
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Re-examining the giant magnetization density in α′′-Fe16N2 with the SCAN+U method
Assa Aravindh Sasikala
Devi
 *a,
Johannes
Nokelainen
bc,
Bernardo
Barbiellini
bc,
Murali
Devaraj
d,
Matti
Alatalo
a and
Arun
Bansil
c
*a,
Johannes
Nokelainen
bc,
Bernardo
Barbiellini
bc,
Murali
Devaraj
d,
Matti
Alatalo
a and
Arun
Bansil
c
aNano and Molecular Systems Research Unit, University of Oulu, P.O. Box 8000, FI-90014, Finland. E-mail: Assa.Sasikaladevi@oulu.fi
bLappeenranta-Lahti University of Technology (LUT), FI-53851 Lappeenranta, Finland
cDepartment of Physics, Northeastern University, Boston, MA 02115, USA
dDepartment of Sciences, Indian Institute of Information Technology Design and Manufacturing, Kurnool, Andhra Pradesh 518002, India
First published on 14th July 2022
Abstract
We present an in-depth discussion of the magnetic ground state of α′′-Fe16N2 within the framework of the density functional theory (DFT). The exchange–correlation effects are treated using a variety of schemes, including the local-spin-density approximation, the generalized-gradient approximation, and the Strongly-Constrained-and-Appropriately-Normed (SCAN) scheme. We also delineate effects of adding an on-site interaction parameter U on the Fe sites. Among all the schemes considered, only SCAN+U is found to capture the surprisingly large magnetization density in α′′-Fe16N2 that has been observed experimentally. Our study shows how the combination of SCAN and self-interaction corrections applied on different Fe sites through the parameter U can reproduce both the correct equilibrium volume and the giant magnetization density of α′′-Fe16N2.
I. Introduction
Permanent magnets for functional applications are currently based largely on the costly rare earths such as Sm, Nd, and Gd and there is great need to find new cost-effective alternatives.1–3 In this connection, magnetic nitrides, such as the iron nitrides, appear to be promising,4 and among these α′′-Fe16N2,5,6 discovered in 1951,7 stands out because it supports a giant magnetization density.8–10 Kim and Takayashi8 have reported a magnetization of 2.58 T and Sugita et al. reported an even higher magnetization of 2.8–3.0 T.11 These large values of the magnetization are surprising because they exceed the maximum magnetization expected from the Slater–Pauling curve,12 which attributes the magnetic saturation of an itinerant ferromagnet solely to the number of empty states in the 3d shell and this number is maximized for the FeCo alloys at a magnetization density of about 2.45 T. Giant magnetization in α′′-Fe16N2 thus implies the emergence of localized magnetic moments in the material. It has been suggested that the giant magnetization of α′′-Fe16N213–15 is not an intrinsic property but that it reflects difficulties of preparing single crystals of Fe16N2 and in performing accurate measurements in multi-phase samples. This topic has been quite controversial: two symposia were held in this connection at the MMM conferences in 1994 and 1996.16 Although Sugita and Jiangs groups reported experiments showing high magnetization, these results were not corroborated by any other group,8,15 and the topic did not draw much attention after 2000. The work of Wang et al.,10 however, which uses many different characterization techniques, including the XMCD, appears to address the issue of giant magnetization. Successful attempts to prepare α′′-Fe16N2 samples have been reviewed by Wang16 and include the synthesis of thick α′′-Fe16N2 foils.17 The most recent X-ray and neutron diffraction results by Hang et al. report saturation magnetizations of 2.6–2.9 T,18 and have been reproduced by a cluster-atom model introduced by Ji et al.19Recent Density Functional Theory (DFT) studies of α′′-Fe16N2 based on the Generalized Gradient Approximation (GGA) corrected by the Hubbard parameter U have been reported by Bhattacharjee et al.,20 Islam et al.,21 and Stoeckl et al.22 These investigations and other DFT+U calculations19,23–27 show, as expected, that the Fe magnetic moment increases with the strength of correlation effects described by U. However, increasing U expands the equilibrium volume when the geometry is optimized,22 so that the magnetization density Ms does not increase beyond the Slater–Pauling limit of 2.45 T.
Here, we show that a DFT+U scheme based on the Strongly-Constrained-and-Appropriately-Normed (SCAN) method28 can avoid the spurious volume expansion encountered in GGA+U, which is also the case for the LSDA+U results of Ji et al.19 SCAN has the satisfactory feature that it satisfies all the 17 exact constraints appropriate to a semilocal functional,28 which is not the case for the GGA.29 For these reasons,30 SCAN has been shown to be systematically superior to GGA and it can be even better than the hybrid functionals,31–33 which present difficulties, for example, in bulk metals34 and cuprate superconductors.35 SCAN+U,36 however, correctly captures the giant magnetization density of α′′-Fe16N2 beyond the Slater–Pauling limit, despite its somewhat larger computational cost compared to GGA+U.
