
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeveraging algorithmic search in quantum chemical reaction path finding†
Atsuyuki
Nakao
a,
Yu
Harabuchi
bcd,
Satoshi
Maeda
 bcd and
Koji
Tsuda
*aef
bcd and
Koji
Tsuda
*aef
aGraduate School of Frontier Sciences, The University of Tokyo, Kashiwa 2778561, Japan. E-mail: tsuda@k.u-tokyo.ac.jp
bInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo 001-0021, Japan
cJST ERATO Maeda Artificial Intelligence for Chemical Reaction Design and Discovery Project, Sapporo 060-0810, Japan
dDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan
eRIKEN Center for Advanced Intelligence Project, Tokyo 103-0027, Japan
fResearch and Services Division of Materials Data and Integrated System, National Institute for Materials Science, Tsukuba 305-0047, Japan
First published on 6th April 2022
Abstract
Reaction path finding methods construct a graph connecting reactants and products in a quantum chemical energy landscape. They are useful in elucidating various reactions and provide footsteps for designing new reactions. Their enormous computational cost, however, limits their application to relatively simple reactions. This paper engages in accelerating reaction path finding by introducing the principles of algorithmic search. A new method called RRT/SC-AFIR is devised by combining rapidly exploring random tree (RRT) and single component artificial force induced reaction (SC-AFIR). Using 96 cores, our method succeeded in constructing a reaction graph for Fritsch–Buttenberg–Wiechell rearrangement within a time limit of 3 days, while the conventional methods could not. Our results illustrate that the algorithm theory provides refreshing and beneficial viewpoints on quantum chemical methodologies.
1 Introduction
In the context of computational chemistry, a chemical reaction is represented as a path from a reactant molecule set to a product molecule set on the potential energy surface (Fig. 1). If n is the total number of atoms involved in the reaction, the potential energy surface resides in a 3n dimensional coordinate space, where each point describes the positions of all atoms. Given two points corresponding to the reactant and the product, reaction path finding refers to the task of finding possible paths between the two points. Conventionally, this task has been accomplished by molecular dynamics or Monte Carlo simulations,1–3 where state transition from the reactant is repeated until it reaches close to the product. To reduce the prohibitive cost of these approaches, automated reaction path search methods have been developed so far.4–12 Single component-artificial force induced reaction (SC-AFIR)13–16 has been successful in elucidating various reactions such as Wöhler's urea synthesis (WUS),17 CO oxidation on the Pt(111) surface,18 and difluoroglycine synthesis.19 In SC-AFIR, transition from one equilibrium structure to another is caused by applying splitting or merging force to two fragments around randomly chosen atoms.13 Despite its successes, it is often the case that SC-AFIR fails to recover known paths of representative reactions within a reasonable time frame (e.g., a few days on a single computational node). | ||
| Fig. 1 Potential energy surface and the corresponding reaction graph. Equilibrium structures are shown as EQ. | ||
In this paper, we leverage algorithmic search for efficient path finding. Algorithmic search algorithms such as A*,20 simulated annealing21 and Monte Carlo tree search22 have been applied to real-world path finding problems such as maze solving, vehicle routing and robot movement scheduling. Among various options, we focus on rapidly-exploring random tree (RRT)23 due to its simplicity and a good track record of successful applications.24–26 Initially, there are a start node (i.e., reactant) and a goal node (i.e., product) in the search space. The search graph is expanded from the start node by alternating the two steps: random node expansion and goal-oriented expansion (Fig. 2). In graph expansion, SC-AFIR is employed to generate adjacent nodes corresponding to neighboring equilibrium structures (denoted as EQs). Finally, our algorithm, termed RRT/SC-AFIR, terminates when a time limit is met.
