
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenchmark ab initio potential energy surface mapping of the F + CH3NH2 reaction
Tímea
Szűcs
 and
Gábor
Czakó
*
and
Gábor
Czakó
*
MTA-SZTE Lendület Computational Reaction Dynamics Research Group, Interdisciplinary Excellence Centre and Department of Physical Chemistry and Materials Science, Institute of Chemistry, University of Szeged, Rerrich Béla tér 1, Szeged H-6720, Hungary. E-mail: gczako@chem.u-szeged.hu
First published on 3rd August 2022
Abstract
This electronic structure study reveals four exothermic and two endothermic reaction pathways of the F + CH3NH2 system: the barrierless hydrogen abstraction from the methyl/amino group (HF + CH2NH2/CH3NH), amino/methyl substitution (NH2 + CH3F and CH3 + NH2F) and hydrogen substitution from the two functional groups (H + CH2FNH2/CH3NHF). The benchmark classical and adiabatic energies are obtained using a high-accuracy composite ab initio approach, where the gold-standard explicitly-correlated coupled cluster method (CCSD(T)-F12b) is applied with the correlation-consistent polarized valence quintuple-zeta F12 basis set (cc-pV5Z-F12) and further additive energy contributions. Considering indispensable post-(T) correlation, core correlation, scalar relativistic, spin–orbit and harmonic zero-point energy corrections, the obtained global minimum of the potential energy surface is the post-reaction CH2NH2⋯HF complex in the product channel. Although each substitution pathway has a high barrier, the energies of amino-substitution and methyl-hydrogen-substitution products are below the energy of the reactants, as well as the submerged-barrier hydrogen-abstraction pathways.
I. Introduction
The F + H2 reaction is one of the most widely studied fundamental chemical processes beyond the H + H2 system.1–6 Unlike H + H2, F + H2 involves a ‘heavy’ atom, making its electronic-structure description more challenging. Nevertheless, a high-level ab initio potential energy surface (PES) was developed for the F + H2 system in the 1990s,3 enabling quantum scattering computations to provide deep insight into the atomic-level state-to-state dynamics. In 2013 Schaefer and co-workers7 asked the question, “What comes after F + H2?”. Of course, the reactions of F with H2O,8 NH3,9 CH4,10,11 and C2H612,13 are more complex due to the larger number of electrons and nuclei making both the electronic structure and nuclear motion parts more challenging. In the past few decades, significant progress has been achieved on these systems as well, advancing our fundamental knowledge on polyatomic reactivity.8–13 Thus, the next step forward could be to replace the molecular reactant with a molecule that has two functional groups, such as, for example, CH3OH and CH3NH2. Theoretical research toward this direction has also been started, especially in the case of the F + CH3OH reaction.14–17In the present study we focus on the F + CH3NH2 system following the early experiments18,19 on the F + CH3ND2 reaction, which measured the HF/DF vibrational and rotational distributions and the theoretical study of Schaefer and co-workers,7 which characterized the two hydrogen-abstraction pathways. What comes after the previous work on the title reaction? First, it is important to note that the previous computations only considered the H-abstraction channels, whereas our recent studies on the F/Cl/Br/I/OH + C2H620,21 and Cl + CH3NH2/CH3CN22,23 reactions revealed several other channels, such as hydrogen and methyl substitution, for the reactions of C2H6, and the picture is even more complex in the cases of CH3NH2 and CH3CN. Thus, in the present study, we plan to characterize all the possible, chemically relevant pathways of the title reaction, thereby providing new qualitative insights into the mechanisms of the F + CH3NH2 process. Furthermore, knowing the challenging electronic structure of the reactions of the fluorine atom, we plan to use the highest technically feasible level of theory to achieve sub-chemical accuracy for the relative energies of the stationary points. In 2013 Schaefer and co-workers7 used the standard CCSD(T) method with the aug-cc-pVnZ [n = 2(D), 3(T), 4(Q)] basis sets; however, this previous work did not consider accelerating the basis-set convergence using the explicitly-correlated CCSD(T)-F12b method, electron correlation beyond CCSD(T), and the effects of core correlation, scalar relativity, and spin–orbit coupling. In the present study, we take all of the above-mentioned corrections into account, thereby anchoring the relative energies of the stationary points of the title reaction. These new benchmark data will guide future PES developments and reaction-dynamics simulations, whose results may be directly compared with experiments. In Section II we describe the details of the composite ab initio computations; the results are presented and discussed in Section III; and the paper ends with our summary and conclusions in Section IV.