II. Computational details
Spin-polarized electronic structure calculations were performed within the framework of the DFT using the projected augmented wave (PAW) method implemented in the Vienna Ab initio Simulation Package (VASP).37,38 The GGA-PBE (Perdew–Burke–Ernzerhof) method29 was used to account for exchange–correlation effects. The effects of gradient corrections to the local-spin-density approximation (LSDA), which are included in the GGA are important for correctly capturing the magnetic phases of pristine Fe.39Strong Coulomb correlation effects are described within the DFT+U scheme as implemented by Dudarev et al.40 The DFT+U scheme is a simplified version of the self-interaction correction,41 where the correction U acts only on the localized 3d states, and often results in splitting the metallic energy bands into upper and lower Hubbard bands descriptive of Mott–Hubbard physics.42–44 We also use the SCAN meta-GGA scheme,28 which has been shown to provide an improved description of correlated materials such as α-Mn,45 cuprates,46–49 nickelates50 spinel LiMn2O4 cathode material,51 iridates,52 and 3d perovskite oxides.53
Within the PAW pseudopotential scheme, we describe Fe and N with 8 and 5 valence electrons, respectively. A plane wave cut-off energy of 600 eV is used.
k-Points within the Brillouin zone were generated using a uniform 8 × 8 × 8 Monkhorst–Pack54 mesh. Electronic energy minimization was performed with a tolerance of 10−6 eV. The conjugate-gradient algorithm was used to relax atomic structures until all residual forces converged to within 0.005 eV Å−1. A Gaussian smearing width of 0.05 eV (FWHM) was applied to the electronic states. In order to obtain more accurate total energies and density of states (DOS), the tetrahedron method with Blöchl corrections55 was used. Collinear spins were used in most calculations but in order to compute the magneto-crystalline anisotropy (MCA), non-collinear calculations were also performed.
III. Results and discussion
Gradient corrections to the LSDA are needed to stabilize the ferromagnetic phase of α-Fe, which has partially filled 3d majority states with a magnetic moment of m = 2.3μB in the self-consistent linearized muffin-tin-orbital (LMTO) calculations.39 Surprisingly, SCAN corrections beyond the GGA can substantially overestimate the magnetic moment of α-Fe as shown by several authors.56–59 Recently, this problem has been cured by introducing a de-orbitalized SCAN potential.60 The de-orbitalization condition is especially important at the Fermi Surface of itinerant ferromagnetic materials as shown by eqn (24) in the paper by Gunnarsson and Lundqvist.61 This constraint has been discussed also by Barbiellini and Bansil.43 However, the SCAN overestimation of the magnetic moment encountered in itinerant ferromagnetic systems does not apply in the present case since the 3d electrons are clearly more localized due to the magnetization density being above the Slater–Pauling limit.Bulk Fe in the α phase assumes BCC structure with the easy-magnetization axis oriented along the (001) axis.62 Insertion of N expands the volume and brings the easy magnetization axis parallel to the c-axis, producing the MCA. Bonding with N also increases the Fe magnetic moments to the order of 3μB, which points to the need for invoking substantial corrections beyond the GGA. The experimental α′′-Fe16N2 crystal structure7,63 is body-centered tetragonal (space group I4/mmm) with lattice parameters of a = b = 5.72 Å and c = 6.29 Å. The experimental structure shown in Fig. 1 can be seen as a distorted version of a 2 × 2 × 2 supercell of BCC Fe with two N atoms occupying the octahedral interstitial positions. The Fe atoms can be divided into three inequivalent Wyckoff sites 8h, 4e and 4d by their coordination with respect to the N atoms. The Fe-8h and 4e atoms form an octahedron around the N atoms (Fig. 1), where the Fe-8h atoms occupy the equatorial sites and the Fe-4e atoms occupy the axial sites. The Fe-4d atoms are not coordinated with the N atoms. This cell size is sufficient for describing the ferromagnetic phase α′′-Fe16N2. However, larger supercells are needed for more complex orders, as is the case for magnetic phases of the cuprates, etc.48
