
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evidence suggesting kinetic unfreezing of water mobility in two distinct processes in pressure-amorphized clathrate hydrates†
Ove
Andersson
 *a,
Paulo H.
B. Brant Carvalho
b,
Ulrich
Häussermann
b and
Ying-Jui
Hsu‡
a
*a,
Paulo H.
B. Brant Carvalho
b,
Ulrich
Häussermann
b and
Ying-Jui
Hsu‡
a
aDepartment of Physics, Umeå University, 901 87 Umeå, Sweden. E-mail: ove.b.andersson@umu.se
bDepartment of Materials and Environmental Chemistry, Stockholm University, 106 91 Stockholm, Sweden
First published on 20th July 2022
Abstract
Type II clathrate hydrates (CHs) with tetrahydrofuran (THF), cyclobutanone (CB) or 1,3-dioxolane (DXL) guest molecules collapse to an amorphous state near 1 GPa on pressurization below 140 K. On subsequent heating in the 0.2–0.7 GPa range, thermal conductivity and heat capacity results of the homogeneous amorphous solid show two glass transitions, first a thermally weak glass transition, GT1, near 130 K; thereafter a thermally strong glass transition, GT2, which implies a transformation to an ultraviscous liquid on heating. Here we compare the GTs of normal and deuterated samples and samples with different guest molecules. The results show that GT1 and GT2 are unaffected by deuteration of the THF guest and exchange of THF with CB or DXL, whereas the glass transition temperatures (Tgs) shift to higher temperatures on deuteration of water; Tg of GT2 increases by 2.5 K. These results imply that both GTs are associated with the water network. This is corroborated by the fact that GT2 is detected only in the state which is the amorphized CH's counterpart of expanded high density amorphous ice. The results suggest a rare transition sequence of an orientational glass transition followed by a glass to liquid transition, i.e., kinetic unfreezing of H2O reorientational and translational mobility in two distinct processes.
1. Introduction
A glass transition (GT), i.e., a transformation from a liquid phase to an amorphous solid on cooling, is an important phenomenon in materials science and technology. In quite a few liquids, with important exceptions such as water, it is possible to study the change from a low-viscosity liquid to a supercooled (metastable) high-viscosity liquid, and the ultimate transformation into glass, an amorphous solid state, which is characterized by a glass transition temperature Tg.1 In addition to the increase of viscosity on cooling, the transition entails a sigmoid-shaped decrease in heat capacity due to kinetic freezing of structural fluctuations on the time scale of an experiment. Normally both translational and reorientational mobility are frozen (almost) concomitantly at GTs in low-molecular-weight liquids, and the two processes cannot be distinguished in macroscopic properties such as the heat capacity. However, in this study we show evidence that water, in a water-rich homogenous solution, is a rare example where these processes are significantly shifted in temperature. The amorphous solid state in which this is observed is formed by pressure collapse of a crystal where moderately polar molecules and water are perfectly homogeneously mixed.Pressure-induced amorphization (PIA), i.e., the transformation of a crystalline material to an amorphous state by collapse of the structure on pressurization, is a feature reported for quite a few materials.2 The most well-known example is the collapse of normal, hexagonal, water ice (ice Ih) on pressurization to 1–1.5 GPa below 140 K to high density amorphous ice (HDA).3 But also clathrate hydrates (CHs), which are ice-like inclusion compounds containing guest molecules in polyhedral cages formed by hydrogen-bonded water molecules,4,5 show PIA; generally, PIA of CHs occurs at slightly higher pressure than that of ice.6–8
CHs typically crystallize in one of three structures denoted: I, II, and H, but PIA has only been reported for the cubic types I9 and II6,7,10–12 CHs. In these structures, the hydrogen-bonded H2O network forms three types of cages: pentagonal dodecahedron (D), tetrakaidecahedral (T), and hexakaidecahedral (H) (inset of Fig. 1). The CHs investigated here, tetrahydrofuran (THF) CH, 1,3-dioxolane (DXL) CH and cyclobutanone (CB) CH crystallize in the type II structure; THF CH is stable at pressures up to about 0.3 GPa for temperatures below 255 K.13,14 Below ca. 140 K, it remains metastable up to a pressure in the range 1–1.5 GPa where it undergoes PIA. The type II unit cell (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), with a lattice constant of 17 Å, consists of 136 water molecules forming 16 D-cages with average radius 3.91 Å, and 8 H-cages of radius 4.73 Å;5 guest molecules reside in the small D-cages, large H-cages, or both types of cages dependent on their sizes. THF, DXL and CB guests all reside in the large cages, which yields an ideal water-rich molar composition of 1
m), with a lattice constant of 17 Å, consists of 136 water molecules forming 16 D-cages with average radius 3.91 Å, and 8 H-cages of radius 4.73 Å;5 guest molecules reside in the small D-cages, large H-cages, or both types of cages dependent on their sizes. THF, DXL and CB guests all reside in the large cages, which yields an ideal water-rich molar composition of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 (H2O).
:17 (H2O).
