
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAb initio molecular dynamics investigation of the co-adsorption of iodine species with CO and H2O in silver-exchanged chabazite
Tarek
Ayadi
 *,
Sébastien
Lebègue
* and
Michael
Badawi
*
*,
Sébastien
Lebègue
* and
Michael
Badawi
*
Laboratoire de Physique et Chimie Théoriques (LPCT, UMR CNRS UL 7019), Université de Lorraine, BP 239, Boulevard des Aiguillettes, 54506 Vandoeuvre-lès-Nancy, Cedex, France. E-mail: tarek.ayadi@univ-lorraine.fr; sebastien.lebegue@univ-lorraine.fr; michael.badawi@univ-lorraine.fr
First published on 10th October 2022
Abstract
In the field of nuclear energy, there is particular interest for the trapping of harmful iodine species (I2 and CH3I) that could be released during a nuclear accident, due to their dangereous impact on the human metabolic processes and the environment. Here, the adsorption of these iodine molecules versus several inhibitory compounds (CO, H2O, CH3Cl and Cl2) in the silver exchanged chabazite zeolite is studied in detail using ab initio molecular dynamics simulations at a realistic temperature and composition. Interestingly, we found that the iodine molecules remain attached to the cations even when the number of water molecules inside the structure is greater than two times the number of cations per cell at T = 413 K. For CO, we found that CH3I is more perturbed than I2 by the presence of this inhibitor. Overall, our results indicate that the silver-exchanged chabazite zeolite is a promising candidate to trap iodine species in the case of a severe nuclear accident.
1 Introduction
The safe storage of nuclear waste is an increasingly important topic, especially after the Fukushima nuclear accident. In particular, iodine species are among the most dangereous radioactive compounds that remain of strong interest and concern:1–3 for public safety, their storage and capture before they reach the environment via some leakages of the nuclear containment is crucially important due to their dangerous impact on the human metabolic processes. However, different molecules are released during a nuclear accident which can inhibit the iodine uptake process: it is expected to find large amounts of steam (40–90%) and other gaseous compounds such as CO (0–20%) and chlorinated species (CH3Cl and Cl2, with lower concentrations).4–7 CO mainly comes from the molten core concrete interactions which start after vessel rupture. Chlorinated compounds (CH3Cl and Cl2) are produced by fuel fission and possible thermal degradation of cables by pyrolysis.6,7 On the other hand, we found very low concentrations of radiotoxic iodine (0.1–1 ppm).8 In this vein, specific filtration devices on the containment venting system are necessary, for instance those based on zeolites, to capture the toxic iodine gas and limit its effect.9–14Zeolites are among the most well-known microporous materials15–18 that have various applications such as catalysts, cleaners, gas filters and agents for the treatment of nuclear waste due to the presence of structured channels with different dimensions able to capture various types of molecules.10,12,19,20 Indeed, zeolites show very high thermal and chemical stabilities, and their large structural and compositional varieties10,12 facilitate the possibility to adjust their properties. Also, the possibility to perform a cationic exchange in the zeolite allows its performance to be tuned by choosing the type of cation to be used to compensate the charge produced in the zeolite structure e.g. by the replacement of Si atoms by Al atoms.18,21,22
Previous studies have shown that silver exchanged zeolites are optimal for the selective trapping of iodine species, compared to Cu, Na, Pb, Tl and Cd exchanged zeolites.11,18,23–27 Through an in-depth experimental study on different Ag-exchanged zeolites (faujasites X and Y, mordenite, *BEA, MFI, and FER) to understand the link between the efficiency of iodine trapping and the chemical and structural parameters, Azambre and Chebbi27 have shown that the formation of AgI precipitates is linked to the performance of zeolites in the capture of the toxic iodine species. Recently, a theoretical study has been carried out by Ayadi et al.28 using density functional theory at T = 0 K to find out the most promising Ag-exchanged zeolitic formulation for the selective trapping of iodine species against several inhibitory molecules (CO, H2O, Cl2 and CH3Cl) in the Ag-exchanged faujasite, mordenite, chabazite and clinoptilolite. They found that CO presents a strong inhibitory effect on the capture of toxic iodine gas for high Si/Al ratios, while Cl2 presents a strong inhibitory effect for low Si/Al ratios. Overall, a systematic comparison has been performed between the different zeolites, including the framework topology and the Si/Al ratios, and it was shown that the Ag-exchanged chabazite structure with a Si/Al ratio of 5 is the most promising zeolitic formulation for the capture of the harmful iodine compounds where no inhibitory effect is observed.
