
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC2 product formation in the CO2 electroreduction on boron-doped graphene anchored copper clusters†
Balázs
Barhács
a,
Ewald
Janssens
 b and
Tibor
Höltzl
*acd
b and
Tibor
Höltzl
*acd
aDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szent Gellért tér 4, H-1111 Budapest, Hungary. E-mail: tibor.holtzl@furukawaelectric.com
bQuantum Solid-State Physics, KU Leuven, Celestijnenlaan 200D, BE-3001 Leuven, Belgium
cELKH-BME Computation Driven Research Group, Budapest University of Technology and Economics, Szent Gellért tér 4, H-1111 Budapest, Hungary
dFurukawa Electric Institute of Technology, Nanomaterials Science Group, Késmárk utca 28/A, H-1158 Budapest, Hungary
First published on 29th August 2022
Abstract
A possible remedy for the increasing atmospheric CO2 concentration is capturing and reducing it into valuable chemicals like methane, methanol, ethylene, and ethanol. However, a suitable catalyst for this process is still under extensive research. Small sized copper clusters have gained attention in recent years due to their catalytic activity in the CO2 reduction reaction. Although C2+ products have a higher economic value, the formation of C1 products was investigated most thoroughly. Graphene is a promising support for small copper clusters in the electrochemical reduction of CO2. It exhibits good mechanical and electrical properties, but the weak interaction between copper and graphene is an issue. Our DFT computations reveal that small Cu clusters on the boron-doped graphene (BDG) support are promising catalysts for the electrochemical reduction of CO2. We found facile reaction pathways towards various C1 (carbon-monoxide, formic acid, formaldehyde, methanol or methane) and C2 (ethanol or ethylene) products on Cu4 and Cu7 clusters on BDG. The reactivity is cluster-size tunable with Cu4 being the more reactive agent, while Cu7 shows a higher selectivity towards C2 products.
Introduction
The continuously increasing atmospheric CO2 concentration due to the combustion of an enormous amount of fossil fuels leads to severe problems of global warming and ocean acidification.1–8 Capturing CO2 and reducing it to useful chemicals like methane, methanol, ethylene, and ethanol is a promising solution to mitigate these problems.8–10 Although CO2 hydrogenation is thermodynamically feasible, in practice, it is kinetically hindered and requires a proper catalyst.11It is well known since the seminal works of Hori et al. that the electrolysis of bicarbonate solution using a copper electrode produces not only hydrogen, but also methane, ethylene, and higher organic compounds.12 Decreasing the catalyst size to the micro- and nanoscale was shown to be a fruitful strategy to increase the catalytic activity. Cu surfaces,13–21 Cu nanoparticles,22–24 and small Cu clusters have been investigated as active catalysts for the CO2 reduction reaction (CO2RR).25–30
Metal clusters are particularly promising due to their very high atom-efficiency. Liu et al. studied the CO2RR on small four atom copper clusters deposited on an alumina surface using both experimental techniques and theoretical methods.27 Cu4 exhibits an excellent catalytic activity with methanol as the main product, while due to the relatively low activation barriers methane is also formed. Later they investigated the cluster size effect in the reaction towards methanol and found a non-monotonous size-dependence: Cu4/Al2O3 is the most reactive followed by Cu20 and Cu3.30 Tao et al. investigated the cluster-size effect on TiO2(110) supported clusters and also found that Cu4 exhibits the highest activity for converting CO2 to methanol.29 Yang et al. studied Fe2O3 supported Cu4 clusters in the same reaction and concluded that Cu4 facilitates the H2 dissociation and spill-over, leading to the reduction of the oxide surface as Fe2+–Cu4, which promotes CO2 activation.31
Depending on the energy source the CO2 reduction reaction (CO2RR) can be activated thermally,32 photocatalytically,9,33 or electrocatalytically,12,34 the latter being particularly promising due to its efficiency. The conductive support for electrocatalysts is highly important with graphene being a promising candidate, not only because of its advantageous mechanical and electronic properties,35,36 but it can also synergistically enhance the activity of the supported catalyst.37–41
Curtiss et al. investigated four atom transition metal clusters and found that they are promising candidates for the CO2 electrochemical reduction. The overpotentials for producing CH4 were in the following order: Co4 < Fe4 < Ni4 < Cu4 < Pt4. They also investigated the effect of a defective graphene support in the case of Cu4 and calculated lower limiting potentials for CH4 production compared to the Cu(111) surface.26
In comparison with C1 species, C2+ species have a higher economic value due to their wider industrial usability,42 thus nowadays the product selectivity is being studied extensively on various catalysts, including metal clusters. Nitrogen doped graphene supported gold clusters were experimentally shown to catalyze the CO2 electroreduction to CO,45 while DFT reaction paths indicate that small transition metal clusters deposited on graphene26 or graphdiyne46,47 can lead to various C1 products. Recent experimental works have shown efficient formation of C2 products on small copper clusters embedded in mesoporus carbon spheres,48 metal organic frameworks49,50 or Cu coordination polymers.51 4,4’-bipyridine (bipy) ligand-protected Cu clusters were synthesized in two different ways from Cu(bipy)Br: one was selective for CH4 production the other for C2 products, mostly ethylene and ethanol.52 The efficient conversion of syngas to C2 products on MoS2 anchored Cu4 clusters was also demonstrated using theoretical methods.25 Xu et al. synthesized a carbon-supported copper catalyst using an amalgamated Cu–Li method and observed high faradaic efficiency of small clusters towards ethanol.43 Su and coworkers investigated CuO catalysts on N-doped carbon nanosheets.44 When applying a potential on the catalyst, Cu2–CuN3 clusters are formed that are the active sites of highly selective ethanol formation, as it was confirmed using operando FTIR experiments and by the reaction paths computed using the DFT method. In the active form of the catalyst three nitrogen atoms surround the cluster in a similar pattern to the one that will be reported in this paper for boron atoms.
