
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence16OSTM10: a new open-shell transition metal conformational energy database to challenge contemporary semiempirical and force field methods†
Arseniy A.
Otlyotov
a,
Andrey D.
Moshchenkov
ab,
Luigi
Cavallo
 *c and
Yury
Minenkov
*ad
*c and
Yury
Minenkov
*ad
aN. N. Semenov Federal Research Center for Chemical Physics RAS, Kosygina Street 4, 119991, Moscow, Russia. E-mail: yury.minenkov@chph.ras.ru
bHigher Chemical College of the Russian Academy of Sciences, Dmitry Mendeleev University of Chemical Technology of Russia, Miusskaya sq. 9, 125047, Moscow, Russia
cKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology, Thuwal, 23955-6900, Saudi Arabia. E-mail: luigi.cavallo@kaust.edu.sa
dJoint Institute for High Temperatures, Russian Academy of Sciences, 13-2 Izhorskaya Street, Moscow, 125412, Russia
First published on 23rd June 2022
Abstract
A new database, 16OSTM10, containing 10 conformations for each of 16 non-multireference realistic-size open-shell transition metal (OSTM) complexes has been developed. Contemporary composite density functional theory (DFT) (PBEh-3c and B97-3c), semiempirical (PM6 and PM7) and the GFNn-xTB/FF family methods were examined against conventional DFT (PBE-D3(BJ), PBE0-D3(BJ), M06 and ωB97X-V) to reproduce the conformational energies. While good performance is observed for the conventional (the average Pearson correlation coefficient is ρ = 0.91) and composite DFT (average ρ = 0.93), semiempirical and force-field methods should still be used with caution for these challenging compounds. The corresponding average ρ values are 0.53 (PM6 and PM7), 0.75 (GFN1-xTB and GFN2-xTB) and 0.62 (GFN-FF). Accounting for the intramolecular dispersion interactions turned out to be crucial for 4 OSTM complexes bearing bulky substituents in close proximity to each other. The influence of the scalar relativistic effects on the conformational energies is negligible for the considered 3d metal species.
1. Introduction
Open-shell (OS) transition metal (TM) complexes are vital precursors for a number of industrial and academic applications. TM compounds act as catalysts in a number of important processes relevant to chemistry1–5 and biology.6 These complexes often possess magnetic anisotropy,7,8 making them applicable as a base for promising information storage devices. To unravel their chemical and physical properties, molecular modeling is often required. The latter is anything but a simple task as, apart from the problems stemming from the complex electronic structures,9–14 many open-shell transition metal complexes bear bulky flexible ligands giving rise to conformational issues that have to be dealt carefully.15–22 The crucial role of a thorough conformational analysis in the investigations of the reaction pathways driven by transition metal catalysts has been revealed in a recent study by Vitek et al.18Contemporary computational chemistry offers a number of methods for conformational sampling/search ranging from cheap force-fields (FF)23–26 to the high-level DFT and ab initio approaches.27 An efficient method should provide desirable accuracy to ensure the realistic PES, but still possess low computational cost to cover a larger part of the conformational space. The development, further tuning and validation of potential energy function approaches require databases containing the spatial structures and relative conformational energies of the relevant compounds. Such datasets have been recently generated for flexible organic molecules28 and closed-shell TM complexes15,16 and used for the systematic investigations of the performance of contemporary semiempirical (SE), force-field and composite DFT methods. The conformational energies obtained with SE methods PM6*/PM7 were found to be in poor agreement with their DFT15,16 and high-level DLPNO-CCSD(T)28 counterparts. A better, yet moderate performance is demonstrated by GFNn-xTB SE methods.15,28 According to Bursch et al.,16 reasonable conformational energies of closed-shell TM complexes can be obtained from single-point energy (SPE) calculations using a composite density functional method (e.g. B97-3c) performed on GFN2-xTB optimized geometries.