Therefore, inspired by these results, we investigate in detail the performance of the silver exchanged chabazite structure with a Si/Al ratio of 5 under more realistic conditions, at T = 413 K and for several molecules at various contents. To this end, we have employed ab initio molecular dynamics which is a cutting-edge technique to describe adsorption phenomena under operando conditions.29–32 During this study, the performance of the chabazite in the adsorption of I2 and CH3I versus CO, H2O, Cl2 and CH3Cl is examined. Then, the effect of water followed by the effect of CO on the capture of the iodine molecules is investigated. Finally, in order to simulate as realistically as possible the experimental conditions, we have studied the effect of CO together with seven molecules of H2O on the interactions between iodine compounds and the chabazite structure.
This paper is organized as follows: first, we describe our computational details and the structural model used, then our results for an Ag-chabazite of Si/Al = 5 are presented and discussed with a focus on the effect of the potentially inhibiting molecules (CO, H2O, Cl2 and CH3Cl), followed by the effect of the number of CO and H2O molecules, and finally we provide the conclusions of our work.
2 Materials and methods
2.1 Computational details
We have performed ab initio calculations by means of density functional theory (DFT) with the projector augmented wave (PAW) method33 as implemented in the Vienna Ab initio Simulation Package (VASP).34 For the exchange and correlation contributions, the Perdew–Burke–Ernzerhof (GGA-PBE) functional35 was employed with a kinetic energy cutoff of 450 eV for the plane wave basis set. The D2 dispersion correction of Grimme was used to describe dispersion interactions.36,37 Also, the rev-vdW-DF2 approximation is used where the revised Becke exchange functional (B86R) is adopted for the exchange functional together with the second version of nonlocal vdW-DF.38 The goal here is to be able to compare the two levels of theory to consolidate our prediction. Regarding the large size of the used unit cell, the calculations were performed with the Γ-point. The convergence parameters were set to 0.02 eV Å−1 for the atomic forces and 10−6 eV for the total energy.Using the electronic parameters described above for static DFT relaxations, ab initio molecular dynamics simulations (AIMD) were performed over a minimum total simulation time of 120 ps which includes 20 ps for the equilibration period and a time step of 1 fs. To avoid fictitious molecular splitting, the mass of the hydrogen atom is increased to 3 which corresponds to the atomic mass of tritium, which allowed us to set the time-step for integration of the equations of motion to a relatively large value of 1 fs. Using a Nosé–Hoover thermostat,39,40 the temperature was set to 413 K (temperature reached at the containment). The convergence parameters were set to 10−5 for the atomic forces.
The interaction energies for the static DFT calculations between the adsorbate molecules and the chabazite have been calculated using Eint = (Ezeolite+X − Ezeolite − NXEX)/NX, where Ezeolite+X is the energy of the zeolite with the adsorbed molecules, Ezeolite is the energy of the clean zeolite, EX is the energy of the isolated molecules in the gaseous phase and NX is the number of molecules inside the zeolite. For AIMD simulations, the internal energy of adsorption was calculated using the equation: Eint = (Ezeolite+X − Ezeolite − NXEX)/NX, where Ezeolite+X, Ezeolite and EX are the average of Ezeolite+X, Ezeolite and EX, respectively.