These examples clearly show that the chemical environment of the clusters plays an important role in the product selectivity, however the detailed effect of the support and the cluster size on the copper cluster catalyzed CO2 electroreduction is not yet fully explored. Here we investigate copper cluster anchoring to boron doped graphene using a nanoflake model and its efficiency for CO2 reduction towards C1 and C2 products.
Computational details
All calculations were carried out using the Q-Chem 5.2 and 5.3 program packages. We used the PBE density functional53 with an additional empirical dispersion correction (Grimme DFT-D2)54 and the def2-TZVP basis set.55 The accuracy of this method was confirmed by CCSD(T)/def2-TZVPPD benchmark computations on small model systems (see the ESI† for the details).The reaction intermediates were fully optimized without constraints, and the solvation free energies were computed in water using the SMD implicit solvent model.56 The reaction free energies were calculated using the Computational Hydrogen Electrode (CHE) model,19 including the solvation free energies, but neglecting the vibrational contributions. A similar method was successfully applied to explore the CO2RR reaction mechanism on metal clusters.26,57 The proton in each reaction step comes from a H+ transport chain in the aqueous electrolyte and is reduced by the electrons from the supporting electrode. The description of the proton–electron transfer is simplified and the barrier corresponding to this reaction is neglected in CHE. In line, recent grand-canonical DFT computations showed that that the barrier corresponding to the proton transfer on a nickel single atom carbon-dioxide electroreduction catalyst is relatively small, namely in the range of a few to a few tens of kJ/mol for CO2 electroreduction.58 As shown in the ESI,† using the method proposed in ref. 58, we found that the protonation can precede the reduction step.
The non-covalent interaction of the clusters and the BDG nanoflake is interpreted based on Complementary Occupied-Virtual Pairs (COVP), computed using Energy Decomposition Analysis based on Absolute Localized Molecular Orbitals (ALMO-EDA).59 Natural atomic charges were obtained from Natural Bond Orbital (NBO) analysis.60 Nucleus-Independent Chemical Shift (NICS) values61 were calculated for the BDG model and compared to those of benzene, as a probe of aromaticity. This method was used previously to quantify the aromaticity and stability of boron and nitrogen-doped graphene nanoflakes.62
Results
Anchoring atoms and doping patterns
The interaction between graphene and copper particles is known to be relatively weak63,64 and chemical modification is necessary for efficient anchoring.Three model features must be selected for the graphene support: the dopant element, the dopant pattern, and the model geometry. We selected boron as the anchoring atom due to its high binding affinity to copper clusters (see the ESI,† for details) and because boron doped graphene allows copper atoms to aggregate into clusters.65 The electrocatalytic activity of pure graphene can be increased by doping with almost any element;41 boron doping is among the few exceptions.66 This ensures that not boron but the anchored metal cluster and its interaction with the support are in this study responsible for the catalytic activity. Joshi et al. investigated boron-doped graphene as a support for IrO2 nanoparticle catalysts for the oxygen evolution reaction and found that boron doping not only increases the stability of the IrO2 nanoparticle–graphene complex but also strengthens binding of the reaction intermediates to the nanoparticle, resulting in increased reactivity.39 Boron doping of copper leads to efficient C2 hydrocarbon production in the CO2RR.67 For the dopant pattern we chose one that was shown in ref. 65. Three boron dopant atoms surround a carbon vacancy, such that each atom bears its formal valency. Finally, we selected finite flake models, motivated by the experimentally limited crystallinity of the doped graphene and, computationally, by the applicability of the well-established quantum chemical wavefunction analysis tools. It is necessary to consider a sufficiently large model system because it is known that copper clusters prefer to bind to the edges of finite polycyclic aromatic hydrocarbons (PAHs).68
Our model system is based on a small graphene nanoflake, where the central carbon atom is removed, and the vacancy is surrounded by boron atoms. The initial graphene nanoflake is a non-Kekulé molecule which has a doublet ground state. If we remove the central carbon atom from the PAH and substitute the surrounding three carbon atoms with boron (Fig. 1), a molecule with a chemical formula of C33H15B3 and a singlet ground state is obtained.
 | ||
| Fig. 1 Construction of a BDG nanoflake model. | ||
The highly negative NICS(1) (the ghost atom is placed 1 Å above the centre of the ring, see the ESI,† for the justification of this choice) value of −8.9 ppm of our nanoflake model compared to −9.8 ppm of that of the benzene shows that the BDG model system is aromatic, suggesting an enhanced stability of this particular doping pattern. The Wiberg bond indices between the carbon atoms are in the range of 1.2–1.3, which is between those of the ideal single and double C–C bonds and close to the ∼1.4 C–C Wiberg bond index of aromatic benzene. The B–C and B–B bonds have bond index values of ∼1.07 and ∼0.4, respectively. The aromaticity of the central ring is consistent with the fully delocalized π orbital (Fig. 2).
