
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A study of the interaction between TMAO and urea in water using NMR spectroscopy†
Mazin
Nasralla
 ,
Harrison
Laurent
,
Daniel L.
Baker
,
Michael E.
Ries
and
Lorna
Dougan
*
,
Harrison
Laurent
,
Daniel L.
Baker
,
Michael E.
Ries
and
Lorna
Dougan
*
School of Physics and Astronomy, University of Leeds, Leeds LS2 9JT, UK. E-mail: l.dougan@leeds.ac.uk
First published on 16th August 2022
Abstract
Trimethylamine N-oxide (TMAO) and urea are small organic biological molecules. While TMAO is known as a protective osmolyte that promotes the native form of biomolecules, urea is a denaturant. An understanding of the impact of TMAO and urea on water structure may aid in uncovering the molecular mechanisms that underlie this activity. Here we investigate binary solutions of TMAO–water, urea–water and ternary solutions of TMAO–urea–water using NMR spectroscopy at 300 K. An enhancement of the total hydrogen bonding in water was found upon the addition of TMAO and this effect was neutralised by a mole ratio of 1-part TMAO to 4-parts urea. Urea was found to have little effect on the strength of water's hydrogen bonding network and the dynamics of water molecules. Evidence was found for a weak interaction between TMAO and urea. Taken together, these results suggest that TMAO's function as a protective osmolyte, and its counteraction of urea, may be driven by the strength of its hydrogen bond interactions with water, and by a secondary reinforcement of water's own hydrogen bond network. They also suggest that the TMAO–urea complex forms through the donation of a hydrogen bond by urea.
1 Introduction
Liquid water is fundamental to biological processes playing an active role in the stability, structure, dynamics and function of biomolecules. Its physical and chemical properties make it an excellent solvent for life.1–4 In the intimate environment of the cell many organisms, particularly those inhabiting denaturing stressful environments, incorporate small organic molecules (protective osmolytes) whose function has been shown to both stabilise the folded state of cellular proteins and RNA,5,6 and control the structure of lipid membranes.7 For example, in the muscle tissue of some fish, including the coelacanth, and elasmobranchii (sharks and rays), the concentration of the denaturant,8,9 urea, reaches ∼400 mM.10 To preserve the functional native form of their cellular proteins, these fish utilise trimethylamine N-oxide (TMAO) at ∼300 mM10,11 to counter urea's denaturing effect. The molecular structures of TMAO and urea are shown in Fig. 1. | ||
| Fig. 1 Trimethylamine N-oxide (TMAO, left), urea (right). Chemical formulae: TMAO–C3H9NO, urea–CH4N2O. TMAO features a zwitterion (N+–O−) with a dipole moment of 4.55 D compared to 1.89 D for water.12 The atoms are coloured: carbon – grey, hydrogen – white, nitrogen – blue and oxygen – red. | ||
The mechanisms by which osmolytes control protein stability have been the subject of many studies, and have been categorised as either enthalpic or entropic in nature.13 Intuitively, research has focused on the protein's hydration layer where competing interactions between solvent, co-solute, and the peptide amide groups have been described6,14 whilst other studies identify the presence of co-solute exclusion zones around the peptide that may provide entropic mechanisms for protein folding.15,16
The TMAO–urea–water system receives particular attention due to its biological relevance, and because of the large differences observed in the free energy of folding between aqueous urea and protein, and aqueous TMAO and protein.17 The TMAO and urea system has been studied using a range of different computational approaches including Molecular Dynamics (MD) simulations and Density Functional Theory Molecular Dynamics (DFT-MD). Classical MD approximate the wave function of matter through parameterised force fields that describe the interaction between bonded and non-bonded particles, whilst DFT-MD use DFT to approximate solutions to the Schrodinger equation and can be computationally demanding. MD provide molecular level insight in respect of the TMAO–urea–water system but as Ganguly et al.12 remark in a comprehensive review, such results are sensitive to the force fields they employ. Using MD and the Kirkwood–Buff theory of solutions, the authors showed that a sensitive balance between the TMAO–water and the TMAO–urea interactions govern osmolyte-induced changes in hydrophobic association in mixed urea–TMAO solutions. The authors cautioned that this balance must be correctly incorporated in force field parameterisation because hydrophobic association can be either enhanced or prevented altogether by slightly increasing or decreasing the osmolyte–water affinity and osmolyte–osmolyte self-affinity of TMAO molecules.’