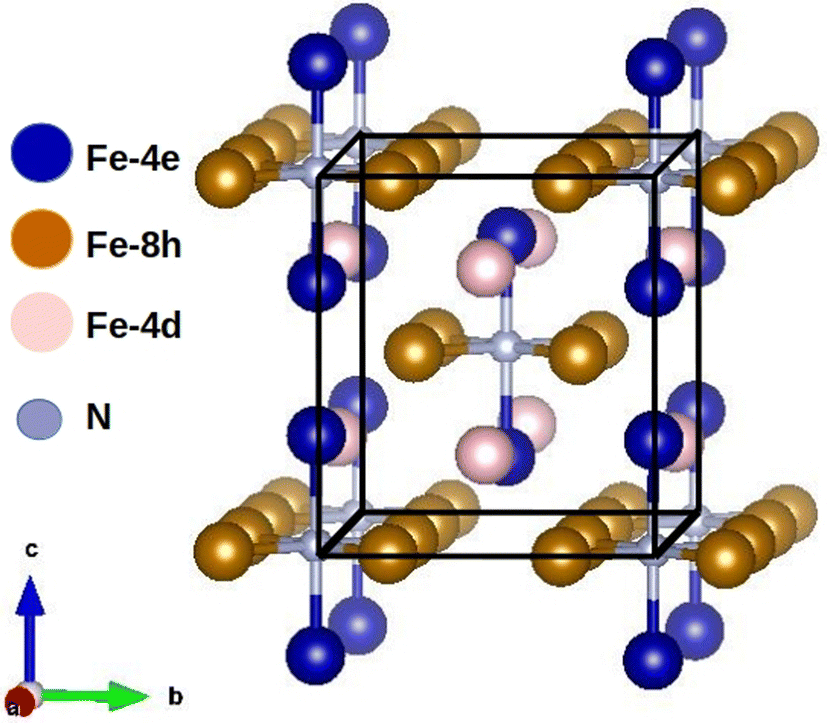 | ||
| Fig. 1 Equilibrium structure of Fe16N2. Fe atoms occupy three inequivalent Wyckoff sites labeled 8h (golden),4e (blue) and 4d (pink). | ||
We first carried out calculations using the fixed experimental volume to ascertain if the giant magnetization might be possible to capture in this way. The results for the LSDA, GGA, GGA+U, SCAN, and SCAN+U corrections are summarized in Table 1. The used U values have been computed by Lai et al.26 using formulae based on screened Coulomb integrals involving the densities of the Fe 3d electrons and have the following values at different atomic sites: Fe-8h: 1.36 eV, Fe-4e: 1.088 eV, and Fe-4d: 3.94 eV. Interestingly, the values of Ms do not exceed the Slater–Pauling limit12 of 2.45 T, even for GGA+U or SCAN, which are expected to improve the description of localized d-states substantially with respect to LSDA and GGA. Only the SCAN+U is found to correctly predict Ms values above the Slater–Pauling limit, thus capturing the subtle balance between tendencies of electrons to form itinerant or localized states in the material.10
| Method | Total moment (μB) | M s (T) |
|---|---|---|
| LSDA | 36.49 | 2.21 |
| GGA | 39.05 | 2.21 |
| GGA+U | 42.75 | 2.42 |
| SCAN | 42.66 | 2.41 |
| SCAN+U | 45.82 | 2.59 |
Next, we fully relaxed all structures in order to understand the connection between the equilibrium volume and magnetic properties. The LSDA produced a total magnetization of 36.49μB per unit cell, which corresponds to an average magnetic moment of 2.28μB per Fe atom. This value is significantly lower than the LSDA-based Fe magnetic moment in BCC Fe, indicating that LSDA encounters problems here in contrast to the case of pristine Fe.39 The equilibrium volume obtained by LSDA (179.24 Å3) is substantially smaller than the experimental value of 205.24 Å3. In view of these shortcomings of the LSDA, we only report the results of our calculations based on the LSDA+U in Table 2.19 The gradient corrections in the GGA improve the volume, but both the volume and the magnetization still remain small compared to the experimental value.18 SCAN yields an increase in the magnetization Ms above the GGA value, but the equilibrium volume slightly shrinks with respect to the GGA. The GGA+U gives volume in excellent agreement with experiment, but it does not improve the magnetization density significantly. Finally, SCAN+U yields reasonable agreement with the experiment both for the volume, magnetization density and the c/a = 1.08 ratio, which is close to the corresponding experimental value of 1.10. The LSDA SCAN+U results in Table 2 thus explain the anomalously large magnetization density. Our calculations in accordance with the LSDA+U method of ref. 19 (see Table 2) are also found to correctly predict the equilibrium volume and the Ms value above the Slater–Pauling limit, but the c/a ratio from LSDA+U is very far from the experimental value. Interestingly, the Heyd, Scuseria and Ernzerhof (HSE) hybrid-functional method31–33 gives a Ms value above the Slater–Pauling limit, but the equilibrium volume is smaller than the experimental value.
