
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The impact of chemical composition of halide surface ligands on the electronic structure and stability of lead sulfide quantum dot materials†
Tamara
Sloboda
 a,
Sebastian
Svanström
b,
Fredrik O. L.
Johansson‡
b,
Erik
Bryngelsson
a,
Alberto
García-Fernández
a,
Andreas
Lindblad
b and
Ute B.
Cappel
*a
a,
Sebastian
Svanström
b,
Fredrik O. L.
Johansson‡
b,
Erik
Bryngelsson
a,
Alberto
García-Fernández
a,
Andreas
Lindblad
b and
Ute B.
Cappel
*a
aDivision of Applied Physical Chemistry, Department of Chemistry, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: cappel@kth.se
bDivision of X-ray Photon Science, Department of Physics and Astronomy, Uppsala University, Box 516, SE-751 20, Uppsala, Sweden
First published on 17th May 2022
Abstract
There is a high fundamental interest in the surface and bulk chemistry of quantum dot (QD) solids, as they have proven to be very promising materials in optoelectronic devices. The choice of surface ligands for quantum dots in solid devices determines many of the film properties, as the ligands influence for example the doping density, chemical stability and charge transport. Lead halide ligands have developed as the main ligand of choice for lead sulfide quantum dots, as they have been shown to passivate quantum dot surfaces and enhance the chemical stability. In this study, we successfully varied the ligand composition on the surface of PbS quantum dot films from pure lead iodide to pure lead bromide and investigated its influence on the chemical and electronic structure of the QD solids using hard X-ray photoelectron spectroscopy (HAXPES). Furthermore, we developed a surface treatment to prevent the surface oxidation of a bulk PbS reference sample. Through measurements of this sample and of lead halide reference samples, we were able to assign the contributions of different chemical bonding to the Pb 4f core level and of different atomic orbitals to the valence band spectral shape of the QD materials. Overall, we found that the valence band edge position was very similar for all different iodide:bromide ratios and that all investigated compositions were able to protect the quantum dot surfaces within solid films from oxidation. However, the ligand composition significantly influences the sample stability under X-rays. The iodide rich QD solids showed the highest stability with very little to no chemical changes over several hours of X-ray exposure, while the bromide rich QD solids changed already within the first hour of exposure.
Introduction
In recent years, quantum dots (QDs) have been extensively studied due to their favorable properties, such as the strong quantum confinement effect, that allows size-dependent bandgap tuning, and cost-effective liquid-phase manufacturing.1–3 As semiconductor nanocrystals, quantum dots are very attractive as materials in thin-film optoelectronic devices in various applications, such as solar cell devices, LEDs, imaging, transistors, sensors, biosensors and detectors.4–13 From a chemical composition perspective, quantum dots can be elemental materials (e.g. Si, Ge, etc.), III–V nanocrystals (InP, InAs), IV–VI materials (PbS, PbSe, PbTe) and II–VI materials mostly based on Zn, Cd and Hg.14 Among these, lead based chalcogenides (especially PbS and PbSe) are perhaps one of the most studied for use in solar cells. As bulk materials at room temperature, these two compounds have narrow bandgaps (0.37 and 0.27 eV for PbS and PbSe, respectively).15 However, due to the large Bohr exciton radius (∼20 nm for PbS and ∼34 nm for PbSe),1 the quantum size effects are observable already at larger crystal sizes (typically 1–20 nm),14,16,17 which allows for engineering of the bandgap over a broad spectral range. The bandgap engineering can be useful in multi-junction solar cell devices, which have a potential to achieve power conversion efficiencies that go beyond current commercial solar cells.18,19 Currently, among the single junction PbS QD solar cells, the highest power conversion efficiencies (above 13%) are achieved using a thin film of PbS QDs with I− and Br− surface ligands as the main absorbing material. These quantum dots have a bandgap of 1.3 eV, achieved by a particle size of approximately 3–4 nm in diameter.10,20 Due to the large surface-to-volume ratio, the properties of the quantum dots are highly dependent on their surface characteristics21 and therefore the choice of ligands for the QDs in a solar cell device is of great importance.Before choosing ligands to be used in a device, the PbS QDs prepared in a typical hot-injection reaction22 are capped with long carbon-chain ligands (e.g. oleic acid, OA). While the function of the long and bulky OA ligands is to keep the QDs stable and separated in solution, in a thin film device these ligands need to be exchanged for shorter ones to allow charge carriers to hop from one dot to another and in this way create a current. Multiple studies show that differences in surface passivation, carrier behavior and defects in QD materials are due to the ligand choice and this can significantly affect the performance of the devices.23–26 In the study of Brown et al., ultraviolet photoelectron spectroscopy and density functional theory calculations were used to show that the ligands can significantly influence the position of the Fermi level in relation to valence and conduction bands.27 Moreover, the ligand choice also influences the stability of the solid film as well as the formation of trap states, as shown in the time-resolved photoluminescence study of Papagiorgis et al.28 Even small surface modifications can drastically enhance the efficiency of absorption as shown in the study of Giansante et al. where they were able to enhance the absorption by 300%.29 Eventually, the search for most optimal ligands moved the research towards inorganic halide ligands. For instance, Bederak et al. have shown that PbS QDs capped with fluoride or chloride anions show a p-type character, which can offer an alternative p-type layer in a quantum dot solar cell device, while bromide and iodide caped ones have an n-type character.30 In another study by Fan et al. it was shown that the chloride and bromide ligands passivate the surface better compared to iodide ligands, which have shown better charge mobility and charge transport.31 Therefore, a mixed halide surface passivation has great potential in achieving the best QD solar cell performances. Different strategies have been tested to determine the best method for the best surface coverage and higher stability and quality of the material. Zhang et al. investigated in situ halide passivation, obtaining devices with 6.5% efficiency,32 while a few years later Ding et al. designed a post-passivation solid state ligand exchange method in which they reached device efficiencies above 12%.26 However, many of these studies use a mixture of chloride, bromide and iodide without a clear explanation of why they chose a certain ratio of these halides. To the best of our knowledge, there are no detailed studies of the effect of systematically varied halide ratios in the PbS capping ligands on the chemical and electronic structure of lead sulfide quantum dots.
The aim of the present study was to investigate the impact of the halide-ligand surface composition on the electronic structure and X-ray stability of the PbS QD by gradually varying the bromide and iodide content on its surface. For this we used hard X-ray photoelectron spectroscopy (HAXPES) as our main tool, which enabled us to investigate the detailed chemical and electronic structure profile of these materials both qualitatively and quantitatively. By choosing two incident photon energies (3000 and 6100 eV), we were able to acquire chemical information from different depths of the sample which will be referred to as the surface and bulk. Furthermore, we used reference compounds to address uncertainties in assigning the chemical states in the Pb core levels. For this, we developed a surface treatment for the synthesis of a PbS reference thin film, which prevents surface oxidation in a similar manner to the halide ligands of quantum dots. In addition to this, the stability of the quantum dots under continuous X-ray illumination was investigated and showed that ligands with a high bromide content are susceptible to degradation by X-rays.
Experimental section
All solvents and reagents in the synthesis of the materials except PbI2 and PbBr2 were purchased from Sigma-Aldrich. PbI2 and PbBr2 were ordered from TCI. MgZnO was synthesized according to a previously reported method33 while the PbS colloidal QDs were synthesized with a few small modifications compared to the reported PbS synthesis method.22Synthetic procedures
PbS QD synthesis was started by adding lead oxide (PbO, 99.99%, 0.933 g), oleic acid (OA, tech. grade 90%, 4.053 g), and 1-octadecene (ODE, tech. grade 90%, 25 ml) to a three-neck round bottom flask creating the reaction mixture. The mixture was first degassed in mild vacuum conditions for 1 h and then heated to 100–110 °C, until all of the PbO was dissolved, and the mixture became a transparent solution. At this moment, nitrogen flow was introduced instead of vacuum and the solution was stirred for approximately 2 h. Simultaneously, the hexamethyldisilathiane ((TMS)2S, synthetic grade, 0.356 g) solution in ODE (10 ml) was prepared in a glovebox. When taken out of the glovebox, the argon in the vial was carefully removed, creating a mild vacuum condition inside the vial, which was then moved into the oven and warmed up to 80 °C for 2 h. After 2 h of stirring, the temperature of the solution was lowered to 90 °C and the hot (TMS)2S solution was quickly injected, giving rise to an instantaneous reaction. The heating source was removed 2–3 minutes after the injection, letting the PbS–OA particles slowly cool down to room temperature. The newly formed PbS–OA quantum dots were washed in two steps with acetone and toluene, dried under vacuum and finally dispersed in octane to a concentration of 50 mg ml−1. The particles synthesized in this way had a diameter of about 3 nm with an optical energy gap of 1.3 eV.The PbS reference (PbS ref) was prepared according to a previously published method34 with an additional surface treatment. First, a precursor solution containing lead acetate and thiourea dissolved in glacial acetic acid is deposited on indium-doped tin oxide (ITO) by spin-coating at 2000 rpm for 30 seconds, followed by thermolysis at ∼200 °C in ambient conditions, resulting in about a 200 nm thick film of lead sulfide. The PbS film was further treated with a methanol solution of tetrabutylammonium iodide (TBAI, 10 mg ml−1) directly after thermolysis (by spin-coating at 2000 rpm for 30 seconds), and washed with methanol to remove the excess TBAI molecules.
HAXPES sample fabrication
The substrate used for sample fabrication was indium-doped tin oxide-coated glass (ITO, ordered from STIGAB Stig Ödlund AB), which was sequentially cleaned with diluted RBS™-25 concentrate, acetone, and ethanol respectively in an ultrasonic bath and dried with air flow. Additionally, the clean ITO was treated with UV-ozone for 20 min directly before deposition of MgZnO nanoparticles. The MgZnO nanoparticles were filtered through a 0.2 μm filter and spin-coated on ITO-substrates at 3000 rpm for 30 s, followed by annealing at 200 °C for 30 min and 300 °C for another 30 min. The PbS QD films were prepared according to a previously reported method with small modifications.35 The concentrated PbS QD solution in octane was diluted to 10 mg ml−1 before being mixed with ligand solution in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. The ligand solution contained a 0.12 M halide salt mixture (e.g. 0.6 M PbI2 and 0.6 M PbBr2 for the sample with a 1:1 iodine to bromine ratio) and 0.04 M ammonium acetate in dimethylformamide (DMF). As soon as the solutions were in contact, they were mixed vigorously for 5 min using a vortex mixer (at room temperature). During this process the PbS QDs moved from the octane to the DMF phase after which the ligand exchange was complete. The octane-OA phase was removed with a pipette and the QD-containing solution was washed twice with octane to remove the OA residues. As the last step, toluene was added to precipitate the QDs, after which they were centrifuged and dried under vacuum for approximately 1h and dispersed in a butylamine:DMF (9:1, vol) mixture so that the final concentration is 200 mg ml−1. This solution is referred to as PbS QD ink. The ink was spin coated at 1800 rpm for 30 s on top of the MgZnO layer and immediately annealed at 70 °C for about 10 min. This procedure gives 250 ± 10 nm thick samples. At this point, the samples for PES were stored in darkness in a desiccator/glovebox for up to one week before measurement.