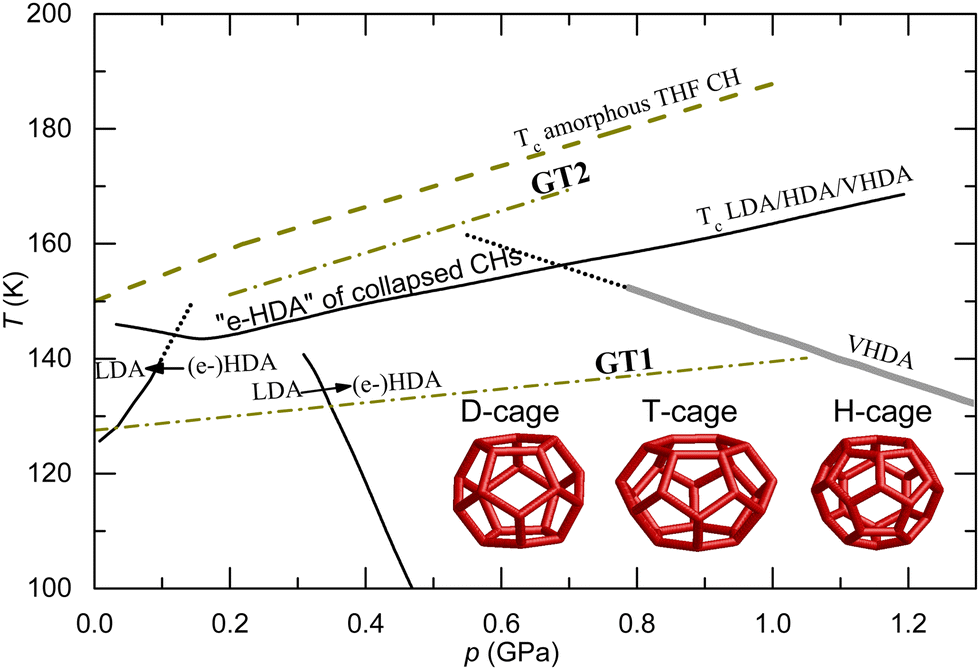 | ||
| Fig. 1 p, T-diagram for pressure amorphized type II clathrate hydrates with tetrahydrofuran or cyclobutanone as guest molecules. GT1 and GT2 lines show glass transition lines for an experimental time scale of ca. 1 s (dash-dot lines).22 The diagram also shows the LDA, HDA and VHDA transition lines of the amorphous states formed after PIA of pure ice;21,23 the (e-)HDA to LDA and (e-)HDA to VHDA lines appear to set a limiting pressure range for GT2 (observed only in the pressure range 0.2–0.7 GPa with diminishing glass transition features above ca. 0.6 GPa). GT1 is observed in both (e-)HDA/VHDA and collapsed CHs whereas GT2 occurs above the crystallization temperature Tc of (e-)HDA/VHDA. The inset shows the three types of cages in crystalline CHs; type II CH consists of 16 D-cages and 8 H-cages. | ||
It has been known for a long time that several different amorphous ice states can be formed via PIA, but the nanostructures of these states are still discussed, and the possibility that the states transform to liquids on heating is disputed. Several descriptions of HDA's nanostructure have been suggested: micro/nano-crystalline15 or amorphous with nano-sized crystalline remnants,16 non-homogeneous amorphous state,17 a “derailed” state along the ice I to ice IV pathway,18 or “an intermediate state in the phase transition from the connected H-bond water network in low pressure ices to the independent and interpenetrating H-bond network of high-pressure ices”,19 and fully amorphous and glass states. The last one is a special case of amorphous states, which is normally obtained by cooling or pressurizing a liquid into its ultraviscous state and further through a glass transition into a solid amorphous state. Originally, the PIA-process of ice was referred to as a pressure-induced melting process, i.e., the produced state would be a liquid or a glass dependent on its glass transition temperature. However, it was later noticed that the amorphization pressure well exceeded that of the (extrapolated) equilibrium melting line and results of simulations suggested that the process was instead due to (non-equilibrium) mechanical collapse;20 now this seems generally accepted and explains subtle structural differences between HDA states formed under different conditions. Recent studies suggest that collapse of hexagonal ice by PIA at low temperatures, e.g. at liquid nitrogen temperatures, produces a heterogeneous state denoted unannealed HDA (u-HDA).21 Heterogeneity is suggested by both neutron scattering results17 and, indirectly, by the subsequent transition behavior,16 which indicates that crystalline nucleation sites remain after the collapse and act as growth centers when the state is heated at low pressures. On heating at high pressures (or PIA at higher temperatures than 77 K) inhomogeneities gradually diminish17 concurrently as the state densifies; the ultimately densified state, which forms on heating to just below the crystallization temperature at pressures above ∼0.8 GPa,21 is referred to as very HDA (VHDA).16 If, instead, HDA (u-HDA or VHDA) is annealed at 130 K at pressures near 0.2 GPa it expands to a state termed expanded HDA (e-HDA), as shown in the temperature–pressure diagram (Fig. 1).21,22 Upon further depressurization of e-HDA, it transforms abruptly to low density amorphous ice (LDA).23
Several studies of CHs have shown remarkable similarities with the behavior of ice. PIA of type II CHs was suggested by Handa et al.6 who detected volume changes reminiscent of that for PIA of ice on isothermal pressurization at liquid nitrogen temperature. However, the sample recrystallized on subsequent pressure decrease so the state could not be recovered at ambient pressure for structural analysis and verification of an amorphous state. In a later study, Suzuki7 stabilized the high-pressure state by temperature cycling to 150 K at 1.5 GPa, before depressurization and recovery of the sample at ambient pressure and liquid nitrogen temperature; the X-ray pattern of the recovered state was amorphous-like. Recent, in situ, neutron scattering investigations have verified PIA in several CHs and also transition and structural similarities between amorphous ices and the water network of pressure-amorphized CHs;8,24 in particular, these studies suggest that the water structures of THF CH amorphs bear great similarities to the structures of HDA and VHDA ices.
In addition to the similarities between the PIA and densification processes of CHs and ice, both VHDA and the corresponding state of CH show one glass transition (GT1) at the same conditions, e.g. 1 GPa and 140 K on a time scale of 1 s. The heat capacity, thermal conductivity and dielectric characteristics of the transition are virtually identical, and because of its occurrence in both VHDA25 and amorphous CHs,11 the transition must be associated with the water network and not with the CH guest. This conclusion is also verified by a strong increase of the dielectric permittivity at the transition,11,12 which cannot be caused by an increased mobility of the relatively weakly polar guest.
A study of the transition behavior of stabilized amorphous CHs at lower pressures has revealed a second glass transition at pressure–temperature conditions slightly above the crystallization line of HDA ice,12 as depicted in the temperature–pressure diagram (Fig. 1).22 Both thermal conductivity and heat capacity results show two glass transitions, GT1 and GT2, on heating in the 0.2–0.7 GPa range. (GT1 is observed also outside this pressure range.) As mentioned, the permittivity increase at GT1 is large but it is thermally weak, with a heat capacity increase of only (3.7 ± 0.4) J (H2O-mol)−1 K−1 for amorphized THF CH and (3.9 ± 0.4) J (H2O-mol)−1 K−1 for amorphized 1,3-dioxolane CH at 1 GPa.11 This is the same as at the GT (GT1) of amorphized ice at 1 GPa; the heat capacity increase at Tg of VHDA, which is partly obscured by crystallization, is estimated to be (3.7 ± 0.4) J mol−1 K−1.25
The second glass transition observed in amorphized CHs (GT2), which occurs in a range slightly above the crystallization boundary of pressure amorphized ice (HDA/VHDA), shows a heat capacity increase of more than 5 times that of GT1 concurrently as the permittivity appears unaffected.12 The origin of this glass transition, unfreezing of guest or water mobility, or a combination, remains uncertain. Therefore, to investigate the origin of GT2, we have studied the four combinations of normal (H) and deuterated (D) samples of tetrahydrofuran clathrate hydrate: THF CH, TDF CH, THF CD and TDF CD to determine the deuterium-induced changes of GT2. Furthermore, we have made a detailed comparison between the GT2s of pressure-amorphized samples of THF CH, CB CH and DXL CH.