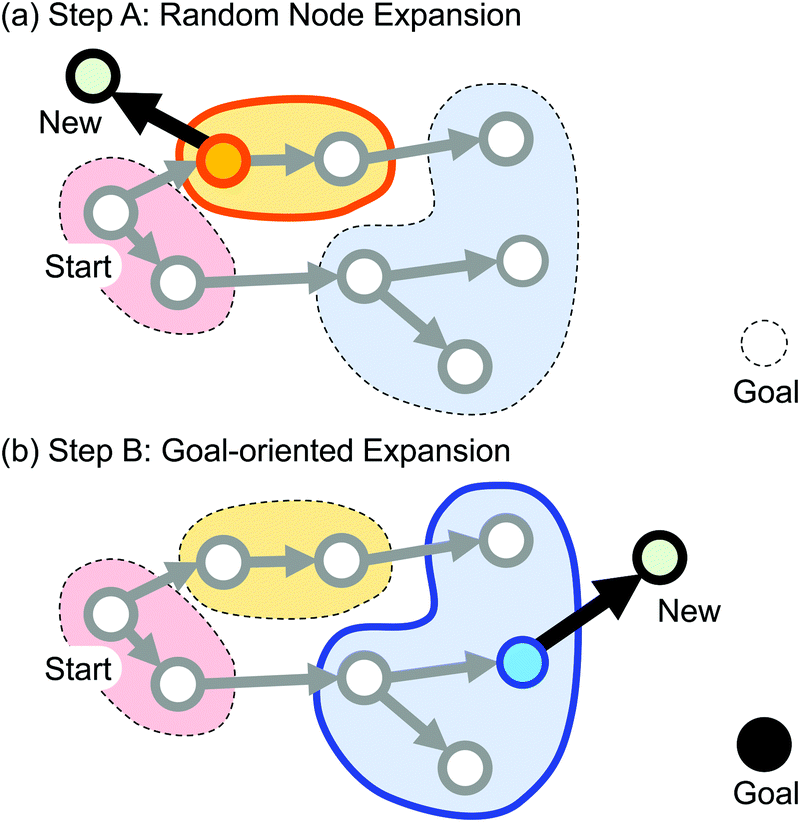 | ||
| Fig. 2 Two steps in RRT/SC-AFIR. In step A, the nodes are clustered according to their connection patterns as shown in red, blue and yellow. A node is selected via cluster-based sampling and expanded with SC-AFIR. In step B, a node is sampled so that those close to the goal node are likely to be chosen. | ||
To meet the challenges from high dimensional and highly constrained conformational spaces, we enhanced RRT with respect to the following two points: (1) fair node sampling and (2) similarity measure of structures. In step A shown in Fig. 2a, a node is randomly selected and expanded. If most of the nodes correspond to similar structures, simple sampling leads to biased exploration. For fairer sampling, the nodes are clustered according to a coarse-grained representation of structures called the connection pattern. A node is selected by the following two-step method. First, a cluster is selected in equal probability. Next, a node in the cluster is randomly selected. In step B shown in Fig. 2b, a node is selected using the similarity between a node and the goal node. Most of the available similarity measures of three-dimensional conformations are developed for comparing two molecules.27 Our equilibrium structure, however, often involves multiple molecules. Since the relative positions of distinct molecules cause only negligible changes in the potential energy, similarity measures of global geometry may fail. To focus on the differences in local geometry, we developed a similarity measure based on greedy match of local atomic environments.
In our computational experiments, RRT/SC-AFIR was evaluated in path finding tasks with respect to Wöhler's urea synthesis (WUS) and Fritsch–Buttenberg–Wiechell rearrangement (FBW). It was compared with two SC-AFIR-based methods: Boltzmann/SC-AFIR15 and kinetics/SC-AFIR.28 The former samples new nodes according to the Boltzmann distribution, while the latter uses kinetics-based navigation. We found that our method was the fastest in finding valid reaction paths that are consistent with the known paths (Fig. 3). Using 96 cores, RRT/SC-AFIR found valid paths of FBW within a time limit of 3 days, while the others could not. Taking statistics of path lengths, it is found that existing SC-AFIR methods tend to generate unreasonably long paths, indicating that RRT was effective in guiding the search towards the goal node.
 | ||
| Fig. 3 Known reaction paths of (a) Wöhler's urea synthesis (WUS) and (b) Fritsch–Buttenberg–Wiechell rearrangement (FBW). | ||
2 Results
2.1 RRT/SC-AFIR
RRT/SC-AFIR is a method to explore the potential energy landscape using a reaction graph, where each node corresponds to an equilibrium structure obtained by density functional theory (DFT) calculations. Initially, there are only two nodes: a start node corresponding to the reactant molecule set and a goal node corresponding to the product molecule set. RRT/SC-AFIR grows the reaction graph by adding nodes and edges to the start node until a given time limit.In algorithmic search, taking balance between exploration (i.e., gathering knowledge with unpurposeful moves) and exploitation (i.e., moving toward the goal) is crucially important.20 If too much weight is put on exploitation, the algorithm is likely to stuck at local minima. Exploration helps it escape from local minima, but too much wandering is also harmful for fast search.