2.2 The chabazite model
Chabazite (CHA) is a three dimensional network of SiO2 composed mainly of four-membered rings (4MRs), double six-ring prisms (6DR) and eight-membered rings (8MRs). The double six-ring prisms (6DR) arranged in layers are connected between each other by tilted four-membered rings (4MRs). The 8MRs function as highly selective doorways with a diameter of 3.8 Å which allow the access to the crystal.41–43 The pore size of CHA zeolites is about 3.7 Å × 4.2 Å44 which makes them able to accommodate the molecules studied in this paper, where the longest molecular axes are approximately 2.7 Å and 2.4 Å for I2 and CH3I,26 respectively. Indeed, the size of the chabazite pore is in the same order as those of other zeolites’ pores such as mordenite 10.2 Å × 8.4 Å (main channels) and 7.6 Å × 5.7 Å (side channel).26 Here, we have studied the silver exchanged chabazite with a Si/Al ratio of 5 (Ag-CHA(5) presented in Fig. 1). In this case, the silver loading is about 23.1 wt%. Indeed, recent experimental studies27 have demonstrated that isolated silver cations can be well inserted into zeolites by cationic exchange even at a high silver loading (up to 23%). | ||
| Fig. 1 Structural model of the chabazite (CHA), with a Si/Al ratio of 5, projected along the c direction. | ||
3 Results and discussion
3.1 Adsorption of the single type of molecule
In Table 1, we present our computed interaction energies for CH3I, I2, CO, H2O, CH3Cl and Cl2 in the chabazite zeolite at T = 0 K and 413 K. According to our calculations, the interaction energy of I2 and CH3I is about −206.3 kJ mol−1 (−221.7 kJ mol−1) and −170.3 kJ mol−1 (−165 kJ mol−1) at T = 0 K with PBE + D2 (rev-vdW-DF2), respectively, and these values decrease to −173.6 kJ mol−1 and −163.7 kJ mol−1, respectively, at a temperature of 413 K. Also, we found that the iodine species are more strongly bound to the zeolite than H2O, CO, Cl2 and CH3Cl, independently of temperature. At T = 0 K, we found with PBE + D2 that the interaction energy of CH3I(I2) is more higher (in terms of the absolute value) than those of H2O, CO, CH3Cl and Cl2 by about 47.4 kJ mol−1 (83.4 kJ mol−1), 40.2 kJ mol−1 (76.2 kJ mol−1), 77.9 kJ mol−1 (114 kJ mol−1) and 33.8 kJ mol−1 (69.8 kJ mol−1), respectively. The same results are obtained also with the nonlocal functional rev-vdW-D2. At a temperature of 413 K, the CH3I (I2) compound is more bound to the chabazite than H2O, CO, Cl2 and CH3Cl, almost with the same quantities of energy obtained at T = 0 K which are 44.6 kJ mol−1 (54.5 kJ mol−1), 47.4 kJ mol−1 (57.3 kJ mol−1), 74.2 kJ mol−1 (84.1 kJ mol−1), and 61.8 kJ mol−1 (71.1 kJ mol−1), respectively.| CH3I | I2 | H2O | CO | Cl2 | CH3Cl | ||
|---|---|---|---|---|---|---|---|
| T = 0 K | PBE + D2 | −170.3 | −206.3 | −122.9 | −130.1 | −92.33 | −136.5 |
| rev-vdW-DF2 | −165 | −221.7 | −116.3 | −130.9 | −106.3 | −113 | |
| T = 413 K | PBE + D2 | −163.7 | −173.6 | −119.1 | −116.3 | −89.5 | −115.3 |