2. Experimental
Tetrahydrofuran THF (99.91% purity by gas chromatography GC, 0.001% water by Karl Fisher), two different batches of Tetrahydrofuran-d8 TDF (both with 99.6 atom% D by NMR, 100% purity by GC and, respectively, 98 ppm and 51 ppm water by Karl Fisher) and D2O (99.90 atom% D by NMR) were purchased from Sigma-Aldrich Chemicals. THF and TDF were mixed with D2O and H2O, Milli-Q® Ultrapure Water Systems, by weighting in THF/TDF concentrations of slightly more than the ideal 1:17 molar concentration to avoid ice in the crystallized samples. The exact concentrations, before loading in the sample cells, were: THF·16.5 H2O (THF CH); TDF·16.1 H2O (TDF CH), and THF·16.5 D2O (THF CD), and TDF·16.1 D2O (TDF CD). During loading of the samples, which takes less than 2 minutes until the sample cell is sealed, evaporation of THF or TDF decreases slightly their concentration. For example, based on measured evaporation rates, the THF concentration of the THF CH sample decreases to about THF·16.6 H2O. A cyclobutanone (99%, Sigma-Aldrich Chemicals) water solution of initial composition CB·16.4 H2O was formed by mixing with pure water (Milli-Q Ultrapure Water Systems), and a 1,3-dioxolane (99.99% by GC, Sigma-Aldrich Chemicals) water solution of initial composition DXL·16.4 H2O was formed by mixing with pure water (Ultrapur water, Merck).
The thermal conductivity κ and the heat capacity per unit volume c were measured using the hot-wire method under high pressure.26 The temperature was measured inside the sample cell by a calibrated Chromel–Alumel thermocouple with an estimated inaccuracy of ±0.5 K. The cell was filled with one of the solutions, sealed with a tightly fitting Teflon lid and mounted into a piston cylinder of 45 mm internal diameter. The whole pressure cylinder device was thereafter transferred to a hydraulic press, which supplied the load, and placed inside a vacuum chamber with a built-in closed helium cycle cryostat equipped with heater.27 Pressure was determined from the ratio of load to piston area, and it was corrected for friction. This correction was determined on increasing pressure in a separate, in situ, experiment using the pressure dependence of the resistance of a manganin wire. The absolute uncertainty in pressure is estimated as ±0.05 GPa at 1 GPa. However, in these studies, it is the imprecision in pressure which is important for the evaluation of the data and it is significantly smaller than the uncertainty; the pressure differs only when the friction differs between the measurement setups. A study of the total friction force by cycling an elastically changing solid (DL-camphor) shows that it corresponds to about 0.04 GPa at 0.4 GPa, or a change in Tg of 1.3 K for GT2 (Fig. 1), and the difference in friction between the runs is only a fraction of the total friction.
Values for κ and c were obtained simultaneously from results of the Ni-wire temperature rise during a 1.4 s heat pulse of about constant power, which raised the temperature of the Ni-wire by about 3.5 K. During the pulse, its resistance was measured versus time, which enabled the temperature rise of the wire to be determined from the known (calibrated) relation between its resistance and temperature. (Because of the low thermal diffusivity of CHs, the heat wave reflected against the Teflon cell wall does not affect the temperature rise of the wire within the short measurement time of 1.4 s.) The analytical solution for the temperature rise was fitted to the data points, thereby yielding κ and c with estimated uncertainties of ±2% and ±5%, respectively.
At low temperatures and in the glass transition range, heating and cooling rates were typically 0.3 K min−1, and the samples were pressurized and depressurized at rates in the 0.15–0.2 GPa h−1 range. Typically, a single heating run to study the glass transition of one sample requires more than 4 days of preparation due to the slow rates and the need to stabilize the amorphized states.
The hot-wire method is a well-established method to study glass transitions. The quantity c(T) shows the typical sigmoid-shaped increase of the specific heat capacity and κ(T) often shows a change (decrease) in the slope (dκ/dT) at Tg on heating. Moreover, due to the transient nature of the method, both κ and c show method-specific features at a thermally pronounced glass transition. These features are known to be consequences of time-dependence in the heat capacity.28,29 It causes a peak in κ and dip in c because κ and c are treated as adjustable time-independent parameters in the fitting of the analytical solution for the temperature rise of the hot-wire (see ESI†).
3. Results
The CHs were formed by freezing the solutions in Teflon sample cells kept at 0.1 MPa, and temperature cycling the samples for more than 3 h at temperatures slightly below the freezing point of 270.5 K for DXL CH,30 273.2 K for CB CH,31 277.4 K for THF CH,32 and the corresponding, slightly higher, freezing temperatures for the deuterated samples.33 The samples were thereafter cooled at a rate of less than 0.6 K min−1 down to 100 K. All samples were fully amorphized by pressurization to a pressure in the range 1–1.3 GPa at a temperature near 130 K and subsequent treatment by heating to 170 K at a pressure near 1 GPa. The latter treatment is required to stabilize the amorphous CH and avoid recrystallization on depressurization. The amorphized CHs were cooled from about 170 K to near 130 K and depressurized from 1 GPa to 0.1 GPa at a temperature in the 129–134 K range. In the 0.1–0.2 GPa range, which here is the conditions of the most significant volume relaxation, samples were kept at (131 ± 1) K and thereafter isothermally repressurized to 0.4 GPa to ensure identical thermal histories of the amorphized states. (In a comparative study between CB and THF CHs samples were relaxed at 145 K and 0.07 GPa.)Fig. 2 shows results for κ on depressurization and repressurization of amorphized CHs. Generally, κ of solid amorphous (and crystalline) states decreases weakly and reversibly on depressurization and increases weakly and reversibly on pressurization following the changes in sample density as the sample elastically expands and contracts. On depressurization, the results show an accelerated decrease of κ in the 0.1 to 0.4 GPa. (For one of the samples, THF CH the depressurization was temporarily halted and the sample was temperature cycled overnight, 132–100–132 K at 0.3 GPa, before continued depressurization.) On repressurization to 0.4 GPa, κ values of all samples deviate significantly from the results measured on pressure decrease. This shows that the samples expand partly non-elastically during depressurization from 1 GPa.34
 | ||
| Fig. 2 Thermal conductivity of amorphized CHs measured on depressurization and subsequent repressurization at ca. 130 K, as indicated by the arrows: THF CH (red open circles) (temperature cycled at 0.3 GPa, see text), TDF CH (black open squares), THF CD (green filled circles) and TDF CD (blue filled squares). The dashed line shows previously unpublished results for κ of VHDA (formed by PIA of pure ice) during depressurization at 130 K; the values for κ are scaled by a factor of 0.8. VHDA and u-HDA are known to transform gradually into expanded HDA on heating at low pressure, e.g. by heating u-HDA to 125 K in the 0.1–0.2 GPa range;21 here the gradual transformation occurs on isothermal depressurization at 130 K. | ||
Fig. 3 shows the results for κ measured on heating at 0.3 K min−1 rate at 0.40 GPa after the samples had first been cooled to 100 K. All CHs show weakly positive, or “glass-like”, temperature dependence of κ from 100 K to about 155 K. This is the normal behavior of κ of amorphous solids. However, on further heating, κ of all samples shows a pronounced (artificial) peak (see ESI† and ref. 29). As discussed in detail previously, this is due to a thermally strong glass transition, i.e., it is associated with a large, time-dependent, increase in the heat capacity.12 Moreover, at a temperature above the peak, κ of all samples changes abruptly and discontinuously simultaneously as an abrupt change in sample temperature, which shows that the samples crystallized exothermically; THF CH crystallized at 166.0 K, TDF CH at 165.4 K, THF CD at 167.7 K, and TDF CD at 167.4 K.
