
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Infrared spectra and structures of protonated amantadine isomers: detection of ammonium and open-cage iminium ions†
Martin Andreas Robert
George
 and
Otto
Dopfer
*
and
Otto
Dopfer
*
Institut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, 10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de
First published on 13th June 2022
Abstract
The protonated form of amantadine (1-C10H15NH2, Ama), the amino derivative of adamantane (C10H16, Ada), is a wide-spread antiviral and anti-Parkinsonian drug and plays a key role in many pharmaceutical processes. Recent studies reveal that the adamantyl cage (C10H15) of Ama can open upon ionization leading to distonic bicyclic iminium isomers, in addition to the canonical nascent Ama+ isomer. Herein, we study protonation of Ama using infrared photodissociation spectroscopy (IRPD) of Ar-tagged ions and density functional theory calculations to characterize cage and open-cage isomers of AmaH+ and the influence of the electron-donating NH2 group on the cage-opening reaction potential. In addition to the canonical ammonium isomer (AmaH+(I)) with an intact adamantyl cage, we identify at least one slightly less stable protonated bicyclic iminium ion (AmaH+(II)). While the ammonium ion is generated by protonation of the basic NH2 group, AmaH+(II) is formally formed by H addition to a distonic bicyclic iminium ion produced upon ionization of Ama and subsequent cage opening.
Introduction
Amantadine (Ama, 1-tricyclo[3.3.1.13,7]decylamine, 1-adamantylamine, 1-aminoadamantane, C16H15NH2) is the amino derivative of adamantane (C10H16, Ada), the parent molecule of the group of diamondoids.1 These nm-sized H-passivated nanodiamonds are rigid, stress-free, and highly stable cycloalkanes and form a fundamental class of saturated sp3-hybridized hydrocarbon molecules,2–4 with (potential) applications in a wide range of disciplines including materials science, molecular electronics, medical sciences, chemical synthesis, and astrochemistry.5–17 For example, radical cations of diamondoids are predicted to be intermediates in substitution reactions in polar solvents.14,18–21 Based on the detection of several amines22 and protonated molecules22–24 in the interstellar medium and the likely presence of diamondoids,22 the occurrence of Ama and its protonated form is also expected.13,25,26Amantadine, particularly in its N-protonated form, belongs to the most important diamondoid derivatives, because of its use in pharmaceutical applications as antiviral and anti-Parkinsonian drug. It is successfully marketed under several brand names, including Symmetrel® (Ama·HCl), Gocovri®, Symadine®, and Osmolex ER®.15,27–30 In all postulated mechanisms, N-protonated Ama (or its dimethylated derivative memantine, Namenda®) is the biochemically active species.31,32 Ama is effective in the prophylaxis and treatment of influenza A infections by preventing the virus from entering the host cell through blocking of the ion channel.33–35 However, due to drug resistance, its use is no longer recommended for influenza.36,37 Although the mechanism of action against Parkinsonism is not yet fully understood, the drug enhances the release of dopamine from nerve endings of brain cells, along with stimulating the norepinephrine response.38–40 In addition to its pharmaceutical use, Ama has antagonistic effects on NMDA (N-methyl-D-aspartate) receptors41,42 and is discussed for treating other diseases such as multiple sclerosis, depression, and cocaine addiction.43–45 In 2013, memantine was in the top 100 best-selling drugs worldwide, with sales exceeding $109.46
In addition to its pharmaceutical use, the diamondoid derivative Ama is also of interest for studying ionization-induced rearrangement processes of the adamantyl cage.47,48 By replacing H with the electron-donating NH2 group at the apical 1-position,42,49 the chemical properties of diamond-like structures are mostly retained but the barrier to cage opening is strongly reduced.47,48 The geometric, vibrational, and electronic properties of Ama are well-known from infrared (IR), Raman, and electron momentum spectroscopy.50–54 Low-resolution electron momentum spectra show that ionization of Ama to the electronic ground state of the cation occurs by electron ejection from the nonbonding N lone pair of the NH2 group (HOMO), with a rough estimate of the vertical ionization energy of 8.6 eV.53 In our recent IR spectroscopic and quantum chemical studies of Ama+Ln clusters (with L = Ar, N2, H2O),47,48,55 we identified, in addition to the canonical nascent Ama+(I) isomer with an intact C10H15 cage, two distonic bicyclic iminium isomers Ama+(II) and Ama+(III) in which the adamantyl cage opens upon ionization. H → NH2 substitution drastically lowers all barriers for the rearrangement processes, because of the strongly p/π-electron donating character of the NH2 group, which leads to high and thus detectable yields for the formation of Ama+(II) and Ama+(III). Starting from the cage structure Ama+(I) produced by vertical ionization of Ama (E0 = 46 kJ mol−1), C–C bond activation opens the adamantyl cage forming the primary radical Ama+(II) (E0 = 87 kJ mol−1) via a boat → chair conversion of one of the cyclohexane rings. In a subsequent 1,2 H-shift accompanied by chair → boat back conversion, the much more stable tertiary radical Ama+(III) is generated (E0 = 0 kJ mol−1). Interestingly, no open-cage isomers were detected in previous IR and optical spectra of Ada+ and its clusters generated in the same type of ion source due to ionization from a different orbital and higher reaction barriers.26,56,57 Instead of ionization of Ama, we characterize herein the effects of protonation using the same IR spectroscopic and quantum chemical tools to determine the structure of protonated amantadine, the impact of the electron-withdrawing NH3+ group on the adamantyl cage of AmaH+, and the possible formation of open-cage isomers. Experimentally, we apply IR photodissociation (IRPD) spectroscopy to mass-selected Ar-tagged AmaH+ ions, whereby the weakly-bonded Ar ligand has essentially no impact on the structural, energetic, vibrational, and chemical properties of AmaH+, as recently demonstrated for the Ama+ radical cation.47,48 Despite its high pharmaceutical importance, no spectroscopic studies are available for isolated AmaH+. Mass spectrometry experiments provide information about its fragmentation and proton affinity.58 Several spectrophotometric, NMR and IR studies of the amantadine hydrochloride salt (Ama·HCl) focus mostly on analytical applications of this drug molecule.54,59–62
2. Experimental and computational techniques
IRPD spectra of AmaH+Ar are recorded in a quadrupole–octupole–quadrupole tandem mass spectrometer coupled to an electron ionization source described elsewhere.63,64 The clusters are produced in a pulsed supersonic plasma expansion by electron and/or chemical ionization (EI/CI) close to the nozzle orifice followed by clustering reactions. The expanding gas mixture is generated by passing a carrier gas mixture of 5% H2/He and Ar in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 through a heated reservoir containing Ama (Sigma Aldrich, >97%, T ∼ 130°) at a stagnation pressure of 5–9 bar. The filaments of the EI source are operated with an offset voltage of 220 V, which determines the upper limit for the kinetic energy of the electrons hitting the molecules in the expansion. A typical mass spectrum of the ion source is shown in the electronic supporting information (Fig. S1, ESI†). One possible production route for AmaH+Ar involves EI of H2 (eqn (1)) followed by exothermic proton transfer reactions (eqn (2) and (3)) to generate AmaH+ and three-body collisions to form the AmaH+Ar clusters (eqn (4)):65,66
:50 through a heated reservoir containing Ama (Sigma Aldrich, >97%, T ∼ 130°) at a stagnation pressure of 5–9 bar. The filaments of the EI source are operated with an offset voltage of 220 V, which determines the upper limit for the kinetic energy of the electrons hitting the molecules in the expansion. A typical mass spectrum of the ion source is shown in the electronic supporting information (Fig. S1, ESI†). One possible production route for AmaH+Ar involves EI of H2 (eqn (1)) followed by exothermic proton transfer reactions (eqn (2) and (3)) to generate AmaH+ and three-body collisions to form the AmaH+Ar clusters (eqn (4)):65,66| H2 + e → H2+ + 2e | (1) |
| H2+ + H2 → H3+ + H | (2) |
| Ama + H3+ → AmaH+ + H2 | (3) |
| AmaH+ + Ar + M → AmaH+Ar + M | (4) |
| Ama + e → Ama+ + 2e | (5) |
| Ama+ + H2 → AmaH+ + H | (6) |
Collision-induced dissociation (CID) experiments are performed to confirm the chemical composition of the produced AmaH+Ar clusters (Fig. S2, ESI†). The generated AmaH+Ar clusters are mass-selected in the first quadrupole and irradiated in an adjacent octupole ion guide with a tuneable IR laser pulse (νIR) emitted from an optical parametric oscillator pumped by a nanosecond Q-switched Nd:YAG laser. The IR radiation is characterized by 10 Hz repetition rate, <4 cm−1 bandwidth, and pulse energies of 2–5 mJ and 0.1–0.5 mJ in the XH stretch (2600–3700 cm−1) and fingerprint ranges (1100–1900 cm−1). Frequency calibration to better than 1 cm−1 is performed using a wavemeter. Resonant vibrational excitation of the clusters leads exclusively to the loss of Ar. The AmaH+ fragment ions are selected by the second quadrupole and monitored by a Daly detector as a function of the laser frequency to obtain the IRPD spectra of AmaH+Ar. Separation of laser-induced dissociation from metastable decay background is achieved by triggering the ion source at twice the repetition rate of the laser and subtracting the signals from alternating triggers. All IRPD spectra are normalized for frequency-dependent variations in the photon flux.