:1 volume ratio. The ligand solution contained a 0.12 M halide salt mixture (e.g. 0.6 M PbI2 and 0.6 M PbBr2 for the sample with a 1:1 iodine to bromine ratio) and 0.04 M ammonium acetate in dimethylformamide (DMF). As soon as the solutions were in contact, they were mixed vigorously for 5 min using a vortex mixer (at room temperature). During this process the PbS QDs moved from the octane to the DMF phase after which the ligand exchange was complete. The octane-OA phase was removed with a pipette and the QD-containing solution was washed twice with octane to remove the OA residues. As the last step, toluene was added to precipitate the QDs, after which they were centrifuged and dried under vacuum for approximately 1h and dispersed in a butylamine:DMF (9:1, vol) mixture so that the final concentration is 200 mg ml−1. This solution is referred to as PbS QD ink. The ink was spin coated at 1800 rpm for 30 s on top of the MgZnO layer and immediately annealed at 70 °C for about 10 min. This procedure gives 250 ± 10 nm thick samples. At this point, the samples for PES were stored in darkness in a desiccator/glovebox for up to one week before measurement.
The PbI2 and PbBr2 reference samples were deposited on top of MgZnO spin-coated on ITO substrates by spin-coating 0.7 M solutions, at a speed of 3000 rpm and annealing at 110 °C.
Material characterization
UV-VIS-NIR absorption and photoluminescence measurements were carried out with an Avantes AvaSpec-UV/Vis/NIR dual channel spectrometer. An Avantes AvaLight HAL-S-Mini 10 W tungsten halogen lamp was used as the excitation source for absorption spectroscopy and for calibration of photoluminescence measurements. An Avantes AvaLight-HPLED-530 LED source with a wavelength of 530 nm was used as the excitation source for photoluminescence.Measurements of the thickness of the PbS thin films were made by means of a profilometer (Veeco Dektak 150).
XRD measurements were run on a PANalytical XPert PRO diffractometer using Cu Kα radiation in a 2θ range between 15 and 65° at room temperature.
HAXPES measurements
HAXPES was carried out at the HIKE end station, KMC-1 beamline at the BESSY II synchrotron facility (Helmholtz Zentrum Berlin, Germany).36 The HIKE end-station provides photon energies from 2000 eV up to 12000 eV. In this experiment, a photon energy of 3000 eV was chosen by selecting the first order light from a Si(111) double crystal monochromator (Oxford Danfysik), whereas 6100 eV was chosen by using Si (311). The analysis chamber was under ultra-high vacuum conditions (1 × 10−8 mbar) and a Scienta R-4000 hemispherical analyzer was used to measure the kinetic energies of the emitted electrons. The measurements were energy calibrated by measuring the Au 4f7/2 level of a gold foil mounted on the manipulator and setting its position to 84.0 eV binding energy, and then further recalibrated internally (discussed below). Measurements were always carried out on fresh sample spots and core level measurements were repeated in a loop to monitor the effects of X-ray irradiation on the spectra.
Data analysis and curve fitting
All spectra were fitted in Matlab, with a number of pseudo-Voigt functions37 representing the number of peaks present in each spectrum and with an appropriate background function (e.g. a slope in fitting of Pb 4f, I 4d and Br 3d, a quadratic function in fitting of S 2p and S 1s, a Shirley background38 in fitting of S 2p and S 1s and a Herrera-Gomez background39 in fitting of Pb 4f and Pb 5d). For the fit of the Pb 4f spectra, two spin–orbit doublets were used, representing on one hand Pb–S and Pb–I and on the other Pb–O and Pb–Br. For S 2p, I 4d and Br 3d, one spin–orbit doublet was used for fitting. The relative intensity and separation of all doublets were fixed to those of the main doublet. The intensity ratio between the Pb 4f7/2 and Pb 4f5/2, I 4d5/2 and I 4d3/2, and Br 3d5/2 and Br 3d3/2 peaks was within the range of 0.6–0.8, and for Pb 4f the separation between the Pb 4f7/2 and Pb 4f 5/2 was fixed to 4.8 ± 0.1 eV. The intensity ratio between S 2p3/2 and S 2p1/2 was fixed to 0.5, and the distance in binding energy to 1.18 eV. C 1s and O 1s spectra were fitted with multiple singlets. The Gaussian width was at 0.9 ± 0.1 eV for the Pb 4f, Pb 5d and S 1s peaks, 0.8 ± 0.1 eV for halide peaks, 0.7 ± 0.1 eV for S 2p, and 1.6 ± 0.1 eV for S 2s, while the Lorentzian contribution was at 0.1 ± 0.05 eV for the Pb 4f, Pb 5d and S 2p peaks, 0.4 ± 0.01 eV for the S 2s peak and 0.2 ± 0.1 eV for the halide peaks. The relative changes in peak positions and intensities could be determined from the fitted parameters. For quantification of the HAXPES data, intensities for each core level were divided by their ionization cross-section.40Results and discussion
Sample characterization
Seven PbS QD samples with different surface ligand compositions were investigated. The ligands consisted of lead halides PbI2 and PbBr2, where the composition of the surface treatment mixture varied from pure lead iodide to pure lead bromide. The precise molar ratios used in the ligand-exchange were 1:0, 5:1, 2:1, 1:1, 1:2, 1:5 and 0:1 PbI2 to PbBr2 and in this manuscript the samples with mixed halide ratios will be referred to in “PbS (x:y)” format and the samples with the pure PbX2 (X = I, Br) as ligands will be referred to as PbS–PbX2 samples. Additionally, reference samples of PbS (non-quantized, referred to as PbS ref), PbI2 (referred to as PbI2 ref) and PbBr2 (referred to as PbBr2 ref) thin films were investigated.
The PbS reference sample showed a significant amount of surface oxidation in the HAXPES spectra, which is observed through a shoulder in the Pb 4f doublet, additional S 1s peaks (assigned to oxidized sulfur) and larger oxygen (O 1s) and carbon (C 1s) peaks (Fig. S1–S3, ESI†). As surface oxidation affects the results of photoelectron spectroscopy and would hinder a clear comparison to the quantum dot samples, we designed a surface treatment inspired by solid-state ligand exchange of quantum dots41,42 that prevented the surface oxidation of the reference film. Namely, the PbS film was treated with a solution of tetrabutylammonium iodide (TBAI) in methanol. The sample prepared in this way showed significantly less surface oxidation in the photoelectron spectra measured with 3000 eV photon energy. Only one clear component of Pb 4f (Fig. S2A, labelled “PbS ref + TBAI”, ESI†) was found and no oxidized S species in the S 1s spectra in Fig. S2B (ESI†). The oxygen (O 1s) and carbon (C 1s) contributions (Fig. S3, ESI†) were also smaller. The formation of the crystalline non-quantized lead sulfide in the sample with the TBAI treatment was confirmed by XRD analysis (Fig. 1A) showing diffraction peaks at the same angles as the diffraction pattern of a single crystal of PbS (planes (111), (200), (220), and (311) in Fig. 1A, black line).43 The TBAI-treated PbS sample was then used as the reference for HAXPES of the PbS quantum dots and is hereafter referred to as PbS ref. Additionally, XRD analysis was performed on a sample of PbS quantum dots with OA ligands deposited on a glass substrate (Fig. 1A, blue line). The same peaks were observed as for the PbS ref confirming the rock salt crystal structure for the quantum dots too. From the positions and full width half maximum (FWHM) of the strongest peaks (e.g. planes (111), (200) and (220)) it is possible to calculate the average crystal grain size in the sample using the Scherrer equation.44 From the fit of the PbS–OA diffraction pattern (Fig. 1B and Table 1), the crystal size was calculated to be 2.8 nm. A more common method for size determination of PbS quantum dots is from the positions of the exciton peak in the absorption spectrum, or in the fluorescence spectrum, which for the QDs in this study were at about 955 nm and 1050 nm, respectively (Fig. S4A, ESI†). The exciton peak in the absorption spectrum, when fitted with a Gaussian distribution, has a center energy (corresponding to the bandgap, Eg) of 1.29 eV with a standard deviation of 0.05 eV. According to the sizing equation developed by Moreels et al.:45
