
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnvironmental effects on the singlet fission phenomenon: a model Hamiltonian-based study
Pablo
Roseiro
 and
Vincent
Robert
*
and
Vincent
Robert
*
Laboratoire de Chimie Quantique, UMR 7177 Université de Strasbourg CNRS, 4 rue Blaise Pascal, 67000, Strasbourg, France. E-mail: vrobert@unistra.fr
First published on 6th June 2022
Abstract
In the screening of compounds for singlet fission, the relative energies of the constitutive units are decisive to fulfil the thermodynamic rules. From a model Hamiltonian constructed on the local spin states of an active chromophore and its environment, it is suggested that embedding greatly influences the energy differences of the active monomer spin states. Even in the absence of charge transfer, the field generated by a singlet environment produces an increase of the [E(S1) − E(S0)]/[E(T1) − E(S0)] critical ratio by up to 6% as compared to the one of a free chromophore. Besides, variations are observed when the intimate electronic structure of the singlet environment is modified. This propensity towards singlet fission is even more pronounced (10%) when the environment is switched to the triplet state. Finally, the embedding is likely to reverse the spin state ordering in the limit of vanishing atomic orbital overlaps. Despite its simplicity, the model stresses the importance of the environment spin nature in the quest for singlet fission candidates, and more generally in spectroscopy analysis.
Introduction
In the field of photophysics, singlet fission continues to attract much interest from many fundamental and applied research groups.1–4 Back to its origin in organic compounds, the conversion of excited singlets into pairs of correlated triplets was revived from its utility in the design of efficient solar cells.5 This phenomenon differs from competing inter-system crossing or multiple excitation generation in inorganic semiconductors where conversion into vibrational energy occurs. Alternant acene-like hydrocarbons are natural target systems; for the excited singlet–triplet, splitting is sizable and connected to a large exchange integral (ca. 1 eV) involving the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), so-called frontier orbitals6–8 (see Scheme 1). Following the three-class classification of organic chromophores, many theoretical efforts have been dedicated to the positioning of the relevant energy levels (first excited singlet S1 and triplet T1 states) to foresee the chemical conditions to stabilize the first excited singlet state S1. Since the T1–S1 energy difference is almost insensitive to the acene size, the so-called energy level matching condition of the singlet fission E(S1) − E(S0) > 2[E(T1) − E(S0)] is attainable by a reduction of the S1–S0 energy gap. This is the thermodynamic condition. | ||
| Scheme 1 Schematic description of the singlet fission phenomenon from the frontier orbital diagram of a class I compound built on A and B chromophores. | ||
In the meantime, the kinetics of the spin-allowed transition was depicted based on electron transfer or energy transfer theories.9–11 A critical parameter that governs singlet fission is the coupling between neighbouring units.12,13 Following this concern, computational studies have also much contributed to the definition of a well-balanced coupling strength and to the identification of the best chromophores.14–18 While too strong an interaction is not favourable for the triplets to separate and move apart, too weak a coupling reduces the rate of singlet fission.19,20 The role of excimer formation in singlet fission still deserves particular attention.21,22 Its formation is a critical step for the generation of pairs of triplets and the energy positioning is traditionally approximated from the monomer values.23–29 Irrespective of the level of theory, wave function-based theory or density functional theory (WFT and DFT, respectively) calculations, such an approximation is considered to be satisfactory for molecular crystals and more questionable in covalent dimers.30–34
However, one may wonder how much the immediate environment of a given monomer may modulate its spectroscopy. In the design of singlet fission compounds, we felt that attention should be paid to the molecular description, and to go beyond the free monomer description. Practically, one would like to stress, if sizable, the influence of the electronic structure of a monomer spin state (so-called environment) on the relative energies of a given neighbouring unit (so-called active chromophore). The importance of the environment is known and was reported in complex systems for which local and collective effects are likely to compete.35,36 Prime examples are spin-crossover compounds where the magnetic response can be modulated under crystallization conditions.37 Evidently, the appropriate definition of the spin nature of the environment in ab initio calculations calls for particular care, not to mention the system size which might make it not tractable from quantum chemistry methods. Our intention is to set the environment characteristics to foresee its impact on the energies of the active chromophore. For a neighbouring closed-shell chromophore, a single configuration might be an acceptable approximation and one strategy would be to freeze the molecular orbitals (MOs) to mimic such a closed-shell environment. However, the presence of a neighbouring open-shell chromophore (e.g. triplet) is more problematic since the electronic structures and spin values of the sub-parts cannot be decided in standard quantum chemistry calculations. Indeed, in a non-relativistic description, the total spin is a good quantum number and any calculation produces spin eigen-states of the full system.