II. Computational details
During the characterization of the potential energy surface, we build upon previous work7,22 and chemical intuition to identify the important stationary points of the F + CH3NH2 reaction. We use the restricted open-shell second-order Møller–Plesset perturbation theory (RMP2)24 and the correlation-consistent aug-cc-pVDZ basis set25 to determine the primary geometries, energies and harmonic vibrational frequencies. In order to refine the structures, further optimizations are performed using the restricted open-shell Hartree–Fock (ROHF)-based unrestricted explicitly correlated coupled-cluster singles, doubles and perturbative triples (CCSD(T)-F12b)26 method accompanied with the aug-cc-pVDZ basis set. To increase the accuracy, based on these structures thus obtained, CCSD(T)-F12b/aug-cc-pVTZ geometry and frequency computations are performed as well as further single-point CCSD(T)-F12b computations with the aug-cc-pVQZ and cc-pVnZ-F12 [n = 2(D), 3(T), 4(Q), 5] basis sets27 being executed by keeping the minima and saddle points fixed at the CCSD(T)-F12b/aug-cc-pVTZ geometries.The most accurate benchmark classical relative energies are obtained in this work using the following expression:
| ΔEclassical = CCSD(T)-F12b/cc-pV5Z-F12 + δ[CCSDT] + δ[CCSDT(Q)] + Δcore + Δrel + ΔSO, | (1) |
To acquire the post-(T) correlation energy contributions, unrestricted CCSD(T),28 CCSDT29 and CCSDT(Q)30 computations – using the unrestricted Hartree–Fock (UHF) reference and the aug-cc-pVDZ basis set – are performed using the MRCC program suite31,32 interfaced to MOLPRO.33 The corrections are calculated as follows:
| δ[CCSDT] = ΔE(CCSDT/aug-cc-pVDZ) − ΔE(CCSD(T)/aug-cc-pVDZ), | (2) |
| δ[CCSDT(Q)] = ΔE(CCSDT(Q)/aug-cc-pVDZ) − ΔE(CCSDT/aug-cc-pVDZ). | (3) |
| Δcore = ΔE(AE-CCSD(T)-F12b/cc-pCVTZ-F12) − ΔE(FC-CCSD(T)-F12b/cc-pCVTZ-F12). | (4) |
| Δrel = ΔE(DK-AE-CCSD(T)/aug-cc-pwCVTZ-DK) − ΔE(AE-CCSD(T)/aug-cc-pwCVTZ). | (5) |
The interacting-states approach37 is used with the Davidson-corrected38 all-electron multi-reference configuration interaction39 (MRCI+Q) method to determine the spin–orbit (SO) coupling effect. The aug-cc-pwCVTZ basis set40 is used and an active space of 5 electrons in the 3 spatial 2p-like fluorine orbitals is applied. During the SO computations the diagonal elements of the 6 × 6 SO matrix are replaced by the Davidson-corrected MRCI energies and the SO eigenstates are obtained by diagonalizing this matrix. Subtracting the non-SO ground-state (non-SO1) energy from the SO ground-state (SO1) energy gives the ΔSO correction:
| ΔSO = SO1(MRCI+Q/aug-cc-pwCVTZ) − non-SO1(MRCI+Q/aug-cc-pwCVTZ). | (6) |
Considering the zero-point energy contributions (ΔZPE) obtained at the CCSD(T)-F12b/aug-cc-pVTZ level of theory, the vibrationally adiabatic relative energies are determined as follows:
| ΔEadiabatic = CCSD(T)-F12b/cc-pV5Z-F12 + δ[CCSDT] + δ[CCSDT(Q)] + Δcore + Δrel + ΔSO + ΔZPE. | (7) |
The computations detailed above are accomplished using the MOLPRO program package.33
III. Results and discussion
1. Benchmark energies and geometries
Our computations reveal the main pathways of the F + CH3NH2 reaction. When a fluorine atom attacks a hydrogen atom at either the methyl or amino group, this gives two different abstraction channels, leading to the HF + CH2NH2 and to HF + CH3NH products, respectively. During a reaction, a hydrogen atom can be substituted as well, resulting in the H + CH2FNH2 or H + CH3NHF products for the two functional groups. The processes for which the attacking atom substitutes NH2 (leading to CH3F + NH2) or CH3 (leading to NH2F + CH3) are also investigated in this study. To illustrate the determined benchmark energies of the stationary points relative to the reactants, we use the schematic energy diagram of the seven examined reaction pathways in Fig. 1. The classical(adiabatic) relative energy values are derived from eqn (1) and (7) and are presented in Table 1. The key structural parameters obtained at the three different levels of theory are shown in Fig. 2, which provide useful information for identifying the stationary structures of the PES.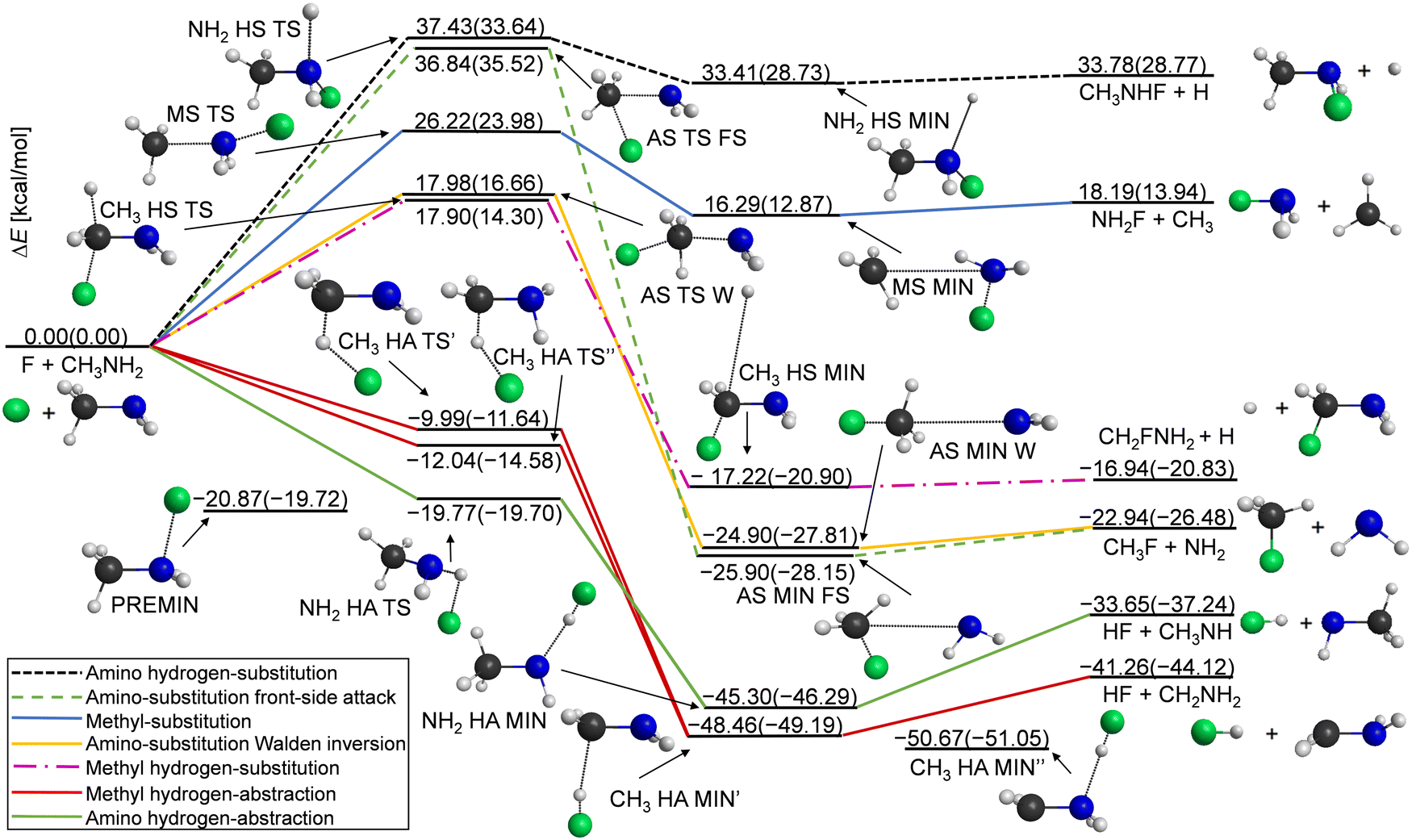 | ||
| Fig. 1 Schematic energy diagram of the F(2P3/2) + CH3NH2 reaction's potential energy surface, illustrating the CCSD(T)-F12b/aug-cc-pVTZ optimized geometries and benchmark classical(adiabatic) relative energies. PREMIN is not connected because it can perform an important role in more pathways. | ||