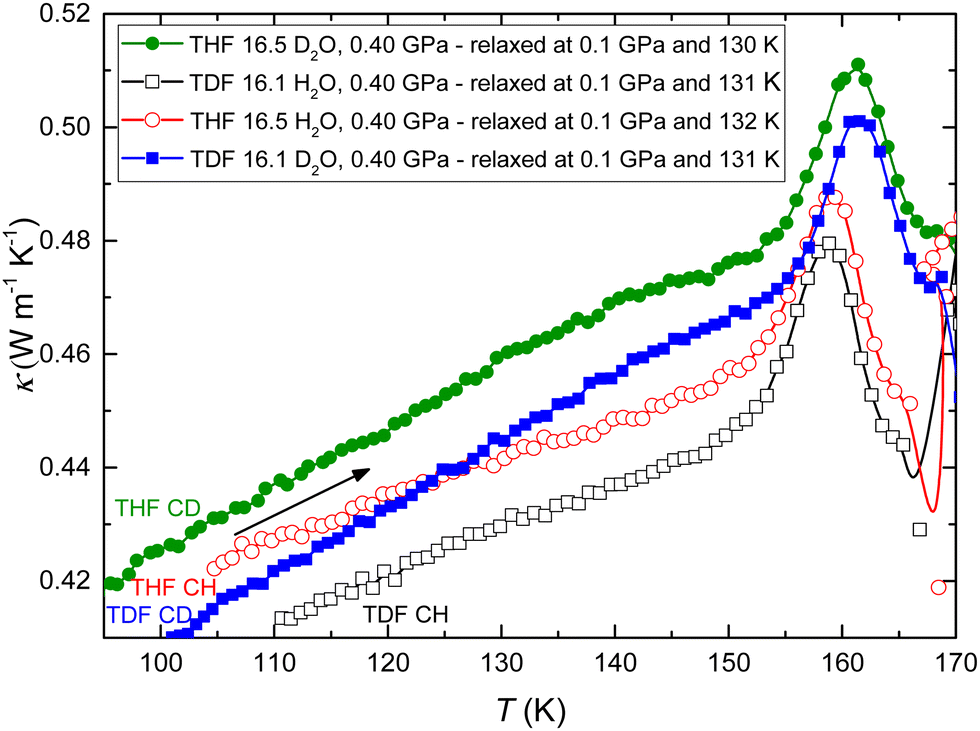 | ||
| Fig. 3 Thermal conductivity of amorphized CHs measured on heating at 0.40 GPa: THF CH (red open circles), TDF CH (black open squares), THF CD (green filled circles) and TDF CD (blue filled squares). | ||
Fig. 4 shows the corresponding changes in c on heating at 0.4 GPa. These results for the excess heat capacity per unit volume Δc were calculated from the measured value for c by subtracting a linear function fitted to c in the temperature range up to 130 K. The results reveal a weak change in dc/dT due to a glass transition, GT1, at temperatures slightly above 130 K.12 THF CH has previously been studied by dielectric spectroscopy and the results show that the weak change in c is associated with a large increase in the dielectric constant; the high temperature state has a relative static permittivity of about 100 at 1 GPa.12 Thus GT1 is thermally weak but the concurrent increase in dielectric permittivity is large. The latter shows that GT1 is associated with the water network and not the relatively weakly polar guest THF (or TDF). On further heating, c shows a large and abrupt increase due to GT2; the increase is shifted to slightly higher temperatures for the D2O samples. The dip in c prior to the increase is artificial and occurs due to the time-dependence in c in the glass transition range, which also causes the artificial peak in κ. Apparently, the previously established glass transition in THF CH, GT1 and GT2, occur in all samples, but the glass transition temperatures are slightly different for the different samples. The heat capacity increase at GT2, based on the two H2O-samples, is (20.9 ± 1.0) J (H2O-mol)−1 K−1, i.e., about 5.5 times larger than that for GT1 at 1 GPa; c of the kinetically unfrozen state at GT2 is ∼1.7 times that of its glassy state.35 As discussed below, this transition shows all the typical characteristics of a glass-liquid transition and we will argue that it is associated with the water network.
 | ||
| Fig. 4 Excess heat capacity per unit volume Δc as a function of temperature on heating at 0.40 GPa: THF CH (solid red line), TDF CH (dash-dot black line), THF CD (short-dashed green line) and TDF CD (dashed blue line). | ||
To investigate if GT2 is affected by the type of guest molecule, we make a detailed comparison of the results for three cases with different guest molecules: THF, CB, and DXL. All show GT2, and those of pressure-amorphized CB and THF CHs are compared in Fig. 5. The temperatures of the peak maxima κ differ only about 0.5 K. The corresponding results for THF and DXL CHs are shown in Fig. 6. Again the glass transition features of GT2 show excellent agreement between the two amorphs; the peak temperature in κ is the same to within 0.2 K.
