
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfrared analysis of catalytic CO2 reduction in hydrogenated germanium
Thierry
de Vrijer
 * and
Arno H. M.
Smets
* and
Arno H. M.
Smets
Photovoltaic Materials and Devices, TU Delft, Mekelweg 4, Delft 2628CD, The Netherlands Photovoltaic Materials and Devices, TU Delft, Mekelweg 4, Delft 2628CD, The Netherlands. E-mail: t.devrijer@tudelft.nl
First published on 5th April 2022
Abstract
The oxidation and carbisation kinetics of porous amorphous and nano-crystalline hydrogenated germanium (a-Ge:H and nc-Ge:H) films exposed to ambient air and deionized water have been studied using vibration modes observed by infrared spectroscopy. Based on infrared analysis, a two-step process of first oxidation by water and secondly carbisation by carbon dioxide (CO2) is proposed that partly mimics the (photo-)catalytic processes in artificial (photo)synthesis. It is shown that water acts like the precursor for oxidation of porous a-Ge:H and nc-Ge:H in the first step. The incorporation of oxygen in a-Ge:H and nc-Ge:H alloys occurs preferentially at Ge-dangling bonds and not at the Ge–Ge back bonds like in hydrogenated silicon alloys (next of kin IV-valence element). The formation of germanium oxide (GeO) tissue at void surfaces locally creates Ge alloys with significantly lower energy levels for the valence band that can align with the half reaction for water reduction. The heterogeneous nature of a-Ge:H and nc-Ge:H oxidation will result in local catalytic generation of electrons and protons. It is proposed that these charge carriers and ions act as precursors for the second-step reaction based on carbisation that includes both the adsorption of CO2 and formation of CO and formaldehyde.
1 Introduction
The Intergovernmental Panel on Climate Change Climate Report 2021 shows that at the end of this century, negative CO2 emissions are required to limit the increase in global temperature to 1.5 °C with reference to the 1850–1900 period.1 The electrochemical reduction of CO2 will likely play an essential role in achieving these negative emissions, as it allows for the synthesis of carbon-based fuels using clean renewable energy and the greenhouse gas CO2 as a feedstock, where traditionally nano-structured metals are used to drive the CO2 reduction reaction (CO2RR).2 Recently, considerable effort has been devoted to the development of heterogeneous catalysts based on earth-abundant materials.2–5The group IV element germanium is such an (relatively) earth-abundant material. The elemental abundance of Ge is ≈1.8 ppm in the earth's crust,6 which makes it more abundant than known conventional catalyst materials (Au, Pt, and Pd) and noble metal complex-based molecular catalysts (Ru, Ir, and Re), although not as abundant as elements used for novel metal complexes (Co, Ni, Fe, Mn, and Cu).3 In this work, infrared evidence is presented based on which the electrochemical reduction of CO2 to CO, formic acid and formaldehyde on hydrogenated (:H) germanium films is proposed. The Ge:H films are processed using plasma-enhanced chemical vapour deposition (PECVD), a technique that is relatively fast, not energy intensive and compatible with the processing of photovoltaic devices. This potentially facilitates the use of Ge:H as nano-particles in solution, or as a photo-electrode in integrated photo-electrochemical or alternative photovoltaic-electrochemical applications.7–9
The post-deposition oxidation by water, and carbisation by CO2, of hydrogenated germanium films are characterized and potential pathways are proposed for the electrochemical reduction of CO2 on Ge:H films. Fundamental insight into the reaction rate and type of reaction are provided by monitoring Fourier transform infrared spectroscopy (FTIR) spectra of Ge:H films stored in ambient conditions and in deionized (DI) water, of amorphous (a-) and nano-crystalline (nc-) samples, of unalloyed and alloyed Ge(Six,Ox):H samples and of Ge:H samples of different material porosity and chemical stability.
2 Experimental section
The films presented in this work are processed on 4 inch, 500 μm thick monocrystalline silicon wafers. The a/nc-Ge:H films were processed in a cascade radio-frequency PECVD reactor that has a circular electrode with a diameter of 160 mm and an electrode spacing of 20 mm. The cascade radio-frequency PECVD reactor is a laminar flow reactor, where germane (GeH4) and molecular hydrogen are used as precursor gases. The a/nc-Six Ge:H and a-GeOx:H films were processed in a different radio-frequency PECVD cluster tool, with a flat 12 cm × 12 cm shower-head electrode. The deposition conditions of the processed films, including deposition times and thickness, are reported in Table 1. After deposition, the wafers were cut into quarter pieces to measure the FTIR spectra as the Ge:H films evolve over time under two storage conditions; in the dark and in DI water.| P RF (mW cm−2) | p (mbar) | T S (°C) | F CO2 or FSiH4 (sccm) | F GeH4 (sccm) | Thickness (nm) | |
|---|---|---|---|---|---|---|
| a-SixGe:H | 20.8 | 3.6 | 180 | 30 | 5.3 | 292.2 |
| nc-SixGe:H | 20.8 | 3.6 | 180 | 30 | 0.4 | 148.1 |
| nc-Ge:H | 99.5 | 3.5 | 200 | 0.5 | 222.6 | |
| a-Ge:H | 24.9 | 4 | 200 | 0.5 | 182.6 | |
| a-GeOx:H | 20.8 | 3.6 | 180 | 20 | 2 | 173.8 |
| a-Ge:H-1 | 49.8 | 5 | 280 | 0.5 | 121.4 | |
| a-Ge:H-2 | 49.8 | 5 | 250 | 0.5 | 85.5 | |
| a-Ge:H-3 | 14.9 | 4 | 290 | 2 | 174.5 | |
| a-Ge:H-4 | 49.8 | 5 | 275 | 1 | 185.6 | |
| a-Ge:H-5 | 74.6 | 4 | 200 | 0.5 | 201.2 | |
The FTIR spectra were obtained using a Thermo Fisher Nicolet 5700 spectrometer. The spectra were fitted using the Fityk freeware.10 The background was subtracted manually. In this work, the subscript “n” is used to indicate bonding configurations with 1, 2 or 3 hydrides, while the subscript “x” is used to indicate non-stoichiometric materials with an unknown, or various, material fractions x.
