
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum molecular simulations of micro-hydrated halogen anions†
Raúl
Rodríguez-Segundo‡
ab,
Alfonso
Gijón
 c and
Rita
Prosmiti
*a
c and
Rita
Prosmiti
*a
aInstitute of Fundamental Physics (IFF-CSIC), CSIC, Serrano 123, 28006 Madrid, Spain. E-mail: rita@iff.csic.es; Tel: +34 915616800
bAtelgraphics S.L., Mota de Cuervo 42, 28043, Madrid, Spain
cMaterials Science Institute of Madrid (ICMM-CSIC), CSIC, Sor Juana Inés de la Cruz 3, 28049 Madrid, Spain
First published on 25th May 2022
Abstract
We report the results of a detailed and accurate investigation focused on structures and energetics of poly-hydrated halides employing first-principles polarizable halide–water potentials to describe the underlying forces. Following a bottom-up data-driven potential approach, we initially looked into the classical behavior of higher-order X−(H2O)N clusters. We have located several low-lying energies, such as global and local minima, structures for each cluster, with various water molecules (up to N = 8) surrounding the halide anion (X− = F−, Cl−, Br−, I−), employing an evolutionary programming method. It is found that the F−–water clusters exhibit different structural configurations than the heavier halides, however independently of the halide anion, all clusters show in general a selective growth with the anion preferring to be connected to the outer shell of the water molecule arrangements. In turn, path-integral molecular dynamics simulations are performed to incorporate explicitly nuclear quantum and thermal effects in describing the nature of halide ion microsolvation in such prototypical model systems. Our data reveal that at low finite temperatures, nuclear quantum effects affect certain structural properties, such as weakening hydrogen bonding between the halide anion and water molecules, with minor distortions in the water network beyond the first hydration shell, indicating local structure rearrangements. Such structural characteristics and the promising cluster size trends observed in the single-ion solvation energies motivated us to draw connections of small size cluster data to the limits of continuum bulk values, toward the investigation of the challenging computational modeling of bulk single ion hydration.
Small clusters act as model systems to explore nuclear quantum effects on the dynamics and thermodynamics of microsolvation, providing important insight into understanding the underlying mechanisms in the solvation process.1–3 Clusters are the primary nanosystems where collective features of macroscopic properties emerge, and understanding theoretically their properties has been the subject of broad conceptual, technological and practical implications.4–7 Research in the field of the clusters aims to establish connections with nanoscale science, as in some cases finite-size system properties evolve smoothly toward the bulk, allowing extrapolation from few-body to many-body behavior.8–15
Single halide ion water aggregates are such examples, and several experimental and theoretical studies have been reported15–48 mainly from IR spectroscopy studies, and classical molecular dynamics (MD) simulations on structural/thermodynamic/spectroscopic aspects, respectively. Electronic structure calculations predicted that all halide ions preferred to reside on the surface of the water clusters, while contradictory results have been obtained from MD simulations, using polarizable and nonpolarizable force-fields, that reveal structures with the chloride ion located in the interior of the water clusters.
As it is well-known, predicting properties at the molecular level often proves difficult, even for small aggregates. To gain useful insights from computer simulations requires going beyond the harmonic approximation, employing more sophisticated force fields for representing the interactions. The potential energy landscape is, in general, a complex function of the atomic coordinates, with several local minima (their number is increasing as the cluster size increases) separated by barriers, with their identification being a serious computational task.
Thus, the traditional manner to proceed cluster simulations is to identify and characterize first the cluster's low energy structures on the potential energy surface (PES), and in turn through proper treatments of the nuclear dynamics to incorporate thermal and quantum effects. However, the latest one is still computationally challenging even for few small molecule sized clusters. Such full dimensional quantum calculations become prohibitive as computational demands increase exponentially with the system size, and path integral formulation approaches49 have offered alternatives to overcome these scaling problems.38,50–54 Just recently PI-AIMD calculations have been reported, highlighting that nuclear quantum effects are likely to play a crucial role in the hydration of chlorine anions, showing that ion hydrogen bonding is weakened in this case,47 while earlier DMC and finite-temperature thermodynamic methodology have revealed that nuclear quantum effects are minuscule in halide–water clusters.45
Given the importance of the underlying interactions in the microsolvation processes of such charged systems, their accurate description is required. The advances in computational software and hardware have made affordable very precise calculations of such finite-size complexes. However, the intriguing interplay between halide–water and water–water interactions in the hydrogen-bonding network and the role of quantum effects on their binding present a real challenge for the theorists. Thus, the present work is directed at studying the energetics and structuring of different finite-size halide–water clusters, through path-integral molecular dynamics (PIMD) simulations, employing the recently developed data-driven i-TTM4 potential models.55 Comparisons with results from classical molecular dynamics (MD) simulations were also presented in order to investigate the impact of temperature over quantum zero-point contributions in both energies and structures.
1 Computational methods, results, and discussion
Computational calculations were carried out using our own codes55–57 for both nuclear classical MD and PIMD simulations, while otherwise, we explicitly reference the corresponding software. Potential optimizations were performed using an evolutionary programming (EP) algorithm,58 while in all cases the DENEB software package59 was employed to generate initial guess configurations and input files, as well as to organize both input and output data files.1.1 Modeling the underlying interactions: a bottom-up data-driven model
Following previous studies on halide–water clusters,55,57,60,61 we have recently reported four families of data-driven PESs, namely i-TTM2, i-TTM3, i-TTM4, and i-MBpol depending on which TTM water model62–65 we choose to describe the water–water interactions in X−(H2O)N systems. Ultimately, a variety of many-body water potential models are available in the literature incorporating explicitly 2-body, 3-body, and even 4-body terms fitted to ab initio data with the remaining n-body terms described by classical induction.63–67 We have chosen to employ the TTM2, TTM3, TTM4 and MB-pol water models available in the literature62–65 in our first-principle, physical and effective many-body bottom-up scheme to represent in an efficient manner the halide–water interactions in X−(H2O)N systems, with X being F, Cl, Br, and I anions. The performance of the resulting halide–water potentials has been assessed by spanning a broad range of representative configurations, from equilibrium, moderate-binding to nonbinding ones, of variable increasing sized clusters up to N= 54, with the i-TTM4 model yielding the best overall results in comparison to the DF-MP2 reference data for all X−(H2O)N clusters. Thus, we decided to use the i-TTM4 potentials in the present calculations.Briefly, the intermolecular anion (X− = F−, Cl−, Br−, I−)–water interactions are represented as a sum of permanent electrostatic, polarization, dispersion and repulsion terms, VX–W = Velec + Vpol + Vdisp + Vrep. For the electrostatic part (charge-charge and induction energy contributions) the reparameterized TTM4-F variant of the Thole-type model was used, employing smeared geometry-dependent point charges and dipoles,64 with the anion polarizabilites calculated at the CCSD(T), as implemented in the Molpro package68 employing even-tempered sets of diffuse functions in correlation with consistent basis sets.60 The dispersion contribution is obtained as a sum of damped Tang-Toennies  terms, with the dispersion C6 coefficients between anion and water atoms adjusted to those computed from DFT/LC-ωPBE calculations using the XDM (exchange-hole dipole moment) model,69 as implemented in postg code,70 while the repulsive term is represented as a sum of Born-Mayer functions over all atoms, fitted to CCSD(T)-F12b energies of the anion–H2O system subtracting the electrostatic and dispersion energy contributions, via an active learning scheme.55
terms, with the dispersion C6 coefficients between anion and water atoms adjusted to those computed from DFT/LC-ωPBE calculations using the XDM (exchange-hole dipole moment) model,69 as implemented in postg code,70 while the repulsive term is represented as a sum of Born-Mayer functions over all atoms, fitted to CCSD(T)-F12b energies of the anion–H2O system subtracting the electrostatic and dispersion energy contributions, via an active learning scheme.55
In Fig. 1 we presented one-dimensional views of the anion–water potentials as a function of the ϕ angle (C2v axis of water molecule and ![[R with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0052_20d1.gif) joining oxygen and anion atoms) for planar H2O-fixed configurations (see upper panel) optimizing the R distance, along the OH of the ion–hydrogen bond keeping the R distance fixed at its equilibrium values (see the middle panel), and as a function of R (see the lower panel). The potential minima represent quasilinear halide–water hydrogen bonds of Cs symmetry, with the dipolar C2v geometry being a saddle point of the potentials, and the F− H2O system exhibits the strongest interaction of all remaining complexes.