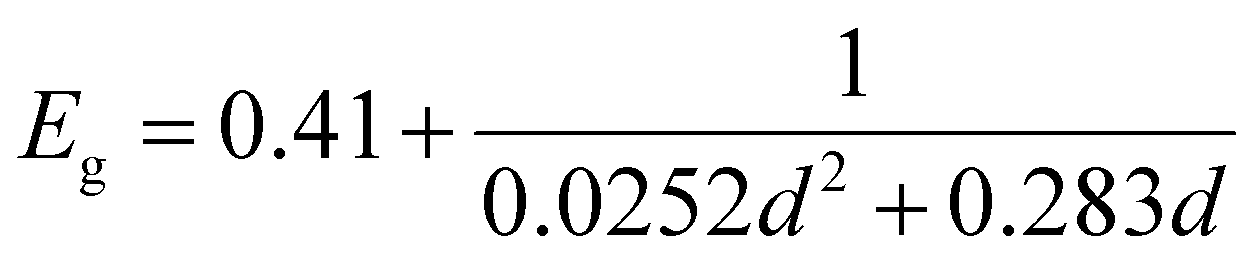 | (1) |
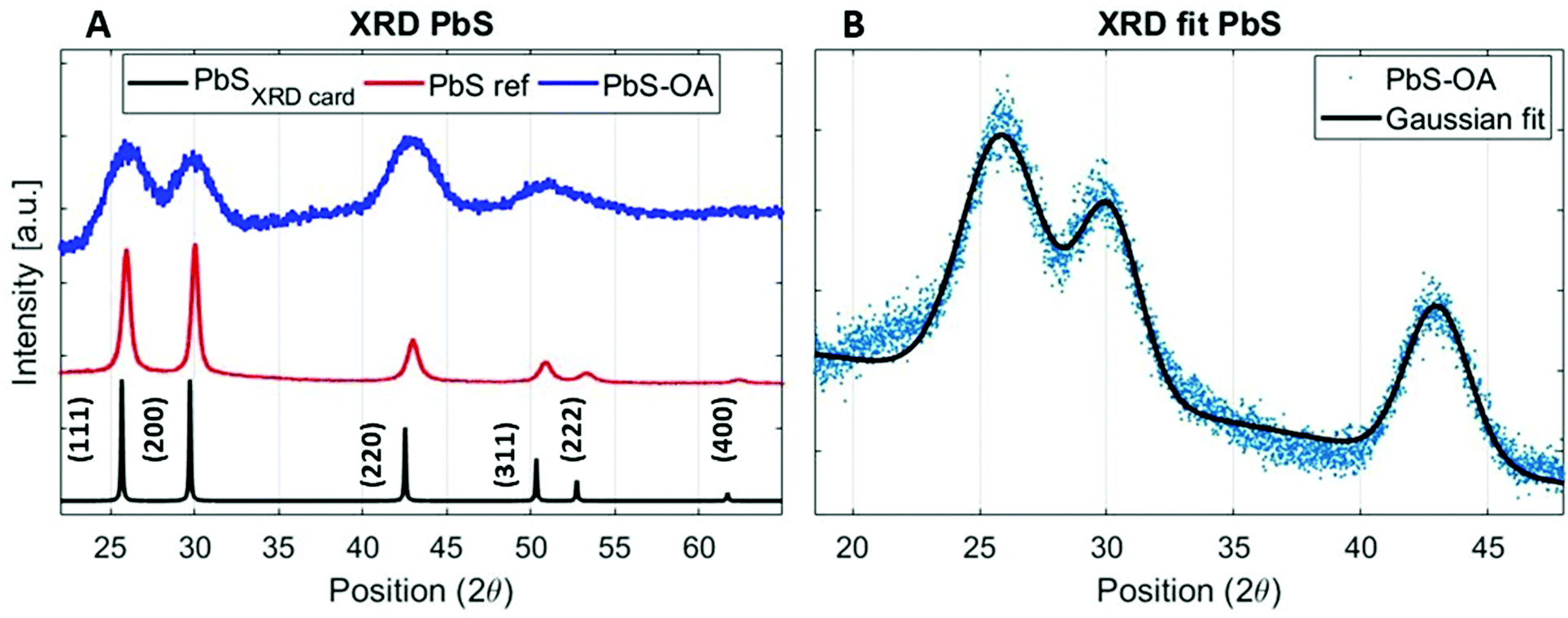 | ||
| Fig. 1 (A)XRD pattern of the PbS ref thin film (PbS ref, red line) and PbS–OA QD thin film (blue line) compared to the XRD pattern of the PbS single crystal (black line) obtained from Materials Data on PbS by Materials Project.40 (B)Gaussian fit of the strongest peaks (111), (200) and (220) of the PbS–OA QD XRD pattern. The XRD measurement was done using a Cu Kα X-ray source. | ||
| Direction | Position XRDcard (2θ) | Position PbS ref (2θ) | Position PbS–OA (2θ) | FWHM PbS ref (2θ) | FWHM PbS–OA (2θ) |
|---|---|---|---|---|---|
| (111) | 25.7 | 25.9 | 25.9 | 0.6 | 3.8 |
| (200) | 29.7 | 30.0 | 30.1 | 0.5 | 2.9 |
| (220) | 42.5 | 43.0 | 43.0 | 0.9 | 2.9 |
PES overview spectra of the quantum dot samples measured at 3000 eV and 6100 eV photon energy (Fig. S5 and S6, ESI†) show core levels as expected from the chemical content (Pb and S core levels from PbS and Br and I core levels from the ligands). In addition to this, the overview spectra show the presence of C 1s and O 1s peaks. This is typical in all PES measurements of ex situ prepared samples, and does not significantly affect the measurements when using high photon energies.41 High resolution core level spectra recorded with a photon energy of 3000 eV for the PbS QD (1:1) sample compared to the reference samples are shown in Fig. 2. The spectra of the reference samples were internally energy calibrated to the anion core levels of the quantum dot samples (to Br 3d, I 4d and S 2p), shown in Fig. 2B–D. Using this calibration, the possible variations in Fermi level position, caused by different doping levels of the materials are avoided. Furthermore, a small amount of charging under X-ray irradiation was observed for the lead halide samples and some of the quantum dot samples, which makes calibration against an external reference unreliable for these samples.
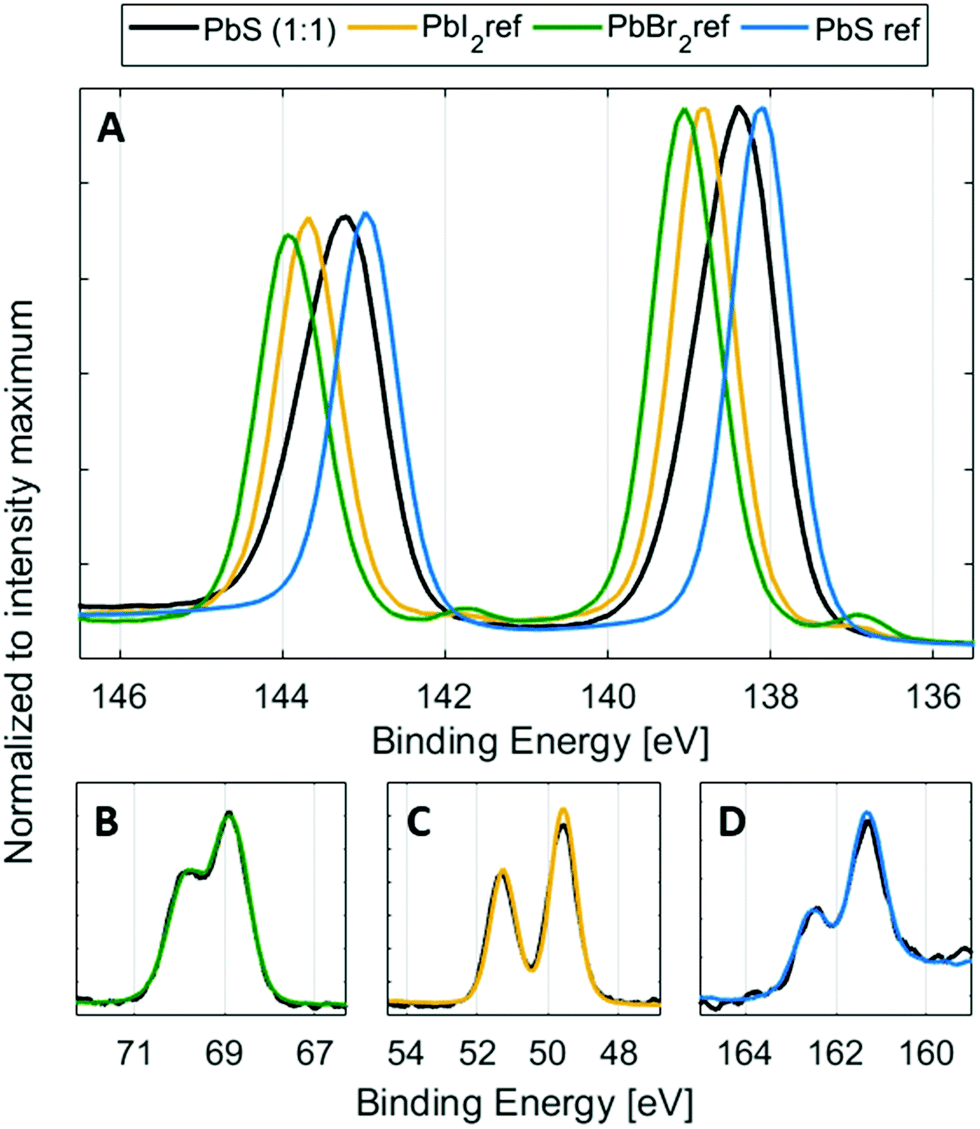 | ||
| Fig. 2 Pb 4f spectra of the PbS (1:1) sample compared to halide reference samples and the PbS reference sample (A) at 3000 eV photon energy, normalized to the intensity maximum of Pb 4f7/2 showing the different positions of the Pb4f peak in different samples. The spectra B, C, and D are normalized to the intensity of Br 3d, I 4d and S 2p, respectively and spectra from the samples shown here have an internal energy calibration to the core levels of the quantum dots, e.g. positions of Br 3d (68.9 eV) (B), I 4d (49.6 eV) (C) and S2p (161.3 eV) (D). | ||
Using this internal calibration, the spectra of the anion core levels clearly overlap for the quantum dot samples and reference samples (Fig. 2B–D), while the Pb 4f spectra show clear differences (Fig. 2A). The lead halide samples show two doublets, where the main one (Pb 4f7/2 at 138.8 eV for PbI2, at 139.0 eV for PbBr2, Fig. S7, ESI†) is assigned to the lead halide bonds. Another lead species is observed at a lower binding energy and can be assigned to metallic Pb46 (Pb 4f7/2 at 137.1 eV, Fig. 2 and Fig. S7, ESI†), which can form during X-ray irradiation of lead halides.47 Such a signal is not observed in the lead sulfide samples during short X-ray illumination times. The PbS ref shows a single Pb 4f doublet with a Pb 4f7/2 peak at 138.1 eV. The PbS quantum dot sample shows a wider Pb 4f doublet with its main intensity at a higher binding energy than for the PbS ref sample and at a lower binding energy than for the lead halide reference samples. The increased width of the peak agrees with the contribution of different chemical environments of Pb atoms to the signal. The reference samples suggest that the bonds should be ordered going from low to high binding energy in the following way: Pb–S, Pb–I and Pb–Br. It should be noted however, that the binding energies in the reference samples are not expected to reflect the exact binding energies in the quantum dot samples, as:
1. The halide anions should be bonded to the surface of the lead sulfide which means that those lead atoms which are bonded to a halide would also have a sulfur atom in their vicinity. They would therefore not appear at the same binding energy as Pb atoms in the lead halide references.
2. Quantum confinement effects cause a difference in the relative binding energies of sulfur and lead core levels between quantum dots and the non-quantized PbS reference (Fig. 2A and Table S1, ESI†).
Additionally, the Pb 4f spectra of the quantum dots can also contain a contribution of Pb–O bonds, if the surface of PbS has oxidized in air.48 The position of Pb–O bonds could vary depending on the type of lead oxide formed. In the case of the surface non-treated PbS ref sample mentioned above, two additional Pb–O peaks were observed, one shifted by 0.7 eV and one shifted by 1.4 eV to higher binding energies compared to the peak from Pb–S. There could therefore be more than four different contributions to the Pb 4f core level of the quantum dot samples. However, due to the small binding energy differences between them (see Table S1, ESI†), it is extremely difficult to create and control a fit model that gives meaningful results in this way. Instead, we created a fit model, where the number of lead species is reduced to two, representing two groups of lead types (see Fig. 3). The first (main) peak is assigned to the combination of the main Pb–S contribution and the Pb–I contribution. The second lead peak is fixed to a position of 0.6 eV higher than the first peak and may contain Pb–Br bonds, different Pb–O bonds and possibly a small amount of Pb–I bonds. In a study by Lobo et al. of oleic acid and trioctylphosphine-capped PbS quantum dots, they similarly assign the main (lower binding energy) peak to Pb atoms in the bulk and the second peak to surface Pb atoms, assigned to Pb–O bonds.49 In our study, the relative intensity of the second peak increases (Fig. S8, ESI†), as the bromide content increases. This confirms that the peak can be attributed mostly to Pb–Br bonds, observed at a higher binding energy than the Pb–I and Pb–S bonds (Fig. 2 and 3) with possible contribution from Pb–O bonds.