3 The oxidation and etching behaviour of Ge:H by water
For a full understanding of the oxidation and carbisation behaviour, the evolution of the a/nc-Ge(Ox,Six):H films over time is monitored. The results are presented in Fig. 1. The vibrational spectra provide some remarkable insight into the chemical stability of the films. For all a/nc-Ge(Ox):H samples, independent of the storage conditions, several distinct vibrational modes appear post-deposition, among which a broad band of vibrations is observed in the 650–1000 cm−1 range. This band of vibrations, not present in the chemically stable a/nc-Ge:H films, was previously identified as a range of Ge–O and potential Ge–C vibrations.11,12 | ||
| Fig. 1 FTIR spectra of a/nc-Ge(Ox,Six):H alloys. All curves are presented after background subtraction. The black solid curves represent measurements at 0 h. The red curves indicate measurements after exposure to the ambient environment and blue curves indicate measurements after exposure to DI water. Insets show specific wavenumber ranges at a reduced vertical scale. Deposition conditions of the films are presented in Table 1. | ||
Notably, while the evolution of the Ge–O vibrations is similar for both storage conditions, i.e. in the dark and in DI water, the evolution of the Ge–H vibrations in water and the ambient environment is dissimilar. For the samples stored in water, Ge–H wagging and stretching vibrations, at ≈560 cm−1 and 1870 cm−1, in the FTIR spectra of a/nc-Ge(Ox):H films, decrease over time. This indicates that the Ge:H phase is etched when submerged in water, assuming that the Ge–H bonds are to a certain extent homogeneously distributed over the film thickness. This etching of the Ge:H films in DI water, and the consequent reduction in film thickness, was confirmed by measurements using spectroscopic ellipsometry (SE). However, the Ge–H vibration intensity of the samples stored in the ambient environment does not change significantly over the observed 1000+ hour period. This can be observed in the FTIR spectra of a-Ge:H-1–2 in Fig. 1, selected from amongst a dozen similar samples; nor did SE measurements indicate a decrease in film thickness. This behaviour leads to two important observations.
The first observation is related to the interaction between water and Ge:H; specifically, the position of the oxygen atom in the amorphous Ge:H phase. The Ge–O vibrations increase without a decrease of the Ge–H vibrations. This means that water will not react with Ge–H bonds and the oxygen atom will not position itself at a site occupied by hydrogen. It also appears unlikely that water will react with a Ge–Ge bond and position itself back-bonded to two Ge-atoms, since Raman spectroscopy measurements did not indicate a significant decrease of Ge–Ge vibrations. Additionally, if we consider the oxidation of Si:H, where oxygen is favourably positioned back-bonded to two Si atoms, distinct SiH2O2 and SiHO3 modes do appear upon the oxidation, positioned up shifted by about 50–250 cm−1 with respect to the SiH2 modes.29–31 Similar O2GeH2 or O3GeH stretching modes are not apparent in the FTIR spectra presented in this work. The absence of these modes and the lack of significant change in the observed Ge–Ge and Ge–H bond intensity, in combination with the relatively poor hydrogen passivation efficiency of dangling bonds in Ge:H with reference to Si:H32–34 and the relatively high dangling bond densities that are expected in these films,12,28 likely indicate that water will react with the Ge-dangling bonds and oxygen will predominantly occupy the Ge-dangling bond position. The different positioning of O in Si:H and Ge:H is schematically shown in Fig. 2. The favorable positioning of O at the Ge-dangling bond site explains the defect passivating behaviour of oxygen in a-Ge:H films, which was observed in earlier work.12 Additionally, it explains why in a-Ge:H, unlike in a-Si:H, H and O incorporation are reportedly competitive.35,36
 | ||
| Fig. 2 Simplified schematic of the favourable position of oxygen in a hydrogenated silicon and germanium structure. Red line represents a dangling bond. | ||
The second important observation is related to the etching behaviour of Ge:H. The a/nc-Ge:H films of Fig. 1 are consumed too fast to allow for a detailed investigation of the etching behaviour of Ge:H over time. For that reason, three additional a-Ge:H samples were processed and their degradation in DI water was monitored at much smaller time increments. In previous research it was observed that the degradation behaviour of Ge:H samples is related to the material density.12,37 The a-Ge:H-3-5 samples are therefore processed at conditions resulting in a-Ge:H films with various porosities and expected chemical stability. The results are shown in Fig. 3. The refractive index at a wavelength of 600 nm (n@600nm) is used as a metric for the material density, with a high n@600nm indicating a dense material.