| Method | Lattice parameters (Å) | V (Å3) | Total moment (μB) | m: individual atom (μB) | M s (T) | ||||
|---|---|---|---|---|---|---|---|---|---|
| a | c | Fe-8h | Fe-4e | Fe-4d | N | ||||
| GGA | 5.67 | 6.24 | 200.61 | 38.71 | 2.36 | 2.15 | 2.83 | −0.06 | 2.25 |
| GGA+U | 5.72 | 6.29 | 205.80 | 42.48 | 2.54 | 2.35 | 3.22 | −0.07 | 2.39 |
| SCAN | 5.68 | 6.14 | 198.09 | 41.83 | 2.57 | 2.42 | 2.92 | −0.05 | 2.45 |
| SCAN+U | 5.75 | 6.19 | 204.66 | 45.51 | 2.75 | 2.63 | 3.27 | −0.05 | 2.58 |
| HSE33 | 5.62 | 6.23 | 200.46 | 45.50 | 2.82 | 2.74 | 2.95 | −0.05 | 2.64 |
| LSDA+U | 5.83 | 6.02 | 204.91 | 46.28 | 3.47 | 2.66 | 1.99 | −0.06 | 2.63 |
The computed highest Fe magnetic moment is located on Fe-4d, while the neutron diffraction experiments18 find that Fe-8h carries the largest magnetization. We now discuss if another SCAN+U correction could possibly improve the description of the magnetic moment distribution on the Fe atoms. Motivated by the agreement between the model of Ji et al.19 and the results of neutron diffraction experiments,18 we have carried out a parametric SCAN+U(Fe-8h) study in which the Hubbard U is only applied to the Fe-8h position. The results are given in Table 3 and show a stronger magnetic moment at the Fe-8h position in better agreement with experiments.18 The Fe-8h magnetic moment increases monotonically from 2.67μB to 3.24μB with increasing U values. The average magnetic moment reaches 2.93μB for U(Fe-8h) value of 4 eV. The Fe-4d site, which is situated outside of the octahedral cluster, preserves magnetic moment of about 2.8μB irrespective of the change in the U(Fe-8h) value. Interestingly, the Fe-4e magnetic moment is the lowest and reaches a minimum of about 2.8μB around U(Fe-8h) = 2 eV. The equilibrium volume and the magnetization density as a function of the Hubbard parameter applied to the Fe-8h sites are presented in Fig. 2. The experimental volume is reached at U(Fe-8h) = 2.5 eV corresponding to a magnetization density of 2.54 T. Fig. 2 shows that the magnetization density saturates at around 2.58 T compared to the limiting value of 2.45 T predicted by the Slater–Pauling theory.12 The local magnetic moments and the average magnetic moment per Fe ion as a function of U(Fe-8h) are presented in Fig. 3. The moments at Fe-8h sites are seen to increase steadily with increasing U. The moments of the Fe-4d atoms, that are located outside the Fe–N clusters, decrease and stabilize with increasing U(Fe-8h). Interestingly, the Fe-4e atoms that lie inside the Fe–N cluster also show the same trend as the Fe-4d atoms. Nevertheless, it is clear that the SCAN+U scheme, which includes self-interaction corrections within the SCAN formalism, predicts average magnetic moment of about 2.8μB per Fe, which is larger than the Slater–Pauling limit for itinerant ferromagnetism.
| U (eV) | Lattice parameters (Å) | V (Å3) | Total moment (μB) | m: individual atom (μB) | M s (T) | ||||
|---|---|---|---|---|---|---|---|---|---|
| a | c | Fe-8h | Fe-4e | Fe-4d | N | ||||
| 0.5 | 5.69 | 6.15 | 199.47 | 42.52 | 2.67 | 2.42 | 2.89 | −0.034 | 2.48 |
| 1.0 | 5.72 | 6.13 | 200.45 | 42.87 | 2.75 | 2.38 | 2.85 | −0.036 | 2.49 |
| 1.5 | 5.73 | 6.13 | 201.11 | 43.24 | 2.84 | 2.34 | 2.82 | −0.040 | 2.50 |