 | ||
| Fig. 2 Contour surface of the π orbital, delocalized over the whole BDG nanoflake. | ||
Single-layer boron-doped graphene can be synthesized using chemical vapor deposition (CVD) from different, boron-containing precursors including triethylborane69 and diborane.70 The band structure of boron-doped graphene was investigated69–71 and the electrical conductivity was measured.71 It was found that even after doping, graphene retains its excellent conductivity. The same is true for the mechanical properties: they do not change significantly with boron doping.72,73 PAHs containing two boron atoms were also successfully synthesized:74 they can be interpreted as boron-doped graphene nanoflakes. This shows that the bottom-up synthesis of boron-doped graphene with a well-defined structure must be feasible. Further discussion on the stability of the BDG nanoflake model is available in the ESI.†
Copper cluster anchoring to BDG nanoflakes
BDG bound copper cluster geometries were generated following the idea of “soft-landing”: the clusters were deposited on BDG with their most stable gas-phase structure, and these structures were subsequently relaxed. This procedure models the typical experimental synthesis process of cluster beam deposition.75 The optimized geometrical structures of the free gas phase and the BDG nanoflake anchored Cun (n = 1–8) clusters with the natural charge of each Cu atom are depicted in Fig. 3. The interaction energies between the copper clusters and the BDG nanoflake and the clusters’ natural charges are given in Table 1. The small copper clusters (similar to the BDG bound single copper atom65) are expected to be thermodynamically less stable than the bulk metal, but the large interaction energies with BDG (Table 1 and Section 3 in the, ESI†) clearly show that clusters synthesized in the gas phase can be soft-landed and immobilized on BDG, which prevents their aggregation.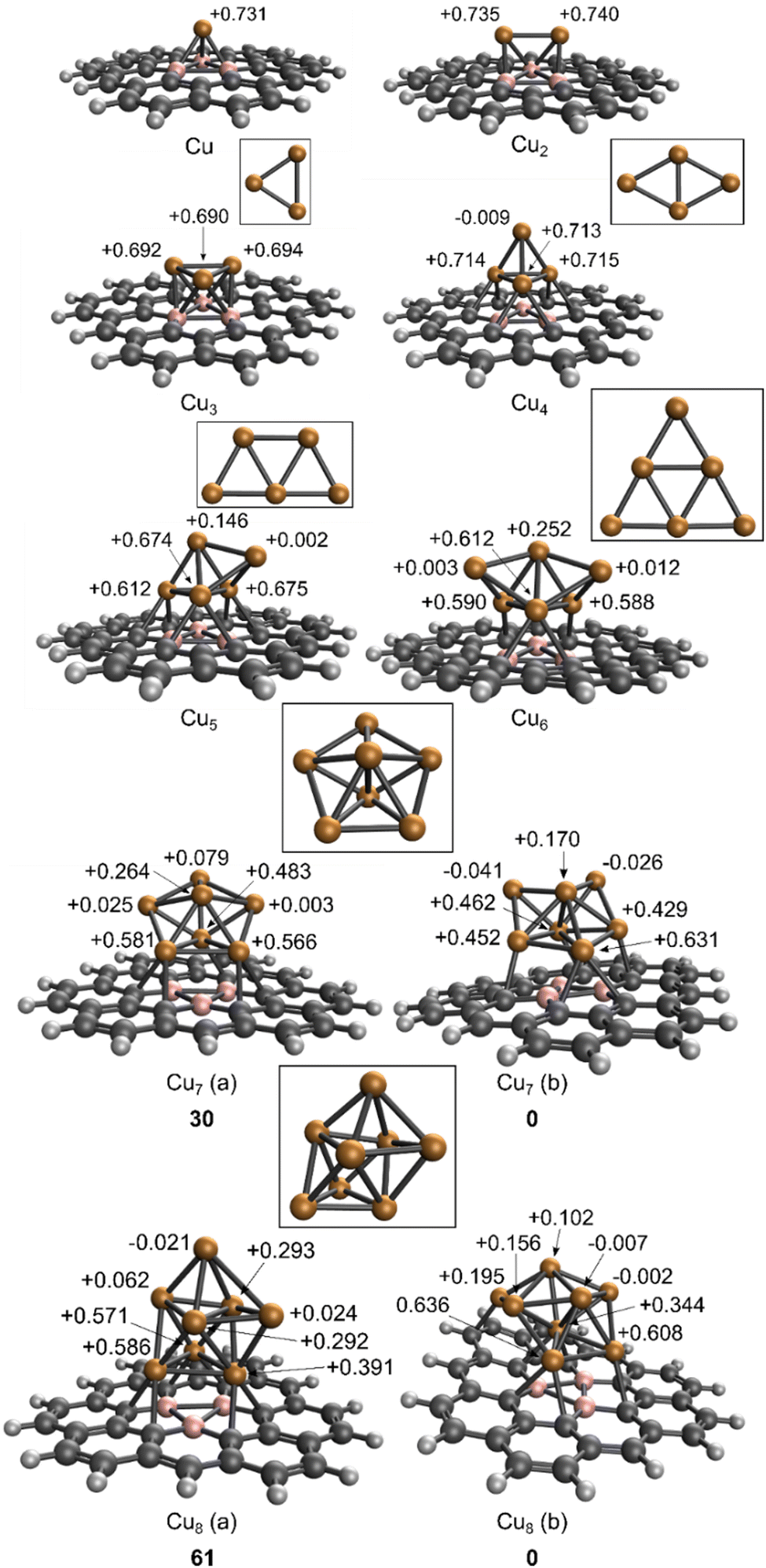 | ||
| Fig. 3 The optimized structures of gas phase Cun (n = 1–8) and BDGCun supported clusters. The natural charges of the different Cu atoms are given in the case of BDG supported clusters. The relative energies of the different isomers are given by bold numbers (in kJ mol−1). | ||
| n | Interaction energy (kJ mol−1) | Cluster natural charge (in units of elementary charge) |
|---|---|---|
| 1 | −326 | 0.731 |
| 2 | −389 | 1.48 |
| 3 | −538 | 2.08 |
| 4 | −507 | 2.13 |
| 5 | −577 | 2.11 |
| 6 | −568 | 2.06 |
| 7(a) | −527 | 2.00 |
| 7(b) | −557 | 2.08 |
| 8(a) | −490 | 1.98 |
| 8(b) | −552 | 2.03 |
The cluster-BDG interaction energy increases from n = 1 to n = 3, while a weaker, non-monotonic size-dependence is observed for larger clusters. The surface anchored Cun clusters exhibit partially positive charges, which saturate at approximately +2 for n = 3. The excess positive charges reside mainly on the boron bound copper atoms (Fig. 3). This observation and the correlation with the increase of cluster natural charges show that the boron-bound copper atoms of the cluster are responsible for the interaction.