Then, we studied the effect of having several molecules of the same type on the capture of the iodine gas and inhibitor compounds (CO and H2O) at T = 413 K. The results are presented in Fig. 2. We found that the number of molecules has an effect on the adsorption of iodine species, in contrast to CO and H2O. For the I2 molecule, the interaction energy decreases from 174 kJ mol−1 to 155 kJ mol−1, and then to 147 kJ mol−1 when n increases to 2 and then to 3, respectively; for the CH3I compound, we found that the interaction energy decreases from 164 kJ mol−1 to 155 kJ mol−1 when n increases to 2, and then to 136 kJ mol−1 when n increases to 3. For the inhibitory molecules (CO and H2O), we found that the effect of the molecule number on the adsorption is rather weak: a decrease by only a few kJ mol−1 is obtained with the increase of n to 2 and 3. The different behavior observed between the iodine species and the inhibitory molecules is related to the size of molecules coupled with their adsorption modes in the zeolite. By looking into the adsorption modes, we found that CH3I is adsorbed on top of two Ag-cations when n = 1, and with the increase in the number of molecules, each CH3I can interact just with a single cation which decreases the interaction energy per molecule (we note that the chabazite contains 4 cations per cell as described above). For I2, we found that the I atom interacts with more than a single cation on each side of the molecule when n = 1, while, with the increase of the number of molecules, each cation in the zeolite can interact with several I2 molecules, which decreases the interaction energy per molecule. On the other hand, the small size of CO and H2O allows them to be adsorbed on top of a single cation, independently of the number of molecules, which explains the additive behavior observed in the interaction energy values when n is greater than 1. The effect of the adsorption modes on the decrease of the interaction energy of iodine species is observed also in a previous study on the adsorption of iodine species in the silver-exchanged mordenite zeolite.14
 | ||
| Fig. 2 The variation of the interaction energies per molecule as a function of the number of molecules (n). | ||
3.2 Effect of water molecules
In this section, we study the competitive sorption of iodine species from water in the chabazite structure. Water is one of the most potent inhibitors of the adsorption of harmful iodine species, where its amount ranges between 40% and 90% during a nuclear accident.4,5,17 We progressively filled the structure by water, molecule by molecule, to study the effect of the number of water molecules on the adsorption of I2 and CH3I. Each molecule added, up to 10 water molecules per unit cell which is in the same order as the water quantity (∼7 mmol g−1) adsorbed in the chabazite observed experimentally,22 with the iodine species is followed by a molecular dynamics simulation of at least 120 ps at 413 K. In Fig. 3, we present the interaction energy per number of water molecules as a function of the number of water molecules. We found that water molecules have a significant effect on the adsorption energies of the iodine species where the interaction energy values per water molecule of CH3I and I2 decrease as the number of water molecules increases. For CH3I, an extreme decrease in the interaction energy by about 140 kJ mol−1 is observed when n increases from 1 to 4, and then, the interaction energy presents a small decrease from 124 kJ mol−1 when n = 4 to 93 kJ mol−1 when n = 6, then to 75 kJ mol−1 when n = 8, and finally to 64.5 kJ mol−1 for n = 10. For the I2 molecule, we also found that the number of water molecules has an effect in the adsorption mechanism, but it is more significant than that observed in the case of CH3I: first, the interaction energy strongly decreases from 422 kJ mol−1 to 148 kJ mol−1 when n increases from 1 to 4; second, Eint decreases by