RRT/SC-AFIR takes the balance by alternating two distinctly different steps (Fig. 2). In step A (random node expansion), a node is chosen randomly and an adjacent node is attached to it. To avoid bias, the nodes are clustered using a labeled graph representation called the connection pattern (see Method 4.1). A set of nodes with an identical connection pattern is summarized as a cluster, where labeled graph isomorphism checking is conducted using the NetworkX python library (https://networkx.org/). A cluster is chosen randomly and a node in the cluster is selected in equal probability. An adjacent node is created by single AFIR-path calculations,29 where an artificial force in either the merging or splitting direction is added to two fragments formed around two randomly-selected atoms. For details about fragment formation, see the ESI.† The AFIR-path calculation finds another equilibrium structure by getting beyond the energy barrier, hence a new node. To keep the search in low energy regions, the following probabilistic filter is applied, i.e., the new node is accepted by the following probability:

In step B (goal-oriented expansion), a node close to the goal node is identified in terms of our similarity measure (see Method 4.2). A cluster is chosen according to the probability proportional to exp(csi), where the similarity to a cluster si is defined as the maximum value among the similarities between the goal and its members, and c is a parameter to control the exploration–exploitation balance. Then, a node is chosen according to the probability proportional to exp(czj) where zj denotes the similarity between the goal and a member. Then, an adjacent node is created by AFIR, and the same probabilistic filter as step A is applied.
2.2 Constructing reaction graphs
We constructed reaction graphs for two reactions: Wöhler's urea synthesis (WUS)30 and Fritsch–Buttenberg–Wiechell rearrangement (FBW).31–33 Forty graphs are constructed in parallel with temperature T set to 40 equally spaced values in [5000; 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000]. The combined reaction graph is then created by assembling all reaction graphs by merging near-identical nodes into one. Nodes in the combined graph have time stamps indicating the time point of creation. In all experiments, parameter c is set to 1. The tasks are processed by 96 cores of Intel Xeon Platinum 9242 CPU (2.3 GHz). After RRT/SC-AFIR finished, we investigated time stamps in the combined graph to figure out the goal time and path connecting time. The former refers to the time point that a node nearly identical to the goal node is found, and the latter refers to the time point that a valid reaction path consistent with the known path (Fig. 3) is found. In addition, the number of calculations of potential energy gradients is recorded.
000]. The combined reaction graph is then created by assembling all reaction graphs by merging near-identical nodes into one. Nodes in the combined graph have time stamps indicating the time point of creation. In all experiments, parameter c is set to 1. The tasks are processed by 96 cores of Intel Xeon Platinum 9242 CPU (2.3 GHz). After RRT/SC-AFIR finished, we investigated time stamps in the combined graph to figure out the goal time and path connecting time. The former refers to the time point that a node nearly identical to the goal node is found, and the latter refers to the time point that a valid reaction path consistent with the known path (Fig. 3) is found. In addition, the number of calculations of potential energy gradients is recorded.