joining oxygen and anion atoms) for planar H2O-fixed configurations (see upper panel) optimizing the R distance, along the OH of the ion–hydrogen bond keeping the R distance fixed at its equilibrium values (see the middle panel), and as a function of R (see the lower panel). The potential minima represent quasilinear halide–water hydrogen bonds of Cs symmetry, with the dipolar C2v geometry being a saddle point of the potentials, and the F− H2O system exhibits the strongest interaction of all remaining complexes.
 | ||
| Fig. 1 1D plots of the X− H2O PESs along planar ϕ angle, ion–hydrogen bond distance and R coordinate (see text). Representative halide–water configurations are also displayed. | ||
For systems containing a halide anion and N water molecules the anion–water potentials are combined via a sum-of-potential scheme with the intramolecular term for the water molecule by the Partridge-Schwenke71 surface, plus the water–water interactions from the fully compatible TTM4 polarizable model potential,64 as  .
.
 with
with  individuals, χi being the Cartesian atomic coordinates and ηi their standard deviation for Gaussian mutation, known as the strategy parameter, that controls the evolution of the population's dispersion in time. The initial coordinates χi with ηi =1 were chosen randomly in the interval (0, Δ), with Δ being a displacement factor that increases the resolution power. Each parent set (χi,ηi) evolves to generate a new population by mutation,58
individuals, χi being the Cartesian atomic coordinates and ηi their standard deviation for Gaussian mutation, known as the strategy parameter, that controls the evolution of the population's dispersion in time. The initial coordinates χi with ηi =1 were chosen randomly in the interval (0, Δ), with Δ being a displacement factor that increases the resolution power. Each parent set (χi,ηi) evolves to generate a new population by mutation,58 and
and  , where
, where  and
and  are Gaussian random numbers with mean zero and standard deviation one generated for each j = 1 − N′ coordinate, while τ′/τ′′ are adjustable parameters, commonly set to
are Gaussian random numbers with mean zero and standard deviation one generated for each j = 1 − N′ coordinate, while τ′/τ′′ are adjustable parameters, commonly set to  and
and  , where N′ is the number of degrees of freedom of each X−(H2O)N cluster. Next, for each individual of the joint parent-child group (
, where N′ is the number of degrees of freedom of each X−(H2O)N cluster. Next, for each individual of the joint parent-child group ( individuals), q (tournament size) opponents are randomly chosen from the
individuals), q (tournament size) opponents are randomly chosen from the  individuals, to compare each other, and the
individuals, to compare each other, and the  individual in each encounter with the lower potential energy become parents for the next generation and so on. Convergence is achieved when the potential energy difference between two consecutive generations is below a threshold value of 0.001 kcal mol−1. Generations with 100 individuals were used, with q = 50, while the displacement of the atom coordinates, governed by a Gaussian function, was initially multiplied by a factor δ0 =0.15, while in every next generation δj was set to 0.85 to control the size of the movement with respect to the previous one.
individual in each encounter with the lower potential energy become parents for the next generation and so on. Convergence is achieved when the potential energy difference between two consecutive generations is below a threshold value of 0.001 kcal mol−1. Generations with 100 individuals were used, with q = 50, while the displacement of the atom coordinates, governed by a Gaussian function, was initially multiplied by a factor δ0 =0.15, while in every next generation δj was set to 0.85 to control the size of the movement with respect to the previous one.
By exploring the potential surface for each X−(H2O)N cluster, we localized various optimized structures, so we additionally performed frequency calculations by analyzing the Hessian eigenvalues properties for characterizing the nature of the localized stationary PES's configurations. In Table S1 (see ESI†) we list the computed energies of various low-energy cluster structures with n up to 8 water molecules, obtained from the EP algorithm58 using the i-TTM4 potential. In turn, in Fig. 2 we summarize the energetics of these low-lying halide–water cluster structures as N increases, together with the global minima structures for F− and I− clusters. For each cluster, we have identified several potential minima, with those lying close in energy to the global minimum being important as they could be accessible when quantum zero-point effects are included. By comparing the minimum energy configurations of the halide–water clusters studied, those of the F− exhibit qualitatively different structural arrangements than the Cl−/Br−/I−, as a consequence of its smaller size and stronger fluoride–water hydrogen bond compared to the water–water ones. Due to the competition between the halide–water and water–water interactions, the stronger F−–water term forces the sequential water molecules to form fluoride–water hydrogen bonds instead of water–water ones in the first hydration shell, contrary to the hydrogen bond arrangements of the heavier Cl−/Br−/I−–water clusters, as it can be clearly seen for the N = 2, 3 and 4 minima structures.
 | ||
| Fig. 2 Schematic representation of optimal low-lying structures and their energetics obtained using the i-TTM4 X−(H2O)N PESs. | ||
Most of these low-lying energy structures correspond to internal rotations of water molecules, with energy differences between them of a few kcal mol−1. Results on the relative stability of such potential minima clearly reflect the intriguing interplay between the ion–water and water–water depending on the accuracy of the potential models employed to represent these underlying interactions. In particular for the F−(H2O)3 cluster we located four low-lying structures within 1.45 kcal mol−1, with the global minimum on the PES at an energy of −67.96 kcal mol−1, while for the F−(H2O)4 and F−(H2O)8 we show seven optimal configurations with the lowest energy of −85.36 and −142.46 kcal mol−1, respectively. In turn, for the Cl−(H2O)8, Br−(H2O)8 and I−(H2O)8 clusters the lowest energy structure corresponds to energies of −115.36, −109.03 and −103.41 kcal mol−1, respectively.
