 water molecules, the cell size was 15.091 Å × 16.437 Å × 36.821 Å. During the course of the simulation, the bottom half of the TiO2 surface was frozen.
The power spectra were calculated for the Fourier transform of the velocity autocorrelation function of all oxygen or hydrogen atoms using the TRAVIS code.69,70 A 900 fs Gaussian windowing function was applied to the correlation function prior to Fourier transformation.
Results and discussion
Experimental data
C stretching vibration of the vinyl group and in the ring. The absorptions observed at 1454, 1439, and 1378 cm−1 for CH3 and CH2 bending vibrations motion, respectively.45,46 In addition, the negative peak at 3656 cm−1 is attributed to the loss of Ti–OH groups due to the hydrogen-bonding interactions with limonene, as a broad peak centered at ∼3447 cm−1 grows in with increasing limonene pressure.
 | ||
| Fig. 1 (a) FTIR spectra of limonene adsorbed on TiO2 surface under dry conditions (<1% RH) as a function of limonene pressure in the spectral range extending from 1200 to 4000 cm−1 (scale bar = 0.1 absorbance units). The equilibrium pressures increase from the light red to black spectra: 3, 6, 11, 20, 31, 42, 75, and 148 mTorr. (b) The ordinate plots the normalized integrated peak area of limonene in the spectral region from 2800 to 3115 cm−1 as function of limonene pressure (mTorr), the right ordinate plots the normalized peak height of the loss of Ti–OH functional groups at 3656 cm−1. | ||
Fig. 1(b) displays the integrated absorbance of the C–H stretching region (ranging from 2800 to 3115 cm−1) and the loss of Ti–OH function groups peak height (3656 cm−1, right ordinate) as function of limonene pressure. It is observed that no apparent increases in the amount of the adsorbed limonene after 60 mTorr as there are no more free Ti–OH functional groups present on the TiO2 surface. This finding indicates that the adsorption of limonene on the hydroxylated TiO2 surface is mainly via the Ti–OH⋯π-electron interaction. Our earlier study shows that the interactions between limonene and hydroxylated SiO2 surface are mainly driven by Si–OH⋯π-hydrogen bonding.45 The desorption free energies of limonene from TiO2 and SiO2 surface are discussed in the computational results section below.
O (ketone) and CC (alkene) stretching vibrations.47 As shown in Fig. 2(b), there is a growth of new band at 1683 cm−1 starting at 8 mTorr. This higher energy shift of 35 cm−1 can be explained by considering the interaction of adsorbed carvone molecules can be through carbonyl bonding with a Ti4+ surface atom on the TiO2 surface.32,47Fig. 2(c) displays the integrated peak area of adsorbed carvone at 1648 cm−1 (ranging from 1560 to 1720 cm−1) and the loss of Ti–OH functional groups peak height at 3656 cm−1 (right ordinate) as function of different equilibrium carvone pressures. It is observed that almost all the free Ti–OH functional groups are occupied by carvone molecules at a low equilibrium pressure of 2 mTorr. As the carvone pressure is changing from 8 mTorr to 18 mTorr with an increase of 10 mTorr, there is about 30% of amount carvone increased on TiO2 surface. In Fig. 1(b), when the limonene pressure is changing from 75 mTorr to 148 mTorr with an increase of 73 mTorr, there is only 10% of amount limonene is increased on TiO2 surface. Once monolayer coverage on the TiO2 surface is reached, the 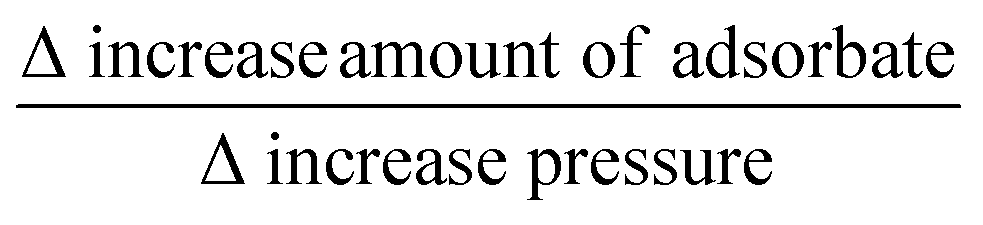 (% mTorr−1) ratios of carvone and limonene are 3 and 0.14, respectively. This observation suggests that the adsorption of carvone on the TiO2 surface is initially via the O–H hydrogen bonding (carbonyl O⋯HO–Ti) and the Ti–OH⋯π-electron interaction; and once all the available Ti–OH groups are occupied in hydrogen bonds, the later adsorption of carvone on TiO2 surface is through a strong carbonyl bonding with a Ti4+ surface atom.47 Although these binding interactions are different, in the discussion above we make the assumption that the increase in intensity is linearly proportional to coverage.
(% mTorr−1) ratios of carvone and limonene are 3 and 0.14, respectively. This observation suggests that the adsorption of carvone on the TiO2 surface is initially via the O–H hydrogen bonding (carbonyl O⋯HO–Ti) and the Ti–OH⋯π-electron interaction; and once all the available Ti–OH groups are occupied in hydrogen bonds, the later adsorption of carvone on TiO2 surface is through a strong carbonyl bonding with a Ti4+ surface atom.47 Although these binding interactions are different, in the discussion above we make the assumption that the increase in intensity is linearly proportional to coverage.