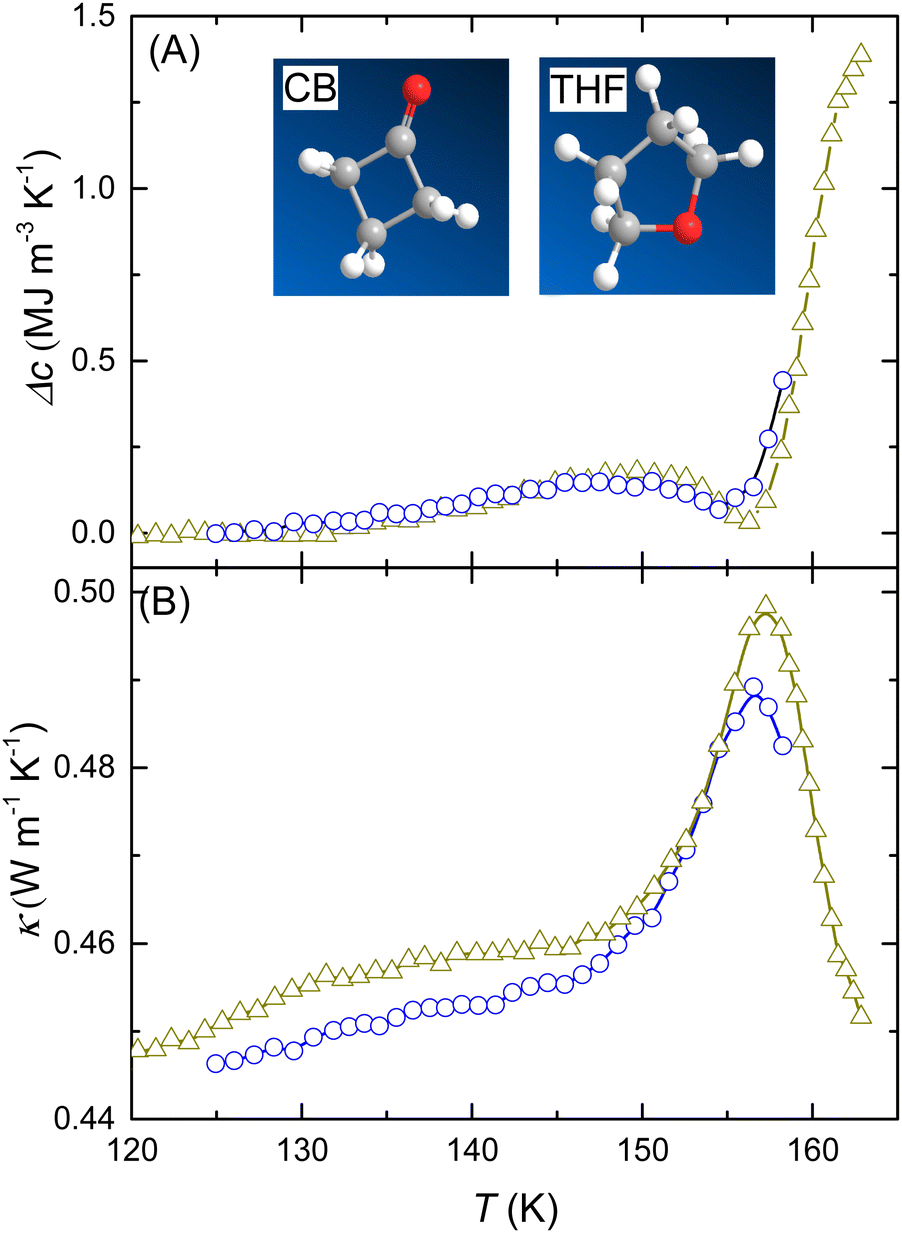 | ||
| Fig. 5 (A) Excess heat capacity per unit volume and (B) thermal conductivity as a function of temperature on heating at 0.40 GPa: THF CH (blue circles) and CB CH (dark yellow triangles). Both pressure amorphized samples had been relaxed at about 145 K and 0.07 GPa prior to cooling and reheating at 0.4 GPa. (The THF CH sample was heated only to 159 K.) | ||
 | ||
| Fig. 6 (A) Excess heat capacity per unit volume and (B) thermal conductivity as a function of temperature on heating at 0.40 GPa: THF CH (red circles) and DXL CH (magenta stars). Both pressure amorphized samples had been relaxed at about 131 K and 0.1 GPa prior to cooling and reheating at 0.4 GPa. (DXL CH crystallized at 162 K; data above the crystallization temperature have been removed.) | ||
4. Discussion
An important purpose of this study was to determine the origin of GT2. As shown in Fig. 5 and 6, GT2 is essentially unaffected by change of guest molecule from THF to CB or DXL. The glass transition temperature of substances typically scales with the melting temperature Tm; empirically it has been found to occur at about 2/3 of Tm. The melting temperatures of THF (165 K), CB (222 K) and DXL (178 K) differ up to 60 K. Moreover, the van der Waals radius of THF is 2.95 Å, whereas that of DXL is about 2.8 Å, which is at the bottom end of the size range for type II CH-formers; the radius of CB, 3.25 Å, is near the top end of those that fit into the H-cage size (4.73 Å at 1 atm). These significant differences suggest that a glass transition, associated with kinetic unfreezing of translational and/or reorientational motions of the guest molecules, should occur at different temperatures. Moreover, the data indicate that the heat capacity rise at Tg is about the same, which again seems unlikely if Tg is due to kinetic unfreezing of guest motions. Consequently, our results suggest that GT2 is unrelated with the guest molecule and, thus, mainly related to the water network. Still, the presence of the guest may modify the GT properties. In order to study this in more detail, we focus on the changes in GT2 of THF CH samples caused by deuteration. We start by discussing the state of the samples in which GT2 occurs.4.1 State of the amorphized clathrate hydrates showing glass transformations; an expanded HDA type of state
In a previous study of THF CH and CB CH, we have established that GT2 could only be detected in the 0.2–0.7 GPa range,12,22 whereas GT1 can be observed also outside this pressure range. On heating below 0.2 GPa, GT2 was not observed; instead the sample crystallized at temperatures near the expected Tg2. Moreover, on isothermal depressurization to near atmospheric pressure at 130 K, κ shows glimpses of an HDA to low density amorphous (LDA) type of transition in THF CH.36 This finding may be due to water enriched sample domains, i.e., domains with a higher water content than the nominal one of the type II CH (THF·17 H2O), caused by sluggish phase separation on heating to above 170 K at high pressure. Here we have therefore avoided heating the samples above 170 K at 1 GPa and also kept the pressure well above the transformation to an LDA type of state at 130 K.Fig. 2 shows that κ of the amorphized CHs decreases significantly in the 0.1–0.4 GPa range on depressurization. The decrease in κ indicates a substantial decrease of density. Moreover, the difference in κ on decrease of pressure and subsequent increase of pressure suggests that the sample expands partly non-elastically on depressurization. One of the samples, THF CH, was temperature cycled overnight between 132 K and 100 K at 0.3 GPa, before continuing the depressurization. Due to the large thermal mass of the vessel, which entails slow initial cooling rate, and the need for stabilization of the temperature on reheating, the sample was kept in the 132–130 K range for 1.3 h at 0.3 GPa. The difference in κ between before and after temperature cycling therefore implies a slow expansion of the sample with time. These characteristics resemble the behavior of amorphous ice produced under similar conditions and thereafter pressure cycled at 130 K, as shown in Fig. 2. The tendency to expand at low pressures seems even more pronounced in the amorphized CHs. Considering the previous results for amorphous ices, the behavior is similar as that at the transformation from VHDA to expanded HDA (e-HDA), which is likely a time- and temperature-dependent process.21 To ensure that the states of the samples were identical upon subsequent heating at 0.4 GPa, we have therefore followed the same pressure–temperature paths for all samples and obtained, as it seems, an e-HDA type of state in the amorphized CHs.