In the inset plot of Fig. 2 we report the incremental energies, defined as ΔE(1) = EN–1 − EN, for each cluster size, as indicators of their stability with respect to its nearest one, for detecting the completion of shell structures. Such energies show quite similar behavior for most Cl−/Br−/I−–water clusters, while significant differences are observed for smaller size F−–water clusters, up to N = 5. For the larger clusters with N = 7 and 8 water molecules the ΔE(1) energies have similar values independently of the halide anion. By analyzing the structures of the clusters as the number of water molecules increase, one can figure out that the halide ions prefer to locate themselves on the cluster surface, with water molecules forming multiple shell structures with 3 (in the case of F−–water) and 4 (in the Cl−/Br−/I−–water cases) molecules in the first hydration shell and 1 to 4 in the second one connected via water–water hydrogen bonds. Such selective growth of these small X−(H2O)N clusters is due to the competition between the weak ion–water and water–water interactions, giving rise to on-surface ion structures. However, we should point out that all structures discussed here correspond to classical optimal structures on the PES of each cluster, and quantum zero-point energy, as well as thermal effects, are expected to cause significant changes in their stability, especially for those lying close in energy.
1.2 Molecular dynamics simulations: the path integral formulation
It is well known the power of the path integral (PI) method as a non-perturbative approach to study finite-temperature properties of quantum many-body systems. The method is based on Feynman's formulation of statistical mechanics, which makes it possible to write the partition function Q in a path integral form.74,75 For a canonical (constant NVT) ensemble of N particles with coordinates r ≡ (r1,…,rN), at temperature T, the partition function is written as | (1) |
 one can sample the Boltzmann statistics of the quantum system through its classical isomorph, while for
one can sample the Boltzmann statistics of the quantum system through its classical isomorph, while for  one falls back onto the classical description.
one falls back onto the classical description.
The internal energy E can be calculated from the partition function via the thermodynamic relation
 | (2) |
 | (3) |
 | (4) |
![[r with combining macron]](https://www.rsc.org/images/entities/b_char_0072_0304.gif) i (center of mass of the replicas of particle i) and employing a path integral version of the virial theorem, we obtain a new kinetic energy estimator for the kinetic energy:76
i (center of mass of the replicas of particle i) and employing a path integral version of the virial theorem, we obtain a new kinetic energy estimator for the kinetic energy:76 | (5) |
The path integral molecular dynamics (PIMD) method consists of integrating the classical motion equations derived from the path integral Hamiltonian to sample the canonical ensemble. Equilibrium properties such as the potential and kinetic energies can be obtained by time-averaging along ring polymer trajectories whose initial conditions are sampled from the Boltzmann distribution. In order to achieve efficient sampling of the canonical ensemble, we combine the ring polymer dynamics with a Bussi-Parrinello77,78 thermostat attached to each degree of freedom. Each coordinate of each particle follows the Langevin dynamics:
 | (6) |
We have performed PIMD simulations at a constant temperature for finite hydrated halide ions with up to 8 water molecules, with the corresponding atomic masses obtained from ref. 79. We used as starting configurations the optimized structures obtained above from the evolutionary algorithm. The equations of motion were integrated using a time step of 1 fs, and a friction parameter of γ = 0.001 a.u., which provided effective sampling. For a given temperature value and cluster size, a typical simulation run consisted of 104 PIMD steps for system equilibration, and up to 2 × 106 steps for the production and calculation of ensemble average properties. In all cases, the associated uncertainties of equilibrium averages were computed with block averaging, as the plateau of the standard error of the mean among the blocks, when increasing the block size.80–82
In Fig. 3 we displayed the computed average total energies, 〈E〉, from such classical MD calculations, while Tables S2 and S4 (ESI†) show all averaged energy components at the temperatures of 10 and 300 K, respectively. One can see that thermal effects affect the averaged energies of the systems, with the values being slightly higher than those of the potential minima, that correspond to zero temperature. As the temperature increases higher energy conformers (such as low-lying local minima) are also populated, contributing to the considerably higher energies obtained. At T = 10 K we found that energy shifts of 0.2–1.0 kcal mol−1 per water molecule, while at T = 300 K they count up to ≈10 kcal mol−1 per water molecule for all X−(H2O)N clusters studied compared to the potential minima values shown in Fig. 2. As expected, thermal effects become important for all halide–water clusters, and they increase as their size and temperature increases. We found that at zero pressure, when the number of water molecules increases they lead to cluster fragmentation, e.g. at T = 300 K and N = 8. As we will discuss next, in comparison with the inclusion of nuclear quantum contributions, such effects are also observable in the structural properties of the clusters.
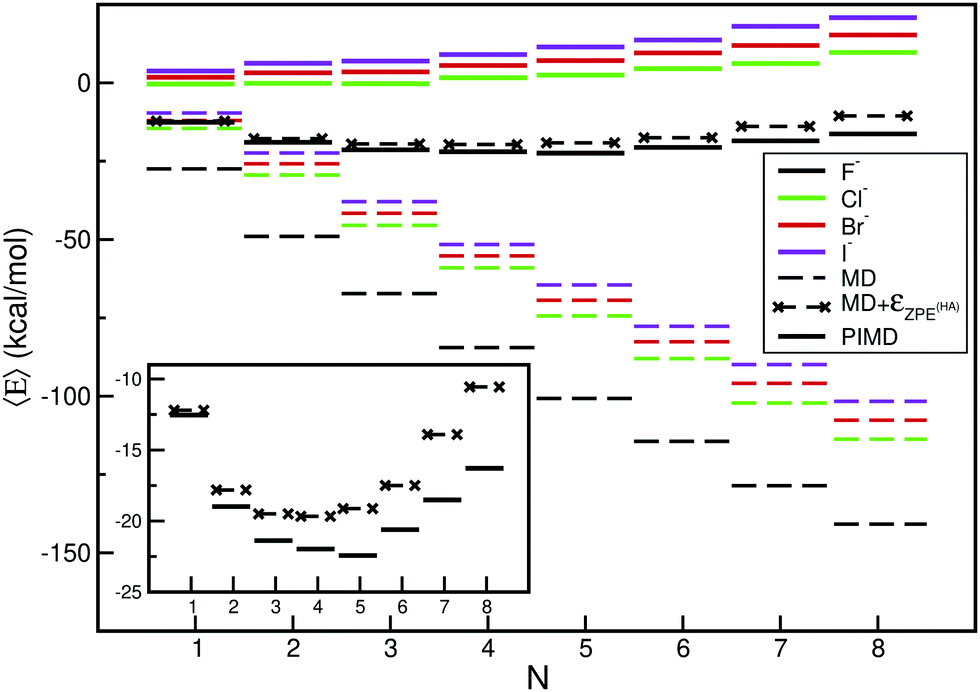 | ||
Fig. 3 Schematic representation of the average total 〈E〉 energy values of the X−(H2O)N=1–8 clusters from the classical MD (dashed lines) and PIMD (solid lines) simulations at T = 10 K. Estimates of the average total 〈E〉 MD energy values corrected by the harmonic  are also shown in the case of F−(H2O)N=1–8 clusters (see text). are also shown in the case of F−(H2O)N=1–8 clusters (see text). | ||
 , with
, with  corresponding to the classical MC simulations, to check the convergence with respect to the finite values of replicas
corresponding to the classical MC simulations, to check the convergence with respect to the finite values of replicas  to be employed. In the left panels of Fig. S1 (ESI†) we display the convergence tests made for the kinetic (〈K〉), potential (〈V〉), and total (〈E〉) energy values as a function of the number of replicas at different temperatures and cluster size. In order to evaluate values at
