
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced water–chromophore electron transfer causes formation of guanosine photodamage†
Mikołaj J.
Janicki
 *a,
Rafał
Szabla
*a,
Jiří
Šponer
bc and
Robert W.
Góra
*a
*a,
Rafał
Szabla
*a,
Jiří
Šponer
bc and
Robert W.
Góra
*a
aFaculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370, Wrocław, Poland. E-mail: mikolaj.janicki@pwr.edu.pl; rafal.szabla@pwr.edu.pl; robert.gora@pwr.edu.pl
bInstitute of Biophysics of the Czech Academy of Sciences, Královopolská 135, 61265 Brno, Czech Republic
cNational Centre for Biomolecular Research, Faculty of Science, Masaryk University, Kamenice 5, 625 00, Brno, Czech Republic
First published on 9th March 2022
Abstract
UV-induced photolysis of aqueous guanine nucleosides produces 8-oxo-guanine and Fapy-guanine, which can induce various types of cellular malfunction. The mechanistic rationale underlying photodestructive processes of guanine nucleosides is still largely obscure. Here, we employ accurate quantum chemical calculations and demonstrate that an excited-state non-bonding interaction of guanosine and a water molecule facilitates the electron-driven proton transfer process from water to the chromophore fragment. This subsequently allows for the formation of a crucial intermediate, namely guanosine photohydrate. Further (photo)chemical reactions of this intermediate lead to the known products of guanine photodamage.
Introduction
Ultraviolet-driven chemical reactions in nucleic acid components could entail irreversible damage to their molecular structure, and consequently may result in undesirable mutagenic processes in biological systems.1–7 Accumulation of such alterations in the genome can induce cellular malfunction, tumorigenesis, and even cell death.8,9 Therefore, understanding the photochemical mechanisms directly enabling the formation of DNA/RNA lesions is a fundamental issue for cellular biology and disease prevention. 8-oxo-guanine and 2,6-diamino-4-oxo-5-formamidopyrimidine (Fapy-guanine) are among the most frequent DNA/RNA photolesions, which are produced upon UV excitation of guanine nucleosides in relative yields strongly dependent on the local chemical environment.10–14 However, despite considerable efforts to scrutinize the molecular mechanisms underlying UV-induced damage of pyrimidine nucleobases,15–21 photochemical processes causing generation of guanine nucleoside lesions (such as 8-oxo-guanine or Fapy-guanine) are still obscure. In this paper, we provide a plausible explanation based on the reinterpretation of available experimental data and our state-of-the-art ab initio calculations.In recent decades, the photodynamics of guanine nucleosides was extensively investigated through time-resolved (TR) spectroscopic measurements that were often supplemented by quantum-chemical calculations.22–28 Joint experimental–theoretical studies demonstrated that the photoinduced dynamics of aqueous guanine nucleosides is characterized by sub-picosecond excited-state lifetimes that were assigned to the population of the 1ππ* excited state. Indeed, the proposed ballistic photorelaxation pathways most likely enable the observed ultrafast deactivation of nucleosides, but this does not explain the origin of guanine photolesions. In addition to the recorded short-lived signals, a complementary longer-lived and weakly-emissive (∼2 ps) excited state has been identified and tentatively assigned to the charge-transfer 1πσ* excited state.22,23 However, the accessibility of such a repulsive state in guanine nucleosides seems rather unlikely as there are no distinctive signatures in the TR absorption spectra22,23 originating from solvated electrons (expected in the range of 600–700 nm) that could be produced through the population of the 1πσ* electronic state.29 Importantly, this puzzling excited state efficiently quenches the strongly emissive 1ππ* states and its lifetime varies in different polar solvents such as water and methanol.22,23
Recent advances in mechanistic studies of aqueous photochemistry of pyrimidine and purine nucleobases30–32 have shown that the dark 1nπ* excited states can serve as a doorway to UV-induced water-to-chromophore electron transfer. This process is enabled by an excited-state interaction of the lone electron pairs of a heteroatom in the aromatic ring and the neighbouring water molecule. Consequently, the photoinduced electron transfer and the subsequent proton transfer may lead to the formation of reactive hydroxyl radicals causing damage to DNA/RNA bases.30–32
Our working hypothesis is that the population of the dark 1nOπ* state in a complex of guanine nucleoside and an explicit water molecule (Guo-H2O, see Fig. 1) is attainable through the excited-state intermolecular C6–O⋯OH2 chalcogen bonding interaction, which triggers the long-sought photochemical pathway to the formation of both 8-oxo-guanosine and Fapy-guanosine.
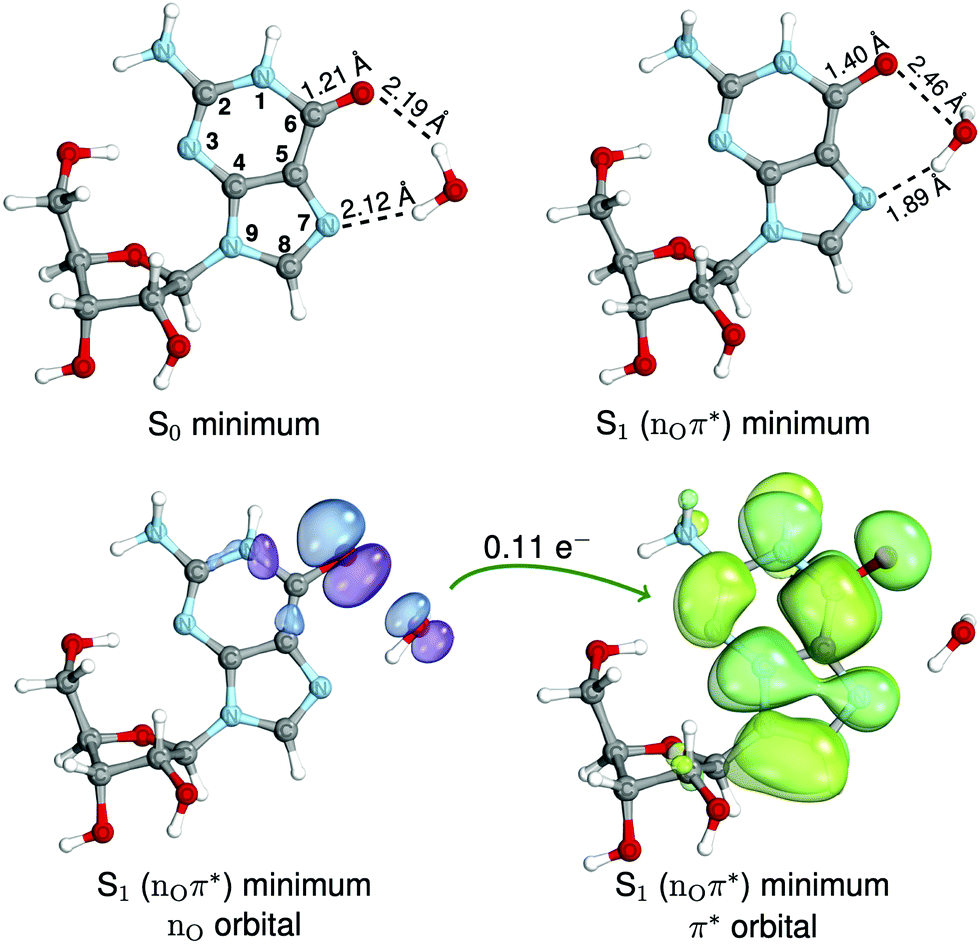 | ||
| Fig. 1 The geometries of the Guo-H2O complex corresponding to the S0 and S1 (nOπ*) minimum-energy structures are shown in the first row. Below, the molecular orbitals involved in the S1 (nOπ*) transition are shown, which demonstrate a charge-transfer character of the process in which 0.11 e− is transferred from the water molecule to the nucleoside. The assumed numbering of atoms and the relevant distances between atoms are indicated. | ||