The e-HDA state of ice transforms distinctly to LDA on isothermal depressurization and gradually to VHDA on pressurization, which provides an explanation for the unusual feature of GT2 being observable only in the 0.2–0.7 GPa range. Nelmes et al. provided a tentative p–T stability diagram (Fig. 1 in ref. 21), which suggests the transformation of e-HDA to LDA and VHDA on isobaric heating would occur below ca. 0.1 GPa and above ca. 0.7 GPa, respectively (Fig. 1). Since the amorphized CHs remain stable to slightly higher temperatures than (pure) e-HDA, we deduce that the lower limit of detecting GT2 (∼0.2 GPa) coincides well with a stability limit for the e-HDA type of state towards an LDA type of state below the crystallization pressure–temperature line. That is, on isobaric heating of the amorphized CHs below 0.2 GPa, it would transform to an LDA type before crystallization on further heating. Although the data for κ did not show features of such transition, it provides an explanation for the abrupt vanishing of GT2 below 0.2 GPa. Moreover, the gradual vanishing of GT2 above ca. 0.6 GPa agrees well with a transformation to a VHDA type of state prior to crystallization. Thus, the pressure range of GT2 (0.2–0.7 GPa) is given a natural explanation by the transition behavior of amorphous ices and, in particular, the stability range of e-HDA. This behavior indicates that GT2 is associated with the water network.
4.2 Effect of deuteration on glass transition 2
In Fig. 3 and 4, it is obvious that Tg2 of the CD-samples is shifted to higher temperatures. Because of the difference in concentrations between the two CH (and the two CD) samples, i.e., the THF:H2O molar ratio of 1:16.5 and the TDF:H2O molar ratio of 1:16.1, we can exclude that the shift in Tg between CH and CD samples is due to slightly different compositions. To quantify the shift, we have superimposed all κ peaks on that for THF CH, by a shift in temperature and κ. The results are depicted in Fig. 7 and show that Tg2 is, within the experimental inaccuracy, unaffected by deuteration of the THF guest, whereas deuteration of H2O and a fully deuterated sample show, respectively, 2.3 K and 2.7 K higher Tg2 than THF CH. (The corresponding temperature shifts obtained by using the temperature at 50% of the total heat capacity increase at Tg2 in Fig. 4 are 2.2 K and 2.6 K.) These results further support that GT2 is associated mainly with the water network. Another weaker indication of this is the difference in the shapes of the κ peaks. The shape of the artificial peak is determined by the activation energy and the distribution of relaxation times;29 the latter is often described by a stretched exponential response function: ϕ(t) = exp[−(t/τ)β], where τ is the relaxation time and β is a stretching parameter with a value in the 0–1 range. As shown in Fig. 7, the shape of the peak changes mainly on deuteration of water. It is also known that the peak maximum in κ, which is slightly dependent on β, occurs near τ = 0.3 s; this provides a useful time-scale of the GT (ESI†).12,29 Furthermore, the relaxation time near the low-temperature onset of the peak is τ = 102 s, i.e., τ typically associated with the glass transition temperature obtained in differential scanning calorimetry.12 Calculations show that the results in this narrow temperature range can be described with an Arrhenius relaxation function: τ = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) exp[Ea/(RT)], where A is a constant, Ea is the activation energy and R is the gas constant (ESI†). The best description of the data in the narrow temperature range 150–165 K was found with Ea = (230 ± 10) kJ mol−1 and (240 ± 10) kJ mol−1 for amorphous CH and CD samples, respectively, with β = 0.42 and 0.39. Based on the effect of deuteration of water, we associate these values with kinetic unfreezing of mainly the water network on heating.
exp[Ea/(RT)], where A is a constant, Ea is the activation energy and R is the gas constant (ESI†). The best description of the data in the narrow temperature range 150–165 K was found with Ea = (230 ± 10) kJ mol−1 and (240 ± 10) kJ mol−1 for amorphous CH and CD samples, respectively, with β = 0.42 and 0.39. Based on the effect of deuteration of water, we associate these values with kinetic unfreezing of mainly the water network on heating.
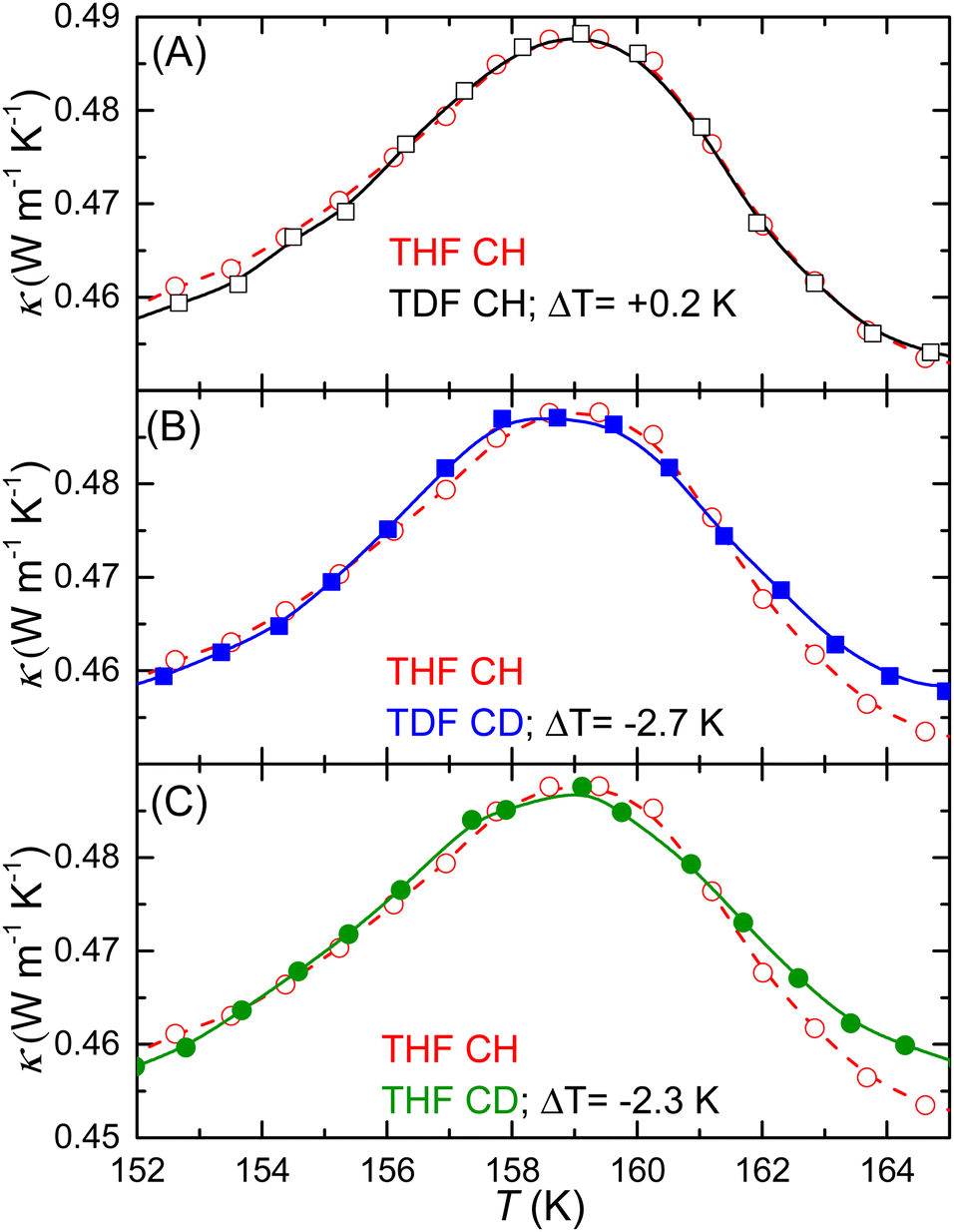 | ||
| Fig. 7 Superposition of the κ peaks to determine the temperature shift of GT2. The results have been shifted manually in T and κ for best fit with the results of THF CH (red circles and dashed lines). (A) TDF CH, (B) TDF CD, and (C) THF CD. | ||
We conclude that the artificial features in κ and c, and the large increase in c at GT2 are typical for glass to liquid transitions. Although a special case of orientationally disordered glasses, plastic crystal phases,37,38e.g. cyclohexanol and cyclooctanol,39,40 show similar characteristics at their orientational glass transition, it seems unlikely that kinetic unfreezing of only reorientational motions of the guest molecules can provide such large increase in c and occur at identical temperatures. In particular, if one considers the low molar content of the guests and the difference in van der Waals radius between guests (15%). In summary, our results of deuterated samples and results after exchange of guest molecules from THF to CB or DXL as well as the pressure range of GT2 provide substantial evidence that GT2 is associated with the water network, and due to a glass to liquid transition. Although we have previously established that GT1's properties: Tg, Ea and the heat capacity rise are unaffected by the presence of the guests,11 one cannot be certain that this is also the case for GT2. However, because of the relatively weak hydrogen bond between, e.g. THF and water in solutions,41 and the invariance of the GT properties with exchange or deuteration of guests (CB, THF and DXL), we argue that the guests mainly affect the crystallization temperature by hampering growth of ice nuclei due to their regular distribution in the water network.
