
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beware of beam damage under reaction conditions: X-ray induced photochemical reduction of supported VOx catalysts during in situ XAS experiments†
Anna
Zabilska
ab,
Adam H.
Clark
 a,
Davide
Ferri
a,
Maarten
Nachtegaal
a,
Oliver
Kröcher
ab and
Olga V.
Safonova
*a
a,
Davide
Ferri
a,
Maarten
Nachtegaal
a,
Oliver
Kröcher
ab and
Olga V.
Safonova
*a
aPaul Scherrer Institute, 5232 Villigen, Switzerland. E-mail: olga.safonova@psi.ch
bÉcole Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland
First published on 25th August 2022
Abstract
In situ X-ray absorption spectroscopy (XAS) is a powerful technique for the investigation of heterogeneous catalysts and electrocatalysts. The obtained XAS spectra are usually interpreted from the point of view of the investigated chemical processes, thereby sometimes omitting the fact that intense X-ray irradiation may induce additional transformations in metal speciation and, thus, in the corresponding XAS spectra. In this work, we report on X-ray induced photochemical reduction of vanadium in supported vanadia (VOx) catalysts under reaction conditions, detected at a synchrotron beamline. While this process was not observed in an inert atmosphere and in the presence of water vapor, it occurred at room temperature in the presence of a reducing agent (ethanol or hydrogen) alone or mixed with oxygen. Temperature programmed experiments have shown that X-ray induced reduction of VOx species appeared very clear at 30–100 °C but was not detected at higher temperatures, where the thermocatalytic ethanol oxidative hydrogenation (ODH) takes place. Similar to other studies on X-ray induced effects, we suggest approaches, which can help to mitigate vanadium photoreduction, including defocusing of the X-ray beam and attenuation of the X-ray beam intensity by filters. To recognize beam damage under in situ/operando conditions, we suggest performing X-ray beam switching (on and off) tests at different beam intensities under in situ conditions.
Introduction
Spectroscopic methods applied under in situ and/or operando conditions are powerful tools for understanding the mechanisms of catalytic reactions including the structure of the catalytic active site.1,2 X-Ray absorption spectroscopy (XAS) is a particularly powerful tool in this respect because it is element-specific and provides information on the electronic and local geometric structure of metal species.3,4 In recent decades, research on catalytic mechanisms has benefited from the development of time-resolved energy-dispersive XAS and quick XAS methods and their proliferation across numerous light sources.5–10 Fast XAS acquisition allows the in situ (operando) experiments to be performed under transient conditions that allows for the detection of active sites directly involved in redox reactions.11–15 While performing in situ/operando investigations, one typically expects to observe only chemically induced transformations; however, an additional reactive component, the X-ray beam, should not be neglected.X-Ray induced damage is a common issue in the X-ray crystallography of proteins16–19 and XAS of biological samples and samples in water.20–24 In such systems, X-ray irradiation may lead to the formation of radicals and electrons, which can cause sample damage including reduction of metal species.16–24 Inorganic water-free systems in this regard are more stable; however, several cases of photo-reduction were reported in X-ray photoelectron spectroscopy (XPS),25,26 where beam damage was enhanced by the use of vacuum. Radiation damage observed during XAS experiments on heterogeneous catalysts has rarely been reported. Newton et al.27 investigated a Cu(II)-containing zeolite in heterogeneously catalyzed methane oxidation to methanol and found that Cu(II) may undergo reduction in the presence of methane or pure helium upon X-ray irradiation. Albrahim et al.28 investigated a highly dispersed RhOx/Al2O3 catalyst by in situ XAS and detected that rhodium clusters undergo reduction even at room temperature in the presence of hydrogen and intense X-rays; in situ heating to 310 °C leads to further agglomeration of clusters, comparable to agglomeration typically observed at 650 °C without exposure to the X-ray beam. Van Schooneveld and DeBeer29 showed that iron in K3[Fe(CN)6] and manganese in KMnO4 can undergo X-ray-induced reduction during XAS measurements; the authors investigated the relationship between the X-ray dose and the extent of metal reduction by measuring these samples at different synchrotron beamlines.
Vanadium-based catalysts are among the most common redox-active systems used in homogeneous and heterogeneous catalyses.30,31 The mechanisms of heterogeneous catalytic reactions of selective oxidation and reduction over vanadium-based catalysts are still being revised by modern in situ and operando X-ray-based techniques, such as XPS and XAS.32–40 There are several publications, which describe X-ray-induced vanadium reduction during XPS experiments.26,41–43 It was suggested that the electron–hole pair generation or Auger decay may lead to holes in the valence band, which from a physical point of view are similar to bond breaking.41 Vanadium reduction in these publications was associated with the loss of lattice oxygen, which is enhanced by ultra-high vacuum,43 whereas the presence of low levels of oxygen in the gas phase prevented or slowed down this process.42
In our previous work, using operando quick XAS,36 we showed that the rate of ethanol oxidation to acetaldehyde over a bilayered 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst (consisting of sub-monolayer VOx species anchored onto a TiOx monolayer supported on SiO2) is kinetically coupled to the V5+ → V4+ redox process. The effect of the X-ray beam on the kinetic data at the catalysis relevant temperatures (above 150 °C) was tested and excluded. We did, however, detect unexpected vanadium reduction below 100 °C, which we associate with X-ray irradiation. This warrants a detailed study of beam-induced vanadium reduction, the results of which are presented here. To understand the nature and the conditions of vanadium photoreduction, we performed a series of experiments showing that below 100 °C the X-ray beam does not affect vanadium speciation in air and in the presence of oxygen, helium, or water vapor. Only a combination of the X-ray beam with a reducing agent, such as ethanol, hydrogen, or an ethanol–oxygen mixture, resulted in the photochemical reduction of vanadium. Herein, we systematically study the X-ray-induced reduction of vanadium and propose approaches to recognize and suppress it.
