
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Collisional energy transfer in the CO–CO system†
Michał
Żółtowski
ab,
Jérôme
Loreau
 c and
François
Lique
*ab
c and
François
Lique
*ab
aLOMC – UMR 6294, CNRS-Université du Havre, 25 rue Philippe Lebon, BP 1123, F-76063 Le Havre, France. E-mail: francois.lique@univ-rennes1.fr
bUniv Rennes, CNRS, IPR (Institut de Physique de Rennes) – UMR 6251, F-35000 Rennes, France
cKU Leuven, Department of Chemistry, B-3001 Leuven, Belgium
First published on 25th April 2022
Abstract
An accurate determination of the physical conditions in astrophysical environments relies on the modeling of molecular spectra. In such environments, densities can be so low (n ≪ 1010 cm−3) that local thermodynamical equilibrium conditions cannot be maintained. Hence, radiative and collisional properties of molecules are needed to correctly model molecular spectra. For comets at large heliocentric distances, the production of carbon monoxide (CO) gas is found to be larger than the production of water, so that molecular excitation will be induced by collisions with CO molecules. This paper presents new scattering calculations for the collisional energy transfer in CO–CO collisions. Using the quantum coupled states approach, cross sections and rate coefficients are provided between the first 37 rotational states of the CO–CO system. Cross sections were calculated for energies up to 800 cm−1, and excitation rate coefficients were derived for temperatures up to 100 K. In comparison with data available in the literature, significant differences were found, especially for the dominant transitions. Due to the high cost of the calculations, we also investigated the possibility of using an alternative statistical approach to extend our calculations both in terms of rotational states and temperatures considered. The use of these new collisional data should help in accurately deriving the physical conditions in CO-dominated comets.
1 Introduction
Comets are valuable sources of information about the evolution of the solar system. Their ice nuclei contain molecules formed at the early stages of planetary formation, and performing spectroscopic observations of the coma, the temporary gaseous atmosphere of a comet, gives insights into the composition of the nucleus. This leads to valuable information about the physical conditions prevailing during planets formation.1 In addition, astronomical models show that various volatile species in Earth's atmosphere, especially noble gases, might have originated from comets.2 Numerous observations of comets have been performed, covering a wide range of wavelengths from ultraviolet, optical, infrared, to radio.1 One of the most significant studies was carried out by the ROSETTA spacecraft, which performed in situ observations of comet 67P/Churyumov–Gerasimenko.3 These observations led to new insights about the composition of cometary ices and atmospheres, which present a huge diversity of molecules including H2O, CO and CO2, CH3OH and CH4.1,4,5Extracting information about the physical conditions and chemical composition of comets, and estimating the abundance of molecules, relies on modeling the observational spectra. The low density conditions in the coma means that the local thermodynamical equilibrium (LTE) is usually not fully achieved, and this modeling requires both radiative and collisional properties of molecules.6,7 While radiative data are analytically available for most of the observed molecules, computationally demanding calculations are required to obtain state-to-state collisional rate coefficients. In cometary atmospheres, H2O, CO, and CO2 are by far the most abundant species, and it is thus crucial to study the mutual collisional excitation of these molecules. In comets, the excitation of molecules is usually dominated by collisions with H2O. Several studies involving collisions with water molecules can be found in the literature, from which the collisional systems of H2O–H2O,8–10 and H2O–CO,7 have the most significant impact for modeling cometary spectra. An important exception is the case of comets at large heliocentric distances, for which the production of gaseous CO is larger than H2O. Hence, the excitation of molecules in the coma of such comets is mainly due to collisions with CO.11–13 Therefore, it is crucial to investigate the excitation of cometary molecules by CO and as a first priority, the mutual interaction of CO molecules to model the physical conditions in these comets.
Rotational energy transfer in CO–CO collisions has been investigated experimentally in a recent study14 that identified unusual pair-correlated excitation mechanisms. Theoretical data for the CO–CO collisional system also exist.15 The calculations were performed by combining the time-independent close-coupling method in the low collisional energy regime with the multi-configuration time-dependent Hartree (MCDTH) approach at higher collisional energies. However, the available rate coefficients only cover transitions from the rotational level j1 = j2 = 0 to levels with  < 4, j1 and j2 being the rotational states of the two colliders. Our goal in the present work is to improve existing data by performing time-independent quantum calculations for rotational energy levels of CO up to, possibly, j1 = j2 = 10 that can be used in modeling of cometary atmospheres.
< 4, j1 and j2 being the rotational states of the two colliders. Our goal in the present work is to improve existing data by performing time-independent quantum calculations for rotational energy levels of CO up to, possibly, j1 = j2 = 10 that can be used in modeling of cometary atmospheres.