Geometric, vibrational, energetic, and electronic properties of Ama, AmaH+, and AmaH+Ar in their electronic ground states are characterized by quantum chemical calculations at the dispersion-corrected B3LYP-D3/cc-pVTZ level.68 This computational approach has already provided reliable results for Ama+Ln clusters and is an efficient compromise between accuracy and computing time.47,55,56 Relative energies and equilibrium binding energies (Ee, De) are corrected for harmonic zero-point vibrational energy to yield E0 and D0. To compensate for the different anharmonicity of the various CH/NH stretch and fingerprint modes, three different scaling factors are employed (0.9732, 0.9618, and 0.9491 for fingerprint and CH and NH stretch modes).55 Computed IR stick spectra are convoluted with Gaussian line profiles (fwhm = 10 cm−1) to facilitate convenient comparison with the measured IRPD spectra. Natural bond orbital (NBO) analysis is employed to evaluate the atomic charge distributions.69,70 We only consider singlet electronic states, because triplets are computed to be very high in energy (E0 ≥ 350 kJ mol−1). Cartesian coordinates and energies of all relevant structures are available in the ESI.†
3. Experimental results
Typical mass spectra of the EI source show the parent Ama+ radical cation (m/z 151), its dehydrogenated and protonated closed-shell ions ([Ama-H]+/AmaH+ at m/z 150/152), and their clusters with Ar, N2 and H2O (Fig. S1, ESI†). Clusters with H2O appear because traces of H2O have been added to the gas inlet system for parallel experiments on AmaH+(H2O)n clusters. In addition, intense fragment ions of Ama+ (F+ at m/z 135, 108, 94, 57) are detected, consistent with the standard EI mass spectrum of Ama, confirming the identity of m/z 151 as Ama+ (Fig. S1, ESI†).54,71–73 Note that the standard EI mass spectrum of Ama is quite different from that of Ada, indicating the strong impact of the NH2 group on the fragmentation process of the adamantyl cage.54 One of the most intense peaks in the mass spectrum of the EI source (m/z 135) reveals the loss of NH3 from AmaH+,58 most likely arising from fragmentation of N-protonated AmaH+ into the very stable 1-adamantyl cation.58 Further fragmentation of m/z 135 down to m/z 93 by elimination of C3H6 is only weakly observed,58 possibly an indication for soft N-protonation of Ama. The CID spectrum of AmaH+Ar shows merely the loss of Ar, so that any significant isobaric mass contamination can be excluded (Fig. S2, ESI†).The IRPD spectrum of AmaH+Ar recorded in the fingerprint (1100–1900 cm−1) and XH stretch ranges (2700–3700 cm−1) is shown in Fig. 1. The positions and widths of the transitions observed are listed in Table 1, along with the proposed vibrational and isomer assignments derived from the B3LYP-D3 calculations. The fingerprint range covers CH2 torsion (τCH2) and wagging (γCH2) modes (A–D), and CH2/NHn bending (βCH2/NHn) and CN stretching (νCN) fundamentals (E–I). The XH stretch range probes aliphatic CHn stretch modes (νCHn) of the Ama+ moiety (J–N), overtone and combinations bands of βNH2/3 and νCN fundamentals (O, P, T), and symmetric and antisymmetric  NH stretch modes of the NH2+ (S, U) and NH3+ groups (Q, R).
NH stretch modes of the NH2+ (S, U) and NH3+ groups (Q, R).
 | ||
| Fig. 1 IRPD spectra of AmaH+Ar in the XH stretch and fingerprint ranges compared to linear IR absorption spectra of Ama+(I–IV) calculated at the B3LYP-D3/cc-pVTZ level (Tables S1 and S2, ESI†). The positions of the transitions observed (A–U) and their vibrational and isomer assignment are listed in Table 1. Relative energies (E0) are given in kJ mol−1. | ||
| Peak | Mode | AmaH+Ar | Isomer |
|---|---|---|---|
| a All values are given in cm−1. b Could be also assigned to the less stable isomers III and IV. | |||
| A | τ CH2 | 1187 (12) | I, IIb |
| B | τ CH2 | 1209 (2) | I, IIb |
| C | γ CH2, τCH2 | 1322 (8) | I, IIb |
| D | γ CH2, τCH2 | 1377 (11) | I, IIb |
| E | γ NH3(umbrella), βCH2 | 1453 (13) | I |
| F | β CH2 | 1474 (6) | I, IIb |
| G | β CH2, βCH3 | 1483 (5) | I, IIb |
| H | β NH3 | 1611 (12) | I |
| I | β NH2 | 1714 (15) | II |
| J | ν CHn | 2853 (10) | II |
| K | ν CHn | 2872 (7) | II |
| L | ν CHn | 2921 (14) | I, IIb |
| M | ν CHn | 2949 (17) | I, IIb |
| N | ν CHn | 2973 (5) | II |
| O | 2βNH3 | 3164 (7) | I |
| P | 2βNH3 | 3185 (5) | I |
| Q |

|
3238 (13) | I |
| R |

|
3317 (21) | I |
| S |

|
3344 (7) | II |
| T | 2βNH2 | 3428 (11) | II |
| U |

|
3451 (10) | II |
To our initial surprise, we observe five bands in the NH stretch range (Q–U) and not just two, which one would expect for Ama protonated at the basic NH2 group. The resulting ammonium ion of AmaH+ with C3v symmetry (RNH3+) has symmetric (a) and doubly degenerate antisymmetric (e) NH stretch modes, whereby the latter may split into two components upon Ar-tagging at the NH3+ group. By comparison with NH4+ and protonated aniline (AnH+) and their complexes with rare gas atoms,67,74 the νNH3 modes of the RNH3+ are expected in the 3150–3400 cm−1 range and may thus be associated with bands Q, R, and possibly S, observed at 3238, 3317, and 3344 cm−1. On the other hand, νNH2 modes of amine cations (RNH2+), such as the NH3+, An+, and the carbenium isomers of AnH+,67,75–77 have their NH stretch frequencies toward higher frequency (3350–3500 cm−1), and such isomers of AmaH+ may thus account for bands U, T, and possibly S observed at 3451, 3428, and 3344 cm−1. Hence, the various intense NH stretch bands suggest the presence of at least two AmaH+ isomers, one expected ammonium ion and one unexpected iminium ion. The larger width of R and its shoulder S may arise from effects of Ar-tagging, giving rise to a splitting upon symmetry reduction and minor shifts upon formation of NH⋯Ar hydrogen bonds (H-bonds). However, Ar-tagging of a single AmaH+ isomer alone cannot account for the rich observed NH stretch spectrum with its large frequency spread.
Similar to Ama+Ar,47 the aliphatic CH stretch range of AmaH+Ar (2800–3000 cm−1) exhibits several bands (J–N), with two strong broader features L and M (2921 and 2949 cm−1), one weak peak N in the blue shoulder of M (2973 cm−1), and two low-frequency bands J and K with medium intensity (2853 and 2872 cm−1). The two very weak bands O and P (3164 and 3185 cm−1) between the CH and NH stretch ranges are probably overtones or combinations bands of βNH2/3 bend modes.
The fingerprint range is dominated by a very intense narrow band E at 1453 cm−1, accompanied by two weaker sharp peaks F and G (1474 and 1483 cm−1) in its blue shoulder. Interestingly, two broader and possibly unresolved features H and I at 1611 and 1714 cm−1 with comparable medium intensity occur in the NH2/3 bending range, confirming the conclusion of the presence of (at least) two isomers drawn from the NH stretch range. Furthermore, one weak transition A (1187 cm−1), one sharp peak B (1209 cm−1), and two bands C and D with similar intensity (1322 and 1377 cm−1) occur in the range of βCH2, γCH2, τCH2, and ρNH2.
In addition to the AmaH+Ar dimer (n = 1), we have also recorded the IRPD spectra of AmaH+Arn with n = 2 and 3 in the XH stretch range. Similar to the case of Ama+Arn,48 the incremental spectral shifts and splittings upon tagging with further Ar ligands are very small and do not provide any additional information about the vibrational and isomer assignment. Hence, we do not discuss them in detail further here. The major information extracted from these IRPD spectra concerns the binding energy of the Ar ligands. The IRPD signal of AmaH+Ar3 is observed exclusively in the AmaH+ monomer fragment channel, providing a rough upper limit of 1000 cm−1 for the Ar binding energy, fully consistent with the results of the B3LYP-D3 calculations detailed in Section 4.
