
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deep sea osmolytes in action: their effect on protein–ligand binding under high pressure stress†
Armin
Kamali
a,
Nisrine
Jahmidi-Azizi
a,
Rosario
Oliva
*b and
Roland
Winter
 *a
*a
aPhysical Chemistry I - Biophysical Chemistry, Department of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn Street 4a, 44227 Dortmund, Germany. E-mail: roland.winter@tu-dortmund.de
bDepartment of Chemical Sciences, University of Naples Federico II, Via Cintia 4, 80126 Naples, Italy. E-mail: rosario.oliva2@unina.it
First published on 24th June 2022
Abstract
Because organisms living in the deep sea and in the sub-seafloor must be able to cope with hydrostatic pressures up to 1000 bar and more, their biomolecular processes, including ligand-binding reactions, must be adjusted to keep the associated volume changes low in order to function efficiently. Almost all organisms use organic cosolvents (osmolytes) to protect their cells from adverse environmental conditions. They counteract osmotic imbalance, stabilize the structure of proteins and maintain their function. We studied the binding properties of the prototypical ligand proflavine to two serum proteins with different binding pockets, BSA and HSA, in the presence of two prominent osmolytes, trimethylamine-N-oxide (TMAO) and glycine betaine (GB). TMAO and GB play an important role in the regulation and adaptation of life in deep-sea organisms. To this end, pressure dependent fluorescence spectroscopy was applied, supplemented by circular dichroism (CD) spectroscopy and computer modeling studies. The pressure-dependent measurements were also performed to investigate the intimate nature of the complex formation in relation to hydration and packing changes caused by the presence of the osmolytes. We show that TMAO and GB are able to modulate the ligand binding process in specific ways. Depending on the chemical make-up of the protein's binding pocket and thus the thermodynamic forces driving the binding process, there are osmolytes with specific interaction sites and binding strengths with water that are able to mediate efficient ligand binding even under external stress conditions. In the binding of proflavine to BSA and HSA, the addition of both compatible osmolytes leads to an increase in the binding constant upon pressurization, with TMAO being the most efficient, rendering the binding process also insensitive to pressurization even up to 2 kbar as the volume change remains close to zero. This effect can be corroborated by the effects the cosolvents impose on the strength and dynamics of hydration water as well as on the conformational dynamics of the protein.
1. Introduction
Efficient binding of ligands to proteins is fundamental to all biochemical processes, including enzymatic reactions, cell signaling, metabolism, DNA replication, transcription and translation.1–3 Further, ligand–protein interactions, including protein–protein interactions, are important targets in medicinal chemistry, for example interactions between antibodies and antigens or between enzymes and small molecule- or peptide-based inhibitors. Hence, elucidating the driving forces involved in ligand binding processes is of particular interest in the biosciences, medicinal chemistry and pharmacology. In most cases, non-covalent bonds, such as electrostatic, H-bonding and hydrophobic interactions, ensure the formation of the protein–ligand complexes, which is often accompanied by conformational changes. However, ligand binding may also be accompanied by hydration changes of the protein and the ligand. In fact, it has been proposed that 'druggable' binding sites are often characterized by ‘unhappy’ high-energy hydration sites, which drives the binding affinity via entropy gain by release of hydration water. However, in some cases, enthalpic hydration contributions may also play a role, e.g. by formation of newly ordered water networks upon binding.4 Further, the activities of the reactants may be affected by the presence of cosolutes in the solution, as for example exemplified by the complex nature of the cellular milieu, affecting both the conformational dynamics and activity coefficients of protein and ligand. Owing to the obvious high complexity of the process, in particular in the cellular environment, many aspects of ligand binding have not been fully explored, yet.Commonly, binding studies of biomolecular systems are carried out at atmospheric pressure. However, besides using temperature variation to reveal enthalpic and entropic contributions to ligand binding (van't Hoff analysis), also complementary pressure dependent studies may be advantageous.5–8 Pressure application enables modulation of intra- and intermolecular interactions (e.g., pressure stabilizes H-bonds and weakens internal salt bridges), and it allows to determine volume changes upon binding with high accuracy (even within fractions of the size of a water molecule), which provides additional information about packing and hydration changes upon ligand binding.8–11 According to the Braun–Le Châtelier principle, pressure favors the state that occupies the smallest possible overall volume.11 Hence, a process that is accompanied by a positive ΔV will be destabilized under pressure, and conversely, a process enhanced by pressure will be accompanied by a negative ΔV. The volume change upon binding, ΔVb, can be obtained by measuring the pressure dependent binding constant, Kb(p), as
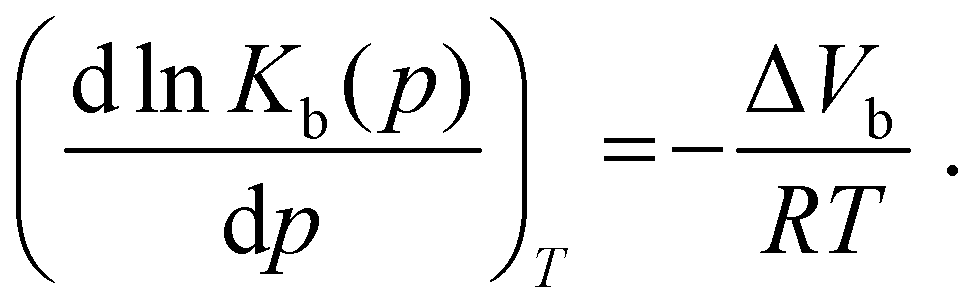 | (1) |
The binding volume, ΔVb, which refers to the difference between the partial molar volumes of the protein–ligand (PL) complex and the uncomplexed state (P + L), can be obtained from absorbance or fluorescence spectroscopic measurements of the pressure dependence of Kb.
Pressure-dependent studies are also of substantial biological interest, in particular for deep-sea biology. The deep oceans on Earth are populated by numerous organisms that are constantly exposed to high hydrostatic pressures (HHP), reaching values even beyond 1000 bar at the deepest ocean trenches (e.g., the Mariana trench at ∼11 km depth) and in sub-seafloor regions.12,13 Hence, knowledge of HHP effects on biological systems is also fundamental for our understanding of life being exposed to such harsh conditions, and of the physical limits of life in general. The molecular effects of pressure on binding equilibria are still largely unknown, however, and the literature on pressure effects on ligand-binding reactions is still scarce.6,7,14–16
Osmolytes, such as carbohydrates or methylamines, play an important role in the regulation and adaptation of the life in deep-sea organisms.17–25 They act as osmoregulators, but they can also stabilize proteins and several have also been found to counteract the deleterious effects of chaotropic cosolutes like urea and HHP on the function of proteins.17,25 Thus, understanding how osmolytes in combination with pressure can impact the formation of complexes between ligands and proteins is of paramount importance for understanding life under extreme conditions.
In this work, we set out to investigate the effects of two prominent osmolytes, trimethylamine-N-oxide (TMAO) and glycine betaine, GB), which play an important role in the regulation and adaptation of life in deep-sea organisms, on the complex formation of a small prototypical aromatic ligand, proflavine, with two proteins, bovine serum albumin (BSA) and, for comparison, human serum albumin (HSA). To this end, pressure dependent fluorescence spectroscopy was applied, supplemented by circular dichroism (CD) spectroscopy and computer simulation (docking) studies. Proflavine (diaminoacridine) belongs to the acriflavine family of compounds, which are also known for their antibacterial and chemotherapeutic potency.26 BSA is a serum albumin protein derived from cows which is often used as a standard in lab experiments. HSA is the most abundant protein in human blood plasma and constitutes about half of the serum proteins. Albumins transport hormones, fatty acids and other compounds, such as small aromatic drugs of interest for medical applications. In addition, many reactions of biological interest involve the interaction of such small ligands with proteins. Hence, BSA and HSA represent a suitable model to study their interaction with a small aromatic ligand and how cellular osmolytes of the type used in this study affect the complex formation. The osmolyte trimethylamine-N-oxide (TMAO) is commonly found in the tissues of marine organisms, for example, in elasmobranches like rays and sharks, and has been found to counteract the deleterious effect of hydrostatic pressure on enzyme activity in deep-sea animals.17,25 TMAO was also found to restore the polymerization ability of tubulin in the presence of urea and upon pressure stress.27,28 It has also been shown to enhance the assembly of the eukaryotic cytoskeletal protein tubulin.29 Glycine betaine (GB) is also a naturally-occurring osmolyte, responsible for maintaining osmotic balance of environmentally stressed cells.22–24 Additionally, like other osmoprotectants, GB can act as a protectant, stabilizing the native structure of proteins and counteracting the denaturing effect of destabilizing cosolutes. Following the widely accepted action mechanism of protein-protecting osmolytes, it is generally believed that the ability of TMAO and GB to protect proteins against denaturation arises from their preferential exclusion from the protein surface, specific interactions between the osmolyte molecules and individual protein backbone units and/or sidechains cannot be excluded, however.18–24 Some studies suggested that TMAO can be slightly attracted to nonpolar groups in proteins.30–33
2. Materials and methods
2.1 Materials
The proteins bovine serum albumin (BSA, molecular weight of 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 463 Da, 583 residues) and human serum albumin (HSA, molecular weight of 66700 Da, 585 residues) in the form of lyophilized powder, proflavine hemisulfate (Pf), the osmolytes trimethylamine-N-oxide (TMAO) and betaine were all purchased from Sigma Aldrich Chemicals (Taufkirchen, Germany). All the sample solutions were prepared in the pressure stable 20 mM Tris–HCl buffer, at the pH of 7.4. Deionized water was used for all buffer and sample preparations.