Table 1 summarizes the goal time and path connecting time and gradient calculations per minute. In WUS, all the three methods found the goal node and the valid reaction path within a time limit of three days. RRT/SC-AFIR found valid reaction paths much faster than the other methods. Comparing kinetics/SC-AFIR and Boltzmann/SC-AFIR, the former was more efficient in finding the goal node, while the latter was better in finding valid paths. In FBW, only RRT/SC-AFIR found valid paths in time.
| Reaction | Method | Goal time (min) | Path connecting time (min) | Gradient calculations per minute |
|---|---|---|---|---|
| RRT | 67 | 67 | 341.3 | |
| WUS | Kinetics | 66 | 776 | 342.8 |
| Boltzmann | 326 | 549 | 343.8 | |
| RRT | 2575 | 2575 | 126.6 | |
| FBW | Kinetics | — | — | 123.4 |
| Boltzmann | — | — | 123.8 |
Fig. 4 shows the distribution of distances from all nodes of the constructed reaction graph to the start node. The distance between nodes is measured by the length of the shortest path. In both reactions, nodes explored by RRT/SC-AFIR are distributed near the start node, while the distributions of Boltzmann/SC-AFIR and kinetics/SC-AFIR cover broad ranges, indicating that they are likely to produce very long reaction paths. This phenomenon is more evident in FBW. Since known reaction paths shown in Fig. 3 consist of three and two steps for WUS and FBW, respectively, the distribution of RRT/SC-AFIR conforms better with the known paths.
 | ||
| Fig. 4 Distance distribution from the start node in the combined reaction graph. (a) Wöhler's urea synthesis (WUS) and (b) Fritsch–Buttenberg–Wiechell rearrangement (FBW). | ||
Boltzmann/SC-AFIR and kinetic/SC-AFIR perform blind search, where the goal is not given a priori. If most feasible products at the present computational level are identical to experimentally validated products, they would find the goal node quickly, but it is not the case in both WUS and FBW as shown in Fig. 5. Since these products are more stable, Boltzmann/SC-AFIR and kinetics/SC-AFIR search around them, leading to unreasonably long paths. A main reason of this gap would be that solvent effects are ignored in the present computational level. These results indicate that, when products are known, RRT/SC-AFIR has a clear advantage in that it is robust against discrepancies between computation and reality.
 | ||
| Fig. 5 Kinetically most feasible products on the potential energy surface of the present computational level. (a) Wöhler's urea synthesis (WUS) and (b) Fritsch–Buttenberg–Wiechell rearrangement (FBW). | ||
3. Conclusion
In this paper, we showed how reaction path finding can be improved by introducing algorithmic search. RRT/SC-AFIR acts as an integrator of quantum chemical calculations and experimental knowledge: it performs efficient search by pulling together the information of the potential energy surface and that of experimentally validated products. Given the difficulty of improving quantum chemical calculations to perfection, this type of integrative approach is more feasible and should be pursued actively. In using RRT/SC-AFIR in real projects, collaboration with chemists is crucially important. To help chemists’ understanding of the reaction, it would be important to enable easy visual inspection of the whole reaction network. In addition, a more powerful search algorithm would be desirable for making reaction graphs more compact and easily comprehensible. RRT itself can be combined with any automated reaction search methods other than the SC-AFIR method, and it is expected to accelerate extracting the desired reaction path from an entire reaction path network.4 Method
4.1 Connection pattern
A connection pattern is a graph generated from an equilibrium structure. Each node in this graph is an atom labeled by its type (i.e., hydrogen, oxygen, etc.). Let rij denote the distance between atoms i and j, and di and dj denote their covalent radii, respectively. An edge is created if and only if rij≤ λ(di + dj), where λ = 1.1 in this paper.4.2 Similarity measure
As a prerequisite, let us introduce an algorithm to compute the minimum value profile V of a matrix A.(1) Make an empty list V.
(2) Let v be the minimum value v = mini,jaij, and i* and j* be the corresponding row and column indices, respectively.
(3) Attach v to the list V and remove row i* and column j*.
(4) Quit if A is empty. Otherwise go to 2.
Similarly, one can define the maximum value profile as well.