Therefore, a model Hamiltonian built on the local spin states of the two H2 sub-systems was considered to complement the current views on molecules with potential applications for singlet fission. A much more sophisticated environment may enrich the description at a cost of a less comprehensive analysis. The model aims at capturing the environment effects using this minimal picture consisting of a single H2 molecule. The relevance of traditional approaches based on isolated chromophores can be examined from this simple H4 model. Following the standard crystal-field theory that relies on a closed-shell structure of the coordination sphere, we wanted to trace the importance of the spin value of the environment and the resulting modifications of the Coulomb and exchange contributions.
Description of the model
Theoretical inspections greatly concentrate on the HOMO and LUMO frontier orbitals.38–41 The S0–S1 transition in popular class I singlet fission chromophores is reasonably described by a HOMO–LUMO electronic transition.42 Thus, the model Hamiltonian is constructed on local spin states of a pair of interacting H2-like molecules (i.e. two electrons in two MOs). Indeed, the most promising candidates are slip-stacked chromophores where the direct mechanism can be optimized.36,43–45 Therefore, charge transfer states which might become competitive were not included in our description by simply constraining the number of electrons on the active chromophore and the environment, a realistic picture when L ≫ l (see Fig. 1). The possibility of mediation by the charge-transfer mechanism was explored elsewhere in covalent dimers.11,31,32 For preliminary inspections, singlet fission is limited to the direct coupling mechanism. In summary, the construction is nothing but a projection of the configuration interaction space into a selection of configurations that preserve the electron numbers and spin states on the subunits. | ||
| Fig. 1 Model system built on the active (A) and environment (E) chromophores with relevant geometrical parameters l = 0.74 Å (fixed intra-chromophore distance) and L (inter-chromophore distance). | ||
The bonding (gA and gE) and anti-bonding (uA and uE) local MOs of the active (A) and environment (E) subunits (see Fig. 1) are easily constructed from a minimal 1s atomic orbital (AO) basis set iA, jA, iE and jE. Then, the configurations are combined into configuration state functions ΓA and ΓE to span the tensorial product space ΓA⊗ΓE. All matrix elements were first analytically calculated as functions of mono-electronic and bi-electronic integrals expressed in the minimal AOs basis set. Evidently, the model Hamiltonian parametrization could be used. However, we preferred to introduce the chemical details of the structure, as any ab initio approach would do, to possibly extend to any realistic singlet fission chromophore. All integrals are distance-dependent and numerically available from the PSI4 suite of programs46 (see Fig. 1).
By construction, the MOs of the active subunit are not orthogonal to the MOs of the environment. In fused-benzene compounds, orthogonality between the carbon-2p AOs becomes numerically acceptable in the π–π interaction regime characterized by ∼2.5 times the carbon–carbon π-bond distance (1.43 Å). Indeed, the 2p carbon AOs σ-overlap is calculated as 0.01 for L = 3.6 Å. At shorter separation distances, the AO overlaps can no longer be neglected, similarly is the hydrogen–1s AO overlap in the H4-model system for distances smaller than L ∼ 2.5l ∼ 1.9 Å. Following the procedure described by Slater,47 the matrix elements were corrected to account for non-orthogonality. Four overlaps must be a priori evaluated between the MOs localized on the A and E subunits. However, SgAuE = SuAgE = 0 for symmetry reasons.