463 Da, 583 residues) and human serum albumin (HSA, molecular weight of 66700 Da, 585 residues) in the form of lyophilized powder, proflavine hemisulfate (Pf), the osmolytes trimethylamine-N-oxide (TMAO) and betaine were all purchased from Sigma Aldrich Chemicals (Taufkirchen, Germany). All the sample solutions were prepared in the pressure stable 20 mM Tris–HCl buffer, at the pH of 7.4. Deionized water was used for all buffer and sample preparations.
2.2 Sample preparation
The Tris–HCl buffer used for all experiments was adjusted to a pH of 7.4 by addition of HCl and filtered with a syringe filter with a cutoff of 0.45 μm. The stock solution of the proteins BSA and HSA were prepared by dissolving the lyophilized powder in Tris–HCl buffer. By measuring the absorbance at 280 nm with a UV-1800 spectrometer from Shimadzu Corporation (Kyoto, Japan), and using a molar extinction coefficient of 43600 M−1 cm−1 for BSA and 35700 M−1 m−1 for HSA,6,34 the exact concentrations of the proteins were determined. The stock solution of proflavine was also prepared by dissolving the proflavine in Tris–HCl buffer and the concentration was determined by diluting a small amount of the stock solution and measuring the absorbance at 444 nm, using a molar extinction coefficient of 33400 M−1 cm−1.7 All samples were prepared by diluting BSA, HSA and proflavine in 20 mM Tris–HCl buffer, pH 7.4, in the absence and presence of 0.5 M betaine or TMAO.
2.3 Steady-state fluorescence spectroscopy
The interaction between proflavine and the proteins was followed by means of steady-state fluorescence spectroscopy using a K2 fluorometer from ISS (Champaign, IL, USA.). First the experiments were performed at ambient temperature (25 °C). The binding isotherms were obtained by recording the fluorescence intensities of proflavine by exciting the solutions at 448 nm and recording the emission intensities at 504 nm. A series of solutions in Tris–HCl buffer, pH 7.4, were prepared, where the concentration of the proteins was varied between 0 and 400 μM while the concentration of proflavine was fixed at 5 μM. The same experiment was also performed in the same buffer containing 0.5 M TMAO or 0.5 M betaine. To determine the binding constants (Kb), a plot of F/F0 (where F and F0 are the fluorescence intensities of proflavine (Pf) in the presence and absence of the proteins, respectively) as a function of the free protein concentration ([P]) were used.35 The experimental data were fitted according to a 1:1 binding model: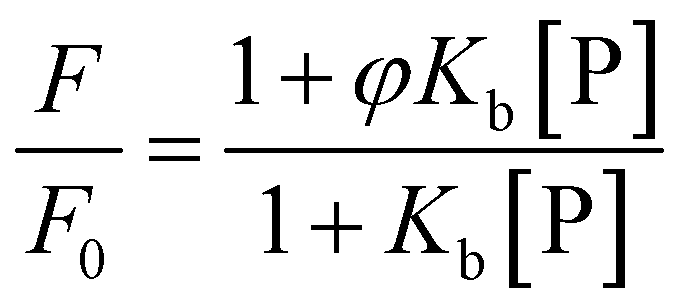 | (2) |
In this equation, φ is the ratio between the quantum yields of complexed and uncomplexed proflavine, i.e., φ = φProflavine–Protein/φProflavine. This parameter is obtained after the fitting procedure. If φ > 1, the binding isotherm will show a positive curvature, indicating that upon binding the intensity of the fluorescence-active species is increasing. Conversely, if φ < 1, the curvature will be negative and the intensity decreases upon complex formation. Kb is the binding constant for the complex formation between proflavine and the protein, defined as Kb = [Pf–P]/([Pf][P]), where [Pf–P], [Pf] and [P] are the concentrations of the complex formed, the free proflavine and the free protein, respectively. Since the free protein concentration is an unknown parameter, from the mass balance, an expression linking the total protein concentration ([P]T) to the free protein concentration, the binding constant and total proflavine concentration ([Pf]T) can be obtained:
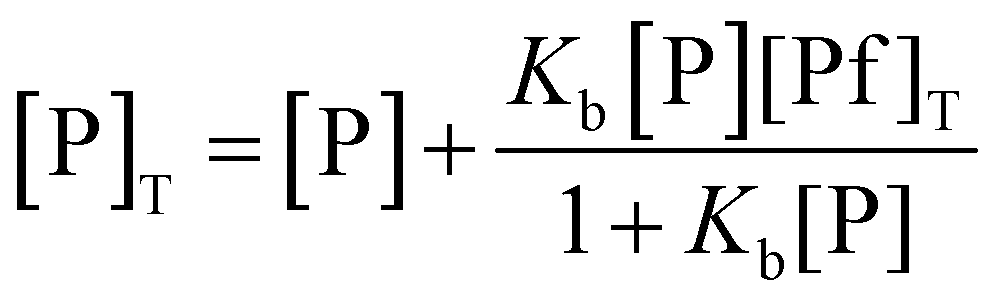 | (3) |
Solving this equation for [P], it is possible to arrive at the final expression linking the free protein concentration to the total protein concentration:
 | (4) |
To obtain the binding isotherm, eqn (4) has to be inserted into eqn (2). To obtain Kb, the experimental data, F/F0vs. [P]T, were fitted by using the above equations until convergence was reached. The data analysis was performed using Origin 2021b software (OriginLab, Northampton, MA, USA).
A high-pressure cell system from ISS with cylindrical quartz cuvettes was used to perform the high hydrostatic pressure measurements. To control the pressure, a manual pump from Nova Werke AG (Illnau-Effretikon, Switzerland) and water as pressurizing fluid was used. The measurements were performed in the pressure range from 1 to 2000 bar. The prepared samples were filled into the cuvette, sealed with DuraSeal™ laboratory stretch film, and then placed into the high-pressure cell. The high-pressure cell was then filled with water and connected to the manual pump through a capillary tube. The plastic film allows the pressure to be transmitted to the sample and prevents it from being released into the water-filled high-pressure cell.