| 2.0 | 5.76 | 6.11 | 202.54 | 43.78 | 2.92 | 2.32 | 2.81 | −0.041 | 2.52 |
| 2.5 | 5.79 | 6.11 | 204.89 | 44.82 | 3.02 | 2.36 | 2.81 | −0.033 | 2.54 |
| 3.0 | 5.83 | 6.13 | 208.03 | 46.03 | 3.12 | 2.43 | 2.85 | −0.021 | 2.57 |
| 3.5 | 5.85 | 6.14 | 210.19 | 46.67 | 3.19 | 2.44 | 2.86 | −0.019 | 2.58 |
| 4.0 | 5.87 | 6.13 | 211.62 | 46.90 | 3.24 | 2.41 | 2.85 | −0.028 | 2.58 |
| 4.5 | 5.89 | 6.14 | 213.05 | 47.21 | 3.28 | 2.39 | 2.85 | −0.036 | 2.58 |
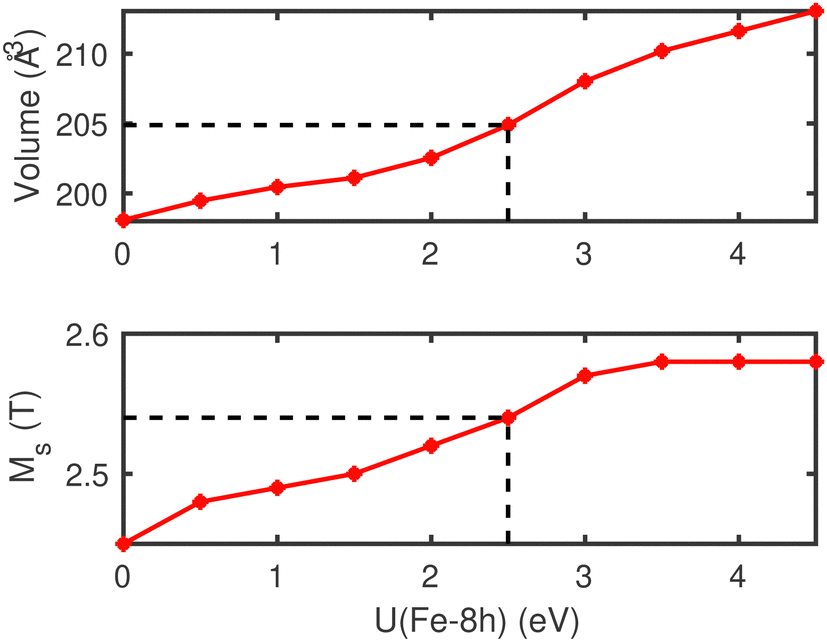 | ||
| Fig. 2 Equilibrium volume and magnetization density using the SCAN+U scheme as a function of U(Fe-8h), where U is applied only to the U(Fe-8h) sites. The computed points are joined by straight lines to guide the eye. The dashed lines mark the values that reproduce the experimental moments and volume. | ||
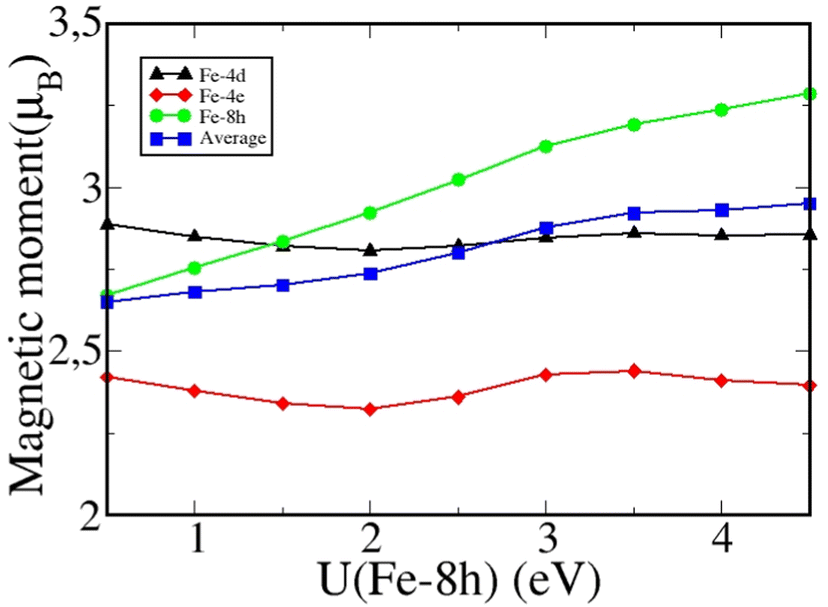 | ||
| Fig. 3 Computed magnetic moments on various Fe sites and the average magnetic moment per unit cell using the SCAN+U scheme as a function of U(Fe-8h), where U is applied only to the Fe-8h sites. | ||
To visualize the electronic structure, we have calculated the density of states (DOS) and the partial DOSs of Fe16N2, see Fig. 4. The three panels of Fig. 4 present results based on GGA, SCAN and SCAN+U(Fe-8h) at U = 2.5 eV. The exchange splitting between the spin-up and spin-down states is seen to increase with the introduction of enhanced correlation effects: for example, the exchange splitting is larger in SCAN compared to the GGA and the addition of U(Fe-8h) further increases the exchange splitting mostly by localizing the eg 3d states of Fe-8h. We have also carried out computations in which spin-orbit coupling effects are included for U(Fe-8h) = 2.5 eV to obtain an MCA energy of 1.3 meV per formula unit, which is consistent with the experimental values reported by Stoeckl et al.22 The magnetic moments were not significantly affected by spin–orbit coupling effects.
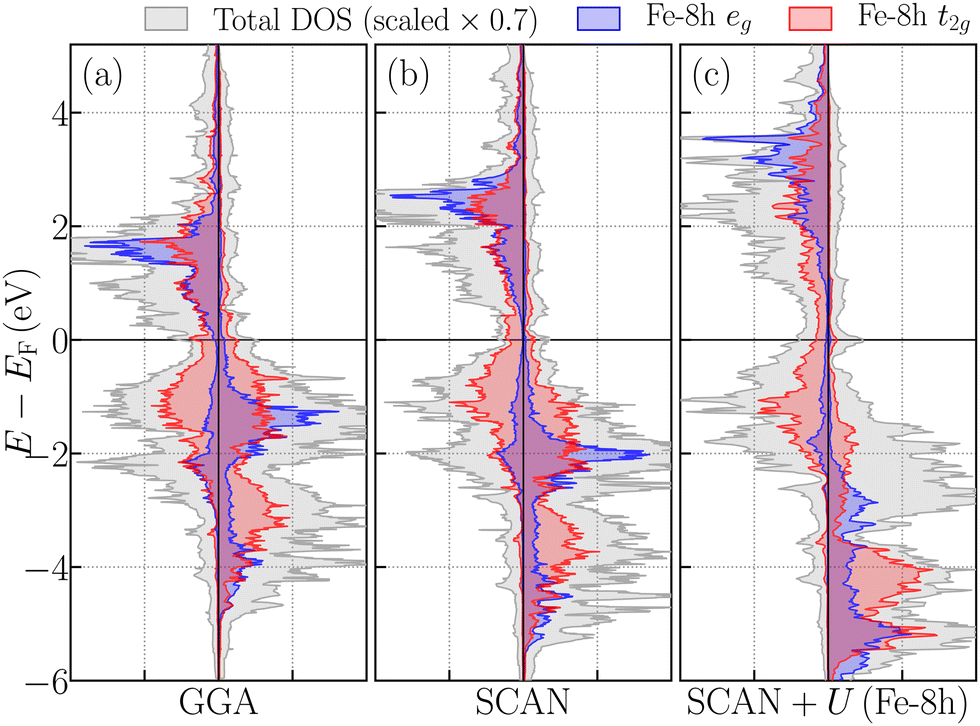 | ||
| Fig. 4 DOS and orbitally-projected and site-resolved partial DOSs (PDOSs) for (a) GGA, (b) SCAN and (c) SCAN+U(Fe-8h), where the Hubbard U is applied only to the Fe-8h sites. The total DOS (scaled by a factor of 0.7) is in gray, the Fe-8h eg PDOS is in blue and the Fe-8h t2g PDOS is in red shading. | ||
Finally, we have performed further SCAN+U calculations in which U is applied uniformly on all Fe sites. Stoeckl et al.22 refer to such a calculation as the baseline case, and the calculations motivated by the results of Ji et al. as the high-moment case because it gave the highest total magnetic moment within their GGA+U scheme. However, Table 4 shows that in the case of SCAN+U both the experimental volume and the giant magnetization density of 2.58 T are recovered for U = 1.5 eV. We have also considered a calculation applying U = 2 eV on Fe 8h and 4e only. The equilibrium volume in this case is 208.55 Å3 and the magnetic moments are 2.953, 2.834 and 2.858μB for 8h, 4e and 4d, respectively. These values are in better agreement with the experimental values given by Hang et al.64 while Ms = 2.59 T remains high. This result highlights the robustness of the SCAN+U scheme: variations in the value of U lead to different combinations of individual magnetic moments, but the experimental average magnetization and volume are obtained in all cases. This is important since the experimental analysis of the magnetic moment distributions remains quite challenging.64,65