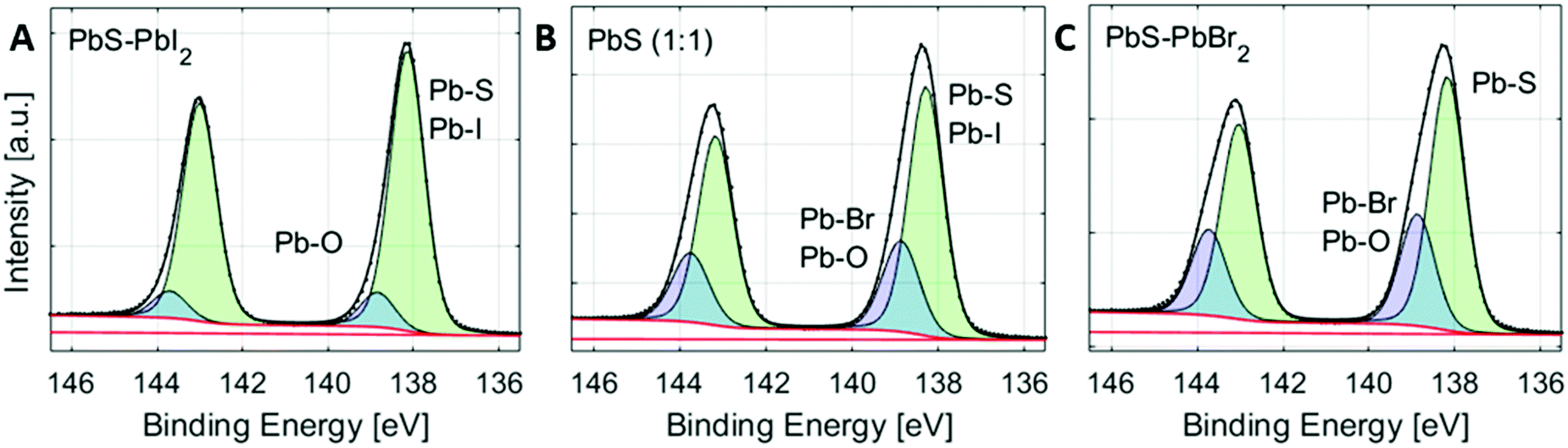 | ||
| Fig. 3 Pb 4f core level spectra including curve fits and assignation for samples PbS–PbI2 (A), PbS (1:1) (B) and PbS–PbBr2 (C) measured at 3000 eV photon energy and internally calibrated to the binding energy of S 2p = 161.3 eV (determined for the PbS (1:1) QD sample. | ||
High-resolution core level spectra of S 2p, Pb 4f, Br 3d and I 4d of the PbS QD samples at 3000 eV incident photon energy are shown in Fig. 4. All spectra were fitted with spin–orbit doublets, calibrated internally to the position of the S 2p core-level in the 1:1 sample and normalized to the total amount of S 2p. The S 2p spectra of all QD samples show similar shapes with only one type of sulfur observed assigned to the lead sulfide bonds, as observed in other studies.49,50 The shape of the Pb 4f spectra varies with halide content as discussed above. However, the positions of the lead core levels relative to the sulfur core levels are similar for all quantum dot samples (Table S1, ESI†). The Br 3d and I 4d spectra in Fig. 4C and Fig. 4D show a clear increase in bromide concentration and decrease in iodide concentration going from the PbS–PbI2 QD sample to the PbS–PbBr2 QD sample. The same trends are observed when using 6100 eV incident photon energy (Fig. S9, ESI†) and the binding energy positions of the core levels measured at this energy also show very similar positions to the ones measured at 3000 eV (Tables S1 and S2, ESI†) which implies that the measurements with the two photon energies give comparable results.
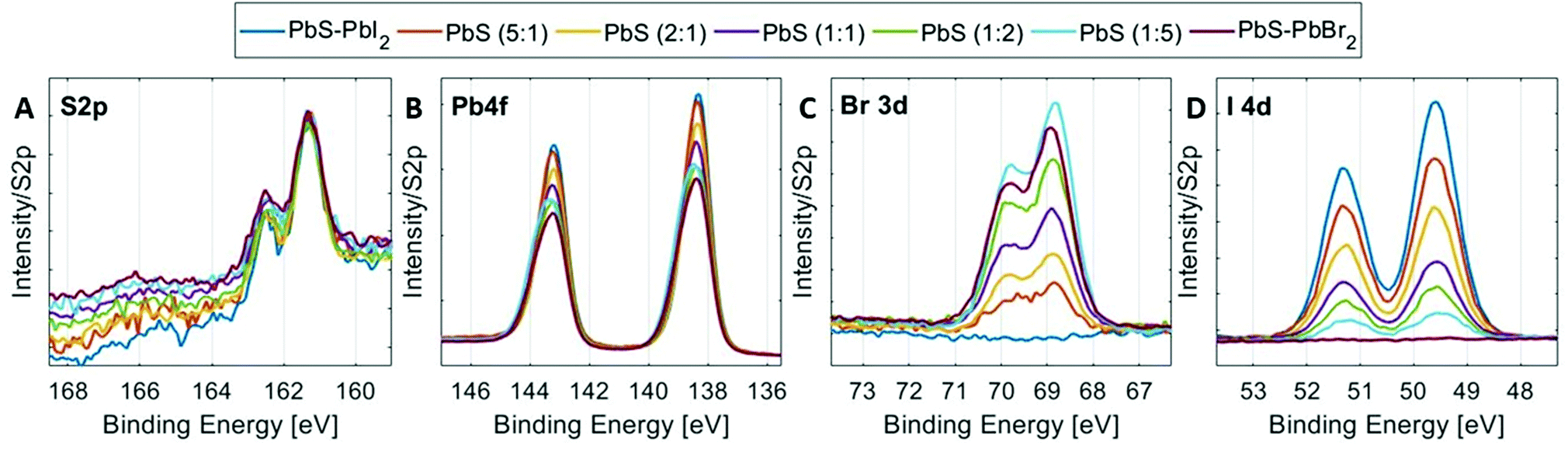 | ||
| Fig. 4 HAXPES spectra of the PbS QD samples. (A) S 2p (B) Pb 4f (C) Br 3d and (D) I 4d regions, normalized to the total S 2p intensity and internally calibrated to S 2p3/2 at 161.3 eV (determined for the PbS (1:1) QD sample). | ||
Relative concentrations of the different elements were estimated from the fitted intensities using photoionization cross sections. The experimental halide ratios in the QD samples are in good agreement with the ratios expected from synthesis, as shown in Fig. 5A. In addition to this, Fig. 5 shows the ratios of total halogen (B) and total sulfur (C) to total lead for all the QD samples at the two photon energies. Due to the low ionization cross-section (Fig. S10, ESI†) and thus low signal-to-noise ratio that would make a quantification comparison with the Br 3d core level less reliable, Br 3p was used instead of the Br 3d core level at 6100 eV. Moreover, in the case of sulfur, there are three options for comparison – S 2p, S 2s and S 1s. One could argue that the S 1s core level is the most convenient one to compare to as it has the highest ionization cross section (Fig. S10, ESI†). However, due to the increased surface sensitivity at this photon energy (see the calculated probing depth values for different core levels51 in Table S3, ESI†), the S 1s is not considered a good choice for quantification at the photon energy of 3000 eV. When it comes to the S 2p core level, Laajalehto et al. show in their study of galena that determination of the S 2p area can be less precise and thus more uncertain due to the vicinity of the strong Pb 4f background.48 Hence, we have decided to use the S 2s core level in the quantification analysis despite the fact that this peak has a much greater FWHM (approx. 1.7 eV), compared to the other sulfur core levels. This could add some uncertainty in the fitting of different sulfur types, but it is not expected to have any significant effect in comparing the total amount of sulfur that is used in such calculations. In addition to this, one should keep in mind that the difference in molecular orbital angular momentum (s, d and f), differences in kinetic energies, and uncertainties in the cross-section all affect the final quantification results to some extent. The relative S/Pb ratios in the QD samples studied here are lower compared to our previous study (0.4 compared to 0.6)41 and the quantification of the PbS ref sample showed a S 2s to Pb 4f ratio of 0.7, which suggests that the ratios found here are lower than the true atomic ratio in the compound and therefore our study focusses on trends rather than the absolute values.
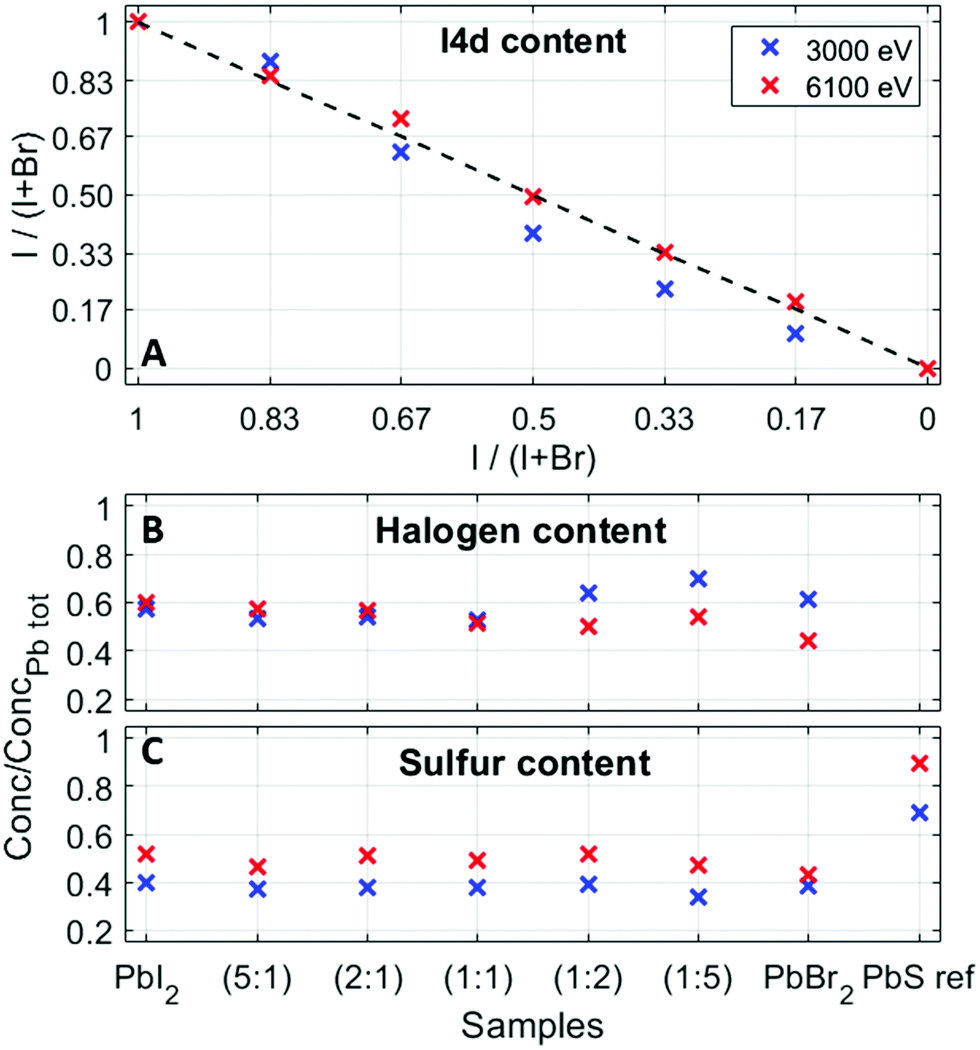 | ||
| Fig. 5 (A) Nominal and experimental values of iodine content, (B) total halogen (Br 3d and I 4d at 3000 eV and Br 3p and I 4d at 6100 eV) to total lead (Pb 4f), and (C) sulfur (S 2s) to total lead (Pb 4f). The uncertainties in the fitted ratios are within the size of the markers. | ||
The ratio of (I 4d + Br 3d)/Pb 4f at 3000 eV shows slightly higher values in Br-rich samples (lowest 0.53; highest 0.72), while the ratio of the (I 4d + Br 3p)/Pb 4f at 6100 eV varies between 0.45 and 0.60, and slightly higher values are observed in the I-rich samples (Fig. 5B). As about 50% of the signal stems from the first quantum dot layer for measurements with 3000 eV photon energy compared to 30% with 6000 eV (Table S3, ESI†), this could suggest that there is an increased number of halides closer to the sample surface in bromide-rich samples. This matches the findings in a study of QD film stability52 and our previous study of electron dynamics in PbS QD thin films,53 where we observed iodide loss at the sample surface over time. It also raises a question of the possibility of halide migration in quantum dot films, which has been observed by photoelectron spectroscopy in lead halide perovskite materials.54 However, as the sulfur to lead ratio (Fig. 5C) is relatively constant at both photon energies, when going from PbS–PbI2 to PbS–PbBr2, one could conclude that the halide ligand variation does not to change the relative amount of sulfur in the quantum dots nor affects the structural properties of the quantum dots. The absolute differences in the ratios at 3000 and 6100 eV can be assigned to the effects discussed above.