By combining DFT-MD, time-resolved infrared spectroscopy, and nuclear magnetic resonance spectroscopy, a previous study suggested that the interaction between TMAO and urea is favoured by hydrophobic association.18
The structure of aqueous urea and the TMAO–urea–water system has previously been studied through neutron diffraction experiments.19–21 When coupled with structural refinement modelling22 these studies produce partial pair distribution functions that reveal molecular structure. They show that urea and water molecules readily substitute themselves into hydrogen bond networks, without changing the extent of these networks but that urea places significant strain on the short range tetrahedral architecture of water.19 They also show that TMAO promotes hydrogen bonding between water molecules, and that TMAO accepts hydrogen bonds from water and urea20,21,23 although the nature and extent of the interaction between TMAO and urea is debated. For example, Meersman et al.20 used neutron diffraction analysis to demonstrate a pair correlation between the oxygen atom of TMAO, and urea hydrogen atoms that featured a mean atomic separation of 1.83 Å that they interpreted to be a hydrogen bond, whilst Hunger et al.24 used dielectric spectroscopy to study the system, and found no evidence for a hydrogen bond.
NMR25 and FTIR spectroscopy18 with MD26 have been used to probe the dynamics of water, and the extent of hydrogen bonding. Sharp et al.27 used IR spectroscopy to measure the effect of TMAO and urea on O–H stretching, and H–O–H bending mode frequencies. They found urea had no impact on water structure, but TMAO red-shifted and blue-shifted the stretching and bending frequencies respectively, implying that TMAO formed strong hydrogen bonds with water, whilst urea substituted for water with little overall effect.
NMR is a powerful technique that probes the chemical environment, and dynamics of nuclei in solution. Palmer et al.25 analysed the structure and activity of a bovine ribonuclease in TMAO–urea–water (urea 0–2 M, TMAO 0–1 M) using 31P and 1H NMR and found that TMAO restored the structure and function of ribonuclease at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TMAO:urea. Furthermore, they concluded that TMAO's effect was mediated through the solvent rather than any direct interaction with the protein.
:1 TMAO:urea. Furthermore, they concluded that TMAO's effect was mediated through the solvent rather than any direct interaction with the protein.
Paul and Patey26 used MD simulations to study the TMAO–urea–water system (urea 7.4–8.6 M, TMAO 0.0–3.7 M) and found that the TMAO–water and TMAO–urea hydrogen bond energies were significant. They conclude that TMAO's counteraction of urea is due to water and urea's preference to solvate TMAO rather than the protein.
In this work we focus on TMAO–urea–water in the absence of a biomolecule, examining the interactions between each component and their impact on the structure and dynamics of water. We have used 1H NMR spectroscopy to explore the interactions between TMAO, urea and water in solutions containing up to 4 moles of TMAO per kg water and 8 moles of urea per kg water. These concentrations were selected as in vitro experimental studies and MD suggest urea denatures proteins progressively from concentrations of ∼1.5 moles of urea per kg water and that TMAO counteracts this denaturation at a urea:TMAO concentration ratio that is between 1:1 and 2:1.8,28
Our results complement previous NMR and diffraction studies as they probe the hydrogen bonding and solute association in the TMAO–urea–water system. Here, we show that urea readily substitutes into water's hydrogen bond network with little overall effect on water's translational and rotational dynamics, whilst TMAO sharply slows water's dynamics and promotes hydrogen bonding in, or with water. We find evidence of a hydrogen bond interaction between urea's hydrogen atoms and the oxygen atom of TMAO, and in investigating the effect of urea concentration on the chemical environment of TMAO's methyl groups we find no evidence of a hydrophobic interaction between TMAO and urea although we do not preclude it. We show that TMAO reinforces the hydrogen bond network of water and interpret this effect as contributing towards TMAO's counteraction of denaturation of biomolecules by urea. We also find that TMAO's interaction with urea is weak, and that this interaction plays a minor part in the counteraction. Our results are consistent with experiments that show that urea's propensity to denature biomolecules is facilitated, in part, by its intrinsic ability to cooperate with water in solvating moieties through a urea–water hydrogen bond network19 that enables access to biomolecules.