4. Computational results and discussion
4.1. AmaH+ isomers I–IV
Starting from neutral Ama and the previously detected Ama+ radical cation isomers (Fig. S3, ESI†), stable structures of AmaH+ are constructed by adding a proton to the basic NH2 group of Ama or a H atom to the C radical centers of the open-cage Ama+ isomers. Protonation of Ama at C is energetically highly unfavourable (when retaining the cage), because it would result in a fivefold coordinated C atom of the cycloalkane. The resulting AmaH+ isomers I–IV are shown in Fig. 2, and their corresponding IR spectra are compared in Fig. 1 to the IRPD spectrum measured for AmaH+Ar (Table S1, ESI†). All relevant energies are listed in Table S2 (ESI†). | ||
| Fig. 2 Calculated equilibrium structures (in Å and degrees) of Ama and AmaH+(I–IV) in their ground electronic state (B3LYP-D3/cc-pVTZ), along with numbering of the C atoms (in red). Relative energies (E0) are given in kJ mol−1. | ||
In the electronic ground state of neutral Ama (1A1, Cs), the NH2 group is attached in a pyramidal configuration (sp3 hybridisation of N) to the C10H15 cage via a C–N single bond (rCN = 1.467 Å). The N–H bonds are relatively short (rNH = 1.015 Å), leading to high-frequency symmetric and antisymmetric NH stretch modes  , with very low IR oscillator strength (3/0.2 km mol−1). In general, the calculated structure and IR spectrum of Ama agree well with previous experimental and computational data.51–54 Protonation of Ama at the NH2 group produces the ammonium isomer I of AmaH+ with C3v symmetry in the 1A1 electronic ground state, with a computed proton affinity (PA = 957 kJ mol−1) in close agreement with the experimental value (PA = 949 kJ mol−1).78 This structure is indeed the global minimum on the AmaH+ potential (E0 = 0 kJ mol−1). N-Protonation of Ama causes a drastic elongation of the N–H bonds by 7 mÅ to 1.022 Å and a slight increase in the NH2 angle to 108.1°, while preserving the pyramidal configuration. The former effect results in massive νNH red shifts of ∼70/60 cm−1 (
, with very low IR oscillator strength (3/0.2 km mol−1). In general, the calculated structure and IR spectrum of Ama agree well with previous experimental and computational data.51–54 Protonation of Ama at the NH2 group produces the ammonium isomer I of AmaH+ with C3v symmetry in the 1A1 electronic ground state, with a computed proton affinity (PA = 957 kJ mol−1) in close agreement with the experimental value (PA = 949 kJ mol−1).78 This structure is indeed the global minimum on the AmaH+ potential (E0 = 0 kJ mol−1). N-Protonation of Ama causes a drastic elongation of the N–H bonds by 7 mÅ to 1.022 Å and a slight increase in the NH2 angle to 108.1°, while preserving the pyramidal configuration. The former effect results in massive νNH red shifts of ∼70/60 cm−1 ( vs. 3214 cm−1,
vs. 3214 cm−1, 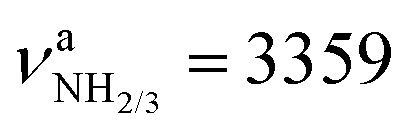 vs. 3301 cm−1, Fig. S4 and Table S3, ESI†). Moreover, the IR intensities of νNH3 are drastically enhanced (by a factor ∼10/500) due to the increased charge on the NH protons (qH = 0.429 e) and thus can readily be detected by IRPD. However, the increase in IR activity is smaller than for Ama+ (factor ∼100/350), for which the νNH2 modes are blue-shifted compared to Ama (Table S4, ESI†). Furthermore, the formation of the third N–H bond produces an additional intense γNH3 umbrella inversion mode at 1441 cm−1 (IγNH3 = 130 km mol−1), which is typical for NH3+ groups.79 The degenerate βNH3 modes (βNH3 = 1611 cm−1, IβNH3 = 39 km mol−1) have almost the same frequency and intensity as the βNH2 mode of Ama (βNH2 = 1609 cm−1, IβNH2 = 34 km mol−1). In contrast to ionization of Ama, protonation lengthens the C–N single bond by 77 mÅ to 1.544 Å and shortens the adjacent C–C bonds (rC1C2 = 1.530 vs. 1.538 Å, rC1C3 = 1.530 vs. 1.544 Å), which is due to the higher positive charge on the NH3+ group (qNH3 = 0.605 e) and its electron-withdrawing character. Moreover, the C–C bonds parallel to the C3 symmetry axis are slightly elongated compared to Ama (rC4C5 = 1.544 vs. 1.538 Å). Almost all C–H bonds contract slightly (ΔrCH = 2 mÅ) resulting in blue shifts of the νCH bands (νCH = 2890 vs. 2899 cm−1, νCH = 2917 vs. 2956 cm−1). The highest occupied molecular orbital (HOMO) has CC σ character and extends over the whole adamantyl cage, while it has no amplitude at the NH3+ group (Fig. S5, ESI†).
vs. 3301 cm−1, Fig. S4 and Table S3, ESI†). Moreover, the IR intensities of νNH3 are drastically enhanced (by a factor ∼10/500) due to the increased charge on the NH protons (qH = 0.429 e) and thus can readily be detected by IRPD. However, the increase in IR activity is smaller than for Ama+ (factor ∼100/350), for which the νNH2 modes are blue-shifted compared to Ama (Table S4, ESI†). Furthermore, the formation of the third N–H bond produces an additional intense γNH3 umbrella inversion mode at 1441 cm−1 (IγNH3 = 130 km mol−1), which is typical for NH3+ groups.79 The degenerate βNH3 modes (βNH3 = 1611 cm−1, IβNH3 = 39 km mol−1) have almost the same frequency and intensity as the βNH2 mode of Ama (βNH2 = 1609 cm−1, IβNH2 = 34 km mol−1). In contrast to ionization of Ama, protonation lengthens the C–N single bond by 77 mÅ to 1.544 Å and shortens the adjacent C–C bonds (rC1C2 = 1.530 vs. 1.538 Å, rC1C3 = 1.530 vs. 1.544 Å), which is due to the higher positive charge on the NH3+ group (qNH3 = 0.605 e) and its electron-withdrawing character. Moreover, the C–C bonds parallel to the C3 symmetry axis are slightly elongated compared to Ama (rC4C5 = 1.544 vs. 1.538 Å). Almost all C–H bonds contract slightly (ΔrCH = 2 mÅ) resulting in blue shifts of the νCH bands (νCH = 2890 vs. 2899 cm−1, νCH = 2917 vs. 2956 cm−1). The highest occupied molecular orbital (HOMO) has CC σ character and extends over the whole adamantyl cage, while it has no amplitude at the NH3+ group (Fig. S5, ESI†).
The AmaH+(II) isomer (1A′, Cs, E0 = 3.0 kJ mol−1) is almost isoenergetic with the global minimum and formally derived from the open-cage Ama+(III) radical cation isomer by adding a H atom at its tertiary C9 radical center. As shown recently, ionization of Ama can trigger a rearrangement reaction, producing via cage opening and subsequent 1,2 H-shift the bicyclic distonic Ama+(III) iminium radical cation (involving a barrier of Vb = 123 kJ mol−1), whose C9 radical center is a good acceptor for a H radical.47,48 Consequently, AmaH+(II) has also an open-cage structure with one CH3 group (C3H3) and a C9H group with its C–H bond oriented toward the nearly planar CNH2+ group (rC9H⋯C1 = 2.450 Å). As a result, the cage opens much more than for Ama+(III) (θC1C9C3 = 155.3° vs. 106.1°, rC1⋯C3 = 4.47 vs. 3.35 Å),47 and the C–C bonds at the former C9 radical center drastically elongate (rC3C9 = 1.482 vs. 1.529 Å, rC9C8 = 1.492 vs. 1.536 Å). Moreover, the C2–C7/C4–C5 bonds are slightly stretched by 9 mÅ to 1.551 Å, whereas the remaining C–C bonds contract (rC1C2 = 1.491 vs. 1.486 Å, rC5C6 = 1.537 vs. 1.532 Å, rC7C8 = 1.543 vs. 1.536 Å). The cyclohexane ring carrying the NH2+ group has chair configuration as in Ama+(III) and AmaH+(I). The C–H bonds of the C atoms adjacent to C9 contract (ΔrCH = 4–8 mÅ), while most of the other C–H bonds elongate (ΔrCH = 1–2 mÅ). The newly formed C9–H bond is the longest bond (rCH = 1.098 Å), which produces an intense νCH mode (νCH = 2848 cm−1) significantly red-shifted from the other νCHn modes. It may thus serve as indicator for this isomer. The N–H bonds of the planar CNH2+ group (sp2 hybridization of N) of the iminium ion are stretched by 3 mÅ to 1.012 Å, while the NH2 bond angle remains almost the same (ΔθNH2 = 0.1°), resulting in a blue shift of  (
( vs. 3358 cm−1) and a slight red shift of
vs. 3358 cm−1) and a slight red shift of  (
( vs. 3433 cm−1) compared to Ama+(III).47 However, the N–H bonds in AmaH+(II) are much shorter than in AmaH+(I), so that its NH2 modes are well separated from the NH3 modes (ΔνNH ∼ 100 cm−1). As with Ama+(III),47 a C
vs. 3433 cm−1) compared to Ama+(III).47 However, the N–H bonds in AmaH+(II) are much shorter than in AmaH+(I), so that its NH2 modes are well separated from the NH3 modes (ΔνNH ∼ 100 cm−1). As with Ama+(III),47 a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond is present in AmaH+(II), whose νCN mode couples with βNH2, resulting in a weak νCN band at 1534 cm−1 and an intense βNH2 band at 1664 cm−1. Compared to Ama+(III), however, the CN double bond is much shorter in AmaH+(II), which leads to blue shifts in both βNH2 and νCN by 34 and 23 cm−1, respectively. Similar to Ama+(III) (qCNH2 = 0.608 e), most of the positive charge in AmaH+(II) is located at the CNH2+ group (qCNH2 = 0.748 e) and the HOMO is localized over the adamantyl framework (Fig. S5, ESI†).