| Stationary points | CCSD(T)-F12b | δ[T]b | δ[(Q)]c | Δ core | Δ rel | Δ SO | Classicalg | Δ ZPE | Adiabatici |
|---|---|---|---|---|---|---|---|---|---|
| cc-pV5Z-F12a | |||||||||
| a CCSD(T)-F12b/cc-pV5Z-F12 relative energies. b CCSDT – CCSD(T) obtained using the aug-cc-pVDZ basis set. c CCSDT(Q) – CCSDT obtained using the aug-cc-pVDZ basis set. d Core correlation correction obtained as the difference between all-electron and frozen-core CCSD(T)-F12b/cc-pCVTZ-F12 relative energies. e Scalar relativistic effect obtained as the difference between DK-AE-CCSD(T)/aug-cc-pwCVTZ-DK and AE-CCSD(T)/aug-cc-pwCVTZ relative energies. f Spin–orbit (SO) corrections obtained as the difference between the SO and non-SO ground-state MRCI+Q/aug-cc-pwCVTZ relative energies. g Benchmark classical relative energies obtained as CCSD(T)-F12b/cc-pV5Z-F12 relative energies + δ[T] (b) + δ[(Q)] (c) + Δcore (d) + Δrel (e) + ΔSO (f). h Zero-point energy (ZPE) corrections obtained at CCSD(T)-F12b/aug-cc-pVTZ. i Benchmark adiabatic relative energies obtained as classical relative energies (g) + ΔZPE (h). | |||||||||
| PREMIN | −20.61 | −0.20 | −0.46 | −0.08 | +0.11 | +0.37 | −20.87 | +1.15 | −19.72 |
| CH3 HA TS′ | −9.24 | −0.66 | −0.52 | −0.12 | +0.20 | +0.34 | −9.99 | −1.65 | −11.64 |
| CH3 HA TS′′ | −11.39 | −0.54 | −0.46 | −0.14 | +0.14 | +0.36 | −12.04 | −2.54 | −14.58 |
| NH2 HA TS | −19.31 | −0.40 | −0.44 | −0.08 | +0.10 | +0.36 | −19.77 | +0.07 | −19.70 |
| AS TS W | 18.37 | −0.60 | −0.62 | +0.47 | +0.01 | +0.36 | 17.98 | −1.32 | 16.66 |
| AS TS FS | 37.72 | −1.08 | −0.69 | +0.53 | +0.00 | +0.37 | 36.84 | −1.32 | 35.52 |
| MS TS | 26.77 | −0.75 | −0.63 | +0.49 | −0.04 | +0.37 | 26.22 | −2.24 | 23.98 |
| CH3 HS TS | 18.30 | −0.42 | −0.58 | +0.12 | +0.11 | +0.37 | 17.90 | −3.60 | 14.30 |
| NH2 HS TS | 37.53 | −0.28 | −0.52 | +0.33 | +0.01 | +0.37 | 37.43 | −3.79 | 33.64 |
| CH3 HA MIN′ | −48.51 | −0.08 | −0.22 | −0.22 | +0.20 | +0.37 | −48.46 | −0.73 | −49.19 |
| CH3 HA MIN′′ | −50.80 | −0.06 | −0.19 | −0.19 | +0.20 | +0.37 | −50.67 | −0.38 | −51.05 |
| NH2 HA MIN | −45.50 | −0.16 | −0.12 | −0.01 | +0.13 | +0.37 | −45.30 | −0.99 | −46.29 |
| AS MIN W | −25.42 | −0.17 | −0.05 | +0.30 | +0.07 | +0.37 | −24.90 | −2.91 | −27.81 |
| AS MIN FS | −26.40 | −0.17 | −0.05 | +0.28 | +0.08 | +0.37 | −25.90 | −2.24 | −28.15 |
| MS MIN | 16.03 | −0.18 | −0.25 | +0.28 | +0.04 | +0.37 | 16.29 | −3.42 | 12.87 |
| CH3 HS MIN | −17.69 | +0.07 | −0.19 | +0.06 | +0.17 | +0.37 | −17.22 | −3.68 | −20.90 |
| NH2 HS MIN | 33.10 | +0.00 | −0.34 | +0.23 | +0.05 | +0.37 | 33.41 | −4.68 | 28.73 |
| CH3NHF + H | 33.47 | +0.00 | −0.34 | +0.23 | +0.05 | +0.37 | 33.78 | −5.00 | 28.77 |
| NH2F + CH3 | 17.91 | −0.16 | −0.24 | +0.28 | +0.04 | +0.37 | 18.19 | −4.25 | 13.94 |
| CH2FNH2 + H | −17.42 | +0.07 | −0.19 | +0.06 | +0.17 | +0.37 | −16.94 | −3.89 | −20.83 |
| CH3F + NH2 | −23.47 | −0.17 | −0.04 | +0.29 | +0.07 | +0.37 | −22.94 | −3.53 | −26.48 |
| HF + CH3NH | −33.92 | −0.16 | −0.11 | +0.07 | +0.10 | +0.37 | −33.65 | −3.59 | −37.24 |
| HF + CH2NH2 | −41.38 | −0.07 | −0.18 | −0.19 | +0.20 | +0.37 | −41.26 | −2.86 | −44.12 |
| CH4 + NHF | 3.77 | −0.38 | −0.19 | +0.35 | −0.05 | +0.37 | 3.87 | −3.84 | 0.03 |
| NH3 + CH2F | −29.96 | −0.15 | −0.11 | +0.07 | +0.15 | +0.36 | −29.65 | −3.12 | −32.77 |
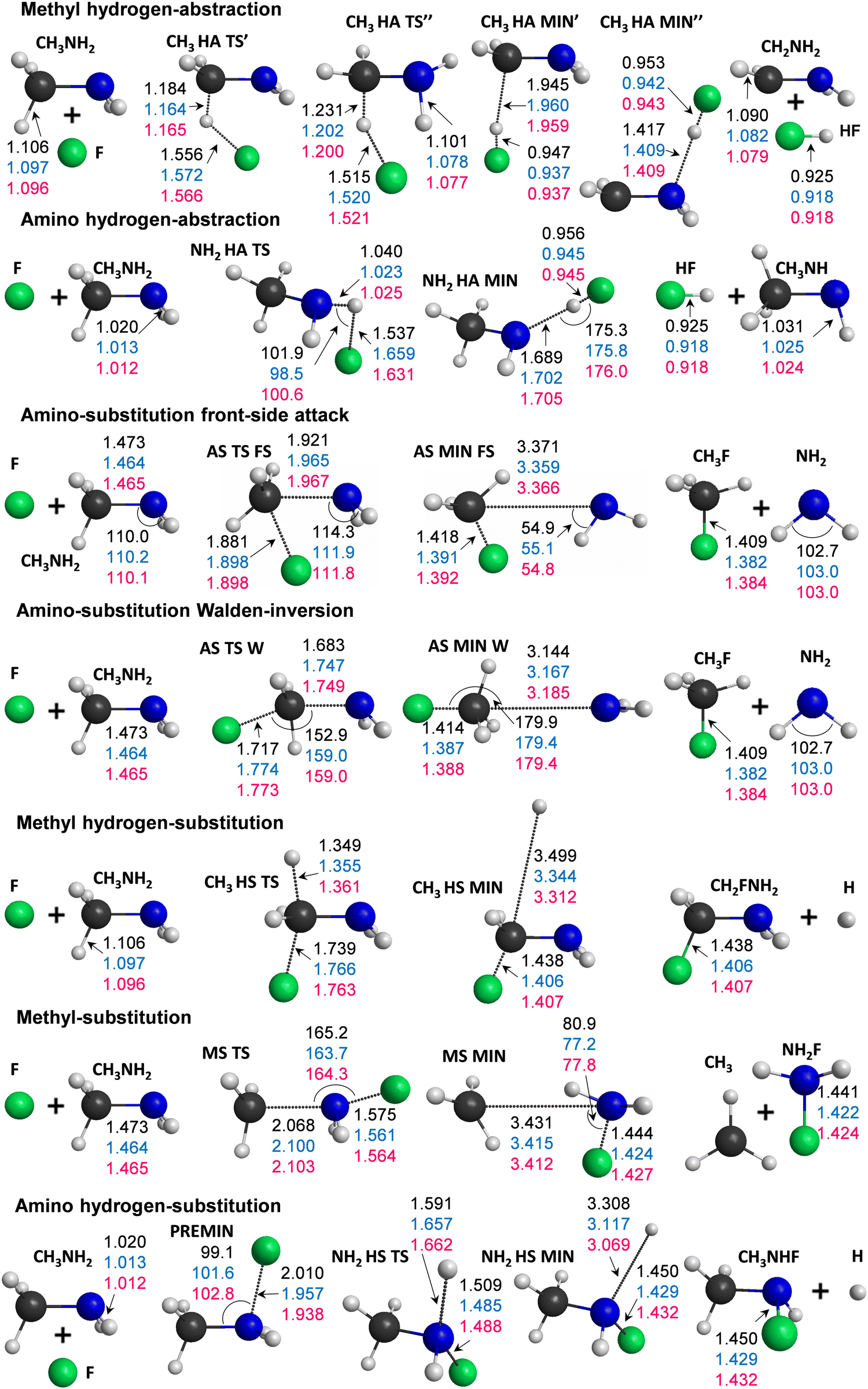 | ||
| Fig. 2 Optimized geometries of the reactants, stationary-points and products, obtained at the CCSD(T)-F12b/aug-cc-pVTZ level of theory with the characteristic distances (Å) and angles (degree) at three levels of theory: MP2/aug-cc-pVDZ (black), CCSD(T)-F12b/aug-cc-pVDZ (blue) and CCSD(T)-F12b/aug-cc-pVTZ (red). | ||
The most exothermic mechanisms are the hydrogen abstraction (HA) reactions, with ΔE(ΔH0) = −41.26(−44.12) kcal mol−1 in the case of hydrogen transfer from the methyl group and ΔE(ΔH0) = −33.65(−37.24) kcal mol−1 for HA from the amino group. These are followed by the substitution pathways as can be seen in Fig. 1. We found that the CH3 HA MIN′′ structure in the product channel is the global minimum of the PES, where the formed HF binds to the nitrogen atom through a 1.409 Å N⋯H distance. We present two transition states in the methyl hydrogen-abstraction channel, where the CH3 HA TS′ has very similar geometrical and energetic parameters to the structure reported by Feng et al.,7 but the classical(adiabatic) relative energy is more negative by 2.05(2.94) kcal mol−1 in the case of TS′′. The TS′ and TS′′ structures differ in bond lengths by a few hundredths of an ångström and in angles, mainly the plane of the C–N–H atoms relative to the plane of the C–H–F atoms. In the entrance channel a reactant-like pre-reaction complex with a substantial dissociation energy (De(D0) = 20.87(19.72) kcal mol−1) is obtained with a C–N–F angle of 102.8°.
