
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA proof-of-concept study of the secondary structure of influenza A, B M2 and MERS- and SARS-CoV E transmembrane peptides using folding molecular dynamics simulations in a membrane mimetic solvent†
Antonios
Kolocouris
 *a,
Isaiah
Arkin
b and
Nicholas M.
Glykos
*c
*a,
Isaiah
Arkin
b and
Nicholas M.
Glykos
*c
aLaboratory of Medicinal Chemistry, Section of Pharmaceutical Chemistry, Department of Pharmacy, National and Kapodistrian University of Athens, Panepistimiopolis, 15771, Greece. E-mail: ankol@pharm.uoa.gr
bDepartment of Biological Chemistry, The Alexander Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Edmond J. Safra Campus Givat-Ram, Jerusalem, 91904, Israel
cDepartment of Molecular Biology and Genetics, Democritus University of Thrace, University Campus, Alexandroupolis, 68100, Greece. E-mail: glykos@mbg.duth.gr
First published on 4th October 2022
Abstract
Here, we have carried out a proof-of-concept molecular dynamics (MD) simulation with adaptive tempering in a membrane mimetic environment to study the folding of single-pass membrane peptides. We tested the influenza A M2 viroporin, influenza B M2 viroporin, and protein E from coronaviruses MERS-Cov-2 and SARS-CoV-2 peptides with known experimental secondary structures in membrane bilayers. The two influenza-derived peptides are significantly different in the peptide sequence and secondary structure and more polar than the two coronavirus-derived peptides. Through a total of more than 50 μs of simulation time that could be accomplished in trifluoroethanol (TFE), as a membrane model, we characterized comparatively the folding behavior, helical stability, and helical propensity of these transmembrane peptides that match perfectly their experimental secondary structures, and we identified common motifs that reflect their quaternary organization and known (or not) biochemical function. We showed that BM2 is organized into two structurally distinct parts: a significantly more stable N-terminal half, and a fast-converting C-terminal half that continuously folds and unfolds between α-helical structures and non-canonical structures, which are mostly turns. In AM2, both the N-terminal half and C-terminal half are very flexible. In contrast, the two coronavirus-derived transmembrane peptides are much more stable and fast helix-formers when compared with the influenza ones. In particular, the SARS-derived peptide E appears to be the fastest and most stable helix-former of all the four viral peptides studied, with a helical structure that persists almost without disruption for the whole of its 10 μs simulation. By comparing the results with experimental observations, we benchmarked TFE in studying the conformation of membrane and hydrophobic peptides. This work provided accurate results suggesting a methodology to run long MD simulations and predict structural properties of biologically important membrane peptides.
Introduction
Viral short open reading frames (sORFs) encoding proteins have a length of 100 amino acids or less.1,2 A major group of viral SEPs encode short single-pass membrane proteins, often with less than 50 amino acids (viral miniproteins),3 which may be advantageous for the virus as they form stable structures to support membrane-related functions at a minimal burden on genome size. Several of these virally encoded sORF proteins oligomerize in cell membranes,1–4 with a known or not known biological function. For example, some of these are viroporins forming hydrophilic pores in host cell membranes, thus altering the physiological properties of the host cells. Viroporins are homo-oligomeric proteins with ion channel pores that are formed by α-helical transmembrane (TM) domains.5Over the past few decades, an increasing number of both cation- and anion-conducting viroporins have been identified and proposed to play central roles in the viral life cycle, in addition to having a huge impact on pathologies in the host.6,7 They have been identified in a vast number of pathogenic viruses. Examples are the homotetrameric influenza A (AM2)8,9 and influenza B M2 (BM2)10 proton channels. Proton channels AM28,11 and BM2,10 with M2TM being the ion channel pore,12 form an active, open state at low pH during endocytosis. In particular, the activation of the M2 tetrameric bundle ultimately leads to the unpacking of the influenza viral genome and to pathogenesis.9,13
The single-pass membrane protein E is a viral sORF encoding protein that has been identified in the severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1 or SARS-CoV), giving rise to the deadly ‘SARS’ epidemic in 2002, SARS-CoV-2, the cause of the ongoing pandemic of COVID-19,14 and Middle East respiratory syndrome coronavirus (MERS-CoV), giving rise to the deadly ‘MERS’ in 2012.14 For the E protein, it has been suggested that it is a viroporin forming a cation-selective channel region across the endoplasmic reticulum Golgi intermediate compartment (ERGIC) membrane that conducts cations, but this came so far from limited experimental data.15–24
The AM2 and BM2 proteins are 97- and 109-residues’ single-pass membrane proteins, respectively, that form homotetramers in membranes.9,25,26 The two proteins AM2 and BM2 share almost no sequence homology except for the HxxxW27,28 sequence motif in the M2TM domain that is essential for channel activity with His acting as a sensor residue for proton conduction and Trp acting as the gate (Fig. 2). At acidic pH, the four His residues are protonated and repel each other and the M2 channel opens and conducts protons.10,11 Their TM domain sequence arrangements are different, i.e., the AM2TM region encompasses residues 22–46 compared to residues 4–33 in BM2TM (Fig. 2). Hence, the unstructured N-terminal segment preceding the TM domain is much longer in AM2. AM2 and BM2 proteins both have relatively large C-terminal cytoplasmic regions. These regions have been suggested to play a role during virus budding29,30 by recruiting the M2 protein to the cell surface and during viral assembly by contributing to the coating of M2 on the viral envelope.31,32 The structure of the homotetrameric AM2TM (Udorn strain, residues 22–46) bundle has been resolved using both solid-state NMR (ssNMR) (PBD IDs 2H95,332L0J34) and X-ray crystallography (PDB ID 4QK735). The structure of the influenza BM2 (residues 1–51) homotetramer has been resolved using ssNMR (PDB ID 6PVR36); this construct contains the TM domain (residues 4–33) with residues 34–43 connecting the TM with the cytoplasmic domain of full-length BM2.37
The shared and conserved virally encoded sORF protein E has been identified in SARS-CoV-1, -2, and MERS-CoV and is highly homologous to SARS-CoV and to MERS-CoV. E is a 75-residue protein that oligomerizes in the cell membrane. It has a TM region of 30 residues8–38 in SARS-CoV-1, -2, and MERS-CoV (SARS ETM and MERS ETM, respectively) with an identical amino acid sequence for SARS-CoV-1 and SARS-CoV-2.38,39 A homopentameric structure for SARS ETM (residues 8–38, PDB ID 7K3G39) has been recently suggested using ssNMR, while previous structures were obtained with solution NMR in micelles (residues 8–65, PDB ID 5 × 29).38 The ssNMR structure39 suggested a rigid and tight pentameric helical bundle, whose pores are occupied by predominantly hydrophobic residues, except for a polar asparagine (Asn15). Three regularly spaced phenylalanines Phe20, Phe23, and Phe26 are at the center of SARS ETM. Using ssNMR at acidic pH and Ca2+ to mimic the ERGIC and lysosomal environment experienced by the E protein in the cell, it has been suggested that the conformational change of two side chain conformations at both Phe20 and Phe26 results in an equilibrium between the closed and open states of ETM,40 with channel opening at acidic pH and in the presence of Ca2+ by increasing the population of the dynamic lipid-facing conformation and water accessibility.
The N-terminus of the CoV channels contains a (E/D/R)8X(G/A/V)10XXhh(N/Q)15 motif (Fig. 2), where h is a hydrophobic residue. The third residue of the motif (G/A/V) is conserved among coronaviruses to be small and flexible, which might permit N-terminus motion. The last residue of the motif is conserved to be either Asn or Gln. At the C-terminal part of the TM segment, the small residues Ala32 and Thr35 are conserved. In contrast to these small (or small and polar) residues, the central portion of the TM domain contains four layers of hydrophobic residues, Leu18, Leu21, Val25, and Leu28, and three Phe20, Phe23, and Phe26 residues at the center of SARS, suggesting an ion channel with a narrow hydrophobic pore of radius ∼2 Å39 that will be much less hydrated compared to AM2TM and BM2TM. Such a pore will permit only a single file of water molecules, thus partially dehydrating any cations that move through the pore, and this seems inconsistent with the suggestion for an ion channel.
It has been shown that in SARS-CoV-1, E mediates the budding and release of progeny viruses and activates the host inflammasome.41 The expression of SARS-CoV-1 viroporins promotes virus replication and virulence,42 and deletion of the protein E gene attenuates the virus, resulting in faster recovery and improved survival in infected mice.17 Thus, the biological activity of protein E represents a determinant for SARS-CoV-1 virulence, mirroring the pathology associated with the severe cases of SARS-CoV-2 infection.