As expected, the clusters preferentially bind to the boron dopant atoms. Comparing the gas-phase geometries to the clusters on BDG, a first noticeable difference is the shape changes from planar gas-phase Cu4, Cu5 and Cu6 to three-dimensional cluster shapes on BDG, with Cu4 on BDG being an almost perfect tetrahedron.
The almost perfect tetrahedral shape of the Cu4 cluster on BDG is remarkable. This cluster donates electrons to the support and obtains an approximately +2 natural charge. In the framework of the Phenomenological Shell Model (PSM):76 the Cu 4s1 electrons are itinerant, which for the neutral Cu4 cluster leads to the 1S2 1P2 electron configuration (cluster orbitals within the PSM are denoted with capital letters). However, there are only two itinerant electrons in the formally di-cationic Cu4 cluster, which corresponds to the 1S2 closed electronic structure; a closed electronic structure with enhanced stability.
Electronic structure analysis of the anchoring
The most relevant COVP pairs of the BDG bound clusters (BDGCun) are investigated. ALMOs are noted with capital letters according to the PSM (S, P, and D) if they belong to the metal cluster, and with Greek letters, commonly used for molecular orbitals, if they belong to the BDG nanoflake. The result for n = 4 is presented in Fig. 4, while analogous results for other cluster sizes are available in the ESI.† According to the PSM, the gas phase Cu4 cluster has a 1S21P2 electronic structure. | ||
| Fig. 4 (a) The most relevant COVP pairs of the BDGCu4 system are depcicted in each column. In the upper line the donor, in the lower line the acceptor orbital is shown. dE refers to the computed charge transfer energy, dQ is the transferred charge in millielectrons. (b) The HOMO of the gas-phase Cu4 cluster and the LUMO of BDG. | ||
The COVP analysis shows that during the anchoring, the P orbital of the cluster donates electrons to the BDG π* orbital (composed partly of the formally empty boron pz atomic orbital). This process involves transfer of approximately half an electron from the cluster to the BDG, clearly highlighting the role of the electron deficient boron atoms. The second relevant donor is the S orbital of the cluster, donating to a σ* orbital of the BDG nanoflake. The charge transfer is much smaller in this case (0.14 e), but the energy contribution is relatively high.
More generally, for n = 3–8 the main charge transfer is due to the electron donation from the clusters’ P orbitals to the π* orbitals of the BDG nanoflake. This π* orbital consists of both the atomic p orbitals of carbon and boron atoms, with higher coefficients for the p orbitals of the three boron atoms. So the ALMO seems to have a delocalized domain in the centre of the ring. This ALMO corresponds formally to the LUMO of the individual BDG nanoflake (Fig. 4b). Likewise, the HOMO of the individual gas phase clusters is a P orbital for all n = 3–8. The second relevant donor ALMO is a cluster S orbital, and the acceptor is a σ* orbital of the boron atoms. It must be noted that the D shell orbital also participates in the chemical bonding.
Overall, we can conclude that the interaction energy of the copper clusters and the BDG nanoflake is mostly due to the significant charge transfer between the two fragments. The cluster donates electrons to the BDG nanoflake. The boron atoms have a significant role in the interaction; they have the highest eigenvalues in the acceptor ALMOs (both π* and σ*).
Descriptors for C2 formation
It was shown recently that the potential-determining, and thus the rate-determining, step of the CO2RR reaction towards C2 products was the C–C coupling through the reductive dimerization of two CO molecules:13,20,21,77where asterisks denote catalyst surface bound species. According to the results of Huang et.al.,22 the reaction energy towards *OCCOH can be used as the reactivity descriptor to estimate the feasibility of CO2RR toward C2 products with various catalysts:


The lower this descriptor is, the higher the CO2RR rate towards C2 products. We computed the reaction energy of this C–C coupling step on all sites of the cluster for BDGCun (n = 3–7), and the reaction energies on the most reactive site of each cluster are listed in Table 2.
| n | ΔEOCCOH (kJ mol−1) |
|---|---|
| 4 | 112 |
| 5 | 29 |
| 6 | 36 |
| 7 | 28 |
This C–C coupling step is always endothermic and is the least favoured for n = 4. For larger clusters (n = 5–7) weak size-dependence is observed. While Cu4 is shown to catalyse the CO2 reduction towards C1 products,26,27,30 this descriptor suggests somewhat lower C2 selectivity. On the other hand, among the investigated cluster sizes, the descriptor has the smallest value in the case of Cu7, implying more facile formation of C2 products. Therefore, for a detailed comparison of the reactivities, we computed the reaction paths towards C1 or C2 products for Cu4 and Cu7 clusters.