 | ||
| Fig. 2 (a) Absorbance FTIR spectra of carvone adsorbed on TiO2 surface under dry conditions (<1% RH) as a function of pressure in the spectral range extending from ca. 1200 to 4000 cm−1 (scale bar = 0.04 absorbance units). (b) The above (a) spectra range extends from 1500 to 1800 cm−1 (scale bar = 0.02 absorbance units). (c) The ordinate plots the normalized integrated peak area of carvone at 1648 cm−1 (ranging from 1560 to 1720 cm−1) as function of carvone pressure (mTorr), the right ordinate plots the normalized peak height of the loss of Ti–OH function groups at 3656 cm−1. The equilibrium pressures are 2, 8, and 18 mTorr, respectively. | ||
| Percent relative humidity (±1%) | Water coverage in monolayers (MLs) (±0.1)a (bare TiO2 surface) | Percentage of organic compound remaining on surface relative to dry conditions (±3%)bc | Water coverage in monolayers (MLs) (±0.1) (with organic compound adsorbed on TiO2 surface)d |
|---|---|---|---|
| a Water coverages determined from Goodman et al.71 b Percent limonene coverages determined from the integrated absorbance of bands from 1415 to 1475 cm−1. c Determined from the integrated absorbance ranging from 1335 to 1490 cm−1 for carvone. d Water coverages estimated from relative ratio of the integrated area of the water bending mode when the organic compound is present relative to the bare surface. | |||
| Limonene | |||
| <1 | <0.1 | 100 | <0.1 |
| 3 | 0.5 | 91 | 0.3 |
| 10 | 1.0 | 74 | 0.5 |
| 18 | 1.4 | 67 | 0.8 |
| 25 | 1.6 | 54 | 1.6 |
| 30 | 1.7 | 30 | 1.7 |
| 51 | 2.0 | 0 | 2.0 |
| Carvone | |||
| <1 | <0.1 | 100 | <0.1 |
| 67 | 2.3 | 75 | 0.6 |
MD simulations data
 | ||
| Fig. 3 (a–c) Shows limonene on non-hydroxylated TiO2 surfaces with increasing levels of hydration, (d–f) shows limonene on hydroxylated surfaces with increasing levels of hydration, (g–i) shows carvone on non-hydroxylated TiO2 surfaces with increasing levels of hydration, (j–l) shows carvone on hydroxylated TiO2 surfaces with increasing levels of hydration. Water molecules engaged in hydrogen bonds (π-hydrogen or OH-hydrogen) with the adsorbate are shown in full color, all other water molecules are shown transparent. Red atoms correspond to O atoms, pink atoms correspond to Ti atoms, white atoms correspond to H atoms and cyan atoms correspond to C atoms. The surfaces have been oriented to easiest see surface/adsorbate interactions. | ||
During the course of the 50 ns simulation, we observed the carvone cyclic ring of carvone and limonene predominantly stays parallel to the surface for the non-hydroxylated 0 ML H2O and 0.5 ML H2O surfaces and the hydroxylated 0 ML H2O surface. In the hydroxylated 0.5 ML H2O surface, the limonene ring was able to freely rotate on the surface. In both the non-hydroxylated and hydroxylated 1 ML H2O surfaces, the limonene ring and carvone ring were able to freely rotate above the surface. On the non-hydroxylated 0 ML H2O surface, we observed that both limonene and carvone stayed between two rows of bridging oxygen atoms for the course of the 50 ns simulation.
A second relevant measure of surface interactions are the formation of hydrogen bonds. As discussed in the introduction, we have studied the importance of hydrogen bonds to promote stabilization between indoor relevant organic compounds and indoor surfaces. We have studied the role of two kinds of hydrogen bonds, π-hydrogen bonds formed between Csp2 carbon centers and surface OH groups and hydrogen bonds formed between the carvone oxygen atom and surface OH groups. To investigate the adsorption of limonene and carvone on TiO2 surface, we have calculated the average number of π-hydrogen bonds during the course of the last 25 ns of the unbiased simulations. π-hydrogen bonds require hydrogen atoms as hydrogen bond donors, so no measurements were made for the non-hydroxylated surface with 0 ML H2O, but the results for the rest of the surfaces are displayed in Table 2.
| Surface | Water (ML) | Hydrogen bond donor | Average number of π-hydrogen bonds | |
|---|---|---|---|---|
| Limonene | Carvone | |||
| Non-hydroxylated | 0.5 | Surface | n. a. | n. a. |
| Water | 0.0049 | 0.0085 | ||
| Non-hydroxylated | 1 | Surface | n. a. | n. a. |
| Water | 0.13 | 0.05 | ||
| Hydroxylated | 0 | Surface | 0.28 | 0.15 |
| Water | n. a. | n. a. | ||
| Hydroxylated | 0.5 | Surface | 0.66 | 0.43 |
| Water | 0.017 | 0.005 | ||
| Hydroxylated | 1 | Surface | 0 | 0 |
| Water | 0.17 | 0.07 | ||
Overall, we see the formation of fewer π-hydrogen bonds with water molecules compared to our similar study of limonene on hydroxylated SiO2 surface with increasing %RH (average of 0.30 for limonene on SiO2 with 1 MLs H2O versus 0.17 for limonene on hydroxylated TiO2 with 1 MLs H2O). This is due to the fact that not only are water–water hydrogen bonds more favorable compared to π-hydrogen bonds, but water hydrogen bonds to TiO2 for either the non-hydroxylated or hydroxylated surface are also quite strong, resulting in fewer water donor π-hydrogen bonds to limonene and carvone Csp2 carbon atoms. We see on the hydroxylated surfaces that there are more surface to adsorbate π-hydrogen bonds, and these interactions occur frequently for the 0 ML H2O and 0.5 ML H2O surface. For the 1 ML H2O surface, Csp2 carbon atoms on the adsorbate are too far away in the water layer to interact with the OH groups on the surface.
Limonene and carvone both have two double bond sites and we found that the endocyclic CC bond always has a higher probability of π-hydrogen bond formation, and this is because the exocyclic CC bond sometimes rotates above the water layer. Finally, there is a clear difference in π-hydrogen bonding probabilities between these two molecules, as the limonene molecule is much more likely to form π-hydrogen bonds compared to the carvone molecule. This does not indicate that limonene has a stronger interaction with the surface, but rather that carvone shows a much lower propensity for π-hydrogen bond formation. This is due to stabilizing hydrogen bonds formed between the carvone oxygen atom and TiO2 surface. Table 3 shows the carvone carbonyl oxygen atom hydrogen bonding statistics.