to be employed. In the left panels of Fig. S1 (ESI†) we display the convergence tests made for the kinetic (〈K〉), potential (〈V〉), and total (〈E〉) energy values as a function of the number of replicas at different temperatures and cluster size. In order to evaluate values at  limit, we employ a quadratic extrapolation expression, such as
limit, we employ a quadratic extrapolation expression, such as  ,52,83 with A and B being the parameters adjusted to the converged total energy values. One can see in Fig. 1 that for the lighter F−(H2O) and F−(H2O)8 clusters, values obtained with
,52,83 with A and B being the parameters adjusted to the converged total energy values. One can see in Fig. 1 that for the lighter F−(H2O) and F−(H2O)8 clusters, values obtained with  for the T = 10 K and 100, 200 and 500 for T = 300 K are fully converged. Thus, we only considered energies from PIMD simulations with
for the T = 10 K and 100, 200 and 500 for T = 300 K are fully converged. Thus, we only considered energies from PIMD simulations with  and 200/500 at 10 and 300 K, respectively, in the quadratic fits for computing the energy values at
and 200/500 at 10 and 300 K, respectively, in the quadratic fits for computing the energy values at  limit for these clusters (as shown by the (cyan) colored lines in the right panel of Fig. S1 (ESI†)). By comparing the results from these two-step extrapolation values with those obtained from the
limit for these clusters (as shown by the (cyan) colored lines in the right panel of Fig. S1 (ESI†)). By comparing the results from these two-step extrapolation values with those obtained from the  at 10 and 300 K, respectively, we decided to consider the latter number of replicas in all computations presented hereafter.
at 10 and 300 K, respectively, we decided to consider the latter number of replicas in all computations presented hereafter.
In Fig. 4 we display representative plots of the total average energy evolution versus the number of steps and time from the NVT PIMD calculations with  at T = 10 K for each X−(H2O)8 and dissociative X− + (H2O)8 systems. One can see that the energy of each system reaches first its thermal equilibrium value, followed then by fluctuations around its corresponding average value. In Fig. 4 we also included snapshots of configurations sampled during the PIMD simulations, for the F−(H2O)8 and I−(H2O)8 clusters at selected step/time. One can see that the clusters show a surface location for the halide anion in the water cluster, and their configurations are significantly extended compared to their compact 4 + 4 minimum energy geometries (see Fig. 2).
at T = 10 K for each X−(H2O)8 and dissociative X− + (H2O)8 systems. One can see that the energy of each system reaches first its thermal equilibrium value, followed then by fluctuations around its corresponding average value. In Fig. 4 we also included snapshots of configurations sampled during the PIMD simulations, for the F−(H2O)8 and I−(H2O)8 clusters at selected step/time. One can see that the clusters show a surface location for the halide anion in the water cluster, and their configurations are significantly extended compared to their compact 4 + 4 minimum energy geometries (see Fig. 2).
 | ||
Fig. 4 Evolution of total energy values during the NVT PIMD simulations for the X−(H2O)8 and corresponding X− + (H2O)8 systems, with  at T = 10 K. at T = 10 K. | ||
All computed 〈E〉 values from the PIMD simulations at T = 10 K are shown in Fig. 3 in comparison with those from the classical MD ones, while in Tables S2 and S5 (ESI†) average potential, kinetic, and total energies together with the corresponding errors are listed for the X−(H2O)N clusters and X− + (H2O)N systems, respectively. One can clearly see in Fig. 3 the impact of nuclear quantum effects in the energetics and binding of such halide–water clusters. The quantum total energies are considerably higher than the classical ones (at the same temperature) due to the zero-point energy (ZPE) effects. By treating each X−(H2O)N cluster in the framework of the harmonic approximation (HA), the corresponding ZPE values are obtained from normal-mode frequency calculation at each cluster's minimum energy structure. In Fig. 3 (see also the inset plot), for all F−(H2O)N clusters, we include these harmonic  estimates to the average 〈E〉 values from the MD simulations at T = 10 K, and as one can see quantum and classical energy values are reaching better agreement.
estimates to the average 〈E〉 values from the MD simulations at T = 10 K, and as one can see quantum and classical energy values are reaching better agreement.
The computed harmonic (HA) and anharmonic ZPE values (from the PIMC simulations at T = 10 K) are given in Table S2 (ESI†) for all clusters studied in comparison with previously reported data from the DMC (T = 0 K) calculations45 for clusters up to N = 5 water molecules. ZPE values of 15.20, 14.55, 14.52 and 14.40 kcal mol−1 are obtained from the present PIMD for the F−(H2O), Cl−(H2O), Br−(H2O) and I−(H2O), respectively, that compare well with earlier and recent reported values from variational quantum calculations available in the literature44,84–86 (see Table S3, ESI†) employing different PESs.60,61,84,85 One can see that the ZPE values for the F−(H2O) cluster show rather larger differences between 13.81 and 15.40 kcal mol−1, highly depending on the underlying potential surface, than those of the heavier halide–H2O clusters, where the ZPE range is significantly smaller, such as 14.33–14.73 kcal mol−1 for the Cl−(H2O), 14.26–14.57 kcal mol−1 for the Br−(H2O), and 14.12–14.40 kcal mol−1 for the I−(H2O). For larger sized clusters, we found that the ZPE energies from the PIMC simulations are in good accord with the reported values from the DMC calculations at T = 0 K45 (see Table S2, ESI†). In particular, due to the potential employed as well as the temperature effects, differences were observed for the smaller sized F−(H2O)N clusters, while smaller differences up to 0.3 kcal mol−1 are found for the heavier X−(H2O)N (X = Cl, Br, I) clusters. Also, upon comparing the anharmonic ZPE values from the PIMC calculations and harmonic  estimates, we observe that anharmonicities and nuclear quantum effects substantially affect these systems, with differences in energies up to 5%.
estimates, we observe that anharmonicities and nuclear quantum effects substantially affect these systems, with differences in energies up to 5%.