All determinants are built in the MS = 0 manifold and the energies are compared to the ones obtained in the absence of the environment subunit. Within our approach restricted to the direct coupling mechanism (i.e. no charge transfer), the singlet state environment SE is constructed as a contraction on the three configuration state functions (see Scheme 2):
 | (1) |
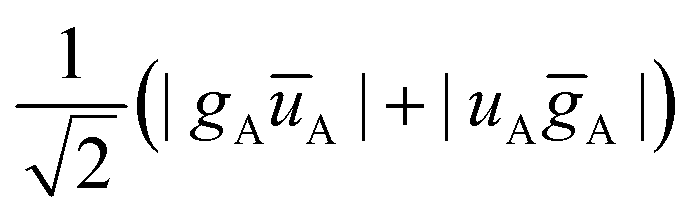 configurations:
configurations: | (2) |
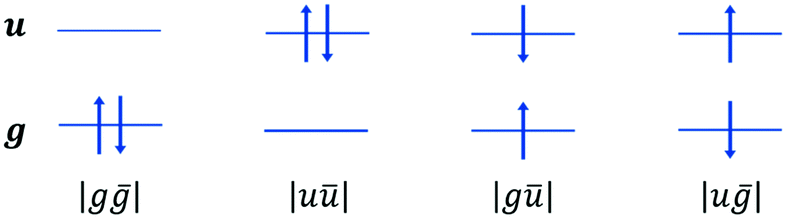 | ||
Scheme 2 Schematic representation of the closed-shell |gḡ|, |uū|, and the open-shell |gū|, |uḡ|, Ms = 0 configurations on a dimer. The singlet and triplet configuration state functions are  and and  , respectively. , respectively. | ||
Let us mention that a SE environment is readily achieved by freezing the occupied and virtual localized MOs of the environment in an ab initio procedure. Evidently, the triplet state consists of a single configuration  , the energy of which is immediately calculated in the SE field.
, the energy of which is immediately calculated in the SE field.
A similar inspection was then performed when the environment is switched into a triplet state TE. However, some particular care must be taken since a global triplet state emerges from the local triplet states TA and TE. Such a triplet can mix in with the triplet state built from the  configuration (i.e. SA⊗TE). The eigenvector analysis evidences the contributions of the singlet and triplet in the active chromophore.
configuration (i.e. SA⊗TE). The eigenvector analysis evidences the contributions of the singlet and triplet in the active chromophore.
In this study, we are primarily concerned with the impact of the environment spin structure on the energy level matching conditions. Thus, the objective is to examine the sensitivity of the low-energy state ordering of an active chromophore A in the field generated by a L-distant environment E. Therefore, the condition for singlet fission reads ρ = [E(SA,1) − E(SA,0)]/[E(TA,1) − E(SA,0)] > 2.
Results and discussion
Let us first concentrate on the active chromophore energy levels in the field of a singlet state environment SE. In the Ms = 0 manifold, the SA⊗SE space is spanned by three determinants. A single determinant built as TA⊗SE defines the local triplet state energy on the active chromophore. Overall, three singlet states SA,i and one single triplet TA,1 energy are evaluated in the presence of a frozen singlet environment. All energies depend on the inter-dimer distance L as well as on the contraction coefficients e1, e2, and e3 (see eqn (1)).In the long-distance regime (L > 3.5 Å), the H2 monomers do not interact, all integrals becoming vanishingly small. The asymptotic limit of the critical ratio ρ = [E(SA,1) − E(SA,0)]/[E(TA,1) − E(SA,0)] (ca. 1.60) corresponds to the value that is usually reported from calculations performed on a single chromophore. However, ρ is very sensitive to the presence of a surrounding partner (see Fig. 2(a)), even in this simplified picture. The nature and amplitude of the dipole–dipole interactions are responsible for this differentiating effect. For interacting π-systems, the ratio between the intramolecular carbon–carbon distance and the packing separation is ca. 2.5. In analogy, one expects that, below L ∼ 2.5l ∼ 1.9 Å, charge transfer contributions should be included between 1s hydrogen AOs and our simplified view would be invalidated. However, even at this low-distance limit, ρ is increased up to 1.70 (Fig. 2(a)).