 | ||
| Fig. 3 Evolution over time of three different a-Ge:H alloys stored in DI water. Samples have various n@600nm, where the high-n@600nm a-Ge:H-3 sample is the most dense and the low-n@600nm a-Ge:H-5 sample is the most porous. Insets show specific wavenumber ranges at a reduced vertical scale. Deposition conditions of the a-Ge:H-3-5 films are reported in Table 1. | ||
The difference in degradation behaviour of the three a-Ge:H films is significant. The a-Ge:H film with the highest material density first shows a decrease of the Ge–H modes at 40 hours and the first Ge–O vibrations only appear at 100 h. For the two more porous samples, post-deposition oxidation has already occurred during the 1–2 minute ambient exposure during the transfer from the PECVD reactor to the FTIR measurement setup. Notably, for the a-Ge:H sample with n@600nm = 4.85, the Ge–O vibrations are decreased after 30 s of etching in DI water. This oxidation and etching behaviour indicates the presence of two distinct regions, a porous top region on top of a dense bulk region, for this particular sample. This observation and the underlying mechanisms are discussed in more detail elsewhere.28 After the initial etching of the porous top region, the Ge–H vibrations gradually decreased with respect to the Ge–O vibrations, and after 25 hours only a fraction of the film thickness remained. It can be observed that the oxidation of the porous a-Ge:H film with n@600nm = 4.63 occurs at a much higher rate.
Fig. 3 yields more insight into the mechanism involved in the porosity-facilitated etching behaviour. A clear trend is observed between the n@600nm of the a-Ge:H films and the intensity and central position of the peak at around 1950–2100 cm−1. This peak is the result of Ge–H stretching and is referred to as the high stretching mode (HSM). The peak at 1875 cm−1 is referred to as the Ge–H low stretching mode (LSM). The HSM is seen to increase with respect to the LSM for decreasing n@600nm, which has been reported in earlier work.28 Compared to hydrogenated silicon alloys,38,39 HSM vibrations are the result of hydrogen bonded to germanium at the surfaces of nanosized voids. A shift of the peak position towards higher wavenumbers, as observed in a-Ge:H-5 with reference to a-Ge:H-3 and a-Ge:H-4, indicates that the vibrating Ge–H bonds reside at the surfaces of yet larger nanosized voids. Not only does the rate of oxidation increase with the HSM/LSM ratio, the HSM is also reduced much more strongly than other bonds. It is likely that when larger voids are present in the material, water can diffuse into the nano-sized voids present in the bulk. With this in-diffusion, the number of reaction sites that water can access is strongly increased. Consequently, the rate of oxidation, as well the etching rate, increases with increasing void fraction. The pores that facilitate the in-diffusion of water are oxidized first, as demonstrated by the strong decrease of the HSM in the first 30 minutes for the sample a-Ge:H-5.
While the oxidation of Ge:H films is observed for both storage conditions, the question remains as to what causes the difference in etching behaviour between the samples stored in the ambient environment and in water. This question has three potential answers. 1. A difference in reactive species. In the ambient environment, O2 could potentially be the source of oxygen, instead of water. This seems unlikely, since the observed relation between density and stability is likely macroscopic by nature. A dense material prevents the in-diffusion of water and thereby bulk oxidation. A dense Ge:H phase, unlike a porous Ge:H phase, has an average bond length small enough to prevent the in-diffusion of H2O but not the in-diffusion of O2.12 Moreover, if a different species was involved in the oxidation reaction, a more distinct difference between Ge–O signatures could be expected. 2. A difference in rate of oxidation, due to a difference in the concentration of H2O in the direct environment of the films. While this cannot be excluded, it seems unlikely to be a dominant factor considering that evidence of etching is observed in a-Ge:H-4-5 in Fig. 3 after seconds, while no such evidence is apparent in a-Ge:H-1-2 in Fig. 1 after 1000+ h. The most likely explanation then is 3. A difference in reaction type. It is likely that the etching reaction is one requiring the simultaneous breaking of multiple Ge-bonds by H2O. The multiple bonds should be broken in quick succession, since a competition will exist between the breaking and reconstruction, or cross-linking, of the Ge-bonds. As H2O will favourably react with the Ge-dangling bond, the etching reaction is unlikely to occur in the absence of abundant water. Alternatively, etching could be the result of a multi-step reaction in which the secondary reaction step, or steps, are not energetically favoured with respect to the first and will therefore only occur if abundant water is present.
4 Carbisation of Ge:H by CO2
In addition to the Ge–O vibrational modes, several distinct vibrations appear post-deposition in the Ge:H spectra. Considering a-Ge:H-4-5 in Fig. 3, the oxidation is accompanied by the appearance, and continuous increase, of C–Hn stretching vibrations in the 2800–3000 cm−1 range. In fact, the appearance of the C–Hn stretching modes can be observed in all films containing a hydrogenated germanium phase.This observation indicates the occurrence of a post-deposition process during which carbon is absorbed on the germanium alloys. Additional information as to the nature of this process is provided by the appearance of several distinct vibrational modes. In Fig. 1, peaks appear at 1100 cm−1, 1230 cm−1, 1660 cm−1 and 1740 cm−1 in the a/nc-Ge(Ox,Six):H spectra. Insets are provided for the a/nc-Six Ge:H samples in Fig. 1, to demonstrate the evolution of the peaks as a function of time. After 10 h the peak at 1740 cm−1 appears, in concert with the C–Hn stretching modes at ≈2850 cm−1 and 2930 cm−1. Following the appearance of these peaks, a vibration appears at 1660 cm−1. Additionally, in the a/nc-Ge(Ox):H spectra in Fig. 1 and in the spectra of a-Ge:H-4 and a-Ge:H-5 in Fig. 3, vibrational modes appear at 1020 cm−1 and 1450 cm−1 that increase over time with reference to the Ge–H modes.