The average structures of these clusters are also obtained and described in terms of radial and angular distributions. In Fig. S2 (ESI†) we plotted such quantum distributions of the X−–O distance (see left panels) and ∠OHO angle (see right panels), respectively, at T = 10 (see solid lines) and 300 K (see dashed/shadow pattern lines). In the radial X−–O distributions at T = 10 K, we first observe the presence of several sharp peaks, and their position is highly dependent on the halide ion and the size of the halide–water cluster. The first set of peaks in the X−–O distributions is shifted to larger distances as the F− is replaced by the heavier Cl−, Br− and I− ions, and more peaks appear as the number of water molecules increases. In turn, the angular ∠OHO distributions show clear peaks shifted towards and backwards in the interval of 100–110° as the size of the cluster increases. By inspecting the radial X−–O distributions information can be gained on the hydration shell formation at low temperatures. One can differentiate four sets of peaks for F−–water clusters, while for the remaining X−–water systems the second and third peaks are overlapped. Representative structures of F−(H2O)8 and I−(H2O)8 clusters are displayed in Fig. 4 and also support the preference of all halide ions to reside on the surface of the water clusters, occupying one of the vertices in the H-bond network, in accord with previous and more recent investigations23,45 employing polarizable force fields. Thus, we could identify the first hydration shell around these halide ions, composed of 3 and 4 water molecules in the case of F− and Cl−/Br−/I− anions, respectively, at rX–O distances of around 2.6 and 3.1/3.5/3.7 Å (see the inset plots of Fig. S2, ESI†).
As the temperature increases at T = 300 K, both classical and quantum radial and angular distributions are found to show similar behavior with the presence of few, low and broad peaks. One can see that at low temperature values quantum effects are appreciable for such small size clusters at both distributions presented, while at high temperatures thermal fluctuations drown out the quantum ones. In Fig. 5 we summarized the evolution of the mean 〈X−O〉 distances computed from the PIMD simulations as a function of the cluster's size, at temperatures of 10 (see solid lines) and 300 K (see dotted lines), together with results obtained from the classical MD calculations at 10 K (see dashed lines). In turn, Fig. S3 (ESI†) shows the dependence of mean 〈HOH〉 angle values obtained from the classical MD (see dashed lines) and PIMD (see solid lines) calculations at temperatures of 10 (see left panel) and 300 K (see right panel) as the number of water molecules increases. We also analysed radial X−–H, as well as O–O and O–H/O*–H (with O or O* and H atoms belonging to the same or different monomers, respectively) radial distributions, and in Fig. 6 we summarize all mean distances and 〈HOH〉 angles for all X−(H2O)8 at T = 10 K, comparing them with those obtained for the pure (H2O)8 water cluster. We should note that the presence of the halide ion affects certain structural properties and changes are mainly observed at the first hydration shell, with the H-bond water network gets weaker leading to an increase in the O–O distance. The analysis of the arrangement of water molecules around the halide ions indicates a compromise between the energetic competition of halide–water and water–water interactions, with the distortion of the water molecules showing only slightly noticeable effects on the O–H, especially in the case of F−, while the O*–H distances are smaller in the cases of F− and Cl−, and slightly larger for Br− and I−, compared to those of the (H2O)8. One can also see shifts of the 〈HOH〉 angle value to smaller bond angles when a halide ion is added. As expected, both X−–O and X−–H distance distributions are highly dependent on the size of the halide ion, and show shifts to larger values as the ion's size increases.
 | ||
| Fig. 5 Mean 〈X−O〉 distances computed from the corresponding classical MD and PIMD radial distributions at T = 10 (dashed and solid lines, respectively) and 300 K (dotted line) as a function of N for the indicated X−(H2O)N=1–8 clusters. | ||
 | ||
| Fig. 6 Mean indicated distances and 〈HOH〉 angle values as obtained from PIMD simulations at T = 10 K for the (H2O)8 and X−(H2O)8 clusters. | ||
Experimental studies on the energetics of ion solvation are available in the literature for halide–water clusters with 1 up to 6, 7, 16 and 60 water molecules, for the F−, Cl−, Br− and I−, respectively, together with experimental single-ion bulk values.10,11,87 Such small cluster size systems are treatable with both theory and experiment. Although all finite cluster sizes considered here are rather small to relate their properties to condensed phase systems, the results obtained indicate promising trends, comparable to the corresponding experimental data, that motivate us to draw connections between the small aqueous halide cluster solvation energies to those of larger ones up to the bulk.
Fig. 7 shows single-ion solvation energies as a function of the cluster size (N−1/3) reported by experiments,10,11,87 in comparison with the ΔEsolv,N values obtained from the present PIMD simulations here. As we can see the patterns in single-ion solvation energetics are similar in both experimental and theoretical values in the small cluster-size regime. Both experimental cluster-ion solvation free energy and the theoretical solvation energy curves show linear dependence vs. cluster size for all X−(H2O)N systems. Although some quantitative differences could be appreciated in the small cluster size regime, where the present computations seem to predict more stable halide–water clusters than the most recent experimental studies,10,11 and less stable ones than the earlier experimental data on stabilization energies on Cl−, Br− and I− water clusters.87
 | ||
| Fig. 7 Single ion solvation energies vs. N1/3, where N is the number of solvating water molecules: ΔEsolv,N from the present NVT PIMD calculations (filled circle lines), and experimental cluster-ion solvation free (ΔG) energies (square/filled diamonds/triangles lines) of the indicated anions from ref. 10, 11 and 87. Linear trends in single-ion solvation energetics in the large cluster size regime are shown with dotted and dotted-dashed lines for the present theoretical and available experimental data, respectively, while experimentally determined single-ion bulk solvation free energies, ΔGsolv,∞ are drawn with long-dashed lines.11 | ||
The computed cluster data energies are extrapolated into the bulk values employing the N−1/3 cluster size dependence,8,88 ΔEsolv,N = ΔEsolv,∞ + ![[scr D, script letter D]](https://www.rsc.org/images/entities/i_char_e523.gif) N−1/3, where ΔEsolv,∞ is the bulk values, and the slope parameter. The dotted lines in Fig. 7 represent such linear fits to the small cluster (up to 8) data, and the computed bulk ΔEsolv,∞ ion solvation energies are −114.53, −73.35, −65.28, and −55.82 kcal mol−1 for the F−, Cl−, Br−, and I−, respectively. One can clearly see that these values are in very good accord with those previously reported10,11 for single-ion bulk solvation free energies of −102.49, −72.71, −66.30, and −57.36 kcal mol−1, for the F−, Cl−, Br−, and I− ions,11 respectively, and −57.81 kcal mol−1 for I− in ref. 10. Furthermore, photoelectron-spectra of Cl−, Br−, and I−, solvated in water clusters formed by 7, 16 and 60 water molecules, respectively, have been also recorded, and have been used to extract the solvent electrostatic stabilization energies of these halide anions clusters.87 By extrapolating their measurements, in the case of the I− surface solvated ion in the bulk, they have concluded that the E∞stab should lie somewhere between 4.74 eV (109.31 kcal mol−1) and 4.37 eV (100.78 kcal mol−1).87 In all cases, the present energies are found between the values experimentally reported in ref. 10, 11 and 87 following the same trend as the results reported in ref. 10 and 11 for the I−, providing estimates based on the cluster size dependence of the single-ion bulk solvation energies in good accord with the experimental ones. The computed bulk solvation energies are lower by 12, and 0.64 kcal mol−1 for the F− and Cl− and higher by 1 and 2 kcal mol−1 for the Br− and I− ions, respectively, compared to experimental values.11 However, we should note that the present simulations were carried out at zero pressure, and specific thermodynamic conditions for the experiments could further affect the energetics. Moreover, we should also mention the influence of possible error cancellations, during the evaluation of the ion–solvation energy differences from the PIMD simulations for the X−(H2O)N and (H2O)N systems, although their effect is fortuitous, and thus difficult to predict.