To investigate whether the added osmolytes are able to bind to proflavine and thereby affect the activity of the ligand, its emission spectra were recorded from samples that did not contain any protein. The spectra were recorded in a wavelength range from 465 to 650 nm and an excitation wavelength of 448 nm was used. The wavelength at the maximum was determined by evaluating the center of mass (CM, in nm), defined as:
 | (5) |
2.4 Circular dichroism spectroscopy
To determine the impact of proflavine binding as well as the osmolytes present in solution on the secondary structure of the proteins, circular dichroism (CD) spectroscopy experiments were performed in the far-UV region (190–260 nm). The CD spectra of 12 μM protein and 120 μM proflavine solution were recorded using a Jasco J-715 spectropolarimeter (Jasco Corporation, Tokyo, Japan) with 0.1 cm path-length quartz cuvettes. The CD spectra in the presence of osmolytes were recorded at a TMAO and betaine concentration of 0.1 M. The instrument parameters were set as follows: scan rate of 50 nm min−1, response of 2 s, and bandwidth of 4 nm. The background blank (neat buffer or osmolyte-containing buffer) was subtracted for each sample. The recorded spectra were all normalized per mole of residue and were the results of three accumulations.2.5 Identification of binding sites
The possible binding site for proflavine to BSA and HSA was identified by means of the CavityPlus software, available at https://www.pkumdl.cn:8000/cavityplus/index.php. Detailed information about this software can also be found in the reference provided.36 For identification of the binding site, the protein structures with the following PDB codes were used: 3V03 for BSA (2.70 Å resolution) and 1BM0 for HSA (2.50 Å resolution). The protein structures with the binding sites obtained were visualized using the VMD (Visual Molecular Dynamics) software.373. Results
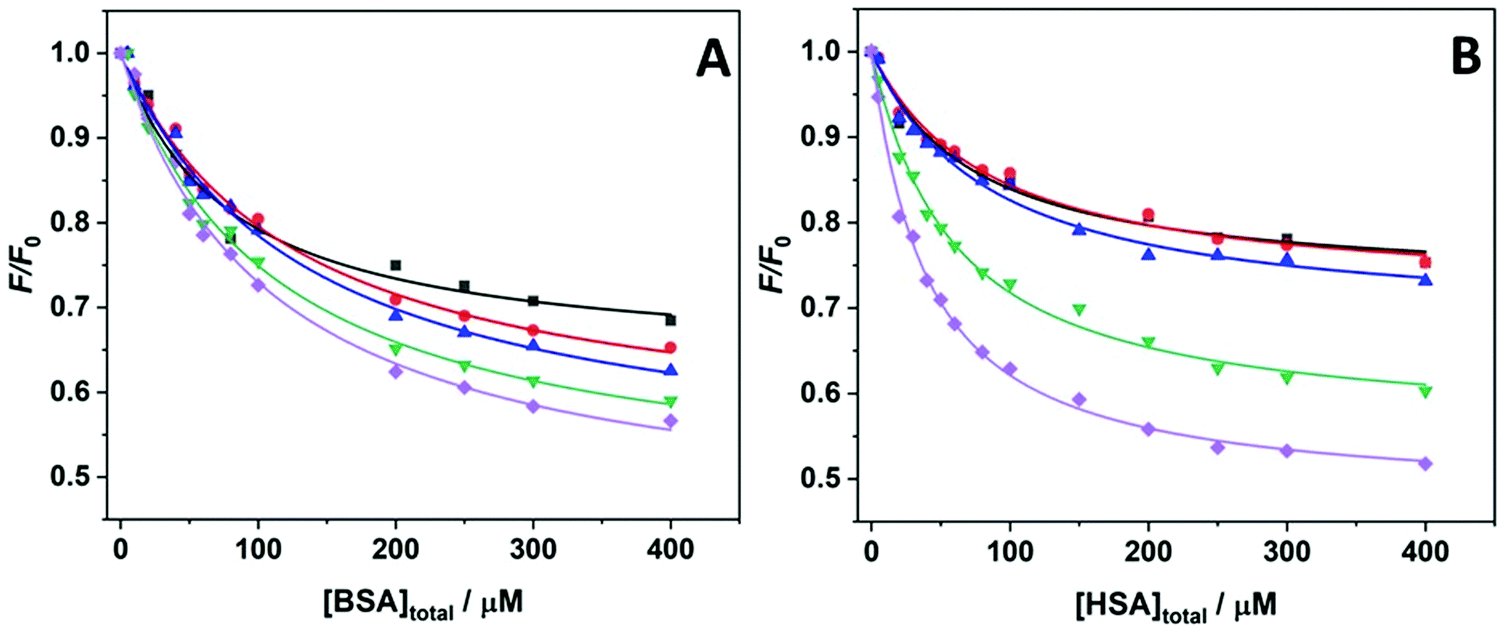
In order to evaluate the impact of the cosolvents TMAO and GB (Fig. 1) on the complex formation between the small aromatic ligand proflavine and the serum proteins BSA and HSA at ambient conditions (T = 25 °C and p = 1 bar), fluorescence titration experiments were carried out. To this end, a series of solutions at fixed concentration of proflavine was prepared while the concentration of the proteins was varied. The extent of binding was determined by following the changes in the fluorescence intensity of proflavine upon excitation at 448 nm, which represents the wavelength maximum of the absorption band of the aromatic molecule. Fig. 1 shows the binding isotherms obtained at ambient conditions for the proflavine binding to BSA (panel A) and to HSA (panel B) in the presence of TMAO and GB, both at 0.5 M concentration. For comparison, the binding isotherms obtained at neat buffer conditions (20 mM Tris–HCl buffer, pH 7.4) are also reported. All the experimental data were fitted with a 1:1 binding model by plotting F/F0vs. the total protein concentration (F and F0 are the fluorescence intensities of proflavine in the presence and in the absence of proteins, respectively). Values of F/F0 < 1 indicate that the intensity of proflavine decreases upon binding. The use of this binding model was justified by the fact that it was previously shown that the stoichiometry of the complex is 1:1 for both proteins.37,38Table 1 summarizes all values of the binding constants.
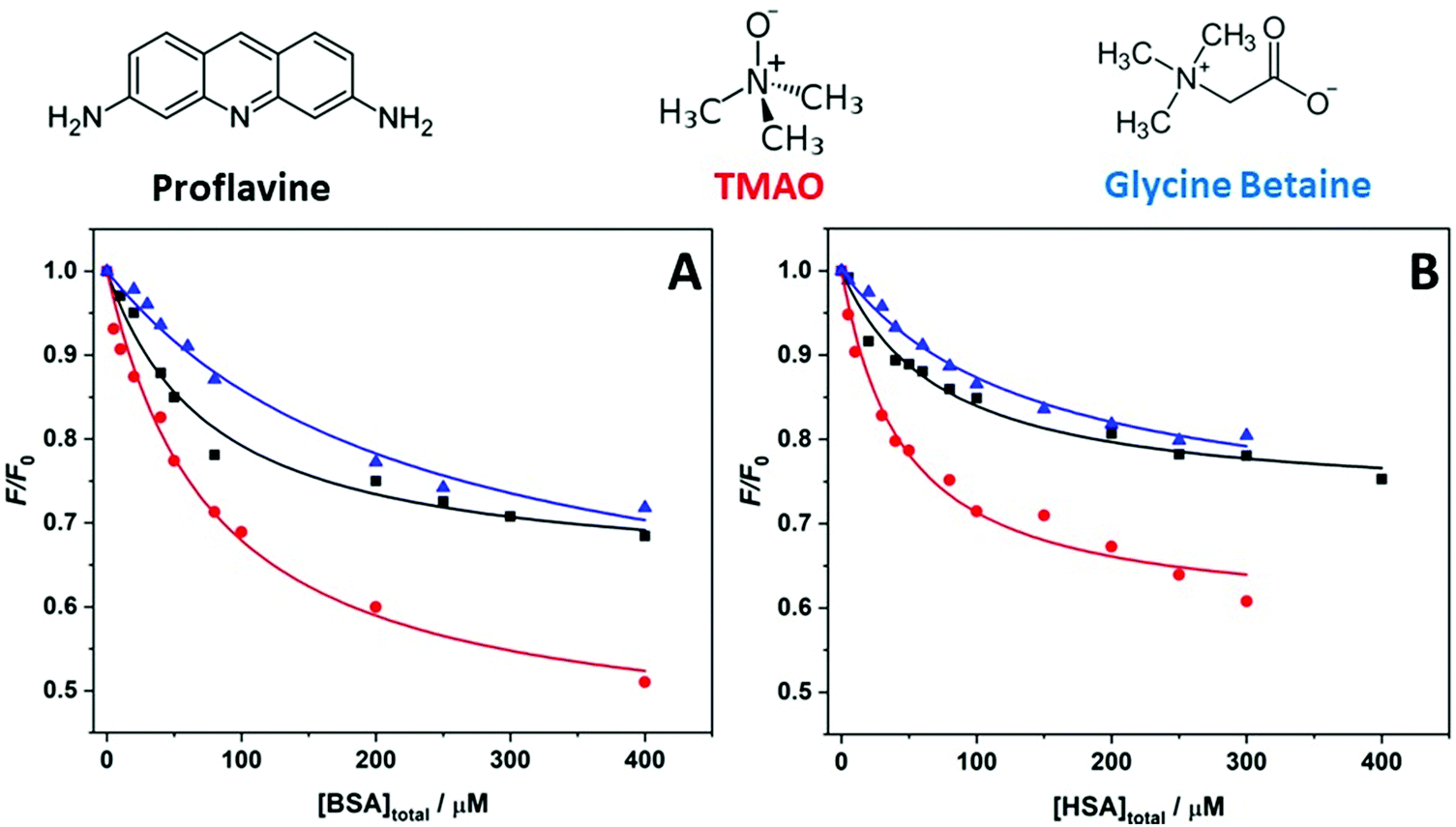 | ||
| Fig. 1 Binding isotherms obtained by means of fluorescence spectroscopy for the complex formation between proflavine and (A) BSA and (B) HSA at the following solution conditions: 20 mM Tris–HCl, pH 7.4 (black squares); 20 mM Tris–HCl, 0.5 M TMAO, pH 7.4 (red circles); 20 mM Tris–HCl, 0.5 M GB, pH 7.4 (blue triangles). The solid lines represent the best fit of experimental data according to a 1:1 binding model. All experiments were performed at the temperature of 25 °C and pressure of 1 bar. | ||
| Proflavine–BSA | Proflavine–HSA | ||
|---|---|---|---|
| Solution conditions | K b/M−1 × 104 | Solution conditions | K b/M−1 × 104 |
| Tris–HCl buffer | 1.6 ± 0.2 | Tris–HCl buffer | 1.4 ± 0.2 |
| +0.5 M TMAO | 1.5 ± 0.4 | +0.5 M TMAO | 2.1 ± 0.3 |
| +0.5 M GB | 0.45 ± 0.07 | +0.5 M GB | 1.2 ± 0.4 |
Complex formation between proflavine and BSA in neat buffer at ambient temperature and pressure conditions is characterized by a binding constant of Kb = (1.6 ± 0.2) × 104 M−1, in very good agreement with previously reported data.38 The addition of the cosolvent TMAO has no significant effect on the binding strength of proflavine to BSA. Within the accuracy of the experiment, the Kb value is the same as the one obtained in buffer-only solution. Surprisingly, the addition of the same concentration of GB has a strong impact on the complex formation, lowering the Kb value by about one order of magnitude. That is, the presence of GB reduces the affinity of proflavine for BSA markedly. For the protein HSA, the binding constant obtained in pure buffer solution is very similar to that for BSA, which is also in agreement with previously reported data.39 In sharp contrast to the results obtained for BSA, the effect of the two cosolvents is different for HSA. In the presence of TMAO, a significant increase of Kb was observed. Instead, addition of GB had no significant effect on the binding characteristics of proflavine to HSA. Of note, the limiting values of F/F0 (at the highest protein concentration) in the presence of the two osmolytes are different with respect to the neat buffer conditions. This observation shows that the relative change in fluorescence intensity of proflavine is dependent on the solvent conditions.