Most viroporins have been identified as virulence factors that lead to viral attenuation when deleted. This attenuation is attributed only in part to their channel activity, but nevertheless, small-molecule channel inhibitors have been explored triggering also the research for the structure determination of the related viroporins. However, a vast majority of channel inhibitors such as amantadine variants or hexamethylene amiloride (HMA)8,43,44 have been developed against AM2.45–49 This is not surprising since AM2 has been the best characterized viroporin to date with an established biological role in viral pathogenesis,13,29,30 combining the most extensive structural investigations conducted without33,35 or with inhibitors acting as pore blockers,43,44,50,51 and has emerged as a validated drug target.43–51 For the other viral sORF encoding proteins, including BM2 and E proteins,19,22 these studies are still in their infancy.
We have previously showed52 using circular dichroism experiments that AM2TM is a stable α-helix and by 1.1 μs MD simulations with adaptive tempering that the AM2TM monomer is dynamic in nature and the region encompassing residues in the C-terminal part17–25 quickly interconverts between an ensemble of various α-helical structures, and less frequently turns and coils, compared to the one α-helix for Ala25. Our results52 from density functional theory (DFT) calculations in this lipophilic peptide showed that this is due to CH⋯O interaction forces between amino acid alkyl side chains and main chain carbonyls, which, although individually weaker than NH⋯O hydrogen bonds, can dissociate and associate easily leading to an ensemble of folded structures observed in folding MD simulations. The CH⋯O interaction forces have a cumulative effect that cannot be ignored and may contribute as much as half of the total hydrogen bonding energy,52 when compared to NH⋯O, to the stabilization of the α-helix in AM2TM. Similar folding forces should characterize α-helical lipophilic peptides.
It has been shown that TFE promotes α-helix formation53–56 and has been applied as a model to study helical peptides using NMR spectroscopy.57–59 Since computational and experimental models for NMR spectroscopy are needed to study hydrophobic peptides in solution, e.g., antimicrobial peptides, amphiphilic, or membrane peptides, we sought this research to explore if simulations in pure TFE can reproduce details of the known experimental structures of peptide monomers. This is a benchmark study applied to four peptides and can also provide a protocol for the investigation of hydrophobic helical peptides.
In this work, we explored comparatively the folding of the TM monomer of the four important virally encoded sORF proteins, i.e., the influenza AM2TM (residues 22–46) and influenza BM2TM (residues 1–31) and the coronaviruses SARS and MERS ETM (residues 8–38), using a total of 50 μs of molecular dynamics simulations with adaptive tempering in TFE, a membrane-mimicking solvent.60,61 Compared to our previous study with AM2TM using 1.1 μs MD simulation,52 we applied here longer simulation times to allow for a more ergodic investigation of the conformational space of the peptides. Although the membrane-mimicking environment60,61 can limit the amount of interpretation that can be based on these folding simulations, making the connection with the peptides’ behavior in the membranes qualitative in nature, it was striking to observe that we obtained accurate measures of structural characteristics that are known for the monomers of these single-pass membrane proteins. These structural features have putative connection to their biological function, suggesting that the methodology used can be useful for structure predictions.
Methods
System setup and MD simulation protocol
The preparation of the systems including the starting peptide structures in the fully extended state together with their solvation and ionization states was performed with the program LEAP from the AMBER tool distribution as previously described in detail.52,62 We followed the dynamics of the peptides’ folding simulations using the program NAMD.63 For all MD simulations, we have used periodic boundary conditions with a cubic unit cell sufficiently large to guarantee a minimum separation between the symmetry-related images of the peptides of at least 16 Å. The simulations were performed in 100% TFE. The parameterization for TFE64 was obtained from the R.E.D. library.65 In all MD simulations, we used the AMBER99SB-STAR-ILDN force field,66–68 which has repeatedly been shown to correctly fold69 numerous peptides70–79 including peptides in mixed organic (TFE/water) solvents.80 For all MD simulations, adaptive tempering81 was applied as implemented in the program NAMD.63 Adaptive tempering is formally equivalent to a single-copy replica exchange folding simulation with a continuous temperature range. For our simulations, this temperature range was from 280 K to 380 K, both inclusive, and was applied to the system through the Langevin thermostat, as described below. The MD simulations’ protocol has also been previously described,52,77–79 and in summary, it is described as follows. The systems were first energy-minimized for 1000 conjugate gradient steps, followed by a slow heating-up phase to a temperature of 320 K (with a temperature step of 20 K) over a period of 32 ps. Subsequently, the systems were equilibrated for 1000 ps under NpT conditions without any restraints, until the volume equilibrated. This was followed by the production NpT runs with the temperature and pressure being controlled using the Nosè–Hoover82 Langevin dynamics83 and Langevin piston barostat84 control methods as implemented using the NAMD program,63 with adaptive tempering being applied through the Langevin thermostat, while the pressure was maintained at 1 atm. The Langevin damping coefficient was set to be 1 ps−1, and the piston's oscillation period was set to be 200 fs, with a decay time of 100 fs. The production runs were performed with the impulse Verlet-I multiple timestep integration algorithm as implemented using NAMD and lasted 10 μs for each of the CoV peptides and approximately 15 μs for each of the the peptides AM2TM and BM2TM, giving a grand total for the four peptides of 50 μs of simulation time.63 The inner timestep was 2.5 fs, with short-range non-bonded interactions being calculated every one step and long-range electrostatic interactions being calculated every two timesteps using the particle mesh Ewald method85 with a grid spacing of approximately 1 Å and a tolerance of 10−6. A cutoff for the van der Waals interactions was applied at 9 Å through a switching function, and SHAKE86 (with a tolerance of 10−8) was used to restrain all bonds involving hydrogen atoms. Trajectories were obtained by saving the atomic coordinates of the whole systems every 1.0 ps.Trajectory analysis
The analysis of the trajectories was performed as previously described.52,77–79 Secondary structure assignments were performed with the program STRIDE.87 All molecular graphic work and figure preparation were performed with the programs VMD,88 RASTER3D,89 PyMol,90 WebLogo,91 and CARMA.92Statistical significance and sufficient sampling
Folding molecular dynamics simulations, especially when performed with an adaptive tempering protocol cover an enormously complex configurational space encompassed by the unfolded state. The implication is that it is essential to quantify the statistical significance and the extent of sampling of the corresponding trajectories before any conclusions can be drawn from them. In this work, we have quantified statistical significance through a recently described probabilistic method, which is based on the application of Good-Turing statistics to estimate how probable it is to observe completely new/unrelated structures if a given simulation was to be extended to longer timescales.93 The results obtained from this method are shown in Fig. 1 for the four trajectories studied in this communication.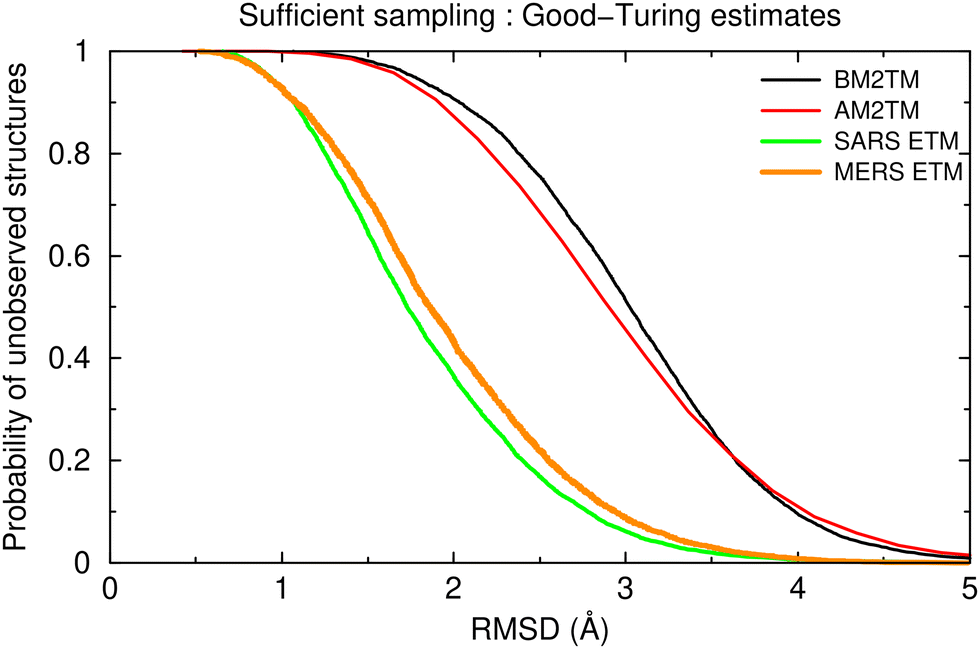 | ||
| Fig. 1 Good-Turing estimates for the probability of unobserved structures as a function of the expected RMSD of these structures from the already known (i.e., observed in the trajectories) peptide structures. See text for a detailed discussion of this figure. | ||
The information about convergence and extent of sampling is contained in these “probability vs. RMSD” diagrams, which show how probable it is to observe a new structure that would differ by more than a given RMSD from all peptide structures already observed in a given trajectory. All curves start with very high probabilities for low RMSD values, indicating that it is very probable to observe structures that differ only slightly from those already observed. The curves asymptotically approach zero for higher RMSD values, and it is the exact form and how quickly they reach low-probability values that inform us just how much structural variability we have not yet observed in our trajectories. For the case examined here, the four peptides are clearly clustered in two sets. The coronavirus-derived peptides (green and orange curves in Fig. 1) fall-off quite quickly to very low probabilities for RMSD values of around 4 Å. What this implies, then, is that if we were to continue the simulation, we would expect almost all new (previously unrecorded) structures to differ by less than ∼4 Å from those already observed. The behavior of the influenza-derived peptides (black and red curves in Fig. 1) is significantly different: the curves fall-off slowly and maintain significant probability values up to ∼5 Å, clearly indicating that a significant volume of the peptides’ configurational space has not yet been sampled in these simulations. Note that it is exactly for this reason that we extended the trajectories for the influenza-derived peptides to 15 μs each (instead of 10 μs for the coronavirus peptides).