Reactions towards C1 products
We first located the most stable electrochemical binding site of CO2, whereafter we systematically investigated the possible reduction pathways. The most feasible reaction paths are depicted in Fig. 5 and 6, while higher energy pathways are available in the ESI.†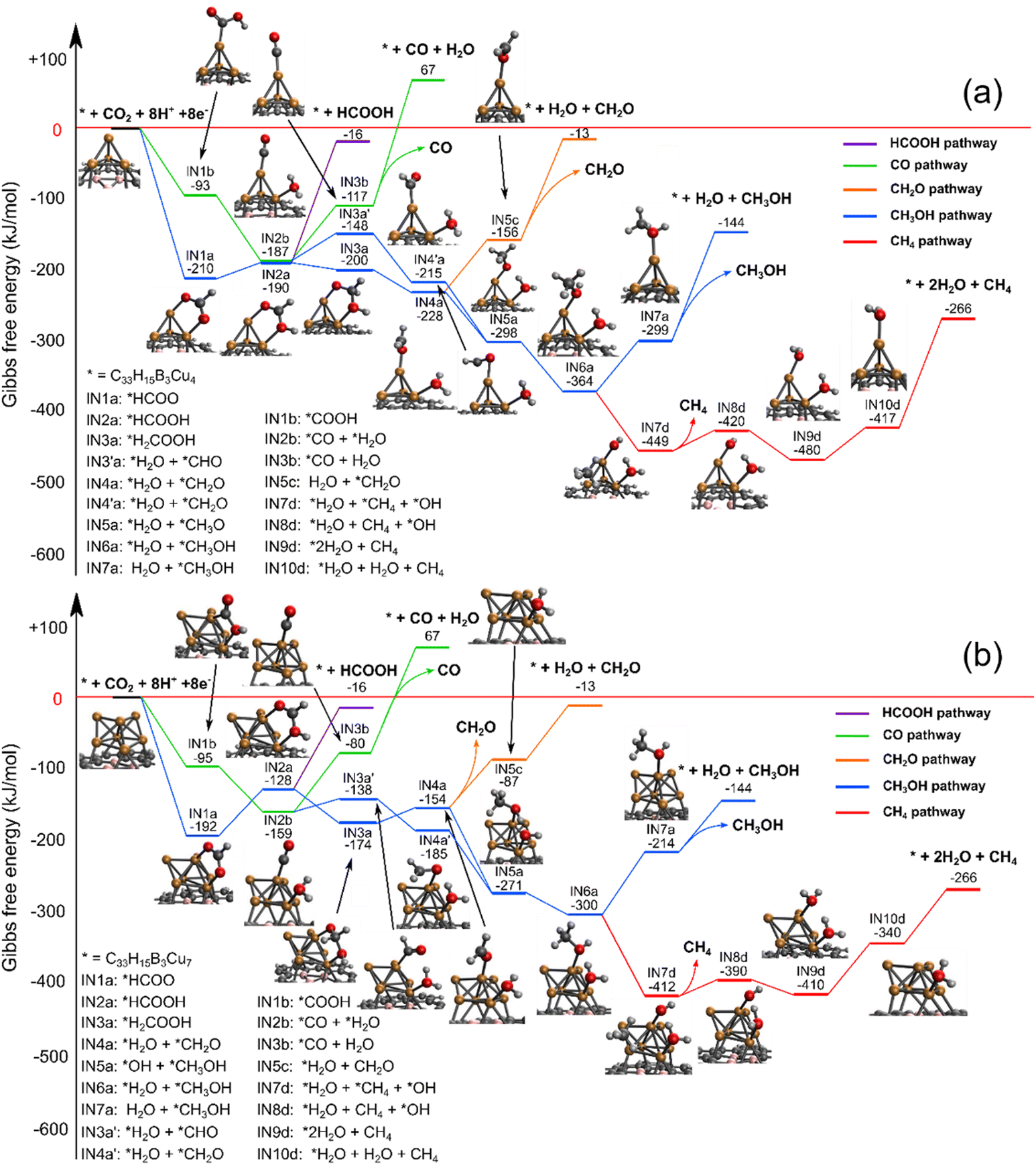 | ||
| Fig. 5 The C1 reaction pathway in an aqueous solution at 298 K, 1 atm using different sized copper cluster catalysts: (a) BDGCu4 and (b) BDGCu7. | ||
 | ||
| Fig. 6 The C2 reaction pathway in an aqueous solution at 298 K, 1 atm using different sized copper cluster catalysts: (a) BDGCu4 and (b) BDGCu7. | ||
Fig. 5 shows the reaction paths towards C1 products using BDGCu4 and BDGCu7 catalysts. The different reaction paths are branching already at the first step, when CO2 is electrochemically adsorbed and reduced to either a carboxyl (*COOH, green line) or a formate group (*HCOO, blue line). It is well accepted14,26,43,58 that in the electrocatalytic process the CO2 adsorption proceeds simultaneously with its reduction. The further reduction of *COOH leads to adsorbed *CO, whose desorption from the cluster is thermodynamically unfavoured. The further hydrogenation of *CO to *CHO (blue line, higher in free energy) leads to *CH2O (formaldehyde) and subsequently to *CH3O. It is interesting to note that the *CH3O intermediate can also be reached by the further consecutive reduction of *HCOO through formic acid (*HCOOH) and *H2COOH. This is in line with previously found reaction paths on deposited Cu4 clusters.27,30 The C–O bond breaks during the further reduction of *H2COOH and formaldehyde (*CH2O) is formed. Desorption of formaldehyde or formic acid is thermodynamically unfavoured. Thus, the further hydrogenation of *CH3O leads to methanol. The desorption of methanol is also an endergonic step. The further reduction of the adsorbed methanol *CH3OH leads to C–O bond breaking, and methane is formed. Methane eliminates easily from the cluster.