The results of our quantum-chemical calculations are supported by comparison with previously recorded time-resolved femtosecond transient absorption spectra obtained for aqueous guanine nucleosides.22,23
Computational methods
To find the most stable structures of nucleosides in an aqueous environment, the conformational space was explored using the Conformer-Rotamer Ensemble Sampling Tool33 (CREST) at the semi-empirical GFN2-xTB34 level of theory. The exploration of the conformational space was performed using the iMTD-GC scheme along with the analytical linearized Poisson–Boltzmann (ALPB) implicit solvent model representing the water solvent. The equilibrium ground-state geometries and harmonic vibrational frequencies of the lowest-energy conformer were then obtained using Kohn–Sham density functional theory (KS-DFT) employing the range-separated hybrid ωB97X-D functional35 and the def2-TZVPP36 basis set. The KS-DFT calculations were performed using the Gaussian 1637 package.Vertical excitation energies were obtained using the spin-component scaled variant38 of the second-order algebraic diagrammatic construction39,40 [SCS-ADC(2)] with the cc-pVTZ basis set41 [SCS-ADC(2)/cc-pVTZ], assuming the equilibrium ground-state structures optimized as described above. The orbital character of each electronic transition was assigned based on natural transition orbitals (NTOs). The estimation of solvent-to-chromophore transferred charge was obtained using the one-electron transition density matrix (1TDM) and the Löwdin style analysis. Tg 1TDM analysis was performed employing the TheoDore42 2.4 package. Excited-state minimum-energy structures were located at the SCS-ADC(2)/cc-pVTZ level of theory. The optimization of minimum-energy crossing points (MECPs) between two electronic states was performed employing the sequential penalty constrained function implemented by Levine et al.43 in the CIOpt package. To locate MECPs, the energies and analytical nuclear gradients for the electronic excited and ground states were computed at the SCS-ADC(2) and SCS-MP2 levels, respectively, with the cc-pVTZ basis set.
To verify excited-state minimum energy structures and the S1/S0 state crossing obtained using the single reference method, we optimized these critical excited-state geometries with the extended multi-state complete active space second-order perturbation theory44,45 (XMS-CASPT2), employing the state-averaged complete active space self-consistent field (SA-CASSCF) method with the cc-pVDZ basis set. To locate the S1 (nOπ*) minimum-energy structure at the XMS-CASPT2 level, the complete active space (CAS) was composed of molecular orbitals (MOs) having natural orbital occupations in the range of 0.02–1.98,46 augmented by nO, σO, and  orbitals to facilitate the C6–O⋯OH2 excited-state interaction in the S1 (nOπ*) state and the possible elongation of the carbonyl bond. Consequently, the active space was built out of one σO, three π and one nO occupied MOs along with three π* and one
orbitals to facilitate the C6–O⋯OH2 excited-state interaction in the S1 (nOπ*) state and the possible elongation of the carbonyl bond. Consequently, the active space was built out of one σO, three π and one nO occupied MOs along with three π* and one  virtual MOs (10 electrons in 9 orbitals, shown in Fig. S10, ESI†). The SA-CASSCF wave function was averaged over four lowest-lying electronic states. The optimization of the S1/S0 minimum-energy conical intersection was performed assuming the CAS constructed of the πO, two π, two nO, and four π* MOs (10 electrons in 9 orbitals, shown in Fig. S11, ESI†). In the latter calculations, we included two lone electron pair nO orbitals in the CAS to allow for an appropriate description of the hydroxyl radical. The SA-CASSCF wave function was averaged over three lowest-lying electronic states. The vertical shift, i.e., the empirical correction applied to the zeroth-order Hamiltonian, was equal to 0.4 Hartree, and the XMS-CASPT2 calculations were conducted using the BAGEL 1.2.047 package. To estimate the emission energy of the S1 (nOπ*) structure, SCS-ADC(2) theory was used alongside the conductor-like screening model (COSMO) as an implicit solvent, assuming the equilibrium solvation limit for the S1 excited state, and SCS-ADC(2) calculations were performed using the TURBOMOLE 7.348 package.
virtual MOs (10 electrons in 9 orbitals, shown in Fig. S10, ESI†). The SA-CASSCF wave function was averaged over four lowest-lying electronic states. The optimization of the S1/S0 minimum-energy conical intersection was performed assuming the CAS constructed of the πO, two π, two nO, and four π* MOs (10 electrons in 9 orbitals, shown in Fig. S11, ESI†). In the latter calculations, we included two lone electron pair nO orbitals in the CAS to allow for an appropriate description of the hydroxyl radical. The SA-CASSCF wave function was averaged over three lowest-lying electronic states. The vertical shift, i.e., the empirical correction applied to the zeroth-order Hamiltonian, was equal to 0.4 Hartree, and the XMS-CASPT2 calculations were conducted using the BAGEL 1.2.047 package. To estimate the emission energy of the S1 (nOπ*) structure, SCS-ADC(2) theory was used alongside the conductor-like screening model (COSMO) as an implicit solvent, assuming the equilibrium solvation limit for the S1 excited state, and SCS-ADC(2) calculations were performed using the TURBOMOLE 7.348 package.