 | ||
| Fig. 2 (a) Critical ratio ρ = [E(SA,1) − E(SA,0)]/[E(TA,1) − E(SA,0)] in the fields of the singlet environment as a function of the inter-dimer distance L. The singlet environment SE consists of a contraction e1 = 0.85, e2 = 0.45, and e3 = 0.27. (b) Upper panel: Variations of the low-lying triplet state energies (eV) for a triplet environment. The ground state consists of a pure singlet on A, and its energy is used as a reference. The excited triplet states are labelled with the dominant contributions on A. The intra-chromophore distance l is set to the equilibrium H2 distance to 0.74 Å. The ρ value variations are given in the lower panel. | ||
As soon as the environment spin state is switched to the triplet, the picture is somewhat modified. Irrespective of the inter-dimer distance, the ground state singlet (reference energy in Fig. 2(b)) is a pure singlet on A dominated by the |gAḡA| configuration. As mentioned before, the excited triplet states result from the mixing between different spin-coupling schemes (SA⊗TE and TA⊗TE). From our numerical inspections, these states labelled as TA,1 and SA,1 in Fig. 2(b) are largely dominated (ca. 98% at L = 2 Å) by the triplet and singlet on A, respectively. Thus, these energies can be compared to the ones obtained from an isolated chromophore picture for which ρ = 1.60. Not only does the SA,1–TA,1 energy difference increases with the decreasing inter-chromophore distance, but also the singlet fission thermodynamic condition ρ > 2 is fulfilled for L values close to 1.9 Å (see Fig. 2(b), lower panel). For comparison, the triplet–singlet energy difference and the ρ value for L = 2.5 Å were calculated as 14.8 eV and 1.61 at the CAS[4,4]SCF level in a minimal 1s AOs basis set, respectively.48 In this regime, the contracted view of the model (see Fig. 2(b) upper panel) reproduces not only the CASSCF energy splitting but also the asymptotic value ρ = 1.60.
Despite its simplicity, our model stresses that the presence of the environment significantly modifies the spin state ordering. As expected, the field generated by the environment depends on its spin state, giving rise to an enhanced singlet fission inclination in the presence of a triplet environment TE.
The observed critical regime of 1.9–2.6 Å in the H4 model agrees with the traditional range of the manifestation of π–stacking interactions in conjugated organic molecules up to a scaling factor based on the intra- and inter-molecular distances. In conclusion, the ρ variations for such hydrogen-based model compounds are in accordance with what is found for carbon-based compounds.
It is known that realistic synthetic systems for singlet fission are acene-like and our model may look at first over-simplistic. First, the hydrogen 1s AO σ-overlap is 0.66 for l = 0.74 Å, whereas the π-overlap between the 2p carbon AOs is reduced down to 0.19 for a typical carbon–carbon distance. Then, the density of states increases with the number of carbon atoms in polyacene. Together, these elements favour a reduction of the HOMO–LUMO gap within the chromophore. In the π-stacking acene arrangements, the inter-chromophore interactions are governed by negligible 2p carbon AO σ-overlap (0.01 for L = 3.6 Å). For all these reasons, we felt that setting all overlap values to zero (intra- and inter-chromophores) in our model would allow in making the contact with synthetic compounds and in analysing the leading mechanisms at work in the hierarchization of the spin states. Importantly, all the one-electron and two-electron energy integrals were maintained as numerically evaluated from PSI4. Different singlet environment structures were generated by varying the amplitudes e1, e2 and e3 (see Fig. 3).
