
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of particle size and composition on the optical and electronic properties of CdO and CdS rocksalt nanoparticles†
Martijn A.
Zwijnenburg

Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: m.zwijnenburg@ucl.ac.uk
First published on 10th August 2022
Abstract
Quantum confinement like behaviour in CdO and CdS nanoparticles is demonstrated through explicit evGW–BSE many-body perturbation theory calculations on 0.6–1.4 nanometre particles of these materials. However, while the lowest optical excited-state, exciton, and the highest occupied and lowest unoccupied quasiparticle states in such nanoparticles are predicted to be delocalised, they are found to be delocalised over the surface of the particle only and not the whole particle volume. The electronic and optical properties of CdO and CdS rocksalt nanoparticles are predicted to differ dramatically from their structurally analogous MgO counterparts, where the lowest exciton and highest occupied and lowest unoccupied quasiparticle states are strongly localised, in contrast. This difference in behaviour between MgO and CdO/CdS is explained in terms of the more polarisable, less ionic, bonding in CdO and CdS. The effect on the optical and fundamental gaps of the particles due to the presence of amine capping agents on the particles’ surface is explored and predicted to be relatively small. However, the highest occupied and lowest unoccupied quasiparticle states are found to consistently shift to more shallow values when increasing the surface density of capping agents. An explanation of this shift, finally, is proposed in terms of the dipole field induced by the aligned dipoles of the capping agents.
Introduction
The effect of nanostructuring a material on its electronic and optical properties is complex. Nanostructures of some materials, such as those of CdO,1–3 CdS,4 CdSe,5,6 PbS7,8 and PbSe,9 display experimentally a significantly blue-shifted optical gap (see Fig. 1) relative to the corresponding bulk material, i.e. the bulk starts absorbing light at longer wavelengths than the nanostructures. In contrast, nanostructures of other materials, such as MgO,10–12 CaO11 and SrO,13 are observed experimentally to have considerably red-shifted optical gaps. The optical gaps of nanostructures of the former materials are also generally observed to shift with particle size,2–9 where the optical gap and the whole absorption spectrum shifts to the blue with decreasing particle size. For nanoparticles of MgO, in contrast, the optical gap of particles does not change with particle size and only the relative intensities of the peaks in the spectrum changes.10 The blue shift is often explained in terms of quantum confinement, where the size of a delocalised exciton, the excited electron–hole pair, is constrained in the nanostructure to a value smaller than in the bulk,14,15 while the red shift is discussed in the experimental literature in terms of the strong localisation of the exciton on surface defect states.10,11,13,16 The exact microscopic picture of how excitons are (de)localised in the nanostructures, how changes in the nature of the chemical bonding in the materials switches between quantum confinement and surface defect localisation, the degree of (de)localisation of free charge carriers, as well as the exact effect of capping agents, is still not well understood. | ||
| Fig. 1 Schematic illustrating the quasiparticle and optical spectra and the definition of the highest occupied and lowest unoccupied quasiparticle states (−IP and −EA), the fundamental gap (ΔF, the gap between the highest occupied and lowest unoccupied quasiparticle states and the energy required to generate a non-interacting electron–hole pair), the optical gap (ΔO, the energy required to generate an interacting electron–hole pair, i.e. an exciton), and the exciton binding energy (EBE, the difference between the fundamental and optical gap). | ||
Materials that in the bulk crystallise in the rocksalt (halite) structure are ideal systems to study to better understand the link between the chemical bonding in materials, nanostructure size and the optical and electronic properties of these nanostructures. In contrast, to materials that crystallise in the zinc blende (sphalerite) and wurtzite structures, nanoparticles of which normally only show a blue-shift, both blue-shifts (CdO, PbS, PbSe) and red-shifts (MgO, CaO, SrO) are observed for materials that crystallise under ambient conditions in the rocksalt structure. Moreover, nanostructures of some of the materials that under ambient conditions crystallise in the wurtzite structure (CdS, CdSe) convert under pressure into the rocksalt structure, which can be subsequently recovered back to ambient conditions.17–19 Another advantage of rocksalt materials as model systems is that even for small nanostructures and even in the absence of capping agents the experimentally relevant nanostructures are generally simple cuts from the bulk crystal structure exposing simple, typical 〈001〉, crystal faces, which is not necessarily true for their wurtzite and zinc blende counterparts. Indeed, global optimisation calculations in the absence of capping agents predict that the global minima of MgO, CaO and SrO are cuts from the rocksalt structure,20–22 while those for ZnS and CdS nanostructures do not resemble the bulk zinc blende or wurtzite structures.23–28
Whilst experimental spectroscopy can clearly demonstrate the effect on the electronic and optical properties of materials of reducing the dimensions of materials to the nanoscale, to elucidate the microscopic origin of these changes one needs to combine experiment with theoretical chemistry calculations. Theoretical chemistry calculations also allow one to compare systems like-for-like, eliminating the effect of differences in nanostructure size (distributions) and the presence of capping agents for some materials experimentally and the absence for others. However, to be useful, the calculations should both be accurate and computationally tractable for nanostructures of at least 1 nm in size. (Time-Dependent) Density Functional Theory ((TD-)DFT) would be the ideal methodology in terms of tractability. However, previous work on MgO29 and TiO230,31 nanostructures demonstrated that the results of TD-DFT calculations on such structures were very sensitive to the exact density functional used, because of the well-known issue32,33 of TD-DFT where charge-transfer (CT) excitations are spuriously stabilised with respect to local, i.e. non-CT, excitations. Well-chosen density functionals with the optimal percentage of exact exchange can reproduce the key features of the experimental spectra including the optical gap (MgO) and the results of high-level quantum chemistry reference calculations (TiO2), but the strong dependency on the percentage of exact exchange makes these calculations more empirical than desirable.
Many-body perturbation theory, solving Hedin's GW34–36 equations followed by the Bethe–Salpeter37–39 equation (GW–BSE), is a promising alternative to (TD-)DFT. While computationally much more expensive it is inherently more accurate than (TD-)DFT and it has become recently tractable to perform GW–BSE calculations on realistic nanostructures due to a combination of methodological advances and increased computer power. By application of partial self-consistency in the GW part of the calculation, evGW, most of the dependency on the specific density functional used in the underlying DFT calculation is removed and evGW–BSE also has been shown40 to avoid most pitfalls of TD-DFT, including the description of CT-states. (ev)GW–BSE, in contrast, to TD-DFT also treats the optical gap, the energy required to generate an interacting exciton, and the fundamental gap, the energy required to generate a non-interacting electron–hole pair, (see Fig. 1) at the same footing allowing for the excitonic character of excited-states to be assessed.
Recently, evGW–BSE was used to study the optical and electronic properties of MgO nanoparticles.41 There it was found that the evGW–BSE predictions of the optical gap of MgO nanoparticles and the characteristic absorption spectrum of such particles agreed well with available experimental data, as well as that evGW–BSE correctly predicts, as discussed above, that the optical gap of MgO nanoparticles does not change with particle size. Here this work is extended to rocksalt nanostructures of two other materials: CdO and CdS.