All of these vibrational modes are the result of different bonding configurations between carbon, hydrogen and oxygen, as indicated in Table 2. The peak at 1020 cm−1 is reportedly the result of the C–Hn wagging vibration,14,15 the peak at 1100 cm−1 is the result of C–Ox stretching,16,17 and the peaks at 1230 cm−1 and 1400 cm−1 are likely the result of C–Hn wagging16,18,19 and bending,17,19–22 respectively. The peak at 1430–1450 cm−1 is the result of (Ox)C–Hn bending,17,20,23,24 while the peaks at 1660 cm−1 and 1740 cm−1 are reportedly the result of different C–Ox16,24,25 and Hn C–Ox26,27 stretching modes.
| Peak position (cm−1) | Suspected bond | Vibration | Ref. |
|---|---|---|---|
| 560–580 | Ge–Hn | W | 11, 12 |
| 650–950 | Ge–Ox | St | 12, 13 |
| 1020 | C–Hn | W | 14, 15 |
| 1100 | C–Ox | St | 16, 17 |
| 1210–1230 | C–Hn | W | 16, 18, 19 |
| 1400 | C–Hn | Be | 17, 19–22 |
| 1430–1450 | (Ox)C–Hn | Be | 17, 20, 23, 24 |
| 1660 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ox Ox |
St | 16, 24, 25 |
| 1740 | HnC–Ox | St | 26, 27 |
| 1875 | Ge–HnLSM | St | 11, 12, 28 |
| 1950–2000 | Ge–HnHSM | St | 11, 12, 28 |
| 2800–3000 | C–Hn | St | 14, 15 |
Since the samples are stored inside an ISO7 cleanroom and exposed to no other source of carbon than the ambient environment, the most likely source of carbon in the post-deposition carbisation process is CO2. All of the CHyOz vibrations listed above, where y and z are discrete numbers, can be attributed to the vibrations of CO, HCHO (formaldehyde), HCOOH (formic acid) and H3COH (methanol). Based on these two observations we propose a post-deposition process involving the electrochemical reduction of CO2. This proposition raises 3 main questions: 1. What is the sequence of events resulting in the electrochemical reduction of CO2 on Ge:H films? 2. What are the potential adsorption mechanisms for CO2 onto Ge:H? 3. What type of CO2RR is taking place on the Ge:H films?
Regarding question 1, the sequence of events resulting in CO2 reduction is as follows. From the FTIR spectra, especially those of a-Ge:H-1-2, it is apparent that the Ge–O vibrations appear before the numerous CHyOz vibrations. The presence of the GeOx:H phase in the heterogeneous a-GeOx:H material is therefore likely a prerequisite for the CO2RR. This prerequisite of the GeOx:H phase could potentially be related to the CO2 adsorption mechanisms, which will be considered in the next section. Alternatively, the requirement of a GeOx:H phase can be understood if we consider that water splitting potentially provides the necessary protons for the CO2RR. Consequently, in order for the CO2RR to occur, the valence band edge should be aligned with the potential for water oxidation and the conduction band edge should be well aligned with the potential for the CO2RR to formaldehyde. For crystalline Ge, the conduction band edge is already well aligned with the CO2RR, as can be observed in Fig. 4. The valence band edge, however, is about 1 eV removed from the potential for water oxidation. As it is unlikely that the valence band edge of Ge:H is positioned 1 eV below that of the crystalline Ge, we propose that the observed CO2RR is the result of a 2-step process. In the first step Ge:H oxidizes in a reaction with water, forming GeOx:H. The band gap energy of GeOx:H increases with oxygen fraction. Several studies show that this difference is mainly caused by a downward shift of the valence band edge with respect to the vacuum level, rather than an upwards shift of the conduction band edge.43,47,52 GeOx:H, therefore, is energetically well suited for the oxidation of water and generation of protons. The second step then involves the adsorption and electrochemical reduction of CO2. Notably, this process, in which the photo-catalytic oxidation of water provides the protons required for the reduction of CO2, resulting in the formation of formaldehyde, or other CHyOz products, is comparable to photosynthesis and therefore often referred to as artificial photosynthesis.7,9,48
 | ||
| Fig. 4 Conduction and valence band edges of Ge materials with reference to the vacuum level and standard hydrogen electrode (SHE). The vacuum level and SHE potential are aligned using the absolute chemical potential of electrons under SHE conditions, of −4.44 eV.9,40,41 Alignment of the conduction and valence band edges, with respect to the vacuum level of Ge based on,40,42 GeO2 I based on,43,44 GeO2 II on43,45,46 and GeO2 III on.47 Band gap energy of a-Ge:H estimated to be in the range of 0.9–1.1 eV.12 a-GeOx:H indicates suspected band alignment upon oxidation of a-Ge:H. Dashed areas of a-Ge(Ox):H materials indicate suspected band edge variations based on the level of hydrogenation and oxidation. The water reduction half reactions at pH = 0 and pH = 7 are indicated by the red lines, and the water oxidation half reaction at pH = 7. The electrochemical redox potentials of six different CO2 reduction pathways, vs. SHE at pH = 7, are indicated by the dashed black lines, including the number of protons and electrons required for the reaction.7,48–51 | ||
Regarding question 2, the potential adsorption mechanisms for CO2 onto Ge:H are as follows. If we consider the sequence of events, and the bonds present in the FTIR spectra prior to the appearance of the CHyOz vibrations, three types of Ge-bonds are available to facilitate CO2 adsorption. These three bonds are the Ge-dangling bond, Ge–H bond and Ge–O bond. These bonds, and their potential roles in facilitating CO2 adsorption, are schematically shown in Fig. 5. Of these potential adsorption mechanisms, those positioned in the red boxes are less likely to occur for various reasons. The adsorption mechanism in box I requires O to back-bond to two Ge-atoms, which is unlikely to be the case in a-Ge:H as established in the previous section. The adsorption mechanism in box II requires H to be shared between Ge and O. Such a reaction would very likely result in a change in the Ge–H vibrational modes, a peak broadening or decrease of the peak intensity, which is not observed in the FTIR spectra. Each of the remaining mechanisms, labeled A–E, are plausible routes for the CO2 adsorption in Ge:H, based on the experimental data presented in this work.