N−1/3, where ΔEsolv,∞ is the bulk values, and the slope parameter. The dotted lines in Fig. 7 represent such linear fits to the small cluster (up to 8) data, and the computed bulk ΔEsolv,∞ ion solvation energies are −114.53, −73.35, −65.28, and −55.82 kcal mol−1 for the F−, Cl−, Br−, and I−, respectively. One can clearly see that these values are in very good accord with those previously reported10,11 for single-ion bulk solvation free energies of −102.49, −72.71, −66.30, and −57.36 kcal mol−1, for the F−, Cl−, Br−, and I− ions,11 respectively, and −57.81 kcal mol−1 for I− in ref. 10. Furthermore, photoelectron-spectra of Cl−, Br−, and I−, solvated in water clusters formed by 7, 16 and 60 water molecules, respectively, have been also recorded, and have been used to extract the solvent electrostatic stabilization energies of these halide anions clusters.87 By extrapolating their measurements, in the case of the I− surface solvated ion in the bulk, they have concluded that the E∞stab should lie somewhere between 4.74 eV (109.31 kcal mol−1) and 4.37 eV (100.78 kcal mol−1).87 In all cases, the present energies are found between the values experimentally reported in ref. 10, 11 and 87 following the same trend as the results reported in ref. 10 and 11 for the I−, providing estimates based on the cluster size dependence of the single-ion bulk solvation energies in good accord with the experimental ones. The computed bulk solvation energies are lower by 12, and 0.64 kcal mol−1 for the F− and Cl− and higher by 1 and 2 kcal mol−1 for the Br− and I− ions, respectively, compared to experimental values.11 However, we should note that the present simulations were carried out at zero pressure, and specific thermodynamic conditions for the experiments could further affect the energetics. Moreover, we should also mention the influence of possible error cancellations, during the evaluation of the ion–solvation energy differences from the PIMD simulations for the X−(H2O)N and (H2O)N systems, although their effect is fortuitous, and thus difficult to predict.
2 Summary and conclusions
The present work is focused on a comprehensive description of the single halide-ion microsolvation in water clusters from a molecular quantum perspective. Small-size X−(H2O)N clusters are used as model microsolutions; they are computationally tractable, and reliable interaction forces from first-principles data-driven methodologies are available for performing the quantum molecular dynamics simulations. Accurate descriptions of the underlying potentials can provide a very useful initial information on solvation shell structure, so the most stable potential configurations for each halide–water cluster were localized via optimizations employing an evolutionary programming algorithm. New insights into the energetics of the clusters as a function of halide ions and the number of solvent molecules highlight the competition between halide–water and water–water intermolecular interactions, that control the stabilization of these low-lying energy conformers. We clearly see that during the initial cluster growth with N > 2 the formation of anion–surface structures is more favored, indicating a preference during the microhydration process.Furthermore, the classical description was completed by explicitly including thermal corrections from CMC computations, and quantum features, such as ZPE values and spatial delocalization, from quantum PIMC simulations. Nuclear quantum effects are presented especially at low temperatures, while at higher temperatures they are overcome by the thermal fluctuations. In fact, the analysis of our quantum simulations data reveals that halides induce minor distortions, varying within the X− series, in the H-bond water network arrangements beyond the first shell hydration shell, indicating that structural rearrangements are strongly localized in the vicinity of the micro-hydrated ions, in line with the available experimental measurements in aqueous solutions. The present results highlight the importance of nuclear quantum effects at certain structural properties, such as the weak H-bonding between anions and solvation shells of water molecules, so the disruption on the solvent water structure is significantly reduced compared to those obtained from classical computations in their absence.
The anisotropy of the underlying PES is reflected in the solvation structure, and this information could serve for describing the transition from micro-hydrated to dissolved anions. The competition between anion–water and water–water structures as cluster size grows can be understood in terms of the strength of each interaction term, with the size of the halide ion determining the energy gain by the attachment of the anion in the water H-bond network. All finite cluster sizes considered in this work are rather small to relate their properties to condensed phase systems, however, it has been shown that there is overall, gradual progress of the cluster properties, which have been examined toward bulk expectations. We observed that the patterns in single-ion solvation energetics show a linear dependence as a function of the size of the clusters. The observed trends are similar in both experiment and computations, with extrapolation of the present cluster energies to bulk values providing estimates in surprisingly good accord with those experimentally reported on single-ion bulk solvation free energies.
However, as such trends are dominated by the ion–solvent interactions as cluster size increases, complementary studies of larger halide–water clusters, as well as on micro-hydration of cations, such as alkali-metals, are particularly interesting to reveal how the transition from ion–solvent to solvent–solvent dominant structure proceeds. Especially, in the latter case, as such cations are not H-bonded to water, potential anharmonicities together with nuclear quantum effects are a priori expected to affect the solvent water structures. The computational cost for nuclear full quantum treatments is getting really high as going to larger dimensional systems. However, benchmark studies for small species could serve for testing other approaches to deal with higher dimensional systems with an increasing number of particles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Eduardo R. Hernández, Dr Daniel Arismendi-Arrieta, and Raquel Yanes-Rodríguez for the useful discussions. The authors acknowledge the “Centro de Calculo del IFF/SGAI-CSIC” and CESGA-Supercomputing centre for allocation of computer time. This work has been supported by Comunidad de Madrid grant No. IND2017/AMB-7696, MINECO grant No. FIS2017-83157-P, MICINN grant No, PID2020-114654GB-I00 and COST Action CA18212(MD-GAS).Notes and references
- Y. Marcus, Chem. Rev., 2009, 109, 1346–1370 CrossRef CAS PubMed.
- P. Jungwirth and P. Cremer, Nat. Chem., 2014, 6, 261–263 CrossRef CAS PubMed.
- Y. Marcus, Ions in Solution and their Solvation, John Wiley & Sons, Inc., Hoboken, New Jersey, 2015 Search PubMed.
- B. C. Garrett, Science, 2004, 303, 1146–1147 CrossRef CAS PubMed.