Well-defined rocksalt MgO nanoparticles, as small as 3 nm in size, can be prepared experimentally in the absence of any capping agents in the gas-phase by chemical vapour synthesis10,11 and flame spray pyrolysis.12 Small, 10–30 nm, rocksalt CdO nanoparticles are synthesized in the literature solvothermally in the presence of phosphine oxide2 or amine3 capping agents, while larger CdO nanostructures are obtained by calcining Cd(OH)2 nanostructures42,43 or directly by chemical vapour deposition.44–46 The preparation of rocksalt CdS nanoparticles involves the application of pressures in excess of 5 GPa to 5–10 nm zinc blende CdS nanoparticles.18,19
Using evGW–BSE differences in the optical and electronic properties of ∼ 1 nm CdO and CdS rocksalt particles (see Fig. 2) are studied and compared with those predicted for similar MgO nanoparticles in previous work. The (de)localisation of the states responsible for the electronic and optical properties of the nanoparticles and how these properties change with particle composition and size is investigated, as is the effect of capping agents on the electronic and optical properties.
 | ||
| Fig. 2 DFT optimised structures of the (CdS)32, (CdS)108 and the (CdS)32(NH2(CH3))4, (CdS)32(NH2(CH3))16 and (CdS)32(NH2(CH3))28 particles. The structures of the CdO particles are similar and shown in the ESI† (Fig. S1). | ||
Methodology
The geometry of the nanoparticles was optimised in DFT calculations, using the B3LYP47–49 density functional in combination with the D4 dispersion correction by Grimme and co-workers50 (D351,52 in the case of MgO structures taken from previous work41) and the def2-SVP or def2-TZVPP basis-sets.53 During the geometry optimisation the symmetry of the nanoparticles was fixed and for the smaller (MX)4 and (MX)32 particles, and selected particles with ligand molecules adsorbed on the surface, harmonic frequency calculations were performed to verify that the optimised structures only display positive frequencies and thus correspond to minima on the particles’ potential energy surface.Single-shot G0W0 and eigenvalue-only self-consistent GW (evGW) calculations, as implemented by Holzer, van Setten, Klopper and co-workers40,54,55 in Turbomole, were performed on the DFT optimised structures, starting from B3LYP orbitals, to predict the quasiparticle spectrum in general and the highest occupied (−IP) and lowest unoccupied (−EA) quasiparticle states in particular, see Fig. 1. These calculations used the def2-SV(P), def2-SVP or def2-TZVPP basis-sets and one of three GW implementations that differ in how the self-energy is calculated; in terms of a spectral representation (SR), by contour deformation (CD), or by analytical continuation (AC). The GW(SR) calculations are the most computationally expensive, scaling as N6, while the GW(AC) calculations are the most computationally tractable, scaling as N4. In the case of GW(AC) only the highest occupied and lowest unoccupied quasiparticle states are explicitly calculated with GW and the rest of the DFT orbitals simply shifted in-line with the shift between DFT and GW for those two states. The results of the different GW calculations are used as input for solving the BSE, again as implemented in Turbomole,56 to obtain vertical excitation energies and oscillator strength values and thus the particles’ optical gap values and the optical (absorption) spectrum. The character of the excited states predicted using BSE is finally analysed in terms of the most prominent natural transition orbitals.57 The same basis-sets are generally used for the geometry optimisation and the BSE–GW calculations, except where explicitly stated to the contrary. Relativistic effects on the quasiparticle spectra are assessed by comparing the results of two-component (2c) GW calculations,55 which include spin–orbit coupling as well as scalar relativistic effects, with normal, non-relativistic, GW calculations.
For the (MX)4 particles the lowest vertical excitation energies including the optical gap are also calculated with LR-CCSD coupled-cluster theory on top of a Hartree–Fock ground-state. These correlated wavefunction calculations use the def2-TZVPP basis-set and are performed as single-point calculations on the DFT optimised geometry.
In the literature −IP and −EA are often referred to as εHOMO and εLUMO, respectively, the energies of the highest occupied and lowest unoccupied (molecular) orbitals. Here −IP and −EA are used to avoid the confusion between the true many-body εHOMO and εLUMO quasiparticle values and the uncorrected energies of the Kohn–Sham orbitals from the DFT calculations, which especially in the case of lowest unoccupied Kohn–Sham orbitals are known to deviate considerably from the true εLUMO.58
All calculations were performed using version 7.5 of the Turbomole code and used a tight integration grid (m5) and tight SCF convergence criteria (scftol and denconv 1 × 10−7). All GW and BSE calculations additionally used the RI-K approximation. Use of symmetry in the GW and BSE calculations was limited to Abelian point groups. Hence all such calculations on particles with Td symmetry were performed in the D2 point group instead. Finally, the relativistic 2c-GW calculations were performed without any use of symmetry.
Results and discussion
Effect of composition – (CdO)32, (CdS)32 and (MgO)32