However, there have been no systematic investigations on the performance of fast SE, FF and composite DFT approaches for the conformational energies of open-shell TM complexes. To fill this gap, we first develop a new database, 16OSTM10, containing 10 energetically and structurally diverse conformations for each of 16 realistic-size open-shell transition metal complexes. Second, we examine well-established DFT methods (PBE-D3(BJ), PBE0-D3(BJ), M06, and ωB97X-V), their recent composite versions (PBEh-3c and B97-3c) and contemporary SE (PM6, PM7, and GFNn-xTB) and FF (GFN-FF) schemes. The influence of scalar relativistic effects and intramolecular dispersion interactions on the conformational energies is discussed. A detailed statistical analysis was performed to find out the relative performance of the tested groups of methods.
Within a broader chemical context, we believe that the 16OSTM10 database together with its previously developed analogues for closed-shell TM complexes15,16 can serve as a prototype for the machine-learning (ML) training tool to yield high-quality conformational energies. Very recently, the use of trained artificial neural networks (ANNs) was demonstrated29 to predict adiabatic spin splitting and ionization potentials of CCSD(T) quality at significantly reduced computational cost.
2. Methods
2.1 Compound selection
The initial structures of transition metal complexes were retrieved from the online version of the Cambridge Structural Database (CSD).30 The pre-selection procedure was based on the following criteria:(1) A compound should contain a first row transition metal and (potentially) possess an open-shell electron configuration.
(2) A compound has at least 5 rotatable bonds implying a conformational manifold.
(3) A compound is of fundamental or applied scientific interest.
2.2 Conformer generation
The spatial structures of all the pre-selected compounds were optimized using the ORCA 5.0.2 suite of programs31,32 at the PBE-D3(BJ)/def2-svp level of theory. Electronic states with relevant multiplicities, namely, 1, 3, 5 for species with an even number of electrons and 2, 4, 6 in the case of an odd number of electrons, were considered. The energies of the optimized structures were re-evaluated at the PBE0-D3(BJ)/def2-tzvp level of theory. Only the species with non-singlet ground electronic states were selected for further processing. As the present study focuses on the applicability of single-reference methods for the calculation of conformational energies, compounds exhibiting significant multireference character were excluded. A well-established T1/T2 diagnostics based on the DLPNO-CCSD(T)/cc-pVDZ calculations was utilized for this purpose. In the case of T1 > 0.025 and/or T2 > 0.15, a compound was considered to be of significant multireference character and excluded from the selection. For the species (FUDNIB, UZEYAA, FIYMEI, LIBLEN, YIKLUC and AJOMIX) for which DLPNO-CCSD(T) turned out not to be accessible, we performed FOD diagnostics33 (see Fig. S1–S6, ESI†). Finally, 16 open-shell transition metal compounds satisfying all the above-mentioned criteria were chosen to form our database. Their structures with CSD names are given in Fig. 1. | ||
| Fig. 1 16OSTM10 database comprising open-shell transition metal complexes with their CSD names. | ||
In the second step, the sets containing 30–35 spatially diverse conformations of the selected compounds were automatically generated using an in-house code. All these structures were pre-optimized using a computationally undemanding approximation combining the standard PBE functional34,35 and double-ζ quality Gaussian-type nuclei-centered λ1 basis sets (their detailed compositions are given in the ESI†)36 as implemented in the Priroda code37 and then checked to exclude possible duplicates. Our experience28 suggests the PBE/λ1 approach to be suitable for the cheap preliminary optimizations. The spatial structures of the survived unique conformations were optimized at the PBE-D3(BJ)/def2-svp level of theory and further used for the calculations of the conformational energies (Section 2.3).