about 51 kJ mol−1 with the increase of n from 4 to 6; and finally, Eint presents a small decrease by a few kJ mol−1 when n increases to 8, and then to 10. It appears that when n = 1, the iodine molecules are adsorbed on top of more than a single cation, while the water molecule is adsorbed on top of an another single cation; when the number of water molecules ranges between 4 and 10, the iodine molecules are adsorbed on top of a single cation and, in some cases, they shared the same cations with water molecules which decreases the interaction energy and explains the quasi-stability in the energy values per water molecule obtained when n is superior to 1. To confirm our explanation, we present in Fig. 4 the last configurations obtained at t = 120 ps where we observe that the cations are shared between iodine molecules and water, and that the adsorption modes are similar when n ranges between 4 and 10, in contrast to n = 1. To better understand the effect of water on the adsorption of iodine species, we have computed the radial distrubution function (RDF) of the Ag–I distances of Ag–ICH3 and Ag–I2 interactions with different numbers of water molecules inside the chabazite structure, see Fig. 4. Without water molecules, the RDFs indicate that the Ag–I distances are about 2.67 Å and 2.65 Å in the case of CH3I and I2, respectively. With the addition of a water molecule with the iodine molecules inside the chabazite, the results show that the presence of water has no effect on the Ag–I distance but a small decrease of the peak intensity is observed. This behavior is also observed with the increase of the number of water molecules to 4. On the other hand, the results obtained for CH3I with n = 6, 8 and 10 present an increase by 0.1 Å for the Ag–I distance, with an appearance of a second weak peak at 3.5 Å for n = 8, and at 4.5 Å for n = 10. This second peak is the result of the movement of CH3I from one cation to another under the effect of the temperature together with water molecules, while the small increase in the Ag–I distance can be a result of the adsorption of the molecules of water with the iodine molecule on top of the same cations. For the I2 molecule, we found that the presence of the H2O molecules also has an effect, depending on the number of molecules, on the interaction of I2 with the chabazite structure. When n = 1, any effect observed in the Ag–I distance, which is about 2.65 Å, is not surprising because I2 and water molecules are adsorbed on the top of different cations as presented in Fig. 5. With the increase of the number of water molecules, we found that the Ag–I distance is around 2.7 Å but with a decrease in the principle peak intensity coupled by an increase in the second peak intensity at 4 Å. Also, we can see that the RDF curves are very similar for n ranging between 4 and 10 which is related to similar adsorption modes as presented in Fig. 5. Indeed, we found that the iodine species remain attached to the cations even when the number of water molecules inside the structure is greater than two-fold the number of cations per cell which means that water is not a strong competitive component against iodine species. These findings are very interesting in the case of a nuclear accident and for the scientists in the field of nuclear energy due to the existence of a high amount of water in the nuclear containment, as described above. | ||
| Fig. 3 Interaction energies as a function of number of water molecules. | ||