In this paper, we present cross sections and rate coefficients for transitions between the first 37 rotational states of the CO–CO system (e.g. states with rotational levels up j1 and j2 ≤ 6, for temperatures up to 100 K). In addition, we explore the applicability of a statistical approach to treat such collisional system based on the statistical adiabatic channel method,16 to obtain data for rotational levels above j1 = j2 = 10. This method was tested and compared with exact quantum calculations and showed excellent agreement especially in low temperatures regime.16–18
Our paper is organized as follows: In Section 2, we present the methodology of our work. Section 3 discusses our results using quantum-mechanical and statistical approaches to scattering calculations. In Section 4, we discuss the implications of our work and summarize our results.
2 Methodology
2.1 Potential energy surface
In addition of the six-dimensional (6D) potential energy surface (PES) of Chen et al.,19 two accurate 4D CO–CO PESs are available in the literature. The most recent PES20 was calculated a few years ago by means of the coupled-cluster approach, and has been tested versus experimental spectroscopic studies,21 showing its high accuracy. However, it is not suitable for low-energy collisional excitation calculations since the long-range (R > 40 a0) part of the potential is missing. We therefore decided to use the PES calculated by Vissers et al.22 This PES was calculated at the coupled-cluster single double and perturbative triples [CCSD(T)] level of theory, with the augmented correlation-consistent triple zeta basis (aug-cc-pVTZ). The CO molecules were treated as rigid rotors with the CO bond length fixed at 2.132 a0. Within the rigid rotor approximation, 4 coordinates (R, θ1, θ2, ϕ) are needed to describe this system. R is the length of the vector R connecting the centers of mass of the two CO monomers, θ1 and θ2 are the polar angles relative to R, and the last angle ϕ is a dihedral angle between the half-planes that contain both CO molecules. Two minima in the PES were found: the first one, which is a local minimum, at R = 6.95 a0, θ1 = 59.63°, θ2 = 120.37° and ϕ = 180.0° with a well depth of V = −124.21 cm−1, and the second one, which is the global minimum of the PES at R = 8.20 a0, θ1 = −134.23°, θ2 = 45.77° and φ = 180.0° with V = −135.53 cm−1. The minima are separated by an energetic barrier which is 72.6 cm−1 higher than the global minimum. The long range part of the PES has been obtained from an extrapolation of the expansion coefficients assuming a Cn/R−n behavior. The validity of the extrapolation has been verified by checking that the coefficients have physical meaning. The PES of Vissers et al.22 was also benchmarked against experimental studies14,21 that demonstrated its high accuracy.2.2 Scattering calculations
All calculations presented in this work were performed using the MOLSCAT (version 14)23 scattering code. =
=  ) than what they should be, and when using the coupled states (CS)25 approach, the cross sections for all transitions randomly overestimate or underestimate the actual results. This would not be a real issue if the CC calculations were feasible since the results can easily be corrected from the double counting.26 However, as will be shown in the following sections, the CC calculations were too CPU consuming and we had to use the approximate CS approach for the calculations.
) than what they should be, and when using the coupled states (CS)25 approach, the cross sections for all transitions randomly overestimate or underestimate the actual results. This would not be a real issue if the CC calculations were feasible since the results can easily be corrected from the double counting.26 However, as will be shown in the following sections, the CC calculations were too CPU consuming and we had to use the approximate CS approach for the calculations.
Hence, in our calculations, we considered both CO molecules as distinguishable molecules, one being the target and one being the projectile. Such an approach is also well suited to astrophysical applications since, in radiative transfer calculations, the notion of colliders and targets is necessarily invoked. Approximate conversion from distinguishable to undistinguishable results will be presented in Section 3.2.
 transitions obtained with a rotational basis that include all levels up to j1 = j2 = jmax
transitions obtained with a rotational basis that include all levels up to j1 = j2 = jmax
| Transition | j max = 7 | j max = 8 | j max = 9 | j max = 10 | j max = 11 |
|---|---|---|---|---|---|
| 00 → 11 | 41.08 | 35.65 | 36.21 | 35.29 | 35.24 |
| 00 → 01 | 40.06 | 36.30 | 30.60 | 29.24 | 28.93 |
| 01 → 11 | 85.24 | 80.16 | 80.86 | 80.49 | 80.23 |
| 02 → 01 | 36.39 | 34.46 | 35.32 | 36.04 | 35.80 |
Fig. 1 presents additional tests performed at total energies of 100 and 500 cm−1. It displays selected cross sections as a function of increasing rotational basis for J = 0. As one can see, at 100 cm−1, the basis set has to include j1 = j2 = 11 for the cross sections to be converged. At 500 cm−1, rotational levels up to j1 = j2 = 15 have to be included in order to numerically converge calculations. Such a rotational basis leads to 2736 coupled channels‡ and would lead to ∼45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 coupled channels for J ≥ 15. With so many coupled channels, calculations using the (almost) exact CC approach are not feasible for large values of J both in terms of memory and CPU time.