First, we consider the predicted quasiparticle and optical absorption spectra of (CdO)32, (CdS)32 and (MgO)32, data for the latter taken from previous work.41 These particles are the smallest rocksalt cuts that preserve most of the symmetry of the infinite material and contain a number of atoms with bulk, 6-fold, coordination. Fig. 3 shows the quasiparticle spectrum around the highest occupied and lowest unoccupied quasiparticle state for the three different particles as calculated using evGW(AC)/def2-TZVPP, data for other method combination can be found in the ESI† (see Tables S1–S4). From Fig. 3 it can be seen that, as expected from the bulk, the fundamental gaps of (CdS)32 and (CdO)32 is much smaller than that of (MgO)32. It is also from clear from Fig. 3 that the highest occupied quasiparticle state for (MgO)32 lies much deeper (−IP value more negative) than for (CdS)32 and (CdO)32 and the lowest unoccupied quasiparticle state much shallower (−EA value less negative). The difference between (CdS)32 and (CdO)32, in contrast is much smaller. (CdS)32 is predicted to have a slightly deeper highest occupied quasiparticle state than (CdO)32, a slightly deeper lowest unoccupied quasiparticle state, and a slightly smaller fundamental gap. The particles also differ in the symmetry of the highest occupied and lowest unoccupied quasiparticle states, A and A in the case of (CdS)32 and T and A in the case of (CdO)32 and (MgO)32. Because the GW calculations are performed in the D2 subgroup instead of the full Td point group, we cannot further distinguish between A1 and A2 or between T1 and T2. However, DFT calculations in the full Td point group suggests that the symmetry of the highest occupied and lowest unoccupied quasiparticle states is A2 and A1 in the case of (CdS)32 and T2 and A1 for (CdO)32. evGW(SR)/def2-SVP and evGW(CD)/def2-TZVPP calculations, see Tables S1 and S2 (ESI†), demonstrate that this difference in the character of the states involved is not an artefact of the fact that in evGW(AC) only the highest occupied and lowest unoccupied quasiparticle states are explicitly calculated with evGW. Finally, the results in Fig. 3 were obtained from non-relativistic calculations. 2c-GW calculations that include spin–orbit coupling on smaller (CdO)4 and (CdS)4 particle, see Tables S1 and S2 (ESI†), suggest that including relativistic effects does not significantly change the predictions.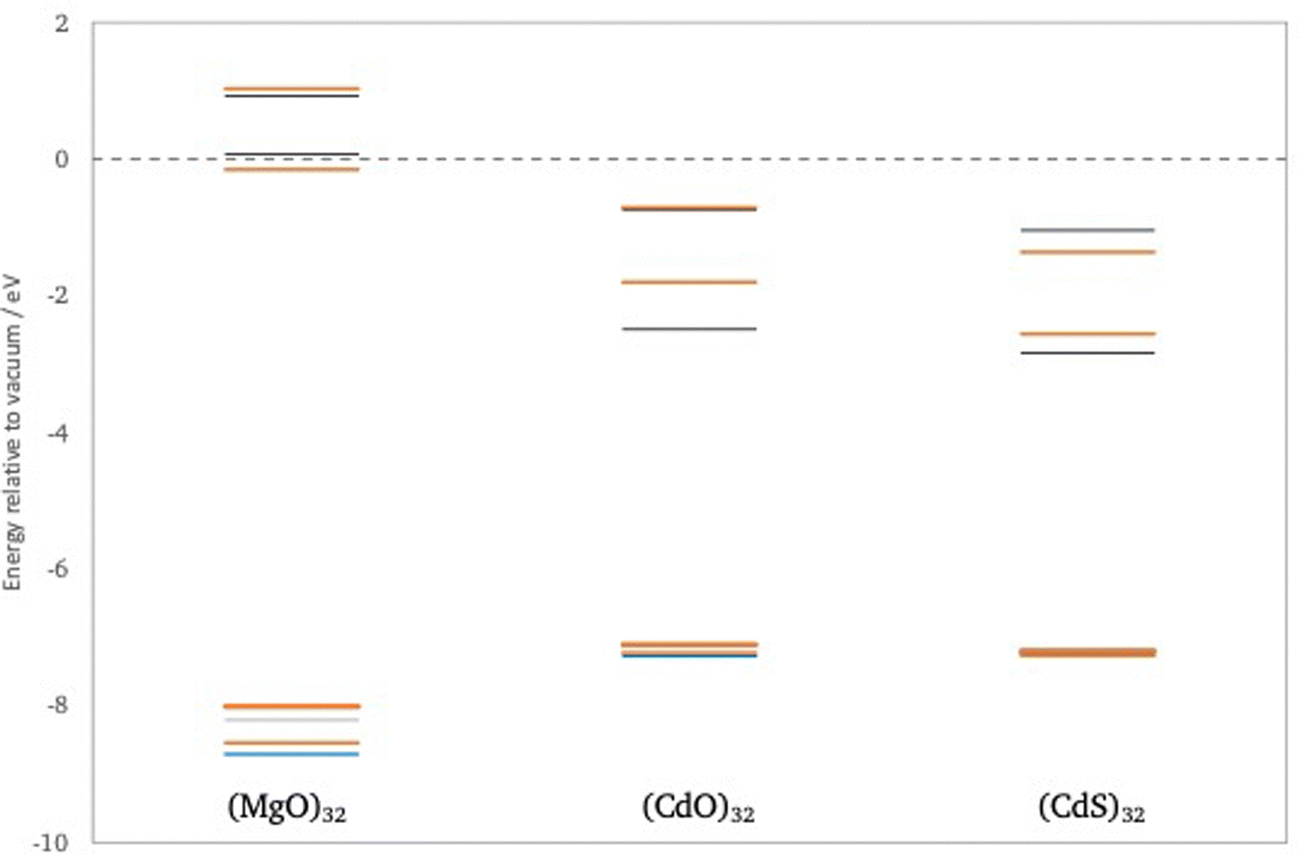 | ||
| Fig. 3 evGW/def2-TZVPP predicted quasiparticle spectra of (MgO)32, (CdO)32 and (CdS)32, showing the four highest occupied and four lowest unoccupied quasiparticle states. Red lines T irrep, blue lines e irrep and grey lines a irrep. | ||
Fig. 4 shows the vertical optical excitation spectrum of (CdO)32, (CdS)32 and (MgO)32 predicted by evGW(AC)-BSE/def2-TZVPP. Not surprisingly the optical excitation spectra of (CdO)32 and (CdS)32 are significantly red-shifted relative to that of (MgO)32. More interestingly, while for (MgO)32 the lowest excitation is dipole allowed, T2 (it is possible to deduce the exact irreducible representation in Td for the optically excited states based on the degeneracy and predicted oscillator strength other than distinguishing between A1 and A2) for (CdO)32 and (CdS)32 only the third and fourth lowest excited states, respectively, are found to be dipole allowed. Dipole forbidden excited states of A1/2 and T1 symmetry in the case of (CdO)32 and A1/2, T1 and E symmetry in the case of (CdS)32, are predicted to lie lower in energy than the lowest energy T2 state. Finally, in all cases the lowest optical excited states are clearly excitonic in character, lying 2–3.5 eV lower in energy than the corresponding fundamental gap values.
 | ||
| Fig. 4 evGW-BSE/def2-TZVPP predicted optical spectra of (MgO)32, (CdO)32 and (CdS)32, shwowing the four lowest energy excited states. Red lines T2 irrep, orange lines T1 irrep, green lines e irrep and grey lines a irrep. | ||
We can analyse the origin of the highest occupied and lowest unoccupied quasiparticle states in terms of the underlying DFT orbitals. Fig. 5 shows those DFT Kohn–Sham orbitals for (CdO)32 and (CdS)32. Compared to MgO data previously reported, where the orbitals corresponding to the highest occupied and lowest unoccupied quasiparticle states are strongly localised on 3-coordinated oxygen corner atoms and the 3-coordinated magnesium corner atoms, respectively, the orbitals for (CdO)32 and (CdS)32 are much more delocalised. Specifically, in the case of (CdO)32 the orbital corresponding to the highest occupied quasiparticle states is delocalised over all the 3- and 4-coordinated oxygen atoms on the surface of the particle and the orbital for the lowest unoccupied quasiparticle state is delocalised over all the cadmium atoms on the particle surface. For (CdS)32 the orbital corresponding to the highest occupied quasiparticle state is delocalised over all the sulfur atoms on the surface of the particle but importantly not the 3-coordinated sulfur atoms, while the orbital for the lowest unoccupied quasiparticle state is delocalised over only the 3- and 4-coordinated the cadmium atoms on the corner and edges of the particle.
 | ||
| Fig. 5 Highest occupied and lowest unoccupied Kohn–Sham orbitals of (CdO)32 (A, left and right, respectively) and (CdS)32 (B, left and right, respectively). The top and bottom rows show the same orbitals but from a different perspective. As the calculation were performed using the D2 instead of the Td point group, the highest occupied orbital for (CdO)32, a triply degenerate T1 orbital in Td, is described as a triplet of orbitals of B1, B2 and B3 symmetry, where only one of these three is shown here (A, left). | ||
We can similarly analyse the character of the optically exited states by plotting the leading natural transition orbitals. Fig. 6 shows the hole and excited electron component of the lowest energy bright T2 optically excited state for (CdO)32 and (CdS)32. For both materials the hole components is essentially delocalised over the surface O/S atoms, while the excited electron component is delocalised over all Cd atoms on the surface in the case of (CdO)32 and the 3-/4-coordinated Cd atoms on the edge of the particle in the case of (CdS)32. This delocalisation contrasts very strongly with the highly localised nature of the same excited state in (MgO)32, which localises on the atoms around the oxygen corners of that particle.