| Surface | Water (ML) | Hydrogen bond donor | Average number of Hydrogen bonds |
|---|---|---|---|
| Non-hydroxylated | 0.5 | Surface | n. a. |
| Water | 0.08 | ||
| Non-hydroxylated | 1 | Surface | n. a. |
| Water | 0.80 | ||
| Hydroxylated | 0 | Surface | 1.57 |
| Water | n. a. | ||
| Hydroxylated | 0.5 | Surface | 2.2 |
| Water | 0.18 | ||
| Hydroxylated | 1 | Surface | 0.0018 |
| Water | 0.76 |
In Table 3, we see few water carvone hydrogen bonds are formed for the non-hydroxylated 0.5 ML H2O surface, which we attribute to the increase in stabilization for water–water and water–surface hydrogen bonds as discussed above. However, we see with 1 ML of water there is an average of around 0.80 hydrogen bonds between carvone and the surrounding water, regardless of the surface hydroxylation. As shown in Table 3, accounting for both π-hydrogen and hydrogen bonds, there are more surface interactions for the carvone molecule compared to those of limonene. This is consistent with our experimental results that indicate carvone has stronger interactions with the TiO2. As we will discuss in detail in the ab initio MD simulation results section, there is an additional surface interaction between the carvone carbonyl oxygen atom and the surface Ti atoms in the non-hydroxylated surface. In this case, we have detected that during the course of the 50 ns non-hydroxylated 0 ML H2O simulation, the carvone carbonyl oxygen is positioned close to surface Ti atoms with an average interaction length of 3.92 Å. More importantly, our ab initio power spectra support the validity of this interaction as there is a red shift in the CO bond length compared to the gas phase carvone spectra. With the presence of water the carvone carbonyl oxygen–Ti surface interaction rapidly decreases, as with 0.5 ML of water, only 8% of the 50 ns trajectory shows nearest neighbor to the carvone carbonyl oxygen is a surface Ti atom. A key benefit of atomistic MD simulations is the ability to study the arrangement of large numbers of molecules. In this case, we can investigate the hydrogen bonding network between water molecules and the TiO2 surface, and how this is disrupted by the adsorption of limonene and carvone molecules. As our molecular snapshots show, water is adsorbed on the surface via hydrogen bonds to surface O atoms and surface OH groups and water molecules are also stabilized by water–water hydrogen bonds. Table 4 shows the extent that limonene and carvone disrupt the hydrogen bonding network by measuring the interactions of water molecules engaged in hydrogen bonding with the adsorbate. In some instances, the water molecule is able to form a hydrogen bond with the adsorbate and also a hydrogen bond with the surrounding water molecules or surface. In other cases, the hydrogen bonding network is “disrupted” as the water molecule is only engaged in a hydrogen bond with the adsorbate. Snapshots of these hydrogen bonding interactions are shown in Fig. S8 (ESI†). These networks are also seen in our ab initio simulations.
| Surface | Water (ML) | Adsorbate | Fraction of bonding waters in water–water bonds | Fraction of bonding waters in water–surface bonds | Fraction of bonding waters only in adsorbate bonds |
|---|---|---|---|---|---|
| Non-hydroxylated | 0.5 | Limonene | 0.50 | 0.47 | 0.03 |
| Carvone | 0.58 | 0.39 | 0.02 | ||
| Non-hydroxylated | 1 | Limonene | 0.80 | 0.17 | 0.03 |
| Carvone | 0.95 | 0.046 | 0.003 | ||
| Hydroxylated | 0.5 | Limonene | 0.32 | 0.678 | 0.002 |
| Carvone | 0.511 | 0.486 | 0.003 | ||
| Hydroxylated | 1 | Limonene | 0.66 | 0.34 | — |
| Carvone | 0.758 | 0.24 | 0.002 |
Table 4 shows the difference in hydrogen bonding networks for the limonene and carvone molecules. Overall, this table shows that limonene forms more π-hydrogen bonds with water molecules engaged in hydrogen bonding with the TiO2 surface. Given the competitive adsorption of water on the TiO2 surface, this suggests why limonene is displaced at high %RH. Contrary, carvone is shown to more often form hydrogen and π-hydrogen bonds with water molecules also in hydrogen bonding with other water molecules. In addition, in the case of the non-hydroxylated surface, limonene is shown to disrupt the hydrogen bond network more compared to that of carvone. This suggests why there is little change in the surface coverage of carvone as a function of %RH. To further quantify the difference in interaction strength, we have calculated the free energy profile (also referred to as potentials of mean force or PMF) for the desorption of the adsorbate from the surface, as shown in Fig. 4. The free energy profiles are placed vertically, such that their minima are at the zero of free energy.
 | ||
| Fig. 4 (a) Potential of mean force for the desorption of limonene and carvone from non-hydroxylated surface calculated. (b) Potential of mean force for the desorption of limonene and carvone from hydroxylated surface. Both are calculated from the classical molecular dynamics simulations. | ||
We first note that these desorption free energies are substantially higher than our previous results for SiO2. This is to be expected as the partial charges of surface Ti and O atoms are much higher compared to those of SiO2. However, the trend of greater free energies of desorption compared to SiO2 are consistent with the experimental measurements of much longer desorption lifetimes for limonene and carvone on the TiO2 surface. As seen in Fig. 4(a), the desorption free energy for carvone on the non-hydroxylated with 0 ML H2O is ∼30 kJ mol−1 higher than limonene. This is consistent with our experimental measurements, and our calculations suggest is due to the close interaction between the carvone oxygen and surface Ti atoms, allowing the carvone molecule to be in closer proximity to the TiO2 surface.
Interestingly, with the addition of 0.5 ML of water for the non-hydroxylated surface, the free energy of desorption is lowered greatly by ∼40 kJ mol−1 for limonene, but the free energy of desorption is slightly higher for carvone. In our previous study of the role of hydration on the desorption of limonene from a hydroxylated SiO2 surface, we found the water molecules displace limonene from the surface, thus lowering the desorption energy.45 While this was the case for limonene in both the non-hydroxylated and hydroxylated surfaces, carvone does not seem to follow this trend for the non-hydroxylated 0.5 ML surface. However, as Table 4 indicates, the carvone molecule does not disrupt the hydrogen bonding network, so the addition of water would not displace carvone from the surface in the same way as limonene. In addition, we can see this trend by investigating the same disruption of the hydrogen bonding network as a function of distance from the surface, which can be seen in Fig. 5. These results are in agreement with our experimental results that show low amounts of carvone are displaced with the addition of water.