Thus, the application of Good-Turing statistics allowed us to quantify the extent of sampling in our trajectories and to differentiate between the two sets of peptides based on the structural uncertainty still remaining. The results clearly indicate that with such large uncertainties, it would be meaningless to even try to quantify differences between the peptides at the atomic level. A lower resolution comparison, for example, at the level of secondary structure stabilities and preferences, is possibly the best that can be achieved with the data available.
We have also calculated the typical (RMSD vs. time) diagrams for all four peptides. These are shown in in Fig. S1 in the ESI† (only Cα atoms have been used for these calculations). The reference structure used for calculating the RMSD was the all-helical experimental-like structure (and not the starting structure since these are folding simulations that were initiated from an unfolded/extended structure). In agreement with the Good-Turing analysis, the pairwise differences between the AM2TM/BM2TM and SARS/MERS are clearly visible also in the RMSD diagrams. The difference in variance and average RMSDs in the case of BM2TM and SARS ETM is also characteristic. To make this even more clear, the diagrams included in Fig, S2 in the ESI† show the distribution of these RMSDs and clearly indicate the differences between the two sets of peptides. For example, the maximum of the distribution is at 3.5 Å for SARS ETM but at 5.2 Å for BM2TM. If we conservatively select 3.5 Å as a cutoff for what we consider to be a native-like peptide structure, we can also obtain conservative estimates of the frequencies of such structures for each of the four peptides studied here. Performing this calculation gave a fraction of 53% native-like peptide stuctures for SARS-ETM and 34% for MERS-ETM but only 17% and 7% for AM2TM and BM2TM, respectively. These estimated frequencies should be considered lower bounds given the malleability of the structure of long isolated α-helices.
Results
Preliminaries: sequence similarity analysis
Fig. 2 shows the amino acid sequences and the corresponding sequence alignment of the four peptides (AM2TM, BM2TM, SARS ETM, and MERS ETM) studied here, highlighting their similarities and differences at the sequence level.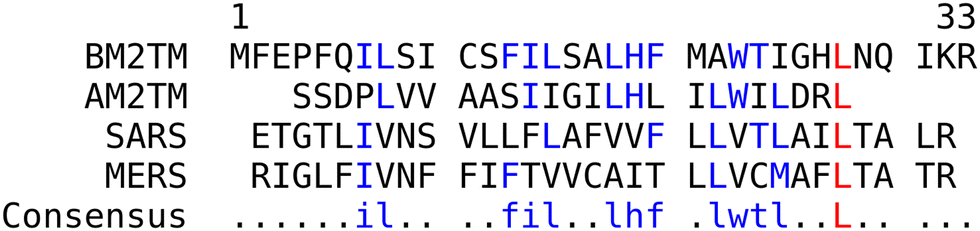 | ||
| Fig. 2 Peptide sequences and alignment for AM2TM (Udorn strain, residues 22–46), BM2TM (residues 1–33), SARS ETM (residues 8–38), and MERS ETM (residues 8–38). Identities and highly similar matches (indicated by a dollar sign in the consensus sequence) are shown in red and pairwise identities are shown in blue. | ||
There are six residues which are pairwise identical between influenza B and A M2TM peptides (three additional residues are similar, i.e., I, L, or V at positions 10, 15, and 25 in the BM2TM numbering scheme, Fig. 2). The two influenza A and B peptides have in common a HxxxW sequence motif that is considered to include the proton filter and primary gate of the proton channels.27,28 The two influenza peptides for the C-terminal half (residues 17–28) share a sequence identity of 33%, which is reduced to 15% when the N-terminal half4–16 is examined. The overall sequence identity between the two peptides is 24%.
The two CoV-derived peptides in their C-terminal half (residues 19–32) share a sequence identity of almost 62%, which drops to 24% for the N-terminal half (residues 2–18). Three additional residues are similar, i.e., I or L at positions 12, 15, and 19.
We also note that there are four residues which are pairwise identical between influenza B and either of the CoV peptides but differ when only the two CoV-derived peptides are compared (these four residues are F13, L15, F20, and T24 in the BM2TM numbering scheme, Fig. 2). This very weak sequence similarity should not be considered to imply the presence of evolutionary relationships between the two families of sequences (AM2TM/BM2TM and SARS/MERS) and no biological implications should be assigned to it. This absence of sequence similarity is also highlighted from the “consensus” sequence, which clearly indicates that there is no detectable sequence similarity between all four peptides.
The simulations indicate the presence of significant differences between the influenza- and coronavirus-derived peptides
Fig. 3 shows the per-residue secondary structure assignment versus simulation time for each of the four peptides studied. Even a cursory examination of this figure clearly shows that there are pronounced differences between the helical propensity and stability of the four structures. | ||
| Fig. 3 Evolution of the per residue secondary structure vs. simulation time. The graphs depict the variation of the per-residue STRIDE-derived secondary structure assignments as a function of simulation time for the four peptides. The color coding is red/magenta → α/310 helical structure, cyan → turns, white → coil, and yellow → β structure. | ||
Although all four peptides do fold to a mostly α-helical structure as expected, it is the SARS peptide that appears to form an exceedingly stable α-helix (noting here that these results have been obtained from adaptive tempering simulations with the temperature ranging from 280 K to 380 K) with second more stable α-helix being the MERS-derived peptide. The influenza-derived peptides, on the other hand, show significant variability–both in helical propensity and helical stability–along the length of their sequences. The influenza-derived peptides appear to show a bipartite organization, with a more stable helical N-terminal half and a less stable and fast interconverting C-terminal half. This bipartite organization is especially noticeable in the case of the BM2TM peptide. The MERS-derived peptide on the other hand shows the opposite pattern, with a mostly stable and well-behaving C-terminal region and a more variable N-terminal part.
The least stably folded of all four peptides is the influenza peptide AM2TM along the length of its sequence. This motif of reduced stability of the AM2TM peptide may be attributed to the presence of a glycine residue at position 13 (corresponding to the G34 in the 98-residue full-M2 protein) since Gly is known to be a helix breaker (it has the lowest helix propensity after proline).
To further quantify these observations, we have calculated the fractional helicity for each residue of each peptide over the whole length of their respective simulations. The results from this calculation are shown in Fig. 4. This figure not only places the previous observations on a solid ground but also highlights salient features that could have been missed in Fig. 3, such as the dip in fractional helicity centered at residue 16 (22 in the ETM numbering scheme; Ala for SARS ETM and Val for MERS ETM) of both CoV-derived peptides. In the paragraphs that follow, we discuss and expand on the different folding behaviors of the four peptides.
 | ||
| Fig. 4 Fractional helicity versus residue. The four graphs depict the per residue fractional helicity over the whole length of the three simulations. The color coding is indicated in the figure legend: orange (MERS ETM), green (SARS ETM), black (BM2TM), and red (AM2TM). The graphs for BM2TM (black) and AM2TM (red) have been translated by one residue to the left to reproduce the sequence alignment shown in Fig. 2. | ||
The SARS ETM peptide forms an exceptionally stable helical structure
Of the four peptides studied here, the SARS-CoV-derived peptide appears to be the fastest and most stable helix-former. Within only ∼250 ns of MD simulation time, an almost complete α-helix was formed. This helical structure persists almost without disruption for the whole 10 μs of the MD simulation. There are some helix-fraying events of the termini (see for example the N-terminal fraying events centered at ∼2.5 μs, 3.8 μs, and 9 μs in Fig. 3), but these do not change the major finding: the SARS-derived peptide is the strongest helix former of the four peptides studied here.There are some salient features of the behavior of the peptide that could have been missed in the data shown in Fig. 3 but are brought forward by the helicity graphs of Fig. 4. Referring to this Fig. 4, there is a small but systematic difference in helicity between the N-terminal half of the peptide (with a helical fraction of ∼0.8) and its C-terminal part (with a helical fraction of ∼0.9). As mentioned before, these two parts are separated by a pronounced dip in helicity centered on residue 16. The same pattern is observed for the MERS-derived peptide. The observed pattern for SARS ETM and MERS ETM of two high-helicity parts separated by a dip in helical content is also in agreement with the sequence alignment of the two peptides shown in Fig. 2. There are two regions showing significant conservation at the sequence level. The first (N-terminal) region encompasses residues 1–14 (ETGTLIVNSVLLFL in the SARS sequence), followed by four variable residues (15–18, AFVV), and then a second (C-terminal) half which again shows significant sequence similarity between SARS ETM and MERS ETM (19–31, FLLVTLAILTALR in the SARS sequence).