 | ||
| Fig. 6 Leading occupied (A, left) and unoccupied (A, right) natural transition orbital for the lowest T2 excitation of (CdO)32. Leading occupied (B, left) and unoccupied (B, right) natural transition orbital for the lowest T2 excitation of (CdS)32. As the calculation were performed using the D2 instead of the Td point group, the triply degenerate T2 excited state is described as a triplet of excited states of B1, B2 and B3 symmetry, the natural transition orbitals of only one is shown here. | ||
Effect of particle size – (CdX)32 and (CdX)108
Next, the effect of particle size on the electronic and optical properties of the CdO and CdS cubic nanoparticles is studied by comparing predictions for the (CdX)32 (4 × 4 × 4) and (CdX)108 (6 × 6 × 6) particles. These calculations were performed using the small def2-SV(P) basis-set on geometries optimised with the def2-SVP basis-set to keep the evGW and especially the BSE calculations for (CdX)108 tractable. Even then we cannot study the bright T2 excitation for (CdO)108 and (CdS)108 as after reducing the symmetry to the D2 point-group this excitation is the second excitation with T symmetry, with the lowest energy T1 excitation, as discussed above, lying lower in energy. While the switch from def2-TZVPP to def2-SV(P) changes the absolute values the relative trends should be well preserved. Fig. 7 show the change with particle size in the quasiparticle spectrum and Fig. 8 and 9 the change in the lowest A1/2 and T1 optically excited states, as calculated with evGW(AC)-BSE/def2-SV(P) (see also Tables S1 and S2, ESI†). The fundamental gap for both CdO and CdS decreases when going from the 4 × 4 × 4 to the 6 × 6 × 6 particle, in line with what is observed for MgO, but also the energies of the lowest A1/2 and T1 optically excited states shifts to the red with particle size. The latter is something which is not observed in the case of MgO, where the energy for the T2 excited state was found to not change when going from (MgO)32 to (MgO)108. Interestingly, in the case of CdO nanoparticles this red shift only clearly occurs for evGW–BSE. G0W0–BSE predicts that the CdO particles behave more MgO like. | ||
| Fig. 7 Change in the highest occupied and lowest unoccupied quasiparticle states with particle size as calculated with evGW(AC)/def2-SV(P), showing the two highest occupied and two lowest unoccupied quasiparticle states. | ||
 | ||
| Fig. 8 Change in the predicted optical spectra of (CdO)n with particle size as calculated with evGW(AC)–BSE/def2-SV(P). Red lines T2 irrep, orange lines T1 irrep and grey lines a irrep. | ||
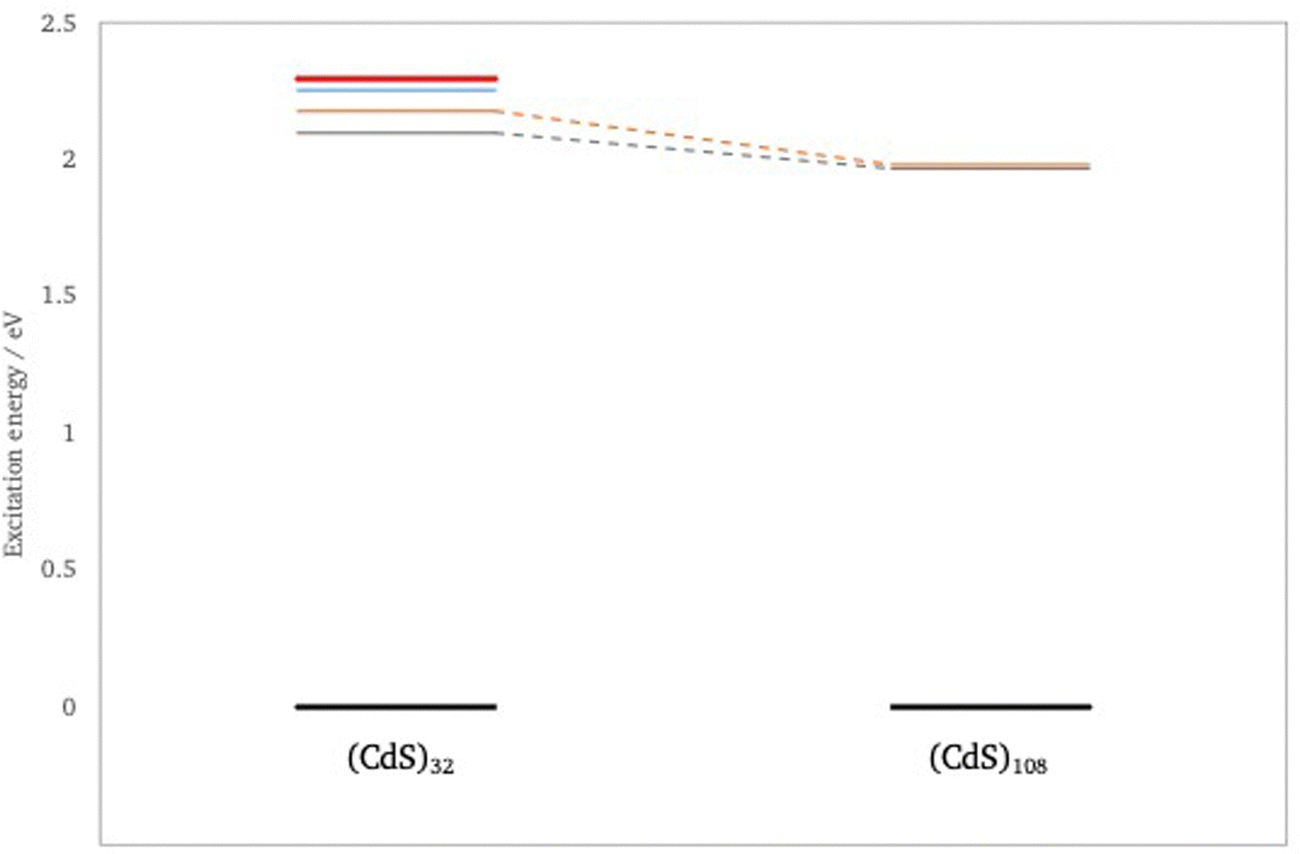 | ||
| Fig. 9 Change in the predicted optical spectra of (CdS)n with particle size as calculated with evGW(AC)–BSE/def2-SV(P). Red lines T2 irrep, orange lines T1 irrep, blue lines e irrep and grey lines a irrep. | ||
Bare versus nanoparticles with capping agents
To study the effect of the presence of capping agents on the surface of the nanoparticle, (CdS)32 and (CdO)32 particles with varying amounts of methylamine (NH2(CH3)) molecules adsorbed on the cadmium atoms on the particle surface were optimised using B3LYP + D4/def2-SVP and their electronic and optical properties predicted using BSE/evGW(AC)/def2-SVP. Specifically, three cases were considered (i) (CdO)32(NH2(CH3))4 and (CdS)32(NH2(CH3))4 particles with four methylamine molecules adsorbed on the cadmium corner atoms, (ii) (CdO)32(NH2(CH3))16 and (CdS)32(NH2(CH3))16 particles with sixteen molecules adsorbed on all the cadmium corner and edge atoms, and (iii) (CdO)32(NH2(CH3))28 and (CdS)32(NH2(CH3))28 particles with twenty-eight molecules adsorbed on all the cadmium atoms on the particle surface. In the case of (CdO)32(NH2(CH3))4 and (CdS)32(NH2(CH3))4 different starting orientations of the adsorbed methylamine molecules were explored to find a low energy configuration. In contrast, for (CdO)32(NH2(CH3))16, (CdS)32(NH2(CH3))16, (CdO)32(NH2(CH3))28, and (CdS)32(NH2(CH3))28 only one or a very small number of configurations was studied. In all cases adsorbing methylamine was found to be reasonably exothermic with adsorption energies per molecule ranging from 160 to 90 kJ per mol per molecule.Adsorbing four methylamine molecules on the cadmium corner atoms of (CdO)32 forming (CdO)32(NH2(CH3))4 results in no significant structural changes in the inorganic part of the nanoparticle. In contrast, adsorption of four methylamine molecules on the cadmium corner atoms of (CdS)32 results in the local geometry around the cadmium atoms the methylamine is adsorbed on becoming more distorted tetrahedral like with S–Cd–S angles around the cadmium corner atoms change from 103° to 112–118°. Similar, the local geometry around the edge sulfur atoms distorts with some of them becoming effectively 3 rather than 4-coordinated when ignoring the ligand. In the case of (CdS)32(NH2(CH3))16 and (CdS)32(NH2(CH3))32 particles similar distortions are observed for the surface cadmium and sulfur atoms. However, the 6-coordinated atoms in the centre of the particles remain 6-coordinated in all cases. Finally, while in (CdO)32(NH2(CH3))4 there is no significant structural changes in the inorganic part of the nanoparticle, for (CdO)32(NH2(CH3))16 and (CdO)32(NH2(CH3))32 any optimisation attempt resulted in structures were at least one cadmium edge atom and an adjacent sulfur edge atom have become 3 rather than 4-coordinated, again ignoring the ligand in the case of the cadmium atom. The structural changes upon adsorption of methylamine on (CdO)32 seem to be driven in part by interaction between amine protons and surface oxygens with N–H O distances less than the sum of the van der Waals radii of hydrogen and oxygen atoms.