2 Method
Stock solutions were prepared by dissolving crystalline samples of TMAO dihydrate and urea (sourced from Sigma Aldrich and Thermo Fischer Scientific respectively) in ultrapure water. These solutions were then further combined with ultrapure water to create mixed solutions of urea, TMAO and water containing 0.2–4.0 moles TMAO per kg water and 0.2–8.0 moles urea per kg water.Each sample was transferred by pipette into a 5 mm NMR tube. Where a spectral measurement was taken, a sealed 1 mm X-ray capillary tube, containing dimethyl sulfoxide (DMSO, Sigma-Aldrich), was inserted into the NMR tube (DMSO generates a reference 1H NMR signal at 2.50 ppm29). Three spectral measurements were made with a Bruker Avance II 400 MHz NMR spectrometer at each concentration and the average result is reported. 1H NMR T1 data were captured with a Magritek Spinsolve 43 MHz NMR spectrometer without a DMSO standard. All measurements were made at 300 K.
The following measurements were taken for each solution mixture: (i) the spectral peak location (ppm) for 1H (water, urea, TMAO), (ii) proton NMR T1 relaxometry (water) and (iii) the diffusion co-efficient of water (D).
2.1 Peak location
In NMR spectroscopy, the spectral location measures the difference in the energy of the nuclide's spin states (ΔE) given by eqn (1), where γ is the magnetogyric ratio of the nuclide and B0 is the effective magnetic field experienced by the nuclide in the sample | (1) |
The spectrometer applies a magnetic field to the sample that is moderated by magnetic fields induced by local electron clouds sensitive to the atom's chemical environment. Hydrogen bond formation has the effect of deshielding the proton from the electron cloud of the bonding orbital, and hence the blue-shift in the spectral peak of the hydrogen proton is a useful indicator of hydrogen bond formation in solution.30–33
In a review of the chemical shift due to van der Waals forces, Diehl et al.34 comment that 'van der Waals effects are large and strongly variable with solvent, solute and even with nuclear site within the solute molecule'. A theoretical discussion of the origins of the chemical shift due to van der Waals forces is beyond the scope of this work and the interested reader is referred to a further review by Homer and Percival.35
2.2 Diffusion coefficient
The diffusion co-efficient is measured using a technique known as pulsed field gradient spin-echo.31–33 A magnetic field with gradient G (mT m−1) is applied parallel to B, which means that as the nuclide moves through the field its Larmor frequency will vary, enabling a measurement of the spatial position of the nuclide to be taken, hence D can be derived from the Stejskal–Tanner expression: | (2) |
2.3 T 1 relaxometry
T 1 relaxometry (also described as longitudinal relaxation) measures the time taken for the z-component of a nuclide's spin to reach (1–2/e) of its maximum net magnetisation, after the nuclide's spin is inverted by an electromagnetic pulse.31–33The period for the spins to return to equilibrium can be derived from eqn (3), where T1 is a time constant, Mz(t) is the nuclear spin magnetization at time t | (3) |
No attempt was made to distinguish the bulk-water correlation time from the hydration shell correlation time of water, so all relaxometry results represent a solution-average correlation time.
3 Results
Fig. 2 reveals differing behaviour in the spectral peak shifts of the hydrogen atoms of TMAO and urea in ternary solutions of TMAO and urea as their concentrations change. Three features are apparent in the behaviour of the 1H urea spectral peaks (blue-lines): (i) an increase in TMAO concentrations generates a blue-shift (shift to higher energy). (ii) At a given TMAO concentration, the blue-shift is greatest in the samples with a lower urea concentration. (iii) The slope between the peak shift and concentration is enhanced at the highest urea concentration.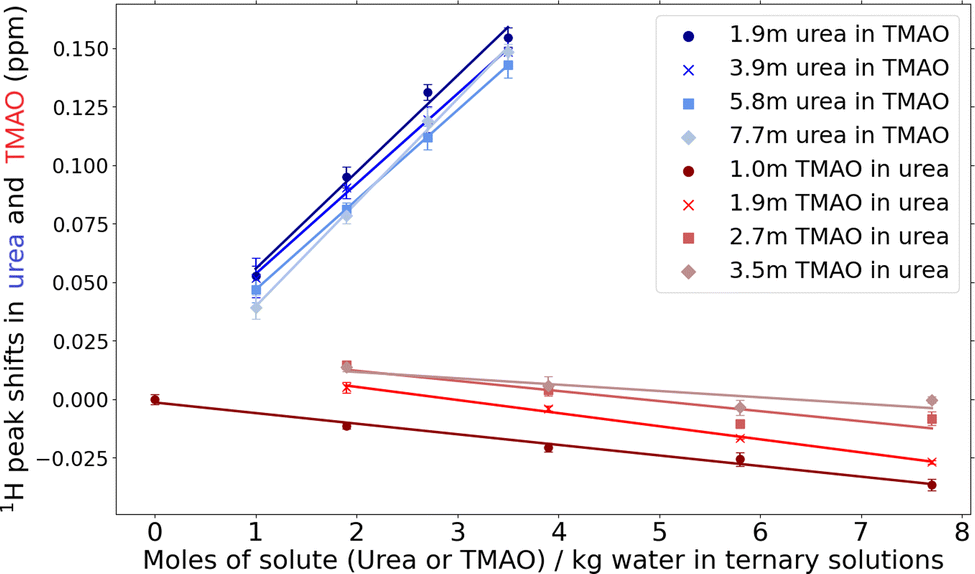 | ||