4.3 Effect of deuteration on glass transition 1
We have used the increase in c to quantify the deuteration-induced change in Tg1. In particular, we used linear fits in the 135–145 K range and calculated the temperature shifts of the functions relative to the function for THF CH as: −0.6 K (at 140 K) for TDF CH, +1.3 K for TDF CD and +1.8 K for THF CD. Assuming that the small changes on deuteration of the guest is simply due to the scatter of the data, the results suggest a shift of Tg1 of about 1.8 K upon deuteration of water. These results corroborate the already compelling evidence from heat capacity and dielectric results that GT1 is associated with the water network.11 The lack of, or only subtle, artificial features in c and κ at GT1, is due to the weak and gradual change in c, as shown quantitatively in the ESI.†The glass transition properties of GT1 are reminiscent of those for proton disordered crystalline ices such as ice Ih and ice V; Tg of ice Ih is about 110 K at 1 atm for a time scale of 103 s42 and that of ice V is about 130 K for a heating rate of 30 K min−1 at 1 atm.43 At both these GTs, the heat capacity rise is weak, e.g. 1.7 J mol−1 K−1 for ice V,43 and the activation energy is low, 22 kJ mol−1 for ice Ih42 and 35 kJ mol−1 for ice V;43 the values at GT1 are up to a factor of two larger, but in comparison to typical values at glass-liquid transitions, these are still relatively small (Ea = 45 kJ mol−1 and the heat capacity rise is 3.7 J mol−1 K−1).11,44 Moreover, the amorphous versus crystalline structure causes a significant change in the proton mobility due to the ice rules.45 These limit the mobility in crystalline ices, but the lack of long-range order and presence of interstitial molecules in HDA and VHDA46 appear to relax the effect of the rules and speed up the proton mobility,44 which affects the glass transition properties. Thus, the similar characteristics suggest that GT1 is the amorphous counterpart of the GTs in crystalline ices, which are associated with kinetic unfreezing of reorientational motions of H2O, or proton mobility.
5. Conclusions
The amorphous, water-rich, solid solutions studied here are formed by pressure collapse of crystals and therefore states in which moderately polar molecules and water are perfectly homogeneously mixed. Despite this, all pressure-amorphized normal and deuterated type II THF CHs, CB CH and DXL CH show two glass transitions on heating in the 0.2–0.7 GPa range. The first, low-temperature, glass transition shows a weak and gradual heat capacity increase and a glass transition temperature similar as that of high density amorphous ice. A concurrent strong increase of the permittivity shows that it is due to the water network and not the THF/TDF, CB or DXL guest. The second glass transition is associated with a large heat capacity increase and it shifts ∼2.5 K to higher temperature upon deuteration of water, whereas it remains unaffected on deuteration of the THF guest and exchange of THF with CB or DXL. This implies that both glass transitions are associated with water; one possibility is an orientational or proton glass transition followed by a glass liquid transition of the water–THF solution. The latter is detected only in the 0.2–0.7 GPa range—the stability limit of expanded high density amorphous ice—which corroborates association with water. Moreover, structural studies have shown that the water network in pressure-amorphized (water-rich) type II CHs bear close similarities to those of the amorphous ices.8,24 It is therefore conceivable that expanded high density amorphous ice undergoes a glass to liquid transition at a temperature near that of glass transition 2 of amorphized CHs, which occurs close to the pressure–temperature crystallization line of expanded HDA. Firm proof of the rare transition sequence, i.e., an orientational glass transition followed by a glass to liquid transition on heating of amorphous ice, may require methods for glass transition detection that allows for rapid heating or, else, methods that probe time-scales longer than ca. 103 s to circumvent crystallization at temperatures prior to detection of the glass to liquid transition. It can also be rewarding to study additional properties of collapsed clathrate hydrates such as density and dynamics to further investigate the second glass transition, which exhibits all typical thermal characteristics of a glass–liquid transition. Confirmation of an orientational glass transition followed by a glass-liquid transition can explain divergent results reported for water's (glass transition) properties. Although water vitrified by rapid cooling at atmospheric pressure is structurally different from high density amorphous ice, it may still show the same glass transition behavior, which implies that low-temperature studies probe kinetic unfreezing of reorientational motions whereas high temperature studies observe liquid properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from Magnus Bergvalls foundation, Stiftelsen Olle Engkvist Byggmästare, Carl Trygger Foundation, and the Swedish Foundation for Strategic Research (SSF) within the Swedish national graduate school in neutron scattering (SwedNess).Notes and references
- M. D. Ediger and P. Harrowell, J. Chem. Phys., 2012, 137, 080901 CrossRef CAS.