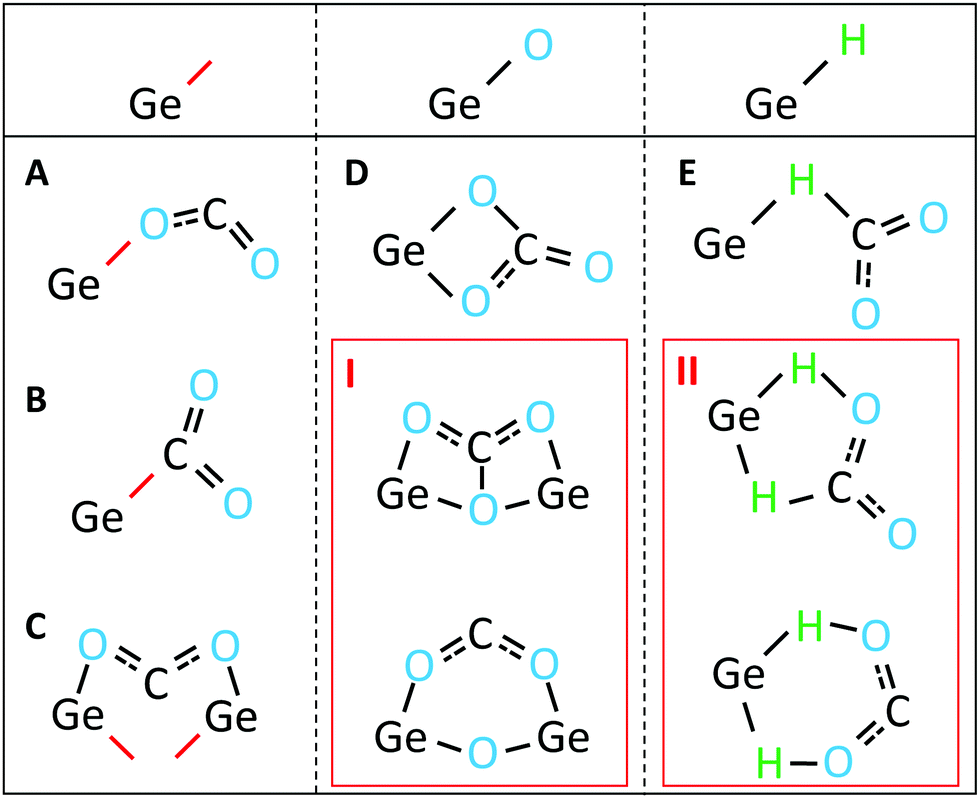 | ||
| Fig. 5 Simplified schematic representation of the potential CO2 adsorption mechanisms facilitated by a Ge-dangling bond (left), Ge–O bond (centre) and Ge–H bond (right). Red lines represent Ge-dangling bonds. Red boxes I and II are unlikely routes and are referenced in the text. | ||
Regarding question 3, the type of CO2RR occurring in Ge:H is as follows. There are 6 pathways for electrochemical CO2 reduction, each resulting in a different reaction product.8,9,48,53 The different pathways require a different number of electrons and protons. The simplest reactions, the reduction of CO2 to formic acid or CO, require 2 electrons and protons. Each successive pathway requires two additional electrons and protons, as indicated in Fig. 4. These are, listed in order of increasing number of charge carriers required, reactions resulting in the formation of CO, formaldehyde, methanol, methane or ethanol.
The peaks observed in the FTIR spectra in this work appear predominantly due to the result of CO, HCHO and HCOOH vibrational modes. Selectivity for a certain reaction product can be the result of the specific energetic alignment of the conduction band edge with a certain reaction. However, in Ge:H this seems unlikely to play a dominant role, since a well-aligned conduction band edge for CO2 reduction to formic acid and CO should also facilitate the CO2RR to methane and ethanol as can be observed in Fig. 4. It is therefore more likely related to the probability of the occurrence of a certain pathway. This probability is directly related to the number of electrons and protons involved in the reaction. The reduction of CO2 to, for example, ethanol, requires the simultaneous availability of 12 protons and 12 electrons. This reaction is therefore less likely to occur than the formation of formic acid, which only requires two of each charge carrier type. Consequently, we would expect the largest fraction of vibrations to be related to the “simplest” reaction pathways, resulting in CO, formic acid and formaldehyde.