The green and blue reaction paths are analogous to the widely accepted ones for methanol formation on various copper surfaces17,18 and nanoparticles.23 The most interesting difference between BDGCu4 and BDGCu7 is that the diverging blue reaction paths cross only in the latter case. Consequently, the reactivity of BDGCu4 differs more from that of Cu surfaces, thus this cluster can open new reaction paths. Also, the different relative free energies of the intermediates imply that the methanol formation is somewhat more favoured in the BDGCu4 than in the BDGCu7 case, which opens up the possibility to tune the product composition by the cluster size.
It was shown for Cu surfaces that methane does not form from the hydrogenation of *CH3O (or *CH3OH). It follows a different pathway, where *COH instead of *CHO is formed from *CO, and after a C–O bond breaking, water and *CHx (x = 0, 1, 2, 3) species are formed resulting in methane. We computed the formation of *COH, however, it is highly endergonic (see the ESI† for details), suggesting thermodynamic blocking of this pathway on small metal clusters.
The left side of Scheme 1 (green background) presents a simplified depiction of the C1 reaction pathways on the BDG supported clusters. Our computations show that on BDG supported small Cu clusters, the main C1 products are methanol and methane with a higher methanol fraction in the case of BDGCu4 than BDGCu7.
 | ||
| Scheme 1 A simplified scheme of the computed reaction pathways towards C1 (green background) and C2 (blue background) products. | ||
Reactions towards C2 products
We systematically investigated the analogous reaction path to those proposed by Kortlever et al.34 and Xiao et al.21 for Cu surfaces towards ethylene and ethanol.The most favoured reaction paths are depicted in Fig. 6. As described above, the most important step in the formation of C2 products is the C–C coupling through the reductive dimerization of two catalyst surface bound *CO molecules, thus the reaction starts with a subsequent partial reduction of two CO2 molecules, which is followed by the reductive dimerization and the formation of *OCCOH.
In a thermal reaction this is always an endothermic step (see the ESI† for the thermally activated reaction pathways). On the other hand, in the electrocatalytic process the solvation makes this step thermodynamically feasible in the case of BDGCu7, thus the solvent even changes the qualitative reactivity. The fact that the C–C coupling is more likely to occur on the larger cluster may have also steric reasons due to the relatively crowded arrangement of the several reactants on the Cu4 cluster.
Following the further hydrogenation of *OCCOH and C–O bond breaking, water dissociates and *CCO forms. Here, the tetrahedral shape of Cu4 on BDG opens, and it recovers only after the desorption of product molecules. *CCO is then further hydrogenated to *CHCO, *CHCHO, and *CH2CHO, where the reaction can continue in two different pathways. We denote the formation of ethanol and ethylene with blue and red lines on the figure, respectively. For the blue path the formation of *CH2CH2O from the *CH2CHO intermediate is more facile than that of *CH3CHO. On Cu4 an adsorbed ethanol *CH3CH2OH is formed directly from *CH3CH2O by taking another hydrogen atom from an adsorbed H2O molecule. On BDGCu7, *CH3CH2O is the most stable intermediate, and *CH3CH2OH formation is slightly endergonic. The dissociation of ethanol from both clusters is an endergonic process, indicated by the reaction free energies of +60 kJ mol−1 for BDGCu4 and +33 kJ mol−1 for BDGCu7. This shows that the ethanol dissociation is more favoured on BDGCu7, but it is thermodynamically not blocked even on BDGCu4. Along the red path, to form ethylene from *CH2CHO, a C–O bond breaking is required after a H+ + e− transfer. The desorption of ethylene from the cluster is always endergonic, thus the further reduction of the adsorbed ethylene to an ethyl group is more likely to occur. From here, the only possibility for ethylene production is the β-elimination step, as it was proposed by Xiao et al.,21 however, we found this step also endergonic (+72 kJ mol−1 for BDGCu4 and +104 kJ mol−1 for BDGCu7). An adsorbed hydrogen atom is left on the cluster, which is needed to form a water molecule with an adsorbed by-product *OH.
As it is described above, we observed small changes of the cluster structures during the reaction. The Cu4 cluster opens during *CCO formation and closes again to its tetrahedral shape shortly after the products (ethanol and ethylene) desorb from the cluster. For Cu7, the distance between the top two Cu atoms changes during the reaction. The small geometry change corresponds to a transition between the more stable pyramidal structure (Cu7(b)) and a hexagonal bipyramidal shape (Cu7(a)).
The computed reaction pathways and reaction free energies clearly show that BDG bound small Cu clusters can catalyse C2 formation towards both ethylene and ethanol. A simplified summary of the C2 reaction pathways is shown in the right part of Scheme 1 (blue background).