| U (eV) | Lattice parameters (Å) | V (Å3) | Total moment (μB) | m: individual atom (μB) | M s (T) | ||||
|---|---|---|---|---|---|---|---|---|---|
| a | c | Fe-8h | Fe-4e | Fe-4d | N | ||||
| 0.5 | 5.70 | 6.15 | 200.33 | 42.89 | 2.64 | 2.53 | 2.95 | −0.037 | 2.49 |
| 1.0 | 5.72 | 6.15 | 201.75 | 43.66 | 2.69 | 2.58 | 2.98 | −0.046 | 2.51 |
| 1.5 | 5.78 | 6.17 | 205.96 | 45.71 | 2.83 | 2.73 | 3.05 | −0.034 | 2.58 |
| 2.0 | 5.81 | 6.19 | 209.42 | 47.09 | 2.92 | 2.82 | 3.11 | −0.041 | 2.62 |
| 2.5 | 5.85 | 6.21 | 212.38 | 47.90 | 2.97 | 2.88 | 3.16 | −0.033 | 2.62 |
| 3.0 | 5.88 | 6.25 | 216.23 | 49.13 | 3.05 | 2.96 | 3.24 | −0.021 | 2.64 |
| 3.5 | 5.91 | 6.28 | 220.19 | 50.15 | 3.10 | 3.04 | 3.30 | −0.019 | 2.65 |
IV. Conclusions
We have presented an in-depth study of magnetism in α′′-Fe16N2 within the framework of the density functional theory (DFT) using a variety of schemes to treat exchange–correlation effects. The experimentally observed giant magnetization density (larger than the Slater–Pauling limit of about 2.45 T) is shown to be captured correctly by the SCAN+U scheme. Predictions of SCAN+U are robust in that magnetization densities surpassing the Slater–Pauling limit as well as the correct equilibrium volume are produced when U is applied either uniformly or unevenly across the various Fe sites. When U is applied only on the Fe-8h sites using the optimal value of 2.5 eV (yields experimental volume), Fe-3d states are localized and the largest magnetic moments on the Fe-8h sites are obtained in agreement with recent neutron diffraction experiments, etc.18 Even when U is applied uniformly on all Fe sites using the corresponding optimal value of 1.5 eV, the total magnetic moment remains high in contrast to the GGA+U results.22 Notably, since U promotes localization of Fe 3d electrons, problems of SCAN with Fe and other itinerant ferromagnets56–59 do not apply to Fe16N2. [Location effects predicted here should be amenable to verification via magnetic Compton scattering experiments.51,66–68] SCAN has been successful in describing elemental Mn, which is a similar non-itinerant transition metal system.45 Finally, we note that since correlation effects weaken directional bonding, DFT generally tends to underestimate the c/a ratio, which could be cured by including spin–orbit interactions and breaking symmetries around the iron atoms.69Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. A. acknowledges support from Academy of Finland grant (311934). Authors gratefully acknowledge CSC-IT, Finland, for computational resources and J. N. acknowledges support from the INERCOM LUT platform. The work at Northeastern University was supported by the US Department of Energy (DOE), Office of Science, Basic Energy Sciences grant number DE-FG02-07ER46352, and benefited from Northeastern University's Advanced Scientific Computation Center (ASCC) and the NERSC supercomputing center through DOE grant number DE-AC02-05CH11231.References
- M. Balintova, M. Holub, N. Stevulova, J. Cigasova and M. Tesarcikova, Chem. Eng. Trans., 2014, 39, 625–630 Search PubMed.