Fig. 10 compares the highest occupied and lowest unoccupied quasiparticle states of (CdS)32, (CdS)32(NH2(CH3))4, (CdS)32(NH2(CH3))16 and (CdS)32(NH2(CH3))28. Fig. S2 in the ESI† shows the same for (CdO)32, (CdO)32(NH2(CH3))4, (CdO)32(NH2(CH3))16 and (CdO)32(NH2(CH3))28. From Fig. 10 and Fig. S2 (and Table S5, ESI†) it is clear that adsorbing methylamine molecules and adsorbing more methylamine molecules consistently shifts both the highest occupied and lowest unoccupied quasiparticle states to much more positive, shallower, values. It also results in an opening of the fundamental gap, and the optical gap, although the change in fundamental and optical gap is much less dramatic than the absolute shift in quasiparticle states. Specifically, as can be seen from Fig. 11 the change in the optical and fundamental gap with the number of capping agents on the particle surface levels off for high surface coverage. Another change, partially caused by a reduction in symmetry, is that for the particles with methylamines adsorbed all singlet vertical excitations are now optically allowed, including the lowest excited state which for the naked (CdO)32 and (CdS)32 particle, as discussed above, is dark for symmetry reasons. evGW–BSE calculations on particles where the ligands have been removed but the geometry of the inorganic part of the particle has not been allowed to relax back, suggest that the majority of the change in the particles’ electronic and optical properties upon adsorption of capping agents is a direct electronic effect of the presence of the ligands rather than the structural distortions caused by their adsorption. Visualising the DFT orbitals also suggests no or limited direct contribution to the highest occupied and lowest unoccupied quasiparticle states by the amine capping agent.
 | ||
| Fig. 10 Change in the highest occupied and lowest unoccupied quasiparticle states of (CdS)32 with number of capping agents on the surface as calculated with evGW(AC)/def2-SVP. Red lines −EA, blue lines −IP, grey lines −IP and −EA of particles. | ||
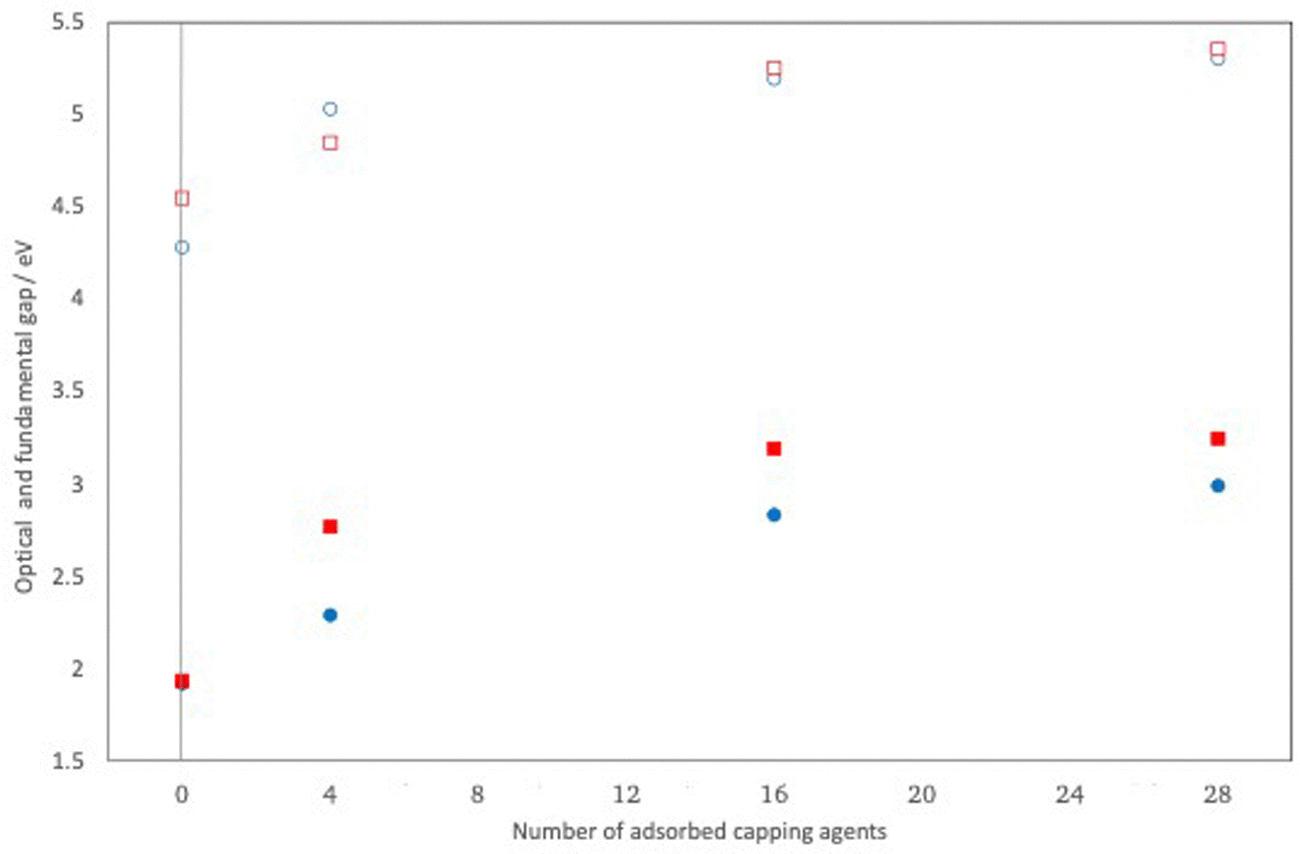 | ||
| Fig. 11 Change in the predicted optical (filled symbols) and fundamental (open symbols) of (CdO)32 (blue symbols) and (CdS)32 (red symbols) upon adsorbing methylamine capping agents on the particle surface as calculated with evGW(AC)–BSE/def2-SVP. | ||
Discussion
Comparing the evGW fundamental gap values predicted for the MgO, CdO and CdS nanoparticles with the fundamental gap (band gap) values predicted using evGW for the corresponding rocksalt bulkphases by Lany59 (8.17 eV, 0.87 eV indirect and 2.0 eV direct, and 1.77 indirect and 3.0 eV direct, respectively), it becomes apparent that while for MgO the fundamental gap predicted for the particles is smaller than that predicted for the bulk for CdO and CdS the opposite is the case. The fundamental gap predicted for the CdO and CdS nanoparticles is larger than that predicted for the bulk. A similar conclusion can be drawn by comparing the predicted fundamental gap for the particles with the experimentally reported fundamental gap values for bulk MgO (7.8 eV)60,61 and CdO (indirect 0.84 eV, direct 2.28 eV).62,63 However, one must be careful with such a comparison as all the calculations ignore the vibronic renormalisation of the fundamental gap and because the experimental measurements on CdO are on heavily doped samples. The apparent fundamental gap measured for a heavily doped sample differs from that of a (hypothetical) undoped sample because of the Moss–Burnstein shift, the fact that the Fermi level for such materials lies in the conduction band and only excitations to above the Fermi level are allowed, and due to the renormalisation of the fundamental gap because of electron–defect interactions.G 0 W 0–BSE calculations by Schleife and co-workers64 predict that the optical gap of bulk CdO lies in between the materials predicted direct and indirect fundamental gap. Combining this prediction with the probably more accurate direct and indirect band gap values from Lany, discussed above, suggests that just as for the fundamental gap the lowest vertical excitations and thus the optical gap of the CdO particles are, as expected in the case of quantum confinement, larger than for the bulk. However, G0W0–BSE calculations for the particles using the same HSE0365 functional to obtain the starting Kohn–Sham orbitals as used by Schleife and co-workers for the bulk, yields an optical gap value that is smaller than the bulk (see Table S6, ESI†). This could be an issue with the underlying G0W0 description, related to the fact that, as discussed above, no optical gap reduction with particle size is observed for CdO particles using G0W0–BSE in contrast to evGW–BSE or might be suggesting that the optical gap of the CdO particles converges to that of the lowest surface instead of bulk exciton. The latter would be in line with the reported experimental optical gap of bulk CdO, 2.1–2.4 eV,66,67 being larger than that predicted for the particles, although as noted above bulk CdO is typically highly defective and to compare with theory the optical gap needs to be extrapolated to zero free electron concentration. The experimental optical gap of CdO nanoparticles capped with amines3 or phosphine oxides2 is similar or slightly larger than that predicted for the smaller capped nanoparticle studied here, although the experimental absorption spectra are rather featureless and hence it is difficult to extract exact values and different authors report wildly different values for similarly sized particles. In contrast, for MgO nanoparticles there is no ambiguity and, as discussed in previous work41 based on GW–BSE calculations and as is know from experiment,10,11 the MgO particles’ optical gap is considerably smaller than that of the bulk.