- H. Bakker, Chem. Rev., 2008, 108, 1456–1473 CrossRef CAS PubMed.
- P. Jena and Q. Sun, Chem. Rev., 2018, 118, 5755–5870 CrossRef CAS PubMed.
- A. Rognoni, R. Conte and M. Ceotto, Chem. Sci., 2021, 12, 2060–2064 RSC.
- J. Jortner, Z. Phys. D Atom Mol. Cl., 1992, 24, 247–275 CrossRef CAS.
- O. Echt, in Convergence of Cluster Properties Towards Bulk Behavior: How Large is Large?, Springer, Dordrecht, 1996 Search PubMed.
- J. V. Coe, J. Phys. Chem. A, 1997, 101, 2055–2063 CrossRef CAS.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. V. Coe and T. R. Tuttle, J. Phys. Chem. A, 1998, 102, 7787–7794 CrossRef CAS.
- R. Ludwig, Angew. Chem., Int. Ed. Engl., 2001, 18, 1808–1827 CrossRef.
- F. N. Keutsch and R. J. Saykally, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10533–10540 CrossRef CAS PubMed.
- G. W.-F. Drake, Springer Handbook of Atomic, Molecular, and Optical Physics, Springer-Verlag New York, New York, 2006 Search PubMed.
- P. K. Gupta, P. Schienbein, J. Daru and D. Marx, J. Phys. Chem. Lett., 2019, 10, 393–398 CrossRef PubMed.
- L. Perera and M. L. Berkowitz, J. Chem. Phys., 1991, 95, 1954–1963 CrossRef CAS.
- L. X. Dang and D. E. Smith, J. Chem. Phys., 1993, 99, 6950 CrossRef CAS.
- L. Perera and M. L. Berkowitz, J. Chem. Phys., 1994, 100, 3085–3093 CrossRef CAS.
- S. J. Stuart and B. J. Berne, J. Phys. Chem., 1996, 100, 11934–11943 CrossRef CAS.
- O. M. Cabarcos, C. J. Weinheimer, J. M. Lisy and S. S. Xantheas, J. Chem. Phys., 1999, 110, 5–8 CrossRef CAS.
- P. Ayotte, S. B. Nielsen, G. H. Weddle, M. A. Johnson and S. S. Xantheas, J. Phys. Chem. A, 1999, 103, 10665–10669 CrossRef CAS.
- J. Kim, H. M. Lee, S. B. Suh, D. Majumdar and K. S. Kim, J. Chem. Phys., 2000, 113, 5259–5272 CrossRef CAS.
- D. J. Tobias, P. Jungwirth and M. Parrinello, J. Chem. Phys., 2001, 114, 7036–7044 CrossRef CAS.
- W. H. Robertson and M. A. Johnson, Ann. Rev. Phys. Chem., 2003, 54, 173–213 CrossRef CAS PubMed.
- R. Ayala, J. M. Martínez, R. R. Pappalardo and E. Sánchez Marcos, J. Chem. Phys., 2003, 119, 9538–9548 CrossRef CAS.
- H. J. Bakker, M. F. Kropman and A. W. Omta, J. Phys.: Condens. Matter, 2005, 17, S3215–S3224 CrossRef CAS.
- C. J. Burnham, M. K. Petersen, T. J.-F. Day, S. S. Iyengar and G. A. Voth, J. Chem. Phys., 2006, 124, 024327 CrossRef PubMed.
- J. D. Smith, R. J. Saykally and P. L. Geissler, J. Am. Chem. Soc., 2007, 129, 13847–13856 CrossRef CAS PubMed.
- X. Huang, S. Habershon and J. M. Bowman, Chem. Phys. Lett., 2008, 450, 253–257 CrossRef CAS.
- E. Guàrdia, I. Skarmoutsos and M. Masia, J. Chem. Theory Comput., 2009, 5, 1449–1453 CrossRef PubMed.
- C. D. Wick and S. S. Xantheas, J. Phys. Chem. B, 2009, 113, 4141–4146 CrossRef CAS PubMed.
- S. Horvath, A. B. McCoy, B. M. Elliott, G. H. Weddle, J. R. Roscioli and M. A. Johnson, J. Phys. Chem. A, 2010, 114, 1556–1568 CrossRef CAS PubMed.
- C. Caleman, J. S. Hub, P. J. van Maaren and D. van der Spoel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6838–6842 CrossRef CAS.
- M. Trumm, Y. O.-G. Martínez, F. Réal, M. Masella, V. Vallet and B. Schimmelpfennig, J. Chem. Phys., 2012, 136, 044509 CrossRef PubMed.
- E. Kamarchik and J. M. Bowman, J. Phys. Chem. Lett., 2013, 4, 2964–2969 CrossRef CAS.
- V. Migliorati, F. Sessa, G. Aquilanti and P. D'Angelo, J. Chem. Phys., 2014, 141, 044509-1–044509-11 CrossRef PubMed.
- C. T. Wolke, F. S. Menges, N. Tötsch, O. Gorlova, J. A. Fournier, G. H. Weddle, M. A. Johnson, N. Heine, T. K. Esser, H. Knorke, K. R. Asmis, A. B. McCoy, D. J. Arismendi-Arrieta, R. Prosmiti and F. Paesani, J. Phys. Chem. A, 2015, 119, 1859–1866 CrossRef CAS PubMed.
- P. E. Videla, P. J. Rossky and D. Laria, J. Phys. Chem. B, 2015, 119, 11783–11790 CrossRef CAS PubMed.
- M. Antalek, E. Pace, B. Hedman, K. O. Hodgson, G. Chillemi, M. Benfatto, R. Sarangi and P. Frank, J. Chem. Phys., 2016, 145, 044318 CrossRef PubMed.
- S. Wang, W. Fang, T. Li, F. Li, C. Sun, Z. Li, Y. Huang and Z. Men, J. Appl. Phys., 2016, 119, 163104 CrossRef.
- S. Chakrabarty and E. R. Williams, Phys. Chem. Chem. Phys., 2016, 18, 25483–25490 RSC.
- N. Yang, C. H. Duong, P. J. Kelleher, M. A. Johnson and A. B. McCoy, Chem. Phys. Lett., 2017, 690, 159–171 CrossRef CAS.
- P. E. Videla, P. J. Rossky and D. Laria, J. Chem. Phys., 2018, 148, 102306 CrossRef PubMed.
- P. Bajaj, X.-G. Wang, T. Carrington and F. Paesani, J. Chem. Phys., 2018, 148, 102321 CrossRef PubMed.
- J. D. Mallory and V. Mandelshtam, J. Phys. Chem. A, 2018, 122, 4167–4180 CrossRef CAS PubMed.
- P. Bajaj, M. Riera, J. K. Lin, Y. E.-M. Montijo, J. Gazca and F. Paesani, J. Phys. Chem. A, 2019, 123, 2843–2852 CrossRef CAS PubMed.