- L. H. Lewis and F. Jiménez-Villacorta, Metall. Mater. Trans., 2013, 44, 2–20 CrossRef CAS.
- R. McCallum, L. Lewis, R. Skomski, M. Kramer and I. Anderson, Annu. Rev. Mater. Res., 2014, 44, 451–477 CrossRef CAS.
- L. Rissanen, M. Neubauer, K. Lieb and P. Schaaf, J. Alloys Compd., 1998, 274, 74–82 CrossRef CAS.
- T. Ogawa, Y. Ogata, R. Gallage, N. Kobayashi, N. Hayashi, Y. Kusano, S. Yamamoto, K. Kohara, M. Doi and M. Takano, et al. , Appl. Phys. Express, 2013, 6, 073007 CrossRef.
- T. Radchenko, O. Gatsenko, V. Lizunov and V. Tatarenko, Prog. Met. Phys., 2020, 21, 580–618 CrossRef CAS.
- K. H. Jack, Proc. R. Soc. A, 1951, 208, 216–224 CAS.
- T. K. Kim and M. Takahashi, Appl. Phys. Lett., 1972, 20, 492–494 CrossRef CAS.
- K. H. Jack, J. Alloys Compd., 1995, 222, 160–166 CrossRef CAS.
- J.-P. Wang, N. Ji, X. Liu, Y. Xu, C. Sanchez-Hanke, Y. Wu, F. De Groot, L. F. Allard and E. Lara-Curzio, IEEE Trans. Magn., 2012, 48, 1710–1717 CAS.
- Y. Sugita, K. Mitsuoka, M. Komuro, H. Hoshiya, Y. Kozono and M. Hanazono, J. Appl. Phys., 1991, 70, 5977–5982 CrossRef CAS.
- J. C. Slater, J. Appl. Phys., 1937, 8, 385–390 CrossRef CAS.
- J. Cadogan, Aust. J. Phys., 1997, 50, 1093–1102 CrossRef CAS.
- J. M. D. Coey, J. Appl. Phys., 1994, 76, 6632–6636 CrossRef CAS.
- M. Takahashi and H. Shoji, J. Magn. Magn. Mater., 2000, 208, 145–157 CrossRef CAS.
- J.-P. Wang, J. Magn. Magn. Mater., 2020, 497, 165962 CrossRef CAS.
- J. Liu, G. Guo, X. Zhang, F. Zhang, B. Ma and J.-P. Wang, Acta Mater., 2020, 184, 143–150 CrossRef CAS.
- X. Hang, M. Matsuda, J. T. Held, K. A. Mkhoyan and J.-P. Wang, Phys. Rev. B, 2020, 102, 104402 CrossRef CAS.
- N. Ji, X. Liu and J.-P. Wang, New J. Phys., 2010, 12, 063032 CrossRef.
- S. Bhattacharjee and S.-C. Lee, Sci. Rep., 2019, 9, 1–9 CrossRef PubMed.
- R. Islam and J. P. Borah, J. Appl. Phys., 2020, 128, 114902 CrossRef CAS.
- P. Stoeckl, P. Swatek and J.-P. Wang, AIP Adv., 2021, 11, 015039 CrossRef CAS.
- Y. Jiang, B. Himmetoglu, M. Cococcioni and J.-P. Wang, AIP Adv., 2016, 6, 056007 CrossRef.
- Y. Shi, Y. Du and G. Chen, Scr. Mater., 2013, 68, 976–979 CrossRef CAS.
- L. Ke, K. D. Belashchenko, M. van Schilfgaarde, T. Kotani and V. P. Antropov, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 024404 CrossRef.
- W. Y. Lai, Q. Q. Zheng and W. Y. Hu, J. Phys. Condens. Matter, 1994, 6, L259 CrossRef CAS.
- S. Ishida, K. Kitawatase, S. Fujii and S. Asano, J. Phys. Condens. Matter, 1992, 4, 765 CrossRef CAS.
- J. Sun, A. Ruzsinszky and J. P. Perdew, Phys. Rev. Lett., 2015, 115, 036402 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- R. Car, Nat. Chem., 2016, 8, 820–821 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- H. Sims, W. Butler, M. Richter, K. Koepernik, E. Sasioglu, C. Friedrich and S. Blügel, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 174422 CrossRef.
- N. Szymanski, V. Adhikari, M. Willard, P. Sarin, D. Gall and S. Khare, J. Appl. Phys., 2019, 126, 093903 CrossRef.
- J. Paier, M. Marsman and G. Kresse, J. Chem. Phys., 2007, 127, 024103 CrossRef PubMed.
- K. Pokharel, C. Lane, J. W. Furness, R. Zhang, J. Ning, B. Barbiellini, R. S. Markiewicz, Y. Zhang, A. Bansil and J. Sun, npj Comput. Mater., 2022, 8, 1–11 CrossRef.