Shape and position of the valence band
Another essential quality of the semiconductor materials are the positions of the valence and conduction band edges as well as the orbital composition of the bands. To determine the exact positions and contributions in the valence band of the quantum dot materials, we recorded the valence band spectra at 3000 eV for the reference and QD samples (Fig. 6). As discussed above, we use an internal calibration for our PE spectra and the valence band spectra shown in Fig. 6 are calibrated to the Pb 5d core level at the position as given in Table S1 (ESI†). When comparing the valence band (VB) spectra of the PbS QD (1:1) sample to those of the reference samples (Fig. 6A), we observe clear differences in the VB feature shape as well as in the position of the valence band maximum (VBM). The VBM is observed at the lowest binding energy for the PbS ref, followed by PbS (1:1), PbI2 and finally PbBr2. From the literature we expect that the main density of the PbS contribution to the valence band should come from the S 3p orbitals closest to the valence band edge and Pb 6s states between 8 and 12 eV,55,56 while in lead halides the main density comes from the Br 4p and I 5p states (between 2 and 5 eV).57–59
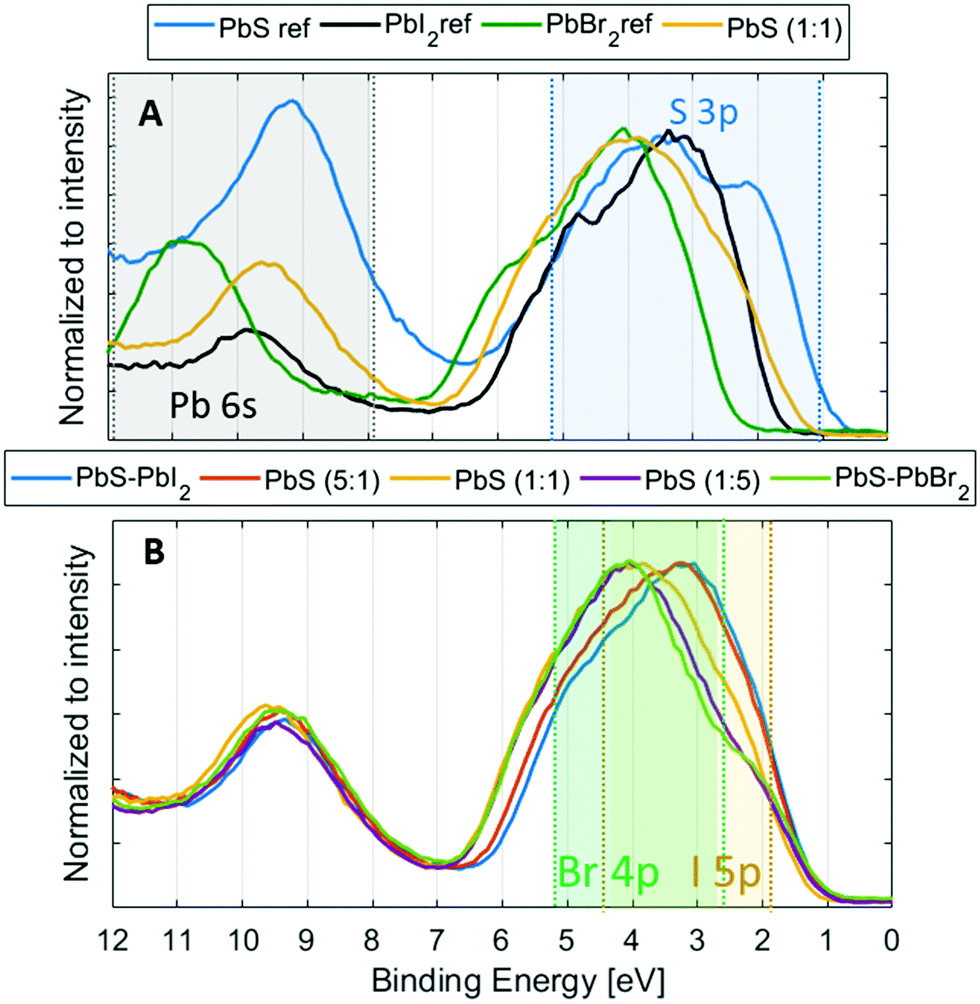 | ||
| Fig. 6 (A) Valence band spectra of PbS (1:1), PbS ref and halide references. (B) Valence band spectra of the QD samples. The S 3p, Pb 6s, Br 4p and I 5p state contributions are highlighted with blue, grey, green and yellow areas, respectively. The spectra in the figure are calibrated to Pb 5d5/2 = 19.5; 19.3; 19.9 and 20.2 eV for QD samples, PbS ref, PbI2 ref and PbBr2 ref, respectively, and normalized to the intensity of the main valence band feature within the range 0–7 eV. | ||
All these contributions should be observed in the valence spectra of the quantum dots and the regions where they are expected to occur are highlighted in Fig. 6. The shape of the VB feature of the QD samples is different from both PbS and halide references (Fig. 6A), and the intensity maximum shifts from about 3 eV to 4 eV as the ligand content in the QDs changes from iodide rich to bromide rich (Fig. 6B). From the reference spectra, the S 3p states should be observed closest to the valence band edge followed by I 5p and Br 4p states and from this we observe that the halide ligands indeed contribute little to the states closest to the valence band edge in the PbS QDs, while the biggest influence on the VBM comes from the S 3p orbital, as previously suggested.57 As a result of this, we observe a clear change in the energy at which the valence band is most intense, but not in the position of the valence band maximum (Fig. 6B). Therefore, the position of the valence band edge relative to the Pb 5d level appears relatively unaffected by the composition of the halide ligands.
When comparing the quantum dot samples to the PbS reference, we observe a clear difference in the energy of the valence band maximum. This means that the energy difference between the S 2p core level and the VBM is lower for the quantum dot sample than for the PbS reference. The quantum confinement, therefore, changes the energy of the valence band edge for the quantum dot sample relative to the PbS reference. The energy of the valence band maximum (EVBM) can be determined by linear or logarithmical extrapolation of the leading edge to where photoemission intensity from the valence band decreases into the background noise. The position of the conduction band (CB) can then indirectly be calculated as the energy difference between the optical bandgap and VB position (Egap − EVBM). Linear and logarithmical extrapolation often give different values, as is also the case in our study (see Table S4, ESI†). For PbS, it has been shown that the linear extrapolation method often gives unrealistic values.60 Due to changes in the density of states coming from the quantum confinement, the difference in values obtained from linear extrapolation and to realistic valence band maxima decreases with increasing quantum confinement. As we are comparing quantum dots of the same size, the same offset should apply, and we can use both linear and logarithmic extrapolation to give upper and lower bounds of the valence band maximum. For the PbS reference sample, the linear extrapolation is likely to lead to a larger percentage overestimation of the binding energy of the VBM (Table S4, ESI†). Indeed, the value of 0.57 eV found is larger than the bandgap of non-quantized PbS (0.37 eV) and it is therefore unrealistic. On the other hand, a value of 0.20 eV is found with logarithmic extrapolation, which might be a more realistic value. When comparing values estimated by the logarithmic and linear extrapolation method (see Fig. 7 and Fig. S11, Table S4, ESI†), the logarithmic method gives EVBM values, which are approximately 0.45 eV lower than those determined by linear extrapolation. However, the values for all QD samples are quite similar (within 0.15 eV of each other). For the quantum dot samples, binding energy shifts of up to 0.1 eV were observed during measurements. The zero binding energy position in Fig. 7 is therefore likely to be within 0.1 eV of the Fermi level in the dark. All quantum dot samples therefore show n-type character, as has been observed previously for iodide- and bromide-capped quantum dots,27,61 although bromide-capped PbS have also shown a p-type character after being exposed to air for several hours in the study bt Ning et al.62 The valence band maximum for the quantum dots with (5:1) and (1:1) ligand composition is at a somewhat higher binding energy than for the other QD samples, suggesting that these potentially are more n-type than the other samples. However, as the energy differences are only within the 0.1 eV range, the previous must be taken with great caution.