Excited-state absorption spectra of S1 minimum-energy structures were simulated based on calculations of 14 excited states, which provided the excitation energies and corresponding oscillator strengths at the SCS-ADC(2)/cc-pVTZ level of theory. Absorption lineshapes, assuming the full width at half-maximum equal to 3000 cm−1, were generated using the GaussSum 3.049 program.
The potential energy (PE) profiles for the singlet electron-driven proton transfer (EDPT) photorelaxation pathway were computed at the SCS-ADC(2)/cc-pVTZ level of theory using the key excited-state structures, namely S2/S1 and S1/S0 MECPs and the S1 minimum-energy structure. The PE profiles between the Franck–Condon region and the S1 minimum were obtained by single-point calculations of intermediate geometries constructed by the image dependent pair potential (IDPP) interpolation50 between the optimized geometries. To show PE profiles between the S1 minimum and S1/S0 MECP, relaxed excited-state geometry optimization was performed constraining the H–O bond length (0.97–1.27 Å) in the N7⋯H–OH moiety at the SCS-ADC(2)/cc-pVTZ level. The IDPP interpolation was also employed to obtain intermediate structures between the optimized constrained S1 geometry, having a H–O bond distance of 1.27 Å, and the S1/S0 MECP. The optimization of ground-state structures of photoproducts was performed using the ωB97X-D functional35 with the def2-TZVPP51 basis set. Relative energies of the photoproducts were determined using the polarizable continuum model (PCM) as an implicit solvent model, assuming their gas-phase optimized geometries. The IDPP interpolations were performed using the Orca 4.2.1.52 package.
Results and discussion
Ground-state structures
Based on the conformational analysis of Guo, performed using the semi-empirical GFN2-xTB method34 (see Fig. S1 in the ESI†), we found that the structure having the syn orientation of the nucleobase and the C2′-endo arrangement of the sugar ring is energetically preferred in an aqueous environment. Our results agree with previous molecular dynamics simulations of guanosine monophosphate that showed dominant population of the syn-like conformer (occurring in 93% of simulations).26 It is worth noting that the syn orientation enables the formation of the OH⋯N3 hydrogen bond between the sugar moiety and the nucleobase (see Fig. 1). This intramolecular hydrogen bond was previously shown to allow for an excited-state forward–backward proton transfer mechanism between the nucleobase and sugar, which could serve as a radiationless deactivation channel of excited purine nucleosides.53–55We added a single explicit water molecule to the selected structure of Guo, to saturate hydrogen bonds with potentially photoreactive sites of the guanine moiety, namely, the C6–O and N7 atoms. Finally, we optimized the ground-state geometry of this Guo-H2O complex (see Fig. 1) using the ωB97X-D/def2-TZVPP method. The resulting equilibrium S0 geometry of the Guo-H2O complex has the water molecule lying in the plane of the aromatic purine ring, to which it is attached by two hydrogen bonds with the carbonyl group (2.19 Å) and the N7 atom (2.12 Å; see Fig. 1). We expect that upon UV excitation, the C6–O⋯H2O hydrogen bond would break and thus facilitate the accessibility of the 1nOπ* dark state.31 It is worth adding that we constructed a similar water complex with deoxyguanosine (dGuo-H2O, see ESI†), and the optimization procedure yielded a virtually identical structure.
Vertical excitation energies
We further computed the vertical excitation energies (shown in Table 1) of the low-lying electronically excited states in the Franck–Condon region of Guo-H2O using the SCS-ADC(2)/cc-pVTZ method. The obtained data show that the absorption of UV photons can result in population of the two locally-excited (LE) 1ππ* states (at 5.13 and 5.76 eV) marked by relatively high oscillator strengths (see Fig. S3 in ESI†). The population of either of the 1ππ* states enables dissipation of the excitation energy through puckering of the aromatic ring and out-of-plane motion of the amino group. These ring-puckering pathways allow for ultrafast non-destructive deactivation to the electronic ground state and are often referred to as ballistic photorelaxation mechanisms.22,26,56,57 However, in the context of this study, we focus on the 1nOπ* excitation at 5.63 eV (Table 1) that is characterized by a transition from the non-bonding lone electron pair molecular orbital located on the carbonyl oxygen atom that is also partially localized on the water molecule (see Fig. S3 in ESI†). The 1nOπ* state in the Franck–Condon region exhibits a moderate charge-transfer (CT) character, associated with the donation of 0.06 e− from the water molecule to the chromophore moiety. We expect that even more pronounced water-to-chromophore electron transfer would occur on the 1nOπ* potential energy (PE) surface due to geometry relaxation. It is worth noting that the photophysical properties of dGuo-H2O (see Table S1 in the ESI†) are entirely consistent with those of the Guo-H2O system.| State/transition | E exc [eV] | f osc | λ [nm] | |
|---|---|---|---|---|
| Guo-H2O | ||||
| S1 |

|
5.13 | 1.71 × 10−1 | 241.7 |
| S2 | nOπ* | 5.63 | 1.37 × 10−4 | 220.2 |
| S3 |

|
5.76 | 3.26 × 10−1 | 215.3 |
| Guo-OH | ||||
| S1 |

|
4.08 | 1.80 × 10−1 | 303.9 |
| S2 | πσ* | 4.23 | 1.95 × 10−2 | 293.1 |
| S3 | πσ* | 4.52 | 1.81 × 10−2 | 274.3 |
Water-to-chromophore electron transfer
To explore the 1nOπ* potential energy surface, we performed excited-state geometry optimization for the Guo-H2O complex. The located S1 (nOπ*) minimum-energy structure (see the upper panel in Fig. 1) shows an intriguing intermolecular chalcogen bonding interaction between the oxygen atoms of the chromophore and the water molecule (C6–O⋯OH2), with an O⋯O distance of 2.46 Å. To the best of our knowledge, such a UV-induced intermolecular interaction between two oxygen atoms has not been reported in the literature so far, although it resembles other excited-state water-to-chromophore contacts reported previously.30,31,58 However, the intramolecular O⋯O chalcogen bonding in the electronic ground state has already been confirmed experimentally by Fellowes et al.59 in oxime. The excited-state chalcogen bonding complex is stabilized through the interaction of the non-bonding orbitals nO located on the carbonyl group and the H2O molecule (see the bottom panel in Fig. 1). The formation of the O⋯O chalcogen contact entails the elongation of the C6–O bond by 0.2 Å and a higher degree of charge transfer (0.11 e− in the S1 minimum) when compared to the Franck–Condon region. In addition, there is a slight pyramidalization of the N1 atom and an out-of-plane distortion of the amino group at the C2 position of the purine ring. We also found the analogous S1 (nOπ*) minimum-energy geometry for dGuo-H2O (see Fig. S4 in ESI†) that closely resembles the charge-transfer mechanism described for Guo-H2O.Interestingly, previous TR infrared experiments revealed that the carbonyl group actively contributes to the energy dissipation process of UV-excited guanine nucleosides and that the C6–O bond vibrations are also coupled to the vibrational modes of the solvent.24,25 Thus, the elongation of the C6–O bond associated with the described chalcogen O⋯O intermolecular interaction (Fig. 1) serves as a reasonable mechanistic rationale for this experimental observation.