2.3 Examined methods for the calculations of conformational energies
A set of computationally efficient quantum-chemical approaches was used to obtain the conformational energies. First, we should note that the high-level DLPNO-CCSD(T1)/CBS method38–40 often used to obtain accurate reference conformational energies turned out to be prohibitively expensive for many species containing up to 200 atoms. As dispersion-corrected DFT methods provide reliable conformational energies,15 these have been used as references. To be unbiased, reference conformational energies were obtained with all-electron triple-ζ def2-tzvp basis sets41 and four DFT functionals including standard PBE34,35 and PBE042 complemented by D3(BJ) Grimme's dispersion correction,43,44 hybrid meta-GGA M0645 and range-separated hybrid ωB97X-V46 functionals. In order to reveal the influence of scalar relativistic effects on the conformational energies we performed additional single-point (SP) calculations utilizing PBE functional and triple-ζ quality Gaussian-type nuclei-centered λ2 basis sets (see the ESI† for their detailed compositions)36 with and without the Dyall scalar relativistic Hamiltonian47 as implemented in the Priroda code.37 This method accounts for the scalar relativistic effects, while spin–orbit coupling is neglected. The impact of dispersion interactions was evaluated by adding –D3(BJ) dispersion energy corrections43,44 to the total PBE/λ2 electronic energies.As practical conformational sampling prioritizes not only accuracy, but also computational efficiency, we have investigated the performance of the recently developed composite methods PBEh-3c48 and B97-3c.49 The cheapest approaches to obtain conformational energies are various semiempirical and force field methods. In the present contribution we examine the standard PM650 and PM751 SE methods as implemented in the MOPAC2016 program52 and contemporary GFNn-xTB/FF53–55 family of methods with use of the xtb 6.4.1 code.56
3. Results and discussion
3.1 Comparison of the relative conformational energies
The quantitative comparison of the conformational energies calculated with different methods was based on the Pearson inter-correlation coefficients:where X and Y are the examined electronic structure methods, Ei are the relative conformational energies, and Ē are the average conformational energies for a given method. The positive ρ values close to 1 indicate a strong correlation between the tested methods, while negative ρ values close to −1 signify an anti-correlation.

Mean absolute error (MAE) was also used to quantify the difference between absolute conformational energies according to the formula:
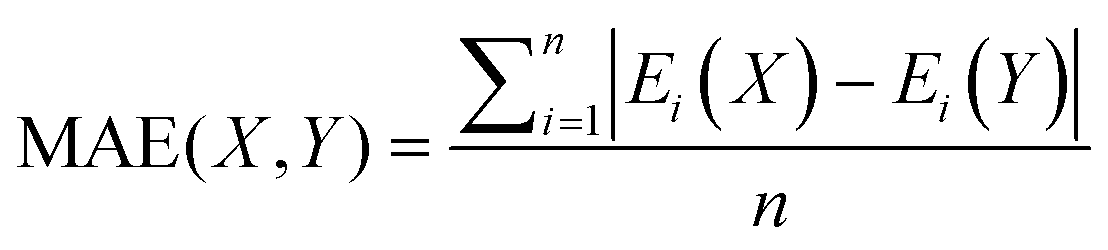
For the conventional DFT functionals (PBE-D3(BJ), PBE0-D3(BJ), M06 and ωB97X-V), the mean Pearson correlation coefficients were calculated as follows: (1) the ρ values for the examined functional and each of the 3 remaining functionals were averaged for each compound; (2) the obtained values were averaged over all 16 OSTM complexes.
In the case of the composite DFT (PBEh-3c and B97-3c), semiempirical methods (PM6 and PM7) and the methods belonging to the GFNn-xTB/FF family (GFN1-xTB, GFN2-xTB and GFN-FF), the mean Pearson coefficients for each compound were obtained by averaging of the four ρ values calculated for the examined method and each of the conventional DFT functionals (PBE-D3(BJ), PBE0-D3(BJ), M06 and ωB97X-V).
MAEs were calculated in the same fashion as the Pearson correlation coefficients for all the examined electronic structure methods.
In the case of the YIKLUC complex, the GFN1-xTB method produced unreasonable results and this set of values was excluded from statistical analysis. A similar problem was previously mentioned for some TM complexes from the compilation of Bursch et al.16
 | ||
| Fig. 2 Mean Pearson correlation coefficients (ρ) for the examined conventional DFT functional and each of the 3 remaining functionals, averaged over 16 OSTM complexes. The solid bars indicate the average ρ values, and the ends of the solid lines at each bar give the lowest and the largest values. | ||
However, several two-coordinate complexes (LUFCIZ, FIYMEI and FUDNIB) remain challenging for the tested conventional methods, revealing low correlation coefficients (ρ = 0.53–0.82) and large (up to 4.5 kcal mol−1) MAEs, and apparently require more sophisticated theoretical treatment.57,58
 | ||
| Fig. 3 Mean Pearson correlation coefficients (ρ) between composite (PBEh-3c and B97-3c) and standard (PBE-D3(BJ), PBE0-D3(BJ), M06 and ωB97X-V) DFT conformational energies. The solid bars indicate the average ρ values, and the ends of the solid lines at each bar give the lowest and the largest values. | ||
The worst correlation for AJOMIX stems from the narrow (0–3.3 kcal mol−1) span in the absolute conformational energies for this compound. The conformational energies for LUFCIZ, FIYMEI and FUDNIB also exhibit a moderate correlation similar to the case of the conventional DFT methods (Section 3.1.1).