To define a similarity between two structures, an atom type is selected first. Suppose there are n and m atoms of the selected type in the first and second structures of interest, respectively (n ≤ m). Let dik denote the distance between atoms i and k in the same structure. Also, let Cij denote a matrix whose k, l element is the difference between dik in the first structure and djl in the second structure. When Vij is the minimum value profile of Cij, the distance between local environments around atoms i and j is defined as

4.3 Computational details
The SC-AFIR searches are done by GRRM2034 interfaced with Gaussian16,35 using the B3LYP functional, LanL2DZ basis set, EmpiricalDispersion=GD3 option, and Int(Grid=FineGrid) option. The calculations are done in the gas phase.This paragraph describes SC-AFIR options used in all the RRT/SC-AFIR, Boltzmann/SC-AFIR, and kinetics/SC-AFIR searches. The algorithm avoiding Hessian calculations is adopted. The searches using the three different methods are initiated from a common initial structure, i.e., a complex among NH+4 + OCN− + 2(H2O) for WUS and a complex between butyllithium and 1-bromo-2-methylpropane for FBW. In the calculations of WUS, all atoms are set as the target atom of the SC-AFIR method. In the case of FBW, lithium, atoms in the CH2 moiety adjacent to lithium, and all atoms in 1-bromo-2-methylpropane are set as the target atom. The model collision energy parameter γ of the AFIR method is set to 500 kJ mol−1. To prevent a molecule from going too far from the reaction centre, a weak force with γ = 100/[N(N − 1)/2] kJ mol−1 is applied to all atom pairs, where N corresponds to the number of atoms in each system. The force induced paths are optimized by the locally updated planes (LUP) method36 to obtain relaxed paths (LUP paths).
During the Boltzmann/SC-AFIR and kinetics/SC-AFIR searches, node clustering is done using the MatchDecScale = 7:0 option of the GRRM20 program.34 The barrier along the LUP paths is used in the kinetics simulations during the kinetics/SC-AFIR searches. In kinetics/SC-AFIR, the initial population 1:0 is given to the initial structure, and a node from which the path calculation is done next is chosen based on the traffic volume of each node obtained by thermal equilibration of 3600 seconds at 200 K, 300 K, and 400 K, where the traffic volume is an index indicating influx/outflux of population to/from each node during the equilibration.28
RRT/SC-AFIR was developed as a stand-alone external code. The GRRM20 program34 equips SubSelectEQ and SubPathsGen options, and these options allow one to develop an SC-AFIR driver without accessing the internal functions of GRRM. More specifically, when these options are provided as SubSelectEQ=code1.exe and SubPathsGen=code2.exe in the GRRM input file, GRRM calls code1.exe and code2.exe when deciding a node from which the path calculation is done next and when choosing a fragment pair to which the artificial force is applied, respectively, and accepts their decisions. In this study, these options were used to develop RRT/SC-AFIR.
Data availability statement
All GRRM inputs of our calculations and all the obtained EQs are available on Zenodo (https://doi.org/10.5281/zenodo.6321866).Author contributions
M. S. and K. T. conceived the idea and designed the research. A. N. and Y. H. implemented the algorithms and conducted the experiments. A. N., Y. H., M. S. and K. T. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Ryo Tamura and Andrejs Tucs for fruitful discussions. This work was supported by JST ERATO JPMJER1903.Notes and references
- J. I. Steinfeld, J. S. Francisco and W. L. Hase, Chemical kinetics and dynamics, Prentice HallUpper Saddle River 2nd edn 1999 Search PubMed.
- D. Frenkel and B. Smit, Understanding molecular simulation: from algorithms to applications, Elsevier, Amsterdam, 2001, vol. 1 Search PubMed.
- D. Wales, Energy landscapes with application to clusters, biomolecules and glasses, Cambridge University PressCambridge, 2003 Search PubMed.
- S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683 RSC.
- L.-P. Wang, A. Titov, R. McGibbon, F. Liu, V. S. Pande and T. J. Martinez, Nat. Chem., 2014, 6, 1044–1048 CrossRef CAS PubMed.
- Z. W. Ulissi, A. J. Medford, T. Bligaard and J. K. Nørskov, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- A. Rodríguez, R. Rodríguez-Fernández, S. A. Vázquez, G. L. Barnes, J. J. P. Stewart and E. Martínez-Núñez, J. Comput. Chem., 2018, 39, 1922–1930 CrossRef PubMed.
- A. L. Dewyer, A. J. Argüelles and P. M. Zimmerman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1354 Search PubMed.
- Y. Kim, J. W. Kim, Z. Kim and W. Y. Kim, Chem. Sci., 2018, 9, 825–835 RSC.