The formation of the O⋯O chalcogen contact in the S1 minimum substantially lowers the vertical energy gap between the S1 (nOπ*) and S0 states, which amounts to merely 2.56 eV in the corresponding S1 minimum. Cheng et al.23 demonstrated that in the photodynamics of guanine nucleosides, the puzzling longer-lived excited state exhibits a very weak broadband emission (420–600 nm) with a maximum at 520 nm. The SCS-ADC(2) S1 (nOπ*) minimum can be associated with the weak emission due to its dipole-forbidden character, and an energy gap of 2.56 eV (484 nm) agrees very well with the experimentally determined fluorescence spectrum. We further validated this result by calculations involving the COSMO solvent model of bulk water. The results of COSMO/SCS-ADC(2)/cc-pVTZ calculations indicate that the S1 minimum-energy structure is only slightly destabilized by ∼0.01 eV in bulk water and that the emission wavelength and the oscillator strength of S1 (nOπ*) amount to 482 nm (or 2.57 eV) and 1.92 × 10−4, respectively. Therefore, we postulate that the previously recorded experimental weak emission band could be another fingerprint of the discussed dark 1nOπ* state.
Excited-state absorption spectra
To further support our hypothesis about the population of the 1nOπ* excited state during the photodynamics of guanine nucleosides, we simulated excited-state absorption spectra (ESA) that are overlayed with experimental transient absorption spectra, taken from previous studies,22,23 and are shown in Fig. 2. For this purpose, we used the S1 (nOπ*) minimum-energy structures of the Guo-H2O complex (Fig. 1) and the analogous complex of deoxyguanosine with one water molecule (dGuo-H2O, Fig. S4 in ESI†).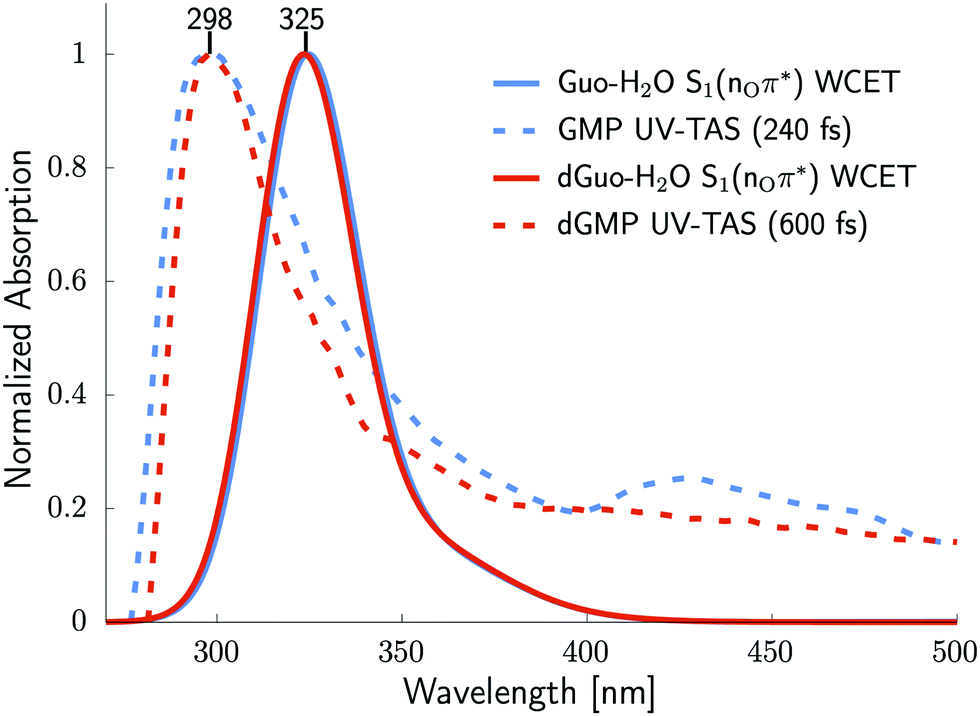 | ||
| Fig. 2 The excited-state absorption (ESA) spectra were simulated using the SCS-ADC(2)/cc-pVTZ method, assuming the S1 (nOπ*) minimum-energy structures for Guo-H2O (solid blue line) and dGuo-H2O (solid red line) obtained at the SCS-ADC(2)/cc-pVTZ level. The digitalized experimental femtosecond transient absorption spectra of aqueous GMP (dashed blue line) and dGMP (dashed red line) recorded at 240 and 600 fs, respectively.22,23 | ||
The experimental and simulated excited-state absorption spectra having maxima at 298 and 325 nm, respectively, exhibit reasonably consistent lineshapes in the range of 290–350 nm, which confirms that the UV-induced O⋯O chalcogen bonding structure may indeed be attainable in the photodynamics of guanine nucleosides.22,23 The discrepancies between the positions and shapes of the simulated and experimental absorption bands are expected and can be ascribed to the following reasons. First, the ESA spectra for the S1 (nOπ*) structures are simulated using model systems containing a single water molecule that do not allow to fully take into account the effects of bulk water on the position of the absorption bands, and thereby the discussed maxima differ by 27 nm. Second, the recorded TA bands possess two different spectral regions, namely below and above 350 nm, which are fingerprints of different photochemical processes. The acquired TA spectrum in the range of 350–500 nm has been previously ascribed to the 1ππ* deactivation channels.22,23 Since our simulated ESA spectra are obtained using only one specific S1 minimum representing a single UV-induced process, it cannot reproduce the entire range of the recorded TA spectrum. Moreover, the 1ππ* state of guanosine that is responsible for the ultrafast ballistic photorelaxation channel would be rather strongly emissive owing to the bright character.22,26,56,57 Accordingly, it seems very unlikely that the puzzling longer-lived excited state having a very weak broadband emission could originate from the ballistic photorelaxation channels.