To reveal a potential influence of the ligand binding and the cosolvents on the structure of the proteins, circular dichroism (CD) spectroscopy experiments were performed.40Fig. 2 shows the CD spectra of BSA and HSA in the absence and in the presence of proflavine under buffer-only conditions. The CD spectrum of BSA in neat buffer is characterized by the presence of two minima, located at about 208 nm and 222 nm, respectively (Fig. 2A). In addition, a positive band centered at 194 nm is visible, in agreement with literature data.6 These spectral features designate a protein that has mainly an α-helical structure.40 However, the intensities of the two minima are not the same, clearly showing that the protein is not 100% folded in a helical structure, some turns as well as unordered regions are also present.41 After addition of the ligand, only very slight changes in the CD spectrum were observed, indicating that the binding of proflavine does not induce significant conformational changes of the secondary structure of the protein. Fig. 2B reports the CD spectra of HSA in the absence and in the presence of proflavine in 20 mM Tris–HCl buffer. The CD spectrum of HSA is very similar to that of BSA, suggesting that the protein adopts also essentially an α-helical fold, consistent with literature data.42 As in the case of BSA, HSA does not change its conformation upon proflavine binding, i.e., the protein retains its major structural features.
 | ||
| Fig. 2 Circular dichroism spectra (mean residue weight ellipticity, [θ]mrw(λ)) of (A) BSA and (B) HSA in the absence (black lines) and in the presence (red lines) of the ligand proflavine in neat buffer conditions. The concentration of the proteins was 12 μM, and the concentration of proflavine was 120 μM. All experiments were performed in 20 mM Tris–HCl buffer, pH 7.4, at ambient temperature (25 °C) and pressure (1 bar). | ||
Fig. S1 (ESI†) displays the CD spectra of the proteins in the presence of both cosolvents and proflavine. It is important to note that TMAO and GB also absorb light in the wavelength region explored. Therefore, although their extinction coefficients are small, it was not possible to record accurate CD spectra at the same relatively high cosolvent concentrations used in the study.43,44 For this reason, CD spectra were recorded at 0.1 M concentration. As expected for these two compatible osmolytes, no significant changes in the spectral features of the BSA/HSA–proflavine complex were observed upon addition of TMAO or GB. Therefore, we can assume that the marked differences observed in some of the Kb-values are not due to a significant conformational change caused by the cosolvents.
To find a possible explanation for the observed changes in the binding constants in the presence of the two cosolvents, fluorescence binding experiments under high hydrostatic pressure conditions (HHP) were performed. The HHP method has proven to be a powerful tool for studying the volumetric properties of biomolecular reactions, such as ligand binding and enzymatic reactions, providing valuable informing on changes in the packing and hydration of the molecules involved in the process.6,11,45 As a representative example, Fig. 3 depicts binding isotherms for the complex formation between proflavine and both proteins in 20 mM Tris–HCl buffer, pH 7.4, in the pressure range between 1 and 2000 bar. The corresponding binding curves in the presence of the osmolytes in the 1–2000 bar pressure range are reported in Fig. S2 (ESI†). The data for the pressure dependence of the binding constant determined for all solution conditions are summarized in Table 2.
 | ||
| Fig. 3 Binding isotherms measured by means of HHP-fluorescence spectroscopy for the complex formation between proflavine and (A) BSA and (B) HSA in 20 mM Tris–HCl buffer, pH 7.4, at the pressures of 1 bar (black squares), 500 bar (red circles), 1000 bar (blue triangles), 1500 bar (green reversed triangles), and 2000 bar (violet diamonds). The solid lines represent the best fit of the experimental data according to a 1:1 binding model. All experiments were performed at the temperature of 25 °C. | ||
| p/bar | Tris–HCl buffer | +0.5 M TMAO | +0.5 M GB |
|---|---|---|---|
| K b/M−1 × 104 | K b/M−1 × 104 | K b/M−1 × 104 | |
| a The first ΔVb value refers to the volume change in the 1–500 bar range. Instead, the second ΔVb value refers to the volume change in the 500–2000 bar range. | |||
| BSA–Proflavine | |||
| 1 | 1.6 ± 0.2 | 1.5 ± 0.4 | 0.45 ± 0.07 |
| 500 | 0.66 ± 0.15 | 1.2 ± 0.5 | 0.52 ± 0.16 |
| 1000 | 0.67 ± 0.15 | 1.4 ± 0.4 | 0.50 ± 0.12 |
| 1500 | 0.84 ± 0.09 | 1.4 ± 0.2 | 0.73 ± 0.23 |
| 2000 | 1.1 ± 0.1 | 1.6 ± 0.1 | 0.76 ± 0.09 |
| ΔVb/mL mol−1 | 43.9 ± 11.4/−9.9 ± 1.9a | −1.4 ± 1.7 | −7.2 ± 1.9 |
| HSA–Proflavine | |||
| 1 | 1.4 ± 0.2 | 2.1 ± 0.3 | 1.2 ± 0.4 |
| 500 | 1.2 ± 0.1 | 2.2 ± 0.5 | 1.0 ± 0.4 |
| 1000 | 1.2 ± 0.1 | 2.0 ± 0.2 | 1.5 ± 0.4 |
| 1500 | 1.8 ± 0.1 | 2.0 ± 0.4 | 1.7 ± 0.4 |
| 2000 | 3.0 ± 0.2 | 2.6 ± 0.5 | 2.0 ± 0.3 |
| ΔVb/mL mol−1 | 4.1 ± 2.0/−22.6 ± 1.4b | −1.7 ± 1.7 | −7.6 ± 2.8 |
The data reported in Table 2 reveal that the effect of HHP on the complex formation between BSA and proflavine is solution dependent. At neat buffer conditions, first an initial decrease of the Kb value was observed, whereas beyond 1500 bar, a significant increase of Kb was seen, the Kb value being still smaller than that at ambient pressure, however. When 0.5 M TMAO was present in the solution, the strength of complex formation was not affected by application of pressure. Surprisingly, in the presence of 0.5 M GB, Kb was initially about a factor of three smaller and increased slightly with pressure, i.e., the application of pressure favored the formation of the complex. A similar scenario was observed for proflavine binding to HSA in neat buffer solution: Kb decreased initially slightly with pressure and increased drastically beyond about 1500 bar, reaching Kb values at 2000 bar that are about twice the value at 1 bar. In the TMAO containing buffer, Kb was not affected by pressure, as was observed with the BSA complex formation. Upon addition of 0.5 M GB to the buffer, Kb increased from 1.2 × 104 M−1 at 1 bar to 2.0 × 104 M−1 at 2000 bar. As in the case of ambient pressure, the limiting values of F/F0 depend on the protein and on the osmolytes present in solution. Under pressure, the limiting values seem to depend also on the strength of complex formation (see Fig. S2 (ESI†) and Table 2).
Measurement of the pressure dependence of the binding constant allows us to determine the volume change upon binding of the ligand. The slope of the plot of ln(Kb) vs. p yields the binding volume, ΔVb, which is defined as the partial molar volume difference between the complexed state and the sum of the proflavine and protein volumes, i.e. ΔVb = Vcomplex – (VProflavine + VProtein). According to the Braun–Le Châtelier principle, the pressure application favors the state that occupies the smallest possible volume.11 Thus, if ΔVb > 0, the complex occupies a larger volume compared to the uncomplexed state, and complex formation is disfavored. Instead, ΔVb < 0 indicates that the complex occupies a smaller volume compared to the uncomplexed state, that is, increasing pressure favors the complex formation. Fig. 4 depicts the plots of ln(Kb) vs. p for the complex formation between proflavine and BSA (panel A) and HSA (panel B). The values of all binding volumes are collected in Table 2. Under buffer-only conditions, ΔVb values were positive in the low-pressure regime (below about 1000 bar) and negative at higher pressure. Instead, in the presence of 0.5 M TMAO, ΔVb was found to be close to zero in both cases, indicating a negligible effect of pressure on complex formation is the presence of this cosolvent. Conversely, when 0.5 M GB was present in the solution, ΔVb of about −7 mL mol−1 was determined for both proteins. This value, even if small, reveals that the pressure is slightly favoring the interaction of the ligand with the serum proteins and that the complexed state occupies a slightly smaller volume with respect to the uncomplexed state. Please recall that the volume change is just a fraction of the volume of one water molecule (the molar volume of water is ∼18 mL mol−1).