Conclusion
In summary, in this work we showed using a nanoflake model that boron doping is a promising method to immobilize small Cun (n = 3–8) clusters on graphene and the resulting system has high catalytic activity in the CO2RR towards both C1 and C2 products. Large binding energies between the boron doped graphene nanoflake and the clusters are due to charge transfer; the Cu clusters donate electrons mainly to the boron-atoms. In this complex, boron-doped graphene is the supporting electrode material and small Cu clusters exhibit catalytic activity in the electrochemical reduction of CO2. The free energies along the possible reaction paths confirm the catalytic activity of BDG supported Cu4 and Cu7 clusters for C1 products. The size dependence is relatively weak but expected to allow the tuning of the methanol/methane product ratio. The investigation of the descriptors towards the C2 products reveals that BDG supported copper clusters are promising catalysts. Detailed reaction paths for the BDG supported Cu4 and Cu7 clusters confirm this and show that BDGCu7 has an increased selectivity towards ethanol and ethylene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the ÚNKP-20-1-I New National Excellence Program of The Ministry of Human Capacities, by the KU Leuven–Budapest University of Technology and Economics joint research funding (CELSA/18/032), by the Research Foundation Flanders (FWO project G.0D56.19N) and by the European Union's Horizon 2020 research and innovation program under grant agreement No. 955650 (CATCHY). T. H. is grateful for the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00642/21/7).References
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. Dubois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed
.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. Ray Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed
- A. Harriman, Philos. Trans. R. Soc., A, 2013, 371, 20110415 CrossRef PubMed
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed
- P. Nejat, F. Jomehzadeh, M. M. Taheri, M. Gohari and M. Z. Muhd, Renewable Sustainable Energy Rev., 2015, 43, 843–862 CrossRef CAS
- W. H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS
- D. Talbi and E. Herbst, Astron. Astrophys., 2002, 386, 1139–1142 CrossRef CAS
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 139, 39–47 CrossRef
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed
- T. Cheng, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2016, 138, 13802–13805 CrossRef CAS PubMed
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pedersen and J. K. Nørskov, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC
- E. Pérez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 3621–3624 CrossRef PubMed
- H. Xiao, T. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed
- Y. Huang, Y. Chen, T. Cheng, L. W. Wang and W. A. Goddard, ACS Energy Lett., 2018, 3, 2983–2988 CrossRef CAS
- D. H. Lim, J. H. Jo, D. Y. Shin, J. Wilcox, H. C. Ham and S. W. Nam, Nanoscale, 2014, 6, 5087–5092 RSC
- X. Zhang, J. X. Liu, B. Zijlstra, I. A. W. Filot, Z. Zhou, S. Sun and E. J. M. Hensen, Nano Energy, 2018, 43, 200–209 CrossRef CAS
- J. Chen, Z. Wang, J. Zhao, L. Ling, R. Zhang and B. Wang, Appl. Surf. Sci., 2021, 540, 148301 CrossRef CAS
- C. Liu, H. He, P. Zapol and L. A. Curtiss, Phys. Chem. Chem. Phys., 2014, 16, 26584–26599 RSC
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. Debartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS
- R. Zhang, M. Peng, T. Duan and B. Wang, Appl. Surf. Sci., 2017, 407, 282–296 CrossRef CAS
- H. Tao, Y. Li, X. Cai, H. Zhou, Y. Li, W. Lin, S. Huang, K. Ding, W. Chen and Y. Zhang, J. Phys. Chem. C, 2019, 123, 24118–24132 CrossRef CAS
- B. Yang, C. Liu, A. Halder, E. C. Tyo, A. B. F. Martinson, S. Seifert, P. Zapol, L. A. Curtiss and S. Vajda, J. Phys. Chem. C, 2017, 121, 10406–10412 CrossRef CAS
- B. Yang, X. Yu, A. Halder, X. Zhang, X. Zhou, G. J. A. Mannie, E. Tyo, M. J. Pellin, S. Seifert, D. Su and S. Vajda, ACS Sustainable Chem. Eng., 2019, 7, 14435–14442 CrossRef CAS
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS
- H. Fei, J. Dong, D. Chen, T. Hu, X. Duan, I. Shakir, Y. Huang and X. Duan, Chem. Soc. Rev., 2019, 48, 5207–5241 RSC
- K. S. Novoselov, V. I. Fal’Ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed
- I. Fampiou and A. Ramasubramaniam, J. Phys. Chem. C, 2012, 116, 6543–6555 CrossRef CAS
- W. X. Ji, C. W. Zhang, F. Li, P. Li, P. J. Wang, M. J. Ren and M. Yuan, RSC Adv., 2014, 4, 55781–55789 RSC