Materials and methods
The majority of experiments described in this work were performed using a model bilayered 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst,44 which we used in our previous work which was focused on the redox activity of vanadium and titanium during thermocatalytic ODH of ethanol.36 This catalyst has close to one monolayer titanium coverage and was prepared as an analog of a widely used V2O5/TiO2 catalyst. The latter one demonstrates outstanding activity in a variety of redox catalytic reactions,45–48 however, it could not be efficiently investigated by time-resolved V K-edge XAS due to almost full X-ray absorption by titanium (the Ti K-edge, 4966 eV, is located just below the V K-edge, 5465 eV). Thus, a 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst was chosen as a compromise: on one hand, its titania loading is sufficient to promote the activity of vanadium species,36,44 and on the other hand, it still allows measuring V K-edge XAS with a time-resolution of 1–10 s.36Materials
The detailed catalyst synthesis and characterization procedures of the 5 wt%V2O5/15 wt%TiO2/SiO2 catalyst can be found elsewhere.36,44 The 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst has a surface density of ca. 4.9 Ti nm−2, which corresponds to monolayer TiOx dispersion on silica.49 The VOx surface density is equal to 1.8 V nm−2, which is below that of monolayer dispersion (8 V nm−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 50). Vanadium surface species are present as dimeric and trimeric VOx species; no crystalline V2O5 was detected by Raman spectroscopy.44
50). Vanadium surface species are present as dimeric and trimeric VOx species; no crystalline V2O5 was detected by Raman spectroscopy.44
For comparison, we also investigated a titania-free 8 wt% V2O5/SiO2 catalyst, which contains ca. 2.3 V nm−2 mostly present as isolated tetrahedral VO4 species.51
V K-edge XAS experiments
The gas flows were controlled using mass-flow controllers (Bronkhorst). To provide ethanol flow, the helium flow (purity 99.997%, 30 mL min−1) passed through a saturator filled with ethanol (purity 99.997%) and kept at 8 °C. The concentration of oxygen was varied by admixing the initial oxygen source (14 or 40 vol% O2 in He) with helium. The total flow passing through the cell was constant in every experiment and was equal to 50 mL min−1.
Before every experiment (unless stated otherwise), the catalyst was pre-treated inside the cell by heating in a flow of 14 vol% O2 in He to 400 °C at 12 °C min−1 and dwelling for 30 min.
Product analysis
During operando investigations, the gas composition at the outlet of the reactor cell was analyzed using a Fourier transform infrared (FT-IR) spectrometer (Bruker Alpha II) equipped with a 70 mm path length cell (95% of the gas is replaced in 15 s) heated to 150 °C to prevent condensation. The absorbance (47 scans per spectrum) was recorded in the 4000–500 cm−1 range at a resolution of 4 cm−1. This allowed a time resolution of the product gas analysis of 1 min, which was particularly important during temperature-programmed experiments. Before every experiment, the background spectrum was recorded in an oxygen flow. To quantify the products, specific spectral regions corresponding to the selected molecules were extracted and analyzed using the multivariate curve resolution alternating least square (MCR-ALS) analysis55 (Fig. S4, ESI†). For quantitative calibration, the spectra of pure compounds with different concentrations were included in the experimental set for the MCR-ALS analysis.Results and discussion
The identification of X-ray beam-induced vanadium reduction
Oxidative dehydrogenation of ethanol over supported VOx catalysts proceeds via the Mars–van Krevelen mechanism.45,56,57 The step of ethanol oxidation by lattice oxygen, which is accompanied by oxygen vacancy formation and metal reduction, is followed by the step of catalyst re-oxidation by molecular oxygen. Mechanistic investigations showed that the step of catalyst re-oxidation by oxygen is faster than the step of catalyst reduction by ethanol. As a result, in the presence of oxygen in the gas feed, the majority of vanadium species are in the highest V5+ oxidation state.36,45,56,58,59 In our previous work,36 using operando quick XAS under steady-state and transient conditions, we quantitatively showed that the rate of ethanol oxidation over a 5 wt%V2O5/15 wt%TiO2/SiO2 catalyst above 150 °C is kinetically coupled to the V5+ to V4+ reduction process. The effect of the X-ray beam on the kinetic data at the catalytic relevant temperatures (above 150 °C) was tested by varying the beam density and based on these tests excluded. Nevertheless, at temperatures below the catalysis relevant conditions, we observed previously unreported beam effects that we focus on in detail in the present work.Fig. 1a shows the evolution of V K-edge XANES spectra of a 5 wt%V2O5/15 wt%TiO2/SiO2 catalyst during an operando temperature-programmed experiment (TPE) in ethanol–oxygen feed, where the fully oxidized catalyst was exposed to an ethanol–oxygen flow (1.6 vol% EtOH, 6.4 vol% O2 in He) while heating from 30 to 250 °C. Fig. 1b shows the rate of acetaldehyde formation detected at the outlet of the cell, which confirmed that the catalyst becomes active at around 120 °C. Unexpected changes in the V K-edge XAS spectra appeared below the temperature of ethanol oxidation, at 30–100 °C (Fig. 1a). In this temperature interval, the pre-edge peak intensity decreased and the V K-edge edge position shifted towards lower energies. At higher temperatures, when the catalyst became active, the spectra were almost reverting to the initial state.
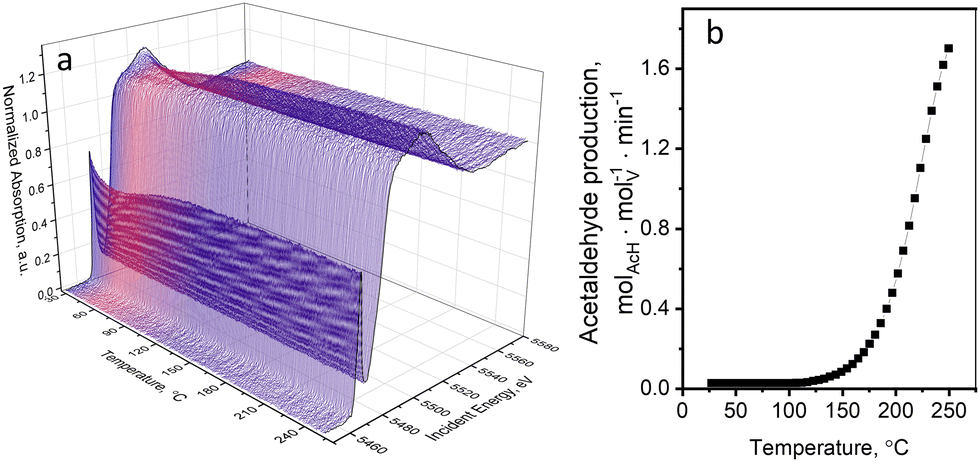 | ||
| Fig. 1 (a) V K-edge XANES spectra of the 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst during TPE in ethanol–oxygen feed (1.6 vol% EtOH, 6.4 vol% O2 in He); and (b) the rate of acetaldehyde production detected by FTIR spectroscopy in the same experiment. | ||
The pre-edge peak in V K-edge XAS spectra is observed due to the dipole-forbidden 1s–3d electron transition, which becomes partially allowed in non-centrosymmetric structures due to 3p–4d orbital mixing and overlap of the V 3d with ligand (O) 2p orbitals.60,61 The pre-edge shape and position depend on the structural parameters such as geometry around vanadium, length of bonds, oxidation state, and type of ligands.61,62 Characteristics of the pre-edge (such as position, height, and intensity) are often used to identify and semi-quantify the oxidation state and geometry of vanadium in unknown compounds.61,63–67 The edge position is another important spectral feature often used in the literature for the determination of the oxidation state.61,66–69 In our previous work, we measured V K-edge spectra of 21 reference compounds with known structure, containing vanadium atoms in different oxidation states and surrounded by oxygen in different local geometries, which are potentially helpful to identify the oxidation state of unknown vanadium structures.36 We showed that using the edge position and the pre-edge surface area (or pre-edge height) one can estimate the oxidation state and the coordination number of vanadium in an unknown state. The edge position and the pre-edge intensity (height and area) generally increase in the order V3+ < V4+ < V5+. The pre-edge intensity, besides, depends on the coordination number of vanadium and decreases in the following order: tetrahedral–pentahedra–octahedra (for details refer to ref. 36 and to the ESI,† Section 3.1). In this work, for the rapid qualitative analysis of the oxidation state of vanadium, we decided to use two descriptors: the pre-edge height and the edge position.