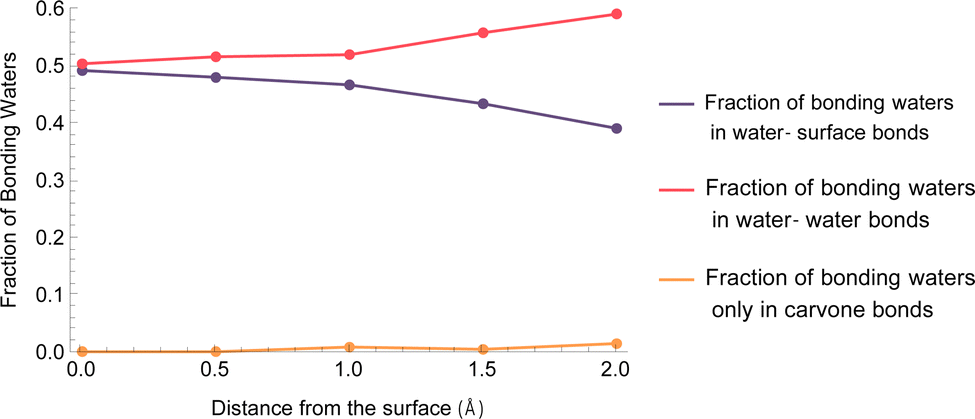 | ||
| Fig. 5 Fraction of water molecules in hydrogen bond interactions with carvone that are also in hydrogen bonds with either the TiO2 surface, surrounding waters, or no other molecules (thus disrupting the hydrogen bonding network) as a function of distance from the surface during the PMF calculation. | ||
An additional component of our study of the non-hydroxylated 0.5 ML H2O surface is that for both limonene and carvone, the potential of mean force curve shows a kink when the adsorbate is 4 Å from the surface. This occurs when the molecule is removed from the water layer, resulting in a shift in free energy. We do not see this happen in the 1 ML H2O surface system as there are more water molecules so the transition from the water layer to gas phase is less abrupt. The geometries of the adsorbate (where the carvone carbonyl oxygen atom sticks down towards the surface) as they move out of the water layer are discussed in more detail in the ESI.†
Fig. 4(b) shows the desorption free energy for the hydroxylated surfaces which are much lower compared to the free energy of desorption for the non-hydroxylated surface, and this is due to the hydroxylation of surface Ti atoms. When incorporating the role of %RH, limonene, which is easily replaced by water at the surface shows a 20 kJ mol−1 decrease in free energy of desorption and carvone shows at 4 kJ mol−1 decrease. We attribute this to hydrogen bonds formed between carvone and the surrounding water molecules. Finally, both surfaces show with 1 ML of water, the free energy of desorption is similar for each adsorbate, converging around 28 kJ mol−1 for limonene and 50 kJ mol−1 for carvone. This further validates our use of two different extrema to describe a real world TiO2 surface.
In order to further investigate the interactions between limonene and carvone on TiO2 surfaces, as well as to provide computational power spectra to compare to our experimental results, ab initio Molecular Dynamics simulations (AIMD) of each adsorbate on TiO2 have been performed. Three TiO2 surfaces have been considered to model the TiO2 surface, as detailed in the methods section. First, we have constructed a non-hydroxylated TiO2 surface with surface Ti atoms, secondly, we have constructed a non-hydroxylated TiO2 surface with 4 water molecules, and thirdly, we have constructed a fully hydroxylated surface. Fig. 6 shows each surface with limonene or carvone.
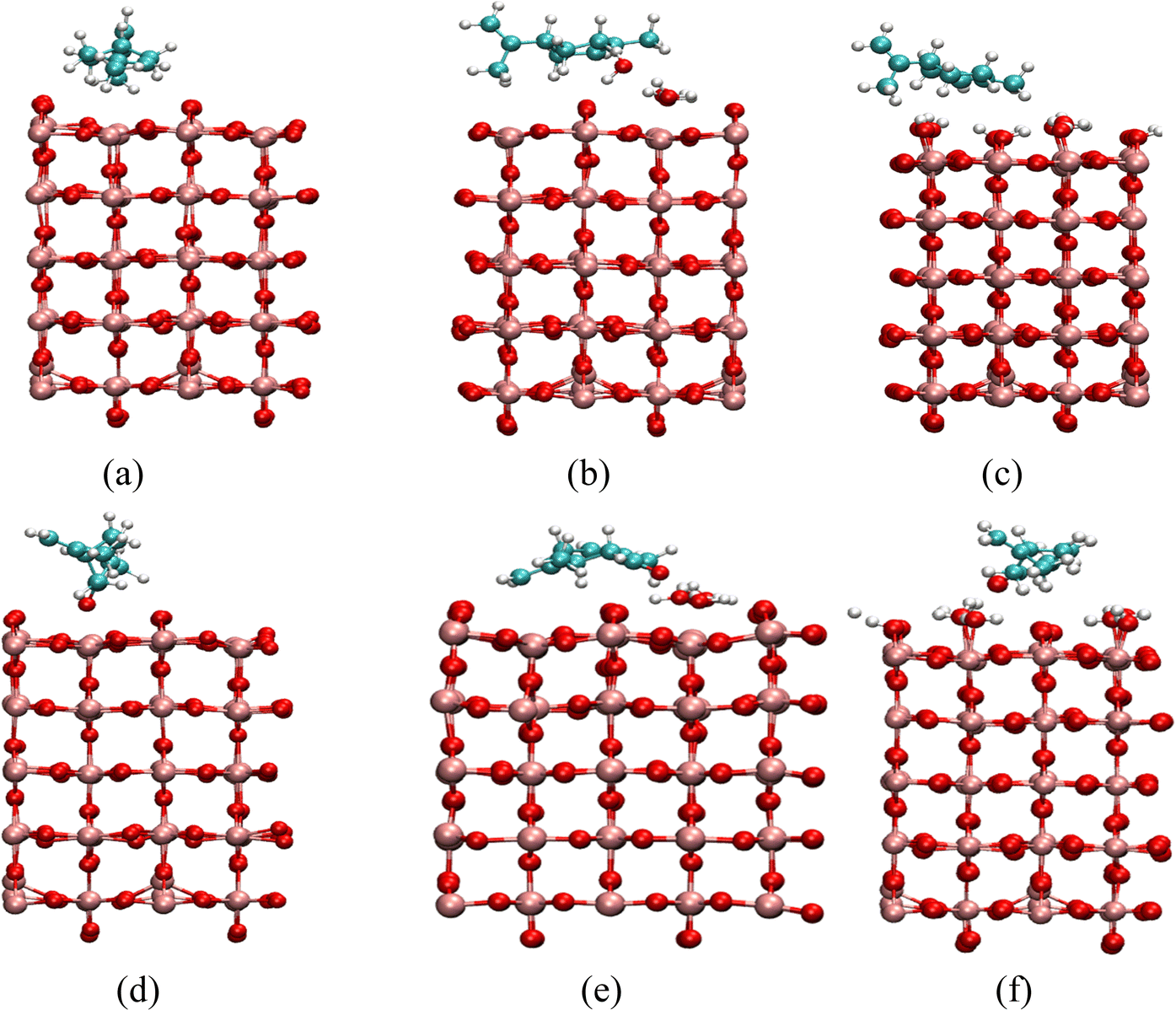 | ||
| Fig. 6 (a–c) Shows limonene on a non-hydroxylated surface with 0 water molecules, on a non-hydroxylated surface with 4 water molecules and hydroxylated surface, (d–f) shows carvone on a non-hydroxylated surface with 0 water molecules, on a non-hydroxylated surface with 4 water molecules and on a hydroxylated surface. Note a slightly larger TiO2 system was used for carvone on the non-hydroxylated surface with 4 water molecules to fully accommodate the carvone and water molecules. Red atoms correspond to O atoms, pink atoms correspond to Ti atoms, white atoms correspond to H atoms and cyan atoms correspond to carbon atoms. | ||
From our simulations, we have constructed the power spectra for the oxygen atoms of each system, as shown in Fig. 7, and the hydrogen atoms of the system, as shown in Fig. 8. Note that the positions and not exact intensities are comparable to experimental data. However, it is reasonable to compare the trends in AIMD power spectra to trends in the experimental FTIR spectra.