- P. Joshi, H. H. Huang, R. Yadav, M. Hara and M. Yoshimura, Catal. Sci. Technol., 2020, 10, 6599–6610 RSC
- H. Xu, W. Chu, W. Sun, C. Jiang and Z. Liu, RSC Adv., 2016, 6, 96545–96553 RSC
- L. Wang, Z. Sofer and M. Pumera, ACS Nano, 2020, 14, 21–25 CrossRef CAS PubMed
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C. J. Sun, T. Li, J. V. Muntean, R. E. Winans, D. J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS
- X. Su, Z. Jiang, J. Zhou, H. Liu, D. Zhou, H. Shang, X. Ni, Z. Peng, F. Yang, W. Chen, Z. Qi, D. Wang and Y. Wang, Nat. Commun., 2022, 13, 1–11 Search PubMed
- L. Jin, B. Liu, P. Wang, H. Yao, L. A. Achola, P. Kerns, A. Lopes, Y. Yang, J. Ho, A. Moewes, Y. Pei and J. He, Nanoscale, 2018, 10, 14678–14686 RSC
- P. Ge, X. Zhai, X. Liu, Y. Liu, X. Yang, H. Yan, G. Ge, J. Yang and Y. Liu, Nanoscale, 2022, 14, 1211–1218 RSC
- J.-C. Liu, H. Xiao, X.-K. Zhao, N.-N. Zhang, Y. Liu, D.-H. Xing, X. Yu, H.-S. Hu and J. Li, CCS Chem., 2022, 1–12 Search PubMed
- Y. Pan, H. Li, J. Xiong, Y. Yu, H. Du, S. Li, Z. Wu, S. Li, J. Lai and L. Wang, Appl. Catal., B, 2022, 121111 CrossRef CAS
- D. H. Nam, O. S. Bushuyev, J. Li, P. de Luna, A. Seifitokaldani, C. T. Dinh, F. P. García De Arquer, Y. Wang, Z. Liang, A. H. Proppe, C. S. Tan, P. Todorović, O. Shekhah, C. M. Gabardo, J. W. Jo, J. Choi, M. J. Choi, S. W. Baek, J. Kim, D. Sinton, S. O. Kelley, M. Eddaoudi and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef CAS PubMed
- D. Yang, S. Zuo, H. Yang, Y. Zhou, Q. Lu and X. Wang, Adv. Mater., 2022, 34, 2107293 CrossRef CAS PubMed
- N. Sakamoto, Y. F. Nishimura, T. Nonaka, M. Ohashi, N. Ishida, K. Kitazumi, Y. Kato, K. Sekizawa, T. Morikawa and T. Arai, ACS Catal., 2020, 10, 10412–10419 CrossRef CAS
- H. Zhang, Y. Yang, Y. Liang, J. Li, A. Zhang, H. Zheng, Z. Geng, F. Li and J. Zeng, ChemSusChem, 2022, 15, e202102010 CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chem. Acc., 1990, 77, 123–141 Search PubMed
- A. v Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
- R. K. Raju, P. Rodriguez and R. L. Johnston, J. Phys. Chem. C, 2019, 123, 14591–14609 CrossRef CAS
- M. D. Hossain, Y. Huang, T. H. Yu, W. A. Goddard and Z. Luo, Nat. Commun., 2020, 11, 2256 CrossRef CAS PubMed
- R. Z. Khaliullin, E. A. Cobar, R. C. Lochan, A. T. Bell and M. Head-Gordon, J. Phys. Chem. A, 2007, 111, 8753–8765 CrossRef CAS PubMed
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Ragué Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed
- A. Akaishi, M. Ushirozako, H. Matsuyama and J. Nakamura, Jpn. J. Appl. Phys., 2017, 57, 0102BA CrossRef
- X. Liu, C. Z. Wang, M. Hupalo, H. Q. Lin, K. M. Ho and M. C. Tringides, Crystals, 2013, 3, 79–111 CrossRef CAS
- H. Valencia, A. Gil and G. Frapper, J. Phys. Chem. C, 2010, 114, 14141–14153 CrossRef CAS
- W. I. Choi, B. C. Wood, E. Schwegler and T. Ogitsu, Adv. Energy Mater., 2015, 5, 1501423 CrossRef
- L. Wang, Z. Sofer, P. Šimek, I. Tomandl and M. Pumera, J. Phys. Chem. C, 2013, 117, 23251–23257 CrossRef CAS
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. de Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed
- U. J. Rangel-Peña, R. L. Camacho-Mendoza, S. González-Montiel, L. Feria and J. Cruz-Borbolla, J. Clust. Sci., 2020, 142, 1–19 Search PubMed
- J. Gebhardt, R. J. Koch, W. Zhao, O. Höfert, K. Gotterbarm, S. Mammadov, C. Papp, A. Görling, H.-P. Steinrück and T. Seyller, Phys. Rev. B, 2013, 87, 155437 CrossRef
- M. Cattelan, S. Agnoli, M. Favaro, D. Garoli, F. Romanato, M. Meneghetti, A. Barinov, P. Dudin and G. Granozzi, Chem. Mater., 2013, 25, 1490–1495 CrossRef CAS
- L. S. Panchakarla, K. S. Subrahmanyam, S. K. Saha, A. Govindaraj, H. R. Krishnamurthy, U. V. Waghmare and C. N. R. Rao, Adv. Mater., 2009, 21, 4726–4730 CAS
- B. Mortazavi and S. Ahzi, Solid State Commun., 2012, 152, 1503–1507 CrossRef CAS
- Z. Dai, G. Wang, Z. Zheng, Y. Wang, S. Zhang, X. Qi, P. Tan, L. Liu, Z. Xu, Q. Li, Z. Cheng and Z. Zhang, Carbon, 2019, 147, 594–601 CrossRef CAS
- C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206–12210 CrossRef CAS PubMed
- A. Yadav, Y. Li, T. W. Liao, K. J. Hu, J. E. Scheerder, O. V. Safonova, T. Höltzl, E. Janssens, D. Grandjean and P. Lievens, Small, 2021, 17, 2004541 CrossRef CAS PubMed
-
T. Höltzl, T. Veszpremi, P. Lievens and M. T. Nguyen, in Aromaticity and metal clusters, ed. P. Kumar, CRC Press, Boca Raton, 2010, ch. 14, pp. 271–296 Search PubMed
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 1795–1800 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01316a |
| This journal is © the Owner Societies 2022 |