000 coupled channels for J ≥ 15. With so many coupled channels, calculations using the (almost) exact CC approach are not feasible for large values of J both in terms of memory and CPU time.
 | ||
| Fig. 1 Excitation cross sections for J = 0 computed at 100 cm−1 (upper panel) and 500 cm−1 (lower panel) as function of the rotational basis. The jmax is equal to highest j1 and j2 levels included in the calculations. Numbers on the plot indicate the total number of channels including in the calculations. | ||
Hence, we explore the possibility of using the CS approximation. In order to evaluate the performance of the CS approach compared to CC, we computed excitation cross sections with a limited basis set containing all rotational levels up to jmax = 7, expecting that the truncation of the rotational basis would have the same effect on CC and CS results. Fig. 2 presents the CC cross sections as a function of the CS ones for selected values of total energy (20, 50, 100, and 150 cm−1). In the low energy regime, where numerous resonances are found, the differences between CC and CS are below a factor of 1.5–2. When the energy increases, as expected, the differences decreases so that the overall agreement is good for energies above 50 cm−1. Such comparison indicates that the CS approach is a reasonable alternative for the CC one in the case of CO–CO collisions. For temperatures below 50 K, the estimated accuracy of the rate coefficients obtained from the CS approach is expected to be better than a factor of 2 and this accuracy is expected to increase with increasing temperature.
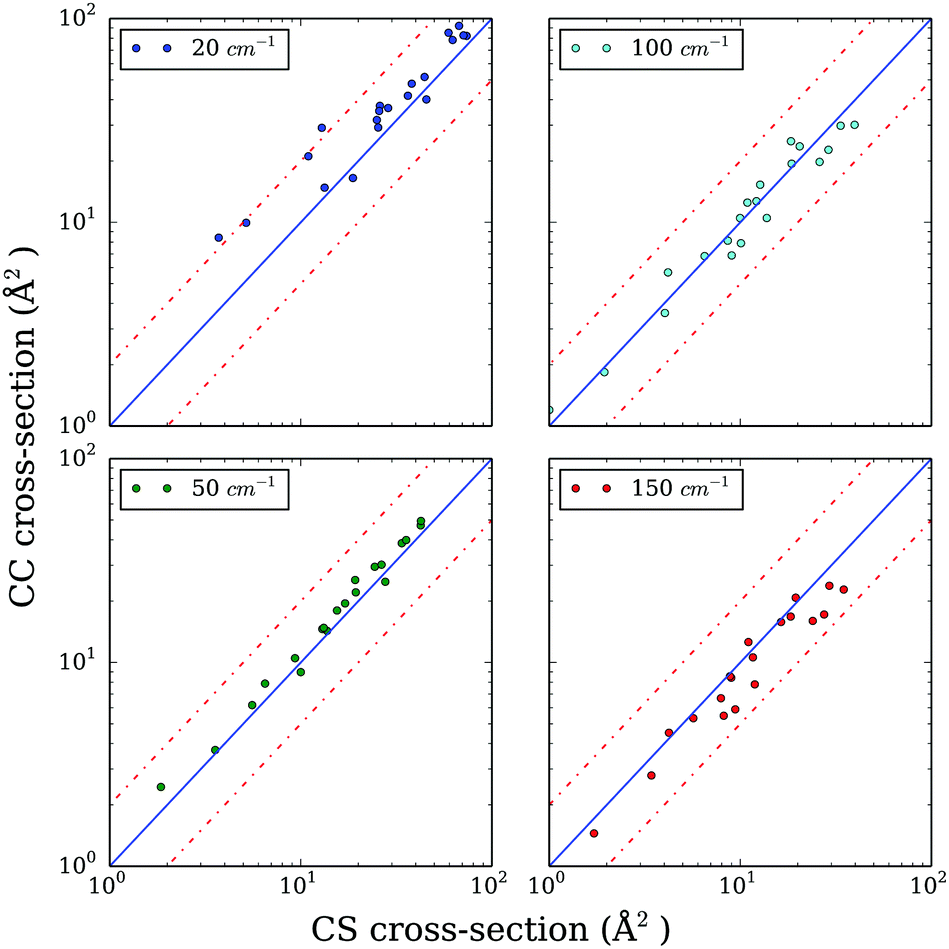 | ||
| Fig. 2 Systematic comparison of selected CS and CC cross sections at 20, 50, 100 and 150 cm−1. The dashed lines represent a factor of 2 of difference. | ||
The coupled equations were then solved using the CS approximation with the log-derivative Airy propagator.27 The STEPS parameter of MOLSCAT was set at 20 in order to obtain a step length of the integrator sufficient to achieve the convergence. The integration was performed for distances between Rmin = 5 a0 and Rmax = 50 a0. The rotational constants of the CO molecules were taken as: Be = 1.931 cm−1, αe = 1.750 × 10−2 cm−1, De = 6.121 × 10−6 cm−1.28 The reduced mass was set at μ = 13.997 u. Excitation cross sections were obtained between rotational states listed in Table 2. The following energy grid was used: in 0–200 cm−1 energy range, we used steps of 1 cm−1, in the 200–300 cm−1 energy range, the energy step was increased to 5 cm−1, and finally in the 300–800 cm−1 energy range, a step of 100 cm−1 was used. The number of total angular momentum J needed to converge calculations vary from 35, at low energy, to 130, at the highest energies. From the computed cross sections, we obtain rate coefficients for temperatures up to 100 K using the following the formula:
 | (1) |
 is the cross section and Ec is collision energy.