 | ||
| Fig. 7 (a) Oxygen power spectra for the non-hydroxylated TiO2 surface, the non-hydroxylated surface with 1 limonene molecule, and the non-hydroxylated surface with 1 carvone molecule. (b) Oxygen power spectra for the non-hydroxylated TiO2 surface with 4 water molecules, the non-hydroxylated surface with 4 water molecules and 1 limonene molecule, and the non-hydroxylated surface with 4 water molecules and 1 carvone molecule. (c) Oxygen power spectra for hydroxylated surface, hydroxylated surface with limonene molecule, and hydroxylated surface with carvone molecule. All intensity values have been rescaled to run from 0 to 1. | ||
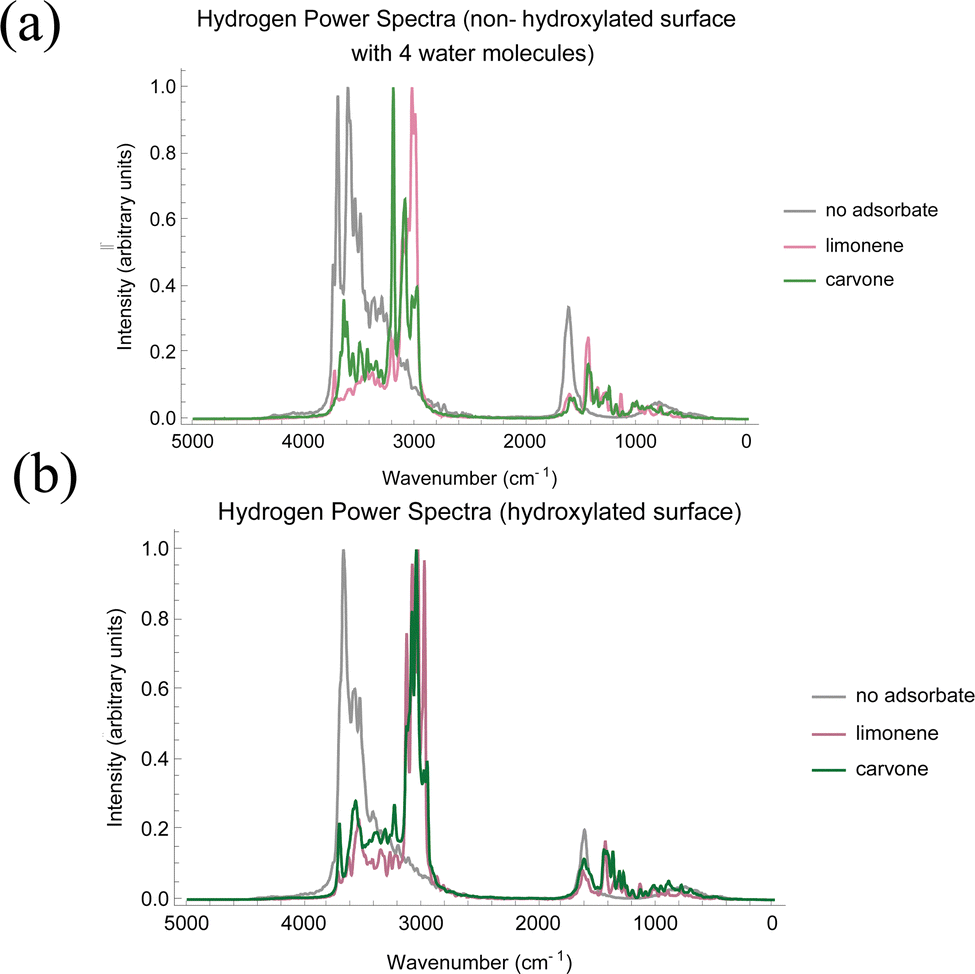 | ||
| Fig. 8 (a) Hydrogen power spectra for the non-hydroxylated TiO2 surface with 4 water molecules, the non-hydroxylated surface with 4 water molecules and 1 limonene molecule, and the non-hydroxylated surface with 4 water molecules and 1 carvone molecule. (b) Hydrogen power spectra for hydroxylated surface, hydroxylated surface with limonene molecule, and hydroxylated surface with carvone molecule. There are no hydrogen atoms on the non-hydroxylated surface, so no hydrogen power spectrum has been computed. All intensity values have been rescaled to run from 0 to 1. | ||
Fig. 7 shows consistency between the oxygen power spectra of all three simulated surfaces. For the carvone carbonyl stretch, we see agreement with our experimental results in Fig. S5 (ESI†), where the carbonyl stretch is at a higher wavenumber with the addition of water. In addition, in the hydroxylated surface, Fig. 7c, we see the presence of both the water stretch at 1620 cm−1 and the carvone carbonyl stretch at 1575 cm−1. This is more clearly seen in Fig. 9.