To further support our interpretation, we simulated the ESA spectrum (see Fig. S7 in the ESI†) for the S1 (nπ*) minimum of Guo in the gas phase located without the explicit water molecule. The absorption maxima (380 and 490 nm) and lineshape for the latter structure strongly differ from the experimental spectra (Fig. 2), and thus it reinforces our hypothesis about the fingerprint of the excited-state chalcogen bonding complex in the recorded transient absorption band.
Furthermore, dynamics of the bleaching band associated with the recovery of the electronic ground state of guanine nucleoside has the same lifetime (∼2.0 ps) as the discussed transient absorption band (290–350 nm), which we assigned to the S1 (nOπ*) structure.22,23 This shows that the 1nOπ* dark state can indeed be responsible for a large part of the photodynamics of guanine nucleosides, thus shedding an entirely new perspective on the aqueous photochemistry of heterocycles having a carbonyl group.
Methanol-to-chromophore electron transfer
In addition to the TR measurements in water, Cheng et al.23 investigated also the photoinduced dynamics of deoxyguanosine in methanol. They observed that the lifetime of the transient absorption band (290–350 nm) and of the weak emission band (at 520 nm) increased to ∼4.2 ps in this medium, when compared to the analogous bands in water.23 This indicates that S1 (nOπ*) minimum is even more stabilized in methanol than in water.To provide a mechanistic rationale for the TR measurements in methanol, we explored the 1nOπ* PE surface for a system containing deoxyguanosine and an explicit CH3OH molecule (dGuo-CH3OH) using the same SCS-ADC(2)/cc-pVTZ level of theory. We located a very similar S1 minimum-energy structure having the C6–O⋯OHCH3 chalcogen bonding interaction between dGuo and the methanol molecule (see Fig. 3 and Fig. S8 in the ESI†). The O⋯O distance at the S1 minimum-energy geometry of dGuo-CH3OH amounts to 2.07 Å (see Fig. S8 in ESI†) and the aromatic purine ring accepts 0.41 e− from the explicit methanol molecule. The excited-state O⋯O chalcogen bond is noticeably shorter by 0.4 Å, and the associated charge transfer from the solvent molecule is roughly twice as large when compared to the S1 minimum of the analogous Guo-H2O complex. Importantly, the excited-state absorption (ESA) spectrum of the S1 (nOπ*) minimum of dGuo-CH3OH (see Fig. S9 in ESI†) closely resembles the simulated ESA presented in Fig. 2 and is consistent with the transient absorption band (in the range of 290–350 nm) recorded in methanol.23 Accordingly, methanol solvent molecules can stabilize the 1nOπ* excited state more strongly than water molecules, and as a result, the associated excited-state lifetime in methanol is doubled. Thus, excited-state chalcogen bonding between deoxyguanosine and methanol is now supported by both experimental23 and theoretical results. It is worth adding that the S1 (nOπ*) minimum of the dGuo-CH3OH complex was also reoptimized using the XMS-CASPT2/SA-CASSCF(10,9)/cc-pVDZ level of theory (see Fig. S8 in ESI†). The latter calculations confirmed the reliability of the excited-state interaction between the methanol molecule and deoxyguanosine.
 | ||
| Fig. 3 The S1 (nOπ*) minimum-energy structure of dGuo-CH3OH found at the SCS-ADC(2)/cc-pVTZ level of theory alongside the delocalized nO and π* molecular orbitals demonstrating a charge-transfer process of 0.41 e− from CH3OH to the nucleobase part. | ||
Electron-driven proton transfer deactivation pathway
To elucidate the role of S1 (nOπ*) minimum (Fig. 1) in the photochemistry of guanine nucleosides, we located the relevant potential energy (PE) surface crossings and key structures at the SCS-ADC(2)/cc-pVTZ level of theory, and we present the associated potential energy profiles in Fig. 4.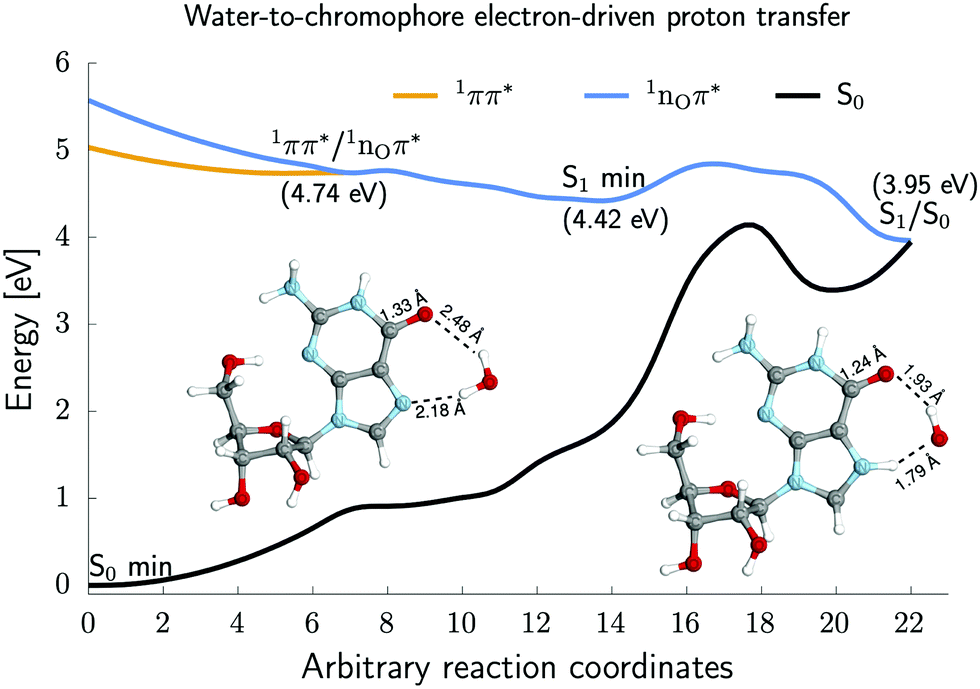 | ||
| Fig. 4 The potential energy profiles (PE) demonstrate a photochemical channel, involving the S2/S1 minimum-energy crossing point (MECP), S1 minimum, and S1/S0 MECP, that enables a water-to-chromophore electron-driven proton transfer (EDPT) mechanism in Guo-H2O. The PE profiles were obtained at the SCS-ADC(2)/cc-pVTZ level of theory. | ||
Upon UV excitation of the 1ππ* excited state (orange line), the system can readily reach the 1ππ*/1nOπ* MECP at 4.74 eV (see the left inset in Fig. 4) that is marked by the elongation of the C6–O carbonyl bond (1.33 Å) as well as weakening of the C6–O⋯H2O hydrogen bond (2.48 Å). Importantly, the located S2/S1 MECP shows that a slight elongation of the carbonyl bond by ∼0.1 Å significantly stabilizes the energy of the 1nOπ* excited state with respect to the Franck–Condon region. The accessibility of S2/S1 MECP enables the population of the 1nOπ* excited state (blue line) and, in turn, the Guo-H2O complex may reach the shallow S1 minimum at 4.42 eV (shown in Fig. 1) in a barrierless manner.