is the cross section and Ec is collision energy.
| Level | j 1 j 2 | Energy (cm−1) | Level | j 1 j 2 | Energy (cm−1) |
|---|---|---|---|---|---|
| 1 | 00 | 0.000 | 20 | 35 | 80.739 |
| 2 | 01 | 3.845 | 21 | 16 | 84.580 |
| 3 | 11 | 7.690 | 22 | 26 | 92.270 |
| 4 | 02 | 11.535 | 23 | 45 | 96.118 |
| 5 | 12 | 15.379 | 24 | 36 | 103.805 |
| 6 | 03 | 23.069 | 25 | 07 | 107.642 |
| 7 | 22 | 23.070 | 26 | 17 | 111.487 |
| 8 | 13 | 26.914 | 27 | 55 | 115.341 |
| 9 | 23 | 34.604 | 28 | 27 | 119.177 |
| 10 | 04 | 38.448 | 29 | 46 | 119.186 |
| 11 | 14 | 42.293 | 30 | 37 | 130.712 |
| 12 | 33 | 46.139 | 31 | 08 | 138.390 |
| 13 | 24 | 49.983 | 32 | 56 | 138.406 |
| 14 | 05 | 57.670 | 33 | 18 | 142.235 |
| 15 | 15 | 61.515 | 34 | 47 | 146.091 |
| 16 | 34 | 61.517 | 35 | 28 | 149.925 |
| 17 | 25 | 69.205 | 36 | 38 | 161.460 |
| 18 | 44 | 76.896 | 37 | 66 | 161.471 |
| 19 | 06 | 80.735 |
3 Results and discussion
Using the methodology described above, we computed state-to-state excitation cross sections for CO–CO collisions. The calculations were performed for energies up to 800 cm−1 and cover transitions between levels up to j1 = j2 = 6. Collisional rate coefficients up to 100 K were derived from these cross sections. Collisional rate coefficients are available as ESI.† All the results presented below have been obtained considering the two CO molecules as distinguishable.3.1 Propensity rules
In Fig. 3, we present selected CO–CO cross sections corresponding to the excitation from the ground state j1j2 = 00 to rotational states in which only one CO molecule is excited. As one can see, the dominant transition is the transition to the = 01 state. The values of the cross sections decrease when
= 01 state. The values of the cross sections decrease when  increases, in agreement with an exponential energy-gap behavior. Obviously, the same propensity rules can be seen when we compare rate coefficients (Fig. 3, lower panel). A similar propensity rule can also be seen when both CO molecules are excited after the collision. Indeed, as one can see on Fig. 4 that presents selected excitation cross sections and rate coefficients from the fundamental j1j2 = 00 state, the dominant transition is the one corresponding to the excitation to the
increases, in agreement with an exponential energy-gap behavior. Obviously, the same propensity rules can be seen when we compare rate coefficients (Fig. 3, lower panel). A similar propensity rule can also be seen when both CO molecules are excited after the collision. Indeed, as one can see on Fig. 4 that presents selected excitation cross sections and rate coefficients from the fundamental j1j2 = 00 state, the dominant transition is the one corresponding to the excitation to the  = 11 state. More generally, the cross sections decrease with the increase of both
= 11 state. More generally, the cross sections decrease with the increase of both  and
and  final levels.
final levels.
 | ||
| Fig. 3 Excitation cross sections as a function of collision energy (upper panel) and the rate coefficients as a function of temperature (lower panel) from the j1j2 = 00 state. | ||
 | ||
| Fig. 4 Excitation cross sections as a function of collision energy (upper panel) and the rate coefficients as a function of temperature (lower panel) from the j1j2 = 00 state. | ||
In Fig. 5, we compare the cross sections and rate coefficients for transitions where only one CO molecule is excited to the ones for transitions where both CO molecules are excited. In contrast to previous findings reported by Ndengué et al.,15 we do not observe propensity rules in favor of pair–pair transitions (j1 = j2 and  ). We found that the magnitude of the cross sections involving excitation of only one CO molecule is close to the one of the cross sections where both CO molecules are excited.