| Fig. 2 The NMR spectral peaks of 1H in urea and 1H in TMAO in 16 mixed samples of TMAO–urea–water. The samples consisted of 4 series of fixed urea concentration where the TMAO concentration varies (blue lines), and 4 series where the TMAO concentration is fixed and the urea concentration is varied (red lines) (Table S1, ESI†). The spectral peaks of hydrogen atoms in urea are blue-shifted as TMAO concentrations increase. The shift is relative to the spectral peak of urea hydrogen atoms in a solution containing 8 moles urea per kg water. The spectral peaks of hydrogen atoms in TMAO are red-shifted as urea concentrations are increased. The shift here is relative to the spectral peak of TMAO hydrogen atoms in a solution containing 1 mole TMAO per kg water. The lines are linear fits to the average of a triplicate of experimental repeats. The error bars represent the standard error. | ||
From these observations we infer that, (i) urea hydrogen atoms are complexing with TMAO either through TMAO's oxygen atom, or with the methyl groups of TMAO. An alternative explanation is that TMAO may significantly strengthen urea–water interactions by enhancing the hydrogen bond acceptance of water molecules.36 This effect though would need to be significant to mitigate the substitution of water by TMAO. (ii) The co-ordination of the TMAO–urea bond is low because at low urea concentrations, a higher proportion of hydrogen atoms in urea appear to be involved in an interaction with TMAO even at higher TMAO concentrations. (iii) The increase in the gradient at high urea concentration suggests that TMAO's affinity for urea is lower than its affinity for water, so that at high concentrations of solute, when water molecules are less common, increased amounts of TMAO are available to complex with urea.
If TMAO and urea are associating through TMAO's methyl groups, then changes in urea concentration should be associated with significant change in the 1H TMAO spectral peak yet Fig. 2 shows minor red shifts (shift to lower energy) that can be seen in the negative gradient of the red lines. This is consistent with substitution of TMAO hydrogen–water oxygen interactions by weaker TMAO hydrogen–urea oxygen interactions, or the interaction of hydration shells in an increasingly concentrated solution. The comparison in the gradients of the red and blue lines is remarkable and suggests that if TMAO and urea are forming a complex then it is through a weak hydrogen bond. This is in agreement with diffraction studies. Meersman et al.20 found a weak hydrogen bonding between TMAO oxygen and urea hydrogen atoms with 1.83 Å bond length (compared to the water–water hydrogen bond length ∼1.78 Å).
Fig. 3 and Fig. S1 (ESI†) present the peak-shift of water's hydrogen atoms as a function of TMAO and urea concentration allowing us to examine the impact of TMAO and urea on hydrogen bonding in water. It shows the spectral peaks of water's hydrogen atoms are aggressively blue-shifted by the addition of TMAO, and quiescently red-shifted by urea. This suggests that the TMAO oxygen atom acts as a hydrogen bond acceptor forming hydrogen bonds with water although we cannot distinguish the proportion of hydrogen bonding between water–water, and water–TMAO. Fig. 3 indicates that ∼4.2 moles of urea per kg water counteracts the observed peak-shift of the water hydrogen atom due to an increase in TMAO concentration of 1 mole TMAO per kg water. This ratio is consistent with diffraction studies20,21 that find that TMAO's oxygen atom accepts ∼3 medium–strong hydrogen bonds from water. These bonds are shorter than those in water (1.71 Å, 1.78 Å at stp), hence the peak shift of water in aqueous TMAO. A separate neutron diffraction study of aqueous urea19 also found
 | ||
| Fig. 3 Peak-shifts of the water hydrogen atom (relative to this spectral peak in pure water) in aqueous urea and TMAO. The lines are linear fits to the average of a triplicate of experimental repeats. The error bars represent the standard error. | ||
‘no marked preference for water or urea to bond either to themselves or to each other. In other words urea and water appear to readily substitute for each other in solution’.