The MERS ETM forms a stable helical structure but has a more flexible N-terminal region compared to SARS ETM
As both Fig. 3 and 4 indicate, the two CoV-derived peptides are quite similar in their folding characteristics and structural behaviors, as would be expected from two peptides sharing a significant sequence similarity. Of the four peptides studied here, the next stronger helix former after the SARS-derived peptide is the MERS-derived peptide. Thus, the same bipartite organization in two (N- and C-terminal) halves each with a high helical content and separated by a region of reduced helicity near the middle of the peptide is also observed in the MERS-derived peptide.The similarity is more pronounced in the second (C terminal) part of the CoV-derived peptides and can be easily identified in Fig. 3, which shows that the helicity of the two CoV-derived peptides is virtually identical in their second half. In the N-terminal part, however, there are differences. This finding is not surprising: the peptide sequences in the C-terminal region (residues 19–32) share a sequence identity of almost 62%, whereas in the N-terminal region (residues 2–18), the identity drops to ∼24%. As can be seen in Fig. 3 (and in Fig. 4), the first half of the N-terminal half of the MERS-derived peptide is highly flexible with residues 1–5 all having an average helical content of less than ∼50%. Why there is such a pronounced difference for the N-terminal residues is not trivial to ascertain as the sequences themselves for the first half of the N-terminal half are closely related (ETGTLIV vs. RIGLFIV for the SARS- and MERS- peptides respectively), with the only consistent difference being the substitution of three hydrophobic residues in MERS ETM (I2, L4, and F9) with three polar residues in SARS ETM (T2, T4, and S9). It has been proved that serines or threonines can stabilize significantly α-helices in membrane peptides through the formation of hydrogen bonding interactions between their hydroxyl side chain and main chain carbonyl of residues i + 3 or/and i + 4.94,95 Here, residues 2, 4, and 9 form hydrogen bonds in SARS ETM with 66.4%, 54.5%, and 62.9% and in MERS ETM with 33.5%, 42.5%, and 67.6% of the 10 μs simulation period.
The influenza AM2TM peptide is mostly α-helical with an α-helix glycine disruptor in the middle of the peptide
The variability in the AM2TM helix propensity along the length of the peptide is shown in Fig. 2 and 3. Generally, this peptide is highly flexible and continuously folds and unfolds between α-helical structures and non-canonical structures, which are mostly turns. There is a discontinuity in the helical content in the middle of the peptide, which coincides with the presence of a glycine residue (Gly13; G34 in the AM2TM numbering scheme), which is known to act as a helix breaker. Although highly flexible along its whole sequence, AM2TM shows a more stable α-helical structure from Leu-5 to Leu-19, while at the C-terminal end of the peptide (W20–L25), the helical fraction is significantly lower. Also, the capping residues have non-helical φ, ψ dihedral angles, although they form helical (i, i + 4) hydrogen bonds.96A shorter (1.1 μs) molecular dynamics simulation performed previously52 had suggested more pronounced differences between the two halves with the C-terminal half (after Gly13, sequence ILHLILWILDRL) being much less helical compared to the N-terminal half (sequence SSDPLVVAASII). The longer 15.5 μs MD simulation described here clearly indicates that the differences in helicity between the two halves are less dramatic than those initially estimated.
The influenza BM2TM peptide is structurally divided into two distinct parts
The BM2TM peptide is significantly different from the three other peptides. It is clearly organized in two structurally distinct parts. The first (N-terminal) region comprises residues 1–15 in the BM2TM numbering. This first half of the peptide demonstrates a strong helix-forming tendency and folds quickly and stably to an α-helix that persists for almost the whole length of the ∼15.5 μs simulation. The fractional helicity of this part is identical (if not somewhat higher) to that of the SARS-derived peptide (Fig. 4).The second (C-terminal) part of the peptide shows a completely different behavior: it is highly flexible and continuously folds and unfolds to transient helical structures interspersed with intervals, where it samples not only non-canonical structures mainly turns but also random coil structures and in few instances β structures (Fig. 3). This flexibility and variability make a pronounced difference in the fractional helicity graphs of Fig. 4: almost all residues of the C-terminal half of the peptide have a helical fraction of less 60%. It should be noted, however, that there are some fine structures present in the helical propensity demonstrated by this C-terminal part. As can be seen from both Fig. 3 and 4, residues 20–24 do show an increased preference for a canonical α-helical structure reaching a helical fraction of ∼0.7 for residue 22. This motif “high helicity → Dip → high helicity” has been observed on all four peptides studied.
Discussion
We performed a total of 50 μs of molecular dynamics simulations with adaptive tempering to study the folding for the transmembrane peptides of the influenza A, B M2, MERS-, and SARS-CoV viroporins in TFE and compared with the experimental results. The AM2TM and BM2TM peptides have amino acid sequences that differ significantly, both between them, as well as with the two CoV-derived peptides studied in this communication (Fig. 2). On the other hand, the two CoV-derived peptides share a high sequence similarity (Fig. 2). Although all peptides are lipophilic, as expected from TM domains, it is worth noting that the AM2TM and BM2TM peptides—which correspond to tetrameric proton channels—are more polar while the two CoV-derived peptides are the most hydrophobic.The results of this study can provide further evidence that TFE can be used as a computational and experimental model to study folding of the studied hydrophobic peptides in a solution medium, allowing long MD simulations. The protocol (methods and analysis) applied here can be used in future studies to explore the character of hydrophobic helical peptides.
The 15.5 μs MD simulation of AM2TM revealed that this peptide is highly flexible and continuously folds and unfolds between α-helical structures and non-canonical structures, which are mostly turns. It seems, however, that AM2TM prefers an α-helical structure from Leu-5 to Leu-19, and only near the C-terminus of the peptide (from W20 to L25), the helicity is lowered. In AM2TM, there is a glycine residue that acts as an α-helix breaker in the middle of the peptide. In the X-ray structure of AM2TM (PDB ID 4QK735) or its ssNMR in the membrane (PBD ID 2H9533), the structure of the peptide in the tetrameric bundle is α-helical and there is a kink at G13 (G34 in the AM2TM numbering scheme) in the middle of the TM domain, which allows the peptide to change conformations and adopt distinct orientations between the C- and N-terminal regions.97 This G13 kink has been observed also in the ssNMR (PDB ID 2H95,332KQT43) or X-ray (PDB ID 6BKK,446US998) drug-bound AM2 structures. This pattern almost exactly matches that in the middle of the membrane. Using ssNMR, it has been shown that in the tetrameric bundle of AM2TM, the helices are flexible with conformational transitions99,100 that enable proton transportation through the channel.
The amino acid sequence of BM2 does not resemble that of the AM2 tetrameric bundle, except for the HxxxW motif, where the proton-selective residue is H19 and the gating residue is W23. BM2 has more polar pore-facing residues,28 whereas AM2 has a more hydrophobic pore. Thus, the aqueous pore of the AM2 ion channel is formed by Val,7 Ala,9 Gly,13 His,16 and Trp,20,35 compared with residues Ser,9 Ser,12 Ser,16 His,19 and Trp23 that line the pore of the four-helix bundle in BM2.28
Although both BM2 and AM2 channels exhibit microsecond-timescale His and Trp sidechain motions, similar to AM2,101 the BM2 peptide lacks the alternating-access hinge motion, but instead, it opens through a scissor motion. Upon activation, BM2 expands its pore along the entire channel, while AM2 constricts its N-terminus but expands its C-terminus. AM2 converts between two conformations: an N terminus-dilated and C-terminus-constricted (Nopen–Cclosed) conformation that is dominant at high pH102,103 and an N terminus-constricted and C-terminus dilated (Nclosed–Copen) conformation that is dominant at low pH.35,104
An interesting question here is which amino acid sequence features cause the alternating-access motion and the asymmetric conductance of AM2 and their absence in BM2 at acidic pH, where His residues are protonated.36 In BM2, the G13(G34) is replaced with S16 (S23), which reinforces the helical backbone and prevents separate motion of the two halves of the BTM helix. Additionally, BM2 has a symmetric HxxxWxxxH motif that is absent in AM2. Therefore, the electrostatic properties of the C-terminal residues in BM2, together with the absence of a central flexible Gly, are consistent with the symmetric backbone scissor motion of BM2 for channel activation and the consequent bidirectional proton conductance. These experimental observations are also consistent with the increased flexibility observed for the C-terminal half of BM2 in our simulations.