). We found that the magnitude of the cross sections involving excitation of only one CO molecule is close to the one of the cross sections where both CO molecules are excited.
 | ||
| Fig. 5 Excitation cross sections as a function of collision energy (upper panel) and rate coefficients as a function of temperature (lower panel) from level j1j2 = 00. The presented transitions show comparisons of transitions where one of the CO molecules is excited with transitions where both of them are excited. | ||
Propensity rules similar to those reported in our work were observed in previous studies of collisions of two identical molecules. For example, in H2–H2 scattering, the cross sections were found to be larger for transitions where only one molecule is excited compared to the transitions where both colliders are excited.29
3.2 Comparison with previous data
As stated in the introduction, theoretical data for the CO–CO collisional system have been published by Ndengué et al.15 It is then of interest to compare our new data with these existing ones. Ndengué et al.15 performed scattering calculations using the PES of Dawes et al.20 The authors combined time-independent quantum CC calculations for energy up to 150 cm−1, with MCTDH calculations for energies above 150 cm−1. In the CC calculations, they included in the rotational basis set CO levels up to j1 = j2 = 7. Using the MCTDH method, the basis was substantially increased due to the lower computational cost. The calculations of Ndengué et al.15 were performed treating the CO molecules as undistinguishable particles. We do not focus here on the differences between collisional cross sections above 150 cm−1 that are most probably due to different scattering methods (quantum time dependent vs. quantum time independent) and that weakly affect the collisional rate coefficients below 100 K.
Fig. 6 and 7 present a comparison of our new collisional data (both the cross sections and rate coefficients) to the ones of Ndengué et al.15 for two selected transitions. Since Ndengué et al.15 considered scattering between undistinguishable particles and performed the calculations with the MOLSCAT code, we also plotted their results for pair–pair transitions divided by two (e.g. discussion in Section 2.2.1). For the purpose of the comparison with Ndengué et al.15 results, we also converted our distinguishable results to undistinguishable ones using the following formulae, that are not strictly exact and based on the assumption that quantum interference effects are negligible30,31§
 | ||
Fig. 6 Comparison between present excitation cross sections as a function of energy and those of Ndengué et al.15 (upper panel) and rate coefficients as a function of temperature (lower panel) for the j1j2 = 00 to  = 01 transition. = 01 transition. | ||
 | ||
Fig. 7 Comparison between present and Ndengué et al.15 excitation cross sections as a function of energy (upper panel) and rate coefficients as a function of temperature (lower panel) for the j1j2 = 00 to 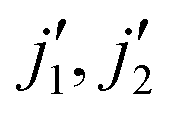 = 11 transition. The solid blue line represents results directly taken from Ndengué et al.,15 the dashed blue line represents the same results divided by a factor of 2, and the red solid line represents our results. = 11 transition. The solid blue line represents results directly taken from Ndengué et al.,15 the dashed blue line represents the same results divided by a factor of 2, and the red solid line represents our results. | ||
• Pair–pair transitions (j1 = j2 and  )
)
τu(j1j2 →  ) = τd(j1j2 → ) = τd(j1j2 →  ) ) | (2) |
• Pair–no-pair transitions (j1 = j2 and  )
)
τu(j1j2 →  ) = τd(j1j2 → ) = τd(j1j2 →  ) + τd(j1j2 → ) + τd(j1j2 →  ) ) | (3) |
• No-pair–pair transitions (j1 ≠ j2 and  )
)
τu(j1j2 →  ) = τd(j1j2 → ) = τd(j1j2 →  ) ) | (4) |
• No-pair–no-pair transitions (j1 ≠ j2 and  )
)
τu(j1j2 →  ) = τd(j1j2 → ) = τd(j1j2 →  ) + τd(j1j2 → ) + τd(j1j2 →  ) ) | (5) |
As one can see, differences between the two sets of data are significant (even when corrected for the possible IDENT MOLSCAT parameter issue). Such differences originate from the different approaches (different CO–CO PESs, different scattering theory and different calculations parameters) used in the scattering studies. It is then of interest to assess the importance of all these different computational aspects in the overall discrepancy between the two sets of data.