Fig. 4 and 5 explore the results of changing TMAO and urea concentrations on the translational and rotational dynamics of water molecules. They show that increasing TMAO concentration substantially slows the dynamics of water molecules whilst increasing the urea concentration marginally retards water's dynamics.
 | ||
| Fig. 4 The diffusion coefficient of the water hydrogen atoms in different concentrations of aqueous TMAO and urea. The lines are linear fits to the data points. | ||
 | ||
| Fig. 5 The proton T1−1 relaxometry for the water hydrogen atoms in water in different concentrations of TMAO and urea. The lines are linear fits to the data points. | ||
These results are consistent with TMAO promoting strong hydrogen bond interactions with water, and urea substituting for water without any significant change in the overall hydrogen bond interaction energy, in agreement with previous studies.19,20 Fig. S2 (ESI†) shows a close correlation between T1 and the diffusion coefficient for water across all concentrations.
4 Discussion
4.1 The effect of TMAO and urea on hydrogen bonding
We have shown that the addition of urea to water has very little effect on the overall extent of hydrogen bonding in water even at 8 moles urea per kg water (Fig. 3 and Fig. S1, ESI†). This implies that urea effectively substitutes itself for water in the hydrogen bond network. This is consistent with an interpretation made by Soper et al.19 of results from their neutron diffraction study of aqueous urea.We found that the addition of TMAO to water induced a blue-shift in the spectral peak of hydrogen atoms in water that may be is indicative of enhanced hydrogen bonding involving water. We could not determine whether this results from strong TMAO–water, and/or stronger water–water interactions. Our results are consistent with neutron diffraction studies and MD that suggest that TMAO forms strong hydrogen bonds with water. These other studies suggest that the TMAO oxygen atoms accept between 2.5 and 3 hydrogen bonds from water. Ganguly et al.,12 Meersman et al.20,21 find that the TMAO oxygen–water oxygen bond length is 1.71 Å (in TMAO–water, 0.05 mole fraction TMAO) whereas the hydrogen bond is 1.78 Å in pure water, suggesting that the TMAO–water bond energy is higher than the water–water equivalent. Meersman et al.20 showed that in a 1:1 TMAO–urea solution there are 2 noticeable effects: the mean hydrogen bond length between water molecules moves from 1.78 Å (pure water) to ∼1.75 Å (TMAO–water), and the number of water–water hydrogen bonds increase. Laurent et al.23 have shown that TMAO counters the perturbation of water's hydrogen bond network by magnesium perchlorate.
IR spectroscopy of aqueous TMAO, utilising the stretching and bending modes of water, also finds TMAO to enhance hydrogen bonding in water.27,37 Most MD simulations find that TMAO strengthens hydrogen bonding in water,12 however Hu et al.38 found no effect.
4.2 The effect of TMAO and urea on water's dynamics
The structural changes that TMAO and urea solutes impose on water strongly influence its dynamics. Increasing solute concentration slows the dynamics of water molecules but the extent to which TMAO and urea perturb water molecules’ dynamics differs greatly (Fig. 4 and 5). We can understand this by considering microviscosity and the Stokes–Einstein relation (eqn (4)), where the diffusion coefficient (D) is a function of thermal energy and fluid viscosity where the radius of the probe particle is r, and kB, T, and η are the Boltzmann constant, temperature and bulk viscosity respectively.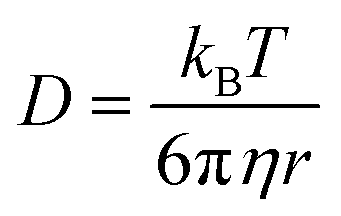 | (4) |
It should be noted that NMR experiments provide a time-averaged response, and cannot distinguish between the behaviour of molecules in the hydration shell and those in the bulk structure of water, however experiments with mid-IR pump probe spectroscopy can probe water in sub-picosecond timescales,40 fast enough to resolve the constant flux of hydrogen bond formation and breakage that occur on a femtosecond timescale.41 Fig. S4 (ESI†) shows a strong correlation between the diffusion rate of water molecules and their rotation rate suggesting that both properties are controlled by the same microviscosity. IR pump probe studies find two populations of water in urea solution, those in the bulk, scarcely slowed at all, and a small population (∼1 water molecule per urea molecule) in the hydration shell of urea that are 6 times slower than those in the bulk.40 This technique provides more granularity, nevertheless the authors arrive at the same conclusion: namely that urea does not change the overall strength of hydrogen bonding in water, and may cooperate with water in solvating hydrophobic residues. Our findings are also in accordance with MD simulations that generally report TMAO slows water molecules’ dynamics.12