The associated charge transfer found for the S1 minimum-energy structure could trigger the electron-driven proton transfer (EDPT) from the water molecule to the N7 atom of guanosine. The EDPT mechanism having an energy barrier of 0.41 eV could then lead to S1/S0 MECP at 3.95 eV (see the right inset in Fig. 4), which allows for radiationless deactivation of guanosine. Notably, an energy barrier of 0.41 eV could be overcome due to excess excitation energy. Thus, the discussed 1nOπ*-mediated photorelaxation channel allows for a water-splitting process leading to the formation of hydrogenated guanosine radical and a hydroxyl (˙OH) radical.
Benchmarking of relevant excited-state structures
Single reference ab initio methods such as SCS-ADC(2) are widely used to investigate photochemical processes, which occur in organic chromophores. However, Marsili et al.60 have demonstrated that the ADC(2)-s method can lead to finding nonphysical S1/S0 surface crossings for carbonyl-containing compounds. To address a potential issue with applying the SCS-ADC(2) method to the excited-state optimization of Guo-H2O, we reoptimized the SCS-ADC(2) S1(nOπ*) minimum and the SCS-ADC(2) S1 (nOπ*)/S0 minimum-energy crossing point using the XMS-CASPT2/SA-CASSCF(10,9)/cc-pVDZ level of theory. The molecular orbitals included in the complete active space for both excited-state structures are shown in the ESI† (Fig. S10 and S11).The superimposed excited-state minimum-energy structures obtained at the SCS-ADC(2) (black) and XMS-CASPT2 (cyan) levels are depicted in Fig. 5. The root-mean-square deviations (RMSDs) evaluated for those structures amount to 0.082 and 0.135 Å for the S1 minimum and S1/S0 surface crossing, respectively. This indicates that the geometries are virtually identical. Thus, the XMS-CASPT2 calculations confirmed that the excited-state structures provided by SCS-ADC(2) method are reliable.
 | ||
| Fig. 5 The superimposed excited-state minimum-energy structures optimized at the SCS-ADC(2) S1 minimum, SCS-ADC(2) S1/S0 surface crossing (black) and XMS-CASPT2/SA-CASSCF(10,9)/cc-pVDZ level (cyan) are presented with the root-mean-square deviation (RMSD) calculated for the corresponding structures. | ||
Regarding the relevant structural parameters, the most significant difference between the S1 minimum-energy geometries is found for the O⋯O intermolecular distances that amount to 2.43 and 2.46 Å at the XMS-CASPT2 and SCS-ADC(2) levels, respectively. Furthermore, the C6–O bond length is shortened to 1.35 Å (by 0.05 Å) at the XMS-CASPT2 level in comparison with the SCS-ADC(2) geometry. Comparing XMS-CASPT2 and SCS-ADC(2) S1/S0 MECP, the noticeable structural difference is a slightly different position of the hydroxyl radical. Since the multireference method (XMS-CASPT2) predicted the excited-state O⋯O intermolecular interaction and confirmed the existence of the electron-driven proton transfer S1/S0 surface crossing, we conclude that the SCS-ADC(2) method is suitable for the description of the electronic structure of our model system.
Photolesions of guanine nucleosides
As described above, the EDPT photorelaxation mechanism generates two reactive radical species close to one another, that is, the ˙OH and GuoH˙ radicals, with the unpaired electron located on the C8 atom in the latter case. These radicals may readily recombine in the hot electronic ground state and produce a guanosine-OH (Guo-OH) photohydrate intermediate, the minimum-energy structure of which lies 1.96 eV (45.2 kcal mol−1) below the EDPT S1/S0 minimum-energy crossing point (Fig. 6 and Fig. S12 in the ESI†). The vertical excitation energies calculated for the Guo-OH photohydrate (see Table 1) show that there is a low-lying 1ππ* locally excited state at 4.08 eV having a substantial oscillator strength, which indicates that the photohydrate can easily absorb longer UV wavelengths (UV-B region) than the parent Guo molecule. In other words, the formation of Guo photohydrate redshifts the UV absorption spectrum of the chromophore and increases the photochemical activity of the molecule since it can be excited with much lower energies. More importantly, Guo-OH is also characterized by the low-lying 1πσ* excited state (at 4.23 eV, Table 1) that could enable the photodetachment of an electron from the intermediate, thus forming a radical cation that was previously proposed as the critical intermediate necessary for the formation of 8-oxo-guanosine.13 Therefore, we propose that Guo-OH photohydrate could be the structural precursor of 8-oxo-guanosine, which is an important photolesion that can be formed in aqueous solution of guanine nucleosides.13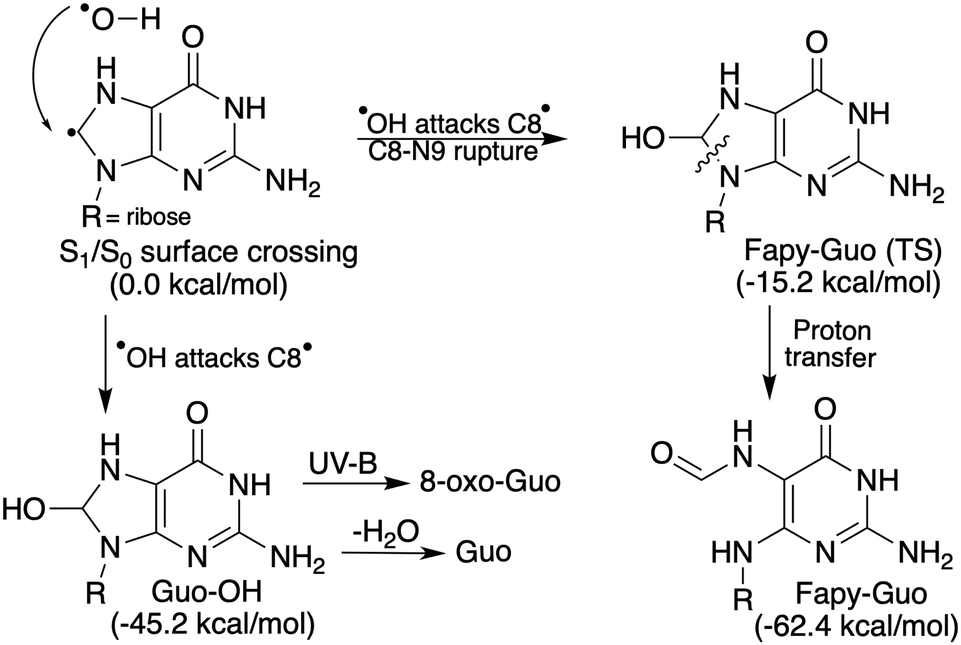 | ||
| Fig. 6 Possible photoproducts generated from the biradical intermediate produced in the EDPT process, along with the relative energies of their ground-state structures with respect to EDPT S1/S0 state crossing. | ||