The BM2TM simulation showed that the N-terminal half of the peptide (comprising residues 1–15) folds quickly and stably to an α-helix with a helical fraction of ∼0.8, which is identical or somewhat higher than the helicity observed in the two CoV-derived peptides. In contrast, the second (C-terminal) part of the peptide with a helical fraction lower than 0.6 is more flexible compared to AM2TM and continuously folds and unfolds to transient α-helical structures, turns and coiled-coil, and instantaneously β-structures (noting, however, that residues 20–24 do demonstrate an increased preference for a canonical α-helical structure).
The SARS-derived peptide appears to be the fastest and most stable helix former with a helical structure that persists almost without disruption for the whole 10 μs of the MD simulation. The N-terminal half of the peptide (with a helical fraction of ∼0.8) is separated by the C-terminal part (with a helical fraction of ∼0.9). Indeed, the first (N-terminal) region encompasses residues 1–14 (ETGTLIVNSVLLFL, corresponding to 8–21 in the SARS ETM sequence), followed by four variable residues 15–18 (AFVV, corresponding to 22–25 in the SARS ETM sequence), and then a second C-terminal half with residues 19–31 which again shows significant sequence similarity (FLLVTLAILTALR, corresponding to 26–38 in the SARS sequence) with a pronounced dip in helicity centered on residue 16 (F, residue 23 in the SARS sequence).
Compared with SARS-CoV-2 ETM, the MERS ETM peptide has a pronounced identical helicity in the second (C-terminal) half, which is not surprising given that the peptide sequences for the region 19–31 share a sequence identity of almost 60%. In contrast, the N-terminal part of the MERS-derived peptide is highly flexible with residues 1–5 all having an average helical content of less than ∼0.5 although also the N-terminal sequences are closely related (ETGTLIV vs. RIGLFIV for the SARS- and MERS-peptides respectively), with the only consistent difference being the substitution of two hydrophobic residues in MERS ETM I2 and L4 (corresponding to I9 and L11 in the MERS ETM sequence) by two threonines in SARS ETM. These threonines can form hydrogen bonding interactions between their hydroxyl side chains and main-chain carbonyls of residues i + 3 or/and i + 4 (when are bifurcated),94,95 stabilizing α-helices.
Fig. 5 shows a collage of representative structures obtained from the trajectories. For each peptide, we showed the representative structure from the top three clusters obtained from a dihedral PCA analysis. The structures are colored according to the RMSF of the respective atoms from the average (for each cluster). For example, a pronounced structural variability can be observed in the AM2TM and BM2TM structures (see also Fig. S1 and S2, ESI†) and a higher RMSF near the middle of the structures as discussed in the other analyses.
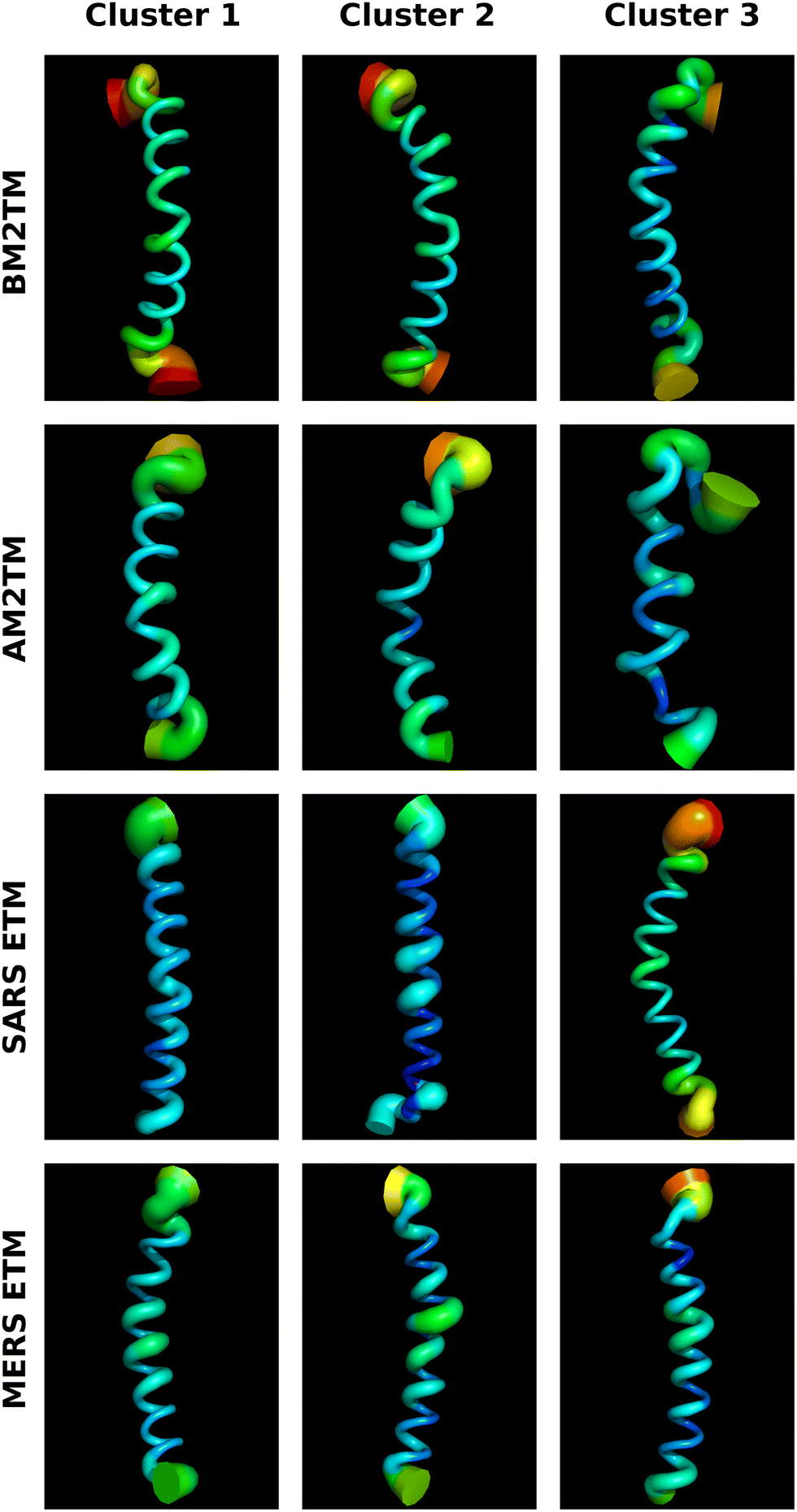 | ||
| Fig. 5 For each peptide is shown a representative structure from the top three clusters obtained from a dihedral PCA analysis. The structures are colored according to the RMSF of the respective atoms from the average structure in each cluster (color scale from blue, green, and yellow to red reflects RMSF lowering). | ||
The peptides’ secondary structure preferences and dynamics reflect features of the quaternary organization of the corresponding proteins and their biological function. The AM2 and BM2 TM tetrameric helical bundles have a higher percentage of polar residues such as His and Ser compared to ETM domains and form wide and hydrated pores of proton channels.35,36,43 The SARS or MERS ETM helical bundles are highly hydrophobic, compact, and rigid. This immobilization suggests that ETM peptides compared to M2TM may interact extensively with lipids while M2TM can form polar ion channel pores. Finally, the helix distortion at residues Phe13–Phe16 (corresponding to Phe20–Phe23 in the SARS ETM sequence) may cause the two halves of the ETM protein to respond semi-independently to environmental factors such as pH, charge, membrane composition, and other viral and host proteins.
At the immobilized C-terminal end of the SARS ETM segment, there are the conserved small residues Ala25 and Ala28 (corresponding to Ala32 and Thr35 in the SARS ETM sequence). In contrast to these small (or small and polar) residues, the central portion of the TM domain contains four layers of hydrophobic residues, Leu11, Leu14, Val18, and Leu21 (corresponding to Leu18, Leu21, Val25, and Leu28 in the SARS ETM sequence) and three regularly spaced phenylalanines Phe13, Phe16, and Phe19 (corresponding to Phe20, Phe23, and Phe26 in the SARS ETM sequence) at the center of ETM, suggesting that if ETM forms an ion channel, it can only form a pore radius to ∼2 Å in the channel's closed state. This narrow pore can permit only a single file of water molecules, thus partially dehydrating any ions that are not forced to move through the pore.