 | ||
| Fig. 4 Radial distribution function of the Ag–I distances of (a) Ag–CH3I and (b) Ag–I2 interactions with different numbers (n) of water molecules inside the chabazite structure. | ||
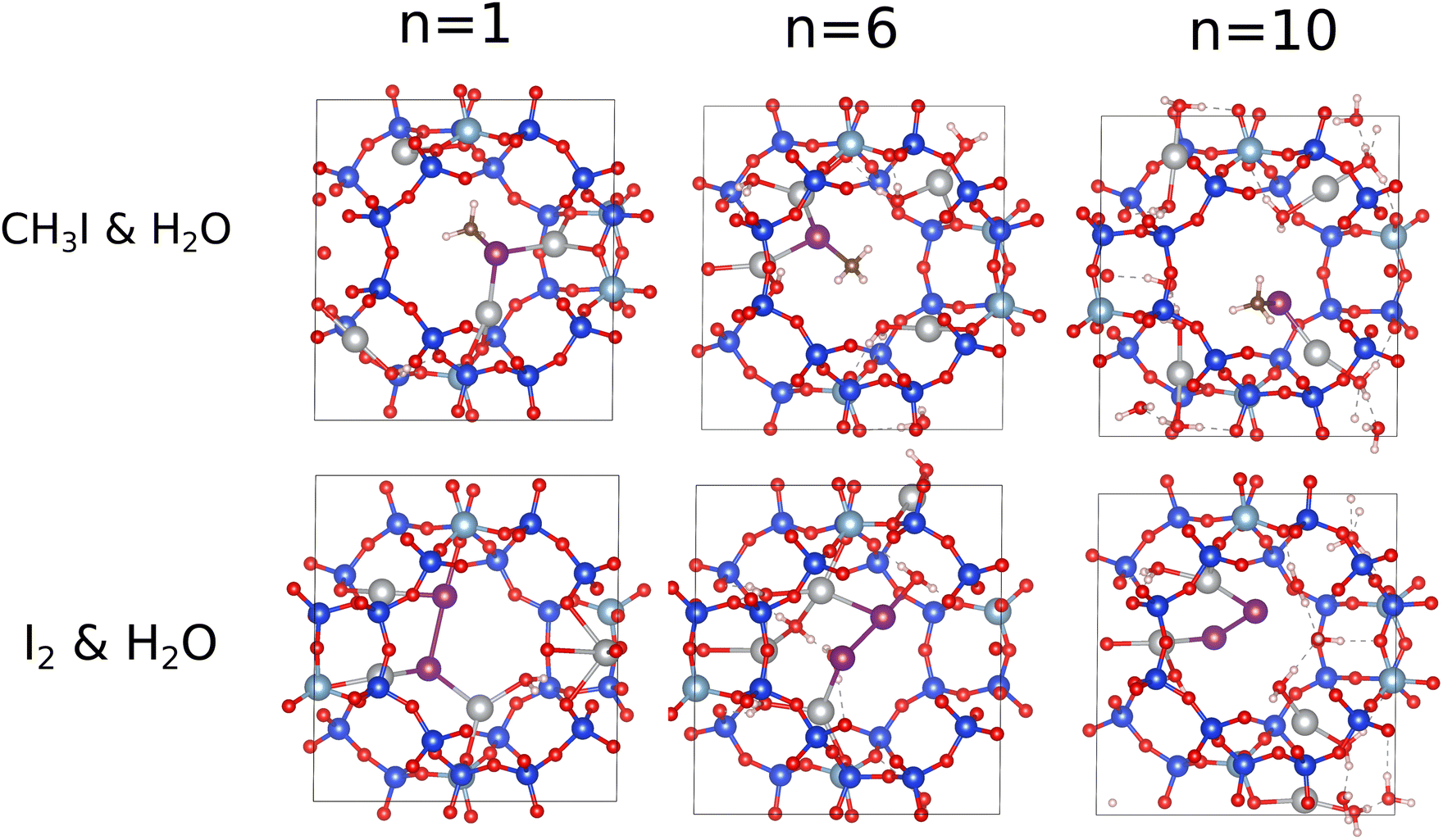 | ||
| Fig. 5 Adsorption modes of CH3I (I2) and water molecules in the chabazite structure. | ||
3.3 Effect of CO molecules
CO is expected to be one of the most potent inhibitors for the trapping of the toxic iodine molecules, where its quantity varies from 0 to 20% during a nuclear accident.4,5,17 Here, we aim to investigate the effect of CO on the interaction of iodine species with zeolite where we put an iodine species together with the CO molecule inside the chabazite. AIMD calculations show that the interaction energy of CO and CH3I is about −257 kJ mol−1, which is less than the sum of the interaction energies of CH3I and CO adsorbed alone by about 23 kJ mol−1. By looking into the RDF calculations of the Ag–I distance presented in Fig. 6 for CH3I, we found that the adsorption of CO in the chabazite has a small effect on the Ag–I distance where it increases from 2.65 Å to 2.73 Å, which means that the interaction between CH3I and the zeolite is weakly disturbed. Otherwise, we found that the interaction energy of I2 and CO is about −295.7 kJ mol−1, while the sum of the interaction energy of I2 and CO adsorbed alone is about −289.9 kJ mol−1. Using RDF analysis, we found that the average Ag–I distance is 2.65 Å in the case of I2, with and without the presence of CO in the zeolite. Here, we observe that CH3I is more perturbed by the presence of CO than I2, where the interaction energy decreases by about 23 kJ mol−1 coupled with an increase in the Ag–I distance in the case of CH3I, compared to a decrease in the interaction energy of I2 by about 5.8 kJ mol−1. Despite the weak effect observed in the case of CH3I, the iodine species remain in close interaction with cations and no deformation in the framework is observed (see Fig. 7). | ||
| Fig. 6 Radial distribution function of the Ag–I distances of (a) Ag–CH3I and (b) Ag–I2 interactions with the CO molecule inside the chabazite structure. | ||
 | ||
| Fig. 7 Adsorption modes of CH3I(I2) and CO in the chabazite structure. | ||
3.4 Capture of iodine species from a realistic feed
We now turn to study the competition between the iodine species and two types of inhibitor compounds (H2O and CO) where we put in the same cage a molecule of iodine species (I2 or CH3I) together with seven molecules of water and a molecule of CO. The choice of the number of molecules is related to the amount of water and CO from nuclear waste. Our calculations at 413 K show that the interaction energy of CH3I(I2), 7 H2O and CO is of −670.4 kJ mol−1 (−626.3 kJ mol−1) which is less than the sum of the interaction energies of CH3I(I2), 7 H2O and CO adsorbed alone which is about −1113.7 kJ mol−1 (−1123.6 kJ mol−1). The RDF calculations of the Ag–I distances for Ag–ICH3 and Ag–I2 interactions show that the presence of water and CO, together with iodine species in the same cage, has no significant effect in the interaction mechanism (see Fig. 8). We found that the mean peak is around 2.8 Å for Ag–CH3I and is around 2.7 Å for the I2 molecule with and without the presence of the inhibitor molecules. With the addition of inhibitor molecules (CO and 7 H2O), we observe the appearance of a second peak at 3.5 Å which corresponds to a movement of the iodine species observed during the molecular dynamics simulations. As the chabazite contains 4 cations, the iodine species have the possibility to move from one cation to another one with temperature. Also, we found that the second peak is more intense with the presence of CO in the case of CH3I. These observations show that CH3I is more perturbed than I2 by the presence of CO as observed in our previous work for faujasite at 0 K.28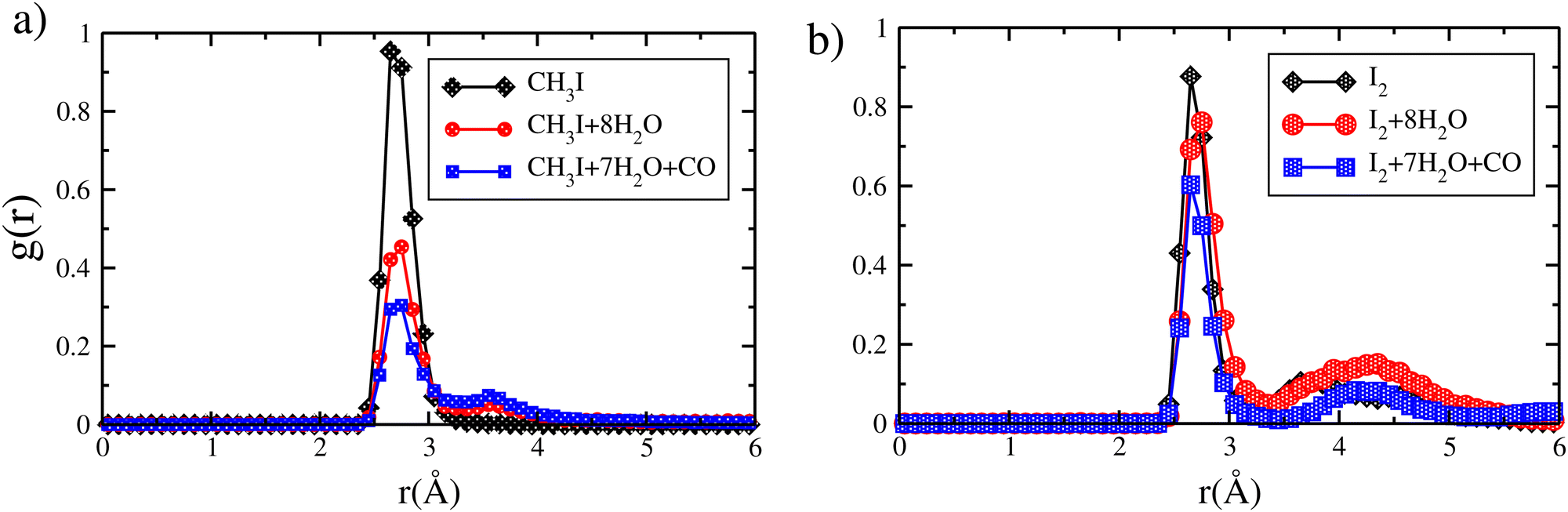 | ||