Fig. 2 (black curves) shows the changes in the pre-edge height and the edge position of each XAS spectrum during ethanol–oxygen TPE. The color bars indicate the ranges of values of the descriptors corresponding to vanadium references in V3+, V4+, and V5+ oxidation states (details in Fig. S12, ESI†). At temperatures relevant to ODH of ethanol, i.e. at 110–250 °C, the pre-edge height and the edge position trends for the catalyst show that vanadium species are mostly in the +5 oxidation state. As expected, during ethanol ODH vanadium re-oxidation is faster than vanadium reduction, thus, in the ethanol–oxygen feed, the resting oxidation state of vanadium in VOx species is close to +5. However, at low temperatures, when acetaldehyde is not detected in the outlet (30–110 °C, Fig. 1b), the pre-edge intensity is lower and the position of the edge shifts towards lower energies. These changes may suggest that vanadium undergoes partial reduction. However, based on the values of the descriptors, which overlap for V5+ and V4+ references (especially for the pre-edge height), these experiments could not exclude that the observed spectral changes are due to an increase in the coordination number of VOx species (see also Fig. S12, ESI†), e.g. due to the coordination of ethanol.
 | ||
| Fig. 2 (a) The pre-edge height and (b) the half-edge step position of the references (indicated by the colored bars) and of the catalyst measured during TPE in ethanol–oxygen feed (1.6 vol% EtOH, 6.4 vol% O2 in He); details are in Fig. S12 (ESI†). | ||
To further test the transformations taking place at low temperatures, we exposed the catalyst to an oxygen-free ethanol-containing feed (1.6 vol% EtOH in He) at 30 °C for two hours. The V K-edge spectra measured in this experiment and the corresponding changes in the V pre-edge height and the edge position are shown in Fig. 3. After two hours of exposure of the catalyst to the X-ray beam, the pre-edge intensity decreased from 0.74 to 0.25 a.u. in the normalized V K-edge XAS spectra, whereas the edge position shifted from 5481 to 5478 eV. Both descriptors suggest strong vanadium reduction, which proceeds beyond V4+, towards V3+. For comparison, the V K-edge spectrum of the catalyst reduced at 400 °C in ethanol (1.6 vol% in He) is also shown in Fig. 3 (dark red curve in (a) and red dot in (b) and (c)). Under these conditions, vanadium is mostly in the +3 oxidation state.36 Interestingly, vanadium reduction of supported VOx species by alcohol (methanol, ethanol) at room temperature was previously reported for VOx/TiO2 catalysts using in situ ambient pressure XPS34,35 and for VOx/SiO2 using high-resolution V K-edge XANES (using the Kβ5,2 emission line).70 In these studies, the authors assumed that vanadium reduction must be related to ethanol desorption, which, however, disturbs the electron balance.
 | ||
| Fig. 3 (a) V K-edge XANES spectra of the 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst and (b) time profiles of the pre-edge intensity and (c) the edge position measured during the ethanol-feeding experiment at 30 °C (1.6 vol% EtOH in He). With dark red color, we show (a) the V K-edge spectrum of the catalyst reduced at 400 °C in ethanol (1.6 vol% EtOH in He) as well as (b) its pre-edge height and (c) half-edge step position. | ||
To verify whether the reduction of vanadium in our catalyst by ethanol occurs at low temperatures, we used in situ diffuse reflectance (DR) UV-Vis spectroscopy, which is a complementary method sensitive to vanadium oxidation state changes in supported VOx catalysts.44 The experimental conditions were very similar to those of the XAS (see the ESI,† for figures and additional discussion, Section 3.2). DR UV-Vis spectroscopy detected no significant vanadium reduction in the ethanol-feeding experiment at 50 °C, which suggests that vanadium reduction detected by XAS is X-ray beam-induced.
We also confirmed that the observed vanadium reduction is beam-induced by an X-ray beam switch-off experiment, which can be applied directly during a XAS beamtime at a synchrotron and does not require additional spectroscopies. Before the experiment, the catalyst was kept in the ethanol–oxygen mixture at 160 °C (in the absence of a beam) to reach steady-state working conditions; at the end, the temperature was adjusted to 50 or kept at 160 °C. Subsequently, we started to record the time-resolved V K-edge XAS spectra under the same reaction conditions, while periodically switching on and off the X-ray beam. The values of the V pre-edge height from such experiments at 50 and 160 °C are shown in Fig. 4; the corresponding changes in the V K-edge position are reported in Fig. S7 (ESI†). The obtained trends indicate vanadium reduction in the presence of the X-ray beam at 50 °C (Fig. 4a), without appreciable catalytic activity. In the absence of the X-ray beam, vanadium is re-oxidized, presumably by oxygen present in the feed. The experiment performed at 160 °C (temperature, at which the catalyst is active in ethanol ODH, Fig. 4b) did not reveal any redox transformations upon exposure to the X-ray beam; the speciation of vanadium before and after X-ray beam switching experiments was identical. Possible beam damage at 160 °C, where the thermocatalytic process of ethanol ODH takes place, was ruled out in our previous work,36 where we performed oxygen cut-off experiments (switching between EtOH + O2 and EtOH feeds) on the same catalyst with different beam sizes (ranging from 150 × 150 μm2 to 500 × 400 μm2). The reversible changes in the pre-edge intensity profiles, related to reversible oxidation and reduction of VOx species, were independent of the X-ray brilliance (Fig. S8, ESI†). At the same time, the redox activity of VOx species was accelerated by temperature,36 which is typical for thermocatalytic processes. This confirms that the X-ray beam used in our experiments affects the vanadium oxidation state only at low temperatures, in the absence of the thermocatalytic process. At higher temperatures, X-ray-induced reduction of vanadium was not detected, which must be associated with much faster chemical processes, taking place under these conditions.