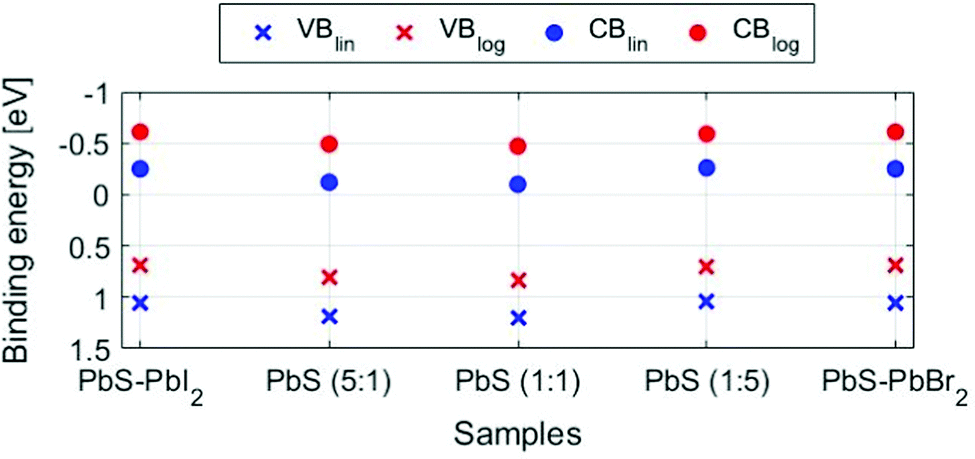 | ||
| Fig. 7 Positions of valence (x) and conduction (●) bands in the QD samples using linear extrapolation (blue dots) and logarithmic extrapolation methods (red dots), where the conduction band is calculated by subtracting the bandgap value (1.3 eV) from the valence band position. The spectra were calibrated internally to the position of the Pb 5d5/2 = 19.5 eV. The uncertainties in the fitted energy positions are within the size of the markers. | ||
Surface oxidation of quantum dot samples
The surface oxidation of PbS QD materials can be investigated with measurements of the S 1s spectra at different photon energies. At 3000 eV incident photon energy, electrons emitted from the S 1s core level have a kinetic energy of approximately 530 eV, giving a mean free path of 1.3 nm for the photoelectrons.51 This means that the measured electrons come from the surface of the film (over 90% of the signal comes from the top quantum dot layer, Table S3, ESI†). At 6100 eV, the kinetic energy of S 1s electrons is approximately 3600 eV and approximately 42% of the signal comes from the top quantum dot layer (Table S3, ESI†). Therefore, the sample surface is probed significantly more with 3000 eV than with 6100 eV for this core level. The S 1s spectra of the QD samples at 3000 eV (Fig. 8A and B) show more than one type of sulfur, while the S 2p spectra at the same energy (Fig. 4A) and the S 1s spectra at 6100 eV (Fig. 8C and D) show only one type of sulfur. The additional sulfur species observed in the S 1s spectra at 3000 eV therefore stem from the sample surface. The singlet at the lowest binding energy (2468.9 eV) is assigned to sulfur bonded to lead in PbS, followed with the singlets of sulfur species bonded to H and C, and at the highest binding energy the sulfur bonded to oxygen,41 which suggests that the surface of the samples is oxidized to some extent. No trend in the amount of surface oxidation in relation to ligand composition was observed (Fig. S12, ESI†), which suggests that the ligand composition does not significantly affect the amount of surface oxidation. Furthermore, no oxidation of the quantum dots is observed toward the bulk of the samples.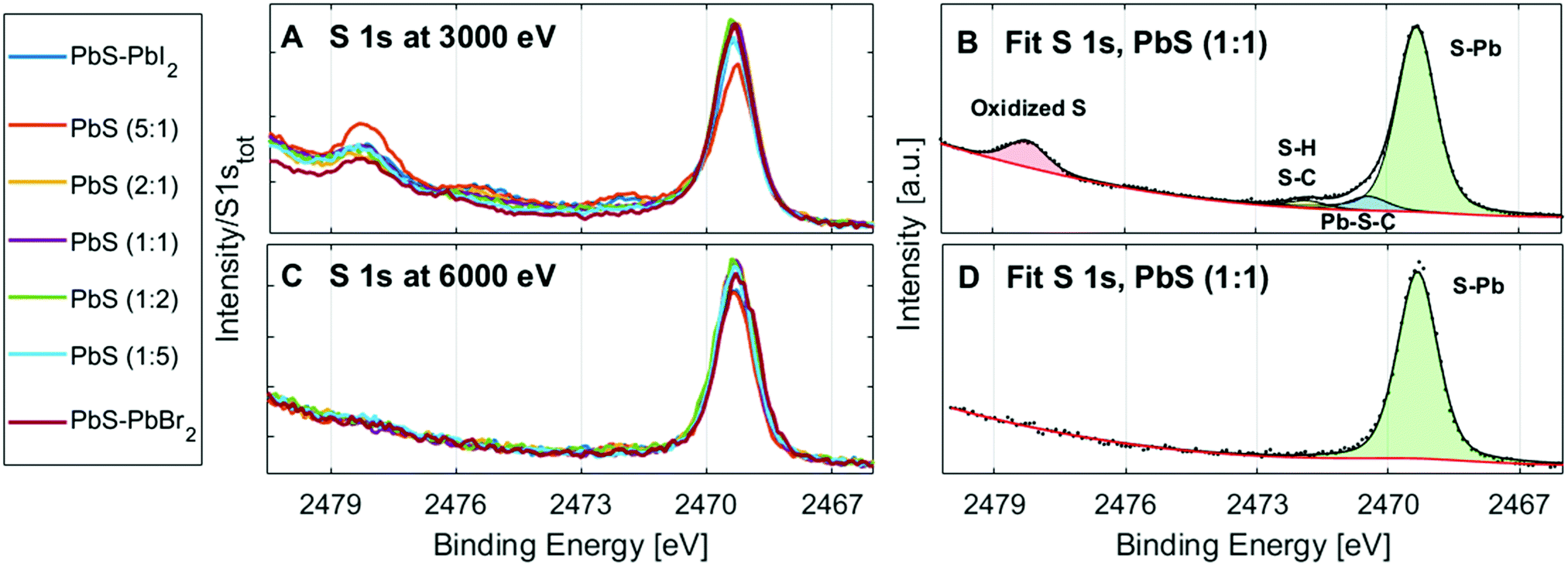 | ||
| Fig. 8 S1s spectra of all the PbS QD samples from the series at 3000 eV (A) and 6100 eV (C), energy calibrated to the PbS (1:1) sample and normalized to the total amount of the S 1s. Curve fits and total fit of the S 1s in the PbS (1:1) QD sample at 3000 eV (B) and 6000 eV (D). | ||
Stability under X-rays
As previously discussed, lead halides can degrade under X-ray radiation47 and this raises a question regarding the X-ray stability of lead halides as surface ligands in PbS quantum dots. Therefore, we investigated how chemical composition on the surface of QD samples changes under longer exposure to X-rays. The spectral evolution of the Pb 4f, I 4d, Br 3d and S 1s core levels during several hours of continuous X-ray illumination at 3000 eV of PbS–PbI2, PbS–PbBr2 and PbS (1:1) QDs is shown in Fig. 9. The measurements were run as a repeating sequence, where the measuring time for each iteration was approximately 30 min. While there are no major changes in the recorded core levels of the PbS-PbI2 and PbS (1:1) samples, the formation of metallic Pb at a lower binding energy is visible for the PbS-PbBr2 QD sample after just a few hours of measurement, as well as the decrease of the Br 3d intensity relative to the Pb 4f intensity in the PbS–PbBr2 QD sample. Similar behavior of PbBr2 was observed in the study of lead halide crystals by Verwey,63 where the mechanism of degradation of lead halides under visible light was described. The study showed that with absorption of light and formation of excitons, the generated holes will move closer to the surface and react with anions in which case halide gas is evolved. The electrons will remain trapped by Pb2+ ions forming in this way areas of metallic Pb according to the reaction:| PbBr2 → Pb0 + Br2(g) | (2) |
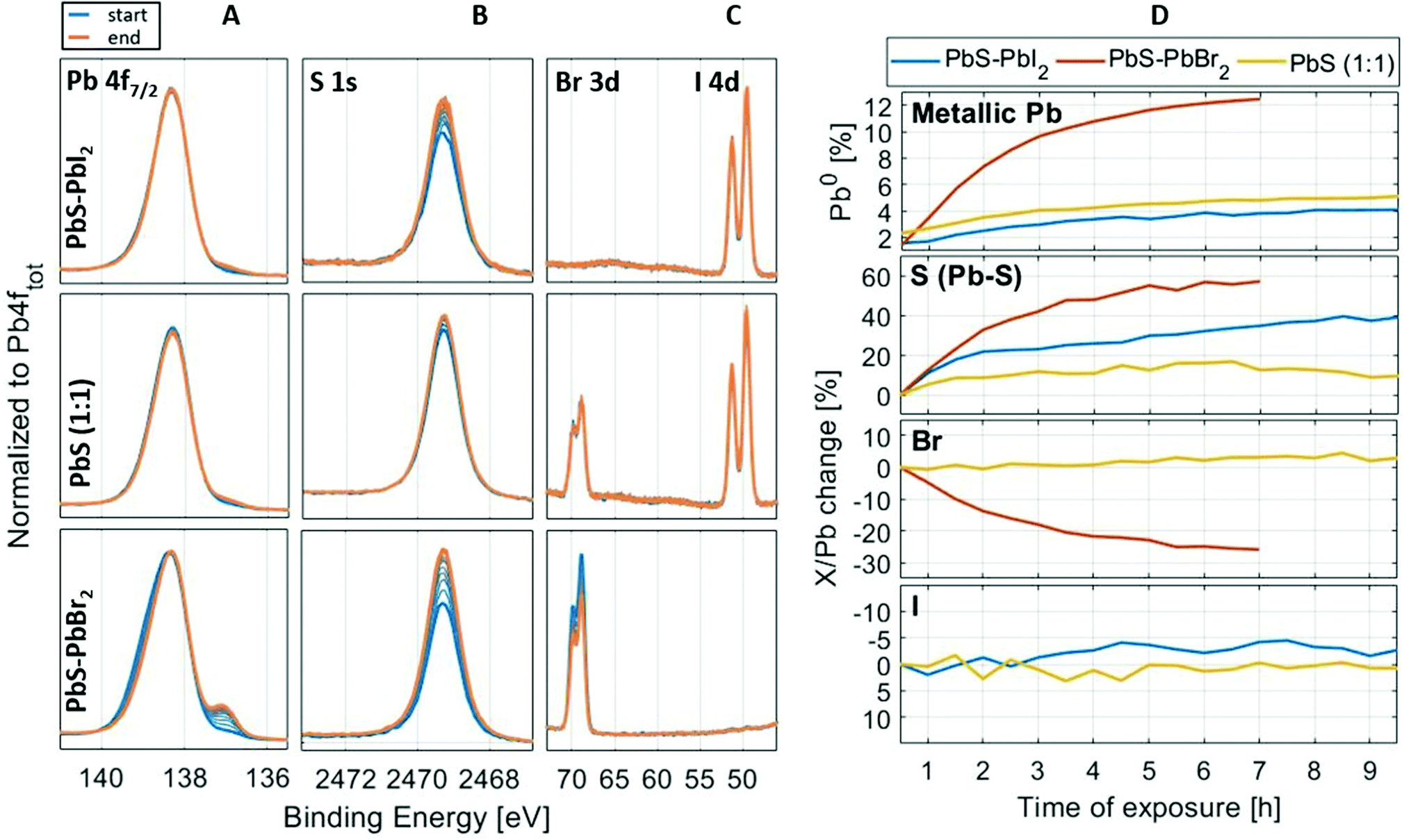 | ||
| Fig. 9 Stability measurements of samples PbS–PbI2, PbS (1:1) and PbS–PbBr2. Panels on the left show the spectral evolution of the Pb 4f7/2 (A column), S 1s (B column), I 4d and Br 3d (C column) core levels, whereas the panels on the right (D column) show the evolution of the metallic Pb (Pb 4f), Pb–S sulfur peak (S 1s) and halogen (Br 3d and I 4d) core level intensities over time, in relation to the total intensities of Pb 4f. The X/Pb stands for the ratio of the core level intensity (S 1s, I 4d or Br 3d) to the total intensity of lead (Pb 4f). The X/Pb change is normalized to the “time zero” ratio (ratio at the beginning of the measurement). The stability measurement took 7–10 h depending on the sample. All spectra are internally energy calibrated (see Fig. 2). | ||
Conclusions
In this study, we successfully synthesized PbS quantum dots with surface ligands in which the composition varied from pure lead iodide to pure lead bromide and investigated their chemical and electronic structure with hard X-ray photoelectron spectroscopy (HAXPES). Quantification of the ligand composition using HAXPES data closely matches the one employed in the synthesis, confirming that we can precisely control the ligand composition of the quantum dot surface. Measurements of reference samples were used to distinguish the contributions of the lead sulfide and ligands to the Pb 4f core level and valence band spectra. In the Pb 4f signal, we observed contributions from lead bonded to sulfur in the QD nanocrystals (Pb–S) and contributions from the surface of the QD nanocrystal (Pb–I, Pb–Br, Pb–O). Using the reference films, we were able to assign these contributions from lowest to highest binding energy in the following sequence: Pb–S, Pb–I, Pb–Br and Pb–O. The reference films also enabled us to assign the contributions of the S 3p, Pb 6s, Br 4p and I 5p states to the position and shape of the valence band in the PbS quantum dots. While the halide content of the ligands determines the overall shape of the valence band spectrum, it has a minimal impact on the position of the valence band edge and the quantum dot materials are n-type for all iodide/bromide ratios. While a small amount of surface oxidation was observed through a surface sensitive measurement of the S 1s core level for all QD samples, no oxidation was observed in more bulk sensitive measurements. The bulk of the QD films therefore remains unchanged and of high purity, and both iodide and bromide ligands are able to effectively prevent oxidation of the quantum dot surfaces within a quantum dot solid. We also showed that surface treatment with iodide was able to prevent the surface oxidation of a reference lead sulfide film.While our results show that the ligand composition did not impact the initial surface oxidation, the sample stability when exposed to X-rays is strongly influenced by the choice of halide. Iodide