| Fig. 8 Radial distribution function of the Ag–I distances of (a) Ag–CH3I and (b) Ag–I2 interactions with the H2O and CO molecules inside the chabazite structure. | ||
4 Conclusion
Using ab initio molecular dynamics simulations at T = 413 K, we have investigated the adsorption mechanism of toxic iodine molecules (CH3I and I2) against several inhibiting molecules (H2O, CO, CH3Cl and Cl2) in the Ag-exchanged chabazite with a Si/Al ratio of 5. We found that this chabazite presents a good performance in the capture of the iodine gas where the smallest difference in the interaction energy between the iodine species and the inhibiting molecule is about 20 kJ mol−1. Then, we studied the effect of water molecules on the adsorption of CH3I and I2: we found that a large decrease in the interaction energies is obtained in the presence of H2O, in contrast to the Ag–I distance where an increase of 0.1 Å is observed in the extreme case. Also, we studied the effect of CO on the trapping of iodine species where we found that I2 is less perturbed by the presence of CO compared to CH3I. Finally, the effect of CO and H2O together on the adsorption of iodine species was investigated: we have shown that there is a small effect in the interaction between CH3I and the zeolite where an increase is observed for the Ag–I distance.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was granted access to the HPC resources of TGCC under the allocations 2021-A0100810433 and A0120810433 provided by GENCI-EDARI and was supported by the French research program ANR-11-RSNR-0013-01 called MiRE (Mitigation of Iodine Releases in the Environment).Notes and references
- J. R. Brian, D. V. John, M. S. Denis, S. M. John and L. J. James, J. Nucl. Mater., 2016, 470, 307–326 CrossRef.
- A. J. González, Health Phys., 2007, 93, 571 CrossRef.
- M. Chebbi, B. Azambre, C. Monsanglant-Louvet, B. Marcillaud, A. Roynette and L. Cantrel, J. Hazard. Mater., 2021, 409, 124947 CrossRef CAS.
- L. E. Herranz, T. Lind, K. Dieschbourg, E. Riera, S. Morandi, P. Rantanen, M. Chebbi and N. Losch, OECD REPORT-RN: 47070575, 2013, 2846.
- D. Jacquemain, S. Guentay, S. Basu, M. Sonnenkalb, L. Lebel, H.-J. Allelein, B. Liebana, B. Eckardt and L. Ammirabile, OECD Report-RN: 45089842, 2014, 174.
- J. Foit, Nucl. Eng. Des., 1997, 170, 73–79 CrossRef CAS.
- A. Auvinen, R. Zilliacus and J. Jokiniemi, Nucl. Technol., 2005, 149, 232–241 CrossRef CAS.
- M. Chebbi, B. Azambre, L. Cantrel, M. Huvé and T. Albiol, Microporous Mesoporous Mater., 2017, 244, 137–150 CrossRef CAS.
- J. Jolley and H. Tompkins, Appl. Surf. Sci., 1985, 21, 288–296 CrossRef CAS.
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897–8899 CrossRef CAS.
- L. N. Rastunov, E. P. Magomedbekov, A. V. Obruchikov and L. A. Lomazova, At. Energy, 2011, 110, 68 CrossRef CAS.
- T. M. Nenoff, M. A. Rodriguez, N. R. Soelberg and K. W. Chapman, Microporous Mesoporous Mater., 2014, 200, 297–303 CrossRef CAS.