The fact that the fundamental gap of the CdO and CdS nanoparticles is larger than that predicted for the bulk and that the optical gap of the CdO nanoparticles might be larger than that predicted for the bulk, combined with the predicted reduction in the fundamental gap and lowest vertical excitation energies of the nanoparticles with particle size, suggests that CdO and CdS nanoparticles, in contrast to their MgO counterparts, display quantum confinement. However, the analysis of the DFT orbitals underlying the highest occupied and lowest unoccupied quasiparticle states and the natural transition orbitals for the excited states, shows that these states are not, as perhaps naively expected, delocalised over the volume of the particle but rather delocalised over the particle surface.
The lowest vertical excitations for particles of all three systems considered correspond to strongly bound excitons. The excitons are most strongly bound in the case of the MgO nanoparticles. The excitons are weaker bound in the CdO nanoparticles and weakest in the CdS nanoparticles, but in all cases the exciton binding energies are still larger than 1500 meV. For all three system studied the exciton binding energies also clearly decrease with particle size.
The differences in the (de)localisation of excitons in MgO, CdO and CdS nanoparticles, as well as differences in the exciton binding energy values, probably find their origin in differences in the bonding in the different materials. MgO is the most ionic of the materials, while the bonding in CdO and CdS is if not more covalent at least less ionic and more polarisable and hence better at screening the electron and hole component of the exciton. Indeed, predictions of the static polarizability of the (MX)32 particles using evGW–BSE yield polarizability values for (CdS)32 and (CdO)32, which are respectively roughly three times (1571 a.u.) and two times (931 a.u.) as large as that for (MgO)32 (502 a.u.). Similarly, calculations of the particles’ ground state atomic charges from the DFT density shows that not only do these charges significantly reduce when going from (MgO)32 to (CdO)32 and (CdS)32 (see Table S7, ESI†), in line with the bonding in the particles becoming less ionic, but also that the magnitude of the difference of the charge on the oxygen/sulfur atoms in the centre and on the corner of the particle increases in the opposite direction, in line with the bonding in the particles becoming more polarisable. It is tempting to interpret the fact that the lowest excitons for CdO and CdS nanoparticles are dark in terms of the fact that the corresponding bulk materials have an indirect band gap, akin to the analysis by Williamson and co-workers68 of the (in)direct gap of quantum dots of III–V materials. However, the fact that the corresponding excitons are not delocalised over the volume of the particle but localised on the surface means that one has to be careful with extrapolating from the bulk.
The presence of capping agents on the surface and increasing the surface coverage of these capping agents clearly and consistently results in upward shift of the highest occupied and lowest unoccupied quasiparticle states. As discussed above, these quasiparticle states do not appear to directly involve the capping agents and the change in these states neither appears to be caused by the structural distortion of the inorganic core of the particles induced by adsorption of the capping agents. Dielectric contrast, where the capping agent changes the screening of the electric field outside the nanoparticles, is naively also unlikely to be the origin of the observed change because both the occupied and unoccupied quasiparticle states shift upwards, although dielectric contrast might be relevant for realistic capping agents with longer alkyl chains. There is also very limited evidence of charge-transfer between the amines and the inorganic core, with e.g. for (CdO)32(NH2(CH3))28 a difference in the NBO charges of the inorganic core in the presence and absence of capping agents of less than 0.05 and a net charge on the collective of capping agents of less than −0.05. Hence the origin of the upward shift of the quasiparticle states is most likely the dipole field of the collected adsorbed capping agents (B3LYP/def2-SVP predicted dipole moment of methylamine: 1.3 Debye). This analysis is supported by the fact that, as expected from a solution to the Poisson equation,69 or at least for planar geometries, that the predicted shift in the quasiparticle states scales approximately linearly with the number of capping agents on the particle surface. This linearity is clearest for the CdS particles, and especially the change in the highest occupied quasiparticle state, see Fig. S3 (ESI†), while the trend appears generally less linear for their CdO counterparts, see Fig. S4 (ESI†). Previous work on PbS quantum dots saw similar shifts in the experimentally measured ionisation potential and work function,70,71 as well as the DFT predicted Kohn–Sham orbitals, when adding and varying the dipole strength of polar capping agents. Moreover, the predicted shifts are also in line with the known effect of the adsorption of (self-assembled) monolayers of polar molecules on the work function of films of bulk semiconductors.69,72–75
Conclusions
CdO and CdS nanoparticles show clear signs of quantum confinement behaviour: the fundamental and optical gaps of the particles decrease with increasing particle size, the predicted fundamental gaps of the particles are larger than that of the corresponding bulk phases, and there is some evidence that the same holds for the optical gap of the CdO particles. However, in contrast to what might be naively expected, the relevant excitonic and quasiparticle states are not delocalised over the whole volume of the nanoparticles but only across the particles’ surface. The predicted delocalisation of excitonic and quasiparticle states and the change in optical gap with particle size contrasts with the strong localisation of all relevant states and the lack of the change in optical gap with particle size for structurally analogous MgO particles. This difference in behaviour is explained in terms of the more polarisable, less ionic, bonding in CdO and CdS. The presence of capping agents is predicted to have a small effect on the particles’ optical and fundamental gap but to result in a dramatic but consistent shift of both the highest occupied and lowest unoccupied quasiparticle state to more shallow values. This shift, finally, can be explained in terms of dipole field of the collected adsorbed capping agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Christof Holzer is kindly acknowledged for discussion. The UK Engineering and Physical Sciences Research Council (EPSRC) is thanked for funding part of this work through grants EP/I004424/1 and EP/N004884/1.References
- W. Dong and C. Zhu, Optical properties of surface-modified CdO nanoparticles, Opt. Mater., 2003, 22, 227–233 CrossRef CAS
.