 | ||
| Fig. 4 The pre-edge height changes during the beam switching experiment performed at (a) 50 and (b) 160 °C. Grey zones illustrate the periods when the X-ray beam was switched off. The feed composition was 1.6 vol% EtOH and 6.4 vol% O2 in He; the beam spot size was 500 × 400 μm2. | ||
Influence of the feed composition
To understand the nature and conditions of X-ray-induced vanadium reduction, we followed the vanadium state in the 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst by V K-edge XAS during TPE experiments in different feed compositions. To identify whether the X-ray beam alone can induce changes in the state of vanadium, we heated the fully oxidized catalyst in a flow of inert helium. In another XAS study, it was reported that copper in Cu-containing zeolites can undergo photoreduction in helium feed during XAS measurements.27 An XPS investigation previously showed that supported V2O5 can undergo reduction to V2O4 and V2O3 under X-ray irradiation due to the loss of oxygen.41–43 The intensity of the pre-edge and the edge position (Fig. 5 and Fig. S9, ESI,† green curves) of the V K-edge XAS spectra measured during TPE in helium did not change significantly over the whole temperature interval (30–400 °C). This suggests that the X-ray beam alone does not cause vanadium reduction but needs a feed that could act as an electron donor. | ||
| Fig. 5 The pre-edge height temperature profiles of the 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst during TPE-XAS experiments in ethanol–oxygen (1.6 vol% EtOH, 6.4 vol% O2 in He, heating rate 5 °C min−1, light-green curve), helium (purity 99.997%, heating rate 5 °C min−1, green curve), water (0.12 vol% in He, red curve), hydrogen (55 vol%, heating rate 10 °C min−1, purple curve) and ethanol (1.6 vol% EtOH in He, heating rate 5 °C min−1, black curve). The beam size is 200 × 400 μm2. | ||
There are several examples in the literature where water as a solvent was identified as a source of electrons for metal reduction during XAS investigations.21–23 In this case, the electrons can be generated in the water radiolysis process:

The thus formed electrons may reduce metal species, whereas radicals and ions may recombine to produce H2O2 and H2.21,71 Even though our feed does not contain a large excess of water during in situ/operando XAS experiments, traces of water can still be present in the feed (from the gas bottle and as a product of catalytic ethanol oxidation). To verify the effect of water, we exposed the catalyst to a water-containing feed (0.12 vol% H2O in He, which corresponds to a water concentration produced over the catalyst at ca. 10% ethanol conversion) during an XAS-TPE experiment. The pre-edge intensity and the half-height edge position profiles are shown in Fig. 5 and Fig. S9, ESI† (red curves). No considerable changes in the pre-edge intensity or the edge position were observed, suggesting that in the presence of water, no X-ray-induced beam reduction takes place.
Another possible source of electrons in the feed, required for vanadium reduction, is ethanol. To see the effect of the X-ray beam in the whole temperature interval, we performed ethanol XAS-TPE. As soon as ethanol reaches the catalyst, vanadium undergoes reduction, which is reflected by a decrease of the pre-edge height and a shift in the edge position (Fig. 5 and Fig. S9, ESI,† respectively, black curve). Upon heating, vanadium continues to reduce, reaching its maximum reduction at ca. 75 °C. A further increase in temperature leads to partial re-oxidation of vanadium, which we associate with oxygen traces present in the feed. We estimated the concentration of oxygen traces in the operando cell of ca. 0.02 vol% under our working conditions based on the residual (background) transformation of ethanol into acetaldehyde over a 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst of 1 × 10−6 molAcH min−1 measured in an ethanol–helium flow of 50 mL min−1 at 160–210 °C using 15–20 mg of the catalyst and considering a complete transformation of oxygen traces into acetaldehyde36 At 120 °C, catalytic ethanol oxidation starts (Fig. 1b), which competes with re-oxidation, and starting from 190 °C, chemical vanadium reduction by ethanol prevails over all other processes. Full vanadium reduction is reached at 350 °C. Thus, by monitoring the pre-edge height measured during ethanol TPE, we can observe both reduction processes: X-ray beam-induced reduction (30–100 °C) and chemical reduction (150–350 °C).
Next, we tested whether in the presence of another reducing agent, i.e. hydrogen, the catalyst may undergo X-ray-induced reduction. The pre-edge intensity and the edge position profiles during hydrogen TPE XAS are shown in Fig. 5 and Fig. S9, ESI† (purple curves). Partial vanadium reduction was observed in the 50–180 °C temperature interval. Also in this case, the traces of oxygen in the feed must have re-oxidized the vanadium species at higher temperatures; starting from 200 °C the oxidation state of vanadium species returned to the initial fully oxidized one. At 400 °C hydrogen was not able to reduce vanadium, in agreement with the observation that reduction takes place at ca. 530 °C.44 Furthermore, the extent of vanadium reduction in the hydrogen TPE was significantly lower than that in ethanol TPE. This correlates to the generally lower ability of hydrogen to reduce this catalyst. To summarize, X-ray beam-induced vanadium reduction occurs only in the presence of reducing agents, such as ethanol or hydrogen.
We additionally tested whether titania (being photoactive72) induces the observed photochemical reduction of vanadium. For this, we performed ethanol TPE-XAS on a titania-free 8 wt% V2O5/SiO2 sample. The V K-edge XAS also revealed a very similar vanadium reduction for the titania-free catalyst in the low-temperature interval (see ESI,† for additional figures and discussion, Section 3.3). This showed that photochemical reduction of vanadium is not related to or promoted by the photoactive titania support.
At low temperatures, no products of ethanol oxidation were detected in the outlet, which may be related to the small amount of the catalyst exposed to the X-rays or to the low sensitivity of the IR-spectrometer used for product analysis. Furthermore, the products could be strongly adsorbed on the catalyst surface.