5 Conclusions
In this work, the first ever evidence of the post deposition appearance of CHyOz vibrational modes in PECVD-processed hydrogenated germanium is presented. It is proposed that the observed carbisation reaction involves the catalytic reduction of CO2 on Ge:H films to products like CO, formic acid and formaldehyde. A two-step process is proposed, in which the reduction of CO2 follows the post-deposition chemical transformation of a-Ge:H to a-GeOx:H upon reaction with water. The reaction rate is demonstrated to be a function of the material porosity.A theory is presented in which the a-GeOx:H phase is essential for energetic alignment of the valence band edge with the water oxidation reaction potential. Protons from water splitting are used for the catalytic CO2 reduction reaction. Moreover, we demonstrate that the carbisation reaction does not result in the consumption of the catalyst material. However, etching of the Ge:H films does occur during the oxidation of Ge:H, only if the film is submerged in deionized water. Many questions remain as to the specific reactions resulting in the etching of the Ge:H phase and the CO2 adsorption reaction. The authors hope to inspire additional computational and experimental research to validate the theories proposed in this work.
Though many questions remain, these findings demonstrate the potential use of a-Ge(Ox):H as an inexpensively processed, relatively earth abundant electro-catalyst or electrode material for photo(voltaic)-electrochemical device applications.
The authors would like to gratefully acknowledge financial support from the Netherlands Organization for Scientific Research (NWO) Solar to Products grant awarded to Arno H. M. Smets and the support provided by Shell International Exploration & Production New Energies Research & Technology Dense Energy Carriers Program.
Conflicts of interest
There are no conflicts to declare.References
- IPCC, Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Tech. rep. (2021).
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Designing materials for electrochemical carbon dioxide recycling, Nat. Catal., 2019, 2(8), 648–658, DOI:10.1038/s41929-019-0306-7.
- Z. Guo, G. Chen, C. Cometto, B. Ma, H. Zhao, T. Groizard, L. Chen, H. Fan, W.-L. Man, S.-M. Yiu, K.-C. Lau, T.-C. Lau and M. Robert, Selectivity control of CO versus HCOO− production in the visible-light-driven catalytic reduction of CO2 with two cooperative metal sites, Nat. Catal., 2019, 2(9), 801–808, DOI:10.1038/s41929-019-0331-6.
- H. Yuan, B. Cheng, J. Lei, L. Jiang and Z. Han, Promoting photocatalytic CO2 reduction with a molecular copper purpurin chromophore, Nat. Commun., 2021, 12(1), 1835, DOI:10.1038/s41467-021-21923-9.
- J.-Y. Kim, D. Hong, J.-C. Lee, H. G. Kim, S. Lee, S. Shin, B. Kim, H. Lee, M. Kim, J. Oh, G.-D. Lee, D.-H. Nam and Y.-C. Joo, Quasi-graphitic carbon shell-induced Cu confinement promotes electrocatalytic CO2 reduction toward C2+ products, Nat. Commun., 2021, 12(1), 3765, DOI:10.1038/s41467-021-24105-9.
- P. Stewart, Abundance of the elements - a new look, Education Chem., 2003, 40(1), 23–24 CAS.
- Y. Y. Lee, H. S. Jung and Y. T. Kang, A review: effect of nanostructures on photocatalytic CO2 conversion over metal oxides and compound semiconductors, J. CO2 Util., 2017, 20, 163–177, DOI:10.1016/j.jcou.2017.05.019.
- L. Liu, S. Wang, H. Huang, Y. Zhang and T. Ma, Surface sites engineering on semiconductors to boost photocatalytic CO2 reduction, Nano Energy, 2020, 75, 104959, DOI:10.1016/j.nanoen.2020.104959.
- J. Feng, H. Huang, S. Yan, W. Luo, T. Yu, Z. Li and Z. Zou, Non-oxide semiconductors for artificial photosynthesis: progress on photoelectrochemical water splitting and carbon dioxide reduction, Nano Today, 2020, 30, 100830, DOI:10.1016/j.nantod.2019.100830.
- M. Wojdyr, Fityk: a general-purpose peak fitting program, J. Appl. Crystallogr., 2010, 43(5), 1126–1128, DOI:10.1107/S0021889810030499.
- W. Paul, Structural, Optical and photoelectronic properties of improved PECVD a-Ge:H, J. Non-Cryst. Solids, 1991, 137&138, 803–808 Search PubMed.
- T. de Vrijer, A. Ravichandran, B. Bouazzata and A. H. Smets, The impact of processing conditions and post-deposition oxidation on the opto-electrical properties of hydrogenated amorphous and nano-crystalline Germanium films, J. Non-Cryst. Solids, 2021, 553, 120507, DOI:10.1016/j.jnoncrysol.2020.120507.
- M. Zacharias and J. Blasing, Preparation of a-GeOx:H alloys: Vibrational, optical, and structural properties, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52(19), 14018–14024 CrossRef CAS PubMed.
- Y. Catherine and G. Turban, Infrared absorption of hydrogenated amorphous Si–C and Ge–C films, Thin Solid Films, 1980, 70(1), 101–104, DOI:10.1016/0040-6090(80)90416-2.
- N. Saito, I. Nakaaki, T. Yamaguchi, S. Yoshioka and S. Nakamura, Influence of deposition conditions on the properties of a-GeC:H and a-Ge:H films prepared by r.f. magnetron sputtering, Thin Solid Films, 1995, 269(1-2), 69–74, DOI:10.1016/0040-6090(95)06671-3.
- E. Abdelrazek, I. Elashmawi, A. El-khodary and A. Yassin, Structural, optical, thermal and electrical studies on PVA/PVP blends filled with lithium bromide, Curr. Appl. Phys., 2010, 10(2), 607–613, DOI:10.1016/j.cap.2009.08.005.