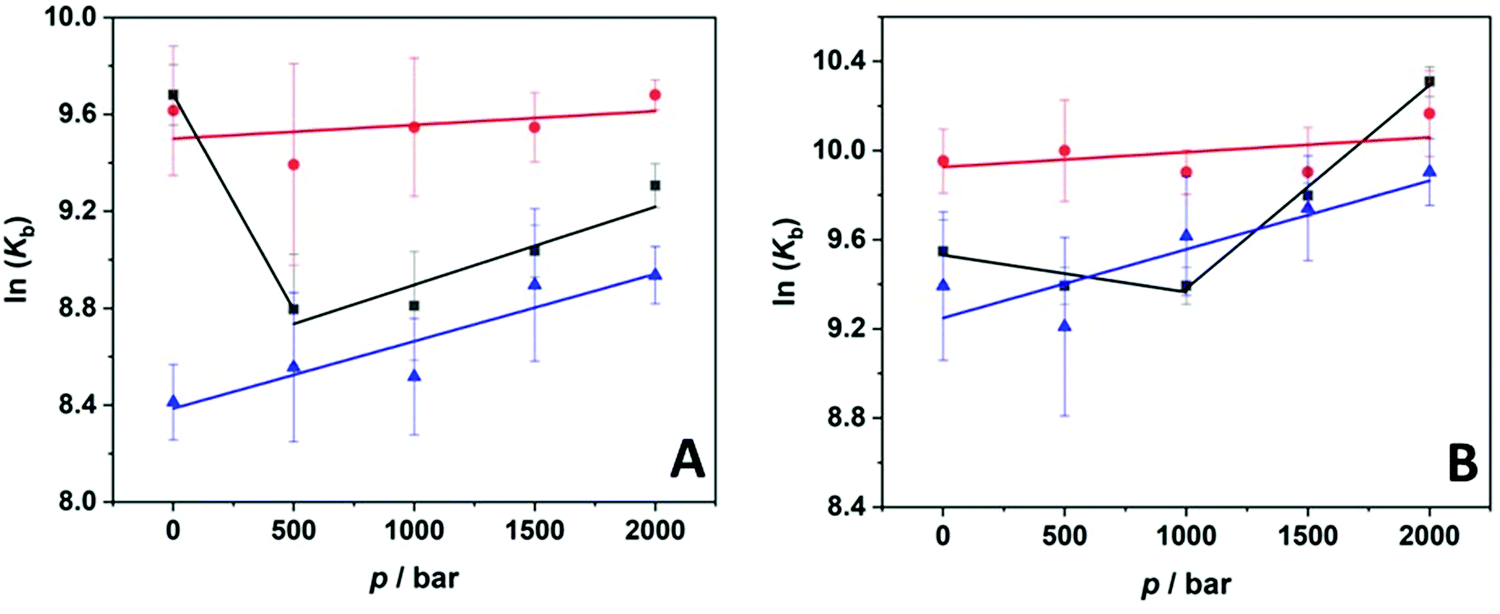 | ||
| Fig. 4 Pressure dependence of the binding constant, Kb, for the complex formation between the ligand proflavine and (A) BSA and (B) HSA in 20 mM Tris–HCl buffer, pH 7.4 (black squares) and in buffer containing 0.5 M TMAO (red circles) and 0.5 M GB (blue triangles) at T = 25 °C. From the slope of ln(Kb) vs. p, the binding volume, ΔVb, was obtained. | ||
Finally, in order to reveal if the two cosolvents have an impact on the microenvironment experienced by the ligand proflavine at high pressures, pressure-dependent fluorescence emission spectra of proflavine in the absence and in the presence of the cosolvents were recorded. The position of the maximum of emission is known to be sensitive to the polarity of the surrounding environment.46,47 Fluorophores located in a more hydrophobic environment show a blue-shifted maximum (lower wavelengths) compared to a more hydrophilic environment. First, we evaluated the center of mass of the proflavine spectrum at neat buffer conditions and in the presence of 0.5 M TMAO and GB at 25 °C and 1 bar. We found that the center of mass (located at 525 nm) did not change in the presence of the two cosolvents, indicating that no direct interaction of the cosolvents with the ligand occurred, in other words, the activity of the ligand did not seem to be significantly affected by the cosolvents. Fig. 5 shows the wavelength shift, Δλ = λp – λp0 for proflavine at neat buffer conditions (20 mM Tris–HCl, pH 7.4) and in the presence of 0.5 M TMAO and 0.5 M GB as a function of pressure. Here, λp and λp0 are the center of mass of the spectra at a given pressure, p, and at 1 bar, respectively; hence, a positive value of Δλ indicates a red shift, a negative value a blue shift. In neat buffer solution, we observed a linear increase of the wavelength maximum of emission, which is typical for an enhancement of water–proflavine interactions in the excited state prompted by the application of pressure, which leads to a compression of water molecules in the hydration shell of the ligand. In the presence of the two cosolvents, a similar pressure shift was observed, revealing that the presence of TMAO and GB is not strongly modifying the ligand's hydration shell or interact with the ligand at all pressure conditions. An interpretation in terms of a pronounced thermodynamic depletion of the osmolytes from the ligand interface cannot be proven by such data, however. In other words, the activity of the ligand can be considered largely unaffected by the presence of the cosolvents even at the high concentration employed here, and this holds true for the whole pressure range covered. Thus, all observed changes in the binding constants are most likely due to changes occurring exclusively in the binding pocket of the proteins.
 | ||
| Fig. 5 Pressure dependence of the wavelength shift, Δλ = λp − λp0, of the maximum of the fluorescence spectrum of proflavine at neat buffer conditions (black squares) and in the presence of 0.5 M TMAO (red circles) and 0.5 M GB (blue triangles). All experiments were performed at 25 °C. | ||
4. Discussion
In this work, the binding characteristics of the model ligand proflavine to two well-known serum proteins, BSA (bovine) and HAS (human), were studied in the presence of two prominent osmolytes, trimethylamine-N-oxide (TMAO) and glycine betaine (GB). They play an important role in the regulation and adaptation of life in deep-sea organisms.17,25 TMAO and GB are able to stabilize proteins, and TMAO has been found to counteract the deleterious effects of urea and high pressure on the stability and function of proteins.17,18,24,48,49 Pressure-dependent measurements were also performed to investigate the intimate nature of the complex formation in relation to hydration and packing changes caused by the presence of the cosolvents. In addition, the pressure dependence of the complex formation was determined to provide insight into the effect hydrostatic pressures has on ligand binding reactions in deep sea organisms. We found that the binding of proflavine to BSA and HSA under pure buffer conditions, i.e., in the absence of the two osmolytes, is characterized by very similar binding constants (∼1.5 × 104 M−1) and a biphasic pressure dependence. Kb initially decreased with pressure and increased again at higher pressures. Interestingly, it was found that pressure always has a stabilizing or even favoring effect on ligand binding in the presence of the two compatible osmolytes.The exact position of proflavine in the BSA and HSA structure is not yet known. A tentative identification of the possible binding site can be achieved based on the 3D structures of the two proteins reported in the protein data bank41,50 using CavityPlus software, which allows robust detection of protein binding sites with 3D structural information of proteins as input36 (Fig. 6). In case of BSA, only one binding pocket was found for proflavine, located in the subdomain IIB, which is in accord with literature data revealing a 1:1 binding stoichiometry for several aromatic ligands.51,52 A single binding site was also reported in the study of Chakraborty and Basu.38 In their study, FRET experiments revealed a high energy transfer efficiency between the two Trp residues and proflavine, indicating that the binding pocket is not too far from the aromatic residues. Inspection of the BSA structure reveals that one Trp residue is localized in the subdomain IIA (Fig. 6, panel A), which is indeed close to the identified binding pocket. For the protein HSA, a single binding cavity was identified as well (Fig. 6, panel B). This pocket is localized between the subdomains IIB and IIIA. This location is consistent with a previous study, using FRET methodology, which determined the distance between the single Trp (located in the subdomain IIA) residue of HSA and proflavine to be ∼2.7 nm.39 The residues present in these cavities (see Table S1 (ESI†) for the list of residues present in the binding pockets of BSA and HSA) reveal different types of potential interactions with proflavine, e.g., hydrogen bonds with its nitrogen atoms, hydrophobic, π–π– as well as π–cation interactions. Furthermore, hydration changes upon ligand binding in the pockets can lead to an entropic contribution to the free energy of ligand binding. Although the extent of these contributions to the ligand binding process may be different for the different sites of the two proteins, the total free energies of binding and thus the magnitude of the binding constants observed are similar.