 | ||
| Fig. 3 Singlet and triplet state energies (eV) as a function of the inter-dimer distance L in the vicinity of a contracted singlet environment SE with the increasing weight on the |uEūE| configuration. (a) e1 = 1 and e2 = e3 = 0, (b) e1 = 0.91 and e2 = e3 = 0.29, and (c) e1 = 0.85, e2 = 0.45, and e3 = 0.27. All AO overlaps are set to zero. Following the Tanabé–Sugano representation, the ground state energy EGS is taken as a reference. Its nature changes for a characteristic L distance which is marked by a vertical dashed line. Horizontal dashed lines represent the spectroscopy of an isolated chromophore. | ||
In the asymptotic limit, the spectroscopy evidently does not depend on the environment singlet structure and exhibits a singlet ground state SA,0. Nevertheless, the absolute energy of the latter is sensitive to the structure of the environment and the ρ value is modulated. By inspecting the different contributions, the leading one arises from the two-electron integral (iAjA, iEjE)†. This a slowly decaying 1/L integral that destabilizes |gAḡAgEḡE| and |uAūAuEūE| configurations, whereas it stabilises |uAūAgEḡE| and |gAḡAuEūE| configurations. As a consequence, the energy of the SA,0 state increases in the presence of a mono-determinantal environment |gEḡE| (e1 = 1, e2 = e3 = 0). Conversely, it is lowered for the |uEūE| environment (e1 = 0, e2 = 1, e3 = 0). For symmetry reasons, the energy of SA,0 is not sensitive to the open-shell environment  (e1 = e2 = 0, e3 = 1). This state of affairs changes for the excited triplet TA,1 and singlet SA,1 states. Indeed, the dominant integral (iAjA, iEjE) has no impact on their energies and the SA,1–TA,1 splitting remains constant and equal to 2KgAuA (exchange integral). As a result of the long-range 1/L potential, the critical ratio ρ is still sensitive to the environment even for L = 20 Å where ρ|gEḡE| = 1.70, ρ|uEūE| = 1.53 and
(e1 = e2 = 0, e3 = 1). This state of affairs changes for the excited triplet TA,1 and singlet SA,1 states. Indeed, the dominant integral (iAjA, iEjE) has no impact on their energies and the SA,1–TA,1 splitting remains constant and equal to 2KgAuA (exchange integral). As a result of the long-range 1/L potential, the critical ratio ρ is still sensitive to the environment even for L = 20 Å where ρ|gEḡE| = 1.70, ρ|uEūE| = 1.53 and  .
.
For L ca. 5 Å, the ρ > 2 regime is reached, in sharp contrast with what was observed previously. When L is further reduced, the energy of the SA,0 state continuously increases, and eventually the nature of the ground state switches to high-spin TA,1. Such an observation somewhat reconsiders the traditional picture “strong field-low spin’’ in metal ion coordination compounds. Interestingly, the switching L distance is shifted to lower values (see Fig. 3 where all AO overlaps are set to zero) as the weight of the |uEūE| configuration increases. This is a reflection of the competing effects of the |gEḡE| and |uEūE| configurations on the stabilization of the SA,0 state.
The examined electronic structure is evidently reminiscent of the one in cyclobutadiene. As seen in Fig. 3, the ground state of the four-electron four-orbital system is the singlet whatever the structure of the singlet environment. This observation is in agreement with the pseudo Jahn–Teller effect manifestation: the triplet square structure is unstable and a distortion to the rectangular geometry leads to a singlet ground state. In contrast, our constrained wave function description favours a triplet ground state (TA,1⊗SE) even for strong deviations from the square geometry. The contracted structure of the E part and the resulting absence of charge transfers are responsible for this behaviour. However, our model highlights the significant impact of the electronic structure of the environment.
Conclusions
The importance of the spin structure of the environment on the energy states of a two-electron active chromophore was examined by building up a model Hamiltonian that affords the splitting into local spin states. The active chromophore state ordering is much affected in the presence of a triplet or a singlet environment. As a consequence, the critical ratio ρ = [E(S1) − E(S0)]/[E(T1) − E(S0)] defined on the excitation energies of the first singlet and triplet excited states may reach the lower bound ρ = 2 value for the singlet fission condition to occur. A simplified description that neglects overlaps leads to an even deeper modification. The ground state is switched from the low-spin singlet to the high-spin triplet when the distance between the active chromophore and the environment is ca. 2.5 Å. This critical distance is governed by the structure of the singlet state environment acting as a field that controls the spin state hierarchisation. Even in the absence of charge transfers between the subunits, our model suggests that spin-dependent environment effects should be taken into account. The simplicity of the H4-based model brings some insights and means of interpretation that should be transferable in the theoretical quest of singlet fission candidates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. R. would like to thank the French minister for the financial support of his PhD grant. We would like to dedicate this scientific work to Professor Bernard Bigot, an enlightening mentor, who passed away in the course of revision of this contribution.Notes and references
- D. Casanova, Chem. Rev., 2018, 118, 7164–7207 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- K. Aryanpour, A. Shukla and S. Mazumdar, J. Phys. Chem. C, 2015, 119, 6966–6979 CrossRef.