The effect of beam brilliance and intensity
The next goal of our investigation was to identify whether X-ray-induced vanadium reduction can be avoided or diminished by defocusing the beam and/or by applying filters. This strategy was successfully used to avoid copper photoreduction observed during catalytic methane oxidation over Cu-containing zeolites.27 The majority of experiments discussed so far (unless stated otherwise) were performed using an X-ray beam with a spot size at the sample position equal to 200 × 400 μm2 (this corresponds to a brilliance of ca. 1.5 × 1012 ph mm−2 s−1). To diminish the rate of the X-ray-induced vanadium reduction, we defocused the incident X-ray beam to a size of 500 × 400 μm2. This reduced the brilliance by a factor of 2.5 while preserving the total flux, and consequently, the quality of the XAS spectra. In Fig. S10 (ESI†), we plotted the pre-edge height profiles obtained during the ethanol TPE experiment with the use of focused and defocused beams. One can see that the extent of vanadium reduction at a low temperature is significantly decreased; however, it is still present. The X-ray beam switching experiments, shown in Fig. 5, were also performed with the use of the defocused beam (500 × 400 μm2), and as we discussed before, vanadium reduction was observed upon X-ray irradiation at 50 °C.We attempted to further decrease the effect of the X-ray beam and find conditions that would allow the investigation of the supported VOx catalysts at low temperature in the presence of a reducing feed. For this, the intensity of the defocused incident beam (500 × 400 μm2) was additionally decreased by a factor of 2–20 by applying aluminum filters of different thicknesses. Before the experiment, the 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst was exposed to the ethanol–oxygen feed in the absence of the X-ray beam for 20 min at 50 °C. This allowed to reach equilibrium. Afterward, the thickest Al-filter (which absorbs 95% of the incident beam) was applied and V K-edge XANES was recorded for 10 min (1200 scans). The same procedure was followed by filters with smaller thicknesses. In Fig. 6, we plotted the pre-edge height values with the use of different filter thicknesses (the V K edge XAS spectra are in Fig. S11, ESI†). The evolution of the pre-edge height in Fig. 6a suggests that vanadium reduction in the low-temperature regime occurred even with a reduced beam intensity. Only the measurement performed with the use of 5% of the full beam intensity (corresponding to a beam brilliance of 3 × 1010 ph mm−2 s−1) did not reveal vanadium reduction. However, by applying such filters, we decrease the total flux (Table S1, ESI†), which leads to a significant decrease in the spectral quality. This can be partially compensated by a longer acquisition time (Fig. 6b). However, despite the 20 times higher number of averaged scans, the resulting spectrum acquired at a lower X-ray intensity loses quality, evidenced by the monochromator glitches appearing in the interval of 5530–5550 eV, which is caused by the different nonlinear responses of the detectors used (i.e. the ionization chamber vs. the PIPS detector).
 | ||
| Fig. 6 (a) Pre-edge height of the V K-edge spectra of the 5 wt% V2O5/15 wt% TiO2/SiO2 catalyst measured in ethanol–oxygen feed (1.6 vol% EtOH, 6.4 vol% O2 in He) at 50 °C with the use of different Al-filters. The percentage numbers indicate the intensity of the beam relative to the beam without filter (intensity of the transmitted beam). (b) Comparison of the quality of a V K-edge XANES spectrum measured with the use of a 5%-transmission filter (averaged within 600 s) and without filter (averaged within 30 s); the beam spot size was 500 × 400 μm2. | ||
Thus, X-ray-induced vanadium photochemical reduction at low temperatures can be significantly diminished by using a defocused X-ray beam, which does not affect the quality of the spectra. The use of filters may further reduce the effect of X-ray irradiation which is necessary when studying rates of reduction and oxidation; however, poor spectral quality may become a limiting factor in efficient time-resolved V K-edge XAS acquisition.
Conclusions
During V K-edge XAS studies at a third-generation synchrotron facility, we detected X-ray-induced vanadium photoreduction, which strongly affected the chemical speciation of the supported VOx species below the temperature of thermocatalytic ethanol oxidation. The variation of the gas feed composition revealed that the photoreduction occurs only in the presence of a reducing agent (ethanol, hydrogen, or ethanol–oxygen mixture) in the gas feed and is particularly evident below 100 °C. The X-ray-induced vanadium photoreduction was also observed on a titania-free 8 wt% V2O5/SiO2 catalyst, showing that photoreduction of vanadium cannot be explained by the photoactive titania. Defocusing the incident beam significantly reduces the photoreduction effect without losing the spectral quality of the XAS data. The use of X-ray filters can further reduce photoreduction; however, it deteriorates the spectral quality, which ultimately can become a limiting factor for probing the vanadium speciation by time-resolved XAS. To exclude beam damage under specific in situ/operando conditions, we suggest performing X-ray beam switching (on and off) tests at different beam intensities.Author contributions
A. Z. and O. S. planned the research. A. Z., A. H. C., and O. V. S performed XAS experiments and contributed to the data analysis. A. Z. under the guidance of D. Ferri performed the UV-Vis experiments and analyzed the results. A. Z. drafted the manuscript. All authors contributed to the scientific discussion and provided corrections to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Swiss National Science Foundation (SNSF) under project 200021_179132. We acknowledge Prof. I. E. Wachs (Lehigh University) for providing the VOx catalysts and Dr V. Sushkevich (PSI), Dr A. Fedorov (ETH Zurich) and Dr G. Smolentsev (PSI) for fruitful discussions. We thank R. J. G. Nuguid, I. Sadykov, and Dr M. Agote-Aran (PSI) for assistance during beamtimes. The Swiss Light Source is acknowledged for granting beamtime at the SuperXAS beamline. A. Z. thanks S. Hitz (PSI) for the technical assistance throughout the study.Notes and references
- A. Chakrabarti, M. E. Ford, D. Gregory, R. Hu, C. J. Keturakis, S. Lwin, Y. Tang, Z. Yang, M. Zhu, M. A. Bañares and I. E. Wachs, A decade+ of operando spectroscopy studies, Catal. Today, 2017, 283, 27–53 CrossRef CAS.
- R. Portela, S. Perez-Ferreras, A. Serrano-Lotina and M. A. Bañares, Engineering operando methodology: Understanding catalysis in time and space, Front. Chem. Sci. Eng., 2018, 12, 509–536 CrossRef.
- J. Sa, in High-Resolution XAS/XES, ed. J. Sa, CRC Press, 2014, pp. 169–194 Search PubMed.
- G. Meitzner, in In-Situ Spectroscopy in Heterogeneous Catalysis, ed. J. F. Haw, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2002, pp. 179–194 Search PubMed.
- O. Müller, M. Nachtegaal, J. Just, D. Lützenkirchen-Hecht and R. Frahm, Quick-EXAFS setup at the SuperXAS beamline for in situ X-ray absorption spectroscopy with 10 ms time resolution, J. Synchrotron Radiat., 2016, 23, 260–266 CrossRef.