On the other hand, the main observed photoproduct upon UV excitation of guanosine in water is Fapy-guanosine (Fapy-Guo).13 Given the above results, this may be explained by the C8–N9 bond rupture (see Fig. 6 and Fig. S12 in ESI†) of the Guo-OH photohydrate and subsequent proton transfer to the N9 atom, which allows for the formation of the more stable Fapy-Guo. Furthermore, the transition-state structure leading to the formation of Fapy-Guo is located 0.66 eV (15.2 kcal mol−1, see Fig. S12 in ESI†) lower than the located S1/S0 minimum-energy crossing point. Consequently, photochemically formed and still vibrationally hot Guo-OH can be readily transformed to Fapy-Guo after passing through this modest energy barrier. It is worth noting that the Fapy-Guo photoproduct is lower in energy than the EDPT S1/S0 surface crossing by 2.71 eV (62.4 kcal mol−1). Interestingly, both Fapy-Guo and Guo-H2O systems are nearly isoenergetic. We expect that Fapy-Guo should be a stable photoproduct that would not undergo a reverse ground-state conversion to Guo, particularly since such a reaction would involve a considerable energy barrier, exceeding 47.2 kcal mol−1 (cf. Fig. S12 in ESI†). The same photoproduct was also detected in the presence of electron scavengers (H3O+ and N2O), suggesting that solvated electrons mediated by the 1πσ* state do not participate in the formation of Fapy-Guo.13 This is consistent with our proposed mechanism for forming guanosine photolesions, which result from a direct water-to-chromophore electron transfer rather than the attachment of free solvated electrons to the chromophore.
Conclusions
In conclusion, we have demonstrated the long-sought plausible photochemical pathway leading to the formation of photolesions of aqueous guanine nucleosides, namely 8-oxo-guanosine and Fapy-guanosine. It involves the formation of an excited-state chalcogen bonded complex with a water molecule that enables the population and stabilization of the dark 1nπ* state. This excited-state complex shows a substantial charge-transfer character, which can entail an electron-driven proton transfer process from a water molecule to the guanine ring leading to the formation of a reactive hydroxyl radical that may damage the canonical structure of guanine. The results of our quantum chemical calculations are strongly supported by reinterpretation of the time-resolved spectra recorded previously for aqueous guanine nucleosides, indicating that the intermolecular chalcogen bonding complex can be easily reached upon UV excitation. The proposed mechanism is the first that explains both the formation of Fapy-Guo as the main photoproduct in an aqueous solution and 8-oxo-guanine as a concomitant minor photoproduct. Furthermore, our results point out that the reactive dark 1nπ* state, which seems to be hardly accessible owing to high excitation energy, can be substantially stabilized beyond the Franck–Condon region. This is in consequence of the formation of a specific and short-lived excited-state interaction that can lower the energy of the nπ* state.Our results also indicate that the SCS-ADC(2) method can be used as a more accurate alternative to the ADC(2)-s approach for studying the photochemical properties of carbonyl-containing compounds. However, we also strongly encourage benchmarking results of this single-reference method using multireference wavefunction methods, such as the XMS-CASPT2 level of theory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. J. J. acknowledges the support of the ‘Diamond Grant’ (0144/DIA/2017/46) from the Polish Ministry of Science and Higher Education, a doctoral scholarship from the National Science Center Poland (2020/36/T/ST4/00564), and the START Fellowship from the Foundation for Polish Science. J. S. acknowledges support by the Czech Science Foundation (21-23718S) and the project SYMBIT reg. number: CZ.02.1.01/0.0/0.0/15_003/0000477 financed by the ERDF.References
- M. L. Kripke, P. A. Cox, L. G. Alas and D. B. Yarosh, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7516–7520 CrossRef CAS PubMed.
- J. S. Taylor, Acc. Chem. Res., 1994, 27, 76–82 CrossRef CAS.
- V. Pagès and R. P. Fuchs, Oncogene, 2002, 21, 8957–8966 CrossRef PubMed.
- R. P. Sinha and D.-P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225–236 CrossRef CAS PubMed.
- J. Cadet, E. Sage and T. Douki, Mutat. Res., 2005, 571, 3–17 CrossRef CAS PubMed.
- J. Cadet and J. R. Wagner, Cold Spring Harbor Perspect. Biol., 2013, 5, a012559 Search PubMed.
- J. Cadet and T. Douki, Photochem. Photobiol. Sci., 2018, 17, 1816–1841 CrossRef CAS PubMed.
- S. P. Jackson, Carcinogenesis, 2002, 23, 687–696 CrossRef CAS PubMed.
- R. Madabhushi, L. Pan and L.-H. Tsai, Neuron, 2014, 83, 266–282 CrossRef CAS PubMed.
- C. E. Crespo-Hernández, S. Flores, C. Torres, I. Negrón-Encarnación and R. Arce, Photochem. Photobiol., 2000, 71, 534–543 CrossRef.
- C. E. Crespo-Hernández and R. Arce, Photochem. Photobiol., 2000, 71, 544–550 CrossRef.
- G. A. Papadantonakis, R. Tranter, K. Brezinsky, Y. Yang, R. B. van Breemen and P. R. LeBreton, J. Phys. Chem. B, 2002, 106, 7704–7712 CrossRef CAS.