- R. E. Merrifield, J. Chem. Phys., 1961, 34, 1835–1839 CrossRef CAS.
- C. J. Bardeen, Annu. Rev. Phys. Chem., 2014, 65, 127–148 CrossRef CAS PubMed.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, J. Chem. Phys., 2013, 138, 114103 CrossRef PubMed.
- D. Beljonne, H. Yamagata, J. L. Brédas, F. C. Spano and Y. Olivier, Phys. Rev. Lett., 2013, 110, 226402 CrossRef CAS PubMed.
- B. Alam, A. F. Morrison and J. M. Herbert, J. Phys. Chem. C, 2020, 124, 24653–24666 CrossRef CAS.
- A. F. Morrison and J. M. Herbert, J. Phys. Chem. Lett., 2017, 8, 1442–1448 CrossRef CAS PubMed.
- N. V. Korovina, S. Das, Z. Nett, X. Feng, J. Joy, R. Haiges, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2016, 138, 617–627 CrossRef CAS PubMed.
- J. C. Johnson and J. Michl, Top. Curr. Chem., 2017, 375, 80 CrossRef PubMed.
- E. Pradhan, J. N. Bentley, C. B. Caputo and T. Zeng, ChemPhotoChem, 2020, 4, 5279–5287 CrossRef CAS.
- S. Sardar, J. Mol. Graphics Modell., 2020, 98, 107608 CrossRef CAS PubMed.
- L. Shen, Y. Chen, X. Li and C. Li, J. Mol. Graphics Modell., 2016, 66, 187–195 CrossRef CAS PubMed.
- M. Zhang, Z. Hua, W. Liu, H. Liu, S. He, C. Zhu and Y. Zhu, J. Mol. Model., 2020, 26, 32 CrossRef CAS PubMed.
- H. L. Stern, A. Cheminal, S. R. Yost, K. Broch, S. L. Bayliss, K. Chen, M. Tabachnyk, K. Thorley, N. Greenham, J. M. Hodgkiss, J. Anthony, M. Head-Gordon, A. J. Musser, A. Rao and R. H. Friend, Nat. Chem., 2017, 9, 1205–1212 CrossRef CAS PubMed.
- N. V. Korovina, J. Joy, X. Feng, C. Feltenberger, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2018, 140, 10179–10190 CrossRef CAS PubMed.
- X. Feng and A. I. Krylov, Phys. Chem. Chem. Phys., 2016, 18, 7751–7761 RSC.
- C. B. Dover, J. K. Gallaher, L. Frazer, P. C. Tapping, A. J. Petty, M. J. Crossley, J. E. Anthony, T. W. Kee and T. W. Schmidt, Nat. Chem., 2018, 10, 305–310 CrossRef CAS PubMed.
- D. Padula, Ö. H. Omar, T. Nematiaram and A. Troisi, Energy Environ. Sci., 2019, 12, 2412–2416 RSC.
- O. El Bakouri, J. R. Smith and H. Ottosson, J. Am. Chem. Soc., 2020, 142, 5602–5617 CrossRef CAS PubMed.
- D. S. Patil, K. C. Avhad and N. Sekar, Comput. Theor. Chem., 2018, 1138, 75–83 CrossRef CAS.
- Y. Gawale and N. Sekar, J. Photochem. Photobiol., B, 2018, 178, 472–480 CrossRef CAS PubMed.
- K. J. Fallon, P. Budden, E. Salvadori, A. M. Ganose, C. N. Savory, L. Eyre, S. Dowland, Q. Ai, S. Goodlett, C. Risko, D. O. Scanlon, C. W. M. Kay, A. Rao, R. H. Friend, A. J. Musser and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 13867–13876 CrossRef CAS PubMed.
- M. Baranac-Stojanović, J. Org. Chem., 2019, 84, 13582–13594 CrossRef PubMed.