4.3 A weak TMAO–urea interaction
Fig. 2 reveals that in a series of aqueous solution of urea, the addition of TMAO deshields the urea hydrogen atoms. It also shows that when urea is added to aqueous TMAO solutions, this leads to a minor red-shift in the 1H spectra of TMAO. Fig. 3 and Fig. S1 (ESI†) show that mixing binary solutions of aqueous TMAO and urea is additive with little observable change in behaviour between the binary and ternary data across the measured concentration range. An alternative interpretation of this additive behaviour is that TMAO and urea associate hydrophobically (Fig. 6(b)) allowing TMAO to hydrogen bond with water. Taken in their entirety, these phenomena are most consistent with the formation of a weak hydrogen bond between the TMAO oxygen atom and urea's hydrogen atoms (Fig. 6(a)) but they do not preclude a hydrophobic association. This interpretation rests to a significant extent on the conclusions of a review of van der Waals forces on chemical shift34 (Section 2.1) which concluded that they are typically large. We observed very minor shifts in the groups susceptible to a hydrophobic association. | ||
| Fig. 6 Two alternate conformations of a TMAO–urea complex are presented here: (a) is hydrogen bonded, and (b) is bound by hydrophobic interactions in the purple zone. The atoms are coloured: carbon – grey, hydrogen – white, nitrogen – blue, and oxygen – red. The hydrogen bond is represented by a dashed black line. Our measurements are most consistent with a weak hydrogen bonded complex. | ||
Xie et al.18 published experimental NMR results which exhibit similar 1H spectral trends (to Fig. 2), albeit in the concentration regime of 0.0–0.5 M urea at 0.35 M TMAO. They concluded that this was evidence for a TMAO–urea interaction mediated through the TMAO methyl groups that they describe as hydrophobic in nature (Fig. 6(b)). They found that at the low concentrations in their study, the peak shifts of the methyl hydrogen atoms of TMAO were as significant as the peak shifts of urea's hydrogen atoms, and a van der Waals interaction through the TMAO methyl group fitted their MD simulations. Our data, at higher solute concentrations show that the peak-shift in the urea hydrogen atoms are more significant. If dispersion interactions operate between the methyl groups of TMAO and urea then a peak-shift should be detectable in the spectral peak of 1H in TMAO on the addition of urea (Section 2.1), however we measured a minimal shift (Fig. 2).
Other MD simulations also find evidence for a TMAO–urea interaction42 and a study with Raman spectroscopy found evidence for an attractive interaction between urea and TMAO.43 However, an experiment using broadband dielectric spectroscopy found no evidence for a TMAO–urea interaction but instead found evidence that the TMAO·3H2O complexes remain stable at high levels of urea concentration.24
4.4 Implications for protein-folding
In Section 1 we commented on the extraordinary control this solute pair exerts on protein folding and the direct and indirect hypotheses proposed to explain this behaviour. Auton and Bolen,44 (Fig. 1) found that of all the known osmolytes, TMAO is the most powerful known stabiliser of folded protein structure. Our results show that TMAO strongly promotes hydrogen bonding in water, and that urea has a negligible impact on the bulk measurements we made of water. We also find evidence for a weak TMAO–urea interaction. These findings have implications for the folding equilibria of proteins. If TMAO makes water 'more hydrophilic' then perhaps the result of this is that at the protein surface, water prefers to hydrogen bond with itself, rather than with the amide groups allowing intra-peptide hydrogen bonds to form, enabling peptide folding.If urea has a neutral effect on the extent of water's hydrogen bond network, then at least two hypotheses follow: (i) urea may then stabilise the unfolded protein through direct interactions with the peptide backbone, or through side chain interactions (thought to be dispersion interactions14), (ii) urea can easily substitute into the water hydrogen bond network, enabling co-operative hydration of hydrophobic entities,19 and also it can participate in the shell of water that surrounds peptide structures enabling it to better access and attack the amide groups on the peptide backbone thought to be the main mechanism by which urea denatures proteins.45