- S. U. Nandanwar, K. Coldsnow, V. Utgikar, P. Sabharwall and D. Eric Aston, Chem. Eng. Sci., 2016, 306, 369–381 CrossRef CAS.
- H. Jabraoui, E. Hessou, S. Chibani, L. Cantrel, S. Lebègue and M. Badawi, Appl. Surf. Sci., 2019, 485, 56–63 CrossRef CAS.
- M. Kitamura, K. Funabashi, M. Kikuchi, H. Yusa, Y. Fukushima and S. Horiuchi, Trans. Am. Nucl. Soc., 1978, 30, https://www.osti.gov/biblio/6537164 Search PubMed.
- D. F. Sava, M. A. Rodriguez, K. W. Chapman, P. J. Chupas, J. A. Greathouse, P. S. Crozier and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 12398–12401 CrossRef CAS.
- S. Chibani, M. Chebbi, S. Lebègue, L. Cantrel and M. Badawi, Phys. Chem. Chem. Phys., 2016, 18, 25574–25581 RSC.
- M. Chebbi, S. Chibani, J.-F. Paul, L. Cantrel and M. Badawi, Microporous Mesoporous Mater., 2017, 239, 111–122 CrossRef CAS.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC.
- E. R. Vance and D. K. Agrawal, J. Mater. Sci., 1982, 17, 1889 CrossRef CAS.
- G. D. Pirngruber, P. Raybaud, Y. Belmabkhout, J. Čejka and A. Zukal, Phys. Chem. Chem. Phys., 2010, 12, 13534–13546 RSC.
- Y. Luo, H. H. Funke, J. L. Falconer and R. D. Noble, Ind. Eng. Chem. Res., 2016, 55, 9749–9757 CrossRef CAS.
- R. T. Jubin, Report Number: ORNL/TM-10477, 1988.
- B. S. Choi, G. I. Park, J. H. Kim, J. W. Lee and S. K. Ryu, Adsorption, 2001, 7, 91 CrossRef CAS.
- Q. Cheng, Z. Li, T. Chu, W. Yang, Q. Zhu, D. He and C. Fang, J. Radioanal. Nucl. Chem., 2015, 303, 1883 CAS.
- S. Chibani, M. Chebbi, S. Lebègue, T. Bucko and M. Badawi, J. Chem. Phys., 2016, 144, 244705 CrossRef PubMed.
- B. Azambre and M. Chebbi, ACS Appl. Mater. Interfaces, 2017, 9, 25194–25203 CrossRef CAS.
- T. Ayadi, M. Badawi, L. Cantrel and S. Lebègue, Mol. Syst. Des. Eng., 2022, 7, 422–433 RSC.
- S. Tan, C. Wang, Y. Foucaud, M. Badawi, H. Guo, K. Sun, G. Yang and S. Mintova, Microporous Mesoporous Mater., 2022, 336, 111853 CrossRef CAS.
- F.-X. Coudert and D. Kohen, Chem. Mater., 2017, 29, 2724–2730 CrossRef CAS.
- Y. Foucaud, J. Lainé, L. O. Filippov, O. Barrès, W. J. Kim, I. V. Filippova, M. Pastore, S. Lebègue and M. Badawi, J. Colloid Interface Sci., 2021, 583, 692–703 CrossRef CAS PubMed.
- J. Lainé, Y. Foucaud, A. Bonilla-Petriciolet and M. Badawi, Appl. Surf. Sci., 2022, 585, 152699 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- T. Bučko, J. Hafner, S. Lebègue and J. G. Ángyán, J. Phys. Chem. A, 2010, 114, 11814–11824 CrossRef PubMed.
- I. Hamada, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 121103 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS.
- M. Abatal, A. R. Ruiz-Salvador and C. H. Norge, Microporous Mesoporous Mater., 2020, 294, 109885 CrossRef CAS.
- Y. Guo, T. Sun, Y. Gu, X. Liu, Q. Ke, X. Wei and S. Wang, Chem. – Asian J., 2018, 13, 3222–3230 CrossRef CAS.
- M. Lee, S. Hong, D. Kim, E. Kim, K. Lim, J. C. Jung, H. Richter, J.-H. Moon, N. Choi, J. Nam and J. Choi, ACS Appl. Mater. Interfaces, 2019, 11, 3946–3960 CrossRef CAS.
| This journal is © the Owner Societies 2022 |