- G. Sai Gautam and E. A. Carter, Phys. Rev. Mater., 2018, 2, 095401 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- B. Barbiellini, E. G. Moroni and T. Jarlborg, J. Phys. Condens. Matter, 1990, 2, 7597 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- M. Cococcioni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- B. Barbiellini and A. Bansil, J. Phys. Chem. Solids, 2005, 66, 2192–2196 CrossRef CAS.
- A. Allerdt, H. Hafiz, B. Barbiellini, A. Bansil and A. E. Feiguin, Appl. Sci., 2020, 10, 2542 CrossRef CAS.
- A. Pulkkinen, B. Barbiellini, J. Nokelainen, V. Sokolovskiy, D. Baigutlin, O. Miroshkina, M. Zagrebin, V. Buchelnikov, C. Lane, R. S. Markiewicz, A. Bansil, J. Sun, K. Pussi and E. Lähderanta, Phys. Rev. B, 2020, 101, 075115 CrossRef CAS.
- C. Lane, J. W. Furness, I. G. Buda, Y. Zhang, R. S. Markiewicz, B. Barbiellini, J. Sun and A. Bansil, Phys. Rev. B, 2018, 98, 125140 CrossRef CAS.
- J. W. Furness, Y. Zhang, C. Lane, I. G. Buda, B. Barbiellini, R. S. Markiewicz, A. Bansil and J. Sun, Commun. Phys., 2018, 1, 11 CrossRef.
- Y. Zhang, C. Lane, J. W. Furness, B. Barbiellini, J. P. Perdew, R. S. Markiewicz, A. Bansil and J. Sun, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 68–72 CrossRef CAS PubMed.
- J. Nokelainen, C. Lane, R. S. Markiewicz, B. Barbiellini, A. Pulkkinen, B. Singh, J. Sun, K. Pussi and A. Bansil, Phys. Rev. B, 2020, 101, 214523 CrossRef CAS.
- R. Zhang, C. Lane, B. Singh, J. Nokelainen, B. Barbiellini, R. S. Markiewicz, A. Bansil and J. Sun, Commun. Phys., 2021, 4, 1–12 CrossRef.
- H. Hafiz, K. Suzuki, B. Barbiellini, Y. Orikasa, S. Kaprzyk, N. Tsuji, K. Yamamoto, A. Terasaka, K. Hoshi and Y. Uchimoto, et al. , Phys. Rev. B, 2019, 100, 205104 CrossRef CAS.
- C. Lane, Y. Zhang, J. W. Furness, R. S. Markiewicz, B. Barbiellini, J. Sun and A. Bansil, Phys. Rev. B, 2020, 101, 155110 CrossRef CAS.
- J. Varignon, M. Bibes and A. Zunger, Phys. Rev. B, 2019, 100, 035119 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef PubMed.
- M. Ekholm, D. Gambino, H. J. M. Jönsson, F. Tasnádi, B. Alling and I. A. Abrikosov, Phys. Rev. B: Condens. Matter Mater. Phys., 2018, 98, 094413 CrossRef CAS.
- Y. Fu and D. J. Singh, Phys. Rev. Lett., 2018, 121, 207201 CrossRef CAS PubMed.
- Y. Fu and D. J. Singh, Phys. Rev. B, 2019, 100, 045126 CrossRef CAS.
- F. Tran, G. Baudesson, J. Carrete, G. K. H. Madsen, P. Blaha, K. Schwarz and D. J. Singh, Phys. Rev. B, 2020, 102, 024407 CrossRef CAS.
- D. Mejía-Rodríguez and S. B. Trickey, Phys. Rev. B, 2019, 100, 041113 CrossRef.
- O. Gunnarsson and B. I. Lundqvist, Phys. Rev. B: Solid State, 1976, 13, 4274–4298 CrossRef CAS.
- J. M. D. Coey and P. Smith, J. Magn. Magn. Mater., 1999, 200, 405–424 CrossRef CAS.
- R. M. Metzger, X. Bao and M. Carbucicchio, J. Appl. Phys., 1994, 76, 6626–6631 CrossRef CAS.
- X. Hang, PhD thesis, University of Minnesota, 2021.
- H. Hiraka, K. Ohoyama, Y. Ogata, T. Ogawa, R. Gallage, N. Kobayashi, M. Takahashi, B. Gillon, A. Gukasov and K. Yamada, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 134427 CrossRef.
- Y. Li, P. A. Montano, B. Barbiellini, P. Mijnarends, S. Kaprzyk and A. Bansil, J. Phys. Chem. Solids, 2007, 68, 1556–1560 CrossRef CAS.
- H. Kobayashi, T. Nagao, M. Itou, S. Todo, B. Barbiellini, P. E. Mijnarends, A. Bansil and N. Sakai, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 104423 CrossRef.
- K. Suzuki, Y. Otsuka, K. Hoshi, H. Sakurai, N. Tsuji, K. Yamamoto, N. Yabuuchi, H. Hafiz, Y. Orikasa and Y. Uchimoto, et al. , Condens. Matter, 2021, 7, 4 CrossRef.
- S. Pittalis, G. Vignale and F. Eich, Phys. Rev. B, 2017, 96, 035141 CrossRef.
| This journal is © the Owner Societies 2022 |