N double bond is present in AmaH+(II), whose νCN mode couples with βNH2, resulting in a weak νCN band at 1534 cm−1 and an intense βNH2 band at 1664 cm−1. Compared to Ama+(III), however, the CN double bond is much shorter in AmaH+(II), which leads to blue shifts in both βNH2 and νCN by 34 and 23 cm−1, respectively. Similar to Ama+(III) (qCNH2 = 0.608 e), most of the positive charge in AmaH+(II) is located at the CNH2+ group (qCNH2 = 0.748 e) and the HOMO is localized over the adamantyl framework (Fig. S5, ESI†).
The significantly less stable AmaH+(III) iminium isomer (1A′, Cs, E0 = 27.4 kJ mol−1) has a similar open-cage structure as AmaH+(II), with one NH2+ and one CH3 group but the C9–H bond pointing away from the CNH2+ group. The cage is less open than in AmaH+(II) (θC1C9C3 = 124.6° vs. 155.3°, rC1⋯C3 = 4.69 vs. 4.47 Å), and the second cyclohexane ring changes from chair to boat configuration. AmaH+(III) can be formed by adding a H atom to either the primary C3 radical center of Ama+(II) or the tertiary C9 radical center of Ama+(III). The C2–C7/C4–C5 bonds and the C7–C8/C5–C10 bonds are elongated compared to AmaH+(II) (rC2C7 = 1.564 vs.1.552 Å, rC7C8 = 1.556 vs. 1.536 Å), while the remaining C–C bonds differ only slightly (rC1C4 = 1.480 vs.1.486 Å, rC5C6 = 1.531 vs. 1.532 Å, rC8C9 = 1.537 vs. 1.537 Å, rC3C9 = 1.527 vs. 1.529 Å). Compared to AmaH+(II), most C–H bonds contract only slightly (ΔrCH = 1–2 mÅ), while the C9–H bond contracts more (ΔrC9H = 5 mÅ), resulting in a blue shift of its νCH mode back to the other νCHn bands (νCH = 2915 vs. 2848 cm−1). The CN double bond in AmaH+(III) is slightly shorter than in AmaH+(II) (rCN = 1.295 vs. 1.297 Å), resulting in a small blue shift of the coupled βNH2 and νCN modes by 8 and 6 cm−1, respectively. The N–H bonds remain at rNH = 1.012 Å with nearly unshifted  modes at 3329/3433 cm−1. Similar to AmaH+(II) (qCNH2 = 0.748 e), most of the positive charge is located at the CNH2+ group (qCNH2 = 0.755 e), and the HOMO is localized over the adamantyl framework (Fig. S5, ESI†).
modes at 3329/3433 cm−1. Similar to AmaH+(II) (qCNH2 = 0.748 e), most of the positive charge is located at the CNH2+ group (qCNH2 = 0.755 e), and the HOMO is localized over the adamantyl framework (Fig. S5, ESI†).
The AmaH+(IV) iminium isomer (1A′, Cs, E0 = 29.5 kJ mol−1) has a similar structure (both differ only in conformation) as the slightly more stable AmaH+(III) minimum and may formally be obtained by addition of an H radical to a radical site of Ama+(II) or Ama+(III). However, it is also possible that AmaH+(IV) is formed by C3-protonation of Ama accompanied by cage opening. The protonation energy for C3-protonation of Ama (927 kJ mol−1) followed by cage opening is only slightly lower than the proton affinity for N-protonation of Ama (PA = 957 kJ mol−1). The AmaH+(IV) structure differs from III mainly by a more closed cage (θC1C9C3 = 73.1° vs. 124.6°, rC1⋯C3 = 3.25 vs. 4.69 Å) and a chair configuration of the second cyclohexane ring. Consequently, most of the C–C bonds are stretched compared to AmaH+(III) (rC1C4 = 1.486 vs.1.480 Å, rC8C9 =1.548 vs. 1.537 Å, rC3C9 = 1.535 vs. 1.527 Å), while the C–C bond lengths of the first amino-cyclohexane ring either decrease or remain the same (rC2C7 = 1.547 vs. 1.565 Å, rC7C8 = 1.539 vs. 1.556 Å, rC5C6 = 1.530 vs. 1.531 Å). The variation in the C–H bond lengths is increased from ΔrCH = 7 mÅ in AmaH+(III) (1.088–1.095 Å) to ΔrCH = 11 mÅ in AmaH+(IV) (1.088–1.099 Å), leading to four better resolved single convoluted bands (νCHn = 2874, 2916, 2948, 2998 cm−1). In particular, the shortened C3–H bond oriented toward the NH2+ group produces a blue shift in the νCH3 mode by ΔνCH3 = 94 cm−1 to 2998 cm−1. The CN double bond is slightly elongated to rCN = 1.296 Å, while the N–H bond lengths remain the same (rNH = 1.012 Å). As a result, the coupled βNH2 and νCN modes are slightly red-shifted to νCN = 1533 cm−1 and βNH2 = 1665 cm−1, while the νNH2 frequencies remain the same  . Again, most of the positive charge is located at the CNH2+ group (qCNH2 = 0.758 e) and the HOMO is localized over the adamantyl framework (Fig. S5, ESI†).
. Again, most of the positive charge is located at the CNH2+ group (qCNH2 = 0.758 e) and the HOMO is localized over the adamantyl framework (Fig. S5, ESI†).
4.2 AmaH+Ar clusters
Inspection of Fig. 1 reveals that the predictions for the three iminium isomers II–IV of AmaH+ are rather similar, while that of the ammonium ion I is rather different, mainly due to the difference in N–H bonding and resulting IR spectra between the NH3+ and NH2+ groups. Prior to detailed comparison to experiment, we briefly consider the impact of Ar-tagging. The effect of Ar on the structural, vibrational, and energetic properties of all AmaH+ isomers is indeed very small, so that the discussion on the AmaH+Ar isomers is kept short. Details of the computed AmaH+Ar structures, energies, charge distributions, and IR spectra are provided in the ESI† (Fig. S6–S10 and Tables S5, S6). In the most stable AmaH+Ar isomers, Ar interacts with both the charged NH2/3+ group and the C10H15/16 moiety to maximize induction and dispersion forces, with low binding energies of D0 = 10.2, 9.8, 6.9, and 8.9 kJ mol−1 for I–IV, respectively. Significantly, the minor differences in the Ar binding energy are too small to affect the energetic order of the monomer ions. The main impact of the weak interaction with Ar is a reduction in symmetry, resulting in a small splitting of the degenerate NH stretching and bending modes of AmaH+(I). The IR spectra calculated for all AmaH+Ar isomers for each monomer (I–IV) show only minimal shifts of less than 11 (I), 4 (II), 7 (III), and 8 cm−1 (IV) in νNH2/3, βNH2/3, γNH3, and νCN upon Ar-tagging, respectively. Similar to Ama+Ar,47,48 the Ar ligands form in the most stable AmaH+Ar isomers weak and strongly nonlinear NH⋯Ar H-bonds (RNH⋯Ar = 2.58–2.86 Å, ϕArHN = 119.8–165.4°), accompanied by additional van der Waals contacts to adjacent CH2/CH3 groups (RCH⋯Ar = 2.96–3.15 Å), yielding D0 = 8.7–10.3 kJ mol−1. As a result, the involved N–H bonds are slightly stretched (by 1 mÅ), while the involved C–H bonds are shortened (by 1 mÅ) and free C–H bonds can be elongated. These structural effects result in minor red and blue shifts in νNH2/3 (<11 cm−1), νCH2 (<6 cm−1) and νCH3 modes (<13 cm−1). Charge transfer from AmaH+ to Ar is rather small (5–9 me). AmaH+(I)–Ar(I) with the ammonium core ion has the strongest and a nearly linear NH⋯Ar H-bond (RNH⋯Ar = 2.58 Å, ϕArHN = 165.4°), with the largest accompanying charge transfer of 9 me. Due to Ar attachment, the two degenerate (3301 cm−1) and
(3301 cm−1) and  (1611 cm−1) modes of AmaH+(I) split into two separate modes in AmaH+(I)–Ar(I),
(1611 cm−1) modes of AmaH+(I) split into two separate modes in AmaH+(I)–Ar(I),  and
and 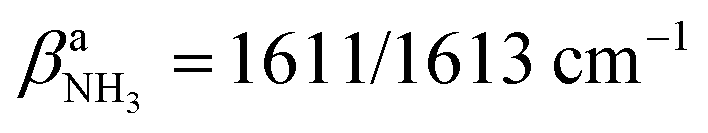 . In AmaH+(II)–Ar(I), Ar binds to the NH2+ and C9H groups, resulting in a blue shift of the corresponding νCH mode from 2847 to 2860 cm−1. In the less stable I-Ar(II–V), II-Ar(II–VI), III-Ar(II–VI), and IV-Ar(II–VI) clusters, Ar binds only to the C10H15/16 cage, with lower binding energies of D0 = 2.0–6.2 kJ mol−1 and negligible effects on the monomer properties.