Amino substitution (AS) and methyl-hydrogen substitution (CH3 HS) are thermodynamically favored like the hydrogen abstractions; however, these substitutions are kinetically not preferred. We could identify front-side attack (FS) and Walden-inversion (W) mechanisms in the case of amino-substitution, where the former has a higher barrier (with 18.86 kcal mol−1) as expected. The classical relative energies of the AS MIN FS and AS MIN W structures in the product channel differ by 1 kcal mol−1, which is in accordance with the very similar lengths of the C–N (3.366/3.185 Å) and C–F (1.392/1.388 Å) bonds. The dipole-polarized-atom force-stabilized CH3 HS MIN complex has the lowest dissociation energy, De(D0) = 0.28(0.07) kcal mol−1, among the product-like complexes.
The most endothermic pathway is hydrogen substitution from the amino group, and it has the highest barrier. The exit-channel minimum of this process is quite shallow, like in the other HS pathway. The methyl substitution is endothermic as well (ΔE(ΔH0) = 18.19(13.94) kcal mol−1), but it has a lower relative energy than the above-mentioned NH2 HS channel (ΔE(ΔH0) = 33.78(28.77) kcal mol−1).
2. Convergence of the relative energies
Depending on the level of theory and the basis sets, the convergence of the relative energies is investigated. To compare the MP2 and CCSD(T)-F12b methods, the difference between the relative energies calculated using the same aug-cc-pVDZ basis set can be a guide. The average absolute difference between the results is 0.80 kcal mol−1, but as shown in Table 2, in some cases the MP2 method can seriously overestimate or underestimate the CCSD(T)-F12b relative energies. An example of the former is the barrier height of AS TS W (by 2.5 kcal mol−1) and the latter occurs in the case of PREMIN (by 6.3 kcal mol−1) or the hydrogen-substitution channels (by 3.9–5.0 kcal mol−1). We can conclude that the CCSD(T)-F12b method is needed to achieve chemical accuracy, where the uncertainty is under 1 kcal mol−1.| Stationary points | MP2 | CCSD(T)-F12b | ||||||
|---|---|---|---|---|---|---|---|---|
| aug-cc-pVDZ | aug-cc-pVDZ | aug-cc-pVTZ | aug-cc-pVQZ | cc-pVDZ-F12 | cc-pVTZ-F12 | cc-pVQZ-F12 | cc-pV5Z-F12 | |
| PREMIN | −27.63 | −21.31 | −20.63 | −20.69 | −20.39 | −20.26 | −20.54 | −20.61 |
| CH3 HA TS′ | −7.33 | −9.14 | −9.39 | −9.38 | −8.07 | −9.00 | −9.21 | −9.24 |
| CH3 HA TS′′ | −11.59 | −11.45 | −11.44 | −11.52 | −10.25 | −11.10 | −11.33 | −11.39 |
| NH2 HA TS | −17.30 | −18.61 | −19.38 | −19.40 | −18.04 | −19.07 | −19.27 | −19.31 |
| AS TS W | 21.28 | 18.81 | 18.26 | 18.27 | 19.52 | 18.62 | 18.41 | 18.37 |
| AS TS FS | 39.88 | 38.19 | 37.53 | 37.66 | 38.77 | 37.93 | 37.76 | 37.72 |
| MS TS | 28.89 | 27.04 | 26.73 | 26.72 | 27.55 | 27.00 | 26.84 | 26.77 |
| CH3 HS TS | 15.37 | 18.57 | 18.30 | 18.22 | 19.68 | 18.67 | 18.38 | 18.30 |
| NH2 HS TS | 37.45 | 37.18 | 37.45 | 37.48 | 38.30 | 37.77 | 37.60 | 37.53 |
| CH3 HA MIN′ | −50.54 | −48.76 | −48.61 | −48.68 | −47.64 | −48.20 | −48.44 | −48.51 |
| CH3 HA MIN′′ | −53.07 | −51.01 | −50.93 | −50.99 | −49.89 | −50.49 | −50.74 | −50.80 |
| NH2 HA MIN | −46.19 | −45.80 | −45.62 | −45.67 | −44.92 | −45.28 | −45.45 | −45.50 |
| AS MIN W | −25.23 | −25.62 | −25.48 | −25.51 | −24.91 | −25.20 | −25.39 | −25.42 |
| AS MIN FS | −26.20 | −26.62 | −26.50 | −26.50 | −25.85 | −26.19 | −26.37 | −26.40 |
| MS MIN | 16.33 | 16.09 | 16.00 | 15.98 | 16.71 | 16.25 | 16.09 | 16.03 |
| CH3 HS MIN | −23.20 | −18.36 | −17.71 | −17.79 | −16.95 | −17.39 | −17.64 | −17.69 |
| NH2 HS MIN | 28.86 | 32.54 | 33.04 | 33.07 | 33.71 | 33.34 | 33.17 | 33.10 |
| CH3NHF + H | 29.10 | 32.97 | 33.41 | 33.40 | 33.98 | 33.73 | 33.61 | 33.47 |
| NH2F + CH3 | 18.45 | 18.26 | 18.01 | 17.89 | 18.54 | 18.13 | 17.96 | 17.91 |
| CH2FNH2 + H | −23.03 | −18.03 | −17.44 | −17.54 | −16.77 | −17.07 | −17.28 | −17.42 |
| CH3F + NH2 | −22.83 | −23.45 | −23.46 | −23.54 | −22.94 | −23.25 | −23.43 | −23.47 |