rich QD samples were the most stable, showing almost no chemical changes over several hours of X-ray exposure, while the bromide rich QD samples already showed changes in the first hour. Future studies should determine whether similar trends can be observed for illumination with photons of different energies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge research funding from the Swedish Research Council (Grant No. VR 2018-04125), the Göran Gustafsson foundation, the Swedish Foundation for Strategic Research (project nr. RMA15-0130), and the Carl Tryggers foundation (Grant No. CTS 18:59). We thank David Fermin and Devendra Tiwari for advice on the synthesis of PbS reference films. Measurements were carried out at the HIKE end-station on the KMC-1 beamline at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin für Materialien und Energie. We would like to thank Roberto Felix Duarte for assistance during the experiment. The research leading to this result has been supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020.References
- F. W. Wise, Lead salt quantum dots: The limit of strong quantum confinement, Acc. Chem. Res., 2000, 33, 773–780 CrossRef CAS PubMed.
- A. L. Rogach, A. Eychmüller, S. G. Hickey and S. V. Kershaw, Infrared-emitting colloidal nanocrystals: Synthesis, assembly, spectroscopy, and applications, Small, 2007, 3, 536–557 CrossRef CAS PubMed.
- J. Y. Kim, O. Voznyy, D. Zhitomirsky and E. H. Sargent, 25th anniversary article: Colloidal quantum dot materials and devices: A quarter-century of advances, Adv. Mater., 2013, 25, 4986–5010 CrossRef CAS PubMed.
- X. Gong, Z. Yang, G. Walters, R. Comin, Z. Ning, E. Beauregard, V. Adinolfi, O. Voznyy and E. H. Sargent, Highly efficient quantum dot near-infrared light-emitting diodes, Nat. Photonics, 2016, 10, 253–257 CrossRef CAS.
- S. Pradhan, F. Di Stasio, Y. Bi, S. Gupta, S. Christodoulou, A. Stavrinadis and G. Konstantatos, High-efficiency colloidal quantum dot infrared light-emitting diodes via engineering at the supra-nanocrystalline level, Nat. Nanotechnol., 2019, 14, 72–79 CrossRef CAS PubMed.
- M. Vijaya Bharathi, S. Maiti, B. Sarkar, K. Ghosh and P. Paira, Water-mediated green synthesis of Pbs quantum dot and its glutathione and biotin conjugates for non-invasive live cell imaging, R. Soc. Open Sci., 2018, 5, 171614 CrossRef CAS PubMed.
- L. Gao, D. Dong, J. He, K. Qiao, F. Cao, M. Li, H. Liu, Y. Cheng, J. Tang and H. Song, Wearable and sensitive heart-rate detectors based on PbS quantum dot and multiwalled carbon nanotube blend film, Appl. Phys. Lett., 2014, 105, 153702 CrossRef.
- A. De Iacovo, C. Venettacci, L. Colace, L. Scopa and S. Foglia, High Responsivity Fire Detectors Based on PbS Colloidal Quantum Dot Photoconductors, IEEE Photonics Technol. Lett., 2017, 29, 703–706 CAS.
- A. Mosahebfard, H. Dehdashti Jahromi and M. H. Sheikhi, Highly Sensitive, Room Temperature Methane Gas Sensor Based on Lead Sulfide Colloidal Nanocrystals, IEEE Sens. J., 2016, 16, 4174–4179 Search PubMed.
- M. Al Mubarok, F. Tri, A. Wibowo, H. Aqoma, N. V. Krishna, W. Lee, D. Y. Ryu, S. Cho, I. H. Jung, S. Y. Jang, F. T.-A. Wibowo, H. Aqoma, N. Vamsi Krishna, W. Lee, D. Y. Ryu, S. Cho, I. H. Jung and S. Y. Jang, PbS-Based Quantum Dot Solar Cells with Engineered I -Conjugated Polymers Achieve 13% Efficiency, ACS Energy Lett., 2020, 5, 3452–3460 CrossRef.
- S. Zheng, J. Chen, E. M.-J. Johansson and X. Zhang, PbS Colloidal Quantum Dot Inks for Infrared Solar Cells, iScience, 2020, 23, 101753 CrossRef CAS PubMed.
- G. L. Wang, K. L. Liu, J. X. Shu, T. T. Gu, X. M. Wu, Y. M. Dong and Z. J. Li, A novel photoelectrochemical sensor based on photocathode of PbS quantum dots utilizing catalase mimetics of bio-bar-coded platinum nanoparticles/G-quadruplex/hemin for signal amplification, Biosens. Bioelectron., 2015, 69, 106–112 CrossRef CAS PubMed.
- Y. Zhang, R. Xu, Q. Kang, Y. Zhang, Q. Wei, Y. Wang and H. Ju, Ultrasensitive Photoelectrochemical Biosensing Platform for Detecting N-Terminal Pro-brain Natriuretic Peptide Based on SnO2/SnS2/mpg-C3N4 Amplified by PbS/SiO2, ACS Appl. Mater. Interfaces, 2018, 10, 31080–31087 CrossRef CAS PubMed.
- D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Prospects of colloidal nanocrystals for electronic and optoelectronic applications, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- S. V. Kershaw, A. S. Susha and A. L. Rogach, Narrow bandgap colloidal metal chalcogenide quantum dots: Synthetic methods, heterostructures, assemblies, electronic and infrared optical properties, Chem. Soc. Rev., 2013, 42, 3033–3087 RSC.
- C. R. Kagan, E. Lifshitz, E. H. Sargent and D. V. Talapin, Building devices from colloidal quantum dots, Science, 2016, 353, aac5523 CrossRef PubMed.
- B. M.-G. and, C. B. Murray and C. R. Kagan, Synthesis and Characterization of Monodisperse Nanocrystals and Close-Packed Nanocrystal Assemblies, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef.
- S. Kashyap, R. Pandey, J. Madan and R. Sharma, Design and Simulation of a-Si:H/PbS Colloidal Quantum Dots Monolithic Tandem Solar Cell for 12% Efficiency, Phys. Status Solidi A, 2020, 217, 1–13 CrossRef.
- L. Yuan, H. Michaels, R. Roy, M. Johansson, V. Öberg, A. Andruszkiewicz, X. Zhang, M. Freitag and E. M.-J. Johansson, Four-Terminal Tandem Solar Cell with Dye-Sensitized and PbS Colloidal Quantum-Dot-Based Subcells, ACS Appl. Energy Mater., 2020, 3, 3157–3161 CrossRef CAS.
- H. Il Kim, S. W. Baek, H. J. Cheon, S. U. Ryu, S. Lee, M. J. Choi, K. Choi, M. Biondi, S. Hoogland, F. P.-G. de Arquer, S. K. Kwon, Y. H. Kim, T. Park and E. H. Sargent, A Tuned Alternating D–A Copolymer Hole-Transport Layer Enables Colloidal Quantum Dot Solar Cells with Superior Fill Factor and Efficiency, Adv. Mater., 2020, 32, 1–7 Search PubMed.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, The surface science of nanocrystals, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed , Nat. Mater., 2016, 15, 364.
- M. A. Hines and G. D. Scholes, Colloidal PbS Nanocrystals with Size-Tunable Near-Infrared Emission: Observation of Post-Synthesis Self-Narrowing of the Particle Size Distribution, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
- N. Ray, N. E. Staley, D. D.-W. Grinolds, M. G. Bawendi and M. A. Kastner, Measuring Ligand-Dependent Transport in Nanopatterned PbS Colloidal Quantum Dot Arrays Using Charge Sensing, Nano Lett., 2015, 15, 4401–4405 CrossRef CAS PubMed.
- L. Hu, A. Mandelis, Z. Yang, X. Guo, X. Lan, M. Liu, G. Walters, A. Melnikov and E. H. Sargent, Temperature- and ligand-dependent carrier transport dynamics in photovoltaic PbS colloidal quantum dot thin films using diffusion-wave methods, Sol. Energy Mater. Sol. Cells, 2017, 164, 135–145 CrossRef CAS.
- D. Becker-Koch, M. Albaladejo-Siguan, V. Lami, F. Paulus, H. Xiang, Z. Chen and Y. Vaynzof, Ligand dependent oxidation dictates the performance evolution of high efficiency PbS quantum dot solar cells, Sustainable Energy Fuels, 2020, 4, 108–115 RSC.