. In AmaH+(II)–Ar(I), Ar binds to the NH2+ and C9H groups, resulting in a blue shift of the corresponding νCH mode from 2847 to 2860 cm−1. In the less stable I-Ar(II–V), II-Ar(II–VI), III-Ar(II–VI), and IV-Ar(II–VI) clusters, Ar binds only to the C10H15/16 cage, with lower binding energies of D0 = 2.0–6.2 kJ mol−1 and negligible effects on the monomer properties.
4.3 Assignment of IRPD spectrum
The minor effects of Ar-tagging on the geometries, energies, and IR spectra of the AmaH+ isomers I–IV justify direct comparison of the latter to the measured IRPD spectrum of AmaH+Ar for vibrational and isomer assignments (Table S1, ESI†). Inspection of Fig. 1 reveals immediately the presence of both ammonium and iminium isomers of AmaH+. As outlined above, the NH stretch and bend ranges are most decisive. While most of the intense features are readily assigned to the most stable ammonium ion I, with clear signatures of the NH3+ group (E, H, Q, R), the strong features I, S, T, and U cannot be explained by this isomer but fit well to the features of the NH2+ group of the iminium isomers II-IV. Specifically, peaks Q and R (3238 and 3317 cm−1) are attributed to the and degenerate
and degenerate  NH stretch modes predicted at 3214 and 3301 cm−1 of I, respectively, while bands E and H (1453 and 1611 cm−1) arise from the γNH3 umbrella (1441 cm−1) and degenerate βNH3 bending modes (1611 cm−1), with maximum, mean, and median deviations of 24, 13, and 14 cm−1, respectively, consistent with the widths of the bands (12–21 cm−1) and the small effect of Ar-tagging (≤5 cm−1). Actually, bands H and R assigned to the doubly degenerate modes are broader than bands E and Q attributed to single transitions, which can readily be explained by splittings of the degenerate modes upon Ar-tagging into two unresolved components with roughly equal intensity. While the predicted splitting for βNH3 is rather small (2 cm−1), that for
NH stretch modes predicted at 3214 and 3301 cm−1 of I, respectively, while bands E and H (1453 and 1611 cm−1) arise from the γNH3 umbrella (1441 cm−1) and degenerate βNH3 bending modes (1611 cm−1), with maximum, mean, and median deviations of 24, 13, and 14 cm−1, respectively, consistent with the widths of the bands (12–21 cm−1) and the small effect of Ar-tagging (≤5 cm−1). Actually, bands H and R assigned to the doubly degenerate modes are broader than bands E and Q attributed to single transitions, which can readily be explained by splittings of the degenerate modes upon Ar-tagging into two unresolved components with roughly equal intensity. While the predicted splitting for βNH3 is rather small (2 cm−1), that for  is larger (13 cm−1) and indeed partly resolved for band R (10 cm−1).
is larger (13 cm−1) and indeed partly resolved for band R (10 cm−1).
Unfortunately, the properties of the NH2+ groups and resulting IR spectra of the iminium ions II–IV are very similar, with differences of ≤2/3 and ≤5 cm−1 for  and βNH2, respectively. Hence, we cannot distinguish between them at the current spectral resolution and consider for the assignment given here the by far most stable of them (II). Following this scenario, bands S and U (3344 and 3451 cm−1) are assigned to
and βNH2, respectively. Hence, we cannot distinguish between them at the current spectral resolution and consider for the assignment given here the by far most stable of them (II). Following this scenario, bands S and U (3344 and 3451 cm−1) are assigned to  (3331 cm−1) and
(3331 cm−1) and  (3435 cm−1) of II, with deviations of 13 and 16 cm−1, respectively. Peak I (1714 cm−1) is then attributed to βNH2 (1664 cm−1) of II which couples strongly with νCN. The somewhat larger deviation of 50 cm−1 between experiment and calculation is probably due to the different anharmonicity of the νCN mode, leading to an improper description of the coupling (including a nonoptimal scaling factor).47 Due to its low intensity, νCN of II is not observed. Band T (3428 cm−1) occurs also in the NH stretch range of the NH2 group and may be attributed to a
(3435 cm−1) of II, with deviations of 13 and 16 cm−1, respectively. Peak I (1714 cm−1) is then attributed to βNH2 (1664 cm−1) of II which couples strongly with νCN. The somewhat larger deviation of 50 cm−1 between experiment and calculation is probably due to the different anharmonicity of the νCN mode, leading to an improper description of the coupling (including a nonoptimal scaling factor).47 Due to its low intensity, νCN of II is not observed. Band T (3428 cm−1) occurs also in the NH stretch range of the NH2 group and may be attributed to a  mode of, for example, III or IV. This option implies that the large splitting between T and U of 23 cm−1 is not reproduced by the calculations for II–IV. An assignment of T and U to different Ar binding sites of II appears also unlikely, because the predicted shifts upon tagging are less than 4 cm−1. A third alternative is an assignment of T to the 2βNH2 overtone, which gains intensity via a classical Fermi resonance with νNH2. Such intense 2βNH2 overtones are also observed in the IRPD spectra of Ama+Ln clusters, and indeed the position of band T is twice the frequency of band I (1714 cm−1), making this interpretation a likely and currently favored scenario.
mode of, for example, III or IV. This option implies that the large splitting between T and U of 23 cm−1 is not reproduced by the calculations for II–IV. An assignment of T and U to different Ar binding sites of II appears also unlikely, because the predicted shifts upon tagging are less than 4 cm−1. A third alternative is an assignment of T to the 2βNH2 overtone, which gains intensity via a classical Fermi resonance with νNH2. Such intense 2βNH2 overtones are also observed in the IRPD spectra of Ama+Ln clusters, and indeed the position of band T is twice the frequency of band I (1714 cm−1), making this interpretation a likely and currently favored scenario.
The assignments of the NH stretch and bend ranges reveal the presence of both ammonium and iminium ions. Their population ratio can roughly be estimated from the observed integrated peak intensities, the computed IR cross sections, and the reasonable assumptions of similar Ar-tagging and photodissociation efficiencies. When considering the isolated peaks H and I, or peaks Q and U, population ratios of 4:1 and 3:1 are obtained for I and II(–IV), i.e., the AmaH+ population and AmaH+Ar spectrum is strongly dominated by the ammonium isomer obtained by simple protonation of Ama at the NH2 group.
With this derived population ratio, we consider now the other spectral ranges, for which clear-cut isomer assignments are less obvious due to similar frequencies and/or spectral congestion. Bands B–D (1209, 1322, 1377 cm−1) are consistent with τCH2 and γCH2 modes of the dominant isomer I with respect to both frequency (1206, 1294, 1359 cm−1) and relative intensity. Similarly, peaks F and G (1474 and 1483 cm−1) can be attributed to βCHn modes of I (1469 cm−1), with its strongest IR active modes at 1463 and 1472 cm−1 (26 and 39 km mol−1). The transitions predicted for the minor isomer II in this spectral range mostly overlap (Table S1, ESI†), and the only isomer-specific transition appears to be band A (1187 cm−1), which has no predicted mode of I but fits to τCH2 of II (1203 cm−1).