 | ||
| Fig. 9 Oxygen power spectra of the carvone carbonyl stretching region. We see red shifting when the carvone molecule is on the surface, and greater red shifting without water/hydroxyl groups. All intensity values have been rescaled to run from 0 to 1 and the spectra for carvone on the TiO2 surfaces has been scaled by 5× for clarity. | ||
According to the hydrogen power spectra, we see a decrease in 3700 cm−1 peak with the addition of adsorbate to the surface, which is in agreement with the Ti–OH loss shown in Fig. 2. This results in red shifting of the hydroxyl/water molecules due to the adsorbate on the surface, which is consistent with our classical MD simulations that show the formation of π-hydrogen and hydrogen bonds on the surface. In addition, in the systems with free water molecules, there was a similar interaction pattern as described in the classical MD simulation section where the adsorbate forms a π-hydrogen or hydrogen bond with a molecule also in a water–water hydrogen bond. Hydrogen bonding statistics for these two surfaces are described in Table 5.
| Surface | Average number of π-hydrogen bonds | Average number of hydrogen bonds | |
|---|---|---|---|
| Limonene | Carvone | Carvone | |
| Non-hydroxylated with 4 water molecules | 0.18 | 0 | 0.98 |
| Hydroxylated | 0.35 | 0.002 | 1.96 |
Table 5 shows consistency with our classical MD simulations where limonene can form more π-hydrogen bonds to the surface and surrounding waters compared to carvone, and that carvone can form many hydrogen bonds via the oxygen atom to the surface and surrounding waters. In our AIMD simulations, we see there is a higher average number of hydrogen bonds for carvone to the hydroxylated surface compared to the classical MD simulations; however, this difference is consistent with our previous studies of carvone on SiO2. For carvone on the non-hydroxylated TiO2 surface with 4 water molecules, there are fewer hydrogen bonds, and this is due to portions of the trajectory where the carvone oxygen atom is not closely interacting with the surrounding water molecules.
For the non-hydroxylated TiO2 surface with 0 water molecules, we see agreement between the trajectories of our classical MD simulations: the limonene molecule is positioned between two rows of bridging oxygen atoms and the carvone carbonyl oxygen atom is positioned very close to a surface Ti atom, in this case within 2.5 Å during the course of the 30 ps simulation. Using AIMD allows us to better probe the carvone carbonyl oxygen–Ti interaction, as Fig. 9 shows the red shift of the carvone carbonyl stretching frequency on the surface.
For the systems with hydrogen atoms, this red shift arises from the carbonyl oxygen forming hydrogen bonds with surrounding hydrogen atoms, at around ∼1575 cm−1. We see a greater red shift for the non-hydroxylated 0H2O surface, at around ∼1525 cm−1, which is consistent with our experimental results (Fig. S5, ESI†) that show a peak at 1648 cm−1 at <1% RH and 1660 cm−1 with 67% RH.
However, since there are no hydrogen atoms in the non-hydroxylated 0H2O surface, yet we still see a red shift, we can attribute this shift to the interaction between the carvone oxygen atom and surface Ti atom. This is highlighted in Fig. 10, which shows the difference in two key distances during the course of the 30 ps simulation. The first distance is between the carvone oxygen atom and Ti atom (the black dashed in line Fig. 10b) and between the Ti atom and the oxygen atom directly below it (the while dashed line in Fig. 10b). When this difference is negative, it corresponds to the carvone carbonyl oxygen atom being closer to the Ti atom then the crystal oxygen atom. This occurs 20% of the time during the course of the simulation. This result indicates the potential reactivity between oxygenated species and the bare TiO2 surface and will be addressed in our future work. By looking at the strength of red shifting for the CO stretch, our AIMD simulations indicate the Ti–O interaction is stronger than the carvone oxygen to water interaction, which is inconsistent with the conclusions of our experimental results. We suggest this is due to the differences between the idealized TiO2 surfaces used for simulations and the real world TiO2 surface.
 | ||
| Fig. 10 Difference between distance (1) and distance (2) during the course of the 30 ps AIMD simulation. | ||
Conclusions
This study investigated the complex interactions of limonene and carvone, indoor relevant organic molecules, with the hydroxylated TiO2 surface under different relative humidity conditions using infrared spectroscopy. The IR absorption spectra show that limonene forms Ti–OH⋯π-hydrogen bonds with the hydroxylated TiO2 surface; and our data suggest that carvone initially forms O–H hydrogen bonds (carbonyl O⋯HO–Ti) in addition to the Ti–OH⋯π-bonds with the hydroxylated TiO2 surface, then a stronger bond between carbonyl group and a Ti4+ surface atom is formed once all the free Ti–OH function groups on the TiO2 surface are occupied. An increase in %RH decreases the adsorption of limonene on TiO2 surface. This is attributed to the completive adsorption of the water coordinated via hydrogen bonding with surface Ti–OH groups. The adsorbed limonene can be completely displaced when the TiO2 surface is exposed to a relative humidity above 50% RH (∼2 MLs H2O). It was found that adsorption strength of carvone on the TiO2 surface is much stronger than that of limonene, which is supported by the evidence that only 25% of carvone is displaced by the adsorbed water. This implies that the carvone adsorption could significantly inhibit the subsequent interactions of other VOCs, such as limonene, on the TiO2 surface.Two kinds of Molecular Dynamics simulations, classical force field-based and ab initio molecular dynamics (AIMD) simulations were performed in order to gain an atomistic perspective of the adsorption of limonene and carvone on TiO2. Due to complications on how to best model the TiO2 surface, two classical force field-based surfaces and three AIMD surfaces were constructed. These surfaces showed consistency between surface interactions present and provided agreement with the experimental conclusions that the adsorption strength of carvone on the TiO2 surface is stronger compared to that of limonene. Our results indicate that limonene forms more π-hydrogen bonds with water molecules engaged in hydrogen bonding with the surface, and due to the competitive adsorption of water molecules, suggests why there is a decrease in adsorption of limonene on TiO2 in surfaces with >1% RH. However, the formation of oxygen hydrogen bonds between carvone and the surrounding water molecules allows carvone to be retained on the TiO2 surface with increasing relative humidity. In addition, our AIMD simulation of a non-hydroxylated TiO2 surface suggests an interaction between the carvone oxygen atom and surface Ti atom that may result in surface reactivity. The results further indicate that increases in relative humidity can significantly decrease photocatalytic oxidation efficiency of limonene due to the adsorption of water on the active sites of TiO2 surface. The role of the adsorbed water in the photocatalytic oxidation of carvone on TiO2 surface needs further examination. Future studies will focus on the TiO2 surface chemistry reaction mechanisms of relevant indoor reactive oxygenated species. There are several critical factors such as relative humidity, temperature, and light will be explored.