 | ||
| Fig. 6 (A) The structure of the protein BSA (pdb code: 3V03) and (B) HSA (pdb code: 1BM0) represented as ribbon diagrams. The identified binding pockets are represented by transparent light grey surfaces. The insets show magnifications of the binding cavities, highlighting the residues forming the cavity (the residues are represented as balls and sticks; the C atoms are colored in cyan, O atoms in red, N atoms in blue, and S atoms in yellow). The different subdomains of the structures are indicated by different colors: IA (blue), IB (red), IIA (orange), IIB (yellow), IIIA (green), and IIIB (grey). | ||
At ambient conditions, the addition of 0.5 M TMAO to the buffer solution was found to have essentially no effect on the complex formation between the ligand and BSA. In contrast, a significant increase in the binding constant was observed when proflavine interacted with HSA. This increase in Kb could be attributed to a conformational change of the binding pocket by TMAO. However, the reported CD data showed that TMAO (as well as GB) had no effect on the secondary structure of HSA (and BSA), though a minor local conformational change cannot be excluded. Another factor that could contribute to the observed result is a change in the activity of the ligand and/or protein by the cosolvent. According to the fluorescence data, a change in ligand activity (coefficient) can most likely be ruled out, since no changes in the emission spectra of proflavine were observed in the presence of the osmolytes (Fig. 5). Another possibility would be a direct interaction of TMAO with certain amino acid residues in the active site of the protein. Indeed, it has been reported that TMAO can form H-bonding interactions with the backbone amide and basic residues such as Lys.49 However, such an effect would have to lead to a decrease in the binding constant. Therefore, another explanation must be operational. TMAO, with its large dipole moment of ∼8 D, can form strong interactions with water molecules by forming H-bonds with at least three of these molecules,18,53 resulting in pronounced depletion of TMAO from protein surfaces (solvophobic effect), in other words, in preferential hydration of the protein surface, which is the basis for the stabilizing effect of TMAO on protein structure. How can this unique property of TMAO affect the ligand binding process?
Complex formation was found to be enthalpy-driven for BSA (−47 kJ mol−1) and accompanied by an unfavorable entropy change of −69 J mol−1 K−1,38 which can be attributed to a loss of conformational entropy of the protein. In contrast, the binding of proflavine to HSA is entropy driven. The entropy change upon binding, ΔSb, is 510 J mol−1 K−1, and the enthalpy change, ΔHb, is 127 kJ mol−1,39i.e., binding is an endothermic process. The large and positive ΔHb value is most likely largely due to the breaking of hydrogen bonds during binding, possibly including those of hydrating water molecules. The observed entropy increase can be attributed to the release of the structured hydration water surrounding the binding pocket to the bulk upon proflavine binding. Consequently, it is plausible to assume that the addition of TMAO promotes the depletion of water molecules at the binding site of HSA, leading to the observed increase in Kb. Such a scenario would be consistent with a lower enthalpy change for complex formation in the presence of TMAO. For this reason, the enthalpy change for such a process in the presence of TMAO (see Fig. S3, ESI†) was estimated by determining the value of Kb at three different temperatures (5, 15, and 25 °C) and applying the van't Hoff relation.54,55 It is important to note that the same approach was used to determine the enthalpy change for the complex formation between BSA, HSA and proflavine under neat buffer conditions reported above. The value of the enthalpy change obtained is 12.9 ± 8.0 kJ mol−1, which is about ten times lower than the value in pure buffer solution. This result indicates that TMAO is able to efficiently modulate the entropic contribution to ligand binding due to its strong hydration propensity. Conversely, the TMAO-induced water loss is less pronounced in BSA because, as suggested by the above thermodynamic parameters, the binding process is enthalpy driven, leading to a negligible effect of TMAO on the complex formation between the ligand and BSA.
In the presence of 0.5 M GB, the binding constant for the interaction of proflavine with HSA does not change compared to the buffer-only conditions. Surprisingly, however, a significant decrease in Kb was observed for the complexation of the ligand with BSA. The reported CD data indicate that no significant conformational changes of BSA occurred in the presence of GB. Thus, a conformational change due to the cosolvent can most likely be excluded as a possible cause for the decrease Kb. GB, like TMAO, is a zwitterionic stabilizer of proteins and is also thought to be readily removed from their surfaces (at high concentrations).23,24,56,57 However, GB is able to establish specific interactions with some amino acid residues of the protein, such as π–cation interactions with aromatic residues and H-bonds with the amide and cationic groups.22–24 A glance at Table S1 (ESI†) shows that the binding cavity of BSA harbors three Lys residues and one Arg residue that are capable of direct interaction with the carboxylate group of GB. Such an interaction could lead to partial occlusion of the binding site, resulting in the observed decrease in Kb value. An inspection of the binding site shown in Fig. 6 shows that not all charged residues could be available to interact with GB. An Arg and Asp residue are close to each other, possibly forming a salt-bridge. According to their location, not all Lys residues seem to be capable to interact with GB, however. By comparison, the binding pocket of HSA has only one Arg residue, rendering the binding pocket less susceptible to GB binding and hampering the binding of proflavine to HSA. Hence, as shown here for the two compatible osmolytes TMAO and GB, osmolytes can have dramatically different effects on the binding of a ligand to these two similar proteins due to their different chemical makeup and the different nature and spatial distribution of the amino acid residues in the binding pocket of the two proteins. A more accurate molecular picture could possibly be obtained employing molecular dynamics simulation studies, which will require suitable force fields that should include both the effects of osmolytes and high-pressure to yield precise volumetric information. Such force fields are probably not yet always available, rendering our site-identification study still a valuable tool for understanding the protein–ligand interaction under such conditions. For TMAO, however, significant progress has been made in the last years to develop pressure dependent force-fields, which reproduce solution thermodynamics and have been successfully applied to explain thermodynamic stabilization of various peptide sequences.33,58
Changes in protein dynamics can also be caused by the solvent.59 Osmolytes can alter the ability of residues to move, rearrange and even lead to changes in the internal protein dynamics. Elastic incoherent neutron scattering (EINS) was recently performed to determine pressure-induced changes in the mean-squared displacement (MSD) of the hydrogen atoms of lysozyme, and to gain insights into the effects of cosolvents, such as TMAO, on the averaged sub-nanosecond dynamics of the protein in the pressure range from ambient to 4 kbar. Al-Ayoubi et al. observed a clear effect of TMAO on the internal hydrogen dynamics, namely a significantly reduced mobility (MSD value), rendering the dynamics of the protein rather pressure insensitive.60,61 Please recall that the average squared volume fluctuations are correlated with the compressibility coefficient, βT, of the system, 〈δV2〉 = kBTVβT, and largest volume fluctuations are generally observed near the active site of proteins.5,62,63 The volume fluctuations of a protein molecule are in fact substantial (∼30 mL mol−1) and comparable in magnitude to the volume change observed for unfolding of proteins.62 Of note, it has recently been found that a change in collective rotational dynamics of water in osmolyte solutions is likely to have also a dominant effect on protein denaturation.21 According to THz spectroscopy analysis, osmolytes that stabilize proteins like TMAO are accompanied by stronger bound hydration water with slower dynamics, leading to higher protein stability (higher protein denaturation temperatures), while the collective rotational dynamics of water is accelerated in the case of denaturant osmolytes like urea.
Pressure modulation allowed us to decipher the volumetric properties of the complex formation in terms of packing (including fluctuations) and hydration changes and the effects of cosolvents on them.64,65 Under pure buffer conditions, a biphasic pressure dependence of lnKb was observed, ΔVb for both proteins is positive in the low-pressure range and becomes negative at higher pressures (above about 1400 bar). Such behavior could originate from structural fluctuations of the protein–ligand complex in the lower pressure-regime, which would contribute positively to the volume change (i.e., the first term in ΔVb = Vcomplex − (VProflavine + VProtein) will dominate the second term) and are expected to decrease with increasing compression. Such effect is expected to be smaller in the presence of compatible osmolytes such as TMAO that are known to lead to a decrease of structural fluctuations (see above), rendering Vcomplex smaller.60 In case of efficient binding, the volume change for the actual binding event contributes generally negatively, and this effect seems to dominate at higher pressures. In fact, in the presence of TMAO, the binding volume for complexation of proflavine with the serum proteins was found to be close to zero, and ΔVb ≈ 0 indicates that under these solution conditions the packing between the ligand and the proteins is almost perfect. Packing defects, i.e., empty micro-voids, would lead to an increase in Vcomplex and hence ΔVb. Positive ΔVb values would also occur upon a significant release of hydrating water molecules into bulk water (leading to an increase of Vcomplex) because the hydration water of proteins is usually about 10% denser compared to pure water, and the compressibility of the hydration layer ∼75% of that of bulk water.64 In the presence of TMAO, such a ΔVb change can be expected to be attenuated as the addition of TMAO to water leads to a small increase in density and an increase in the hydrogen bonding strength of the solvent.53 In the presence of 0.5 M GB, the binding volume of proflavine to both proteins was found to be slightly negative (ΔVb = −7.4 mL mol−1). Such negative values can be explained by a small decrease in void volume upon binding,65 for example, due to decreased fluctuations of the protein framework caused by complex formation, resulting in a small decrease in the overall volume of the system.