In the CH stretch range, isomer-specific assignments are not as straightforward due to the similar frequencies of νCHn of I and II, but bands K (2872 cm−1) and M (2949 cm−1) may be assigned to I (νCHn = 2899 and 2956 cm−1) and bands J (2853 cm−1), L (2921 cm−1), and N (2973 cm−1) to II (νCHn = 2848, 2917, and 2977 cm−1) with maximum, mean, and median deviations of 27, 9, and 5 cm−1. In particular, peak J (2853 cm−1) agrees well with the blue-shifted νCH mode of II (2847 cm−1), which is another argument for observing II, because none of the other isomers have modes predicted in this frequency range.
The weak bands O and P (3164 and 3185 cm−1) cannot readily be explained by fundamentals of I–IV and thus are likely overtones and combination bands of βNH2 and/or νCN. Similar bands have been observed previously for Ama+Ar (3151 cm−1) and AnH+Ar (3175 cm−1).47,67 In analogy to assigning T as overtone of H of isomer II, O and P are attributed to overtones of the two components of H (1611 cm−1). In this scenario, the splitting of the degenerate fundamental upon Ar-tagging is better resolved for the overtone. The corresponding expected overtone of E falls in the rich νCH range (2906 cm−1) and is not easily identified.
4.4 Potential energy diagram and production routes
The analysis of the IRPD spectrum of AmaH+Ar by B3LYP-D3 calculations reveals the unambiguous spectroscopic fingerprints of (at least) two different AmaH+ isomers in the supersonic plasma expansion: the dominant ammonium isomer (I) and the minor iminium isomer (II). The similar iminium ions III and IV cannot be completely ruled out because they have similar predicted IR spectra as II. However, due to their higher relative energy (E0 ≥ 27 kJ mol−1), their formation is considered unlikely (although kinetic trapping may lead to their significant population). Assuming that only I and II are observed, their population ratio is derived in the range 3:1 to 4:1.
In an effort to rationalize possible production routes for I and II, we consider the potential energy diagram for Ama, and the various Ama+ and AmaH+ isomers in Fig. 3. The ionization process of Ama and the formation of the three isomers I–III of Ama+ by vertical ionization and structural rearrangement reactions (cage opening and 1,2, H-shift) have been discussed in detail previously (Fig. S3, ESI†).47 In a bare Ar expansion of Ama, the population ratio of I–III was determined as 10:3:7, although this may change somewhat in a H2/Ar/He plasma employed here. Starting from Ama, two main reaction pathways are possible. The first route involves N-protonation of Ama most likely by exothermic proton transfer from H3+ (eqn (3)). The second route involves EI/CI of Ama, and subsequent H-addition to isomers I–III of Ama+, most likely by H-abstraction from H2 (eqn (5) and (6)), although internal energy resulting from EI/CI is required to overcome the endothermicity of this reaction. The ammonium ion I of AmaH+ can be produced by either route, namely N-protonation of Ama or H2 reacting with Ama+(I). On the other hand, the iminium ions II–IV of AmaH+ are most likely formed via the second route, involving EI/CI of Ama and H2 reacting with Ama+(II,III). However, C-protonation of Ama followed by cage opening, which would lead to AmaH+(IV), cannot be completely ruled out, because of the comparable protonation energy of 927 kJ mol−1. Moreover, protonation of Ama with H3+ leading to AmaH+(I) is highly exothermic and thus may also produce II–IV by cage opening.
 | ||
| Fig. 3 Potential energy surfaces (minima and transition states) of AmaH+ (blue), Ama+ (red), and Ama (green) for the protonation and ionization pathways including cage-opening processes. Ionization energy (IE) and proton affinity (PA) values are given in kJ mol−1. | ||
In AmaH+(I), N-protonation at the N lone pair elongates the C–N bond and contracts the adjacent C1–C3 bonds, thereby preventing cage opening with an intact NH3+ group. The calculated proton affinity for N-protonation is in good agreement with experiment (PA = 957 vs. 949 kJ mol−1).78 Starting from the AmaH+(I) global minimum, a 1,2 H-shift from the NH3+ group to the C3H2 group can occur, which opens the cage and forms AmaH+(IV) at E0 = 29.5 kJ mol−1. However, the energy barrier for this rearrangement process at TS(I–IV) is rather high (Vb = 263 kJ mol−1), making this process unlikely. Nonetheless, from AmaH+(IV) the cage can open further via TS(IV–III) at a very low barrier (Vb = 14 kJ mol−1) to form AmaH+(III) at E0 = 27.4 kJ mol−1 (III and IV differ only in their conformation). This rearrangement is accompanied by a chair to boat conversion of the cyclohexane ring. A rearrangement from AmaH+(III) to AmaH+(II) is rather improbable, since this process would require most of the structure (C6H8NH2) to flip once around the C8–C10 axis, which probably involves a high barrier. Because of this substantial rearrangement, the associated barrier for TS(III–II) could not be calculated. Hence, we assume that the detected AmaH+(II) isomer cannot be formed via the protonation pathway and therefore must be formed via the ionization route.
Unlike N-protonation of Ama, ionization leads to a contraction of the C–N bond and a drastic elongation of the C1–C3 bond in Ama+(I) (E0 = 45.9 kJ mol−1) due to the removal of an electron from the N lone pair and the increased delocalization of this orbital into the adamantyl cage. The computed adiabatic ionization energy of 7.9 eV agrees well with the estimated vertical value (8.6 eV),53 and both are well below the available maximum electron energy of the EI process (220 eV). As a result, part of the nascent Ama+(I) population may overcome the energy barrier at TS(I–II) (Vb = 54.1 kJ mol−1, E0 = 100.0 kJ mol−1) for opening the cage. This process generates the bicylic distonic Ama+(II) iminium ion (E0 = 87.4 kJ mol−1) with a primary CH2˙ radical center. A much more stable tertiary CR3˙ radical center is then formed in the most stable distonic bicyclic iminium ion Ama+(III) (E0 = 0) by 1,2 H-shift from C9H to CH2˙ via Ama+(TS(II–III)) (Vb = 81.6 kJ mol−1). This tertiary CR3˙ radical center is the very attractive for H-atom attachment leading to the formation of AmaH+(II). As Ama+(III) has already been detected in high abundance in the Ar plasma expansion of Ama,47 this formation mechanism of the low-energy AmaH+(II) isomer (E0 = 3.0 kJ mol−1) is highly plausible.
6. Concluding remarks
In conclusion, we characterize herein the structural, vibrational, and energetic properties of AmaH+ ions by Ar-tagging IRPD spectroscopy and dispersion-corrected B3LYP-D3 calculations. To this end, the AmaH+ ions are generated in a supersonic plasma expansion generated by electron/chemical ionization of a molecular beam containing Ama, He, and Ar. Significantly, the obtained IRPD spectrum of AmaH+Ar provides the first spectroscopic characterization of the structure of this pharmaceutically active ion. The influence of Ar is shown to be negligible by B3LYP-D3 calculations and the evolution of IRPD spectra recorded for AmaH+Arn with n = 1–3. The IRPD spectra cover the structure-sensitive NH stretch and bend ranges, which reveal the presence of two types of isomers. The dominant population corresponds to N-protonated AmaH+, which is the global minimum on the computed potential energy surface (isomer I, E0 = 0) and the relevant structure for pharmaceutical applications in solution. In addition to this ammonium ion, a smaller population is assigned to the only slightly less stable bicyclic iminium ion (isomer II, E0 = 3 kJ mol−1), which is probably produced by ionization of Ama, followed by opening of the adamantyl cage and subsequent addition of H. A study on AmaH+(H2O)n clusters using similar experimental and computational techniques is underway to characterize the impact of microhydration on the structure and reactivity of this fundamental biomolecular ion.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by Deutsche Forschungsgemeinschaft (grant DO 729/8). We thank P. R. Schreiner for valuable discussions about the structure and reactivity of diamondoid cations.References
- S. Landa and V. Machacek, Adamantane, a new hydrocarbon extracted from petroleum, Collect. Czech. Chem. Commun., 1933, 5, 1 CrossRef CAS.
- J. E. Dahl, S. G. Liu and R. M. K. Carlson, Isolation and structure of higher diamondoids, nanometer-sized diamond molecules, Science, 2003, 299, 96–99 CrossRef CAS PubMed.
- R. Schleyer and P. von, A simple preparation of adamantane, J. Am. Chem. Soc., 1957, 79, 3292 CrossRef.
- R. C. Fort and P. v R. Schleyer, Adamantane: Consequences of the diamondoid structure, Chem. Rev., 1964, 64, 277–300 CrossRef CAS.
- J. E. P. Dahl, J. M. Moldowan, Z. Wei, P. A. Lipton, P. Denisevich, R. Gat, S. Liu, P. R. Schreiner and R. M. K. Carlson, Synthesis of higher diamondoids and implications for their formation in petroleum, Angew. Chem., Int. Ed., 2010, 49, 9881–9885 CrossRef CAS PubMed.
