
The solution structures and relative stability constants of lanthanide–EDTA complexes predicted from computation†

Abstract
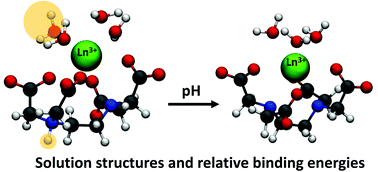
Ligand selectivity to specific lanthanide (Ln) ions is key to the separation of rare earth elements from each other. Ligand selectivity can be quantified with relative stability constants (measured experimentally) or relative binding energies (calculated computationally). The relative stability constants of EDTA (ethylenediaminetetraacetic acid) with La3+, Eu3+, Gd3+, and Lu3+ were predicted from relative binding energies, which were quantified using electronic structure calculations with relativistic effects and based on the molecular structures of Ln–EDTA complexes in solution from density functional theory molecular dynamics simulations. The protonation state of an EDTA amine group was varied to study pH ∼7 and ∼11 conditions. Further, simulations at 25 °C and 90 °C were performed to elucidate how structures of Ln–EDTA complexes varying with temperature are related to complex stabilities at different pH conditions. Relative stability trends are predicted from computation for varying Ln3+ ions (La, Eu, Gd, Lu) with a single ligand (EDTA at pH ∼11), as well as for a single Ln3+ ion (La) with varying ligands (EDTA at pH ∼7 and ∼11). Changing the protonation state of an EDTA amine site significantly changes the solution structure of the Ln–EDTA complex resulting in a reduction of the complex stability. Increased Ln–ligand complex stability is correlated to reduced structural variations in solution upon an increase in temperature.
 Please wait while we load your content...
Please wait while we load your content...