Conflicts of interest
There are no conflicts to declare.References
- Y. Finkel, N. Stern-Ginossar and M. Schwartz, Viral Short ORFs and Their Possible Functions, Proteomics, 2018, 18(10), 1700255 CrossRef PubMed.
- S. J. Opella, Relating structure and function of viral membrane-spanning miniproteins, Curr. Opin. Virol., 2015, 12, 121–125 CrossRef CAS PubMed.
- D. DiMaio, Viral Miniproteins, Annu. Rev. Microbiol., 2014, 68(1), 21–43 CrossRef CAS PubMed.
- N. Urakova, M. Frese, R. N. Hall, J. Liu, M. Matthaei and T. Strive, Expression and partial characterisation of rabbit haemorrhagic disease virus non-structural proteins, Virology, 2015, 484, 69–79 CrossRef CAS PubMed.
- J. L. Nieva, V. Madan and L. Carrasco, Viroporins: structure and biological functions, Nat. Rev. Microbiol., 2012, 10(8), 563–574 CrossRef CAS PubMed.
- J. To, W. Surya and J. Torres, Targeting the Channel Activity of Viroporins. in: Advances in Protein Chemistry and Structural Biology, 2016. p. 307–355. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1876162315001005 Search PubMed.
- C. Scott and S. Griffin, Viroporins: Structure, function and potential as antiviral targets, J. Gen. Virol., 2015, 96(8), 2000–2027 CrossRef CAS PubMed.
- C. Wang, K. Takeuchi, L. H. Pinto and R. A. Lamb, Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block, J. Virol., 1993, 67(9), 5585–5594 CrossRef CAS PubMed.
- R. A. Lamb, S. L. Zebedee and C. D. Richardson, Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface, Cell, 1985, 40(3), 627–633 CrossRef CAS PubMed.
- J. A. Mould, R. G. Paterson, M. Takeda, Y. Ohigashi, P. Venkataraman and R. A. Lamb, et al., Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes, Dev. Cell, 2003, 5, 175–184 CrossRef CAS PubMed.
- L. H. Pinto, L. J. Holsinger and R. A. Lamb, Influenza virus M2 protein has ion channel activity, Cell, 1992, 69(3), 517–528 CrossRef CAS PubMed.
- L. J. Holsinger, D. Nichani, L. H. Pinto and R. A. Lamb, Influenza A virus M2 ion channel protein: a structure-function analysis, J. Virol., 1994, 68(3), 1551–1563 CrossRef CAS PubMed.
- R. A. Lamb, K. J. Holsinger and L. H. Pinto, The influenza A virus M2 ion channel protein and its role in the influenza virus life cycle. in: E. Wimmer, ed. Cellular Receptors for Animal Viruses, Cold Spring Harbor Laboratory Press, Plainview, NY: Cold Spring Harbor Press, 1994. p. 303–21 Search PubMed.
- Y. Yang, F. Peng, R. Wang, K. Guan, T. Jiang and G. Xu, et al., The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China, J. Autoimmun., 2020, 109, 102434 CrossRef CAS PubMed.
- L. Wilson, C. Mckinlay, P. Gage and G. Ewart, SARS coronavirus E protein forms cation-selective ion channels, Virology, 2004, 330(1), 322–331 CrossRef CAS PubMed.
- J. L. Nieto-Torres, C. Verdiá-Báguena, J. M. Jimenez-Guardeño, J. A. Regla-Nava, C. Castaño-Rodriguez and R. Fernandez-Delgado, et al., Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome, Virology, 2015, 485, 330–339 CrossRef CAS PubMed.
- J. L. Nieto-Torres, M. L. DeDiego, C. Verdiá-Báguena, J. M. Jimenez-Guardeño, J. A. Regla-Nava and R. Fernandez-Delgado, et al., Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. M. R. Denison, ed. PLoS Pathog. 2014;10(5):e1004077. Available from: https://dx.plos.org/10.1371/journal.ppat.1004077 Search PubMed.
- C. Verdiá-Báguena, V. M. Aguilella, M. Queralt-Martín and A. Alcaraz, Transport mechanisms of SARS-CoV-E viroporin in calcium solutions: Lipid-dependent Anomalous Mole Fraction Effect and regulation of pore conductance, Biochim. Biophys. Acta, Biomembr., 2021, 1863(6), 183590 CrossRef PubMed.
- T. L. Toft-Bertelsen, M. G. Jeppesen, E. Tzortzini, K. Xue, K. Giller and S. Becker, et al., Amantadine inhibits known and novel ion channels encoded by SARS-CoV-2 in vitro, Commun. Biol., 2021, 4(1), 1–10 CrossRef PubMed.
- C. Verdiá-Báguena, J. L. Nieto-Torres, A. Alcaraz, M. L. DeDiego, J. Torres and V. M. Aguilella, et al., Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids, Virology, 2012, 432(2), 485–494 CrossRef PubMed.
- D. Cabrera-Garcia, R. Bekdash, G. W. Abbott, M. Yazawa and N. L. Harrison, The envelope protein of SARS-CoV-2 increases intra-Golgi pH and forms a cation channel that is regulated by pH, J. Physiol., 2021, 599(11), 2851–2868 CrossRef CAS PubMed.
- J. Torres, U. Maheswari, K. Parthasarathy, L. Ng, D. X. Liu and X. Gong, Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein, Protein Sci., 2007, 16(9), 2065–2071 CrossRef CAS PubMed.
- B. Xia, X. Shen, Y. He, X. Pan, F.-L. Liu and Y. Wang, et al., SARS-CoV-2 envelope protein causes acute respiratory distress syndrome (ARDS)-like pathological damages and constitutes an antiviral target, Cell Res., 2021, 31(8), 847–860 CrossRef CAS PubMed.
- C. Verdiá-Báguena, J. L. Nieto-Torres, A. Alcaraz, M. L. Dediego, L. Enjuanes and V. M. Aguilella, Analysis of SARS-CoV e protein ion channel activity by tuning the protein and lipid charge, Biochim. Biophys. Acta, Biomembr., 2013, 1828(9), 2026–2031 CrossRef PubMed.
- R. J. Sugrue and A. J. Hay, Structural characteristics of the M2 protein of influenza a viruses: Evidence that it forms a tetrameric channel, Virology, 1991, 180(2), 617–624 CrossRef CAS PubMed.
- L. J. Holsinger and R. Alams, Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds, Virology, 1991, 183(1), 32–43 CrossRef CAS PubMed.
- J. Hu, R. Fu, K. Nishimura, L. Zhang, H. X. Zhou and D. D. Busath, et al., Histidines, heart of the hydrogen ion channel from influenza A virus: Toward an understanding of conductance and proton selectivity, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(18), 6865–6870 CrossRef CAS PubMed.
- C. Ma, C. S. Soto, Y. Ohigashi, A. Taylor, V. Bournas and B. Glawe, et al., Identification of the pore-lining residues of the BM2 ion channel protein of influenza B virus, J. Biol. Chem., 2008, 283(23), 15921–15931 CrossRef CAS PubMed.
- G. P. Leser and R. A. Lamb, Lateral Organization of Influenza Virus Proteins in the Budozone Region of the Plasma Membrane. D. S. Lyles, ed. J. Virol. 2017;91(9):e02104–e02116. Available from: https://journals.asm.org/doi/10.1128/JVI.02104-16 Search PubMed.
- K. L. Roberts, G. P. Leser, C. Ma and R. A. Lamb, The Amphipathic Helix of Influenza A Virus M2 Protein Is Required for Filamentous Bud Formation and Scission of Filamentous and Spherical Particles, J. Virol., 2013, 87(18), 9973–9982 CrossRef CAS PubMed.
- B. J. Chen, G. P. Leser, D. Jackson and R. A. Lamb, The Influenza Virus M2 Protein Cytoplasmic Tail Interacts with the M1 Protein and Influences Virus Assembly at the Site of Virus Budding, J. Virol., 2008, 82(20), 10059–10070 CrossRef CAS PubMed.
- M. Imai, K. Kawasaki and T. Odagiri, Cytoplasmic Domain of Influenza B Virus BM2 Protein Plays Critical Roles in Production of Infectious Virus, J. Virol., 2008, 82(2), 728–739 CrossRef CAS PubMed.
- J. Hu, T. Asbury, S. Achuthan, C. Li, R. Bertram and J. R. Quine, et al., Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from influenza A virus, Biophys. J., 2007, 92(12), 4335–4343 CrossRef CAS PubMed.