- C. Ding, F. Liu, Y. Zhang, S. Hayase, T. Masuda, R. Wang, Y. Zhou, Y. Yao, Z. Zou and Q. Shen, Passivation Strategy of Reducing Both Electron and Hole Trap States for Achieving High-Efficiency PbS Quantum-Dot Solar Cells with Power Conversion Efficiency over 12%, ACS Energy Lett., 2020, 5, 3224–3236 CrossRef CAS.
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulović, Energy level modification in lead sulfide quantum dot thin films through ligand exchange, ACS Nano, 2014, 8, 5863–5872 CrossRef CAS PubMed.
- P. Papagiorgis, D. Tsokkou, K. Gahlot, L. Protesescu, A. Manoli, F. Hermerschmidt, C. Christodoulou, S. A. Choulis, M. V. Kovalenko, A. Othonos and G. Itskos, Exciton–ligand interactions in PbS quantum dots capped with metal chalcogenides, J. Phys. Chem. C, 2020, 124, 27848–27857 CrossRef CAS.
- C. Giansante, I. Infante, E. Fabiano, R. Grisorio, G. P. Suranna and G. Gigli, ‘darker-than-black’ PbS quantum dots: Enhancing optical absorption of colloidal semiconductor nanocrystals via short conjugated ligands, J. Am. Chem. Soc., 2015, 137, 1875–1886 CrossRef CAS PubMed.
- D. Bederak, D. M. Balazs, N. V. Sukharevska, A. G. Shulga, M. Abdu-Aguye, D. N. Dirin, M. V. Kovalenko and M. A. Loi, Comparing Halide Ligands in PbS Colloidal Quantum Dots for Field-Effect Transistors and Solar Cells, ACS Appl. Nano Mater., 2018, 1, 6882–6889 CrossRef CAS PubMed.
- J. Z. Fan, N. T. Andersen, M. Biondi, P. Todorović, B. Sun, O. Ouellette, J. Abed, L. K. Sagar, M. J. Choi, S. Hoogland, F. P.-G. de Arquer and E. H. Sargent, Mixed Lead Halide Passivation of Quantum Dots, Adv. Mater., 2019, 31, 1–8 Search PubMed.
- J. Zhang, J. Gao, E. M. Miller, J. M. Luther and M. C. Beard, Diffusion-Controlled Synthesis of PbS and PbSe Quantum Dots with in Situ Halide Passivation for Quantum Dot Solar Cells, ACS Nano, 2014, 8, 614–622 CrossRef CAS PubMed.
- X. Zhang, P. K. Santra, L. Tian, M. B. Johansson, H. Rensmo and E. M.-J. Johansson, Highly Efficient Flexible Quantum Dot Solar Cells with Improved Electron Extraction Using MgZnO Nanocrystals, ACS Nano, 2017, 11, 8478–8487 CrossRef CAS PubMed.
- D. Tiwari and D. J. Fermin, Textured PbI2 photocathodes obtained by gas phase anion replacement, Electrochim. Acta, 2017, 254, 223–229 CrossRef CAS.
- A. Fischer, L. Rollny, J. Pan, G. H. Carey, S. M. Thon, S. Hoogland, O. Voznyy, D. Zhitomirsky, J. Y. Kim, O. M. Bakr and E. H. Sargent, Directly Deposited Quantum Dot Solids Using a Colloidally Stable Nanoparticle Ink, Adv. Mater., 2013, 25, 5742–5749 CrossRef CAS PubMed.
- F. Schaefers, M. Mertin and M. Gorgoi, KMC-1: A high resolution and high flux soft x-ray beamline at BESSY, Rev. Sci. Instrum., 2007, 78, 123102 CrossRef CAS PubMed.
- T. Ida, M. Ando and H. Toraya, Extended pseudo-Voigt function for approximating the Voigt profile, J. Appl. Crystallogr., 2000, 33, 1311–1316 CrossRef CAS.
- D. A. Shirley, High-resolution x-ray photoemission spectrum of the valence bands of gold, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- A. Herrera-Gomez, M. Bravo-Sanchez, F. S. Aguirre-Tostado and M. O. Vazquez-Lepe, The slope-background for the near-peak regimen of photoemission spectra, J. Electron Spectrosc. Relat. Phenom., 2013, 189, 76–80 CrossRef CAS.
- J. H. Scofield, Theoretical Photoionization Cross Sections from 1 to 1500 keV, Livermore, Calif.: Lawrence Livermore Laboratory, University of California, Springfield, Va, 1973, vol. 5 Search PubMed.
- X. Zhang, U. B. Cappel, D. Jia, Q. Zhou, J. Du, T. Sloboda, S. Svanström, F. O.-L. Johansson, A. Lindblad, E. Giangrisostomi, R. Ovsyannikov, J. Liu, H. Rensmo, J. M. Gardner and E. M.-J. Johansson, Probing and Controlling Surface Passivation of PbS Quantum Dot Solid for Improved Performance of Infrared Absorbing Solar Cells, Chem. Mater., 2019, 31, 4081–4091 CrossRef CAS.
- C. H.-M. Chuang, P. R. Brown, V. Bulović and M. G. Bawendi, Improved performance and stability in quantum dot solar cells through band alignment engineering, Nat. Mater., 2014, 13, 796–801 CrossRef CAS PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, The materials project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef.
- P. Scherrer, Kolloidchemie Ein Lehrbuch, Springer Berlin Heidelberg, Berlin, Heidelberg, 1912, pp. 387–409 Search PubMed.
- I. Moreels, K. Lambert, D. Smeets, D. De Muynck, T. Nollet, J. C. Martins, F. Vanhaecke, A. Vantomme, C. Delerue, G. Allan and Z. Hens, Size-dependent optical properties of colloidal PbS quantum dots, ACS Nano, 2009, 3, 3023–3030 CrossRef CAS PubMed.
- NIST X-ray Photoelectron Spectroscopy (XPS) Database, Version 3.5, https://srdata.nist.gov/xps/, accessed 9 August 2021.
- S. Svanström, A. Garcia-Fernandez, T. Sloboda, J. T. Jacobsson, H. Rensmo and U. B. Cappel, X-ray stability and degradation mechanism of lead halide perovskites and lead halides, Phys. Chem. Chem. Phys., 2021, 12479–12489 RSC.
- K. Laajalehto, R. S.-C. Smart, J. Ralston and E. Suoninen, STM and XPS investigation of reaction of galena in air, Appl. Surf. Sci., 1993, 64, 29–39 CrossRef CAS.
- A. Lobo, T. Möller, M. Nagel, H. Borchert, S. G. Hickey and H. Weller, Photoelectron spectroscopic investigations of chemical bonding in organically stabilized PbS nanocrystals, J. Phys. Chem. B, 2005, 109, 17422–17428 CrossRef CAS PubMed.
- J. A. Leiro, K. Laajalehto, I. Kartio and M. H. Heinonen, Surface core-level shift and phonon broadening in PbS(100), Surf. Sci., 1998, 412–413, 3–8 Search PubMed.
- S. Tanuma, C. J. Powell and D. R. Penn, Calculations of Electron Inelastic Mean Free Paths, Surf. Interface Anal., 1993, 21, 165–176 CrossRef.
- E. Bryngelsson, MSc thesis, KTH Royal Institute of Technology, 2019.
- T. Sloboda, S. Svanström, F. O.-L. Johansson, A. Andruszkiewicz, X. Zhang, E. Giangrisostomi, R. Ovsyannikov, A. Föhlisch, S. Svensson, N. Mårtensson, E. M.-J. Johansson, A. Lindblad, H. Rensmo and U. B. Cappel, A method for studying pico to microsecond time-resolved core-level spectroscopy used to investigate electron dynamics in quantum dots, Sci. Rep., 2020, 10, 1–14 CrossRef PubMed.
- S. Svanström, T. J. Jacobsson, T. Sloboda, E. Giangrisostomi, R. Ovsyannikov, H. Rensmo and U. B. Cappel, Effect of halide ratio and Cs + addition on the photochemical stability of lead halide perovskites, J. Mater. Chem. A, 2018, 6, 22134–22144 RSC.
- C. Zha, C. Ji, J. Zhang, L. Shen, X. Zhang, S. Dong and B. Ningzhong, Facet engineering of monodisperse PbS nanocrystals with shape- and facet-dependent photoresponse activity, RSC Adv., 2016, 6, 107151–107157 RSC.
- K. Lonsdale and M. Chemistry, Effects of reduced dimensionality on the electronic structure and defect chemistry of semiconducting hybrid organic – inorganic PbS solids, Proc. R. Soc. A, 2011, 467, 1970–1985 CrossRef.
- B. Wang, H. Xia, Z. Zhang, J. Yang, R. Patterson, S. Huang, S. Shrestha and G. Conibeer, Ab initio calculation of halide ligand passivation on PbSe quantum dot facets, RSC Adv., 2016, 6, 104699–104707 RSC.
- T. Matsukawa and T. Ishii, Valence-Band X-ray Photoemission of PbI2 and CdI2, J. Phys. Soc. Jpn., 1976, 41, 1285–1290 CrossRef CAS.
- M. G. Mason and L. J. Gerenser, X-ray photoemission studies of the lead halide valence bands, Chem. Phys. Lett., 1976, 40, 476–480 CrossRef CAS.
- E. M. Miller, D. M. Kroupa, J. Zhang, P. Schulz, A. R. Marshall, A. Kahn, S. Lany, J. M. Luther, M. C. Beard, C. L. Perkins and J. Van De Lagemaat, Revisiting the Valence and Conduction Band Size Dependence of PbS Quantum Dot Thin Films, ACS Nano, 2016, 10, 3302–3311 CrossRef CAS PubMed.
- R. W. Crisp, D. M. Kroupa, A. R. Marshall, E. M. Miller, J. Zhang, M. C. Beard and J. M. Luther, Metal Halide Solid-State Surface Treatment for High Efficiency PbS and PbSe QD Solar Cells, Sci. Rep., 2015, 5, 1–6 Search PubMed.
- Z. Ning, O. Voznyy, J. Pan, S. Hoogland, V. Adinolfi, J. Xu, M. Li, A. R. Kirmani, J. Sun, J. Minor, K. W. Kemp, H. Dong, L. Rollny, A. Labelle, G. Carey, B. Sutherland, I. Hill, A. Amassian, H. Liu, J. Tang, O. M. Bakr and E. H. Sargent, Air-stable n-type colloidal quantum dot solids, Nat. Mater., 2014, 13, 4–10 CrossRef PubMed.
- J. F. Verwey, Time and intensity dependence of the photolysis of lead halides, J. Phys. Chem. Solids, 1970, 31, 163–168 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional HAXPES data and curve fits of HAXPES data, absorption and photoluminescence spectra, tables of BE positions of the measured core levels and valence band maxima. See DOI: https://doi.org/10.1039/d2cp01050j |
| ‡ Current address: Sorbonne Université, UMR CNRS 7588, Institut des Nanosciences de Paris, F-75005 Paris, France. |
| This journal is © the Owner Societies 2022 |