 | ||
| Fig. 4 Mean Pearson correlation coefficients (ρ) between semiempirical (PM6 and PM7) and standard (PBE-D3(BJ), PBE0-D3(BJ), M06 and ωB97X-V) DFT conformational energies. The solid bars indicate the average ρ values, and the ends of the solid lines at each bar give the lowest and the largest values. | ||
 | ||
| Fig. 5 Mean Pearson correlation coefficients (ρ) between GFNn-xTB/FF and standard (PBE-D3(BJ), PBE0-D3(BJ), M06 and ωB97X-V) DFT conformational energies. The solid bars indicate the average ρ values, and the ends of the solid lines at each bar give the lowest and the largest values. | ||
 | ||
| Fig. 6 Pearson correlation coefficients (ρ) for the examined methods: the left and right sides of the boxes correspond to the first (Q1) and third (Q3) quartiles, respectively. The vertical solid line inside each box gives the median ρ value. The whiskers give the lowest and largest ρ values for each method. | ||
 | ||
| Fig. 7 Relative time for 1 SCF iteration performed with different methods for a conformation of LIBLEN. The logarithmic scale is used. | ||
Very fast semiempirical and force-field methods should be used with caution and proper validation for a target compound as they are characterized by large interquartile Q1/Q3 ranges as compared to the conventional and composite DFT (Fig. 6). Comparison of the NDDO PM6/7 methods with their tight-binding GFN counterparts in Fig. 6 illustrates a clear progress in semiempirical method development, still leaving them computationally affordable.
3.2 Impact of scalar relativistic effects on the conformational energies
For the 3d-metal species considered in this work relativistic effects start to be relevant.59 In order to quantify their influence on the conformational energies, we performed separate calculations with and without the Dyall scalar-relativistic Hamiltonian (see Section 2.3). Accounting for the relativistic effects slightly (less than 0.4 kcal mol−1) changes the absolute values of the relative conformational energies, but not the ranking of the conformations (ρ = 1.00) for all the considered compounds. This conclusion is true with and without a posteriori D3(BJ) dispersion correction added to the DFT energies.3.3 Influence of dispersion interactions on the conformational energies
Intramolecular dispersion interactions play a crucial role for many compounds of the 16OSTM10 compilation (Fig. 8). It becomes immediately clear from the selected correlation coefficients between PBE/λ2 and PBE-D3(BJ)/λ2 energies: ρ = −0.03 (AVIXIO), ρ = 0.36 (OQOQOB), ρ = 0.43 (FIYMEI), ρ = 0.57 (FUDNIB). Low correlation for these compounds can be explained by a major impact of the non-covalent interactions between their bulky substituents (see Fig. 1) located in the close proximity to each other. Overall, the obtained results indicate that conformational analysis performed without accounting for the dispersion interactions can lead to even qualitatively erroneous results. | ||
| Fig. 8 Pearson correlation coefficients for the conformational energies obtained with (PBE-D3(BJ)/λ2) and without (PBE/λ2) accounting for the intramolecular dispersion interactions. | ||
4. Conclusions