- E. Fonda, A. Rochet, M. Ribbens, L. Barthe, S. Belin and V. Briois, The SAMBA quick-EXAFS monochromator: XAS with edge jumping, J. Synchrotron Radiat., 2012, 19, 417–424 CrossRef CAS.
- V. Briois, C. La Fontaine, S. Belin, L. Barthe, T. Moreno, V. Pinty, A. Carcy, R. Girardot and E. Fonda, ROCK: The new Quick-EXAFS beamline at SOLEIL, J. Phys.: Conf. Ser., 2016, 712, 012149 CrossRef.
- B. Bornmann, J. Kläs, O. Müller, D. Lützenkirchen-Hecht and R. Frahm, The quick EXAFS setup at beamline P64 at PETRA III for up to 200 spectra per second, AIP Conf. Proc., 2019, 2054, 040008 CrossRef.
- C. W. Pao, J. L. Chen, J. F. Lee, M. C. Tsai, C. Y. Huang, C. C. Chiu, C. Y. Chang, L. C. Chiang and Y. S. Huang, The new X-ray absorption fine-structure beamline with sub-second time resolution at the Taiwan Photon Source Pao Chih-Wen, J. Synchrotron Radiat., 2021, 28, 930–938 CrossRef CAS.
- S. Pascarelli, O. Mathon, T. Mairs, I. Kantor, G. Agostini, C. Strohm, S. Pasternak, F. Perrin, G. Berruyer, P. Chappelet, C. Clavel and M. C. Dominguez, The Time-resolved and Extreme-conditions XAS (Texas) facility at the European Synchrotron Radiation Facility: The energy-dispersive X-ray absorption spectroscopy beamline ID24, J. Synchrotron Radiat., 2016, 23, 353–368 CrossRef CAS PubMed.
- M. Zabilskiy, V. L. Sushkevich, D. Palagin, M. A. Newton, F. Krumeich and J. A. van Bokhoven, The unique interplay between copper and zinc during catalytic carbon dioxide hydrogenation to methanol, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- J. Imbao, J. A. van Bokhoven, A. Clark and M. Nachtegaal, Elucidating the mechanism of heterogeneous Wacker oxidation over Pd-Cu/zeolite Y by transient XAS, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- A. Marberger, A. W. Petrov, P. Steiger, M. Elsener, O. Kröcher, M. Nachtegaal and D. Ferri, Time-resolved copper speciation during selective catalytic reduction of NO on Cu-SSZ-13, Nat. Catal., 2018, 1, 221–227 CrossRef CAS.
- R. Kopelent, J. A. Van Bokhoven, J. Szlachetko, J. Edebeli, C. Paun, M. Nachtegaal and O. V. Safonova, Catalytically Active and Spectator Ce3+ in Ceria-Supported Metal Catalysts, Angew. Chem., Int. Ed., 2015, 54, 8728–8731 CrossRef CAS PubMed.
- O. V. Safonova, A. Guda, Y. Rusalev, R. Kopelent, G. Smolentsev, W. Y. Teoh, J. A. van Bokhoven and M. Nachtegaal, Elucidating the Oxygen Activation Mechanism on Ceria-Supported Copper-Oxo Species Using Time-Resolved X-ray Absorption Spectroscopy, ACS Catal., 2020, 10, 4692–4701 CrossRef CAS.
- J. M. Holton, A beginner's guide to radiation damage, J. Synchrotron Radiat., 2009, 16, 133–142 CrossRef CAS PubMed.
- E. F. Garman, Radiation damage in macromolecular crystallography: What is it and why should we care?, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 339–351 CrossRef CAS PubMed.
- R. B. G. Ravelli and S. M. McSweeney, The ‘fingerprint’ that X-rays can leave on structures, Structure, 2000, 8, 315–328 CrossRef CAS.
- C. Nave, Radiation damage in protein crystallography, Radiat. Phys. Chem., 1995, 45, 483–490 CrossRef CAS.
- G. N. George, I. J. Pickering, M. Jake Pushie, K. Nienaber, M. J. Hackett, I. Ascone, B. Hedman, K. O. Hodgson, J. B. Aitken, A. Levina, C. Glover and P. A. Lay, X-ray-induced photo-chemistry and X-ray absorption spectroscopy of biological samples, J. Synchrotron Radiat., 2012, 19, 875–886 CrossRef CAS PubMed.
- J. G. Mesu, A. M. Beale, F. M. F. De Groot and B. M. Weckhuysen, Probing the influence of X-rays on aqueous copper solutions using time-resolved in situ combined video/X-ray absorption near-edge/ultraviolet-visible spectroscopy, J. Phys. Chem. B, 2006, 110, 17671–17677 CrossRef CAS PubMed.
- J. G. Mesu, A. M. J. Van Der Eerden, F. M. F. De Groot and B. M. Weckhuysen, Synchrotron radiation effects on catalytic systems as probed with a combined in-situ UV-Vis/XAFS spectroscopic setup, J. Phys. Chem. B, 2005, 109, 4042–4047 CrossRef CAS PubMed.
- M. Kubin, J. Kern, M. Guo, E. Källman, R. Mitzner, V. K. Yachandra, M. Lundberg, J. Yano and P. Wernet, X-ray-induced sample damage at the Mn L-edge: A case study for soft X-ray spectroscopy of transition metal complexes in solution, Phys. Chem. Chem. Phys., 2018, 20, 16817–16827 RSC.
- S. D. Maleknia, M. Brenowitz and M. R. Chance, Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry, Anal. Chem., 1999, 71, 3965–3973 CrossRef CAS PubMed.
- P. Froment, M. J. Genet and M. Devillers, Surface reduction of ruthenium compounds with long exposure to an X-ray beam in photoelectron spectroscopy, J. Electron Spectrosc. Relat. Phenom., 1999, 104, 119–126 CrossRef CAS.
- S. Süzer, XPS Investigation of X-Ray-Induced Reduction of Metal Ions, Appl. Spectrosc., 2000, 54, 1716–1718 CrossRef.
- M. A. Newton, A. J. Knorpp, J. Meyet, D. Stoian, M. Nachtegaal, A. H. Clark, O. V. Safonova, H. Emerich, W. van Beek, V. L. Sushkevich and J. A. van Bokhoven, Unwanted effects of X-rays in surface grafted copper(II) organometallics and copper exchanged zeolites, how they manifest, and what can be done about them, Phys. Chem. Chem. Phys., 2020, 22, 6826–6837 RSC.