- C. E. Crespo-Hernández and R. Arce, J. Photochem. Photobiol., B, 2004, 73, 167–175 CrossRef PubMed.
- M. Gomez-Mendoza, A. Banyasz, T. Douki, D. Markovitsi and J.-L. Ravanat, J. Phys. Chem. Lett., 2016, 7, 3945–3948 CrossRef CAS PubMed.
- D. B. Hall, R. E. Holmlin and J. K. Barton, Nature, 1996, 382, 731–735 CrossRef CAS PubMed.
- W. J. Schreier, T. E. Schrader, F. O. Koller, P. Gilch, C. E. Crespo-Hernandez, V. N. Swaminathan, T. Carell, W. Zinth and B. Kohler, Science, 2007, 315, 625–629 CrossRef CAS PubMed.
- K. V. Nguyen and C. J. Burrows, J. Am. Chem. Soc., 2011, 133, 14586–14589 CrossRef CAS PubMed.
- R. Szabla, J. Campos, J. E. Šponer, J. Šponer, R. W. Góra and J. D. Sutherland, Chem. Sci., 2015, 6, 2035–2043 RSC.
- D. B. Bucher, C. L. Kufner, A. Schlueter, T. Carell and W. Zinth, J. Am. Chem. Soc., 2016, 138, 186–190 CrossRef CAS PubMed.
- R. Szabla, H. Kruse, P. Stadlbauer, J. Šponer and A. L. Sobolewski, Chem. Sci., 2018, 9, 3131–3140 RSC.
- R. Szabla, M. Zdrowowicz, P. Spisz, N. J. Green, P. Stadlbauer, H. Kruse, J. Šponer and J. Rak, Nat. Commun., 2021, 12, 3018 CrossRef PubMed.
- V. Karunakaran, K. Kleinermanns, R. Improta and S. A. Kovalenko, J. Am. Chem. Soc., 2009, 131, 5839–5850 CrossRef CAS PubMed.
- C. C.-W. Cheng, C. Ma, C. T.-L. Chan, K. Y.-F. Ho and W.-M. Kwok, Photochem. Photobiol. Sci., 2013, 12, 1351–1365 CrossRef CAS PubMed.
- D. A. McGovern, G. W. Doorley, A. M. Whelan, A. W. Parker, M. Towrie, J. M. Kelly and S. J. Quinn, Photochem. Photobiol. Sci., 2009, 8, 542–548 CrossRef CAS PubMed.
- J. B. Nielsen, J. Thøgersen, S. K. Jensen, S. B. Nielsen and S. R. Keiding, Phys. Chem. Chem. Phys., 2011, 13, 13821–13826 RSC.
- S. F. Altavilla, J. Segarra-Martí, A. Nenov, I. Conti, I. Rivalta and M. Garavelli, Front. Chem., 2015, 3, 29 Search PubMed.
- S. E. Krul, S. J. Hoehn, K. J. Feierabend and C. E. Crespo-Hernández, J. Chem. Phys., 2021, 154, 075103 CrossRef CAS PubMed.
- J. Ortín-Fernández, J. González-Vázquez, L. Martínez-Fernández and I. Corral, Molecules, 2022, 27, 989 CrossRef PubMed.
- T. A. A. Oliver, Y. Zhang, A. Roy, M. N. R. Ashfold and S. E. Bradforth, J. Phys. Chem. Lett., 2015, 6, 4159–4164 CrossRef CAS PubMed.
- M. Barbatti, J. Am. Chem. Soc., 2014, 136, 10246–10249 CrossRef CAS PubMed.
- R. Szabla, H. Kruse, J. Šponer and R. W. Góra, Phys. Chem. Chem. Phys., 2017, 19, 17531–17537 RSC.
- X. Wu, J. Ehrmaier, A. L. Sobolewski, T. N. V. Karsili and W. Domcke, Phys. Chem. Chem. Phys., 2019, 21, 14238–14249 RSC.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian-16 Revision B.03, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- A. Hellweg, S. A. Grün and C. Hättig, Phys. Chem. Chem. Phys., 2008, 10, 4119 RSC.
- C. Hättig, Adv. Quantum Chem., 2005, 50, 37–60 CrossRef.
- A. Dreuw and M. Wormit, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 82–95 CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- B. G. Levine, J. D. Coe and T. J. Martínez, J. Phys. Chem. B, 2008, 112, 405–413 CrossRef CAS PubMed.
- K. Andersson, P. A. Malmqvist, B. O. Roos, A. J. Sadlej and K. Wolinski, J. Phys. Chem., 1990, 94, 5483–5488 CrossRef CAS.
- T. Shiozaki, W. Györffy, P. Celani and H.-J. Werner, J. Chem. Phys., 2011, 135, 081106 CrossRef PubMed.
- V. Veryazov, P. Å. Malmqvist and B. O. Roos, Int. J. Quantum Chem., 2011, 111, 3329–3338 CrossRef CAS.
- T. Shiozaki, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1331 Search PubMed.
- TURBOMOLE V7.3 2018, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- N. M. O’boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- S. Smidstrup, A. Pedersen, K. Stokbro and H. Jónsson, J. Chem. Phys., 2014, 140, 214106 CrossRef PubMed.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 Search PubMed.
- D. Tuna, A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2014, 118, 122–127 CrossRef CAS PubMed.
- D. Tuna and W. Domcke, Phys. Chem. Chem. Phys., 2016, 18, 947–955 RSC.
- L. Zhao, G. Zhou, B. Jia, G. Teng, K. Zhan, H. Zheng, J. Luo and B. Liu, J. Photochem. Photobiol., A, 2020, 401, 112753 CrossRef CAS.
- L. Serrano-Andrés, M. Merchán and A. C. Borin, J. Am. Chem. Soc., 2008, 130, 2473–2484 CrossRef PubMed.
- B. Heggen, Z. Lan and W. Thiel, Phys. Chem. Chem. Phys., 2012, 14, 8137–8146 RSC.
- M. J. Janicki, R. Szabla, J. Šponer and R. W. Góra, Chem. Phys., 2018, 515, 502–508 CrossRef CAS.
- T. Fellowes, B. L. Harris and J. M. White, Chem. Commun., 2020, 56, 3313–3316 RSC.
- E. Marsili, A. Prlj and B. F. E. Curchod, Phys. Chem. Chem. Phys., 2021, 23, 12945–12949 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Computational procedures, conformational analysis, and characteristics of excited-state crucial points. See DOI: 10.1039/d2cp00801g |
| This journal is © the Owner Societies 2022 |