5. Concluding remarks
In conclusion, we have shown that pressure-dependent studies can provide valuable new information about thermodynamic properties of ligand-binding reactions. Unlike temperature, the effect of pressure on a ligand-binding reaction depends solely on the volume change in the course of the interaction, since the thermal energy remains constant, and therefore the recorded pressure changes can be used to determine accurate volumetric data of complexation reactions. The magnitude and sign of the binding volume provide valuable information about packing properties, structural fluctuations, the interaction mechanism, and hydration changes of the protein and ligand, which are of particular importance when cosolvents are present in the solution. Because organisms living in the deep sea must cope with hydrostatic pressures of up to 1000 bar or more, their biomolecular processes, including ligand-binding reactions, must be tuned to keep the associated volume changes low in order to function efficiently. As we have seen, such adaptation can be achieved by the presence of particular cosolvents.Next to improving protein stability by introducing stabilizing mutations in the sequence of their amino acids, nearly all organisms use organic solutes, such as polyols (e.g., glucose, sorbitol, trehalose), amino acids (e.g., glycine), and methylamines (e.g., TMAO, GB, beta-hydroxybutyrate) to protect their cells against adverse conditions to counteract osmotic imbalance and to stabilize proteins against adverse environmental conditions.25,66 They have been shown to significantly increase not only the temperature stability, but also the pressure stability (+1.5 … 2.5 kbar M−1) of proteins.67,68 Here, we show that the osmolytes are also able to modulate ligand binding processes, and this in a specific manner. Depending on the chemical make-up of the binding sites and hence the thermodynamic forces (ΔHbvs. ΔSb) driving the binding process, osmolytes with different binding properties (solvophobic vs. specific binding to surface groups) and interaction strengths with water (kosmotropic vs. chaotropic) may be chosen. In the case of proflavine binding to BSA and HSA, addition of both cosolvents leads to an increase of the binding strength, Kb, upon pressurization, with TMAO being the most efficient cosolvent, rendering the binding process also quite insensitive to pressurization even up to the 2 kbar level by keeping the volume change, ΔVb, close to zero. This effect may be corroborated by the effects cosolvents impose on the strength and dynamics of hydration water as well as on the conformational dynamics of the protein, for example by decreasing fluctuations of the protein framework as caused by TMAO–water solutions, resulting in the dynamics of the protein becoming rather pressure insensitive.60
To conclude, enhancing our understanding of the role of cosolvents in ligand binding processes will not only help understanding adaptation mechanisms of organisms thriving in high-pressure environments and other harsh geological settings, but will also have important ramifications for regulating binding affinities in biotechnological and pharmaceutical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC-2033 – Projektnummer 390677874.References
- Protein-Ligand Interactions, in Methods and Principles in Medicinal Chemistry Series, ed. H. Gohlke, Wiley-VCH, Weinheim, 2012, vol. 53 Search PubMed.
- X. Du, Y. Li, Y.-L. Xia, S.-M. Ai, J. Liang, P. Sang, X.-L. Ji and S.-Q. Liu, Insights into protein-ligand interactions: Mechanisms, models, and methods, Int. J. Mol. Sci., 2016, 17, 144 CrossRef PubMed.
- E. Di Cera, Mechanisms of ligand binding, Biophys. Rev., 2020, 1, 011303 CrossRef CAS PubMed.
- J. F. Darby, A. P. Hopkins, S. Shimizu, S. M. Roberts, J. A. Brannigan, J. P. Turkenburg, G. H. Thomas, R. E. Hubbard and M. Fischer, Water networks can determine the affinity of ligand binding to proteins, J. Am. Chem. Soc., 2019, 141, 15818–15826 CrossRef CAS PubMed.
- D. J. Wilton, R. Kitahara, K. Akasaka, M. J. Pandya and M. P. Williamson, Pressure-dependent structure changes in Barnase on ligand binding reveal intermediate rate fluctuations, Biophys. J., 2009, 97, 1482–1490 CrossRef CAS PubMed.
- R. Oliva, S. Banerjee, H. Cinar, C. Ehrt and R. Winter, Alteration of protein binding affinities by aqueous two-phase systems revealed by pressure perturbation, Sci. Rep., 2020, 10, 8074 CrossRef CAS PubMed.
- R. Oliva, N. Jahmidi-Azizi, S. Mukherjee and R. Winter, Harnessing pressure modulation for exploring ligand binding reactions in cosolvent solutions, J. Phys. Chem. B, 2021, 125, 539–546 CrossRef CAS PubMed.
- High Pressure Bioscience: Basic Concepts, Applications and Frontiers, in Subcellular Biochemistry, ed. K. Akasaka, Springer, Dordrecht, 2015 Search PubMed.
- J. L. Silva, A. C. Oliveira, T. C. R. G. Vieira, G. A. P. de Oliveira, M. C. Suarez and D. Foguel, High-pressure chemical biology and biotechnology, Chem. Rev., 2014, 114, 7239–7267 CrossRef CAS PubMed.
- K. Heremans and L. Smeller, Protein structure and dynamics at high pressure, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1998, 1386, 353–370 CrossRef CAS.
- R. Winter, Interrogating the structural dynamics and energetics of biomolecular systems with pressure modulation, Annu. Rev. Biophys., 2019, 48, 441–463 CrossRef CAS PubMed.
- I. Daniel, P. Oger and R. Winter, Origins of life and biochemistry under high-pressure conditions, Chem. Soc. Rev., 2006, 35, 858 RSC.
- F. Meersman, I. Daniel, D. H. Bartlett, R. Winter, R. Hazael and P. F. McMillan, High-pressure biochemistry and biophysics, Rev. Mineral. Geochem., 2013, 75, 607–648 CrossRef CAS.
- S. Marchal, R. Lange, P. Tortora and C. Balny, High pressure as a tool for investigating protein–ligand interactions, J. Phys.: Condens. Matter, 2004, 16, S1271–S1275 CrossRef CAS.
- Z. Toleikis, V. A. Sirotkin, G. Skvarnavičius, J. Smirnovienė, C. Roumestand, D. Matulis and V. Petrauskas, Volume of Hsp90 protein-ligand binding determined by fluorescent pressure shift assay, densitometry and NMR, J. Phys. Chem. B, 2016, 120, 9903–9912 CrossRef CAS PubMed.
- A. Levin, S. Cinar, M. Paulus, J. Nase, R. Winter and C. Czeslik, Analyzing protein-ligand and protein-interface interactions using high pressure, Biophys. Chem., 2019, 252, 106194 CrossRef CAS PubMed.
- P. H. Yancey, Cellular responses in marine animals to hydrostatic pressure, J. Exp. Zool., Part A, 2020, 333, 398–420 CrossRef CAS PubMed.
- D. R. Canchi and A. E. García, Cosolvent effects on protein stability, Annu. Rev. Phys. Chem., 2013, 64, 273–293 CrossRef CAS PubMed.
- M. A. Schroer, Y. Zhai, D. C. Wieland, C. J. Sahle, J. Nase, M. Paulus, M. Tolan and R. Winter, Exploring the piezophilic behavior of natural cosolvent mixtures, Angew. Chem., Int. Ed., 2011, 50, 11413–11416 CrossRef CAS PubMed.
- K. Julius, J. Weine, M. Berghaus, N. König, M. Gao, J. Latarius, M. Paulus, M. Schroer, M. Tolan and R. Winter, Water-mediated protein-protein interactions at high pressures are controlled by a deep-sea osmolyte, Phys. Rev. Lett., 2018, 121, 038101 CrossRef CAS PubMed.
- M. Hishida, R. Anjum, T. Anada, D. Murakami and M. Tanaka, Effect of osmolytes on water mobility correlates with their stabilizing effect on proteins, J. Phys. Chem. B, 2022, 126, 2466–2475 CrossRef CAS PubMed.
- Y. L. Shek and T. V. Chalikian, Interactions of glycine betaine with proteins: insights from volume and compressibility measurements, Biochemistry, 2013, 52, 672–680 CrossRef CAS PubMed.
- A. Acharyya, D. Shin, T. Troxler and F. Gai, Can glycine betaine denature proteins?, Phys. Chem. Chem. Phys., 2020, 22, 7794–7802 RSC.
- E. J. Guinn, L. M. Pegram, M. W. Capp, M. N. Pollock and M. T. Record Jr, Quantifying why urea is a protein denaturant, whereas glycine betaine is a protein stabilizer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16932–16937 CrossRef CAS PubMed.
- A. Macdonald, Life at High Pressure: In the Deep Sea and Other Environments, Springer, Switzerland, 2021 Search PubMed.
- W. A. Denny, Acridine derivatives as chemotherapeutic agents, Curr. Med. Chem., 2002, 9, 1655–1665 CrossRef CAS PubMed.
- M. Gao, C. Held, S. Patra, L. Arns, G. Sadowski and R. Winter, Crowders and cosolvents - major contributors to the cellular milieu and efficient means to counteract environmental stresses, ChemPhysChem, 2017, 18, 2951–2972 CrossRef CAS PubMed.