Author contributions
Hanyu Fan made experimental measurements; Elianna S. Frank performed theoretical simulations. Vicki H. Grassian (oversaw all experimental aspects) and Douglas J. Tobias (oversaw all theoretical aspects) participated in the analysis of results and conclusions and are corresponding authors. All authors contributed to either the writing and/or editing of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the support of the Alfred P. Sloan Foundation (G-2020-12675 to VHG; G-2019-12306 to DJT).References
- N. E. Klepeis, W. C. Nelson, W. R. Ott, J. P. Robinson, A. M. Tsang, P. Switzer, J. V. Behar, S. C. Hern and W. H. Engelmann, J. Exposure Sci. Environ. Epidemiol., 2001, 11, 231–252 CrossRef CAS PubMed.
- J. Wu, Y. Alipouri, H. Luo and L. Zhong, J. Hazard. Mater., 2022, 421, 126766 CrossRef CAS PubMed.
- E. Gallego, X. Roca, J. F. Perales and X. Guardino, J. Environ. Sci., 2009, 21, 333–339 CrossRef CAS.
- V. V. Tran, D. Park and Y.-C. Lee, Int. J. Environ. Res. Public Health, 2020, 17, 2927 CrossRef PubMed.
- I. Rivas, J. Fussell, F. Kelly and X. Querol, Indoor Air Pollution, 2019, 1–34, 10.1039/9781788016179-00001.
- C. J. Weschler and N. Carslaw, Environ. Sci. Technol., 2018, 52, 2419–2428 CrossRef CAS PubMed.
- V. Davamani, D. Mohan, E. Parameswari, S. Arulmani, P. Sebastian and I. Tamilselvan, Int. Res. J. Pure Appl. Chem., 2020, 40–61, DOI:10.9734/irjpac/2020/v21i930197.
- J. P. D. Abbatt and C. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 25–48 RSC.
- EPA, https://www.epa.gov/indoor-air-quality-iaq/introduction-indoor-air-quality, in 2021.
- WHO, https://www.who.int/news-room/fact-sheets/detail/household-air-pollution-and-health, in 2021.
- S. Wang, H. M. Ang and M. O. Tade, Environ. Int., 2007, 33, 694–705 CrossRef CAS PubMed.
- C. H. Ao and S. C. Lee, Chem. Eng. Sci., 2005, 60, 103–109 CrossRef CAS.
- B. Guieysse, C. Hort, V. Platel, R. Munoz, M. Ondarts and S. Revah, Biotechnol. Adv., 2008, 26, 398–410 CrossRef CAS PubMed.
- P. J. Asilevi, P. Boakye, S. Oduro-Kwarteng, B. Fei-Baffoe and Y. A. Sokama-Neuyam, Sci. Rep., 2021, 11, 22830 CrossRef CAS.
- Y. Huang, S. S. Ho, Y. Lu, R. Niu, L. Xu, J. Cao and S. Lee, Molecules, 2016, 21, 56 CrossRef PubMed.
- H. Chen, C. E. Nanayakkara and V. H. Grassian, Chem. Rev., 2012, 112, 5919–5948 CrossRef CAS PubMed.
- D. B. Collins and D. K. Farmer, Environ. Sci. Technol., 2021, 55, 12172–12179 CrossRef CAS PubMed.
- Z. Shayegan, C.-S. Lee and F. Haghighat, Chem. Eng. J., 2018, 334, 2408–2439 CrossRef CAS.
- H. Yu, K. Zhang and C. Rossi, Indoor Built Environ., 2007, 16, 529–537 CrossRef CAS.
- A. Rabajczyk, M. Zielecka, W. Klapsa and A. Dziechciarz, Materials, 2021, 14, 2161 CrossRef CAS PubMed.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- A.-I. Gopalan, J.-C. Lee, G. Saianand, K.-P. Lee, P. Sonar, R. Dharmarajan, Y.-L. Hou, K.-Y. Ann, V. Kannan and W.-J. Kim, Nanomaterials, 2020, 10, 1854 CrossRef CAS.
- L. Frazer, Environ. Health Perspect., 2001, 109, A174–A177 CrossRef CAS.
- F. Thevenet, L. Olivier, F. Batault, L. Sivachandiran and N. Locoge, Chem. Eng. J., 2015, 281, 126–133 CrossRef CAS.
- H. O. Seo, D. Kim, K.-D. Kim, E. J. Park, C. Sim and Y. D. Kim, Adsorption, 2013, 19, 1181–1187 CrossRef CAS.
- A. H. Mamaghani, F. Haghighat and C.-S. Lee, Appl. Catal., B, 2017, 203, 247–269 CrossRef CAS.
- A. P. Ault, V. H. Grassian, N. Carslaw, D. B. Collins, H. Destaillats, D. J. Donaldson, D. K. Farmer, J. L. Jimenez, V. F. McNeill, G. C. Morrison, R. E. O’Brien, M. Shiraiwa, M. E. Vance, J. R. Wells and W. Xiong, Chem, 2020, 6, 3203–3218 CAS.
- Y. Liu, A. G. Bé, V. W. Or, M. R. Alves, V. H. Grassian and F. M. Geiger, Cell Rep. Phys. Sci., 2020, 1, 100256 CrossRef.
- A. H. Mamaghani, F. Haghighat and C.-S. Lee, Build. Environ., 2021, 189, 107518 CrossRef.