| HF + CH3NH | −34.16 | −33.77 | −33.80 | −33.98 | −33.46 | −33.72 | −33.87 | −33.92 |
| HF + CH2NH2 | −42.75 | −41.02 | −41.19 | −41.45 | −40.58 | −41.06 | −41.32 | −41.38 |
| CH4 + NHF | 6.31 | 4.04 | 3.87 | 3.79 | 4.07 | 3.88 | 3.80 | 3.77 |
| NH3 + CH2F | −29.71 | −29.77 | −29.89 | −30.05 | −29.16 | −29.65 | −29.92 | −29.96 |
Furthermore, this study reveals the basis-set effect of the results determined using the CCSD(T)-F12b method, comparing the aug-cc-pVnZ (n = D, T, Q) and the cc-pVnZ-F12 (n = D, T, Q, 5) basis sets. In Fig. 3 the relative energies, which are obtained using the above basis sets, are presented relative to the CCSD(T)-F12b/cc-pV5Z-F12 data. In the case of the aug-cc-pVDZ and aug-cc-pVTZ basis sets, the geometry optimizations are carried out, whereas in the other cases single-point computations are performed at the CCSD(T)-F12b/aug-cc-pVTZ geometries. With the increasing basis set, the average absolute deviations between the CCSD(T)-F12b/aug-cc-pVnZ and corresponding CCSD(T)-F12b/cc-pVnZ-F12 relative energies are decreasing, as 0.80 (n = D), 0.29 (n = T) and 0.15 (n = Q) kcal mol−1. If the results that are achieved using the cc-pV5Z-F12 basis set – used for benchmark values – are examined, as opposed to the results obtained with the mentioned other basis sets, we can establish that the root-mean-square deviation (RMSD) is less for the comparison of aug-cc-pVDZ and cc-pV5Z-F12 (RMSD = 0.37 kcal mol−1), than cc-pVDZ-F12 vs. cc-pV5Z-F12 (RMSD = 0.82 kcal mol−1). The inference is similar in the case of the aVTZ – V5Z-F12 and the VTZ-F12 – V5Z-F12 differences, where RMSDs are 0.09 and 0.26 kcal mol−1, respectively. The VQZ-F12 – V5Z-F12 difference is the least, with RMSD = 0.07 kcal mol−1. Thus, the CCSD(T)-F12b/cc-pV5Z-F12 relative energies are clearly basis-set-converged within 0.1 kcal mol−1.
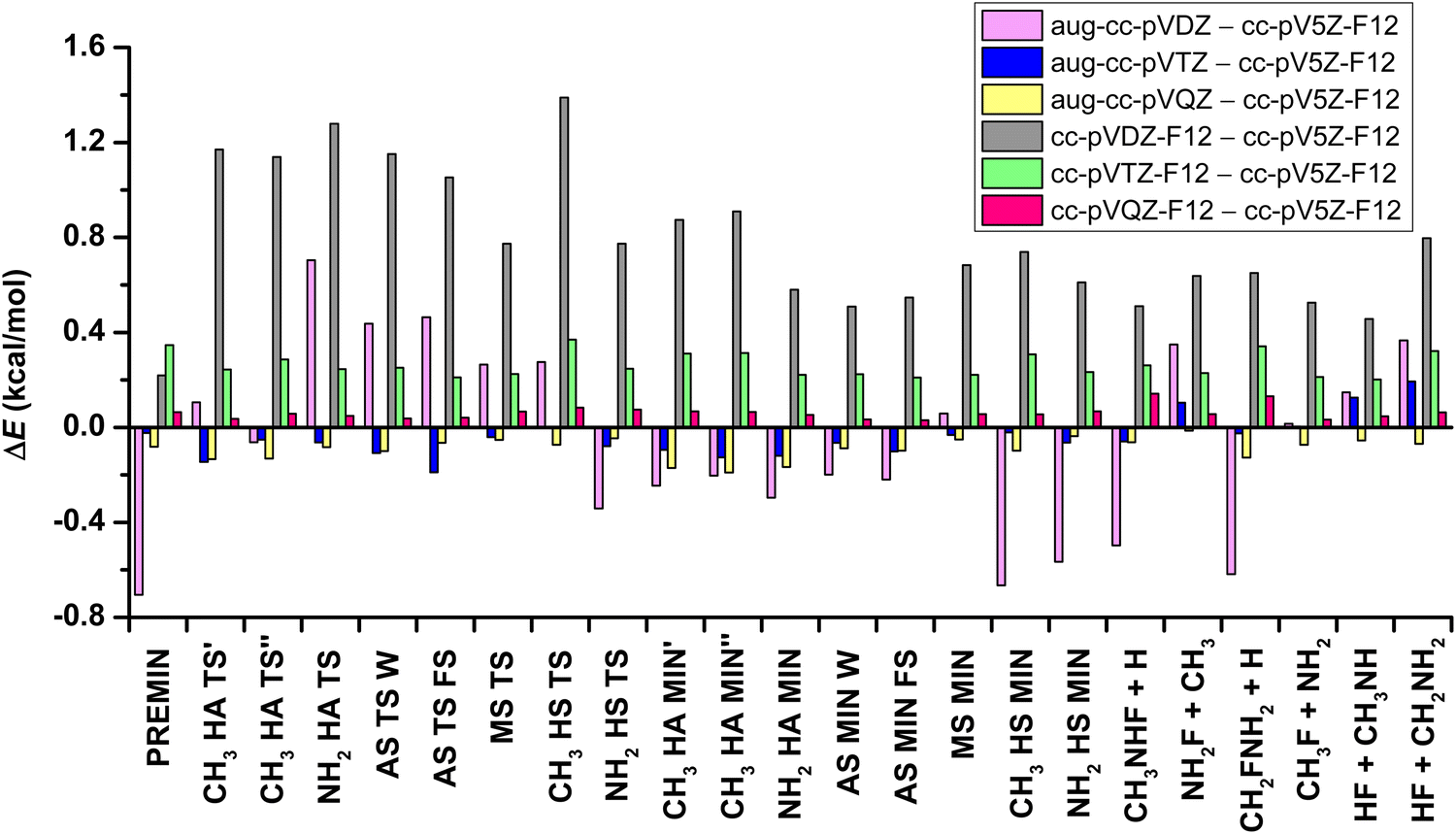 | ||
| Fig. 3 Difference between the relative energies of the stationary points and products, obtained at the CCSD(T)-F12b level of theory with the aug-cc-pVnZ (n = D, T, Q) and the cc-pVnZ-F12 (n = D, T, Q) basis sets compared with the cc-pV5Z-F12 basis set. | ||
3. Energy contributions
One of the goals of this electronic-structure study is to achieve subchemical accuracy in the relative energies of the characterized structures, thus auxiliary energy corrections must be computed. As we can read from Table 1, the post-CCSD(T) corrections are negative contributions with a few exceptions. The four cases, where δ[CCSDT] is not negative, occur for the hydrogen-substitution exit-channel complexes and products. All of the absolute post-CCSD(T) corrections are in the range of 0.00 to 1.08 kcal mol−1 in the case of δ[CCSDT] and go from 0.04 to 0.69 kcal mol−1 for δ[CCSDT(Q)], showing that these effects are not negligible. The largest post-(T) correlation effects are found for PREMIN and the TSs, which may have slight multi-reference characters as the T1 diagnostic41 values between 0.02 and 0.05 also show. Nevertheless, we think that the neglected post-(Q) effects must be small, and the present correlation treatment is converged within about 0.1–0.2 kcal mol−1, even for the TSs. For the exit-channel complexes and products the T1 values are always below 0.02, indicating that the use of single-reference wave functions is clearly adequate in these cases.The sum of the energy contributions of the core correlation and scalar relativistic effect is in the range of −0.02 to 0.52 kcal mol−1. These corrections play the most significant role in the case of the functional-group-substitution pathways, the finding of which is supported by the fact that Δcore + Δrel = 0.52 kcal mol−1 for the AS TS FS, whereas this value is only −0.01 kcal mol−1 for the CH3 HA TS′′. As this latter value shows, for the stationary points and products of the hydrogen-abstraction channels, these effects almost cancel each other.