- M. Ghosh and C. N. R. Rao, Solvothermal synthesis of CdO and CuO nanocrystals, Chem. Phys. Lett., 2004, 393, 493–497 CrossRef CAS
- T. Xaba, M. J. Moloto, M. A. Malik and N. Moloto, The influence of temperature on the formation of cubic structured CdO nanoparticles and their thin films from bis(2-hydroxy-1-naphthaldehydato)cadmium(II) complex via thermal decomposition technique, J. Nanotechnol., 2017, 2017, 8317109 Search PubMed
- W. W. Yu and X. Peng, Formation of high-quality CdS and Other II–VI semiconductor nanocrystals in noncoordinating solvents: Tunable reactivity of monomers, Angew. Chem., Int. Ed., 2002, 41, 2368–2371 CrossRef CAS PubMed
- C. B. Murray, D. J. Norris and M. G. Bawendi, Synthesis and characterization of nearly monodisperse CdE (E = sulfur, selenium, tellurium) semiconductor nanocrystallites, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS
- X. Peng, J. Wickham and A. P. Alivisatos, Kinetics of II–VI and III–V colloidal semiconductor nanocrystal growth:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) “Focusing” of size distributions, J. Am. Chem. Soc., 1998, 120, 5343–5344 CrossRef CAS
“Focusing” of size distributions, J. Am. Chem. Soc., 1998, 120, 5343–5344 CrossRef CAS - M. A. Hines and G. D. Scholes, Colloidal PbS nanocrystals with size-tunable near-infrared emission: Observation of post-synthesis self-narrowing of the particle size distribution, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS
- L. Cademartiri, E. Montanari, G. Calestani, A. Migliori, A. Guagliardi and G. A. Ozin, Size-dependent extinction coefficients of PbS quantum dots, J. Am. Chem. Soc., 2006, 128, 10337–10346 CrossRef CAS PubMed
- C. B. Murray, S. Sun, W. Gaschler, H. Doyle, T. A. Betley and C. R. Kagan, Colloidal synthesis of nanocrystals and nanocrystal superlattices, IBM J. Res. Dev., 2001, 45, 47–56 CAS
- S. Stankic, M. Müller, O. Diwald, M. Sterrer, E. Knözinger and J. Bernardi, Size-dependent optical properties of MgO nanocubes, Angew. Chem., Int. Ed., 2005, 44, 4917–4920 CrossRef CAS PubMed
- S. Stankic, J. Bernardi, O. Diwald and E. Knözinger, Optical surface properties and morphology of MgO and CaO nanocrystals, J. Phys. Chem. B, 2006, 110, 13866–13871 CrossRef CAS PubMed
- T. Schwab, M. Niedermaier, K. Aicher, M. S. Elsässer, G. A. Zickler and O. Diwald, Always cubes: A comparative evaluation of gas phase synthesis methods and precursor selection for the production of MgO nanoparticles, Open Ceram., 2021, 6, 100104 CrossRef
- S. Stankic, J. Bernardi, O. Diwald and E. Knözinger, Photoexcitation of local surface structures on strontium oxide grains, J. Phys. Chem. C, 2007, 111, 8069–8074 CrossRef CAS
- L. E. Brus, Electron–electron and electron–hole interactions in small semiconductor crystallites: The size dependence of the lowest excited electronic state, J. Chem. Phys., 1984, 80, 4403–4409 CrossRef CAS
- L. Brus, Electronic wave functions in semiconductor clusters: Experiment and theory, J. Phys. Chem., 1986, 90, 2555–2560 CrossRef CAS
-
S. Coluccia, Surface structure and energy transfer on oxides investigated by photoluminescence spectroscopy, in Studies in Surface Science and Catalysis, ed. M. Che and G. C. Bond, Elsevier, 1985, vol. 21, pp. 59–70 Search PubMed
- K. Jacobs, J. Wickham and A. P. Alivisatos, Threshold size for ambient metastability of rocksalt CdSe nanocrystals, J. Phys. Chem. B, 2002, 106, 3759–3762 CrossRef CAS
- R. Martín-Rodríguez, J. González, R. Valiente, F. Aguado, D. Santamaría-Pérez and F. Rodríguez, Reversibility of the zinc blende to rock-salt phase transition in cadmium sulfide nanocrystals, J. Appl. Phys., 2012, 111, 063516 CrossRef
- L. Meng, J. M. D. Lane, L. Baca, J. Tafoya, T. Ao, B. Stoltzfus, M. Knudson, D. Morgan, K. Austin, C. Park, P. Chow, Y. Xiao, R. Li, Y. Qin and H. Fan, Shape dependence of pressure-induced phase transition in CdS semiconductor nanocrystals, J. Am. Chem. Soc., 2020, 142, 6505–6510 CrossRef CAS PubMed
- C. Roberts and R. L. Johnston, Investigation of the structures of MgO clusters using a genetic algorithm, Phys. Chem. Chem. Phys., 2001, 3, 5024–5034 RSC
- M. Chen, A. R. Felmy and D. A. Dixon, Structures and stabilities of (MgO)N nanoclusters, J. Phys. Chem. A, 2014, 118, 3136–3146 CrossRef CAS PubMed
- S. G. E. T. Escher, T. Lazauskas, M. A. Zwijnenburg and S. M. Woodley, Synthesis target structures for alkaline earth oxide clusters, Inorganics, 2018, 6, 29 CrossRef
- E. Spanó, S. Hamad and C. R. A. Catlow, Computational evidence of bubble ZnS clusters, J. Phys. Chem. B, 2003, 107, 10337–10340 CrossRef
- E. Spanó, S. Hamad and C. R. A. Catlow, ZnS bubble clusters with onion-like structures, Chem. Commun., 2004, 864–865 RSC
- S. Hamad, C. R. A. Catlow, E. Spanó, J. M. Matxain and J. M. Ugalde, Structure and properties of ZnS nanoclusters, J. Phys. Chem. B, 2005, 109, 2703–2709 CrossRef CAS
- A. Burnin, E. Sanville and J. J. BelBruno, Experimental and computational study of the ZnnSn and ZnnSn+ clusters, J. Phys. Chem. A, 2005, 109, 5026–5034 CrossRef CAS PubMed
- E. Sanville, A. Burnin and J. J. BelBruno, Experimental and computational study of small (N = 1–16) stoichiometric zinc and cadmium chalcogenide clusters, J. Phys. Chem. A, 2006, 110, 2378–2386 CrossRef CAS PubMed
- M. A. Zwijnenburg, Optical excitations in stoichiometric uncapped ZnS nanostructures, Nanoscale, 2011, 3, 3780–3787 RSC
- M. C. C. Wobbe, A. Kerridge and M. A. Zwijnenburg, Optical excitation of MgO nanoparticles; a computational perspective, Phys. Chem. Chem. Phys., 2014, 16, 22052–22061 RSC
- E. Berardo, H.-S. Hu, S. A. Shevlin, S. M. Woodley, K. Kowalski and M. A. Zwijnenburg, Modeling excited states in TiO2 Nanoparticles: On the accuracy of a TD-DFT based description, J. Chem. Theory Comput., 2014, 10, 1189–1199 CrossRef CAS
- E. Berardo, H.-S. Hu, H. J. J. van Dam, S. A. Shevlin, S. M. Woodley, K. Kowalski and M. A. Zwijnenburg, Describing excited state relaxation and localization in TiO2 nanoparticles using TD-DFT, J. Chem. Theory Comput., 2014, 10, 5538–5548 CrossRef CAS PubMed
- D. J. Tozer, R. D. Amos, N. C. Handy, B. O. Roos and L. Serrano-Andres, Does density functional theory contribute to the understanding of excited states of unsaturated organic compounds?, Mol. Phys., 1999, 97, 859–868 CrossRef CAS
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, Excitation energies in density functional theory: An evaluation and a diagnostic test, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed
- L. Hedin, New method for calculating the one-particle green's function with application to the electron-gas problem, Phys. Rev., 1965, 139, A796–A823 CrossRef
- F. Aryasetiawan and O. Gunnarsson, Thegwmethod, Rep. Prog. Phys., 1998, 61, 237–312 CrossRef CAS
- D. Golze, M. Dvorak and P. Rinke, The GW compendium: A practical guide to theoretical photoemission spectroscopy, Front. Chem., 2019, 7 Search PubMed
- E. E. Salpeter, H. A. Bethe and A. Relativistic, Equation for bound-state problems, Phys. Rev., 1951, 84, 1232–1242 CrossRef
- G. Strinati, Application of the Green’s functions method to the study of the optical properties of semiconductors, Riv. Nuovo Cimento (1978–1999), 1988, 11, 1–86 CrossRef CAS