The partition of TMAO from biomolecular surfaces to bulk solutions generates a depletion force13,46 that underlies TMAO's ability to shape proteins and lipids. This behaviour has its origins in the amphiphilic nature of the TMAO molecule that has been demonstrated in this work through the starkly different chemical shifts induced, we propose, by the TMAO dipole in urea and in TMAO's methyl groups by urea (Fig. 2). The behaviour of these molecules in solution, the effects they have in water and in the conformation of biomolecules are fundamental to biochemistry. Through studying the mechanisms by which these changes are effected we may learn what are the boundaries of life, how it evolves in stressed environments,47 and how we may better manage and control protein-folding conditions.48
5 Conclusion
We show that on changing the concentration of a mixed TMAO and urea solution, the NMR spectral shift of 1H in water behaves as if the solution were the sum of its two binary component parts. We also show that in mixed urea–TMAO–water, the 1H NMR spectral shift of urea/vs. TMAO concentration change is much greater than the 1H NMR spectral shift of TMAO/vs. urea concentration change. The most likely conclusion then is that TMAO and urea form a weak hydrogen bond in aqueous solution.Data availability
The data associated with this paper are openly available from the University of Leeds Data Repository: https://doi.org/10.5518/1196.Author contributions
Mazin Nasralla completed the experiments, analysis and wrote the first draft of the paper. Daniel Baker provided training and support for the operation of the NMR instrument. Harrison Laurent provided technical guidance. Michael Ries provided guidance on the NMR methodology. Lorna Dougan conceived, led and supervised the project. All authors contributed to the production of the final manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the reviewers for their helpful comments on the manuscript. This work was supported by the Engineering Physical Sciences Research Council (EPSRC), UK through a grant to L. Dougan EP/P02288X/1 and through an EPSRC DTA studentship to M. Nasralla.Notes and references
- A. Szent-Györgyi, Perspect. Biol. Med., 1971, 14, 239–249 CrossRef PubMed.
- M. C. Bellissent-Funel, A. Hassanali, M. Havenith, R. Henchman, P. Pohl, F. Sterpone, D. Van Der Spoel, Y. Xu and A. E. Garcia, Chem. Rev., 2016, 116(13), 7673–7697 CrossRef CAS PubMed.
- E. Brini, C. J. Fennell, M. Fernandez-Serra, B. Hribar-Lee, M. Lukšič and K. A. Dill, Chem. Rev., 2017, 117, 12385–12414 CrossRef CAS PubMed.
- P. Ball, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13327–13335 CrossRef CAS PubMed.
- K. A. Dill, S. B. Ozkan, M. S. Shell and T. R. Weikl, Annu. Rev. Biophys., 2008, 37, 289–316 CrossRef CAS PubMed.
- R. Gilman-Politi and D. Harries, J. Chem. Theory Comput., 2011, 7, 3816–3828 CrossRef CAS PubMed.
- S. Sukenik, S. Dunsky, A. Barnoy, I. Shumilin and D. Harries, Phys. Chem. Chem. Phys., 2017, 19, 29862–29871 RSC.
- A. Wallqvist, D. G. Covell and D. Thirumalai, J. Am. Chem. Soc., 1998, 120, 427–428 CrossRef CAS.
- J. Pan, D. Thirumalai and S. A. Woodson, J. Mol. Biol., 1997, 273, 7–13 CrossRef CAS PubMed.
- P. K. Pang, R. W. Griffith and J. W. Atz, Integr. Comp. Biol., 1977, 17, 365–377 CAS.
- P. Yancey, M. Clark, S. Hand, R. Bowlus and G. Somero, Science, 1982, 217, 1214–1222 CrossRef CAS PubMed.
- P. Ganguly, J. Polák, N. F. Van Der Vegt, J. Heyda and J. E. Shea, J. Phys. Chem. B, 2020, 124, 6181–6197 CrossRef CAS PubMed.
- S. Sukenik, L. Sapir, R. Gilman-Politi and D. Harries, Faraday Discuss., 2013, 160, 225–237 RSC.
- L. Hua, R. Zhou, D. Thirumalai and B. J. Berne, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16928–16933 CrossRef CAS PubMed.
- D. Hall and A. P. Minton, Biochim. Biophys. Acta, Proteins Proteomics, 2003, 1649, 127–139 CrossRef CAS PubMed.
- S. S. Cho, G. Reddy, J. E. Straub and D. Thirumalai, J. Phys. Chem. B, 2011, 115, 13401 CrossRef CAS PubMed.
- M. Auton and D. W. Bolen, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15065 CrossRef CAS PubMed.
- W. J. Xie, S. Cha, T. Ohto, W. Mizukami, Y. Mao, M. Wagner, M. Bonn, J. Hunger and Y. Nagata, Chem, 2018, 4, 2615–2627 CAS.
- A. K. Soper, E. W. Castner and A. Luzar, Biophys. Chem., 2003, 105, 649–666 CrossRef CAS PubMed.