- J. Xu, Z. Sun, C. Zhang, M. DelloStritto, D. Lu, M. L. Klein and X. Wu, Phys. Rev. Mater., 2021, 5, L012801 CrossRef CAS.
- J. P. Joseph, P. Heindel and S. S. Xantheas, J. Chem. Theory Comput., 2021, 17, 2200–2216 CrossRef PubMed.
- D. Ceperley, Rev. Mod. Phys., 1995, 67, 279 CrossRef CAS.
- S. Habershon, G. S. Fanourgakis and D. E. Manolopoulos, J. Chem. Phys., 2008, 129, 074501 CrossRef PubMed.
- R. P. de Tudela, P. Barragán, R. Prosmiti, P. Villarreal and G. Delgado-Barrio, J. Phys. Chem. A, 2011, 115, 2483–2488 CrossRef CAS PubMed.
- R. Pérez de Tudela, P. Barragán, A. Valdés and R. Prosmiti, J. Phys. Chem. A, 2014, 118, 6492–6500 CrossRef PubMed.
- M. Ceriotti, W. Fang, P. G. Kusalik, R. H. McKenzie, A. Michaelides, M. A. Morales and T. E. Markland, Chem. Rev., 2016, 116, 7529–7550 CrossRef CAS PubMed.
- A. Gijón and E. R. Hernández, Quantum simulations of neutral water clusters and singly-charged water cluster anions, 2022 DOI:10.48550/arXiv.2203.00375.
- R. Rodríguez-Segundo, D. J. Arismendi-Arrieta and R. Prosmiti, Molecules, 2022, 27, 1654 CrossRef PubMed.
- A. Gijón-Gijón, PhD thesis, Materials Science Institute of Madrid, ICMM-CSIC, 2021 Search PubMed.
- R. Rodríguez-Segundo, D. Arismendi-Arrieta and R. Prosmiti, A bottom-up approach for ion–water interactions: From clusters to bulk, 2019, Chapter 8, pp. 205–225 Search PubMed.
- M. Iwamatsu, Comput. Phys. Commun., 2001, 142, 214–218 CrossRef CAS.
- DENEB 1.30 beta: the Nanotechnology Software by Atelgraphics, 2020, https://www.atelgraphics.com.
- D. J. Arismendi-Arrieta, M. Riera, P. Bajaj, R. Prosmiti and F. Paesani, J. Phys. Chem. B, 2016, 120, 1822–1832 CrossRef CAS PubMed.
- P. Bajaj, A. W. Götz and F. Paesani, J. Chem. Theory Comput., 2016, 12, 2698–2705 CrossRef CAS PubMed.
- G. S. Fanourgakis and S. S. Xantheas, J. Phys. Chem. A, 2006, 110, 4100–4106 CrossRef CAS PubMed.
- G. S. Fanourgakis and S. S. Xantheas, J. Chem. Phys., 2008, 128, 074506 CrossRef PubMed.
- C. J. Burnham, D. J. Anick, P. K. Mankoo and G. F. Reiter, J. Chem. Phys., 2008, 128, 154519-1–154519-20 CrossRef PubMed.
- G. R. Medders, V. Babin and F. Paesani, J. Chem. Theory Comput., 2014, 10, 2906–2910 CrossRef CAS PubMed.
- Y. Wang, X. Huang, B. Shepler, B. Braams and J. Bowman, J. Chem. Phys., 2011, 134, 094509 CrossRef PubMed.
- A. Nandi, C. Qu, P. L. Houston, R. Conte, Q. Yu and J. M. Bowman, J. Phys. Chem. Lett., 2021, 12, 10318–10324 CrossRef CAS PubMed.
- H.-J. Werner, P. Knowles, G. Knizia, R. ManbySchütz and others. MOLPRO, version 2012.1, a package of ab initio programs, 2012, see http://www.molpro.net.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 122, 154104 CrossRef PubMed.
- A. Otero-de-la Roza and E. R. Johnson, J. Chem. Phys., 2013, 138, 204109-1–204109-13 Search PubMed.
- H. Partridge and D. W. Schwenke, J. Chem. Phys., 1997, 106, 4618–4639 CrossRef CAS.
- D. Fogel, Evolutionary Computation: Toward a New Philosophy of Machine Intelligence, IEEE Press-Wiley, Hoboken, New Jersey, 2006 Search PubMed.
- N. Alharzali, R. Rodríguez-Segundo and R. Prosmiti, Phys. Chem. Chem. Phys., 2021, 23, 7849–7859 RSC.
- M. Tuckerman, Statistical Mechanics: Theory and Molecular Simulation, Oxford University Press Inc., New York, 2010 Search PubMed.
- M. J. Gillan, in The Path-Integral Simulation of Quantum Systems, ed. C. R. A. Catlow, S. C. Parker and M. P. Allen, Springer Netherlands, Dordrecht, 1990, pp. 155–188 Search PubMed.
- M. F. Herman, E. J. Bruskin and B. J. Berne, J. Chem. Phys., 1982, 76, 5150–5155 CrossRef CAS.
- M. Ceriotti, M. Parrinello, T. E. Markland and D. E. Manolopoulos, J. Chem. Phys., 2010, 133, 124104 CrossRef PubMed.
- G. Bussi and M. Parrinello, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 75, 056707 CrossRef PubMed.
- CIAAW. Atomic weights of the elements 2019, https://www.nist.gov/pml/atomic-weights-and-isotopic-compositions-relative-atomic-masses, 2019, 2020-09-09.
- H. Flyvbjerg and H. G. Petersen, J. Chem. Phys., 1989, 91, 461–466 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Second Edition, Oxford University Press, Oxford, 2017 Search PubMed.
- A. Grossfield, P. N. Patrone, D. R. Roe, A. J. Schultz, D. Siderius and D. M. Zuckerman, LiveCoMS, 2018, 1, 5067 Search PubMed.
- R. Zillich and K. Whaley, J. Phys. Chem. A, 2007, 111, 7489–7498 CrossRef CAS PubMed.
- J. Sarka, D. Lauvergnat, V. Brites, A. G. Császár and C. Léonard, Phys. Chem. Chem. Phys., 2016, 18, 17678–17690 RSC.
- J. Rheinecker and J. Bowman, J. Chem. Phys., 2006, 125, 133206 CrossRef PubMed.
- X.-G. Wang and T. Carrington, J. Chem. Phys., 2014, 140, 204306 CrossRef PubMed.
- G. Markovich, S. Pollack, R. Giniger and O. Cheshnovsky, J. Chem. Phys., 1994, 101, 9344–9353 CrossRef CAS.
- G. Makov and A. Nitzan, J. Phys. Chem., 1994, 98, 3459–3466 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01396g |
| ‡ Doctoral Programme in Theoretical Chemistry and Computational Modelling, Doctoral School, Universidad Autónoma de Madrid, Spain. |
| This journal is © the Owner Societies 2022 |