The differences induced from the use of different PESs is found to be minor. Indeed, we performed test calculations using the Vissers et al.22 PES and calculations parameters reported by Ndengué et al.15 (for energies up to 50 cm−1) and we found that the differences were lower than 10% on the average despite the fact that the resonances seen in the excitation cross sections were slightly shifted. We expect that the difference would be even lower in the case of rate coefficients, where the cross sections are averaged over a thermal distribution of collisional energies.
The impact of the use of the CS vs. CC scattering approach was evaluated in the Section 2. Indeed, Fig. 2 showed that the CC and CS results can differ by up to a factor of 1.5–2 at low collision energies as can be observed when comparing our results to the results from Ndengué et al.15 (Fig. 6 and 7). However, it was also found that the agreement between CC and CS results increases with increasing collision energies. Such an improvement is not seen here.
This can probably be explained this by the size of the rotational basis set used in the two quantum time independent calculations. Ndengué et al.15 used a rotational basis containing all levels with j1 and j2 up to 7 that clearly does not allow full convergence of the collisional cross sections (e.g.Table 1), the non convergence of the results obviously increasing with increasing energies. Indeed, in order to fully converge our scattering calculations, we used basis j1 = j2 = 15 and a significant part of the difference between the two sets of data above 100 cm−1 can be explained by the lack of convergence of the Ndengué et al.15 calculations with respect to the rotational basis.
3.3 Statistical approach
As we reported, the accuracy of our data for temperatures below 50 K is within a factor of 1.5–2. The pure CC calculations being not feasible, alternative approaches were therefore explored in order to improve accuracy. We decided to investigate the validity of statistical methods to check whether the accuracy of our rate coefficients could be improved in the low temperatures regime and whether the calculations could be extended to transitions involving higher rotational levels. We used the Statistical Adiabatic Chanel Model (SACM) presented in the work of Loreau et al.16,32 The significant advantage of this method is that the calculations are performed only for one value of the energy since the adiabatic curves are energy-independent. In addition, we could use a much smaller rotational basis set to reach convergence of the cross sections (jmax = 6 was enough to obtain converged calculations for transitions between levels up to j1 = j2 = 5).We calculated cross sections for transitions between rotational levels up to j1 = j2 = 5, and rate coefficients for temperatures up to 100 K. In Fig. 8, we show a systematic comparison between rate coefficients obtained using SACM and CS methods. Previous use of the SACM method showed that it works well at low temperatures and starts to deviate from accurate results when the temperature increases.16 However, in our case, the opposite behavior is observed. As one can see from Fig. 8, at 20 K, the differences between the two sets of data are above a factor of 3 for numerous transitions while the agreement is increasing with increasing temperature.
 | ||
| Fig. 8 Systematic comparison of SACM and CS rate coefficients, at temperatures of 20, 50, and 100 K, for all transitions calculated in this work (using the CS method). The blue line represents exact results, red dashed lines represent a factor of 2 difference, and black dashed lines represent a factor of 3 difference. | ||
We have to keep in mind that we compare here two approximate methods. Therefore, we calculated cross sections for a few energies using the almost exact CC method including in the rotational basis levels up to j1 = j2 ≤ 10. Fig. 9 and 10 show an example of comparison of the CS, SACM and the CC cross sections. Generally, we observed that the results obtained with the CS method underestimate the cross sections, while the SACM results slightly overestimate the cross sections compared to the CC ones in the low energy regime. However, both the CS and SACM results stays within a factor of 2 compared to the CC results; therefore, we cannot clearly determine which method is more accurate. In the intermediate region of energy (30 ≤ Ec ≤ 70 cm−1), all three methods agrees reasonably well, thereby explaining the fairly good agreement seen in Fig. 8 for temperatures of 50 and 100 K. Important deviations start to occur at higher energies. Indeed, we observe a substantial decrease in the cross sections obtained with the SACM method, while the CC and CS results stay in good agreement. We presume that this decrease of cross sections is due to the dense distribution of the accessible (open) channels and also to the fact that the well depth of the CO–CO PES is not large enough for statistical approaches to work above the very low energy regime.16
 | ||
Fig. 9 Comparison of the cross section obtained with the CC, CS and statistical method for the transition j1j2 = 00 to  = 01 as a function of energy. = 01 as a function of energy. | ||
 | ||
Fig. 10 Comparison of the cross section obtained with the CC, CS and statistical method for the transition j1j2 = 00 to  = 11 as a function of energy. = 11 as a function of energy. | ||
We conclude that the rate coefficients obtained with the SACM method may be useful for temperatures up to 100 K. The good agreement between SACM and CS rate coefficients observed at a temperature of 100 K is rather fortuitous and difference above 100 K can become very significant because of the strong deviation between quantum and SACM results at high collisional energies. Therefore, we are not able to extend the rate coefficients to higher temperatures and hence higher rotational levels (populated only at high temperature) using the SACM method.