- M. Sharma, M. Yi, H. Dong, H. Qin, E. Peterson and D. D. Busath, et al., Insight into the Mechanism of the Influenza A Proton Channel from a Structure in a Lipid Bilayer, Science, 2010, 330(6003), 509–512 CrossRef CAS PubMed.
- J. L. Thomaston, M. Alfonso-Prieto, R. A. Woldeyes, J. S. Fraser, M. L. Klein and G. Fiorin, et al., High-resolution structures of the M2 channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(46), 14260–14265 CrossRef CAS PubMed.
- V. S. Mandala, A. R. Loftis, A. A. Shcherbakov, B. L. Pentelute and M. Hong, Atomic structures of closed and open influenza B M2 proton channel reveal the conduction mechanism, Nat. Struct. Mol. Biol., 2020, 27(2), 160–167 CrossRef CAS PubMed.
- S. Watanabe, M. Imai, Y. Ohara and T. Odagiri, Influenza B Virus BM2 Protein Is Transported through the trans- Golgi Network as an Integral Membrane Protein, J. Virol., 2003, 77(19), 10630–10637 CrossRef CAS PubMed.
- W. Surya, Y. Li and J. Torres, Structural model of the SARS coronavirus E channel in LMPG micelles, Biochim. Biophys. Acta, Biomembr., 2018, 1860(6), 1309–1317 CrossRef CAS PubMed.
- V. S. Mandala, M. J. McKay, A. A. Shcherbakov, A. J. Dregni, A. Kolocouris and M. Hong, Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers, Nat. Struct. Mol. Biol., 2020, 27(12), 1202–1208 CrossRef CAS PubMed.
- J. Medeiros-Silva, K. H Somberg N, J. Wang H, S. McKay M, J. Mandala V and A. Dregni, et al., pH- and Calcium-Dependent Aromatic Network in the SARS-CoV-2 Envelope Protein, J. Am. Chem. Soc., 2022, 144(15), 6839–6850 CrossRef CAS PubMed.
- D. Schoeman and B. C. Fielding, Coronavirus envelope protein: current knowledge, Virol. J., 2019, 16(1), 69 CrossRef PubMed.
- C. Castaño-Rodriguez, J. M. Honrubia, J. Gutiérrez-Álvarez, M. L. DeDiego, J. L. Nieto-Torres and J. M. Jimenez-Guardeño, et al., Role of Severe Acute Respiratory Syndrome Coronavirus Viroporins E, 3a, and 8a in Replication and Pathogenesis. M. R. Denison, ed. Mbio, 2018;9(3). Available from: https://journals.asm.org/doi/10.1128/mBio.02325-17 Search PubMed.
- S. D. Cady, K. Schmidt-Rohr, J. Wang, C. S. Soto, W. F. DeGrado and M. Hong, Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers, Nature, 2010, 463(7281), 689–692 CrossRef CAS PubMed.
- J. L. Thomaston, N. F. Polizzi, A. Konstantinidi, J. Wang, A. Kolocouris and W. F. Degrado, Inhibitors of the M2 proton channel engage and disrupt transmembrane networks of hydrogen-bonded waters, J. Am. Chem. Soc., 2018, 140(45), 15219–15226 CrossRef CAS PubMed.
- N. Kolocouris, G. B. Foscolos, A. Kolocouris, P. Marakos, N. Pouli and G. Fytas, et al., Synthesis and Antiviral Activity Evaluation of Some Aminoadamantane Derivatives, J. Med. Chem., 1994, 37(18), 2896–2902 CrossRef CAS PubMed.
- E. Torres, R. Fernández, S. Miquet, M. Font-Bardia, E. Vanderlinden and L. Naesens, et al., Synthesis and anti-influenza a virus activity of 2,2-dialkylamantadines and related compounds, ACS Med. Chem. Lett., 2012, 3(12), 1065–1069 CrossRef CAS PubMed.
- A. Kolocouris, C. Tzitzoglaki, F. B. Johnson, R. Zell, A. K. Wright and T. A. Cross, et al., Aminoadamantanes with persistent in vitro efficacy against H1N1 (2009) influenza A, J. Med. Chem., 2014, 57(11), 4629–4639 CrossRef CAS PubMed.
- J. J. Wang, Y. Wu, C. Ma, G. Fiorin, L. H. Pinto and R. A. Lamb, et al., Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(4), 1315–1320 CrossRef CAS PubMed.
- J. Wang, C. Ma, G. Fiorin, V. Carnevale, T. Wang and F. Hu, et al., Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2, J. Am. Chem. Soc., 2011, 133(32), 12834–12841 CrossRef CAS PubMed.
- C. Tzitzoglaki, A. Wright, K. Freudenberger, A. Hoffmann, I. Tietjen and I. Stylianakis, et al., Binding and Proton Blockage by Amantadine Variants of the Influenza M2WT and M2S31N Explained, J. Med. Chem., 2017, 60(5), 1716–1733 CrossRef CAS PubMed.
- A. K. Wright, P. Batsomboon, J. Dai, I. Hung, H. X. Zhou and G. B. Dudley, et al., Differential Binding of Rimantadine Enantiomers to Influenza A M2 Proton Channel, J. Am. Chem. Soc., 2016, 138(5), 1506–1509 CrossRef CAS PubMed.
- I. Stylianakis, A. Shalev, S. Scheiner, M. P. Sigalas, I. T. Arkin and N. Glykos, et al., The balance between side-chain and backbone-driven association in folding of the α-helical influenza A transmembrane peptide, J. Comput. Chem., 2020, 41(25), 2177–2188 CrossRef CAS PubMed.
- A. Kentsis and T. R. Sosnick, Trifluoroethanol Promotes Helix Formation by Destabilizing Backbone Exposure: Desolvation Rather than Native Hydrogen Bonding Defines the Kinetic Pathway of Dimeric Coiled Coil Folding, Biochemistry, 1998, 37(41), 14613–14622 CrossRef CAS PubMed.
- P. Luo and R. L. Baldwin, Mechanism of Helix Induction by Trifluoroethanol: A Framework for Extrapolating the Helix-Forming Properties of Peptides from Trifluoroethanol/Water Mixtures Back to Water, Biochemistry, 1997, 36(27), 8413–8421 CrossRef CAS PubMed.
- B. Chaubey, A. Dey, A. Banerjee, N. Chandrakumar and S. Pal, Assessment of the Role of 2,2,2-Trifluoroethanol Solvent Dynamics in Inducing Conformational Transitions in Melittin: An Approach with Solvent 19 F Low-Field NMR Relaxation and Overhauser Dynamic Nuclear Polarization Studies, J. Phys. Chem. B, 2020, 124(28), 5993–6003 CrossRef CAS PubMed.
- J. Vymětal, L. Bednárová and J. Vondrášek, Effect of TFE on the Helical Content of AK17 and HAL-1 Peptides: Theoretical Insights into the Mechanism of Helix Stabilization, J. Phys. Chem. B, 2016, 120(6), 1048–1059 CrossRef PubMed.
- R. C. Neuman and J. T. Gerig, Interactions of 2,2,2-trifluoroethanol with melittin, Magn. Reson. Chem., 2009, 47(11), 925–931 CrossRef CAS PubMed.
- L. S. Ramirez, J. Pande and A. Shekhtman, Helical Structure of Recombinant Melittin, J. Phys. Chem. B, 2019, 123(2), 356–368 CrossRef CAS PubMed.
- L. Ramirez, A. Shekhtman and J. Pande, Nuclear Magnetic Resonance-Based Structural Characterization and Backbone Dynamics of Recombinant Bee Venom Melittin, Biochemistry, 2018, 57(19), 2775–2785 CrossRef CAS PubMed.
- A. M. S. Duarte, C. P. M. Van Mierlo and M. A. Hemminga, Molecular dynamics study of the solvation of an α-helical transmembrane peptide by DMSO, J. Phys. Chem. B, 2008, 112(29), 8664–8671 CrossRef CAS PubMed.
- Y. Yu, J. Wang, Q. Shao, J. Shi and W. Zhu, The effects of organic solvents on the folding pathway and associated thermodynamics of proteins: A microscopic view, Sci. Rep., 2016, 6, 19500 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo and K. M. Merz, et al., The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26(16), 1668–1688 CrossRef CAS PubMed.
- L. Kalé, R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy and N. Krawetz, et al., NAMD2: Greater Scalability for Parallel Molecular Dynamics, J. Comput. Phys., 1999, 151, 283–312 CrossRef.
- R. Chitra and P. E. Smith, Properties of 2, 2, 2-trifluoroethanol and water mixtures, J. Chem. Phys., 2001, 114(1), 426–435 CrossRef CAS.