- P. R. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Overcoming lability of extremely long alkane carbon–carbon bonds through dispersion forces, Nature, 2011, 477, 308 CrossRef CAS PubMed.
- W. L. Yang, J. D. Fabbri, T. M. Willey, J. R. I. Lee, J. E. Dahl, R. M. K. Carlson, P. R. Schreiner, A. A. Fokin, B. A. Tkachenko and N. A. Fokina, Monochromatic electron photoemission from diamondoid monolayers, Science, 2007, 316, 1460–1462 CrossRef CAS PubMed.
- D. F. Blake, F. Freund, K. F. M. Krishnan, C. J. Echer, R. Shipp, T. E. Bunch, A. G. Tielens, R. J. Lipari, C. J. D. Hetherington and S. Chang, The nature and origin of interstellar diamond, Nature, 1988, 332, 611 CrossRef CAS PubMed.
- R. S. Lewis, E. Anders and B. T. Draine, Properties, detectability and origin of interstellar diamonds in meteorites, Nature, 1989, 339, 117 CrossRef CAS.
- J.-Y. Raty and G. Galli, Ultradispersity of diamond at the nanoscale, Nat. Mater., 2003, 2, 792 CrossRef CAS PubMed.
- P. R. Schreiner, N. A. Fokina, B. A. Tkachenko, H. Hausmann, M. Serafin, J. E. P. Dahl, S. Liu, R. M. K. Carlson and A. A. Fokin, Functionalized nanodiamonds: Triamantane and [121] tetramantane, J. Org. Chem., 2006, 71, 6709–6720 CrossRef CAS PubMed.
- K. Gerzon, E. V. Krumkalns, R. L. Brindle, F. J. Marshall and M. A. Root, The adamantyl group in medicinal agents. I. Hypoglycemic N-arylsulfonyl-N′-adamantylureas, J. Med. Chem., 1963, 6, 760–763 CrossRef CAS PubMed.
- H. Schwertfeger, A. A. Fokin and P. R. Schreiner, Diamonds are a chemist's best friend: diamondoid chemistry beyond adamantane, Angew. Chem., Int. Ed., 2008, 47, 1022–1036 CrossRef CAS PubMed.
- A. A. Fokin and P. R. Schreiner, Selective alkane transformations via radicals and radical cations: Insights into the activation step from experiment and theory, Chem. Rev., 2002, 102, 1551–1594 CrossRef CAS PubMed.
- L. Wanka, K. Iqbal and P. R. Schreiner, The lipophilic bullet hits the targets: Medicinal chemistry of adamantane derivatives, Chem. Rev., 2013, 113, 3516–3604 CrossRef CAS PubMed.
- O. Pirali, M. Vervloet, J. E. Dahl, R. M. K. Carlson, A. Tielens and J. Oomens, Infrared spectroscopy of diamondoid molecules: New insights into the presence of nanodiamonds in the interstellar medium, Astrophys. J., 2007, 661, 919 CrossRef CAS.
- L. Landt, K. Klünder, J. E. Dahl, R. M. K. Carlson, T. Möller and C. Bostedt, Optical response of diamond nanocrystals as a function of particle size, shape, and symmetry, Phys. Rev. Lett., 2009, 103, 47402 CrossRef PubMed.
- A. A. Fokin, P. R. Schreiner, P. A. Gunchenko, S. A. Peleshanko, T. y E. Shubina, S. D. Isaev, P. V. Tarasenko, N. I. Kulik, H.-M. Schiebel and A. G. Yurchenko, Oxidative single-electron transfer activation of σ-bonds in aliphatic halogenation reactions, J. Am. Chem. Soc., 2000, 122, 7317–7326 CrossRef CAS.
- M. Mella, M. Freccero, T. Soldi, E. Fasani and A. Albini, Oxidative functionalization of adamantane and some of its derivatives in solution, J. Org. Chem., 1996, 61, 1413–1422 CrossRef CAS.
- A. A. Fokin, B. A. Tkachenko, P. A. Gunchenko, D. V. Gusev and P. R. Schreiner, Functionalized nanodiamonds part I. An experimental assessment of diamantane and computational predictions for higher diamondoids, Chem. – Eur. J., 2005, 11, 7091–7101 CrossRef CAS PubMed.
- P. A. Gunchenko, A. A. Novikovskii, M. V. Byk and A. A. Fokin, Structure and transformations of diamantane radical cation: Theory and experiment, Russ. J. Org. Chem., 2014, 50, 1749–1754 CrossRef CAS.
- B. A. McGuire, 2018 census of interstellar, circumstellar, extragalactic, protoplanetary disk, and exoplanetary molecules, Astrophys. J., Suppl. Ser., 2018, 239, 17 CrossRef CAS.
- J. Cernicharo, B. Tercero, A. Fuente, J. L. Domenech, M. Cueto, E. Carrasco, V. J. Herrero, I. Tanarro, N. Marcelino and E. Roueff, Detection of the ammonium ion in space, Astrophys. J., Lett., 2013, 771, L10 CrossRef.
- E. E. Etim, P. Gorai, A. Das and E. Arunan, Interstellar protonated molecular species, Adv. Space Res., 2017, 60, 709–721 CrossRef CAS.
- T. Henning and F. Salama, Carbon in the Universe, Science, 1998, 282, 2204–2210 CrossRef CAS PubMed.
- P. B. Crandall, D. Müller, J. Leroux, M. Förstel and O. Dopfer, Optical spectrum of the adamantane radical cation, Astrophys. J., 2020, 900, L20 CrossRef CAS.
- W. L. Davies, R. R. Grunert, R. F. Haff, J. W. McGahen, E. M. Neumayer, M. Paulshock, J. C. Watts, T. R. Wood, E. C. Hermann and C. E. Hoffmann, Antiviral activity of 1-adamantanamine (amantadine), Science, 1964, 144, 862–863 CrossRef CAS PubMed.
- R. S. Schwab, A. C. England, D. C. Poskanzer and R. R. Young, Amantadine in the treatment of Parkinson's disease, JAMA, 1969, 208, 1168–1170 CrossRef CAS PubMed.
- G. Hubsher, M. Haider and M. S. Okun, Amantadine: The journey from fighting flu to treating Parkinson disease, Neurology, 2012, 78, 1096–1099 CrossRef CAS PubMed.
- K. Spilovska, F. Zemek, J. Korabecny, E. Nepovimova, O. Soukup, M. Windisch and K. Kuca, Adamantane – A lead structure for drugs in clinical practice, Curr. Med. Chem., 2016, 23, 3245–3266 CrossRef CAS PubMed.
- J. W. Johnson and S. E. Kotermanski, Mechanism of action of memantine, Curr. Opin. Pharmacol., 2006, 6, 61–67 CrossRef CAS PubMed.
- C. G. Parsons, A. Stöffler and W. Danysz, Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system–too little activation is bad, too much is even worse, Neuropharmacology, 2007, 53, 699–723 CrossRef CAS PubMed.
- J. S. Oxford and A. Galbraith, Antiviral activity of amantadine: A review of laboratory and clinical data, Pharmacol. Ther., 1980, 11, 181–262 CrossRef CAS PubMed.
- J. Hu, R. Fu and T. A. Cross, The chemical and dynamical influence of the anti-viral drug amantadine on the M2 proton channel transmembrane domain, Biophys. J., 2007, 93, 276–283 CrossRef CAS PubMed.
- I. V. Chizhmakov, F. M. Geraghty, D. C. Ogden, A. Hayhurst, M. Antoniou and A. Hay, J. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells, J. Physiol., 1996, 494, 329–336 CrossRef PubMed.
- WHO, WHO | Summary of influenza antiviral susceptibility surveillance findings, 2010–2011, retrieved 2019.
- A. S. Monto and N. H. Arden, Implications of viral resistance to amantadine in control of influenza A, Clin. Infect. Dis., 1992, 15, 362–367 CrossRef CAS PubMed.
- R. P. Grelak, R. Clark, J. M. Stump and V. G. Vernier, Amantadine-dopamine interaction: possible mode of action in Parkinsonism, Science, 1970, 169, 203–204 CrossRef CAS PubMed.
- L.-O. Farnebo, K. Fuxe, M. Goldstein, B. Hamberger and U. Ungerstedt, Dopamine and noradrenaline releasing action of amantadine in the central and peripheral nervous system: a possible mode of action in Parkinson's disease, Eur. J. Pharmacol., 1971, 16, 27–38 CrossRef CAS PubMed.
- P. F. von Voigtlander and K. E. Moore, Dopamine: Release from the brain in vivo by amantadine, Science, 1971, 174, 408–410 CrossRef CAS PubMed.
- J. Kornhuber, M. Weller, K. Schoppmeyer and P. Riederer, Amantadine and memantine are NMDA receptor antagonists with neuroprotective properties, J. Neural Transm., 1994, 43, 91–104 CAS.