A new database, 16OSTM10, containing 10 conformations for each of 16 realistic-size open-shell transition metal complexes has been developed. Contemporary composite DFT, semiempirical and force-field methods have been examined against a set of conventional DFT methods (PBE-D3(BJ), PBE0-D3(BJ), M06 and ωB97X-V) in reproducing the relative conformational energies of the PBE-D3(BJ)/def2-svp optimized spatial structures. Similar to their closed-shell analogues, open-shell transition metal complexes remain challenging for PM6/PM7 semiempirical methods exhibiting the lowest Pearson correlation coefficients with the standard DFT methods. Significantly better performance was achieved for the GFN2-xTB semiempirical method, but still with particular failures for some compounds. The conformational energies obtained with the composite DFT methods B97-3c and PBEh-3c correlate well with their standard DFT counterparts. The recommendation to use the B97-3c approach for the reasonable single-point energies made in ref. 16 for the closed-shell species can be transferred to the open-shell ones.Accounting for the relativistic effects results in a slight shift of the absolute conformational energies, but has no influence on the ranking of the conformations. The influence of the intramolecular dispersion interactions on the conformational energies is much more pronounced.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the grant of the President of the Russian Federation (“The development of an efficient approach for prediction of the properties of the open-shell organometallic compounds”, project MK-176.2022.1.3). L. C. gratefully acknowledges the financial support from King Abdullah University of Science and Technology (KAUST). For computer time, this research used the resources of the Supercomputing Laboratory at King Abdullah University of Science and Technology (KAUST) in Thuwal, Saudi Arabia and the Joint Supercomputer Center of RAS in Moscow, Russia.Notes and references
- F. Allouche, D. Klose, C. P. Gordon, A. Ashuiev, M. Wörle, V. Kalendra, V. Mougel, C. Copéret and G. Jeschke, Low-Coordinated Titanium(III) Alkyl—Molecular and Surface—Complexes: Detailed Structure from Advanced EPR Spectroscopy, Angew. Chem., Int. Ed., 2018, 57, 14533–14537 CrossRef CAS PubMed.
- Z. H. Qi and J. Ma, Dual Role of a Photocatalyst: Generation of Ni(0) Catalyst and Promotion of Catalytic C-N Bond Formation, ACS Catal., 2018, 8, 1456–1463 CrossRef CAS.
- J. Yang and T. D. Tilley, Efficient hydrosilylation of carbonyl compounds with the simple amide catalyst [Fe{N(SiMe3)2}2], Angew. Chem., Int. Ed., 2010, 49, 10186–10188 CrossRef CAS PubMed.
- A. Galindo, DFT Studies on the Mechanism of the Vanadium-Catalyzed Deoxydehydration of Diols, Inorg. Chem., 2016, 55, 2284–2289 CrossRef CAS.
- L. J. Clouston, V. Bernales, R. K. Carlson, L. Gagliardi and C. C. Lu, Bimetallic Cobalt-Dinitrogen Complexes: Impact of the Supporting Metal on N2 Activation, Inorg. Chem., 2015, 54, 9263–9270 CrossRef CAS.
- A. Decker and E. I. Solomon, Dioxygen activation by copper, heme and non-heme iron enzymes: Comparison of electronic structures and reactivities, Curr. Opin. Chem. Biol., 2005, 9, 152–163 CrossRef CAS.
- P. C. Bunting, M. Atanasov, E. Damgaard-Møller, M. Perfetti, I. Crassee, M. Orlita, J. Overgaard, J. Van Slageren, F. Neese and J. R. Long, A linear cobalt(II) complex with maximal orbital angular momentum from a non-Aufbau ground state, Science, 2018, 362, eaat7319 CrossRef CAS PubMed.
- W. A. Merrill, T. A. Stich, M. Brynda, G. J. Yeagle, J. C. Fettinger, R. De Hont, W. M. Reiff, C. E. Schulz, R. D. Britt and P. P. Power, Direct spectroscopic observation of large quenching of first-order orbital angular momentum with bending in monomeric, two-coordinate Fe(II) primary amido complexes and the profound magnetic effects of the absence of Jahnand- and Renner-Teller distortions, J. Am. Chem. Soc., 2009, 131, 12693–12702 CrossRef PubMed.
- W. Jiang, N. J. Deyonker and A. K. Wilson, Multireference character for 3d transition-metal-containing molecules, J. Chem. Theory Comput., 2012, 8, 460–468 CrossRef CAS.
- K. Pierloot, Transition metals compounds: Outstanding challenges for multiconfigurational methods, Int. J. Quantum Chem., 2011, 111, 3291–3301 CrossRef CAS.