- M. Albrahim, C. Thompson, D. Leshchev, A. Shrotri, R. R. Unocic, J. Hong, A. S. Hoffman, M. J. Meloni, R. C. Runnebaum, S. R. Bare, E. Stavitski and A. M. Karim, Reduction and Agglomeration of Supported Metal Clusters Induced by High-Flux X-ray Absorption Spectroscopy Measurements, J. Phys. Chem. C, 2021, 125, 11048–11057 CrossRef CAS.
- M. M. van Schooneveld and S. DeBeer, A close look at dose: Toward L-edge XAS spectral uniformity, dose quantification and prediction of metal ion photoreduction, J. Electron Spectrosc. Relat. Phenom., 2015, 198, 31–56 CrossRef CAS.
- I. E. Wachs, Catalysis science of supported vanadium oxide catalysts, Dalt. Trans., 2013, 42, 11762–11769 RSC.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Catalytic Applications of Vanadium: A Mechanistic Perspective, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS PubMed.
- V. V. Kaichev, Y. A. Chesalov, A. A. Saraev and A. M. Tsapina, A Mechanistic Study of Dehydrogenation of Propane over Vanadia-Titania Catalysts, J. Phys. Chem. C, 2019, 123, 19668–19680 CrossRef CAS.
- T. V. Andrushkevich, V. V. Kaichev, Y. A. Chesalov, A. A. Saraev and V. I. Buktiyarov, Selective oxidation of ethanol over vanadia-based catalysts: The influence of support material and reaction mechanism, Catal. Today, 2017, 279, 95–106 CrossRef CAS.
- V. V. Kaichev, G. Y. Popova, Y. A. Chesalov, A. A. Saraev, D. Y. Zemlyanov, S. A. Beloshapkin, A. Knop-Gericke, R. Schlögl, T. V. Andrushkevich and V. I. Bukhtiyarov, Selective oxidation of methanol to form dimethoxymethane and methyl formate over a monolayer V2O5/TiO2 catalyst, J. Catal., 2014, 311, 59–70 CrossRef CAS.
- V. V. Kaichev, Y. A. Chesalov, A. A. Saraev, A. Y. Klyushin, A. Knop-Gericke, T. V. Andrushkevich and V. I. Bukhtiyarov, Redox mechanism for selective oxidation of ethanol over monolayer V2O5/TiO2 catalysts, J. Catal., 2016, 338, 82–93 CrossRef CAS.
- A. Zabilska, A. H. Clark, B. M. Moskowitz, I. E. Wachs, Y. Kakiuchi, C. Copéret, M. Nachtegaal, O. Kröcher and O. V. Safonova, Redox Dynamics of Active VOx Sites Promoted by TiOx during Oxidative Dehydrogenation of Ethanol Detected by Operando Quick XAS, JACS Au, 2022, 2, 762–776 CrossRef CAS PubMed.
- L. H. Vieira, L. G. Possato, T. F. Chaves, J. J. Lee, T. P. Sulmonetti, C. W. Jones and L. Martins, Insights into Redox Dynamics of Vanadium Species Impregnated in Layered Siliceous Zeolitic Structures during Methanol Oxidation Reactions, ChemCatChem, 2020, 12, 141–151 CrossRef CAS.
- W. C. Vining, J. Strunk and A. T. Bell, Investigation of the structure and activity of VO x/CeO 2/SiO 2 catalysts for methanol oxidation to formaldehyde, J. Catal., 2012, 285, 160–167 CrossRef CAS.
- W. Wu, K. Ding, J. Liu, T. Drake, P. Stair and E. Weitz, Methanol Oxidation to Formate on ALD-Prepared VOx/θ -Al 2 O 3 Catalysts: A Mechanistic Study, J. Phys. Chem. C, 2017, 121, 26794–26805 CrossRef CAS.
- A. Iglesias-Juez, M. V. Martínez-Huerta, E. Rojas-García, J. M. Jehng and M. A. Banares, On the Nature of the Unusual Redox Cycle at the Vanadia Ceria Interface, J. Phys. Chem. C, 2018, 122, 1197–1205 CrossRef CAS.
- S. P. Chenakin, R. P. Silvy and N. Kruse, Effect of X-rays on the surface chemical state of Al2O 3, v2O5, and aluminovanadate oxide, J. Phys. Chem. B, 2005, 109, 14611–14618 CrossRef CAS PubMed.
- S. Koust, B. N. Reinecke, K. C. Adamsen, I. Beinik, K. Handrup, Z. Li, P. G. Moses, J. Schnadt, J. V. Lauritsen and S. Wendt, Coverage-dependent oxidation and reduction of vanadium supported on anatase TiO2(101), J. Catal., 2018, 360, 118–126 CrossRef CAS.
- J. Mendialdua, R. Casanova and Y. Barbaux, XPS studies of V2O5, V6O13, VO2 and V2O3, J. Electron Spectrosc. Relat. Phenom., 1995, 71, 249–261 CrossRef CAS.
- X. Gao, S. R. Bare, J. L. G. Fierro and I. E. Wachs, Structural Characteristics and Reactivity/Reducibility Properties of Dispersed and Bilayered V2O5/TiO 2/SiO 2 Catalysts, J. Phys. Chem. B, 1999, 103, 618–629 CrossRef CAS.
- B. Beck, M. Harth, N. G. Hamilton, C. Carrero, J. J. Uhlrich, A. Trunschke, S. Shaikhutdinov, H. Schubert, H. J. Freund, R. Schlögl, J. Sauer and R. Schomäcker, Partial oxidation of ethanol on vanadia catalysts on supporting oxides with different redox properties compared to propane, J. Catal., 2012, 296, 120–131 CrossRef CAS.
- M. A. Bañares, M. V. Martínez-Huerta, X. Gao, J. L. G. Fierro and I. E. Wachs, Dynamic behavior of supported vanadia catalysts in the selective oxidation of ethane. In situ Raman, UV-Vis DRS and reactivity studies, Catal. Today, 2000, 61, 295–301 CrossRef.
- G. Deo and I. E. Wachs, J. Catal., 1994, 146, 323–334 CrossRef CAS.
- M. D. Amiridis, I. E. Wachs, G. Deo, J. M. Jehng and D. S. Kim, Reactivity of V2O5 catalysts for the selective catalytic reduction of NO by NH3: Influence of vanadia loading, H2O, and SO2, J. Catal., 1996, 161, 247–253 CrossRef CAS.