The relativistic spin–orbit (SO) interactions in halogen atoms such as F may cause energy lowering in open-shell systems, and we have to take them into account in order to achieve chemical accuracy. The non-relativistic 2P ground state (non-SO) of the fluorine atom splits to the ground 2P3/2 state, with a lower energy by 1/3ε, and to the excited 2P1/2 state (SO3), with a higher energy by 2/3ε regarding the non-SO state, where ε is the SO splitting between the 2P3/2 and 2P1/2 states. While the F atom approaches the CH3NH2 molecule, the former fourfold degenerate state splits into two twofold degenerate states: the SO ground (SO1) and SO excited (SO2) states. Furthermore, three twofold degenerate states form as a result of the splitting of the non-SO ground state, resulting in a non-SO ground (non-SO1) and two non-SO excited states (non-SO2 and non-SO3). The potential energy curves of these six states, for the cases where the fluorine atom approaches the methylamine from seven different directions, are investigated and detailed in Subsection III.4. The SO-coupling-instigated energy decrease of the F atom is calculated as the difference between the asymptotic limits of the SO1 and non-SO1 potential energies. In general, this results in a SO correction of 0.37 kcal mol−1, whereas ΔSO is 0.34/0.36 kcal mol−1 for hydrogen-abstraction transition states and AS TS W, the effect of which will be further discussed below.
The energy contribution values, as mentioned above, do not exceed 1 kcal mol−1 (one exception), whereas the effect of the relative-energy shifting caused by the harmonic zero-point energy (ZPE) is more significant. For the reactant-like PREMIN and NH2 HA TS the ZPE correction is positive, but in all other cases ΔZPE is negative. The absolute correction for the products is larger than for the TS or MIN complexes, due to the decrease in the number of vibrational degrees of freedom in the product channels, or, for example, for the H-substitution pathways, due to the replacement of the high-frequency C–H/N–H stretching modes with lower-frequency C–F/N–F bonds. For example, the NH2 HS TS has a −3.79 kcal mol−1 ZPE correction, the NH2 HS MIN has −4.68 kcal mol−1, and this effect is −5.00 kcal mol−1 for the CH3NHF + H products.
Considering of all the energy corrections mentioned so far, we can compare the computed benchmark adiabatic relative energies (obtained using eqn (7)) with the experimental data, which are provided by the Active Thermochemical Tables (ATcT).42 The 0 K reaction enthalpy is determined by extracting the enthalpy of formation from the ATcT database for the reactants and products for the following three reaction pathways: hydrogen abstraction from the methyl group (CH3 HA) and from the amino group (NH2 HA) and for amino substitution (AS), where ΔH0 = −43.98 ± 0.11, −37.22 ± 0.12 and −26.10 ± 0.08 kcal mol−1, respectively. By comparison, the present benchmark ab initio ΔH0 values are −44.12, −37.24 and −26.48 kcal mol−1 in order. In the case of NH2 HA, the agreement between the experimental and theoretical data is outstanding, where the deviation is only 0.02 kcal mol−1. Although for the other two pathways the differences are not within the ATcT uncertainty determined via Gaussian propagation law, they are nevertheless well below the 1 kcal mol−1 chemical accuracy.
4. Potential energy curves of the F⋯CH3NH2 system
As described in the previous subsection, three SO and three non-SO states play key roles in describing the F⋯CH3NH2 interaction. Representative potential energy curves, relative to the ground non-SO1 asymptotic limit, are shown in Fig. 4A, where the fluorine atom approaches the methylamine molecule perpendicularly to the C–N bond (CN2). At the reaction asymptote, where the inter-fragment distance is large, the previously-mentioned SO correction of about 0.37 kcal mol−1 can be seen as the difference between the energy of SO1 and non-SO1, but as the reactants approach each other, this effect drops to zero. Fig. 4C shows this SO1 – non-SO1 difference for the seven approaching directions as indicated in Fig. 4D. The decrease in energy is not steep; it starts around at 4 Å and lasts until about 2 Å, but when the fluorine atom attacks methylamine from the side of the amino group (CN1, N1, N3), the decay starts at around 5 Å.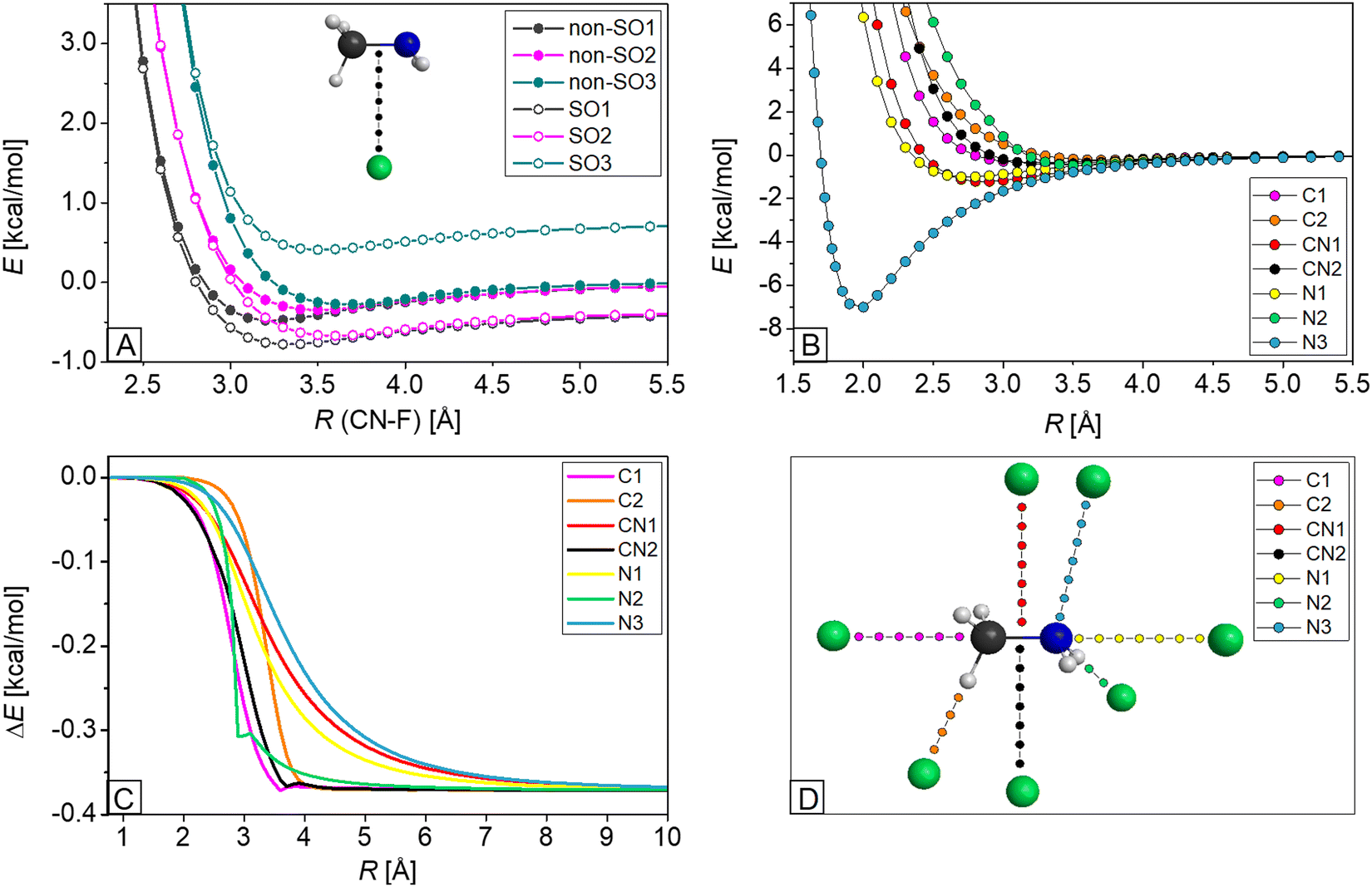 | ||
| Fig. 4 (A) Potential energy curves of six states in the case of CN2 as indicated in panel D; (B and C) distance-dependence of the energy of the SO1 state (B) and E(SO1) – E(non-SO1) (C) for the seven orientations indicated in panel D; and (D) seven different separation orientations of the F atom and methylamine, which is kept frozen in its equilibrium geometry, where fluorine approaches the C atom in line with the C–N bond (C1) or in line with a C–H bond (C2), the C–N bond perpendicularly from two sites (CN1 and CN2), the N atom in line with the C–N bond (N1), in line with the N–H bond (N2), or from the same orientation as in PREMIN (N3). Potential energies are obtained at the MRCI+Q/aug-cc-pwCVTZ level of theory. | ||
To investigate the reactive SO1 state corresponding to the different orientations of separation, we illustrate the potential curves of this state in Fig. 4B. The van der Waals wells are shallow with depths of 0.2–0.5 kcal mol−1 in the region of 3.0–3.5 Å, when fluorine approaches the C or N atom from the hydrogen (C2, N2), the C atom along the line of the C–N bond (C1) or the C–N bond perpendicularly (CN2). The wells are deeper for the cases where F approaches methylamine from the more polar amino group in line with (N1) or perpendicularly to (CN1) the C–N bond, and are the deepest (−7.0 kcal mol−1 at 2 Å distance) when the angle of the approach is equal to the C–N–F angle obtained in PREMIN (N3). Here it is important to note that MRCI+Q seriously underestimates the well depth of PREMIN as the benchmark value is 20.87 kcal mol−1 (see Fig. 1) and the geometry effect corresponding to fixed vs. relaxed CH3NH2 is only 0.1 kcal mol−1. These findings indicate that dynamical electron correlation, even beyond single and double excitations, is important for describing the F⋯CH3NH2 interaction. To further support this conclusion, we report that the PREMIN complex is unbound at the ROHF level, and CCSD-F12b underestimates the CCSD(T)-F12b well depth by 3.7 kcal mol−1. These extremely large correlation effects are not unprecedented as the barrier height of the F + CH4 reaction is 9.41, 1.80, and 0.55 kcal mol−1 at the ROHF, CCSD, and CCSD(T) levels of theory, respectively.10