- F. Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong and N. Grivel, et al., The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building, Phys. Chem. Chem. Phys., 2010, 12(28), 7821–7839 RSC.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Comparison of multiple amber force fields and development of improved protein backbone parameters, Proteins: Struct., Funct., Genet., 2006, 65(3), 712–725 CrossRef CAS PubMed.
- L. Wickstrom, A. Okur and C. Simmerling, Evaluating the performance of the FF99SB force field based on NMR scalar coupling data, Biophys. J., 2009, 97(3), 853–856 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis and R. O. Dror, et al., Improved side-chain torsion potentials for the Amber ff99SB protein force field, Proteins: Struct., Funct., Bioinf., 2010, 78(8), 1950–1958 CrossRef CAS PubMed.
- R. B. Best and G. Hummer, Optimized Molecular Dynamics Force Fields Applied to the Helix–Coil Transition of Polypeptides, J. Phys. Chem. B, 2009, 113(26), 9004–9015 CrossRef CAS PubMed Available from:.
- P. S. Georgoulia and N. M. Glykos, Using J-coupling constants for force field validation: Application to hepta-alanine, J. Phys. Chem. B, 2011, 115(51), 15221–15227 CrossRef CAS PubMed.
- P. S. Georgoulia and N. M. Glykos, On the foldability of tryptophan-containing tetra-and pentapeptides: An exhaustive molecular dynamics study, J. Phys. Chem. B, 2013, 117(18), 5522–5532 CrossRef CAS PubMed.
- K. K. Patapati and N. M. Glykos, Order through disorder: Hyper-mobile C-terminal residues stabilize the folded state of a helical peptide. A molecular dynamics study, PLoS One, 2010, 5(12), e15290 CrossRef CAS PubMed.
- I. Patmanidis and N. M. Glykos, As good as it gets? Folding molecular dynamics simulations of the LytA choline-binding peptide result to an exceptionally accurate model of the peptide structure, J. Mol. Graphics Modell., 2013, 41, 68–71 CrossRef CAS PubMed.
- A. M. Razavi and V. A. Voelz, Kinetic Network Models of Tryptophan Mutations in β-Hairpins Reveal the Importance of Non-Native Interaction, J. Chem. Theory Comput., 2015, 11(6), 2801–2812 CrossRef CAS PubMed.
- R. B. Best, W. W. Zheng and J. Mittal, Balanced protein-water interactions improve properties of disordered proteins and non-specific protein association, J. Chem. Theory Comput., 2014, 10(11), 5113–5124 CrossRef CAS PubMed.
- S. Piana, J. L. Klepeis and D. E. Shaw, Assessing the accuracy of physical models used in protein-folding simulations: Quantitative evidence from long molecular dynamics simulations, Curr. Opin. Struct. Biol., 2014, 24(1), 98–105 CrossRef CAS PubMed.
- A. P. Serafeim, G. Salamanos, K. K. Patapati and N. M. Glykos, Sensitivity of Folding Molecular Dynamics Simulations to even Minor Force Field Changes, J. Chem. Inf. Model., 2016, 56(10), 2035–2041 CrossRef CAS PubMed.
- P. I. Koukos and N. M. Glykos, Folding molecular dynamics simulations accurately predict the effect of mutations on the stability and structure of a vammin-derived peptide, J. Phys. Chem. B, 2014, 118(34), 10076–10084 CrossRef CAS PubMed.
- A. S. Baltzis and N. M. Glykos, Characterizing a partially ordered miniprotein through folding molecular dynamics simulations: Comparison with the experimental data, Protein Sci., 2016, 25(3), 587–596 CrossRef CAS PubMed.
- T. Adamidou, K.-O. Arvaniti and N. M. Glykos, Folding Simulations of a Nuclear Receptor Box-Containing Peptide Demonstrate the Structural Persistence of the LxxLL Motif Even in the Absence of Its Cognate Receptor, J. Phys. Chem. B, 2018, 122(1), 106–116 CrossRef CAS PubMed.
- C. Zhang and J. Ma, Enhanced sampling and applications in protein folding in explicit solvent, J. Chem. Phys., 2010, 132(24), 244101 CrossRef PubMed.
- D. J. Evans and B. L. Holian, The Nose-Hoover thermostat, J. Chem. Phys., 1985, 83(8), 4069–4074 CrossRef CAS.
- J. A. Lzaguirre, D. P. Catarello, J. M. Wozniak and R. D. Skeel, Langevin stabilization of molecular dynamics, J. Chem. Phys., 2001, 114(5), 2090–2098 CrossRef.
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, Constant pressure molecular dynamics simulation: The Langevin piston method, J. Chem. Phys., 1995, 103(11), 4613–4621 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23(3), 327–341 CrossRef CAS.
- D. Frishman and P. Argos, Knowledge-based protein secondary structure assignment, Proteins: Struct., Funct., Bioinf., 1995, 23(4), 566–579 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- E. A. Merritt and D. J. Bacon, Raster3D: Photorealistic Molecular Graphics, Methods Enzymol., 1997, 277, 505–524 CAS.
- The PyMOL molecular graphics system, Version 1.8, Schrödinger LLC, 2015 Search PubMed.
- G. E. Crooks, G. Hon, J. M. Chandonia and S. E. Brenner, WebLogo: A sequence logo generator, Genome Res., 2004, 14(6), 1188–1190 CrossRef CAS PubMed.
- N. M. Glykos, Carma: A Molecular Dynamics Analysis Program, J. Comput. Chem., 2006, 27(14), 1765–1768 CrossRef CAS PubMed.
- P. I. Koukos and N. M. Glykos, On the Application of Good-Turing Statistics to Quantify Convergence of Biomolecular Simulations, J. Chem. Inf. Model., 2014, 54(1), 209–217 CrossRef CAS PubMed Available from:.
- E. S. Feldblum and I. T. Arkin, Strength of a bifurcated H bond, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4085–4090 CrossRef CAS PubMed.
- E. S. Brielle and I. T. Arkin, Quantitative Analysis of Multiplex H-Bonds, J. Am. Chem. Soc., 2020, 142(33), 14150–14157 CrossRef CAS PubMed.
- D. A. E. Cochran, Effect of the N1 residue on the stability of the alpha-helix for all 20 amino acids, Protein Sci., 2001, 10, 463–470 CrossRef CAS PubMed.
- S. D. Cady, C. Goodman, C. D. Tatko, W. F. DeGrado and M. Hong, Determining the orientation of uniaxially rotating membrane proteins using unoriented samples: A 2H, 13C, and 15N solid-state NMR investigation of the dynamics and orientation of a transmembrane helical bundle, J. Am. Chem. Soc., 2007, 129(17), 5719–5729 CrossRef CAS PubMed.
- J. L. Thomaston, M. L. Samways, A. Konstantinidi, C. Ma, Y. Hu and H. E. Bruce Macdonald, et al., Rimantadine Binds to and Inhibits the Influenza A M2 Proton Channel without Enantiomeric Specificity, Biochemistry, 2021, 60(32), 2471–2482 CrossRef CAS PubMed.
- L. C. Watkins, R. Liang, J. M. J. Swanson, W. F. DeGrado and G. A. Voth, Proton-Induced Conformational and Hydration Dynamics in the Influenza A M2 Channel, J. Am. Chem. Soc., 2019, 141(29), 11667–11676 CrossRef CAS PubMed.
- W. Luo and M. Hong, Conformational changes of an ion channel detected through water - Protein interactions using solid-state NMR spectroscopy, J. Am. Chem. Soc., 2010, 132(7), 2378–2384 CrossRef CAS PubMed.
- F. Hu, W. Luo and M. Hong, Mechanisms of Proton Conduction and Gating in Influenza M2 Proton Channels from Solid-State NMR, Science, 2010, 330(6003), 505–508 CrossRef CAS PubMed.
- F. Hu, W. Luo, S. D. Cady and M. Hong, Conformational plasticity of the influenza A M2 transmembrane helix in lipid bilayers under varying pH, drug binding, and membrane thickness, Biochim. Biophys. Acta, Biomembr., 2011, 1808(1), 415–423 CrossRef CAS PubMed.
- L. B. Andreas, M. Reese, M. T. Eddy, V. Gelev, Q. Zhe Ni and E. A. Miller, et al., Structure and Mechanism of the Influenza A M218–60 Dimer of Dimers, J. Am. Chem. Soc., 2015, 137(47), 14877–14886 CrossRef CAS PubMed.
- R. Acharya, V. Carnevale, G. Fiorin, B. G. Levine, A. L. Polishchuk and V. Balannik, et al., Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(34), 15075–15080 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2 – RMSD (Ca carbons) of peptides tested vs. time and distribution of the RMSD (Ca carbons) over calculated snapshots for the peptides tested from folding dynamics. See DOI: https://doi.org/10.1039/d2cp02881f |
| This journal is © the Owner Societies 2022 |