- F. Batault, F. Thevenet, V. Hequet, C. Rillard, L. Le Coq and N. Locoge, Chem. Eng. J., 2015, 264, 197–210 CrossRef CAS.
- M. Nagao and Y. Suda, Langmuir, 1989, 5, 42–47 CrossRef CAS.
- M. Singh, N. Zhou, D. K. Paul and K. J. Klabunde, J. Catal., 2008, 260, 371–379 CrossRef CAS.
- M. R. Alves, Y. Fang, K. J. Wall, V. Vaida and V. H. Grassian, J. Phys. Chem. A, 2019, 123, 7661–7671 CrossRef CAS PubMed.
- M. R. Nimlos, E. J. Wolfrum, M. L. Brewer, J. A. Fennell and G. Bintner, Environ. Sci. Technol., 1996, 30, 3102–3110 CrossRef CAS.
- T. N. Obee and R. T. Brown, Environ. Sci. Technol., 1995, 29, 1223–1231 CrossRef CAS.
- R. M. Alberici and W. F. Jardim, Appl. Catal., B, 1997, 14, 55–68 CrossRef CAS.
- L. Zhong, C.-S. Lee and F. Haghighat, J. Hazard. Mater., 2012, 243, 340–349 CrossRef CAS.
- L. Huang, E. S. Frank, M. Shrestha, S. Riahi, D. J. Tobias and V. H. Grassian, Environ. Sci. Technol., 2021, 55, 6623–6630 CrossRef CAS PubMed.
- A. K. Boulamanti and C. J. Philippopoulos, Atmos. Environ., 2009, 43, 3168–3174 CrossRef CAS.
- A. K. Boulamanti, C. A. Korologos and C. J. Philippopoulos, Atmos. Environ., 2008, 42, 7844–7850 CrossRef CAS.
- G. Rubasinghege and V. H. Grassian, Chem. Commun., 2013, 49, 3071–3094 RSC.
- D. Vildozo, R. Portela, C. Ferronato and J.-M. Chovelon, Appl. Catal., B, 2011, 107, 347–354 CrossRef CAS.
- L. Zhang, W. Anderson, S. Sawell and C. Moralejo, Chemosphere, 2007, 68, 546–553 CrossRef CAS PubMed.
- Q. Geng, Q. Guo and X. Yue, Ind. Eng. Chem. Res., 2010, 49, 4644–4652 CrossRef CAS.
- E. S. Frank, H. Fan, M. Shrestha, S. Riahi, D. J. Tobias and V. H. Grassian, J. Phys. Chem. A, 2020, 124, 10592–10599 CrossRef CAS PubMed.
- Y. Fang, P. S. J. Lakey, S. Riahi, A. T. McDonald, M. Shrestha, D. J. Tobias, M. Shiraiwa and V. H. Grassian, Chem. Sci., 2019, 10, 2906–2914 RSC.
- H. Fan, E. S. Frank, P. S. J. Lakey, M. Shiraiwa, D. J. Tobias and V. H. Grassian, J. Phys. Chem. C, 2022, 126, 6267–6279 CrossRef CAS.
- G. Munuera and F. S. Stone, Discuss. Faraday Soc., 1971, 52, 205–214 RSC.
- C. Zhang and P. J. D. Lindan, J. Chem. Phys., 2004, 121, 3811–3815 CrossRef CAS.
- P. J. D. Lindan, N. M. Harrison and M. J. Gillan, Phys. Rev. Lett., 1998, 80, 762–765 CrossRef CAS.
- L. A. Harris and A. A. Quong, Phys. Rev. Lett., 2004, 93, 086105 CrossRef PubMed.
- L.-M. Liu, C. Zhang, G. Thornton and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 161415 CrossRef.
- J. Margineda and N. J. English, Chem. Phys. Lett., 2020, 743, 137164 CrossRef CAS.
- Materials Project. https://Materialsproject.Org/Materials/Mp-2657/.
- E. G. Brandt and A. P. Lyubartsev, J. Phys. Chem. C, 2015, 119, 18110–18125 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- LAMMPS Molecular Dynamics Simulator. https://lammps.sandia.gov/index.html (accessed Feb 25, 2020).
- H. C. Andersen, J. Comput. Phys., 1983, 52, 24–34 CrossRef CAS.
- R. E. Isele-Holder, W. Mitchell and A. E. Ismail, J. Chem. Phys., 2012, 137, 174107 CrossRef PubMed.
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- A. Grossfield, “WHAM: the weighted histogram method”, version 2.0.9.1. https://membrane.urmc.rochester.edu/?page_id=126 (accessed Mar 11, 2020).
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. D. Kühne, M. Iannuzzi, M. D. Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2003, 118, 4365–4369 CrossRef CAS.
- P. Pulay, J. Comput. Chem., 1982, 3, 556–560 CrossRef CAS.
- M. Thomas, M. Brehm, R. Fligg, P. Vöhringer and B. Kirchner, Phys. Chem. Chem. Phys., 2013, 15, 6608–6622 RSC.
- M. Brehm and B. Kirchner, J. Chem. Inf. Model., 2011, 51, 2007–2023 CrossRef CAS PubMed.
- A. L. Goodman, E. T. Bernard and V. H. Grassian, J. Phys. Chem. A, 2001, 105, 6443–6457 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The ESI contains 7 figures and 2 tables including: Fig. S1. FTIR spectra of gas-phase limonene; Fig. S2. FTIR spectra of gas-phase carvone; Fig. S3. FTIR spectra of limonene on SiO2 surface; Fig. S4 and S5. FTIR spectra of limonene and carvone under different RH conditions; Fig. S6. FTIR spectra of water on TiO2 surface under different pressures; Fig. S7. Density profiles for limonene on each surface; Fig. S8. Snapshots of Hydrogen Bonding Interactions; Fig. S9. Discussion of non-hydroxylated 0.5 ML kink in PMF; Tables S1 and S2. List of simulations for all classical molecular dynamics and all ab initio molecular dynamics. See DOI: https://doi.org/10.1039/d2cp03021g |
| ‡ Hanyu Fan and Elianna S. Frank contributed equally to this work. |
| This journal is © the Owner Societies 2022 |