5. Comparing the F + CH3NH2 and the Cl + CH3NH2 reactions
As we published in our previous work,22 in the reaction of Cl + CH3NH2 there are six reaction channels, as in the title reaction: two hydrogen-abstraction pathways from the two functional groups and are four substitution pathways, where the halogen atom substitutes the amino/methyl group or one hydrogen of these. When methylamine reacts with the fluorine atom, the reaction channels are more exothermic than in the case of reaction with chlorine; however, in both cases the CH3 HA products have the lowest relative energy and the NH2 HS products have the highest. The global minimum of the vibrationally adiabatic PES belongs to the CH3 HA MIN′′ complex for the two reactions (−51.05/−15.56 kcal mol−1 for F/Cl + methylamine), but while in case of the F + CH3NH2 system it is the classical minimum as well (−50.67 kcal mol−1), in the case of Cl + CH3NH2 classically the global minimum belongs to the PREMIN structure (−15.30 kcal mol−1). We have found that the CH3F/CH3Cl + NH2 and CH2ClNH2 + H products can be formed by both Walden-inversion and front-side attack mechanisms; however, we have not found the front-side attack pathway for the formation of the CH2FNH2 + H products. These two channels are exothermic in the title reaction, but in the case of Cl + CH3NH2 they are endothermic.The two additional kinetically hindered product channels are also investigated in the title reaction, where two bonds are required to break and form, leading to NH3 + CH2F and CH4 + NHF. The products of the former channel have a ΔE(ΔH0) = −29.65(−32.77) kcal mol−1 relative energy and those of the latter have a ΔE(ΔH0) = 3.87(0.03) kcal mol−1, whereas the corresponding values for the Cl + CH3NH2 system are −3.70(−8.20) and 14.24(9.82) kcal mol−1, respectively.22 In both reactions, the exothermic NH3 formation is thermodynamically competitive with H-abstractions; however, the former may have a much higher barrier due to the two bond cleavages required to form the NH3 + CH2F/CH2Cl products.
The calculated SO interactions, which lower the asymptote of the reactants by 0.84 kcal mol−1 and 0.37 kcal mol−1 for the chlorine and fluorine reactions, respectively, show good agreement with the measured SO splittings (εexpt)43 of these atoms, as εexpt/3 = 0.84 (Cl) kcal mol−1 and 0.39 (F) kcal mol−1. Considering the different separation orientations of the reactants, it can be seen for both systems that the deepest van der Waals wells of the potential energy curves occur when the halogen atom approaches the amino group of methylamine with the angle of C–N–Cl/F obtained at the PREMIN geometry. The depth is shallower if the F/Cl atom attacks the molecule at the side of the hydrogens or at the methyl group in line with the C–N bond. The distance dependence of the SO – non-SO ground-state energy difference for the different approaching orientations is quite similar for the two systems.
IV. Summary and conclusions
In addition to the previously-studied7 exothermic hydrogen-abstraction pathways of the F + CH3NH2 reaction, the hydrogen and functional-group substitutions are investigated as well in the present theoretical study. Transition states and post-reaction complexes are identified in all the reaction channels. The methyl-hydrogen abstraction has the lowest adiabatic energy among the exothermic channels (ΔH0 = −44.12 kcal mol−1), which is followed by amino-hydrogen abstraction, amino-substitution, and methyl-hydrogen-substitution with reaction enthalpies of −37.24, −26.48, and −20.83 kcal mol−1, in ascending energy order. Amino substitution can proceed via Walden-inversion and front-side-attack mechanisms, where the latter has a higher classical barrier height by 18.86 kcal mol−1, although the energies of the post-reaction minima are close to each other. The two thermodynamically unfavored endothermic pathways are methyl substitution and amino-hydrogen substitution. The global minimum of the potential energy surface corresponds to the complex CH3 HA MIN′′ in the product channel, where the formed HF binds to the N atom of the CH2NH2 fragment. A pre-reaction minimum has been found in the entrance channel, which has a deeper energy (−19.72 kcal mol−1 adiabatically) than all saddle points, and thus it may play an important steering role in all the reaction pathways.The above-indicated subchemically accurate benchmark energy values are obtained by taking into account further energy contributions besides the CCSD(T)-F12b/cc-pV5Z-F12 single-point energies obtained at the CCSD(T)-F12b/aug-cc-pVTZ geometries. As we have already seen in the case of the reaction between chlorine and methylamine,22 the zero-point energy correction is the most significant among the energy contributions determined by us. By considering the post-(T) correlation (which mainly affects the energy of the TS structures and often by more than 1 kcal mol−1), the core electron correlation and scalar relativistic effects (which are usually negligible to reach chemical accuracy but are often needed if subchemical accuracy is desired), and the SO correction of about 0.37 kcal mol−1, we are able to achieve excellent agreement with the experimental results for the three product channels, where data are available in the ATcT database.