- A. L. Tchougreeff and M. B. Darkhovskii, in Progr. Theor. Chem. Phys., Springer, Dordrecht, 2006, pp. 451–505 Search PubMed.
- I. B. Bersuker, Electronic Structure and Properties of Transition Metal Compounds: Introduction to the Theory, John Wiley and Sons, 2nd edn, 2010 Search PubMed.
- M. Atanasov and C. A. Daul, Modeling properties of molecules with open d-shells using density functional theory, C. R. Chim., 2005, 8, 1421–1433 CrossRef CAS.
- J. Wang, S. Manivasagam and A. K. Wilson, Multireference Character for 4d Transition Metal-Containing Molecules, J. Chem. Theory Comput., 2015, 11, 5865–5872 CrossRef CAS.
- Y. Minenkov, D. I. Sharapa and L. Cavallo, Application of Semiempirical Methods to Transition Metal Complexes: Fast Results but Hard-to-Predict Accuracy, J. Chem. Theory Comput., 2018, 14, 3428–3439 CrossRef CAS PubMed.
- M. Bursch, A. Hansen, P. Pracht, J. T. Kohn and S. Grimme, Theoretical study on conformational energies of transition metal complexes, Phys. Chem. Chem. Phys., 2021, 23, 287–299 RSC.
- K. Munkerup, M. Thulin, D. Tan, X. Lim, R. Lee and K. W. Huang, Importance of thorough conformational analysis in modelling transition metal-mediated reactions: Case studies on pincer complexes containing phosphine groups, J. Saudi Chem. Soc., 2019, 23, 1206–1218 CrossRef CAS.
- A. K. Vitek, T. M. E. Jugovic and P. M. Zimmerman, Revealing the Strong Relationships between Ligand Conformers and Activation Barriers: A Case Study of Bisphosphine Reductive Elimination, ACS Catal., 2020, 10, 7136–7145 CrossRef CAS.
- J. Bartol, P. Comba, M. Melter and M. Zimmer, Conformational searching of transition metal compounds, J. Comput. Chem., 1999, 20, 1549–1558 CrossRef CAS.
- M. Besora, A. A. C. Braga, G. Ujaque, F. Maseras and A. Lledós, The importance of conformational search: A test case on the catalytic cycle of the Suzuki-Miyaura cross-coupling, Theor. Chem. Acc., 2011, 128, 639–646 Search PubMed.
- D. Suárez, N. Díaz and R. Lõpez, A combined semiempirical and DFT computational protocol for studying bioorganometallic complexes: Application to molybdocene-cysteine complexes, J. Comput. Chem., 2014, 35, 324–334 CrossRef.
- D. Suárez and N. Díaz, Molecular modeling of bioorganometallic compounds: Thermodynamic properties of molybdocene-glutathione complexes and mechanism of peptide hydrolysis, Chem. Phys. Chem., 2015, 16, 1646–1656 CrossRef PubMed.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- A. K. Rappé, K. S. Colwell and C. J. Casewit, Application of a Universal Force Field to Metal Complexes, Inorg. Chem., 1993, 32, 3438–3450 CrossRef.
- P. O. Norrby and P. Brandt, Deriving force field parameters for coordination complexes, Coord. Chem. Rev., 2001, 212, 79–109 CrossRef CAS.
- P. Comba and R. Remenyi, Inorganic and bioinorganic molecular mechanics modeling - The problem of the force field parameterization, Coord. Chem. Rev., 2003, 238–239, 9–20 CrossRef CAS.
- D. Troya, Reactivity Consequences of Conformational Isomerism in 1-Propanol, J. Phys. Chem. A, 2019, 123, 1044–1050 CrossRef CAS PubMed.
- D. I. Sharapa, A. Genaev, L. Cavallo and Y. Minenkov, A Robust and Cost-Efficient Scheme for Accurate Conformational Energies of Organic Molecules, Chem. Phys. Chem., 2019, 20, 92–102 CAS.