- A. A. Spasov, T. V. Khamidova, L. I. Bugaeva and I. S. Morozov, Adamantane derivatives: Pharmacological and toxicological properties, Pharm. Chem. J., 2000, 34, 1–7 CrossRef CAS.
- T. J. Murray, Amantadine therapy for fatigue in multiple sclerosis, Can. J. Neurol. Sci., 1985, 12, 251–254 CrossRef CAS PubMed.
- K. M. Kampman, J. R. Volpicelli, A. I. Alterman, J. Cornish and C. P. O’Brien, Amantadine in the treatment of cocaine-dependent patients with severe withdrawal symptoms, Am. J. Psychiatry, 2000, 157, 2052–2054 CrossRef CAS PubMed.
- J. Kornhuber, W. Retz and P. Riederer, Slow accumulation of psychotropic substances in the human brain. Relationship to therapeutic latency of neuroleptic and antidepressant drugs?, J. Neural Transm. Suppl, 1995, 46, 315–324 CAS.
- F. Weber and G. Sedelmeier, Top 200 drugs by worldwide sales 2013, Nachr. Chem., 2014, 62, 997 CrossRef.
- M. A. R. George and O. Dopfer, Infrared spectrum of the amantadine cation: Opening of the diamondoid cage upon ionization, J. Phys. Chem. Lett., 2022, 13, 449–454 CrossRef PubMed.
- M. A. R. George and O. Dopfer, Opening of the diamondoid cage upon ionization probed by infrared spectra of the amantadine cation solvated by Ar, N2, and H2O, Chem. – Eur. J., 2022 DOI:10.1002/chem.202200577.
- J. C. Garcia, J. F. Justo, W. V. M. Machado and L. V. C. Assali, Structural, electronic, and vibrational properties of amino-adamantane and rimantadine isomers, J. Phys. Chem. A, 2010, 114, 11977–11983 CrossRef CAS PubMed.
- L. Rivas, S. Sanchez-Cortes, J. Stanicova, J. V. Garcia-Ramos and P. Miskovsky, FT-Raman, FTIR and surface-enhanced Raman spectroscopy of the antiviral and antiparkinsonian drug amantadine, Vib. Spectrosc., 1999, 20, 179–188 CrossRef CAS.
- C. Parlak and Ö. Alver, A density functional theory investigation on amantadine drug interaction with pristine and B, Al, Si, Ga, Ge doped C60 fullerenes, Chem. Phys. Lett., 2017, 678, 85–90 CrossRef CAS.
- P. C. Yates, T. J. Hill and M. Kaur, A molecular modelling study of the anti-viral agent amantadine and its derivatives, THEOCHEM, 1992, 258, 299–313 CrossRef.
- I. V. Litvinyuk, Y. Zheng and C. E. Brion, Electron momentum spectroscopy study of amantadine: Binding energy spectra and valence orbital electron density distributions, Chem. Phys., 2000, 261, 289–300 CrossRef CAS.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, National Institute of Standards and Technology, NIST Standards and Technology, Gaithersburg, MD, https://webbook.nist.gov (accessed 2022-02-01) Search PubMed.
- M. A. R. George, F. Buttenberg, M. Förstel and O. Dopfer, Microhydration of substituted diamondoid radical cations of biological relevance: infrared spectra of amantadine+-(H2O)n=1–3 clusters, Phys. Chem. Chem. Phys., 2020, 22, 28123–28139 RSC.
- M. A. R. George, M. Förstel and O. Dopfer, Infrared spectrum of the adamantane+-water cation: Hydration-induced C–H bond activation and free internal water rotation, Angew. Chem., Int. Ed., 2020, 12098–12104 CrossRef CAS PubMed.
- A. Patzer, M. Schütz, T. Möller and O. Dopfer, Infrared spectrum and structure of the adamantane cation: Direct evidence for Jahn–Teller distortion, Angew. Chem., Int. Ed., 2012, 51, 4925–4929 CrossRef CAS PubMed.
- M. Štícha, R. L. Chayrov and I. G. Stankova, Understanding the fragmentation pathways of carbocyclic derivatives of amino acids by using electrospray ionization tandem mass spectrometry, Anal. Lett., 2019, 52, 2069–2076 CrossRef.
- J. W. Turczan and T. Medwick, NMR analysis of pharmaceuticals XII: Determination of amantadine hydrochloride in soft gelatin capsules and syrups, J. Pharm. Sci., 1974, 63, 425–427 CrossRef CAS PubMed.
- H. A. Omara and A. S. Amin, Spectrophotometric microdetermination of anti-Parkinsonian and antiviral drug amantadine HCl in pure and in dosage forms, Arab. J. Chem, 2011, 4, 287–292 CrossRef CAS.
- A. M. Mahmoud, N. Y. Khalil, I. A. Darwish and T. Aboul-Fadl, Selective spectrophotometric and spectrofluorometric methods for the determination of amantadine hydrochloride in capsules and plasma via derivatization with 1,2-naphthoquinone-4-sulphonate, Int. J. Anal. Chem., 2009, 810104 Search PubMed.
- Y. Dou, Y. Sun, Y. Ren, P. Ju and Y. Ren, Simultaneous non-destructive determination of two components of combined paracetamol and amantadine hydrochloride in tablets and powder by NIR spectroscopy and artificial neural networks, J. Pharm. Biomed., 2005, 37, 543–549 CrossRef CAS PubMed.
- O. Dopfer, IR spectroscopy of microsolvated aromatic cluster ions: Ionization-induced switch in aromatic molecule–solvent recognition, Z. Phys. Chem., 2005, 219, 125–168 CrossRef CAS.
- O. Dopfer, Spectroscopic and theoretical studies of CH3+-Rgn clusters (Rg = He, Ne, Ar): From weak intermolecular forces to chemical reaction mechanisms, Int. Rev. Phys. Chem., 2003, 22, 437–495 Search PubMed.
- T. Oka, Interstellar H3+, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12235–12242 CrossRef CAS PubMed.
- O. Dopfer, D. Roth, R. V. Olkhov and J. P. Maier, Infrared spectrum and ab initio calculations of the CH3CNH+-H2 ionic complex, J. Chem. Phys., 1999, 110, 11911–11917 CrossRef CAS.
- F. M. Pasker, N. Solcà and O. Dopfer, Spectroscopic identification of carbenium and ammonium isomers of protonated aniline (AnH+): IR spectra of weakly bound AnH+–Ln clusters (L = Ar, N2), J. Phys. Chem. A, 2006, 110, 12793–12804 CrossRef CAS PubMed.
- M. J. Frisch; et al., Gaussian 09, version D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- E. D. Glendening; J. K. Badenhoop; A. E. Reed; J. E. Carpenter; J. A. Bohmann; C. M. Morales; C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- R. J. Waltman and A. C. Ling, Mass spectrometry of diamantane and some adamantane derivatives, Can. J. Chem., 1980, 58, 2189–2195 CrossRef CAS.
- K. K. Khullar, C. L. Bell and L. Bauer, Mass spectrometry of 1-substituted adamantanes. Effect of functional groups on the primary fragmentation pathways, J. Org. Chem., 1973, 38, 1042–1044 CrossRef CAS.
- Z. Dolejšek, S. Hala, V. Hanuš and S. Landa, Adamantane and its derivatives. VIII. Mass spectra of derivatives of adamantane formed by substitution at C(1), Collect. Czechoslov. Chem. Commun., 1966, 31, 435–449 CrossRef.
- O. Dopfer, S. A. Nizkorodov, M. Meuwly, E. J. Bieske and J. P. Maier, Microsolvation of the ammonium ion in argon: Infrared spectra of NH4+-Arn complexes (n = 1–7), Int. J. Mass Spectrom., 1997, 167, 637–647 CrossRef.
- N. Solcà and O. Dopfer, Interaction between aromatic amine cations and quadrupolar ligands: Infrared spectra of aniline+–(N2)n (n = 1–5) complexes, J. Phys. Chem. A, 2002, 106, 7261–7270 CrossRef.
- O. Dopfer, Microsolvation of the ammonia cation in argon: I. Ab initio and density functional calculations of NH3+–Arn (n = 0–5), Chem. Phys., 2002, 283, 63–84 CrossRef CAS.
- O. Dopfer, N. Solcà, R. V. Olkhov and J. P. Maier, Microsolvation of the ammonia cation in argon: II. IR photodissociation spectra of NH3+–Arn (n = 1–6), Chem. Phys., 2002, 283, 85–110 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, Evaluated gas phase basicities and proton affinities of molecules: An update, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- T. Khuu, N. Yang and M. A. Johnson, Vibrational spectroscopy of the cryogenically cooled O- and N-protomers of 4-aminobenzoic acid: Tag effects, isotopic labels, and identification of the E, Z isomer of the O-protomer, Int. J. Mass Spectrom., 2020, 457, 116427 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01947g |
| This journal is © the Owner Societies 2022 |