This study provides guidance for the development of a global analytical ab initio potential energy surface required for the first-principles investigation of reaction dynamics, thereby enabling a comparison with the results obtained via experiments, and gaining deeper atomic insight into the mechanisms of the F + CH3NH2 reaction.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the National Research, Development and Innovation Office – NKFIH, K-125317; the Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT; Project no. TKP2021-NVA-19, provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-NVA funding scheme; and the Momentum (Lendület) Program of the Hungarian Academy of Sciences for financial support.References
- G. C. Schatz and A. Kuppermann, J. Chem. Phys., 1975, 62, 2502 CrossRef.
- G. C. Schatz, J. M. Bowman and A. Kuppermann, J. Chem. Phys., 1975, 63, 674 CrossRef CAS.
- K. Stark and H.-J. Werner, J. Chem. Phys., 1996, 104, 6515 CrossRef CAS.
- D. E. Manolopoulos, J. Chem. Soc., Faraday Trans., 1997, 93, 673 RSC.
- L. Che, Z. Ren, X. Wang, W. Dong, D. Dai, X. Wang, D. H. Zhang, X. Yang, L. Sheng, G. Li, H.-J. Werner, F. Lique and M. H. Alexander, Science, 2007, 317, 1061 CrossRef CAS PubMed.
- W. Dong, C. Xiao, T. Wang, D. Dai, X. Yang and D. H. Zhang, Science, 2010, 327, 1501 CrossRef CAS PubMed.
- H. Feng, W. Sun, Y. Xie and H. F. Schaefer III, Chem. Phys. Chem., 2013, 14, 896 CrossRef CAS PubMed.
- B. Fu, X. Shan, D. H. Zhang and D. C. Clary, Chem. Soc. Rev., 2017, 46, 7625 RSC.
- L. Tian, Y. Zhu, H. Song and M. Yang, Phys. Chem. Chem. Phys., 2019, 21, 11385 RSC.
- G. Czakó, B. C. Shepler, B. J. Braams and J. M. Bowman, J. Chem. Phys., 2009, 130, 084301 CrossRef PubMed.
- J. F. Castillo, F. J. Aoiz, L. Banares, E. Martinez-Nunez, A. Fernandez-Ramos and S. Vazquez, J. Phys. Chem. A, 2005, 109, 8459 CrossRef CAS PubMed.
- J. Espinosa-Garcia, J. C. Corchado, M. Garcia-Chamorro and C. Rangel, Phys. Chem. Chem. Phys., 2018, 20, 19860 RSC.
- D. Papp and G. Czakó, J. Chem. Phys., 2020, 153, 064305 CrossRef CAS PubMed.
- M. L. Weichman, J. A. DeVine, M. C. Babin, J. Li, L. Guo, J. Ma, H. Guo and D. M. Neumark, Nat. Chem., 2017, 9, 950 CrossRef CAS PubMed.
- D. Lu, J. Li and H. Guo, Chem. Sci., 2019, 10, 7994 RSC.
- D. Lu and J. Li, Theor. Chem. Acc., 2020, 139, 157 Search PubMed.
- M. Zhang, Y. Zhu and J. Li, Chin. J. Chem. Phys., 2022, 35, 153 CrossRef CAS.
- R. G. Macdonald, J. J. Sloan and P. T. Wassell, Chem. Phys., 1979, 41, 201 CrossRef CAS.
- S. Wategaonkar and D. W. Setser, J. Chem. Phys., 1987, 86, 4477 CrossRef CAS.
- D. Papp, B. Gruber and G. Czakó, Phys. Chem. Chem. Phys., 2019, 21, 396 RSC.
- B. Gruber and G. Czakó, Phys. Chem. Chem. Phys., 2020, 22, 14560 RSC.
- T. Szűcs and G. Czakó, Phys. Chem. Chem. Phys., 2021, 23, 10347 RSC.
- P. Tóth, T. Szűcs and G. Czakó, J. Phys. Chem. A, 2022, 126, 2802 CrossRef PubMed.
- R. D. Amos, J. S. Andrews, N. C. Handy and P. J. Knowles, Chem. Phys. Lett., 1991, 185, 256 CrossRef CAS.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef.
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- K. A. Peterson, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2008, 128, 084102 CrossRef PubMed.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS.
- J. Noga and R. J. Bartlett, J. Chem. Phys., 1987, 86, 7041 CrossRef CAS.
- M. Kállay and J. Gauss, J. Chem. Phys., 2005, 123, 214105 CrossRef PubMed.
- M. Kállay, P. R. Nagy, D. Mester, Z. Rolik, G. Samu, J. Csontos, J. Csóka, B. P. Szabó, L. Gyevi-Nagy, B. Hégely, et al. MRCC, a quantum chemical program suite, see http://www.mrcc.hu.
- M. Kállay, P. R. Nagy, D. Mester, Z. Rolik, G. Samu, J. Csontos, J. Csóka, P. B. Szabó, L. Gyevi-Nagy and B. Hégely, et al. , J. Chem. Phys., 2020, 152, 074107 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, et al., Molpro, version 2015.1, a package of ab initio programs, see https://www.molpro.net Search PubMed.
- J. G. Hill, S. Mazumder and K. A. Peterson, J. Chem. Phys., 2010, 132, 054108 CrossRef PubMed.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89 CAS.
- W. A. de Jong, R. J. Harrison and D. A. Dixon, J. Chem. Phys., 2001, 114, 48 CrossRef CAS.
- A. Berning, M. Schweizer, H.-J. Werner, P. J. Knowles and P. Palmieri, Mol. Phys., 2000, 98, 1823 CrossRef CAS.
- S. R. Langhoff and E. R. Davidson, Int. J. Quantum Chem., 1974, 8, 61 CrossRef CAS.
- K. R. Shamasundar, G. Knizia and H.-J. Werner, J. Chem. Phys., 2011, 135, 054101 CrossRef CAS PubMed.
- K. A. Peterson and T. H. Dunning, Jr., J. Chem. Phys., 2002, 117, 10548 CrossRef CAS.
- T. J. Lee and P. R. Taylor, Int. J. Quantum Chem., 1989, S23, 199 Search PubMed.
- B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.122r of the Thermochemical Network (2021); available at http://ATcT.anl.gov.
- The National Institute of Standards and Technology (NIST), Handbook of Basic Atomic Spectroscopic Data, https://www.nist.gov/pml/data/handbook/.
| This journal is © the Owner Societies 2022 |