- A. Tawansi, A. Oraby, H. Zidan and M. Dorgham, Effect of one-dimensional phenomena on electrical, magnetic and ESR properties of MnCl2 -filled PVA films, Phys. B: Condens. Matter, 1998, 254(1-2), 126–133, DOI:10.1016/S0921-4526(98)00414-1.
- I. Vivaldo, M. Moreno, A. Torres, R. Ambrosio, P. Rosales, N. Carlos, W. Calleja, K. Monfil and A. Benítez, A comparative study of amorphous silicon carbide and silicon rich oxide for light emission applications, J. Lumin., 2017, 190, 215–220, DOI:10.1016/j.jlumin.2017.05.048.
- S. Ray, D. Das and A. Barua, Infrared vibrational spectra of hydrogenated amorphous silicon carbide thin films prepared by glow discharge, Solar Energy Mater., 1987, 15, 45–57 CrossRef CAS.
- I. Omkaram, R. Sreekanth Chakradhar and J. Lakshmana Rao, EPR, optical, infrared and Raman studies of VO2+ ions in polyvinylalcohol films, Phys. B: Condens. Matter, 2007, 388(1-2), 318–325, DOI:10.1016/j.physb.2006.06.134.
- M. Martin, Characteristic IR Band Positions.
- H. Jamali, R. Mozafarinia and A. Eshaghi, Evaluation of chemical and structural properties of germanium-carbon coatings deposited by plasma enhanced chemical vapor deposition, J. Alloys Compd., 2015, 646, 360–367, DOI:10.1016/j.jallcom.2015.06.091.
- N. Ojha, A. Bajpai and S. Kumar, Enriched oxygen vacancies of Cu2O/SnS2/SnO2 heterostructure for enhanced photocatalytic reduction of CO2 by water and nitrogen fixation, J. Colloid Interface Sci., 2021, 585, 764–777, DOI:10.1016/j.jcis.2020.10.056.
- H. Zhang, Y. Li, J. Wang, N. Wu, H. Sheng, C. Chen and J. Zhao, An unprecedent hydride transfer pathway for selective photocatalytic reduction of CO2 to formic acid on TiO2, Appl. Catal., B, 2021, 284, 119692, DOI:10.1016/j.apcatb.2020.119692.
- S. W. Pyo, C. Manianglung and Y. S. Ko, In-situ IR study on stability of amine-impregnated CO2 adsorbents to acidic gases, Catal. Today, 2020, 352, 198–203, DOI:10.1016/j.cattod.2020.01.036.
- L. Wang, H. Tan, L. Zhang, B. Cheng and J. Yu, In-situ growth of few-layer graphene on ZnO with intimate interfacial contact for enhanced photocatalytic CO2 reduction activity, Chem. Eng. J., 2021, 411, 128501, DOI:10.1016/j.cej.2021.128501.
- C. Pirim and L. Krim, A neon-matrix isolation study of the reaction of non-energetic H-atoms with CO molecules at 3 K, Phys. Chem. Chem. Phys., 2011, 13(43), 19454, 10.1039/c1cp21835b.
- T. de Vrijer, J. E. van Dingen, P. J. Roelandschap, K. Roodenburg and A. H. Smets, Improved PECVD processed hydrogenated germanium films through temperature induced densification, Mater. Sci. Semicond. Process., 2022, 138, 106285, DOI:10.1016/j.mssp.2021.106285.
- M. Niwano, J. Kageyama, K. Kurita, K. Kinashi, I. Takahashi and N. Miyamoto, Infrared spectroscopy study of initial stages of oxidation of hydrogen-terminated Si surfaces stored in air, J. Appl. Phys., 1994, 76(4), 2157–2163, DOI:10.1063/1.357627.
- A. H. M. Smets, T. Matsui and M. Kondo, High-rate deposition of microcrystalline silicon p–i–n solar cells in the high pressure depletion regime, J. Appl. Phys., 2008, 104(3), 034508, DOI:10.1063/1.2961334.
- A. Bronneberg, A. Smets, M. Creatore and M. van de Sanden, On the oxidation mechanism of microcrystalline silicon thin films studied by Fourier transform infrared spectroscopy, J. Non-Cryst. Solids, 2011, 357(3), 884–887 CrossRef CAS.
- W. Paul, D. Paul, B. von Roedern, J. Blake and S. Oguz, Preferential Attachment of H in Amorphous Hydrogenated Binary Semiconductors and Consequent Inferior Reduction of Pseudogap State Density, Phys. Rev. Lett., 1981, 46(15), 1016–1020 CrossRef CAS.
- S. K. Estreicher and D. M. Maric, What is so strange about hydrogen interactions in germanium?, Phys. Rev. Lett., 1993, 70(25), 3963–3966, DOI:10.1103/PhysRevLett.70.3963.
- A. Bhaduri, P. Chaudhuri, S. Vignoli and C. Longeaud, Correlation of structural inhomogeneities with transport properties in amorphous silicon germanium alloy thin films, Solar Energy Mater. Solar Cells, 2010, 94(9), 1492–1495, DOI:10.1016/j.solmat.2010.02.043.
- G. Lucovsky, S. S. Chao, J. Yang, J. E. Tyler, R. C. Ross and W. Czubatyj, Chemical bonding of hydrogen and oxygen in glow-discharge - deposited thin films of a -Ge:H and a -Ge:(H,O), Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31(4), 2190–2197, DOI:10.1103/PhysRevB.31.2190.