- P. Richet and P. Gillet, Eur. J. Mineral., 1997, 9, 907–933 CrossRef CAS.
- O. Mishima, L. D. Calvert and E. Whalley, Nature, 1984, 310, 393–395 CrossRef CAS.
- D. W. Davidson, in Water, A comprehensive treatise, ed. F. Franks, Plenum Press, New York, 1973, p. 115 Search PubMed.
- E. D. Sloan, Clathrate Hydrates of Natural Gas, Dekker, New York, 2nd edn, 1998 Search PubMed.
- Y. P. Handa, J. S. Tse, D. D. Klug and E. Whalley, J. Chem. Phys., 1991, 94, 623–627 CrossRef CAS.
- Y. Suzuki, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 1–4 Search PubMed.
- P. H. B. Brant Carvalho, A. Mace, C. L. Bull, N. P. Funnell, O. Andersson and U. Häussermann, J. Chem. Phys., 2019, 150, 204506 CrossRef PubMed.
- C. A. Tulk, D. D. Klug, J. J. Molaison, A. M. Dos Santos and N. Pradhan, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 1–8 CrossRef.
- R. G. Ross and P. Andersson, Can. J. Chem., 1982, 60, 881–892 CrossRef CAS.
- O. Andersson and A. Inaba, J. Phys. Chem. Lett., 2012, 3, 1951–1955 CrossRef CAS.
- O. Andersson and U. Häussermann, J. Phys. Chem. B, 2018, 122, 4376–4384 CrossRef CAS PubMed.
- S. R. Gough and D. W. Davidson, Can. J. Chem., 1971, 49, 2691–2699 CrossRef CAS.
- M. Zakrzewski, D. D. Klug and J. A. Ripmeester, J. Incl. Phenom. Mol. Recognit. Chem., 1994, 17, 237–247 CrossRef CAS.
- G. P. Johari, Phys. Chem. Chem. Phys., 2000, 2, 1567–1577 RSC.
- J. Stern and T. Loerting, Sci. Rep., 2017, 7, 1–10 CrossRef CAS.
- M. M. Koza, T. Hansen, R. P. May and H. Schober, J. Non Cryst. Solids, 2006, 352, 4988–4993 CrossRef CAS.
- J. J. Shephard, S. Ling, G. C. Sosso, A. Michaelides, B. Slater and C. G. Salzmann, J. Phys. Chem. Lett., 2017, 8, 1645–1650 CrossRef CAS PubMed.
- C. Lin, X. Yong, J. S. Tse, J. S. Smith, S. V. Sinogeikin, C. Kenney-Benson and G. Shen, Phys. Rev. Lett., 2017, 119, 1–6 CAS.
- J. S. Tse, J. Chem. Phys., 1992, 96, 5482–5487 CrossRef CAS.
- R. J. Nelmes, J. S. Loveday, T. Strässle, C. L. Bull, M. Guthrie, G. Hamel and S. Klotz, Nat. Phys., 2006, 2, 414–418 Search PubMed.
- O. Andersson, P. H. B. Brant Carvalho, Y.-J. Hsu and U. Häussermann, Chem. Phys., 2019, 151, 014502 Search PubMed.
- O. Mishima, J. Chem. Phys., 1994, 100, 5910–5912 CrossRef CAS.
- P. H. B. Brant Carvalho, P. I. R. Moraes, A. A. Leitão, O. Andersson, C. A. Tulk, J. Molaison, A. P. Lyubartsev and U. Häussermann, RSC Adv., 2021, 11, 30744–30754 RSC.
- O. Andersson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11013–11016 CrossRef CAS PubMed.
- B. Håkansson, P. Andersson and G. Bäckström, Rev. Sci. Instrum., 1988, 59, 2269–2275 CrossRef.
- O. Andersson, B. Sundqvist and G. Bäckström, High Press. Res., 1992, 10, 599–606 CrossRef.
- N. O. Birge and S. R. Nagel, Phys. Rev. Lett., 1985, 54, 2674–2677 CrossRef CAS PubMed.
- O. Andersson, Int. J. Thermophys., 1997, 18, 195–208 CrossRef CAS.
- T. Yonekura, O. Yamamuro, T. Matsuo and H. Suga, Thermochim. Acta, 1995, 266, 65–77 CrossRef CAS.
- B. Morris and D. W. Davidson, Can. J. Chem., 1971, 49, 1243–1251 CrossRef CAS.
- O. Yamamuro, M. Oguni, T. Matsuo and H. Suga, J. Phys. Chem. Solids, 1988, 49, 425–434 CrossRef CAS.
- C. Y. Jones, J. S. Zhang and J. W. Lee, J. Thermodyn., 2010, 1–6 Search PubMed.
- Due to reversal of friction, results on pressurization generally differ from results on depressurization, but the difference observed here appears to be outside the range that can be caused by friction. A test on an elastically changing solid, DL-camphor, showed a maximum pressure hysteresis of 0.15 GPa at 130 K.
- Here we have used the estimate that the pressure amorphized CH has 4% larger density than HDA,12 combined with the measured density of expanded HDA of 1.18 g cm−3 at 0.4 GPa and 130–140 K.23.
- O. Andersson and Y. Nakazawa, J. Phys. Chem. B, 2015, 119, 3846–3853 CrossRef CAS PubMed.
- J. N. Sherwood, The Plastically Crystalline State, Wiley, Chichester, 1979 Search PubMed.
- N. G. Parsonage and L. A. K. Staveley, Disorder in Crystals, Oxford, Clarendon, 1978 Search PubMed.
- O. Andersson, R. G. Ross and G. Bäckström, Mol. Phys., 1989, 66, 619–635 CrossRef CAS.
- O. Andersson and R. G. Ross, Mol. Phys., 1990, 71, 523–539 CrossRef CAS.
- M. J. Shultz and T. H. Vu, J. Phys. Chem. B, 2015, 119, 9167–9172 CrossRef CAS PubMed.
- O. Yamamuro, M. Oguni, T. Matsuo and H. Suga, J. Phys. Chem. Solids, 1987, 48, 935–942 CrossRef CAS.
- C. G. Salzmann, I. Kohl, T. Loerting, E. Mayer and A. Hallbrucker, Phys. Chem. Chem. Phys., 2003, 5, 3507–3517 RSC.
- O. Andersson and A. Inaba, Phys. Rev B., 2006, 74, 184201 CrossRef.
- J. D. Bernal and R. H. Fowler, J. Chem. Phys., 1933, 1, 515–548 CrossRef CAS.
- J. L. Finney, D. T. Bowron, A. K. Soper, T. Loerting, E. Mayer and A. Hallbrucker, Phys. Rev. Lett., 2002, 89, 205503 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01993k |
| ‡ Present address: Asian School of the Environment, Nanyang Technological University, Singapore. |
| This journal is © the Owner Societies 2022 |