- F. Meersman, D. Bowron, A. K. Soper and M. H. Koch, Biophys. J., 2009, 97, 2559–2566 CrossRef CAS PubMed.
- F. Meersman, D. Bowron, A. K. Soper and M. H. J. Koch, Phys. Chem. Chem. Phys., 2011, 13, 13765–13771 RSC.
- A. K. Soper, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 104204 CrossRef.
- H. Laurent, A. K. Soper and L. Dougan, Phys. Chem. Chem. Phys., 2020, 22, 4924–4937 RSC.
- J. Hunger, N. Ottosson, K. Mazur, M. Bonn and H. J. Bakker, Phys. Chem. Chem. Phys., 2015, 17, 298–306 RSC.
- H. R. Palmer, J. J. Bedford, J. P. Leader and R. A. Smith, J. Biol. Chem., 2000, 275, 27708–27711 CrossRef CAS PubMed.
- S. Paul and G. N. Patey, J. Am. Chem. Soc., 2007, 129, 4476–4482 CrossRef CAS PubMed.
- K. A. Sharp, B. Madan, E. Manas and J. M. Vanderkooi, J. Chem. Phys., 2001, 114, 1791–1796 CrossRef CAS.
- R. F. Greene and C. N. Pace, J. Biol. Chem., 1974, 249, 5388–5393 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- P. Charisiadis, V. G. Kontogianni, C. G. Tsiafoulis, A. G. Tzakos, M. Siskos and I. P. Gerothanassis, Molecules, 2014, 19, 13643 CrossRef PubMed.
- P. Hore, Nuclear Magnetic Resonance, Oxford University Press, Oxford, 2nd edn, 1995 Search PubMed.
- J. B. Lambert, E. P. Mazzola and C. D. Ridge, Nuclear Magnetic Resonance Spectroscopy: An Introduction to Principles, Applications, and Experimental Methods, Wiley, 2nd edn, 2018 Search PubMed.
- T. Claridge, High-Resolution NMR Techniques in Organic Chemistry, Elsevier, 3rd edn, 2016 Search PubMed.
- P. Diehl, E. Fluck and R. Kosfeld, van der Waals Forces and Shielding Effects, Berlin, Heidelberg, 1975, pp. 5–7 Search PubMed.
- J. Homer and C. C. Percival, J. Chem. Soc., Faraday Trans. 2, 1984, 80, 1–29 RSC.
- B. J. Bennion and V. Daggett, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6433–6438 CrossRef CAS PubMed.
- M. Freda, G. Onori and A. Santucci, J. Phys. Chem. B, 2001, 105, 12714–12718 CrossRef CAS.
- C. Y. Hu, G. C. Lynch, H. Kokubo and B. Montgomery Pettitt, Proteins: Struct., Funct., Bioinf., 2010, 78, 695–704 CrossRef CAS PubMed.
- A. B. Goins, H. Sanabria and M. N. Waxham, Biophys. J., 2008, 95, 5362–5373 CrossRef CAS PubMed.
- Y. L. Rezus and H. J. Bakker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18417–18420 CrossRef CAS PubMed.
- D. Laage, G. Stirnemann, F. Sterpone and J. T. Hynes, Acc. Chem. Res., 2012, 45, 53–62 CrossRef CAS PubMed.
- P. Ganguly, N. F. Van Der Vegt and J. E. Shea, J. Phys. Chem. Lett., 2016, 7, 3052–3059 CrossRef CAS PubMed.
- S. G. Zetterholm, G. A. Verville, L. Boutwell, C. Boland, J. C. Prather, J. Bethea, J. Cauley, K. E. Warren, S. A. Smith, D. H. Magers and N. I. Hammer, J. Phys. Chem. B, 2018, 122, 8805–8811 CrossRef CAS PubMed.
- M. Auton and D. W. Bolen, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15065–15068 CrossRef CAS PubMed.
- N. Steinke, R. J. Gillams, L. C. Pardo, C. D. Lorenz and S. E. McLain, Phys. Chem. Chem. Phys., 2016, 18, 3862–3870 RSC.
- S. Asakura and F. Oosawa, J. Chem. Phys., 2004, 22, 1255 CrossRef.
- T. Lamitina, C. G. Huang and K. Strange, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12173–12178 CrossRef CAS PubMed.
- S. Sukenik, L. Sapir and D. Harries, J. Chem. Theory Comput., 2015, 11, 5918–5928 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp02475f |
| This journal is © the Owner Societies 2022 |