- Z. Liu, L. Lin, Q. Jia, Z. Cheng, Y. Jiang, Y. Guo and J. Ma, J. Chem. Inf. Model., 2021, 61, 1066–1082 CrossRef CAS PubMed.
- J. C. Johnson, A. J. Nozik and J. Michl, Acc. Chem. Res., 2013, 46, 1290–1299 CrossRef CAS PubMed.
- C. M. Mauck, Y. J. Bae, M. Chen, N. Powers-Riggs, Y.-L. Wu and M. R. Wasielewski, ChemPhotoChem, 2018, 2, 223–233 CrossRef CAS.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120–1125 CrossRef CAS PubMed.
- T. Zeng, J. Phys. Chem. Lett., 2016, 7, 4405–4412 CrossRef CAS PubMed.
- V. I. Minkin, A. G. Starikov and A. A. Starikova, J. Phys. Chem. A, 2021, 125, 6562–6570 CrossRef CAS PubMed.
- L. Wang, Y. Olivier, O. V. Prezhdo and D. Beljonne, J. Phys. Chem. Lett., 2014, 5, 3345–3353 CrossRef CAS PubMed.
- N. Renaud, P. A. Sherratt and M. A. Ratner, J. Phys. Chem. Lett., 2013, 4, 1065–1069 CrossRef CAS PubMed.
- J. Olguín and S. Brooker, Coord. Chem. Rev., 2011, 255, 203–240 CrossRef.
- M. E. Sandoval-Salinas and D. Casanova, ChemPhotoChem, 2021, 5, 282–293 CrossRef CAS.
- A. Thomas, C. Ji, B. Siddlingeshwar, P. U. Manohar, F. Ying and W. Wu, Phys. Chem. Chem. Phys., 2021, 23, 1050–1061 RSC.
- K. Tanaka, D. Sakamaki and H. Fujiwara, Chem. – Eur. J., 2021, 27, 4430–4438 CrossRef CAS PubMed.
- X. Feng, A. V. Luzanov and A. I. Krylov, J. Phys. Chem. Lett., 2013, 4, 3845–3852 CrossRef CAS.
- E. A. Buchanan, Z. Havlas and J. Michl, in Advances in Quantum Chemistry, ed. J. R. Sabin and E. J. Brändas, Academic Press, 2017, vol. 75, pp. 175–227 Search PubMed.
- K. Kolata, T. Breuer, G. Witte and S. Chatterjee, ACS Nano, 2014, 8, 7377–7383 CrossRef CAS PubMed.
- K. M. Felter and F. C. Grozema, J. Phys. Chem. Lett., 2019, 10, 7208–7214 CrossRef CAS PubMed.
- A. Zaykov, P. Felkel, E. A. Buchanan, M. Jovanovic, R. W. A. Havenith, R. K. Kathir, R. Broer, Z. Havlas and J. Michl, J. Am. Chem. Soc., 2019, 141, 17729–17743 CrossRef CAS PubMed.
- D. G. A. Smith, L. A. Burns, A. C. Simmonett, R. M. Parrish, M. C. Schieber, R. Galvelis, P. Kraus, H. Kruse, R. D. Remigio, A. Alenaizan, A. M. James, S. Lehtola, J. P. Misiewicz, M. Scheurer, R. A. Shaw, J. B. Schriber, Y. Xie, Z. L. Glick, D. A. Sirianni, J. S. O’Brien, J. M. Waldrop, A. Kumar, E. G. Hohenstein, B. P. Pritchard, B. R. Brooks, H. F. SchaeferIII, A. Y. Sokolov, K. Patkowski, A. E. DePrinceIII, U. Bozkaya, R. A. King, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, J. Chem. Phys., 2020, 152, 184108 CrossRef CAS PubMed.
- J. C. Slater, Quantum theory of molecules and solids: Volume 1: Electronic structure of molecules, McGraw-Hill, 1963 Search PubMed.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. F. Galván, N. Ferré, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. Å. Malmqvist, T. Müller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Martí, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed.
Footnote |
†  |
| This journal is © the Owner Societies 2022 |