- X. Gao, S. R. Bare, J. L. G. Fierro, M. A. Banares and I. E. Wachs, Preparation and in-situ spectroscopic characterization of molecularly dispersed titanium oxide on silica, J. Phys. Chem. B, 1998, 102, 5653–5666 CrossRef CAS.
- I. E. Wachs, Raman and IR studies of surface metal oxide species on oxide supports: Supported metal oxide catalysts, Catal. Today, 1996, 27, 437–455 CrossRef CAS.
- X. Gao, S. R. Bare, B. M. Weckhuysen and I. E. Wachs, In Situ Spectroscopic Investigation of Molecular Structures of Highly Dispersed Vanadium Oxide on Silica under Various Conditions, J. Phys. Chem. B, 1998, 102, 10842–10852 CrossRef CAS.
- A. H. Clark, P. Steiger, B. Bornmann, S. Hitz, R. Frahm, D. Ferri and M. Nachtegaal, Fluorescence-detected quick-scanning X-ray absorption spectroscopy, J. Synchrotron Radiat., 2020, 27, 681–688 CrossRef CAS PubMed.
- A. H. Clark, J. Imbao, R. Frahm and M. Nachtegaal, ProQEXAFS: A highly optimized parallelized rapid processing software for QEXAFS data, J. Synchrotron Radiat., 2020, 27, 551–557 CrossRef PubMed.
- G. L. Chiarello, M. Nachtegaal, V. Marchionni, L. Quaroni and D. Ferri, Adding diffuse reflectance infrared Fourier transform spectroscopy capability to extended X-ray-absorption fine structure in a new cell to study solid catalysts in combination with a modulation approach, Rev. Sci. Instrum., 2014, 85, 074102 CrossRef PubMed.
- J. Jaumot, A. de Juan and R. Tauler, MCR-ALS GUI 2.0: New features and applications, Chemom. Intell. Lab. Syst., 2015, 140, 1–12 CrossRef CAS.
- B. Kilos, A. T. Bell and E. Iglesia, Mechanism and site requirements for ethanol oxidation on vanadium oxide domains, J. Phys. Chem. C, 2009, 113, 2830–2836 CrossRef CAS.
- H. Nair, J. E. Gatt, J. T. Miller and C. D. Baertsch, Mechanistic insights into the formation of acetaldehyde and diethyl ether from ethanol over supported VOx, MoOx, and WOx catalysts, J. Catal., 2011, 279, 144–154 CrossRef CAS.
- J. L. Bronkema, D. C. Leo and A. T. Bell, Mechanistic Studies of Methanol Oxidation to Formaldehyde on Isolated Vanadate Sites Supported on High Surface Area Anatase, J. Phys. Chem. C, 2007, 111, 14530–14540 CrossRef CAS.
- L. J. Burcham, X. Gao, I. E. Wachs and G. Deo, In situ IR, Raman, and UV-Vis DRS spectroscopy of supported vanadium oxide catalysts during methanol oxidation, Top. Catal., 2000, 11–12, 85–100 CrossRef.
- T. Yamamoto, Assignment of pre-edge peaks in K-edge x-ray absorption spectra of 3d transition metal compounds: electric dipole or quadrupole?, X-Ray Spectrom., 2008, 37, 572–584 CrossRef CAS.
- J. Wong, F. W. Lytle, R. P. Messmer and D. H. Maylotte, K-edge absorption spectra of selected vanadium compounds, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 5596–5610 CrossRef CAS.
- J. A. Rees, A. Wandzilak, D. Maganas, N. I. C. Wurster, S. Hugenbruch, J. K. Kowalska, C. J. Pollock, F. A. Lima, K. D. Finkelstein and S. DeBeer, Experimental and theoretical correlations between vanadium K-edge X-ray absorption and K β emission spectra, J. Biol. Inorg. Chem., 2016, 21, 793–805 CrossRef CAS PubMed.
- G. Giuli, E. Paris, J. Mungall, C. Romano and D. Dingwell, V oxidation state and coordination number in silicate glasses by XAS, Am. Mineral., 2004, 89, 1640–1646 CrossRef CAS.
- S. R. Sutton, J. Karner, J. Papike, J. S. Delaney, C. Shearer, M. Newville, P. Eng, M. Rivers and M. D. Dyar, Vanadium K edge XANES of synthetic and natural basaltic glasses and application to microscale oxygen barometry, Geochim. Cosmochim. Acta, 2005, 69, 2333–2348 CrossRef CAS.
- P. Chaurand, J. Rose, V. Briois, M. Salome, O. Proux, V. Nassif, L. Olivi, J. Susini, J. L. Hazemann and J. Y. Bottero, New methodological approach for the vanadium K-edge X-ray absorption near-edge structure interpretation: Application to the speciation of vanadium in oxide phases from steel slag, J. Phys. Chem. B, 2007, 111, 5101–5110 CrossRef CAS PubMed.
- G. Silversmit, J. A. van Bokhoven, H. Poelman, A. M. J. van Der Eerden, G. B. Marin, M. F. Reyniers and R. De Gryse, The structure of supported and unsupported vanadium oxide under calcination, reduction and oxidation determined with XAS, Appl. Catal. A Gen., 2005, 285, 151–162 CrossRef CAS.
- A. N. Mansour, P. H. Smith, W. M. Baker, M. Balasubramanian and J. McBreen, In situ XAS investigation of the oxidation state and local structure of vanadium in discharged and charged V2O5 aerogel cathodes, Electrochim. Acta, 2002, 47, 3151–3161 CrossRef CAS.
- C. Rossignol and G. Ouvrard, General behavior upon cycling of LiNiVO4 as battery electrode, J. Power Sources, 2001, 97–98, 491–493 CrossRef CAS.
- D. A. McKeown, I. S. Muller, K. S. Matlack and I. L. Pegg, X-ray absorption studies of vanadium valence and local environment in borosilicate waste glasses, Mater. Res. Soc. Symp. – Proc., 2002, 713, 547–554 CrossRef CAS.
- Y. Izumi, K. Konishi, T. Miyajima and H. Yoshitake, Photo-oxidation over mesoporous V-TiO2 catalyst under visible light monitored by vanadium Kβ5,2-selecting XANES spectroscopy, Mater. Lett., 2008, 62, 861–864 CrossRef CAS.
- J. A. LaVerne and S. M. Pimblott, New mechanism for H2 formation in water, J. Phys. Chem. A, 2000, 104, 9820–9822 CrossRef CAS.
- A. Fujishima, T. Rao and D. Tryk, Titanium dioxide photocatalysis Akira, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp02721f |
| This journal is © the Owner Societies 2022 |