- C. Rosin, P. H. Schummel and R. Winter, Cosolvent and crowding effects on the polymerization kinetics of actin, Phys. Chem. Chem. Phys., 2015, 17, 8285–8540 RSC.
- P. H. Schummel, C. Anders, M. Jaworek and R. Winter, Cosolvent and crowding effects on the temperature- and pressure-dependent dissociation process of the α/β-tubulin heterodimer, ChemPhysChem, 2019, 20, 1098–1109 CrossRef CAS PubMed.
- P. Ganguly, J. Polaók, N. F. A. van der Vegt, J. Heyda and J.-E. Shea, Protein stability in TMAO and mixed urea-TMAO solutions, J. Phys. Chem. B, 2020, 124, 6181–6197 CrossRef CAS PubMed.
- J. Mondal, D. Halverson, I. T. S. Li, G. Stirnemann, G. C. Walker and B. J. Berne, How osmolytes influence hydrophobic polymer conformations: A unified view from experiment and theory, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9270–9275 CrossRef CAS PubMed.
- Y.-T. Liao, A. C. Manson, M. R. DeLyser, W. G. Noid and P. S. Cremer, Trimethylamine N-oxide stabilizes proteins via a distinct mechanism compared with betaine and glycine, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2479–2484 CrossRef CAS PubMed.
- A. Folberth, J. Polák, J. Heyda and N. F. A. van der Vegt, Pressure, peptides, and a piezolyte: Structural analysis of the effects of pressure and trimethylamine-N-oxide on the peptide solvation shell, J. Phys. Chem. B, 2020, 124, 6508–6519 CrossRef CAS PubMed.
- C. Leggio, L. Galantini and N. V. Pavel, About the albumin structure in solution: cigar expanded form versus heart Normal shape, Phys. Chem. Chem. Phys., 2008, 10, 6741–6750 RSC.
- K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, Wiley, New York, 1987 Search PubMed.
- Y. Xu, S. Wang, Q. Hu, S. Gao, X. Ma, W. Zhang, Y. Shen, F. Chen, L. Lai and J. Pei, CavityPlus: a web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction, Nucleic Acids Res., 2018, 46, W374–W379 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- B. Chakraborty and S. Basu, Interaction of BSA with proflavin: a spectroscopic approach, J. Lumin., 2009, 129, 34–39 CrossRef CAS.
- E. Gökoğlu, F. Kıpçak and Z. Seferoğlu, Studies on the interactions of 3,6-diaminoacridine derivatives with human serum albumin by fluorescence spectroscopy, Luminescence, 2014, 29, 872–877 CrossRef PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, How to study proteins by circular dichroism, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A.-J. Stewart, M. Chruszcz and W. Minor, Structural and immunologic characterization of bovine, horse, and rabbit serum albumins, Mol. Immunol., 2012, 52, 174–182 CrossRef CAS PubMed.
- T. R. C. Guizado, Analysis of the structure and dynamics of human serum albumin, J. Mol. Model., 2014, 20, 2450 CrossRef PubMed.
- S. A. Fahmy, F. Ponte, I. M. Fawzy, E. Sicilia and H. M. E.-S. Azzazy, Betaine host–guest complexation with a calixarene receptor: enhanced in vitro anticancer effect, RSC Adv., 2021, 11, 24673–24680 RSC.
- T. Kubota, M. Yamakawa and I. Tanaka, Ultraviolet absorption spectra of trimethylamine N-oxide, J. Mol. Spectrosc., 1966, 20, 226–232 CrossRef CAS.
- R. Oliva, S. Banerjee, H. Cinar and R. Winter, Modulation of enzymatic activity by aqueous two-phase systems and pressure – rivalry between kinetic constants, Chem. Commun., 2020, 56, 395–398 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence: Principles and Applications, Wiley, Weinheim, 2nd edn, 2013 Search PubMed.
- Z. Su and C. L. Dias, Individual and combined effects of urea and trimethylamine N-oxide (TMAO) on protein structures, J. Mol. Liq., 2019, 293, 111443 CrossRef CAS.
- S. S. Cho, G. Reddy, J. E. Straub and D. Thirumalai, Entropic stabilization of proteins by TMAO, J. Phys. Chem. B, 2011, 115, 13401–13407 CrossRef CAS PubMed.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Crystal structure of human serum albumin at 2.5 Å resolution, Protein Eng., Des. Sel., 1999, 12, 439–446 CrossRef CAS PubMed.
- F. Macii and T. Biver, Spectrofluorimetric analysis of the binding of a target molecule to serum albumin: tricky aspects and tips, J. Inorg. Biochem., 2021, 216, 111305 CrossRef CAS PubMed.
- L. Fielding, S. Rutherford and D. Fletcher, Determination of protein-ligand binding affinity by NMR: observations from serum albumin model systems, Magn. Reson. Chem., 2005, 43, 463–470 CrossRef CAS PubMed.
- C. Hölzl, P. Kibies, S. Imoto, R. Frach, S. Suladze, R. Winter, D. Marx, D. Horinek and S. M. Kast, Design principles for high-pressure force fields: Aqueous TMAO solutions from ambient to kilobar pressures, J. Chem. Phys., 2016, 144, 144104 CrossRef PubMed.
- N. Jahmidi-Azizi, S. Gault, C. S. Cockell, R. Oliva and R. Winter, Ions in the deep subsurface of Earth, Mars, and icy moons: Their effects in combination with temperature and pressure on tRNA–ligand binding, Int. J. Mol. Sci., 2021, 22, 10861 CrossRef CAS PubMed.
- K. E. Van Holde, W. C. Johnson and P. S. Ho, Principles of Physical Biochemistry, Pearson/Prentice Hall, Upper Saddle River, New Jersey, 2nd edn, 2006 Search PubMed.
- S. Cozzolino, R. Oliva, G. Graziano and P. Del Vecchio, Counteraction of denaturant-induced protein unfolding is a general property of stabilizing agents, Phys. Chem. Chem. Phys., 2018, 20, 29389–29398 RSC.
- M. M. Santoro, Y. Liu, S. M. A. Khan, L. X. Hou and D. W. Bolen, Increased thermal stability of proteins in the presence of naturally occurring osmolytes, Biochemistry, 1992, 31, 5278–5283 CrossRef CAS PubMed.
- C. Hölzl, P. Kibies, S. Imoto, R. Frach, S. Suladze, R. Winter, D. Marx, D. Horinek and S. M. Kast, Design principles for high–pressure force fields: Aqueous TMAO solutions from ambient to kilobar pressures, J. Chem. Phys., 2016, 144, 144104 CrossRef PubMed.
- J. C. Smith, P. Tan, L. Petridis and L. Hong, Dynamic neutron scattering by biological systems, Annu. Rev. Biophys., 2018, 47, 335–354 CrossRef CAS PubMed.
- S. R. Al-Ayoubi, P. H. Schummel, M. Golub, J. Peters and R. Winter, Influence of cosolvents, self-crowding, temperature and pressure on the sub-nanosecond dynamics and folding ability of lysozyme, Phys. Chem. Chem. Phys., 2017, 19, 14230–14237 RSC.
- M. Erlkamp, J. Marion, N. Martinez, C. Czeslik, J. Peters and R. Winter, Influence of pressure and crowding on the sub-nanosecond dynamics of globular proteins, J. Phys. Chem. B, 2015, 119, 4842–4848 CrossRef CAS PubMed.
- A. Copper, A. Thermodynamic fluctuations in protein molecules, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 2740–2741 CrossRef PubMed.
- A. Cooper, Protein fluctuations and the thermodynamic uncertainty principle, Prog. Biophys. Mol. Biol., 1984, 44, 181–214 CrossRef CAS PubMed.
- T. V. Chalikian and R. B. Macgregor, On empirical decomposition of volumetric data, Biophys. Chem., 2019, 246, 8–15 CrossRef CAS PubMed.
- N. Jahmidi-Azizi, R. Oliva, S. Gault, C. S. Cockell and R. Winter, The effects of temperature and pressure on protein-ligand binding in the presence of Mars-relevant salts, Biology, 2021, 10, 687 CrossRef CAS PubMed.
- E. Bremer and R. Krämer, Responses of microorganisms to osmotic stress, Annu. Rev. Microbiol., 2019, 73, 323–334 CrossRef PubMed.
- H. Herberhold, C. Royer and R. Winter, Effects of chaotropic and kosmotropic cosolvents on the pressure-induced unfolding and denaturation of proteins: an FT-IR study on staphylococcal nuclease, Biochemistry, 2004, 43, 3336–3345 CrossRef CAS PubMed.
- C. Krywka, C. Sternemann, M. Paulus, M. Tolan, C. Royer and R. Winter, Effect of osmolytes on pressure-induced unfolding of proteins: a high-pressure SAXS study, ChemPhysChem, 2008, 9, 2809–2815 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01769e |
| This journal is © the Owner Societies 2022 |