- C. A. Grambow, A. Jamal, Y.-P. Li, W. H. Green, J. Zádor and Y. V. Suleimanov, J. Am. Chem. Soc., 2018, 140, 1035–1048 CrossRef CAS PubMed.
- G. N. Simm, A. C. Vaucher and M. Reiher, J. Phys. Chem. A, 2019, 123, 385–399 CrossRef CAS PubMed.
- R. Van de Vijver and J. Zador, Comput. Phys. Commun., 2020, 248, 106947 CrossRef CAS.
- S. Maeda, T. Taketsugu and K. Morokuma, J. Comput. Chem., 2014, 35, 166–173 CrossRef CAS PubMed.
- S. Maeda, Y. Harabuchi, M. Takagi, T. Taketsugu and K. Morokuma, Chem. Rec., 2016, 16, 2232–2248 CrossRef CAS PubMed.
- S. Maeda, Y. Harabuchi, M. Takagi, K. Saita, K. Suzuki, T. Ichino, Y. Sumiya, K. Sugiyama and Y. Ono, J. Comput. Chem., 2018, 39, 233–251 CrossRef CAS PubMed.
- S. Maeda and Y. Harabuchi, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, e1538 CAS.
- Y. Sumiya and S. Maeda, Chem. Lett., 2019, 47–50 CrossRef CAS.
- K. Sugiyama, Y. Sumiya, M. Takagi, K. Saita and S. Maeda, Phys. Chem. Chem. Phys., 2019, 21, 14366–14375 RSC.
- T. Mita, Y. Harabuchi and S. Maeda, Chem. Sci., 2020, 11, 7569–7577 RSC.
- S. Russel and P. Norvig, Artificial Intelligence: A Modern Approach, Third Edition, Pearson EducationLondon, 2016 Search PubMed.
- S. Kirkpatrick, C. D. Gelatt and M. P. Vecchi, Science, 1983, 220, 671–680 CrossRef CAS PubMed.
- C. B. Browne, E. Powley, D. Whitehouse, S. M. Lucas, P. I. Cowling, P. Rohlfshagen, S. Tavener, D. Perez, S. Samothrakis and S. Colton, IEEE Trans. Comput. Intell. AI Games, 2012, 4, 1–43 Search PubMed.
- S. M. LaValle Planning algorithms Cambridge university Press Cambridge 2006 Search PubMed.
- I. Al-Bluwi, M. Vaisset, T. Siméon and J. Cortés, BMC Struct. Biol., 2013, 13, 1–14 CrossRef PubMed.
- S. Kirillova, J. Cortés, A. Stefaniu and T. Siméon, Proteins Struct. Funct. Bioinfomat., 2008, 70, 131–143 CrossRef CAS PubMed.
- L. Jaillet, J. Cortés and T. Siméon, IEEE Trans. Robot., 2010, 26, 635–646 Search PubMed.
- A. G. Maldonado, J. Doucet, M. Petitjean and B.-T. Fan, Mol. Divers., 2006, 10, 39–79 CrossRef CAS PubMed.
- Y. Sumiya and S. Maeda, Chem. Lett., 2020, 553–564 CrossRef CAS.
- S. Maeda and K. Morokuma, J. Chem. Theory Comput., 2011, 7, 2335–2345 CrossRef CAS PubMed.
- F. Wöhler, Ann. Phys. Chem., 1828, 87, 253–256 CrossRef.
- P. Fritsch, Justus Liebigs Ann. Chem., 1894, 279, 319–323 CrossRef.
- W. P. Buttenberg, Justus Liebigs Ann. Chem., 1894, 279, 324–337 CrossRef.
- H. Wiechell, Justus Liebigs Ann. Chem., 1894, 279, 337–344 CrossRef.
- S. Maeda, Y. Harabuchi, Y. Sumiya, M. Takagi, K. Suzuki, K. Sugiyama, Y. Ono, M. Hatanaka, Y. Osada, T. Taketsugu, K. Morokuma and K. Ohno, GRRM20, Hokkaido University, Sapporo, Japan, 2020 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams- Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Revision C.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- C. Choi and R. Elber, J. Chem. Phys., 94, 751–760 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01079h |
| This journal is © the Owner Societies 2022 |