4 Conclusions
Using the potential of Vissers et al.,22 we presented the first complete quantum scattering calculations of CO–CO rotational excitation between first 37 rotational energy states of CO molecules. We obtained state-to-state collisional rate coefficients for temperatures up to 100 K. We estimated that our data below 50 K are accurate within a factor of 1.5–2 and that the accuracy increases with increasing temperature.Significant differences were found between our results and those previously reported by Ndengué et al.15 Transitions towards “pair” rotational levels (with j1 = j2) seem to be overestimated by a factor 2. For temperatures above 50 K, we believe that our results are more accurate than the previous ones. Our convergence tests show that the basis used in the previous study was insufficient. At the same time, we are confident that the basis size used in our work allows us to converge calculations over the whole energy regime considered. The question about the accuracy of the rate coefficients below 50 K remain. Even though the previous calculations were performed using the CC method, their results were only partially converged due to the small basis set. On the contrary, our calculations were converged. However, we used the CS approximation, which was estimated to provide accurate results within a factor of 2 difference.
We tried to improve our data by investigating the possibility of the statistical approach to the CO–CO scattering. The results we obtained do not lead to a clear answer. The agreement was excellent for part of the transitions in the low energy regime. For other transitions, differences of a factor of ∼2 were observed between SACM and partly converged CC calculations. In addition, for all the considered transitions, we observed a sudden decrease of cross section above 60–70 cm−1. This behavior can be expected when the collision energy becomes comparable to the well depth of the PES, but further investigation on similar systems would be needed.
The CS calculations will be continued using the methodology reported in this work. We will extend data for transitions between levels up to j1 = j2 = 10. The impact of our data on the astrophysical models, in particular of cometary atmospheres, will be presented in a separate article.
Data availability
The data that support the findings of this study are available within the article and its online ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. L. and M. Ż. acknowledge financial support from the European Research Council (Consolidator Grant COLLEXISM, grant agreement 811363), the Institut Universitaire de France and the Programme National “Physique et Chimie du Milieu Interstellaire” (PCMI) of CNRS/INSU with INC/INP co-funded by CEA and CNES. We wish to acknowledge the support from the CINES/GENCI for awarding us access to the OCCIGEN supercomputer within the A0090411036 project. M. Ż. acknowledges that calculations have been carried out at the Wroclaw Centre for Networking and Supercomputing, Grant 218 (PSZ). M. Ż. thanks B. Desrousseaux, C. Bop, S. Demes, A. Godard Palluet and P. Pirlot for fruitful discussions. J. L. acknowledges support from Internal Funds KU Leuven through Grant 19-00313. M. Ż. acknowledges B. Desrousseaux, A. Godard-Palluet, P. Pirlot, C. Bop, S. Demesand and M. Gueguen for their help and support during this work.Notes and references
- A. Cochran, A.-C. Levasseur-Regourd, M. Cordiner, E. Hadamcik, J. Lasue, A. Gicquel, D. Schleicher, S. Charnley, M. Mumma, L. Paganini, D. Bockelee-Morvan, N. Biver and Y.-J. Kuan, Space Sci. Rev., 2015, 197, 9–46 CrossRef CAS.
- B. Marty, K. Altwegg, H. Balsiger, A. Bar-Nun, D. V. Bekaert, J.-J. Berthelier, A. Bieler, C. Briois, U. Calmonte, M. Combi, J. D. Keyser, B. Fiethe, S. A. Fuselier, S. Gasc, T. I. Gombosi, K. C. Hansen, M. Hässig, A. Jäckel, E. Kopp, A. Korth, L. L. Roy, U. Mall, O. Mousis, T. Owen, H. Rème, M. Rubin, T. Sémon, C.-Y. Tzou, J. H. Waite and P. Wurz, Science, 2017, 356, 1069–1072 CrossRef CAS PubMed.
- K.-H. Glassmeier, H. Boehnhardt, D. Koschny, E. Kührt and I. Richter, Space Sci. Rev., 2007, 128, 1–21 CrossRef.
- M. J. Mumma and S. B. Charnley, Annu. Rev. Astron. Astrophys., 2011, 49, 471–524 CrossRef CAS.
- D. Bockelée-Morvan and N. Biver, Philos. Trans. R. Soc., A, 2017, 375, 20160252 CrossRef PubMed.
- E. Roueff and F. Lique, Chem. Rev., 2013, 113, 8906–8938 CrossRef CAS PubMed.
- A. Faure, F. Lique and J. Loreau, Mon. Not. R. Astron. Soc., 2020, 493, 776–782 CrossRef CAS.