- X. Blase, I. Duchemin, D. Jacquemin and P.-F. Loos, The Bethe–Salpeter equation formalism: From physics to chemistry, J. Phys. Chem. Lett., 2020, 11, 7371–7382 CrossRef CAS PubMed
- X. Gui, C. Holzer and W. Klopper, Accuracy assessment of GW starting points for calculating molecular excitation energies using the Bethe–Salpeter formalism, J. Chem. Theory Comput., 2018, 14, 2127–2136 CrossRef CAS PubMed
- M. A. Zwijnenburg, The effect of particle size on the optical and electronic properties of magnesium oxide nanoparticles, Phys. Chem. Chem. Phys., 2021, 23, 21579–21590 RSC
- J. Li, Y. Ni, J. Liu and J. Hong, Preparation, conversion, and comparison of the photocatalytic property of Cd(OH)2, CdO, CdS and CdSe, J. Phys. Chem. Solids, 2009, 70, 1285–1289 CrossRef CAS
- V. Eskizeybek, A. Avcı and M. Chhowalla, Structural and optical properties of CdO nanowires synthesized from Cd(OH)2 precursors by calcination, Cryst. Res. Technol., 2011, 46, 1093–1100 CrossRef CAS
- X. Liu, C. Li, S. Han, J. Han and C. Zhou, Synthesis and electronic transport studies of CdO nanoneedles, Appl. Phys. Lett., 2003, 82, 1950–1952 CrossRef CAS
- T. Terasako, T. Fujiwara, M. Yagi and S. Shirakata, Various shapes of ZnO and CdO nanostructures grown by atmospheric-pressure chemical vapor deposition, Jpn. J. Appl. Phys., 2011, 50, 01BJ15 CrossRef
- T. Terasako, T. Fujiwara, Y. Nakata, M. Yagi and S. Shirakata, Structural and optical properties of CdO nanostructures prepared by atmospheric-pressure CVD, Thin Solid Films, 2013, 528, 237–241 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Development of the colle-salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed
- A. D. Becke, Density-functional thermochemistry. III the role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch,
Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, A generally applicable atomic-charge dependent london dispersion correction, J. Chem. Phys., 2019, 150, 154122 CrossRef
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- M. J. van Setten, F. Weigend and F. Evers, The GW-method for quantum chemistry applications: Theory and implementation, J. Chem. Theory Comput., 2013, 9, 232–246 CrossRef CAS PubMed
- C. Holzer and W. Klopper, Ionized, electron-attached, and excited states of molecular systems with spin–orbit coupling: Two-component GW and Bethe–Salpeter implementations, J. Chem. Phys., 2019, 150, 204116 CrossRef PubMed
- K. Krause and W. Klopper, Implementation of the Bethe–Salpeter equation in the turbomole program, J. Comput. Chem., 2017, 38, 383–388 CrossRef CAS PubMed
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS
- E. J. Baerends, O. V. Gritsenko and R. van Meer, The kohn–sham gap, the fundamental gap and the optical gap: The physical meaning of occupied and virtual Kohn–Sham orbital energies, Phys. Chem. Chem. Phys., 2013, 15, 16408–16425 RSC
-
S. Lany, Polymorphism,
Band-Structure, Band-Lineup, and Alloy Energetics of the Group II Oxides and Sulfides MgO, ZnO, CdO, MgS, ZnS, CdS, SPIE, 2014, vol. 8987, p. 89870K Search PubMed
- D. M. Roessler and W. C. Walker, Electronic Spectrum and Ultraviolet Optical Properties of Crystalline MgO, Phys. Rev., 1967, 159, 733–738 CrossRef CAS
- R. Schmidt-Grund, A. Carstens, B. Rheinländer, D. Spemann, H. Hochmut, G. Zimmermann, M. Lorenz, M. Grundmann, C. M. Herzinger and M. Schubert, Refractive indices and band-gap properties of rocksalt MgxZn1−xO (0.68 ⩽ X ⩽ 1), J. Appl. Phys., 2006, 99, 123701 CrossRef
- F. P. Koffyberg, Thermoreflectance spectra of CdO: Band gaps and band-population effects, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 4470–4476 CrossRef CAS
- P. H. Jefferson, S. A. Hatfield, T. D. Veal, P. D. C. King, C. F. McConville, J. Zúñiga–Pérez and V. Muñoz–Sanjosé, Bandgap and effective mass of epitaxial cadmium oxide, Appl. Phys. Lett., 2008, 92, 022101 CrossRef
- A. Schleife and F. Bechstedt,
Ab initio description of quasiparticle band structures and optical near-edge absorption of transparent conducting oxides, J. Mater. Res., 2012, 27, 2180–2189 CrossRef CAS
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125, 224106 CrossRef
- A. Segura, J. F. Sánchez-Royo, B. García-Domene and G. Almonacid, Current underestimation of the optical gap and burstein-moss shift in CdO thin films: A consequence of extended misuse of A2-versus-hν plots, Appl. Phys. Lett., 2011, 99, 151907 CrossRef
- S. K. V. Farahani, V. Muñoz-Sanjosé, J. Zúñiga-Pérez, C. F. McConville and T. D. Veal, Temperature dependence of the direct bandgap and transport properties of CdO, Appl. Phys. Lett., 2013, 102, 022102 CrossRef
- A. J. Williamson, A. Franceschetti, H. Fu, L. W. Wang and A. Zunger, Indirect band gaps in quantum dots made from direct-gap bulk materials, J. Electron. Mater., 1999, 28, 414–425 CrossRef CAS
- E. Zojer, T. C. Taucher and O. T. Hofmann, The Impact of dipolar layers on the electronic properties of organic/inorganic hybrid interfaces, Adv. Mater. Interfaces, 2019, 6, 1900581 CrossRef
- P. K. Santra, A. F. Palmstrom, J. T. Tanskanen, N. Yang and S. F. Bent, Improving performance in colloidal quantum dot solar cells by tuning band alignment through surface dipole moments, J. Phys. Chem. C, 2015, 119, 2996–3005 CrossRef CAS
- D. M. Kroupa, M. Vörös, N. P. Brawand, B. W. McNichols, E. M. Miller, J. Gu, A. J. Nozik, A. Sellinger, G. Galli and M. C. Beard, Tuning colloidal quantum dot band edge positions through solution-phase surface chemistry modification, Nat. Commun., 2017, 8, 15257 CrossRef CAS PubMed
- M. Bruening, E. Moons, D. Yaron-Marcovich, D. Cahen, J. Libman and A. Shanzer, Polar Ligand Adsorption Controls Semiconductor Surface Potentials, J. Am. Chem. Soc., 1994, 116, 2972–2977 CrossRef CAS
- J. Krüger, U. Bach and M. Grätzel, Modification of TiO2 heterojunctions with benzoic acid derivatives in hybrid molecular solid-state devices, Adv. Mater., 2000, 12, 447–451 CrossRef
- G.-H. Kim, F. P. García de Arquer, Y. J. Yoon, X. Lan, M. Liu, O. Voznyy, Z. Yang, F. Fan, A. H. Ip, P. Kanjanaboos, S. Hoogland, J. Y. Kim and E. H. Sargent, High-efficiency colloidal quantum dot photovoltaics via robust self-assembled monolayers, Nano Lett., 2015, 15, 7691–7696 CrossRef CAS PubMed
- R. Wick-Joliat, T. Musso, R. R. Prabhakar, J. Löckinger, S. Siol, W. Cui, L. Sévery, T. Moehl, J. Suh, J. Hutter, M. Iannuzzi and S. D. Tilley, Stable and tunable phosphonic acid dipole layer for band edge engineering of photoelectrochemical and photovoltaic heterojunction devices, Energy Environ. Sci., 2019, 12, 1901–1909 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Tables with predicted quasiparticle energies and excitation energies, figure showing the different (CdO)n and (CdO)n(NH2(CH3))m structures, and plots of the change in the highest occupied and lowest unoccupied quasiparticle states of (CdO)32 with number of capping agents on the surface and the change in the highest occupied and lowest unoccupied quasiparticle states with number of capping agents adsorbed. See DOI: https://doi.org/10.1039/d2cp01342h |
| This journal is © the Owner Societies 2022 |