- B. Schröder, A. Annen, T. Drüsedau, H. Freistedt, P. Deák and H. Oechsner, Influence of oxygen incorporation on the properties of magnetron sputtered hydrogenated amorphous germanium films, Appl. Phys. Lett., 1993, 62(16), 1961–1963, DOI:10.1063/1.109504.
- T. de Vrijer, B. Bouazzata, A. Ravichandran, J. van Dingen, P. Roelandschap, K. Roodenburg, S. Roerink, F. Saitta, T. Blackstone and A. Smets, Opto-electrical properties of group IV alloys: the inherent challenges of processing hydrogenated germanium, Adv. Sci., 2022 DOI:10.1002/advs.202200814.
- A. H. M. Smets, W. M. M. Kessels and M. C. M. van de Sanden, Vacancies and voids in hydrogenated amorphous silicon, Appl. Phys. Lett., 2003, 82(10), 1547–1549, DOI:10.1063/1.1559657.
- A. H. M. Smets and M. C. M. van de Sanden, Relation of the Si-H stretching frequency to the nanostructural Si-H bulk environment, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76(7), 073202, DOI:10.1103/PhysRevB.76.073202.
- C. G. van de Walle and J. Neugebauer, Universal alignment of hydrogen levels in semiconductors insulators and solutions, Nature, 2003, 423, 626–628 CrossRef CAS PubMed.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, 2D Transition-Metal-Dichalcogenide-Nanosheet-Based Composites for Photocatalytic and Electrocatalytic Hydrogen Evolution Reactions, Adv. Mater., 2016, 28(10), 1917–1933, DOI:10.1002/adma.201503270.
- C. Van de Walle, J. Weber and A. Janotti, Role of hydrogen at germanium/dielectric interfaces, Thin Solid Films, 2008, 517(1), 144–147, DOI:10.1016/j.tsf.2008.08.071.
- J. F. Binder, P. Broqvist and A. Pasquarello, Charge trapping in substoichiometric germanium oxide, Microelectron. Eng., 2011, 88(7), 1428–1431, DOI:10.1016/j.mee.2011.03.133.
- A. Ohta, H. Nakagawa, H. Murakami, S. Higashi and S. Miyazaki, Photoemission Study of Ultrathin GeO2/Ge Heterostructures Formed by UV-O3 Oxidation, e-J. Surf. Sci. Nanotechnol., 2006, 4, 174–179, DOI:10.1380/ejssnt.2006.174.
- M. Perego, G. Scarel, M. Fanciulli, I. L. Fedushkin and A. A. Skatova, Fabrication of GeO2 layers using a divalent Ge precursor, Appl. Phys. Lett., 2007, 90(16), 162115, DOI:10.1063/1.2723684.
- M. Yang, R. Q. Wu, Q. Chen, W. S. Deng, Y. P. Feng, J. W. Chai, J. S. Pan and S. J. Wang, Impact of oxide defects on band offset at GeO2/Ge interface, Appl. Phys. Lett., 2009, 94(14), 142903, DOI:10.1063/1.3115824.
- B. L. Ong, S. W. Ong, A. Rusydi and E. S. Tok, Important roles of native-oxides on the electronic band offsets at Ge-oxide/Ge(0 0 1) heterojunction in ambient environment, Appl. Surf. Sci., 2020, 530, 147256, DOI:10.1016/j.apsusc.2020.147256.
- I. I. Alkhatib, C. Garlisi, M. Pagliaro, K. Al-Ali and G. Palmisano, Metal-organic frameworks for photocatalytic CO2 reduction under visible radiation: A review of strategies and applications, Catal. Today, 2020, 340, 209–224, DOI:10.1016/j.cattod.2018.09.032.
- R. R. Ikreedeegh and M. Tahir, A critical review in recent developments of metal-organic-frameworks (MOFs) with band engineering alteration for photocatalytic CO2 reduction to solar fuels, J. CO2 Util., 2021, 43, 101381, DOI:10.1016/j.jcou.2020.101381.
- D. Ješić, D. Laši Jurković, A. Pohar, L. Suhadolnik and B. Likozar, Engineering photocatalytic and photoelectrocatalytic CO2 reduction reactions: Mechanisms, intrinsic kinetics, mass transfer resistances, reactors and multi-scale modelling simulations, Chem. Eng. J., 2021, 407, 126799, DOI:10.1016/j.cej.2020.126799.
- S. Kreft, D. Wei, H. Junge and M. Beller, Recent advances on TiO2-based photocatalytic CO2 reduction, Energy Chem., 2020, 2(6), 100044, DOI:10.1016/j.enchem.2020.100044.
- S. Fadida, M. Eizenberg, L. Nyns, S. Van Elshocht and M. Caymax, Band alignment of Hf–Zr oxides on Al2O3/GeO2/Ge stacks, Microelectron. Eng., 2011, 88(7), 1557–1559, DOI:10.1016/j.mee.2011.03.075.
- L. F. Garay-Rodríguez, L. M. Torres-Martínez and E. Moctezuma, Photocatalytic evaluation of composites of Ba3Li2Ti8O20-CuO in the reduction of CO2 to formaldehyde under visible light irradiation, J. Photochem. Photobiol., A, 2018, 361, 25–33, DOI:10.1016/j.jphotochem.2018.05.003.
| This journal is © the Owner Societies 2022 |