- C. Boursier, B. Mandal, D. Babikov and M. L. Dubernet, Mon. Not. R. Astron. Soc., 2020, 498, 5489–5497 CrossRef CAS.
- G. Buffa, O. Tarrini, F. Scappini and C. Cecchi-Pestellini, Astrophys. J., Suppl. Ser., 2000, 128, 597–601 CrossRef CAS.
- A. Semenov and D. Babikov, J. Phys. Chem. A, 2017, 121, 4855–4867 CrossRef CAS PubMed.
- D. Bockelée-Morvan, J. Crovisier, M. J. Mumma and H. Weaver, in Comets II, ed. M. Festou, H. U. Keller and H. A. Weaver, Univ. Arizona Press, Tucson, 2005, pp. 391–423 Search PubMed.
- D. Bodewits, J. W. Noonan, P. D. Feldman, M. T. Bannister, D. Farnocchia, W. M. Harris, J.-Y. Li, K. E. Mandt, J. W. Parker and Z.-X. Xing, Nat. Astron., 2020, 4, 867–871 CrossRef.
- M. A. Cordiner, S. N. Milam, N. Biver, D. Bockelée-Morvan, N. X. Roth, E. A. Bergin, E. Jehin, A. J. Remijan, S. B. Charnley, M. J. Mumma, J. Boissier, J. Crovisier, L. Paganini, Y.-J. Kuan and D. C. Lis, Nat. Astron., 2020, 4, 861–866 CrossRef.
- Z.-F. Sun, M. C. van Hemert, J. Loreau, A. van der Avoird, A. G. Suits and D. H. Parker, Science, 2020, 369, 307 CrossRef CAS PubMed.
- S. A. Ndengué, R. Dawes and F. Gatti, J. Phys. Chem. A, 2015, 119, 7712–7723 CrossRef PubMed.
- J. Loreau, F. Lique and A. Faure, Astrophys. J., 2018, 853, L5 CrossRef.
- M. Konings, B. Desrousseaux, F. Lique and J. Loreau, J. Chem. Phys., 2021, 155, 104302 CrossRef CAS PubMed.
- B. Desrousseaux, M. Konings, J. Loreau and F. Lique, Phys. Chem. Chem. Phys., 2021, 23, 19202–19208 RSC.
- J. Chen, J. Li, J. M. Bowman and H. Guo, J. Chem. Phys., 2020, 153, 054310 CrossRef CAS PubMed.
- R. Dawes, X.-G. Wang and T. Carrington, J. Phys. Chem. A, 2013, 117, 7612–7630 CrossRef CAS PubMed.
- L. A. Surin, D. N. Fourzikov, T. F. Giesen, S. Schlemmer, G. Winnewisser, V. A. Panfilov, B. S. Dumesh, G. W. M. Vissers and A. van der Avoird, J. Phys. Chem. A, 2007, 111, 12238–12247 CrossRef CAS PubMed.
- G. W. M. Vissers, P. E. S. Wormer and A. van der Avoird, Phys. Chem. Chem. Phys., 2003, 5, 4767–4771 RSC.
- J. Hutson and S. Green, CCP6, Daresbury, 1994.
- A. M. Arthurs, A. Dalgarno and D. R. Bates, Proc. R. Soc. London, Ser. A, 1960, 256, 540–551 Search PubMed.
- P. McGuire and D. J. Kouri, J. Chem. Phys., 1974, 60, 2488–2499 CrossRef CAS.
- J. P. Fonfria, A. Ramos, F. Thibault, G. Tejeda, J. M. Fernandez and S. Montero, J. Chem. Phys., 2007, 127, 134305 CrossRef CAS PubMed.
- M. H. Alexander and D. E. Manolopoulos, J. Chem. Phys., 1987, 86, 2044–2050 CrossRef CAS.
- A. L. Floch, Mol. Phys., 1991, 72, 133–144 CrossRef.
- S. Green, J. Chem. Phys., 1975, 62, 2271–2277 CrossRef CAS.
- W. M. Huo and S. Green, J. Chem. Phys., 1996, 104, 7572–7589 CrossRef CAS.
- J. Perez-Rios, G. Tejeda, J. M. Fernandez, M. I. Hernandez and S. Montero, J. Chem. Phys., 2011, 134, 174307 CrossRef CAS PubMed.
- J. Loreau, A. Faure and F. Lique, J. Chem. Phys., 2018, 148, 244308 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01065h |
| ‡ Considering the exchange symmetry, the numbers of channels would still be 1496. |
| § Note that there is not a clear convergence of the conversion factor to be used (e.g. discussion in Perez-Rios et al.31) and we do not recommend to use our estimated undistinguishable observables for experiments analysis. |
| This journal is © the Owner Societies 2022 |