- C. Duan, D. B. K. Chu, A. Nandy and H. J. Kulik, Detection of multi-reference character imbalances enables a transfer learning approach for virtual high throughput screening with coupled cluster accuracy at DFT cost, Chem. Sci., 2022, 13, 4962–4971 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge structural database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Software update: The ORCA program system—Version 5.0, WIREs Comput. Mol. Sci., 2022, e1606 Search PubMed.
- S. Grimme and A. Hansen, A Practicable Real-Space Measure and Visualization of Static Electron-Correlation Effects, Angew. Chem., Int. Ed., 2015, 54, 12308–12313 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Erratum: Generalized gradient approximation made simple (Physical Review Letters (1996) 77 (3865)), Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- D. N. Laikov, A new class of atomic basis functions for accurate electronic structure calculations of molecules, Chem. Phys. Lett., 2005, 416, 116–120 CrossRef CAS.
- D. N. Laikov and Y. A. Ustynyuk, PRIRODA-04: A quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing, Russ. Chem. Bull., 2005, 54, 820–826 CrossRef CAS.
- C. Riplinger and F. Neese, An efficient and near linear scaling pair natural orbital based local coupled cluster method, J. Chem. Phys., 2013, 138, 34106 CrossRef.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, Natural triple excitations in local coupled cluster calculations with pair natural orbitals, J. Chem. Phys., 2013, 139, 134101 CrossRef.
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, Sparse maps - A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory, J. Chem. Phys., 2016, 144, 24109 CrossRef.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- N. Mardirossian and M. Head-Gordon, ωb97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924 RSC.
- K. G. Dyall, An exact separation of the spin-free and spin-dependent terms of the Dirac-Coulomb-Breit Hamiltonian, J. Chem. Phys., 1994, 100, 2118–2127 CrossRef CAS.
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. Hansen, Consistent structures and interactions by density functional theory with small atomic orbital basis sets, J. Chem. Phys., 2015, 143, 54107 CrossRef PubMed.
- J. G. Brandenburg, C. Bannwarth, A. Hansen and S. Grimme, B97-3c: A revised low-cost variant of the B97-D density functional method, J. Chem. Phys., 2018, 148, 064104 CrossRef PubMed.
- J. J. P. Stewart, Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS.
- J. J. P. Stewart, Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters, J. Mol. Model., 2013, 19, 1–32 CrossRef CAS.
- J. J. P. Stewart, MOPAC2016; Stewart Computational Chemistry, Colorado Springs, CO, USA, 2016. https://openmopac.net (accessed March 25, 2022) Search PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1-86), J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB - An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Spicher and S. Grimme, Robust Atomistic Modeling of Materials, Organometallic, and Biochemical Systems, Angew. Chem., Int. Ed., 2020, 59, 15665–15673 CrossRef CAS.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Extended tight-binding quantum chemistry methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 CAS.
- M. Atanasov, D. Ganyushin, D. A. Pantazis, K. Sivalingam and F. Neese, Detailed Ab initio first-principles study of the magnetic anisotropy in a family of trigonal pyramidal iron(II) pyrrolide complexes, Inorg. Chem., 2011, 50, 7460–7477 CrossRef CAS PubMed.
- M. Atanasov, J. M. Zadrozny, J. R. Long and F. Neese, A theoretical analysis of chemical bonding, vibronic coupling, and magnetic anisotropy in linear iron(ii) complexes with single-molecule magnet behavior, Chem. Sci., 2013, 4, 139–156 RSC.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, Energy-adjusted ab initio pseudopotentials for the first row transition elements, J. Chem. Phys., 1986, 86, 866–872 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Tabulated values forming the basis of Fig. 2–8, T1/T2 diagnostic values, FOD plots (Fig. S1–S6), Cartesian coordinates (Å) of PBE-D3(BJ)/def2-svp optimized spatial structures, and total single-point conformational energies: M06/def2-tzvp, PBE0-D3(BJ)/def2-tzvp, PBE-D3(BJ)/def2-tzvp, ωB97X-V/def2-tzvp, PBEh-3c, B97-3c, PM6, PM7, GFN1-xTB, GFN2-xTB, and GFN-FF. See DOI: https://doi.org/10.1039/d2cp01659a |
| This journal is © the Owner Societies 2022 |